

Abstract
Objective.
Although oral methotrexate (MTX) remains the anchor drug for rheumatoid arthritis (RA), up to 50% of patients do not achieve a clinically adequate outcome. In addition, there is a lack of prognostic tools for treatment response prior to drug initiation. This study was undertaken to investigate whether interindividual differences in the human gut microbiome can aid in the prediction of MTX efficacy in new-onset RA.
Methods.
We performed 16S ribosomal RNA gene and shotgun metagenomic sequencing on the baseline gut microbiomes of drug-naive patients with new-onset RA (n = 26). Results were validated in an additional independent cohort (n = 21). To gain insight into potential microbial mechanisms, we conducted ex vivo experiments coupled with metabolomics analysis to evaluate the association between microbiome-d riven MTX depletion and clinical response.
Results.
Our analysis revealed significant associations of the abundance of gut bacterial taxa and their genes with future clinical response (q < 0.05), including orthologs related to purine and MTX metabolism. Machine learning techniques were applied to the metagenomic data, resulting in a microbiome-based model that predicted lack of response to MTX in an independent group of patients. Finally, MTX levels remaining after ex vivo incubation with distal gut samples from pretreatment RA patients significantly correlated with the magnitude of future clinical response, suggesting a possible direct effect of the gut microbiome on MTX metabolism and treatment outcomes.
Conclusion.
Taken together, these findings are the first step toward predicting lack of response to oral MTX in patients with new-onset RA and support the value of the gut microbiome as a possible prognostic tool and as a potential target in RA therapeutics.
INTRODUCTION
Despite multiple advances in the understanding of rheumatoid arthritis (RA) pathogenesis and in the development of therapeutics (1), oral methotrexate (MTX) remains the mainstay of therapy (2,3). MTX is a dihydrofolate reductase inhibitor and is considered a disease-modifying drug since it ameliorates symptoms and prevents joint destruction. Importantly, only up to 50% of patients will have a clinically adequate response when the drug is administered as monotherapy (2,4). The reasons for this discrepancy in clinical outcomes are not clearly understood, although one possibility may relate to interindividual differences in the bioavailability of oral MTX, which is known to be highly variable (range 20–80%) (5). This is of utmost relevance since MTX is well tolerated, safe, and has a significantly lower cost compared to newer biologic therapies (6), making it first-line therapy in RA worldwide (7,8).
Despite decades of study, the interindividual variation in MTX response cannot be accurately predicted by host biomarkers (9–14), and the determination of responder status requires a lengthy trial, creating a window for joint damage to accrue. More recently, several groups have characterized the dependence of immunomodulatory therapies on the gut microbiome and its utility as a predictor of clinical response (15–20). Accordingly, it is possible that the bioavailability and/or subsequent response to MTX could at least be partially driven by differences in the microbial species, genes, enzymes, and/or metabolites found within the gastrointestinal tracts of RA patients. The premise for this hypothesis is supported by prior work in rodents, where both germ-free housing and antibiotic depletion significantly decreased intestinal absorption and metabolism of MTX (21,22). However, our understanding of the role of the human gut microbiome in RA treatment remains limited (23). In this study, we sought to address these knowledge gaps by determining if the pretreatment gut microbiome is associated with prediction of drug efficacy and whether human bacterial communities could directly metabolize MTX.
PATIENTS AND METHODS
Patients.
Consecutive patients from the New York University Langone Medical Center, Lutheran Hospital, Staten Island and Mount Sinai School of Medicine rheumatology clinics and offices were screened for RA. Eligible patients with active, new-onset RA were included in the study (see Supplementary Methods, available on the Arthritis & Rheumatology website a http://onlinelibrary.wiley.com/doi/10.1002/art.41622/abstract).
Study design.
All patients with new-onset RA (n = 26 for the training cohort and n = 21 for the validation cohort) were recruited using established protocols (24). Clinical and demographic characteristics of the training and validation cohorts are shown in Supplementary Tables 1 and 2, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41622/abstract). Biologic samples and metadata were obtained before treatment with MTX and folic acid and 1, 2, and 4 months after therapy initiation. A responder to MTX was defined a priori as any patient with new-onset RA with an improvement in the Disease Activity Score in 28 joints (DAS28) (25) of ≥1.8 by month 4 after initiation of MTX monotherapy. Biologic samples from 20 RA patients who subsequently either never initiated MTX or were prospectively prescribed other medications were analyzed as controls (see Supplementary Methods).
Microbiome sequencing and analysis.
We performed 16S ribosomal RNA (16S rRNA) and shotgun sequencing analysis as described in detail in the Supplementary Methods. The 16S rRNA sequencing led to the identification of each taxa, operational taxonomic unit (OTU), and ribosomal sequence variant (RSV) present in a given sample. Shotgun sequencing led to the identification of KEGG modules, pathways, and gene orthologs. Sequencing data generated during the study are available at the NCBI Sequence Read Archive (accession #PRJNA682730). Data include accession codes and unique alphanumeric identifiers associated with raw data.
Identification of a microbiome-based model to predict response to MTX.
Features (i.e., OTUs, RSVs, and KEGG orthologs [KOs]) for model development were selected by applying the Boruta algorithm (26) to the samples from the training cohort (Supplementary Methods and https://github.com/scher-lab). A random forests model was fitted using the features identified, and the accuracy of the model was evaluated in an independent validation cohort of patients with new-onset RA (Supplementary Methods) and in a control cohort of RA patients who subsequently either never initiated MTX or were prospectively prescribed other medications.
Ex vivo incubation of fecal samples.
Fecal samples were incubated ex vivo with MTX, and the remaining levels of MTX were measured by nuclear magnetic resonance (NMR) spectroscopy or liquid chromatography mass spectroscopy (LC-MS) (see Supplementary Methods).
Statistical analysis.
The DESeq2 algorithm and the false discovery rate were applied to identify differences in the abundance of microbiome features, while Bray-Curtis distance-based permutational multivariate analysis of variance (PERMANOVA) was used to detect overall differences in the gut microbiome (Supplementary Methods). Spearman’s correlation test was used to detect associations between continuous variables using GraphPad Prism version 6.0. P values less than 0.05 and q values less than 0.05 were considered significant.
RESULTS
Pretreatment gut microbial community structure differentiates clinical response to MTX.
We first investigated whether the pretreatment gut microbial community structure could differentiate clinical response to MTX in patients with new-onset RA. We collected stool samples from a training cohort of 26 patients with new-onset RA (Supplementary Figure 1 and Supplementary Tables 1 and 2, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41622/abstract). All patients enrolled received oral MTX at standard-of-care dosing (average 20 mg/week, range 15–25 mg). Fecal samples were obtained within 48 hours prior to treatment initiation. We then classified patients as either MTX responders (39% of the cohort) or MTX nonresponders (61% of the cohort) based on a stringent definition of clinical response (improvement in DAS28 of ≥1.8 and no need for adding a biologic drug) at month 4 after therapy initiation (7).
Using 16S rRNA gene sequencing, we first analyzed differences in microbial diversity between MTX nonresponders (n = 16) and MTX responders (n = 10). Patients who responded to therapy had significantly lower microbial diversity at the OTU level (P < 0.05 by Wilcoxon’s 2-sided test) (Figure 1a), with a similar trend observed for richness (Figure 1a). A significant difference in overall gut microbial community structure was also observed between groups based on abundance of OTUs and other taxonomic levels (principal components analysis based on Bray-Curtis distance; P < 0.05 by PERMANOVA) (Figures 1b and c). Subsequently, we applied the DESeq2 algorithm to test for differential abundance in bacterial groups that could be driving this separation. At the phylum level, we detected a higher abundance of Firmicutes and lower abundance of Bacteroidetes in nonresponders (P < 0.05, q = 0.08) (Supplementary Figure 2, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41622/abstract) and, consequently, a higher Firmicutes:Bacteroidetes ratio in nonresponders (P < 0.05 by Wilcoxon’s 2-sided test) (Figure 1d). In addition, samples from nonresponders to MTX showed a higher abundance of the Euryarchaeota phylum (q < 0.05 by DESeq2) (Supplementary Figure 2), unclassified Clostridiales/Clostridiales incertae sedis XIII family, and Escherichia/Shigella genera (q < 0.05 by DESeq2) (Supplementary Figure 2). We also detected differences in 5 low abundance taxa (median abundance per group <0.01%) (Supplementary Table 3, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41622/abstract).
Figure 1.
