

Abstract
Ferroptosis, a form of regulated cell death driven by iron-dependent phospholipid peroxidation, has been implicated in multiple diseases including cancer1–3, degenerative disorders4, and organ ischemia-reperfusion injury (IRI)5,6. Here, by using a genome-wide CRISPR-Cas9 screening, we identified that the enzymes involved in the distal cholesterol biosynthesis play pivotal yet opposing roles in regulating ferroptosis via dictating the level of 7-dehydrocholesterol (7-DHC), an intermediate metabolite of the distal cholesterol biosynthesis that is synthesized by sterol C5-desaturase (SC5D) and metabolized by 7-DHC reductase (DHCR7) for cholesterol synthesis. We found that the pathway components including MSMO1, CYP51A1, EBP and SC5D act as potential suppressors of ferroptosis, whereas DHCR7 functions as a pro-ferroptotic gene. Mechanistically, 7-DHC dictates ferroptosis surveillance via utilizing the conjugated diene to exert its anti-phospholipid autoxidation function and shields plasma and mitochondria membranes from phospholipid autoxidation. Importantly, blocking the biosynthesis of endogenous 7-DHC by pharmacological targeting EBP induces ferroptosis and inhibits tumour growth, whereas increasing the 7-DHC level via inhibiting DHCR7 effectively promotes cancer metastasis and attenuates the progression of kidney ischemia-reperfusion injury (IRI), supporting a critical function of this axis in vivo. In conclusion, our data reveal an unexpected role of 7-DHC serving as a natural anti-ferroptotic metabolite and suggest that pharmacological manipulation of 7-DHC levels is a promising therapeutic strategy for cancer and IRI.
Ferroptosis, a unique modality of regulated cell death, has garnered extensive attention due to its critical role in various patho(physio)logical conditions including cancer1–3, neurodegenerative disorders4 and organ IRI5,6. A crucial hallmark of ferroptosis is iron-dependent phospholipid peroxidation7,8, which contributes to membrane damage9. Cells possess intrinsic defensive systems to protect themselves from phospholipid (PL) peroxidation. The cyst(e)ine-GSH- glutathione peroxidase 4 (GPX4) axis is the main pathway suppressing ferroptosis by detoxifying phospholipid hydroperoxide (PLOOH) into phospholipid alcohol (PLOH)10. Alternative protection mechanisms include (1) that mediated by radical-trapping antioxidant (RTA) activity, as exemplified by ferroptosis suppressor protein 1 (FSP1)/coenzyme Q10 (CoQ10, also known as ubiquinone-10)11,12, dihydroorotate dehydrogenase (DHODH)/CoQ1013, and GTP cyclohydrolase 1 (GCH1)/ tetrahydrobiopterin (BH4)14,15 axes, and (2) the MBOAT1/2-mediated cellular phospholipid remodeling16.
7-DHC is a metabolic intermediate of the distal cholesterol biosynthesis (CB) pathway and is converted to cholesterol by DHCR717. As an unsaturated sterol, 7-DHC contains a 5,7-diene in its B-ring and can function as a potent H-atom donor to fatty acid peroxyl radicals18–20. Yet, apart from the cytotoxicity of 7-DHC caused by its derived oxysterols to neuron and retinal cells20–22, the biological role of 7-DHC remains elusive, although mutation of DHCR7 is a signature of Smith-Lemli Opitz syndrome (SLOS)23 and has been found in Burkitt’s lymphoma patients24. Our current study identified an unexpected role of 7-DHC in protecting cells from phospholipid peroxidation and ferroptosis, which highlights the important role of the distal cholesterol biosynthesis pathway in patho(physio)logical conditions, such as cancer and IRI.
Distal CB pathway regulates ferroptosis
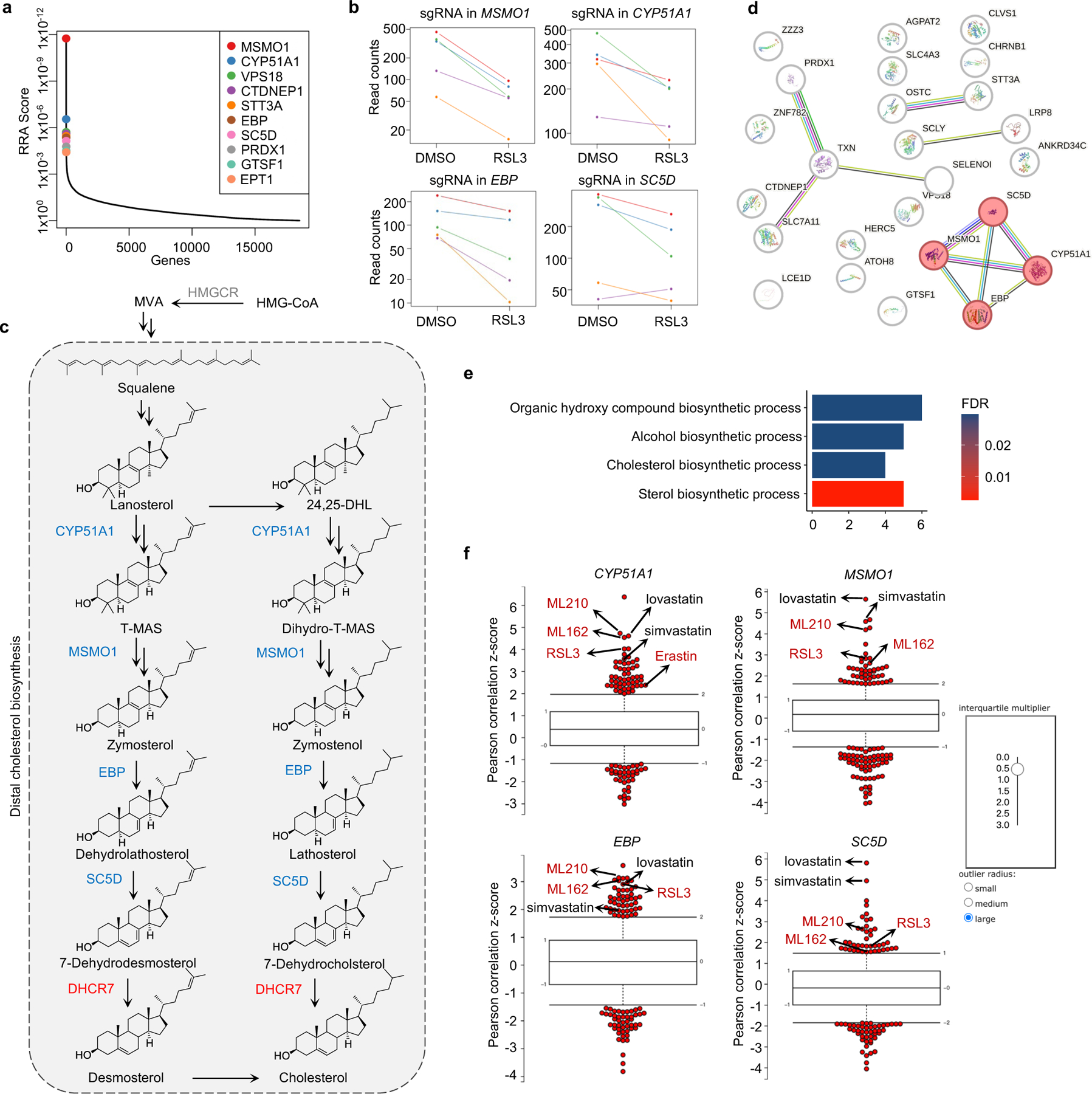
To identify novel ferroptosis regulatory mechanisms, we performed a genome-wide CRISPR-Cas9-based screening using HEK293T cells, a transformed human embryonic kidney cell line with moderate sensitivity to GPX4 inhibitor 1S,3R-RSL3 (hereafter RSL3). We introduced a whole genome sgRNA library targeting 1854325 genes into HEK293T cells and treated cells with RSL3 or DMSO for 8 days (Fig. 1a). By analyzing sgRNAs enriched or depleted in survived RSL3-treated cells in comparison with DMSO control, we identified several known ferroptosis suppressors, such as SLC7A1126, FSP111,12 and GCH114,15 in our anti-ferroptotic group (i.e., sgRNAs targeting these genes were depleted), indicating the robustness of our screening system (Fig. 1b). Interestingly, multiple genes involved in distal cholesterol biosynthesis, including MSMO1, CYP51A1, EBP and SC5D, were also identified as potential suppressors of ferroptosis (Fig. 1b, c, Extended Data Fig. 1a–c). Further enrichment analysis of our screening data by STRING27 or Gene Ontology showed that the cluster of cholesterol synthesis was top-ranked (Extended Data Fig. 1d, e), supporting a potential role of the distal cholesterol biosynthesis pathway in ferroptosis. We also analyzed the database of Cancer Therapeutics Response Portal (CTRP)28 and found that expression of CYP51A1, MSMO1, EBP and SC5D was positively correlated with the resistance to several ferroptosis inducers, including ML210, ML162 and RSL3 (Extended Data Fig. 1f). These data together prompted us to hypothesize that these genes for distal cholesterol biosynthesis may have a potential anti-ferroptosis function.
Fig. 1|. The genes involved in distal cholesterol synthesis differentially regulate ferroptosis.
