

Abstract
The greatest challenge in cancer therapy is to eradicate cancer cells with minimal damage to normal cells. Targeted therapy has been developed to meet that challenge, showing a substantially increased therapeutic index compared with conventional cancer therapies. Antibodies are important members of the family of targeted therapeutic agents because of their extraordinarily high specificity to the target antigens. Therapeutic antibodies use a range of mechanisms that directly or indirectly kill the cancer cells. Early antibodies were developed to directly antagonize targets on cancer cells. This was followed by advancements in linker technologies that allowed the production of antibody-drug conjugates (ADCs) that guide cytotoxic payloads to the cancer cells. Improvement in our understanding of the biology of T cells led to the production of immune checkpoint-inhibiting antibodies that indirectly kill the cancer cells through activation of the T cells. Even more recently, bispecific antibodies were synthetically designed to redirect the T cells of a patient to kill the cancer cells. In this Review, we summarize the different approaches used by therapeutic antibodies to target cancer cells. We discuss their mechanisms of action, the structural basis for target specificity, clinical applications and the ongoing research to improve efficacy and reduce toxicity.
Introduction
In the past century, potent therapeutic approaches such as chemo-therapy and radiation therapy have been developed to treat cancer. Unfortunately, these approaches often lack sufficient specificity to allow high enough doses required to eradicate cancer cells without causing intolerable toxicity. Monoclonal antibodies can provide the level of specificity needed for a substantially enlarged therapeutic window, with some antibodies even able to discriminate between two antigens that differ by a single amino acid or by a posttranslational modi-fication1,2. The development of several seminal technologies enabled the use of antibodies for therapeutic purposes. First, the hybridoma system that was developed in the 1970s allowed for the production and selection of highly specific mouse monoclonal antibodies targeting human antigens. Second, the ability to graft a human antibody con-stant region to a mouse antibody variable region generates chimeric antibodies (Fig. 1) with better therapeutic efficacy and fewer adverse effects. Third, the use of transgenic mouse models and phage display systems in the 1990s enabled the generation of fully human antibodies against cancer targets. With these technological advancements and increased clinical demands, the past two and a half decades have seen an explosion of new antibody-based therapeutics. Since 1997, more than fifty antibody-based therapeutics for oncology applications have received approval by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA)3 (Supplementary Table 1). In addition to antibody-based therapeutics, antibody fragments are also used to generate chimeric antigen receptors (CARs) for T cell engi-neering4,5. In this Review, we focus on antibody-based therapeutics approved by the FDA and EMA and on antibodies that are progressing through preclinical and clinical development. We discuss the evolution of methods that were successively used for the generation of chimeric, humanized and human antibodies. We detail the range of formats such as monospecific antibodies, immune checkpoint inhibitors, bispecific antibodies and conjugated antibodies that are being developed for targeting cancer cells. Finally, we summarize the emerging research being conducted to meet the current challenges in cancer-targeting antibody development.
Fig. 1 ∣. Antibody components.

a, Antibodies consist of two identical light chains and heavy chains that are held together by disulfide bonds and resemble a Y-shaped structure. Each light and heavy chain contains a variable (VL and VH) domain responsible for antigen binding and constant (CL and CH) domains that determine the half-life and effector function of the antibody. Enzymatic processing can break up antibodies into two fragments named fragment antigen binding (Fab) and fragment crystallizable (Fc). The light and heavy chain variable regions together make up the fragment variable (Fv), the smallest fragment that retains antigen-binding capacity. Manufactured Fv fragments are joined together by a flexible peptide linker to form a single chain named single-chain variable fragment (scFv). Antibodies are grouped as mouse, chimeric, humanized and human based on the amount of peptide sequence derived from each species. aBelantamab mafodotin was withdrawn but may gain re-approval based on ongoing trials. Teclistamab (a combination of a humanized and a human antibody) is considered as a humanized antibody for this figure. CDRs, complementarity-determining regions.
Antibody generation
A range of techniques are used to identify therapeutic monoclonal antibodies or antibody fragments targeting tumour antigens. Such techniques include immunizations in rodents with the target antigen, display technologies such as phage or yeast display, single B cell cloning from humans and transgenic mice, and identifying single-domain antibodies (named nanobodies) from camelids6–8. Further develop-ment of antibodies of clinical utility involves antibody engineering enabling them to be progressively more compatible with the human immune system.
Chimeric and humanized antibodies
Monoclonal antibodies have traditionally been produced by using the hybridoma technology developed by Cesar Milstein and Georges Kohler in 1975 (ref. 9). In this process, B cells are isolated from mouse spleens or lymph nodes after immunization with a specific antigen and then fused with a myeloma cell line such as Sp2/0 to form hybrid cells called hybridoma cell lines. The hybridoma clones are usually screened by enzyme-linked immunosorbent assay (ELISA) for protein binding and flow cytometry for cell binding. The hybridoma technol-ogy generated a range of mouse monoclonal antibodies robustly and cost-effectively. However, mouse antibodies infused in humans generate a human anti-mouse antibody (HAMA) immune response limiting their efficacy and increasing adverse effects10. Consequently, the majority of mouse antibodies that received regulatory approval were later withdrawn (Fig. 1). Blinatumomab (a bispecific T cell engager targeting CD19), which uses two mouse-derived single-chain variable fragment (scFv) formats and lacks a fragment crystallizable (Fc) segment, is the only murine antibody that gained widespread adoption in the clinic.
To reduce the induction of an immune response in patients, the constant region of mouse antibodies was replaced with their human counterparts to produce chimeric antibodies (Fig. 1). Rituximab (anti- CD20) and cetuximab (anti-epidermal growth factor receptor (EGFR)) (Table 1) are among the first chimeric antibodies that were generated by attaching variable regions of mouse antibodies 2B8 (ref. 11) and 225 (ref. 12) to the human immunoglobulin G1 (IgG1) heavy chain and human kappa light chain constant regions. However, chimeric antibodies with mouse variable regions can still be recognized as foreign, leading to a human anti-chimeric antibody (HACA) immune response that may clear the therapeutic antibody13. This may limit the repetitive applica-tion of chimeric antibodies and impede their clinical development. To further reduce an unwanted immune response, the human content of mouse monoclonal antibodies were increased by a process developed in the 1980s known as ‘humanization’14. Humanization involves graft-ing only the complementarity-determining regions (CDRs) of a mouse antibody into the framework region of a human antibody (Fig. 1). Tras- tuzumab was one of the first humanized antibodies to be developed by transplanting the CDRs of the mouse human epidermal growth factor receptor 2 (HER2) antibody mumAb4D5, into a human antibody frame-work15, and a similar strategy was utilized to develop obinutuzumab (anti-CD20), pembrolizumab (anti-programmed cell death protein 1 (PD1)) and atezolizumab (anti-PD1 ligand 1 (PDL1)) (Table 1).
Table 1 ∣.
Monospecific antibodies approved or nearing approval by the FDA and EMA
| Target | Antibody | Isotype | Format | Antibody modification | Major indications |
|---|---|---|---|---|---|
| Mechanism of action: cancer killing by direct cytotoxicity (receptor blocking, ADCC, ADCP and CDC) | |||||
| CD20 | Rituximab | IgG1 | Chimeric | NA | Lymphomas |
| Obinutuzumab | Humanized | Afucosylated (increase ADCC) | |||
| Ofatumumab | Human | NA | |||
| Her2 | Trastuzumab | IgG1 | Humanized | NA | HER2+ breast cancer (all), gastric cancer (trastuzumab only) |
| Margetuximab | Chimeric | L235V; F243L; R292P; Y300L; P396L (increase ADCC) | |||
| Pertuzumab | Humanized | NA | |||
| EGFR | Cetuximab | IgG1 | Chimeric | NA | HNC, colorectal cancer |
| Necitumumab | Human | NA | Lung cancer | ||
| Panitumumab | IgG2 | Human | NA | Colorectal cancer | |
| VEGF | Bevacizumab | IgG1 | Humanized | NA | Colorectal cancer, lung cancer, glioblastoma, RCC, ovarian and fallopian tube cancer, HCC |
| VEGFR2 | Ramucirumab | IgG1 | Human | NA | Gastroesophageal cancer, lung cancer, HCC |
| CD52 | Alemtuzumab | IgG1 | Humanized | NA | Chronic lymphocytic leukaemia |
| GD2 | Dinutuximab | IgG1 | Chimeric | NA | Neuroblastoma |
| Naxitamab | Humanized | NA | |||
| CD38 | Daratumumab | IgG1 | Human | NA | Multiple myeloma |
| Isatuximab | Chimeric | NA | |||
| SLAMF7 | Elotuzumab | IgG1 | Humanized | NA | Multiple myeloma |
| CCR4 | Mogamulizumab | IgG1 | Humanized | Afucosylation (increase ADCC) | Lymphoma |
| CD19 | Tafasitamab | IgG1 | Humanized | S239D and I332E (increase ADCC) | Lymphoma |
| Mechanism of action: cancer killing by T cell activation (immune checkpoint inhibition) | |||||
| CTLA4 | Ipilimumab | IgG1 | Human | NA | Melanoma, RCC, lung cancer, HCC, oesophageal cancer, colorectal cancer (MSI-H) |
| Tremelimumab | IgG2 | Human | NA | HCC, lung cancer | |
| PD1 | Nivolumab | IgG4 | Human | S228P (prevent Fab arm exchange with wild-type IgG4) | Melanoma, lung cancer, mesothelioma, RCC, Hodgkin lymphoma, SCC, urothelial cancer, colorectal cancer (MSI-H), HCC, gastric and oesophageal cancer |
| Pembrolizumab | Humanized | S228P | Melanoma, lung cancer, HNC, Hodgkin lymphoma, PMBCL, MSI-H cancers, gastric and oesophageal cancers, cervical cancer, HCC, MCC, RCC, endometrial cancer, SCC, TNBC | ||
| Cemiplimab | Humanized | S228P | SCC, BCC, lung cancer | ||
| Dostarlimab | Humanized | S228P | Endometrial cancer, MSI-H/dMMR solid cancers | ||
| Retifanlimab | Humanized | S228P | MCC | ||
| Tislelizumab | Humanized | S228P and R409K, and E233P, F234V, L235A and D265A (reduce FcγR binding) | Oesophageal squamous cell carcinoma | ||
| Toripalimab | Humanized | S228P | Nasopharyneal carcinoma, oesophageal squamous cell carcinoma | ||
| Sintilimab (pending approval) | Human | S228P | NA | ||
| Serplulimab (pending approval) | Human | S228P | NA | ||
| PDL1 | Atezolizumab | IgG1 | Humanized | N297A (aglycosylated Fc to decrease ADCC) | Lung cancer, HCC, melanoma, ASPS |
| Avelumab | Human | NA | MCC, urothelial cancer, RCC | ||
| Durvalumab | Human | L234F/L235E/P331S (Fc disabled to decrease ADCC) | Lung cancer, biliary cancer, HCC | ||
| Cosibelimab (pending approval) | Human | NA | NA | ||
| Sugemalimab (pending approval) | IgG4 | Human | S228P | NA | |
| LAG-3 | Relatlimab | IgG4 | Human | S228P | Melanoma (in combination with nivolumab) |
ADCC, antibody-dependent cellular cytotoxicity; ADCP, antibody-dependent cellular phagocytosis; ASPS, alveolar soft part sarcoma; BCC, basal cell cancer; CDC, complement-dependent cytotoxicity; dMMR, deficient mismatch repair; HCC, hepatocellular carcinoma; HNC, head and neck cancer; IgG, immunoglobulin G; MCC, Merkel cell carcinoma; MSI-H, microsatellite instability-high; NA, not available; PMBCL, primary mediastinal large B cell lymphoma; RCC, renal cell cancer; SCC, squamous cell cancer; TNBC, triple-negative breast cancer.
Besides mouse hybridoma technology, rabbit hybridoma16 has been developed for producing rabbit monoclonal antibodies against tumour antigens such as mesothelin. The CDR grafting methodology has been adapted to humanize rabbit monoclonal antibodies such as YP218 targeting mesothelin for clinical development17. The majority of therapeutic antibodies approved by the regulatory agencies have been generated using the hybridoma method followed by engineering into chimeric or humanized forms and are extensively used for cancer therapy18 (Fig. 1).
Human antibodies
The final step towards generating fully human antibodies was made pos-sible with the help of two techniques developed in the 1990s, the human antibody phage display19 and the human antibody expressing trans-genic mouse models20. To generate transgenic mice producing human antibodies, human Ig loci or variable regions are inserted into the mouse genome, along with the disruption of the mouse Ig genes20. B cells are isolated from the mice immunized with a target antigen for single B cell cloning and sequencing. The transgenic mouse platforms XenoMouse (Abgenix/Amgen), VelocImmune (Regeneron) and HuMab (Medarex/ Bristol Myers Squibb) yielded nine approved cancer-targeting human antibodies: daratumumab (anti-CD38), ipilimumab (anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA4)), nivolumab (anti-PD1), ofatumumab (anti-CD20), panitumumab (anti-EGFR), durvalumab (anti-PDL1), cemiplimab (anti-PD1), tisotumab (anti-tissue factor (TF)) and relatlimab (anti-lymphocyte activation gene 3 (LAG3))21 (Table 1). Successful clinical development of these antibodies led to the rapid rise of multiple transgenic mouse platforms such as KyMouse (Kymab), OmniRat (Ligand), H2L2 Mouse (Harbour Antibodies), Trianni Mouse (Trianni Inc/AbCellera) and RenMab (Biocytogen).
Display technologies provide an alternate platform for human antibody development. Using phage display, human scFv or fragment antigen binding (Fab) fragments (Fig. 1) can be isolated from a large library with a diversity exceeding 1010. Phage display technology com-monly uses the M13 filamentous phage to express antibody fragments such as scFv fused to the pill coat protein of the phage22. The phage library is screened with a target antigen immobilized on a microtiter plate or beads. Nonspecific phage binders are washed away and the remaining specific binders are harvested for the next round of screen-ing. Normally, three to five rounds of screening are needed for isolation of specific binders. This process is called phage panning, which mim-ics immune selection in vitro. Newer methods using next-generation sequencing facilitate rapid identification of rare binders23. The phage display systems from Dyax produced three fully human antibodies that gained regulatory approval: ramucirumab (anti-vascular endothelial growth factor receptor 2 (VEGFR2)), necitumumab (anti-EGFR) and avelumab (anti-PDLl) (Table 1). In addition, multiple phage display- derived human antibodies generated by Dyax, Cambridge Antibody Technology, MorphoSys and others are now undergoing clinical tri-als24. Both transgenic mouse and phage display technologies have their unique set of advantages. In general, transgenic mouse platforms tend to produce antibodies with more desirable biophysical properties and better performance in clinical trials25–27, whereas phage display allows the selection of antibodies targeting a specific epitope28,29. In addition to phage display, cell surface display (yeast30, bacteria31 and mammalian32), ribosome display and mRNA display33 technologies have been used for antibody generation. Yeast and mammalian cell surface displays allow for recombinant antibody fragments to be expressed on the cell surface of eukaryote cells. This enables the isolation of antigen- specific cells by flow cytometry and the discovery of high-affinity binders. Sintilimab, a human IgG4 antibody specific for PD1 (ref. 34) (Table 1), was isolated by yeast cell surface display and is under FDA regulatory review for approval.
Comparing chimeric, humanized and human antibodies
The goal of chimeric, humanized, or fully human antibody production is to reduce the immune response against the antibody that carries the risk of neutralization and adverse reactions (for example, infu-sion reactions and anaphylaxis) with repeated administration. A large analysis of human clinical trials with various antibody therapeutics has shown a progressive decline in immunogenicity with the use of chimeric, humanized, and human antibodies, respectively10. However, the study did not examine if the lower immunogenicity results in supe-rior tumour regression or patient survival. Several FDA-approved and EMA-approved chimeric, humanized and human antibodies target com-mon cancer-antigens such as CD20, HER2, and EGFR albeit at different epitopes and with different binding characteristics (Table 1). Phase III clinical trials comparing the antibodies targeting CD20 (rituximab vs. obinutuzumab35,36 or rituximab vs. ofatumumab37,38), HER2 (trastu- zumab vs. margetuximab39) and EGFR (cetuximab vs. panitumumab40) have demonstrated similar overall survival and similar rates of therapy- related treatment discontinuation, indicating equivalent therapeutic efficacy. However, a substantial HAMA immune response has impeded the development of mouse antibodies targeting the disialoganglioside GD2 (ref. 41) and a strong HACA immune response was observed with the use of rituximab in patients with autoimmune disorders13. Con-cern over HACA immune responses coupled with technical advance-ments that simplified humanized and human antibody generation have increased utilization of these two formats. Antibodies entering current clinical trials are either humanized or fully human products (Fig. 1 and Supplementary Table 2) and based on the previous trial outcomes, the humanized and human antibodies tend to demonstrate similar therapeutic efficacies in patients.
Different formats of antibody therapies
Antibody therapeutics can be segregated into three major formats based on their structures and mechanisms of function - monospecific antibodies, bispecific antibodies, and antibodies conjugated to payloads (such as drugs, toxins or radioactive isotopes) (Fig. 2).
Fig. 2 ∣. Antibody formats and mechanisms of action.

a, On the basis of structure and mechanism of action, therapeutic antibodies can be grouped in three different formats: monospecific antibodies, bispecific antibodies, and drug-conjugated, toxin-conjugated or radioisotope-conjugated antibodies. b, Monospecific antibodies bind antigens on cancer cells leading to cell death by a variety of mechanisms, which include disruption of survival signals from growth factor receptors (such as human epidermal growth factor receptor 2 (HER2)), activation of immune cells (such as natural killer (NK) cell-mediated killing by antibody-dependent cellular cytotoxicity (ADCC) and macrophage-mediated killing by antibody-dependent cellular phagocytosis (ADCP)), and through activation of the complement cascade (complement-dependent cytotoxicity (CDC)). The immune checkpoint-blocking antibodies bind to and activate immune cells such as T cells leading to immune-mediated cancer cell death. Bispecific antibodies bind two disparate antigens. Most bispecific antibodies are designed to bind T cells (T cell engagers) and cancer cells, and redirect the T cells to kill the cancer cells. The non-T cell-engaging bispecifics bind to two different antigens on the cancer cell surface, leading to direct cancer cell killing. Antibody–drug conjugates (ADCs), immunotoxins and radioisotope-conjugated antibodies carry a toxic payload that enhances the ability of the antibody to kill the cancer cell. BCMA, B cell maturation antigen; EGFR, epidermal growth factor receptor; gp100, glycoprotein 100; GPRC5D, G-protein-coupled receptor family C group 5 member D; FcγR, Fc γ-receptor; PD1, programmed cell death protein 1; PDL1, PD1 ligand 1; scFv, single-chain variable fragment; TCR, T cell receptor.
Monospecific antibody formats
The monospecific antibody format involves full-length immunoglobu-lins that bind to a target antigen. Among the five immunoglobulin isotypes (IgG, IgM, IgA, IgE and IgD), only IgG binds to the neonatal Fc receptor (FcRn), leading to a long half-life (approximately 21 days)42,43. Cancer-targeting antibodies use the IgG isotype to take advantage of this extended half-life and are often dosed every 21 days. The majority of FDA-approved and EMA-approved antibodies, and the antibodies in development utilize the monospecific IgG antibody format (see Fig. 1 for the basic structure of the naturally occurring IgG antibody). The IgG antibody exists as four subclasses (IgG1, IgG2, IgG3 and IgG4) and most therapeutic antibodies utilize the IgG1 subclass. The target antigens of the monospecific antibody format are cell-surface proteins, mostly growth factor receptors overexpressed in solid cancers (for example, HER2, which is overexpressed in breast, gastric and gastroesophageal cancers44,45; EGFR, which is overexpressed in colon cancers46; and MET, which is overexpressed in lung cancers47). For haematological malignan-cies, the antibodies usually target the cell-surface glycoproteins (also called cluster of differentiation (CD) markers) expressed by different immune cell subsets (for example, CD19 and CD20 in B cell malignan-cies; CD52 and CC-motif chemokine receptor 4 (CCR4; also known as CD194) in T cell malignancies; CD38 and B cell maturation antigen (BCMA; also known as TNFRSF17) in plasma cell malignancies). The targets and their cognate antibodies are listed in Table 1.
