

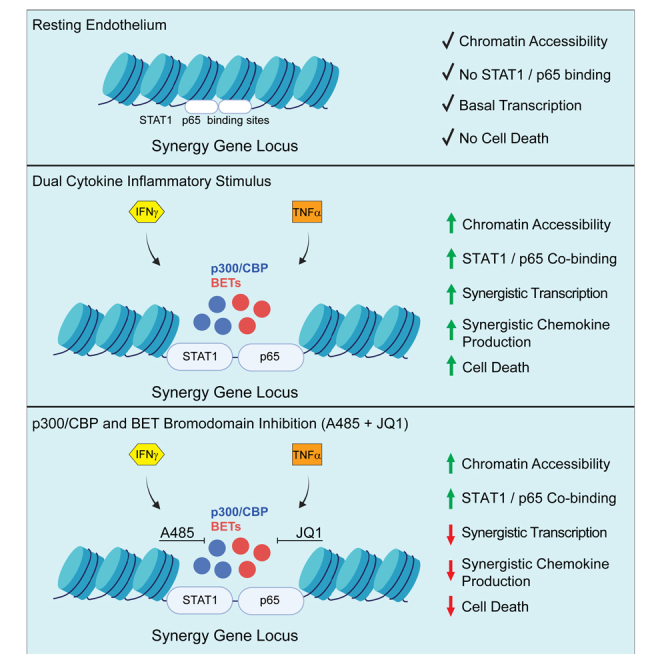
Summary
Combinatorial signaling by proinflammatory cytokines synergizes to exacerbate toxicity to cells and tissue injury during acute infections. To explore synergism at the gene-regulatory level, we investigated the dynamics of transcription and chromatin signaling in response to dual cytokines by integrating nascent RNA imaging mass spectrometry, RNA sequencing, amplification-independent mRNA quantification, assay for transposase-accessible chromatin using sequencing (ATAC-seq), and transcription factor profiling. Costimulation with interferon-gamma (IFNγ) and tumor necrosis factor alpha (TNFα) synergistically induced a small subset of genes, including the chemokines CXCL9, -10, and -11. Gene induction coincided with increased chromatin accessibility at non-coding regions enriched for p65 and STAT1 binding sites. To discover coactivator dependencies, we conducted a targeted chemogenomic screen of transcriptional inhibitors followed by modeling of inhibitor dose-response curves. These results identified high efficacy of either p300/CREB-binding protein (CBP) or bromodomain and extra-terminal (BET) bromodomain inhibitors to disrupt induction of synergy genes. Combination p300/CBP and BET bromodomain inhibition at half-maximal inhibitory concentrations (subIC50) synergistically abrogated IFNγ/TNFα-induced chemokine gene and protein levels.
Subject areas: Components of the immune system, Cell biology, Systems biology, Genomics, Transcriptomics, Chemogenomics
Graphical abstract
Highlights
-
•
IFNγ/TNFα synergistically induce a subset of proinflammatory transcripts
-
•
STAT1/p65 co-bind at enhanced accessible elements in response to IFNγ/TNFα
-
•
Synergistic transcription is vulnerable to combined CBP/p300 and BET inhibition
-
•
Open chromatin and STAT1/p65 binding are unaffected by CBP/p300 and BET inhibition
Components of the immune system; Cell biology; Systems biology; Genomics; Transcriptomics; Chemogenomics
Introduction
Acute inflammation triggered by infectious microorganisms is a tightly regulated process essential to ensure human health. As part of this response, an array of cytokines released by the innate and adaptive immune system mobilize host cellular defenses with the goal of clearing invading pathogens and resolving tissue damage. However, in a subset of infections including severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), cytokine storm develops featuring massive release of proinflammatory cytokines that promote progressive cell death, organ damage, and heightened mortality in humans.1 In the particular case of severe SARS-CoV-2 illness, high circulating levels of interferon-gamma (IFNγ) and tumor necrosis factor alpha (TNFα) predominate.2 At a cellular level, the combination of IFNγ and TNFα signaling causes PANoptosis, which if unchecked over time ultimately causes shock and death at the organism level.2
Gene regulation is one important facet of control in cytokine responses. Signaling elicited by IFNγ and TNFα promotes rapid transcription of proinflammatory genes by activating two master transcription factors (TFs): signal transducer and activator of transcription-1 (STAT1) and p65/nuclear factor-kappa B (NF-κB), respectively. In the absence of inflammatory stimuli, STAT1 and p65 reside as inactive proteins in the cytosol.3,4 IFNγ or TNFα signal transduction cascades prompt phosphorylation and rapid translocation of each TF to the nucleus where they bind unique sequence-specific regulatory DNA elements, assemble coregulatory complexes including mediator and histone remodeling factors, and alter chromatin conformation, thereby driving RNA polymerase II (Pol II)-dependent transcription of inflammatory genes.5
Transcriptional synergy—when induction of a gene by two or more stimuli is greater than the additive effects on expression induced by each stimulus—is a special case in gene control.6 Synergy has been described for multiple combinations of cytokines and extracellular signals including IFNγ/TNFα,2,7,8,9 interleukin-4/lipopolysaccharide,10 and IFNβ1/IFNγ.11 As a general property, synergy can occur in many cell types, though the specific gene expression programs induced may differ depending on cell lineage, developmental stage, tissue context, and cytokine combinations.2,7,10,12,13,14 At least two non-mutually exclusive models have been proposed to explain how synergy emerges: 1) TF cooperativity, in which DNA binding affinity of one TF is enhanced by local occupancy of another TF, leading to non-linear output of gene expression as a consequence of the combined TF action to recruit Pol II,6 or 2) kinetic transcription in which individual TFs stimulate distinct steps in the Pol II transcriptional cycle (i.e., initiation or elongation), which augments overall Pol II processivity.15,16
In addition to intrinsic transactivation functions, both STAT1- and p65-dependent transcription are potentiated via a dynamic interplay with chromatin-associated coactivators. The p300/CREB-binding protein (CBP) family of histone acetyltransferases physically interact with the transactivation domain (TAD) of STAT1 via their TAZ2 domains, increasing STAT1 acetylation in the TAD and thereby increasing DNA binding and transactivation activity.17,18 p300/CBP also associates with p65 via its Rel homology and C-terminal TADs.19,20 This interaction promotes reversible acetylation of p65 and histones locally, which increases transcriptional output.21 Specific acetylation of p65 on lysine residue 310 recruits BRD4, a member of the BET bromodomain-containing protein family via its bromodomain, an acetyl-lysine reader motif.22 Once recruited, BRD4 forms super enhancers (SEs) that drive dynamic increases in transcription of proinflammatory genes through recruitment of positive elongation factor-b (PTEF-b), which along with BRD4 phosphorylates Pol II on its C-terminal domain and releases Pol II from promoter-proximal pausing.23,24,25 Altering IFNγ- or TNFα-dependent transcriptional activation in certain types of cancers and SARS-CoV-2 through inhibition of p300/CBP and BET bromodomain proteins may also have clinical significance.26,27,28,29 Thus, chromatin-dependent signaling plays critical roles in communicating many of the transcriptional functions of STAT1 and p65 to Pol II. However, comparatively less is known about how chromatin coactivators contribute to synergism of gene expression and the associated cellular phenotypes induced by IFNγ and TNFα.
Vascular endothelium—the single-cell thick layer lining blood vessels—plays a critical role in cardiovascular health. In addition to homeostatic control of systemic blood pressure,30 thrombosis,31 and metabolism,32 endothelial cells (ECs) also serve as innate immune-like cells that transduce proinflammatory cytokine signals including IFNγ and TNFα, present antigen through major histocompatibility complexes, and regulate leukocyte trafficking to sites of injury or infection.33,34,35,36 As such, ECs control the tempo, magnitude, and propagation of inflammation locally and systemically. In this study we investigated transcriptional synergy and its dependencies during costimulation of human aortic ECs (HAECs) with IFNγ/TNFα. By integrating stable isotope quantification of nascent RNA, RNA sequencing (RNA-seq), and non-amplified mRNA analyses, we identified a small subset of proinflammatory genes including the chemokines CXCL9, -10, and -11 that are synergistically induced by IFNγ/TNFα in 1 h. This synergism occurred even at non-saturating levels of cytokines. Prolonged IFNγ/TNFα costimulation increased chemokine protein levels and cell death. At a chromatin level, this resulted in formation of increased sites of accessibility in non-coding regions of synergistically induced genes and co-binding of STAT1 and p65. Cotreatment of HAECs with low-dose inhibitors targeting p300/CBP and BET bromodomain proteins synergistically abrogated chemokine gene induction and protein production. Thus, we demonstrate that the rapid induction of synergistic transcription by IFNγ/TNFα mediated by STAT1 and p65 is especially vulnerable to combinatorial disruption of chromatin-dependent signaling.
Results
IFNγ/TNFα costimulation of HAECs induces nascent RNA production and synergistic expression of proinflammatory chemokine genes
We first examined the functional consequences of combined IFNγ/TNFα signaling on global RNA regulation by utilizing multi-isotope imaging mass spectrometry (MIMS). MIMS leverages nanoscale secondary ion mass spectrometry to measure isotope ratios at suborganelle resolution Figure 1C). Incorporation of a stable isotope tracer is quantitatively captured by an increase in the isotopic ratio above the natural background ratio.37 We have previously used thymidine tracers to measure DNA synthesis and cell division.38,39 Here, we used 13C-thymidine to saturate DNA labeling in proliferating HAECs (label 14 days) and map intranuclear DNA architecture, followed by 15N-uridine pulse labeling concurrent with cytokine stimulation to capture changes in global nascent RNA synthesis (Figure 1B). Costimulation of HAECs with IFNγ/TNFα augmented 15N-uridine labeling that was detectable in the nucleus at 1 h and in the cytoplasm at 1 and 4 h. This effect was not observed in HAEC exposed to each individual cytokine stimulus (Figures 1D, S1A, and S1B). These data identify that the dynamic cellular response to INFγ/TNFα costimulation involves a rapid surge in global RNA synthesis, suggestive of a distinct regulatory interaction between IFNγ and TNFα at the level of gene expression.
Figure 1.

IFNγ/TNFα costimulation of HAECs induces nascent RNA production and synergistic expression of proinflammatory chemokine genes
(A) Schematic depicting treatment groups and experimental time points in HAECs.
(B) Schematic of experimental approach for MIMS experiments.
(C) MIMS images of HAEC stimulated with IFNγ/TNFα for 1 (top) or 4 h (bottom) versus unstimulated cells. 32S images reveal cellular and nuclear contours (left column). Hue saturation intensity images are used to visually represent isotope ratio measurements and in turn map 13C-thymidine labeling of DNA (13C/12C, middle) and 15N-uridine labeling of nascent RNA (15N/14N, right). The lower bound of the color scale (blue) is set to natural background (13C = 1.1%; 15N = 0.37%) and the upper bound of the scale is set to reveal differential labeling expressed as % above natural background. Scale bars = 5 μm. See also Figure S1B.
(D) Nuclear (top) and cytoplasmic (bottom) 15N-uridine labeling was quantified at the 1 and 4-h timepoints after stimulus and expressed as % above natural background. Each dot represents a different cell/nucleus with mean ± SD. One-way ANOVA, Dunnett’s test used for 4-h time point; Kruskal-Wallis/Dunn’s nonparametric test used for 1-h time point. ∗p < 0.05 ∗∗p < 0.01. Yellow dots correspond to the cells shown in the images in Figure 1C.
(E) Heatmap of averaged differential gene expression in unstimulated HAECs and HAECs treated with IFNγ (50 ng/mL), TNFα (25 ng/mL) or IFNγ/TNFα (n = 4 per condition).
(F) Scatterplot of difference in expression calculated by log2(FC) of IFNγ/TNFα (i.e., costimulated) minus the sum of the log2FC (IFNγ) plus log2FC (TNFα) (n = 4 per condition). Color indicates outlier status. All log2FC were calculated with cytokine vs. unstimulated cells.
(G) Scatterplot of ranked log2(FC) difference between IFNγ/TNFα minus the sum of the FC observed in IFNγ plus FC observed in TNFα. Green color indicates gene expression greater in IFNγ/TNFα, blue color indicates gene expression is less in IFNγ/TNFα than the sum of the FC observed in IFNγ plus FC in TNFα (n = 4 per condition).
Despite the quantitative and spatial power of MIMS to measure nascent RNA in cell compartments, it cannot resolve gene-level 15N-uridine signal needed to determine changes of individual transcripts in the nucleus. As such, we next determined the differential gene expression patterns in HAECs after 1 h stimulation with maximal concentrations of IFNγ, TNFα, or both using unbiased RNA-seq. We chose 1 h to focus on primary transcriptional effects and thereby avoid the secondary waves of gene regulation that arise during more prolonged cytokine stimulation. Differential gene expression analysis using pairwise comparisons of each cytokine vs. unstimulated cells identified the induction of 43 genes with IFNγ, 218 genes with TNFα, and 277 genes with IFNγ/TNFα, using a threshold of 2-fold change and adjusted p value <0.05 (Figures S1C–S1E). The synergy conditioned overlapped almost completely with TNFα stimulation at this time point. To investigate synergy-specific gene targets, we next examined the pattern of changes in expression using all genes upregulated by any cytokine by at least 2-fold. We row-normalized the data for each gene and compared across each treatment group (Figure 1E). This analysis revealed a group of 72 genes that were upregulated in the synergy treatment specifically, as compared to IFNγ or TNFα alone (Figure 1E). To determine whether the induction of these genes was different from the individual cytokine stimulations, we performed an outlier analysis. We compared the difference between the measured fold change in IFNγ/TNFα vs. unstimulated cells and the sum of the fold change for IFNγ vs. unstimulated plus TNFα vs. unstimulated cells (Figure 1F, top). This scatterplot revealed a small group of genes with very large differences, indicating fold changes for synergy conditions that far exceeded the predicted fold changes if the regulation was purely linear between the cytokine treatment groups. By graphing the gene density distribution, it was clear overall the fold changes followed a Gaussian (i.e., Normal) distribution with the vast majority of genes displaying no differences between cytokine groups and hence a difference score near zero (Figure 1F, bottom). This observation enabled us to identify genes whose fold changes clearly fell outside this normal distribution, thereby representing statistical outliers. Notably, we observed outliers in both positive (green) and negative (purple) directions, indicating expression of certain genes changed less than additively compared to each cytokine alone (purple dots), while the magnitude of change in expression of other genes was clearly much greater than predicted compared to individual cytokine results (green dots, n = 65). Formal ranking of these difference scores identified that the chemokines CXCL9, -10, and -11 were among the most extreme outliers in terms of positive gene induction/fold change (Figure 1G). Based on these outlier analyses, we concluded that these genes represented a distinct group that was induced synergistically by dual cytokines.