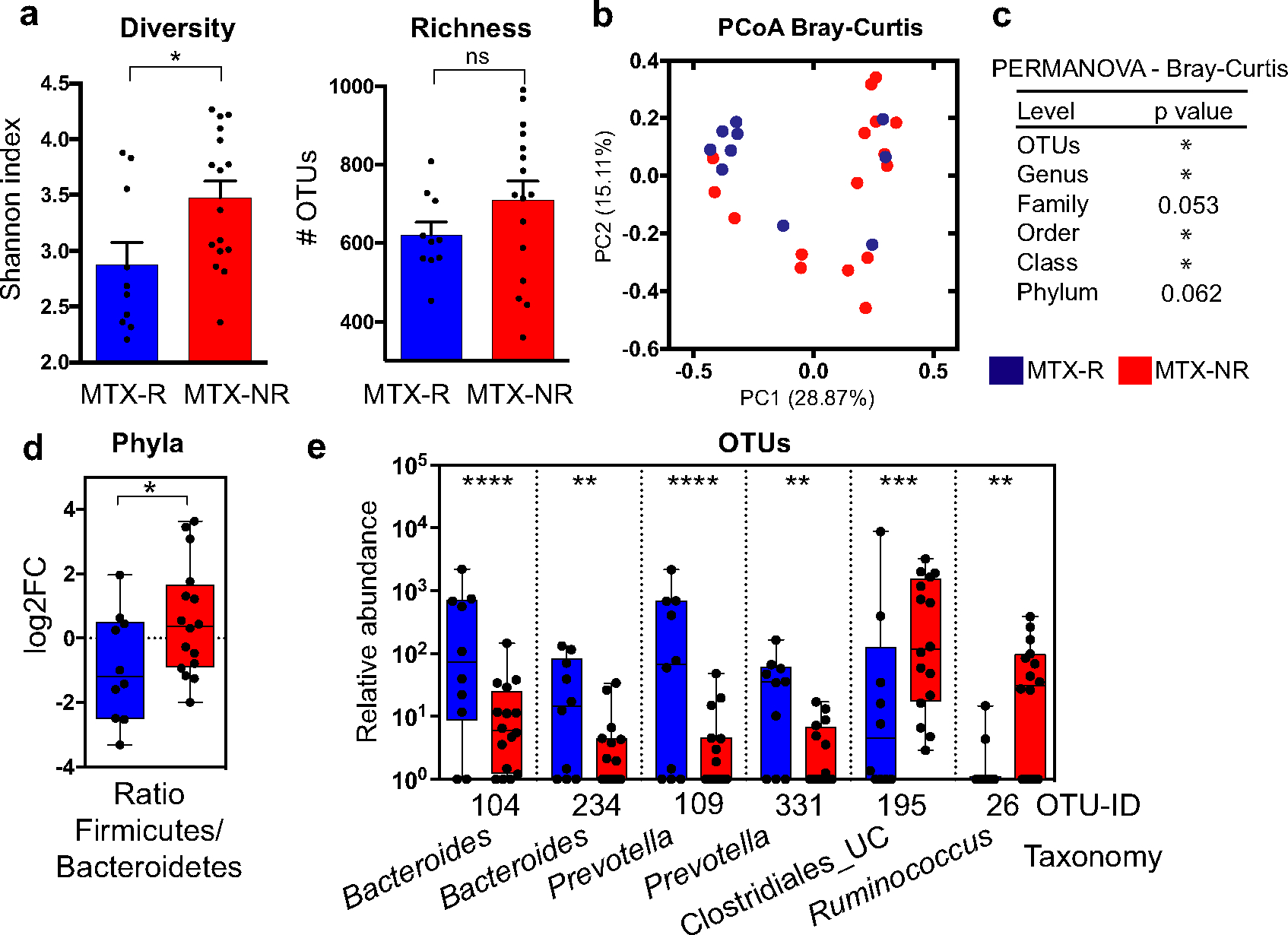
Pretreatment gut microbial diversity and taxa in a training cohort of patients with new-onset rheumatoid arthritis (RA) who responded to methotrexate (MTX-R) and patients with new-onset RA who did not respond to MTX (MRX-NR). a, Diversity (Shannon index) and richness (number of operational taxonomic units [OTUs]) of pretreatment microbiota in responders and nonresponders to MTX. Bars show the mean ± SEM. Symbols represent individual patients (n = 10–16 per group). * = P < 0.05. NS = not significant. b, Principal components analysis (PcoA) of samples from responders and nonresponders to MTX based on their pretreatment microbiota composition at the OTU level, using Bray-Curtis distance. PC1 = principal component 1. c, Significant differences in gut microbial community structure between responders and nonresponders to MTX at the indicated taxonomic levels, determined by Bray-Curtis distance–based permutational multivariate analysis of variance (PERMANOVA). * = P < 0.05. d, Firmicutes:Bacteroidetes ratio in responders and nonresponders to MTX. * = P < 0.05 by Wilcoxon’s 2-tailed test. FC = fold change. e, Significantly different relative abundance (counts per 105) of OTUs in responders to MTX versus nonresponders to MTX (q < 0.05 by DESeq2). Only OTUs with a median abundance >0.01% in ≥1 group are shown. Significantly different low abundance OTUs (q < 0.05 by DESeq2) are shown in Supplementary Table 3, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41622/abstract. In d and e, data are shown as box plots. Boxes represent the 25th to 75th percentiles. Lines inside the boxes represent the median. Whiskers indicate maximum and minimum values. Symbols represent individual patients (n = 10–16 per group). UC = unclassified. ** = P < 0.01; *** = P < 0.001; **** = P < 0.0001.
Consistent with the lower Firmicutes:Bacteroidetes ratio in responders to MTX, we found that several OTUs from the Bacteroides and Prevotella genera (Bacteroidetes phylum) were significantly more abundant in this group (q < 0.05, by DESeq2) (Figure 1e), with a concomitant decrease in OTUs from the order Clostridiales and the genus Ruminococcus (Firmicutes phylum) (q < 0.0 by DESeq2) (Figure 1e). Differences in 14 additional low abundant OTUs were also detected between groups (Supplementary Table 3). Analysis of RSVs revealed similar results (Supplementary Figure 3 and Supplementary Table 3).
MTX nonresponders have consistent differences in gut microbial gene abundance relative to responders.
Although we identified some differences in gut microbial community structure between patient groups, 16S rRNA sequencing can only approximate metabolic capacity, in particular for genes related to drug metabolism (27,28). We therefore performed shotgun sequencing to define the bacterial metagenome and gene abundance of the pretreatment gut microbiome in our training cohort. An average of 1.7 × 109 bp per sample were obtained after quality filtering (Supplementary Table 4, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41622/abstract), which allowed us to functionally annotate gut microbial genes into a total of 6,356 KOs. Although the percentage of open-reading frames annotated using the KEGG database was similar between groups (mean ± SD 27.64 ± 0.01 in responders versus 27.87 ± 0.01 in nonresponders; P = 0.78 by 2-sided t-test), the metagenome from pretreatment fecal samples separated most MTX responders from MTX nonresponders (principal components analysis; P < 0.05 by PERMANOVA) (Figure 2a). Moreover, our analysis identified 7 microbial modules that differed significantly between groups (q < 0.05 by DESeq2) (Supplementary Figure 4 and Supplementary Table 5, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41622/abstract), indicating a major clustering by metabolic and biosynthetic potential.
Figure 2.
