

Abstract
Early in tumorigenesis, a DNA damage‐response network is activated in preneoplastic cells that delays or prevents cancer. Activation of the Chk2 G2/M checkpoint kinase and loss of fragile histidine triad (Fhit) tumor suppressor expression increase cellular susceptibility to DNA‐damaging ‘oncogenic’ stressors, particularly in precursor or precancerous lesions. To understand the mechanism of oral carcinogenesis, we assessed the association between phosphorylated Chk2 (pChk2) and Fhit expression in oral squamous cell carcinoma. Loss of Fhit expression was an early event during oral carcinogenesis, whereas a decrease in the number of pChk2‐positive cells was associated with tumor progression. Although tyrosine 114 is known to be essential to Fhit's tumor‐suppressing activity, both wild‐type and tyrosine 114 mutant Fhit increased the population of subG1 DNA‐containing HSC‐3 OSCC cells with elevated pChk2 levels. In particular, when cells were exposed to ionizing radiation, pChk2 levels were upregulated dramatically, as were those of its downstream target Cdc25C. Knockdown of Fhit with FHIT small interfering RNA diminished the ionizing radiation‐induced Chk2 phosphorylation in HEK293 cells. Furthermore, Fhit‐deficient mice demonstrated a decrease in the number of pChk2‐positive cells not only in dysplastic lesions but also in N‐nitrosobenzylamine‐induced papilloma of the forestomach, suggesting that lack of Fhit expression and the resultant defects of the ataxia telangiectasia mutated–Chk2 pathway can cause a difference in the incidence of N‐nitrosobenzylamine‐induced forestomach lesions. These findings suggest that Fhit plays a key role in the regulation of the ataxia telangiectasia mutated–Chk2 DNA damage response during oral carcinogenesis. (Cancer Sci 2008; 99: 524–530)
During the process of tumorigenesis, accumulation of genetic or epigenetic changes allows evasion of antiproliferative and cell death‐inducing mechanisms, which promotes transformation and clonal expansion of cells.( 1 ) The incipient ‘oncogenic’ stress, involving hypoxia, nutrient starvation, UV, ionizing radiation (IR), and chemicals, causes DNA damage in cells in which the mechanism for elimination of such hazardous cells is already compromised in the initial step of human multistep carcinogenesis.( 2 , 3 , 4 ) In normal cells, double‐strand and single‐strand DNA breaks lead to the activation of checkpoint responses and subsequent cell cycle arrest in the G1/S and G2/M phases, or to apoptosis. Two members of the phosphatidylinositol 3‐kinase‐related serine/threonine protein kinase family, ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3‐related (ATR), are key molecules in controlling the DNA‐damage checkpoint response.( 5 , 6 ) ATM responds very rapidly to low levels of DNA damage, leading to a conformational change that stimulates autophosphorylation,( 5 ) and ATR responds primarily to damage that causes bulky DNA adducts and stalled replication forks.( 6 ) Of the many downstream targets of ATM and ATR, the tumor suppressors Chk1 and Chk2 have been suggested to play important roles in the regulation of the G1/S and G2/M checkpoint responses, respectively.( 7 , 8 , 9 )
Although cancer‐associated CHK1 mutation is extremely rare,( 10 ) CHK2 mutations occur in a number of sporadic cancers and in a subset of cases of Li–Fraumeni syndrome.( 11 , 12 ) Chk2 is a serine/threonine protein kinase that phosphorylates a number of proteins involved in the DNA damage response, such as Cdc25C, Brca1, and p53, all of which can contribute to cell cycle arrest in the G2/M phase, apoptosis, and DNA repair.( 13 ) New insights into the biological role of Chk2 in multistep carcinogenesis have been demonstrated using clinical specimens from different stages of human tumors: phosphorylated (p) Chk2 expression was upregulated in precursor or precancerous lesions, whereas defects in the ATM–Chk2 pathway might allow cell proliferation, survival, genomic instability, and tumor progression.( 14 , 15 )
The fragile histidine triad (FHIT) gene spans the most active common chromosome fragile site in humans, FRA3B (3p14.2), which is often involved in biallelic loss, genomic rearrangement, and cytogenetic abnormalities in human malignancies.( 16 , 17 ) Deletions at the FHIT locus, abnormal FHIT transcripts, promoter hypermethylation, and the resultant loss of Fhit expression are detected frequently in most types of human cancer; therefore, Fhit is believed to be a tumor suppressor.( 18 , 19 , 20 , 21 ) Indeed, Fhit‐deficient mice exhibit increased susceptibility to spontaneous tumors and N‐nitrosomethylbenzylamine (NMBA)‐induced tumors,( 22 , 23 ) and restoration of the FHIT gene in human carcinoma cells suppresses cell growth and induces caspase‐dependent apoptosis in vitro and in vivo.( 24 , 25 , 26 ) In addition, deficiency of Fhit alleles increases resistance to DNA‐damaging stressors, such as mitomycin C (MMC) and UVC lights.( 27 ) Fhit mediation of the DNA‐damage checkpoint response has been proposed based on a variety of evidence: (1) Fhit‐deficient cells show stronger G1/S and G2/M checkpoint responses caused by the ATR–Chk1 pathway when the cells are exposed to IR;( 28 ) and (2) Fhit effectively increases expression of Hus1, a member of the Rad9–Rad1–Hus1 DNA damage‐response complex,( 29 ) which results in enhancement of Chk1 phosphorylation levels in primary human fibroblasts.( 30 ) However, little is known about the function of Fhit in the Chk2‐mediated checkpoint response in G2/M phase.
