

Change in cell-free DNA methylomes and fragmentomes after just two cycles of pembrolizumab were associated with progression-free and overall survival in patients with various metastatic cancers, without requiring matched tumor tissue.
Abstract
Early kinetics of circulating tumor DNA (ctDNA) in plasma predict response to pembrolizumab but typically requires sequencing of matched tumor tissue or fixed gene panels. We analyzed genome-wide methylation and fragment-length profiles using cell-free methylated DNA immunoprecipitation and sequencing (cfMeDIP-seq) in 204 plasma samples from 87 patients before and during treatment with pembrolizumab from a pan-cancer phase II investigator-initiated trial (INSPIRE). We trained a pan-cancer methylation signature using independent methylation array data from The Cancer Genome Atlas to quantify cancer-specific methylation (CSM) and fragment-length score (FLS) for each sample. CSM and FLS are strongly correlated with tumor-informed ctDNA levels. Early kinetics of CSM predict overall survival and progression-free survival, independently of tumor type, PD-L1, and tumor mutation burden. Early kinetics of FLS are associated with overall survival independently of CSM. Our tumor-naïve mutation-agnostic ctDNA approach integrating methylomics and fragmentomics could predict outcomes in patients treated with pembrolizumab.
Significance:
Analysis of methylation and fragment length in plasma using cfMeDIP-seq provides a tumor-naive approach to measure ctDNA with results comparable with a tumor-informed bespoke ctDNA. Early kinetics within the first weeks of treatment in methylation and fragment quantity can predict outcomes with pembrolizumab in patients with various advanced solid tumors.
This article is featured in Selected Articles from This Issue, p. 897
INTRODUCTION
Immune-checkpoint blockade (ICB) including programmed death 1 (PD-1) inhibition has emerged as a standard therapeutic strategy for many tumor types such as lung, melanoma, and head and neck cancers, among others (1–3). However, even in melanoma, 40% to 70% of patients demonstrate primary resistance (4). To better identify patients likely to benefit from ICB, various predictive genomic biomarkers have been identified, including microsatellite instability (MSI; ref. 5), tumor mutation burden (TMB; ref. 6), expression of specific genes such as programmed death ligand 1 (PD-L1), and multigene-expression signatures (7, 8). A few have been validated across cancer types such as MSI or high TMB and are currently tumor-agnostic biomarkers for pembrolizumab. In addition to baseline molecular features, another promising strategy is monitoring the early response to treatment or the emergence of resistance (9). Noninvasive biomarkers such as circulating tumor DNA (ctDNA) could enable on-treatment response assessment and early treatment adaptation (10). We have previously demonstrated in the INSPIRE study (an investigator-initiated phase II pan-cancer study in patients treated with pembrolizumab every 3 weeks) that a decrease in ctDNA by cycle 3 was predictive of response to ICB and longer survival (9, 11). Moreover, patients with ctDNA clearance, defined as undetectability during treatment, demonstrated the longest survival, confirming ctDNA as a promising surrogate marker of treatment efficacy in patients treated with ICB. This previous report utilized a tumor-informed approach, wherein ctDNA was monitored using a bespoke panel that tracked 16 specific variants for each individual patient identified from whole-exome sequencing (WES) of tumor tissue. However, this assay may not be feasible in all patients as tumor tissue may not always be accessible or suitable for genomic analysis.
Cell-free methylated DNA immunoprecipitation and sequencing (cfMeDIP-seq) involves selective antibody enrichment of cell-free DNA (cfDNA) that contains 5-methylcytosine and avoids degradative bisulfite conversion of DNA (12). Methylation is a stable, cell-type–specific marker that has been used to quantify tissue-specific cfDNA (13). Cancers are known to harbor characteristic hypermethylation, especially in CpG islands, making enrichment-based methylation profiling a promising strategy for estimating ctDNA levels. Studies in various tumor types have demonstrated the ability of cfMeDIP-seq to facilitate tumor-naïve estimation of ctDNA levels (14–16), which could overcome the abovementioned limitation of tissue availability. cfMeDIP-seq also enables the quantification of cfDNA fragment lengths. DNA fragments released by tumors into circulation are known to have a characteristic fragment-length distribution with a higher proportion of short fragments (<150 bp) than cfDNA derived from normal tissue (17, 18). This enables the calculation of a tumor-specific score based on fragment lengths, providing another promising biomarker of treatment response orthogonal to methylation (19, 20). Previous attempts have been made to boost cancer early detection using multimodal analysis of ctDNA including bisulfite conversion with targeted deep sequencing (21), cfDNA fragmentation patterns and parallel gene panel (18), and whole-genome sequencing and cfMeDIP-seq (22). Here, we attempt an integrative methylation and fragmentation analysis from a single data type, cfMeDIP-seq, to monitor the response to ICB.
In this study, we performed a joint tumor-naïve analysis of cfDNA methylation and fragmentation. We conducted this analysis in blood plasma specimens from the INSPIRE study and correlated ctDNA abundance as estimated by cfMeDIP-seq against the previously used tumor-informed bespoke mutation-based method. We demonstrate that early changes in tumor-naïve ctDNA methylation and fragmentation can predict clinical benefit and survival in patients treated with pembrolizumab.
RESULTS
Characteristics of the Cohort
From a total of 106 patients treated with pembrolizumab at 200 mg every 3 weeks in INSPIRE (NCT02644369), cfMeDIP-seq was performed using 204 blood samples taken at various time points from 87 patients (Supplementary Fig. S1A and S1B). This included 85 samples at baseline and 56 at or just prior to cycle 3, and 55 patients were analyzed at both time points. Other time points were selected based on temporal proximity to radiologic response and immune-related adverse events as well as sample availability. A CONSORT diagram is provided in Fig. 1A. Characteristics of the patients included in the analysis as well as response rate are summarized in Table 1. The largest cohort was cohort B (triple-negative breast cancer, TNBC, n = 22), followed by cohort C (high-grade serous ovarian cancer, HGSOC, n = 21), cohort A (head and neck squamous cell carcinoma, HNSCC, n = 19), and cohort D (melanoma, n = 12). For cohort E (mixed solid tumors, MST, n = 13), tumor types with more than one patient enrolled and potential benefit on immunotherapy were selected (Merkel cell carcinoma, n = 6; microsatellite instability-high tumor, n = 4; and other head and neck cancer including nasopharyngeal cancer, n = 3). Because the event rate exceeded 50%, median overall survival (OS) and median follow-up time were equivalent at 11.5 (range, 0.600–64.4) months. Median follow-up among surviving patients was 59.8 (range, 46.7–64.4) months. Median progression-free survival (PFS) was 1.90 (range, 0.300–63.5) months. Median OS and PFS for each cohort are summarized in Table 1 and Supplementary Fig. S2A and S2B.
Figure 1.

Overview of the study and the analysis of cell-free methylomes and fragmentomes. A, CONSORT diagram for cfMeDIP-seq analysis within the INSPIRE study. B, We performed cfMeDIP-seq in a cohort of patients with various solid tumors treated with pembrolizumab. We computed CSM scores and fragment-length scores through joint analysis of the methylome and fragmentome. C, CSM and (D) FLS were significantly higher in samples from patients with cancer than those from normal controls, by Wilcoxon rank-sum tests. Boxplots show median and quartiles.
Table 1.
Overview of the cohort included for cfMeDIP-seq analysis.
| Characteristic | A: Head and neck, n = 19a | B: Breast, n = 22a | C: Ovarian, n = 21a | D: Melanoma, n = 12a | E: Other, n = 13a |
|---|---|---|---|---|---|
| Gender | |||||
| Female | 3 (16%) | 22 (100%) | 21 (100%) | 7 (58%) | 5 (38%) |
| Male | 16 (84%) | 0 (0%) | 0 (0%) | 5 (42%) | 8 (62%) |
| Age at diagnosis | 59 (56, 63) | 52 (43, 61) | 55 (49, 64) | 58 (55, 67) | 60 (48, 71) |
| Prior surgery | 9 (47%) | 16 (73%) | 13 (62%) | 9 (75%) | 8 (62%) |
| Prior radiotherapy | 16 (84%) | 18 (82%) | 6 (29%) | 3 (25%) | 9 (69%) |
| Prior lines of systemic treatment | |||||
| 0 | 1 (5.3%) | 0 (0%) | 0 (0%) | 7 (58%) | 6 (46%) |
| 1 | 10 (53%) | 4 (18%) | 1 (4.8%) | 4 (33%) | 3 (23%) |
| 2 | 4 (21%) | 10 (45%) | 5 (24%) | 1 (8.3%) | 3 (23%) |
| 3 or more | 4 (21%) | 8 (36%) | 15 (71%) | 0 (0%) | 1 (7.7%) |
| Prior immunotherapy | 0 (0%) | 0 (0%) | 2 (9.5%) | 3 (25%) | 0 (0%) |
| Age at pembrolizumab | 62 (58, 67) | 56 (46, 67) | 61 (52, 68) | 65 (57, 71) | 62 (49, 72) |
| Responder (CR or PR) | 4 (21%) | 1 (4.5%) | 0 (0%) | 8 (67%) | 4 (31%) |
| Progression | 12 (63%) | 19 (86%) | 20 (95%) | 4 (33%) | 9 (69%) |
| Death | 17 (89%) | 21 (95%) | 19 (90%) | 4 (33%) | 11 (85%) |
| Median OS (months) | 8.7 | 8.8 | 16.2 | Not reached | 17.8 |
| Median PFS (months) | 3.2 | 1.7 | 1.8 | Not reached | 3.9 |
a n (%); median (IQR).