Transcriptional synergy elicited by IFNγ/TNFα signaling requires STAT1 and p65
IFNγ and TNFα signal transduction pathways are known to activate STAT1 and p65/NF-κB, respectively. Crosstalk between IFNγ and TNFα has also been reported between these two pathways.40 Thus, we first examined the impact of individual cytokine stimulation on STAT1 or p65 nuclear translocation—a key step in the activation of these TFs. Subcellular localization of each TF was tracked over time in response to cytokine stimulation using immunofluorescence. IFNγ or TNFα each stimulated movement of STAT1 or p65, respectively, into the nucleus within 15 min (Figures 2A and 2B). This shift persisted over 1 and 4 h (Figures S2A and S2B). Costimulation with IFNγ/TNFα had similar effects on STAT1 and p65 translocation as compared to individual cytokines.
Figure 2.

Transcriptional synergy elicited by IFNγ/TNFα signaling requires STAT1 and p65
(A) Representative photomicrographs of STAT1 (green) and p65 (red) immunofluorescence in TeloHAECs treated with vehicle or combined IFNγ/TNFα for 15 min. Nuclei are indicated by DAPI staining (blue color). All samples were imaged at 10X magnification on Zeiss confocal microscope. Scale bar = 100 μm.
(B) Boxplots for quantification of images in (A). Kruskal-Wallis test was performed followed by a pairwise Wilcoxon test to determine statistical differences between treatment groups. Box indicates first quartile (top), median (middle, orange), and third quartile (bottom), whiskers indicate minium and maxium values in the range. ∗∗∗p value <0.001; ∗∗∗∗p-value <0.0001.
(C and D) Western blots of STAT1 and p65 in STAT1 KO (C) or RELA KO (D). ACTB is a loading control.
(E–G) Bar graphs of CXCL9 (E), CXCL10 (F), and CXCL11 (G) expression in STAT1 KO HAECs unstimulated or treated with IFNγ/TNFα. Data shown are mean ± SD (n = 3 per condition). ∗∗p values <0.01; p value <0,0001.
(H–J) Bar graphs of CXCL9 (G), CXCL10 (H), and CXCL11 (I) expression in RELA KO HAECs unstimulated or treated with IFNγ/TNFα. Data shown are mean ± SD (n = 3 per condition). ∗∗∗p value < 0.001; ∗∗∗∗p value <0,0001.
To further investigate the specific roles of STAT1 and p65 in regulating synergy, we used CRISPR-Cas9 to generate stable knockout (KO) lines of each TF in HAECs. Electroporation of cells with Cas9-Ribonucleoprotein complexes containing single guide RNAs (sgRNAs) targeting each TF achieved 92% and 94% KO of STAT1 and p65, respectively, at the protein level (Figures 2C and 2D). A non-target (NT) sgRNA was included as a negative control for comparison. Given the near-complete KO effect, we used these polyclonal lines to assess TF dependency in cytokine-mediated gene regulation. STAT1 KO resulted in a 98%, 97%, and 97% decrease in the maximal induction of CXCL9, -10, and -11 (Figures 2E–2G, the fold change values are provided in Table S1), respectively, while p65 KO reduced maximal activation of CXCL9, -10, and -11 by 92%, 97%, and 85%, respectively (Figures 2H–2J; the fold change values provided in Table S1). It is possible that cytokines can alter mRNA stability.41 To address this point, we performed cytokine washout experiments. ECs were treated with dual cytokines for 1 h and then switched to media without cytokine for 3 h. RNA was harvested at 4 h for all conditions. By qPCR, transcripts were decreased by 50%–80%, as compared to 4-h synergy condition, suggesting that stabilization of RNA could not explain all observed effects (Figures S2F–S2I). Notably, several other TFs including interferon regulatory factors (IRFs) are induced by IFNγ and TNFα (Figure 1E) albeit sub-additively. However, the observed synergistic response did not require new protein synthesis, as determined by cycloheximide cotreatment during cytokine stimulation, suggesting that newly induced TF protein expression was not critical for gene induction (Figure S2J). Collectively, these data demonstrate that both STAT1 and p65 are required for the transcriptional synergy response by IFNγ/TNFα.
Synergistic induction of proinflammatory genes is not dependent on saturation of IFNγ or TNFα signaling
The experiments described utilized saturating concentrations of IFNγ and TNFα, which raises a key question: does synergy occur simply as a function of maximal activation of p65 and STAT1? Furthermore, RNA-seq results are semi-quantitative and fold change measurements can be misleadingly large when baseline levels of gene expression are low. To circumvent these issues, we leveraged the NanoString platform that directly counts mRNA transcripts quantitatively without RNA amplification, thereby avoiding potential analytical challenges arising with relative expression changes determined by RNA-seq (Figure 3A). We designed a custom NanoString probe set that included 3 housekeepers (GUSB, HPRT1, and NOL7), five genes that are putatively synergistic (CXCL9, CXCL10, CXCL11, CX3CL1, and IL32) and two genes that are responsive to IFNγ or TNFα only (SOCS3 and VCAM1 or SELE, respectively), based on the RNA-seq data. The gene expression changes elicited by each cytokine were first investigated by generating a 12-point dose-response curve for IFNγ or TNFα treatment in HAECs. (Figure S3A). From these data the half-maximal effective concentration (EC50) for each cytokine was extrapolated: EC50 for IFNγ = 7.8 ng/mL and EC50 for TNFα = 0.39 ng/mL. Based on these analyses, HAECs were then treated with IFNγ and/or TNFα at maximal, EC50, or concentrations 16-fold lower than EC50, and transcript counts for genes were measured in our probe set. Synergistic induction of CXCL9, -10, and -11 was evident at concentrations as low as 0.78 ng/mL of IFNγ and 0.39 ng/mL of TNFα (Figures 3B–3G). Similar patterns of synergy were observed with all concentrations of TNFα when combined with maximal IFNγ (Figures S3B–S3J). We observed no synergistic induction of SELE or SOCS3 at any combination of IFNγ and TNFα (Figures 3H–3K). These results indicate that STAT1 cooperates with p65 to synergistically induce genes at sub-saturating levels of cytokine activation.
Figure 3.

Synergistic induction of proinflammatory genes is not dependent on saturation of IFNγ or TNFα signaling
(A) Schematic of NanoString workflow, identification of EC50 for IFNγ and TNFα (Figure S3A), and experimental design of dual-cytokine dose-response experiments.
(B, D, F, H, and J) Heatmap of CXCL9 (B), CXCL10 (D), CXCL11 (F), SELE (H), and SOCS3 (J) expression in response to maximal concentrations, EC50 and 16-Fold < EC50 of cytokine responsive genes of IFNγ and/or TNFα (n = 4 per condition).
(C, E, G, I, and K) Bar plot of normalized CXCL9 (C), CXCL10 (E), CXCL11 (G), SELE (I), and SOCS3 (K) transcript counts in Figures 2B–2D, 2F, 2H, and 2J. Bars indicate mean ± SD (n = 4 per condition). One-way ANOVAs were performed followed by Tukey’s post hoc analysis to determine statistical differences between treatment groups. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p ≤ 0.0001.
Prolonged IFNγ/TNFα costimulation functionally synergizes to increase chemokine protein and cell death in HAECs
The dynamic changes in chemokine transcripts induced by INFγ/TNFα motivated us to examine whether protein levels also change. As these chemokines serve critical biological roles in immune cell activation when released from cells, we performed enzyme-linked immunosorbent assays (ELISAs) to quantify the secreted proteins in the supernatant of HAECs.42 Baseline levels of CXCL9, -10 or -11 were below the limit of detection in the ELISA (lower limit: CXCL9 = 31.2 pg/mL, CXCL10 = 7.8 pg/mL, CXCL11 = 62.5 pg/mL). IFNγ stimulation for 8 h increased CXCL9 to 35 pg/mL and CXCL10 to 315.99 pg/mL, while CXC11 protein levels remained less than assay (Figures 4A–4C). No induction for any chemokine was detected in response to TNFα, which is consistent with the absence of an effect on gene expression (Figures 4A–4C). Following IFNγ/TNFα costimulation, multiplicative increases in the protein levels of CXCL9 (97.3 pg/mL), CXCL10 (27,097 pg/mL), and CXCL11 (711 pg/mL) occurred relative to the levels following either cytokine alone (Figures 4A–4C). These results align with transcript data in which CXCL10 had the highest transcript counts on NanoString and the highest reads per kilobase million (RPKMs) in RNA-seq, followed by CXCL11 and then CXCL9 (Figures 1F, 3B–3G, and S3B–S3J).
Figure 4.

Prolonged IFNγ/TNFα costimulation functionally synergizes to increase chemokine protein and cell death in HAECs
(A–C) Bar plots of secreted protein for CXCL9 (A) CXCL10 (B) and CXCL11 (C). Data shown are mean ± SD (n = 3 per condition).
(D) Bar plot of absorbance at 600nm as an indicator of cell viability as measured by an MTT assay in unstimulated HAECs and HAECs treated with IFNγ, TNFα, or IFNγ/TNFα. Data shown are mean ± SD (n = 3 per condition).
(E) Table containing means of viable cell counts ± SD in HAECs treated with vehicle or cytokines for 48 h (n = 3). One-way ANOVA followed by a Tukey’s post hoc analysis was utilized to determine statistical significance for cell count. p-value comparisons are listed underneath the table.
For (A–D) one-way ANOVAs were performed followed by a Tukey’s post hoc analysis to determine statistical differences between treatment groups. ∗∗∗∗p ≤ 0.0001.
Previous work has demonstrated that prolonged (>24 h) IFNγ/TNFα signaling results in death of multiple cell types including ECs via PANoptosis.2,43,44 To test this idea in our model, we compared cell counts and viability of unstimulated HAECs versus cells stimulated with IFNγ, TNFα, or IFNγ/TNFα cytokines for 48 h. In unstimulated HAECs, counts doubled over the course of the experiment, consistent with normal growth and doubling times of these cells. Single cytokine-treated cells also proliferated, but to a lesser degree. By contrast, IFNγ/TNFα stimulation decreased total counts compared to starting values, culminating in 39% cell death relative to other groups cells (Figure 4E). The combination of decreased overall count and viability in the IFNγ/TNFα-stimulated cells reveals that prolonged dual cytokine stimulation is toxic in HAECs (Figure 4E). Overall, these data demonstrate that more prolonged IFNγ/TNFα stimulation drives large increases in these chemokine proteins that are known to play key roles in systemic inflammatory responses, and the overall activation ultimately culminates in significant cell death.
Dual cytokine stimulation results in the formation of accessible elements and recruitment of STAT1 and p65 at the CXCL9, -10, and -11 locus
After determining changes in gene, protein, and cell phenotypes in response to IFNγ/TNFα costimulation, we next investigated the chromatin landscapes in HAECs. ATAC-seq was performed in HAECs stimulated with or without IFNγ, TNFα, or IFNγ/TNFα for 1 h (n = 2 for each condition), and reads were filtered on nucleosomal-free regions (<150 bp). All four conditions had similar numbers of peaks (unstimulated = 186,612 peaks, IFNγ = 154,313 peaks, TNFα = 154,252 peaks, and IFNγ/TNFα = 168,185 peaks). The overall distribution of ATAC-seq peaks at promoter-transcriptional start sites (TSS), 5′-untranslated region (UTR), 3′-UTR, intragenic, or intergenic regions did not differ between groups (Figures 5A and S4A–S4D).
Figure 5.

Dual-cytokine stimulation results in the formation of de novo accessible elements at the CXCL9, -10, and -11 locus and recruitment of STAT1 and p65
(A) Pie chart of genomic localization of ATAC-seq peaks as a percentage of the total peaks. Unstimulated (upper left), IFNγ (upper right), TNFα (lower left), and IFNγ/TNFα (lower right) (n = 2 per condition).
(B) Stacked bar plot depicting number of enhanced (dark purple) or de novo (light purple) accessible regions in peaks differential to IFNγ/TNFα.
(C) Rank plot of log2(RPKM) ATAC-seq signal in SSAEs.
(D) Position weight matrix plots of the top motifs found in the IFNγ/TNFα-specific differential peaks.
(E) Signal plots of p65 or STAT1 motif density distribution in differential peaks in each cytokine condition.
(F) Pie charts of genomic localization of CUT&RUN peaks for FLAG-STAT1 and FLAG-p65. (FLAG) indicate the antibody used for CUT&RUN.
(G and H) Position weight matrix plots of the top motif found in the STAT1 peaks (G) and p65 peaks (H) in IFNγ/TNFα stimulated HAECs.
(I) Scatterplots of STAT1 signal (top, orange) and p65 signal (bottom, blue) vs. ATAC signal at SSAEs. R value indicates Pearson correlation coefficient.