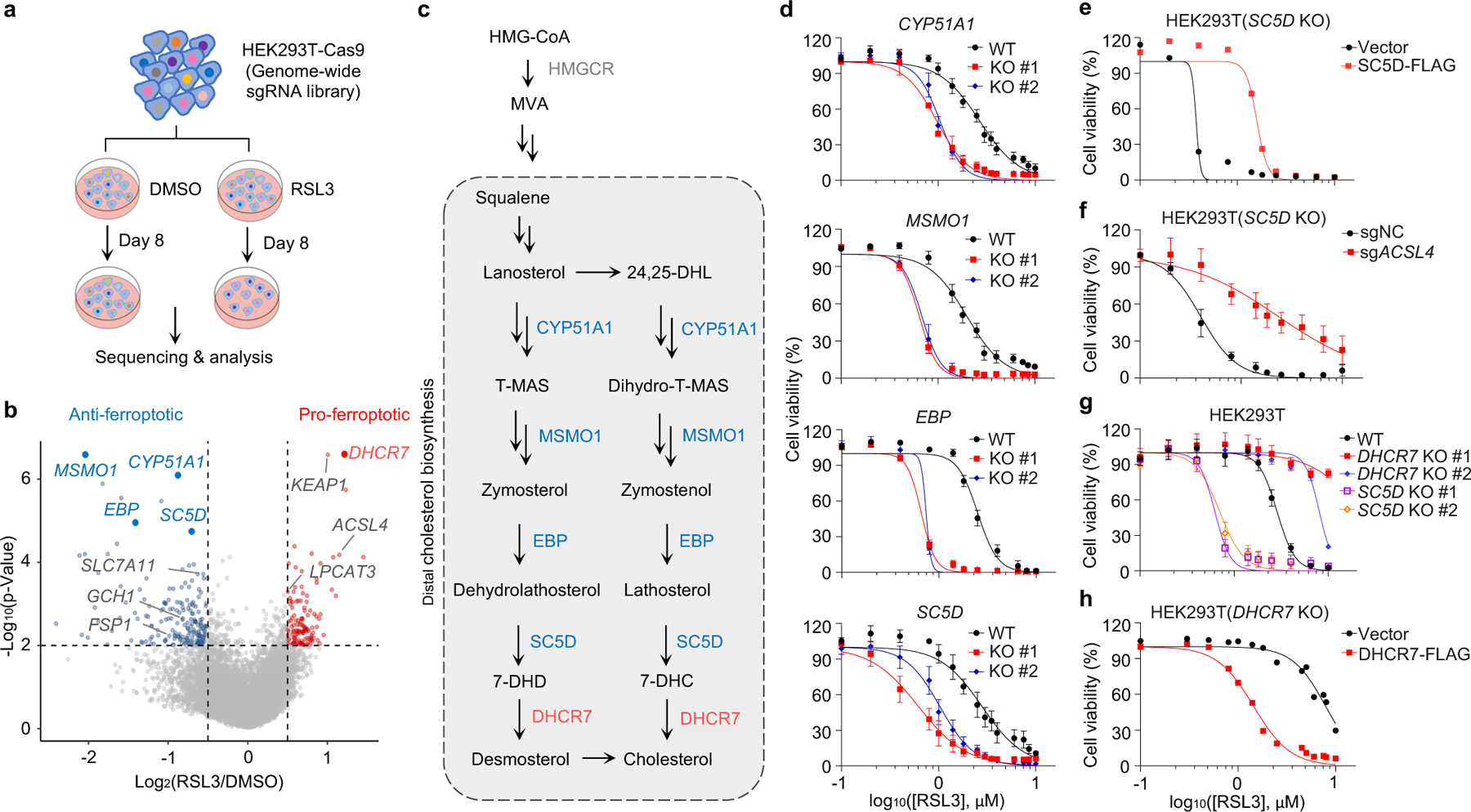
a, CRISPR screening workflow for identification of ferroptosis regulators. b, Volcano plot of top-ranked genes identified in RSL3-treated HEK293T-Cas9 cells. Genes that meet the cutoff criteria (P<0.01 and LFC>0.5 or <-0.5) are indicated as follows: genes (grey) represent known ferroptosis regulators and genes (blue and red) are involved in cholesterol biosynthesis. The p-Value and LFC are generated by the MAGeCK-test module using modified robust ranking aggregation (α-RRA) analysis. Top screen hits are shown in red (pro-ferroptotic) or blue (anti-ferroptotic). c, Schema of distal cholesterol biosynthesis pathway. d, Cell viability of HEK293T cells expressing sgRNAs targeting negative control (WT), CYP51A1 (CYP51A1 KO), MSMO1 (MSMO1 KO), EBP (EBP KO), or SC5D (SC5D KO), after treatment of indicated concentrations of RSL3 for 6–8 h. e, Cell viability of SC5D KO HEK293T cells expressing vector or SC5D cDNA after treatment of RSL3 for 6–8 h. f, Cell viability of SC5D KO HEK293T cells expressing sgRNAs targeting negative control or ACSL4 after treatment of RSL3 for 6–8 h. g, Cell viability of HEK293T cells expressing sgRNAs targeting negative control, DHCR7 (DHCR7 KO), or SC5D (SC5D KO) after treatment of RSL3 for 6–8 h. h, Cell viability of DHCR7 KO HEK293T cells expressing vector or DHCR7 cDNA after treatment of RSL3 for 10–12 h. Data are mean ± s.d. of n=3 biological replicates (d, f, g). For d-h, n=3 biological replicates (d, f, g) and n=2 biological replicates (e, h) are representative of three independent experiments.
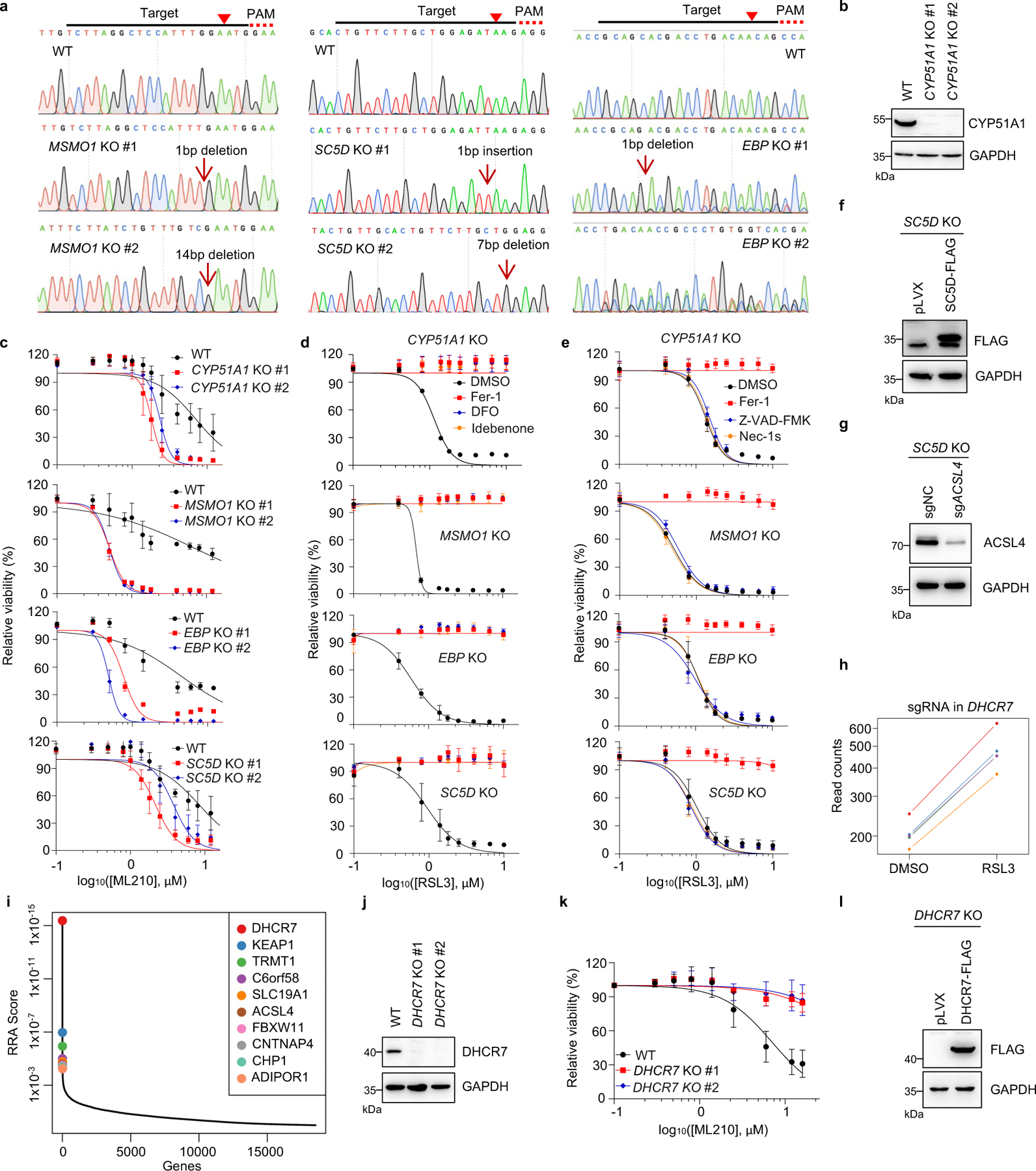
Next, we confirmed that individual deletion of CYP51A1, MSMO1, EBP and SC5D in HEK293T cells increased the susceptibility to ferroptosis induced by RSL3 and ML210 (Fig. 1d, Extended Data Fig. 2a–c). This lethal effect was effectively blocked by ferroptosis inhibitors deferoxamine (DFO), ferrostatin-1 (Fer-1) and idebenone (Ide), but not by apoptosis inhibitor (ZVAD-FMK) or necroptosis inhibitor Nec-1s (Extended Data Fig. 2d, e). Re-expression of SC5D reversed the severity of ferroptosis induced by RSL3 in SC5D KO cells (Fig. 1e, Extended Data Fig. 2f). Furthermore, deletion of the essential pro-ferroptotic gene ACSL4, which is required for incorporation of polyunsaturated fatty acids (PUFA) into phospholipids29, largely abolished the effect of SC5D deficiency on RSL3-induced cell death (Fig. 1f, Extended Data Fig. 2g). Together, these results indicate a common anti-ferroptosis activity possessed by the key players in distal cholesterol biosynthesis, including CYP51A1, MSMO1, EBP and SC5D.
