

Abstract
Gain-of-function (GoF) variants in KCNT1 channels cause severe, drug-resistant forms of epilepsy. Quinidine is a known KCNT1 blocker, but its clinical use is limited due to severe drawbacks. To identify novel KCNT1 blockers, a homology model of human KCNT1 was built and used to screen an in-house library of compounds. Among the 20 molecules selected, five (CPK4, 13, 16, 18, and 20) showed strong KCNT1-blocking ability in an in vitro fluorescence-based assay. Patch-clamp experiments confirmed a higher KCNT1-blocking potency of these compounds when compared to quinidine, and their selectivity for KCNT1 over hERG and Kv7.2 channels. Among identified molecules, CPK20 displayed the highest metabolic stability; this compound also blocked KCNT2 currents, although with a lower potency, and counteracted GoF effects prompted by 2 recurrent epilepsy-causing KCNT1 variants (G288S and A934T). The present results provide solid rational basis for future design of novel compounds to counteract KCNT1-related neurological disorders.
Introduction
Potassium (K+) channels represent a broad and structurally heterogeneous class of voltage- and/or ligand-gated ion channels contributing to complex cellular functions ranging from neuronal excitability control to cell proliferation and survival. Among K+ channel genes, KCNT1 belongs to the Slo family1 and encodes for sodium (Na+)- and chloride (Cl–)-gated K+ channel KCNT1 subunits (also known as KNa1.1, Slo2.2), or sequence like a calcium-activated K+ channel (SLACK).2,3 KCNT1 subunits may form homo- or heteromeric channels with highly homologous KCNT2 subunits (encoded by the KCNT2 gene; also known as KNa1.2, Slick, or Slo2.1) contributing to the Na+-dependent K+ currents (IKNa). In central and peripheral neurons, IKNa mediates a large part of the slow hyperpolarisation (sAHP) which regulates action potentials (APs) firing;4,5 additional roles for IKNa are the regulation of cardiac electrical activity and arrhythmogenesis,6 auditory signal transduction,7 and pain sensing.8,9IKNa has been also proposed to protect against hypoxic damage in cardiomyocytes and neurons.10,11
Functional KCNT1 subunits display six transmembrane α-helices, with the S5–S6 loop forming the pore and selectivity filter, and two regulation of conductance of potassium (RCK) domains located intracellularly which critically control ion-dependent gating.12
Mutations in KCNT1 cause severe, drug-resistant rare forms of childhood epilepsy13 such as Epilepsy of Infancy with Migrating Focal Seizures (EIMFS) and Autosomal Dominant Sleep-Related Hypermotor (Hyperkinetic) Epilepsy (ADSHE).14−16 Additional phenotypes associated with KCNT1 variants include Ohtahara syndrome,17 West syndrome,18 and Lennox-Gastaut syndrome.18 Notably, patients affected by KCNT1-related encephalopathies also display developmental plateauing or regression and psychiatric and intellectual disabilities.19 Collectively, these developmental epileptic encephalopathies (DEE) are classified among the rare or orphan diseases.20 Intriguingly, mutations in KCNT2 have also been recently demonstrated to cause developmental and/or epileptic encephalopathy.21−23
More than 60 distinct pathogenic variants have been reported in KCNT1, the vast majority of which prompt gain-of-function (GoF) effects on KCNT1 channels, resulting in enhanced potassium currents when studied in heterologous expression systems in vitro.24,25 Different molecular mechanisms leading to enhanced channel function have been identified, including: (1) changes in Na+ sensitivity;25 (2) increased channel cooperative gating;26 (3) suppression of subconductance states;14 (4) changes in protein kinase C regulation;14 and (5) altered interactions with binding partners.27,28
Seizures in patients with KCNT1-related epilepsy, particularly those with EIMFS or DEE phenotypes, are often highly refractory to pharmacological therapy.29 Given the GoF effects prompted by most pathogenic KCNT1 variants, KCNT1 blockers have been proposed as possible precision therapy in patients affected by KCNT1-related diseases.30 In particular, the antiarrhythmic quinidine (QND), a low-potency KCNT1 blocker (ca. 80–100 μM IC50),31,32 has been used, resulting in heterogeneous anticonvulsant efficacy, ranging from positive responses to a lack of efficacy or excessive toxicity.33 Several factors, including the natural history and severity of the underlying disease, the specific molecular defect, the age of symptom onset, and quinidine therapy initiation, in one with drug-specific pharmacokinetic and pharmacodynamic factors, might provide plausible explanations for such heterogeneity.34 In addition, the drug’s well-known ability to prolong the QT interval on the ECG35 severely limits the maximum administrable dose of QND.33,36,37
QND arrhythmogenic potential is likely due to its nonspecific block of several ion channels in the heart, with Kv11.1 (hERG) channel playing a dominant role;38 notably, when tested in vitro, QND showed a 100-fold greater potency in blocking hERG channels39,40 when compared to KCNT1 channels. In addition to QND, other cardiac cation channel modulators such as the antianginal bepridil32 and the antiarrhythmic clofilium41 have been proposed as KCNT1 inhibitors, but none of these drugs significantly overcomes QND pitfalls and limitations.
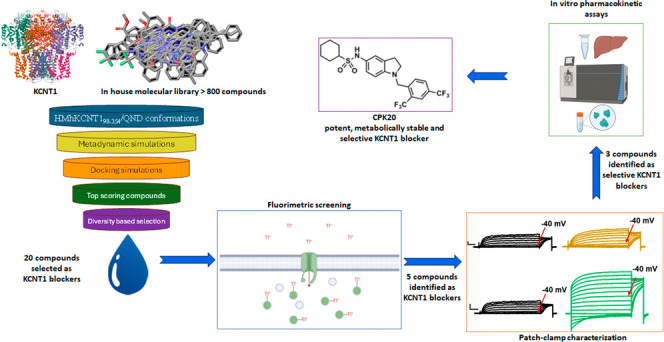
The unsatisfactory responses to QND, as well as the severe refractoriness to conventional antiseizure medications of patients with KCNT1-related epilepsies, highlight the urgent need for novel drugs targeting KCNT1 channels. As a consequence, the results from several drug discovery campaigns for KCNT1 blockers have been recently described, together with few available chemotypes (Figure 1A).42−46 However, limited support has been provided for a structure-based design of KCNT1 blockers, mainly due to the paucity of information about the structural determinants driving the binding of these molecules to the target protein, especially in the context of human channels.
Figure 1.

(A) Molecular structures of some previously identified KCNT1 blockers: VU0606170,42 VU0935685,46 Compound 31,43 BC12,44 and BC13.44 (B) The in silico workflow that led to the selection of the 20 compounds to be experimentally screened.
Building on these premises, in the present work, we describe our strategy to develop novel KCNT1 blockers. An in-house molecular library of >800 compounds was virtually screened for binding at the QND binding site in a homology model of human KCNT1; 20 compounds were selected based on their chemical diversity among the ones predicted as the best binders (Figure 1B). These compounds were functionally investigated for their KCNT1-blocking ability by a thallium (Tl+)-based fluorimetric assay. In this assay, 5 compounds showed a KCNT1-blocking potency up to 35 times higher than QND; the ability of these 5 compounds to potently block KCNT1-mediated currents was confirmed by manual patch-clamp. The same technique was also used to validate the binding site, and to investigate the selectivity profile of the 5 molecules over hERG and Kv7.2 potassium channels targets, leading to the identification of 3 compounds (CPK16, 18, and 20) showing high KCNT1 selectivity with no activity on hERG channels. Studies assessing the metabolic stability of each of those three compounds highlighted that CPK20 displayed the best pharmacokinetic profile. Finally, CPK20 was found to block preferentially KCNT1 vs KCNT2 channels, and to exert a potent blocking effect over clinically-relevant pathogenic KCNT1 variants, thus revealing its potential as target therapy for patients affected by KCNT1-related epilepsies.
Collectively, the present results, pursued by complementary in silico, synthetic and in vitro pharmacological approaches expand the current knowledge concerning structure–activity relationship of KCNT1 blockers, and reveal new chemotypes worthy of further investigation for their optimal characteristics in terms of potency, selectivity, and in vitro pharmacokinetics.
Results and Discussion
In-House Library Virtual Screening
No experimental structure of human KCNT1 was available when we started our KCNT1 inhibitors discovery program, but mutagenesis experiments located the QND binding site in the intracellular pore vestibule and suggested F346 as a critical residue for QND binding.44,47 We therefore built a homology model of human KCNT198–354 (HMhKCNT198–354) by mean of Prime48 using the Cryo-EM structure of chicken Slo2.2 in an open conformation.12 Thus, we performed a dynamic docking49 of QND into the intracellular pore vestibule of HMhKCNT198–354 to search for the most favorable HMhKCNT198–354/QND complex conformations. Briefly, QND was randomly placed into the intracellular pore vestibule and the system was submitted to a metadynamics simulation in which two collective variables, i.e., ligand RMSD and distance between the centers of mass of the ligand and of the four F346 residues, were defined and biased to enhance the conformational sampling. From the simulation, the 50 lowest-energy HMhKCNT198–354/QND conformations were retrieved and used to compose a docking target ensemble that was used to screen in silico our in-house compounds database by mean of a stepwise docking protocol (see the Experimental Section, Figure 1B).
The docking-predicted bound conformations were ranked by XP GScore, and all docking poses with a score > −10 were discarded, leading to a final hits list of 241 docked compounds with scores ranging from −13.432 to −10.536. Finally, a diversity-based selection was carried out on the top scoring 241 compounds using MOLPRINT2D fingerprints and Soergel distances (see the Experimental Section), and 20 compounds (Table S1) were finally selected for the biological tests (Figure 1B).
Chemistry
The chemical synthesis of compounds CPK1, 4, 6, 9, 11–17, 19, and 20 has been previously described,50−57 as also reported in Table S1. The remaining compounds have been synthesized as reported below.
The synthesis of compound CPK2 was performed as described in Scheme 1. l-Tryptophan methyl ester was solubilized in methanol and added with 4-(2-cyclohexylethoxy)benzaldehyde (2a), which has been previously synthesized by reaction of 4-hydroxybenzaldehyde with (2-bromoethyl)cyclohexane under Williamson ether synthesis conditions. The mixture was stirred at room temperature for 3 h, then, reductive amination was accomplished by adding NaBH4. In this way, intermediate 2b was obtained in 76% yield. Reaction of 2b with benzyl bromide, using triethylamine as catalyst under microwave irradiation gave final compound CPK2 in 83% yield.
Scheme 1. Synthesis of Final Compound CPK2.
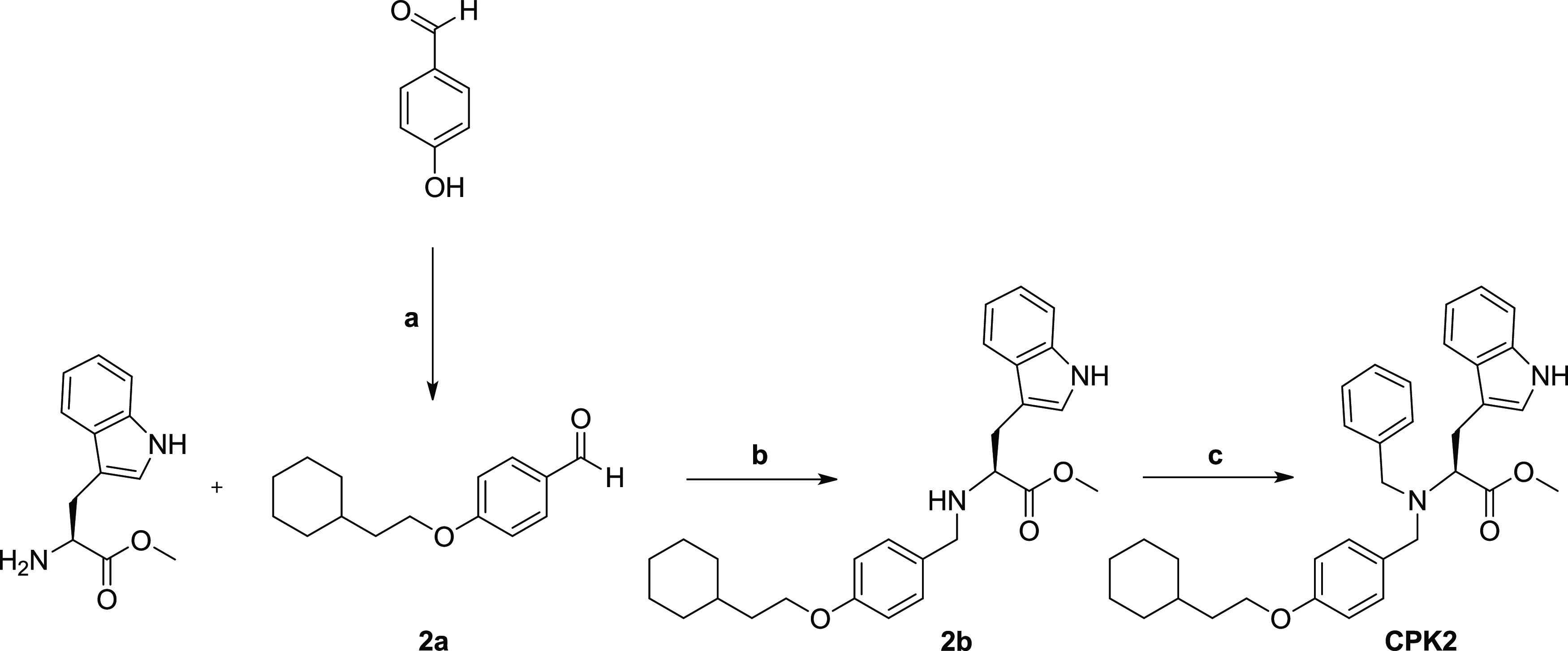
Reagents and conditions: (a) (2-Bromoethyl)cyclohexane, potassium tert-butoxide, DMF, 130 °C, overnight; (b) MeOH dry, 3 h, RT then NaBH4, 1 h, RT; (c) benzyl bromide, DIPEA, 100 °C, μW, 20 min.
The synthesis of compound CPK3 was performed as described in Scheme 2. Boc-l-tyrosine methyl ester was reacted with benzyl bromide in the presence of potassium tert-butoxide to give the corresponding benzyl ether 3a. Deprotection of the tert-butoxycarbonyl group of 3a using trifluoroacetic acid (TFA) in dichloromethane (DCM) and triisopropylsilane (TIS) as radical scavenger, led to the amine 3b. Intermediate 3b was reacted with 4-amino-1-Boc-piperidine and triphosgene to provide urea 3c in 76% yield. Refluxing 3c in methanol under acidic conditions led to the final hydantoin compound CPK3 in 67% yield
Scheme 2. Synthesis of Final Compound CPK3.
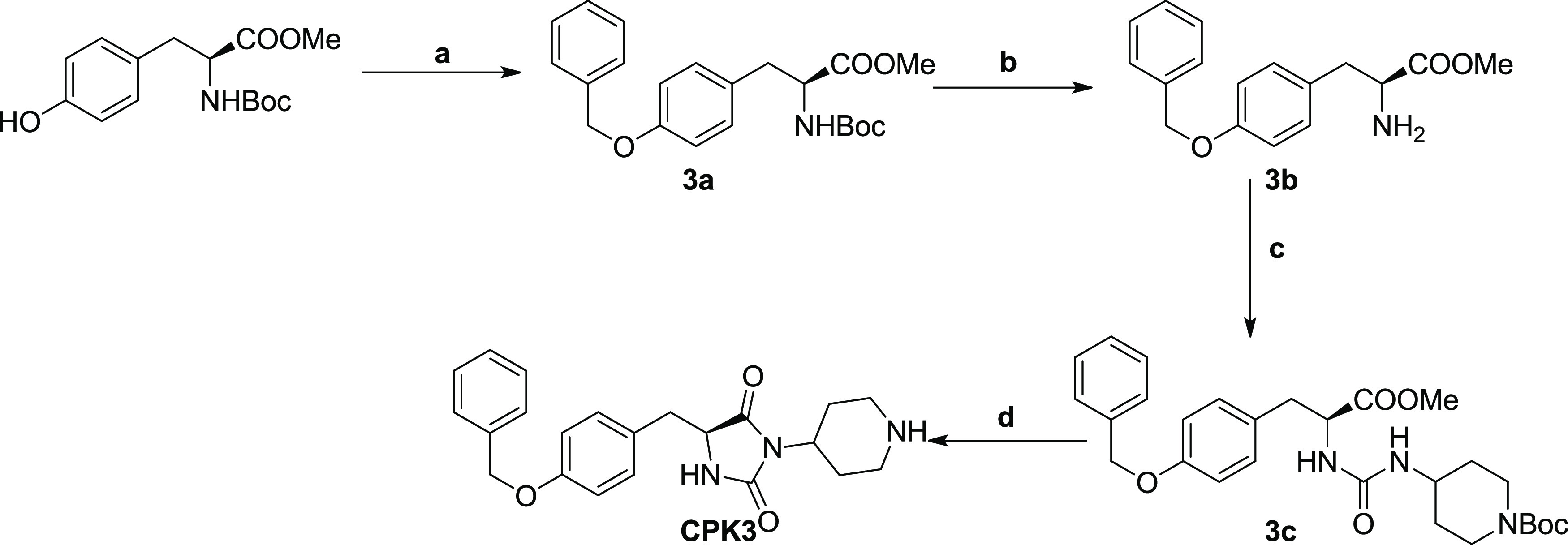
Reagents and conditions: (a) Benzyl bromide, potassium tert-butoxide, DMF, 130 °C, overnight; (b) TFA/DCM (1/3, v/v), triisopropylsilane, 3 h, RT; (c) triphosgene, 4-amino-1-Boc-piperidine, TEA (to pH 8), THF, reflux, 1 h; (d) MeOH/HCl 2 M (1/1, v/v), reflux, 3 h.
Compounds CPK5 and CPK10 were synthesized as depicted in Scheme 3. For the synthesis of CPK5, tert-butyl piperidin-4-ylcarbamate was reacted with 4-chlorobenzoyl chloride, using N,N-diisopropylethylamine (DIPEA) as catalyst. The resulting intermediate 5a was deprotected from the Boc group using TFA/TIS mixture, as described above, giving 5b. Compound 5b was coupled with Boc-l-Tyr(2-Br-Z)–OH using HOBt and HBTU as coupling agents and DIPEA as base to provide final compound CPK5 in 64% yield. Final compound CPK10 was synthesized following similar synthetic procedures. In this case, the tert-butyl piperidin-4-ylcarbamate was initially reacted with 4-chlorobenzenesulfonyl chloride to give the sulfonamide 10a which was deprotected as described for 5a. The resulting compound (10b) was coupled with Boc-l-Tyr(tBu)–OH using the above-described coupling protocol, to give intermediate 10c. The tert-butyl protecting group was removed from 10c using 20% TFA in DCM. The intermediate 10c thus obtained was finally reacted with 4-bromobenzyl bromide using potassium tert-butoxide as base to give final compound CPK10 in 82% yield.
Scheme 3. Synthesis of Final Compounds CPK5 and CPK10.
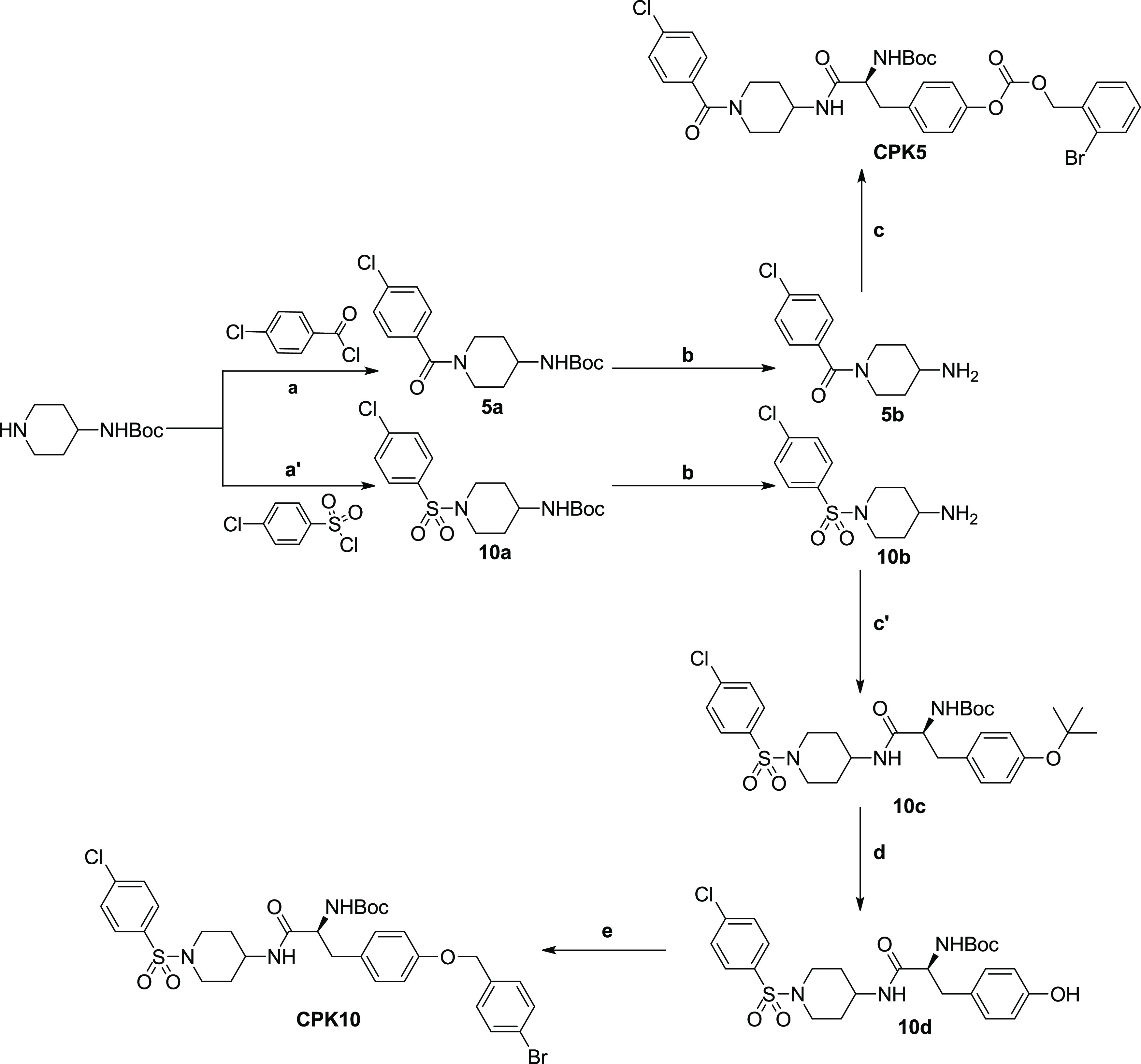
Reagents and conditions: (a) 4-Chlorobenzoyl chloride, DIPEA, DCM, 2 h, RT; (a′) 4-chlorobenzenesulfonyl chloride, DIPEA, DCM, 2 h, RT; (b) TFA/DCM (1/3, v/v), triisopropylsilane, 3 h, RT; (c) Boc-l-Tyr(2-Br-Z)–OH, HOBt, HBTU, DIPEA, DCM/DMF (4/1, v/v), RT, overnight; (c′) Boc-l-Tyr(tBu)–OH, HOBt, HBTU, DIPEA, DCM/DMF (4/1, v/v), RT, overnight; (d) TFA/DCM (1/5, v/v), triisopropylsilane, 2 h, RT; (e) 4-bromobenzyl bromide, potassium tert-butoxide, DMF, 130 °C, overnight.