(J) Gene tracks of ATAC-seq and CUT&RUN (C&R) for STAT1 and p65 at the CXCL9, -10, and -11 locus in HAECs at baseline and in response to IFNγ, TNFα or both (red). N = 2 per condition. Replicates are shown in two shades of color on the same track. Y axis is rpm/bp. Gray boxes indicate SSAEs co-bound by STAT1 and/or p65; asterisks indicate SSAEs with no TF co-binding; arrows indicate STAT1 and/or p65 binding at ATAC-seq sites that are not categorized as SSAEs.
(K) Gene track zoomed in on the CXCL10 and -11 intergenic enhancer showing the close proximity of STAT1 and p65 co-binding at this location centered on the ATAC-seq SSAE.
We next investigated dynamic changes in chromatin accessibility in response to cytokines. When compared to unstimulated cells, statistically significant increases in regions of open chromatin were evident in all cytokine groups: IFNγ (5,105 peaks), TNFα (9,778 peaks), and IFNγ/TNFα (13,738 peaks) using a fold change threshold of 1.5 and p-value <0.0001 (Figures S4E–S4G). The total number of differential peaks suggested considerable overlap in accessible elements. Our goal was to determine whether any unique chromatin features could be identified in HAECs treated with IFNγ/TNFα. Thus, we next performed 3 pairwise analyses: IFNγ/TNFα versus 1) unstimulated, 2) IFNγ-stimulated, or 3) TNFα-stimulated HAECs. 658 peaks were significantly enriched in synergy conditions compared to IFNγ or TNFα, of which 375 were enhanced (i.e., increased compared to individual cytokine) and 283 were de novo sites (i.e., not present in single cytokine) (Figures 5B and S4E–S4G). We designated these 658 regions as synergy-specific accessible elements (SSAEs). Ranking of SSAEs by signal identified several sites in the CXCL9, -10, and -11 locus that were among the top peaks in the IFNγ/TNFα group (Figure 5C). Motif enrichment analysis confirmed that STAT1 and p65 were among the most enriched TF motifs in these regions (Figure 5D).
TF cooperativity is one mechanism posited to explain transcriptional synergy.6 To evaluate this in ECs, we generated histograms of p65 and STAT1 motif density centered on ATAC-seq summits. Both STAT1 and p65 motifs were enriched in the differential ATAC sites detected in IFNγ and TNFα groups, respectively. The p65 profile was more pronounced than STAT1 in the IFNγ/TNFα group (Figure 5E), consistent with the RNA-seq results, which showed TNFα drove a large proportion of the changes in gene expression with synergy. There was also a strong alignment between the summit of the peak and the apex of STAT1 and/or p65 motif (Figure 5E). The pattern of p65 and STAT1 motifs was distinctive in the IFNγ/TNFα differential peaks, in which they were spaced apart by ∼20 bp periodicity. Both IFNγ and TNFα can direct the formation of SEs—exceptionally large regions of cis-regulatory DNA that are densely bound by TFs.23,45 However, in this dataset the overall ATAC signal, peak length, and signal density at distal elements and gene bodies were not different between all cytokine-stimulated groups (Figures S4H and S4I); and most regions had similar chromatin accessibility in single- and dual-cytokine conditions (Figure S5A). The overall correlation between SSAEs and RNA-seq was weak, but there was a statistically significant increase in ATAC-seq signal at genes that were deemed outliers from our computational analysis (Figure S5B) and 46% of outlier genes (30 of 65) had an SSAE within 500 kb of the transcriptional start site (Figure S5C).
Based on the chromatin accessibility predictions, we next directly mapped the DNA binding of STAT1 and p65 in response to IFNγ/TNFα. We first attempted “cleavage under targets and release under nuclease” (CUT&RUN) using antibodies for the endogenously expressed proteins, but the results were inconsistent. To overcome this technical problem, we engineered stable HAEC lines to express either N-terminal 3xFLAG-STAT1 or 3xFLAG-p65 via lentivirus in STAT1 or p65 single KO cells, respectively, which we had already validated in Figure 2B. The protein expression levels of the FLAG-tagged STAT1 and p65 were similar to wild-type cells (i.e., STAT1+/p65+ cells) and were equally responsive to dual-cytokine stimulation (Figures S5D and S5E). Anti-FLAG CUT&RUN identified clear recruitment and binding of STAT1 (3056 sites) and p65 (4355 sites) in response to IFNγ/TNFα as compared to unstimulated cells. Genomic localization of the TFs was distributed among intergenic, intragenic, and TSS regions (Figure 5F), and the top motif in 3xFLAG-STAT1 cells or 3xFLAG-p65 cells was STAT1 (Figure 5G) or p65 (Figure 5H), respectively. Co-binding analysis showed that 17% of de novo ATAC-seq sites were co-bound by one or both TFs; this percentage increased to 31% when using only the enhanced SSAEs. Overall, there was a moderate correlation between TF signal and SSAEs (Figure 5I). At the CXCL9, -10, and -11 locus there was clear evidence of co-binding at 3 SSAEs (Figure 5J, gray boxes). Two other sites were co-bound by STAT1 and p65, with clear ATAC-seq signal, which were not classified as SSAEs (Figure 5J, arrows). Notably, close spacing of STAT1 and p65 binding at some of these co-bound regions was consistent with the ATAC motif predictions (Figure 5K). To examine the interdependency of STAT1 and p65 recruitment to this region, we next performed CUT&RUN in cells with KO of both TFs that were then reconstituted with either 3xFLAG-STAT1 or 3xFLAG-p65. As before, protein expression was similar to wild-type levels (Figure S5F). Following dual-cytokine stimulation, overall binding of p65 and STAT1 was predominantly unaffected genome-wide with only 48 STAT1 sites (1.6%) and 293 p65 sites (6.7%) decreased after KO of the opposite TF (Figure S5G). Most regions including at canonical IFNγ (SOCS3) and TNFα (CCL2) enhancers were not altered (Figures S5H and S5I). At the CXCL9, -10, and -11 locus, STAT1 recruitment did not change with p65 KO; however, p65 recruitment at the CXCL9 and -10 promoters and intergenic sites between CXCL9 and -10 and CXCL10 and -11 was completely lost in cells lacking endogenous STAT1 (Figure S5J arrows). These data demonstrate that STAT1 is a critical determinant of p65 recruitment to select genes in IFNγ/TNFα-induced transcriptional synergy.
Synergistically induced genes harbor transcriptional dependencies on p300/CBP and BET-bromodomain-containing proteins
Changes in accessibility implicate chromatin regulators in these synergy transcriptional responses. Indeed, the STRING database of protein-protein interactions for STAT1 and p65 confirms interactions between coactivators including BRD4—a member of the BET bromodomain-containing protein family—and p300/CBP as well as multiple histone modifiers (deacetylases, acetyltransferases, and methyltransferases) (Figures S6A and S6B).To explore coactivator dependencies more systematically, we developed a targeted chemogenomic miniscreen using 27 small-molecules inhibitors targeting coactivators, histone-modifying enzymes, nucleosome-remodeling proteins, components of the general transcriptional machinery, and key nodes in the IFNγ or TNFα signaling pathways. HAECs were stimulated with IFNγ/TNFα in the presence or absence of each compound for 1 h. Following RNA extraction, CXCL9, -10, SELE, and SOCS3 mRNA levels were measured by real-time qPCR. As expected, small-molecule inhibitors of IFNγ-JAK-STAT (Ruxolitinib) or TNFα signaling (BAY) completely abrogated SOCS3 and SELE expression, revealing fidelity of the gene responses to each cytokine pathway (Figure 6A). The screen further identified ten small-molecule inhibitors that more selectively reduced induction of CXCL9 and CXCL10 compared to IFNγ- or TNFα-responsive genes (SOCS3 or SELE, respectively) (Figure 6A, red). Of these more selective inhibitors, four directly targeted the BET bromodomain protein family (BRD2, 3 ,4) and three targeted the p300/CBP family (Figure 6A). Surprisingly, targeted inhibition of BET bromodomain 1 (BD1 = GSK778) but not bromodomain 2 (BD2 = GSK046) selectively inhibited CXCL9 and -10 (Figure 6A), while BD2 had much less of an effect overall. This result differs from prior reports that implicate BD2 specifically in signal-responsive transcription.46 Inhibition of p300/CBP lysine acetyltransferase (KAT) activity (A-485) or the p300/CBP bromodomain (GNE-781) as well as targeted protein degradation of p300/CBP (dCBP-1) all resulted in more selective inhibition of CXCL9 and -10 induction (Figure 6A).
Figure 6.

Synergistically induced genes harbor transcriptional dependencies on p300/CBP and BET bromodomain-containing proteins
(A) Bar plots of gene expression of CXCL9 (top), CXCL10 (2nd from top), SELE (2nd from bottom), and SOCS3 (bottom) comparing untreated cells versus cells costimulated with IFNγ/TNFα+Vehicle or maximal concentrations (10μM) of transcriptional inhibitors. For AU-15330 and FHT1 samples were pretreated at 1 μM for 1 h before adding cytokine. Data shown are mean ± SD (n = 3 per condition).
(B–E) Dose-response curves for A-485 (B), JQ1 (C), THZ1 (D), and YKL-5-124 (E) in HAECs stimulated with IFNγ/TNFα and inhibitors (1 h). Graphs show % maximal expression (y axis) for CXCL9 (red line), CXCL10 (yellow line) and SELE (green line) vs. log10 inhibitor dose (x axis). Data are reported as mean ± SD (n = 4 per observation).
(F–H) Tables of biochemical parameters calculated from dose-response curves for each gene from (B–E).
Guided by the known STAT1 and p65 protein-interaction profiles (Figures S6A and S6B), we focused on the inhibitors for p300/CBP lysine acetyltransferase function (A-485) and the BET-family of bromodomain-containing proteins (JQ1, pan-BET bromodomain inhibitor) to perform more in-depth biochemical modeling. Eight-point dose-response curves were generated for A-485 and JQ1 and determination of the baseline inhibition (I0), IC50, maximal inhibition (Imax), and Hill coefficients (HCs) of each gene in response to each inhibitor (Figures 6B and 6C–6H). Since both p300/CBP and BRD4 are known to play essential roles in transcription more broadly, we wanted to ensure that the effects were not resulting simply from generalized inhibition of all transcription. As such we included additional dose-response curves for THZ1 (CDK-7, -12, and -13 inhibitor) and YKL-5-124 (CDK7 inhibitor) for comparisons and to assist in the biochemical modeling of unique transcriptional dependencies for CXCL9 and CXCL10—two of the exemplary synergy genes (Figures 6D and 6E). While there were no differences in the A-485 and JQ1 IC50 data when comparing synergistically induced genes with SELE (CXCL9 68.5 nM; 12 nM, CXCL10 152 nM; 11 nM, SELE 57.6 nM; 13 nM), there were differences in the Imax values with CXCL9 (0.19%, 1.53%) and CXCL10 (3.53%, 9.55%), nearly completely inhibited at high concentrations of A-485 and JQ1, respectively (Figures 6F and 6G). By comparison, Imax values for SELE (17.06%, 29.96%) revealed decreased efficacy of JQ1 (Figure 6H). In contrast to these results, THZ1 and YKL-5-124 strongly inhibited all genes measured based on Imax values: THZ1:(CXCL9 0.31%, CXCL10 0.80%, SELE 1.77%) and YKL-5-124: (CXCL9 7.20%, CXCL10 9.27%, SELE-10.05%) (Figures 6D–6H). These data suggest that the observed effects of A-485 and JQ1 may not be caused by global effects on transcription.
The Imax data identified that p300/CBP KAT and BET bromodomain inhibition possessed high efficacy to abrogate CXCL9 and -10 induction. To determine the sensitivity of gene induction to these inhibitors, we determined the HCs—indicative of the extent of cooperativity of a drug or inhibitor to alter a stimulus-coupled response. HCs were calculated by generating a 4-parameter logistic fit on dose-response data for CXCL9, CXCL10, and SELE induction by IFNγ/TNFα during A-485 or JQ1 cotreatment. These calculations revealed that CXCL9 and CXCL10 display ultrasensitivity to A-485, indicated by higher HC values (HC = 2.25 (CXCL9), = 2.13 (CXCL10), = 1.76 (SELE)) and JQ1: HC = 2.63 (CXCL9), = 2.42 (CXCL10), = 1.53 (SELE) (Figures 6F–6H)).
We next assessed how treatment of HAECs with A-485 or JQ1 functionally impacts the secretion of CXCL9, -10 and -11 protein. HAECs were cotreated with IFNγ/TNFα and maximal concentrations of A-485 (2,000 nM) or JQ1 (500 nM) for 8 h. A-485 completely abrogated CXCL9 and CXCL11 induction and inhibited CXCL10 protein levels by 79.19% (Figures S6C–S6E). CXCL9 induction was completely inhibited after cotreatment with JQ1 while CXCL10 and CXCL11 displayed a 79.64% and 85.91% reduction, respectively, in protein levels in the presence of JQ1. These effects on protein suggested that A-485 or JQ1 might ameliorate other effects of IFNγ/TNFα synergy. To test this idea, cells were costimulated with IFNγ/TNFα and varying concentrations of A-485 or JQ1 for 48 h, after which the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide) assay was performed to assess cell death. We observed a significant decrease in viability after 48 h of IFNγ/TNFα treatment, which is in line with our previous data and published literature (Figure 4E).2 A-485 and JQ1 both partially rescued cell viability in a concentration-dependent manner. The lowest concentration for a detectable effect was 125 nM for A-458 and 62.5 nM for JQ1 (Figure S6F). Collectively, these data provide evidence that A-485 or JQ1 partially inhibited the toxic effects of IFNγ/TNFα dual stimulation in HAECs.
p300/CBP or BET bromodomain inhibition does not alter chromatin accessibility or p65 and STAT1 recruitment to chromatin
Due to the strong inhibitory effects of A-485 and JQ1 on synergy genes, we investigated how they affect chromatin accessibility and genomic recruitment of STAT1 and p65, respectively. As compared to vehicle-treated cells, the distribution of ATAC-seq signal in inhibitor-treated HAECS was similar genome-wide (Figure 7A). Using all ATAC-seq sites in vehicle-treated samples, the signal alignment in the inhibitor-treated cells overlapped almost identically compared to vehicle (Figure 7B). When focusing only on SSAEs, there was a small decrease in signal in the A-485-treated group compared to JQ1 or vehicle, suggesting a modest effect of p300/CBP inhibition at SSAE sites, though not complete loss of accessibility (Figure 7C).
