

Abstract
Introduction
Antibodies are significant agents in the immune system and have proven to be effective in treating bacterial infections. With the advancement of antibody engineering in recent decades, antibody therapy has evolved widely.
Aim
This review aimed to investigate a new method as a therapeutic platform for the treatment of bacterial infections and explore the novel features of this method in conferring pathogen specificity to broad‐spectrum antibiotics.
Material and Methods
A literature review was conducted addressing the following topics about antibody–antibiotic conjugates (AACs): (1) structure and mechanism of action; (2) clinical effectiveness; (3) advantages and disadvantages.
Result
Antibody conjugates are designed to build upon the progress made in the development of monoclonal antibodies for the treatment of diseases. Despite the growing emergence of antibiotic resistance among pathogenic bacteria worldwide, novel antimicrobials have not been sufficiently expanded to combat the global crisis of antibiotic resistance. A recently developed strategy for the treatment of infectious diseases is the use of AACs, which are specifically activated only in host cells.
Conclusion
A novel therapeutic AAC employs an antibody to deliver the antibiotic to the bacteria. The AACs can release potent antibacterial components that unconjugated forms may not exhibit with an appropriate therapeutic index. This review highlights how this science has guided the design principles of an impressive AAC and discusses how the AAC model promises to enhance the antibiotic effect against bacterial infections.
Keywords: antibodies, antibody‐based therapy, immune system, pharmacokinetic
Structure and action mechanism of Antibody‐Antibiotic Conjugates (AAC).
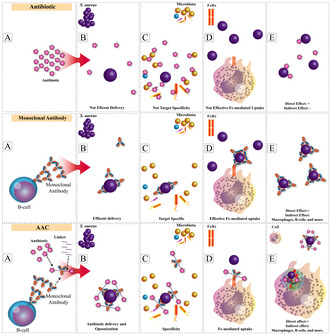
1. Introduction
Historically, two patents in 1998 and 1994 developed a novel approach using antibody–antibiotic conjugates (AAC) that were covalently connected for the prevention of and treatment for bacterial sepsis. The first research performed on AAC, conducted in 1989, focused on plaque‐forming or caries‐inducing bacteria, particularly Streptococcus mutans. Upgrades to these inventions were described between 2002 and 2012 [1, 2]. In 2014 and 2015, Genentech Inc. developed three inventions related to antiwall teichoic (WTA) antibodies and their conjugates, including clindamycin, novobiocin, retapamulin, daptomycin, GSK‐2140944, CG‐400549, sitafloxacin, teicoplanin, triclosan, naphthyridone, linezolid, doxorubicin, ampicillin, vancomycin, imipenem, doripenem, gemcitabine, dalbavancin, rifamycin‐type antibiotics, and azithromycin [2, 3]. Despite the existence of many potent bactericidal antibiotics to eradicate difficult‐to‐treat intracellular bacteria, they have been unsuccessful in clinical practice because of weak pharmacokinetic actions or high toxicity. The data [3, 4] demonstrate that arming antibodies with exclusive bactericidal antibiotics can result in the introduction of novel therapies. The AAC technology promises to enhance antibiotic efficacy against difficult‐to‐treat intracellular bacteria [4] (Figure 1). In 2015, Lehar et al. [4] developed a combination of the immunoconjugate and chemotherapy strategy known as AAC (THIOMAB; Genentech, South San Francisco, CA, USA) against Staphylococcus aureus. Interestingly, this exciting and novel therapeutic technology uses a monoclonal anti‐S. aureus antibody against WTA antigens to opsonize the bacteria, which is conjugated to a highly efficacious antibiotic, ensuring efficient eradication of intracellular bacteria and ultimately treating the infection. Research data revealed that an AAC against Pseudomonas aeruginosa can locally enhance the concentration of arylomycin in phagocytic cells, resulting in the clearance of intracellular bacteria [5]. A number of in vitro and in vivo experiments have demonstrated the potent and efficacious bactericidal ability of AAC technology against intracellular reservoirs of methicillin‐resistant S. aureus (MRSA), which is a difficult‐to‐treat invasive bacterial infection, compared to standard vancomycin treatment [5, 6, 7]. The in vivo data showed that prophylaxis with monoclonal anti‐MRSA‐specific unconjugated antibodies (against WTA antigens) was unsuccessful in preventing infection and eradicating MRSA after intravenous administration in mice. These findings align with clinical results that indicate anti‐staphylococcal antibodies specific to surface motifs are ineffective in eradicating infectious bacteria in low birth weight infants [8, 9]. Staben et al. [5] developed a concise targeted drug delivery system through the traceless synthesis of tertiary and heteroaryl amines, utilizing a cathepsin‐cleavable linker, as a novel linker connection to an effective rifamycin derivative in the AAC strategy. This strategy efficiently killed intracellular reservoirs of S. aureus associated with bacteremia in an in vivo mouse model [4, 10]. Additionally, the AAC strategy is expected to exhibit promising pharmacokinetics, such as long half‐lives, and reduced off‐target toxicity [11]. Recently, Deng et al. [12] provided insights into the pharmacokinetic actions of DSTA4637A, a novel THIOMAB composed of a monoclonal THIOMAB IgG1 connected via a protease‐cleavable linker to dmDNA31, a rifamycin‐class antibiotic. They administered total antibody, antibody‐conjugated dmDNA31, and unconjugated dmDNA31 intravenously in preclinical models of S. aureus infection in rats and cynomolgus monkeys. They administered total antibody, antibody‐conjugated dmDNA31, and unconjugated dmDNA31 intravenously in preclinical models. An integrated semimechanistic pharmacokinetic model for total antibody and antibody‐conjugated dmDNA31 was effectively extended and was able to accurately depict the complex pharmacokinetics of DSTA4637A in preclinical models. Previously published data [13, 14, 15] utilizing multiple bioanalytical methodologies suggested that minimal physiologically based pharmacokinetic modeling and a cross‐species scaling strategy provide useful means to facilitate the understanding and translation of DSTA4637A disposition from animal models to humans. In the AAC strategy, the selected antibiotics should maintain bactericidal activity in the acidic environment of the phagolysosome to eradicate intracellular bacteria. Interestingly, in vitro data [9] have demonstrated that AAC not only kills nonreplicating intracellular bacteria but also significantly reduces cell‐to‐cell transfer of bacteria. However, although AAC therapy was effective even in the presence of competing antibodies targeting surface motifs of S. aureus, only human clinical research can determine whether this technology can effectively opsonize nonspecific IgG‐coated S. aureus and eliminate the bacteria [9]. In this study, we investigate how insights into the pathophysiology of various infections, such as S. aureus, were incorporated into the design of an AAC. We also discuss the potential of this new method as a therapeutic platform for the treatment of bacterial infections and explore the novel features of this method in conferring pathogen specificity to broad‐spectrum antibiotics. This new method may enable the revival of the use of antibiotics that were previously abandoned because of toxicity or poor pharmacokinetics, as well as addressing antibiotic resistance observed in certain infections.
FIGURE 1.
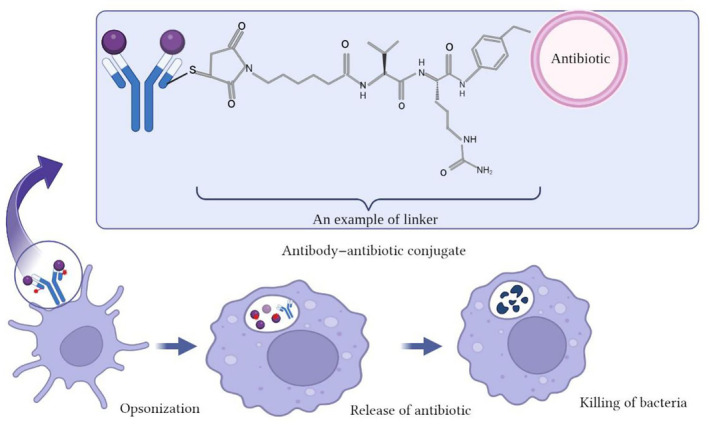
Mechanism of killing bacteria by antibody–antibiotic conjugates (AAC). AAC binds bacteria and the host cells internalize AAC‐bound bacteria. In the next Step, fusion occurs with the phagolysosome where lysosomal cathepsins cleave the VC linker, releasing AAC. Then unconjugated AAC kills the intracellular bacteria.