Derivatives CPK7 and CPK8 were obtained following the procedures described in Scheme 4. The final compound CPK7 was obtained by a single-step procedure involving indole and 4-phenylbenzyl iodide that were subjected to an ultrasound-catalyzed reaction with NaH and dichloromethane, used as methylene source.58 For the synthesis of CPK8, Fmoc-Arg(Pbf)–OH was reacted by Weinreib procedure to obtain the N,O-dimethylhydroxylamide 8a, that was reduced by the use of lithium aluminum hydride and then further reacted with l-Cys-OEt in basic medium to give the thiazolidine 8b. Reaction of 8b with benzylamine and triphosgene provided hydantoin derivative 8c, which, upon the deprotection step using TFA/TIS, gave final compound CPK8 in 74% yield.
Scheme 4. Synthesis of Final Compound CPK7 and CPK8.

Reagents and conditions: (a) Sodium hydride, 4-(iodomethyl)-1,1′-biphenyl, dichloromethane, CH3CN, ultrasounds, 50 °C, 2 h; (b) N,O-dimethylhydroxylamine, HOBt, HBTU, DIPEA, DCM/DMF (4/1, v/v), RT, overnight; (c) LiAlH4 (1 M in THF), THF dry, N2, 0 °C, 10 min; (d) l-Cys-OEt, NaHCO3, EtOH, overnight; (e) benzylamine, triphosgene, TEA, THF, reflux, 1 h; (f) TFA/DCM (1/3, v/v), triisopropylsilane, RT, 3 h.
The synthesis of compound CPK18 was performed as described in Scheme 5. HOBt/HBTU-mediated coupling of l-Boc-tryptophan with benzylamine led to intermediate 18a. Removal of the Boc protecting group by TFA afforded 18b, which was subjected to the same reductive amination protocol described above, using 4-phenoxybenzaldehyde as a reactant, to give the final compound CPK18 in 72% yield.
Scheme 5. Synthesis of Final Compound CPK18.
Reagents and conditions: (a) Benzylamine, HOBt, HBTU, DIPEA, DCM/DMF (4/1, v/v), RT, overnight; (b) TFA/DCM (1/3, v/v), triisopropylsilane, 3 h, RT; (c) 4-phenoxybenzaldehyde, MeOH dry, 3 h, RT then NaBH4, 1 h, RT.
In Vitro Screening of the CPK Library Using a Fluorescence-Based Assay
The ability of the 20 in silico selected molecules to block KCNT1 channels was subsequently tested using a fluorescence-based medium-throughput assay. For this purpose, a fluorescence assay based on the thallium (Tl+) permeability of potassium (K+) channels was performed in CHO cells stably-expressing hKCNT1 channels. The influx of Tl+ through these channels was measured using a fluorescent Tl+-sensitive dye (FluxOR);59 the fluorescent signal generated upon Tl+ exposure was continuously measured for 30 s. The slope of the recorded fluorescence curves in the first 5 s after Tl+ exposure was used to quantify drug effects. While untransfected CHO cells only showed minimal changes in the fluorescent signal upon Tl+ exposure, CHO cells stably transfected with KCNT1, instead, displayed a marked increase in fluorescence; the slopes of the curves were 0.0008 ± 0.0002 and 0.011 ± 0.003 for untransfected and KCNT1 stably transfected CHO cells, respectively. In the latter cells, Tl+ fluorescence signal was further enhanced (resulting in a slope of 0.041 ± 0.006) by a 15 min preincubation with 10 μM of the dibenzoxazepine antipsychotic drug loxapine (LOX), a well-known KCNT1 opener (Figure 2A);60 by contrast, no increase in Tl+ fluorescence (slope of 0.0007 ± 0.0006) could be detected with 10 μM LOX in untransfected CHO cells, strongly suggesting that the fluorescence signal was specifically due to the expression of KCNT1 channels (Figure 2A–D). Concentration–response experiments revealed that LOX (0.1–30 μM) increased the slope of the fluorescent signal with an EC50 of 4.0 ± 1.7 μM (Figure 2B), a potency similar to that previously measured with whole-cell patch-clamp recordings.60 In the presence of 10 μM LOX, QND (1–1000 μM) reduced the slope of the fluorescent signal in a concentration-dependent manner, with an IC50 of 147 ± 31 μM (Figure 2C), a value consistent with those reported using electrophysiological techniques.31,34,44 The Z′ factor calculated in hKCNT1-transfected CHO cells was >0.5, indicative of a robust and reproducible assay.61 Using this assay, all 20 compounds selected from the CPK library based on the in silico screening were tested at a 10 μM concentration for their KCNT1-blocking ability. The results obtained revealed that five compounds (CPK4, 13, 16, 18, and 20) markedly reduced the fluorescent signal (Figure 2D); their potency, calculated from concentration–response experiments (Figure 2E) was 12–35 times higher when compared to QND. Indeed, IC50’s of 5.5 ± 2.35, 8.7 ± 1.45, 12.2 ± 2.60, 3.4 ± 0.70, and 6.4 ± 1.61 μM for CPK4, 13, 16, 18, and 20, respectively, were estimated (Figure 2E).
Figure 2.
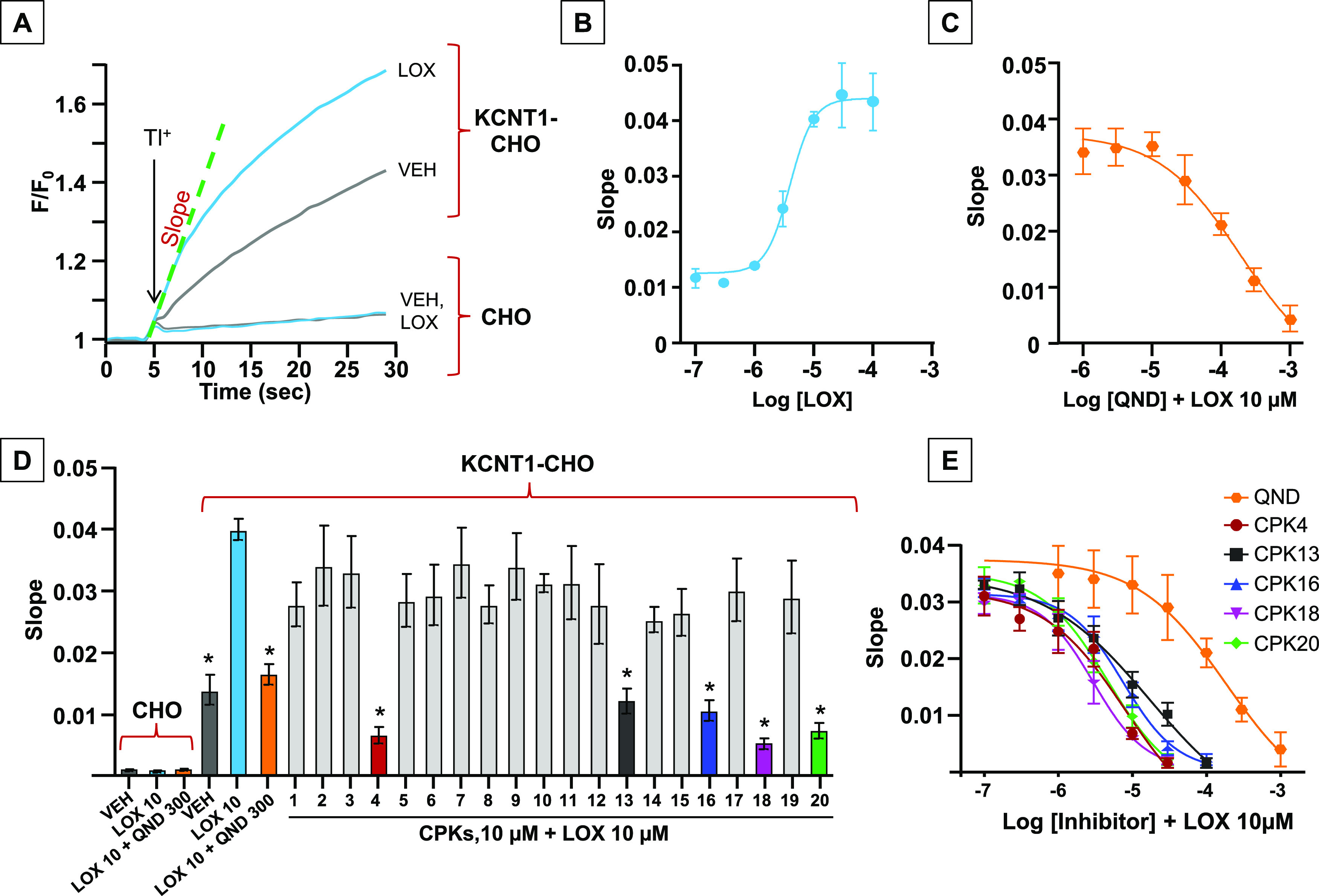
In vitro screening of the CPK library using the fluorescence-based assay FluxOR. (A) Representative curves describing the FluxOR fluorescent signals generated in stably KCNT1-transfected CHO cells and in untransfected CHO cells after incubation with vehicle (VEH) (gray curve) or LOX 10 μM (light blue curve). (B, C) Concentration–response curves of LOX (B) and QND (C) in stably KCNT1-transfected CHO cells. Solid lines represent fits of the experimental data to the four-parameter logistic equation used to estimate EC50/IC50 values. (D) Average FluxOR fluorescence signals obtained in stably KCNT1-transfected CHO cells and in untransfected CHO cells upon incubation with vehicle (VEH) (gray), LOX 10 μM (LOX, light blue), QND at 300 μM (orange), or with CPKs compounds, each at a concentration of 10 μM. QND and CPKs incubation was followed by incubation with LOX 10 μM. * indicates values significantly different (p < 0.05) from LOX (n = 5–13). (E) Concentration–response curves of QND (orange), CPK4 (red), CPK13 (black), CPK16 (blue), CPK18 (magenta), and CPK20 (green) in stably KCNT1-transfected CHO cells. Solid lines represent fits of the experimental data to the four-parameter logistic equation used to estimate IC50 values (n = 5).
Molecular modeling studies were conducted to get insight into the KCNT1/inhibitor interaction. Initially, these studies took advantage of the homology model (HMhKCNT198–354) described above, since the experimentally determined structure of the human KCNT1 channel was not yet available. A description of the predicted bound conformations on the homology model is given in Supporting Information (Figures S43 and S44). While our work was in progress, the EM structure of human KCNT1 in complex with the C23 inhibitor (a structural analogue of compound 31, Figure 1A) was released,62 thus we decided to update our results using this new structure (EMhKCNT1110–354). Induced-fit docking and Molecular Dynamics (MD) simulations of QND and the human KCNT1 EM structure (EMhKCNT1110–354) suggested that QND may bind in two different docking poses. Indeed, a 960 ns-long MD simulation showed QND bouncing between two poses (Figure 3C). In the first and most populated one (Figure 3C, green-lined area; Figure 3A) QND bound in between two S6 helices belonging to two adjacent KCNT1 subunits, interacting with residues L339, P343, L342 and making π–π and π-cation interactions with F346. In the second bound conformation (Figure 3C, red-lined area; Figure 3B) QND is instead flipped upside down and shifted toward the pore helix (p-helix), where it interacted with the backbone of F312 via direct H-bond and with the hydroxyl of T314 via water-mediated H-bond. A π–π stacking interaction was observed between the side chain of F312 and the QND aromatic moiety. The main EMhKCNT1110–354/QND interaction observed during the whole MD trajectory is summarized in Figure 3D.
Figure 3.

(A, B) Predicted bound conformations of QND. KCNT1 subunits are depicted in gray, gold, white, and blue cartoons and sticks, while QND is represented in yellow sticks. Direct H-bonds are represented as magenta dashed lines, water-mediated H-bonds as orange dashed lines, π–π stacking interactions as green dashed lines, and π-cation interactions as red dashed lines. In (A, B), for reference, F312 and F346 are always shown as sticks and the experimental bound conformation of C23 is shown in white transparent sticks. (C) RMSD of QND as a function of MD simulation time. (D) Quindine/KCNT1 interaction diagram. Only residues interacting with the ligand for at least 144 out of 960 ns of MD simulation are shown. Residues are colored according to the following scheme: cyan, polar; green, hydrophobic; gray, water molecule. Gray halos highlight solvent exposure. H-bonds are represented by magenta arrows (dashed when side-chain atoms are involved, solid in the case of backbone atoms involvement); green solid lines represent π–π stacking interactions; red solid lines represent π-cation interactions.
While QND mainly relies on the interaction with S6, p-loop, and p-helix residues to stabilize its binding to KCNT1, molecular mechanics simulations of compounds CPK4, CPK13, CPK16, CPK18, and CPK20 suggested that additional interactions may occur between inhibitors and S5 residues or with a pocket lined by S6, S5 and p-helix residues, which was made accessible upon rotation of the Cα–Cβ dihedral angle of F312. Among the CPK series compounds, only CPK4, CPK18, and CPK20 are predicted to stably wedge into the S6/S5/P-helix pocket (Figures 4 and 5). In particular, CPK4 sticks deeply into the pocket, interacting with residues M334 and I335 and making a hydrogen bond with the side chain of T311. Among the other residues that are supposed to interact significantly with CPK4, we found S6 residue L281 and S6 residues F312, A338, and L342 (Figures 4A and 5A). CPK18 phenoxy moiety sticks into the S6/S5/p-helix pocket where it interacts with S6 residues A338 and M334. Multiple interactions are established between the ligand and F312, i.e., two π–π stacking interactions with the residue side chain and a hydrogen bond with the main chain. Additional interactions are predicted with S6 residues F346 and with two L339 residues from two adjacent KCNT1 subunits (Figures 4D and 5D). Analogously to CPK4 and CPK18, CPK20 interacts with A338 from the S6/S5/p-helix pocket. It makes a hydrogen bond between its sulfonamide NH and the main chain carbonyl oxygen of F312, and its bis(trifluoromethyl)phenyl group stacks between F346 and P343 from the adjacent channel subunit. Additional interactions were predicted between the ligand and S5 L274 and L278 (Figures 4E and 5E). With the obvious exception of those involving residues accessible only from the S6/S5/p-helix pocket, CPK13 and CPK16 share many interactions similar to CPK4, CPK18, and CPK20. Compound CPK13 is predicted to interact with S313 via H-bond and its hydroxynaphthyl moiety is sandwiched between F346, with which a π–π stacking interaction is established, and P343 from the adjacent subunit. Further interactions are predicted between the ligand and L278 and L342, and with L339 from the adjacent subunit (Figures 4B and 5B). CPK16 is predicted to make water-mediated hydrogen bonds with the backbone oxygens of F312 and S313, while other significant contacts are found with L278, L339 (from the adjacent monomer), and F346 (Figures 4C and 5C).
Figure 4.

Docking poses of CPK4 (A), CPK13 (B), CPK16 (C), CPK18 (D), and CPK20 (E). KCNT1 subunits are depicted in gray, gold, white, and blue cartoons and sticks, while ligands are represented as yellow sticks. Direct H-bonds are represented as magenta dashed lines, water-mediated H-bonds as orange dashed lines, and π–π stacking interactions as green dashed lines. In every panel, for reference, F312 and F346 are always shown as sticks and the experimental bound conformation of C23 is shown in white transparent sticks.
Figure 5.

Ligand interaction diagrams of CPK4 (A), CPK13 (B), CPK16 (C), CPK18 (D), and CPK20 (E) in complex with EMhKCNT1110–354. Only residues interacting with the ligand for at least 72 out of 480 ns of MD simulation are shown. Residues are colored according to the following scheme: Cyan, polar; green, hydrophobic; gray, water molecule. Gray halos highlight solvent exposure. H-bonds are represented by magenta arrows (dashed when side-chain atoms are involved, solid in the case of backbone atoms involvement); green solid lines represent π–π stacking interactions.
Electrophysiological Validation of Selected CPK Compounds as Novel KCNT1 Blockers by Whole-Cell Patch-Clamp
Given the higher efficacy and potency showed by compounds CPK4, 13, 16, 18, or 20 when compared to QND as KCNT1 blockers in the fluorescence-based assay, their effects on KCNT1 currents were further investigated using whole-cell patch-clamp electrophysiological experiments, the gold-standard assay for ion channel modulators, in CHO cells transiently transfected with hKCNT1 cDNA. While no voltage-gated K+ currents could be detected in untransfected CHO cells (data not shown), cells transfected with hKCNT1 generated strong outward currents upon application of 3s voltage ramps from −90 to +60 mV; KCNT1 currents were only slightly reduced (35.0 ± 3.7%, n = 7) by perfusion with 10 μM QND, but almost fully blocked by 1 mM of the same alkaloid (98.0 ± 0.3%, n = 6), as previously reported (Figure 6A).31
Figure 6.

Pharmacological characterization of QND and CPK compounds on KCNT1 channels. (A–F) Representative current traces recorded upon exposure to the voltage protocol shown in (A) in CHO cells expressing KCNT1 channels recorded in control solution (CTL), upon perfusion with 10 μM or 1 mM QND (QND; A), 10 μM of the indicated CPK compounds (B–F), or upon drug washout (W). Current scale: 1 nA; time scale: 100 ms. (G–L) Time course of current decrease and recovery in CHO cells expressing KCNT1 channels in the absence or presence of the indicated compounds. (M) Quantification of maximal currents measured in cells expressing KCNT1 channels at +60 mV in experiments like those shown in (A–F) in the presence of the indicated compounds (*=p < 0.05 vs CTL; **=p < 0.05 vs QND). (N) Time constants of the activation kinetics (τon) in seconds for all tested compounds (*=p < 0.05 vs QND; **=p < 0.05 vs CPK4; ***=p < 0.05 vs CPK16).
By contrast, all five CPK compounds highlighted as potent KCNT1 blockers in the fluorescence-based assay, as 10 μM blocked KCNT1 currents almost completely (current blockade was of 90.6 ± 2.0, 96.2 ± 1.5, 89.1 ± 1.2, 97.4 ± 0.7, or 90.5 ± 1.2% for CPK4, CPK13, CPK16, CPK18, or CPK20, respectively; n = 10–18; Figure 6B–F,M), confirming their higher potency than QND. Notably, while blocking kinetics for QND were fast, those of all five novel KCNT1 blockers were significantly slower. Indeed, blocking time constants (τon) were 1.7 ± 0.1 s for QND (1 mM) and 49.0 ± 9.6, 17.7 ± 2.3, 30.8 ± 4.8, 15.2 ± 2.6, or 16.6 ± 1.8 s for CPK4, CPK13, CPK16, CPK18, or CPK20, respectively (each at 10 μM) (Figure 6N). Furthermore, while QND blockade was almost completely reversible upon drug washout (current recovered was 84.7 ± 6.1%), the extent of current recovered upon removal from the perfusion bath of CPK4, CPK13, CPK16, CPK18, or CPK20 was much lower, being 30.4 ± 10.0, 15.7 ± 4.8, 49.3 ± 4.7, 55.3 ± 15.9, or 25.0 ± 6.9%, respectively, after 8 min of drug washout (Figure 6G–L). Finally, in addition to the extent of current recovery, also KCNT1 current recovery kinetics were markedly slower in CPK4-, CPK13-, CPK16-, CPK18, or CPK20-exposed cells when compared to those of QND (Figure 6G–L). Altogether, the electrophysiological data shown confirm the higher potency of CPK4, 13, 16, 18, and 20 when compared to QND in blocking KCNT1 currents.