Figure 7.

p300/CBP or BET bromodomain inhibition does not alter chromatin accessibility or p65 and STAT1 recruitment to chromatin
(A) Pie charts of genomic localization of ATAC-seq peaks cotreated with IFNγ/TNFα with A-485 or JQ1.
(B) Signal alignment plots of ATAC-seq costimulated with IFNγ/TNFα along with Vehicle, A-485 (2 μM) or JQ1 (500 nM), binned on all peaks present in IFNγ/TNFα + Vehicle.
(C) Signal alignment plots of ATAC-seq cotreated with IFNγ/TNFα and Vehicle, A-485 (2 μM) or JQ1 (500 nM), binned on SSAEs only.
(D) Heatmap alignments of STAT1 (left side) or p65 (right side) signal in Vehicle (red), A-485 (orange) or JQ1-treated (green) samples. All samples were stimulated with IFNγ/TNFα. All samples were aligned to the Synergy + Vehicle condition.
(E) Gene tracks of CXCL9, 10, and 11 locus showing ATAC-seq, p65 CUT&RUN (C&R) and STAT1 CUT&RUN in synergy conditions cotreated with Vehicle (Red), A-485 (orange) or JQ1 (green). Tracks for unstimulated cells are shown at the top in black. N = 2 replicates. Replicates are shown in two shades of color on the same track. Gray box indicates ATAC-seq site lost with A-485. Asterisks indicate ATAC SSAEs without TF binding that are lost with A-485 or JQ1 treatment.
To assess the effect of p300/CBP or BET bromodomain inhibition on genomic localization of each TF, we performed CUT&RUN using 3XFLAG-STAT1 or 3XFLAG-p65 cells cotreated with IFNγ/TNFα with or without A-485 or JQ1. Signal alignments were superimposed when comparing the groups aligned on IFNγ/TNFα vehicle (Figure 7D) as well as genomic distribution of peaks (Figures S7A and S7B). Differential peak analysis via HOMER did reveal that 23 out of 3,057 peaks (0.75%) were decreased with A-485 and 9 out of 3,057 (0.29%) decreased with JQ1 for STAT1; for p65 a decrease in 52 out of 4,356 peaks (1.22%) with A-485 and 234 out of 4,256 peaks (5.37%) with JQ1 were detected. All together, these results suggest that global recruitment of p65 and STAT1 was unaffected by the presence of A-485 or JQ1, consistent with prior reports that these small-molecules target coactivators without affecting TFs. Examination of the CXCL9, -10, and -11 locus specifically demonstrated that both p65 and STAT1 were recruited in response to dual cytokines and overlapped multiple ATAC-seq sites including SSAEs (Figure 7E) except for p65 binding at the CXCL9 promoter (Figure 7E, gray boxes). The remainder of p65 and STAT1 occupancy profiles were insensitive to either A-485 or JQ1 (Figure 7E). Similar results were observed at SELE and SOCS3 (Figures S7C and S7D). With respect to chromatin accessibility, two SSAEs that were not co-bound by STAT1 or p65 were decreased with A-485, and one with JQ1. These sites were located at the 3′ UTRs of CXCL10 and CXCL11, perhaps indicative of the overall transcriptional effects of A-485 and JQ1 (Figure 7E, asterisks). These data indicate that the mechanism of transcriptional inhibition by A-485 and JQ1 does not involve complete displacement of STAT1 or p65 from cis-regulatory DNA at these regions.
Synergy genes are sensitive to combined p300/CBP and BET bromodomain inhibition
Considering the differences in HCs for A-485 and JQ1 and their distinct mechanisms of action, we next hypothesized that cotreatment with lower doses of A-485 and JQ1 in combination could selectively inhibit synergism. To test this idea, we costimulated cells with IFNγ/TNFα along with A-485 and/or JQ1 at concentrations ranging from the IC50 or below and measured gene expression with NanoString to capture quantitative transcript counts. Existing methods for determining synergistic effects of drug combinations are not uniform. Web-based tools operate using arbitrary thresholds for synergism, and source code is not always publicly available to evaluate fitness in specific experimental models. To overcome these limitations, we developed and tested a general linearized model (GLM) to determine if the A-485 and JQ1 combinations synergistically inhibited chemokine induction. A Poisson regression model demonstrates that the multiplicative interaction term between A-485 and JQ1 successfully fits the data for CXCL9 and CXCL11, but not for CXCL10 (Figures 8A–8C; Table S2). This indicates that A-485 and JQ1 have a negative multiplicative effect on CXCL9 and -11 but not -10. Further examination of the model demonstrated that the negative effect observed with A-485 alone increases per experimental dose of JQ1 and vice versa. This is highlighted in Table S2, where, in the absence of A-485, CXCL9 expression would be predicted to decrease by 3.5% per experimental dose of JQ1; however at 62 nM of A-485 the per experimental dose effect of JQ1 increased to 11.8%, demonstrating synergistic effect (Table S2). Based on this GLM analysis, both CXCL9 and CXCL11 demonstrated synergistic inhibition with A-485 and JQ1, while CXCL10 was resistant to dual-inhibitor treatment at these lower, subIC50 concentrations (Figures 8A–8C).
Figure 8.

Synergy genes are sensitive to combined p300/CBP and BET Bromodomain inhibition
(A–C) 3D bar plots of normalized transcript counts for CXCL9 (A), CXCL10 (B), and CXCL11 (C) with the indicated amounts of A-485 and/or JQ1 (n = 3 per condition).
(D–F) Bar plots listing supernatant concentrations of CXCL9 (D) CXCL10 (E) and CXCL11 (F) in IFNγ/TNFα + Vehicle-treated cells or with costimulation across various concentrations of A-485 and/or JQ1. A one-way ANOVA followed by Dunnett’s test was performed to assess statistical significance. Data shown are mean ± SD (n = 3 per condition). ∗p < 0.05 ∗∗∗∗p ≤ 0.0001.
Given the GLM results, we tested whether A-485 or JQ1 at or below IC50 could inhibit the production of secreted CXCL9, -10, and -11. HAECs were cotreated with IFNγ/TNFα and either A-485 or JQ1 for 8 h before supernatants were harvested and ELISAs was performed. These data revealed a strong trend toward synergistic inhibition as A-485 (62.5 nM) and JQ1 (15.6 nM) costimulation resulted in a 74.72% inhibition compared to A-485 (62.5 nM) alone (49.28% inhibition) and JQ1 (15.6 nM) alone (7.81% inhibition) (Figure 8D). CXCL10 was resistant to dual inhibition with A-485 and JQ1 (Figure 8E). Dual inhibition with A-485 and JQ1 did synergistically inhibit CXCL11 protein as A-485 (62.5 nM) and JQ1 (15.6 nM) costimulation resulted in a 59% reduction in protein levels, whereas A-485 (62.5 nM) alone or JQ1 (15.6 nM) alone resulted in 37% or no inhibition, respectively (Figure 8F). Together these data demonstrate that targeting p300/CBP and BET bromodomain proteins resulted in the inhibition of functional responses following IFNγ/TNFα costimulation. Importantly subIC50 concentrations were able to achieve specific inhibition of both transcript and protein levels for CXCL9 and CXCL11.
Discussion
Transcriptional synergy—more-than-additive induction of a gene by two or more TFs—is an important mechanism for driving robust gene expression programs in development.47 In disease contexts, excess inflammation often arises via cytokine-mediated activation of signal-responsive TFs that dynamically induce proinflammatory programs. In particular, synergy elicited by IFNγ/TNFα is associated with severe SARS-CoV-2 illness.2 Here, we characterized the synergistic transcriptional response governed by IFNγ-STAT1 and TNFα-p65, as well as the changes in chromatin landscape and the chromatin coactivators involved.
Synergism arising from IFNγ and TNFα costimulation has previously been described in multiple cell types including ECs using candidate gene approaches.2,8 Here, unbiased RNA-seq identified that transcriptional synergism between IFNγ-STAT1 and TNFα−p65/NF-κB occurs rapidly within 1 h at a small subset of all cytokine-induced genes. Using stable isotope imaging with MIMS, we demonstrate that nascent RNA production is also detectable within 1 h in the nucleus, consistent with new transcription. Our outlier analysis classifying the chemokines CXCL9, -10, and -11 as synergistic aligns with other studies that have observed similar induction in cultured HAECs, umbilical vein ECs, and cultured atherosclerotic plaques.48,49 Notably, we did not detect the synergistic induction of other previously reported genes such as CCL5, IL-8, and HLA-A.8,9,13 Compared to the 1-h duration presented here, these other studies stimulated cells with IFNγ/TNFα over longer periods of time, which could lead to secondary induction of other gene programs via action of TFs including IRFs. An additional explanation for the synergistic response is cytokine-induced changes in RNA stability that lead to increases in transcript counts, as previously shown for chemokine genes.41 No mRNA induction of RNA-binding proteins was evident by RNA-seq in response to IFNγ/TNFα stimulation (ELAVL1/HuR or LUBAC complex), and transcripts decreased significantly with cytokine washout. Our work focused on an early time point and primary transcriptional response to avoid some of these secondary effects. By first analyzing global transcriptomes with outlier analysis and not candidate genes, we identified that most genes in HAECs were induced to a similar degree by TNFα or combined IFNγ/TNFα, suggesting that TNFα responses dominate in ECs generally, as compared to IFNγ. However, the fact that synergistic transcription was restricted to a small fraction of all potential genes (n = 65) reinforces the concept that synergy is both tightly regulated and initially constrained.
One important concept in synergy is saturation. Studies of signal-responsive transcription commonly utilize high cytokine concentrations without defining the concentration-dependent effects on TF action vis-a-vis target gene output. Determination of the individual cytokine dose responses for IFNγ- and TNFα-regulated genes with NanoString enabled us to examine the thresholds for and magnitudes of transcriptional synergy even at non-saturating levels of STAT1 or p65 activity—as inferred from the maximal gene expression induced by IFNγ or TNFα, respectively. We found that synergy still occurs, albeit to a lesser degree, even at lower cytokine concentrations. Signaling crosstalk at the level of kinase cascades emanating from each receptor does exist between IFNγ and TNFα, which could impact TF activation in nuanced ways. We did not detect any evidence for crosstalk at the level of nuclear translocation, as IFNγ and TNFα only induced translocation of STAT1 and p65, respectively. However, more subtle changes in phosphorylation or other posttranslational modifications could impact DNA binding, transactivation, or coactivator interactions, leading to greater gene induction even at lower cytokine concentrations. IFNγ has also previously been shown to change looping at the CXCL9, -10, and -11 locus in murine macrophages.50 Thus, it is possible that changes in chromatin confirmation driven by IFNγ can bring STAT1- and p65-regulated enhancers into closer physical proximity, amplifying the transactivation activity and inducing the synergistic response observed. Overall, the data establish that transcriptional synergy requires STAT1 and p65 and occurs across a range of their full transactivation potential.
Cooperativity is a prevailing model used to explain how TFs drive transcriptional synergy. Indeed, prior work dissecting a CXCL9 promoter fragment with heterologous reporter constructs demonstrated that the number, orientation, and spacing of STAT1 and p65 binding sites have direct consequences on transactivation.7,51 While these prior data clearly support the cooperativity model, the focus on the promoter and lack of chromatinized DNA or genomic context limited the generalizability. Our paired ATAC-seq and TF CUT&RUN datasets suggest that cooperative binding does occur at the endogenous CXCL9, -10, and -11 locus, as we identified multiple STAT1 and p65 motifs in accessible chromatin that were co-bound by both TFs. Several of these binding events occurred at intergenic regions, representing previously unrecognized distal enhancers. Furthermore, recruitment of p65 was significantly reduced in STAT1 KO cells, but not vice versa, suggesting a possible example of dynamic STAT1-assisted loading of p65, which has been described with inflammatory signaling in the hepatic acute phase response.52,53 In that context, p65 activation primed hepatocytes for STAT3 binding. Loss of p65 DNA occupancy due to increased STAT1 acetylation has been reported54; STAT1 can also interact directly with p300/CBP, which in turn acetylates p65 and thereby enhances p65 nuclear signaling. This hypothesis could explain why loss of STAT1 alters DNA binding of p65 but requires additional experimental validation in our system.21,55 Overall, the work herein identifies novel p65 and STAT1 functional interactions that drive synergistic gene induction in ECs.