2. Antibacterial Antibodies
Anthim (obiltoxaximab) is an injectable drug used for the treatment of pulmonary anthrax, consisting of an antibody conjugated to an appropriate antibiotic, usually ciprofloxacin. This drug was approved by the FDA as the first Hu‐mAb. Since conducting human studies to evaluate the effectiveness of Anthim for infections caused by Bacillus anthracis would be unethical, and conducting trials in the field is not feasible, the drug was developed under the Animal Rule. This rule allows drugs to be approved on the basis of animal efficacy studies [16]. Raxibacumab, which targets the protective antigen (PA) component of anthrax toxin, was also approved by the FDA [17]. Zinplava (bezlotoxumab), an antibacterial antibody used to treat Clostridium difficile infections in adults, has also received FDA approval [18]. Furthermore, several antibacterial antibodies, including actozumab, edobacumab, and aurograb, have entered phase III clinical trials and have shown successful results against C. difficile, Escherichia coli, and S. aureus infections, respectively [19]. Monoclonal antibodies can act against bacterial virulence factors, such as bacterial toxins, or exhibit bactericidal activity [20]. Recently, other virulence factors such as the Type III secretion system, pili, and outer membrane transporters have been evaluated. Unlike exotoxins, antigens located on the bacterial membrane can be targets for antibodies that not only neutralize the antigens but also exhibit a bactericidal effect [21]. Ideally, the target antigen should be abundant and accessible for proper antibody binding, while also being restricted to specific bacteria for an optimal effect [22]. The main advantage of monoclonal antibodies (mAbs) is their high specificity and favorable selection of the target antibody, which can reduce side effects and minimize selection pressure for cross‐resistance. However, there are still some issues to consider, such as cost and administration methods [23]. A recent setback in the production of antibodies may have dampened the enthusiasm of researchers and investors who were considering Hu‐mAbs for antibacterial development. Research and production of antibodies are costly, and some recent failures have tempered expectations [24]. Table 1 provides a list of antibacterial antibodies, the diseases they are used against, their targets, advantages, and disadvantages.
TABLE 1.
List of antibacterial antibodies, their used disease, targets, advantages, and disadvantages.
| Antibacterial antibodies | Disease | Target | Advantages | Disadvantages |
|---|---|---|---|---|
| Anthim (obiltoxaximab) | Pulmonary anthrax | Toxin |
Easy to use Specific diagnosis Low toxicity No damage to microbiota |
High cost Low variety |
| Raxibacumab | Anthrax | Toxin | ||
| Zinplava (bezlotoxumab) | Clostridium difficile infections | C. difficile toxin B | ||
| Actozumab | C. difficile infections | C. difficile toxin A | ||
| Edobacumab | Escherichia coli infections | Lipid A | ||
| Nebacumab | E. coli infections | Lipid A | ||
| Aurograb | Staphylococcus aureus infections | Grfa (lipoprotein) |
3. Antibody
Antibodies, also known as immunoglobulins (Ig), are major soluble proteins in the mammalian immune system that bind to foreign bodies [1]. The structure of an antibody contains two arms of antigen‐binding fragments (Fab) at their tips, which have identical antigen‐binding sites, and one crystallizable fragment (Fc) linked to the Fabs by a flexible joint [25]. Igs are primarily produced by plasma cells and employed by the immune system to neutralize pathogenic bacteria. They have the capability to bind to a nearly infinite repertoire of molecules with exquisite specificity and high affinity [25, 26]. This concept also aligns with the notion of complementarity in antibody–antigen recognition, analogous to the “lock and key” fit proposed by Fischer for enzymes [27]. An antigen is a molecule that can specifically bind to an antibody through the variable region of the Fab [27]. The main function of the humoral immune system is to generate antibodies, often through differentiated B cells called plasma cells. The interaction between B cells and T helper cells is crucial for the full activation of the B cell and subsequent antibody production in response to antigen binding [28]. An antibody consists of two heavy chains (H) and two light chains (L). Each chain contains an N‐terminal variable domain (VH and VL in the heavy and light chains, respectively), whereas the remaining portion has a C‐terminal constant domain (CL, CH1, CH2, CH3, and CH4) used to determine the Ig class [29, 30]. The antigen‐binding fragments can be formed by combining different types of the five main heavy chain classes or isotypes. These isotypes, authorized by antibodies, are categorized into five classes: IgA, IgD, IgE, IgG, and IgM [25, 30]. IgG1 and IgG3 isotypes are involved in antibody‐dependent cellular cytotoxicity. IgG3 is not engineerable because of isotypic variation and its short half‐life. The most commonly used isotypes in this field are IgG1, IgG2, and IgG4 [31]. Although the structure of all antibodies is very similar, a small region at the N‐terminus exhibits significant variability, resulting in the existence of a diverse array of antibodies with slightly different N‐terminal structures. This variable region is known as the hypervariable area. Each of these regions can bind to multiple antigens [27]. This extensive diversity in the antigen‐binding fragments (paratope) allows the immune system to recognize a wide range of antigens [27, 30]. Antibodies are potent components of the immune system and have proven to be effective in treating bacterial infections. In recent decades, there has been a significant progress in antibody engineering, leading to extensive research on antibody therapy for various diseases [14]. The emergence of monoclonal antibody technology [32] and its application in antibody production [30] aims to provide improved therapeutic options [33]. The development of antibody conjugates has also benefited from advancements in monoclonal antibody production [34].
Historically, mouse antibodies were utilized to create antibody–drug conjugates (ADCs). However, to minimize immune responses in humans, murine chimeric antibodies (65% human), humanized antibodies (95% human), or fully human antibodies (100% human) are now employed. When selecting an antibody, the biological activity of the Fc domain, which can interact with Fc receptor (FcR)‐bearing cells, should be taken into consideration. Proper design and production of monoclonal antibodies play a crucial role in creating effective antibody–drug conjugates. Currently, human IgG1 is the preferred isotype for generating ADCs because of its ability to stimulate both antibody‐dependent cellular cytotoxicity (ADCC) and complement‐dependent cytotoxicity [33, 35].
When considering conjugated antibodies, it is important to mention that the target of the antibody should be abundant and highly expressed both in vitro and during infection. For instance, β‐N‐acetylglucosamine cell wall teichoic acid in S. aureus fulfills these criteria as it displays 50,000 binding sites [4, 35].
As a result, numerous studies have been conducted in this field, and various antibodies are extensively utilized for the treatment of immunodeficiencies, cancers, multiple sclerosis, rheumatoid arthritis, and psoriasis. Additionally, several antibodies are currently undergoing clinical evaluation for the treatment of bacterial infections.
4. AAC Mechanism of Action
Until now, unfortunately, the production of new drugs has been a slow and time‐consuming process in the healthcare system. The promising use of antibody–antibiotic combinations and advancements in molecular science, coupled with the indifference of the pharmaceutical industry toward new antibacterial agents, can be attributed to the lack of economic incentives and challenging regulatory requirements. These factors hinder the development and progress of new therapeutic agents in this field [36]. The Centers for Disease Control and Prevention (CDC) and the World Health Organization (WHO) have declared antimicrobial resistance (AMR) a “global public health concern” that necessitates the development of new drugs to combat drug‐resistant pathogens [37]. Before the advent of antibiotics, antibodies served as the first effective line of defense against bacteria. The groundbreaking discoveries of Emil von Behring and Shibasaburo Kitasato in 1893 demonstrated the passive transfer of antibodies through the blood of infected animals, providing immunity against tetanus and diphtheria. This paved the way for the development of sheep protection serum as the initial step toward creating a therapeutic treatment for humans [12]. A recent report from the UN General Council in New York on September 21, 2016, highlighted the global rise of pathogens that have become highly resistant to antibiotics. However, the development of novel antibiotics or alternative therapeutic options has not kept pace to effectively combat these drug‐resistant pathogens [38].