In the present study, we assessed the expression of pChk2 and Fhit in the process of human oral carcinogenesis involving dysplasia, oral squamous cell carcinoma (OSCC) in situ, and invasive OSCC to investigate the timing that appears to cause dysfunction of the G2/M checkpoint and loss of Fhit expression. The role of Fhit in the regulation of Chk2 phosphorylation was also investigated using an adenoviral gene‐delivery system.
Materials and Methods
Cell lines and tissue samples. Three HSC‐3 human OSCC and HEK293 human embryonic kidney cell lines were maintained in RPMI‐1640 and Dulbecco's modified Eagle's medium (DMEM), respectively supplemented with 10% fetal bovine serum. Irradiation of cells was done by exposing cells to X‐rays (150 kV, 40 mA, 2 mm Al filter). Cells were cultured at 37°C in an atmosphere of 5% CO2 and 95% air for 1 h after IR, and then collected for western blot and flow cytometric analysis. A total of 65 oral squamous epithelial lesions (10 normal epithelia, 25 dysplasias, five OSCC in situ, and 35 invasive OSCC) surgically removed at Kobe University Hospital (Kobe, Japan) from 2000 to 2006 were used. The patients did not have any presurgical chemotherapy or radiotherapy and informed consent was obtained from all patients. All of the specimens were fixed in 10% formalin and embedded in paraffin. Histological examination was carried out according to the General Rules for Clinical Studies on Head and Neck Cancer.( 31 ) Dysplastic and papilloma lesions developed in the forestomach of Fhit+/+ and Fhit−/– mice were kindly provided by Professor Kay Huebner (Ohio State University, Columbus, OH, USA). These mice were given intragastric doses of NMBA (Ash Stevens, Detroit, MI, USA), 2 mg/kg bodyweight, a total of six times over a 3‐week period. At autopsy 15 weeks after NMBA treatment, whole stomachs were removed and opened longitudinally.( 22 )
Immunohistochemistry. Immunohistochemistry was carried out using the streptavidin–biotin–peroxidase method with a LSAB kit (Dako, Carpinteria, CA, USA). Deparaffinized and rehydrated sections were autoclaved in a citrate buffer (pH 6.0) to retrieve antigenicity. After blocking endogenous peroxidase with 0.3% H2O2 and non‐specific binding sites with 3% bovine serum albumin (BSA), antisera against pATM (Cell Signaling, Beverly, MA, USA), pChk2 (Cell Signaling), Chk2 (Santa Cruz, Santa Cruz, CA, USA), Fhit,( 22 ) p53 (Santa Cruz), Ki‐67 (Dako), and pCdc25C (Cell Signaling) was applied to sections as the primary antibody. Subsequently, sections were incubated with biotinylated goat antimouse or antirabbit IgG and streptavidin conjugated to horseradish peroxidase (HRP). Chromogenic fixation was carried out by immersing the sections in a solution of 3,3‐diaminobenzidine tetrahydrochloride. The sections were then counterstained with Mayer's hematoxylin. Immunoreactivities of pChk2 and Chk2, Fhit, p53, and pCdc25C were graded according to the number of stained cells and the staining intensity in individual cells as follows: negative, almost no positive cells or <20% of tumor cells showed weak immunoreactivity; positive, >20% of tumor cells showed moderate or intense immunoreactivity. The Ki‐67 labeling index was determined by counting cells with intense immunoreactivity (%).
Microsatellite analysis. For DNA extraction, the tumor areas and corresponding normal oral mucosa from the deparaffinized specimens were scraped using a sterile needle and placed in a microtube containing extraction buffer and proteinase K. The microsatellite marker D3S1234 at the FHIT locus (FRA3B) was analyzed. The forward primers were fluorescein labeled with 6‐carboxy‐fluorescein (6‐FAM). Polymerase chain reaction products were electrophoresed in ABI PRISM 310 Genetic Analyzer along with GeneScan‐500 (ROX) molecular weight standard (Applied Biosystems, Foster City, CA, USA). The size of the polymerase chain reaction product was analyzed using GeneScan software (Applied Biosystems). Scoring of loss of heterozygosity (LOH)/allelic imbalance (AI) was carried out by manual inspection of the GeneScan output. A ratio of peak heights of alleles between normal and cancer DNA of 1.5 was used to define LOH/AI, as described elsewhere.( 32 , 33 )
Gene transduction with FHIT‐expressing adenoviruses. Adenoviruses carrying human recombinant wild‐type (Ad‐FHIT‐wt) and tyrosine 114 (Y114) mutant FHIT (Ad‐FHIT‐Y114F) were used.( 34 , 35 ) Briefly, wild‐type and Y114 mutant FHIT cDNA were cloned into the transfer vector pAdenoVator‐CMV5‐GFP (Qbiogene, Carlsbad, CA, USA). After recombination with the construct AdenoVator ΔE1/E3, the resulting vectors were transfected into HEK293 cells to package viruses. Single viral plaques were isolated and recombinant wild‐type and mutant Fhit expression was determined by western blot analysis. Green fluorescent protein (GFP)‐expressing adenovirus (Ad‐GFP) was used as a non‐specific control for gene transfer (Qbiogene). Virus titers were determined by absorbance measurement, multiplicity of infection (MOI) test, and the tissue culture inhibition dose (ID)50 method. Cells were incubated with adenoviral aliquots at a MOI of 25 for 4 h before the addition of culture medium (>25 × volume of virus inoculum).