A kinetic model of transcription has also been proposed to explain synergism. Two TFs acting on different steps of the transcription cycle (e.g., initiation and elongation) can promote dynamic and greater than multiplicative increases in Pol II processivity.15,56 Kinetic and cooperativity models are not mutually exclusive, and the contribution of each to synergy is difficult to deconstruct in endogenous gene contexts. However, a key prediction from both models is that chromatin-dependent signaling to Pol II via transcriptional coactivators plays a strong role. Yet, it was not clear at the outset of this study whether synergy genes might be especially resilient to coactivator disruption. The targeted inhibitor screen of coregulators identified selective vulnerabilities to p300/CBP and BET bromodomain inhibition. The selectivity of p300/CBP was evident targeting not only KAT function (A-485), but also the bromodomain (GNE-781) and p300/CBP degradation (dCBP). These results implicate coactivator recruitment, physical scaffolding, and acetylation by p300/CBP all as important determinants of synergy at the chemokine locus. In preclinical models, BET bromodomain inhibitors reduced inflammation associated with sepsis and cytokine storm.29,57 The role of BRD4 in control of transcriptional pause release via recruitment of the P-TEFb complex is well established and represents an important general mechanism for how BET bromodomain inhibitors disrupt signal-dependent pathologic transcription.58,59,60 More recent evidence also indicates that BRD4 inhibition alters cytokine-induced recruitment of Pol II to the CXCL10 promoter in macrophages, suggesting that BRD4 can contribute to transcriptional initiation in certain signaling contexts as well.61 Furthermore, p300/CBP-dependent acetylation of p65 recruits BRD4 to TFs and histones via its bromodomains, leading to proinflammatory gene induction.22 SE-associated genes are also vulnerable to BET bromodomain inhibition.23 But, we did not detect differences in ATAC fragment size or signal, suggesting dynamic SE formation does not explain our results. One limitation is that H3K27-acetyl or BRD4 genome-wide profiling is needed to make this determination definitively. Neither p300/CBP nor BET bromodomain inhibition affected recruitment of STAT1 or p65. As such, the underlying mechanisms for inhibiting transcriptional synergy are likely stemming specifically from coactivator-dependent signaling from the TFs to the transcriptional machinery. When considered in totality, the known convergence of p300/CBP and BET bromodomain proteins on STAT1 and p65 pathways provides several candidate mechanisms to explain the sensitivity of synergy genes to each of these coactivators.
Dual p300/CBP and BET bromodomain inhibitors are currently in clinical development for cancer therapeutics.28 The rationale for this approach is, in part, based on the crosstalk between histone acetyltransferases and BET proteins. Here, we developed and tested a GLM framework to statistically determine if two inhibitors display synergistic inhibition on gene induction. This statistical approach provides rigor to data analysis when considering combination inhibitor therapies. Using a GLM identified that the combination of low doses (IC50 or lower) of p300/CBP (A-485) and BET bromodomain (JQ1) inhibitors can synergize to achieve inhibition of proinflammatory genes. At the protein level, these combinations also inhibited CXCL9 and CXCL11 protein secretion but had no impact on CXCL10. The reasons for resistance of CXCL10 protein to low-dose dual inhibition are unclear. Overall, given that IFNγ-STAT1 and TNFα−p65 signaling are implicated in acute and chronic diseases of inflammation (e.g., psoriasis, atherosclerosis, and cytokine storm syndromes including SARS-CoV-2), the transcriptional vulnerabilities identified herein suggest that low-dose inhibitor therapies could be a strategy for selectively targeting proinflammatory pathways, while avoiding global gene dysregulation and dose-limiting toxicities.
Limitations of the study
All experiments were performed using immortalized ECs which were grown in static tissue culture conditions, differing from their native environment of laminar flow and shear stress. It remains unclear if the p65 and STAT1 binding observed in our CUT&RUN experiments uniquely occurs in the combined cytokine condition since we did not perform CUT&RUN on cells stimulated with individual cytokines. In addition, our work in cell culture may not fully capture the transcriptional responses that occur in vivo. Lastly, transcriptional coactivator binding profiles were not done in this index study. Occupancy maps of p300/CBP and individual BET bromodomain proteins will likely shed new light on underlying mechanisms controlling transcriptional synergy.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| p65 (Rabbit) | Active Motif (39369) | RRID:AB_2793231 |
| Beta Actin Mouse (Monoclonal) | ThermoFisher (Clone 15G5A/E2) (MA1-140) (Lot: TK276375) | RRID:AB_2536844 |
| STAT1 (Rabbit) | Cell Signaling Technology (14995) (Lot:4) | RRID:AB_2716280 |
| Goat anti-Rabbit Alexa Fluor™ 594 | Thermo Fisher Scientific (A-11037) (Lot: 1777945) | RRID:AB_2534095 |
| Goat anti-mouse Alexa Fluor™ 488 | Thermo Fisher Scientific (A-21121) (Lot: 1704461) | RRID:AB_2535764 |
| Rabbit IgG | Diagenode (C15410206) (Lot: RIG001AP ) | RRID:AB_2722554 |
| Rabbit monoclonal p65 (D14E12) | Cell Signaling Technology (8242) (Lot: 8242S) | RRID:AB_10859369 |
| Rabbit monoclonal STAT1 (D4Y6Z) | Cell Signaling Technology (14995) (Lot: 4) | RRID:AB_2716280 |
| Goat anti-Rabbit Alexa Fluor™ 647 | ThermoFisher (A-21244) (Lot: 2247991) | RRID:AB_2535812 |
| Mouse anti-FLAG antibody M2 | Sigma Aldrich (F3165) (Lot: SLCJ3741 ) | RRID:AB_259529 |
| Mouse IgG | Diaganode (C15400001) | RRID:AB_2722553 |
| Chemicals, peptides, and recombinant proteins | ||
| Interferon Gamma (IFNγ) | PeproTech | 300–02 |
| Tumor Necrosis Factor Alpha (TNFα) | PeproTech | 300-01A-100UG |
| BAY 11-7085 | Med Chem Express | HY-10257 |
| THZ1 | Med Chem Express | HY-80013 |
| Triptolide | Med Chem Express | HY-32735 |
| Flavopiridol | Med Chem Express | HY-10005 |
| BMS-345541 | Med Chem Express | HY-10519 |
| THZ531 | Med Chem Express | HY-103618 |
| Ruxolitinib | Med Chem Express | HY-50856 |
| Vorinostat | Med Chem Express | HY-10221 |
| Trichostatin A | Med Chem Express | HY-15144 |
| dBET6 | Med Chem Express | HY-112588 |
| YKL-5-124 | Med Chem Express | HY-101257 |
| dCBP-1 | Med Chem Express | HY-134582 |
| GNE-781 | Med Chem Express | HY-108696 |
| JQ1 | Med Chem Express | HY-13030 |
| I-BET151 | Med Chem Express | HY-13235 |
| iBET-BD2 | Med Chem Express | HY-136571 |
| α-Amanitin | Med Chem Express | HY-19610 |
| Mocetinostat | Med Chem Express | HY-12164 |
| JIB-04 | Med Chem Express | HY-13953 |
| I-CBP112 | Med Chem Express | HY-19541 |
| iBET-BD2 | Med Chem Express | HY-136571 |
| A-366 | Med Chem Express | HY-12583 |
| GSK-J1 | Med Chem Express | HY-15648 |
| GSK-J4 | Med Chem Express | HY-15648B |
| Cycloheximide | Sigma Aldrich | C4859 |
| A-485 | Med Chem Express | HY-107455 |
| AU-15330 | Med Chem Express | HY-145388 |
| FHT 2344 | Tocris Biotechne | 7644/2 |
| AltR S.p. HiFi Cas9 Nuclease V3 | IDT | 1081061 |
| 13C-Thymidine | Cambridge Isotope Laboratories | CLM-3647-PK |
| 15N-Uridine | Cambridge Isotope Laboratories | NLM-812-PK |
| Biotin-conjugated Concanavalin A | Sigma Aldrich | C2272 |
| Myone Streptavidin T1 DynaBeads | ThermoFisher | 65601 |
| Critical commercial assays | ||
| Illumina Tagment DNA TDE1 Enzyme and Buffer Kit | Illumina | 20034197 |
| Human CXCL9/MIG Quantikine ELISA Kit | Bio-Techne | DCX900 |
| Human CXCL10/IP -10 Quantikine ELISA Kit | Bio-Techne | DIP100 |
| Human CXCL11/I-TAC Quantikine ELISA Kit | Bio-Techne | DCX110 |
| nCounter Standard Master Kit | NanoString | NAA-AKIT-192 |
| nCounter Standard Prep Pack | NanoString | NAA-PPCK-048 |
| nCounter Standard Prep Plates | NanoString | NAA-PPLT-048 |
| nCounter Standard Cartridges | NanoString | NAA-CART-048 |
| 8 well chamber slides | Chemglass Life Sciences | CGN-3530-008 |
| Vascular Cell Basal Medium | ATCC | PCS-100-030 |
| Endothelial Cell Growth Kit-VEGF | ATCC | PCS-100-041 |
| ProLong Gold antifade reagent with DAPI | Invitrogen | P36931 Lot: 2501098 |
| Cell Proliferation Kit I (MTT) | Roche | 11465007001 |
| Pure-link RNA Mini kit | Invitrogen, ThermoFisher | 12183025 |
| NEBNext® Poly(A) mRNA Magnetic Isolation Module | NEB | E7490 |
| NuPage 4–12% Bis Tris 12 well gels | Invitrogen | NP0322BOX |
| Kaleidoscope Protein ladder | BioRad | 1610375 |
| DNA Clean & Concentrator-5 | Zymo Research | D4004 |
| Fugene HD | Fugene | HD-1000 |
| TransDux Max | Systems Biosciences | LV860A-1 Lot:230331-003 |
| PEG-IT Virus Precipitation solution | Systems Biosciences | LV825A-1 |
| Universal Mycoplasma Detection Kit | ATCC | 30-1012K |
| iScript RT-qPCR Sample Prep Reagent | Bio-Rad | 1708899 |
| iTaq™ Universal SYBR® Green One-Step Kit | BioRad | 1725150 |
| Deposited data | ||
| ATAC-seq, CUT&RUN, and RNA-seq data | NCBI GEO | GSE232168 |
| Experimental models: Cell lines | ||
| TeloHAEC | ATCC (CRL-4052) | RRID:CVCL_Z065 |
| 293T/17 | ATCC (CRL-11268) | RRID:CVCL_1926 |
| Oligonucleotides | ||
| STAT1 cRNA (AGTGGTTAGAAAAGCAAGAC ) | IDT | NA |
| STAT1 cRNA (TTCCCTATAGGATGTCTCAG ) | IDT | NA |
| p65 cRNA (AGCGCCCCTCGCACTTGTAG ) | IDT | NA |
| Alt-R CRISPR-Cas9 Negative Control crRNA | IDT | 1072544 |
| Alt-R CRISPR-Cas9 tracrRNA | IDT | 1073191 |
| CXCL9 For PCR Primer (GCTGGTTCTGATTGGAGTGC ) | IDT | NA |
| CXCL9 Rev PCR Primer (GAACAGCGACCCTTTCTCAC ) | IDT | NA |
| CXCL10 For PCR Primer (CTGTACGCTGTACCTGCATCA ) | IDT | NA |
| CXCL10 Rev PCR Primer (CAACACGTGGACAAAATTGG ) | IDT | NA |
| CXCL11 For PCR Primer (GAGTGTGAAGGGCATGGCTA ) | IDT | NA |
| CXCL11 Rev PCR Primer (GGGGAAGCCTTGAACAACTG ) | IDT | NA |
| SELE For PCR Primer (GAGGTGCAGCAAGAAGAAGCTTCG ) | IDT | NA |
| SELE Rev PCR Primer (CACTGAAGCCAGGGTCACAC ) | IDT | NA |
| Nol7 For PCR Primer (CGGAAGCGAGAGAAGAGGAG ) | IDT | NA |
| Nol7 Rev PCR Primer (CGCTTCCTCTTCTCCTTCAG ) | IDT | NA |
| HPRT1 For PCR Primer (CTTTGCTGACCTGCTGGA ) | IDT | NA |
| HPRT1 Rev PCR Primer (TGTCCCCTGTTGACTGGT ) | IDT | NA |
| Software and algorithms | ||
| Bowtie2 | Langmead et al.62 | NA |
| HOMER | Heinz et al.63 | NA |
| MACS2 | Zhang et al.64 | NA |
| DESeq2 | Love et al.65 | NA |
| Custom NanoString Analysis Code | This study | https://github.com/jonathanbrown484/synergy |
| Custom GLM Code | This study | https://github.com/jonathanbrown484/synergy |
| Other | ||
| 3xFLAG-STAT1 Plasmid | Addgene | 220323 |
| 3xFLAG-RELA Plasmid | Addgene | 220324 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jonathan D. Brown (jonathan.d.brown@vumc.org).
Materials availability
Plasmids generated in this study have been deposited to Addgene (3xFLAG-STAT1 = 220323, 3xFLAG-RELA = 220324).
Data and code availability
-
•
ATAC-sequencing, CUT&RUN, and RNA-sequencing datasets generated in this manuscript have been deposited at NCBI GEO under the accession number: GSE232168 and are publicly available.
-
•
Custom NanoString analysis code and GLM for determining synergistic inhibition are publicly available at Github:/jonathanbrown484/synergy
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
Experimental model and study participant details
H-TERT-immortalized human aortic endothelial cells (TeloHAEC) (ATCC: CRL-4052) (RRID:CVCL_Z065) were purchased from ATCC. These cells were extracted from a 23-year old female donor and have been immortalized via stable expression of the human Telomerase catalytic subunit hTERT. No information about race or ancestry is provided by ATCC. Cells were tested for mycoplasma contamination using the Universal Mycoplasma Detection Kit (ATCC: 30-1012K).
Method details
ATAC-seq experimental
Cells were treated with media lacking hydrocortisone for 16–20 h before stimulation. Cells were unstimulated or stimulated for 1 h IFNγ, TNFα, or IFNγ/TNFα (n = 2 per condition) before cells were lifted from the plate. 50,000 cells were used to perform ATAC-seq as previously described.66 0.1% BSA was added to cells to assist with pelleting.
ATAC-seq computational
ATAC-seq fastq files were processed using the standard ENCODE pipeline. Reads were filtered to contain reads which were less than 150bp to obtain information from nucleosomal free reads (NFRs).