Additionally, we evaluated the top-ranked candidate genes from the pro-ferroptotic group of our whole genome screen (i.e., sgRNAs targeting these genes were enriched). Intriguingly, DHCR7, encoding the terminal enzyme of cholesterol synthesis by converting 7-DHC to cholesterol, was ranked first in this group, even higher than the known ferroptosis mediators such as ACSL429, LPCAT330, KEAP131 (Fig. 1b, Extended Data Fig. 2h, i). We confirmed that DHCR7 deficiency strongly suppressed ferroptosis induced by RSL3 or ML210 (Fig. 1g, Extended Data Fig. 2j, k). In addition, re-expression of DHCR7 reversed the ferroptosis resistance in DHCR7 KO cells (Fig. 1h, Extended Data Fig. 2l).
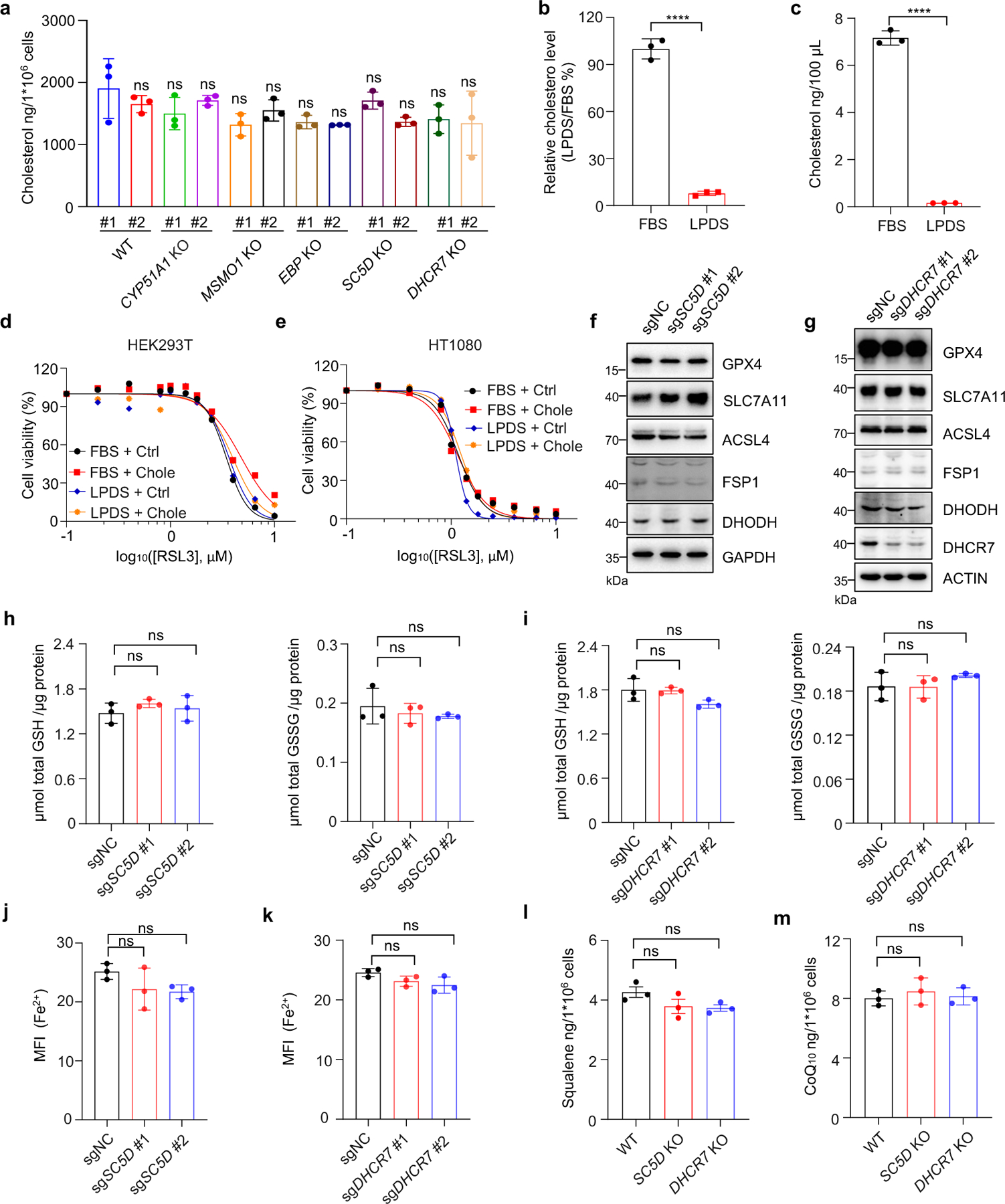
We next examined whether cholesterol is involved in ferroptosis regulation. For this purpose, we assessed the cholesterol levels of individual distal cholesterol biosynthesis genes deficient cells cultured in normal media. Our data showed that individual deletion of CYP51A1, MSMO1, EBP, SC5D and DHCR7 exhibited minor effects on cholesterol levels (Extended Data Fig. 3a). Furthermore, direct addition of cholesterol to the cell culture medium containing either normal serum (FBS) or lipoprotein deficient serum (LPDS) had little effect on cell viability (Extended Data Fig. 3b–e). Taken together, our data suggest that cholesterol does not directly regulate ferroptosis, which is consistent with the previous study32.
We further investigated whether the ablation of distal cholesterol biosynthesis genes could affect the known ferroptosis regulators. Our data showed that deletion of either SC5D or DHCR7 had neither effect on the protein levels of known ferroptosis regulators, including GPX4, FSP1, DHODH, SLC7A11 and ACSL4 (Extended Data Fig. 3f, g) nor the levels of GSH, Fe2+ (Extended Data Fig. 3h–k) and anti-ferroptotic metabolites CoQ10 and squalene (Extended Data Fig. 3l, m). These results suggest that loss of DHCR7 protects cells by an unknown mechanism that might be responsible for the differential roles of distal cholesterol biosynthesis genes in ferroptosis regulation.
7-DHC suppresses ferroptosis
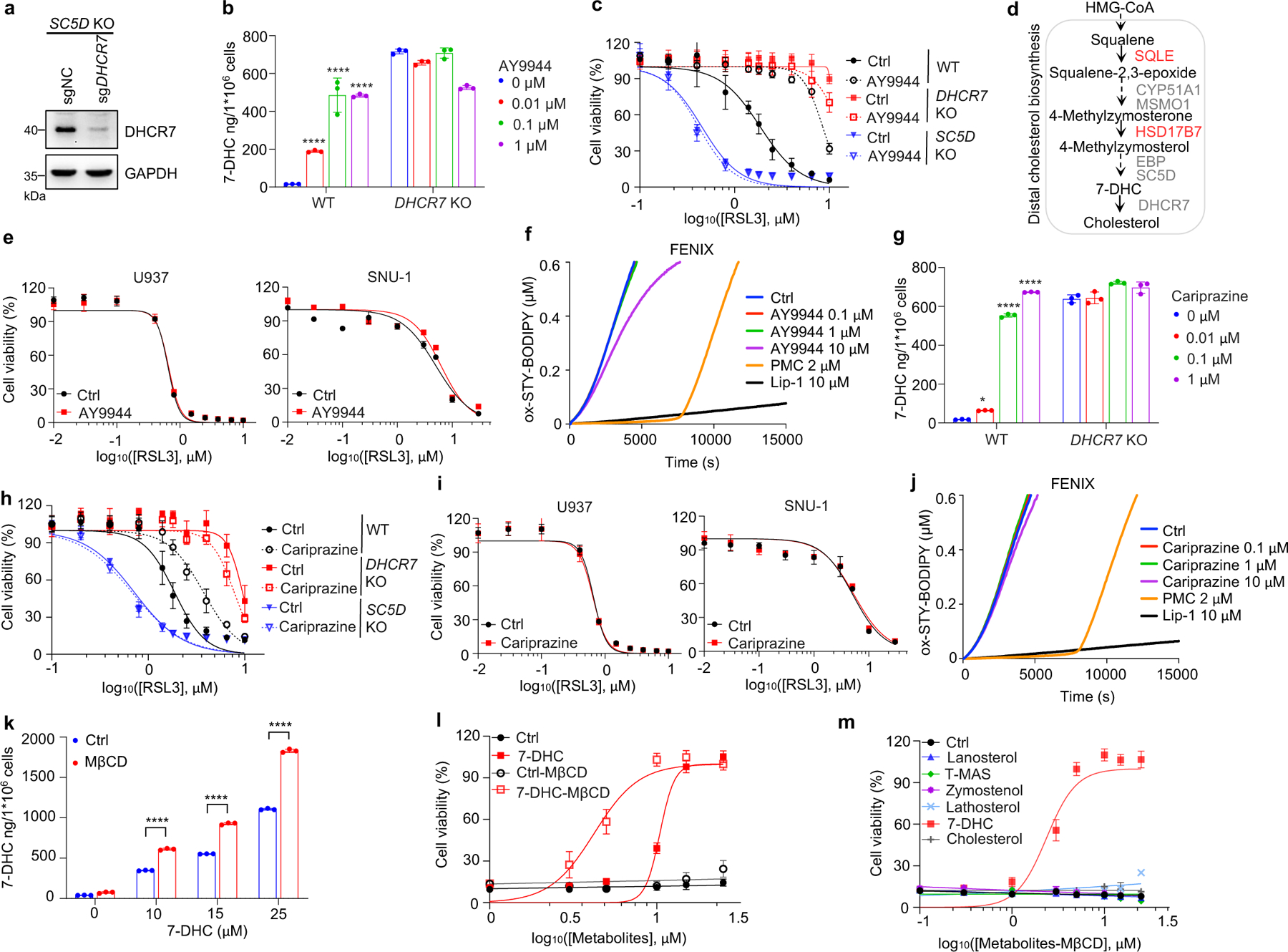
Next, we explored the mechanism by which distal cholesterol biosynthesis genes differentially regulate ferroptosis. It is known that 7-DHC is the major intermediate metabolite that may be accumulated by deletion of DHCR7, but abolished by deletion of genes in upstream distal cholesterol biosynthesis17. We confirmed that the level of 7-DHC was markedly increased by DHCR7 deletion and decreased by individual deletion of CYP51A1, MSMO1, EBP and SC5D (Fig. 2a). This information led us to hypothesize that the abundance of 7-DHC may be responsible for the differential roles of distal cholesterol biosynthesis genes in ferroptosis regulation. To test this hypothesis, we blocked the 7-DHC synthesis in DHCR7 KO cells by deleting SC5D, the immediate upstream enzyme for 7-DHC synthesis and found that deletion of SC5D significantly reversed the suppressive effect of DHCR7 deficiency on ferroptosis (Fig. 2b, c). In contrast, depletion of DHCR7 failed to suppress ferroptosis in SC5D deficient cells (Fig. 2d, Extended Data Fig. 4a). These data suggest that 7-DHC abundance is essential for their differential functions in ferroptosis.