Tumor-Naive Plasma Methylomes and Fragmentomes Accurately Estimate ctDNA Levels
To quantify ctDNA abundance without guidance from matched tumor tissue, we undertook an integrative analysis of methylation and fragment-length profiles by quantifying cancer-specific methylation (CSM) and fragment-length score (FLS) for each sample (Fig. 1B). To do this, we first independently generated a set of regions hypermethylated in cancer using 450K methylation array data from The Cancer Genome Atlas (TCGA) Pan-cancer Atlas (PanCanAtlas). We extended a previously described approach (16) to identify differentially methylated CpGs (DMC) using 1,350 tumors from 27 cancer types, then excluded any DMCs methylated in cfMeDIP-seq of 100 normal control samples donated for two cancer early detection studies (ref. 22, medRxiv: 2023.01.30.23285027). Next, we removed sites methylated in peripheral blood leukocytes (PBL) based on 30 previous samples profiled by conventional MeDIP-seq (16) and an atlas of cell-type–specific methylation profiled by methylation arrays (13). This yielded a final immune-depleted signature of 200 DMCs. Consistent with the frequent observation that hypermethylation in cancer often occurs in CpG-dense regions, 184 (92%) of the DMCs were in CpG islands, and 13 (6.5%) were in shores. The CSM score was computed by summing methylation probabilities across these signature regions for each sample (e.g., see Supplementary Fig. S3). We also computed this signature in independent publicly available data sets from whole-genome bisulfite sequencing of human breast cancer, esophageal cancer, and adjacent normal esophagus, which confirmed that the signature was indeed hypermethylated in cancer relative to normal tissue (Supplementary Fig. S4A–S4C).
To calculate FLS, we first computed the sample-specific fragment-length histogram of all read pairs mapping at least one of the aforementioned CSM signature regions. We then determined each sample's relative similarity to cancer versus normal fragment-length profiles by computing the mean log cancer-to-normal fragment-length frequency ratio using a previously published method and reference data (19). This results in FLS > 0 if the fragment-length profile more closely resembles ctDNA. We also checked for characteristic cancer-associated fragmentomic features. The ratio of short to long fragments across the genome in 5 Mb windows was more variable in cancers relative to normal controls, with a 30.3% (range, −41.1 to 227.9) higher median standard deviation across windows (Supplementary Fig. S5), consistent with previous findings (18). We identified variably fragmented regions in cancer versus normal by performing two-sided Ansari–Bradley dispersion tests and merging neighboring regions using Comb-p (23). This revealed 10 regions with significantly more fragment-length variability across cfDNA samples from patients with cancer than in normal controls, spanning 0.910 Gb, or 31.7% of the autosomal genome (Supplementary Fig. S5 SA–S5C). Unsupervised nonnegative matrix factorization (NMF) analysis revealed signatures of short fragment length (24) and a higher fraction of fragment ends residing within the nucleosome core (25) in the cfDNA of patients with cancer (Supplementary Fig. S6A–S6C). Together, these findings confirm that cfMeDIP-seq fragment-length profiles demonstrate cancer-distinguishing characteristics similar to those observed previously in plasma whole-genome sequencing studies (18, 24, 25).
We compared CSM and FLS between normal controls and patients with cancer at baseline, cycle 3, and later cycles (Fig. 1C and D). Compared with normal controls (median CSM = 0.0418), CSM was higher in patients with cancer at baseline (median 1.47, Wilcoxon rank-sum P < 0.001), cycle 3 (median 0.151, P < 0.001), and later cycles (median 0.0745, P = 0.001). For FLS, compared with normal controls (median FLS = −0.213), FLS was higher in patients with cancer at baseline (median = 0.151, P < 0.001), cycle 3 (median = −0.0803, P < 0.001), and later cycles (median = −0.110, P = 0.005).
Higher CSM and FLS were each independently associated with cancer in a logistic regression model that included 85 patients with cancer at baseline and 100 normal controls (Fig. 2A). We then computed a unified cfMeDIP-seq score by calculating the log odds of cancer based on both CSM and FLS (Fig. 2B) and regressed this score against ctDNA estimates from a tumor-informed bespoke assay (Signatera by Natera), which we refer to here as cancer mutation concentration (CMC) and were previously reported (11). Briefly, a custom panel based on matched tumor exomes was designed specifically to detect and quantify 16 mutations in plasma samples for each patient, and the resulting CMCs are reported as the mean number of tumor molecules observed per mL of plasma (MTM/mL). A total of 75 patients had data for both CMC and cfMeDIP-seq at baseline and 53 at cycle 3.
Figure 2.

Joint analysis of cell-free methylomes and fragmentomes enables accurate estimation of ctDNA abundance. A, We fit a logistic regression model with cancer vs. noncancer as the response variable using cfMeDIP-seq data from 85 blood samples from patients with baseline advanced cancer and 100 normal controls. CSM and FLS were each independently associated with cancer. B, The predictions of the logistic regression model can be interpreted as a joint cfMeDIP-seq score corresponding to the log odds of a sample arising from a patient with cancer. C, Using this score, we found that a threshold of −1.754 best identified cases which had undetectable ctDNA based on targeted deep sequencing of cancer-specific mutations. D, cfMeDIP-seq scores correlated with tumor-informed CMC determined by tumor-informed bespoke array (SignateraTM) in all cohorts at baseline (SB) and cycle 3 (C3B). E, cfMeDIP-seq scores vary dynamically across time points and decrease more in patients who exhibit PR or CR to pembrolizumab.
Because we previously found CMC clearance to be an informative threshold for treatment response and outcomes, we aimed to determine whether there was a cfMeDIP-seq level that accurately corresponded with CMC undetectability. cfMeDIP-seq discriminates samples by CMC detectability with an area under the receiver-operator characteristic (AUROC) of 0.902 (Fig. 2C). We found that a cancer log-odds threshold on the joint cfMeDIP-seq score (incorporating both CSM and FLS) of −1.75 maximized the F1 accuracy. 95.8% of samples that fell below this threshold were likely to have demonstrated clearance by CMC, and of the samples that were undetectable on CMC, 54.5% fell below this threshold.
Next, we examined the quantitative correlation of cfMeDIP-seq with CMC. We found that cancer log odds from cfMeDIP-seq demonstrated strong and statistically significant correlations with CMC within every individual cohort and time point (every Spearman correlation coefficient >0.6, each P < 0.015, Fig. 2D). Taken together, these data suggest that ctDNA levels can be accurately estimated using a tumor-naïve approach by integrating cell-free methylomes and fragmentomes. We also assessed the association of baseline radiologically assessed tumor burden (as evaluated by the sum of diameters of RECIST v1.1 target lesions) with baseline CMC and the joint cfMeDIP-seq scores, as well as the CSM and FLS scores separately (Supplementary Fig. S7). Although each of these metrics was positively associated with tumor burden, none of these associations were strong, with only CMC demonstrating a borderline significant association (Spearman rho = 0.203, P = 0.048).
Finally, we examined kinetics across the course of treatment (Fig. 2E). From baseline to cycle 3, the cancer log odds decreased significantly more in responders than nonresponders, with a median change of −51.1% (range, −99.5 to 58.3) in patients with complete response (CR) or partial response (PR) and −2.77% (range, −75.74 to 191.34) in those with stable disease (SD) or progressive disease (PD; two-sided Wilcoxon rank-sum test P = 0.002). These results suggest that early changes in CSM and FLS may be associated with objective response.
Early Change in CSM Predicts Immunotherapy Outcomes
We next turned our attention to the association of cfMeDIP-seq with survival outcomes after pembrolizumab treatment, analyzing CSM and FLS, first independently and then together in a joint survival model. We performed survival analysis using Cox proportional hazards models and reported hazard ratios and P values adjusted for the cohort. First, we assessed CSM at single time points—baseline and cycle 3—splitting both at the median and by quartiles (Supplementary Fig. S8). At baseline (n = 85), below-median CSM was associated with favorable OS [adjusted HR (aHR) = 0.57 (0.33–0.98), P = 0.044] and trended nonsignificantly toward favorable PFS [aHR = 0.63 (0.37–1.08), P = 0.091]. At cycle 3 (n = 56), below-median CSM was significantly associated with favorable PFS [aHR = 0.51 (0.26–0.99), P = 0.047] and OS [aHR = 0.45 (0.20–0.99), P = 0.047], but was no longer significant in a multivariable Cox model incorporating PD-L1 expression and log TMB (Supplementary Fig. S9). Similarly, CMC was predictive at individual time points when adjusted only for cohort (Supplementary Fig. S10). In multivariable analysis (MVA), these associations remained statistically significant at baseline, but not at cycle 3 (Supplementary Fig. S11).
We next examined the kinetics of CSM. We computed ΔCSM as the difference in log-adjusted CSM from baseline to cycle 3 (Fig. 3A). We hypothesized that change in ctDNA levels from baseline to pre-cycle 3 would be associated with both OS and PFS, as per the primary finding from our previous publication utilizing a tumor-informed bespoke mutational assay (11). We performed this analysis in 53 patients assayed by both cfMeDIP-seq (CSM score) and bespoke ctDNA (CMC level) at both baseline and cycle 3, excluding one patient with undetectable CMC at both time points. A decrease in CSM from baseline to cycle 3 was associated with prolonged OS [aHR = 0.40 (0.21–0.75); P = 0.005] and PFS [aHR = 0.35 (0.18–0.69); P = 0.003; Fig. 3B]. Similarly, ΔCMC was associated with OS [aHR = 0.45 (0.23–0.87); P = 0.018] and PFS [aHR = 0.45 (0.24–0.85); P = 0.014]. In MVA incorporating cohort, PD-L1 expression, and log TMB, ΔCSM remained an independent predictive biomarker for OS [aHR = 0.45 (0.23–0.88); P = 0.019] and PFS [aHR = 0.37 (0.19–0.74), P = 0.005; Fig. 3C]. ΔCMC was also an independent predictive biomarker in MVA but only for PFS [aHR = 0.51 (0.27–0.97), P = 0.04; Supplementary Fig. S12].
Figure 3.