Mechanism of action.
Antibody binding to the cancer cell leads to cancer cell death by a variety of mechanisms. Directly blocking the survival signal from growth factors and blocking angiogenesis lead-ing to disruption of tumour blood supply are two major mechanisms of solid tumour regression for these antibodies48. By contrast, the CD markers targeted in haematological malignancies generally do not have a substantial role in promoting cancer cell survival, even though some studies have shown that antibodies targeting CD20 or CD52 can induce caspase-independent programmed cell death49,50. Instead, the antibodies induce cytotoxicity through the recruitment and activation of immune effector cells. Each IgG antibody subclass51 lends differ-ent effector functions to the antibody. The IgG1 subclass Fc segment has a strong affinity to the activating Fcy receptors (FcyRs; FcyRI, FcyRIIa, FcyRIIIa and FcyRIIIb) expressed by macrophages and natural killer (NK) cells51–53. The FcyR binding results in macrophage and NK cell-mediated antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP) that directly kill the cancer cells (Fig. 2). The two earliest antibodies to receive FDA approval were rituximab targeting CD20 expressed by malignant B cells and trastuzumab targeting HER2 overexpressed by breast cancer cells and both of these antibodies use the common IgG1 isotype. This ability of IgG1 to induce ADCC and ADCP ultimately leads to the death of the cancer cells and can be further enhanced by Fc engineering54. In support of this mechanism of action, FcyR polymorphisms such as patients harbouring homozygous 158 valine/valine in FcyRIIIa alleles demonstrated higher responses to rituximab55 and trastuzumab56. The IgG1 subclass also binds the complement protein C1q leading to cancer cell death by complement-dependent cytotoxicity (CDC)57. Although most cancer-targeting antibodies that directly mediate cancer cell death preferentially use the IgG1 subclass (Table 1), one exception is panitumumab, an IgG2 EGFR antibody. IgG2 antibodies only weakly activate NK cells and complement, and instead, mediate cancer cell death by recruiting myeloid cells58,59. At present, the marketed mono-clonal antibodies are predominantly composed of IgG1. Besides effec-tor functions, the IgG subclass selection is based on structural stability, circulation half-life, experience with manufacturing and regula-tory approval, absence of immunogenicity or unanticipated side effects, and the availability of a particular IgG subclass in a company’s development portfolio.
Studies of the Fab fragment of trastuzumab in complex with the HER2 extracellular domain have provided the first structural basis of the therapeutic mechanism. Trastuzumab binds the fourth subdomain of HER2, blocking its ability to homodimerize and in turn, reducing cell proliferation60 (Protein Data Bank (PDB): 1N8Z; Fig. 3a). Advancement in structural techniques led to the first cryogenic electron microscopy (cryo-EM) structure of HER2 in complex with the two HER2-targeting antibodies trastuzumab and pertuzumab61. In contrast to trastuzumab, pertuzumab inhibits ligand-induced heterodimerization of HER2 with HER3 by interacting with the second subdomain of HER2, disrupting downstream signalling (Fig. 3a). The trastuzumab and pertuzumab binding epitopes are located ~60 angstrom (A) apart and the HER2- trastuzumab-pertuzumab complex structure guided the design of the bispecific antibody zanidatamab that interacts with both HER2 epitopes and has improved therapeutic efficacy62.
Fig. 3 ∣. The structural basis of antibody–antigen interactions.

a, Human epidermal growth factor receptor 2 (HER2)-specific antibodies bind different HER2 epitopes. Cryogenic electron microscopy (cryo-EM) structure of the HER2 extracellular domain (ECD) in complex with trastuzumab and pertuzumab fragment antigen binding (Fab) domains (PDB ID: 8Q6J). b, The cryo-EM structure of a CD20 homodimer in complex with two rituximab Fab domains (PDB ID: 6VJA) demonstrates that each CD20 molecule engages a rituximab Fab. Rituximab promotes clustering of CD20 by forming large supramolecular complexes via cross-linking CD20 dimers. c, Programmed cell death protein 1 (PD1)-specific antibodies bind different PD1 epitopes. Both the pembrolizumab Fab (orange and gold; PDB ID: 5GGS) and the nivolumab Fab (magenta and pink; PDB ID: 5GGR) overlap with the PD1 ligand 1 (PDL1) (white) binding site on PD1 (teal), preventing PD1–PDL1 interactions. Surface representations are shown for all protein molecules. d, Alignment of the PD1–PDL2 (PDB ID: 6UMT) and the PDL1 antibody atezolizumab (PDB ID: 5XXY) structures. The structural analysis suggests that the PDL2 residue Trp100 fits in a pocket inside PD1 and aids PD1-PDL2 binding. The same residue in PDL2 (Trp100) perturbs atezolizumab binding to PDL2. e, The complex structures of ipilimumab (PDB ID: 5TRU) and tremelimumab (PDB ID: 5GGV) Fab domains with cytotoxic T lymphocyte-associated antigen 4 (CTLA4) revealed similar binding epitopes that have a large buried surface area effectively outcompeting the binding of the natural ligand, CD80 and CD86. f, The p53(R175H) peptide–major histocompatibility complex (MHC) binding antibody binds parallel to the peptide binding cleft within the MHC molecule (PDB ID: 6W51). By contrast, the p53(R175H)-specific T cell receptor (TCR) binds perpendicular to the peptide binding cleft (PDB ID: 6VQO).
Structural biology techniques have also been utilized to under-stand the therapeutic mechanism of rituximab; specifically, how rituximab promotes clustering of CD20. In the initial high-resolution structures, it appeared that CD20 forms a homodimer, and a Fab frag-ment of rituximab binds to the extracellular region of each CD20 mono-mer (PDB: 6VJA; Fig. 3b). The distance between the two Fab fragments of rituximab suggested a single rituximab IgG molecule could not bind the CD20 homodimer. Additional electron microscopy studies using full- length IgG rituximab have provided the first evidence that rituximab cross-links two CD20 homodimers resulting in a large supramolecular complex (Fig. 3b) establishing the binding mechanism of rituximab63.
Monospecific antibody Fc engineering.
The majority of IgG1 antibody effector functions are mediated by the Fc domain. Decades of research have increased our understanding of Fc domain interactions with dif-ferent Fc receptors and assisted in engineering antibodies with desir-able effector functions. Common Fc domain modifications that have shown increased activity in preclinical studies include afucosylation (removing fucose from the Fc region to increase FcyRIIIa binding) or amino acid substitutions of key residues (such as S239D and I332E that increase binding to Fc receptors), both leading to enhanced ADCC and ADCP64,65. Two antibodies in clinical use have an afucosylated Fc (obinutuzumab targeting CD20 and mogamulizumab targeting CCR4) to augment their immune effector function (Table 1). Randomized clini-cal trials (the GALLIUM and GOYA studies) have evaluated the benefit of rituximab (CD20 antibody without an Fc modification) or obinutu-zumab (Fc afucosylated CD20 antibody) in different lymphoma sub-types. In the GOYA study, both patient groups had similar outcomes36. The GALLIUM study has shown superior progression-free survival (PFS) in patients receiving obinutuzumab35, but it has failed to demonstrate overall survival benefit, an important benchmark in oncology. In addi-tion, the GALLIUM study was also criticized for using a higher dose of obinutuzumab compared with rituximab which may have confounded the PFS results. Two antibodies with amino acid substitutions in the Fc domain to increase ADCC have received FDA approval: HER2 antibody margetuximab and CD19 antibody tafasitamab (Table 1). A phase III trial has compared margetuximab with trastuzumab (Fc unmodified HER2 antibody) in patients with breast cancer. Although early analysis has shown a reduction in cancer progression with margetuximab66, the overall survival was identical in both groups in the final analysis39. On the basis of these clinical trial outcomes, the benefit of Fc-engineered antibodies to treat patients with cancer remains unclear.
Immune checkpoint inhibitors
In the past decade, an entirely different group of monospecific antibod-ies that target immune cell regulatory checkpoints have shown remark-able clinical efficacy for patients with cancer. The 11 FDA-approved and EMA-approved immune checkpoint-inhibiting antibodies are now being used for the treatment of more than twenty different types of can-cer, including lung cancer, melanoma, renal cell cancer, head and neck squamous cell cancers (Fig. 4a,b) and several more of these inhibitory antibodies are expected to gain approval in the near future (Table 1). Immune checkpoint inhibitors demonstrate response rates between 20–30% in most cancer types67. However, certain malignancies such as Hodgkin lymphoma and skin cancers67, microsatellite instability-high (MSI-H) cancers68,69, cancers with elevated PDL1 expression70, or high mutational burden (>10 mutations per megabase)71, and some cancers associated with viruses72 have substantially higher response rates.
Fig. 4 ∣. The treatment effect of T cells reinvigorated or redirected against cancer cells with immune checkpoint inhibitors or bispecific antibodies.

a,b, Fluorodeoxyglucose (FDG)-positron emission tomography (PET) images (a) and haematoxylin and eosin (H&E)-stained tumour sections (b) from an individual with head and neck squamous cell carcinoma (HNSCC) involving the border of the left side of the tongue. The patient received treatment with the immune checkpoint inhibitors nivolumab and ipilimumab and experienced a substantial reduction in tumour burden. The on-treatment H&E section shows keratinous debris (KD) and surrounding multinucleated giant cells (arrows) and the on-treatment FDG-PET image shows a reduction in FDG uptake at the border of the tongue. c, CT scan of a patient with B cell lymphoma, before and 4 weeks after treatment with the bispecific antibody T cell engager targeting CD19, blinatumomab. This patient had a partial response to blinatumomab. Arrows point to involved lymph node tumours in the mediastinum. d, Bone marrow biopsy sample from another patient with B cell lymphoma before and 15 days after treatment with blinatumomab. Tumour cells are in blue (haematoxylin stain) and T cells are in brown (CD3 stain). Parts a and b are adapted from ref. 292, Springer Nature. Parts c and d are reprinted with permission from ref. 101, AAAS.
Mechanism of action.
The immune checkpoint-blocking antibodies inhibit the pathways negatively regulating T cells, thereby reinvigor-ating the cytotoxic T cells to kill cancer cells (Fig. 2). Among the more than 20 immune checkpoints being investigated in clinical trials, the three proteins or pathways targeted by therapeutic antibodies that have received FDA or EMA approval are CTLA4, PD1-PDL1 and LAG3. The mechanisms of action of immune checkpoint inhibitors have been extensively discussed in other review articles73,74 and so are only briefly mentioned here. Expression of CTLA4, PD1 and LAG3 is induced after T cell stimulation with the primary purpose of limiting the extent of T cell activation. PD1 interacts with PDL1 and PDL2, which are often overex-pressed on the surface of cancer cells. PD1 engagement with its ligands leads to the recruitment of a tyrosine phosphatase, that dephospho- rylates signalling molecules downstream of the T cell receptor (TCR). This blocks TCR-mediated T cell proliferation (signal 1) and dampens T cell responses against cancer cells, enabling tumours to evade the immune system74. CTLA4 competes with the T cell co-stimulatory receptor CD28 for binding to CD80 and CD86 on antigen-presenting cells (APCs). CTLA4 engagement with these ligands blocks CD28 co-receptor signalling (signal 2), thereby suppressing T cell activa-tion and impairing the immune response against cancer73. Similarly, LAG3 expression negatively regulates T cell and NK cell functions when LAG3 binds to major histocompatibility complex (MHC) class II (ref. 75) molecules or fibrinogen-like protein 1 (FGL1)76. Antibodies that disrupt these interactions allow enhanced T cell activation by lower-ing the activation threshold74, enabling ‘re-invigoration’ of exhausted T cells77,78 and recruiting new T cell clones into tumours79. Immune checkpoint inhibitors may also have distinct effects on regulatory T (Treg) cells, which are a subset of T cells that suppress immune activity within the tumour microenvironment. Some studies using mouse mod-els and human ex vivo models suggest that PD1 and CTLA4 blockade can preferentially deplete Treg cells and enhance antitumour immu-nity80,81. However, such Treg cell depletion has not been observed in patients treated with immune checkpoint inhibitors82 and therefore, overwhelming reactivation of cytotoxic T cells rather than Treg cell inactivation may be driving immune checkpoint inhibitor-mediated tumour regressions in patients83,84.
The PD1 blocking antibodies are by far the most widely used immune checkpoint inhibitors. The seven approved PD1 blocking antibodies (nivolumab, pembrolizumab, cemiplimab, dostarlimab, retifanlimab, tislelizumab and toripalimab) and the two currently in clinical trials (Table 1) use the IgG4 format, which cannot efficiently activate the complement cascade and has weaker Fc receptor binding compared with the IgG1 isotype. Thus, the IgG4 format probably pro-tects PDl-expressing effector T cells from being inadvertently killed via ADCC or CDC. A similar IgG4 format is also used by the LAG3 targeting antibody relatlimab. All IgG4 antibodies carry the S228P mutation to prevent Fab arm exchange (Table 1). Of the approved PD1 antibodies, only the full-length structure of pembrolizumab has been determined. The structure revealed a CH2 domain rotation within the Fc domain completely exposing the glycan85. Compared with other reported Fc domain structures, this CH2 domain conformation was a new depiction of the molecular flexibility of these IgG subclasses and could be further investigated to understand how this conformation contributes to the weaker Fc receptor binding. By contrast, the PDL1 and CTLA4 blocking antibodies use an IgG1 format. Although atezolizumab and durvalumab use a modified Fc domain that limits FcR-mediated effector function, avelumab and ipilimumab use an unmodified Fc domain that retains CDC and ADCC activity. Although there are no direct comparisons between PD1 and PDL1 blocking antibodies in clinical trials for their potency, a meta-analysis of clinical trials with PD1 pathway-blocking antibodies has concluded that PD1 antibodies are somewhat more effective than PDL1 antibodies86, possibly because PD1 antibodies block interactions of PD1 with both PDL1 and PDL2. Curiously, avelumab, which uses the IgG1 format with a functioning Fc region failed to exhibit benefit in patients with lung, ovarian, gastric, and head and neck can-cers87–90 despite success demonstrated by other immune checkpoint inhibitors in patients with these same cancer types. Thus, the use of the IgG1 format and its potential to mediate the killing of effector T cells may contribute to its suboptimal trial results.
The structural basis of recognition for several immune checkpoint inhibitor antibodies described herein has been determined in complex with their target proteins91,92. The structures revealed the diversity in epitope interfaces, buried surface areas of the complexes, and con-formational changes of the target protein upon antibody binding. The two PD1 blocking antibodies, pembrolizumab (PDB ID: 5GGS) and nivolumab (PDB ID: 5GGR), bind PD1 at different epitopes despite each antibody extensively overlapping with the PDL1 ligand-binding site91,93,94 (Fig. 3c). The crystal structures of PDL1 targeting antibodies (atezolizumab, durvalumab, avelumab) in complex with PDL1 and PDL2 identified a key residue in PDL2 (Trp100) that hinders binding of anti-PDL1 antibodies to PDL2 and provided a mechanism of selectivity between PDL1 and PDL2 (refs. 92,95,96) (PDB ID: 5XXY) (Fig. 3d). The corresponding residue in PDL1 is an alanine allowing the binding of PDL1 to atezolizumab. In contrast to PD1-binding antibodies, the two CTLA4 targeting antibodies, ipilimumab (PDB ID: 5XJ3) and tremeli-mumab (PDB ID: 5GGV), share similar binding epitopes effectively com-peting with the natural ligand, CD80 and CD86 (refs. 91,97) (Fig. 3e). The larger buried surface area (~600 A2) of the antibody complexes with CTLA4 compared with CD86 contributes to this mechanism of action.
Immune checkpoint inhibitor toxicities.
Immune checkpoint-blocking antibodies mediate cancer cell killing through general immune activa-tion, which can sometimes be mis-directed against healthy tissues. Immune checkpoint-inhibiting antibodies have a very distinct spec-trum of adverse effects referred to as immune-related adverse events (irAEs). These irAEs encompass various manifestations, including dermatological, gastrointestinal, hepatic, endocrine, pulmonary, neurological, and cardiac events98. Although infrequent, severe and potentially fatal toxicities may occasionally arise as a result of immune checkpoint inhibition99. In many instances, temporary immunosup-pression with glucocorticoids, tumour necrosis factor (TNF) antago-nists, mycophenolate mofetil, or other immunoregulatory agents is required to manage irAEs98.
Bispecific antibody formats
The second format includes the diverse category of bispecific antibod-ies100. Unlike monospecific antibodies, bispecific antibodies bind two different antigens or epitopes. The antigens can be localized either on the same target cell or on different cells. The bispecific antibodies targeting two different cells are mostly T cell engagers crosslinking a cancer cell with an effector T cell; thus, they are named T cell engager (TCE) bispecific antibodies. Upon crosslinking, the effector T cell is activated to kill the bound target cancer cell by releasing cytotoxic granules and lymphokines (Fig. 2). Another class of bispecific antibod-ies engages disparate antigens expressed by the same target cell, such as two distinct growth factor receptors. Such bispecific antibodies kill target cells by blocking proliferation signals through the target growth factor receptors, and by activating NK cells and macrophages against the cancer cells.
Bispecific T cell engagers.
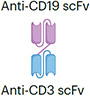
The regulatory approval of blinatumomab in 2014 led to the explosive growth of this category of antibody-based therapeutics. The prototype TCE bispecific blinatumomab is a small 54-kDa fusion protein and is about one-third the size of an IgG anti-body (Table 2). Blinatumomab is composed of the minimum elements required for a bispecific antibody; a cancer-targeting scFv (anti-CD19) joined by a glycine-serine peptide linker with a T cell binding scFv (anti-CD3). Blinatumomab engages B cells with the anti-CD19 scFv, and simultaneously connects and activates T cells through the anti-CD3 scFv, resulting in B cell death (Fig. 2). Blinatumomab demonstrated activity in a range of B cell malignancies101 (Fig. 4c,d) and received approval for the treatment of B cell precursor acute lymphoblastic leukaemia (B-ALL). The basic design of blinatumomab was adopted by the subsequent TCE bispecific antibodies that gained regulatory approvals (Fig. 5a). Pharmaceutical companies often trademark unique TCE formats that use distinct architectures and methods to assemble the bispecific antibodies. The common formats include bispecific T cell engager (BiTE) (Amgen), Duobody (Genmab), DART (MacroGenics) and Xmab (Xencor), with blinatumomab using the BiTE format (reviewed here102). Tebentafusp is a unique TCE bispecific format named ImmTAC that links an affinity-enhanced TCR targeting the glycoprotein-100 (gp100)-human leukocyte antigen (HLA)-A02 complex expressed in melanoma cells with an anti-CD3 scFv103. Tebentafusp demonstrated superior overall survival in patients with metastatic uveal melanoma, leading to its approval in 2022 (ref. 104). In the past 2 years, six new bispecific antibodies targeting haematological malignancies received regulatory approval. These include teclistamab and elranatamab (BCMAxCD3), and talquetamab (G protein-coupled receptor family C group 5 member D (GPRC5D)xCD3) for multiple myeloma as well as mosunetuzumab, epcoritamab and glofitamab (CD20xCD3) for B cell lymphomas (Table 2). However, approvals of TCE bispecific antibod-ies have lagged in solid tumours. Yet, a surge in clinical trials with TCE bispecific antibodies targeting solid malignancies has provided hope that this strategy will increase its breadth of utility. The recent report of a positive phase I trial with a TCE bispecific antibody targeting delta-like protein 3 (DLL3) in small-cell lung cancer is encouraging and suggests that the solid tumour barrier will be broken105.
Table 2 ∣.