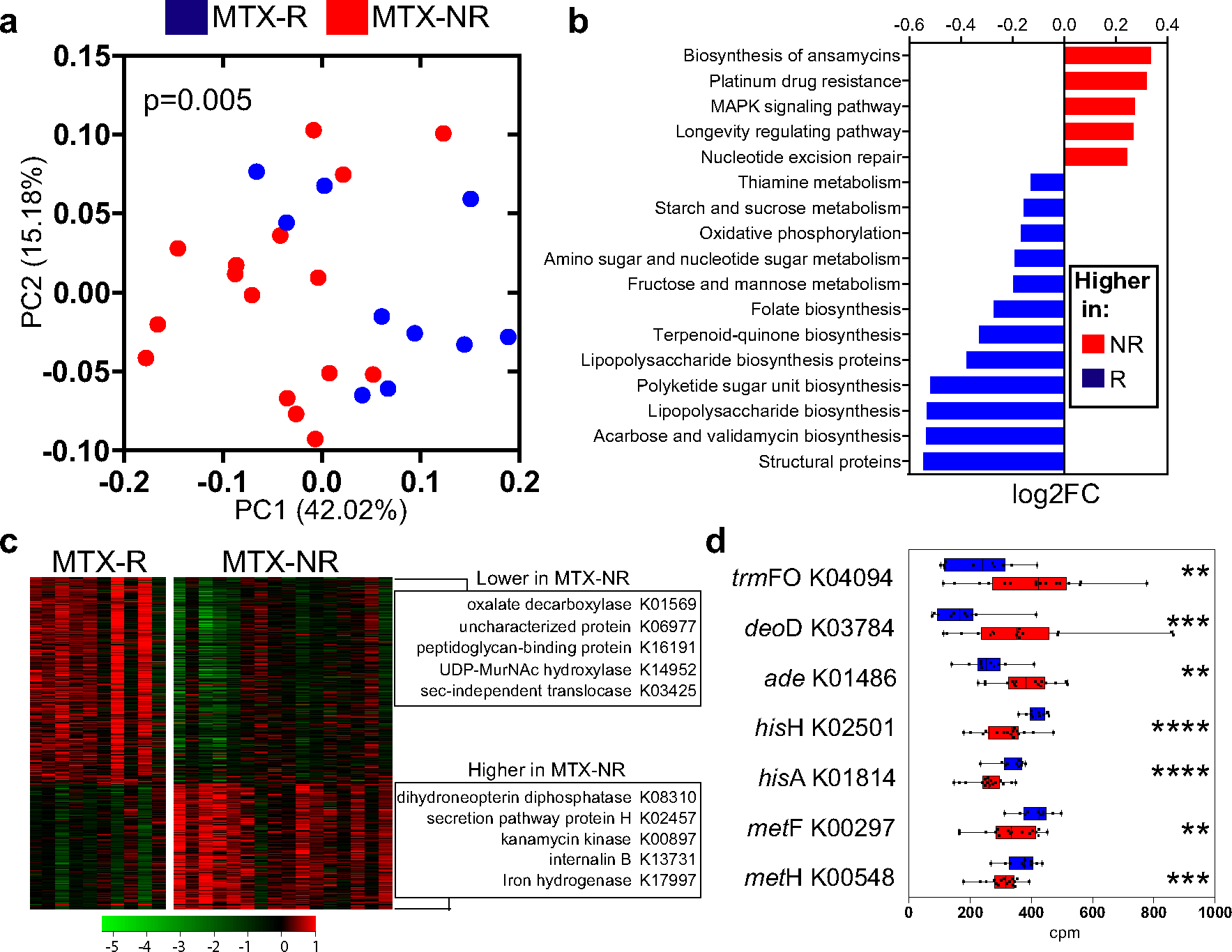
Differential bacterial pathways and gene orthologs in the pretreatment microbiomes in a training cohort of patients with new-onset RA who responded to MTX and patients with new-oset RA who did not respond to MTX. a, Principal components analysis of samples from responders and nonresponders to MTX based on the relative abundance of KEGG orthologs (KOs), using Bray-Curtis distance. Significant differences in gene family abundance were determined by PERMANOVA. b, Significantly different microbial pathways (q < 0.01 by DESeq2) identified in the pretreatment microbiomes of nonresponders and responders to MTX. The relative abundance (log2 fold change) is shown for each pathway. Other significant pathways (q < 0.05 by DESeq2) are shown in Supplementary Table 5, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41622/abstract. c, Heatmap showing 462 significantly different KOs in the gut microbiome of responders versus nonresponders to MTX (q < 0.05 by DESeq2). The KOs with the highest fold change difference for each group are indicated. Colors in the heatmap represent the KO abundance deviation from the median corrected by group size (see Patients and Methods). d, Relative abundance (in counts per million [cpm]) of pretreatment intestinal microbiome–derived KOs that significantly differed between responders and nonresponders to MTX and have previously been implicated in purine metabolism and/or MTX biotransformation (in either mammalian or bacterial cells). Data are shown as box plots. Boxes represent the 25th to 75th percentiles. Lines within the boxes represent the median. Whiskers indicate the maximum and minimum values. Symbols represent individual patients (n = 10–16 per group). ** = P < 0.01; *** = P < 0.001; **** = P < 0.0001. See Figure 1 for other definitions.
At the microbial pathway level, 26 features were significantly increased in MTX nonresponders, including the MAPK signaling pathway (ko04016), DNA replication (ko03030/3032), fatty acid degradation (ko00071), and ABC transporters (ko02010) (Figure 2b and Supplementary Table 5) (q < 0.05 by DESeq2). In contrast, 28 pathways were diminished in the MTX nonresponder group (Figure 2b), including those associated with lipopolysaccharide biosynthesis (ko01005) and either carbohydrate/vitamin metabolism (e.g., fructose/mannose [ko00051] and thiamine [ko00730]) or biosynthetic pathways, most notably folate biosynthesis (ko00790).
In total, 462 KOs separated MTX nonresponders from MTX responders (q < 0.05 by DESeq2) (Figure 2c and Supplementary Table 5), with 86 of these orthologs showing at least a 2-f old difference between groups (Supplementary Table 5). Some of the top genes with higher effect sizes encode for bacterial structural proteins (e.g., peptidoglycan-binding protein) or enzymes (e.g., oxalate decarboxylase) (Figure 2c and Supplementary Figure 5, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41622/abstract).
In addition, we detected significant differences in the abundance of multiple KOs encoding enzymes that may be indicative of potential changes in the bacterial metabolism of MTX, folate, and/or other molecules that have been linked, at least in mammalian cells, to an inadequate response to MTX (Figure 2d and Supplementary Table 5). A notable example was trmFO (K04094), an enzyme that leads to increased levels of tetrahydrofolate, which was found to be significantly higher in MTX nonresponders. Other differentiating KOs that may be of importance include adenine deaminase (K01486) and purine nucleoside phosphorylase (K03784), which were also increased in nonresponders. In contrast, several genes that encode for enzymes that can potentially lead to higher production of aminoimidazole carboxamide ribonucleotide (AICAR; an intermediate molecule of downstream MTX effects), including hisH (K02501) and hisA (K01814), were decreased in nonresponders. Other genes known to be involved in the intracellular folate/MTX pathway (e.g., metF/MTHFR [K00297] and metH/MTR [K00548]) were also significantly lower in nonresponders.
Taken together, these results indicate that the gut microbiome of patients with new-onset RA who respond favorably to MTX is distinct from that of nonresponders to MTX, prompting us to hypothesize that the pretreatment microbiome could be used to predict clinical nonresponse.
The pretreatment new-onset RA gut microbiome enables robust machine learning–based prediction of MTX response.
Because the gut metagenomes of MTX nonresponders and MTX responders were significantly different prior to treatment initiation, we sought to build a microbiome-based model that could predict clinical response to the drug. We first applied the Boruta algorithm (26) to our training cohort in order to identify metagenomic features relevant to the development of a predictive model, yielding a total of 38 Boruta-confirmed KOs (Figure 3a). Of those, and although most of them have no known implication in the folate/MTX pathways, the KO with the highest discriminative score (thiamine-phosphate diphosphorylase) is indeed involved in the metabolism of thiamine (a byproduct of which can interfere with MTX transport), while 2 other confirmed KOs encode for enzymes that may be involved in MTX response (i.e., hisH and hisA).
Figure 3.