ATAC-seq differential peak calling
To make comparisons across 4 conditions the following was performed: First, BAM files produced by the ENCODE pipeline were converted to SAM files using samtools view -h -o output.sam input.bam.67 Tag directories were produced for each replicate using the makeTagDirectory function from the HOMER package.63 For each set of replicates, the tag directories were merged producing one tag directory per condition.63 Differential peaks were called using IFNγ/TNFα peaks, desired tag directories for each desired pairwise comparison using the getDifferentialPeaks function from HOMER with F- 1.5.63 The function annotatePeaks.pl from HOMER was used to create an annotated normalized signal matrix for each differential comparison with options ‘hg38’, ‘-tbp 1’, ‘-rpkm’, and ‘-strand both’.63 To obtain peaks that are unique to the IFNγ/TNFα condition specifically, pairwise comparisons between IFNγ/TNFα and the three other conditions (unstimulated, IFNγ, or TNFα) were performed. The results were then filtered to peaks that reached a p-value of less than 0.0001in all three comparisons, which indicates higher accessibility in the synergy condition compared to all other treatment conditions.
ATAC-seq motif signal plots
Localization of p65 and STAT1 motifs relative to the summit of each peak was determined using annotatePeaks.pl peakfile.txt hg38 -size 150 -hist 15 -m p65.motif STAT1.motif.63 The resultant histogram was utilized to produce line graphs. ATAC-seq signal plots for IFNγ/TNFα+DMSO (Vehicle), IFNγ/TNFα+A-485, and IFNγ/TNFα+JQ1 were produced using annotatePeaks.pl -size 2000 -hist 10 -d tag_directories > out_file.63
ATAC-seq motif enrichment
bed2fasta function from MEMEsuite was utilized to produce fasta files from BED4 formatted peak files.68 AME from MEMEsuite was utilized to determine motif enrichment in peaks that are unique in the IFNγ/TNFα condition using the following script ame -control --shuffle-- --oc output --method ranksum --scoring avg fasta_file.fasta consensus_pwms.meme.28,69,70 Families of motifs were utilized instead of individual motifs to avoid representing and performing statistical analysis on redundant motifs. Families of motifs were classified via non-redundant TF motif clustering as previously described.70
ATAC-seq peak size, signal, and normalized signal
Peaks were binned by the genomic annotation provided by the annotatePeaks command in HOMER.63 Peaks with the intergenic annotation were considered distal elements with all other annotations binned as gene body. Peak size and signal were obtained from the annotated peak files and utilized to generate the normalized signal which is signal/peak size.
Cell culture
hTERT-immortalized human aortic endothelial (TeloHAEC) (ATCC: CRL-4052) (RRID:CVCL_Z065) cells were subcultured using Vascular Cell Basal Medium (ATCC: PCS-100-030) supplemented with Endothelial Cell Growth Kit-VEGF (ATCC: PCS-100-041). Cells were cultured until p25 and media was changed every 3 days. Cells were changed into media lacking hydrocortisone 16–20 h before stimulation.
Cell viability experiment
200,000 cells were plated in media lacking hydrocortisone. 16 h post plating they were stimulated with 50 ng/mL IFNγ and 25 ng/mL TNFα for 48 h (n = 3 per condition). Cells were then harvested and cell viability was assessed via staining with Trypan Blue and the total number of viable and dead cells were counted.
CRIPSR KO generation
crRNAs were designed for RELA and STAT1 using the CRISPOR tool.71 Ribonucleoprotein (RNP) assembly and CRISPR knockout was performed as previously described.72 In brief crRNA and tracrRNA were mixed 1:1 and incubated at 95°C for 5 min then cooled at RT for 10 min before AltR S.p. HiFi Cas9 Nuclease V3 (IDT 1081061) was added to gRNA. The RNP complex was incubated at 30°C for 20 min, then electroporated into HAECs using the NEON Electroporation system with settings: 1500V, 20 ms pulse width x 2 pulses. Cells were electroporated with RNPs containing 2 gRNAs (for STAT1), and 1 gRNA (for p65), or 1 non-target gRNA (see key resources table for sequences). Knockout of STAT1 and/or p65 was confirmed via Western Blotting.
Cloning and lentivirus production
To clone 3xFLAG-STAT1 and 3xFLAG-p65 constructs into lentivirus gateway transfer vector we synthesized gBlocks (IDT) with attB sites and a stop codon at the end of each TF cDNA reading frame. We performed BP and LR cloning per manufacturer protocol (Gateway Cloning ThermoFisher) using pDONR221 for the donor vector and pLEX305-C-dTAG (Addgene #91798) as the destination vector. Of note, the destination vector first required that the antibiotic selection be changed from Puromycin to Blasticidin (BSD gene), because TELO-HAECs have a puromycin resistance gene present due to expression of Telomerase used to immortalize the cells. We changed Puromycin to BSD by restriction enzyme cloning with a gBlock containing compatible KpnI and HpaI sites flanking the BSD gene. After gateway cloning the 3xFLAG constructs, we transformed NEB10-beta cells at 30°C for 24 h. Clones were sequence verified. For lentivirus production, 293T/17 cells were transfected with 20 μg of the transfer plasmid, 15 μg of psPAX2 packaging plasmid (addgene 12260) and 5 μg of pMD2.G envelope plasmid (addgene 12259) with Fugene HD (3:1 ratio) in high glucose DMEM with 10% FBS with no antibiotics. 48 h after transfection supernatant was harvested, dead cells cleared with centrifugation at 2000 rpm and then filtered with 0.45 micron syringe filter. Virus was precipitated with PEG-IT (Systems Biosciences) per manufacturer protocol for 16 h at 4°C, concentrated 100-fold in sterile PBS and then aliquoted for storage at −80. For infection of HAECs, 10 μL of virus were used, based on cell titration studies of p65 and STAT1 protein expression. Cells were selected with blasticidin (3 μg/mL) for 7–10 days prior to validation studies and experiments.
CUT&RUN
CUT&RUN was performed with a few modifications for transcription factor enrichment as previously described.73 In brief, adherent ECs were trypsinized, counted and 1e6 cells per IP were fixed in 0.1% formaldehyde for 1 min then quenched with 125 mM glycine for 1 min at room temperature. Cells were bound to 10uL of activated concanavalin A conjugated streptavidin (MyOne T1, Thermo 65601) beads per IP as previously described.74 Beads were washed with wash buffer containing (20mM HEPES pH 7.5, 150mM NaCl, 0.5mM Spermidine, 0.1% BSA Protease and Phosphatase Inhibitors and 1% BSA). Cells were permeabilized in Wash buffer supplemented with 0.01% digitonin, 0.1% Tween and 0.1% NP-40 for 10 min. After permeabilization, immunoprecipitation was performed with 0.7 μg of antibody per IP in Digitionin-Wash buffer supplemented with 2mM EDTA. Samples were incubated overnight on a nutator at 4°C. Samples next washed in the Dig-wash buffer twice and then incubated with pAG-MNase (MOLOX, 700 ng/mL) for 1 h at 4°C to bind pAG-MNase, then washed with Dig-wash buffer twice. DNA Cleavage reaction was done with 100mM CaCL2 for 30 min while samples were submerged in ice. Cleavage was terminated with STOP buffer with EDTA/EGTA and chromatin released by gentle heating of samples at 37°C for 30 min. Samples were decrosslinked with 1% SDS and 250 μg/mL proteinase K for 4 h in a thermomixer at 65°C. DNA was then purified via phenol-chloroform-isoamyl alcohol extraction and Ethanol precipitation. Sequencing libraries were generated using the NEBNext Ultra II DNA Library Prep Kit (NEB #E7645) with transcription factor modifications.73 PCR program to enrich for short fragments as follows: Step 1: 45 s at 98°C. Step 2: 15 s at 98°C. Step 3: 10 s at 60°C. Step 4: Repeat Steps 2–3 for a total of 14 cycles. Step 5: 1 min at 72°C final extension.
CUT&RUN computational
Reads were trimmed using trimmomatic and then mapped to hg38 using bowtie2. Duplicate reads were discarded and peaks were called using HOMER as previously described.63 In short peaks were called using the IFNγ/TNFα + Veh peaks and tag directories for each desired pairwise comparison. Peaks were called using getDifferentialPeaks – IFNγ/TNFα + Veh peaks -d tag directories. Motif enrichment was performed as described above for ATAC-seq.
Difference and outlier detection
To identify potentially non-additive effects of joint treatment, for each gene differentially expressed between IFNγ/TNFα-treated group and untreated group, the fold change difference was calculated by the following approach. Firstly, we calculated the log2(fold change) between the IFNγ/TNFα-treated group and the untreated group. Next, we computed the sum of the log2(fold changes) for the TNFα-treated group and the IFNγ-treated group, each compared to the untreated group. Both of the above results were used to calculate the combined difference value expressed as (IFNγ/TNFα – (TNFα + IFNγ)). Next, the outliers within these difference values were detected using the interquartile range (IQR) method. The criterion for classifying a value as an outlier was based on two conditions: either the difference value exceeded the highest quantile cutoff by 1.5 times the IQR or it fell below the lowest quantile cutoff by 1.5 times the IQR. The functions IQR and quantile are from the package stats included in R.
ELISAs
70,000 cells were plated into a 24 well plate in media lacking hydrocortisone for 16–20 h before stimulation. Cells were stimulated with cytokines or cytokines + inhibitors for 8 h before supernatant was harvested (n = 3 per condition). Supernatant was centrifuged for 5 min at 500g; supernatants were then stored at −80C until use. ELISAs were performed per manufacturer’s instructions (CXCL9: Bio-techne (DCX900) CXCL10: Bio-techne (DIP100) CXCL11: Bio-techne (DCX110)). All supernatants for CXCL9 and CXCL11 were run undiluted. Supernatants for CXCL10 in the IFNγ/TNFα, IFNγ/TNFα +DMSO, IFNγ/TNFα +62nM A-485, IFNγ/TNFα +15.6nM JQ1, IFNγ/TNFα +62nM A-485 15.6nM JQ1, IFNγ/TNFα +31nM A-485, IFNγ/TNFα +7.8nM JQ1, and IFNγ/TNFα +31nM A-485 7.8nM JQ1 conditions were diluted 1:240. Supernatants for CXCL10 in the IFNγ/TNFα +2000nM A-485 and IFNγ/TNFα +500nM JQ1 were diluted 1:120.
GLM computational
NanoString data was processed as described below. The averaged normalized transcript counts were utilized to generate a general linearized model (GLM) utilizing a Poisson regression model. This GLM included variables to examine the ability of A-485 alone, JQ1 alone or the multiplicative interaction between both inhibitors to explain the observed experimental data. The ability of these models to fit the experimental data was assessed via an ANOVA using Wald statistics. Estimated marginal means utilizing the multiplicative interaction term were calculated using the emmeans package in R for each gene. Trends at each dose of A-485 or JQ1 were calculated using emmtrends in R on the output from the emmeans function described above.
Hill coefficient modeling and IC50 calculations
Hill coefficient modeling and subsequent IC50 calculations were performed using the hillfit python package with the bottom_param = False and log_x = True.
Immunofluorescence
30,000 cells were plated into 8-well chamber slides (Chemglass Life Sciences CGN-3530-008). Cells were incubated in media lacking hydrocortisone for 16–20 h before being stimulated for 5, 15, 60, or 240 min. Cells were fixed with 4% formaldehyde for 15 min at RT and washed 3x with PBS. Cells were permeabilized for 10 min with methanol at −20C, washed 3x with PBS and blocked for 1 h at RT. Antibodies were added at the dilution of 1:6000, 1:1000, and 1:200 (IgG: Rabbit IgG Diagenode C15410206 Lot: RIG001AP RRID:AB_2722554, p65: Rabbit monoclonal p65 Cell Signaling Technology D14E12 Lot: 8242S RRID:AB_10859369 and STAT1: Rabbit monoclonal STAT1 Cell Signaling Technology 1495S Lot: 4 RRID:AB_2716280) respectively and antibodies were incubated overnight at 4C. Secondary antibody (ThermoFisher Goat anti-Rabbit Alexa Fluor 647 A21244 Lot: 2247991 RRID:AB_2535812) was added at a dilution of 1:500 and incubated for 1 h at RT. DAPI stain (ProLong Gold antifade reagent with DAPI Invitrogen: P36931 Lot: 2501098) was added to slides and cover slips were applied. Images were captured on Zeiss LSM880 Confocal Microscope 10X magnification. Scale bars indicate 100 micron.
Multi-isotope imaging mass spectrometry (MIMS)
Stable isotope tracers for DNA synthesis (13C-thymidine) and RNA synthesis (15N-uridine) were purchased from Cambridge Isotope Laboratories. HAEC were cultured in 13C-thymidine at a concentration of 50 (μM) for 14 days. At the final passage prior to the experiment, cells were seeded onto silicon wafers in culture dishes. 15N-uridine (50 μM) was added for 1 or 4 h with cytokines, then washed with PBS and fixed with 4% paraformaldehyde for 15 min. The cells were then washed with PBS, subjected to serial ethanol dehydration (50%, 70%, 90%, 95%, 100%), and air dried. Samples were then analyzed with a NanoSIMS 50L instrument (CAMECA), using previously published analytical methods.38,39,75 13C-thymidine labeling was measured by the 13C12C−/12C2− ratio and 15N-uridine was measured by the 12C15N−/12C14N− ratio as described previously.38,39,75 The instrument was also tuned to capture 32S−. Image files were visualized and analyzed with a custom plugin to ImageJ: OpenMIMS 3.0: https://github.com/BWHCNI/OpenMIMS.75 32S− and 13C12C−/12C2− ratio images were used to guide manual selection of regions of interest (ROIs) corresponding to the entire area of the nucleus and the cytoplasm (excluding the nucleus). The corresponding isotope ratios were then extracted representing the mean of all pixels contained within each respective ROI. Isotope ratio data are displayed in the manuscript (Figure 1) as hue saturation intensity (HSI) images. The lower bound of the scale (blue) was set at natural background (e.g., for 15N-uridine data a lower bound of 0 is equivalent to the natural background of 0.37% = no labeling and an upper bound of 100 corresponds to a ratio of 0.74%). For representative images, the upper bound of the scale was set to demonstrate regional differences in labeling. Importantly, the underlying quantitative data are unmodified by changes in the scaling of displayed images.