Bispecific antibodies approved or nearing approval by the FDA and EMA
| Target | Antibody | Structure | Format | Major indications | Adverse effects |
|---|---|---|---|---|---|
| T cell engagers | |||||
| CD19xCD3 | Blinatumomab |
|
Lacks Fc (short half-life ~2 h) ~54 kDa |
B cell precursor ALL | CRS Neurotoxicity Infection Cytopenia TLS |
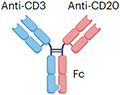
| CD20xCD3 | Mosunetuzumab |
|
IgG1 Fc (half-life extension) N297G mutation (aglycosylation and reduced ADCC) ~146 kDa |
Lymphoma | CRS Neurotoxicity Infection Cytopenia Embryo–fetal toxicity |
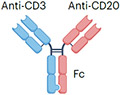
| Epcoritamab |
|
IgG1 Fc (half-life extension) L234F, L235E, D265A mutation (reduced ADCC) ~146 kDa |
Lymphoma | CRS Neurotoxicity Infection Cytopenia TLS |
|
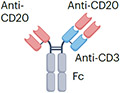
| Glofitamab |
|
Two anti-CD20 scFvs linked with one anti-CD3 scFv IgG1 Fc (half-life extension) P329G, L234A, L235A mutation (reduced ADCC) ~194 kDa |
Lymphoma | CRS Neurotoxicity Infection Cytopenia |
|
| Imvotamab (in clinical trial) |
|
IgM pentamer (ten CD20-binding sites) Albumin fusion (half-life extension) ~960 kDa |
Lymphoma | CRS Cytopenia Hypophosphatemia |
|
| BCMAxCD3 | Teclistamab |
|
IgG4 Fc (half-life extension) S228P (hinge stabilization), and F234A and L235A mutation (reduced FcγR binding) ~143 kDa |
Multiple myeloma | CRS Neurotoxicity Infection Cytopenia Hepatotoxicity Embryo–fetal toxicity |
| Elranatamab |
|
IgG2 Fc (half-life extension) ~145 kDa |
Multiple myeloma | CRS Neurotoxicity Cytopenia |
|
| GPRC5DxCD3 | Talquetamab |
|
IgG4 Fc (half-life extension) S228P (hinge stabilization), and F234A and L235A mutation (reduced FcγR binding) ~147 kDa |
Multiple myeloma | CRS Neurotoxicity Cytopenia Skin rash Hepatotoxicity Embryo–fetal toxicity |
| GP100xCD3 | Tebentafusp |

|
Lacks Fc, MW above renal filtration cut-off (half-life 6–8 h) ~75–77 kDa |
Melanoma | CRS Skin rash Hepatotoxicity Embryo–fetal toxicity |
| Receptor blockers | |||||
| EGFRxcMET | Amivantamab |
|
IgG1 (half-life extension) Fc afucosylation (enhanced ADCC) K409R, F405L mutation (for Fab exchange) ~146 kDa |
Lung cancer | Skin rash Stomatitis Muscle pain Cytopenia Electrolyte abnormality Embryo–fetal toxicity |
| Her2xHer2 | Zanidatamab (in clinical trial) |
|
Two anti-HER2 scFvs (bind two distinct HER2 epitopes) IgG1 (half-life extension) ~125 kDa |
HER2+ cancers | Diarrhoea Infusion reaction Cardiac failure |
| PD1xCTLA4 | Volrustomig MEDI5752 (in clinical trial) |
|
IgG1 (half-life extension) L234F, L235E, P331S mutation (reduced FcγR binding) ~145 kDa (estimated) |
Multiple solid tumours | Checkpoint inhibitor-associated irAEs: Diarrhoea Thyroid disorders Skin rash Hepatotoxicity |
ADCC, antibody-dependent cellular cytotoxicity; ALL, acute lymphoblastic leukaemia; CRS, cytokine release syndrome; Ig, immunoglobulin; irAEs, immune-related adverse events; IV, intravenous; kDa, kilodalton; scFv, single-chain variable fragment; TLS, tumour lysis syndrome. Fc-bearing bispecific antibodies also carry Fc mutations that enable pairing of the heterogenous heavy chains (not shown).
Fig. 5 ∣. Timeline of the development of bispecific antibodies and conjugated antibodies.

a, Timeline of bispecific antibodies. b, Timeline of drug-conjugated, immunotoxin-conjugated and radioactive isotope-conjugated antibodies. aCatumaxomab, an epithelial cell adhesion molecule (EpCAM)xCD3 bispecific antibody, was approved by the European Medicines Agency (EMA) in 2009 for the treatment of malignant ascites but was subsequently withdrawn by the manufacturer for commercial reasons. ADC, antibody–drug conjugate; ALL, acute lymphoblastic leukaemia; B-ALL, B cell precursor acute lymphoblastic leukaemia; BCMA, B cell maturation antigen; AML, acute myeloid leukaemia; CEA, carcinoembryonic antigen; EGFR, epidermal growth factor receptor; FDA, Food and Drug Administration; gp100, glycoprotein 100; HCL, hairy cell leukaemia; MRD, minimal residual disease; NHL, non-Hodgkin lymphoma; NSCLC, non-small-cell lung cancer; OS, overall survival; PFS, progression-free survival; TCR, T cell receptor.
Bispecific antibodies targeting disparate antigens on the same cell.
Bispecific antibodies have also been designed to bind two distinct antigens or epitopes on target cancer cells without engaging effector T cells. Their anticancer effect is mediated by blocking two prolifera-tion signalling pathways, thus maximizing the antitumour activity. In addition to their receptor-blocking activities, these bispecific antibod-ies may also be engineered to contain a functioning lgG1 Fc domain, enabling them to kill cancer cells through non-T cell-based immune effector pathways such as ADCC, ADCP and CDC. Amivantamab was the first receptor-blocking bispecific antibody targeting EGFR and MET (Table 2) expressed by cancer cells and received regulatory approval for the treatment of non-small-cell lung cancers with exon 20 inser-tion mutations106. Amivantamab blocks signalling through EGFR and MET pathways that drive a subset of lung cancers more potently than the combination of single-receptor binding antibodies107. In addi-tion, the lgG1 Fc domain of amivantamab was engineered to have low fucose levels, which enhances FcyRllla binding and NK cell-mediated ADCC107. A similar bispecific design was adopted by zanidatamab that binds two distinct HER2 epitopes with each Fab arm (Table 2). The arms target HER2 subdomain 2 and subdomain 4, the respective binding sites for pertuzumab and trastuzumab, the HER2-targeting antibod-ies approved for the treatment of HER2+ breast cancers108. Preclinical studies have demonstrated that zanidatamab enhanced HER2 cluster-ing, receptor downregulation and increased CDC-mediated HER2+ cancer cell killing when compared with trastuzumab, pertuzumab or the combination of trastuzumab and pertuzumab109. In a phase I trial, zanidatamab showed encouraging activity against a range of HER2+ solid tumours110.
Bispecific antibody Fc engineering.
Blinatumomab, has a short ~2-h serum half-life as the small polypeptide can be rapidly cleared by the kidneys and lacks an Fc domain required for FcRn-mediated recy-cling. Because of its short half-life, blinatumomab is administered by a continuous intravenous infusion to maintain consistent serum concentrations. Continuous infusion setups require additional health care resources, increase costs, and are less convenient for patients compared with intermittent infusions. The bispecific antibodies that gained regulatory approval after blinatumomab have higher molecu-lar weights with the majority possessing an Fc domain that prolongs half-life (Table 2). Specifically, the molecular weight of tebentafusp being ~75 kDa is above the cut-off for renal filtration and therefore, is not cleared rapidly by the kidneys. However, tebentafusp lacks an Fc domain and thus possesses a relatively short half-life of 7.5 h. Mosunetu- zumab, teclistamab, epcoritamab, glofitamab and elranatamab include either an IgG1, IgG2 or IgG4 Fc domain (Table 2), which increases their molecular weight over that of tebentafusp (~145 kDa) and enables FcRn binding. These modifications extend the serum half-life and allow intermittent (weekly) dosing. Addition of such Fc domains tends to be included in all upcoming bispecific antibody designs. Because the IgG1 Fc segment has a strong affinity for Fcy receptors on macrophages and NK cells, the TCE bispecific antibodies with an IgG1 Fc segment can potentially crosslink T cells (through the anti-CD3 scFv) with macro-phages or NK cells (via the IgG1 Fc segment), causing unintended T cell killing by macrophages or NK cells and vice versa. Amino acid substitutions (such as N297G in mosunetuzumab or P329G, L234A and L235A in glofitamab) have been introduced in the Fc domain to silence FcyR binding and reduce inadvertent immune cell killing (Table 2). TCE bispecific antibodies using IgG2 or IgG4 Fc domains may not require such Fc domain silencing owing to their weaker interactions with FcyRs.
Bispecific antibody toxicities.
The TCE bispecific antibodies share similar adverse effects as they use a common anticancer mechanism of target cell killing through T cell activation (Table 2). The levels of toxicity are somewhat related to the total body burden of cells (tumour or normal) bearing the target of the TCE bispecific antibody. TCE bispe-cific antibodies induce systemic inflammation characterized by fever, and varying degrees of hypoxia, hypotension and occasionally multi-organ failure, collectively known as cytokine release syndrome (CRS)111. Neurotoxicity is another unique adverse effect associated with TCE bispecific antibodies and manifests as confusion and tremors, along with alterations of speech and behaviour111. The pathophysiology of CRS and neurotoxicity is incompletely understood and is probably related to elevated levels of cytokines such as interferon γ (IFNy), interleukin-6 (IL-6), IL-1 and IL-10. Patients receiving TCE bispecific antibodies require close monitoring and may require immunosup-pression with glucocorticoids or anti-cytokine agents such as the IL-6 neutralizing antibody tocilizumab. In addition, several strategies have been implemented to prevent or reduce CRS and neurotoxicity including gradual up-titration of the TCE bispecific antibody dose, prophylactic glucocorticoids, and using chemotherapy to reduce the tumour burden before initiation of the TCE bispecific antibody treatment. Receptor-blocking bispecific antibodies are incapable of T cell activation and thus do not induce CRS or neurotoxicity that is typically observed with the TCE bispecific antibodies. The adverse effects of receptor-blocking bispecific antibodies are similar to their monospecific antibody counterparts such as skin rashes induced by amivantamab (also seen with EGFR-targeting antibodies cetuximab and panitumumab) and heart failure induced by zanidatamab (also seen with the HER2-targeting antibody trastuzumab) (Table 2).
Conjugated antibody formats
The third major format involves antibodies linked with toxic payloads such as cytotoxic drugs (antibody-drug conjugates (ADCs)), bacterial or plant toxins (immunotoxins), or radioactive isotopes, which aug-ment the ability of the antibody to kill cancer cells. Within this group, ADCs are by far the most extensively used format, whereas toxin- conjugated and radioisotope-conjugated antibodies have yet to achieve widespread adoption (Fig. 5b).
Antibody-drug conjugates.
ADCs are constructed by linking a tumour-targeting antibody to a cytotoxic drug (Fig. 2). The binding of ADC molecules to the cell-surface antigen leads to their internaliza-tion followed by the release of the cytotoxic drug inside the cell. This allows selective delivery of the cytotoxic drug to cancer cells while sparing most of the healthy tissues. Key components of an ADC include a tumour-targeting antibody, a cytotoxic drug and a linker connect-ing the antibody to the cytotoxic drug (Table 3). The success of ADCs depends on the optimal selection of these key components, along with the conjugation method used to attach the linker to the antibody which often determines the drug-antibody ratio (DAR).
Table 3 ∣.
Antibody–drug conjugates approved or nearing approval by the FDA and EMA
| Drug | ADC | Linker | DAR | Target | Major indications | Adverse effects |
|---|---|---|---|---|---|---|
| Microtubule inhibitors | ||||||
| MMAE | Brentuximab vedotin (ADCETRIS, Seagen) | mc-VC-PABC (cleavable) | 4.0 | CD30 | Hodgkin lymphoma, CD30+ PTCL | Peripheral neuropathy Cytopenia Embryo–fetal toxicity Opportunistic infections |
| Polatuzumab vedotin (POLIVY, Roche/Seagen) | 3.8 | CD79a | Diffuse large B cell lymphoma | Peripheral neuropathy Cytopenia Hepatotoxicity Embryo–fetal toxicity Opportunistic infections |
||
| Enfortumab vedotin (PADCEV, Astellas/Seagen) | 3.8 | Nectin-4 | Metastatic urothelial cancer | Peripheral neuropathy Embryo–fetal toxicity Ocuiar disorders Pneumonitis Stevens–Johnson syndrome |
||
| Tisotumab vedotin (TIVDAK, Genmab/Seagen) | 4.0 | TF | Cervical cancer | Peripheral neuropathy Pneumonitis Embryo–fetal toxicity Haemorrhage |
||
| MMAF | Belantamab mafodotin (BLENREP, GSK/Seagen)a | mc linker (non-cleavable) | 4.0 | BCMA | Multiple myeloma | Vision loss Embryo–fetal toxicity |
| DM1 | Ado-trastuzumab emtansine (KADCYLA, Roche) | SMCC (non-cleavable) | 3.5 | HER2 | HER2-positive, metastatic breast cancer | Interstitial lung disease Peripheral neuropathy Cytopenia Heart failure |
| DM4 | Mirvetuximab soravtansine (ELAHERE, ImmunoGen) | Sulfo-SPDB disulfide linker (cleavable) | 3.5 | FRα | Ovarian, fallopian tube or primary peritoneal cancer | Vision loss Pneumonitis Peripheral neuropathy Embryo–fetal toxicity |
| DNA binders | ||||||
| N-Acetyl-calicheamicin | Gemtuzumab ozogamicin (MYLOTARG, Pfizer) | Hydrazone (cleavable) | 2.0–3.0 | CD33 | CD33+ AML | Fatal hepatotoxicity Embryo–fetal toxicity Thrombocytopenia and bleeding |
| Inotuzumab ozogamicin (BESPONSA, Pfizer) | 6.0 | CD22 | CD22+ ALL | Fatal hepatotoxicity Embryo–fetal toxicity Cytopenia and bleeding QT prolongation |
||
| PBD (SG3199) | Loncastuximab tesirine (ZYNLONTA, ADC therapeutics) | Valine–alanine dipeptide (cleavable) | 2.3 | CD19 | Diffuse large B cell lymphoma | Effusion and oedema Cytopenia Embryo–fetal toxicity |
| Duocarmycin | Trastuzumab duocarmazine (Byondis/Synthon pending approval) | Valine–citruline (cleavable) | 2.8 | HER2 | HER2-positive breast cancer | Conjunctivitis and dry skin Heart failure |
| Topoisomerase inhibitors | ||||||
| DXd | Trastuzumab deruxtecan (ENHERTU, Daiichi Sankyo/AstraZeneca) | Glycine–glycine–phenylalanine–glycine (GGFG) tetrapeptide (cleavable) | 8.0 | HER2 | HER2-positive breast cancer, metastatic HER2-positive solid tumoursb | Interstitial lung disease Cytopenia Embryo–fetal toxicity Heart failure |
| SN38 | Sacituzumab govitecan (TRODELVY, Gilead) | CL2A (cleavable) | 7.6 | TROP2 | TNBC | Cytopenia Diarrhoea |
DM1 and DM4 are derivatives of maytansine 1. DXd is a derivative of Exatecan used for ADC. SN38 is an active metabolite of irinotecan. ADC, antibody–drug conjugate; ALL, acute lymphoblastic leukaemia; AML, acute myeloid leukaemia; BCMA, B cell maturation antigen; DAR, drug–antibody ratio; FRα, folate receptor α; GSK, GlaxoSmithKline; HER2, human epidermal growth factor receptor 2; mc, maleimidocaproyl; mc-VC-PABC, maleimidocaproyl-valine-citrulline-p-aminobenzoyloxycarbonyl; MMAE, monomethyl auristatin E; MMAF, monomethyl auristatin-F; PBD, pyrrolobenzodiazepine; PTCL, peripheral T cell lymphoma; SMCC, succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate; TF, tissue factor; TNBC, triple-negative breast cancer. aIn 2023, the FDA revoked the biological license and the EMA recommended against the renewal of marketing authorization of belantamab mafodotin, as the confirmatory DREAMM-3 trial failed to demonstrate an improvement in progression-free survival. Ongoing trial suggests that belantamab may regain approval. bTrastuzumab deruxtecan received FDA approval for HER2-low breast cancer (immunohistochemistry score 1+) and in all solid tumours with high HER2 expression (immunohistochemistry score 3+).
Most ADCs use a humanized or human IgGl as the tumour-targeting antibody (except for brentuximab, which uses a chimeric IgGl, and gemtuzumab and inotuzumab, which use a humanized lgG4)112,113. As alluded to above, the popularity of using IgGl is owing to its long plasma half-life of ~21 days (for example, compared with the half-life of lgG3, which is ~7 days)114, and its ability to bind Fc receptors leading to enhanced target cell killing by ADCC and ADCP (for example, compared with lgG2 and lgG4, which are less efficient at ADCC and ADCP)115. Two ADCs, gemtuzumab and inotuzumab, use lgG4, which has a lower affinity for FcyRII and FcyRlll, thus limiting ADCP, along with a possible reduction in toxicity owing to diminished nonspecific uptake of the ADC into immune cells through the Fc receptor.
The majority of the linkers connect the cytotoxic drug to the anti-body at random lysine or cysteine residues on the lgG1 antibody back-bone. An effective linker minimizes the early release of the cytotoxic drug in the bloodstream while facilitating the controlled release of the active drug at preferred targeted locations. Linkers are broadly clas-sified as cleavable and non-cleavable. Ten out of the twelve approved ADCs use a cleavable linker such as a peptide linker, hydrazone linker, disulfide linker or the CL2A linker (Table 3). One of the first linkers developed for drug attachment was a cleavable linker using hydrazone bonds. This linker was used to attach the antitumour antibiotic cali- cheamicin to the ADCs gemtuzumab and inotuzumab. The hydrazone bonds were designed to break down under acidic conditions inside the lysosomes of the target cells to release calicheamicin. However, hydra-zine bonds can undergo hydrolysis in plasma causing the unintended release of drug molecules leading to systemic toxicity116. A second type of cleavable linker is the peptide linker that requires selective cleav-age mediated by cathepsin B inside the lysosomes117. Four ADCs use the mc-VC-PABC dipeptide linker, first developed by Seagen to generate the ADC brentuximab, which uses a CD30-targeting antibody linked to the microtubule inhibitor monomethyl auristatin E (MMAE)118 (Table 3). Brentuximab in combination with chemotherapy showed remarkable efficacy in patients with several different lymphoma subtypes and is considered standard of care first-line therapy in Hodgkin lymphoma and CD30+ peripheral T cell lymphomas119,120. The success of bren-tuximab led to the widespread adoption of the mc-VC-PABC dipeptide linker. Seagen collaborated with Roche, Astellas and Genmab to use the mc-VC-PABC dipeptide linker to develop CD79B, nectin-4 and TF targeting ADCs, respectively. A third type of cleavable linker is disulfide linkers, wherein linker cleavage is mediated by glutathione present at high concentrations inside the cell121. ADCs with disulfide linkers include mirvetuximab, which demonstrated survival benefit in a phase lll trial of patients with ovarian cancer122. ln contrast to cleavable linkers, non-cleavable linkers are resistant to various mechanisms of break-down. The payload is released after degradation of the antibody inside the lysosomes of target cells. Two ADCs, trastuzumab emtansine and belantamab mafodotin use non-cleavable linkers for drug attachment. Preclinical studies have shown low toxicity to non-target cells owing to increased plasma stability of non-cleavable linkers123,124. However, cleavable and non-cleavable linkers have not been directly compared in clinical trials and the advantage of one over the other remains unclear.