Association of CSM and CMC with survival outcomes in patients with advanced cancer treated with pembrolizumab. Circulating tumor DNA was quantified using a methylation-based approach (CSM) and a bespoke tumor-informed mutation sequencing approach (CMC). A, Methylation probabilities were summed across 200 cancer-specific sites, curated based on independent analysis of methylation array data from The Cancer Genome Atlas. ΔCSM was calculated based on the change in CSM from SB to C3B. Changing regions are shown in the heat map, alongside final ΔCSM values. B, Decrease in CSM and CMC from baseline to cycle 3 are each associated with improved OS and PFS. C, In multivariable Cox analyses, ΔCSM was a significant, independent predictor of PFS and OS, adjusted for cohort, PD-L1 expression, and tumor mutation burden. D, A decrease in either CSM or CMC was associated with improved survival, whereas increase in both metrics identified patients with particularly poor outcome.
To assess the role for joint assessment of methylation-based and mutation-based ctDNA estimates, we examined ΔCSM and ΔCMC in combination. CSM and CMC changed in the same direction in 38 patients and in opposite directions in 15 patients. Of the 15 patients with discordant changes, 9 had very low CSM and/or CMC scores below 1.0, and three of the remaining six had minor shifts in CMC (<50% change from baseline; Supplementary Table S1). The combination of ΔCSM and ΔCMC appeared to identify a subgroup with particularly poor outcome characterized by increase in both CSM and CMC (Fig. 3D). We show in a post-hoc analysis (Supplementary Fig. S13) that a decrease in either CSM or CMC was sufficient to result in a significant improvement in OS [aHR = 0.30 (0.14–0.61), P = 0.001] and PFS [aHR = 0.33 (0.16–0.69), P = 0.003].
Change in FLS Is Associated with Immunotherapy Outcomes
The FLS captures characteristic cancer-specific fragmentation patterns within our CSM windows using a previously established formula, detailed in the Methods section (19). FLS was correlated with CSM (Spearman ρ = 0.655, P < 0.001; Supplementary Fig. S14) and demonstrated a median absolute percent change of 67.7% between baseline and cycle 3. Higher FLS represents more cancer-like fragmentation profiles, and we thus hypothesized that lower FLS and decreasing FLS during pembrolizumab would be associated with favorable outcomes. PFS and OS were indeed prolonged in patients with below-median cycle 3 FLS, but these differences were not statistically significant when adjusted for cohort (Supplementary Fig. S15). A decrease in FLS from baseline to cycle 3 was significantly associated with improved OS [aHR = 0.40 (0.20–0.77), P = 0.006; Fig. 4A], which remained independently predictive in MVA that included cohort, PD-L1, and TMB (Supplementary Fig. S16). There was a nonsignificant trend toward improved PFS in patients with decreased FLS [aHR = 0.51 (0.26–1.00), P = 0.0503; Fig. 4A].
Figure 4.

CSM and FLS are jointly predictive of survival outcomes in patients with advanced cancer receiving pembrolizumab. A, We computed FLS based on the relative similarity of fragmentomic profiles to reference cancer and normal fragment-length histograms. A decrease in FLS from baseline to pre-cycle 3 is associated with prolonged OS, with a nonstatistically significant trend for prolonged PFS. B, In a joint model of CSM and FLS, Kaplan–Meier plots show that patients with a decrease in each biomarker from baseline to cycle 3 are associated with improved OS and PFS. C, Multivariable analyses confirm that both CSM and FLS are independently predictive of OS, whereas only CSM achieves statistical significance for PFS.
Cell-Free Methylomes and Fragmentomes Jointly Predict Treatment Outcomes
We next assessed whether early changes in CSM and FLS were jointly predictive of outcomes. Kaplan–Meier curves were consistent with decreases in CSM and FLS each being associated with favorable PFS and OS (Fig. 4B). An MVA including ΔCSM, ΔFLS, and cohort confirmed that both ΔCSM and ΔFLS were significant independent predictors of OS, whereas only ΔCSM was a significant predictor of PFS (Fig. 4C). This supports the complementary use of methylation and fragmentation data from the same cfMeDIP-seq assay for response prediction.
In our previous study, we showed that clearance of CMC at any on-treatment time point was associated with exceptional response to pembrolizumab (11). However, for CSM and FLS, there is not yet an established threshold below which ctDNA can be said to be undetectable. As mentioned previously, we trained a logistic regression model with CSM and FLS as predictors and cancer versus control as the response variable. The output values of the final logistic regression model represent predicted log odds of cancer, incorporating information from both CSM and FLS. A score below −1.75 was associated with undetectability by CMC.
By this definition, 11 cases were identified to have achieved clearance, 10 by cycle 3 and one by cycle 6. Median follow-up for these cases was 59.5 (range, 37.6–64.4) months. Of these, nine (82%) were in agreement with clearance criteria by CMC (Fig. 5A and B). The two patients with cfMeDIP-seq clearance who did not clear by CMC both remained alive at the last follow-up of 59.5 and 60.5 months (Fig. 5C). Both were in cohort C (ovarian cancer). One patient (INS-C-020) progressed after 1.8 months with a 26% increase in target lesions according to RECIST 1.1 criteria, whereas the other patient (INS-C-018) had a 25% decrease in target lesion size and achieved the longest period of SD in this study for a patient with ovarian cancer before progression (10.5 months). Conversely, two patients achieved clearance by CMC but not cfMeDIP-seq. However, both of these cases had low cfMeDIP-seq scores just slightly above the clearance threshold (INS-A-019 with HNSCC cycle 3 score = −1.13, INS-D-012 with melanoma cycle 3 score = −1.63). INS-A-019 had a 56% reduction in target lesions and passed away after 37.6 months without progression. INS-D-012 had a 97% reduction in target lesions and remained alive at last follow-up (61.0 months) without evidence of progression. In aggregate, the cases with clearance identified by this definition demonstrated exceptional PFS and OS (Fig. 5D).
Figure 5.

CSM and FLS estimate ctDNA clearance status, identifying patients with durable response to pembrolizumab. The log odds of cancer were computed using a logistic regression model with CSM and FLS as independent variables. A threshold of below −1.754 was determined to identify patients with clearance. A, By this criterion, 11 patients cleared ctDNA based on cfMeDIP-seq, of which 9 also had undetectable ctDNA based on CMC from targeted deep sequencing. B, Patients with clearance demonstrated persistently low cfMeDIP-seq scores. The two patients with clearance by CMC but not cfMeDIP-seq (INS-A-019 and INS-D-012) both had low cfMeDIP-seq scores near the clearance threshold. C, The clinical course and radiologic size of index tumors of all 13 patients with clearance by either method are shown. Vertical lines show the median and maximum PFS of patients within each cohort. D, Patients meeting the cfMeDIP-seq clearance criteria demonstrated strikingly favorable PFS and OS.
DISCUSSION
To our knowledge, this is the first reported analysis of methylated cfDNA in patients with advanced cancer during treatment with ICB. This is also one of the first direct comparisons between a tumor-informed mutation-based approach and a tumor-naïve methylation-based approach to ctDNA analysis. Moreover, this is the first analysis to examine the integration of methylomic and fragmentomic scores from a single assay to refine ctDNA quantification and response prediction. Our results suggest that analysis of the cfDNA methylome and fragmentome assayed by cfMeDIP-seq yields promising tumor-naïve predictive biomarkers. Tumor-naive CSM and FLS were strongly correlated with tumor-informed, mutation-based ctDNA quantification. Change in CSM from baseline to cycle 3 predicted both PFS and OS whereas change in FLS predicted OS, each independently of cohort, TMB, and PD-L1 status. Lastly, we proposed a reasonable threshold for clearance within our data, which was concordant with undetectable CMC, and identified a set of exceptional responders. This clearance threshold requires validation in independent cohorts. Taken together, these findings suggest that prediction of response to pembrolizumab can be refined through the integration of methylome and fragmentome analysis and represents a possible alternative to tumor-informed, mutation-based approaches.
We previously reported that early ctDNA kinetics are predictive of pembrolizumab outcomes (11). However, this approach requires WES of the tumor, thus excluding patients without available tumor tissue and potentially leading to delays in obtaining results. cfMeDIP-seq is a tumor-naïve approach that could overcome these limitations. Because methylation is highly cell-type specific (26), the use of methylation also bypasses challenges in tumor-naïve ctDNA profiling caused by nontumoral mutations arising from clonal hematopoiesis (27). Unlike clonal hematopoiesis, methylation in PBLs is largely conserved and nonrandom and can be reliably filtered out. We also observed differences in outcomes according to CSM at cycle 3 approaching statistical significance in MVA that warrant further study. This single time-point readout may allow simplification by omitting baseline ctDNA profiling in resource-constrained settings. The earliest time-point on-treatment for ctDNA analysis in our study was cycle 3 day 1 of pembrolizumab (i.e., around weeks 6–7 on-treatment). It aligns with recent work that suggests this is the optimal time point in patients treated with pembrolizumab in non–small cell lung cancer, where ctDNA using a mutation-based assay was measured every cycle (28). However, another study in non–small cell lung cancer has suggested that the optimal time point to detect ctDNA decrease may be later (i.e., cycle 4 in patients treated with a combination of immunotherapy and chemotherapy; ref. 29). The lack of a standardized on-treatment analysis time point is a challenge in the implementation of ctDNA in clinical practice. Moreover, an earlier time point could be more informative as it would precede radiologic assessments and could potentially be helpful in making early treatment decisions. The kinetics of ctDNA prior to cycle 3 and whether a different time point could also predict clinical outcome with our approach warrant further investigation.
Methylation is thought to be a stable epigenetic marker and differential methylation, especially CpG island hypermethylation, is prevalent among cancers (30). This makes methylation a promising candidate biomarker for developing pan-cancer signatures. Our independent signature was capable of segregating cancer from noncancer, was highly correlated with CMC, and could identify most cases of clearance. To maximize the potential for clinical translation of methylation biomarkers across technologies, We also used both array and PBL MeDIP-seq data to filter PBL-derived background, a crucial step because as much as 80% of cfDNA is known to be PBL derived (13, 16). Filtering out additional healthy tissue types is also an option, but CpGs become increasingly likely to overlap tumor-specific hypermethylation as filtering is extended to solid tissues.