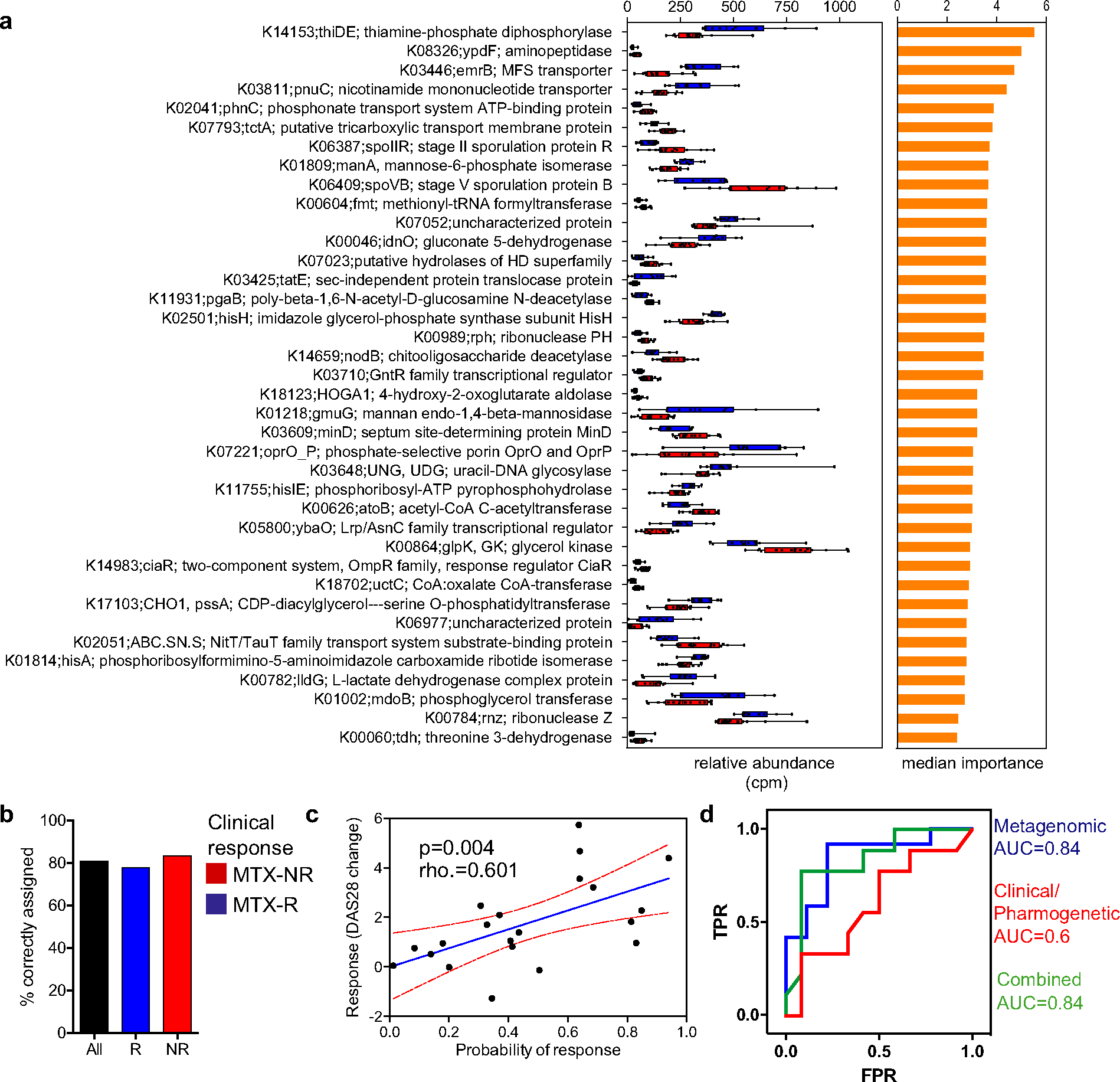
Pretreatment microbiota KEGG orthologs (KOs) as predictors of response to MTX treatment. a, KOs confirmed by the Boruta algorithm (n = 38) that discriminated between responders and nonresponders to MTX in a training cohort. Relative abundance (in counts per million [cpm]) (left) and median importance in a random forests model (right) are shown for each KO. In the left panel, data are shown as box plots. Boxes represent the 25th to 75th percentiles. Lines within the boxes represent the median. Whiskers indicate the maximum and minimum values. Symbols represent individual patients (n = 10–1 6 per group). b, Proportion of patients from a validation cohort who were correctly assigned to each group using a threshold of probability of response of 0.5 (those with a probability of response of >0.5 were considered responders; those with a probability of response of <0.5 were considered nonresponders). c, Correlation between actual (observed) response to MTX (based on change in Disease Activity Score in 28 joints [DAS28] at month 4 after treatment initiation) and predicted probability of response according to the metagenome-based model in the validation cohort (rho = 0.601; P < 0.05 by Spearman’s 2-sided rank correlation test). The blue line shows the mean linear regression; red lines indicate 95% confidence intervals. Symbols represent individual patients (n = 21). d, Comparison of the predictive potential of different models. A random forest model was built using the Boruta-selected gene orthologs (metagenomic model), clinical-pharmacogenetic variables (see Supplementary Methods, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41622/abstract), and a combination of both. The area under the curve (AUC) obtained with each model is shown. TPR = true-positive rate; FPR = false-positive rate (see Figure 1 for other definitions).
All 38 Boruta-selected KOs were included as predictors in a random forest model that was fitted using data exclusively from our training cohort. We then tested the predictive capability of this model in a new validation cohort of 21 patients with new-onset RA (with demographic characteristics similar to those of the training cohort) (Supplementary Tables 1 and 2), which yielded a high discriminative performance (area under the curve [AUC] 0.84). This translated into 80% of the patients being correctly classified (83.3% as nonresponders and 78% as responders) (Figure 3b). Consistent with this predictive capacity, a significant positive correlation was detected between the observed clinical improvement at month 4 and the probability of response provided by the model (rho = 0.601, P = 0.004 by Spearman’s rank correlation test) (Figure 3c). To study whether these findings could be rather reflective of microbial surrogates of clinical variables, we evaluated the correlations of the probability of response with both the observed baseline disease activity and the disease activity after MTX treatment. We found only a modest correlation with the former (rho = 0.45, P = 0.039) and no correlation with the latter (rho = −0.29, P = 0.19). Further, we observed a correlation between baseline disease activity and change in DAS28 in this population of patients with new-onset RA with high disease activity prior to MTX treatment (rho = 0.34, P = 0.018). Taken together, these results suggest that the predictive capacity of the model is mostly related to the change in disease activity, although the possibility that a few gene orthologs may concomitantly behave as a proxy for systemic inflammation cannot be ruled out.
The predictive capacity of the model was maintained or even increased when the number of features included in the model were reduced to as few as 12 KOs, based on the importance score assigned by the Boruta algorithm (Supplementary Figure 6 and Supplementary Table 6, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41622/abstract Methods). As expected, the predictive power was enhanced (89% of patients correctly classified; AUC 0.94) when considering only those patients with the highest probability score of belonging to either group (i.e., probabilities of response ≤0.2 or ≥0.8). A similar prediction outcome was obtained when analyzing sequences from a different platform (i.e., MiSeq), further validating the potential utility of our tool (Supplementary Figure 7, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41622/abstract).
To expand on its clinical applicability, we tested the model in yet another group of RA patients (n = 20) who were either prescribed different antirheumatic drugs (i.e., conventional synthetic disease-modifying antirheumatic drugs [DMARDs] or biologic agents) or were not started on any medications at all (Supplementary Tables 2 and 7, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41622/abstract). In these patients, clinical response could not be predicted using the microbiome-based model that included 38 KOs (i.e., only 50% of the patients were correctly assigned to their respective group), suggesting that the potential clinical utility of the model is restricted to RA patients who are both drug-naive and exposed directly to MTX, but not to other drugs.
We also applied the Boruta algorithm to the OTU-level data set (Supplementary Figure 8, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41622/abstract) and confirmed 13 features with predictive potential. However, a model based on these 13 OTUs did not satisfactorily classify patients from the validation cohort (AUC 0.63) (Supplementary Figures 8A–D, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41622/abstract). A similar result was obtained when using RSV data (AUC 0.72) (Supplementary Figures 8E–H, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41622/abstract).
Although previous studies have suggested that clinical-pharmacogenetic variables at baseline can also predict response to MTX therapy (29), we were not able to validate those findings in our new-onset RA cohort (Table 1). In order to compare the potential prediction capability of metagenomic variables (i.e., gene orthologs, KOs) to that of clinical-pharmacogenetic variables, the Boruta algorithm was applied after combining both sets of features. Notably, the Boruta algorithm only selected gene orthologs as predictors (results not shown). We then built a random forest model containing exclusively the clinical-pharmacogenetic features, which failed to predict response to therapy (AUC 0.6) (Figure 3d). Moreover, when clinical-pharmacogenetic variables were added to the random forest model based on gene orthologs, the prediction potential did not differ from the one obtained with a model containing only metagenomic features (AUC 0.84) (Figure 3d). Taken together, these results indicate that the model based on microbiome features can determine response to MTX, while the clinical-pharmacogenetic features do not add to its predictive potential.
Table 1.
Prediction power of the clinical- pharmacogenetic and metagenomic- based models for predicting response to MTX in patients with new- onset RA in our validation cohort*
| Clinical-pharmacogenetic model | Metagenomic-based model | |
|---|---|---|
|
| ||
| Patients classified as MTX nonresponders or MTX responders | 48.9 | 100 |
| True- negative rate | 69 | 83.3 |
| True- positive rate | 0 | 78 |
Values are the percent. MTX = methotrexate; RA = rheumatoid arthritis.
Gut bacteria derived from MTX nonresponders differentially deplete MTX ex vivo, and remaining drug levels correlate with decreased clinical response.
In order to gain further mechanistic insights into whether the gut microbiome of patients with new-onset RA may directly mediate differences in clinical response by affecting MTX metabolism, we performed ex vivo studies using 2 independent metabolomic platforms.
We first incubated human stool samples from 22 patients with new-onset RA (n = 9 MTX responders and n = 13 MTX nonresponders) with MTX (100 μg/ml, 220 μM) ex vivo for 72 hours (see Patients and Methods). MTX and bacteria-produced downstream metabolites were measured in the supernatant using NMR spectroscopy. A total of 28 NMR signals (variable-size regions) were integrated in the NMR spectra (Supplementary Table 8, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41622/abstract). Subsequently, orthogonal partial least squares (OPLS) analyses were performed to minimize the potential contribution of variability between samples and to facilitate the identification of the NMR signals most relevant in the separation between the groups, as previously described (30).