MTT assay
5,000 cells were plated into a 96 well plate in media lacking hydrocortisone (n = 3 per condition). MTT assay was performed per manufacturer’s instructions (Cell Proliferation Kit I (MTT) Roche 11465007001). In brief cells were stimulated for 48 h before MTT assay labeling reagent was added. MTT assay labeling reagent incubated for 4 h at 37C and 5% CO2 after which MTT solubilization reagent was added. Plates were allowed to incubate overnight before absorption was measured at 550nm.
NanoString experimental
20,000 cells were plated into 96 well plate in media lacking hydrocortisone 16–20 h before stimulation. Cells were stimulated for 1 h before they were harvested with iScript RT-qPCR Sample Prep Reagent (Bio-Rad 1708899). Lysates were transferred to PCR strips and placed at −80C or processed immediately after harvesting. NanoString hybridization was performed per manufacturer’s instructions with a custom probe set. In short A and B probes were resuspended at 5nM and 25nM respectively probes were then mixed and added to 7uL of sample lysate. Samples hybridized at 67°C for 24 h. Samples were quick spun down and then pooled and loaded on the NanoString nCounter Max prep station.
NanoString computational
The output of each.RCC file was processed to produce a file containing transcript counts for each gene of interest mapped to the position in the 96 well plate. The data was normalized in the following manner: first differences in hybridization were normalized by generating the geometric mean of the positive control samples within each column. The geometric mean was divided by the positive control of each well to generate the scaling factor which was applied to all genes in in the corresponding well, this was repeated to normalize all samples in a given column after which the geometric mean was calculated for the next column and the process was repeated again.
Next the samples were normalized to account for plex set hybridization variability by generating the geometric mean for each gene in the calibration column. A scaling factor was generated by multiplying the geometric mean of all calibration samples by 1 over the transcript count for a given gene in the calibration sample in a specific plex set. This scaling factor was applied to all transcript measurements in the specific plex set and was repeated for each gene that was measured and for all plex sets.
Housekeeper normalization was done by obtaining the geometric means of all the housekeeper genes at each position in the plex set. Next the arithmetic mean of all geometric means is calculated and used to generate a scaling factor dividing the arithmetic mean of all housekeeper geometric means by the geometric mean of all house keeper genes in a given position. This scaling factor is then applied to all transcripts in that corresponding position.
One step, real-time qPCR
10,000 TELOHAECs were plated in a 96 well plate containing media without hydrocortisone 16–20 h before stimulation. Cells were stimulated for 1 h before they were harvested with iScript RT-qPCR Sample Prep Reagent (Bio-Rad 1708899). qPCR was performed per manufacturer’s instructions using 1uL of lysate and the iTaq Universal SYBR Green One-Step Kit (Bio-Rad 1725151).
RNA-seq
75,000 cells were plated in media lacking hydrocortisone for 16–20 h. Cells were simulated for 1 h before cells were harvested using Pure-link lysis buffer containing 10% beta-mercaptoethanol. Lysate was processed immediately using the Pure-link RNA Mini kit (Invitrogen, ThermoFisher 12183025) or stored at −80C. RNA was isolated following the manufacturer’s instructions for the Pure-link RNA Mini kit (Invitrogen, ThermoFisher 12183025). RNA-seq libraries were generated using NEBNext Poly(A) mRNA Magnetic Isolation Module (NEB E7490) per manufacturer’s instructions. Paired end 150bp sequencing was performed on an Illumina NovaSeq 6000 machine targeting 20,000,000 reads per sample.
RNA-seq computational analysis
Reads were trimmed to remove adapter sequences using Cutadapt (v2.10)76 and aligned to the Gencode GRCh38.p13 genome using STAR (v2.7.8a).77 Gencode v38 gene annotations were provided to STAR to improve the accuracy of mapping. Quality control on both raw reads and adaptor-trimmed reads was performed using FastQC (v0.11.9) (www.bioinformatics.babraham.ac.uk/projects/fastqc). featureCounts (v2.0.2)78 was used to count the number of mapped reads to each gene. Heatmap3 was used for cluster analysis and visualization.79 Significantly differential expressed genes with absolute fold change ≥ 2 and FDR adjusted p value ≤ 0.05 were detected by DESeq2 (v1.30.1).65
Western Blot
200,000 TELO-HAECs were harvested and pelleted by centrifugation at 500g for 5 min at 4C. Cells were lysed via the addition of whole cell lysis buffer and passed through a 28-gauge insulin needle 10 times. Samples were rested on ice for 10 min before being centrifuged at 21,000g for 7 min at 4C. Supernatant was transferred to new tube and protein concentration determined via BCA assay. 22.5 μg of protein was loaded into NuPage 4–12% Bis Tris 12 well gels (Invitrogen NP0322BOX). Kaleidoscope Protein ladder (BioRad 1610375) was diluted 1:1 in 1x LDS and gel was run at 120V for 2 h in 1x MOPS buffer. Gel was transferred to a nitrocellulose membrane at 30V for 90 min in 10% methanol transfer buffer. After transfer the membrane was transferred to block for 1 h at RT on rocker. p65 blot was performed by diluting p65 (Rabbit Active Motif 39369 RRID:AB_2793231) 1:2500, beta actin mouse (Monoclonal (Clone 15G5A/E2) ThermoFisher MA1140 Lot: TK276375 RRID:AB_2536844) was diluted 1:2500. STAT1 blot was performed by diluting Rabbit STAT1 (Cell Signaling Technology 14995S Lot:4 RRID:AB_2716280) diluted 1:1000 and beta actin mouse (Monoclonal (Clone 15G5A/E2) ThermoFisher MA1140 Lot: TK276375 RRID:AB_2536844) was diluted 1:2500. Anti-FLAG antibody (Sigma Aldrich, F3165) was used at a final concentration of 1 μg/mL. Membranes were washed 3× in 0.1% TBS-T and secondary antibodies were diluted 1:1000 (Goat anti-Rabbit) and 1:1000 (Goat anti-mouse) and incubated for 1 h while rocking. After secondary antibody, membranes were again washed 3× in 0.1% TBST then imaged on LICOR Odyssey. Densitometry was performed using Image Studio Lite from LI-COR Biosciences.
Quantification and statistical analysis
For assessment of statistical significance both R and Prism were utilize. Details on the statistical tests performed for individual experiments can be found in the figure legend of each respective figure. Any p-value less than 0.05 was determined to be statistically significant in this study. For RNA-seq and ATAC-seq adjusted p-values were used.
Acknowledgments
We thank Angela Jones and the other members of the team at the Vanderbilt genomics core (VANTAGE). This work was supported by the National Institutes of Health under the grants R01HL146654 and R01HL160970. Graphical depictions of experimental workflows and graphical summary of the manuscript were created with Biorender.com.
Author contributions
R.C.B. and J.D.B. designed all experiments, analyzed all data (except MIMS), and wrote the manuscript. L.M.D. and D.P.B. performed western blots and ATAC-seq cell preparation and assisted with NanoString experiments. M.E.F. and E.E.C. cloned the 3xFLAG-STAT1 and p65 constructs. C.G. and M.L.S. performed MIMS experiments and analyzed data for MIMS. L.K.S. and Q.S. performed RNA-seq analysis and assisted with ATAC-seq analysis.
Declaration of interests
The authors declare no competing interests.
Published: May 16, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.110011.
Supplemental information
References
- 1.Chousterman B.G., Swirski F.K., Weber G.F. Cytokine storm and sepsis disease pathogenesis. Semin. Immunopathol. 2017;39:517–528. doi: 10.1007/s00281-017-0639-8. [DOI] [PubMed] [Google Scholar]
- 2.Karki R., Sharma B.R., Tuladhar S., Williams E.P., Zalduondo L., Samir P., Zheng M., Sundaram B., Banoth B., Malireddi R.K.S., et al. Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell. 2021;184:149–168.e17. doi: 10.1016/j.cell.2020.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baltimore D. NF-κB is 25. Nat. Immunol. 2011;12:683–685. doi: 10.1038/ni.2072. [DOI] [PubMed] [Google Scholar]
- 4.Majoros A., Platanitis E., Kernbauer-Hölzl E., Rosebrock F., Müller M., Decker T. Canonical and Non-Canonical Aspects of JAK-STAT Signaling: Lessons from Interferons for Cytokine Responses. Front. Immunol. 2017;8:29. doi: 10.3389/fimmu.2017.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hill C.S., Treisman R. Transcriptional regulation by extracellular signals: mechanisms and specificity. Cell. 1995;80:199–211. doi: 10.1016/0092-8674(95)90403-4. [DOI] [PubMed] [Google Scholar]
- 6.Carey M., Lin Y.S., Green M.R., Ptashne M. A mechanism for synergistic activation of a mammalian gene by GAL4 derivatives. Nature. 1990;345:361–364. doi: 10.1038/345361a0. [DOI] [PubMed] [Google Scholar]
- 7.Ohmori Y., Schreiber R.D., Hamilton T.A. Synergy between interferon-gamma and tumor necrosis factor-alpha in transcriptional activation is mediated by cooperation between signal transducer and activator of transcription 1 and nuclear factor kappaB. J. Biol. Chem. 1997;272:14899–14907. doi: 10.1074/jbc.272.23.14899. [DOI] [PubMed] [Google Scholar]
- 8.Johnson D.R., Pober J.S. HLA class I heavy-chain gene promoter elements mediating synergy between tumor necrosis factor and interferons. Mol. Cell Biol. 1994;14:1322–1332. doi: 10.1128/mcb.14.2.1322-1332.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barker J.N., Jones M.L., Mitra R.S., Crockett-Torabe E., Fantone J.C., Kunkel S.L., Warren J.S., Dixit V.M., Nickoloff B.J. Modulation of keratinocyte-derived interleukin-8 which is chemotactic for neutrophils and T lymphocytes. Am. J. Pathol. 1991;139:869–876. [PMC free article] [PubMed] [Google Scholar]
- 10.Czimmerer Z., Halasz L., Daniel B., Varga Z., Bene K., Domokos A., Hoeksema M., Shen Z., Berger W.K., Cseh T., et al. The epigenetic state of IL-4-polarized macrophages enables inflammatory cistromic expansion and extended synergistic response to TLR ligands. Immunity. 2022;55:2006–2026.e6. doi: 10.1016/j.immuni.2022.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peng T., Zhu J., Hwangbo Y., Corey L., Bumgarner R.E. Independent and cooperative antiviral actions of beta interferon and gamma interferon against herpes simplex virus replication in primary human fibroblasts. J. Virol. 2008;82:1934–1945. doi: 10.1128/JVI.01649-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yuan H., Corbi N., Basilico C., Dailey L. Developmental-specific activity of the FGF-4 enhancer requires the synergistic action of Sox2 and Oct-3. Genes Dev. 1995;9:2635–2645. doi: 10.1101/gad.9.21.2635. [DOI] [PubMed] [Google Scholar]
- 13.Marfaing-Koka A., Devergne O., Gorgone G., Portier A., Schall T.J., Galanaud P., Emilie D. Regulation of the production of the RANTES chemokine by endothelial cells. Synergistic induction by IFN-gamma plus TNF-alpha and inhibition by IL-4 and IL-13. J. Immunol. 1995;154:1870–1878. [PubMed] [Google Scholar]
- 14.Elsarrag S., Lin C.Y., Hodges C.H., Martin J.F., Zechiedrich L. Baylor College of Medicine. Quantitative and Computational Biosciences Graduate Program. 2021. Mapping Transcription Factor Control of Inflammatory Regulation. [Google Scholar]
- 15.Herschlag D., Johnson F.B. Synergism in transcriptional activation: a kinetic view. Genes Dev. 1993;7:173–179. doi: 10.1101/gad.7.2.173. [DOI] [PubMed] [Google Scholar]
- 16.Martinez-Corral R., Park M., Biette K.M., Friedrich D., Scholes C., Khalil A.S., Gunawardena J., DePace A.H. Transcriptional kinetic synergy: A complex landscape revealed by integrating modeling and synthetic biology. Cell Syst. 2023;14:324–339.e7. doi: 10.1016/j.cels.2023.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang J.J., Vinkemeier U., Gu W., Chakravarti D., Horvath C.M., Darnell J.E., Jr. Two contact regions between Stat1 and CBP/p300 in interferon gamma signaling. Proc. Natl. Acad. Sci. USA. 1996;93:15092–15096. doi: 10.1073/pnas.93.26.15092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wojciak J.M., Martinez-Yamout M.A., Dyson H.J., Wright P.E. Structural basis for recruitment of CBP/p300 coactivators by STAT1 and STAT2 transactivation domains. EMBO J. 2009;28:948–958. doi: 10.1038/emboj.2009.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mukherjee S.P., Behar M., Birnbaum H.A., Hoffmann A., Wright P.E., Ghosh G. Analysis of the RelA:CBP/p300 Interaction Reveals Its Involvement in NF-κB-Driven Transcription. PLoS Biol. 2013;11 doi: 10.1371/journal.pbio.1001647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerritsen M.E., Williams A.J., Neish A.S., Moore S., Shi Y., Collins T. CREB-binding protein/p300 are transcriptional coactivators of p65. Proc. Natl. Acad. Sci. USA. 1997;94:2927–2932. doi: 10.1073/pnas.94.7.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen L., Fischle W., Verdin E., Greene W.C. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science. 2001;293:1653–1657. doi: 10.1126/science.1062374. [DOI] [PubMed] [Google Scholar]