The tumour cell-killing process is carried out by the cytotoxic drug attached to a tumour-targeting antibody. The drug is usually a small molecule with high cell-killing potency (half-maximal inhibitory concentration (lC50) < 5 nM) that induces cell death by one of three mechanisms - direct DNA damage, disruption of the microtubule network or inhibition of topoisomerase activity113,125 (Table 3). The currently used drugs have widely varying lC50 from 5 nM (for Dxd, a topoisomerase inhibitor) to 5pM (for pyrrolobenzodiazepines (PBDs), a class of DNA damaging agents). However, the potency of an ADC depends on both the lC50 of the drug and the DAR. Thus, the ADCs lon- castuximab tesirine with the potent PBD payload and a relatively low DAR ~2.3 and trastuzumab deruxtecan with a less potent payload Dxd but a relatively high DAR ~8.0 were both capable of inducing complete tumour regression in several in vivo models126,127. The benefits of an ADC with a high DAR coupled with a novel linker design have also been observed in clinical trials. Trastuzumab emtansine and trastuzumab deruxtecan share an identical HER2-targeting antibody. However, although trastuzumab emtansine carries the microtubule inhibitor DM1 (lC50 of 2 nM, DAR 3.5) conjugated by a non-cleavable linker128, trastuzumab deruxtecan utilizes the drug Dxd (lC50 of 5 nM, DAR 8.0) conjugated with a cleavable linker126. ln a phase lll trial comparing the two ADCs, trastuzumab deruxtecan showed a higher response rate (79.7% vs 34.2%) and overall survival (94.1% vs 85.9% at 12 months)129. ln addition, trastuzumab deruxtecan prolonged survival in patients with breast cancers that express low levels of HER2 (ref. 130), making it the first ADC to be effective in HER2-low breast cancer. More recently, fam- trastuzumab deruxtecan demonstrated tumour regressions in a range of solid tumours with high HER2 expression and received the first FDA approval for a tumour-agnostic HER2-directed therapy.
ADC toxicities.
ADCs are thought to be targeted agents and better tolerated than conventional cytotoxic chemotherapies. However, most patients will experience some form of toxicity with the use of ADCs. Toxicities that are shared by most ADCs include infusion reac-tions, cytopenias, infections, elevated liver enzymes, gastrointestinal symptoms (diarrhoea, vomiting and constipation) and embryo-fetal toxicity (Table 3). Certain toxicities are associated with the use of specific payloads such as peripheral neuropathy with microtubule inhibitors (MMAE, DM1, DM4) and hepatotoxicity with calicheamicin. Other toxicities are shared across ADCs that target a common antigen. For instance, the HER2-targeting ADCs can cause heart failure and mediate interstitial lung disease.
Antibody-toxin conjugates or immunotoxins.
Immunotoxins have two components, a targeting antibody or Fv of an antibody, and a cytotoxic protein usually derived from bacterial or plant-based toxins. The targeting antibody or Fv binds to the target cell, allowing selec-tive delivery of the toxin. The toxins are derived from bacteria such as Pseudomonas exotoxin A (PE) and diphtheria toxin (DT) or from plants such as ricin131-132. In theory, any targeting antibody used to generate an ADC can be used in an immunotoxin. However, despite 12 ADC approv-als in the past several years, only one immunotoxin, moxetumomab pasudotox, was approved by the FDA and the EMA for the treatment of hairy cell leukaemia (Figs. 2 and 5b). Moxetumomab is a fusion protein combining a CD22-binding Fv with a truncated Pseudomonas exotoxin A (PE38)133 developed by I. Pastan at the National Cancer Institute (NCI)134. Upon entering the target cell, the PE38 toxin binds to elongation factor 2 (EF2) and blocks protein synthesis. Moxetumomab demonstrated a high complete response rate of 41% in a clinical trial of patients with hairy cell leukaemia135, and it gained regulatory approval in 2018. How-ever, moxetumomab production was discontinued in 2023 owing to low clinical uptake. Moxetuzumab and other investigational immunotoxins faced several challenges including high immunogenicity causing loss of therapeutic efficacy and a narrow therapeutic window leading to toxicities such as capillary leak syndrome (CLS) and haemolytic uremic syndrome (HUS)136. The low adoption of moxetumomab was also prob-ably owing to the complexity of administration that included toxicity prophylaxis, pretreatment and posttreatment hydrations, the need for safety monitoring to avoid adverse effects and the availability of alternate therapies in hairy cell leukaemia. Although several clinical trials in the past have evaluated the efficacy of immunotoxins targeting BCMA, mesothelin and other cancer-associated antigens131,132, none are currently in regulatory review nor expected to receive approval in the near future.
Antibody-radioisotope conjugates.
Antibody-radioisotope conju-gates consist of a targeting antibody linked to a radioisotope. The radio-isotope emits α-particles or β-particles causing DNA-strand breaks in the target cell resulting in cell death. The antibody-radioisotope conjugates do not require internalization to induce cell death, which offers unique advantages over ADCs and immunotoxins includ-ing bystander effects. α-Particles are large positively charged particles that comprise two protons and two neutrons, and have a relatively short effective range of −50–100 μm (ref. 137). By contrast, β-particles are small negatively charged electrons (−8,000 times smaller than α-particles) with a longer effective range of around 0.5–10 mm but with less DNA-damaging energy138. Examples of β-particle emitters include iodine-131, lutetium-177 and yttrium-90. The two FDA-approved and/or EMA-approved antibody-radioisotope conjugates, 90Y-ibritumomab and 131I-tositumomab, attach β-particle-emitting radionuclide to a CD20-targeting antibody (Figs. 2 and 5b) for the treatment of B cell lymphomas. 90Y-ibritumomab and 131I-tositumomab demonstrated an overall response rate of 65–80% with a complete response rate of 20–30% in clinical trials139,140. Both studies have observed some predict-able treatment-related adverse effects including prolonged and severe cytopenias from exposure ofhealthy bone marrow to radiation, hypothy-roidism from radioactive iodine in the case of 131I-tositumomab, and sec-ondary malignancies such as myelodysplastic syndromes (MDS) and leukaemia. Despite the high response rates, the drugs did not achieve widespread clinical use and only 75 patients received 131I-tositumomab in 2012 (ref. 141). As a result, 131I-tositumomab was voluntarily with-drawn in 2013. 90Y-ibritumomab remains available for patients with relapsed or refractory low-grade B cell lymphomas, but reports suggest only a small number of patients are receiving this therapy142. The limited number of approvals and low clinical adoptions are probably because of the requirement of a multidisciplinary team of medical oncologists, radiation oncologists, pharmacologists and physicists to develop and deploy the molecules in the clinic143. Few centres have such capabilities and as a result, the use of antibody-radioisotope conjugates has lagged behind that of ADCs.
Challenges and future perspectives
Drug development in oncology is an arduous process with a success rate of 3–7% (refs. 144,145). Data is lacking for the success rate of antibody-based therapeutics in oncology specifically, but −18% of thera-peutic antibodies (for all indications including oncology) that enter phase I trials proceed to drug launch146. These low numbers reflect the many obstacles encountered during drug development. Therapeutic antibody development is a resource-intensive multistep operation. It requires the generation of number of antibodies against a target antigen followed by the selection of a few lead candidates with ideal binding and biophysical properties27. The lead antibodies are then tested in vivo for efficacy using mouse models, for toxicity and pharmacokinetics using non-human primates followed by first-in-human clinical trials. Multiple antibodies demonstrating promising efficacy in mouse models have failed clinical development owing to toxicity either in non-human primates147 or in first-in-human trials148. Despite these hurdles, several antibodies described below are showing encouraging preclinical and clinical data and may change patient care in the coming years.
Novel targets
Solid tumours account for the majority of new patients with cancer. A significant percentage of patients with solid tumours present with metastatic disease requiring systemic therapy, which is rarely curative. Thus, metastatic lung, colon, pancreas, breast, prostate, liver and bile duct cancers are collectively responsible for the majority of all cancer deaths (Fig. 6). However, compared with haematological malignancies, the development of antibodies is lagging for solid tumours primarily because of the lack of targetable antigens. The different lymphoma subtypes account for ~3% of all cancer deaths and have five tumour antigens (CD19, CD20, CD79b, CD30, CCR4 and PD1) targeted by thera-peutic antibodies. By contrast, lung cancers responsible for ~21% of cancer deaths have only one targetable tumour antigen (EGFR-MET). In addition, cancers of the pancreas (~8% of cancer deaths), prostate (~6% of cancer deaths) and brain (~3% of cancer deaths) completely lack FDA-approved and/or EMA-approved therapeutic antibodies. This dis-parity is largely because lymphomas and myelomas arise from normal B cells, which express distinct targetable antigens (Fig. 6). In addition, normal B cells can be eradicated without intolerable consequences. Unfortunately, target antigen discovery has yielded few leads in solid tumours. Therefore, new target identification may need to involve single-cell sequencing of large numbers of patient samples to find antigens that are differentially expressed in cancer and normal tissue149. In addition, recent clinical trials are showing encouraging results for antibodies targeting claudins150,151, HER3 (ref. 152) fibroblast growth fac-tor receptor 2B (FGFR2B)153, DLL3 (ref. 154), connective tissue growth factor (CTGF; also known as CCN2)155 and six-transmembrane epithelial antigen of the prostate 1 (STEAP1)156. The antibody targets currently in phase II/III clinical trials are listed in Fig. 6 and Supplementary Table 2. Lastly, an innovative method to circumvent the lack of tumour-specific antigens is combinatorial targeting of multiple antigens. Most cancers lack expression of a truly specific antigen that is absent in healthy tis-sue. Thus, targeting antigen combinations that are co-expressed only in the tumours, but not in healthy tissue, may provide a viable therapeutic pathway157. Preclinical studies utilizing Boolean logic AND gates that require two or three targets to activate cytotoxic mechanisms have demonstrated tumour regression158–160, but they are yet to be tested in clinical trials.
Fig. 6 ∣. Antibody targets in common solid and haematological cancers.

These graphs show the percentage of deaths from the top 15 cancer types in the USA versus the number of unique antigens being targeted by approved antibodies and antibodies in late-phase clinical trials. The antigen targets are displayed across from each cancer tissue. Note that non-small-cell and small-cell lung cancers are included under ‘Lung’; Hodgkin lymphoma, non-Hodgkin lymphoma, chronic lymphocytic leukaemia (CLL) and central nervous system (CNS) lymphoma are included under ‘Lymphoma’; and acute myeloid leukaemia (AML), acute lymphoblastic leukaemia (ALL), chronic myeloid leukaemia (CML) and other leukaemias are included under ‘Leukaemia’. Microsatellite instability-high (MSI-H) cancers irrespective of tissue type are eligible for immune checkpoint inhibitor therapy, and HER2-positive solid tumours are eligible for HER2-directed therapy with trastuzumab deruxtecan, and are not included in the analysis. Lutetium Lu 177 vipivotide tetraxetan for prostate cancer was not included because the targeting moiety is not an antibody. Data for the percentage of cancer deaths and the number of approved targets were obtained from the https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/2023-cancer-facts-figures.html and the Antibody Society, respectively. aProgrammed cell death protein 1 (PD1) therapy is approved only in Hodgkin lymphoma. bB7-H3 is considered a tumour-associated antigen and a possible immune checkpoint antigen.
Some cell surface proteoglycans such as chondroitin sulfate and heparan sulfate are important modulators of signalling pathways in cancer and other human diseases161. Chondroitin sulfate proteoglycans have been shown to promote tumour vascularization and modulate signal transduction pathways related to tumour growth, and they are expressed in multiple cancer types162. Glypicans are glycosylphosphati- dylinositol (GPI)-anchored cell surface HSPGs163,164. Several glypicans such as GPC1, GPC2 and GPC3 are overexpressed in cancers, indicating they are potential tumour-associated antigens165. Antibodies and CAR T cells targeting GPC1 (refs. 166,167), GPC2 (refs. 168–170) and GPC3 (refs. 171–174) have been developed for treating pancreatic cancer, neuroblastoma and liver cancer, respectively.
Tumour-specific antigens.
Most currently approved antibody-based therapeutics target tumour-associated antigens or tissue-specific antigens. Tumour-associated antigens, such as HER2 are overexpressed by cancer cells but also expressed by a subset of normal tissues albeit at lower levels. The difference in the expression levels between cancer and normal tissues provides a therapeutic window. Tissue-specific antigens such as CD20 are expressed at similar levels in cancer cells (B cell lymphomas) and normal cells (B cells). Therapies targeting such tissue-specific antigens aim to eliminate both normal and cancer cells, as depletion of the normal B cells is clinically tolerable. Thera-pies targeting CD19 (refs. 101,175,176), CD20 (refs. 11,177,178), CD38 (ref. 179) and BCMA180,181 for cancers of the B cell lineage represent the most successful examples of tissue-specific therapeutic antibodies. For cancers arising out of less dispensable tissues such as T cells, TCR β-chain constant domain (TRBC)-specific and TCR β-chain variable domain (TRBV)-specific antibodies targeting clonal variations in TCRs have been developed to target all clonal cancer cells, but only a fraction of the normal T cells, thus preserving enough of the normal tissue to carry out its functions182–184. Some emerging antibody targets include tumour-specific antigens, that are expressed exclusively by cancer cells and not by normal cells. Leading examples of tumour-specific antigens are neoantigens, which are mutant peptides presented by MHC molecules. The mutant peptides are short proteolytic products of proteins carrying amino acid changes resulting from mutations (point mutations, small insertion/deletions, frameshift mutations, fusions and splice variants) that are hallmarks of cancer cells. Public neoantigens derived from mutant cancer driver genes such as BRAF, RAS, PIK3CA, TP53 and isocitrate dehydrogenase (IDH), provide tar-gets exquisitely unique to cancer cells and can be exploited to benefit patients28,29,185,186. These public neoantigens are currently being tar-geted by bispecific antibodies and are showing encouraging activity towards cancers harbouring TP53 and RAS mutations in preclinical studies28,29,187. Multiple efforts have described the structural basis for recognition of neoantigens by antibodies29,186,188. Crystal structures of the p53(R175H) mutant peptide-MHC have been determined in complex with both p53(R175H)-specific TCRs and antibodies (Fig. 3f). The TCRs shared a pronounced ‘canonical’ perpendicular binding orientation to the peptide-MHC molecule, skewed to the site of the mutation189. By contrast, the p53(R175H) neoantigen-specific antibody uses a non-canonical parallel binding model along the length of the peptide29 (Fig. 3f). Moreover, the measured affinity of the antibody for the p53(R175H) neoantigen is −100-fold higher than that of the corresponding TCR. It is possible that the different mode of binding contributes to the specificity and high affinity of the antibody.
Intracellular antigens.
Antibodies are unable to permeate through the cell membrane and thus can only target cell-surface antigens. Intracellular antigens can be presented on the cell surface in associa-tion with MHC molecules making them targetable. In this way, teben- tafusp targets the intracellular antigen gp100), which is overexpressed in melanoma and presented on the cell surface in association with HLA-A*02:01 (refs. 103,104). Antibodies are now being developed to target similar cancer-specific overexpression of intracellular pro-teins such as human telomerase reverse transcriptase (hTERT), MUC1, NY-ESO1, TCR gamma alternate reading frame protein (TARP), p53, WT1 and preferentially expressed antigen of melanoma (PRAME)190 with PRAME targeting showing encouraging preliminary results in clinical trials191.
Antigens in the tumour microenvironment.
Cancers develop within a complex mixture of immune cells, fibroblasts and extracellular matrix, which is referred to as the tumour microenvironment. Early attempts at antibody-based targeting of cancer-associated fibroblasts192,193 or the extracellular matrix194,195 failed to yield clinical benefit. Current studies are examining the role of immune cells within the tumour microenvironment such as tumour-associated macrophages (TAMs), dendritic cells, and myeloid-derived suppressor cells (MDSCs) that are known to drive resistance to immune checkpoint-inhibitors196. Thus, depleting or blocking the function of these immunosuppres-sive cells may render the tumour sensitive to immune checkpoint inhibitors. The early-phase clinical trials have demonstrated the safety and feasibility of antibody-mediated inhibition of TAMs by targeting macrophage colony-stimulating factor 1 receptor (CSF1R)197, trig-gering receptor expressed on myeloid cells 1 (TREM1)198 and TREM2 (ref. 199). Preclinical studies also suggest that augmenting the func-tion of dendritic cells within the tumour microenvironment using the agonistic CD40 monoclonal antibody can enhance T cell mediated tumour regression200. Clinical trials in patients with pancreatic cancers with anti-CD40 were well-tolerated by patients and have demonstrated enhanced T cell activation and infiltration into the tumours201,202. The T cell immunoglobulin domain and mucin domain 3 (TIM3) receptor is expressed by a range of immune cells such as lymphocytes, dendritic cells, monocytes and macrophages and TIM-3 antagonistic antibod-ies can similarly promote tumour killing by modulating the tumour microenvironment. TIM-3 antagonistic antibodies in combination with currently approved immune checkpoint inhibitors demonstrated a manageable safety profile in patients with solid cancers203,204. All the antibodies that counter the immune suppressive state within the tumour microenvironment are now being tested in phase II/III trials that will determine their ultimate utility.
Targeting low-density antigens
The currently approved ADCs and bispecific antibodies target rela-tively abundant antigens such as CD19, CD20 and BCMA, which have about 1,000 to 50,000 copies per cancer cell205–209. The major chal-lenge for targeting neoantigens stems from their low abundance on the cancer cell surface, with some cells expressing 1–10 copies of the target antigen28,29. In addition to neoantigens, effective targeting of tumour-associated or tissue-specific antigens can suffer from their variable cell surface densities as well. Furthermore, therapeutic pres-sure can also select for cancer cell clones with a low expression of target antigens. Preclinical studies have shown that bispecific antibodies were able to kill cancer cells expressing as low as 1–10 antigens per cell29 whereas ADCs require 1,000 copies or above for cell killing210. Unmodified full-length antibodies probably require an even higher target antigen expression211. Targeting such low-density antigens remains a challenge and optimized antibody engineering are mak-ing it possible to effectively target neoantigens at single-digit levels in preclinical studies28,29.
Novel immune checkpoint inhibitors
The approval of ipilimumab in 2011 heralded the age of immunother-apy. Immune checkpoint blockers have the unprecedented ability to induce long-term remissions in patients with some relapsed solid tumours, which has led to a flurry of clinical trials. The FDA and EMA have approved 11 different versions of CTLA4, PD1 and PDL1 targeting antibodies for more than 65 different indications across more than 20 cancer types212–214 (Table 1). After the initial success of CTLA4, PD1 and PDL1 targeting, a host of additional immune checkpoints such as LAG3, T cell immunoreceptor with immunoglobulin and ITIM domains (TIGIT), V-domain immunoglobulin suppressor of T cell activation (VISTA), TIM3, PVRIG, natural killer group 2A (NKG2A) and OX40 (refs. 215–217) (Supplementary Table 2) were explored for their ability to induce remissions in a range of cancer types218. Except for relatlimab targeting LAG3 in melanoma219, all others have yet to demonstrate the therapeutic activity in phase III trials required for regulatory approval. Similarly, a macrophage checkpoint inhibitor magrolimab that blocks CD47 showed encouraging activity in early-phase trials, but phase III trials in patients with MDS and acute myeloid leukaemia (AML) were discontinued owing to lack of efficacy220,221. The current performance of the novel immune checkpoint blockers being below expectations led the pharmaceutical industries to focus on the PD1-PDL1 space with five new agents entering regulatory review in addition to the nine existing antibodies (Table 1). Despite the success with PD1-PDL1 targeting, only a minority (~20%) of patients respond to immune checkpoint block-ers and an even smaller fraction (~13%) achieve durable remissions222. In addition, current immune checkpoint inhibitors are associated with considerable complications, wherein ~1% are fatal and ~40% are chronic toxicities223. Thus, we need to look beyond PD1-PDL1 inhibition. This will require a change in the direction of research in cancer biology and the antibody-drug approval methods by the regulatory agencies. We should prioritize basic and translational research in novel immune checkpoint-modulating agents such as B and T lymphocyte attenu-ator (BTLA)224, B7-H3 (also known as CD276)225, CD27 (ref. 226) and 4–1BB (ref. 227), instead of revisiting the much-explored PD1-PDL1 axis and combination therapies. Regulatory agencies may consider revising the approval process, enabling the new PD1-PDL1 blockers to receive approval for all existing indications after demonstrating equivalent efficacy and toxicity profiles with competing products at an initial phase III trial.
Novel antibody formats
IgM antibodies.
All full-length therapeutic antibodies use the IgG isotype. Preclinical studies and early-phase trials are exploring the pentameric IgM format which has ten antigen-binding sites compared with two binding sites available in the IgG format. This provides higher binding avidity compared with IgG antibodies targeting the same epitope228–230. An example is IGM-8444, a high avidity pentameric IgM agonistic antibody targeting death receptor 5 (DR5), which has demonstrated tumour regression in mouse models229, and has now entered phase I trials231.