Because of the highly novel nature of this cohort and approach, there does not exist true independent validation data. For this reason, we opted to generate an independent pan-cancer methylation signature from TCGA data instead of extracting disease-specific signatures from the cfMeDIP-seq data, as has often been done previously (15, 16, 31). This allowed us to treat the study cohort as the first validation set for the signature, and the successful transfer of this signature from methylation arrays to cfMeDIP-seq supports its generalizability. An obvious limitation of using methylation data from TCGA is the relatively lower coverage of 450K methylation arrays compared with, for example, whole-genome bisulfite sequencing. However, we chose to use TCGA data as they are generated using standardized protocols, a harmonized analysis pipeline, and stringent quality standards across a large variety of histologies, and no comparable whole-genome methylation data set yet exists. There is a pressing need for high-quality, harmonized genome-wide reference methylation data in cancer and normal cells, which could further improve prediction by expanding beyond methylation array probe sites. This added resolution could significantly boost sensitivity, for example, by identifying signatures of longer uniformly methylated regions across which a cancer methylation signal could be integrated (26).
This study reveals the potential of multimodal liquid biopsy analysis for treatment response monitoring in cancer. We demonstrated how a single assay could yield complementary methylomic and fragmentomic metrics, which may in turn be complementary with mutational analysis. Indeed, the separate mutational assay may also become unnecessary as emerging methods and assays are poised to enable simultaneous inference of mutations, methylation, fragmentation, and even gene expression from a single noninvasive assay (18, 32–34). In our study, ΔCSM and ΔCMC yielded different but complementary predictions of survival outcomes, and the poorest responding patients demonstrated increases in both. In 28.3% of cases, CSM and CMC changed in different directions, but most had low baseline values or minor shifts. Moreover, FLS was independently prognostic alongside CSM. These important early findings may help to inform future strategies for noninvasive biomarker integration. The main advantage of a tumor-naïve assay is that it could be applicable to more patients, as WES of the tumor is not needed. We only performed the analysis of this assay in patients treated with anti–PD-1 antibody, and we have not yet explored whether these changes in CSM and FLS could also be seen with other systemic treatments (i.e., chemotherapy or targeted therapy). Changes in CSM and FLS may also be predictive of clinical outcome with other therapies, and further validation is warranted.
Limitations of our study include a small sample size (204 samples from 87 patients), as not all of the plasma samples collected during the prospective INSPIRE study were available, especially for pre-cycle 3 samples. This may have limited the statistical significance of some findings, especially in the analysis of individual time points. However, the association of CSM/FLS with outcomes is consistently more significant with the change from baseline to cycle 3, suggesting that kinetics rather than cross-sectional time points may be more informative. Although INSPIRE is a prospective clinical study, the analysis presented here was performed retrospectively, after the clinical trial was completed. The heterogeneity of tumor types is also a limitation, but also potentially a strength as we aimed to generate a pan-cancer signature. Although we showed that changes in CSM and FLS may predict outcomes across cancer types, these changes may be more informative in those tumor types where anti–PD-1 antibody is usually more effective (i.e., melanoma or head and neck) compared with those cancers with less response to ICB (breast, ovarian, or some of the mixed tumors). We primarily focused on changes from baseline to cycle 3, which was the earliest on-treatment time point available in this study. However, the most optimal time point for estimating the response to ICB remains an open question. Normal control cfMeDIP-seq data were chosen, which were processed in the same laboratory as the cancer samples, and may harbor some sex-bias, as 72 of the 100 normal controls were healthy females from a breast cancer early detection study (medRxiv: 2023.01.30.23285027). A limitation of the cfMeDIP-seq assay is the inability to accurately call hypomethylation due to a lack of DNA pulldown, which may make it difficult to distinguish between tissues of origin (26). Instead, we focused on detecting hypermethylation, which is a distinguishing feature across cancers (30). Lastly, rigorous analysis of the detection limits, such as using a dilution series, was not within the scope of this study, and has been described previously (14).
In summary, this study represents the first combined use of tumor-naïve cell-free methylomes and fragmentomes from a single assay, cfMeDIP-seq, to monitor cancer burden in response to pembrolizumab. Although a priori prediction of immunotherapy response remains challenging, our results support the idea that an on-treatment dynamic biomarker can provide an accurate readout of treatment effectiveness early in the course of treatment with ICB. This approach could enable earlier response assessment, allowing for prompt redirection to next-line treatment options in nonresponders. It also shows promise for a truly minimally invasive disease monitoring solution in the future that does not rely on tumor tissue availability. We anticipate that these early translational results will help guide the design of future interventional studies leveraging dynamic biomarkers for treatment adaptation.
METHODS
Study Design
The INSPIRE study (Investigator-initiated Phase II Study of Pembrolizumab Immunological Response Evaluation, NCT02644369) included a total of 106 patients with advanced solid tumors from March 21, 2016, to May 9, 2018. It was approved by the University Health Network research ethics board (REB ID: 15-9828). The primary aim of this phase II investigator-initiated study was to analyze potential pharmacodynamic biomarkers of response to pembrolizumab. This clinical trial was approved by the Research Ethics Board at the University Health Network and was conducted in accordance with the Declaration of Helsinki. Adult patients provided written informed consent and were accrued onto five parallel cohorts consisting of HNSCC (cohort A), triple-negative breast cancer (cohort B), high-grade serous ovarian cancer (cohort C), malignant melanoma (cohort D), and mixed solid tumor (cohort E). Inclusion and exclusion criteria have been summarized previously (11, 35). Treatment was conducted at Princess Margaret Cancer Centre with pembrolizumab 200 mg administered intravenously every 3 weeks. Clinical data as well as baseline biomarkers were recorded in every patient. Biomarker data for PD-L1 expression and TMB (from WES) were unchanged from our previous publications (9, 11). The cutoff date for clinical outcomes was December 6, 2021.
Blood Collection and Processing
Peripheral blood plasma was collected at baseline and at the beginning of every three cycles during treatment. At each collection time point, 30 mL of peripheral blood was collected in EDTA tubes. Plasma was separated from the cell pellet within 2 hours of collection and aliquoted for storage at −80°C. cfDNA was purified from clarified plasma using the QIAamp Circulating Nucleic Acid Kit (Qiagen). PBL genomic DNA was extracted using the AllPrep DNA/RNA/miRNA Universal Extraction Kit (Qiagen). cfDNA was quantified using the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific) and then processed by the Translational Genomics Laboratory. Following previous studies utilizing these specimens, we reviewed the remaining stock of cfDNA available. A subset of cfDNA samples were chosen for analysis, with the aim to maximize baseline and cycle 3 pairs with matched data from bespoke ctDNA analysis and to maintain a balanced representation across the five cohorts.
Bespoke Mutational Analysis of cfDNA
CMC data were generated using the Signatera assay as previously described (11). The same values were used as in the present study, without modification. Briefly, 16 confident cancer-specific variants were identified in tissue exome sequencing, and primers were designed for each (36). Multiplexed targeted PCR and amplicon deep sequencing were performed on an Illumina platform. For each of the 16 target mutations, the variant allele fraction (VAF) was determined, and absolute ctDNA levels (MTM per mL) in the plasma were calculated by normalizing VAF by the plasma volume used for each sample, yielding the CMC value.
cfMeDIP-seq Library Construction and Sequencing
cfMeDIP-seq libraries were constructed from 10 ng input cfDNA using a multiplexed protocol adapted from Shen and colleagues (12). Precapture libraries were synthesized using a modified KAPA Hyper Prep Kit protocol. cfDNA and Arabidopsis internal control DNA were end repaired, A-tailed, and adapter ligated, with the inclusion of a unique molecular identifier (UMI). 5-Methylcytosine antibody (Diagenode Mag MeDIP kit) was used to selectively enrich cfDNA by immunoprecipitation via a modified manufacturer's protocol, prior to library amplification and sequencing. Libraries were sequenced on Illumina NovaSeq 6000 or NextSeq550 platforms using V1 and V2 chemistry and reagents, respectively, targeting a read depth of 60 million clusters (120 million paired-end reads). Actual median read depth was 80.0 (range, 9.9–176.2) million clusters.
Alignment and Quality Control
Sequencing libraries were validated for quality prior to deep sequencing via pre-analytic sequencing on the MiSeq platform to a minimum read depth of 10,000 clusters, 150 bp × 8 bp × 8 bp × 150 bp. cfMeDIP libraries are assessed for CpG relH and CpG GoGe enrichment, AT dropout, and methylated Arabdopsis enrichment relative to control library. cfMeDIP-seq output sequences were analyzed using a reproducible Snakemake (v6.13.1; ref. 37) pipeline that includes all steps from post-processing to calculation of methylation probabilities, available at https://github.com/pughlab/cfMeDIP-seq-analysis-pipeline. First, UMIs were extracted from read sequences into headers using UMI-tools (v1.0.1; ref. 38). Adapter sequences were removed using “Trim Galore!” (v0.6.7) RRID:SCR_011847 (39). Next, FASTQ files were aligned to the iGenomes human reference genome hg38 (http://igenomes.illumina.com.s3-website-us-east-1.amazonaws.com/Homo_sapiens/UCSC/hg38/Homo_sapiens_UCSC_hg38.tar.gz) using the bwa_mem aligner (v0.7.17-r1188; ref. 40). UMI-tools was used to remove PCR duplicates, informed by unique molecular identifiers (38). FastQC (v0.11.9) output was inspected pre- and post-alignment, and post-alignment QC was also performed using Qualimap (v2.2.2; ref. 41).
Normal Control cfMeDIP-seq
Two cohorts of normal control cfMeDIP-seq data totaling 100 samples were chosen because both cohorts were sequenced in the same laboratory as the cancer data. The first cohort included 72 healthy women enrolled in the Ontario Health Study (42) who had been screened and known to be negative for breast cancer over the course of numerous years. The study methodology is currently available in a pre-print form (medRxiv: 2023.01.30.23285027) and data were kindly provided by the Awadalla lab. The second cohort included 8 males and 20 females who were profiled as normal controls for a multicenter early detection study of hereditary cancer susceptibility syndromes (22).