Among the NMR signals integrated in the spectra, 10 regions were found to be relevant (variable importance on projection [VIP] value > 1) when comparing the MTX responders and MTX nonresponders (percentage of variability described in the first component R2 [Y] = 0.664; predictive value Q2 [Y] = 0.35) (Figure 4a), and 10 of them were found to be relevant (VIP value > 1) in the OPLS model when DAS28 was used as a continuous discriminant variable (R2 [Y] = 0.73, Q2 [Y] = 0.386) (Figure 4b and Supplementary Table 9, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41622/abstract). We then analyzed changes in the levels of those metabolites corresponding to signals with a VIP value >1 from the OPLS model using DAS28 as a continuous discriminant variable (the best-fitting model as determined by the R2 and Q2 values). This analysis revealed that 2 of the features found to be relevant for discriminating between samples were compatible with MTX NMR signals (Supplementary Table 9). Higher intensities of these MTX signals in the supernatant were positively correlated with response to treatment (greater change in DAS28 4 months after treatment initiation) (rho = 0.619, P = 0.002 by Spearman’s rank correlation) (Figure 4c).
Figure 4.
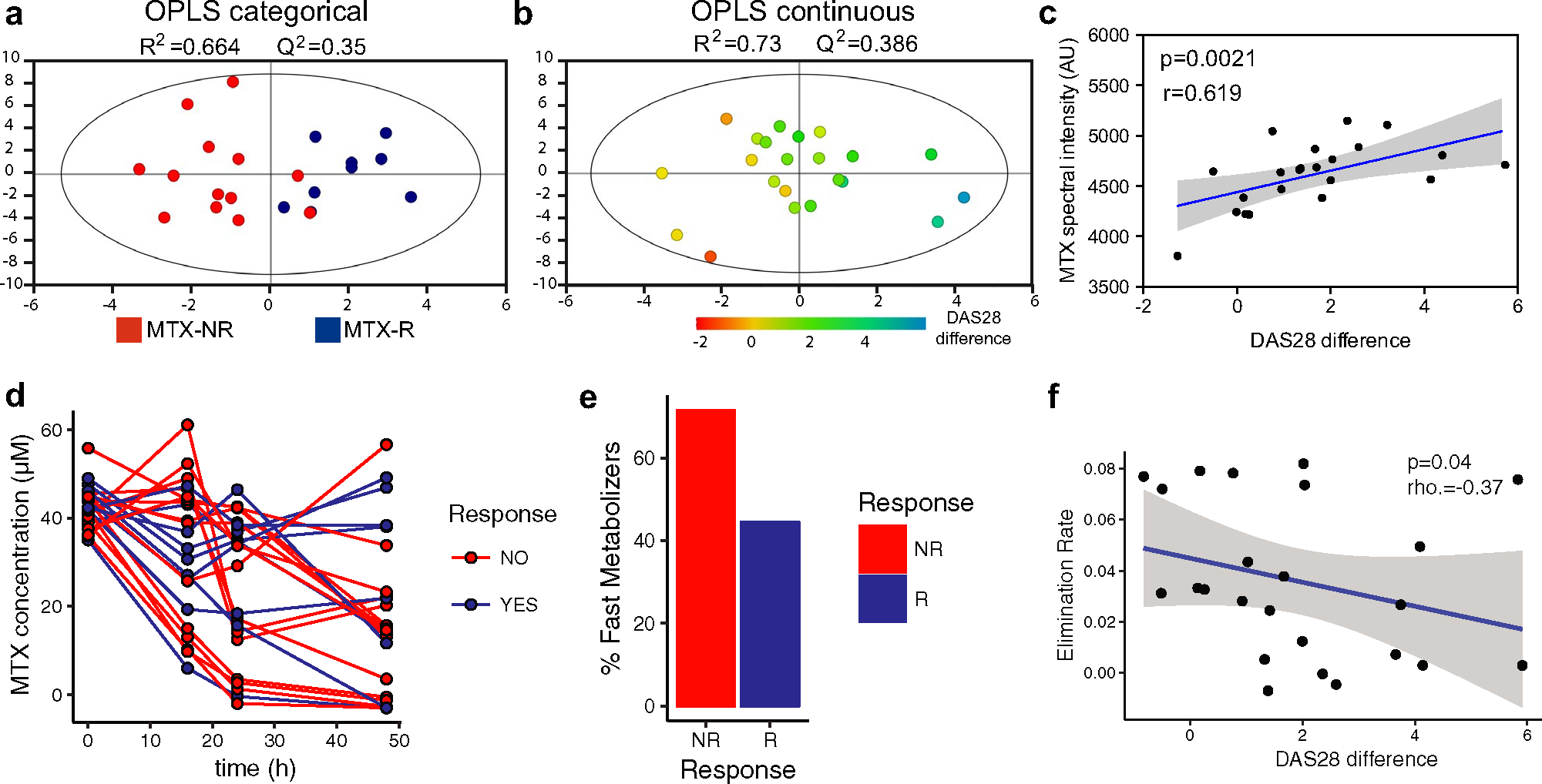
Differential ex vivo depletion of MTX by gut microbiomes from patients with new-onset RA, and correlation between remaining drug concentrations and future clinical response. Fecal samples from responders and nonresponders to MTX were incubated with MTX (100 μg/ml) at a temperature of 37°C under anaerobic conditions for 72 hours (a–c) or 48 hours (d–f). Abundance of MTX was measured in the supernatant using nuclear magnetic resonance (NMR) spectroscopy (a–c) or liquid chromatography mass spectroscopy (LC-MS) (d–f). a, Clustering of samples from nonresponders to MTX (red; n = 13) separately from samples from responders to MTX (blue; n = 9), determined by orthogonal partial least squares (OPLS) analysis based on the integration of 28 signals identified on the NMR spectra. b, OPLS model of the correlation of change in the Disease Activity Score in 28 joints (DAS28) with change in NMR signals. c, Significant correlation of the mean abundance (spectral intensity in AU) of the 2 peaks corresponding to MTX (B8_6200 and B1_9825) with future clinical response (DAS28) in patients with new-onset RA (rho = 0.619, P < 0.05 by Spearman’s 2-s ided rank correlation test). The blue line shows the mean linear regression; shading indicates 95% confidence intervals. d, Levels of MTX at the indicated time points after incubation with fecal samples from RA patients (n = 30). Most patients whose fecal microbiota rapidly depleted MTX did not have an adequate response to treatment. e, Proportion of nonresponders and responders to MTX who were fast metabolizers. f, Significant correlation of MTX elimination rate (slope of a linear fit to a semi-log plot of MTX concentration versus time) with future clinical response (DAS28 score) in patients with new-onset RA (rho = −0.37, P = 0.04 by Spearman’s 1-sided rank correlation test). See Figure 1 for other definitions.
We next validated the NMR-based results using targeted LC-MS. This analysis facilitated a more specific measurement of the MTX concentration available upon incubation with different fecal microbiomes. Pretreatment samples from 30 patients with new-onset RA were incubated ex vivo with MTX (100 μg/ml) for 48 hours. Supernatants were taken at 0, 16, 24, and 48 hours prior to LC-MS analysis, followed by quantification of MTX concentration at each time point for each sample. We first analyzed the ability of fecal microbiomes to metabolize MTX ex vivo, and found, as expected, a high interindividual variability (Figure 4d). While the microbiome of some patients was able to rapidly reduce the levels of MTX, the concentration of MTX was not modified in others (percent conversion ranging from 0 to 100). The samples that diminished MTX levels faster were mostly from MTX nonresponders (71% of nonresponder samples showed >50% reduction in MTX levels by 48 hours), while those samples that did not substantially alter drug quantity were mostly from MTX responders (56% of responder samples showed <50% MTX reduction at 48 hours) (Figure 4e). We next applied linear regression on log-transformed MTX concentrations to determine the elimination rate of MTX for each individual sample and observed that the elimination rate was significantly and negatively associated with future, observed clinical response (rho = −0 .37, P = 0.04 by Spearman’s 1-sided correlation test) (Figure 4f).
Taken together, these data provide a plausible mechanistic explanation for the association between the pretreatment gut microbiome and drug response, where patients’ communities enriched for gut bacteria capable of efficiently metabolizing and/or depleting MTX are associated with worsened clinical outcomes.
DISCUSSION
Although several efficacious therapies have recently been developed, the field of rheumatology lacks tools to help clinicians and their patients decide early on which drugs are most likely to be beneficial. In RA, prior models based on clinical-pharmacogenetic features could not be generalized or validated to predict MTX outcomes (31–33). Consequently, the current state of clinical care for new-onset RA is to initiate MTX regardless (34), turning the therapeutic decision-making process effectively aleatory. This approach is most problematic for those patients who fail to respond within the early therapeutic window of opportunity (35–39).