Novel bispecific formats.
The TCE bispecific format continues to evolve by improving efficacy while reducing adverse effects. More than 200 bispecific antibodies are now in clinical development with the majority targeting solid tumours using the TCE approach232. Glofitamab (a CD20xCD3 bispecific antibody for lymphoma)233 and xaluritamig (also known as AMG 509) (a STEAP1xCD3 bispecific antibody for pros-tate cancer)156 have both shown efficacy in clinical trials and share a unique 2:1 T cell-engaging bispecific format with bivalent binding to the target and monovalent binding to CD3. This format may provide increased tumour cell killing capacity over a traditional 1:1 target-to- CD3 binding design234,235 (Table 2). The IgM format TCE bispecific anti-bodies have a more impressive 10:1 target-to-CD3 binding ratio owing to the use of the pentameric IgM228,230,236 (Table 2). The high target-to- CD3 binding ratio designs allow high-avidity target binding, potentially engendering increased target cytotoxicity, while reducing cytokine secretion. A phase I trial of imvotamab (also known as IGM-2323), a CD20xCD3 IgM bispecific antibody, has demonstrated several complete responses in patients with advanced B cell malignancies237.
Bispecific immune checkpoint blockers.
The bispecific format is also being adopted for the simultaneous inhibition of two immune checkpoints to enhance T cell activation beyond what can be achieved with a combination of two monospecific immune checkpoint blockers. Bispecific immune checkpoint blockers targeting PD1xLAG3 (ref. 238), PD1xCTLA4 (refs. 239–241) (Table 2) and PDL1xCTLA4 (ref. 242) are showing encouraging efficacy in early-phase clinical trials. Although the initial group of bispecific immune checkpoint blockers has focused on inhibiting the clinically established immune checkpoints, preclinical studies are investigating whether similar bispecific immune checkpoint blockers can induce antitumour immunity by targeting novel immune receptors such as OX40 (ref. 243), TIGIT244 and C-type lectin domain family 9 member A (CLEC9A)245.
Trispecific antibodies.
Trispecific antibodies possess the capability to concurrently interact with three distinct antigens100. This func-tionality enables trispecific antibodies to interact with two distinct cancer-associated antigens, such as CD19 and CD20 in the context of B cell malignancies, while simultaneously engaging a cytotoxic T cell through binding to CD3 using the third binding domain. This innova-tive approach serves to mitigate therapy resistance arising from the loss of a singular antigen, such as CD19 or CD20, as the cancer cells must concurrently downregulate both antigens to evade killing246. Alternatively, trispecific antibodies are designed to interact with two T cell receptors, such as CD3 and CD28, thereby delivering a more potent activating signal against the cancer cell247.
Nanobodies.
One area of research that may lead to an important paradigm change in antibody engineering is the development of single-domain antibodies (also called nanobodies). The nanobod-ies, predominantly derived from camelid heavy chain antibodies, are small, easy to produce, stable and capable of penetrating bur-ied sites in tumour or viral antigens. Nanobodies are usually isolated from camelid-derived phage display libraries166,248,249, and recently from transgenic mice such as RenNano and nanomice250,251. Nanobodies have a long CDR3 loop that can penetrate cavities on the surface of an antigen252. The first nanobody-based CAR T cell targeting BCMA has received FDA approval253 and demonstrated improved efficacy when compared with antibody-based CAR T cells254. Nanobody-derived drugs targeting CD19 or CD20 and HER2 are currently being tested in clinical trials for patients with lymphomas and breast cancer, respectively255. Whether nanobodies will provide improved efficacy over current antibody-based drugs remains to be seen.
Activatable antibodies.
The tumour microenvironment often exhib-its a lower pH, harbours active protease enzymes and possesses a particular combination of antigens that are lacking in normal tissues. These unique properties are exploited to selectively target cancer cells with activatable antibodies that gain or lose function based on changes to their environmental conditions256. The addition of histi-dine residues at certain positions within the antibody sequence ena-bles pH-dependent binding to its target antigen. This principle was used to generate a HER2-targeting antibody with increased binding to HER2 expressed within the acidic microenvironment of solid tumours compared with normal tissue257. Similar engineering led to increased antibody dissociation from target antigens (HER2 and CTLA4) within acidic lysosomes, thereby promoting antigen re-cycling and presen-tation at the cell membrane, and enhancing antitumour efficacy258,259. Activatable antibodies also use protease-cleavable masking moieties that block the epitope-binding site. Cleavage of the masking moie-ties within the tumour microenvironment enables antibody-antigen binding and antitumour activity. Antibodies with such masking mechanisms are often called probody therapies and are being used to improve the targeting of PDL1, CTLA4, EGFR, CD166 and CD71 in a range of solid and haematological cancers260–263. Lastly, cancer cells often display a unique combination of antigens that enables discrimi-nation between cancer and non-cancerous tissues157. A combination of antibodies or antibody-like peptides are being designed to enable combinatorial antigen targeting of CD45+HLA-A2+ cancers arising after haploidentical bone marrow transplantations, and EGFR+HER2+ breast cancers159,160,264.
Novel conjugated antibodies
Improving ADCs.
Studies are now focusing on improving the com-ponents of an ADC (the targeting antibody, linker, and drug payload) to increase ADC efficacy. Preclinical studies suggest that the large molecular weights of IgG antibodies (~150 kDa) may reduce tissue penetration265. Thus, ADC activity may be improved with the use of antibodies lacking Fc segments266 or by replacing antibodies with small tumour-targeting peptides267. Modulating linker designs and increasing DAR may enhance therapeutic benefits. For example, an ADC targeting the cell surface glycoprotein TROP2 (sacituzumab govitecan) with high DAR was the first ADC to improve survival in patients with triple-negative breast cancer268. The innovative tetrapeptide linker developed by Daiichi Sankyo, used in trastuzumab deruxtecan, masks the hydrophobicity of the payload DXd and allows uniform attachment of a large number of drugs while maintaining favourable pharmacoki-netics and minimizing premature drug release into plasma126. This linker-payload combination is now being adopted by several ADCs that are either nearing approval (datopotamab deruxtecan and patritumab deruxtecan) or in clinical trials (ifinatamab deruxtecan)269. Rinatabart sesutecan, which targets folate receptor α (FRa), also uses a novel cleavable linker to mask even more hydrophobic payloads, in this case exatecan, a topoisomerase-1 inhibitor, thus allowing the generation of more potent ADCs269. The ADCs in late clinical trials are listed in Sup-plementary Table 2. It is expected that such linker-payload combina-tions could enable better HER2-targeting in patients with lung and colon cancers270, which have a low response rate to the HER2 targeting ADC trastuzumab deruxtecan271,272. Finally, novel drug payloads such as protein degraders, RNA polymerase inhibitors, BCL-xL inhibitors, Toll-like receptor (TLR) agonists and stimulator of interferon genes (STING) agonists are expected to improve ADCs by using novel killing mechanisms112,125. Improvements in linker and conjugation techniques have helped drive the higher clinical adoption of ADCs over the other conjugation formats such as toxin or radioisotope conjugates or the bispecific TCE formats273,274. Clinical trials are now underway to test novel ADCs either as single agents or in combinations with chemo-therapies or immunotherapies with several of these yielding promising results so far275–278.
Immunotoxin conjugates.
A distinct limitation of immunotoxins is the generation of antitoxin immune responses that reduce half-life and efficacy. Current work is focusing on reducing the immunogenicity of the Pseudomonas exotoxin by removing epitopes that are recognized by host immune cells. Such modified immunotoxins may improve efficacy and allow immunotoxins to target a range of malignancies. The generation of interleukin-toxin conjugates is another development in this area and exploits the overexpression of interleukin receptors in cancer cells279. The interleukin-toxin conjugates behave similarly to antibody-toxin conjugates. Tagraxofusp, a cytotoxin targeting interleukin-3 receptor subunit alpha (IL3RA; also known as CD123) consisting of IL-3 fused to diphtheria toxin was shown to be effective in patients with blastic plasmacytoid dendritic cell neoplasm (BPDCN)280. Similarly, denileukin diftitox (withdrawn in 2014 to enable manufac-turing improvements), an IL-2R-targeting cytotoxin consisting of IL-2 fused to diphtheria toxin was effective in patients with cutaneous T cell lymphoma (CTCL)281. However, these IL-toxin conjugates are not considered antibody-based drugs as they use a receptor ligand instead of a scFv for tumour targeting.
Radioisotope conjugates.
There has been some renewed interest in radioisotope conjugates after the success of small molecule- conjugated and peptide-conjugated radioisotopes in solid tumours. Conjugating lutetium-177 to a small molecule that binds prostate- specific membrane antigen (PSMA), or to a peptide analogue that binds somatostatin receptor demonstrated remarkable tumour regression in patients with prostate cancer282 and neuroendocrine tumours283, respectively, leading to FDA approvals. Such tumour-targeting small molecule-radioisotope or peptide-radioisotope conjugates behave similarly to antibody-radioisotope conjugates but have distinct struc-tures and pharmacokinetics. Clinical trials are now underway to test peptide-radioisotope conjugates targeting carbonic anhydrase IX (CAIX) in patients with renal cell cancers284 and antibody-radioisotope conjugates targeting GD2 in patients with brain tumours, CD33 in patients with AML, PSMA in patients with prostate cancer, and CD45 and CD37 in patients with haematological malignancies138,285. Addi-tional approaches involve conjugating tumour-targeting antibodies to α-particle emitters such as actinium-225 (225Ac)286. α-Particles have higher DNA-damaging energy coupled with a shorter range and may increase tumour-specific cytotoxicity while limiting adverse effects from irradiation to surrounding healthy tissues138.
Selecting the optimal format
Currently approved antibody-based therapeutics target tumour- associated antigens that are expressed by cancer cells and by a sub-set of normal cells. The antibodies require a therapeutic window that allows cancer cell killing while limiting off-tumour toxicities to normal cells. A judiciously selected antibody format is required to harness this therapeutic window, and the use of an improper format may be ineffective owing to reduced therapeutic efficacy or unacceptable toxicity. Although definitive quantifiable studies and head-to-head comparisons are lacking, it is generally accepted that cell killing by unmodified antibodies requires high antigen expression (>10,000 copies per cell), whereas ADCs (>1,000 copies per cell) and TCE bispe-cific antibodies or CAR T cells (>10–100 copies per cell) can kill lower antigen-expressing cells. Thus, switching from an unmodified antibody to an ADC or TCE bispecific antibody format for a given target can increase efficacy but may also cause a concomitant increase in toxicity. This effect was exemplified with HER2 targeting. HER2 is expressed by a subset of breast and gastric cancers and a range of normal human tissues287,288. HER2 targeting with naked antibodies (trastuzumab and pertuzumab) and later by an ADC (trastuzumab emtansine) was well- tolerated by patients and led to tumour regression in patients with a high level of HER2 expression44,289. More recently, trastuzumab deruxte- can, a HER2-targeting ADC with high DAR, induced tumour regression even in cancers with low HER2 expression that were unresponsive to the previous HER-targeting antibodies130. However, anti-HER2 CAR T cells that further lower the HER2-targeting threshold caused fatal respiratory failure because of on-target, off-tumour toxicity to the low HER2-expressing cells in the lungs290. The lethal toxicity of the anti- HER2 CAR T cells raises the possibility that HER2-targeting TCE bispe-cific antibodies, which can target similarly low-level HER2-expressing cells, may also carry unacceptable toxicities. Thus, HER2-targeting by the bispecific antibody zanidatamab adopted the dual HER2 binding strategy instead of the TCE approach (Table 2) and has demonstrated efficacy without the lethal toxicity110. Thus, ‘clean’ targets with minimal off-tumour expression in critically important tissues (such as CD19) and low-density targets (such as peptide-MHC molecules) are opti-mal for bispecific TCE-based therapies. Targets with some level of off- tumour expression in non-dispensable tissues (such as HER2) may be best targeted by unmodified antibodies, ADCs or non-TCE bispecific antibodies.
It should be noted that the off-tumour toxicity and efficacy of a particular design are also target and drug payload-dependent. For example, DLL3 is expressed by a subset of patients with small-cell lung cancer291. In this case, the clinical development of rovalpituzumab tesirine, a DLL3-targeting ADC, was halted owing to a high level of toxic-ity related to the PBD payload and a lack of survival benefit. However, a phase I trial has indicated that DLL3 targeting by the TCE bispecific antibody tarlatamab leads to tumour regression and is well-tolerated by patients105,154. Ultimately, the chosen target and its antigen density may critically inform the optimal therapeutic format.
Concluding remarks
In summary, antibody-based therapeutics hold great promise in the oncology space owing to the level of precision an antibody can provide in binding to its target. Major challenges remain, but new targets and a variety of novel designs and engineering strategies are expected to provide solutions to many of these challenges. The explosion of more effective antibody-based therapeutics receiving regulatory approval will probably continue to follow the trend shown in the past two and a half decades.
Supplementary Material
Acknowledgements
S.P. M.F.K., C.B., N.P. and S.Z. were supported by The Virginia and D.K. Ludwig Fund for Cancer Research, the Lustgarten Foundation for Pancreatic Cancer Research, The Commonwealth Fund, the Bloomberg-Kimmel Institute for Cancer Immunotherapy, and the National Institutes of Health (NIH) Cancer Center Support grant P30 CA006973. S.P. was supported by NCI grant K08CA270403, the Leukaemia Lymphoma Society Translation Research Program award, the American Society of Hematology Scholar award, and the Swim Across America Translational Cancer Research Award. M.F.K. was supported by NIH/NIAID grant 1R21AI176764–01, the Jerome Greene Foundation, the Cupid Foundation, the Lupus Research Alliance Lupus Innovation Award, the Rheumatology Research Foundation Investigator Award, the Harrington Discovery Institute Harrington Scholar-Innovator Award, the Sol Goldman MS Research Program, and the Bisciotti Foundation Translational Fund. C.B. was supported by NCI grant R37 CA230400. M.H. was supported by the Intramural Research Program of NIH, NCI and Center for Cancer Research (Z01 BC010891, ZIA BC010891 and ZIC BC 011891).
Glossary
- Anaphylaxis
A severe and potentially life-threatening reaction owing to exposure to an allergen such as an antibody or other medication. Common symptoms of anaphylaxis include swelling of the face and throat, difficulty in breathing, an increase in heart rate, a drop in blood pressure and loss of consciousness.
- Antibody-dependent cellular cytotoxicity
(ADCC). A mechanism through which antibodies bind to target cells followed by recruitment of immune cells such as NK cells and macrophages to kill the target cells. The immune cells secrete cytotoxic granules (perforins and granzymes), and induce FAS signalling leading to target cell death.
- Antibody-dependent cellular phagocytosis
(ADCP). A mechanism through which antibodies bind to target cells, which in turn stimulates immune cells such as macrophages to engulf and degrade the target cells.
- B cell cloning
Isolation and expansion of single B cells that produce the desired monoclonal antibodies, to obtain the antibody-coding sequence.
- Bystander effects
With antibody-drug conjugates (ADCs), refers to a phenomenon wherein neighbouring cells near the target cancer cell are killed by the released cytotoxic payload. This effect can enhance the overall potency of the ADC by causing a broader destruction of cancer cells beyond the primary target cell.
- Capillary leak syndrome
Condition characterized by the leakage of fluid from small blood vessels (capillaries) into surrounding tissues. This leakage leads to a decrease in blood volume and can result in low blood pressure along with oedema (swelling) in various parts of the body, including the lungs, and organ failure.
- Complementarity-determining regions
(CDRs). Specific regions within the antibody heavy and light chain variable domains that bind to the target antigen.
- Complement-dependent cytotoxicity
(CDC). A mechanism through which antibodies bind to the target cell followed by activation of the complement system, leading to lysis of the target cells.
- Cytokine release syndrome
(CRS). Systemic inflammation characterized by a constellation of symptoms such as fever, hypotension and hypoxia and mediated by the release of multiple cytokines from the immune cells of patients. CRS is a typical adverse effect observed with the use of T cell engager (TCE) bispecific antibodies.
- Cytopenias
A reduction in the number of circulating blood cells, such as red blood cells (erythrocytes), white blood cells (leukocytes) and/or platelets (thrombocytes). Cytopenias can be caused by several factors including exposure to drugs or antibodies that hinder the growth of new cells.
- Drug-antibody ratio
(DAR). The number of drugs attached to each antibody in an ADC.
- Fragment antigen binding
(Fab). An antibody which consists of two identical Fab fragments and one Fc fragment. Each Fab fragment is responsible for binding to a specific antigen. The Fab fragment is obtained by cleaving the antibody at specific sites using enzymes, such as papain or pepsin.
- Fragment crystallizable
(Fc). Fc fragment interacts with various immune cells through Fc receptors and with complement proteins that contribute to the immune response generated by the antibody. Each antibody class and subclass has unique Fc regions. Understanding the Fc fragment is crucial in the design of therapeutic antibodies because modifications to this region can impact the pharmacokinetics, effector functions and therapeutic efficacy of an antibody.
- Haemolytic uremic syndrome
A rare but serious condition that is characterized by the combination of haemolytic anaemia (destruction of red blood cells), thrombocytopenia (low platelet count) and acute kidney injury. It can be mediated by bacterial infections (such as Escherischia coii) or exposure to drugs and antibodies.
- Hydrophobicity
Refers to the property of being repelled by water. Hydrophobic substances are insoluble or poorly soluble in water. The hydrophobicity of the payload can affect the overall stability of the ADC. Highly hydrophobic payloads may lead to aggregation or destabilization of the ADC structure, potentially impacting its efficacy and safety.
- Microsatellite instability-high
(MSI-H). Cells with mismatch-repair deficiency resulting in high mutation burden and altered microsatellite (tract of repetitive DNA) sequences. MSI-H cancers are associated with a higher response to immune checkpoint- inhibiting antibodies.
- Monoclonal antibodies
Identical antibodies that bind to a specific part of the target antigen (epitope) and are derived from single clones of immune cells (such as B cells, plasma cells or hybridoma cells).
- Myelodysplastic syndromes
A group of disorders characterized by abnormal production and maturation of blood cells in the bone marrow. In myelodysplastic syndromes, the bone marrow fails to produce enough healthy blood cells, leading to low levels of red blood cells (anaemia), white blood cells (leukopenia) and platelets (thrombocytopenia).
- Neonatal Fc receptor
(FcRn). A receptor expressed by vascular endothelial cells and immune cells, which binds to the Fc portion of IgG antibodies. IgG antibody binding to FcRn leads to receptor-mediated internalization and recycling of the IgG, which is responsible for the long IgG half-life (about 21 days) in circulation.
- Peripheral neuropathy
A potential side effect that can occur owing to the cytotoxic payload component of the ADC affecting the peripheral nerves. Peripheral neuropathy caused by ADCs can manifest as numbness, tingling, burning sensations or pain in the hands, feet or other extremities.
- Public neoantigens
A public neoantigen is derived from a mutated protein and is found in multiple individuals with the same type of cancer. This shared characteristic makes public neoantigens particularly important in cancer immunotherapy because therapies targeting these common neoantigens can benefit a broad patient population. Common public neoantigens include BRAFV600E, KRASG2D, KRASG12C and TP53R175H By contrast, private neoantigens are unique to an individual patient with cancer. Targeting private neoantigens requires the development of personalized therapies such as custom cancer vaccines and T cell-based therapies.
- Single-chain variable fragment
(scFv). An engineered antibody fragment composed of variable regions of the heavy and light chains combined into a single peptide chain by a linker. The scFv retains the ability to bind specifically to a target antigen, similar to a full-size antibody. The advantages of scFv include its smaller size (−25 kDa), which facilitates easier production and manipulation.
- Single-domain antibodies
Also known as nanobodies, are antibodies derived from camelids that consist of only a variable heavy domain and as a result have a relatively low molecular weight (~15 kDa), hence the name nanobody. By contrast, human antibodies consist of variable heavy and light domains and have higher molecular weights (a full-length IgG antibody is −150 kDa and an scFv is - 25 kDa).