Western blot analysis. Samples were extracted in cell lysis buffer containing 50 mM Tris‐HCl (pH 7.4), 125 mM NaCl, 0.1% Triton X‐100, 5 mM ethylenediaminetetraacetic acid, 1% protease inhibitor cocktail (Sigma, St Louis, MO, USA), and 1% phosphatase inhibitor cocktail (Sigma). Total protein (20 µg) was separated on polyacrylamide gels and transferred to Hybond C membrane (GE Healthcare, Piscataway, NJ, USA). Membranes were probed with antisera against Fhit,( 22 ) pChk2, Chk2, p53, pCdc25C, and β‐actin (Sigma). After probing with the appropriate secondary antirabbit or antimouse IgG conjugated to HRP (GE Healthcare), signals were detected using a chemiluminescence substrate.
Immunocytochemistry and flow cytometric analysis. HSC‐3 and HEK293 cells were grown on sterile chamber glass slides. Cells were washed with phosphate‐buffered saline (PBS) containing 0.1% NaN3 and then fixed with 4% paraformaldehyde. After blocking with 1% BSA–PBS, cells were incubated with anti‐pChk2 antibody, followed by incubation with Texas Red‐conjugated antimouse IgG antibody (Invitrogen, Carlsbad, CA, USA). The nuclei were stained with 4′,6‐diamino‐2‐phenylindole. The fluorescence images were obtained using an immunofluorescence microscope. To analyze cellular DNA content in the presence or absence of adenoviral gene delivery, cells were collected and fixed in 70% methanol, treated with RNase A, and stained with propidium iodide. The analysis was carried out with a FACS Caliber cytometer (BD Biosciences, San Jose, CA, USA). Experiments were repeated three times.
RNA interference analyses. Knockdown of Fhit expression by transfection of small interfering RNA (siRNA) was carried out. For the RNA interference analyses, human FHTI‐specific siRNA (siFHIT; sense, 5′‐GGA AGG CUG GAG ACU UUC ACA GGA AAG‐3′, and antisense, 5′‐UUC CUG UGA AAG UCU CCA GCC UUC CAU‐3′) was designed and synthesized according to a previous report,( 36 ) as well as control scrambled siRNA (siScramble). HSC‐3 cells seeded in 10‐cm dishes (1 × 106/dish) were transfected with each siRNA and Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Knockdown of Fhit expression was confirmed by western blot analysis.
Results
Loss of Fhit expression and disruption of the ATM–Chk2 pathway during oral carcinogenesis. To confirm activation of Chk2 in preneoplastic lesions and inactivation of Chk2 in advanced OSCC,( 14 , 15 ) we first explored the phosphorylation status of Chk2 and loss of Fhit expression in the development of OSCC (Fig. 1a,b). Chk2 expression was relatively retained in oral epithelial lesions throughout oral multistep tumorigenesis (normal, 100%; dysplasia, 78%; OSCC in situ, 76%; invasive OSCC, 68%), whereas positive immunoreactivity against pChk2 was elevated in dysplasia and OSCC in situ (normal, 0%; dysplasia, 48%; OSCC in situ, 60%) but fell when cells acquired invasive potential (invasive OSCC, 25%). Loss of Fhit expression occurred in accordance with tumor progression (normal, 0%; dysplasia, 28%; OSCC in situ, 58%; invasive OSCC, 63%), as well as abnormal nuclear accumulation of the p53 tumor suppressor (normal, 0%; dysplasia, 27%; OSCC in situ, 60%; invasive OSCC, 63%). In particular, all of the invasive OSCC cases demonstrated LOH and AI at the FHIT locus microsatellite marker D3S1234 (Fig. 1c).
Figure 1.
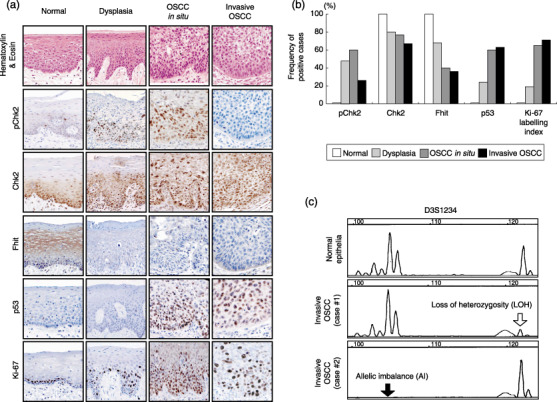
Disruption of the ataxia telangiectasia mutated (ATM)–Chk2 pathway in human oral epithelial lesions. (a) Representative illustrations of immunohistochemistry on the expression of phosphorylated (p) Chk2, Chk2, fragile histidine triad (Fhit), p53, and Ki‐67 in human squamous epithelial lesions (normal epithelia, dysplasia, oral squamous cell carcinoma [OSCC]in situ, and invasive OSCC) Original magnification, ×100. (b) Summary of immunohistochemical analyses. (c) Microsatellite analysis at D3S1234 of the FHIT locus. Loss of heterozygosity (white arrow) and allelic imbalance (black arrow) are shown.