Effects of Selected CPK Compounds on hERG and Kv7.2 Currents
Pro-arrhythmic effects are among the most relevant safety concerns for antiarrhythmics such as QND,39,63 as well as for many other drug classes.38,64 Blockade of hERG potassium channels involved in the early phase (IKr) of the ventricular action potential repolarization, is a major cause for pro-arrhythmic effects associated with QT prolongation and torsade des pointes in humans.38 It is widely known that hERG channels interact with several distinct chemotypes, often at concentrations close to those needed for therapeutic efficacy. Indeed, several reports estimate that about 45% of the new molecular entities synthesized as potential therapeutic agents possess intrinsic hERG blockade activity.65,66 Statistics are even worse when potassium channel modulators are designed, considering that more than 60% of these molecules show hERG-blocking activity that falls below the 30-fold selectivity window, that is considered the safety limit.67QND itself is a rather nonselective blocker of several ion channels; in addition to KCNT1 currents, it is known to block heterologously expressed hERG channels with an IC50’s of ca. 0.5–1 μM,39,40 values about 100 times lower than those for KCNT1 currents.31,34,44 In the past few years, research aiming to develop new KCNT1 blockers has led to the discovery of new potent inhibitors (Figure 1A), with heterogeneous hERG inhibitory actions; VU0606170,42 for example, blocks 40% of hERG current at 10 μM, BC12 and BC1344 block 15–20% of hERG current at 10 μM, and compound 3143 inhibits hERG with IC50 of 11.9 μM; the only notable exception is compound VU0935685, described as completely inactive on hERG at 10 μM in a fluorescence-based assay.46 To assess whether CPK compounds, similarly to QND, also inhibited hERG currents, hERG cDNA was transiently transfected in CHO cells and hERG currents recorded with whole-cell patch-clamp. hERG currents were activated using a series of 1 s voltage steps from a holding potential of −80 to +60 mV in 10 mV increments, followed by an isopotential step of 140 ms to −100 mV, to record tail currents. Current inhibition by QND or CPK molecules, each tested at 10 μM, was quantified at −100 mV. 10 μM QND significantly inhibited hERG channels by 74 ± 4% (n = 6; Figure 7A,B). In contrast, CPK16, CPK18, and CPK20, always at 10 μM, were devoid of hERG-blocking activity (n = 6–7); for this reason, these 3 compounds underwent additional functional testing. By contrast, CPK4, and CPK13 inhibited hERG currents by approximately 30–40% at the same concentration (n = 6, Figure 7A,B); thus, their functional investigation was not pursued any further. In silico studies suggest that a plausible structural motif for hERG blockade is represented by a basic nitrogen center flanked by aromatic or hydrophobic groups.68−70 Moreover, experimental studies confirmed that, although the positive charge on the nitrogen is not strictly necessary to block hERG, nitrogen basicity might play a major role in hERG-blocking potency.71 In this context, the results obtained in our study are not surprising, since the KCNT1 antagonists herein reported do not present basic nitrogens (CPK16 and CPK20) or their nitrogens have low pKa values (CPK4, CPK13, and CPK18—Jaguar predicted pKa values: 4.48, 5.76, and 5.28, respectively)72 compared to the nitrogens of hERG blockers such as quinidine, astemizole, cisapride, and dofetilide. Moreover, it has been reported that F656, a residue that is strongly involved in coordinating the inhibitor binding in hERG, is at the equivalent position in the S6 segment as F346 in KCNT1.44 This is likely the reason why, CPK13, exerting a direct π–π stacking interaction with F346 is also the most potent hERG inhibitor of the series. Thus, our pharmacological data appear consistent with available structure–activity relationships for hERG blockade. In addition to hERG, we also investigated the potential blockade by our CPK compounds of the currents carried by other voltage-gated potassium channels. In particular, we focused on Kv7.2 channels encoded by the KCNQ2 gene. Kv7.2 subunits provide a fundamental contribution to the M-current, a widespread neuronally expressed voltage-gated K+ current which sets the resting membrane potential and plays a critical role in controlling neuronal excitability;73 variants in the KCNQ2 gene are responsible for mostly neonatal-onset epileptic and neurodevelopmental disorders with widely heterogeneous phenotypic presentations.74 It has been reported that retigabine, the prototype Kv7 activator, exerts anticonvulsant effect in several in vitro and in vivo seizure models, and has been approved as adjunctive therapy of partial onset seizures in humans.75,76 Since retigabine was withdrawn from the market due to safety reasons, in our previous work51 we designed a library of retigabine analogues, comprising also CPK20 which only showed weak agonistic activity on Kv7.2 currents in vitro.51 Therefore, to investigate the Kv7.2 blocking ability of QND and selected CPK compounds, electrophysiological experiments were performed in CHO cells transiently expressing Kv7.2 channels. Surprisingly, QND was able to block Kv7.2 channels with an IC50 of about 20 μM, a potency similar to that calculated in KCNT1 channels in our patch-clamp experiments (15.8 ± 2.3 μM, see below) and even higher than that reported in the literature.31,34,44 More importantly, while the present experiments confirmed the slight (10%) Kv7.2-activating properties of CPK20, they also revealed that CPK18 and CPK16 were unable to activate Kv7.2, confirming their selectivity for KCNT1 channels (Figure 7C,D).
Figure 7.
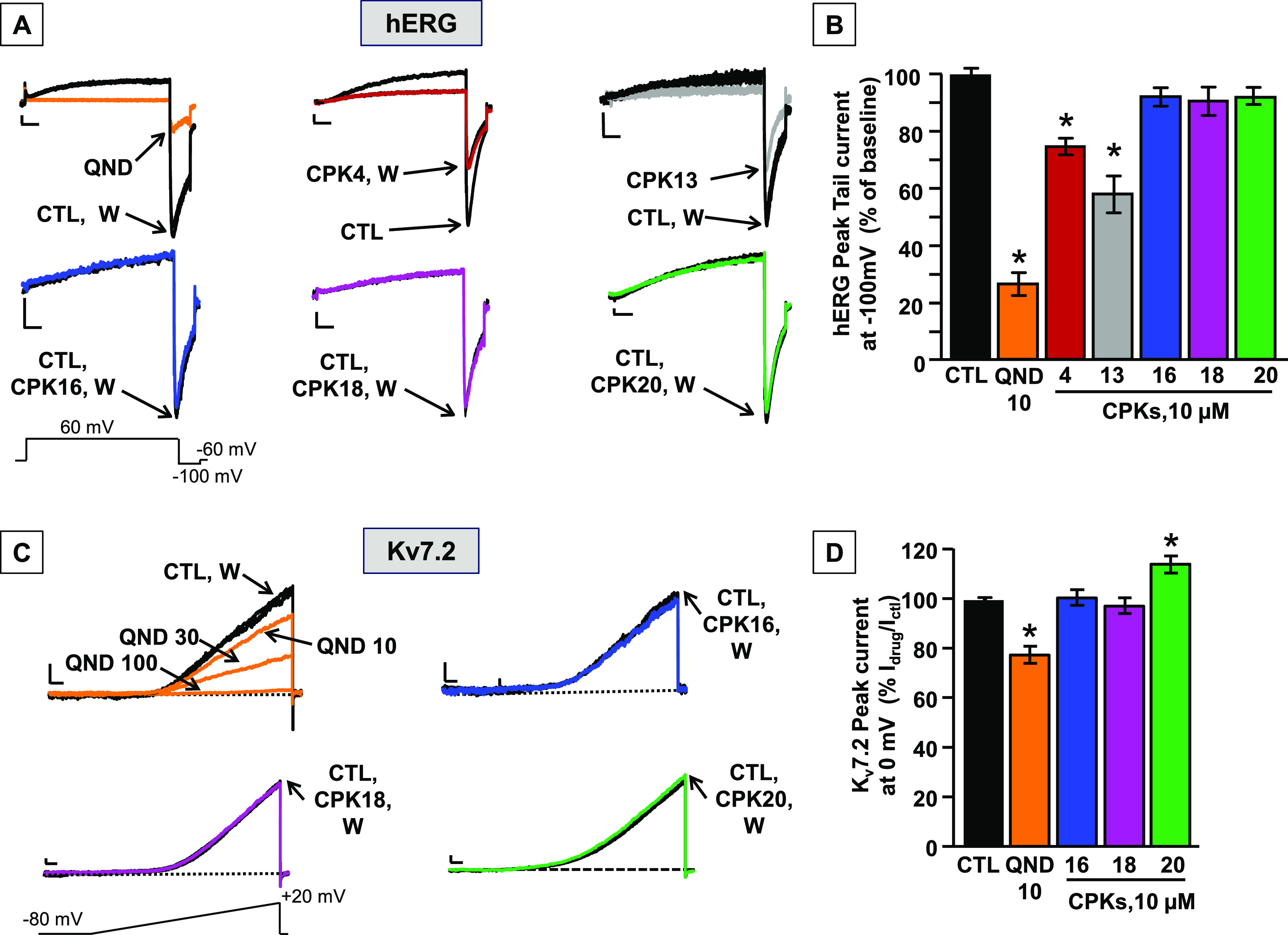
Effects of QND and CPK compounds on hERG and Kv7.2 channels. (A) Representative whole-cell current traces from hERG channels activated by the indicated ramp protocol recorded in control conditions and upon exposure to 10 μM quinidine (QND), 10 μM of the indicated CPK compounds, or upon drug washout (W). Current scale: 100 pA; time scale: 500 ms. (B) Quantification of the effects of the indicated compounds on hERG currents; data are expressed as the ratio between current amplitude at −100 mV in the presence and absence of 10 μM drugs (Idrug/ICTL); control value was calculated as the ratio between current amplitude at −100 mV at the beginning and after 1 min of perfusion with extracellular solution. Each data point is expressed as the mean ± SEM of at least three cells recorded in at least two independent transfections. * indicates values significantly different (p < 0.05) from control. (C) Representative whole-cell current traces from Kv7.2 channels activated by the indicated ramp protocol recorded in control conditions and upon exposure to 10–30–100 μM QND, 10 μM of the indicated CPK compounds, or upon drug washout (W). Current scale: 200 pA; time scale: 200 ms. (D) Quantification of the effects of the indicated compounds on Kv7.2 currents; data are expressed as the ratio between current amplitude at 0 mV in the presence and absence of 10 μM drugs (Idrug/ICTL); control value was calculated as the ratio between current amplitude at 0 mV at the beginning and after 1 min of perfusion with extracellular solution. Each data point is expressed as the mean ± EM of at least three cells recorded in at least two independent transfections. * indicates values significantly different (p < 0.05) from control.
Mutagenesis Experiments Confirm the In Silico Clues
To validate our docking and MD predictions, the effects of compounds CPK16, 18, and 20 in blocking KCNT1 channels carrying the F346S mutation were evaluated by patch-clamp experiments in transiently transfected CHO cells. Currents carried by KCNT1 F346S channels were 3.7-fold higher when compared to wild-type channels (at +60 mV, current densities were 239.9 ± 39.5 or 884.2 ± 138.9 pA/pF for wild-type or mutant KCNT1 channels, respectively; n = 9–14; p < 0.05). In addition, Boltzmann analysis of the G/V curves revealed a significant hyperpolarizing shift in activation gating for KCNT1 F346S when compared to wild-type KCNT1 channels (V1/2 was −47.9 ± 2.1 or −19.5 ± 2.7 mV, respectively; p < 0.05); by contrast, slope (k) values were unchanged (24.4 ± 1.6 or 21.8 ± 1.9 mV/efold, respectively). These functional changes are consistent with a strong gain-of-function (GoF) effect exerted by the F346S mutation on KCNT1 currents (Figure S45). As previously described,44 KCNT1 F346S currents showed a 10-fold reduced sensitivity to QND when compared to wild-type channels; in fact, at +60 mV, QND IC50’s were 15.8 ± 2.3 or 137.2 ± 60.5 μM for KCNT1 or KCNT1 F346 channels, respectively (n = 21–26; p < 0.05). The higher potency calculated for QND in this set of experiments is likely due to the fact that data were calculated at very positive potentials (+60 mV), thus favoring state- and voltage-dependent blockade by this alkaloid; nonetheless, these results confirm the crucial role for F346 in QND binding (Figure 8A). Similarly to QND, IC50 values for KCNT1 F346S current blockade by both CPK16 and CPK20 (each tested at 0.1–10.000 nM) were slightly but statistically significantly higher when compared to wild-type KCNT1 currents. The IC50 for CPK16 in KCNT1 or KCNT1 F346S currents were 0.20 ± 0.08 or 0.51 ± 0.06 μM, respectively (n = 5–45; p < 0.05), whereas those for CPK20 were 0.21 ± 0.02 μM or 1.93 ± 0.82 μM, respectively (n = 4–35; p < 0.05) (Figure 8B,D). For both compounds, no significant changes in the Hill slope of the concentration–response curve could be detected between KCNT1 and KCNT1 F346S channels (data not shown). By contrast, no statistically significant change could be detected in CPK18 blocking potency between KCNT1 or KCNT1 F346S currents (IC50 values were 0.08 ± 0.06 or 0.16 ± 0.04 μM, respectively; n = 5–81; p > 0.05, Figure 8C). However, when comparing drug effects at very low concentrations (10 nM), a much reduced blocking efficacy could be detected for CPK18 in KCNT1 F346S channels, leading to a steeper concentration–response curve; in fact, a statistically significant increase in the Hill slope could be detected (slope values were −0.43 ± 0.11 or −0.97 ± 0.21, in KCNT1 or KCNT1 F346S currents, respectively; n = 5–81; p < 0.05). Exemplary traces for the effects of CPK16, CPK18, and CPK20 on currents carried by KCNT1 and KCNT1 F346S channels are shown in Figure S46. Altogether, these mutagenesis results provide strong experimental support for the in silico prediction: CPK compounds appear less potent in blocking mutant KCNT1 F346S currents compared to wild-type KCNT1 currents suggesting that the residue F346 plays a role in CPK16 and CPK20 binding to KCNT1 channels pore, as similarly observed for QND. By contrast, the smaller differences observed for the effects prompted by the CPK18 compound when tested on wild-type or mutant channels could be due to the observation that this is the only derivative showing an interaction network at the binding site not involving the F346 residue (Figure 5E).
Figure 8.
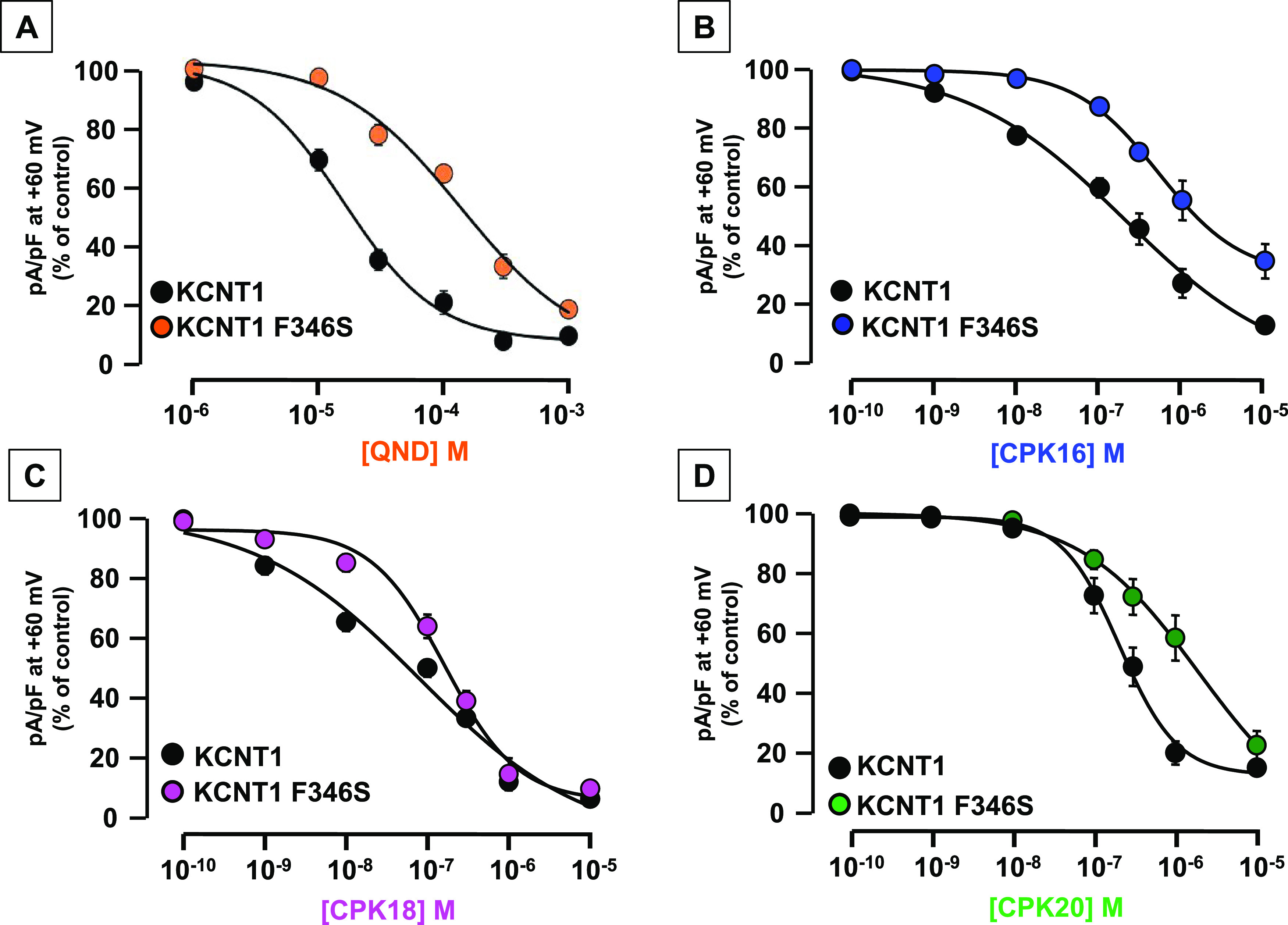
Concentration–response curves for KCNT1 and KCNT1 F346S inhibition current by (A) QND, (B) CPK16, (C) CPK18, and (D) CPK20. Current density after exposure to each drug concentration was expressed as % of the control current; normalized data were fitted to the following binding isotherm: y = max/(1 + x/EC50)n, where x is the drug concentration and n is the Hill coefficient. Each data point is the mean ± SEM of 3–26 (for QND), 5–45 (for CPK16), 5–81 (for CPK18), or 4–35 (for CPK20) determinations.
In Vitro Pharmacokinetics of CPK16, CPK18, and CPK20
The selection of a candidate drug is based on a delicate equilibrium among adequate target potency, optimized pharmacokinetic properties, and favorable safety profiles. This balance ensures the appropriate dosage and dosing regimen while minimizing the risks of drug–drug interactions and adverse effects. By employing high-throughput metabolic stability screening and rapid metabolite identification, medicinal chemists can strategically design and synthesize compounds characterized by low clearance. This strategic approach aims to reduce both drug dosage requirements and dosing frequency. To these aims, we initially evaluated the stability of compounds CPK16, CPK18, and CPK20 after incubation with human plasma for up to 120 min. Our results revealed high stability for the compounds CPK18 and CPK20 with not less than 80% compound recovery. On the other hand, compound CPK16 showed extremely low stability: after 120 min in contact with plasma, liquid chromatography–mass spectrometry (LC–MS) analysis revealed the complete loss of its molecular integrity (corresponding to 0% compound recovery, data not shown). Thus, CPK16 was excluded from the following stability assay. Hepatic stability of CPK18 and CPK20 was assessed after incubation with human liver microsomes (HLMs). HLMs consist of endoplasmic reticulum-localized enzymes, including cytochrome P450s (CYPs), flavin monooxygenases (FMOs), and some phase II enzymes, e.g., certain uridine 5′-diphospho-glucuronosyltransferase (UDP-glucuronosyltransferase, UGTs) isoforms and epoxide hydrolase (EH) enzymes. HLMs require fortification with appropriate cofactors such as NADPH for CYPs and FMOs and UDP-GlcUA for UGTs enzymes.
In our assay, we followed the loss of the test compounds over time under CYP-UGT-mediated metabolic pathways. The extent of hepatic metabolism allowed us to determine different pharmacokinetic parameters such as in vitrot1/2, CLintin vitro, and CLintin vivo. In vitrot1/2 and CLint of test compounds after liver microsomes incubation were calculated according to “well stirred” model.77 Predicted CLintin vivo values were determined using human physiologically-based scaling factor (PBSF). Our results highlighted that compounds CPK18 and CPK20 are categorized as high (CLintin vivoCPK18 < 80 mL min–1 kg–1) and low clearance compounds (CLintin vitroCPK20 < 1 mL min–1 kg–1), respectively (Figure S47).78 Considering the high hepatic susceptibility of compound CPK18 (Figure S48A), in the next step of our study we investigated the metabolites generated after its incubation with HLMs. For this purpose, a strategy integrating high-resolution MS/MS data and advanced processing algorithms in the Compound Discoverer software was employed. The metabolites were tentatively characterized by their accurate mass, fragmentation pattern, and retention times.
LC-MS/MS analysis highlighted that the main metabolite products were mono-oxidized (Figure S48B) and glucuronide (Figure S48C) derivatives. In detail, M1-M6-Ox metabolites presented the same precursor ion [M – H]+ at m/z 492 (C31H29N3O3) and showed a 16 Da mass difference to the parent compound, indicating a single oxidation reaction (Figure S49A–F). In detail, the fragment ions of M1-, M2- and M5-Ox at m/z 199 (C13H11O2, Rt: 2.33, 2.34, and 3.31 min, Figure S49A,B,E), M3- and M4-Ox at m/z 146 (C9H8ON, Rt: 2.43 and 2.51 min, Figure S49C,D) and M6-Ox at m/z 357 (C23H21O2N2, Rt: 3.63 min Figure S49F) indicated a possible oxidation of oxydibenzene, indole ring and aliphatic linker, respectively. It is interesting to note a significant increase in the quantity of oxidized derivatives after a 15 min incubation with HLMs, followed by a time-dependent decrease in metabolite formation. This phenomenon may be ascribed to a conjugation reaction catalyzed by the UGT enzymes, involving the addition of glucuronic acid to the hydroxyl functional groups introduced by cytochrome P450 (CYP) enzymes. Consequently, this process leads to a time-dependent increase in glucuronate derivative (M1-M3-Ox-Glu, C37H37N3O9, m/z 668, Figure S50A–C). Particularly, the fragment ions of M1-Ox-Glu (Rt: 2.13 min) at m/z 199 (C13H11O2) and 375 (C19H19O8) indicated a possible glucuronidation of phenoxy phenol moieties (Figure S50A). Similarly, the fragment ions of M2-Ox-Glu (Rt: 2.28 min) at m/z 146 (C9H8ON) and 322 (C15H16O7N) suggested a possible glucuronidation of the indole ring (Figure S50B). The M3-Ox-Glu metabolites (Rt: 2.97 min) exhibited a fragment ion at m/z 533 (C29H29O8N2) likely indicating the conjugation of glucuronic acid to the aliphatic linker (Figure S50C). Furthermore, the ability of compounds CPK18 and CPK20 to bind to plasma proteins was determined. Both compounds exhibited high binding affinity to plasma proteins (99%).
Effects of CPK20 in Blocking KCNT2 and Mutant KCNT1 Channels Carrying Pathogenic Gain-of-Function Variants
Given that CPK20 showed the best pharmacokinetic profile, being endowed with a remarkable metabolic stability, this compound was selected for these experiments. To expand the selectivity profile of CPK20 to channels encoded by other members of the Slo gene family,1 concentration–response curves were performed by patch-clamp electrophysiology in HEK cells to assess the ability of this compound to block currents carried by KCNT2 channels. As shown in Figure 9A,B, CPK20 also blocks KCNT2 currents, though with a lower potency when compared to KCNT1. In fact, the IC50’s for the current block, calculated at +60 mV, were, respectively, 0.20 ± 0.02 and 1.27 ± 0.52 μM for KCNT1 and KCNT2 channels, respectively (n = 44; p < 0.05); moreover, a statistically significant change in the Hill slope could be observed (k values were −1.09 ± 0.14 mV/efold or −0.57 ± 0.09 mV/efold in KCNT1 or KCNT2 currents, respectively; p < 0.05). Despite the lack of structural information on KCNT2 channels, this result is consistent with the strong degree of conservation in KCNT2 of the residues previously identified as potentially involved in CPK20 binding to KCNT1 (see Figure 5). Noteworthy, a stronger inhibitory potency for KCNT1 versus KCNT2 channels has been also shown by clofilium41 and compound 31,43 whereas no KCNT2 blocking effect (up to 10 μM) was shown by VU0606170 when tested using a Tl+ flux assay.42
Figure 9.