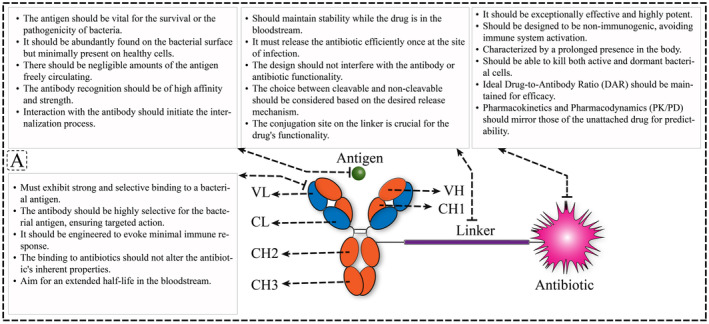
In response to the failure of traditional antibiotics in treating bacterial infections, a novel form of treatment known as AAC has been recently developed. AAC specifically targets and delivers antibiotics directly to host cells, enabling targeted delivery of antibiotics to bacteria [4]. AAC is a form of ADC that has already demonstrated successful application in cancer therapy [1, 39, 40, 41, 42]. An example in Gram‐negative bacteria, Kajihara et al. reported in 2021 in this field on P. aeruginosa, in which this bacterium was observed to cause life‐threatening infections associated with widespread antibiotic failure and resistance. In this study, it is reported that the antibiotic G2637, an analog of arylomycin, targets bacterial signal peptidase Type I, which has moderate potency against P. aeruginosa bacteria. In this study, it was hypothesized that an AAC could increase its activity by colocalizing P. aeruginosa bacteria with high local concentrations of G2637 antibiotic in the intracellular environment of phagocytes. Using a novel advanced screening technology for hybridomas that recognize intact bacteria, a monoclonal antibody 26F8 was selected that binds to the lipopolysaccharide O antigen region on the surface of P. aeruginosa bacteria. Finally, this study suggests that an anti‐P. aeruginosa AAC can locally concentrate the antibiotic and target P. aeruginosa inside phagocytes, providing new therapeutic options for antibiotics with relatively active or pharmacokinetic profiles or have adverse toxicity [43]. One example of a Gram‐positive AAC is DSTA4637A, a modern THIOMAB antibody–antibiotic conjugate (TAC) with potential therapeutic efficacy against hard‐to‐treat S. aureus infections [14]. The study of this novel AAC form comprises an anti‐S. aureus antibody conjugated to a highly potent antibiotic that only becomes active upon release into the proteolytic environment of the phagolysosome [4]. DSTA4637S is another novel AAC designed to target intracellular S. aureus, as standard surveillance antibiotics are insufficient for treatment [44]. Refer to Figure 2 for the mechanism of action of DSTA4637S. Generally, an AAC consists of three different components: (i) antibody, (ii) antibiotic, and (iii) linker (Figure 3). In the AAC design, the combination of the antibody and the antibiotic should be carefully selected to maximize efficiency (Figure 4A) [1]. The antibody chosen for AAC should meet several criteria. For instance, the primary role of antibodies is to deliver the payload to bacteria (Figure 4B). The antigen to which the antibody binds should be well‐characterized for selection, with a consistently abundant amount expressed on the bacterial surface at all stages of infection and not present on mammalian cells [45]. Additionally, the absence of high levels of soluble antigen and the need for specificity toward the target cell antigen should be considered (Figure 4C) [44]. The pharmacokinetic properties of antibodies are also crucial for maintaining the antibody structure while binding the necessary number of antibiotics and ensuring resistance to antibiotics such as β‐lactam and other active enzymes in the cell wall [46]. An antibody against β‐GlcNAc‐WTA (IgG1 subtype), isolated from patients with S. aureus infection, was selected for AAC as it met the aforementioned criteria. This monoclonal antibody specifically binds to the β‐GlcNAc residue on the ribitol phosphate wall unit [34]. Consequently, novel antibody‐based therapies are currently being developed to provide improved treatment options.
FIGURE 2.
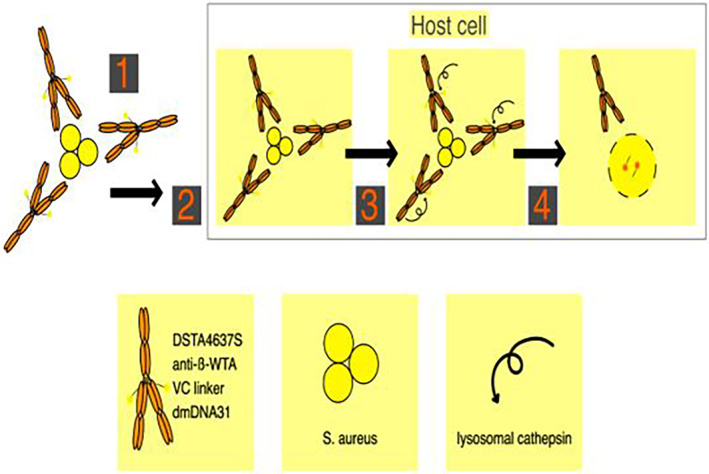
Antibody–antibiotic conjugates (AAC) mechanism for killing Staphylococcus aureus. DSTA4637S mechanism for killing intracellular S. aureus. Step 1, DSTA4637S binds S. aureus. Step 2, host cells internalize DSTA4637S‐bound S. aureus. Step 3, fusion occurs with the phagolysosome where lysosomal cathepsins cleave the VC linker, releasing dmDNA31. Step 4, unconjugated dmDNA31 kills the intracellular bacteria.
FIGURE 3.
Antibody–antibiotic conjugates (AAC) structure.
FIGURE 4.
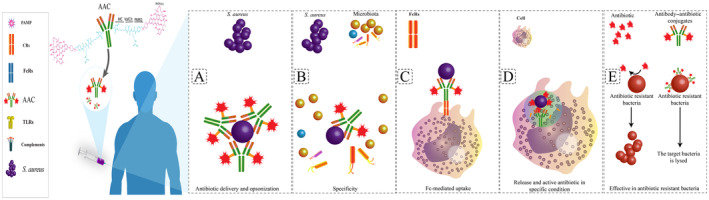
Advantages and overall structure of antibody–antibiotic conjugates (AAC). (A) Antibiotic delivery and opsonization: AACs enable precise delivery of antibiotics to target bacteria, ensuring efficient opsonization for enhanced phagocytosis. (B) Specificity: AACs exhibit high specificity by targeting surface antigens on bacteria, minimizing off‐target effects on host cells. (C) Fc‐mediated uptake: AACs utilize Fc receptors (FcR) to facilitate uptake by immune cells, enhancing the clearance of intracellular bacteria. (D) Release and active antibiotic in specific conditions: Within intracellular environments like phagolysosomes, AACs release active antibiotics, ensuring effective bacterial eradication. (E) Effective in antibiotic‐resistant bacteria: AACs demonstrate efficacy even against antibiotic‐resistant bacterial strains, addressing the challenge of antibiotic resistance.
The antibacterial activity of AAC is primarily defined by the antibiotic, and the ideal antibiotic in this combination should meet several criteria [18]. These antibiotics should exhibit bactericidal activity in the nanomolar range or lower. They should possess functional groups that can bind to monoclonal antibodies (mAbs). Additionally, antibiotics should be soluble and stable under physiological conditions (Figure 4D) [47]. DSTA4637S is composed of a human mAb of the IgG1 subtype conjugated to a new rifamycin‐class antibiotic (4‐dimethylaminopiperidino‐hydroxybenzoxazino rifamycin [dmDNA31]) via a protease‐cleavable linker. The newly developed dmDNA31 antibiotic demonstrates potent bactericidal effects against S. aureus infections [44]. The rifamycin class of antibiotics was chosen for DSTA4637 because of their highly efficient delivery with limited antigen copies, sustained antibacterial activity at low pH in phagolysosomes, and their ability to combat intracellular pathogens [4]. AAC utilizes highly potent antibiotics that are stable and, when conjugated to mAbs, are nonimmunogenic. The antibody–antibiotic linker is designed to release the active drug only when the mAb has reached the target site [48]. The antibiotics used should be conjugatable agents and retain antibacterial activity in the acidic pH of the lysosomal environment, while also having a long half‐life as a free drug within mammalian cells [4]. Although the choice of antibody and antibiotic in AAC design is important, selecting ideal target‐specific antibodies and potent payloads based on the type of infection, the linker plays a key role in the AAC design by binding the antibiotics to monoclonal antibodies (Figure 4E) [49].