Wild‐type and Y114F mutant FHIT enhance phosphorylation of Chk2 in response to IR. Mutations that compromise the ATM–Chk2 pathway are some of the most important events in human carcinogenesis.( 14 ) To examine disruption of this DNA damage‐response network in human OSCC cells, we next assessed the levels of pChk2 expression when cells were exposed to IR. In HEK293 cells, IR (10 Gy)‐induced upregulation of pChk2 expression was detected; however, in HSC‐3 cells, no induction of pATM and pChk2 was detected (Fig. 2A).
Figure 2.
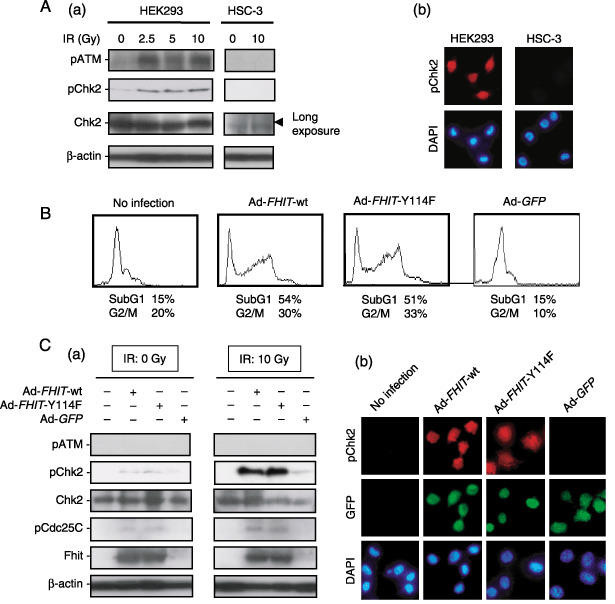
Restoration of the fragile histidine triad (FHIT) gene enhances the ataxia telangiectasia mutated (ATM)–Chk2 pathway in HSC‐3 human oral squamous cell carcinoma (OSCC) cells. (A) Ionizing radiation (IR)‐induced phosphorylated (p) ATM and pChk2 levels in HEK293 cells but not HSC‐3 cells. (a) Low levels of Chk2 expression in HSC‐3 cells were detected by long exposure. (b) Induction of pChk2 was confirmed in HEK293 human embryonic kidney cells by immunofluorescence. (B) Flow cytometric analysis of HSC‐3 cells infected with adenoviruses carrying wild‐type (Ad‐FHIT‐wt) and Y114 mutant FHIT (Ad‐FHIT‐Y114F) at multiplicity of infection (MOI) 25. Green fluorescent protein (GFP)‐expressing adenovirus (Ad‐GFP) was also used as a control. (C) The pChk2 levels were upregulated by Ad‐FHIT‐wt and Ad‐FHIT‐Y114F (MOI 25) in the presence or absence of IR (10 Gy). (a) Infection with Ad‐FHIT‐wt and Ad‐FHIT‐Y114F restored pChk2 and pCdc25C levels, particularly when the cells were exposed to IR (10 Gy). (b) Immunofluorescence of pChk2 expression when HSC‐3 cells were exposed to IR (10 Gy).
Ishii et al. have shown that stabilization of the Hus1 protein by Fhit results in upregulation of pChk1 levels in human primary fibroblasts, although this Fhit‐mediated regulation of checkpoint proteins is disrupted in carcinoma cells.( 30 ) We therefore examined the role of Fhit in the regulation of Chk2 phosphorylation status. An experiment involving adenoviral gene delivery was conducted to determine whether restoration of the FHIT gene could alter pChk2 levels in HSC‐3 (Fhit‐negative) OSCC cells. We previously demonstrated that substitution of Y114 with phenylalanine diminished the tumor suppressor activity of Fhit in H1299 and A549 human non‐small‐cell lung carcinoma cells; downregulation of the Akt–survivin pathway was induced by wild‐type Fhit but not by Y114 mutant Fhit proteins, suggesting that Y114 is essential for Fhit function.( 34 ) However, in HSC‐3 cells, both Ad‐FHIT‐wt and Ad‐FHIT‐Y114F increased the cell population with subG1 DNA content that were in G2/M phase (Fig. 2B). Although restoration of the FHIT gene and exposure to IR did not increase pATM expression levels, we found that Ad‐FHIT‐wt and Ad‐FHIT‐Y114F weakly upregulated levels of pChk2 expression, as well as that of pCdc25C, and that Fhit‐mediated upregulation of pChk2 levels was enhanced dramatically by exposure to IR (Fig. 2C). As reported previously,( 27 , 28 ) we also confirmed enhancement of pChk1 levels by restoration of the FHIT gene in HSC‐3 cells when cells were exposed to IR (data not shown).