Using a combination of 16S rRNA gene and metagenome sequencing, we report for the first time that the pretreatment microbiome can differentiate response to oral MTX in a cohort of patients with new-onset RA. We found that overall bacterial diversity is distinct between patients who respond to MTX and those who do not. Although we found differences at higher taxonomic levels, these differences could not be explained by specific relative expansion/contraction at lower taxonomic hierarchies (i.e., OTUs and RSVs) (Supplementary Figure 9, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41622/abstract), suggesting that group(s) of microbes or bacterial functions, rather than dominant species, may be associated with or implicated in clinical response. Consistent with this finding, metagenomic sequencing enabled improved predictions of clinical response. Notably, based on the abundance of 38 KOs, we were able to predict lack of response to MTX in the majority of patients from an independent, validation cohort. This indicates that our metagenome-based classifier constitutes a potentially valuable tool for decision-making in newly diagnosed RA (Figure 5).
Figure 5.
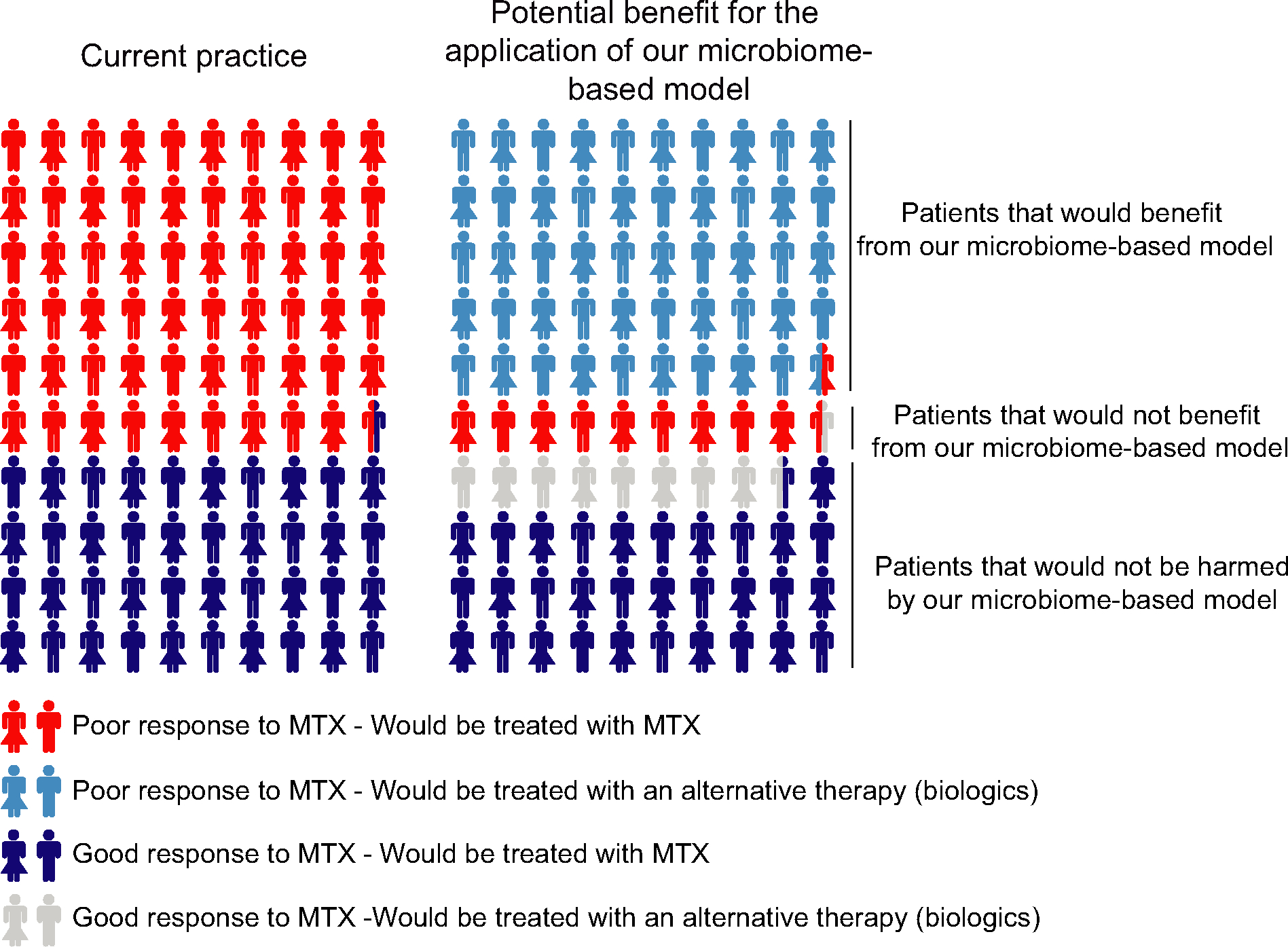
Illustration showing the proportions of patients with rheumatoid arthritis (RA) who would benefit from our microbiome-based model. Left, Percentage of patients with new-onset RA (based on our cohort) with observed poor (red) or good (dark blue) response to methotrexate (MTX) therapy at 4 months. Right, Visual representation of how treatment decision-making could potentially be improved using our microbiome-based model of treatment response. Five of 6 patients with new-onset RA (light blue) predisposed to not respond adequately to MTX would have benefited from the model (i.e., nonresponders to MTX would have been correctly classified as such and could have been treated earlier with alternative, more efficacious therapies). Similarly, all of those patients with new-onset RA predisposed to respond to MTX would have been treated adequately with either MTX (75%; dark blue) or erroneously, but still adequately, with alternative, efficacious therapies (25%; gray). This benefit would have been at the expense of 1 of 10 patients with new-onset RA who our model predicts would be responders to MTX when they are actually observed nonresponders (red).
To our knowledge, only one other group has used microbiome features to predict MTX response in RA (23). However, that study focused primarily on the oral microbiome as a predictor and mostly on RA patients with longstanding, established, treated disease, who are known to have a markedly distinct microbiome from patients with new-onset RA (40). In addition, predictors of response to MTX in the prior study were based on the abundance of metagenomic species rather than specific gene orthologs.
Nevertheless, we note limitations to our model, which may prevent its immediate applicability. First, although our cohorts were heterogeneous in nature (i.e., patients derived from various ethnic backgrounds and clinics), they were limited in number and, therefore, the tool should still be tested in expanded, distinct RA populations. Second, we chose a change in DAS28 of 1.8 to enhance the stringency of our outcome of response. In doing so, however, this approach led us to consider some patients to be MTX nonresponders even though they had a probability of response very close to that of MTX responders (i.e., near 0.5). This result is not unexpected, since the threshold used to classify response was high (i.e., a change in DAS28 of 1.2–1.8 is still considered moderate to good response in practice) and because of the dynamic nature of DAS28 as a continuous measurement. Nevertheless, the predictive capacity of this metagenomic model is on par with those observed in other chronic diseases and cancer (16,41), and outperforms both a previous clinical pharmacogenetic-based approach in RA (29) (Table 1) and the current clinical practice in early disease (Figure 5). Although prediction could not be enhanced by incorporating clinical-pharmacogenetic features (Figure 3d), it is possible that the discriminating power of the model may be increased by adding other previously studied predictors of MTX efficacy (42). Third, our study focused exclusively on oral MTX and could not address how microbiome features associate with response to parenteral MTX. Although of interest, the clinical relevance of such an approach may be limited since: a) MTX undergoes enterohepatic circulation, and b) the overall use of subcutaneous MTX, even when more effective, is exceedingly limited (43). In addition, and based on our results, our model may not be applicable to other DMARDs/oral small molecules or other biologic therapies (Supplementary Table 7, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41622/abstract). This finding is consistent with work from other groups suggesting that specific models may be required for different medications (41,44).
Importantly, using shotgun sequencing, we identified microbial-derived gene orthologs in pretreatment samples from patients with new-onset RA, which enabled us to study microbiome-based features that characterize response to MTX. Many of the pathways and genes that were significantly distinct between groups were linked to nonspecific bacterial structure and physiology. However, other gene orthologs were related to known MTX metabolic pathways, at least in mammalian cells. For example, the microbiome of MTX nonresponders showed increased abundance of genes encoding for adenine deaminase and purine-nucleoside phosphorylase. These enzymes are involved in the purine metabolism pathway and catalyze reactions leading to the production of hypoxanthine, a purine derivative known to rescue cells from MTX cytotoxicity (45,46). This could potentially allow for a higher incorporation of MTX by intestinal bacteria and further reduce drug bioavailability. Conversely, a relative decrease in genes that encode for enzymes implicated in the accumulation of AICAR (i.e., hisH and hisA) was detected in MTX nonresponders. This finding is relevant because MTX exerts many of its functions through the accumulation of (mammalian) intracellular AICAR, which in turn leads to the release of extracellular adenosine, which modulates many of the immune-mediated effects of MTX. A second series of genes encoding for enzymes involved in intracellular folate/MTX disposition, at least in humans (47), were also significantly lower in the microbiome of MTX nonresponders, including metH and metF.