One important feature of linkers is that they should possess biorthogonal properties, remain stable in plasma, and efficiently release the active antibiotic during externalization [50]. The linker should not adversely affect the antibiotics or antibodies [51]. Another consideration is the hydrophobicity of the linker [52]. Binding a hydrophobic linker to a hydrophobic payload can promote aggregation and compromise the stability of the AAC. Various techniques have been widely employed to enhance their physicochemical properties because of their usefulness. The predominant classes of linkers include cleavable and noncleavable linkers [34]. Cleavable linkers, such as hydrazone, are specifically designed to remain stable at neutral pH [34]. Noncleavable linkers, often containing thioether bonds, are resistant to proteolysis and offer better stability than cleavable linkers [53]. Another important consideration is the choice of the conjugation method between the antibiotic and the antibody, which can involve chemical and enzymatic conjugation methods [34]. Today, methods for FDA‐approved ADC generation have been extensively studied under in vivo and in vitro conditions, including native cysteine, THIOMA, transglutaminase, and unnatural AA [43, 44]. These novel methods for site‐specific drug attachment lead to more homogeneous conjugates and enable control over the site of drug attachment [11, 44]. The underlying strategy of these new methods involves modifying cysteine residues to achieve greater conjugate uniformity and allows for examination of the drug attachment site. Such modifications can have a significant impact on in vivo results and the therapeutic index [41].
Currently, one of the novel AAC methods being considered as a potential treatment for severe bacterial infections is the THIOMAB antibody–antibiotic conjugate, which serves as a model for engineered antibodies with passive cysteine residues located at specific sites on the antibody. This design allows antibiotics to be conjugated with a specified stoichiometry without disrupting interchain disulfide bonds (Figure 4F) [4, 49, 51].
The study by Lehar et al. [4] demonstrated that the binding of TAC to the surface antigen of S. aureus leads to changes in bacterial structures. Initially, the binding of TAC in the bloodstream is inactive because of the covalent bond of dmDNA31. However, when AAC‐tagged bacteria reach the intracellular environment, host proteases present in acidic endo‐ or phagolysosomes, such as cathepsin‐B or plasmin, cleave the linker and activate the antibiotics. Since multiple TAC molecules can potentially be linked to a single bacterium, the antibiotics can be released within the intracellular environment, effectively eliminating labeled bacteria and bystander killing of untagged binding bacteria [14, 20].
The mechanism of action of AAC involves the internalization of extracellular bacteria and the killing of intracellular bacteria through the intracellular release of active antibiotics. AAC is initially a biologically inactive compound as the covalently bound antibiotic agent remains inactive. The high affinity of the antibody to a highly abundant surface antigen on the bacteria ensures rapid opsonization or labeling of circulating bacteria with AAC. The bacteria–AAC complex is then taken up by both phagocytic and nonphagocytic cells through opsonophagocytosis and the endogenous host entry mechanism of bacteria. Upon the detachment of AAC‐tagged bacteria within acidic endolysosomes or phagolysosomes, cathepsins released in these intracellular vesicles can cleave the linker and release the active antibiotic.
The entry of AAC‐tagged bacteria into cells and subsequent intracellular release of active antibiotics may eliminate both tagged bacteria and untagged commensals. This mechanism allows the AAC molecule to target sites where bacteria are poorly treated with conventional antibiotics. Indeed, in vitro studies have shown that AAC‐tagged S. aureus efficiently kills all tested cell types [4]. The great advantages of mAbs include the optimal selection of antibody targets and their broad specificity, resulting in fewer off‐target effects and reduced selective pressure for cross‐resistance. However, challenges remain regarding the cost of production and systemic administration [54].
5. Challenge of AAC
Although numerous studies in this field have demonstrated the many advantages associated with the use of antibacterial monoclonal antibodies, significant limitations exist. Because of the high specificity of monoclonal antibodies in binding to their target pathogens, it is ideally necessary to identify the bacterial species causing the infection before initiating treatment. One of the main disadvantages of such techniques has been shown in studies to be the use of high temperature and/or low pH, which may greatly affect the physicochemical properties of peptides. This feature may also affect the production of peptibodies toward bacterial infections. Nevertheless, the use of expression‐ and structure‐simple proteomics, which relies on the use of ultrafast cleaved proteins to produce protein α‐thioesters, has been proposed as one promising strategy to overcome these limitations [55].
This aspect is particularly relevant in severe infections among hospitalized patients, which are primarily caused by Gram‐negative microorganisms that are resistant to multiple drugs. In clinical practice, patients with suspected infections often receive multiple empiric antimicrobial treatments before the initial microbiological test results become available. Empiric treatment typically relies on broad‐spectrum antibiotics that offer coverage against the various bacterial species causing infection in such cases [56]. Therefore, the development and implementation of diagnostics enabling rapid species identification directly from clinical samples greatly facilitate the use of monoclonal antibody‐based therapies in these circumstances. Currently, several techniques for the swift identification of bacterial pathogens and the determination of antimicrobial susceptibility profiles in infections are being developed.
ADCs pose intricate manufacturing challenges due to their dual‐component nature, involving antibodies (mAb) and cytotoxic payload linkers (LTs), each demanding distinct good manufacturing practice (GMP) processes. The production of LTs involves intricate chemistries and safety concerns when handling potent cytotoxins, limiting the pool of capable manufacturers. The conjugation step, crucial for attaching LTs to mAbs, introduces additional hurdles. Removal of unconjugated mAb and excess cytotoxins is essential for generating the bulk drug product. Purification methods like hydrophobic‐interaction chromatography (HIC) are often employed, particularly as LT addition increases hydrophobicity. The purification strategy must carefully consider which fractions, obtained through techniques like tangential flow filtration (TFF) and chromatographic separations, constitute the final drug product. mAb biomanufacturing involves distinct steps, including cell line and process development, production scale‐up, downstream processing, filling, labeling, and clinical packaging, often executed by multiple service providers requiring effective coordination by the product sponsor [57].
6. Absorption, Distribution, Metabolization, and Elimination
In addition to the mentioned benefits, other factors that contribute to the effectiveness of these treatments should also be considered. One such factor is the disposition of a pharmaceutical compound within an organism, which is characterized by absorption, distribution, metabolization, and elimination (ADME) [58]. For protein‐based drugs, parenteral administration is preferred because of limitations in the oral route [59]. Therefore, intravenous (IV) or subcutaneous (SC) administration is the preferred route for mAb administration [60]. Additionally, attention should be given to the distribution of these factors. The size and polarity of ADCs, as well as the fenestration size and membrane thickness of blood vessels and tissues, influence their distribution [61]. Although the delivery procedures for ADCs and mAbs are similar, the carried mass of cytotoxic agents may further affect their delivery. Conjugated agents can modify the binding affinity and internalization efficiency of the antibody component, which may impact the delivery across binding site barriers. Hydrophobic drugs can permeate the membrane and distribute outside of target cells, whereas hydrophilic drugs are typically restricted to cells expressing antigens [58, 62].
ADC breakdown occurs nonspecifically through proteolysis via macrophage uptake in various tissues, including the skin, muscle, and liver [63]. These cells can absorb antibodies through nonspecific pinocytosis and degrade them through lysosomal proteolysis. The ADC can also be cleared through specific mechanisms. For cell‐targeted antibodies, target‐mediated clearance occurs when the antibody binds to the target cell, internalizes, and degrades. The antibodies can also bind to Fc‐gamma receptors (FcγRs) expressed on cells of the mononuclear phagocytic system. This pathway may be particularly important for ADCs that target circulating or secreted proteins or form larger immune complexes, as larger complexes tend to bind rapidly and avidly to FcγRs [64].