Knockdown of Fhit expression diminishes phosphorylation of Chk2 in vitro and in vivo. Conversely, in order to evaluate the effect of Fhit on pChk2 levels, knockdown of Fhit expression with siRNA was carried out in human embryonic kidney HEK293 cells. Effective depletion of endogenous Fhit by siFHIT but not siScramble was confirmed 48 h after transfection (Fig. 3A). We therefore treated the cells with exposure to IR 48 h after siRNA transfection. HEK293 cells exhibited induction of pChk2 by exposure to IR in both the time‐course and dose‐dependent experiments (Fig. 3B, upper panels). Interestingly, knockdown of Fhit expression by siFHIT transfection effectively inhibited IR‐induced Chk2 phosphorylation (Fig. 3B, middle panels), which was not suppressed by siScramble transfection (Fig. 3B, lower panels). Transient depletion of endogenous Fhit did not alter the levels of Chk2 expression in these experiments.
Figure 3.
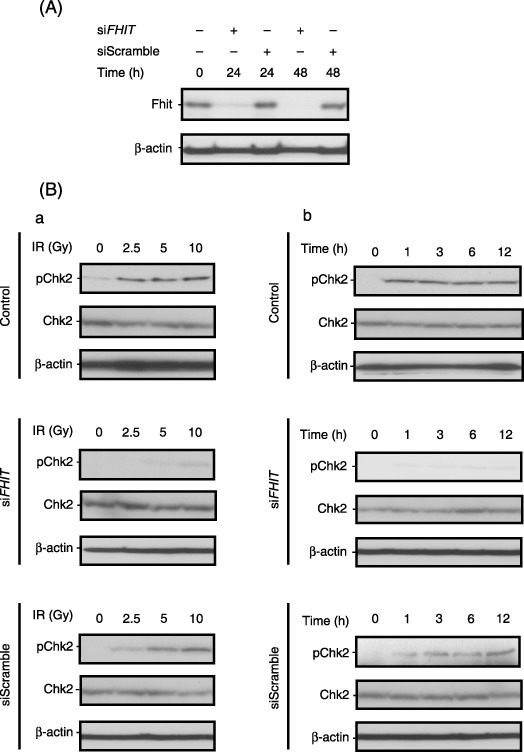
Depletion of fragile histidine triad (Fhit) by small interfering RNA (siRNA) downregulates Chk2 phosphorylation in response to ionizing radiation (IR) in HEK293 cells. (A) Knockdown of Fhit expression by transfection of FHIT‐specific siRNA (siFHIT). (B) Depletion of Fhit expression decreased pChk2 levels in response to IR. Forty‐eight hours after siRNA transfection (10 nM), cells were exposed to IR at a different condition: (a) time course (0–12 h) and (b) dose‐dependent (0–10 Gy) analyses were conducted.
Homozygous deletion of Fhit alleles increased susceptibility to the development of NMBA‐induced tumors in the mouse forestomach,( 22 ) the frequency of papillomas in the Fhit−/– forestomach (12 of 15 [80%] cases), and was significantly higher than that in the Fhit+/+ forestomach (five of 15 [33%] cases).( 35 ) Finally, we assessed the possibility that loss of Fhit alleles may diminish the Chk2‐mediated DNA‐damage response in vivo, and thereby cause the differences in tumor susceptibility between Fhit−/– and Fhit+/+ mice. In Fhit+/+ mice, treatment with NMBA increased the number of pChk2‐ and pCdc25C‐positive cells not only in the non‐neoplastic epithelia but also in NMBA‐induced papilloma lesions; nevertheless, induction of neither pChk2 nor pCdc25C was detected in Fhit−/– mice (Fig. 4).
Figure 4.
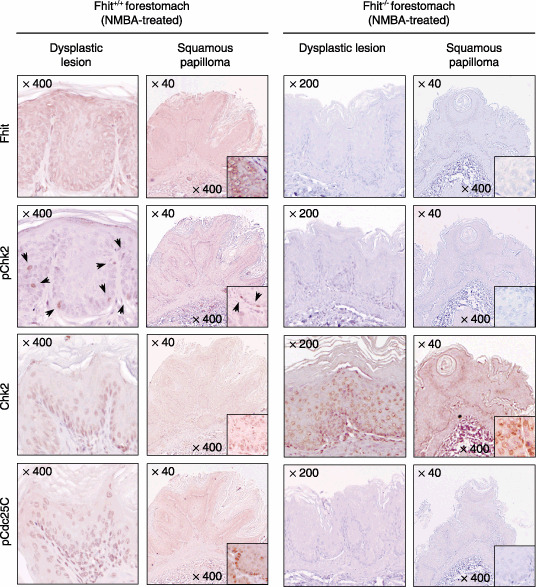
Deficiency of fragile histidine triad (Fhit) decreases phosphorylated (p) Chk2‐positive cells in N‐nitrosomethyl benzylamine (NMBA)‐induced murine forestomach tumors. Immunohistochemistry was carried out with antibodies against Fhit, pChk2, Chk2, and pCdc25C. Arrow heads indicate the pChk2‐positive cells.