Overall, many more KOs differentiate MTX nonresponders from MTX responders than OTUs and RSVs. It is quite possible that this relates to the established functional redundancy between bacterial species and strains (48,49). In the case of RA, it is conceivable that comparable metagenomes implicated in MTX transformation could functionally converge through several combinations of distinct taxa in any given patient. In fact, many KOs related to purine metabolism and/or MTX biotransformation pathways that significantly differentiated MTX nonresponders from responders were imputed to different taxa (Supplementary Figure 10, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41622/abstract). Taken together, our results further underscore the importance of gut metagenomic characterization for studies that aim to develop a functional view of drug metabolism (18).
While the metagenomic sequencing data suggest that human gut bacteria possess the metabolic potential to act on MTX, we demonstrated experimentally that MTX is depleted by human gut bacteria ex vivo. Although previous studies in mice suggested that the gut microbiome is necessary for the biotransformation of MTX (21,22), they failed to demonstrate that human gut bacteria are capable of directly metabolizing MTX (50). Using 2 independent analytical platforms, our data show for the first time that human gut microbiota derived from patients with new-onset RA differentially deplete MTX ex vivo. We further found that, when incubated with fecal samples, MTX levels measured in the supernatant correlate with future clinical response. Although a comparison between the MTX depletion rate and the probability of response given by the metagenome-based model did not show a significant correlation (P > 0.05), this may reflect the fact that most gene orthologs included in the model do not seem to have a direct role in MTX depletion (Figure 3a). One possibility is that these KOs could be considered solely as biomarkers/predictors of MTX response. Another complementary explanation is that they could be influencing MTX response but independently of drug metabolism (e.g., priming the immune response to enhance systemic drug activity). Intriguingly, however, abundance of some KOs whose function may involve reduction in MTX levels (i.e., trmFO and deoD) (Figure 2d) was in fact significantly correlated with MTX depletion ex vivo (P < 0.05) (Supplementary Figure 11, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41622/abstract), suggesting a potential direct effect of the microbiome on MTX metabolism. Future studies should be performed in order to confirm whether the microbiome can directly metabolize MTX in vivo, prime the immune system to enhance response, or both.
In summary, we have characterized, for the first time, the potential clinical value of the pretreatment microbiome as a predictor of early response to MTX in drug-naive patients with new-onset RA. Our work suggests that the intestinal metagenome could be exploited in the development of biomarkers of response, either by high-throughput sequencing or through simplified (e.g., polymerase chain reaction–based) precision medicine approaches. Finally, our results open the possibility of rationally designing microbiome-modulating strategies to improve oral absorption of MTX and its downstream immune effects, inform clinical decision-making, or both.
Supplementary Material
ACKNOWLEDGMENTS
We extend special thanks to Ann Rupel for insights and comments on the manuscript as well as Rhina Medina and Luz Alvarado for assistance in the acquisition, deidentification, and storage of biologic samples. We are indebted to Parvathy Girija and Adriana Heguy for technical assistance as well as the staff of the NYU Genome Technology Center for help with 16S rRNA sequencing. We thank Jose Clemente for his assistance with sequencing methods. We thank the staff of the Servicio de Secuenciación y Bioinformática (FISABIO) for help with the shotgun sequencing.
Supported in part by the NIH (grants 5T32-AR-007304-37, TR-001871, and 1K08-AR-073930 to Dr. Nayak, grants R01-HL-122593 and R01-AR-074500 to Dr. Turnbaugh, and grants R03-AR-072182 and R01-AR-074500 to Dr. Scher), the Rheumatology Research Foundation (grant AWD00003947 to Dr. Scher), the Searle Scholars Program (grant SSP-2016–1352), the Spanish Ministry of Economy and Competitiveness (grant SAF2017–89229-R to Drs. Puchades-Carrasco and Pineda-Lucena and grant SAF2017–90083-R to Dr. Ubeda), the University of California San Francisco Breakthrough Program for Rheumatoid Arthritis-Related Research, and the Arthritis Foundation Center of Excellence. Dr. Flor-Duro’s work was supported by an FPI Fellowship from the Spanish Ministry of Economy and Competitiveness. Drs. Puchades-Carrasco and Pineda-Lucena’s work was supported in part by the European Regional Development Fund and Generalitat Valenciana Consellería de Sanidad Universal y Salud Pública. Dr. Turnbaugh’s work was supported by the Burroughs Wellcome Fund Investigators in the Pathogenesis of Infectious Disease award, Chan Zuckerberg Biohub, and the Damon Runyon Cancer Research Foundation (Nadia’s Gift grant DRR-42–16). Dr. Ubeda’s work was supported in part by Conselleria d’Innovació, Universitats, CiènciaI Societat Digital (grant AICO/2019/266). Dr. Scher’s work was supported in part by the Riley Family Foundation, the Snyder Family Foundation, the Hagedorn Foundation, and the NYU Langone Judith and Stewart Colton Center for Autoimmunity.
Footnotes
Dr. Izmirly has received consulting fees from GlaxoSmithKline (less than $10,000). Dr. Turnbaugh has received consulting fees and/or honoraria from Kaleido, Seres, SNIPRbiome, uBiome, and Whole Biome (less than $10,000 each). Dr. Ubeda has received consulting fees from Vedanta Biosciences (less than $10,000). Dr. Scher has received consulting fees from UCB, Janssen, Bristol Myers Squibb, AbbVie, Pfizer, and Novartis (less than $10,000 each) and from Sanofi (more than $10,000). No other disclosures relevant to this article were reported.
REFERENCES
- 1.McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med 2011;365:2205–19. [DOI] [PubMed] [Google Scholar]
- 2.Detert J, Bastian H, Listing J, Weiss A, Wassenberg S, Liebhaber A, et al. Induction therapy with adalimumab plus methotrexate for 24 weeks followed by methotrexate monotherapy up to week 48 versus methotrexate therapy alone for DMARD-naive patients with early rheumatoid arthritis: HIT HARD, an investigator-initiated study. Ann Rheum Dis 2013;72:844–50. [DOI] [PubMed] [Google Scholar]
- 3.Favalli EG, Biggioggero M, Meroni PL. Methotrexate for the treatment of rheumatoid arthritis in the biologic era: still an “anchor” drug? [review]. Autoimmun Rev 2014;13:1102–8. [DOI] [PubMed] [Google Scholar]
- 4.Emery P, Breeveld FC, Hall S, Durez P, Chang DJ, Robertson D, et al. Comparison of methotrexate monotherapy with a combination of methotrexate and etanercept in active, early, moderate to severe rheumatoid arthritis (COMET): a randomised, double-blind, parallel treatment trial. Lancet 2008;372:375–82. [DOI] [PubMed] [Google Scholar]
- 5.Van Roon EN, van de Laar MA. Methotrexate bioavailability. Clin Exp Rheumatol 2010;28:S27–32. [PubMed] [Google Scholar]
- 6.Lee J, Pelkey R, Gubitosa J, Henrick MF, Ganz ML. Comparing healthcare costs associated with oral and subcutaneous methotrexate or biologic therapy for rheumatoid arthritis in the United States. Am Health Drug Benefits 2017;10:42–9. [PMC free article] [PubMed] [Google Scholar]
- 7.Singh JA, Saag KG, Bridges SL Jr, Akl EA, Bannuru RR, Sullivan MC, et al. 2015 American College of Rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Rheumatol 2016;68:1–26. [DOI] [PubMed] [Google Scholar]
- 8.Smolen JS, Landewé R, Bijlsma J, Burmester G, Chatzidionysiou K, Dougados M, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis 2017;76:960–77. [DOI] [PubMed] [Google Scholar]
- 9.Halilova KI, Brown EE, Morgan SL, Bridges SL Jr, Hwang MH, Arnett DK, et al. Markers of treatment response to methotrexate in rheumatoid arthritis: where do we stand? Int J Rheumatol 2012;2012:978396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dervieux T, Furst D, Lein DO, Capps R, Smith K, Walsh M, et al. Polyglutamation of methotrexate with common polymorphisms in reduced folate carrier, aminoimidazole carboxamide ribonucleotide transformylase, and thymidylate synthase are associated with methotrexate effects in rheumatoid arthritis. Arthritis Rheum 2004;50:2766–74. [DOI] [PubMed] [Google Scholar]
- 11.Dervieux T, Greenstein N, Kremer J. Pharmacogenomic and metabolic biomarkers in the folate pathway and their association with methotrexate effects during dosage escalation in rheumatoid arthritis. Arthritis Rheum 2006;54:3095–103. [DOI] [PubMed] [Google Scholar]