Fig. 2|. 7-DHC suppresses ferroptosis.
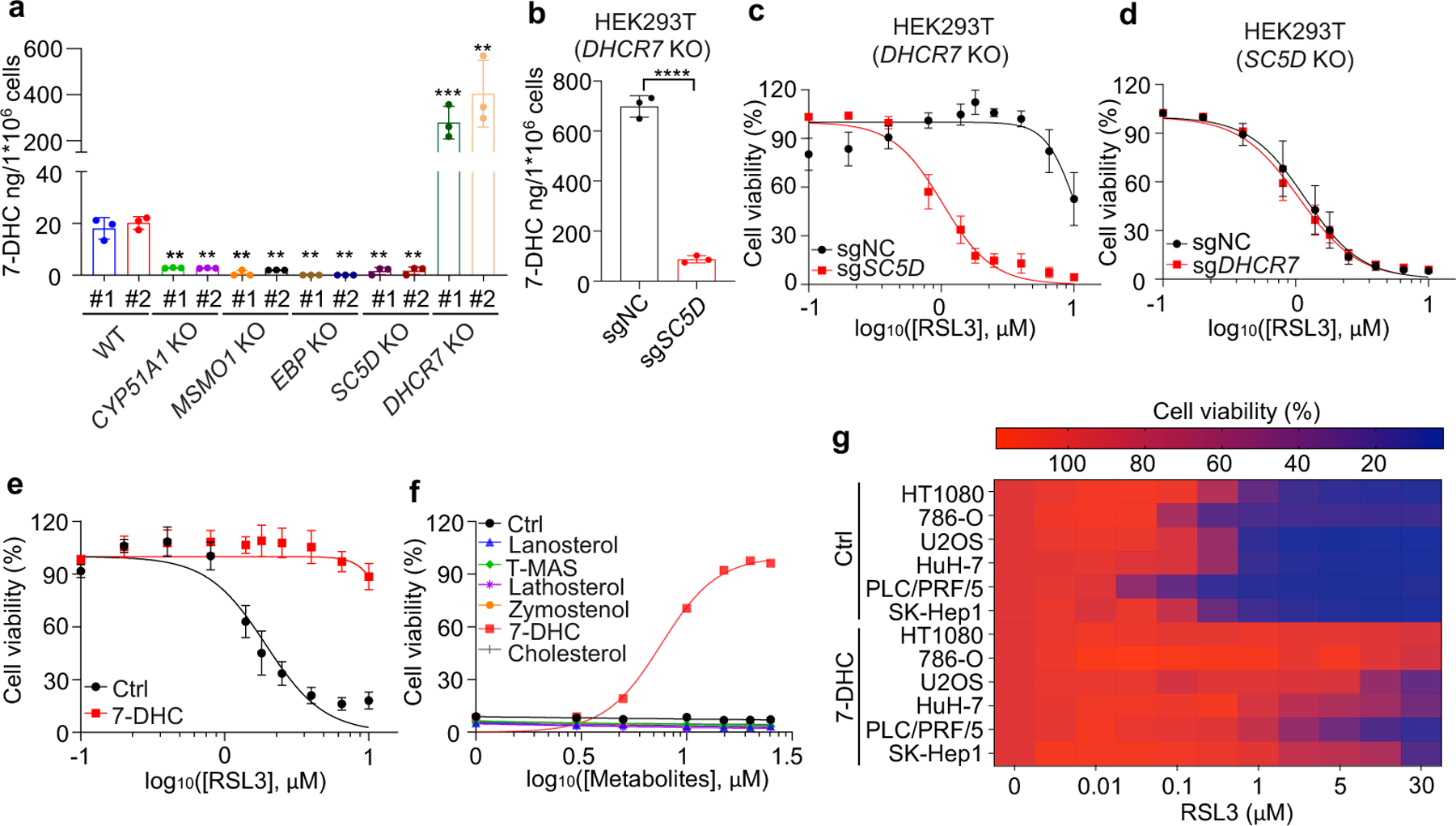
a, Levels of 7-DHC in WT and CYP51A1, MSMO1, EBP, SC5D, DHCR7 KO HEK293T cells b, Levels of 7-DHC in DHCR7 KO HEK293T cells expressing sgRNAs targeting negative control (sgNC) and SC5D (sgSC5D). c, Cell viability of DHCR7 KO HEK293T cells expressing sgRNAs targeting negative control (sgNC) and SC5D (sgSC5D) treated with RSL3 for 6–8 h. d, Cell viability of SC5D KO HEK293T cells expressing sgRNAs targeting negative control (sgNC) and DHCR7 (sgDHCR7) treated with RSL3 for 6–8 h. e, Cell viability of HEK293T cells treated with RSL3 for 6–8 h after pretreatment of 7-DHC (25 μM) for 24 h. f, Cell viability of HEK293T cells treated with RSL3 (5 μM) for 8 h after pretreatment of indicated concentrations of different sterols for 24 h. n=2 biological replicates. g, Heat map (data summarized from Extended Data Fig. 5a) depicting the dose-dependent toxicity of RSL3 in various human cancer cell lines, 7-DHC treatment vs. the ethanol control (Ctrl). Data are representative of two (a, b) and three (c-g) independent experiments. Data are mean ± s.d. of n=3 biological replicates (a-e), statistical analysis was performed using one-way analysis of variance (ANOVA) (a) or unpaired two-tailed t-tests (b); **P < 0.01; ***P < 0.001; ****P < 0.0001
To delve into the protective role of 7-DHC against ferroptosis, we elevated 7-DHC level by treating cells with DHCR7 selective inhibitor AY994433. Our data showed that AY9944 was able to effectively increase 7-DHC level in WT, but not in DHCR7 KO cells (Extended Data Fig. 4b), confirming its on-target efficacy. Importantly, AY9944 significantly suppressed ferroptosis in WT, but not in SC5D or DHCR7 deficient cells (Extended Data Fig. 4c). In addition, little effect of AY9944 treatment was detected in U937 and SNU-1 cells, which are defective of 7-DHC biosynthesis due to HSD17B7 or SQLE deficiency34 (Extended Data Fig. 4d, e). Notably, AY9944 exhibited little radical trapping antioxidant (RTA) activity at the concentration that we used to treat cells by an in vitro FENIX assay (Extended Data Fig. 4f). The anti-ferroptosis property of 7-DHC in ferroptosis was further confirmed by using cariprazine, an FDA approved drug which can increase 7-DHC level via targeting DHCR733 (Extended Data Fig. 4g–j). Altogether, these data indicate that the accumulation of 7-DHC is responsible for the differential roles of distal cholesterol biosynthesis genes in ferroptosis regulation.
We next evaluated whether 7-DHC is a general suppressor of ferroptosis. Our data showed that delivery of 7-DHC by direct supplement or MβCD-coated methods into cells significantly increased the intracellular 7-DHC levels (Extended Data Fig. 4k) and enabled a robust protection against ferroptosis (Fig. 2e, Extended Data Fig. 4l). In contrast, other intermediates such as lanosterol (substrate of CYP51A1), T-MAS (substrate of MSMO1), zymostenol (substrate of EBP) and lathosterol (substrate of SC5D)17 displayed little effect on cell viability (Fig. 2f, Extended Data Fig. 4m). The inhibitory effect of 7-DHC on ferroptosis triggered by different inducers was also evidenced in a larger panel of cell lines (Fig. 2g, Extended Data Fig. 5a, b). Consistently, inhibition of DHCR7 by its inhibitor AY9944 or cariprazine suppressed the ferroptosis in various types of cancer cells (Extended Data Fig. 5c, d). Collectively, these data indicate that 7-DHC is a potent ferroptosis suppressor and suggest that the genes controlling its intracellular abundance may dictate the sensitivity of ferroptosis.