Discussion
Fragile histidine triad is a member of the histidine triad nucleotide‐binding protein superfamily, and encodes a diadenosine polyphosphate hydrolase.( 16 , 17 ). However, Fhit hydrolase activity is not required for its tumor‐suppressing activity because Fhit‐H96N, a hydrolase ‘dead’ mutant in which histidine 96 is substituted with asparagine, also suppresses tumorigenicity in vitro and in vivo.( 36 ) Instead, the Src‐family tyrosine kinase phosphorylation target Y114 is necessary for inhibiting the Akt–survivin pathway and the resultant induction of caspase‐dependent cell death.( 34 ) However, as shown in the present study, Fhit‐Y114F also has tumor‐suppressing activity in HSC‐3 cells with restoration of the Chk2‐mediated checkpoint response against IR. A large fraction of Fhit‐deficient human cancer cells and murine kidney cells survived treatment with MMC and UVC light compared to matched Fhit‐positive cells.( 27 ) A possible role of Fhit at the G1/S checkpoint has been proposed in the regulation of Chk1 activity;( 27 , 28 , 30 ) however, little is known about Fhit function at the G2/M checkpoint.
In the present study, we showed disruption of the Chk2‐mediated G2/M checkpoint pathway in HSC‐3 cells, which was restored by both wild‐type and Y114F mutant Fhit expression. Furthermore, we also detected disruption of the ATM–Chk2 checkpoint pathway in HSC‐3 cells: no induction of pATM and pChk2 was detected in HSC‐3 cells even when cells were exposed to IR. Combined with the results of our immunohistochemical analyses, these findings suggest that downregulation of Chk2‐modulated DNA damage response by loss of Fhit is one of the most important events in oral carcinogenesis, and that Y114 is not necessary for Fhit‐mediated Chk2 activation. Interestingly, restoration of the FHIT gene with Ad‐FHIT‐wt and Ad‐FHIT‐Y114F dramatically enhanced pChk2 levels in response to IR, suggesting that Fhit's ability for modulation of Chk2 phosphorylation might be strengthened when cells have DNA damage without pATM. In addition, homozygous deletion of Fhit alleles caused the difference in tumor susceptibility between Fhit−/– and Fhit+/+ mice with a significant difference in the number of pChk2‐ and pCdc25C‐positive cells, suggesting that lack of Fhit expression and the resultant defects of the ATM–Chk2 pathway cause the difference in the incidence of NMBA‐induced forestomach lesions.( 22 , 35 ) Recently, the biological significance of phosphorylated Fhit at Y114 has been reported as a proteasome‐mediated degradation form; the subsequent reduction in Fhit protein levels allows transmission of the Src‐induced mitogenic signal.( 37 ) A better understanding of the impact of Fhit function on the control of Chk2 activity awaits further study.
The present study was conducted based on the hypothesis that double‐strand DNA breakage caused by DNA replication stress or IR is an initial event in oral carcinogenesis, that is, breakage at the FHIT locus, the most common fragile site of the human genome FRA3B, and the resultant loss of Fhit expression may trigger subsequent downregulation of Chk2 activity, genomic instability, and selective pressure for p53 mutations.( 14 , 15 ) Conversely, Durkin et al. investigated the effects of the depletion of the cell cycle checkpoint kinases Chk1 and Chk2 on common fragile site stability including that of the FHIT locus, and found that deficiency of Chk1, but not Chk2, accelerates destabilization of chromosomes and specific common fragile sites.( 38 ) These findings raise the issue of the correlation between Chk2 and Fhit in human carcinogenesis. Indeed, they demonstrate that depletion of Chk2 using siRNA methods did not increase genetic alterations at common fragile sites. Therefore, further investigation will be required to prove our hypothesis that genetic or epigenetic alteration at the FHIT locus and resultant loss of Fhit expression might be an initial event during oral carcinogenesis, subsequently inducing breakage of the DNA checkpoint mechanism.
According to the results in the present study, we propose a model for Fhit‐mediated oral carcinogenesis (Fig. 5). The initial DNA damage can be a trigger for genetic alterations at the FHIT locus and resultant loss of Fhit expression. Also, the occurrence of a DNA‐damage response can be ascertained by monitoring pChk2. However, because loss of the Fhit protein decreases pChk2 levels, consequent Chk2 inactivation accelerates genetic instability and subsequent genetic alterations in oncogenes and tumor suppressor genes (e.g. p53); activation of oncogenes and inactivation of the tumor suppressor genes may contribute to the subsequent progression and development of OSCC cells. Understanding the relationship between Fhit and pChk2 might provide useful information not only for early detection of premalignant lesions of the oral cavity but also for the assessment of patients with a high risk of recurrent OSCC and the development of new strategies for radiotherapy and chemotherapy.
Figure 5.