7. Clinical Effectiveness of AAC
To date, numerous studies have investigated the efficacy of AACs against various bacterial pathogens in vitro. However, animal and human studies have only examined the effectiveness of AAC against S. aureus, which causes several important bacterial infections in humans, along with complications such as endocarditis, pyelonephritis, and osteomyelitis [65]. Vancomycin and nafcillin are standard antibiotics used to treat S. aureus infections [66]. Unfortunately, these antibiotics have shown limited effectiveness in treating invasive S. aureus infections. S. aureus can seek refuge inside phagocytes, making it inaccessible to antibiotics. Therefore, a therapeutic agent is needed to target intracellular S. aureus in invasive cases to achieve better clinical outcomes [67]. To fulfill this purpose, a THIOMAB antibody–antibiotic conjugate (TAC) called DSTA4637S has been developed. TAC is composed of THIOMAB (a monoclonal IgG1 antibody recognizing S. aureus), dmDNA31 (a rifamycin‐class antibiotic), and a protease‐cleavable linker. TAC specifically binds to cells containing the bacterium using its antibody fragment. Once inside the cell, TAC is broken down by cellular proteases, releasing antibiotics to kill the bacterium [4]. There are limited in vivo studies that have evaluated the efficacy of TAC against S. aureus infections. Notably, a well‐known study conducted by Lehar et al. [4] found that TAC is more effective than vancomycin in vivo for treating intracellular S. aureus infections. In this study, mice were first treated with unconjugated anti‐MRSA antibodies (anti‐WTA antibodies) or intravenous immunoglobulin (IGIV). After 1 h, the mice were intravenously infected with wild‐type USA300 MRSA. The study revealed that prophylactic treatment with unconjugated antibodies was unable to prevent or eliminate S. aureus infection, whereas vancomycin successfully eliminated detectable bacteria. Vancomycin treatment was found to be ineffective when administered several hours after infection. This failure of vancomycin in the model could be attributed to the bacteria escaping into the host cell and the antibiotic's inability to enter the host cell. Subsequently, mice were treated with a single dose of TAC 24 h after infection. It was observed that the single‐dose TAC was significantly more effective than twice‐daily vancomycin therapy. The efficacy of TAC was found to rely on the release of the antibiotic, as TAC produced with a noncleavable linker was not effective in vivo. Although rifampicin lacks sufficient lipophilicity to penetrate the macrophage membrane, TAC can easily accumulate to lethal concentrations [4]. In a study by Deng et al. [12], the pharmacokinetics (PK) of DSTA4637A (a liquid formulation of DSTA4637S) and its unconjugated antibody MSTA3852A were characterized in rats and monkeys. Similarly, Zhou et al. [14] conducted a study in 2016 to characterize the pharmacokinetics (PK) and pharmacodynamics (PD) of TAC (DSTA4637A) in mice. These studies measured the systemic concentrations of total antibody (tAb), TAC (DSTA4637A), and unconjugated antibiotic to describe the PK of TAC (DSTA4637A) in nonclinical settings. The results showed that after intravenous injection, DSTA4637A can rapidly distribute to organs such as the heart, kidney, and bone, and it is cleared from the body 2–3 times faster than tAb. DSTA4637A substantially reduced the bacterial load in the heart, kidney, and bones on Days 7 and 14 after injection. Plasma concentrations of the unconjugated antibiotic (dmDNA31) remained low (<4 ng/mL) regardless of the dose. These studies have provided valuable insights into the PK/PD of DSTA4637A in preclinical species, its human PK/PD, and its clinical translatability [12, 14]. These studies utilized colony‐forming unit (CFU) measurements but had certain limitations and difficulties, which did not fully demonstrate the impact of TAC on disease progression in each individual animal. Therefore, another study employed the bioluminescence approach to investigate the antibacterial dynamics of TAC alone or in combination with vancomycin using a mouse model infected with stably luminescent S. aureus bacteria [67]. In this study, it was observed that after injection of the stably luminescent S. aureus, whole‐body bioluminescence increased whereas body weight and survival rate decreased. The researchers reported that vancomycin administered twice a day suppressed bacterial growth, but once treatment was discontinued, bacterial growth resumed in these animals. Interestingly, a single dose of TAC was sufficient to rapidly reduce bioluminescence intensity and bacterial growth for up to 19 days. Furthermore, the combination of TAC and vancomycin exhibited a greater reduction compared to vancomycin alone [67].
The first and only study to date investigating the efficacy of TAC in humans was conducted by Peck et al. [44]. This phase 1 trial analyzed the safety, pharmacokinetics, and immunogenicity of DSTA4637S in healthy volunteers. 30 healthy volunteers aged 18–65 years (both male and female) were randomly divided into five cohorts, receiving single intravenous (i.v.) doses of 5, 15, 50, 100, and 150 mg/kg of DSTA4637S or placebo (4 active:2 placebo), and followed for 85 days after dosing. The authors reported that there were no subject withdrawals, serious or severe adverse effects, clinically meaningful or dose‐related changes in laboratory parameters, or vital signs. The study found that the pharmacokinetics of plasma DSTA4637S conjugate and serum DSTA4637S total antibody were proportional to the dose. Systemic exposure to unconjugated dmDNA31 was low. They also observed no DSTA4637S‐induced antidrug antibody responses. Finally, they showed that DSTA4637S has a proper safety and pharmacokinetic profile, supporting its future development as a novel therapeutic for S. aureus infections [44].
8. Advantages and Disadvantages of AAC
The AAC platform can revive antibacterials that lack appropriate characteristics as unconjugated drugs. The pharmacokinetics of an antibacterial can be significantly enhanced by conjugating it to an IgG1, as demonstrated by the effective AAC against S. aureus (DSTA4637S). AACs utilize antibodies as carriers that selectively deliver antibiotics to the target, followed by efficient internalization and subsequent intracellular drug release. Because of the linked antibodies, AACs can target antigen‐positive cells even more effectively than antibiotics alone, while having fewer side effects on other host cells [68]. In fact, the AAC strategy combines the antibacterial effect of antibiotics with the target‐specific attachment of antibodies, resulting in superior pharmacokinetic properties associated with antibodies, such as slow clearance and a long half‐life. AACs offer several advantages in combating bacterial infections, including antibiotic‐resistant pathogens. Firstly, AACs enable precise delivery of antibiotics to target bacteria, ensuring efficient opsonization for enhanced phagocytosis [69]. This targeted delivery minimizes off‐target effects on host cells, resulting in high specificity, and permits long‐term therapy of persistent infections with an AAC with minimal antibiotic side effects. In addition, this property of AAC transforms a broad‐spectrum antibiotic into a pathogen‐specific antibiotic, resulting in a reduction of dysbiosis in the gut microbiota and related disorders such as Clostridioides difficile infection (CDI), inflammatory bowel disease (IBD), obesity, and atherosclerosis [1]. Additionally, AACs utilize Fc receptors (FcR) to facilitate uptake by immune cells, enhancing the clearance of intracellular bacteria. Within intracellular environments like phagolysosomes, AACs release active antibiotics, ensuring effective bacterial eradication [69]. Another potential application of AAC might be for bacterial biofilms, where an antibody that recognizes specific antigens on the biofilm can concentrate AACs at these sites and consequently release a high local concentration of active antibiotics at the biofilm [1, 3]. Furthermore, AACs demonstrate efficacy even against antibiotic‐resistant bacterial strains, addressing the challenge of antibiotic resistance [48]. These advantages make AACs a promising therapeutic platform for combating bacterial infections, particularly in the context of antibiotic resistance [48]. By specifically targeting bacteria and delivering antibiotics directly to the site of infection, AACs can improve the effectiveness of drugs while reducing the selection pressure of antibiotics [3]. This targeted approach may help overcome resistance and reduce adverse effects associated with high drug dosages [48]. Overall, AACs offer a multifaceted approach to tackling bacterial infections, providing targeted delivery, enhanced clearance of bacteria, and efficacy against antibiotic‐resistant strains. Although the high cost of AACs compared with antibiotics is one of the major disadvantages of this strategy, its future use is predictable through further research aimed at reducing costs and the probability of the emergence of antibiotic‐resistant infections [1, 68, 70]. In comparison with native protein therapeutics (e.g., cytokines and antibodies), biotherapeutics such as AACs face new challenges related to their stability, catabolism, and elimination because of their novelty and complexity [71].