- 22.Huang B., Yang X.D., Zhou M.M., Ozato K., Chen L.F. Brd4 coactivates transcriptional activation of NF-kappaB via specific binding to acetylated RelA. Mol. Cell Biol. 2009;29:1375–1387. doi: 10.1128/MCB.01365-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brown J.D., Lin C.Y., Duan Q., Griffin G., Federation A., Paranal R.M., Bair S., Newton G., Lichtman A., Kung A., et al. NF-κB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol. Cell. 2014;56:219–231. doi: 10.1016/j.molcel.2014.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Devaiah B.N., Lewis B.A., Cherman N., Hewitt M.C., Albrecht B.K., Robey P.G., Ozato K., Sims R.J., Singer D.S. BRD4 is an atypical kinase that phosphorylates serine2 of the RNA polymerase II carboxy-terminal domain. Proc. Natl. Acad. Sci. USA. 2012;109:6927–6932. doi: 10.1073/pnas.1120422109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marshall N.F., Price D.H. Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J. Biol. Chem. 1995;270:12335–12338. doi: 10.1074/jbc.270.21.12335. [DOI] [PubMed] [Google Scholar]
- 26.Hogg S.J., Motorna O., Kearney C.J., Derrick E.B., House I.G., Todorovski I., Kelly M.J., Zethoven M., Bromberg K.D., Lai A., et al. Distinct modulation of IFNγ-induced transcription by BET bromodomain and catalytic P300/CBP inhibition in breast cancer. Clin. Epigenetics. 2022;14:96. doi: 10.1186/s13148-022-01316-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hogg S.J., Motorna O., Cluse L.A., Johanson T.M., Coughlan H.D., Raviram R., Myers R.M., Costacurta M., Todorovski I., Pijpers L., et al. Targeting histone acetylation dynamics and oncogenic transcription by catalytic P300/CBP inhibition. Mol. Cell. 2021;81:2183–2200.e13. doi: 10.1016/j.molcel.2021.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spriano F., Gaudio E., Cascione L., Tarantelli C., Melle F., Motta G., Priebe V., Rinaldi A., Golino G., Mensah A.A., et al. Antitumor activity of the dual BET and CBP/EP300 inhibitor NEO2734. Blood Adv. 2020;4:4124–4135. doi: 10.1182/bloodadvances.2020001879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mills R.J., Humphrey S.J., Fortuna P.R.J., Lor M., Foster S.R., Quaife-Ryan G.A., Johnston R.L., Dumenil T., Bishop C., Rudraraju R., et al. BET inhibition blocks inflammation-induced cardiac dysfunction and SARS-CoV-2 infection. Cell. 2021;184:2167–2182.e22. doi: 10.1016/j.cell.2021.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Godo S., Shimokawa H. Endothelial Functions. Arterioscler. Thromb. Vasc. Biol. 2017;37:e108–e114. doi: 10.1161/ATVBAHA.117.309813. [DOI] [PubMed] [Google Scholar]
- 31.Wang M., Hao H., Leeper N.J., Zhu L., Early Career Committee Thrombotic Regulation From the Endothelial Cell Perspectives. Arterioscler. Thromb. Vasc. Biol. 2018;38:e90–e95. doi: 10.1161/ATVBAHA.118.310367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luk C., Haywood N.J., Bridge K.I., Kearney M.T. Paracrine Role of the Endothelium in Metabolic Homeostasis in Health and Nutrient Excess. Front. Cardiovasc. Med. 2022;9 doi: 10.3389/fcvm.2022.882923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muller W.A. Mechanisms of leukocyte transendothelial migration. Annu. Rev. Pathol. 2011;6:323–344. doi: 10.1146/annurev-pathol-011110-130224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adamski J., Ma Z., Nozell S., Benveniste E.N. 17beta-Estradiol inhibits class II major histocompatibility complex (MHC) expression: influence on histone modifications and cbp recruitment to the class II MHC promoter. Mol. Endocrinol. 2004;18:1963–1974. doi: 10.1210/me.2004-0098. [DOI] [PubMed] [Google Scholar]
- 35.Hirschberg H., Bergh O.J., Thorsby E. Antigen-presenting properties of human vascular endothelial cells. J. Exp. Med. 1980;152:249s–255s. [PubMed] [Google Scholar]
- 36.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 37.Gyngard F., Steinhauser M.L. Biological explorations with nanoscale secondary ion mass spectrometry. J. Anal. At. Spectrom. 2019;34:1534–1545. doi: 10.1039/c9ja00171a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Steinhauser M.L., Bailey A.P., Senyo S.E., Guillermier C., Perlstein T.S., Gould A.P., Lee R.T., Lechene C.P. Multi-isotope imaging mass spectrometry quantifies stem cell division and metabolism. Nature. 2012;481:516–519. doi: 10.1038/nature10734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guillermier C., Doherty S.P., Whitney A.G., Babaev V.R., Linton M.F., Steinhauser M.L., Brown J.D. Imaging mass spectrometry reveals heterogeneity of proliferation and metabolism in atherosclerosis. JCI Insight. 2019;4 doi: 10.1172/jci.insight.128528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qi X.F., Kim D.H., Yoon Y.S., Jin D., Huang X.Z., Li J.H., Deung Y.K., Lee K.J. Essential involvement of cross-talk between IFN-gamma and TNF-alpha in CXCL10 production in human THP-1 monocytes. J. Cell. Physiol. 2009;220:690–697. doi: 10.1002/jcp.21815. [DOI] [PubMed] [Google Scholar]
- 41.Hao S., Baltimore D. The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat. Immunol. 2009;10:281–288. doi: 10.1038/ni.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Metzemaekers M., Vanheule V., Janssens R., Struyf S., Proost P. Overview of the Mechanisms that May Contribute to the Non-Redundant Activities of Interferon-Inducible CXC Chemokine Receptor 3 Ligands. Front. Immunol. 2017;8:1970. doi: 10.3389/fimmu.2017.01970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Freeman A.J., Vervoort S.J., Michie J., Ramsbottom K.M., Silke J., Kearney C.J., Oliaro J. HOIP limits anti-tumor immunity by protecting against combined TNF and IFN-gamma-induced apoptosis. EMBO Rep. 2021;22 doi: 10.15252/embr.202153391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Woznicki J.A., Saini N., Flood P., Rajaram S., Lee C.M., Stamou P., Skowyra A., Bustamante-Garrido M., Regazzoni K., Crawford N., et al. TNF-alpha synergises with IFN-gamma to induce caspase-8-JAK1/2-STAT1-dependent death of intestinal epithelial cells. Cell Death Dis. 2021;12:864. doi: 10.1038/s41419-021-04151-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kang K., Park S.H., Chen J., Qiao Y., Giannopoulou E., Berg K., Hanidu A., Li J., Nabozny G., Kang K., et al. Interferon-gamma Represses M2 Gene Expression in Human Macrophages by Disassembling Enhancers Bound by the Transcription Factor MAF. Immunity. 2017;47:235–250.e4. doi: 10.1016/j.immuni.2017.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gilan O., Rioja I., Knezevic K., Bell M.J., Yeung M.M., Harker N.R., Lam E.Y.N., Chung C.W., Bamborough P., Petretich M., et al. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immunoinflammation. Science. 2020;368:387–394. doi: 10.1126/science.aaz8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Okawa S., Saltó C., Ravichandran S., Yang S., Toledo E.M., Arenas E., Del Sol A. Transcriptional synergy as an emergent property defining cell subpopulation identity enables population shift. Nat. Commun. 2018;9:2595. doi: 10.1038/s41467-018-05016-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mehta N.N., Teague H.L., Swindell W.R., Baumer Y., Ward N.L., Xing X., Baugous B., Johnston A., Joshi A.A., Silverman J., et al. IFN-gamma and TNF-alpha synergism may provide a link between psoriasis and inflammatory atherogenesis. Sci. Rep. 2017;7 doi: 10.1038/s41598-017-14365-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kandhaya-Pillai R., Yang X., Tchkonia T., Martin G.M., Kirkland J.L., Oshima J. TNF-α/IFN-γ synergy amplifies senescence-associated inflammation and SARS-CoV-2 receptor expression via hyper-activated JAK/STAT1. Aging Cell. 2022;21 doi: 10.1111/acel.13646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Platanitis E., Gruener S., Ravi Sundar Jose Geetha A., Boccuni L., Vogt A., Novatchkova M., Sommer A., Barozzi I., Müller M., Decker T. Interferons reshape the 3D conformation and accessibility of macrophage chromatin. iScience. 2022;25 doi: 10.1016/j.isci.2022.103840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ohmori Y., Hamilton T.A. The interferon-stimulated response element and a kappa B site mediate synergistic induction of murine IP-10 gene transcription by IFN-gamma and TNF-alpha. J. Immunol. 1995;154:5235–5244. [PubMed] [Google Scholar]
- 52.Voss T.C., Hager G.L. Dynamic regulation of transcriptional states by chromatin and transcription factors. Nat. Rev. Genet. 2014;15:69–81. doi: 10.1038/nrg3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goldstein I., Paakinaho V., Baek S., Sung M.H., Hager G.L. Synergistic gene expression during the acute phase response is characterized by transcription factor assisted loading. Nat. Commun. 2017;8:1849. doi: 10.1038/s41467-017-02055-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kramer O.H., Knauer S.K., Greiner G., Jandt E., Reichardt S., Guhrs K.H., Stauber R.H., Bohmer F.D., Heinzel T. A phosphorylation-acetylation switch regulates STAT1 signaling. Genes Dev. 2009;23:223–235. doi: 10.1101/gad.479209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Haspel R.L., Darnell J.E., Jr. A nuclear protein tyrosine phosphatase is required for the inactivation of Stat1. Proc. Natl. Acad. Sci. USA. 1999;96:10188–10193. doi: 10.1073/pnas.96.18.10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Scholes C., DePace A.H., Sánchez Á. Combinatorial gene regulation through kinetic control of the transcription cycle. Cell Syst. 2017;4:97–108.e109. doi: 10.1016/j.cels.2016.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nicodeme E., Jeffrey K.L., Schaefer U., Beinke S., Dewell S., Chung C.W., Chandwani R., Marazzi I., Wilson P., Coste H., et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468:1119–1123. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Anand P., Brown J.D., Lin C.Y., Qi J., Zhang R., Artero P.C., Alaiti M.A., Bullard J., Alazem K., Margulies K.B., et al. BET bromodomains mediate transcriptional pause release in heart failure. Cell. 2013;154:569–582. doi: 10.1016/j.cell.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang Z., Yik J.H.N., Chen R., He N., Jang M.K., Ozato K., Zhou Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell. 2005;19:535–545. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 60.Hargreaves D.C., Horng T., Medzhitov R. Control of inducible gene expression by signal-dependent transcriptional elongation. Cell. 2009;138:129–145. doi: 10.1016/j.cell.2009.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chan C.H., Fang C., Yarilina A., Prinjha R.K., Qiao Y., Ivashkiv L.B. BET bromodomain inhibition suppresses transcriptional responses to cytokine-Jak-STAT signaling in a gene-specific manner in human monocytes. Eur. J. Immunol. 2015;45:287–297. doi: 10.1002/eji.201444862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based Analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Grandi F.C., Modi H., Kampman L., Corces M.R. Chromatin accessibility profiling by ATAC-seq. Nat. Protoc. 2022;17:1518–1552. doi: 10.1038/s41596-022-00692-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bailey T.L., Johnson J., Grant C.E., Noble W.S. The MEME Suite. Nucleic Acids Res. 2015;43:W39–W49. doi: 10.1093/nar/gkv416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McLeay R.C., Bailey T.L. Motif Enrichment Analysis: a unified framework and an evaluation on ChIP data. BMC Bioinf. 2010;11:165. doi: 10.1186/1471-2105-11-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vierstra J., Lazar J., Sandstrom R., Halow J., Lee K., Bates D., Diegel M., Dunn D., Neri F., Haugen E., et al. Global reference mapping of human transcription factor footprints. Nature. 2020;583:729–736. doi: 10.1038/s41586-020-2528-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Concordet J.P., Haeussler M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018;46:W242–W245. doi: 10.1093/nar/gky354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Layden H.M., Eleuteri N.A., Hiebert S.W., Stengel K.R. A protocol for rapid degradation of endogenous transcription factors in mammalian cells and identification of direct regulatory targets. STAR Protoc. 2021;2 doi: 10.1016/j.xpro.2021.100530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu N., Hargreaves V.V., Zhu Q., Kurland J.V., Hong J., Kim W., Sher F., Macias-Trevino C., Rogers J.M., Kurita R., et al. Direct Promoter Repression by BCL11A Controls the Fetal to Adult Hemoglobin Switch. Cell. 2018;173:430–442.e17. doi: 10.1016/j.cell.2018.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fujiwara Y., Tanno Y., Sugishita H., Kishi Y., Makino Y., Okada Y. Preparation of optimized concanavalin A-conjugated Dynabeads® magnetic beads for CUT&Tag. PLoS One. 2021;16 doi: 10.1371/journal.pone.0259846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guillermier C., Poczatek J.C., Taylor W.R., Steinhauser M.L. Quantitative imaging of deuterated metabolic tracers in biological tissues with nanoscale secondary ion mass spectrometry. Int. J. Mass Spectrom. 2017;422:42–50. doi: 10.1016/j.ijms.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 77.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 79.Zhao S., Guo Y., Sheng Q., Shyr Y. Advanced heat map and clustering analysis using heatmap3. BioMed Res. Int. 2014;2014 doi: 10.1155/2014/986048. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
ATAC-sequencing, CUT&RUN, and RNA-sequencing datasets generated in this manuscript have been deposited at NCBI GEO under the accession number: GSE232168 and are publicly available.
-
•
Custom NanoString analysis code and GLM for determining synergistic inhibition are publicly available at Github:/jonathanbrown484/synergy
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.