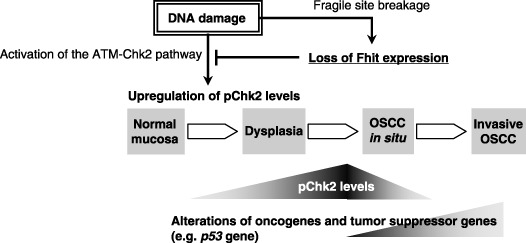
Proposed model for downregulation of the ataxia telangiectasia mutated (ATM)–Chk2 pathway by loss of fragile histidine triad (Fhit) during oral carcinogenesis. DNA damage increases the incidence of loss of Fhit expression and loss of heterozygosity and allelic imbalance at the FHIT locus, simultaneously upregulating phosphorylated (p) Chk2 levels in the initial step of carcinogenesis. Loss of Fhit expression decreases pChk2 levels, resulting in a defect of the ATM–Chk2 DNA‐response mechanism and accumulation of genetic alterations in oncogenes and tumor suppressor genes in the development and progression of oral squamous cell carcinoma (OSCC).
Acknowledgments
This work was supported in part by a Grant‐in‐Aid for Scientific Research (C‐19590347) from the Japan Society for Promotion of Science, and by the Terry Fox Run Foundation for Cancer Research.
References
- 1. Lowe SW, Cepero E, Evan G. Intrinsic tumor suppression. Nature 2004; 432: 307–15. [DOI] [PubMed] [Google Scholar]
- 2. Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature 1998; 396: 643–9. [DOI] [PubMed] [Google Scholar]
- 3. Graeber TG, Peterson JF, Tsai M, Monica K, Fornace AJ Jr, Giaccia AJ. Hypoxia induces accumulation of p53 protein, but activation of a G1‐phase checkpoint by low‐oxygen conditions is independent of p53 status. Mol Cell Biol 1994; 14: 6264–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Routhier EL, Donover PS, Prendergast GC. hob1+, the fission yeast homolog of Bin1, is dispensable for endocytosis or actin organization, but required for the response to starvation or genotoxic stress. Oncogene 2003; 22: 637–48. [DOI] [PubMed] [Google Scholar]
- 5. Hammond EM, Dorie MJ, Giaccia AJ. ATR/ATM targets are phosphorylated by ATR in response to hypoxia and ATM in response to reoxygenation. J Biol Chem 2003; 278: 12 207–13. [DOI] [PubMed] [Google Scholar]
- 6. Cliby WA, Roberts CJ, Cimprich KA et al . Overexpression of a kinase‐inactive ATR protein causes sensitivity to DNA‐damaging agents and defects in cell cycle checkpoints. EMBO J 1998; 17: 159–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ahn JY, Schwarz JK, Piwnica‐Wrms H, Canman CE. Threonine 68 phosphorylation by ataxia telangiectasia mutated is required for efficient activation of Chk2 in response to ionizing radiation. Cancer Res 2000; 60: 5934–6. [PubMed] [Google Scholar]
- 8. Matsuoka S, Huang M, Elledge SJ. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 1998; 282: 1893–7. [DOI] [PubMed] [Google Scholar]
- 9. Helt CE, Cliby WA, Keng PC, Bambara RA, O’Reilly MA. Ataxia telangiectasia mutated (ATM) and ATM and Rad3‐related protein exhibit selective target specificities in response to different forms of DNA damage. J Biol Chem 2005; 280: 1186–92. [DOI] [PubMed] [Google Scholar]
- 10. Menoyo A, Alazzouzi H, Espin E, Armengol M, Yamamoto H, Schwartz S Jr. Somatic mutations in the DNA damage‐response genes ATR and CHK1 in sporadic stomach tumors with microsatellite instability. Cancer Res 2001; 61: 7727–30. [PubMed] [Google Scholar]
- 11. Matsuoka S, Nakagawa T, Matsuda A, Haruki N, Elledge SJ, Takahashi T. Reduced expression and impaired kinase activity of a Chk2 mutant identified in human lung cancer. Cancer Res 2001; 61: 5362–5. [PubMed] [Google Scholar]
- 12. Staalesen V, Falck J, Geisler S et al . Alternative splicing and mutation status of CHEK2 in stage III breast cancer. Oncogene 2004; 23: 8535–44. [DOI] [PubMed] [Google Scholar]
- 13. Peng CY, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica‐Worms H. Mitotic and G2 checkpoint control: regulation of 14‐3‐3 protein binding by phosphorylation of Cdc25C on serine‐216. Science 1997; 277: 1501–5. [DOI] [PubMed] [Google Scholar]
- 14. Bartkova J, Horejsi Z, Koed K et al . DNA damage response as a candidate anti‐cancer barrier in early human tumorigenesis. Nature 2005; 434: 864–70. [DOI] [PubMed] [Google Scholar]
- 15. Gorgoulis VG, Vassiliou LV, Karakaidos P et al . Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005; 434: 907–13. [DOI] [PubMed] [Google Scholar]
- 16. Ohta M, Inoue H, Cotticelli MG et al . The FHIT gene, spanning the chromosome 3p14.2 fragile site and renal carcinoma‐associated t(3;8) breakpoint, is abnormal in digestive tract cancers. Cell 1996; 84: 587–97. [DOI] [PubMed] [Google Scholar]