Considering that AAC consists of three components: antibiotic, linker, and antibody, the production is very complex and specific [72]. Linkers are made of complex chemicals that sometimes require safety considerations when working with strong compounds. Therefore, the number of manufacturers capable of producing linkers is limited [72, 73]. The conjugation step required to attach the linker to the monoclonal antibody (mAb) presents another production hurdle. mAb bioproduction involves cell line and process development for the recombinant protein, followed by large‐scale production and downstream processing of clinical material [73, 74]. The production of AAC involves several general steps, including mAb design and synthesis, linker design and synthesis, conjugation, drug synthesis and production (including purification, formulation, filtration, filling, and lyophilization), and labeling and packaging of vials. Therefore, the AAC supply chain is highly complex and may involve coordination among five different companies, all of whose activities must be coordinated by the sponsor [72, 73, 74].
9. The Future of Antibody–Antibiotic Conjugates
Since ancient times, the use of antibiotics has significantly improved the treatment for bacterial infections. However, the increase in drug‐resistant pathogens and treatment failure has impacted the study of innovative strategies [75]. As a result, peptides have emerged as a promising alternative to antibiotics in the next step. Peptides possess several properties, such as potency, very low toxicity, high biological diversity, and unique mechanisms of action (targeting the bacterial membrane and/or cytoplasm), which differ from the performance of conventional antibiotics. These properties can potentially contribute to a new era of antimicrobials by reducing bacterial resistance [76]. However, the main disadvantage of peptides in their clinical use is related to their physiological stability. Peptidomimetics have been investigated in several studies to overcome this limitation, as various chemical and physical modifications can be applied to increase proteolytic stability, bioavailability, and improve pharmacokinetics [3]. Nevertheless, the results indicate that the production of peptidomimetics may not be effective enough to further enhance the application of peptides. Therefore, similar to drugs and antibiotics, peptides can also be conjugated with antibodies to improve their physicochemical properties and create peptides that are sufficiently effective in therapy [75].
AAC has the ability to release potent antibacterial compositions, which may not be achieved by unconjugated drugs because of poor pharmacokinetic parameters and a narrow therapeutic index. Weak antibiotic concentrations and recurrent diseases have been associated with the ability of various bacterial pathogens to survive within host cells. According to the results of a phase 1 randomized, single‐ascending‐dose study, the current AAC system shows promise in enhancing the antibiotic effect against these infections.
10. Conclusion
One of the problems currently facing bacterial infections is antibiotic resistance, which leads to treatment failure. As a potential treatment for severe bacterial infections, a novel ADC method called AAC is being considered. AAC is composed of three parts: (i) antibody, (ii) antibiotic, and (iii) linker. Many classical antibiotics have a narrow therapeutic index and poor pharmacokinetic parameters against intracellular bacteria because of limited intracellular penetration. The triggered release of antibiotics from AACs can improve their pharmacokinetic parameters and therapeutic index. AACs aim to expand the therapeutic index of conventional antibiotics by utilizing the targeting specificity of antibodies to enhance the delivery efficiency of antibiotics to infected cells. Phase 1 randomized, single‐ascending‐dose studies have shown that the AAC system holds promise in increasing the effectiveness of antibiotics against dormant bacteria.
Author Contributions
R.G. and B.T. conceived, designed, and supervised the study. A.D. and M.A. contributed to data collection, interpretation, and final approval of data for the work. S.D. and S.Y. developed the first and final drafts of the manuscript. M.S. and E.K. developed the second draft of the manuscript. All figures and tables were designed and checked by T.D. and A.D. All authors reviewed and contributed to the revisions and finalized the drafts.
Ethics Statement
This study was approved by the Ethics Committee (code number: IR.BHN.REC.1402.014) of Behbahan Faculty of Medical Sciences.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding: This study was supported by Behbahan Faculty of Medical Sciences (grant number: 401088).
Roya Ghanavati and Behrouz Taheri contributed equally to this work.
Contributor Information
Roya Ghanavati, Email: r.ghanavati@behums.ac.ir, Email: qanavati.r@gmail.com.
Behrouz Taheri, Email: taheri.b1980@gmail.com.
Data Availability Statement
All relevant data are included in the manuscript.
References
- 1. Mariathasan S. and Tan M.‐W., “Antibody–Antibiotic Conjugates: A Novel Therapeutic Platform Against Bacterial Infections,” Trends in Molecular Medicine 23, no. 2 (2017): 135–149. [DOI] [PubMed] [Google Scholar]
- 2. Ricciardi B. F., Muthukrishnan G., Masters E., Ninomiya M., Lee C. C., and Schwarz E. M., “ Staphylococcus aureus Evasion of Host Immunity in the Setting of Prosthetic Joint Infection: Biofilm and Beyond,” Current Reviews in Musculoskeletal Medicine 11 (2018): 389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cavaco M., Castanho M. A., and Neves V., “The Use of Antibody–Antibiotic Conjugates to Fight Bacterial Infections,” Frontiers in Microbiology 13 (2022): 835677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lehar S. M., Pillow T., Xu M., et al., “Novel Antibody–Antibiotic Conjugate Eliminates Intracellular S. aureus ,” Nature 527, no. 7578 (2015): 323–328. [DOI] [PubMed] [Google Scholar]
- 5. Staben L. R., Koenig S. G., Lehar S. M., et al., “Targeted Drug Delivery Through the Traceless Release of Tertiary and Heteroaryl Amines From Antibody–Drug Conjugates,” Nature Chemistry 8, no. 12 (2016): 1112–1119. [DOI] [PubMed] [Google Scholar]
- 6. Pillow T. H., “Novel Linkers and Connections for Antibody–Drug Conjugates to Treat Cancer and Infectious Disease,” Pharmaceutical Patent Analyst 6, no. 1 (2017): 25–33. [DOI] [PubMed] [Google Scholar]
- 7. Wagner E. K. and Maynard J. A., “Engineering Therapeutic Antibodies to Combat Infectious Diseases,” Current Opinion in Chemical Engineering 19 (2018): 131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shah P. S. and Kaufman D. A., “Antistaphylococcal Immunoglobulins to Prevent Staphylococcal Infection in Very Low Birth Weight Infants,” Cochrane Database of Systematic Reviews, no. 2 (2009): CD006449. [DOI] [PubMed] [Google Scholar]
- 9. Shopsin B., Kaveri S. V., and Bayry J., “Tackling Difficult Staphylococcus aureus Infections: Antibodies Show the Way,” Cell Host & Microbe 20, no. 5 (2016): 555–557. [DOI] [PubMed] [Google Scholar]
- 10. Chetchotisakd P. and Anunnatsiri S., “Linezolid in the Treatment of Disseminated Nontuberculous Mycobacterial Infection in Anti‐Interferon‐[Gamma] Autoantibody‐Positive Patients,” Southeast Asian Journal of Tropical Medicine and Public Health 45, no. 5 (2014): 1125–1131. [PubMed] [Google Scholar]