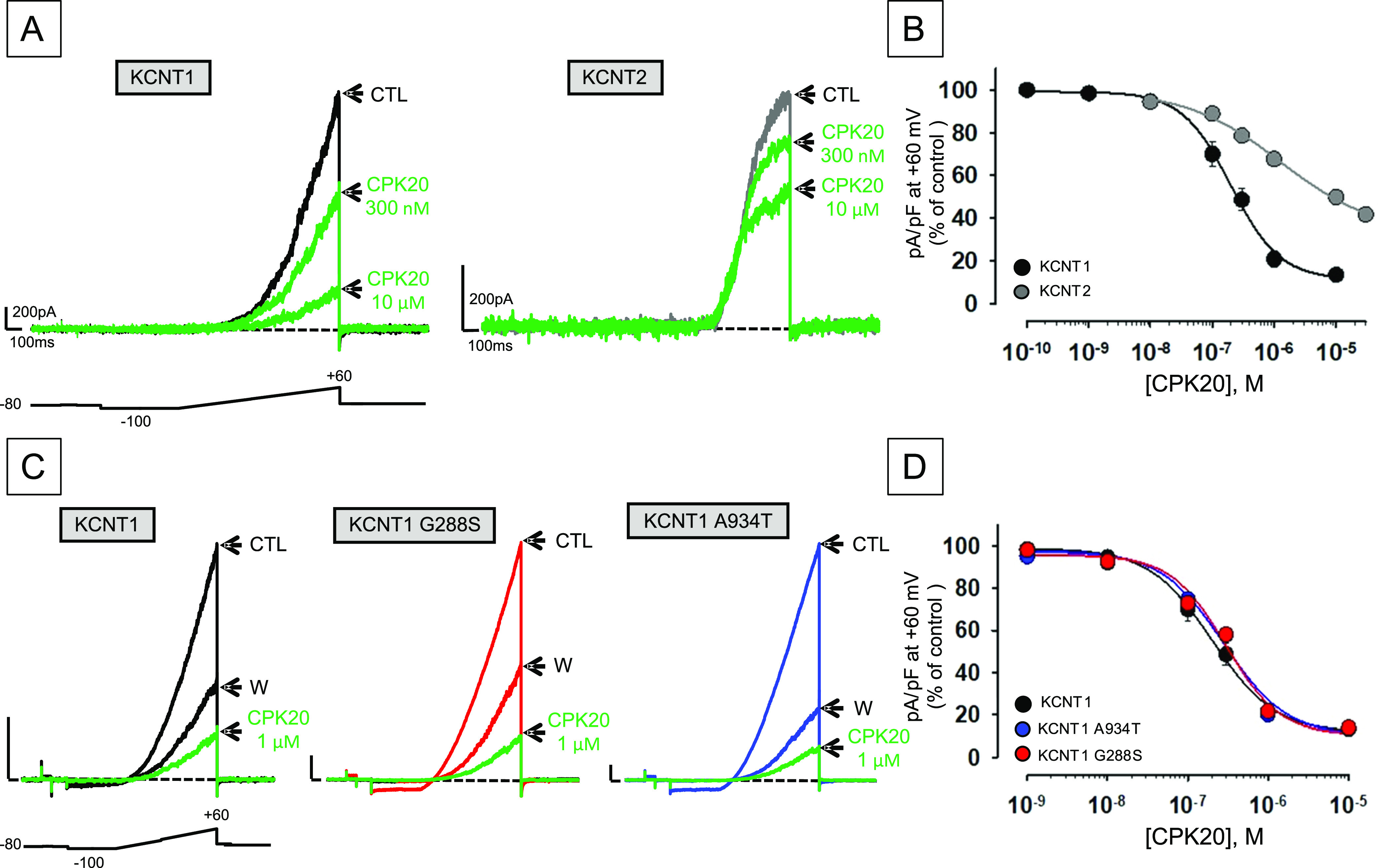
Effect of CPK20 on KCNT2 and KCNT1 channels incorporating pathogenic GoF variants. (A, B) Representative current traces (A) and concentration–response curves for inhibition by CPK20 (B) from CHO or HEK cells expressing KCNT1 or KCNT2 channels, as indicated. Current values in control solution (CTL), upon perfusion with the indicated concentrations of CPK20, or upon drug washout (W), were measured at the end of the depolarizing pulse and normalized data were fitted to the following binding isotherm: y = max/(1 + x/EC50)n, where x is the drug concentration and n is the Hill coefficient. Current scale: 200 pA; time scale: 100 ms. Each data point is the mean ± SEM of at least 6 independent determinations. (C, D) Representative current traces (C) and concentration–response curves for inhibition by CPK20 (D) from CHO cells expressing KCNT1, KCNT1 G288S, or KCNT1 A934T channels, as indicated. Current scale: 1 nA; time scale: 100 ms. Data were recorded and analyzed as described for (A, B).
As described in the Introduction Section, the largest majority of disease-causing KCNT1 variants show GoF effects. Considering that patients carrying these variants are often refractory to canonical anticonvulsants and that no safe and effective targeted treatment exists for KCNT1-related disorders, additional in vitro experiments were performed to investigate whether the new molecular entities herein described as potent KCNT1 blockers could counteract the GoF effects prompted by two recurrent pathogenic variants in KCNT1, namely, G288S31,79 and A934T,14,18 both found in EIMFS-affected patients. Notably, at least for QND, in vitro response in these assays may predict, in some but not in all patients, clinical response.80 Concentration–response curves showed no statistically significant difference in the blocking potency of CPK20 among wild-type KCNT1, KCNT1 G288S, and KCNT1 A934T channels (Figure 9C,D). In fact, the IC50’s for the current block calculated at +60 mV were 0.20 ± 0.02, 0.30 ± 0.07, and 0.28 ± 0.07 μM for wild-type KCNT1, KCNT1 G288S, and KCNT1 A934T channels, respectively (n = 28–44; p > 0.05). These data suggest that CPK20 might represent a potential therapeutic option for patients carrying KCNT1 variants showing GoF features; future experiments in relevant animal disease models will investigate whether CPK20, or other CPK compounds herein described, might show anticonvulsant activity.
Conclusions
We herein describe the identification of two different chemotypes acting as KCNT1 antagonists, namely, CPK18 and 20, thereby expanding the current armamentarium of KCNT1 channel modulators. Despite their large differences in terms of chemical structures and target binding, the two chemotypes described share some common basic features, mainly represented by a potent inhibition of KCNT1 currents and a lack of inhibition of hERG channels, being pivotal for further developments. In addition to the results herein described, further hit-to-lead development is mandatory, focused both on pharmacokinetic and pharmacodynamic optimization procedures. Moreover, the binding hypothesis raised in our silico studies should be further challenged by the combined use of cryo-EM and mutagenesis experiments; these future steps will provide solid rational basis for the future design of CPK18 and CPK20 analogues, for the treatment of KCNT1-related neurological disorders.
Experimental Section
General
All reagents and solvents used were purchased from Merck Italia (Milan, Italy), unless otherwise noted. Reactions were carried out with magnetic stirring in round-bottomed flasks excepting for microwave-assisted and ultrasound-assisted reactions that were conducted using glass vials and a microwave closed-vessel apparatus (CEM discover 2.0, CEM Co.) or a Bandelin Sonorex Digital 10P ultrasonic bath with a frequency of 60 Hz and power of 240 W (Bandelin Electronic, Germany), respectively. Oven-dried glassware under nitrogen stream and freshly distilled solvents were used to perform moisture-sensitive reactions. During the synthetic procedures, no unexpected or unusually high safety hazards were encountered. Thin layer chromatography (TLC) analysis of reaction mixtures was performed over precoated glass silica gel plates (F254, 0.25 mm, VWR International), while crude products were purified by an automated flash chromatography system (Isolera Spektra one, Biotage, Sweden) using preloaded silica gel cartridges (SNAP KP-Sil, Biotage). NMR spectra were recorded on a Bruker Avance 400 MHz apparatus, at room temperature. Chemical shifts are reported in δ values (ppm) relative to internal Me4Si, while J values are reported in hertz (Hz). The following abbreviations are used to describe the 1H NMR peaks: s (singlet), d (doublet), dd (doublet doublet), t (triplet), q (quadruplet), m (multiple), and bs (broad singlet). High resolution-MS (HR-MS) analysis was conducted using the LTQ-Orbitrap-XL-ETD mass spectrometer (Thermo Scientific, Bremen, Germany), through an electrospray source. The purity of final compounds was assessed by ultra-high performance liquid-chromatography (UHPLC) analyses, performed on a Jasco Extrema LC 4000 (Jasco, Japan) consisting of an LC-Net CG cable controller, quaternary flow pump system PU-4285, a DG-4000–04 degasser, a UV-4075 detector, and an AS-4250 autosampler. Purity assessment UHPLC runs were carried out on an EVO C18 150 mm × 2.1 mm × 2.6 μm (100 Å) column (Phenomenex, Bologna, Italy). The optimal mobile phase consisted of 0.1% HCOOH/H2O v/v (A) and 0.1% HCOOH/ACN v/v (B). Analysis was performed in gradient elution as follows: 0–10.00 min, 5–95% B; 10–12.00 min, 95–95% B; 12–15.00 min, isocratic to 5% B. Flow rate was 0.5 mL min-1. The injection volume was set at 5 μL.
Chromatograms were monitored at 254 nm. Final compounds always showed a purity >95% as assessed by the software ChromNav (Jasco) using the European Pharmacopeia criteria. The corresponding chromatograms are reported in the Supporting Information. CD experiments were performed on an 810-Jasco spectropolarimeter using a quartz cuvette with a path length of 1 mm, a measurement range from 190 to 290 nm (far UV), and a temperature of 25 °C. An average of 4 scans were performed with 10 nm/min scan speed, 4 s response time, and 2 nm bandwidth. The processed curves of the compounds were obtained by using Spectra Analysis tool of Jasco software. The CD curves were corrected for the solvent contribution by subtraction of CD reference spectrum and then final CD spectra were obtained after baseline correction and binomial smoothing.
(5S,10aR)-5-(4-Fluorophenyl)-7,8-dihydroxy-2-[3-(trifluoromethyl)phenyl]-10,10a-dihydroimidazo[1,5-b]isoquinoline-1,3(2H,5H)-dione (CPK1)
The compound was synthesized according to the procedure previously described.50 NMR and mass spectra were in accordance with those reported, while UHPLC traces are reported in the Supporting Information.
4-(2-Cyclohexylethoxy)benzaldehyde (2a)
0.5 equiv of 4-hydroxybenzaldehyde was dissolved in DMF, added with 1.5 equiv of potassium tert-butoxide and 1.5 equiv of (2-bromoethyl)cyclohexane, and refluxed and warmed to 130 °C overnight. After cooling to room temperature, the organic phase was diluted with dichloromethane, washed with 1 N NaOH (3 × 20 mL), extracted, dried over Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography using n-hexane/ethyl acetate (ratio 4/1, v/v) as mobile phase. Rf: 0.49. Whitish oil (75% yield). 1H NMR (400 MHz, CDCl3): δ: 1.03 (dd, 2H, CH2, J′ = 10.2 Hz, J″ = 15.1 Hz); 1.21–1.38 (m, 3H, CH, CH2); 1.73–1.85 (m, 8H, CH2); 2.21 (bs, 1H, NH); 3.18–3.29 (m, 2H, CH2); 3.66–3.69 (m, 4H, CH, CH3); 3.76 (t, 1H, CH, J = 6.3 Hz); 3.82 (d, H, CH, J = 12.8 Hz); 4.01 (t, 2H, CH2, J = 6.5 Hz); 6.84 (d, 2H, aryl, J = 8.1 Hz); 6.97 (s, 1H, aryl); 7.14–7.33 (m, 4H, aryl); 7.32 (d, 2H, aryl, J = 8.0); 7.62 (d, 1H, aryl, J = 7.8 Hz); 8.50 (s, 1H, NH). HR-MS m/z calcd for C15H20O2 [(M + H)]+: 233.1536; found 233.1545.
Methyl [4-(2-Cyclohexylethoxy)benzyl]-l-tryptophanate (2b)
l-Tryptophan methyl ester (1.0 equiv) was dissolved in dry MeOH at room temperature under positive nitrogen flow. To this solution, an amount of 1.5 equiv of 4-(2-cyclohexylethoxy)benzaldehyde was added and the mixture was stirred at room temperature for 3 h. Then, 3 equiv of NaBH4 were added portionwise and the mixture was stirred for further 1 h. The mixture was evaporated under vacuum and diluted with ethyl acetate, then 1 N NaOH was added. The organic phase was separated, extracted, dried over Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography using n-hexane/ethyl acetate (ratio 1/4, v/v) as eluent. Rf: 0.45. White powder (76% yield). 1H NMR (400 MHz, CDCl3): δ: 1.03 (dd, 2H, CH2, J′= 10.2 and J″ = 21.1 Hz); 1.21–1.38 (m, 3H, CH, CH2); 1.73–1.85 (m, 8H, CH2); 2.21 (bs, 1H, NH); 3.18–3.29 (m, 2H, CH2); 3.66–3.69 (m, 4H, CH, CH3); 3.76 (t, 1H, CH, J = 6.3 Hz); 3.82 (d, H, CH, J = 12.8 Hz); 4.01 (t, 2H, CH2, J = 6.5 Hz); 6.84 (d, 2H, aryl, J = 8.1 Hz); 6.97 (s, 1H, aryl); 7.14–7.33 (m, 4H, aryl); 7.32 (d, 2H, aryl, J = 8.0); 7.62 (d, 1H, aryl, J = 7.8 Hz); 8.50 (s, 1H, NH). HR-MS m/z calcd for C27H34N2O3 [(M + H)]+: 435.2642; found 435.2666.
(S)-Methyl 2-{Benzyl[4-(2-cyclohexylethoxy)benzyl]amino}-3-(1H-indol-3-yl)propanoate (CPK2)
To a solution of intermediate 2b dissolved in DCM, 1.5 equiv of benzyl bromide and 1.5 equiv of DIPEA were added. The reaction was conducted under μW, at 100 °C, for 20 min. The resulting mixture was washed with water (3 × 20 mL), dried over anhydrous Na2SO4, filtered, concentrated, and purified by column chromatography using DCM/ethyl acetate (ratio 4/1, v/v) as mobile phase. Rf: 0.42. White powder (83% yield). 1H NMR (400 MHz, CD3OD): δ: 1.00 (dd, 2H, CH2, J′ = 11.7 and J″ = 20.1 Hz); 1.19–1.36 (m, 4H, CH2); 1.50–1.59 (m, 1H, CH); 1.65 (dd, 2H, CH2, J′ = 6.6 and J″ = 12.1 Hz); 1.70–1.80 (m, 4H, CH2); 3.05 (dd, 1H, CH2, J′ = 5.7 and J″ = 14.1 Hz); 3.30 (d, 1H, CH, J = 9.0 Hz); 3.45 (dd, 2H, CH2, J′ = 13.4 and J″ = 20.1 Hz); 3.67 (s, 3H, CH3); 3.79 (dd, 1H, CH, J′ = 6.0 and J″ = 9.0 Hz); 3.85 (d, 1H, CH, J = 13.4 Hz); 3.95–4.02 (m, 3H, CH, CH2); 4.86 (s, 2H, CH2); 6.67 (d, 2H, aryl, J = 8.6 Hz); 6.82 (d, 1H, aryl, J = 6.2 Hz); 6.90 (s, 1H, aryl); 6.99–7.05 (m, 2H, aryl); 7.15 (d, 2H, aryl, J = 8.6 Hz); 7.21–7.31 (m, 6H, aryl). 13C NMR (100 MHz, CD3OD): δ: 14.1, 22.7, 26.3, 26.6, 31.6, 33.4, 34.6, 36.7, 50.9, 54.1, 54.5, 61.2, 66.0, 110.9, 112.3, 114.2, 118.8, 119.2, 121.8, 122.7, 126.9, 127.5, 128.2, 128.8, 129.9, 131.2, 136.1, 139.8, 158.3, 173.0. HR-MS m/z calcd for C34H40N2O3 [(M + H)]+: 525.3112; found 525.3086.
Methyl (S)-3-[4-(Benzyloxy)phenyl]-2-[(tert-butoxycarbonyl)amino]propanoate (3a)
Obtained from Boc-l-tyrosine methyl ester and benzyl bromide as reported for 2a. FC in n-hexane/ethyl acetate (ratio 4/1, v/v). Yellowish powder (58% yield). 1H NMR (400 MHz, CDCl3): δ: 1.44 (s, 9H, CH3); 2.78–2.83 (m, 1H, CH); 3.04–3.08 (m, 1H, CH); 3.74 (s, 3H, CH3); 4.02 (t, 1H, CH, J = 8.7 Hz); 5.01 (s, 2H, CH2); 6.88 (d, 2H, aryl, J = 8.6 Hz); 7.07 (d, 2H, aryl, J = 8.5 Hz); 7.28–7.41 (m, 5H, aryl). HR-MS m/z calcd for C22H27NO5 [(M + H)]+: 386.1962; found 386.1990.
Methyl (S)-2-Amino-3-[4-(benzyloxy)phenyl]propanoate (3b)
The N-Boc-protected intermediate 3a (1.0 equiv) was dissolved in a mixture of TFA/DCM (ratio 1/3, v/v), and triisopropylsilane (0.25 equiv) was added. The reaction was stirred at room temperature for 3 h. Then, a solution of K2CO3 (2N) was added. The mixture was diluted with dichloromethane, and the organic phase was extracted, dried over Na2SO4, filtered, and concentrated under vacuum. The crude product was precipitated from MeOH/diethyl ether. Yellowish powder (76% yield). 1H NMR (400 MHz, CDCl3): δ: 2.76–2.85 (m, 1H, CH); 3.01–3.05 (m, 1H, CH); 3.69 (s, 3H, CH3); 3.99 (t, 1H, CH, J = 8.7 Hz); 5.01 (s, 2H, CH2); 6.82 (d, 2H, aryl, J = 8.6 Hz); 7.00 (d, 2H, aryl, J = 8.5 Hz); 7.28–7.38 (m, 5H, aryl). HR-MS m/z calcd for C17H19NO2 [(M + H)]+: 269.1416; found 269.1432.
tert-Butyl (S)-4-{3-[3-(4-(Benzyloxy)phenyl)-1-methoxy-1-oxopropan-2-yl]ureido}piperidine-1-carboxylate (3c)
To a solution of intermediate 3b (1.0 equiv) dissolved in THF, 0.4 equiv of triphosgene and 1.2 equiv of 4-amino-1-Boc-piperidine were added. The pH was adjusted to 8 by addition of TEA, and the mixture was refluxed for 1 h. After cooling to room temperature, the mixture was diluted with dichloromethane and washed with water (3 × 20 mL). The organic phase was extracted, dried over Na2SO4, filtered, and concentrated under vacuum. The crude product was purified by flash chromatography using n-hexane/ethyl acetate (ratio 2/3, v/v) as eluent. Rf: 0.50. Yellowish oil (52% yield). 1H NMR (400 MHz, CDCl3): δ: 0.90 (t, 1H, CH, J = 7.0 Hz); 1.21–1.30 (m, 3H, CH, CH2); 1.46 (s, 9H, CH3); 1.79–1.91 (dd, 2H, CH2, J′ = 14.2 and J″ = 15.2 Hz); 2.76–2.85 (m, 2H, CH2); 3.01 (t, 1H, CH, J = 6.0 Hz); 3.67–3.72 (m, 4H, CH, CH3, J = 12.8 Hz); 3.99 (bs, 2H, CH2); 4.69–4.74 (m, 1H, NH); 5.03 (s, 2H, CH2); 6.90 (d, 2H, aryl, J = 8.6 Hz); 7.03 (d, 2H, aryl, J = 8.5 Hz); 7.31–7.43 (m, 5H, aryl). HR-MS m/z calcd for C28H37N3O6 [(M + H)]+: 512.2755; found 512.2778.
(S)-5-[4-(Benzyloxy)benzyl]-3-(piperidin-4-yl)imidazolidine-2,4-dione (CPK3)
A solution of urea intermediate 3c was dissolved in a mixture of MeOH/HCl 2 M (ratio 1/1, v/v) and refluxed for 3 h. After the completion of the reaction, monitored by TLC, the organic phase was washed with 1 N NaOH, dried over anhydrous Na2SO4, filtered, concentrated, and purified by flash chromatography using ethyl acetate/MeOH (ratio 4/1, v/v) as mobile phase. Rf: 0.44. White powder (67% yield). 1H NMR (400 MHz, CDCl3): δ: 1.19–1.23 (m, 1H, CH2a); 1.35–1.38 (m, 1H, CH2b); 1.90 (bs, 2H, CH2); 2.04–2.14 (m, 1H, CH); 2.51 (t, 2H, CH2, J = 12.6 Hz); 2.80 (dd, 1H, CH2a, J′ = 7.2 and J″ = 14.1 Hz); 3.02–3.07 (m, 3H, CH2 and CH2b); 3.78–3.85 (m, 1H, CH); 4.06 (dd, 1H, CH, J′ = 3.9 and J″ = 7.2 Hz); 4.96 (s, 2H, CH2); 5.97 (bs, 1H, NH); 6.82 (d, 2H, aryl, J = 8.5 Hz); 7.02 (d, 2H, aryl, J = 8.5 Hz); 7.25–7.34 (m, 5H, aryl). 13C NMR (100 MHz, CDCl3): δ: 29.6, 29.7, 36.9, 46.0, 49.5, 57.6, 70.0, 115.1, 127.1, 127.4, 128.0, 128.6, 130.6, 136.9, 157.0, 158.1, 173.1. HR-MS m/z calcd for C22H25N3O3 [(M + H)]+: 380.1969; found 380.1944.