Calculation of Methylation Probability
The genome was divided into contiguous 300 bp bins, and coverage was computed within each. The coverage in any given bin is the sum of the fractions of fragments covering that bin. For example, a fragment that falls entirely within a 300 bp bin counts as 1 coverage unit, whereas a 150 bp fragment with 50 bp overlapping a bin would contribute 1/3 of a coverage unit. To compute methylation probabilities, coverage was corrected for CpG density. In conventional MeDIP-seq experiments, binding affinity is known to rise with increasing CpG density. We modeled the coverage as a mixture of two negative binomial random variables, representing the methylated and unmethylated components, respectively. The unmethylated component is modeled as yu ∼ NegBinom(μ = αo + α1g, θu), where g ∈ [0,1] is the GC content of the bin. The parameters α0, α1, and θu were inferred by fitting a negative binomial generalized linear model to the bins with zero CpGs. The methylated component is modeled as ym ∼ NegBinom(μ = β0 + β1c, θm), where c ∈ Z, c ≥ 1 is the number of CpGs in the bin. This component was fit using an expectation maximization (EM) procedure. The steps of EM are as follows. (i) Make an initial guess for β0, β1, and θm. (ii) Calculate the methylation probability of each bin as 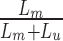 where the methylated likelihood is
where the methylated likelihood is 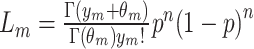 and the unmethylated likelihood
and the unmethylated likelihood 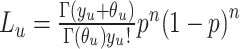 using the alternative parametrization of the negative binomial where
using the alternative parametrization of the negative binomial where 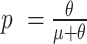 . (iii) Update estimates of β0, β1, and θm, by negative binomial regression across all bins inferred to be methylated, where Lm > Lu. (iv) Repeat, starting from step (ii) (calculating likelihoods), until convergence. This procedure yields a probability of methylation for every bin.
. (iii) Update estimates of β0, β1, and θm, by negative binomial regression across all bins inferred to be methylated, where Lm > Lu. (iv) Repeat, starting from step (ii) (calculating likelihoods), until convergence. This procedure yields a probability of methylation for every bin.
Generating an Independent Cancer Methylation Signature
We generated an independent pan-cancer signature using tissue methylation array data, by extending the approach proposed by Burgener and colleagues (16) to a much larger set of tumor types. 450K methylation array IDAT files from TCGA PanCanAtlas were downloaded and post-processed using ChAMP (v2.21.1; ref. 43). Twenty-seven cancer types were included, with the criteria that each data set contained at least 50 independent primary tumors profiled by 450K methylation array. To avoid class imbalances, 50 tumors of each cancer type were sampled at random. DMCs were identified between every pair of cancer types using limma, using an adjusted P value threshold of <0.05. We selected DMCs hypermethylated at minimum 2-fold over at least two thirds of all other TCGA cohorts. We then filtered out any CpGs with a mean normal control methylation probability >0.01 or maximum methylation probability >0.1 among cfMeDIP-seq data from normal controls. Next, we undertook filtering to remove CpGs in regions methylated in PBLs, the greatest normal tissue contributor of cfDNA. First, CpGs were restricted to those overlapping the PBL-depleted regions derived from prior PBL MeDIP-seq (16). To further suppress peripheral immune signal, we used data from tissue-specific 450K methylomes (13) downloaded from the Gene-Expression Omnibus (GEO, RRID:SCR_005012; GSE122126) and included only the 200 CpGs with the lowest mean beta value among blood cell types, resulting in all probes having mean blood cell beta value below 0.086. To check the cross-assay generalizability of this signature, we downloaded publicly available data from whole-genome bisulfite sequencing of esophageal squamous cell carcinoma and adjacent normal tissue (GSE149608; ref. 44) and breast cancer (GSE186747; ref. 45). We computed the signature within these data sets using the sum of beta values overlapping the signature sites, averaged within each window.
Calculation of CSM Score
The CSM score was computed as the arithmetic sum of methylation probabilities from all bins overlapping the immune-depleted cancer signature DMCs. Supplementary Fig. S3 provides examples depicting how CSM is computed from methylation probabilities.
NMF of Fragment Lengths
NMF was used to compute signatures of cancer- and normal-derived fragment-length frequencies. Global fragment-length profiles were computed using CollectInsertSizeMetrics from Picard (v2.27.4; RRID:SCR_006525; ref. 46). To focus on the mononucleosome peak, only fragments with lengths between 30 and 250 bp were considered. Rank 2 NMF was performed on the matrix of fragment-length counts across patients using the NMF package in R as previously described (25).
Nucleosome Footprinting
We used a previously reported method that assesses the position of fragment ends relative to nucleosome occupancy sites (19, 46). The distance from each read start position to the nearest expected nucleosome center was calculated. The frequency of each distance was tabulated, and the 100 most variable distances were considered as salient features. Rank 2 NMF was performed using these features in the same manner as described above for fragment lengths.
Calculation of FLS
Fragments overlapping the TCGA-derived CSM signature were subset, and their fragment-length histograms were computed. We obtained previously computed frequencies of fragment lengths associated with fragments derived from cancer and normal cells (19). Using these reference data, every fragment length was assigned a cancer-associated score equivalent to the log2 difference between the frequencies of cancer and normal fragments of that length. Next, the sequence alignment map of each cfMeDIP-seq sample was filtered to include only fragments overlapping the CSM signature sites. Every fragment was assigned its own cancer-associated score depending on its length, and the FLS for the sample was calculated as the mean of these scores.
Statistical Analysis
All measurements in plasma were blinded to the clinical outcomes data. The primary outcome was OS and PFS since the start of the clinical trial. Our main hypothesis was that the change from baseline to pre-cycle 3 in methylated cfDNA, ∆CSM, and/or fragment-length scores, ∆FLS, could predict OS and PFS. The sample size was not calculated. Descriptive statistics were used to summarize patients and clinical characteristics. Median and range were used for continuous variables, whereas frequency and percentage for categorical variables. Correlation between CSM and CMC was calculated using Spearman correlation coefficients. The effect of baseline and pre-cycle 3 CSM on OS and PFS was analyzed using Kaplan–Meier curves comparing below- and above-median CSM. All survival analyses were undertaken with a Cox proportional hazards model incorporating the parameter(s) of interest as well as the study cohort as a categorical variable. The adjusted hazards ratios after adjusting for the cohort are reported, as well as their 95% confidence intervals and associated P values. Multivariable Cox models were used to assess the impact of ∆CSM and ∆CMC, while adjusting for cohort, PDL-1 status, and TMB. Results were considered statistically significant if the P value was ≤0.05. All statistical analyses were performed using R (v4.2.1, https://www.r-project.org/; RRID:SCR_001905).
Data Availability
Raw cfMeDIP-seq reads from patients in the INSPIRE cohort are deposited at the European Genome-Phenome Archive under Study ID EGAS00001003280. Sequencing reads for the normal control data from the Ontario Health Study can be requested at https://www.ontariohealthstudy.ca/for-researchers/data-access-forms-and-templates/.
Code Availability
cfMeDIP-seq analysis is packaged for reuse as a Snakemake pipeline and can be found at https://github.com/pughlab/cfMeDIP-seq-analysis-pipeline. Fragmentomic analysis pipelines can be found at https://github.com/pughlab/fragmentomics. In the interest of reproducibility and transparency, the complete code and associated data assets used to generate all figures and reported numeric values for this paper can be found in a unified R markdown file at https://github.com/pughlab/paper-inspire-cfmedip or in the form of a Code Ocean compute capsule, which can be found at https://codeocean.com/capsule/3574944/tree/v1.
Supplementary Material
CMC and CSM early kinetics discordant cases
Overview of the timepoints included in cfMeDIP-seq analysis for each patient. (A) Every sample included by patient and timepoint. (B) Numbers of samples analyzed per cohort and timepoint.
Overall survival (OS) and progression free survival (PFS) in included patients by cohort. (A) Kaplan-meier curves are shown indicating the OS and PFS of patients in five histology-specific cohorts. (B) Forest plot of the hazard ratios for each cohort in a Cox proportional hazards model, with Cohort A as the reference level.
Examples of cancer-specific methylation score calculation. Cancer-specific methylation scores were computed using the sum of inferred absolute methylation values for all reads overlapping an independently-trained cancer-specific signature. Here, we show examples illustrating how absolute methylation levels are inferred from coverage depths in 300 bp bins while adjusting for the density of CpGs.
Validation of the 200 CpG signature using publicly available WGBS data. A 200 CpG signature was generated using 450K array data from TCGA PanCanAtlas. We validated this signature in publicly available data from WGBS of esophageal squamous cell carcinoma and adjacent normal tissue (GSE149608), as well as breast cancers (GSE186747). (A) A heatmap of methylation beta values of our signature sites demonstrates that the methylation signature demonstrated hypermethylation in both esophageal and breast cancers relative to adjacent normal esophageal tissue. (B) A scatter plot of mean beta values, with one point for each signature window is shown. This confirms that any differentially methylated windows were generally hypermethylated in tumor relative to normal. (C) Methylation signature scores were computed by summing the beta values across all sites for each sample. This verified that tumors demonstrated higher scores than adjacent normal tissue.
Cell-free DNA from cancer patients demonstrates greater fragment length variability. (A) Genome wide fragment lengths were computed and averaged within 5 megabase windows. Normalized short-to-long fragment ratios were calculated. Shaded confidence intervals represent 80% and 95% of the data. (B) The variance of mean fragment lengths across patients were compared and the log fold change between cancer and normal variances are shown for each window. Windows with significantly different variance between cancer and normal were identified using non-parametric Ansari-Bradley tests. Neighbouring windows were combined using Comb-p into regions. This revealed greater variability in cancer sample fragment lengths spanning 56.2% of the autosomal genome. (C) Beeswarm plot of fragment length variability. The median cancer window had 30.3% [range: -41.1, 228.] more fragment length variability in samples from cancer patients than normal controls.