- 11. Wang‐Lin S. X. and Balthasar J. P., “Pharmacokinetic and Pharmacodynamic Considerations for the Use of Monoclonal Antibodies in the Treatment of Bacterial Infections,” Antibodies 7, no. 1 (2018): 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deng R., Zhou C., Li D., et al., “Preclinical and Translational Pharmacokinetics of a Novel THIOMAB™ Antibody–Antibiotic Conjugate Against Staphylococcus aureus ,” MAbs 11 (2019): 1162–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Drake P. M. and Rabuka D., “Recent Developments in ADC Technology: Preclinical Studies Signal Future Clinical Trends,” BioDrugs 31, no. 6 (2017): 521–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhou C., Lehar S., Gutierrez J., et al., “Pharmacokinetics and Pharmacodynamics of DSTA4637A: A Novel THIOMAB™ Antibody Antibiotic Conjugate Against Staphylococcus aureus in Mice,” MAbs 8 (2016): 1612–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang‐Lin S. X., Zhou C., Kamath A. V., et al., “Minimal Physiologically‐Based Pharmacokinetic Modeling of DSTA4637A, A Novel THIOMAB™ Antibody Antibiotic Conjugate Against Staphylococcus aureus, in a Mouse Model,” MAbs 10 (2018): 1131–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nagy C. F., Leach T. S., Hoffman J. H., Czech A., Carpenter S. E., and Guttendorf R., “Pharmacokinetics and Tolerability of Obiltoxaximab: A Report of 5 Healthy Volunteer Studies,” Clinical Therapeutics 38, no. 9 (2016): 2083–2097.e7. [DOI] [PubMed] [Google Scholar]
- 17. Tsai C.‐W. and Morris S., “Approval of Raxibacumab for the Treatment of Inhalation Anthrax Under the US Food and Drug Administration “Animal Rule”,” Frontiers in Microbiology 6 (2015): 1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wilcox M. H., Gerding D. N., Poxton I. R., et al., “Bezlotoxumab for Prevention of Recurrent Clostridium Difficile Infection,” New England Journal of Medicine 376, no. 4 (2017): 305–317. [DOI] [PubMed] [Google Scholar]
- 19. Troisi M., Marini E., Abbiento V., Stazzoni S., Andreano E., and Rappuoli R., “A New Dawn for Monoclonal Antibodies Against Antimicrobial Resistant Bacteria,” Frontiers in Microbiology 13 (2022): 1080059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vacca F., Sala C., and Rappuoli R., “Monoclonal Antibodies for Bacterial Pathogens: Mechanisms of Action and Engineering Approaches for Enhanced Effector Functions,” Biomedicine 10, no. 9 (2022): 2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nagy E., Nagy G., Power C. A., Badarau A., and Szijártó V., “Anti‐Bacterial Monoclonal Antibodies,” Recombinant Antibodies for Infectious Diseases 1053 (2017): 119–153. [DOI] [PubMed] [Google Scholar]
- 22. Lu L. L., Suscovich T. J., Fortune S. M., and Alter G., “Beyond Binding: Antibody Effector Functions in Infectious Diseases,” Nature Reviews Immunology 18, no. 1 (2018): 46–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bebbington C. and Yarranton G., “Antibodies for the Treatment of Bacterial Infections: Current Experience and Future Prospects,” Current Opinion in Biotechnology 19, no. 6 (2008): 613–619. [DOI] [PubMed] [Google Scholar]
- 24. Zurawski D. V. and McLendon M. K., “Monoclonal Antibodies as an Antibacterial Approach Against Bacterial Pathogens,” Antibiotics 9, no. 4 (2020): 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lapidoth G., Parker J., Prilusky J., and Fleishman S. J., “AbPredict 2: A Server for Accurate and Unstrained Structure Prediction of Antibody Variable Domains,” Bioinformatics 35, no. 9 (2019): 1591–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Almagro J. C., Teplyakov A., Luo J., et al., “Second Antibody Modeling Assessment (AMA‐II),” Proteins: Structure, Function, and Bioinformatics 82, no. 8 (2014): 1553–1562. [DOI] [PubMed] [Google Scholar]
- 27. Wang W., Singh S., Zeng D. L., King K., and Nema S., “Antibody Structure, Instability, and Formulation,” Journal of Pharmaceutical Sciences 96, no. 1 (2007): 1–26. [DOI] [PubMed] [Google Scholar]
- 28. Esteve‐Turrillas F. A., Armenta S., Garrigues S., and de la Guardia M., “Smart Sorption Materials in Green Analytical Chemistry,” in Green Analytical Chemistry, vol. 22, eds. Płotka‐Wasylka J. and Namieśnik J. (Singapore: Springer, 2019), 167–202. [Google Scholar]
- 29. Chiu M. L., Goulet D. R., Teplyakov A., and Gilliland G. L., “Antibody Structure and Function: The Basis for Engineering Therapeutics,” Antibodies 8, no. 4 (2019): 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Davies D. R. and Chacko S., “Antibody Structure,” Accounts of Chemical Research 26, no. 8 (1993): 421–427. [Google Scholar]
- 31. Vidarsson G., Dekkers G., and Rispens T., “IgG Subclasses and Allotypes: From Structure to Effector Functions,” Frontiers in Immunology 5 (2014): 520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Swindells M. B., Porter C. T., Couch M., et al., “abYsis: Integrated Antibody Sequence and Structure—Management, Analysis, and Prediction,” Journal of Molecular Biology 429, no. 3 (2017): 356–364. [DOI] [PubMed] [Google Scholar]
- 33. Hayat S. M. G. and Sahebkar A., “Antibody–Drug Conjugates: Smart Weapons Against Cancer,” Archives of Medical Science 16, no. 5 (2019): 1257–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tsuchikama K. and An Z., “Antibody–Drug Conjugates: Recent Advances in Conjugation and Linker Chemistries,” Protein & Cell 9, no. 1 (2018): 33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Baah S., Laws M., and Rahman K. M., “Antibody–Drug Conjugates—A Tutorial Review,” Molecules 26, no. 10 (2021): 2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aslam B., Wang W., Arshad M. I., et al., “Antibiotic Resistance: A Rundown of a Global Crisis,” Infection and Drug Resistance 11 (2018): 1645–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ventola C. L., “The Antibiotic Resistance Crisis: Part 1: Causes and Threats,” Pharmacy and Therapeutics 40, no. 4 (2015): 277–283. [PMC free article] [PubMed] [Google Scholar]
- 38. Gautam S., Kim T., Lester E., Deep D., and Spiegel D. A., “Wall Teichoic Acids Prevent Antibody Binding to Epitopes Within the Cell Wall of Staphylococcus aureus ,” ACS Chemical Biology 11, no. 1 (2016): 25–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yaghoubi S., Gharibi T., Karimi M. H., et al., “Development and Biological Assessment of MMAE‐Trastuzumab Antibody–Drug Conjugates (ADCs),” Breast Cancer 28 (2021): 216–225. [DOI] [PubMed] [Google Scholar]
- 40. Abdollahpour‐Alitappeh M., Lotfinia M., Bagheri N., et al., “Trastuzumab‐Monomethyl Auristatin E Conjugate Exhibits Potent Cytotoxic Activity In Vitro Against HER2‐Positive Human Breast Cancer,” Journal of Cellular Physiology 234, no. 3 (2019): 2693–2704. [DOI] [PubMed] [Google Scholar]
- 41. Abdollahpour‐Alitappeh M., Hashemi Karouei S. M., Lotfinia M., Amanzadeh A., and Habibi‐Anbouhi M., “A Developed Antibody–Drug Conjugate Rituximab‐vcMMAE Shows a Potent Cytotoxic Activity Against CD20‐Positive Cell Line,” Artificial Cells, Nanomedicine, and Biotechnology 46, no. suppl 2 (2018): 1–8. [DOI] [PubMed] [Google Scholar]