- 12.Angelis-Stoforidis P, Vajda FJ, Christophidis N. Methotrexate polyglutamate levels in circulating erythrocytes and polymorphs correlate with clinical efficacy in rheumatoid arthritis. Clin Exp Rheumatol 1999;17:313–20. [PubMed] [Google Scholar]
- 13.Stamp LK, O’Donnell JL, Chapman PT, Zhang M, James J, Frampton C, et al. Methotrexate polyglutamate concentrations are not associated with disease control in rheumatoid arthritis patients receiving long-term methotrexate therapy. Arthritis Rheum 2010;62:359–68. [DOI] [PubMed] [Google Scholar]
- 14.Danila MI, Hughes LB, Brown EE, Morgan SL, Baggott JE, Arnett DK, et al. Measurement of erythrocyte methotrexate polyglutamate levels: ready for clinical use in rheumatoid arthritis? Curr Rheumatol Rep 2010;12:342–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Routy B, Le Chatelier E, Derosa L, Duong CP, Alou MT, Daillere R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018;359:91–7. [DOI] [PubMed] [Google Scholar]
- 16.Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML, et al. The commensal microbiome is associated with anti-P D-1 efficacy in metastatic melanoma patients. Science 2018;359:104–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-P D-1 immunotherapy in melanoma patients. Science 2018;359:97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spanogiannopoulos P, Bess EN, Carmody RN, Turnbaugh PJ. The microbial pharmacists within us: a metagenomic view of xenobiotic metabolism [review]. Nat Rev Microbiol 2016;14:273–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koppel N, Rekdal VM, Balskus EP. Chemical transformation of xenobiotics by the human gut microbiota [review]. Science 2017;356:eaag2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zimmermann M, Zimmermann-Kogadeeva M, Wegmann R, Goodman AL. Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature 2019;570:462–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valerino DM, Johns DG, Zaharko DS, Oliverio VT. Studies of the metabolism of methotrexate by intestinal flora. I. Identification and study of biological properties of the metabolite 4-amino-4-deoxy-N10-methylpteroic acid. Biochem Pharmacol 1972;21:821–31. [DOI] [PubMed] [Google Scholar]
- 22.Zaharko DS, Bruckner H, Oliverio VT. Antibiotics alter methotrexate metabolism and excretion. Science 1969;166:887–8. [DOI] [PubMed] [Google Scholar]
- 23.Zhang X, Zhang D, Jia H, Feng Q, Wang D, Liang D, et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nature Med 2015;21:895–905. [DOI] [PubMed] [Google Scholar]
- 24.Isaac S, Scher JU, Djukovic A, Jimenez N, Littman DR, Abramson SB, et al. Short- and long-term effects of oral vancomycin on the human intestinal microbiota. J Antimicrob Chemother 2017;72:128–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prevoo ML, van ‘t Hof MA, Kuper HH, van Leeuwen MA, van de Putte LB, van Riel PL. Modified disease activity scores that include twenty-eight–joint counts: development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum 1995;38:44–8. [DOI] [PubMed] [Google Scholar]
- 26.Kursa MB, Rudnicki WR. Feature selection with the Boruta package. J Stat Softw 2010;36:1–13. [Google Scholar]
- 27.Bisanz JE, Spanogiannopoulos P, Pieper LM, Bustion AE, Turnbaugh PJ. How to determine the role of the microbiome in drug disposition. Drug Metab Dispos 2018;46:1588–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spanogiannopoulos P, Turnbaugh PJ. Broad collateral damage of drugs against the gut microbiome [review]. Nat Rev Gastroenterol Hepatol 2018;15:457–8. [DOI] [PubMed] [Google Scholar]
- 29.Wessels JA, van der Kooij SM, le Cessie S, Kievit W, Barerra P, Allaart CF, et al. A clinical pharmacogenetic model to predict the efficacy of methotrexate monotherapy in recent-onset rheumatoid arthritis. Arthritis Rheum 2007;56:1765–75. [DOI] [PubMed] [Google Scholar]
- 30.Jacob D, Deborde C, Lefebvre M, Maucourt M, Moing A. NMRProcFlow: a graphical and interactive tool dedicated to 1D spectra processing for NMR-based metabolomics. Metabolomics 2017;13:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li X, Hu MC, Li WP, Gu L, Chen MJ, Ding HH, et al. The association between reduced folate carrier-1 gene 80G/A polymorphism and methotrexate efficacy or methotrexate related-toxicity in rheumatoid arthritis: a meta-analysis. Int Immunopharmacol 2016;38:8–15. [DOI] [PubMed] [Google Scholar]
- 32.Taylor JC, Bongartz T, Massey J, Mifsud B, Spiliopoulou A, Scott IC, et al. Genome-w ide association study of response to methotrexate in early rheumatoid arthritis patients. Pharmacogenomics J 2018;18:528–38. [DOI] [PubMed] [Google Scholar]
- 33.Eektimmerman F, Allaart CF, Hazes JM, Badhar MB, den Broeder AA, Fransen J, et al. Validation of a clinical pharmacogenetic model to predict methotrexate nonresponse in rheumatoid arthritis patients. Pharmacogenomics 2019;20:85–93. [DOI] [PubMed] [Google Scholar]
- 34.Smolen JS, Landewé RB, Bijlsma JW, Burmester GR, Dougados M, Kerschbaumer A, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann Rheum Dis 2020;79:685–99. [DOI] [PubMed] [Google Scholar]
- 35.Boers M Understanding the window of opportunity concept in early rheumatoid arthritis [editorial]. Arthritis Rheum 2003;48:1771–4. [DOI] [PubMed] [Google Scholar]
- 36.O’Dell JR. Treating rheumatoid arthritis early: a window of opportunity? [editorial]. Arthritis Rheum 2002;46:283–5. [DOI] [PubMed] [Google Scholar]
- 37.Nell VP, Machold KP, Eberl G, Stamm TA, Uffmann M, Smolen JS. Benefit of very early referral and very early therapy with disease-modifying anti-rheumatic drugs in patients with early rheumatoid arthritis. Rheumatology (Oxford) 2004;43:906–14. [DOI] [PubMed] [Google Scholar]
- 38.Finckh A, Liang MH, van Herckenrode CM, de Pablo P. Long-term impact of early treatment on radiographic progression in rheumatoid arthritis: a meta-analysis. Arthritis Care Res (Hoboken) 2006;55:864–72. [DOI] [PubMed] [Google Scholar]
- 39.Farragher TM, Lunt M, Fu B, Bunn D, Symmons DP. Early treatment with, and time receiving, first disease-modifying antirheumatic drug predicts long-term function in patients with inflammatory polyarthritis. Ann Rheum Dis 2010;69:689–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scher JU, Ubeda C, Artacho A, Attur M, Isaac S, Reddy SM, et al. Decreased bacterial diversity characterizes the altered gut microbiota in patients with psoriatic arthritis, resembling dysbiosis in inflammatory bowel disease. Arthritis Rheumatol 2015;67:128–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Doherty MK, Ding Tao, Koumpouras C Telesco SE, Monast C, Das A, et al. Fecal microbiota signatures are associated with response to ustekinumab therapy among Crohn’s disease patients. mBio 2018;9:e02120–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peres RS, Liew FY, Talbot J, Carregaro V, Oliveira RD, Almeida SL, et al. Low expression of CD39 on regulatory T cells as a biomarker for resistance to methotrexate therapy in rheumatoid arthritis. Proc Natl Acad Sci U S A 2015;112:2509–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Curtis JR, Zhang J, Xie F, Beukelman T, Chen L, Fernandes J, et al. Use of oral and subcutaneous methotrexate in rheumatoid arthritis patients in the United States. Arthritis Care Res (Hoboken) 2014;66:1604–11. [DOI] [PubMed] [Google Scholar]
- 44.Ananthakrishnan AN, Luo C, Yajnik V, Khalili H, Garber JJ, Stevens BW, et al. Gut microbiome function predicts response to anti-integrin biologic therapy in inflammatory bowel diseases. Cell Host Microbe 2017;21:603–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Refsum H, Christensen B, Djurhuus R, Ueland PM. Interaction between methotrexate, “rescue” agents and cell proliferation as modulators of homocysteine export from cells in culture. J Pharmacol Exp Ther 1991;258:559–66. [PubMed] [Google Scholar]
- 46.Howell SB, Mansfield SJ, Taetle R. Thymidine and hypoxan-thine requirements of normal and malignant human cells for protection against methotrexate cytotoxicity. Cancer Res 1981;41:945–50. [PubMed] [Google Scholar]
- 47.Qiu Q, Huang J, Shu X, Fan H, Zhou Y, Xiao C. Polymorphisms and pharmacogenomics for the clinical efficacy of methotrexate in patients with rheumatoid arthritis: a systematic review and meta-analysis. Sci Rep 2017;7:44015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature 2012;489:220–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature 2009;457:480–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stewart MJ, Watson ID, Farid YY, Skellern GG. An investigation into the source of the deglutamated metabolites of methotrexate in patients treated with high dose infusions. Ann Clin Biochem 1986;23:210–5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.