7-DHC suppresses PL peroxidation
We next addressed the mechanism by which 7-DHC suppressed ferroptosis. As phospholipid (PL) peroxidation is the hallmark of ferroptosis7, we evaluated whether 7-DHC could affect phospholipid peroxidation in cells by staining with BODIPY 581/591 C11, a selected probe monitoring lipid peroxidation35. Our data showed that 7-DHC treatment significantly blocked lipid peroxidation induced by RSL3 in both HEK293T and HT1080 cells (Fig. 3a, Extended Data Fig. 6a–c). Notably, accumulation of endogenous 7-DHC by deletion of DHCR7 reduced lipid peroxidation, while deletion of MSMO1, CYP51A1, EBP and SC5D enhanced lipid peroxidation (Fig. 3b, Extended Data Fig. 6d), which was associated with a decrease in 7-DHC level (Fig. 2a). Furthermore, our targeted oxidative lipidomic analysis showed that RSL3-induced phospholipid peroxidation was markedly reduced upon DHCR7 deficiency (Extended Data Fig. 6e). Additionally, the intrinsic level of most phospholipids did not change significantly upon DHCR7 deficiency (Extended Data Fig. 6f, g). These data together indicate that 7-DHC can effectively protect cells from phospholipid peroxidation.
Fig. 3|. 7-DHC shields plasma and mitochondria membranes from autoxidation.
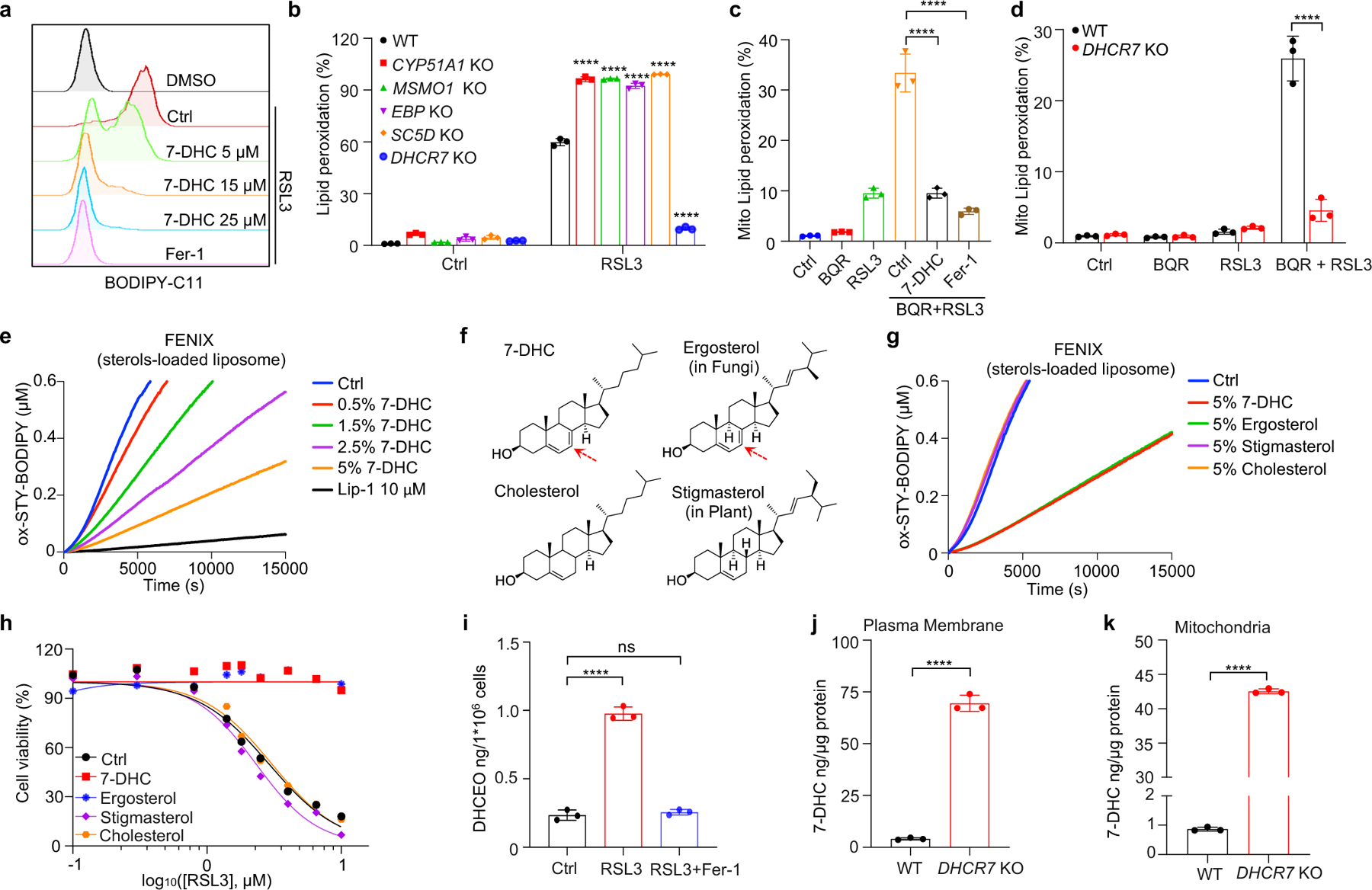
a, Lipid peroxidation assessment of HEK293T cells treated with RSL3 (2.5 μM) for 2 h with pretreatment of indicated concentrations of 7-DHC or Fer-1 (1 μM) for 24 h. b, Lipid peroxidation assessment of WT and CYP51A1, MSMO1, EBP, SC5D, DHCR7 KO HEK293T treated with RSL3 (2.5 μM) for 1 h. c. Mitochondrial lipid peroxidation assessment of HEK293T cells treated with RSL3 (1 μM) and BQR (200 μM) for 1 h with pretreatment of 7-DHC (25 μM) or Fer-1 (1 μM) for 24 h. d, Mitochondrial lipid peroxidation assessment of WT and DHCR7 KO HEK293T cells treated with RSL3 (1 μM) and BQR (200 μM). e, Representative autoxidation of STY-BODIPY (1 μM) and EggPC liposome containing indicated percentage of 7-DHC respectively. f, Chemical structures of 7-DHC, ergosterol, cholesterol and stigmasterol. Red arrows point to 5, 7-diene. g, Representative autoxidation of STY-BODIPY (1 μM) and EggPC liposome containing 5% 7-DHC, 5% ergosterol, 5% stigmasterol and 5% cholesterol respectively. h, Cell viability of HEK293T cells treated with RSL3 for 6–8 h after pretreatment of 7-DHC (25 μM), ergosterol (25 μM), stigmasterol (25 μM) or cholesterol (25 μM) for 24 h. n=2. i, Levels of DHCEO in HEK293T cells treated with RSL3 (5 μM) or plus Fer-1(1 μM) for 4 h. j, k, The concentration of 7-DHC of plasma membrane (j) and mitochondria (k) in WT and DHCR7 KO HEK293T cells. Data are representative of two (e, g, j, k) and three (a-d, and h, i) independent experiments. Data are mean ± s.d. of n=3 biological replicates (a-d and i-k). Statistical analysis was performed using two-way ANOVA (b and d) or one-way ANOVA (c and i) or unpaird two-tailed t-tests (j and k); ****P < 0.0001, ns, not significant.
Recent evidence has highlighted the vital role of mitochondrial lipid peroxidation in regulating ferroptosis13,35,36. We thus accessed whether 7-DHC could also block mitochondrial lipid peroxidation. Consistent with the previous report13, mitochondrial lipid peroxidation was significantly increased by combined inactivation of GPX4 and DHODH with their inhibitors, RSL3 and brequinar (BQR). Importantly, 7-DHC treatment markedly blocked mitochondrial lipid peroxidation (Fig. 3c, Extended Data Fig. 6h). Moreover, mitochondrial lipid peroxidation was aggravated by SC5D deletion but blocked by DHCR7 deletion (Fig. 3d, Extended Data Fig. 6i).
We next explored how 7-DHC suppressed phospholipid peroxidation. As 7-DHC contains a unique conjugated 5,7-diene in the B-ring compared to other sterols (Extended Data Fig. 6j) and has been reported as a highly oxidizable lipid19,20,37, we hypothesized that 7-DHC may possess ability to prevent phospholipid autoxidation. To test this possibility, we performed a FENIX assay38 and found that 7-DHC, but not other sterols such as lanosterol, T-MAS, zymostenol and lathosterol involved in distal cholesterol biosynthesis, showed strong activity to block the autoxidation of egg phosphatidylcholine in a dose-dependent manner (Extended Data Fig. 6k, l). Consistent with previous report20, pre-loading 7-DHC in liposomes markedly increased its activity to prevent phospholipid autoxidation (Fig. 3e). Notably, a comparison of 7-DHC with other analog sterols including ergosterol48, stigmasterol, as well as cholesterol showed that only 7-DHC and ergosterol possessed strong activity to block phospholipid autoxidation and to inhibit ferroptosis (Fig. 3f–h, Extended Data Fig. 6m). Analyzing the structure of the sterols in distal cholesterol biosynthesis and analogs of 7-DHC showed that only 7-DHC and ergosterol contain a 5, 7-diene, which may function as a H-atom donor to fatty ester (Fig. 3f, Extended Data Fig. 6j), indicating that the conjugated diene in the B-ring is important for 7-DHC to suppress ferroptosis. Consistently, 7-dehydrodemosterol (7-DHD), another sterol that shares the same ring structure as 7-DHC in distal cholesterol biosynthesis (Extended Data Fig. 1c), also strongly inhibits ferroptosis (Extended Data Fig. 6n). Further, induction of ferroptosis significantly decreased the level of 7-DHC (Extended Data Fig. 6o), while increased its oxidized product DHCEO (Fig. 3i, Extended Data Fig. 6p); and 7-DHC treatment neither affected the expressions of the ferroptotic related genes including GPX4, FSP1, SLC7A11 and ACSL4 (Extended Data Fig. 6q, r) nor activated the cholesterol homeostasis regulator SREBP2 as well as its downstream genes, such as SCD1, ACSL1 or transferrin (TF)39 (Extended Data Fig. 6s, t).