- 42. Najminejad Z., Dehghani F., Mirzaei Y., et al., “Clinical Perspective: Antibody–Drug Conjugates (ADCs) for the Treatment of HER2‐Positive Breast Cancer,” Molecular Therapy 31 (2023): 1874–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kajihara K. K., Pantua H., Hernandez‐Barry H., et al., “Potent Killing of Pseudomonas aeruginosa by an Antibody–Antibiotic Conjugate,” mBio 12, no. 3 (2021): e0020221, 10.1128/mbio.00202-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Peck M., Rothenberg M. E., Deng R., et al., “A Phase 1, Randomized, Single‐Ascending‐Dose Study to Investigate the Safety, Tolerability, and Pharmacokinetics of DSTA4637S, an Anti‐Staphylococcus aureus Thiomab Antibody–Antibiotic Conjugate, in Healthy Volunteers,” Antimicrobial Agents and Chemotherapy 63, no. 6 (2019): e02588‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kurokawa K., Jung D.‐J., An J.‐H., et al., “Glycoepitopes of Staphylococcal Wall Teichoic Acid Govern Complement‐Mediated Opsonophagocytosis Via Human Serum Antibody and Mannose‐Binding Lectin,” Journal of Biological Chemistry 288, no. 43 (2013): 30956–30968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Brown S., Xia G., Luhachack L. G., et al., “Methicillin Resistance in Staphylococcus aureus Requires Glycosylated Wall Teichoic Acids,” National Academy of Sciences of the United States of America 109, no. 46 (2012): 18909–18914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cavaco M., Castanho M., and Neves V., “Peptibodies: An Elegant Solution for a Long‐Standing Problem,” Biopolymers 110 (2017): e23095. [DOI] [PubMed] [Google Scholar]
- 48. Yu L., Shang Z., Jin Q., et al., “Antibody–Antimicrobial Conjugates for Combating Antibiotic Resistance,” Advanced Healthcare Materials 12, no. 1 (2023): 2202207. [DOI] [PubMed] [Google Scholar]
- 49. Khongorzul P., Ling C. J., Khan F. U., Ihsan A. U., and Zhang J., “Antibody–Drug Conjugates: A Comprehensive Review,” Molecular Cancer Research 18, no. 1 (2020): 3–19. [DOI] [PubMed] [Google Scholar]
- 50. Negash K. H., Norris J. K., and Hodgkinson J. T., “Siderophore–Antibiotic Conjugate Design: New Drugs for Bad Bugs?” Molecules 24, no. 18 (2019): 3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Behrens C. R. and Liu B., “Methods for Site‐Specific Drug Conjugation to Antibodies,” MAbs 6, no. 1 (2014): 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Su D. and Zhang D., “Linker Design Impacts Antibody–Drug Conjugate Pharmacokinetics and Efficacy via Modulating the Stability and Payload Release Efficiency,” Frontiers in Pharmacology 12 (2021): 687926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Birrer M. J., Moore K. N., Betella I., and Bates R. C., “Antibody–Drug Conjugate‐Based Therapeutics: State of the Science,” Journal of the National Cancer Institute 111, no. 6 (2019): 538–549. [DOI] [PubMed] [Google Scholar]
- 54. McConnell M. J., “Where Are We With Monoclonal Antibodies for Multidrug‐Resistant Infections?” Drug Discovery Today 24, no. 5 (2019): 1132–1138. [DOI] [PubMed] [Google Scholar]
- 55. Shimamoto G., Gegg C., Boone T., and Queva C., “Peptibodies: A Flexible Alternative Format to Antibodies,” MAbs 4 (2012): 586–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Metersky M. L. and Kalil A. C., “Management of Ventilator‐Associated Pneumonia: Guidelines,” Clinics in Chest Medicine 39, no. 4 (2018): 797–808. [DOI] [PubMed] [Google Scholar]
- 57. Beck A., Goetsch L., Dumontet C., and Corvaïa N., “Strategies and Challenges for the Next Generation of Antibody–Drug Conjugates,” Nature Reviews Drug Discovery 16, no. 5 (2017): 315–337. [DOI] [PubMed] [Google Scholar]
- 58. Lucas A. T., Price L. S. L., Schorzman A. N., et al., “Factors Affecting the Pharmacology of Antibody–Drug Conjugates,” Antibodies 7, no. 1 (2018): 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bruno B. J., Miller G. D., and Lim C. S., “Basics and Recent Advances in Peptide and Protein Drug Delivery,” Therapeutic Delivery 4, no. 11 (2013): 1443–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhao L., Ji P., Li Z., Roy P., and Sahajwalla C. G., “The Antibody Drug Absorption Following Subcutaneous or Intramuscular Administration and Its Mathematical Description by Coupling Physiologically Based Absorption Process With the Conventional Compartment Pharmacokinetic Model,” Journal of Clinical Pharmacology 53, no. 3 (2013): 314–325. [DOI] [PubMed] [Google Scholar]
- 61. Tabrizi M., Bornstein G. G., and Suria H., “Biodistribution Mechanisms of Therapeutic Monoclonal Antibodies in Health and Disease,” The AAPS Journal 12, no. 1 (2010): 33–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Roberts J. A., Pea F., and Lipman J., “The Clinical Relevance of Plasma Protein Binding Changes,” Clinical Pharmacokinetics 52, no. 1 (2013): 1–8. [DOI] [PubMed] [Google Scholar]
- 63. Ferri N., Bellosta S., Baldessin L., Boccia D., Racagni G., and Corsini A., “Pharmacokinetics Interactions of Monoclonal Antibodies,” Pharmacological Research 111 (2016): 592–599. [DOI] [PubMed] [Google Scholar]
- 64. Lux A., Yu X., Scanlan C. N., and Nimmerjahn F., “Impact of Immune Complex Size and Glycosylation on IgG Binding to Human FcγRs,” Journal of Immunology 190, no. 8 (2013): 4315–4323. [DOI] [PubMed] [Google Scholar]
- 65. Ortines R. V., Wang Y., Liu H., et al., “Efficacy of a Multimechanistic Monoclonal Antibody Combination Against Staphylococcus aureus Surgical Site Infections in Mice,” Antimicrobial Agents and Chemotherapy 63, no. 8 (2019): e00346‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Liu C., Bayer A., Cosgrove S. E., et al., “Clinical Practice Guidelines by the Infectious Diseases Society of America for the Treatment of Methicillin‐Resistant Staphylococcus aureus Infections in Adults and Children,” Clinical Infectious Diseases 52, no. 3 (2011): e18–e55. [DOI] [PubMed] [Google Scholar]
- 67. Zhou C., Cai H., Baruch A., et al., “Sustained Activity of Novel THIOMAB Antibody–Antibiotic Conjugate Against Staphylococcus aureus in a Mouse Model: Longitudinal Pharmacodynamic Assessment by Bioluminescence Imaging,” PLoS One 14, no. 10 (2019): e0224096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Liu R., Wang R. E., and Wang F., “Antibody–Drug Conjugates for Non‐Oncological Indications,” Expert Opinion on Biological Therapy 16 (2016): 591–593. [DOI] [PubMed] [Google Scholar]
- 69. Romski M. and Sevcik R. A., “Augmentative Communication and Early Intervention: Myths and Realities,” Infants & Young Children 18, no. 3 (2005): 174–185. [Google Scholar]
- 70. Forte N., Chudasama V., and Baker J. R., “Homogeneous Antibody–Drug Conjugates via Site‐Selective Disulfide Bridging,” Drug Discovery Today: Technologies 30 (2018): 11–20. [DOI] [PubMed] [Google Scholar]
- 71. Tibbitts J., Canter D., Graff R., Smith A., and Khawli L. A., “Key Factors Influencing ADME Properties of Therapeutic Proteins: A Need for ADME Characterization in Drug Discovery and Development,” MAbs 8, no. 2 (2016): 229–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sifniotis V., Cruz E., Eroglu B., and Kayser V., “Current Advancements in Addressing Key Challenges of Therapeutic Antibody Design, Manufacture, and Formulation,” Antibodies 8, no. 2 (2019): 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Alexander P., Weitzel K., Linderholm A., Haskins W. E., Ghone S., and Chamow S. M., Manufacturing Challenges of Therapeutic Antibody–Drug Conjugates: Bioprocess International 2023, https://www.bioprocessintl.com/sponsored‐content/manufacturing‐challenges‐of‐therapeutic‐antibody‐drug‐conjugates.
- 74. Scanlan C., Zhao J., Rogers M., et al., Antibody‐Drug Conjugates: Manufacturing Challenges and Trends (ADC Review, 2017). 10.14229/jadc.2017.21.03.001. [DOI] [Google Scholar]
- 75. Mood E. H., Goltermann L., Brolin C., et al., “Antibiotic Potentiation in Multidrug‐Resistant Gram‐Negative Pathogenic Bacteria by a Synthetic Peptidomimetic,” ACS Infectious Diseases 7, no. 8 (2021): 2152–2163. [DOI] [PubMed] [Google Scholar]
- 76. Zhang Q.‐Y., Yan Z.‐B., Meng Y.‐M., et al., “Antimicrobial Peptides: Mechanism of Action, Activity and Clinical Potential,” Military Medical Research 8 (2021): 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are included in the manuscript.