1-[1-(4-Fluorobenzyl)indolin-5-yl]-3-neopentylurea (CPK4)
The compound was synthesized according to the procedure previously described.51 NMR and mass spectra were in accordance with those reported, while UHPLC traces are reported in the Supporting Information.
tert-Butyl [1-(4-Chlorobenzoyl)piperidin-4-yl]carbamate (5a)
To a solution of 4-(N-Boc-amino)piperidine (1.0 equiv) in dichloromethane, 4-chlorobenzoyl chloride (2.0 equiv) and DIPEA (2.0 equiv) were added. The mixture was stirred at room temperature for 2 h. Then, the organic phase was washed with a saturated solution of NaHCO3 (3 × 20 mL), and a solution of citric acid (10% w/w, 3 × 20 mL), dried on Na2SO4, filtered, and concentrated in vacuo. Intermediate 5a was purified by flash chromatography using n-hexane/ethyl acetate (ratio 8/2, v/v) as mobile phase. Rf: 0.46. Whitish oil (87% yield). 1H NMR (400 MHz, CDCl3): δ: 1.43 (s, 9H, CH3); 1.49–1.55 (m, 2H, CH2); 1.95–2.03 (d, 2H, CH2, J = 9.9 Hz); 3.02 (bs, 2H, CH2); 3.42 (bs, 1H, CH); 3.56–3.68 (m, 2H, CH2); 4.62 (t, 2H, CH2, J = 11.0 Hz); 7.32 (d, 2H, aryl, J = 8.4 Hz); 7.58 (d, 2H, aryl, J = 8.7 Hz). HR-MS m/z calcd for C17H23ClN2O3 [(M + H)]+: 339.1470; found 339.1482.
(4-Aminopiperidin-1-yl)(4-chlorophenyl)methanone (5b)
Intermediate 5b was synthesized starting from 5a as described for 3b. Precipitated from MeOH/diethyl ether. White powder (90% yield). 1H NMR (400 MHz, CDCl3): δ: 1.38–1.45 (m, 2H, CH2); 1.84 (d, 2H, CH2, J = 10.5 Hz); 2.45 (m, 2H, CH2); 2.63–2.68 (m, 1H, CH); 3.67 (d, 2H, CH2, J = 11.9 Hz); 7.28 (d, 2H, aryl, J = 8.3 Hz); 7.50 (d, 2H, aryl, J = 8.3 Hz). HR-MS m/z calcd for C12H15ClN2O [(M + H)]+: 239.0946; found 239.0955.
tert-Butyl (S)-{3-[4-({[(2-Bromobenzyl)oxy]carbonyl}oxy)phenyl]-1-{[1-(4-chlorobenzoyl)piperidin-4-yl]amino}-1-oxopropan-2-yl}carbamate (CPK5)
0.1 equiv of intermediate 5b were dissolved in DCM/DMF (ratio 4/1, v/v) and added with HOBt (1.2 equiv), HBTU (1.2 equiv), DIPEA (2.4 equiv), and Boc-L-Tyr(2-Br-Z)–OH (1.2 equiv) and stirred at room temperature overnight. Then, the organic phase was diluted with dichloromethane and washed with water (3 × 20 mL), a saturated solution of NaHCO3 (3 × 20 mL), and a solution of citric acid (10% w/w, 3 × 20 mL). The combined organic layer was extracted, dried over Na2SO4, filtered, and concentrated under vacuum. The crude product was purified by flash chromatography using ethyl acetate as mobile phase. Rf: 0.47. White powder (64% yield). 1H NMR (400 MHz, CDCl3): δ: 1.36 (s, 9H, CH3); 1.50–1.86 (m, 4H, CH2); 2.88–3.08 (m, 4H, CH2); 3.56 (bs, 1H, CH2a); 3.84–3.87 (m, 1H, CH); 4.15 (d, 1H, CH, J = 6.6 Hz); 4.40 (bs, 1H, CH2b); 4.96 (bs, 1H, NH); 5.28 (s, 2H, CH2); 5.60 (d, 1H, NH, J = 6.2 Hz); 7.06 (d, 2H, aryl, J = 8.0 Hz); 7.14–7.19 (m, 3H, aryl); 7.23–7.32 (m, 5H, aryl); 7.43 (d, 1H, aryl, J = 7.2 Hz); 7.55 (d, 1H, aryl, J = 7.9 Hz). 13C NMR (100 MHz, CDCl3): δ: 28.3, 29.7, 37.9, 46.5, 53.4, 56.1, 69.7, 77.2, 121.0, 121.3, 135.5, 127.7, 128.4, 128.8, 130.2, 130.4, 130.5, 133.0, 134.1, 134.6, 135.8, 150.2, 153.4, 169.3, 170.3. HR-MS m/z calcd for C37H37BrClN3O7 [(M + H)]+: 716.1576; found 716.1549.
N-{2-Amino-4-[(4-fluorophenethyl)amino]phenyl}heptanamide (CPK6)
The compound was synthesized according to the procedure previously described.52 NMR and mass spectra were in accordance with those reported, while UHPLC traces are reported in the Supporting Information.
Bis[1-([1,1′-biphenyl]-4-ylmethyl)-1H-indol-3-yl]methane (CPK7)
Indole (1.0 equiv) was dissolved in acetonitrile (5 mL) in a falcon tube (10 mL) and added with sodium hydride (2.0 equiv), 4-(iodomethyl)-1,1′-biphenyl (1.5 equiv), and dichloromethane (3 equiv). The mixture was introduced in an ultrasonic bath setting the temperature at 50 °C and irradiating for 2 h. Then, the reaction was diluted with dichloromethane, washed with citric acid (10% w/w, 3 × 20 mL), extracted, filtered, dried over Na2SO4, and evaporated in vacuo. CPK7 was obtained after flash chromatography, using n-hexane/ethyl acetate (ratio 4.5/0.5, v/v) as eluent. Rf: 0.46. Whitish oil (48% yield). 1H NMR (400 MHz, CDCl3): δ: 4.22 (s, 2H, CH2); 5.20 (s, 4H, CH2); 6.89 (s, 2H, aryl); 7.01 (t, 2H, aryl, J = 7.7 Hz); 7.07–7.12 (m, 6H, aryl); 7.20 (d, 4H, aryl, J = 6.4 Hz); 7.22 (s, 2H, aryl); 7.25 (d, 4H, aryl, J = 7.2 Hz); 7.33 (t, 4H, aryl, J = 7.7 Hz); 7.40–7.45 (m, 8H, aryl); 7.58 (d, 2H, aryl, J = 11.8 Hz). 13C NMR (100 MHz, CDCl3): δ: 21.3, 49.6, 109.6, 115.0, 118.9, 119.5, 121.7, 126.5, 127.0, 127.1, 127.3, 127.4, 128.3, 128.8, 136.9, 140.5, 140.7. HR-MS m/z calcd for C43H34N2 [(M + H)]+: the compound does not ionize.
(9H-Fluoren-9-yl)methyl (S)-{1-[Methoxy(methyl)amino]-1-oxo-5-{2-[(2,2,4,6,7-pentamethyl-2,3-dihydrobenzofuran-5-yl)sulfonyl]acetimidamido}pentan-2-yl}carbamate (8a)
Synthesized from Fmoc-l-Arg(Pbf)–OH and N,O-dimethylhydroxylamine as described for CPK5. FC in n-hexane/ethyl acetate (ratio 6/4, v/v). Rf: 0.45. Whitish oil (67% yield). 1H NMR (400 MHz, CDCl3): δ: 1.26–1.33 (m, 2H, CH2); 1.40–1.46 (m, 2H, CH2); 2.01 (s, 6H, CH3); 2.45 (s, 6H, CH3); 2.53 (s, 3H, CH3); 2.72 (s, 2H, CH2); 3.21 (d, CH3); 3.66 (s, 3H, CH3); 4.11 (t, 1H, CH, J = 6.5 Hz); 4.26 (d, 2H, CH2, J = 8.2 Hz); 4.62 (t, 1H, NH, J = 6.8 Hz); 5.85 (bs, 1H, CH); 6.15 (t, 1H, NH, J = 6.9 Hz); 7.21 (d, 2H, aryl, J = 7.6 Hz); 7.27 (t, 2H, aryl, J = 7.5 Hz); 7.52 (t, 2H, aryl, J = 8.0 Hz); 7.67 (d, 2H, aryl, J = 8.0 Hz). HR-MS m/z calcd for C37H46N4O7S [(M + H)]+: 691.3160, found 691.3188.
Ethyl (4R)-2-{(S)-1-[({[9H-Fluoren-9-yl]methoxy}carbonyl)amino]-4-{3-[(2,2,4,6,7-pentamethyl-2,3-dihydrobenzofuran-5-yl)sulfonyl]guanidino}butyl}thiazolidine-4-carboxylate (8b)
Intermediate 8a (1.0 equiv) was dissolved in dry THF and mixed at 0 °C under nitrogen atmosphere. To this solution, 2.5 equiv of LiAlH4 (1 M in THF) were added and the reaction was mixed at 0 °C for further 10 min. Then, the crude was diluted with DCM, washed with a solution of citric acid (10% w/w), extracted, dried over anhydrous sodium sulfate, filtered, and concentrated. The obtained intermediate, without further purification, was dissolved in ethanol, then 1.2 equiv of l-Cys-OEt and 1.2 equiv of NaHCO3 were added and the reaction was mixed at room temperature overnight. The solvent was removed in vacuo, and the crude was diluted with DCM and washed with a saturated solution of NaHCO3 (3 × 20 mL). The combined organic layer was dried over anhydrous sodium sulfate, filtered, concentrated, and purified by flash chromatography using n-hexane/ethyl acetate (ratio 1/1, v/v) to furnish intermediate 8b as a colorless oil (48% yield). Rf: 0.53. 1H NMR (400 MHz, CDCl3): δ: 1.13 (t, 3H, CH3, J = 6.8 Hz); 1.38–1.46 (m, 2H, CH2); 1.49–1.57 (m, 2H, CH2); 1.97 (s, 6H, CH3); 2.06 (s, 3H, CH3); 2.41 (s, 3H, CH3); 2.48 (s, 3H, CH3); 2.86 (s, 2H, CH2); 3.05–3.12 (m, 2H, CH2); 3.72 (t, 1H, CH, J = 6.6 Hz); 3.85–3.89 (m, 1H, CH2a); 4.07 (q, 2H, CH2, J = 6.7 Hz); 4.11–4.15 (m, 3H, CH2b and CH); 4.41 (d, 2H, CH2, J = 6.3 Hz); 4.47 (d, 1H; CH, J = 6.8 Hz); 7.18 (t, 2H, aryl, J = 8.1 Hz); 7.27 (t, 2H, aryl, J = 7.8 Hz); 7.55 (t, 2H, aryl, J = 8.0 Hz); 7.68 (d, 2H, aryl, J = 8.2 Hz). HR-MS m/z calcd for C39H49N5O7S2 [(M + H)]+: 764.3146; found 764.3168.
(9H-Fluoren-9-yl)methyl {(S)-1-(3S,7aR)-6-Benzyl-5,7-dioxotetrahydro-1H,3H-imidazo[1,5-c]thiazol-3-yl-4-{3-[(2,2,4,6,7-pentamethyl-2,3-dihydrobenzofuran-5-yl)sulfonyl]guanidino}butyl}carbamate (8c)
Intermediate 8b (1.0 equiv) was dissolved in THF and added with 0.4 equiv of triphosgene, 1.2 equiv of benzylamine, and 1.2 equiv of TEA, and the mixture was refluxed for 1 h. After cooling to room temperature, the solvent was evaporated, the residue reconstituted in dichloromethane, and washed with water (3 × 20 mL). The organic phase was extracted, dried over Na2SO4, filtered, and concentrated under vacuum. Flash chromatography using n-hexane/ethyl acetate (ratio 2/3, v/v) as eluent yielded intermediate 8c (35% yield). Yellowish powder. Rf: 0.48. 1H NMR (400 MHz, CDCl3): δ: 1.48–1.57 (m, 2H, CH2); 1.62–1.68 (m, 1H, CH2a); 1.72–1.77 (m, 1H, CH2b); 2.10 (s, 6H, CH3); 2.52 (s, 6H, CH3); 2.60 (s, 3H, CH3); 2.94 (s, 2H, CH2); 2.97–3.03 (m, 1H, CH2a); 3.23 (dd, 1H, CH2b, J′ = 6.9 and J″ = 9.9 Hz); 3.32–3.40 (m, 2H, CH2); 4.05–4.11 (m, 1H, CH); 4.15–4.17 (m, 2H, CH2); 4.34 (t, 1H, CH, J = 7.1 Hz); 4.41 (d, 2H, CH2, J = 7.6 Hz); 4.77 (t, 1H, CH, J = 6.5 Hz); 5.34 (t, 1H, CH, J = 5.8 Hz); 5.65 (d, 1H, NH, J = 7.1 Hz); 7.21–7.29 (m, 7H, aryl); 7.39 (t, 2H, aryl, J = 7.2 Hz); 7.61 (t, 2H, aryl, J = 7.5 Hz); 7.76 (d, 2H, aryl, J = 8.0 Hz). HR-MS m/z calcd for C45H50N6O7S2 [(M + H)]+: 851.3255; found 851.3255.
(9H-Fluoren-9-yl)methyl [(R)-1-((3S,7aR)-6-Benzyl-5,7-dioxotetrahydro-1H,3H-imidazo[1,5-c]thiazol-3-yl)-4-guanidinobutyl]carbamate (CPK8)
1.0 equiv of 8c was dissolved in a mixture of TFA/DCM (ratio 1/3, v/v), and triisopropylsilane (TIS, 0.25 equiv) was added. The reaction mixture was stirred at room temperature for 3 h. After the completion of the reaction, monitored by TLC, a solution of K2CO3 (2 N) was added. The mixture was diluted with dichloromethane, and the organic layer was extracted, dried over Na2SO4, filtered, and concentrated in vacuo. CPK8 was obtained as a white powder without further purification step. Yield 74%. 1H NMR (400 MHz, CD3OD): δ: 1.41–1.57 (m, 2H, CH2); 1.66–1.69 (m, 1H, CH2a); 1.77–1.80 (m, 1H, CH2b); 3.01 (dd, 1H, CH2a, J′ = 8.3 and J″ = 10.6 Hz); 3.13–3.21 (m, 2H, CH2); 3.30–3.37 (m, 1H, CH2b); 3.78 (t, 1H, CH, J = 7.1 Hz); 4.23 (t, 1H, CH, J = 6.1 Hz); 4.39 (dd, 1H, CH2a, J′ = 6.5 and J″ = 10.6 Hz); 4.47 (t, 1H, CH2b, J = 7.8 Hz); 4.55–4.63 (m, 3H, CH2 and CH); 5.25 (d, 1H, CH, J = 6.8 Hz); 7.26–7.42 (m, 9H, aryl); 7.66 (d, 2H, aryl, J = 7.0 Hz); 7.79–7.82 (m, 2H, aryl). 13C NMR (100 MHz, CD3OD): δ: 24.9; 28.5; 31.8; 40.6; 42.2; 55.2; 65.0; 66.1; 66.9; 119.6; 124.7; 124.75; 126.8; 127.4; 127.6; 127.7; 128.3; 135.8; 141.3; 143.8; 157.2; 157.5; 158.6; 171.4. HR-MS m/z calcd for C32H34N6O4S [(M + H)]+: 599.2435; found 599.2418.
(2S′) and (2R′)-N-{3-[1-(Cyclohexanecarbonyl)-5-methyl-2,4′-dioxospiro[indoline-3,2′-thiazolidin]-3′-yl]propyl}-2,3,4,5,6-pentafluorobenzamide (CPK9)
The compound was synthesized according to the procedure previously described.53 NMR and mass spectra were in accordance with those reported, while UHPLC traces are reported in the Supporting Information.
tert-Butyl {1-[(4-Chlorophenyl)sulfonyl]piperidin-4-yl}carbamate (10a)
Obtained from 4-(N-Boc-amino)piperidine and 4-chlorobenzenesulfonyl chloride following the same procedure reported for 5a. FC n-hexane/ethyl acetate 4/1. Rf: 0.44. Whitish powder (85% yield). 1H NMR (400 MHz, CDCl3): δ: 1.43 (s, 9H, CH3); 1.49–1.55 (m, 2H, CH2); 2.00 (d, 2H, CH2, J = 10.8 Hz); 2.48 (t, 2H, CH2, J = 11.4 Hz); 3.42 (bs, 1H, CH); 3.70 (d, 2H, CH2, J = 10.2 Hz); 4.43 (bs, 1H, NH); 7.53 (d, 2H, aryl, J = 8.5 Hz); 7.71 (d, 2H, aryl, J = 8.6 Hz). HR-MS m/z calcd for C16H23ClN2O4S [(M + H)]+: 375.1140; found 375.1132.
1-[(4-Chlorophenyl)sulfonyl]piperidin-4-amine (10b)
Synthesized from 11 following the same procedure described for 5b. Precipitated from MeOH/diethyl ether. White powder (90% yield). 1H NMR (400 MHz, CDCl3): δ: 1.37–1.47 (m, 2H, CH2); 1.85 (d, 2H, CH2, J = 10.5 Hz); 2.44 (t, 2H, CH2, J = 11.8 Hz); 2.63–2.69 (m, 1H, CH); 3.65 (d, 2H, CH2, J = 11.9 Hz); 7.49 (d, 2H, aryl, J = 8.3 Hz); 7.69 (d, 2H, aryl, J = 8.3 Hz). HR-MS m/z calcd for C16H23ClN2O4S [(M + H)]+: 275.0616; found 275.0634.
tert-Butyl (S)-{3-[4-(tert-Butoxy)phenyl]-1-{[1-((4-chlorophenyl)sulfonyl)piperidin-4-yl]amino}-1-oxopropan-2-yl}carbamate (10c)
Synthesized from 10b and Boc-l-Tyr(tBu)–OH as described for CPK5. FC in n-hexane/ethyl acetate (ratio 3/2, v/v). Whitish oil (55% yield). 1H NMR (400 MHz, CDCl3): δ: 1.31 (s, 9H, CH3); 1.43 (s, 9H, CH3); 1.48–1.53 (m, 2H, CH2); 1.78 (d, 1H, CH, J = 8.2 Hz); 1.88 (d, 1H, CH, J = 11.0 Hz); 2.38–2.51 (m, 2H, CH2); 2.85–2.91 (m, 1H, CH); 2.98–3.03 (m, 1H, CH); 3.61–3.67 (m, 3H, CH, CH2); 4.17 (t, 2H, CH2, J = 11.8 Hz); 5.76 (d, 1H, NH, J = 7.8 Hz); 6.76 (d, 2H, aryl, J = 8.2 Hz); 7.02 (d, 2H, aryl, J = 8.2 Hz); 7.54 (d, 2H, aryl, J = 8.5 Hz); 7.69 (d, 2H, aryl, J = 8.6 Hz). HR-MS m/z calcd for C29H40ClN3O6S [(M + H)]+: 594.2399; found 594.2381.
tert-Butyl (S)-{1-[(1-[(4-Chlorophenyl)sulfonyl)piperidin-4-yl]amino]-3-(4-hydroxyphenyl)-1-oxopropan-2-yl}carbamate (10d)
Intermediate 10c was dissolved in a mixture of TFA/DCM (ratio 1/5, v/v), and triisopropylsilane (TIS, 0.25 equiv) was added. The reaction was stirred at room temperature for 2 h. After the completion of the reaction, a solution of K2CO3 (2N) was added. Then, the mixture was diluted with dichloromethane, and the organic layer was extracted, dried over Na2SO4, filtered, and concentrated in vacuo. The obtained intermediate was used in the next step without further purification. FC in n-hexane/ethyl acetate (ratio 1/4, v/v). Rf: 0.48. Whitish powder (77% yield). 1H NMR (400 MHz, CDCl3): δ: 1.42 (s, 9H, CH3); 1.50–1.54 (m, 2H, CH2); 1.77 (d, 1H, CH, J = 8.2 Hz); 1.90 (d, 1H, CH, J = 11.0 Hz); 2.39–2.52 (m, 2H, CH2); 2.87–2.93 (m, 1H, CH); 2.99–3.05 (m, 1H, CH); 3.63–3.69 (m, 3H, CH, CH2); 4.19 (t, 2H, CH2, J = 11.8 Hz); 5.77 (d, 1H, NH, J = 7.8 Hz); 6.78 (d, 2H, aryl, J = 8.2 Hz); 7.01 (d, 2H, aryl, J = 8.2 Hz); 7.55 (d, 2H, aryl, J = 8.5 Hz); 7.68 (d, 2H, aryl, J = 8.6 Hz). HR-MS m/z calcd for C29H40ClN3O6S [(M + H)]+: 538.1773; found 538.1789.
tert-Butyl (S)-{3-[4-((4-Bromobenzyl)oxy)phenyl]-1-{[1-((4-chlorophenyl)sulfonyl)piperidin-4-yl]amino}-1-oxopropan-2-yl}carbamate (CPK10)
Obtained from 10d and 4-bromobenzyl bromide as reported for 2a. FC in n-hexane/ethyl acetate (ratio 6/4, v/v). Rf: 0.42. White powder (82% yield). 1H NMR (400 MHz, CDCl3): δ: 1.17–1.26 (m, 3H, CH and CH2); 1.33 (s, 9H, CH3); 1.55 (bs, 1H, CH); 1.73 (d, 1H, CH2a, J = 11.8 Hz); 1.82 (d, 1H, CH2b, J = 11.8 Hz); 2.31–2.39 (bs, 2H, CH2); 2.85 (dd, 1H, CH2a, J′ = 7.5 and J″ = 13.6 Hz); 2.93 (dd, 1H, CH2b, J′ = 5.8 and J″ = 13.6 Hz); 3.54–3.60 (bs, 3H, CH2 and CH); 4.06–4.12 (m, 1H, CH, J = 6.8 Hz); 4.91 (bs, 1H, NH); 5.04 (s, 2H, CH2); 5.56 (d, 1H, NH, J = 7.7 Hz); 6.81 (d, 2H, aryl, J = 8.1 Hz); 7.02 (d, 2H, aryl, J = 8.0 Hz); 7.43–7.49 (m, 4H, aryl); 7.59 (t, 4H, aryl, J = 7.9 Hz). 13C NMR (100 MHz, CDCl3): δ: 28.3, 29.7, 31.2, 37.6, 45.1, 45.7, 56.2, 69.2, 115.0, 125.6, 127.4, 129.0, 129.5, 130.4, 134.7, 139.5, 141.0, 157.5, 170.6. HR-MS m/z calcd for C33H37ClF3N3O6S [(M + H)]+: 696.2116; found 696.2094.
N,N′-Dibenzyl-2-{2-[(2,4-dinitrophenyl)thio]-1H-indol-3-yl}ethanamine (CPK11)
The compound was synthesized according to the procedure previously described.54 NMR and mass spectra were in accordance with those reported, while UHPLC traces are reported in the Supporting Information.