- Topoisomerase
Enzymes that maintain proper function and stability of DNA by cleaving DNA to relieve torsional strain and supercoiling occurring owing to processes such as DNA replication. Topoisomerase inhibitors disrupt this ability to maintain DNA and cause cell death.
- Tumour antigens
Proteins and other antigenic molecules expressed on the surface of tumour cells that can be targeted by therapeutic antibodies.
Related links
- American Cancer Society: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/2023-cancer-facts-figures.html
- Antibody Society: https://www.antibodysociety.org/resources/approved-antibodies/
Footnotes
Competing interests
The Johns Hopkins University has filed patent applications related to technologies described in this paper on which S.P., M.F.K., N.P., D.M.P. and S.Z. are listed as inventors. M.H. is an inventor on NIH patents in antibody and cell therapies and may receive blinded royalties from the NIH. N.P. is a founder of Thrive Earlier Detection, an Exact Sciences company. N.P. is a consultant to Thrive Earlier Detection. N.P. and S.Z. hold equity in Exact Sciences. N.P. and S.Z. are founders of and/or consultants to and own equity in ManaT Bio., Neophore and Personal Genome Diagnostics. N.P. holds equity in Haystack Oncology and CAGE Pharma. N.P. is a consultant to Vidium. S.P. owns equity in Gilead, is a consultant to Merck and received payment from IQVIA. M.F.K. is a consultant to Argenx, Atara Biotherapeutics, Revel Pharmaceuticals, Sana Biotechnology and Sanofi. S.Z. has a research agreement with BioMed Valley Discoveries, Inc. C.B. is a consultant for Depuy-Synthes, Bionaut Labs, Galectin Therapeutics, Haystack Oncology and Privo Technologies. C.B. is a co-founder of OrisDx and Belay Diagnostics. D.M.P. reports grant and patent royalties through institution from BMS, a grant from Compugen, stock from Trieza Therapeutics and Dracen Pharmaceuticals, and founder equity from Potenza; is a founder of and consultant to and owns equity in ManaT Bio; is a consultant for Aduro Biotech, Amgen, Astra Zeneca (Medimmune/Amplimmune), Bayer, DNAtrix, Dynavax Technologies Corporation, Ervaxx, FLX Bio, Rock Springs Capital, Janssen, Merck, Tizona and Immunomic-Therapeutics; is on the scientific advisory board of Five Prime Therapeutics, Camden Nexus II, WindMil; and is on the board of directors for Dracen Pharmaceuticals. K.M.W. and S.B.G. are employees of Merck & Co., Inc. at the time of submission and may have stock ownership in Merck & Co., Inc., Rahway, NJ, USA.
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41568-024-00690-x.
Data availability
The data used to make Figs. 1 and 6 are available from the American Cancer Society and the Antibody Society.
References
- 1.Sternberger LA & Sternberger NH Monoclonal antibodies distinguish phosphorylated and nonphosphorylated forms of neurofilaments in situ. Proc. Natl Acad. Sci. USA 80, 6126–6130 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stark SE & Caton AJ Antibodies that are specific for a single amino acid interchange in a protein epitope use structurally distinct variable regions. J. Exp. Med 174, 613–624 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Antibody therapeutics approved or in regulatory review in the EU or US. Antibody Society www.antibodysociety.org/resources/approved-antibodies (2023). The Antibody Society is an association that supports research and development of antibody-based drugs and maintains an updated list of antibodies approved by the FDA and EMA.
- 4.June CH & Sadelain M Chimeric antigen receptor therapy. N. Engl. J. Med 379, 64–73 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Labanieh L & Mackall CL CAR immune cells: design principles, resistance and the next generation. Nature 614, 635–648 (2023). [DOI] [PubMed] [Google Scholar]
- 6.Carter P. Improving the efficacy of antibody-based cancer therapies. Nat. Rev. Cancer 1, 118–129 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Weiner GJ Building better monoclonal antibody-based therapeutics. Nat. Rev. Cancer 15, 361–370 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ho M. Inaugural editorial: searching for magic bullets. Antib. Ther 1, 1–5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kohler G & Milstein C Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256, 495–497 (1975). This seminal paper describes the hybridoma technology that enabled the production of monoclonal antibodies. Kohler and Milstein received the Nobel Prize for this work.
- 10.Hwang WY & Foote J Immunogenicity of engineered antibodies. Methods 36, 3–10 (2005). [DOI] [PubMed] [Google Scholar]
- 11. Reff ME et al. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood 83, 435–445 (1994). This paper describes the generation of rituximab, the first chimeric antibody that demonstrated significant tumour reduction and later became the standard of care for the treatment of patients with B cell lymphomas.
- 12.Goldstein NI, Prewett M, Zuklys K, Rockwell P & Mendelsohn J Biological efficacy of a chimeric antibody to the epidermal growth factor receptor in a human tumor xenograft model. Clin. Cancer Res 1, 1311–1318 (1995). [PubMed] [Google Scholar]
- 13.Looney RJ et al. B cell depletion as a novel treatment for systemic lupus erythematosus: a phase I/II dose-escalation trial of rituximab. Arthritis Rheum. 50, 2580–2589 (2004). [DOI] [PubMed] [Google Scholar]
- 14. Jones PT, Dear PH, Foote J, Neuberger MS & Winter G Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature 321, 522–525 (1986). This paper describes the process of CDR grafting that enabled the production of humanized antibodies. The majority of cancer antibodies utilize the humanized antibody format.
- 15.Carter P. et al. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc. Natl Acad. Sci. USA 89, 4285–4289 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spieker-Polet H, Sethupathi P, Yam PC & Knight KL Rabbit monoclonal antibodies: generating a fusion partner to produce rabbit-rabbit hybridomas. Proc. Natl Acad. Sci. USA 92, 9348–9352 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang YF & Ho M Humanization of rabbit monoclonal antibodies via grafting combined Kabat/IMGT/Paratome complementarity-determining regions: rationale and examples. MAbs 9, 419–429 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parray HA et al. Hybridoma technology a versatile method for isolation of monoclonal antibodies, its applicability across species, limitations, advancement and future perspectives. Int. Immunopharmacol 85, 106639 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McCafferty J, Griffiths AD, Winter G & Chiswell DJ Phage antibodies: filamentous phage displaying antibody variable domains. Nature 348, 552–554 (1990). This paper describes the early phage display technologies that enabled the selection of antibodies against a range of antigens including cancer antigens. G. Smith and G. Winter received the Nobel Prize for their work on phage display-based antibody production.
- 20. Lonberg N. et al. Antigen-specific human antibodies from mice comprising four distinct genetic modifications. Nature 368, 856–859 (1994). This paper describes the development of transgenic mouse models that led to the production of fully human antibodies.
- 21.Lu RM et al. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci 27, 1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Winter G, Griffiths AD, Hawkins RE & Hoogenboom HR Making antibodies by phage display technology. Annu. Rev. Immunol 12, 433–455 (1994). [DOI] [PubMed] [Google Scholar]
- 23.Lu S. et al. The rapid and highly parallel identification of antibodies with defined biological activities by SLISY. Nat. Commun 14, 17 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alfaleh MA et al. Phage display derived monoclonal antibodies: from bench to bedside. Front. Immunol 11, 1986 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Almagro JC, Daniels-Wells TR, Perez-Tapia SM & Penichet ML Progress and challenges in the design and clinical development of antibodies for cancer therapy. Front. Immunol 8, 1751 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Booth B. Human antibody discovery: of mice and phage. Forbes https://www.forbes.com/sites/brucebooth/2017/05/11/human-antibody-discovery-of-mice-and-phage/?sh=1f76520c7f26 (2017). [Google Scholar]
- 27.Jain T. et al. Biophysical properties of the clinical-stage antibody landscape. Proc. Natl Acad. Sci. USA 114, 944–949 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Douglass J. et al. Bispecific antibodies targeting mutant RAS neoantigens. Sci. Immunol 6, eabd5515 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hsiue EH et al. Targeting a neoantigen derived from a common TP53 mutation. Science 371, eabc8697 (2021). This paper describes the generation of bispecific antibodies targeting the most common oncogenic mutation (R175H) in the tumour suppressor protein p53.
- 30.Boder ET, Midelfort KS & Wittrup KD Directed evolution of antibody fragments with monovalent femtomolar antigen-binding affinity. Proc. Natl Acad. Sci. USA 97, 10701–10705 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Georgiou G. et al. Display of heterologous proteins on the surface of microorganisms: from the screening of combinatorial libraries to live recombinant vaccines. Nat. Biotechnol 15, 29–34 (1997). [DOI] [PubMed] [Google Scholar]
- 32.Ho M, Nagata S & Pastan I Isolation of anti-CD22 Fv with high affinity by Fv display on human cells. Proc. Natl Acad. Sci. USA 103, 9637–9642 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lipovsek D & Pluckthun A In-vitro protein evolution by ribosome display and mRNA display. J. Immunol. Methods 290, 51–67 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Wang J. et al. Durable blockade of PD-1 signaling links preclinical efficacy of sintilimab to its clinical benefit. MAbs 11, 1443–1451 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marcus R. et al. Obinutuzumab for the first-line treatment of follicular lymphoma. N. Engl. J. Med 377, 1331–1344 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Vitolo U. et al. Obinutuzumab or rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated diffuse large B-cell lymphoma. J. Clin. Oncol 35, 3529–3537 (2017). [DOI] [PubMed] [Google Scholar]
- 37.van Imhoff GW et al. Ofatumumab versus rituximab salvage chemoimmunotherapy in relapsed or refractory diffuse large B-cell lymphoma: the ORCHARRD study. J. Clin. Oncol 35, 544–551 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Maloney DG et al. A phase 3 randomized study (HOMER) of ofatumumab vs rituximab in iNHL relapsed after rituximab-containing therapy. Blood Adv. 4, 3886–3893 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rugo HS et al. Margetuximab versus trastuzumab in patients with previously treated HER2-positive advanced breast cancer (SOPHIA): final overall survival results from a randomized phase 3 trial. J. Clin. Oncol 41, 198–205 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Price TJ et al. Panitumumab versus cetuximab in patients with chemotherapy-refractory wild-type KRAS exon 2 metastatic colorectal cancer (ASPECCT): a randomised, multicentre, open-label, non-inferiority phase 3 study. Lancet Oncol. 15, 569–579 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Furman WL Monoclonal antibody therapies for high risk neuroblastoma. Biologics 15, 205–219 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peter HH et al. Targeting FcRn for immunomodulation: benefits, risks, and practical considerations. J. Allergy Clin. Immunol 146, 479–491.e5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roopenian DC & Akilesh S FcRn: the neonatal Fc receptor comes of age. Nat. Rev. Immunol 7, 715–725 (2007). [DOI] [PubMed] [Google Scholar]
- 44. Slamon DJ et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med 344, 783–792 (2001). This is a groundbreaking clinical trial demonstrating the survival benefit of the HER2-targeting antibody trastuzumab in patients with breast cancer. Slamon received the Lasker–DeBakey award for his work related to HER2 targeting in breast cancer.
- 45.Bang YJ et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 376, 687–697 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Cunningham D. et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N. Engl. J. Med 351, 337–345 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Camidge DR et al. Phase I study of 2- or 3-week dosing of telisotuzumab vedotin, an antibody-drug conjugate targeting c-Met, monotherapy in patients with advanced non-small cell lung carcinoma. Clin. Cancer Res 27, 5781–5792 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Redman JM, Hill EM, AlDeghaither D & Weiner LM Mechanisms of action of therapeutic antibodies for cancer. Mol. Immunol 67, 28–45 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mone AP et al. Alemtuzumab induces caspase-independent cell death in human chronic lymphocytic leukemia cells through a lipid raft-dependent mechanism. Leukemia 20, 272–279 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Meyer S. et al. New insights in type I and II CD20 antibody mechanisms-of-action with a panel of novel CD20 antibodies. Br. J. Haematol 180, 808–820 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Vidarsson G, Dekkers G & Rispens T IgG subclasses and allotypes: from structure to effector functions. Front. Immunol 5, 520 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Salfeld JG Isotype selection in antibody engineering. Nat. Biotechnol 25, 1369–1372 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Yu J, Song Y & Tian W How to select IgG subclasses in developing anti-tumor therapeutic antibodies. J. Hematol. Oncol 13, 45 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang X, Mathieu M & Brezski RJ IgG Fc engineering to modulate antibody effector functions. Protein Cell 9, 63–73 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weng WK & Levy R Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J. Clin. Oncol 21, 3940–3947 (2003). [DOI] [PubMed] [Google Scholar]
- 56.Musolino A. et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J. Clin. Oncol 26, 1789–1796 (2008). [DOI] [PubMed] [Google Scholar]
- 57.Golay J & Taylor RP The role of complement in the mechanism of action of therapeutic anti-cancer mAbs. Antibodies 9, 58 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schneider-Merck T et al. Human IgG2 antibodies against epidermal growth factor receptor effectively trigger antibody-dependent cellular cytotoxicity but, in contrast to IgG1, only by cells of myeloid lineage. J. Immunol 184, 512–520 (2010). [DOI] [PubMed] [Google Scholar]
- 59.Rosner T. et al. Immune effector functions of human IgG2 antibodies against EGFR. Mol. Cancer Ther 18, 75–88 (2019). [DOI] [PubMed] [Google Scholar]
- 60. Cho HS et al. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 421, 756–760 (2003). This study of early structural biology has improved our understanding of trastuzumab binding to the HER2 extracellular subdomain 4 to inhibit HER2 function.
- 61.Hao Y, Yu X, Bai Y, McBride HJ & Huang X Cryo-EM structure of HER2-trastuzumab-pertuzumab complex. PLoS ONE 14, e0216095 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weisser NE et al. An anti-HER2 biparatopic antibody that induces unique HER2 clustering and complement-dependent cytotoxicity. Nat. Commun 14, 1394 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rouge L. et al. Structure of CD20 in complex with the therapeutic monoclonal antibody rituximab. Science 367, 1224–1230 (2020). [DOI] [PubMed] [Google Scholar]
- 64.Liu R, Oldham RJ, Teal E, Beers SA & Cragg MS Fc-engineering for modulated effector functions-improving antibodies for cancer treatment. Antibodies 9, 64 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Nimmerjahn F, Vidarsson G & Cragg MS Effect of posttranslational modifications and subclass on IgG activity: from immunity to immunotherapy. Nat. Immunol 24, 1244–1255 (2023). This review describes the Fc domain modifications that alter IgG antibody effector functions.
- 66.Rugo HS et al. Efficacy of margetuximab vs trastuzumab in patients with pretreated ERBB2-positive advanced breast cancer: a phase 3 randomized clinical trial. JAMA Oncol. 7, 573–584 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ribas A & Wolchok JD Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Le DT et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med 372, 2509–2520 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Le DT et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413 (2017). This paper demonstrates the utility of the PD1 antibody, pembrolizumab, in a range of cancer types with mismatch repair deficiency. This led to the FDA approval of pembrolizumab in any cancer with mismatch repair deficiency which was the first tissue agnostic drug approval in oncology.
- 70.Gandhi L. et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N. Engl. J. Med 378, 2078–2092 (2018). [DOI] [PubMed] [Google Scholar]
- 71.Marabelle A. et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 21, 1353–1365 (2020). [DOI] [PubMed] [Google Scholar]
- 72.Nghiem PT et al. PD-1 blockade with pembrolizumab in advanced Merkel-cell carcinoma. N. Engl. J. Med 374, 2542–2552 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sharma P. et al. The next decade of immune checkpoint therapy. Cancer Discov. 11, 838–857 (2021). [DOI] [PubMed] [Google Scholar]
- 74. Chamoto K, Yaguchi T, Tajima M & Honjo T Insights from a 30-year journey: function, regulation and therapeutic modulation of PD1. Nat. Rev. Immunol 23, 682–695 (2023). Together with Sharma et al. (2021), this review describes the mechanism of immune checkpoint-inhibiting antibodies in cancer. Allison and Honjo jointly received the Nobel Prize for their discoveries of a cancer therapy by inhibition of the immune checkpoint PD1.
- 75.Huard B. et al. Characterization of the major histocompatibility complex class II binding site on LAG-3 protein. Proc. Natl Acad. Sci. USA 94, 5744–5749 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang J. et al. Fibrinogen-like protein 1 is a major immune inhibitory ligand of LAG-3. Cell 176, 334–347.e12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Blackburn SD, Shin H, Freeman GJ & Wherry EJ Selective expansion of a subset of exhausted CD8 T cells by alphaPD-L1 blockade. Proc. Natl Acad. Sci. USA 105, 15016–15021 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Im SJ et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yost KE et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med 25, 1251–1259 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Romano E. et al. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc. Natl Acad. Sci. USA 112, 6140–6145 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim MJ et al. Deletion of PD-1 destabilizes the lineage identity and metabolic fitness of tumor-infiltrating regulatory T cells. Nat. Immunol 24, 148–161 (2023). [DOI] [PubMed] [Google Scholar]
- 82.Sharma A. et al. Anti-CTLA-4 immunotherapy does not deplete FOXP3+ regulatory T cells (Tregs) in human cancers. Clin. Cancer Res 25, 1233–1238 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kamada T. et al. PD-1+ regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc. Natl Acad. Sci. USA 116, 9999–10008 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kumagai S. et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat. Immunol 21, 1346–1358 (2020). [DOI] [PubMed] [Google Scholar]
- 85.Scapin G. et al. Structure of full-length human anti-PD1 therapeutic IgG4 antibody pembrolizumab. Nat. Struct. Mol. Biol 22, 953–958 (2015). [DOI] [PubMed] [Google Scholar]
- 86.You W. et al. A network meta-analysis comparing the efficacy and safety of anti-PD-1 with anti-PD-L1 in non-small cell lung cancer. J. Cancer 9, 1200–1206 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Barlesi F. et al. Avelumab versus docetaxel in patients with platinum-treated advanced non-small-cell lung cancer (JAVELIN Lung 200): an open-label, randomised, phase 3 study. Lancet Oncol. 19, 1468–1479 (2018). [DOI] [PubMed] [Google Scholar]
- 88.Lee NY et al. Avelumab plus standard-of-care chemoradiotherapy versus chemoradiotherapy alone in patients with locally advanced squamous cell carcinoma of the head and neck: a randomised, double-blind, placebo-controlled, multicentre, phase 3 trial. Lancet Oncol. 22, 450–462 (2021). [DOI] [PubMed] [Google Scholar]
- 89.Monk BJ et al. Chemotherapy with or without avelumab followed by avelumab maintenance versus chemotherapy alone in patients with previously untreated epithelial ovarian cancer (JAVELIN Ovarian 100): an open-label, randomised, phase 3 trial. Lancet Oncol. 22, 1275–1289 (2021). [DOI] [PubMed] [Google Scholar]
- 90.Bang YJ et al. Phase III, randomised trial of avelumab versus physician’s choice of chemotherapy as third-line treatment of patients with advanced gastric or gastro-oesophageal junction cancer: primary analysis of JAVELIN Gastric 300. Ann. Oncol 29, 2052–2060 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lee JY et al. Structural basis of checkpoint blockade by monoclonal antibodies in cancer immunotherapy. Nat. Commun 7, 13354 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lee HT et al. Molecular mechanism of PD-1/PD-L1 blockade via anti-PD-L1 antibodies atezolizumab and durvalumab. Sci. Rep 7, 5532 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee HT, Lee SH & Heo YS Molecular interactions of antibody drugs targeting PD-1, PD-L1, and CTLA-4 in immuno-oncology. Molecules 24, 1190 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lin DY et al. The PD-1/PD-L1 complex resembles the antigen-binding Fv domains of antibodies and T cell receptors. Proc. Natl Acad. Sci. USA 105, 3011–3016 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu K. et al. Structural basis of anti-PD-L1 monoclonal antibody avelumab for tumor therapy. Cell Res. 27, 151–153 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tang S & Kim PS A high-affinity human PD-1/PD-L2 complex informs avenues for small-molecule immune checkpoint drug discovery. Proc. Natl Acad. Sci. USA 116, 24500–24506 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.He M. et al. Remarkably similar CTLA-4 binding properties of therapeutic ipilimumab and tremelimumab antibodies. Oncotarget 8, 67129–67139 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Brahmer JR et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol 36, 1714–1768 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang DY et al. Fatal toxic effects associated with immune checkpoint inhibitors: a systematic review and meta-analysis. JAMA Oncol. 4, 1721–1728 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Runcie K, Budman DR, John V & Seetharamu N Bi-specific and tri-specific antibodies — the next big thing in solid tumor therapeutics. Mol. Med 24, 50 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Bargou R. et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 321, 974–977 (2008). This paper describes the first-in-human clinical trial of the CD19-targeting bispecific antibody blinatumomab, which was the first bispecific antibody to achieve widespread adoption in the clinic and subsequently set the stage for the widespread adoption of the bispecific antibody format.