- 17. Huebner K, Garrison PN, Barnes LD, Croce CM. The role of the FHIT/FRA3B locus in cancer. Annu Rev Genet 1998; 32: 7–31. [DOI] [PubMed] [Google Scholar]
- 18. Croce CM, Sozzi G, Huebner K. Role of FHIT in human cancer. J Clin Oncol 1999; 17: 1618–24. [DOI] [PubMed] [Google Scholar]
- 19. Huebner K, Croce CM. FRA3B and other common fragile sites: the weakest links. Nat Rev Cancer 2001; 1: 214–21. [DOI] [PubMed] [Google Scholar]
- 20. Ishii H, Zanesi N, Vecchione A et al . Regression of upper gastric cancer in mice by FHIT gene delivery. FASEB J 2003; 17: 1768–70. [DOI] [PubMed] [Google Scholar]
- 21. Kuroki T, Trapasso F, Yendamuri S et al . Allele loss and promoter hypermethylation of VHL, RAR‐β, RASSF1A, and FHIT tumor suppressor genes on chromosome 3p in esophageal squamous cell carcinoma. Cancer Res 2003; 63: 3724–8. [PubMed] [Google Scholar]
- 22. Fong LY, Fidanza V, Zanesi N et al . Muir‐Torre‐like syndrome in Fhit‐deficient mice. Proc Natl Acad Sci USA 2000; 97: 4742–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zanesi N, Fidanza V, Fong LY et al . The tumor spectrum in FHIT‐deficient mice. Proc Natl Acad Sci USA 2001; 98: 10 250–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dumon KR, Ishii H, Vecchione A et al . Fragile histidine triad expression delays tumor development and induces apoptosis in human pancreatic cancer. Cancer Res 2001; 61: 4827–36. [PubMed] [Google Scholar]
- 25. Ishii H, Dumon KR, Vecchione A et al . Effect of adenoviral transduction of the fragile histidine triad gene into esophageal cancer cells. Cancer Res 2001; 61: 1578–84. [PubMed] [Google Scholar]
- 26. Roz L, Gramegna M, Ishii H, Croce CM, Sozzi G. Restoration of fragile histidine triad (FHIT) expression induces apoptosis and suppresses tumorigenicity in lung and cervical cancer cell lines. Proc Natl Acad Sci USA 2002; 99: 3615–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ottey M, Han SY, Druck T et al . Fhit‐deficient normal and cancer cells are mitomycin C and UVC resistant. Br J Cancer 2004; 91: 1669–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hu B, Han S‐Y, Wang X et al . Involvement of the Fhit gene in the ionizing radiation‐activated ATR/CHK1 pathway. J Cell Physiol 2005; 202: 518–23. [DOI] [PubMed] [Google Scholar]
- 29. Sancar A, Lindsey‐Boltz LA, Unsal‐Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 2004; 73: 39–85. [DOI] [PubMed] [Google Scholar]
- 30. Ishii H, Mimori K, Inoue H et al . Fhit modulates the DNA damage checkpoint response. Cancer Res, 2006; 66: 11 287–92. [DOI] [PubMed] [Google Scholar]
- 31. Japan Society for Head and Neck Cancer . The General Rules for Clinical Studies on Head and Neck Cancer, 4th edn. Tokyo: Kanehara, 2005. [Google Scholar]
- 32. Marsh DJ, Zheng Z, Zedenius J et al . Differential loss of heterozygosity in the region of the Cowden locus within 10q22–23 in follicular thyroid adenomas and carcinomas. Cancer Res 1997; 57: 500–3. [PubMed] [Google Scholar]
- 33. Dacic S, Ionescu DN, Finkelstein S, Yousem SA. Patterns of allelic loss of synchronous adenocarcinomas of the lung. Am J Surg Pathol 2005; 29: 897–902. [DOI] [PubMed] [Google Scholar]
- 34. Semba S, Trapasso F, Fabbri M et al . Fhit modulation of the Akt–survivin pathway in lung cancer cells: Fhit‐tyrosine 114 (Y114) is essential. Oncogene 2006; 25: 2860–72. [DOI] [PubMed] [Google Scholar]
- 35. Semba S, Han S‐Y, Qin HR et al . Biological functions of mammalian Nit1, the counterpart of the invertebrate NitFhit Rosetta stone protein, a possible tumor suppressor. J Biol Chem 2006; 281: 28 244–54. [DOI] [PubMed] [Google Scholar]
- 36. Siprashvili Z, Sozzi G, Barnes LD et al . Replacement of Fhit in cancer cells suppresses tumorigenicity. Proc Natl Acad Sci USA 1997; 94: 13 771–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bianchi F, Magnifico A, Olgiati CE et al . FHIT‐proteasome degradation caused by mitogenic stimulation of the EGF receptor family in cancer cells. Proc Natl Acad Sci USA 2006; 103: 18 981–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Durkin SG, Arlt MF, Howlett NG, Glover TW. Depletion of CHK1, but not CHK2, induces chromosomal instability and breaks at common fragile sites. Oncogene 2006; 25: 4381–8. [DOI] [PubMed] [Google Scholar]