Non-negative matrix factorization identifies characteristic cancer-associated signatures of shorter fragment lengths and greater nucleosome core occupancy. (A) Genome-wide fragment lengths were used as features in a two-component non-negative matrix factorization analysis. This revealed a longer and a shorter component. The weight of the shorter was elevated in cell-free DNA of cancer patients relative to normal controls. (B) The distances of fragment ends to nucleosome centers were also used as features in two-component non-negative matrix factorization. This revealed two components with different proportions of intra-nucleosomal fragment ends. The signature with more intra-nucleosomal fragment ends was elevated in the cell-free DNA of cancer patients relative to normal controls. (C) A heatmap showing the localization of fragment ends within the nucleosome core, meaning within 50 bp of nucleosome peaks. Arranging by group and total cfMeDIP-seq score, we observe that those with higher estimated ctDNA levels demonstrated a higher fraction of read ends terminating within the nucleosome core.
Association of tumor burden with cancer mutation concentration (CMC), as well as cancer-specific methylation (CSM) and fragment length score (FLS). We computed CMC from personalized tumor-informed mutation arrays. We also computed CSM and FLS from methylation and fragmentomic analysis respectively of data from cell-free methylated DNA immunoprecipitation and sequencing (cfMeDIP-seq) assays. Coefficients and p-values were computed using Spearman correlations.
Predicting survival outcomes using cancer-specific methylation (CSM) scores at baseline and cycle 3 of pembrolizumab. We computed CSM scores across the trial cohort. At both baseline and cycle 3, patients were split into above- and below-median groups. Survival outcomes are shown, with hazard ratios and p-values adjusted for cohort using a Cox model. PFS analysis at cycle 3 excludes one patient who progressed before the collection of the cycle 3 sample.
Multivariable analysis of survival using cancer-specific methylation (CSM) scores at baseline and cycle 3 of pembrolizumab. We computed CSM scores across the trial cohort. At both baseline and cycle 3, patients were split into above- or below-median groups. Survival outcomes are shown in a multivariable analysis including cohort, PD-L1 expression, and tumor mutation burden (TMB).
Predicting survival outcomes using cancer mutation concentration (CMC) at baseline and cycle 3 of pembrolizumab. CMC was determined using a tumor-informed bespoke approach across the trial cohort. At both baseline and cycle 3, patients were split into above- or below-median groups. Survival outcomes are shown, with hazard ratios and p-values adjusted for cohort using a Cox model. PFS analysis at cycle 3 excludes one patient who progressed before the collection of cycle 3 sample.
Multivariable analysis of OS and PFS using cancer mutation concentration (CMC) at baseline and cycle 3 of pembrolizumab. CMC was determined using a bespoke targeted approach across the trial cohort. At both baseline and cycle 3, patients were split into above- or below-median groups. Survival outcomes are shown in a multivariable analysis including cohort, PD-L1 expression, and tumor mutation burden (TMB).
Multivariate Cox analysis of change in cancer mutation concentration (CMC) from baseline to cycle 3 of pembrolizumab. Covariates include cohort, PD-L1 expression, and tumor mutation burden
Increase in both ctDNA metrics identifies a subgroup with particularly poor outcome. Post-hoc analysis of ΔCSM and ΔCMC together demonstrates that a decrease in either metric was sufficient to result in significantly improved PFS and OS, whereas increase in both metrics identified a group with particularly poor outcomes. PFS analysis at cycle 3 excludes one patient who progressed before the collection of cycle 3 sample.
Cancer-specific methylation (CSM) and fragment length scores (FLS) are moderately correlated. CSM and FLS scores were computed for each sample. Correlation testing between FLS and log-adjusted CSM was performed using Spearman's method.
Predicting survival outcomes using fragment length score (FLS) at baseline and cycle 3 of pembrolizumab. FLS was determined as the mean of the log2 transformed cancer-to-normal ratio of the length of each fragment in a given cfMeDIP-seq sample. At both baseline and cycle 3, patients were split into above- or below-median groups. Survival outcomes are shown, with hazard ratios and p-values adjusted for cohort using a Cox model. PFS analysis at cycle 3 excludes one patient who progressed before the collection of cycle 3 sample.
Multivariate Cox analysis of the change in fragment length score (FLS) from baseline to cycle 3 of pembrolizumab. Covariates in clude cohort, PD-L1 expression, and tumor mutation burden.
Acknowledgments
We would like to thank patients and their families for their participation and contributions to the INSPIRE clinical trial. Major funding support for the project was made possible by the Princess Margaret Cancer Foundation, Ontario Institute for Cancer Research, and Terry Fox Research Institute. We thank Merck Canada Inc., Kirkland, QC, Canada for contributing the study drug for the clinical trial. E.Y. Stutheit-Zhao was supported by a Cancer Research Institute Irvington Postdoctoral Fellowship and the Princess Margaret Cancer Centre Global Oncology Program. E. Sanz-Garcia was supported by the Hold'em for Life Fellowship from the University of Toronto. L.L. Siu holds the BMO Chair in Precision Cancer Genomics. T.J. Pugh holds the Canada Research Chair in Translational Genomics and is supported by a Senior Investigator Award from the Ontario Institute for Cancer Research and the Gattuso-Slaight Personalized Cancer Medicine Fund. We gratefully acknowledge the individuals from the Princess Margaret Tumor Immunotherapy Program (pm-tumorimmunotherapyprogram.ca), including the Administrative (Kendra Ross, Helen Chow, Sawako Elston, and Aileen Trang), Correlatives (Vanessa Speers, Sevan Hakgor, Amanda Giesler, and Koosha Vakilli), and the Immune Profiling Team (Marcus Butler, Ben Wang, Derek Clouthier, Valentin Sotov, Diana Gray, Scott Lien, Diane Liu, Mark Camacho, Darya Lemiashkova, Emily Enfante, Tyler Redublo, Hobin Seo, and Arianne Galindez) for coordinating, receiving, processing, and biobanking tumor and blood samples. We thank members of the Princess Margaret MeDIP-seq working group for helpful discussion around analysis methods, particularly Emma Bell, Samantha Wilson, Althaf Singhawansa, Ian Smith, Nick Cheng, Ming Han, and Sasha Main. We thank the staff of the Princess Margaret Genomics Centre (www.pmgenomics.ca), UHN Bioinformatics, and HPC Core (https://bhpc.uhnresearch.ca). This study was conducted with the support of the Ontario Institute for Cancer Research's Genomics Program (http://genomics.oicr.on.ca) and Translational Genomics Laboratory, a joint initiative between the Princess Margaret Cancer Centre and the Ontario Institute for Cancer Research. These programs were enabled through funding provided by the Government of Ontario and the Princess Margaret Cancer Foundation. We extend particular thanks to staff members of the Ontario Institute for Cancer Research closely involved in this project, including Morgan Taschuk, Lawrence Heisler, and Richard Jovelin. Additional infrastructure support from the Canada Foundation for Innovation, Leaders Opportunity Fund (CFI no. 32383 and no. 38401); Ontario Ministry of Research and Innovation, Ontario Research Fund Small Infrastructure Program; and the Ontario Institute for Cancer Research.
Footnotes
Note: Supplementary data for this article are available at Cancer Discovery Online (http://cancerdiscovery.aacrjournals.org/).
Authors’ Disclosures
E. Sanz-Garcia reports grants from Novartis outside the submitted work. A.R. Abdul Razak reports grants from Merck during the conduct of the study; grants from Adaptimmune, Bayer, GlaxoSmithKline, Medison, Inhibrx, research funding from Deciphera, Karyopharm Therapeutics, Pfizer, Roche/Genentech, Bristol Myers Squibb, MedImmune, Amgen, GlaxoSmithKline, Blueprint Medicines, Merck, AbbVie, Adaptimmune, Iterion Therapeutics, Neoleukin Therapeutics, Daiichi Sankyo, Symphogen, Rain Therapeutics, and Boehringer Ingelheim and personal fees from Boehringer Ingelheim, Merck, ELi-Lilly, and Medison outside the submitted work. A. Spreafico reports grants and personal fees from Merck, BMS, grants from Novartis, Symphogen, AstraZeneca/Medimmune, Bayer, Surface Oncology, Janssen Oncology/Johnson&Johnson, Roche, Regeneron, Alkermes, ArrayBiopharma/Pfizer, GSK, NuBiyota, Oncorus, Treadwell, Amgen, ALX Oncology, Genentech, Seagen, Servier, Incyte, and Gilead outside the submitted work. P.L. Bedard reports grants from BMS, Sanofi, Merck, Lilly, AZ, GSK, BIcara Therapeutics, Zymeworks, Medicenna, Pfizer, Amgen, Bayer, LegoChem, Roche/Genentech, and Gilead outside the submitted work; and Uncompensated advisory for Gilead, Repare, Janssen, Zyemworks, Roche/Genentech, Repare, and Zymeworks. A.R. Hansen reports other support from Janssen, BMS, Advancell, Roche-Genetech, and GSK, personal fees and other support from MSD, Pfizer, and Astellas outside the submitted work. S. Lheureux reports grants from Merck during the conduct of the study; grants and personal fees from Astra-Zeneca, GSK, Roche, Merck, personal fees from Eisai and Schrodinger, and grants from Repare Therapeutics outside the submitted work. J. Burgener reports other support from Adela outside the submitted work. S.V. Bratman reports personal fees from Adela and EMD Serono and grants from AstraZeneca outside the submitted work; in addition, S.V. Bratman has a patent for cfDNA methylation analysis pending and licensed to Adela, a patent for methylated cfDNA fragmentation analysis pending and licensed to Adela, and a patent for cfDNA mutation analysis issued, licensed, and with royalties paid from Roche. T.J. Pugh reports grants and personal fees from AstraZeneca, personal fees from Chrysalis Biomedical Advisors, Merck, SAGA Diagnostics, and grants from Roche/Genentech outside the submitted work. L.L. Siu reports personal fees from Merck and grants from Merck during the conduct of the study; personal fees from AstraZeneca/Medimmune, Roche, Voronoi, Health Analytics, Oncorus, GlaxoSmithKline, Seattle Genetics, Arvinas, Navire, Janpix, Relay Therapeutics, Daiichi Sankyo/UCB Japan, Janssen, Hoopika, InteRNA, Tessa Therapeutics, Sanofi, Amgen, Agios, and Treadwell Therapeutics; grants from Bristol Myers Squibb, Genentech/Roche, GlaxoSmithKline, Novartis, Pfizer, AstraZeneca, Boehringer Ingelheim, Bayer, Amgen, Astellas Pharma, Shattucks Lab, Symphogen, AVID pharmaceuticals, Mirati Therapeutics, Intensity Therapeutics, and Karyopharm Therapeutics outside the submitted work. No disclosures were reported by the other authors.