As oxidation of PUFA tails within membrane phospholipids, instead of that of free PUFAs, drives ferroptosis40, we examined whether 7-DHC can function as a suppressor for phospholipid peroxidation on mitochondrial and plasma membrane. Our data showed that 7-DHC could be readily detected on both mitochondria and plasma membranes, where 7-DHC was greatly accumulated upon DHCR7 deficiency (Fig. 3j, k). Unexpectedly, we found that DHCR7 could be readily detected on both plasma membrane (Extended Data Fig. 7a) and mitochondria (Extended Data Fig. 7b, c). A previous study indicated that DHCR7 C380 could be carbonylated by lipid-derived electrophiles (LDEs), which are generated during lipid peroxidation41. This prompted us to examine whether DHCR7 activity is regulated during ferroptosis. We confirmed that DHCR7 was indeed carbonylated at C380 during ferroptosis in a time dependent manner (Extended Data Fig. 7d–i). Interestingly, our in vitro assay showed that carbonylation promoted the enzyme activity of DHCR7 (Extended Data Fig. 7j, k). Structural alignment between human DHCR7 predicted by AlphaFold2 and the crystal structure of delta14-sterol reductase from Methylomicrobium alcaliphilum (Protein Data Bank (PDB): 4QUV; Extended Data Fig. 7l) showed that C380 is localized close to the NADPH-binding pocket of DHCR7 (Extended Data Fig. 7l, m), suggesting that modification at C380 may affect NADPH-binding affinity as well as the catalytic activity of DHCR7. Together, these data suggest that the anti-ferroptotic effect of 7-DHC may be dynamically regulated at different stage of ferroptosis and might be more relevant at the early stage of ferroptosis (Extended Data Fig. 7n).
7-DHC regulates tumour ferroptosis
Although accumulating evidence indicate that targeting ferroptosis could be a promising therapeutic strategy for cancers2,3,11,13, the vulnerability to ferroptosis varies largely in different malignancies2,11. Thus, we examined whether pharmacological blocking 7-DHC synthesis could be exploited for cancer therapy by using TASIN-30, a selective inhibitor targeting EBP42, an upstream enzyme for 7-DHC synthesis. We confirmed that treatment with TASIN-30 markedly reduced the level of 7-DHC in cancer cells (Fig. 4a, Extended Data Fig. 8a). Furthermore, TASIN-30 treatment markedly increased the susceptibility of various cancer cells to ferroptosis (Fig. 4b, Extended Data Fig. 8b). The effect of TASIN-30 on ferroptosis via targeting 7-DHC synthesis was confirmed using EBP and DHCR7 KO cells (Extended Data Fig. 8c). We also assessed whether blocking 7-DHC synthesis could trigger ferroptosis in cancer cells. As 7-DHC is synthesized by SC5D but consumed by DHCR7, we searched for cell lines whose fitness may be dependent on 7-DHC by analyzing the Project Score database43 (https://score.depmap.sanger.ac.uk/) and found that the fitness of 42 cancer cell lines is largely dependent on SC5D, but not DHCR7 (Fig. 4c). Among them, SU-DHL-8 was identified as the top-scoring cell line (Fig. 4c, Extended Data Fig. 8d), suggesting the essential role of 7-DHC in its fitness. Notably, we confirmed that SU-DHL-8 cells contained high level of 7-DHC (~110 ng/million cells) (Fig.4f). Importantly, blocking 7-DHC biosynthesis by TASIN-30 alone markedly triggered ferroptosis in SU-DHL-8 cells, even in the absence of any other ferroptosis inducers, indicating that the endogenous 7-DHC is essential to protect SU-DHL-8 cells against ferroptosis (Fig. 4d–f, Extended Data Fig. 8e). Altogether, these data indicate the importance of 7-DHC in anti-ferroptosis in a wide range of cancer types. In particular, we demonstrate 7-DHC is an essential prerequisite for the survival of certain type of cancer cells.
Fig. 4|. Targeting 7-DHC biosynthesis regulates cancer cell sensitivity to ferroptosis.
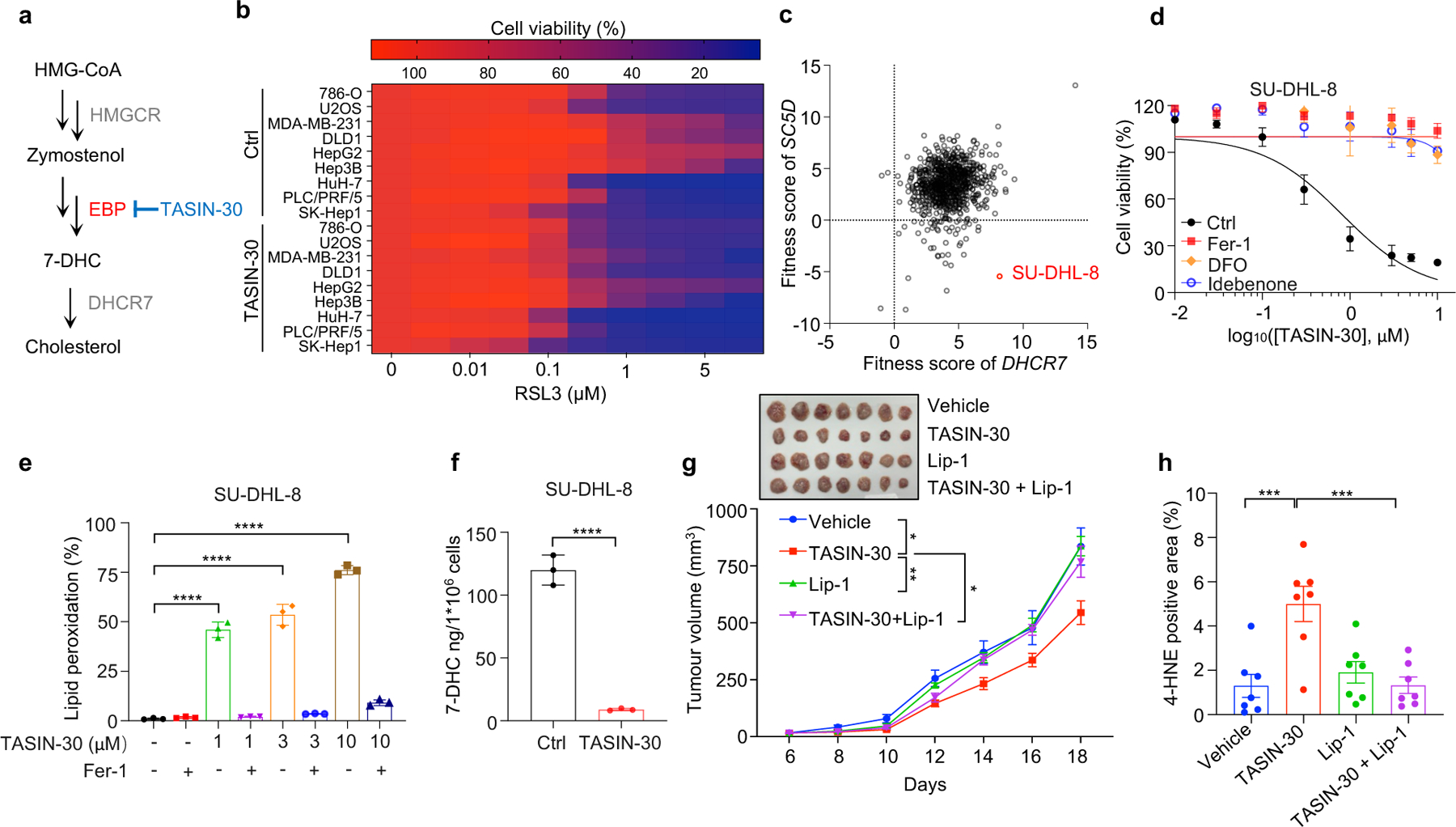
a, Schema of cholesterol biosynthesis pathway. TASIN-30 is an inhibitor of EBP. b, Heat map (data summarized from Extended Data Fig. 8b) depicting the dose-dependent toxicity of RSL3 in various human cancer cell lines (human kidney cancer cell line 786-O; human osteosarcoma cell line U2OS; human breast cancer cell line MDA-MB-231; human colon cancer cell line DLD1; human hepatocellular carcinoma cell lines HepG2, Hep3B, HuH-7, PLC/PRF/5 and SK-Hep1), TASIN-30 treatment vs. the DMSO control (Ctrl). c, The fitness scores of SC5D and DHCR7 in 810 cancer cell lines were explored by Project Score Database. Fitness score is a quantitative evaluation of the cell viability effect elicited by CRISPR-Cas9-mediated cell inactivation. d, Cell viability of SU-DHL-8 cells treated with indicated concentrations of TASIN-30 for 24 h after pretreatment of Fer-1 (1 μM), DFO (5 μM) or Idebenone (5 μM). e, Lipid peroxidation assessment of SU-DHL-8 cells treated with indicated concentrations of TASIN-30 for 16 h with pretreatment of Fer-1 (1 μM). f, Levels of 7-DHC in SU-DHL-8 cells treated with TASIN-30 (3 μM) for 24 h, both cells were cultured with Fer-1 (1 μM). g, Tumour growth rates of SU-DHL-8 xenografts with indicated treatments over time. n=7 in each group. h, Quantification of immunohistochemical 4-HNE staining in SU-DHL-8 xenografts with indicated treatments. n=7 in each group. Data are representative of three (b, d, e) and two (f, g) independent experiments. For d-f, data are mean ± s.d. of n=3 biological replicates, statistical analysis was performed one-way ANOVA (e and f). For g, h, data plotted are mean ± s.e.m., statistical analysis was performed two-way ANOVA (g and h) ;*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
We next accessed whether the anti-ferroptotic feature of 7-DHC could be a therapeutic vulnerability by using a preclinical tumour animal model. To this end, we examined the effect of TASIN-30 on the tumour growth of SU-DHL-8 in xenografts in mice. Our data showed that TASIN-30 injection not only markedly reduced the 7-DHC levels in tumour tissues (Extended Data Fig. 8f), but also inhibited tumour growth (Fig. 4g, Extended Data Fig. 8g) without significant effect on body weight of mice (Extended Data Fig. 8h). Co-injection of liproxstatin-1 abolished the tumour-suppressive effect induced by TASIN-30, suggesting that TASIN-30 inhibited tumour growth in a ferroptosis dependent manner. The induction of ferroptosis in tumour by TASIN-30 was further confirmed by detecting 4-hydroxynonenal (4-HNE), a product of lipid peroxidation, which is commonly used as a surrogate marker of ferroptosis (Fig. 4h, Extended Data Fig. 8i). Altogether, these data suggest that pharmacological impediment to 7-DHC biosynthesis may be a potent strategy to evoke ferroptosis for treatment cancer, especially those with high level of 7-DHC.