N-[1-(4-Fluorobenzoyl)indolin-5-yl]cyclohexanesulfonamide (CPK12)
The compound was synthesized according to the procedure previously described.51 NMR and mass spectra were in accordance with those reported, while UHPLC traces are reported in the Supporting Information.
1-{[(2-(1H-Indol-3-yl)ethyl]amino)methyl}naphthalen-2-ol (CPK13)
The compound was synthesized according to the procedure previously described.55 NMR and mass spectra were in accordance with those reported, while UHPLC traces are reported in the Supporting Information.
N-{[3-({[2-([1,1′-Biphenyl]-4-yl)ethyl](methyl)amino}methyl)-1-methyl-1H-indol-5-yl]methyl}-4-fluoroaniline (CPK14)
The compound was synthesized according to the procedure previously described.56 NMR and mass spectra were in accordance with those reported, while UHPLC traces are reported in the Supporting Information.
(3S,7aR)-1′-(4-Chlorobenzoyl)-5′-methyl-6-(3,4,5-trimethoxybenzyl)-1H-spiro[imidazo[1,5-c]thiazole-3,3′-indoline]-2′,5,7(6H,7aH)-trione (CPK15) and (2′S,4′R)-Ethyl 5-bromo-3′-(4-chlorobenzoyl)-2-oxospiro[indoline-3,2′-thiazolidine]-4′-carboxylate (CPK16)
The compounds were synthesized according to the procedure previously described.57 NMR and mass spectra were in accordance with those reported, while UHPLC traces are reported in the Supporting Information.
(R)- and (S)-1-Phenyl-2-(1-phenyl-3,4-dihydro-1H-pyrido[3,4-b]indol-2(9H)-yl)ethenone (CPK17)
The compound was synthesized according to the procedure previously described.54 NMR and mass spectra were in accordance with those reported, while UHPLC traces are reported in the Supporting Information.
(S)-tert-Butyl[1-(benzylamino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl]carbamate (18a)
Synthesized as described for CPK5, starting from Boc-l-Trp-OH and benzylamine.
FC in n-hexane/ethyl acetate (ratio 7/3, v/v). Rf = 0.45. Yellowish oil (80% yield). 1H NMR (400 MHz, CD3OD): δ: 1.40 (s, 9H, CH3); 3.10 (dd, 1H, CH2a, J′ = 6.6 and J″ = 13.8 Hz); 3.25 (dd, 1H, CH2b and J′ = 6.9, J″ = 14.4 Hz); 4.19–4.23 (m, 1H, CH); 4.34–4.41 (m, 2H, CH2); 7.02–7.06 (m, 5H, aryl); 7.12 (t, 1H, aryl, J = 7.1 Hz); 7.20–7.25 (m, 2H, aryl); 7.37 (d, 1H, aryl, J = 8.1 Hz); 7.62 (d, 1H, aryl, J = 7.8 Hz). HR-MS m/z calcd for C23H28N3O3 [(M + H)]+: 394.2125; found 394.2118.
(S)-2-Amino-N-benzyl-3-(1H-indol-3-yl)propanamide (18b)
Intermediate 18b was obtained starting from 18a, following the same procedure described for 3b. White powder (95% yield). 1H NMR (400 MHz, CD3OD): δ: 3.05 (dd, 1H, CH2a, J′ = 5.8, J″ = 10.4 Hz); 3.20 (dd, 1H, CH2b, J′ = 6.0 and J″ = 10.4 Hz); 3.69 (t, 1H, CH, J = 5.2 Hz); 4.25 (d, 1H, CH2a, J = 12.6 Hz); 4.35 (d, 1H, CH2b, J = 12.6 Hz); 7.02–7.07 (m, 5H, aryl); 7.12 (t, 1H, aryl, J = 7.2 Hz); 7.20–7.26 (m, 2H, aryl); 7.39 (d, 1H, aryl, J = 8.2 Hz); 7.64 (d, 1H, aryl, J = 8.0 Hz). HR-MS m/z calcd for C18H20N3O [(M + H)]+: 294.1601; found 294.1608.
(S)-N-Benzyl-3-(1H-indol-3-yl)-2-[(4-phenoxybenzyl)amino]propanamide (CPK18)
CPK18 was synthesized starting from 18b and 4-phenoxybenzaldehyde, following the same procedure applied to 2b. FC in n-hexane/ethyl acetate (ratio 1/4, v/v). Rf = 0.44. Colorless oil (72% yield). 1H NMR (400 MHz, CDCl3): δ: 3.04 (dd, 1H, CH2a, J′ = 9.0 and J″ = 14.4 Hz); 3.41 (dd, 1H, CH2b, J′ = 3.7 and J″ = 14.5 Hz); 3.55–3.59 (m, 2H, CH); 3.69 (d, 1H, CH, J= 13.3 Hz); 4.48 (d, 1H, CH2, J = 5.7 Hz); 6.83 (d, 2H, aryl, J = 8.2 Hz); 6.94–6.99 (m, 5H, aryl); 7.10–7.16 (m, 2H, aryl); 7.22 (d, 2H, aryl, J = 7.2 Hz); 7.29–7.40 (m, 6H, aryl); 7.61 (bs, 1H, aryl); 7.68 (d, 1H, aryl, J = 7.8 Hz); 8.15 (bs, 1H, NH). 13C NMR (100 MHz, CDCl3): δ: 29.0, 43.1, 52.1, 62.4, 111.2, 111.5, 118.8, 118.9, 119.8, 122.4, 122.9, 123.2, 127.4, 127.5, 127.7, 128.7, 129.3, 129.7, 134.2, 138.5, 156.2, 157.3, 174.0. HR-MS m/z calcd for C31H29N3O2 [(M + H)]+: 476.2333; found 476.2356.
(1R,3S)-N-(4-Fluorobenzyl)-1-(4-fluorophenyl)-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (CPK19)
The compound was synthesized according to the procedure previously described.50 NMR and mass spectra were in accordance with those reported, while UHPLC traces are reported in the Supporting Information.
N-{1-[2,4-Bis(trifluoromethyl)benzyl]indolin-5-yl}cyclohexanesulfonamide (CPK20)
The compound was synthesized according to the procedure previously described.51 NMR and mass spectra were in accordance with those reported, while UHPLC traces are reported in the Supporting Information.
hKCNT198–354 Model Building
The sequence of human KCNT1 (accession code: Q5JUK3) was downloaded from UniProt81 and the 98–354 region was extracted and aligned, using ClustalW,82 to the Cryo-EM structure of chicken Slo2.2 in open conformation. (PDB ID: 5u70).12 Alignment metrics resulted as follows: score = 450 bits (1158); query cover = 100%; e-value = 3e-150; identities = 225/257 (88%); positives = 240/257 (93%); gaps = 0/257 (0%). hKCNT198–354 was then modeled as a tetramer by homology using the knowledge-based method implemented in Prime.83,84 Side chains were optimized, with the exception of those belonging to conserved residues, whose rotamers were retained from the template structure. Residues not obtained from the templates were minimized. The tetramer was then submitted to the Protein Preparation utility85 to cap the protein termini by ACE and NMA residues, to optimize the H-bond network, and to finally relax the system by a short, restrained minimization that was stopped when the convergence criterion (0.30 Å RMSD for heavy atoms) was reached.
Ligands Preparation
Quinidine structure was downloaded from PubChem (PubChem CID 441074)86 and together with the in-house library was submitted to the LigPrep tool87 to generate low-energy three-dimensional (3D) structures and account for ionization and tautomerization states at the pH range of 6–8.
Metadynamics Simulations
Quinidine was randomly placed into the intracellular pore vestibule of the hKCNT198–354 tetramer and the simulated environment was set and run through Desmond.88 The complex was inserted into a POPC bilayer based on the coordinates of the 5u70 entry from the OPM database.89 Solvation was treated explicitly using the TIP3P water molecule.90 The system was neutralized by Na+ and Cl– ions, which were added to a final concentration of 0.15 M. OPLS 200591 was used as force field. Two collective variables were defined for the metadynamics simulation: CV1—quinidine RMSD (repulsive Gaussian potential width: 0.1 Å); CV2—distance between the centers of mass of quinidine and of the four F346 residues (repulsive Gaussian potential width: 0.05 Å—walled at 10 Å). Prior to the metadynamics simulation, the system was equilibrated using the default stepwise protocol for membrane systems distributed with Desmond. After system equilibration, a 192 ns-long metadynamics simulation was run in the NPγT ensemble, using a Nosé–Hoover chain thermostat (300 K) and a Martyna–Tobias–Klein barostat (1 atm). Time steps were set to 2, 2, and 6 fs for bonded, near, and far interactions, respectively. The repulsive Gaussian potential was added every 0.18 ps and its height was set to 0.01 kcal/mol. The 50 lowest-energy conformations from the metadynamics simulation were used to compose a docking target ensemble.
Virtual Screening
For each of the 50 above-mentioned target structures, a cubic Glide docking grid was centered on the centroid of the four F346 residues and its volume was set to 64,000 Å3, requiring the diameter midpoint of each docking ligand to stay within a smaller, nested 27,000 Å3 cubic grid. For each target structure, docking simulations were carried out using previously reported stepwise protocols92,93 in which the ligands were first docked flexibly using Glide SP,94,95 and each docking pose was then refined and rescored using Glide XP.96 All of the docking poses were then sorted by docking score retaining, for each ligand, only the top scoring docking result, regardless of its ionization and tautomeric state and of the target structure it was retrieved by. The top scoring 241 compounds, i.e., those showing a docking score < −10, were then advanced to the final selection task. Docking scores of the top scoring 241 compounds ranged from −13.432 to −10.536.
Diversity-Based Selection
By means of the Canvas software,97,98 MOLPRINT2D 64-bit binary fingerprints were generated for the above-described 241 top scoring compounds list. Fingerprints were used to calculate a Soergel distance matrix that in turn was used to select 20 diverse compounds by the sphere exclusion (size = 0.5) method.
EMhKCNT1110–354 Preparation
Residues 110–354 from PDB entry 8HKQ,62 containing the six transmembrane helices of KCNT1, were submitted in tetramer form to the Protein Preparation utility85 to cap the protein termini by ACE and NMA residues, to optimize the H-bond network and to finally relax the system by a short, restrained minimization that was stopped when the convergence criterion (0.30 Å RMSD for heavy atoms) was reached.
Induced-Fit Docking on EMhKCNT1110–354
Extended sampling protocol of Schrodinger induced-fit docking99 was used to dock QND and CPK series compound into the prepared EMhKCNT1110–354 structure. The docking space for the first stage was centered on the experimental bound conformation of the native ligand and defined as a (30 Å)3 cubic box containing a smaller, nested (20 Å)3 cubic box where the ligand diameter midpoint was required to stay. In the second docking stage, the inner box volume was instead set to (125 Å)3. No constraints were applied. Ring conformations were sampled in a 2.5 kcal/mol energy window. Residues within 5 Å of ligands poses were refined using implict membrane treatment based on the coordinates of the 5u70 entry from the OPM database.89 The best scoring predicted complex for each ligand was advanced to the MD studies.
Molecular Dynamics Simulations of Ligand/HMhKCNT198–354 and Ligand/EMhKCNT1110–354 Complexes
The best scoring ligand/KCNT1 complexes from the virtual screening and from the induced-fit docking were set and simulated using Desmond.88 The complexes were inserted into a POPC bilayer based on the coordinates of the 5u70 entry from the OPM database.89 Solvation was treated explicitly using the TIP3P water model.90 The systems were neutralized by Na+ and Cl– ions, which were added to a final concentration of 0.15 M. OPLS 200591 was used as force field. Prior to the production stages, the systems were equilibrated using the default stepwise protocol for membrane systems distributed with Desmond. After system equilibration, molecular dynamics simulations were run in the NPγT ensemble, using a Nosé–Hoover chain thermostat (300 K) and a Martyna–Tobias–Klein barostat (1 atm). Time steps were set to 2, 2, and 6 fs for bonded, near, and far interactions, respectively. In the production stages, ligand-bound HMhKCNT198–354 structures were simulated for 120 ns, QND/EMhKCNT1110–354 complexes for 960 ns, and CPKs/EMhKCNT1110–354 complexes for 480 ns.
Generation of Stable Cell Lines and Fluorescence-Based Assay
Transfection
The KCNT1-stable cell line was generated using a plasmid-based PiggyBac transposon system. cDNA for KCNT1 was cloned into the PiggyBac expression vector (pB-CMV-MCS-EF1α-RedPuro, System Biosciences) and transfected, together with a plasmid encoding for the transposase (System Biosciences), into CHO cells using Lipofectamine 2000 (Invitrogen) as indicated in the protocol provided by the seller company.
Clone Selection and Characterization
An initial screening of the clones was based on two selection markers: puromycin resistance and red fluorescence. Puromycin (Sigma) at a concentration of 4 μg/mL was added to the medium 24 h after transfection. After 6 days in plate, clones were serially diluted into a 96-well. After a week, cells showing the strongest red fluorescence were selected, trypsinized (Trypsin 0.25%, Gibco), and plated into Petri dishes, obtaining seven different clones. Cells were grown in DMEM supplemented with 10% FBS, 50 U/mL Pen-Strep, 2 mM l-glutamine (all purchased from Gibco), and 4 μg/mL Puromycin (Sigma) in a humidified atmosphere at 37 °C with 5% CO2. A subsequent screening was made using the electrophysiological patch clamp technique to identify a clone exhibiting biophysical properties similar to those shown by CHO cells transiently transfected with the KCNT1 cDNA. Transiently transfected cells generated a voltage-dependent K+ selective current with a current density at +40 mV of 38.0 ± 14.8 pA/pF (n = 5). Among five clones, clone number 4, showing a current density at +40 mV of 29.4 ± 7.6 pA/pF (n = 8, p < 0.05), was selected.
Fluorescence-Based Assay
The selected clone stably expressing KCNT1 channels was grown in DMEM supplemented with 10% FBS, 50 U/mL Pen-Strep, 2 mM l-glutamine (all purchased from Gibco), 4 μg/mL puromycin (Sigma) in a humidified atmosphere at 37 °C with 5% CO2. To perform the FluxOR II Green Potassium Ion Channel Assay (Invitrogen), the cells were seeded in 96-well Biocoat Poly-d-Lysine Cellware White/Clear Plate (Corning) at a density of 1.6 × 104 cells/well in 80 μL of medium. 24 h after seeding, the FluxOR Assay was performed, following the “Wash method”, and the “modified Assay Buffer”, as described in the manufacturer’s protocol. Cells were incubated with KCNT1 blockers (QND and CPKs) diluted in modified Assay Buffer, for 15 min, followed by incubation of 15 min with LOX 10 μM, at room temperature. Then, the plate was moved into a microplate reader (FLUOstar Optima) to read the plate, setting the excitation filter at 485 nm and the emission filter at 520 nm; “Stimulus buffer” (2 mM Tl+) prepared as indicated in the protocol was automatically added to wells after 5 s of recording; the plate was read every 1 s, for 30 s. The results were analyzed using Optima Data Analysis and Microsoft Excel.
Electrophysiological Experiments
Cell Culture and Transfection
Chinese Hamster ovary (CHO) or human embryonic kidney (HEK) cells were grown in Petri dishes in DMEM (Dulbecco’s minimum Eagle’s medium) supplemented with 10% fetal bovine serum (FBS) decomplemented at 56 °C for 30 min, 1% l-glutamine (2 mM in 0.85% NaCl), 1% penicillin (50 U/mL), and streptomycin (50 μg/mL) in a humidified atmosphere at 37 °C with 5% CO2. Channel subunits were expressed by transient transfection, using plasmids containing cDNAs encoding for human KCNT1, human hERG, or human Kv7.2 in CHO cells; by contrast, those encoding for human KCNT2 was expressed in HEK cells. For transient transfection, Lipofectamine 2000 (Invitrogen, Milan, Italy) was used, as described.100 Total cDNA in the transfection mixture was kept constant at 4 μg for electrophysiological experiments and a plasmid encoding the Enhanced Green Fluorescent Protein (eGFP) was used as a transfection marker.
Mutagenesis
Mutations were engineered in a plasmid containing the cDNA for a myc-DDK tagged human isoform 2 (Q5JUK3–2) of KCNT1 (RC214820; Origene, Rockville, MD) by Quick-change mutagenesis, as described.101 Mutant vectors were all verified by Sanger sequencing.
Whole-Cell Electrophysiology
Macroscopic currents from transiently transfected CHO or HEK cells were recorded at room temperature (20–22 °C) 24 h after transfection, with an Axopatch 200B amplifier (Molecular Devices, Union City, CA) using the whole-cell configuration of the patch-clamp technique. The pipet (intracellular) solution contained (in mM): 130 KCl, 10 NaCl, 10 HEPES, 5 EGTA, 5 Mg-ATP, pH 7.3–7.4 with HCl (ATP was omitted when KCNT2 currents were measured). Extracellular solution composition, as well as data acquisition and analysis were as previously described.34 For hERG and Kv7.2 channels recording the extracellular solution contained (in mM) 138 NaCl, 2 CaCl2, 5.4 KCl, 1 MgCl2, 10 glucose, and 10 HEPES, at pH 7.4, with NaOH. The pipet (intracellular) solution contained (in mM) 140 KCl, 2 MgCl2, 10 EGTA, 10 HEPES, 5Mg2+-ATP, at pH 7.3–7.4, with KOH. Current densities (expressed in pA/pF) were calculated as peak K+ currents at all tested membrane potentials divided by cell capacitance (C). QND and CPK compounds (Sigma-Aldrich, Milan, Italy) were dissolved in DMSO (final vehicle concentration ≤0.3%). In each experiment, the same volume of DMSO used to dissolve each drug to be tested was added to the control solution. Drugs were perfused (each cell was exposed to only one drug concentration, to avoid cumulative block) using a fast solution exchange system.100
Statistics
Whole-Cell Electrophysiology
Statistically, significant differences in electrophysiological data were evaluated with the Student t test, or with ANOVA followed by the Student–Newman–Keuls test when multiple groups were compared, with the threshold set at p < 0.05. Data were analyzed using the SigmaPlot 12.3 for Windows (Systat Software, Inc., San Jose, CA). Values are expressed as the mean ± standard error of the mean (SEM) of at least three cells recorded in at least three independent transfections.
Fluorescence-Based Assay
Assay robustness was determined according to the Z′ factor.61
where SD is the standard deviation of triplicate in a single experiment, AV is the average of triplicate in a single experiment, LOX is loxapine at a concentration of 10 μM; Vehicle is the Assay buffer prepared as indicated in the FluxOR protocol +0.1% DMSO.
Only experiments resulting in a Z′ > 0.5 were considered. Data shown (Figure 2B–E) were obtained from at least three independent experiments. Values are expressed as the mean ± SEM. Slope of the fluorescent curves was calculated from point at second 5 to point at second 15 (as shown in Figure 3A) in Microsoft Excel. Data were analyzed using the GraphPad Prism 8.0.2 (GraphPad Software, LaJolla, CA). Statistically, significant differences in the initial slope of the curves were evaluated through ordinary one-way ANOVA, multiple comparisons were corrected with the Dunnett test. The threshold of p < 0.05 is indicated in figures as asterisk. The slope values of the curves, were plotted versus log(concentration) of the compound, fitted to a four-parameter logistic equation, and EC50 values were calculated with SigmaPlot (version 12.3). Indicated EC50/IC50 values are the mean of at least three independent experiments ± standard error of the mean (SEM).
Instrumentation and Analytical Conditions for Pharmacokinetic Analyses
LC-MS/MS analyses were performed on a Vanquis UHPLC system connected online to an Orbitrap Exploris 120 mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) equipped with a heated electrospray ionization probe (HESI II). All additives and mobile phases were LC/MS grade and purchased from Merck (Milan, Italy).
The separation was performed on a Kinetex 2.6 μm Evo C18 100 Å column (50 mm × 2.1 mm, Phenomenex, Bologna, Italy) employing as mobile phases: (A) H2O and (B) ACN, both acidified with 0.1% formic acid with the following gradient: 0.01–5.00 min, 5–85% B; isocratic to 85% B for 1 min; 6–6.50 min, 85–5% B; then 1.5 min for ri-equilibration analytical column. The flow rate and column oven were set at 0.4 mL min–1 and 45 °C, respectively.
The ESI was operated in positive mode. The MS was calibrated by Thermo Pierce FlexMix calibration solutions in both polarities. Full MS parameters: Orbitrap Resolution: 120,000; Scan Range (m/z): 100–1500; RF Lens (%): 70; Normalized AGC Target (%): 200; Maximum Injection Time (ms): 200. Data-dependent MS/MS: Orbitrap Resolution 15,000; Isolation Window (m/z): 2; Collision Energy Type: Normalized; HCD Collision Energy (%): 30. Ion source parameters: Sheath gas pressure: 60 arbitrary units; auxiliary gas flow: 15 arbitrary units; Sweep Gas: 2 arbitrary units; Ion Transfer Tube Temp (°C): 300; Vaporizer Temp (°C): 300; Spray voltage, +3.4 kV, −3.0 kV.
For the calibration curves, the primary stock solutions (10 mM) were prepared in DMSO. The intermediate stock solutions (0.1 mM) and the working standard solutions were prepared by serial dilution of the stock solutions in methanol to obtain necessary concentrations (Table S2). Tolbutamide was used as the internal standard (IS, 1 μM). Quantitation of compounds CPK18 and CPK20 was performed using linear regression of the response ratios (peak area analyte/peak area IS) obtained from the calibration curve to calculate the corresponding amount.
The method was validated in terms of specificity and selectivity, LOD, and LOQ (Table S2). Specificity and selectivity parameters were evaluated by analyzing blank matrix samples from different batches to assess the potential interference of endogenous components in the samples. Chromatograms of these blank matrix samples were compared with chromatograms of matrix samples spiked with a very low concentration of the compound of interest. Limits of detection (LOD) and quantification (LOQ) were calculated by using the standard deviation (SD) and the slope of the calibration curve, multiplied by 3.3 and 10, respectively.
Human Plasma Stability
The plasma stability of compounds CPK16, CPK18, and CPK20 was evaluated. Briefly, plasma is warmed to 37 °C for 10 min, mixed, and centrifuged to pellet any aggregated protein. Plasma is equilibrated to 37 °C and biotransformation is initiated by the addition of compound solution and mixing. At each specified time point (0, 30, 60, 90, and 120 min), test compounds were removed into 200 μL ice-cold methanol to stop degradation. Internal standard was added during the quenching phase. Finally, the concentration of test compound was monitored by LC-MS. The percentage of test compound remaining (relative to the 0 min time point) at the individual time points is reported (Figure S47). All experiments were performed in duplicate. Procaine was used as control.