Authors’ Contributions
E.Y. Stutheit-Zhao: Conceptualization, resources, software, formal analysis, investigation, visualization, methodology, writing–original draft. E. Sanz-Garcia: Conceptualization, resources, data curation, investigation, visualization, methodology, writing–original draft. Z. Liu: Data curation, formal analysis, visualization, writing–review and editing. D. Wong: Software, formal analysis, investigation, visualization, writing–review and editing. K. Marsh: Resources, investigation, methodology, writing–review and editing. A.R. Abdul Razak: Resources, investigation, writing–review and editing. A. Spreafico: Resources, investigation, writing–review and editing. P.L. Bedard: Resources, investigation, methodology. A.R. Hansen: Resources, investigation, writing–review and editing. S. Lheureux: Resources, investigation, writing–review and editing. D. Torti: Investigation, project administration, writing–review and editing. B. Lam: Resources, methodology, writing–review and editing. S. Yang: Resources, investigation, writing–review and editing. J. Burgener: Software, methodology, writing–review and editing. P. Luo: Methodology, writing–review and editing. Y. Zeng: Methodology, writing–review and editing. N. Cheng: Methodology, writing–review and editing. P. Awadalla: Methodology, writing–review and editing. S.V. Bratman: Investigation, methodology, writing–review and editing. P.S. Ohashi: Funding acquisition, methodology, writing–review and editing. T.J. Pugh: Conceptualization, resources, supervision, funding acquisition, investigation, methodology, writing–original draft. L.L. Siu: Conceptualization, resources, supervision, funding acquisition, investigation, writing–original draft.
References
- 1. Mok TSK, Wu Y-L, Kudaba I, Kowalski DM, Cho BC, Turna HZ, et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): a randomised, open-label, controlled, phase 3 trial. Lancet 2019;393:1819–30. [DOI] [PubMed] [Google Scholar]
- 2. Robert C, Ribas A, Schachter J, Arance A, Grob J-J, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol 2019;20:1239–51. [DOI] [PubMed] [Google Scholar]
- 3. Harrington KJ, Burtness B, Greil R, Soulières D, Tahara M, de Castro G, et al. Pembrolizumab with or without chemotherapy in recurrent or metastatic head and neck squamous cell carcinoma: updated results of the Phase III KEYNOTE-048 study. J Clin Oncol 2023;41:790–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gide TN, Wilmott JS, Scolyer RA, Long GV. Primary and acquired resistance to immune checkpoint inhibitors in metastatic melanoma. Clin Cancer Res 2018;24:1260–70. [DOI] [PubMed] [Google Scholar]
- 5. Diaz LA, Shiu K-K, Kim T-W, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab versus chemotherapy for microsatellite instability-high or mismatch repair-deficient metastatic colorectal cancer (KEYNOTE-177): final analysis of a randomised, open-label, phase 3 study. Lancet Oncol 2022;23:659–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marabelle A, Fakih M, Lopez J, Shah M, Shapira-Frommer R, Nakagawa K, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol 2020;21:1353–65. [DOI] [PubMed] [Google Scholar]
- 7. Liu X, Guo C-Y, Tou F-F, Wen X-M, Kuang Y-K, Zhu Q, et al. Association of PD-L1 expression status with the efficacy of PD-1/PD-L1 inhibitors and overall survival in solid tumours: a systematic review and meta-analysis. Int J Cancer 2020;147:116–27. [DOI] [PubMed] [Google Scholar]
- 8. Bareche Y, Kelly D, Abbas-Aghababazadeh F, Nakano M, Esfahani PN, Tkachuk D, et al. Leveraging big data of immune checkpoint blockade response identifies novel potential targets. Ann Oncol 2022;33:1304–17. [DOI] [PubMed] [Google Scholar]
- 9. Cindy Yang SY, Lien SC, Wang BX, Clouthier DL, Hanna Y, Cirlan I, et al. Pan-cancer analysis of longitudinal metastatic tumors reveals genomic alterations and immune landscape dynamics associated with pembrolizumab sensitivity. Nat Commun 2021;12:5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sanz-Garcia E, Zhao E, Bratman SV, Siu LL. Monitoring and adapting cancer treatment using circulating tumor DNA kinetics: Current research, opportunities, and challenges. Sci Adv 2022;8:eabi8618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bratman SV, Yang SYC, Iafolla MAJ, Liu Z, Hansen AR, Bedard PL, et al. Personalized circulating tumor DNA analysis as a predictive biomarker in solid tumor patients treated with pembrolizumab. Nat Cancer 2020;1:873–81. [DOI] [PubMed] [Google Scholar]
- 12. Shen SY, Burgener JM, Bratman SV, De Carvalho DD. Preparation of cfMeDIP-seq libraries for methylome profiling of plasma cell-free DNA. Nat Protoc 2019;14:2749–80. [DOI] [PubMed] [Google Scholar]
- 13. Moss J, Magenheim J, Neiman D, Zemmour H, Loyfer N, Korach A, et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat Commun 2018;9:5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shen SY, Singhania R, Fehringer G, Chakravarthy A, Roehrl MHA, Chadwick D, et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 2018;563:579–83. [DOI] [PubMed] [Google Scholar]
- 15. Nuzzo PV, Berchuck JE, Korthauer K, Spisak S, Nassar AH, Abou Alaiwi S, et al. Detection of renal cell carcinoma using plasma and urine cell-free DNA methylomes. Nat Med 2020;26:1041–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Burgener JM, Zou J, Zhao Z, Zheng Y, Shen SY, Huang SH, et al. Tumor-naïve multimodal profiling of circulating tumor DNA in head and neck squamous cell carcinoma. Clin Cancer Res 2021;27:4230–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mouliere F, Chandrananda D, Piskorz AM, Moore EK, Morris J, Ahlborn LB, et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci Transl Med 2018;10:eaat4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cristiano S, Leal A, Phallen J, Fiksel J, Adleff V, Bruhm DC, et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 2019;570:385–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vessies DCL, Schuurbiers MMF, van der Noort V, Schouten I, Linders TC, Lanfermeijer M, et al. Combining variant detection and fragment length analysis improves detection of minimal residual disease in postsurgery circulating tumour DNA of stage II-IIIA NSCLC patients. Mol Oncol 2022;16:2719–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peneder P, Bock C, Tomazou EM. LIQUORICE: detection of epigenetic signatures in liquid biopsies based on whole-genome sequencing data. Bioinform Adv 2022;2:vbac017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nguyen THH, Lu Y-T, Le VH, Bui VQ, Nguyen LH, Pham NH, et al. Clinical validation of a ctDNA-based assay for multi-cancer detection: an interim report from a Vietnamese longitudinal prospective cohort study of 2795 participants. Cancer Invest 2023;41:232–48. [DOI] [PubMed] [Google Scholar]
- 22. Wong D, Luo P, Oldfield LE, Gong H, Brunga L, Rabinowicz R, et al. Early cancer detection in Li-Fraumeni syndrome with cell-free DNA. Cancer Discov 2024;14:104–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pedersen BS, Schwartz DA, Yang IV, Kechris KJ. Comb-p: Software for combining, analyzing, grouping and correcting spatially correlated P-values. Bioinformatics 2012;28:2986–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Renaud G, Nørgaard M, Lindberg J, Grönberg H, De Laere B, Jensen JB, et al. Unsupervised detection of fragment length signatures of circulating tumor DNA using non-negative matrix factorization. eLife 2022;11:e71569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vanderstichele A, Busschaert P, Landolfo C, Olbrecht S, Coosemans A, Froyman W, et al. Nucleosome footprinting in plasma cell-free DNA for the pre-surgical diagnosis of ovarian cancer. npj Genom Med 2022;7:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Loyfer N, Magenheim J, Peretz A, Cann G, Bredno J, Klochendler A, et al. A DNA methylation atlas of normal human cell types. Nature 2023;613:355–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krebs MG, Malapelle U, André F, Paz-Ares L, Schuler M, Thomas DM, et al. Practical considerations for the use of circulating tumor DNA in the treatment of patients with cancer: a narrative review. JAMA Oncol 2022;8:1830–9. [DOI] [PubMed] [Google Scholar]
- 28. Anagnostou V, Ho C, Nicholas G, Juergens RA, Sacher A, Fung AS, et al. ctDNA response after pembrolizumab in non-small cell lung cancer: phase 2 adaptive trial results. Nat Med 2023;29:2559–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pellini B, Madison RW, Childress MA, Miller ST, Gjoerup O, Cheng J, et al. Circulating tumor DNA monitoring on chemo-immunotherapy for risk stratification in advanced non-small cell lung cancer. Clin Cancer Res 2023;29:4596–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nishiyama A, Nakanishi M. Navigating the DNA methylation landscape of cancer. Trends Genet 2021;37:1012–27. [DOI] [PubMed] [Google Scholar]
- 31. Nassiri F, Chakravarthy A, Feng S, Shen SY, Nejad R, Zuccato JA, et al. Detection and discrimination of intracranial tumors using plasma cell-free DNA methylomes. Nat Med 2020;26:1044–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhou Q, Kang G, Jiang P, Qiao R, Lam WKJ, Yu SCY, et al. Epigenetic analysis of cell-free DNA by fragmentomic profiling. Proc Natl Acad Sci U S A 2022;119:e2209852119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zviran A, Schulman RC, Shah M, Hill STK, Deochand S, Khamnei CC, et al. Genome-wide cell-free DNA mutational integration enables ultra-sensitive cancer monitoring. Nat Med 2020;26:1114–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vaisvila R, Ponnaluri VKC, Sun Z, Langhorst BW, Saleh L, Guan S, et al. Enzymatic methyl sequencing detects DNA methylation at single-base resolution from picograms of DNA. Genome Res 2021;31:1280–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Clouthier DL, Lien SC, Yang SYC, Nguyen LT, Manem VSK, Gray D, et al. An interim report on the investigator-initiated phase 2 study of pembrolizumab immunological response evaluation (INSPIRE). J Immunother Cancer 2019;7:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Coombes RC, Page K, Salari R, Hastings RK, Armstrong A, Ahmed S, et al. Personalized detection of circulating tumor DNA antedates breast cancer metastatic recurrence. Clin Cancer Res 2019;25:4255–63. [DOI] [PubMed] [Google Scholar]