Recent studies showed that melanoma cells in the bloodstream can suffer from excess of oxidative stress which renders metastasizing melanoma cells more vulnerable to ferroptosis44. We therefore examined whether 7-DHC could protect cancer cells against oxidative stress in blood by examining its effect on the metastatic capacity of B16F10 melanoma cells, which contain low concentrations 7-DHC (~ 5–10 ng/million cells) (Extended Data Fig. 9a). Our data showed that pre-treatment of 7-DHC increased 7-DHC level (to ~70 ng/million cells) and significantly protected B16F10 cells from ferroptosis (Extended Data Fig. 9a, b). Importantly, 7-DHC treatment promoted B16F10 lung metastasis (Extended Data Fig. 9c, d). Consistently, Dhcr7 deletion attenuated RSL3-induced ferroptosis (Extended Data Fig. 9e–g) and significantly increased the metastasis without affecting the tumour growth. (Extended Data Fig. 9h–k). It is worth noting that TASIN-30 pretreatment effectively promoted the ferroptosis and blocked metastasis of B16F10 in Dhcr7 deletion cells (Extended Data Fig. 9l–o), suggesting that endogenous concentration of 7-DHC may be a critical checkpoint to dictate the sensitivity of TASIN-30 both in vivo and in vitro. Direct injection of AY9944 to mice failed to promote metastasis (Extended Data Fig. 9p, q), suggesting that high level of 7-DHC in cancer cells, but not in host, can augment melanoma to overcome oxidative stress in vivo and aggravate metastasis seeding in the lung. At this stage, we cannot rule out the possibility that metastasis promoted by 7-DHC may be via a more complex mechanism, in addition to ferroptosis inhibition.
7-DHC protects kidney from IRI
Ferroptosis has been implicated as the major form of cell death in ischemia-reperfusion injury (IRI)5,6. We thus examined whether increasing 7-DHC level in vivo could protect kidney against IRI. As direct injection of 7-DHC was inapplicable in vivo (Extended Data Fig. 10a), we chose to elevate the endogenous level of 7-DHC via targeting its catalyzing enzyme, DHCR7 (Fig. 5a). Our data showed that pre-injection of AY9944 significantly increased the levels of 7-DHC in mouse kidney and serum (Fig. 5b, Extended Data Fig. 10b). Importantly, pre-injection of AY9944 was able to protect the kidney from IRI, assessed by blood urea nitrogen (BUN) and serum creatinine (SCr) (Fig. 5c, d). The protection was further confirmed by histopathological analysis (Fig. 5e, f). Consistently, pre-injection of AY9944 significantly reduced ferroptosis indicated by the reduction of 4-HNE levels (Fig. 5g, h). Moreover, the comparable protection effect was detected by using another DHCR7 inhibitor, cariprazine (Extended Data Fig. 10c–o). Taken together, these results illustrate that accumulation of endogenous 7-DHC can suppress ferroptosis in vivo and targeting DHCR7 may be a promising therapeutic approach for IRI.
Fig. 5|. 7-DHC attenuates ischemia-reperfusion injury in vivo.
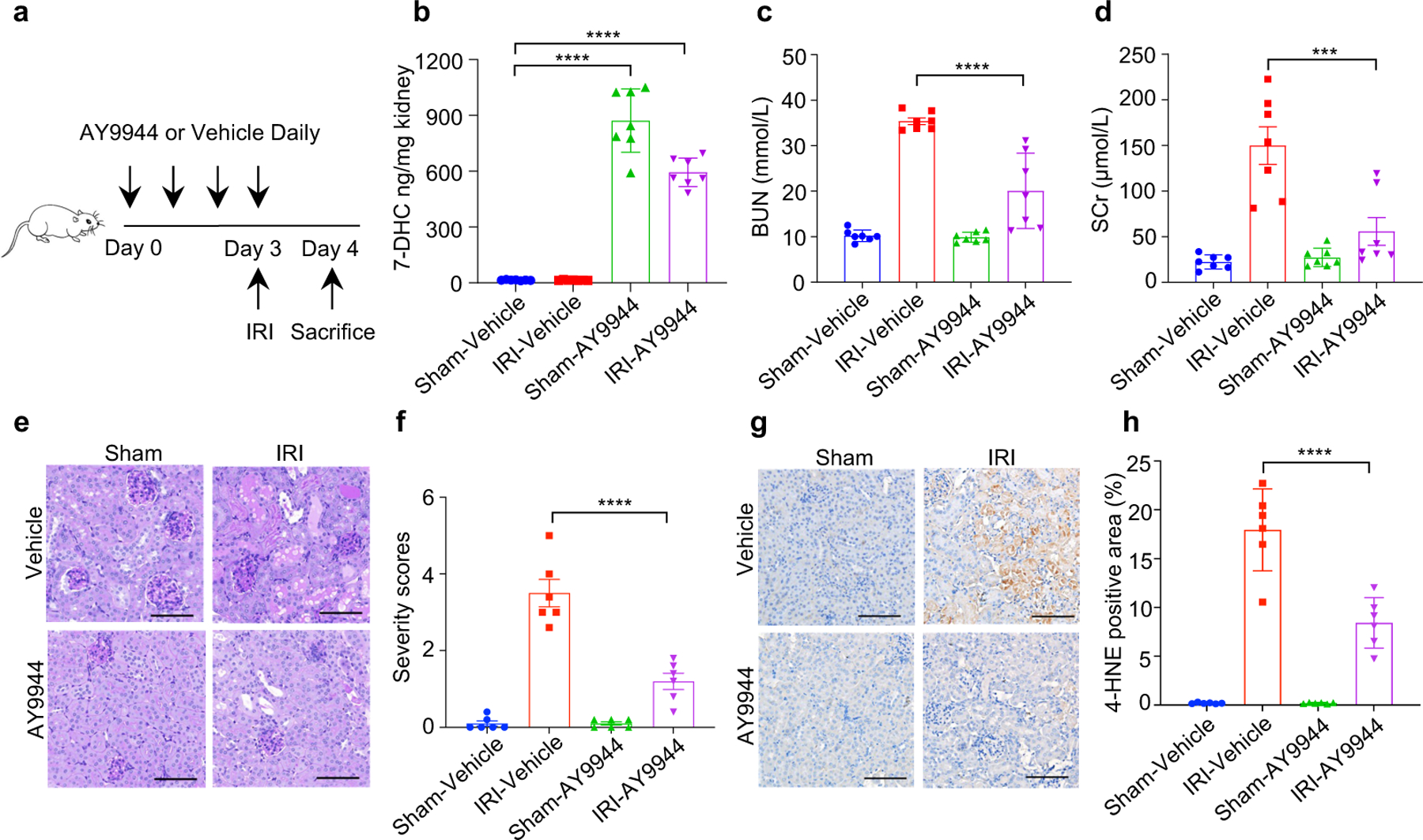
a, Workflow of kidney ischemia–reperfusion injury (IRI) model with pretreatment of AY9944 via intraperitoneal injection once daily for four times before IRI at the dose of 25 mg/kg. b, Levels of 7-DHC in kidneys of mice IRI model. n=7 in each group. c, d, The concentration of blood urea nitrogen (BUN) (c) and serum creatinine (SCr) (d) of mice kidney IRI model. n=7 in each group. e, f, Representative PAS staining (e) and quantification of renal damage (f) from mice kidney IRI model. n=6 in each group. Scale bar, 100 μm. g, h, Representative immunohistochemical staining (g) and quantification (h) of 4-HNE in kidneys from mice IRI model. n=6 in each group. Scale bar, 100 μm. For b-h, data are representative of two independent experiments and data plotted are mean ± s.e.m., statistical analysis was performed two-way ANOVA, ***P < 0.001, ****P < 0.0001.
Discussion
As a metabolic intermediate, 7-DHC has been shown to possess anti-viral activity45,46; yet high level of 7-DHC is toxic to neurons and retina-derived cell types due to its derived oxysetrols21,22,47. However, the patho(physio)logical function of 7-DHC remains largely unknown. Our current study provides clear evidence demonstrating that 7-DHC is a potent anti-ferroptotic metabolite. 7-DHC protects cells from phospholipid peroxidation on both plasma membrane and mitochondria via diverting the peroxidation pathway from phospholipids, thus mitigating ferroptosis (Extended Data Fig. 10p). We showed that ergosterol, an analog of 7-DHC found in S. cerevisiae, can also suppress ferroptosis. Presumably, common chemical property shared by these unsaturated sterols, particularly their possession of conjugated dienes, enables radical trapping for ferroptosis inhibition47,48. Our study also indicates that peroxidation of different lipids, such as phospholipids and sterols, may have distinct functions during ferroptosis. Furthermore, it warrants further investigation whether various metabolites of 7-DHC, derived by oxidation and other chemistry, are involved in ferroptosis regulation.