In Vitro Drug Metabolism Using Human Liver Microsomes
For the liver microsomal stability assay, 25 μL of 5 mg/mL human (CD-1) microsomes (HLM, Thermo Fisher Scientific, Bremen, Germany) were preincubated with alamethicin. The reaction was initiated by adding 50 μL of a mixture containing NADPH 10 mM and UDPGA 10 mM. The incubation was carried out at 37 °C for 15, 30, 45, and 60 min in a Thermomixer comfort (Eppendorf, Hamburg, Germany).
The reaction was stopped by the addition of 200 μL of ice-cold methanol containing IS. Subsequently, samples were centrifuged at 14,000 rpm at 4 °C for 7 min (Eppendorf microcentrifuge 5424, Hamburg, Germany). The supernatants were collected and injected in LC-MS. The control at 0 min was established by adding the organic solvent immediately after incubation with microsomes. Testosterone, 7-hydroxyl-coumarin, and 3-(α-acetonylbenzyl)-4-hydroxycoumarin were employed as positive controls. A negative control was prepared by incubating for up to 60 min without UDPGA/NADPH cofactors. The extent of metabolism is quantified as a percentage of the parent compound turnover. The results were expressed in terms of in vitro microsome half-lives (t1/2), in vitro intrinsic clearance (CLintin vitro), and intrinsic in vivo clearance (CLintin vivo). The in vitro half-lives (t1/2) were calculated using the expression t1/2 = 0.693/b, where b is the slope found in the linear fit of the natural logarithm of the fraction remaining of the parent compound vs incubation time. In vitro intrinsic clearance was calculated as CLintin vitro = (1000) × (0.693/t1/2)/0.5. The intrinsic in vitro clearance was scaled to the intrinsic in vivo clearance (CLintin vivo) using human physiology-based scaling factor (PBSF): CLintin vivo = CLintin vitro × PBSF (microsome protein/gram liver: 32 × gram liver/kg b.w.: 25.7).101 All experiments were performed in duplicate. The identification of metabolites generated after liver microsomes incubation was conducted using a node-based processing workflow custom-built in Compound Discoverer software v.3.3 (Thermo Fisher Scientific). The workflow subtracted chemical background using blank samples, performed retention time alignment (0.2 min), and detected expected compounds and biotransformation products (mass tolerance: 5 ppm) with resolution-aware isotope pattern matching. Mass defect filter and Fragment ion search (FISh) Scoring were applied. Each annotation was further manually reviewed considering HCD spectra, molecular formula, isotopic pattern, and FISh coverage.
Plasma Protein Binding
A Rapid Equilibrium Dialysis Plate (Pierce RED, Thermo Scientific) was used to determine the percentage of compound binding to plasma proteins.
In the RED plate, potassium phosphate buffer (350 μL, 0.1 M, pH 7.4, 37 °C) was placed in every white well and each plasma/compound (10 μM) mixture was added to red wells. The RED Plate was sealed and incubated at 37 °C on an orbital shaker at 300 rpm for 4 h. 50 μL each of postdialysis samples from the buffer and the plasma chambers were pipetted into separate microcentrifuge tubes. To create a uniform matrix, 50 μL of plasma was added to the buffer sample, and an equal volume of buffer was added to the collected plasma/sample. Precooled internal standard was added to the assay plate to quench the reaction. The compounds were quantified by LC-MS/MS. The percentage of free and bound compounds was calculated using the following equations: % free = (concentration buffer chamber/concentration plasma chamber) × 100, and % bound = 100 – % free.102 All experiments were performed in triplicate. Atenolol was used as positive control.
Acknowledgments
This project received funding from the Italian Ministry for University and Research (MUR) in the frame of the Next Generation EU National Recovery and Resilience Plan for the following projects: (1) PRIN2022PNRR, project P2022YYTKF—Targeting rare epileptic encephalopathies: design and preclinical development of KCNT1 modulators—TELOS to C.O., A.B., and I.G.-M.; (2) PRIN2022PNRR, project P2022FJXY5—KCNT potassium channels variants in severe epilepsies: identification of new pathogenic mechanisms and assessment of novel molecular and pharmacological strategies as precision-medicine approaches to M.V.S. and F.M.; (3) PRIN2022PNRR, project P2022ZANRF—Pharmacological targeting G protein-modulated potassium channels in a genetic model of absence epilepsy to M.T.; M.T. received funding from MUR in the frame of the Next Generation EU National Recovery and Resilience Plan, within the project “A multiscale integrated approach to the study of the nervous system in health and disease” (MNESYS), Mission 4 “Education and research”, Component 2 “From research to firms”, Investment line 1.3 “Enlarged partnerships extended to universities, research centres, enterprises and funding research projects”. The present work was also supported by MUR for the project PRIN2022, 2022M3KJ4N – Kv7 channels in pain: pathophysiological mechanisms, peripheral biomarkers, and treatment opportunities to M.T., and by the following Ricerca Finalizzata (RF) Projects from the Italian Ministry of Health (MoH): RF-2019-12370491 to M.T., and PNRR-MR1-2022-12376528 to M.T. and M.V.S., C.O. received funding from the University of Salerno (FARB2023 project ORSA238219). C.O. and M.T. also received funding from the Regione Campania, within PON FESR 2021–2027 fundings, with the project “Approcci innovativi allo studio di patogenesi, modelli, terapie e percorsi assistenziali in malattie rare neuropsichiatriche, neuromuscolari e sensoriali—NEURORARE”. NVIDIA Corporation is gratefully acknowledged for its support with the donation of the GPUs used for this research to CO.
Glossary
Abbreviations List
- ACE
acetyl group
- ACN
acetonitrile
- ADSHE
autosomal dominant sleep-related hypermotor (hyperkinetic) epilepsy
- APs
action potentials
- CHO
Chinese Hamster ovary
- Cryo-EM
cryo-electron microscopy
- DEE
developmental epileptic encephalopathies
- DIPEA
N,N-diisopropylethylamine
- DMEM
Dulbecco’s modified Eagle’s medium
- EGTA
ethylene glycol tetraacetic acid
- EH
epoxide hydrolase
- EIMFS
epilepsy of Infancy with migrating focal seizures
- FBS
fetal bovine serum
- FC
flash chromatography
- FISh
fragment ion search
- FMOs
flavin monooxygenases
- HBTU
2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- HCD
higher-energy collisional dissociation
- HEPES
N-2-hydroxyethylpiperazine-N-2-ethanesulfonic acid
- HESI II
heated electrospray ionization probe
- HLMs
human liver microsomes
- HOBt
hydroxybenzotriazole
- IKNa
Na+-dependent K+ currents (IKNa)
- KCTN
potassium sodium-activated channel subfamily T
- Kv
voltage-gated potassium channels
- LOD
limits of detection
- LOQ
limits of quantification
- LOX
loxapine
- NMA
N-methylacetamide
- NP γT
constant value of surface tension γ
- AGC
automatic gain control
- OPM
orientations of proteins in membranes
- Pbsf
pre-B cell growth stimulating factor human
- POPC
1-palmitoyl-2-oleoyl-glycero-3-phosphocholine
- QND
quinidine
- RCK
regulation of conductance of potassium
- RED
rapid equilibrium dialysis
- sAHP
slow hyperpolarisation
- TIP3P
transferable intermolecular potential with 3 points
- TIS
triisopropylsilane
- VEH
vehicle
- μW
microwaves
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.4c00268.
Chemical structures and characterizing chemical-physical parameters of the tested molecules (Table S1); NMR and CD spectra of the newly synthesized compounds (Figures S1–S22); HPLC traces of all of the tested compounds (Figures S23–S42); docking poses of CPK4 (A), CPK13 (B), CPK16 (C), CPK18 (D), CPK20 (E) and quinidine (F) on HMhKCNT198–354 (Figure S43); ligand interaction diagrams of CPK4 (A), CPK13 (B), CPK16 (C), CPK18 (D), CPK20 (E) and quinidine (F) in complex with HMhKCNT198–354 (Figures S44); method validation parameters for LC/MS quantitation of test compounds (Table S2); functional and pharmacological properties of KCNT1 F346S channels (Figure S45); pharmacological characterization of selected CPK compounds on KCNT1 or KCNT1 F346S channels (Figure S46); representative graph showing the linear regression of the natural logarithm of % remaining parent compounds CPK19 and CPK20 (Figure S47); compound CPK18 metabolic fate as determined by LC analysis (Figure S48); molecular structures of compound CPK18 main oxidized metabolites as determined by LC-MS/MS analysis (Figure S49); molecular structures of compound CPK18 main glucuronic metabolites as determined by LC-MS/MS analysis (Figure S50) (PDF)
Molecular formula strings (CSV)
EMhKCNT1_CPK4 (PDB)
EMhKCNT1_CPK13 (PDB)
EMhKCNT1_CPK16 (PDB)
EMhKCNT1_CPK18 (PDB)
EMhKCNT1_CPK20 (PDB)
EMhKCNT1_QND_pose1 (PDB)
EMhKCNT1_QND_pose2 (PDB)
HMhKCNT1_CPK4 (PDB)
HMhKCNT1_CPK13 (PDB)
HMhKCNT1_CPK16 (PDB)
HMhKCNT1_CPK18 (PDB)
HMhKCNT1_CPK20 (PDB)
HMhKCNT1_QND (PDB)
Author Contributions
○ N.I., L.C., T.C., and G.P. contributed equally to this work. All authors approved the final manuscript.
The authors declare no competing financial interest.
Special Issue
Published as part of Journal of Medicinal Chemistry virtual special issue “Many Faces of Medicinal Chemistry”.
Supplementary Material
References
- Yuan A.; Santi C. M.; Wei A.; Wang Z.-W.; Pollak K.; Nonet M.; Kaczmarek L.; Crowder C. M.; Salkoff L. The Sodium-Activated Potassium Channel Is Encoded by a Member of the Slo Gene Family. Neuron 2003, 37 (5), 765–773. 10.1016/S0896-6273(03)00096-5. [DOI] [PubMed] [Google Scholar]
- Kaczmarek L. K. Slack, Slick, and Sodium-Activated Potassium Channels. ISRN Neurosci. 2013, 2013, 354262 10.1155/2013/354262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J.; Lv Y.-T.; Zhao X.-Y.; Wang J.-J.; Shen Z.-S.; Li J.; Zhang F.-F.; Liu J.; Wang X.-H.; Xu Y.; et al. Identification of Sodium- and Chloride-Sensitive Sites in the Slack Channel. J. Neurosci. 2023, 43 (15), 2665–2681. 10.1523/JNEUROSCI.1365-22.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschetti S.; Lavazza T.; Curia G.; Aracri P.; Panzica F.; Sancini G.; Avanzini G.; Magistretti J. Na+-Activated K+ Current Contributes to Postexcitatory Hyperpolarization in Neocortical Intrinsically Bursting Neurons. J. Neurophysiol. 2003, 89 (4), 2101–2111. 10.1152/jn.00695.2002. [DOI] [PubMed] [Google Scholar]
- Liu X.; Leung L. S. Sodium-Activated Potassium Conductance Participates in the Depolarizing Afterpotential Following a Single Action Potential in Rat Hippocampal CA1 Pyramidal Cells. Brain Res. 2004, 1023 (2), 185–192. 10.1016/j.brainres.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Kuret A.; Ehinger R.; Bischof H.; Luczak A.; Bausch A.; Zhou X.; Malli R.; Ruth P.; Lukowski R. Cardioprotective Actions of the Sodium-Activated Potassium Channel Slack (aka Slo2.2, KNa1.1 or KCNT1). Eur. Heart J. 2020, 41, ehaa946.3646 10.1093/ehjci/ehaa946.3646. [DOI] [Google Scholar]
- Yang B.; Desai R.; Kaczmarek L. K. Slack and Slick KNaChannels Regulate the Accuracy of Timing of Auditory Neurons. J. Neurosci. 2007, 27 (10), 2617–2627. 10.1523/JNEUROSCI.5308-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F.; Wang X.; Ostertag E. M.; Nuwal T.; Huang B.; Jan Y.-N.; Basbaum A. I.; Jan L. Y. TMEM16C Facilitates Na+-activated K+ Currents in Rat Sensory Neurons and Regulates Pain Processing. Nat. Neurosci. 2013, 16 (9), 1284–1290. 10.1038/nn.3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R.; Bausch A. E.; Kallenborn-Gerhardt W.; Stoetzer C.; Debruin N.; Ruth P.; Geisslinger G.; Leffler A.; Lukowski R.; Schmidtko A. Slack Channels Expressed in Sensory Neurons Control Neuropathic Pain in Mice. J. Neurosci. 2015, 35 (3), 1125–1135. 10.1523/JNEUROSCI.2423-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee A.; Kaczmarek L. For K Channels, Na Is the New Ca. Trends Neurosci. 2005, 28 (8), 422–428. 10.1016/j.tins.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Ruffin V. A.; Gu X. Q.; Zhou D.; Douglas R. M.; Sun X.; Trouth C. O.; Haddad G. G. The Sodium-Activated Potassium Channel Slack Is Modulated by Hypercapnia and Acidosis. Neuroscience 2008, 151 (2), 410–418. 10.1016/j.neuroscience.2007.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hite R. K.; MacKinnon R. Structural Titration of Slo2.2, a Na + -Dependent K + Channel. Cell 2017, 168 (3), 390–399.e311. 10.1016/j.cell.2016.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonardi C. M.; Heyne H. O.; Fiannacca M.; Fitzgerald M. P.; Gardella E.; Gunning B.; Olofsson K.; Lesca G.; Verbeek N.; Stamberger H.; et al. KCNT1-Related Epilepsies and Epileptic Encephalopathies: Phenotypic and Mutational Spectrum. Brain 2021, 144 (12), 3635–3650. 10.1093/brain/awab219. [DOI] [PubMed] [Google Scholar]
- Barcia G.; Fleming M. R.; Deligniere A.; Gazula V.-R.; Brown M. R.; Langouet M.; Chen H.; Kronengold J.; Abhyankar A.; Cilio R.; et al. De Novo Gain-of-Function KCNT1 Channel Mutations Cause Malignant Migrating Partial Seizures of Infancy. Nat. Genet. 2012, 44 (11), 1255–1259. 10.1038/ng.2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heron S. E.; Smith K. R.; Bahlo M.; Nobili L.; Kahana E.; Licchetta L.; Oliver K. L.; Mazarib A.; Afawi Z.; Korczyn A.; et al. Missense Mutations in the Sodium-Gated Potassium Channel Gene KCNT1 Cause Severe Autosomal Dominant Nocturnal Frontal Lobe Epilepsy. Nat. Genet. 2012, 44 (11), 1188–1190. 10.1038/ng.2440. [DOI] [PubMed] [Google Scholar]
- Rubboli G.; Plazzi G.; Picard F.; Nobili L.; Hirsch E.; Chelly J.; Prayson R. A.; Boutonnat J.; Bramerio M.; Kahane P.; et al. Mild Malformations of Cortical Development in Sleep-Related Hypermotor Epilepsy Due to KCNT1Mutations. Ann. Clin. Transl. Neurol. 2019, 6 (2), 386–391. 10.1002/acn3.708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin H. C.; Kim G. E.; Pagnamenta A. T.; Murakami Y.; Carvill G. L.; Meyer E.; Copley R. R.; Rimmer A.; Barcia G.; Fleming M. R.; et al. Clinical Whole-Genome Sequencing in Severe Early-Onset Epilepsy Reveals New Genes and Improves Molecular Diagnosis. Hum. Mol. Genet. 2014, 23 (12), 3200–3211. 10.1093/hmg/ddu030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohba C.; Kato M.; Takahashi N.; Osaka H.; Shiihara T.; Tohyama J.; Nabatame S.; Azuma J.; Fujii Y.; Hara M.; et al. De Novo KCNT1Mutations in Early-Onset Epileptic Encephalopathy. Epilepsia 2015, 56 (9), e121–e128. 10.1111/epi.13072. [DOI] [PubMed] [Google Scholar]
- Lim C. X.; Ricos M. G.; Dibbens L. M.; Heron S. E. KCNT1Mutations in Seizure Disorders: the Phenotypic Spectrum and Functional Effects. J. Med. Genet. 2016, 53 (4), 217–225. 10.1136/jmedgenet-2015-103508. [DOI] [PubMed] [Google Scholar]
- McTague A.; Howell K. B.; Cross J. H.; Kurian M. A.; Scheffer I. E. The Genetic Landscape of the Epileptic Encephalopathies of Infancy and Childhood. Lancet Neurol. 2016, 15 (3), 304–316. 10.1016/S1474-4422(15)00250-1. [DOI] [PubMed] [Google Scholar]
- Gururaj S.; Palmer E. E.; Sheehan G. D.; Kandula T.; Macintosh R.; Ying K.; Morris P.; Tao J.; Dias K.-R.; Zhu Y.; et al. A De Novo Mutation in the Sodium-Activated Potassium Channel KCNT2 Alters Ion Selectivity and Causes Epileptic Encephalopathy. Cell Rep. 2017, 21 (4), 926–933. 10.1016/j.celrep.2017.09.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosino P.; Soldovieri M. V.; Bast T.; Turnpenny P. D.; Uhrig S.; Biskup S.; Döcker M.; Fleck T.; Mosca I.; Manocchio L.; et al. De Novo Gain-of-Function Variants in KCNT2 as a Novel Cause of Developmental and Epileptic Encephalopathy. Ann. Neurol. 2018, 83 (6), 1198–1204. 10.1002/ana.25248. [DOI] [PubMed] [Google Scholar]
- Cioclu M. C.; Mosca I.; Ambrosino P.; Puzo D.; Bayat A.; Wortmann S. B.; Koch J.; Strehlow V.; Shirai K.; Matsumoto N.; et al. KCNT2-Related Disorders: Phenotypes, Functional, and Pharmacological Properties. Ann. Neurol. 2023, 94 (2), 332–349. 10.1002/ana.26662. [DOI] [PubMed] [Google Scholar]
- Rychkov G. Y.; Shaukat Z.; Lim C. X.; Hussain R.; Roberts B. J.; Bonardi C. M.; Rubboli G.; Meaney B. F.; Whitney R.; Møller R. S.; et al. Functional Effects of Epilepsy Associated KCNT1Mutations Suggest Pathogenesis via Aberrant Inhibitory Neuronal Activity. Int. J. Mol. Sci. 2022, 23 (23), 15133 10.3390/ijms232315133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinckley C. A.; Zhu Z.; Chu J.; Gubbels C.; Danker T.; Cherry J. J.; Whelan C. D.; Engle S. J.; Nguyen V. Functional Evaluation of Epilepsy-Associated KCNT1 Variants in Multiple Cellular Systems Reveals a Predominant Gain of Function Impact on Channel Properties. Epilepsia 2023, 64 (8), 2126–2136. 10.1111/epi.17648. [DOI] [PubMed] [Google Scholar]
- Kim G. E.; Kronengold J.; Barcia G.; Quraishi I. H.; Martin H. C.; Blair E.; Taylor J. C.; Dulac O.; Colleaux L.; Nabbout R.; Kaczmarek L. Human Slack Potassium Channel Mutations Increase Positive Cooperativity between Individual Channels. Cell Rep. 2014, 9 (5), 1661–1672. 10.1016/j.celrep.2014.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming M. R.; Brown M. R.; Kronengold J.; Zhang Y.; Jenkins D. P.; Barcia G.; Nabbout R.; Bausch A. E.; Ruth P.; Lukowski R.; et al. Stimulation of Slack K+ Channels Alters Mass at the Plasma Membrane by Triggering Dissociation of a Phosphatase-Regulatory Complex. Cell Rep. 2016, 16 (9), 2281–2288. 10.1016/j.celrep.2016.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Brown M. R.; Hyland C.; Chen Y.; Kronengold J.; Fleming M. R.; Kohn A. B.; Moroz L. L.; Kaczmarek L. K. Regulation of Neuronal Excitability by Interaction of Fragile X Mental Retardation Protein with Slack Potassium Channels. J. Neurosci. 2012, 32 (44), 15318–15327. 10.1523/JNEUROSCI.2162-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole B. A.; Clapcote S. J.; Muench S. P.; Lippiat J. D. Targeting KNa1.1 Channels in KCNT1-Associated Epilepsy. Trends Pharmacol. Sci. 2021, 42 (8), 700–713. 10.1016/j.tips.2021.05.003. [DOI] [PubMed] [Google Scholar]
- Milligan C. J.; Li M.; Gazina E. V.; Heron S. E.; Nair U.; Trager C.; Reid C. A.; Venkat A.; Younkin D. P.; Dlugos D. J.; et al. KCNT1Gain of Function in 2 Epilepsy Phenotypes Is Reversed by Quinidine. Ann. Neurol. 2014, 75 (4), 581–590. 10.1002/ana.24128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo F.; Ambrosino P.; Guacci A.; Chetta M.; Marchese G.; Rocco T.; Soldovieri M. V.; Manocchio L.; Mosca I.; Casara G.; et al. Characterization of Two De Novo KCNT1Mutations in Children with Malignant Migrating Partial Seizures in Infancy. Mol. Cell. Neurosci. 2016, 72, 54–63. 10.1016/j.mcn.2016.01.004. [DOI] [PubMed] [Google Scholar]
- Yang B.; Gribkoff V.; Pan J.; Damagnez V.; Dworetzky S.; Boissard C.; Bhattacharjee A.; Yan Y.; Sigworth F.; Kaczmarek L. Pharmacological Activation and Inhibition of Slack (Slo2.2) Channels. Neuropharmacology 2006, 51 (4), 896–906. 10.1016/j.neuropharm.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Fitzgerald M. P.; Fiannacca M.; Smith D. M.; Gertler T. S.; Gunning B.; Syrbe S.; Verbeek N.; Stamberger H.; Weckhuysen S.; Ceulemans B.; et al. Treatment Responsiveness in KCNT1-Related Epilepsy. Neurotherapeutics 2019, 16 (3), 848–857. 10.1007/s13311-019-00739-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilena R.; DiFrancesco J. C.; Soldovieri M. V.; Giacobbe A.; Ambrosino P.; Mosca I.; Galli M. A.; Guez S.; Fumagalli M.; Miceli F.; et al. Early Treatment with Quinidine in 2 Patients with Epilepsy of Infancy with Migrating Focal Seizures (EIMFS) Due to Gain-of-Function KCNT1Mutations: Functional Studies, Clinical Responses, and Critical Issues for Personalized Therapy. Neurotherapeutics 2018, 15 (4), 1112–1126. 10.1007/s13311-018-0657-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik M.; Camm A. J. Evaluation of Drug-Induced QT Interval Prolongation. Drug Saf. 2001, 24 (5), 323–351. 10.2165/00002018-200124050-00001. [DOI] [PubMed] [Google Scholar]
- Mullen S. A.; Carney P. W.; Roten A.; Ching M.; Lightfoot P. A.; Churilov L.; Nair U.; Li M.; Berkovic S. F.; Petrou S.; Scheffer I. E. Precision Therapy for Epilepsy due to KCNT1Mutations. Neurology 2018, 90 (1), e67–e72. 10.1212/WNL.0000000000004769. [DOI] [PubMed] [Google Scholar]
- Xu D.; Chen S.; Yang J.; Wang X.; Fang Z.; Li M. Precision Therapy with Quinidine of KCNT1-Related Epileptic Disorders: A systematic review. Br. J. Clin. Pharmacol. 2022, 88 (12), 5096–5112. 10.1111/bcp.15479. [DOI] [PubMed] [Google Scholar]
- Finlayson K.; Witchel H. J.; McCulloch J.; Sharkey J. Acquired QT Interval Prolongation and HERG: Implications for Drug Discovery and Development. Eur. J. Pharmacol. 2004, 500 (1–3), 129–142. 10.1016/j.ejphar.2004.07.019. [DOI] [PubMed] [Google Scholar]
- Po S. S.; Wang D. W.; Yang I. C.-H.; Johnson J. P.; Nie L.; Bennett P. B. Modulation of HERG Potassium Channels by Extracellular Magnesium and Quinidine. J. Cardiovasc. Pharmacol. 1999, 33 (2), 181–185. 10.1097/00005344-199902000-00002. [DOI] [PubMed] [Google Scholar]
- Paul A. A.; Witchel H. J.; Hancox J. C. Inhibition of the Current of Heterologously Expressed HERG Potassium Channels by Flecainide and Comparison with Quinidine, Propafenone and Lignocaine. Br. J. Pharmacol. 2002, 136 (5), 717–729. 10.1038/sj.bjp.0704784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaerke D. A.; Tejada M.; Stolpe; Meinild Clofilium Inhibits Slick and Slack Potassium Channels. Biol.: Targets Ther. 2012, 6, 465–470. 10.2147/BTT.S33827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitznagel B. D.; Mishra N. M.; Qunies A. a. M.; Prael F. J.; Du Y.; Kozek K. A.; Lazarenko R. M.; Denton J. S.; Emmitte K. A.; Weaver C. D. VU0606170, a Selective Slack Channels Inhibitor, Decreases Calcium Oscillations in Cultured Cortical Neurons. ACS Chem. Neurosci. 2020, 11 (21), 3658–3671. 10.1021/acschemneuro.0c00583. [DOI] [PubMed] [Google Scholar]
- Griffin A. M.; Kahlig K. M.; Hatch R. J.; Hughes Z. A.; Chapman M. L.; Antonio B.; Marron B. E.; Wittmann M.; Martinez-Botella G. Discovery of the First Orally Available, Selective KNa1.1 Inhibitor: In Vitro and In Vivo Activity of an Oxadiazole Series. ACS Med. Chem. Lett. 2021, 12 (4), 593–602. 10.1021/acsmedchemlett.0c00675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole B. A.; Johnson R. M.; Dejakaisaya H.; Pilati N.; Fishwick C. W. G.; Muench S. P.; Lippiat J. D. Structure-Based Identification and Characterization of Inhibitors of the Epilepsy-Associated KNa1.1 (KCNT1) Potassium Channel. iScience 2020, 23 (5), 101100 10.1016/j.isci.2020.101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qunies A. M.; Emmitte K. A. Small-Molecule Inhibitors of Slack Potassium Channels as Potential Therapeutics for Childhood Epilepsies. Pharm. Pat. Anal. 2022, 11 (2), 45–56. 10.4155/ppa-2022-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qunies A. M.; Spitznagel B. D.; Du Y.; Weaver C. D.; Emmitte K. A. Design, synthesis, and biological evaluation of a novel series of 1,2,4-oxadiazole inhibitors of SLACK potassium channels: Identification of in vitro tool VU0935685. Bioorg. Med. Chem. 2023, 95, 117487 10.1016/j.bmc.2023.117487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTague A.; Nair U.; Malhotra S.; Meyer E.; Trump N.; Gazina E. V.; Papandreou A.; Ngoh A.; Ackermann S.; Ambegaonkar G.; et al. Clinical and Molecular Characterization of KCNT1-Related Severe Early-Onset Epilepsy. Neurology 2018, 90 (1), e55–e66. 10.1212/WNL.0000000000004762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger Release 2022–3: Prime; Schoredinger LLC: New York, 2023.