- 37. Köster J, Rahmann S. Snakemakea scalable bioinformatics workflow engine. Bioinformatics 2012;28:2520–2. [DOI] [PubMed] [Google Scholar]
- 38. Smith TS, Heger A, Sudbery I. UMI-tools: Modelling sequencing errors in unique molecular identifiers to improve quantification accuracy. Genome Res 2017;27:491–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 2011;17:10–12. [Google Scholar]
- 40. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence Alignment/Map format and SAMtools. Bioinformatics 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. García-Alcalde F, Okonechnikov K, Carbonell J, Cruz LM, Götz S, Tarazona S, et al. Qualimap: evaluating next-generation sequencing alignment data. Bioinformatics 2012;28:2678–9. [DOI] [PubMed] [Google Scholar]
- 42. Kirsh VA, Skead K, McDonald K, Kreiger N, Little J, Menard K, et al. Cohort profile: the Ontario Health Study (OHS). Int J Epidemiol 2022;dyac156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Morris TJ, Butcher LM, Feber A, Teschendorff AE, Chakravarthy AR, Wojdacz TK, et al. ChAMP: 450k chip analysis methylation pipeline. Bioinformatics 2014;30:428–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cao W, Lee H, Wu W, Zaman A, McCorkle S, Yan M, et al. Multi-faceted epigenetic dysregulation of gene expression promotes esophageal squamous cell carcinoma. Nat Commun 2020;11:3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Guillen KP, Fujita M, Butterfield AJ, Scherer SD, Bailey MH, Chu Z, et al. A human breast cancer-derived xenograft and organoid platform for drug discovery and precision oncology. Nat Cancer 2022;3:232–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ulz P, Thallinger GG, Auer M, Graf R, Kashofer K, Jahn SW, et al. Inferring expressed genes by whole-genome sequencing of plasma DNA. Nat Genet 2016;48:1273–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
CMC and CSM early kinetics discordant cases
Overview of the timepoints included in cfMeDIP-seq analysis for each patient. (A) Every sample included by patient and timepoint. (B) Numbers of samples analyzed per cohort and timepoint.
Overall survival (OS) and progression free survival (PFS) in included patients by cohort. (A) Kaplan-meier curves are shown indicating the OS and PFS of patients in five histology-specific cohorts. (B) Forest plot of the hazard ratios for each cohort in a Cox proportional hazards model, with Cohort A as the reference level.
Examples of cancer-specific methylation score calculation. Cancer-specific methylation scores were computed using the sum of inferred absolute methylation values for all reads overlapping an independently-trained cancer-specific signature. Here, we show examples illustrating how absolute methylation levels are inferred from coverage depths in 300 bp bins while adjusting for the density of CpGs.
Validation of the 200 CpG signature using publicly available WGBS data. A 200 CpG signature was generated using 450K array data from TCGA PanCanAtlas. We validated this signature in publicly available data from WGBS of esophageal squamous cell carcinoma and adjacent normal tissue (GSE149608), as well as breast cancers (GSE186747). (A) A heatmap of methylation beta values of our signature sites demonstrates that the methylation signature demonstrated hypermethylation in both esophageal and breast cancers relative to adjacent normal esophageal tissue. (B) A scatter plot of mean beta values, with one point for each signature window is shown. This confirms that any differentially methylated windows were generally hypermethylated in tumor relative to normal. (C) Methylation signature scores were computed by summing the beta values across all sites for each sample. This verified that tumors demonstrated higher scores than adjacent normal tissue.
Cell-free DNA from cancer patients demonstrates greater fragment length variability. (A) Genome wide fragment lengths were computed and averaged within 5 megabase windows. Normalized short-to-long fragment ratios were calculated. Shaded confidence intervals represent 80% and 95% of the data. (B) The variance of mean fragment lengths across patients were compared and the log fold change between cancer and normal variances are shown for each window. Windows with significantly different variance between cancer and normal were identified using non-parametric Ansari-Bradley tests. Neighbouring windows were combined using Comb-p into regions. This revealed greater variability in cancer sample fragment lengths spanning 56.2% of the autosomal genome. (C) Beeswarm plot of fragment length variability. The median cancer window had 30.3% [range: -41.1, 228.] more fragment length variability in samples from cancer patients than normal controls.
Non-negative matrix factorization identifies characteristic cancer-associated signatures of shorter fragment lengths and greater nucleosome core occupancy. (A) Genome-wide fragment lengths were used as features in a two-component non-negative matrix factorization analysis. This revealed a longer and a shorter component. The weight of the shorter was elevated in cell-free DNA of cancer patients relative to normal controls. (B) The distances of fragment ends to nucleosome centers were also used as features in two-component non-negative matrix factorization. This revealed two components with different proportions of intra-nucleosomal fragment ends. The signature with more intra-nucleosomal fragment ends was elevated in the cell-free DNA of cancer patients relative to normal controls. (C) A heatmap showing the localization of fragment ends within the nucleosome core, meaning within 50 bp of nucleosome peaks. Arranging by group and total cfMeDIP-seq score, we observe that those with higher estimated ctDNA levels demonstrated a higher fraction of read ends terminating within the nucleosome core.
Association of tumor burden with cancer mutation concentration (CMC), as well as cancer-specific methylation (CSM) and fragment length score (FLS). We computed CMC from personalized tumor-informed mutation arrays. We also computed CSM and FLS from methylation and fragmentomic analysis respectively of data from cell-free methylated DNA immunoprecipitation and sequencing (cfMeDIP-seq) assays. Coefficients and p-values were computed using Spearman correlations.
Predicting survival outcomes using cancer-specific methylation (CSM) scores at baseline and cycle 3 of pembrolizumab. We computed CSM scores across the trial cohort. At both baseline and cycle 3, patients were split into above- and below-median groups. Survival outcomes are shown, with hazard ratios and p-values adjusted for cohort using a Cox model. PFS analysis at cycle 3 excludes one patient who progressed before the collection of the cycle 3 sample.
Multivariable analysis of survival using cancer-specific methylation (CSM) scores at baseline and cycle 3 of pembrolizumab. We computed CSM scores across the trial cohort. At both baseline and cycle 3, patients were split into above- or below-median groups. Survival outcomes are shown in a multivariable analysis including cohort, PD-L1 expression, and tumor mutation burden (TMB).
Predicting survival outcomes using cancer mutation concentration (CMC) at baseline and cycle 3 of pembrolizumab. CMC was determined using a tumor-informed bespoke approach across the trial cohort. At both baseline and cycle 3, patients were split into above- or below-median groups. Survival outcomes are shown, with hazard ratios and p-values adjusted for cohort using a Cox model. PFS analysis at cycle 3 excludes one patient who progressed before the collection of cycle 3 sample.
Multivariable analysis of OS and PFS using cancer mutation concentration (CMC) at baseline and cycle 3 of pembrolizumab. CMC was determined using a bespoke targeted approach across the trial cohort. At both baseline and cycle 3, patients were split into above- or below-median groups. Survival outcomes are shown in a multivariable analysis including cohort, PD-L1 expression, and tumor mutation burden (TMB).
Multivariate Cox analysis of change in cancer mutation concentration (CMC) from baseline to cycle 3 of pembrolizumab. Covariates include cohort, PD-L1 expression, and tumor mutation burden
Increase in both ctDNA metrics identifies a subgroup with particularly poor outcome. Post-hoc analysis of ΔCSM and ΔCMC together demonstrates that a decrease in either metric was sufficient to result in significantly improved PFS and OS, whereas increase in both metrics identified a group with particularly poor outcomes. PFS analysis at cycle 3 excludes one patient who progressed before the collection of cycle 3 sample.
Cancer-specific methylation (CSM) and fragment length scores (FLS) are moderately correlated. CSM and FLS scores were computed for each sample. Correlation testing between FLS and log-adjusted CSM was performed using Spearman's method.
Predicting survival outcomes using fragment length score (FLS) at baseline and cycle 3 of pembrolizumab. FLS was determined as the mean of the log2 transformed cancer-to-normal ratio of the length of each fragment in a given cfMeDIP-seq sample. At both baseline and cycle 3, patients were split into above- or below-median groups. Survival outcomes are shown, with hazard ratios and p-values adjusted for cohort using a Cox model. PFS analysis at cycle 3 excludes one patient who progressed before the collection of cycle 3 sample.
Multivariate Cox analysis of the change in fragment length score (FLS) from baseline to cycle 3 of pembrolizumab. Covariates in clude cohort, PD-L1 expression, and tumor mutation burden.
Data Availability Statement
Raw cfMeDIP-seq reads from patients in the INSPIRE cohort are deposited at the European Genome-Phenome Archive under Study ID EGAS00001003280. Sequencing reads for the normal control data from the Ontario Health Study can be requested at https://www.ontariohealthstudy.ca/for-researchers/data-access-forms-and-templates/.