- 102. Labrijn AF, Janmaat ML, Reichert JM & Parren P Bispecific antibodies: a mechanistic review of the pipeline. Nat. Rev. Drug Discov 18, 585–608 (2019). This detailed review describes the different formats of bispecific antibodies.
- 103.Liddy N. et al. Monoclonal TCR-redirected tumor cell killing. Nat. Med 18, 980–987 (2012). [DOI] [PubMed] [Google Scholar]
- 104.Nathan P. et al. Overall survival benefit with tebentafusp in metastatic uveal melanoma. N. Engl. J. Med 385, 1196–1206 (2021). [DOI] [PubMed] [Google Scholar]
- 105.Ahn MJ et al. Tarlatamab for patients with previously treated small-cell lung cancer. N. Engl. J. Med 389, 2063–2075 (2023). [DOI] [PubMed] [Google Scholar]
- 106.Park K. et al. Amivantamab in EGFR exon 20 insertion-mutated non-small-cell lung cancer progressing on platinum chemotherapy: initial results from the CHRYSALIS phase I study. J. Clin. Oncol 39, 3391–3402 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Moores SL et al. A novel bispecific antibody targeting EGFR and cMet is effective against EGFR inhibitor-resistant lung tumors. Cancer Res. 76, 3942–3953 (2016). [DOI] [PubMed] [Google Scholar]
- 108.Dixit S, Abraham L, Weiser N & Gold MR Super-resolution imaging studies of zanidatamab: providing insights into its bispecific mode of action. Cancer Res. 81, abstr. 1032 (2021). [Google Scholar]
- 109.Weiser NE et al. The bispecific antibody zanidatamab’s (ZW25’s) unique mechanisms of action and durable anti-tumor activity in HER2-expressing cancers. Cancer Res. 81, abstr. 1005 (2021). [Google Scholar]
- 110.Meric-Bernstam F. et al. Zanidatamab, a novel bispecific antibody, for the treatment of locally advanced or metastatic HER2-expressing or HER2-amplified cancers: a phase 1, dose-escalation and expansion study. Lancet Oncol. 23, 1558–1570 (2022). [DOI] [PubMed] [Google Scholar]
- 111.Falchi L, Vardhana SA & Salles GA Bispecific antibodies for the treatment of B-cell lymphoma: promises, unknowns, and opportunities. Blood 141, 467–480 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Tsuchikama K, Anami Y, Ha SYY & Yamazaki CM Exploring the next generation of antibody-drug conjugates. Nat. Rev. Clin. Oncol 21, 203–223 (2024). This review focuses on the current state and future directions of ADCs in oncology.
- 113.Fu Z, Li S, Han S, Shi C & Zhang Y Antibody drug conjugate: the “biological missile” for targeted cancer therapy. Signal Transduct. Target. Ther 7, 93 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Stapleton NM et al. Competition for FcRn-mediated transport gives rise to short half-life of human IgG3 and offers therapeutic potential. Nat. Commun 2, 599 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Natsume A, Niwa R & Satoh M Improving effector functions of antibodies for cancer treatment: enhancing ADCC and CDC. Drug Des. Dev. Ther 3, 7–16 (2009). [PMC free article] [PubMed] [Google Scholar]
- 116.Flygare JA, Pillow TH & Aristoff P Antibody-drug conjugates for the treatment of cancer. Chem. Biol. Drug Des 81, 113–121 (2013). [DOI] [PubMed] [Google Scholar]
- 117.Hingorani DV et al. Monomethyl auristatin antibody and peptide drug conjugates for trimodal cancer chemo-radio-immunotherapy. Nat. Commun 13, 3869 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Francisco JA et al. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 102, 1458–1465 (2003). This study describes the generation of a CD30-targeting ADC using a peptide cleavable linker. The ADC was later named brentuximab, and it is considered standard first-line therapy for patients with Hodgkin lymphoma and other CD30+ lymphomas. Multiple approved ADCs targeting other cancer antigens now use this drug–linker combination.
- 119.Ansell SM et al. Overall survival with brentuximab vedotin in stage III or IV Hodgkin’s lymphoma. N. Engl. J. Med 387, 310–320 (2022). [DOI] [PubMed] [Google Scholar]
- 120.Horwitz S. et al. The ECHELON-2 trial: 5-year results of a randomized, phase III study of brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma. Ann. Oncol 33, 288–298 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhang D. et al. Catalytic cleavage of disulfide bonds in small molecules and linkers of antibody-drug conjugates. Drug Metab. Dispos 47, 1156–1163 (2019). [DOI] [PubMed] [Google Scholar]
- 122.Moore KN et al. Mirvetuximab soravtansine in FRα-positive, platinum-resistant ovarian cancer. N. Engl. J. Med 389, 2162–2174 (2023). [DOI] [PubMed] [Google Scholar]
- 123.Kovtun YV et al. Antibody-drug conjugates designed to eradicate tumors with homogeneous and heterogeneous expression of the target antigen. Cancer Res. 66, 3214–3221 (2006). [DOI] [PubMed] [Google Scholar]
- 124.Oflazoglu E. et al. Potent anticarcinoma activity of the humanized anti-CD70 antibody h1F6 conjugated to the tubulin inhibitor auristatin via an uncleavable linker. Clin. Cancer Res 14, 6171–6180 (2008). [DOI] [PubMed] [Google Scholar]
- 125.Conilh L, Sadilkova L, Viricel W & Dumontet C Payload diversification: a key step in the development of antibody-drug conjugates. J. Hematol. Oncol 16, 3 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Ogitani Y. et al. DS-8201a, a novel HER2-targeting ADC with a novel DNA topoisomerase I inhibitor, demonstrates a promising antitumor efficacy with differentiation from T-DM1. Clin. Cancer Res 22, 5097–5108 (2016). This paper describes the generation of tetrapeptide linker that masks the hydrophobicity of the payload and allows attachment of a large number of hydrophobic payloads without affecting antibody pharmacokinetics. This linker–drug combination is now being used in multiple ADCs that are nearing regulatory approval.
- 127.Zammarchi F. et al. ADCT-402, a PBD dimer-containing antibody drug conjugate targeting CD19-expressing malignancies. Blood 131, 1094–1105 (2018). [DOI] [PubMed] [Google Scholar]
- 128.Lewis Phillips GD et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 68, 9280–9290 (2008). [DOI] [PubMed] [Google Scholar]
- 129. Cortes J. et al. Trastuzumab deruxtecan versus trastuzumab emtansine for breast cancer. N. Engl. J. Med 386, 1143–1154 (2022). This phase III clinical trial directly compares two HER2-targeting ADCs, showing the benefit of the ADC trastuzumab deruxtecan, which carries the payload DXd attached by a cleavable linker and possesses a high DAR.
- 130.Modi S. et al. Trastuzumab deruxtecan in previously treated HER2-low advanced breast cancer. N. Engl. J. Med 387, 9–20 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kreitman RJ & Pastan I Immunotoxins: from design to clinical application. Biomolecules 11, 1696 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Li M, Mei S, Yang Y, Shen Y & Chen L Strategies to mitigate the on- and off-target toxicities of recombinant immunotoxins: an antibody engineering perspective. Antib. Ther 5, 164–176 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Kreitman RJ & Pastan I Antibody fusion proteins: anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clin. Cancer Res 17, 6398–6405 (2011). This review of immunotoxins shows the development of the first cancer-targeting immunotoxin moxetumomab that received FDA approval for the treatment of hairy cell leukaemia.
- 134.Lin P, Qi J & Liu W Expert’s views and perspectives: an interview with distinguished investigator Dr. Ira Pastan at the National Cancer Institute at NIH. Antib. Ther 3, 163–166 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kreitman RJ et al. Moxetumomab pasudotox in relapsed/refractory hairy cell leukemia. Leukemia 32, 1768–1777 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kreitman RJ & Pastan I Contextualizing the use of moxetumomab pasudotox in the treatment of relapsed or refractory hairy cell leukemia. Oncologist 25, e170–e177 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kim YS & Brechbiel MW An overview of targeted alpha therapy. Tumour Biol. 33, 573–590 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Parakh S, Lee ST, Gan HK & Scott AM Radiolabeled antibodies for cancer imaging and therapy. Cancers 14, 1454 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Witzig TE et al. Randomized controlled trial of yttrium-90-labeled ibritumomab tiuxetan radioimmunotherapy versus rituximab immunotherapy for patients with relapsed or refractory low-grade, follicular, or transformed B-cell non-Hodgkin’s lymphoma. J. Clin. Oncol 20, 2453–2463 (2002). [DOI] [PubMed] [Google Scholar]
- 140.Kaminski MS et al. Pivotal study of iodine I 131 tositumomab for chemotherapy-refractory low-grade or transformed low-grade B-cell non-Hodgkin’s lymphomas. J. Clin. Oncol 19, 3918–3928 (2001). [DOI] [PubMed] [Google Scholar]
- 141.Prasad V. The withdrawal of drugs for commercial reasons: the incomplete story of tositumomab. JAMA Intern. Med 174, 1887–1888 (2014). [DOI] [PubMed] [Google Scholar]
- 142.Alhaj Moustafa M. et al. Real world long-term follow-up experience with yttrium-90 ibritumomab tiuxetan in previously untreated patients with low-grade follicular lymphoma and marginal zone lymphoma. Clin. Lymphoma Myeloma Leuk 22, 618–625 (2022). [DOI] [PubMed] [Google Scholar]
- 143.Sgouros G, Bodei L, McDevitt MR & Nedrow JR Radiopharmaceutical therapy in cancer: clinical advances and challenges. Nat. Rev. Drug Discov 19, 589–608 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Dowden H & Munro J Trends in clinical success rates and therapeutic focus. Nat. Rev. Drug Discov 18, 495–496 (2019). [DOI] [PubMed] [Google Scholar]
- 145.Wong CH, Siah KW & Lo AW Estimation of clinical trial success rates and related parameters. Biostatistics 20, 273–286 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Smietana K, Siatkowski M & Moller M Trends in clinical success rates. Nat. Rev. Drug Discov 15, 379–380 (2016). [DOI] [PubMed] [Google Scholar]
- 147.Pham E. et al. Preclinical assessment of a MUC12-targeted BiTE (bispecific T-cell engager) molecule. Mol. Cancer Ther 20, 1977–1987 (2021). [DOI] [PubMed] [Google Scholar]
- 148.Kebenko M. et al. A multicenter phase 1 study of solitomab (MT110, AMG 110), a bispecific EpCAM/CD3 T-cell engager (BiTE(R)) antibody construct, in patients with refractory solid tumors. Oncoimmunology 7, e1450710 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.DeFrancesco L. Cartography biosciences and nested therapeutics: diamonds in the rough. Nat. Biotechnol 10.1038/d41587-023-00013-9 (2023). [DOI] [PubMed] [Google Scholar]
- 150.Mullard A. Claudin-18.2 attracts the cancer crowd. Nat. Rev. Drug Discov 22, 683–686 (2023). [DOI] [PubMed] [Google Scholar]
- 151.Shah MA et al. Zolbetuximab plus CAPOX in CLDN18.2-positive gastric or gastroesophageal junction adenocarcinoma: the randomized, phase 3 GLOW trial. Nat. Med 29, 2133–2141 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Janne PA et al. Efficacy and safety of patritumab deruxtecan (HER3-DXd) in EGFR inhibitor-resistant, EGFR-mutated non-small cell lung cancer. Cancer Discov. 12, 74–89 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wainberg ZA et al. Bemarituzumab in patients with FGFR2b-selected gastric or gastro-oesophageal junction adenocarcinoma (FIGHT): a randomised, double-blind, placebo-controlled, phase 2 study. Lancet Oncol. 23, 1430–1440 (2022). [DOI] [PubMed] [Google Scholar]
- 154.Paz-Ares L. et al. Tarlatamab, a first-in-class DLL3-targeted bispecific T-cell engager, in recurrent small-cell lung cancer: an open-label, phase I study. J. Clin. Oncol 41, 2893–2903 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Picozzi V. et al. Gemcitabine/nab-paclitaxel with pamrevlumab: a novel drug combination and trial design for the treatment of locally advanced pancreatic cancer. ESMO Open 5, e000668 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kelly WK et al. Xaluritamig, a STEAP1 x CD3 XmAb 2+1 immune therapy for metastatic castration-resistant prostate cancer: results from dose exploration in a first-in-human study. Cancer Discov. 14, 76–89 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Dannenfelser R. et al. Discriminatory power of combinatorial antigen recognition in cancer T cell therapies. Cell Syst. 11, 215–228.e5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kloss CC, Condomines M, Cartellieri M, Bachmann M & Sadelain M Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat. Biotechnol 31, 71–75 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Banaszek A. et al. On-target restoration of a split T cell-engaging antibody for precision immunotherapy. Nat. Commun 10, 5387 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Oostindie SC et al. Logic-gated antibody pairs that selectively act on cells co-expressing two antigens. Nat. Biotechnol 40, 1509–1519 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Iozzo RV & Schaefer L Proteoglycan form and function: a comprehensive nomenclature of proteoglycans. Matrix Biol. 42, 11–55 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ilieva KM et al. Chondroitin sulfate proteoglycan 4 and its potential as an antibody immunotherapy target across different tumor types. Front Immunol. 8, 1911 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Filmus J, Capurro M & Rast J Glypicans. Genome Biol. 9, 224 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ho M & Kim H Glypican-3: a new target for cancer immunotherapy. Eur. J. Cancer 47, 333–338 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Li N, Gao W, Zhang YF & Ho M Glypicans as cancer therapeutic targets. Trends Cancer 4, 741–754 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Li N. et al. The IgG4 hinge with CD28 transmembrane domain improves V(H)H-based CAR T cells targeting a membrane-distal epitope of GPC1 in pancreatic cancer. Nat. Commun 14, 1986 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kato D. et al. GPC1 specific CAR-T cells eradicate established solid tumor without adverse effects and synergize with anti-PD-1 Ab. eLife 9, e49392 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Li N, Fu H, Hewitt SM, Dimitrov DS & Ho M Therapeutically targeting glypican-2 via single-domain antibody-based chimeric antigen receptors and immunotoxins in neuroblastoma. Proc. Natl Acad. Sci. USA 114, E6623–E6631 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Bosse KR et al. Identification of GPC2 as an oncoprotein and candidate immunotherapeutic target in high-risk neuroblastoma. Cancer Cell 32, 295–309.e12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Li N. et al. CAR T cells targeting tumor-associated exons of glypican 2 regress neuroblastoma in mice. Cell Rep. Med 2, 100297 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Phung Y, Gao W, Man Y-G, Nagata S & Ho M High-affinity monoclonal antibodies to cell surface tumor antigen glypican-3 generated through a combination of peptide immunization and flow cytometry screening. mAbs 4, 592–599 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Li D. et al. Persistent polyfunctional chimeric antigen receptor T cells that target glypican 3 eliminate orthotopic hepatocellular carcinomas in mice. Gastroenterology 158, 2250–2265.e20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Nakano K. et al. Anti-glypican 3 antibodies cause ADCC against human hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun 378, 279–284 (2009). [DOI] [PubMed] [Google Scholar]
- 174.Shi D. et al. Chimeric antigen receptor-glypican-3 T-cell therapy for advanced hepatocellular carcinoma: results of phase I trials. Clin. Cancer Res 26, 3979–3989 (2020). [DOI] [PubMed] [Google Scholar]
- 175. Loffler A. et al. A recombinant bispecific single-chain antibody, CD19 x CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood 95, 2098–2103 (2000). This paper describes the production of large quantities of a stable bispecific antibody targeting CD19 on B cell lymphomas. The CD19-targeting bispecific antibody was later named blinatumomab and was the first successful bispecific antibody that gained widespread adoption for cancer treatment.
- 176. Kantarjian H. et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N. Engl. J. Med 376, 836–847 (2017). This phase III clinical trial establishes the survival benefit of blinatumomab therapy over chemotherapy in patients with ALL.
- 177.Maloney DG et al. Phase I clinical trial using escalating single-dose infusion of chimeric anti-CD20 monoclonal antibody (IDEC-C2B8) in patients with recurrent B-cell lymphoma. Blood 84, 2457–2466 (1994). [PubMed] [Google Scholar]
- 178.Coiffier B. et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N. Engl. J. Med 346, 235–242 (2002). [DOI] [PubMed] [Google Scholar]
- 179.de Weers M. et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J. Immunol 186, 1840–1848 (2011). [DOI] [PubMed] [Google Scholar]
- 180.Carpenter RO et al. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin. Cancer Res 19, 2048–2060 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Frerichs KA et al. Preclinical activity of JNJ-7957, a novel BCMAxCD3 bispecific antibody for the treatment of multiple myeloma, is potentiated by daratumumab. Clin. Cancer Res 26, 2203–2215 (2020). [DOI] [PubMed] [Google Scholar]
- 182.Nichakawade TD et al. TRBC1-targeting antibody–drug conjugates for the treatment of T cell cancers. Nature 10.1038/s41586-024-07233-2 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Paul S. et al. TCR β chain-directed bispecific antibodies for the treatment of T cell cancers. Sci. Transl. Med 10.1126/scitranslmed.abd3595 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Maciocia PM et al. Targeting the T cell receptor β-chain constant region for immunotherapy of T cell malignancies. Nat. Med 23, 1416–1423 (2017). [DOI] [PubMed] [Google Scholar]
- 185. Pearlman AH et al. Targeting public neoantigens for cancer immunotherapy. Nat. Cancer 2, 487–497 (2021). This review describes the prevalence of targetable public neoantigens in cancer.