- Gioia D.; Bertazzo M.; Recanatini M.; Masetti M.; Cavalli A. Dynamic Docking: A Paradigm Shift in Computational Drug Discovery. Molecules 2017, 22 (11), 2029 10.3390/molecules22112029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Sarno V.; Giovannelli P.; Medina-Peris A.; Ciaglia T.; Di Donato M.; Musella S.; Lauro G.; Vestuto V.; Smaldone G.; Di Matteo F.; et al. New TRPM8 Blockers Exert Anticancer Activity over Castration-Resistant Prostate Cancer Models. Eur. J. Med. Chem. 2022, 238, 114435 10.1016/j.ejmech.2022.114435. [DOI] [PubMed] [Google Scholar]
- Ostacolo C.; Miceli F.; Di Sarno V.; Nappi P.; Iraci N.; Soldovieri M. V.; Ciaglia T.; Ambrosino P.; Vestuto V.; Lauritano A.; et al. Synthesis and Pharmacological Characterization of Conformationally Restricted Retigabine Analogues as Novel Neuronal Kv7 Channel Activators. J. Med. Chem. 2020, 63 (1), 163–185. 10.1021/acs.jmedchem.9b00796. [DOI] [PubMed] [Google Scholar]
- Musella S.; Carotenuto L.; Iraci N.; Baroli G.; Ciaglia T.; Nappi P.; Basilicata M. G.; Salviati E.; Barrese V.; Vestuto V.; et al. Beyond Retigabine: Design, Synthesis, and Pharmacological Characterization of a Potent and Chemically Stable Neuronal Kv7 Channel Activator with Anticonvulsant Activity. J. Med. Chem. 2022, 65 (16), 11340–11364. 10.1021/acs.jmedchem.2c00911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerqua I.; Musella S.; Peltner L. K.; D’Avino D.; Di Sarno V.; Granato E.; Vestuto V.; Di Matteo R.; Pace S.; Ciaglia T.; et al. Discovery and Optimization of Indoline-Based Compounds as Dual 5-LOX/sEH Inhibitors: In Vitro and In Vivo Anti-Inflammatory Characterization. J. Med. Chem. 2022, 65 (21), 14456–14480. 10.1021/acs.jmedchem.2c00817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertamino A.; Iraci N.; Ostacolo C.; Ambrosino P.; Musella S.; Di Sarno V.; Ciaglia T.; Pepe G.; Sala M.; Soldovieri M. V.; et al. Identification of a Potent Tryptophan-Based TRPM8 Antagonist With in Vivo Analgesic Activity. J. Med. Chem. 2018, 61 (14), 6140–6152. 10.1021/acs.jmedchem.8b00545. [DOI] [PubMed] [Google Scholar]
- Bertamino A.; Ostacolo C.; Ambrosino P.; Musella S.; Di Sarno V.; Ciaglia T.; Soldovieri M. V.; Iraci N.; Fernandez Carvajal A.; de la Torre-Martinez R.; et al. Tryptamine-Based Derivatives as Transient Receptor Potential Melastatin Type 8 (TRPM8) Channel Modulators. J. Med. Chem. 2016, 59 (5), 2179–2191. 10.1021/acs.jmedchem.5b01914. [DOI] [PubMed] [Google Scholar]
- Ostacolo C.; Di Sarno V.; Lauro G.; Pepe G.; Musella S.; Ciaglia T.; Vestuto V.; Autore G.; Bifulco G.; Marzocco S.; et al. Identification of an Indol-Based Multi-Target Kinase Inhibitor through Phenotype Screening and Target Fishing Using Inverse Virtual Screening Approach. Eur. J. Med. Chem. 2019, 167, 61–75. 10.1016/j.ejmech.2019.01.066. [DOI] [PubMed] [Google Scholar]
- Gomez-Monterrey I.; Bertamino A.; Porta A.; Carotenuto A.; Musella S.; Aquino C.; Granata I.; Sala M.; Brancaccio D.; Picone D.; et al. Identification of the Spiro(oxindole-3,3′-thiazolidine)-Based Derivatives as Potential p53 Activity Modulators. J. Med. Chem. 2010, 53 (23), 8319–8329. 10.1021/jm100838z. [DOI] [PubMed] [Google Scholar]
- Ostacolo C.; Di Sarno V.; Musella S.; Ciaglia T.; Vestuto V.; Pepe G.; Merciai F.; Campiglia P.; Monterrey I. M. G.; Bertamino A. An Efficient Approach to Aromatic Aminomethylation Using Dichloromethane as Methylene Source. Front. Chem. 2019, 7, 568 10.3389/fchem.2019.00568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beacham D. W.; Blackmer T.; O’ Grady M.; Hanson G. T. Cell-Based Potassium Ion Channel Screening Using the FluxOR Assay. SLAS Discovery 2010, 15 (4), 441–446. 10.1177/1087057109359807. [DOI] [PubMed] [Google Scholar]
- Biton B.; Sethuramanujam S.; Picchione K. E.; Bhattacharjee A.; Khessibi N.; Chesney F.; Lanneau C.; Curet O.; Avenet P. The Antipsychotic Drug Loxapine Is an Opener of the Sodium-Activated Potassium Channel Slack (Slo2.2). J. Pharmacol. Exp. Ther. 2012, 340 (3), 706–715. 10.1124/jpet.111.184622. [DOI] [PubMed] [Google Scholar]
- Zhang J.-H.; Chung T. D. Y.; Oldenburg K. R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. SLAS Discovery 1999, 4 (2), 67–73. 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Liu S.; Fan J.; Yan R.; Huang B.; Zhou F.; Yuan T.; Gong J.; Huang Z.; Jiang D. Structural Basis of Human Slo2.2 Channel Gating and Modulation. Cell Rep. 2023, 42 (8), 112858 10.1016/j.celrep.2023.112858. [DOI] [PubMed] [Google Scholar]
- Ross D. L.; Cooper M. J.; Koo C. C.; Skinner M. P.; Davis L. M.; Richards D. A.; Uther J. B. Proarrhythmic Effects of Antiarrhythmic Drugs. Med. J. Aust. 1990, 153 (1), 37–47. 10.5694/j.1326-5377.1990.tb125462.x. [DOI] [PubMed] [Google Scholar]
- Taglialatela M.; Castaldo P.; Pannaccione A.; Giorgio G.; Annunziato L. Human Ether-a-gogo Related Gene (HERG) K Channels as Pharmacological Targets. Biochem. Pharmacol. 1998, 55 (11), 1741–1746. 10.1016/S0006-2952(98)00002-1. [DOI] [PubMed] [Google Scholar]
- Aronov A. M. Common Pharmacophores for Uncharged Human Ether-a-go-go-Related Gene (hERG) Blockers. J. Med. Chem. 2006, 49 (23), 6917–6921. 10.1021/jm060500o. [DOI] [PubMed] [Google Scholar]
- Bains W.; Basman A.; White C. HERG Binding Specificity and Binding Site Structure: Evidence from a Fragment-Based Evolutionary Computing SAR Study. Prog. Biophys. Mol. Biol. 2004, 86 (2), 205–233. 10.1016/j.pbiomolbio.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Down K.; Amour A.; Baldwin I. R.; Cooper A. W. J.; Deakin A. M.; Felton L. M.; Guntrip S. B.; Hardy C.; Harrison Z. A.; Jones K. L.; et al. Optimization of Novel Indazoles as Highly Potent and Selective Inhibitors of Phosphoinositide 3-Kinase δ for the Treatment of Respiratory Disease. J. Med. Chem. 2015, 58 (18), 7381–7399. 10.1021/acs.jmedchem.5b00767. [DOI] [PubMed] [Google Scholar]
- Yamakawa Y.; Furutani K.; Inanobe A.; Ohno Y.; Kurachi Y. Pharmacophore Modeling for hERG Channel Facilitation. Biochem. Biophys. Res. Commun. 2012, 418 (1), 161–166. 10.1016/j.bbrc.2011.12.153. [DOI] [PubMed] [Google Scholar]
- Cavalli A.; Poluzzi E.; De Ponti F.; Recanatini M. Toward a Pharmacophore for Drugs Inducing the Long QT Syndrome: Insights from a CoMFA Study of HERG K+ Channel Blockers. J. Med. Chem. 2002, 45 (18), 3844–3853. 10.1021/jm0208875. [DOI] [PubMed] [Google Scholar]
- Stoyanova-Slavova I. B.; Slavov S. H.; Buzatu D. A.; Beger R. D.; Wilkes J. G. 3D-SDAR Modeling of hERG Potassium Channel Affinity: A Case Study in Model Design and Toxicophore Identification. J. Mol. Graphics Modell. 2017, 72, 246–255. 10.1016/j.jmgm.2017.01.012. [DOI] [PubMed] [Google Scholar]
- Cavalli A.; Buonfiglio R.; Ianni C.; Masetti M.; Ceccarini L.; Caves R.; Chang M. W. Y.; Mitcheson J. S.; Roberti M.; Recanatini M. Computational Design and Discovery of “Minimally Structured” hERG Blockers. J. Med. Chem. 2012, 55 (8), 4010–4014. 10.1021/jm201194q. [DOI] [PubMed] [Google Scholar]
- Bochevarov A. D.; Harder E.; Hughes T. F.; Greenwood J. R.; Braden D. A.; Philipp D. M.; Rinaldo D.; Halls M. D.; Zhang J.; Friesner R. A. Jaguar: A High-Performance Quantum Chemistry Software Program with Strengths in Life and Materials Sciences. Int. J. Quantum Chem. 2013, 113 (18), 2110–2142. 10.1002/qua.24481. [DOI] [Google Scholar]
- Soldovieri M. V.; Miceli F.; Taglialatela M. Driving With No Brakes: Molecular Pathophysiology of Kv7 Potassium Channels. Physiology 2011, 26 (5), 365–376. 10.1152/physiol.00009.2011. [DOI] [PubMed] [Google Scholar]
- Nappi P.; Miceli F.; Soldovieri M. V.; Ambrosino P.; Barrese V.; Taglialatela M. Epileptic channelopathies caused by neuronal Kv7 (KCNQ) channel dysfunction. Pflügers Arch. - Eur. J. Physiol. 2020, 472 (7), 881–898. 10.1007/s00424-020-02404-2. [DOI] [PubMed] [Google Scholar]
- Taglialatela M.; Barrese V.; Miceli; Soldovieri M.; Ambrosino P.; Iannotti F.; Cilio R. Neuronal Potassium Channel Openers in the Management of Epilepsy: Role and Potential of Retigabine. Clin. Pharmacol.: Adv. Appl. 2010, 2, 225–236. 10.2147/CPAA.S15369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miceli F.; Soldovieri M. V.; Martire M.; Taglialatela M. Molecular Pharmacology and Therapeutic Potential of Neuronal Kv7-Modulating Drugs. Curr. Opin. Pharmacol. 2008, 8 (1), 65–74. 10.1016/j.coph.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Pang K. S.; Han Y. R.; Noh K.; Lee P. I.; Rowland M. Hepatic Clearance Concepts and Misconceptions: Why the Well-Stirred Model is Still Used Even Though it is not Physiologic Reality?. Biochem. Pharmacol. 2019, 169, 113596 10.1016/j.bcp.2019.07.025. [DOI] [PubMed] [Google Scholar]
- McNaney C. A.; Drexler D. M.; Hnatyshyn S. Y.; Zvyaga T. A.; Knipe J. O.; Belcastro J. V.; Sanders M. An Automated Liquid Chromatography-Mass Spectrometry Process to Determine Metabolic Stability Half-Life and Intrinsic Clearance of Drug Candidates by Substrate Depletion. Assay Drug Dev. Technol. 2008, 6 (1), 121–129. 10.1089/adt.2007.103. [DOI] [PubMed] [Google Scholar]
- Ishii A.; Shioda M.; Okumura A.; Kidokoro H.; Sakauchi M.; Shimada S.; Shimizu T.; Osawa M.; Hirose S.; Yamamoto T. A Rrecurrent KCNT1Mutation in two Sporadic Cases with Malignant Migrating Partial Seizures in Infancy. Gene 2013, 531 (2), 467–471. 10.1016/j.gene.2013.08.096. [DOI] [PubMed] [Google Scholar]
- Mikati M. A.; Jiang Y.; Carboni M.; Shashi V.; Petrovski S.; Spillmann R.; Milligan C. J.; Li M.; Grefe A.; McConkie A.; et al. Quinidine in the Treatment of KCNT1-Positive Epilepsies. Ann. Neurol. 2015, 78 (6), 995–999. 10.1002/ana.24520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A.; Martin M.-J.; Orchard S.; Magrane M.; Ahmad S.; Alpi E.; Bowler-Barnett E. H.; Britto R.; Bye-A-Jee H.; Cukura A.; et al. UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51 (D1), D523–D531. 10.1093/nar/gkac1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J. D.; Higgins D. G.; Gibson T. J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22 (22), 4673–4680. 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson M. P.; Pincus D. L.; Rapp C. S.; Day T. J. F.; Honig B.; Shaw D. E.; Friesner R. A. A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins: Struct., Funct., Bioinf. 2004, 55 (2), 351–367. 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- Jacobson M. P.; Friesner R. A.; Xiang Z.; Honig B. On the Role of the Crystal Environment in Determining Protein Side-chain Conformations. J. Mol. Biol. 2002, 320 (3), 597–608. 10.1016/S0022-2836(02)00470-9. [DOI] [PubMed] [Google Scholar]
- Madhavi Sastry G.; Adzhigirey M.; Day T.; Annabhimoju R.; Sherman W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput.-Aided Mol. Des. 2013, 27 (3), 221–234. 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- Kim S.; Chen J.; Cheng T.; Gindulyte A.; He J.; He S.; Li Q.; Shoemaker B. A.; Thiessen P. A.; Yu B.; et al. PubChem 2023 update. Nucleic Acids Res. 2023, 51 (D1), D1373–D1380. 10.1093/nar/gkac956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger Release 2019–1: LigPrep; Schrödinger, LLC: New York, NY, 2019.
- Bowers K. J.; Chow D. E.; Xu H.; Dror R. O.; Eastwood M. P.; Gregersen B. A.; Klepeis J. L.; Kolossvary I.; Moraes M. A.; Sacerdoti F. D.. et al. In Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters, ACM/IEEE SC 2006 Conference (SC’06); IEEE, 2006.
- Lomize M. A.; Pogozheva I. D.; Joo H.; Mosberg H. I.; Lomize A. L. OPM Database and PPM Web Server: Resources for Positioning of Proteins in Membranes. Nucleic Acids Res. 2012, 40 (D1), D370–D376. 10.1093/nar/gkr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79 (2), 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Jorgensen W. L.; Maxwell D. S.; Tirado-Rives J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118 (45), 11225–11236. 10.1021/ja9621760. [DOI] [Google Scholar]
- Barreca M. L.; Iraci N.; Manfroni G.; Gaetani R.; Guercini C.; Sabatini S.; Tabarrini O.; Cecchetti V. Accounting for Target Flexibility and Water Molecules by Docking to Ensembles of Target Structures: The HCV NS5B Palm Site I Inhibitors Case Study. J. Chem. Inf. Model. 2014, 54 (2), 481–497. 10.1021/ci400367m. [DOI] [PubMed] [Google Scholar]
- Astolfi A.; Iraci N.; Sabatini S.; Barreca M.; Cecchetti V. p38α MAPK and Type I Inhibitors: Binding Site Analysis and Use of Target Ensembles in Virtual Screening. Molecules 2015, 20 (9), 15842–15861. 10.3390/molecules200915842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47 (7), 1739–1749. 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Halgren T. A.; Murphy R. B.; Friesner R. A.; Beard H. S.; Frye L. L.; Pollard W. T.; Banks J. L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47 (7), 1750–1759. 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Murphy R. B.; Repasky M. P.; Frye L. L.; Greenwood J. R.; Halgren T. A.; Sanschagrin P. C.; Mainz D. T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein–Ligand Complexes. J. Med. Chem. 2006, 49 (21), 6177–6196. 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- Duan J.; Dixon S. L.; Lowrie J. F.; Sherman W. Analysis and Comparison of 2D Fingerprints: Insights Into Database Screening Performance Using eight Fingerprint Methods. J. Mol. Graphics Modell. 2010, 29 (2), 157–170. 10.1016/j.jmgm.2010.05.008. [DOI] [PubMed] [Google Scholar]
- Sastry M.; Lowrie J. F.; Dixon S. L.; Sherman W. Large-Scale Systematic Analysis of 2D Fingerprint Methods and Parameters to Improve Virtual Screening Enrichments. J. Chem. Inf. Model. 2010, 50 (5), 771–784. 10.1021/ci100062n. [DOI] [PubMed] [Google Scholar]
- Sherman W.; Day T.; Jacobson M. P.; Friesner R. A.; Farid R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 2006, 49, 534–553. 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- Ambrosino P.; Soldovieri M. V.; De Maria M.; Russo C.; Taglialatela M. Functional and Biochemical Interaction Between PPARα Receptors and TRPV1 Channels: Potential Role in PPARα Agonists-Mediated Analgesia. Pharmacol. Res. 2014, 87, 113–122. 10.1016/j.phrs.2014.06.015. [DOI] [PubMed] [Google Scholar]
- Miceli F.; Soldovieri M. V.; Ambrosino P.; De Maria M.; Migliore M.; Migliore R.; Taglialatela M. Early-Onset Epileptic Encephalopathy Caused by Gain-of-Function Mutations in the Voltage Sensor of Kv7.2 and Kv7.3 Potassium Channel Subunits. J. Neurosci. 2015, 35 (9), 3782–3793. 10.1523/JNEUROSCI.4423-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Zhu F.; Hammill J.; Holbrook G.; Yang L.; Freeman B.; White K. L.; Shackleford D. M.; O’Loughlin K. G.; Charman S. A.; et al. Selecting an Anti-Malarial Clinical Candidate from two Potent Dihydroisoquinolones. Malar. J. 2021, 20 (1), 107 10.1186/s12936-021-03617-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.