- 186.Hwang MS et al. Structural engineering of chimeric antigen receptors targeting HLA-restricted neoantigens. Nat. Commun 12, 5271 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Hattori T. et al. Creating MHC-restricted neoantigens with covalent inhibitors that can be targeted by immune therapy. Cancer Discov. 13, 132–145 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Wright KM et al. Hydrophobic interactions dominate the recognition of a KRAS G12V neoantigen. Nat. Commun 14, 5063 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Wu D, Gallagher DT, Gowthaman R, Pierce BG & Mariuzza RA Structural basis for oligoclonal T cell recognition of a shared p53 cancer neoantigen. Nat. Commun 11, 2908 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Trenevska I, Li D & Banham AH Therapeutic antibodies against intracellular tumor antigens. Front. Immunol 8, 1001 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Hamid O. et al. 728O results from phase I dose escalation of IMC-F106C, the first PRAME × CD3 ImmTAC bispecific protein in solid tumors. Ann. Oncol 33, S875 (2022). [Google Scholar]
- 192.Hofheinz RD et al. Stromal antigen targeting by a humanised monoclonal antibody: an early phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie 26, 44–48 (2003). [DOI] [PubMed] [Google Scholar]
- 193.Scott AM et al. A phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin. Cancer Res 9, 1639–1647 (2003). [PubMed] [Google Scholar]
- 194.Benson AB III et al. A phase II randomized, double-blind, placebo-controlled study of simtuzumab or placebo in combination with gemcitabine for the first-line treatment of pancreatic adenocarcinoma. Oncologist 22, 241–e15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Hecht JR et al. A phase II, randomized, double-blind, placebo-controlled study of simtuzumab in combination with FOLFIRI for the second-line treatment of metastatic KRAS mutant colorectal adenocarcinoma. Oncologist 22, 243–e23 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Bejarano L, Jordao MJC & Joyce JA Therapeutic targeting of the tumor microenvironment. Cancer Discov. 11, 933–959 (2021). [DOI] [PubMed] [Google Scholar]
- 197.Gomez-Roca C. et al. Anti-CSF-1R emactuzumab in combination with anti-PD-L1 atezolizumab in advanced solid tumor patients naive or experienced for immune checkpoint blockade. J. Immunother. Cancer 10, e004076 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Ira Seth W. et al. A phase 1a dose-escalation study of PY159, a monoclonal antibody targeting TREM1 (triggering receptor expressed on myeloid cells 1). J. Clin. Oncol 41, 2523 (2023).36809028 [Google Scholar]
- 199.Amita P. et al. A phase 1a dose-escalation study of PY314, a TREM2 (triggering receptor expressed on macrophages 2) targeting monoclonal antibody. J. Clin. Oncol 10.1200/JCO.2022.40.16_suppl.2648 (2022). [DOI] [Google Scholar]
- 200.Byrne KT & Vonderheide RH CD40 stimulation obviates innate sensors and drives T cell immunity in cancer. Cell Rep. 15, 2719–2732 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Byrne KT et al. Neoadjuvant selicrelumab, an agonist CD40 antibody, induces changes in the tumor microenvironment in patients with resectable pancreatic cancer. Clin. Cancer Res 27, 4574–4586 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.O’Hara MH et al. CD40 agonistic monoclonal antibody APX005M (sotigalimab) and chemotherapy, with or without nivolumab, for the treatment of metastatic pancreatic adenocarcinoma: an open-label, multicentre, phase 1b study. Lancet Oncol. 22, 118–131 (2021). [DOI] [PubMed] [Google Scholar]
- 203.Curigliano G. et al. Phase I/Ib clinical trial of sabatolimab, an anti-TIM-3 antibody, alone and in combination with spartalizumab, an anti-PD-1 antibody, in advanced solid tumors. Clin. Cancer Res 27, 3620–3629 (2021). [DOI] [PubMed] [Google Scholar]
- 204.Kim HR et al. Cobolimab with dostarlimab and docetaxel in patients with advanced non-small cell lung cancer (NSCLC): COSTAR lung. J. Thorac. Oncol 10.1016/j.jtho.2022.07.183 (2022). [DOI] [Google Scholar]
- 205.Spiegel JY et al. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: a phase 1 trial. Nat. Med 27, 1419–1431 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Bluhm J. et al. CAR T cells with enhanced sensitivity to B cell maturation antigen for the targeting of B cell non-Hodgkin’s lymphoma and multiple myeloma. Mol. Ther 26, 1906–1920 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Friedman KM et al. Effective targeting of multiple B-cell maturation antigen-expressing hematological malignances by anti-B-cell maturation antigen chimeric antigen receptor T cells. Hum. Gene Ther 29, 585–601 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Golay J. et al. CD20 levels determine the in vitro susceptibility to rituximab and complement of B-cell chronic lymphocytic leukemia: further regulation by CD55 and CD59. Blood 98, 3383–3389 (2001). [DOI] [PubMed] [Google Scholar]
- 209.Teeling JL et al. The biological activity of human CD20 monoclonal antibodies is linked to unique epitopes on CD20. J. Immunol 177, 362–371 (2006). [DOI] [PubMed] [Google Scholar]
- 210.Perez HL et al. Antibody-drug conjugates: current status and future directions. Drug Discov. Today 19, 869–881 (2014). [DOI] [PubMed] [Google Scholar]
- 211.Sharma P & Kranz DM Recent advances in T-cell engineering for use in immunotherapy. F1000Res 10.12688/f1000research.9073.1 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Cancer Research Institute. Approval timeline of active immunotherapies. Cancer Research Institute; https://www.cancerresearch.org/regulatory-approval-timeline-of-active-immunotherapies (2024). [Google Scholar]
- 213.Johnson PC, Gainor JF, Sullivan RJ, Longo DL & Chabner B Immune checkpoint inhibitors — the need for innovation. N. Engl. J. Med 388, 1529–1532 (2023). [DOI] [PubMed] [Google Scholar]
- 214.Revisiting checkpoint blockade. Nat. Biotechnol 40, 981 (2022). [DOI] [PubMed] [Google Scholar]
- 215.Postel-Vinay S. et al. First-in-human phase I study of the OX40 agonist GSK3174998 with or without pembrolizumab in patients with selected advanced solid tumors (ENGAGE-1). J. Immunother. Cancer 11, e005301 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Davis EJ et al. First-in-human phase I/II, open-label study of the anti-OX40 agonist INCAGN01949 in patients with advanced solid tumors. J. Immunother. Cancer 10, e004235 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Andre P. et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell 175, 1731–1743.e13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Qin S. et al. Novel immune checkpoint targets: moving beyond PD-1 and CTLA-4. Mol. Cancer 18, 155 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Tawbi HA et al. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N. Engl. J. Med 386, 24–34 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Gilead. Gilead statement on the discontinuation of magrolimab study in AML with TP53 mutations. Gilead; https://www.gilead.com/news-and-press/company-statements/gilead-statement-on-the-discontinuation-of-magrolimab-study-in-aml-with-tp53-mutations (2023). [Google Scholar]
- 221.Gilead. Gilead to discontinue phase 3 ENHANCE study of magrolimab plus azacitidine in higher-risk MDS. Gilead; https://www.gilead.com/news-and-press/press-room/press-releases/2023/7/gilead-to-discontinue-phase-3-enhance-study-of-magrolimab-plus-azacitidine-in-higher-risk-mds (2023). [Google Scholar]
- 222.Zhao B, Zhao H & Zhao J Efficacy of PD-1/PD-L1 blockade monotherapy in clinical trials. Ther. Adv. Med. Oncol 12, 1758835920937612 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Johnson DB, Nebhan CA, Moslehi JJ & Balko JM Immune-checkpoint inhibitors: long-term implications of toxicity. Nat. Rev. Clin. Oncol 19, 254–267 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Cheng Ying et al. Phase I/II combination study of tifcemalimab with toripalimab in patients with refractory extensive stage small cell lung cancer (ES-SCLC). J. Clin. Oncol 41, 8579 (2023). [Google Scholar]
- 225.Shenderov E. et al. Neoadjuvant enoblituzumab in localized prostate cancer: a single-arm, phase 2 trial. Nat. Med 29, 888–897 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Ansell SM et al. Safety and activity of varlilumab, a novel and first-in-class agonist anti-CD27 antibody, for hematologic malignancies. Blood Adv. 4, 1917–1926 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Claus C, Ferrara-Koller C & Klein C The emerging landscape of novel 4–1BB (CD137) agonistic drugs for cancer immunotherapy. MAbs 15, 2167189 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Elizabeth Budde M. et al. Preliminary results of a phase 1 dose escalation study of the first-in-class IgM based bispecific antibody Igm-2323 (anti-CD20 x anti-CD3) in patients with advanced B-cell malignancies. Blood 136, 45–46 (2020). [Google Scholar]
- 229.Wang BT et al. Multimeric anti-DR5 IgM agonist antibody IGM-8444 is a potent inducer of cancer cell apoptosis and synergizes with chemotherapy and BCL-2 inhibitor ABT-199. Mol. Cancer Ther 20, 2483–2494 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Li K, Yun R, Chai M, Yakkundi P & Rosete R Novel CD38xCD3 bispecific IgM T cell engager, IGM-2644, potently kills multiple myeloma cells though complement and T cell dependent mechanisms. Cancer Res. 83, 2959 (2023). [Google Scholar]
- 231.Wang B. et al. Anti-DR5 agonist IgM antibody IGM-8444 combined with SMAC mimetic birinapant induces strong synergistic tumor cytotoxicity. Cancer Res. 82, 1068 (2022). [Google Scholar]
- 232. Klein C, Brinkmann U, Reichert JM & Kontermann RE The present and future of bispecific antibodies for cancer therapy. Nat. Rev. Drug Discov 10.1038/s41573-024-00896-6 (2024). This review focuses on the current state and future directions of bispecific antibodies in oncology.
- 233.Dickinson MJ et al. Glofitamab for relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med 387, 2220–2231 (2022). [DOI] [PubMed] [Google Scholar]
- 234.Bacac M. et al. CD20-TCB with obinutuzumab pretreatment as next-generation treatment of hematologic malignancies. Clin. Cancer Res 24, 4785–4797 (2018). [DOI] [PubMed] [Google Scholar]
- 235.Nolan-Stevaux O. et al. AMG 509 (xaluritamig), an anti-STEAP1 XmAb 2+1 T-cell redirecting immune therapy with avidity-dependent activity against prostate cancer. Cancer Discov. 14, 90–103 (2024). [DOI] [PubMed] [Google Scholar]
- 236.Li G. et al. Novel CD123xCD3 bispecific IgM antibody, IGM-2537, potently induces T-cell mediated cytotoxicity of acute myeloid leukemia cells with minimal cytokine release. Cancer Res. 83, 2933 (2023). [Google Scholar]
- 237.Genevive HH et al. Pharmacodynamics and biomarker correlates of imvotamab (IGM-2323), the first-in-class CD20xCD3 bispecific IgM antibody with dual mechanisms of action, in patients with advanced B cell malignancies. Blood 140, 6436–6438 (2022). [Google Scholar]
- 238.Luke JJ et al. The PD-1- and LAG-3-targeting bispecific molecule tebotelimab in solid tumors and hematologic cancers: a phase 1 trial. Nat. Med 29, 2814–2824 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Dovedi SJ et al. Design and efficacy of a monovalent bispecific PD-1/CTLA4 antibody that enhances CTLA4 blockade on PD-1+ activated T cells. Cancer Discov. 11, 1100–1117 (2021). [DOI] [PubMed] [Google Scholar]
- 240.Gao X. et al. Safety and antitumour activity of cadonilimab, an anti-PD-1/CTLA-4 bispecific antibody, for patients with advanced solid tumours (COMPASSION-03): a multicentre, open-label, phase 1b/2 trial. Lancet Oncol. 24, 1134–1146 (2023). [DOI] [PubMed] [Google Scholar]
- 241.Chen B. et al. A phase Ib/II study of cadonilimab (PD-1/CTLA-4 bispecific antibody) plus anlotinib as first-line treatment in patients with advanced non-small cell lung cancer. Br. J. Cancer 130, 450–456 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Li Q. et al. The anti-PD-L1/CTLA-4 bispecific antibody KN046 in combination with nab-paclitaxel in first-line treatment of metastatic triple-negative breast cancer: a multicenter phase II trial. Nat. Commun 15, 1015 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Kvarnhammar AM et al. The CTLA-4 x OX40 bispecific antibody ATOR-1015 induces anti-tumor effects through tumor-directed immune activation. J. Immunother. Cancer 7, 103 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Lee Karin et al. Preclinical studies support clinical development of AZD2936, a monovalent bispecific humanized antibody targeting PD-1 and TIGIT. J. Immunother. Cancer 10, A489 (2023). [Google Scholar]
- 245.Shapir Itai Y. et al. Bispecific dendritic-T cell engager potentiates anti-tumor immunity. Cell 187, 375–389.e18 (2024). [DOI] [PubMed] [Google Scholar]
- 246.Zhao L. et al. A novel CD19/CD22/CD3 trispecific antibody enhances therapeutic efficacy and overcomes immune escape against B-ALL. Blood 140, 1790–1802 (2022). [DOI] [PubMed] [Google Scholar]
- 247.Wu L. et al. Trispecific antibodies enhance the therapeutic efficacy of tumor-directed T cells through T cell receptor co-stimulation. Nat. Cancer 1, 86–98 (2020). [DOI] [PubMed] [Google Scholar]
- 248.Pardon E. et al. A general protocol for the generation of nanobodies for structural biology. Nat. Protoc 9, 674–693 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Li D. et al. Camel nanobody-based B7-H3 CAR-T cells show high efficacy against large solid tumours. Nat. Commun 14, 5920 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Xu J. et al. Nanobodies from camelid mice and llamas neutralize SARS-CoV-2 variants. Nature 595, 278–282 (2021). This paper describes the generation of a transgenic mouse (nanomouse), which expresses camelid variable heavy domain of heavy chain (VHH) genes. Such transgenic mouse platforms enable the production of nanobodies from mice.
- 251.Hu Y. et al. RenNano® mice: a heavy-chain-only antibody platform for the generation of nanobody therapeutics. Cancer Res. 83, LB210 (2023). [Google Scholar]
- 252.De Genst E. et al. Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc. Natl Acad. Sci. USA 103, 4586–4591 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Berdeja JG et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet 398, 314–324 (2021). [DOI] [PubMed] [Google Scholar]
- 254.Munshi NC et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N. Engl. J. Med 384, 705–716 (2021). [DOI] [PubMed] [Google Scholar]
- 255.Jovcevska I & Muyldermans S The therapeutic potential of nanobodies. BioDrugs 34, 11–26 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Lucchi R, Bentanachs J & Oller-Salvia B The masking game: design of activatable antibodies and mimetics for selective therapeutics and cell control. ACS Cent. Sci 7, 724–738 (2021). This review describes the recent advancements in activatable antibodies in cancer and other diseases.
- 257.Sulea T. et al. Structure-based engineering of pH-dependent antibody binding for selective targeting of solid-tumor microenvironment. MAbs 12, 1682866 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Kang JC et al. Engineering a HER2-specific antibody-drug conjugate to increase lysosomal delivery and therapeutic efficacy. Nat. Biotechnol 37, 523–526 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Zhang Y. et al. Hijacking antibody-induced CTLA-4 lysosomal degradation for safer and more effective cancer immunotherapy. Cell Res. 29, 609–627 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Desnoyers LR et al. Tumor-specific activation of an EGFR-targeting probody enhances therapeutic index. Sci. Transl. Med 5, 207ra144 (2013). [DOI] [PubMed] [Google Scholar]
- 261.Autio KA, Boni V, Humphrey RW & Naing A Probody therapeutics: an emerging class of therapies designed to enhance on-target effects with reduced off-tumor toxicity for use in immuno-oncology. Clin. Cancer Res 26, 984–989 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Boni V. et al. Praluzatamab ravtansine, a CD166-targeting antibody-drug conjugate, in patients with advanced solid tumors: an open-label phase I/II Trial. Clin. Cancer Res 28, 2020–2029 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Boustany LM et al. A probody T cell-engaging bispecific antibody targeting EGFR and CD3 inhibits colon cancer growth with limited toxicity. Cancer Res. 82, 4288–4298 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Lajoie MJ et al. Designed protein logic to target cells with precise combinations of surface antigens. Science 369, 1637–1643 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Xu S. Internalization, trafficking, intracellular processing and actions of antibody-drug conjugates. Pharm. Res 32, 3577–3583 (2015). [DOI] [PubMed] [Google Scholar]
- 266.Saunders KO Conceptual approaches to modulating antibody effector functions and circulation half-life. Front. Immunol 10, 1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Brandl F, Busslinger S, Zangemeister-Wittke U & Pluckthun A Optimizing the anti-tumor efficacy of protein-drug conjugates by engineering the molecular size and half-life. J. Control. Release 327, 186–197 (2020). [DOI] [PubMed] [Google Scholar]
- 268.Bardia A. et al. Sacituzumab govitecan in metastatic triple-negative breast cancer. N. Engl. J. Med 384, 1529–1541 (2021). [DOI] [PubMed] [Google Scholar]
- 269.Senior M. Cancer-targeting antibody-drug conjugates drive dealmaking frenzy. Nat. Biotechnol 42, 362–366 (2024). [DOI] [PubMed] [Google Scholar]
- 270.Weng W. et al. Antibody-exatecan conjugates with a novel self-immolative moiety overcome resistance in colon and lung cancer. Cancer Discov. 13, 950–973 (2023). [DOI] [PubMed] [Google Scholar]
- 271.Li BT et al. Trastuzumab deruxtecan in HER2-mutant non-small-cell lung cancer. N. Engl. J. Med 386, 241–251 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Siena S. et al. Trastuzumab deruxtecan (DS-8201) in patients with HER2-expressing metastatic colorectal cancer (DESTINY-CRC01): a multicentre, open-label, phase 2 trial. Lancet Oncol. 22, 779–789 (2021). [DOI] [PubMed] [Google Scholar]
- 273.Esfandiari A, Cassidy S & Webster RM Bispecific antibodies in oncology. Nat. Rev. Drug Discov 21, 411–412 (2022). [DOI] [PubMed] [Google Scholar]
- 274.do Pazo C, Nawaz K & Webster RM The oncology market for antibody-drug conjugates. Nat. Rev. Drug Discov 20, 583–584 (2021). [DOI] [PubMed] [Google Scholar]
- 275.Criscitiello C, Morganti S & Curigliano G Antibody-drug conjugates in solid tumors: a look into novel targets. J. Hematol. Oncol 14, 20 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Michael LW et al. Zilovertamab vedotin targeting of ROR1 as therapy for lymphoid cancers. NEJM Evid. 10.1056/EVIDoa2100001 (2021). [DOI] [PubMed] [Google Scholar]
- 277.Saura Manich C. et al. LBA15 — primary outcome of the phase III SYD985.002/TULIP trial comparing [vic-]trastuzumab duocarmazine to physician’s choice treatment in patients with pre-treated HER2-positive locally advanced or metastatic breast cancer. Ann. Oncol 32, S1283–S1346 (2021). [Google Scholar]
- 278.Powles TB et al. LBA6 EV-302/KEYNOTE-A39: Open-label, randomized phase III study of enfortumab vedotin in combination with pembrolizumab (EV+P) vs chemotherapy (Chemo) in previously untreated locally advanced metastatic urothelial carcinoma (la/mUC). Ann. Oncol 34, S1340 (2023). [Google Scholar]
- 279.Antignani A. et al. Targeting receptors on cancer cells with protein toxins. Biomolecules 10.3390/biom10091331 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Pemmaraju N. et al. Tagraxofusp in blastic plasmacytoid dendritic-cell neoplasm. N. Engl. J. Med 380, 1628–1637 (2019). [DOI] [PubMed] [Google Scholar]
- 281.Saleh MN et al. Antitumor activity of DAB389IL-2 fusion toxin in mycosis fungoides. J. Am. Acad. Dermatol 39, 63–73 (1998). [DOI] [PubMed] [Google Scholar]
- 282.Sartor O. et al. Lutetium-177-PSMA-617 for metastatic castration-resistant prostate cancer. N. Engl. J. Med 385, 1091–1103 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Strosberg J. et al. Phase 3 trial of (177)Lu-dotatate for midgut neuroendocrine tumors. N. Engl. J. Med 376, 125–135 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Michael SH et al. First-in-human safety, imaging and dosimetry of [68Ga]Ga-DPI-4452, a novel CA IX-targeting peptide, in patients with clear cell renal cell carcinoma. J. Clin. Oncol 10.1200/JCO.2024.42.4_suppl.37 (2024). [DOI] [Google Scholar]
- 285.Dolgin E. Radioactive drugs emerge from the shadows to storm the market. Nat. Biotechnol 36, 1125–1127 (2018). [DOI] [PubMed] [Google Scholar]
- 286.Sathekge MM et al. Actinium-225-PSMA radioligand therapy of metastatic castration-resistant prostate cancer (WARMTH Act): a multicentre, retrospective study. Lancet Oncol. 25, 175–183 (2024). [DOI] [PubMed] [Google Scholar]
- 287.Slamon DJ et al. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235, 177–182 (1987). [DOI] [PubMed] [Google Scholar]
- 288.Rubin I & Yarden Y The basic biology of HER2. Ann. Oncol 12, S3–8 (2001). [DOI] [PubMed] [Google Scholar]
- 289.Verma S. et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med 367, 1783–1791 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Morgan RA et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther 18, 843–851 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Saunders LR et al. A DLL3-targeted antibody-drug conjugate eradicates high-grade pulmonary neuroendocrine tumor-initiating cells in vivo. Sci. Transl. Med 7, 302ra136 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Vos JL et al. Neoadjuvant immunotherapy with nivolumab and ipilimumab induces major pathological responses in patients with head and neck squamous cell carcinoma. Nat. Commun 12, 7348 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data used to make Figs. 1 and 6 are available from the American Cancer Society and the Antibody Society.