Importantly, our study has strong therapeutic implications. We provide compelling evidence indicating that inhibiting 7-DHC biosynthesis is a potential strategy for the treatment of cancers, especially those with high level of 7-DHC or harboring DHCR7 mutation, such as Burkitt’s lymphoma24. Conversely, our study also suggests that elevating 7-DHC levels by pharmacologically inhibiting DHCR7 may improve the clinical outcomes of IRI patients. As a panel of FDA approved drugs have been shown to inhibit DHCR733, this possibility is readily testable in clinical settings for treating chronic IRI patients.
Methods
Cell culture studies
All cells were cultured in a 37 °C humidified incubator with 5% CO2. Human embryonic kidney 293 cells HEK293T, human fibrosarcoma cell line HT1080, human hepatocellular carcinoma cell lines (SK-Hep1, HepG2, HuH-7, Hep3B, PLC/PRF/5), human kidney cancer cell lines (RCC4, Caki-1), human breast cancer cell line MDA-MB-231, were cultured in DMEM (Multicell) with 10% FBS (Gibco) and 1% penicillin-streptomycin. Human kidney cancer cell line 786-O, human colon cancer cell line DLD1, human gastric carcinoma cell line SNU-1, human osteosarcoma cell line U2OS, human lymphoma cell line U937, human DLBCL cell line SU-DHL-8, were cultured in RPMI1640 (Multicell) with 10% FBS (Gibco) and 1% penicillin-streptomycin. Mouse melanoma cell line B16F10 was cultured in DMEM (Multicell) with 10% FBS (Gibco) and 1% penicillin-streptomycin. All the cell line were kindly provided by Cell Bank/Stem Cell Bank, Chinese Academy of Sciences, except for RCC4, SU-DHL-8 that were obtained from ATCC, and MDA-MB-231 was a gift from Dr. Yao Sun at department of Implantology, School and Hospital of Stomatology, Tongji University. All cell lines tested negative for mycoplasma contamination.
Genome-wide CRISPR screen and data analysis HEK293T-Cas9 cell line generation
HEK293T cells were infected with lentivirus expressing Cas9 and blasticidin S deaminase. After one week of drug selection by blasticidin S, cells were sorted and seeded into 96-well plate by FACS. Subclones were analyzed by immunoblotting.
Genome-wide CRISPR screen in HEK293T-Cas9
The experiment was carried out as previously described25,49. Briefly, the screening was performed by using the human genome-wide lentiviral sgRNA library (Addgene, 1000000095). The library was split into three sub-libraries, containing 10 sgRNAs per gene. Two libraries have ∼90,000 gRNAs each (hL1nC9 and hL2nC9), and one library (hL3nC9) has ∼5000 gRNAs. Libraries of hL1nC9 and hL3nC9 were selected to produce a high titer genome-wide lentiviral sgRNA library and transfected HEK293T-Cas9 cells at a multiplicity of infection (MOI) of 0.3, selected with 1μg/mL puromycin for 7 days. After selection, 5 × 107 cells were treated with either DMSO or 2.5 μM RSL3. At day 8, 1 × 107 cells were collected from the surviving population of RSL3-treated and untreated samples. Genomic DNA was extracted and sgRNA sequences were amplified by PCR followed by Next Generation Sequencing processed by GENEWIZ.
Analysis of CRISPR screen data
MAGeCK-VISPR50 were used for the whole process, including read mapping, sgRNA annotation, and comparison of the conditions with MAGeCK RRA algorithm. The principle of data analysis was consulted with previous study51. Briefly, sgRNAs with fewer than 20 reads in the DMSO treatment group were omitted from the RRA algorithm analyses. Genes from both positive selection and negative selection were selected with P-value cut-off of 0.01. For the volcano plot, genes targeted by fewer than three distinct sgRNAs were omitted, and the pos-P-value from RRA algorithm was used when LFC>0 and the neg-P-value RRA algorithm was used when LFC<0.T. The analysis data was provided in Supplementary Table 1. STRING analysis and GO term enrichment analyses were conducted using STRING (https://string-db.org). The volcano plot of CRISPR screen data and the column chart of GO term enrichment was visualized using Hiplot52 (https://hiplot.com.cn).
Generation of CRISPR–Cas9 genome-edited cell lines
For CYP51A1, MSMO1, EBP, SC5D or DHCR7 knockout HEK293T cell lines and Dhcr7 knockout B16F10 cell lines, one or two sgRNAs targeting each gene were cloned into PX458 for subclone construction, or cloned into lentiCRISPR-V2 for lentivirus production and pooled generation. For subclone construction, HEK293T cells were cultured in 6-well tissue culture plates for 12 h and then transfected with the corresponding PX458 vector. After transfection for 72 h, GFP-positive cells were sorted by FACS and seeded into 96-well plates followed by genotyping validation. For lentivirus production, the sgRNA-expressing and lentiviral packaging vectors (psPAX2 and pMD2.G) were co-transfected into HEK293T cells using PEI in a 1:3 ratio. The viral supernatant was collected 48 h after transfection and filtered with a 0.45 μm filter. HEK293T cells were infected with lentivirus containing 8 µg/mL polybrene. Infected cells were selected with 1 μg/mL puromycin after 48 h post-infection and propagated for further analysis. Guide RNA sequences are provided in Supplementary Table 2.
Overexpression plasmid construct and cell line generation
In brief, cDNA of DHCR7 and SC5D were cloned into pLVX expression vector with a 4 × Flag on the C-terminal, DHCR7-WT and DHCR7-C380A were also cloned into pcDNA3.1 expression vector with a 4 × Flag on the C-terminal. For stably overexpression cell line generation, pLVX empty vector or target gene constructs together with psPAX2 and pMD2.G lentiviral packaging vectors, were co-transfected into HEK293T cells, viral supernatant was collected and infected target cell lines. After 48 h infection, puromycin was used to select the infected cells.
CellTiter-Glo assay for cell viability analysis
To evaluate cell viability, cells were seeded into 96-well plate at a density of 3000–20000 cells/well (depending on cell size). After 15 h, cells were treated with different reagents or metabolites including RSL3 (Selleck, S8155), ML210 (MCE, S0788), Erastin (Sigma, E7781), Brequinar (MCE, HY-108325), Ferrostatin-1 (Selleck, S7243), Liproxstatin-1 (Selleck, S7699), AY9944 (APExBIO, A8658), Cariprazine (MCE, HY-14763), Idebenone (Selleck, S2605), Z-VAD-FMK (Selleck, S7023), Nec-1s (Selleck, S8641). For metabolite treatment, 7-DHC (Avanti, 700066), Lanosterol (Avanti, 700063), T-MAS (Avanti, 700073), Lathosterol (Avanti, 700069), Zymostenol (Avanti, 700118), Cholesterol (Sangon, C3045), Ergosterol (MCE, HY-N0181), Stigmasterol (MCE, HY-N0131) were treated for indicated time before ferroptosis induction. For LPDS pretreatment, cells were seeded in 96-well plate at a density of 3000–8000 cells/well (depending on cell size) and cultured with 10% LPDS supplemented DMEM. Cells were treated with AY9944 or cariprazine for 48 h as indicated concentration, then treated with RSL3 to induce ferroptosis. Cellular ATP levels were quantified using CellTiter-Glo (Promega, G7573) on a multi-plate reader (TECAN, iControl software). Unless otherwise indicated, relative viability evaluated by cellular ATP was normalized to the corresponding untreated condition. The mean and s.d. were presented with at least three biological replicates. Non-linear regression models were used to simplify curve fitting by GraphPad Prism 9 and the IC50 values were summarized in Supplementary Table 3.
Lipid peroxidation and mitochondrial lipid peroxidation measurement
Cells were seeded in 24-well or 48-well tissue culture plates and treated as shown in the figures. BODIPY 581/591 C11 dye (Invitrogen, D3861) for lipid peroxidation (total) was added at a concentration of 5 μM and MitoPeDPP dye (Dojindo, M466) for mitochondrial lipid peroxidation was added at a concentration of 0.5 μM for 30 min at 37 °C, then cells were harvested by trypsinization and washed once with HBSS, followed by resuspension in HBSS and analyzed by flow cytometry on BD FACS verse or Guava easyCyte. Initial cell population gating(FSC and SSC)was drawn to exclude debris as shown in the Supplementary figure 1. The signal from oxidized C11 (FITC channel) and oxidized MitoPeDPP was monitored and calculated. A minimum of 5 × 103 cells were analyzed for each sample, and each experiment was independently performed at least three times. Representative experimental results were indicated, data collected and analysis was performed using the BD FACSuite software, guava 3.1.1 software, and FlowJo 10 software.
Preparation of lipoprotein-deficient serum (LPDS)
The experiment was carried out as previously described53. In brief, the frozen serum was thawed at room temperature, and the density of serum was adjusted by adding 101 g of KBr into 300 mL serum. The mixture was stirred at room temperature until the KBr was dissolved. Then the serum solution was added to a 50 mL syringe fitted with a 14G needle. 40 mL of serum solution was loaded into an ultracentrifuge tube. Tubes were spun at 250,000 × g and decelerated slowly at 10 °C for 24 h. After ultracentrifugation, the fraction was collected and dialysis in a beaker containing 5 L of 150 mM NaCl. After dialysis, LPDS was adjusted and filter-sterilized via a 0.22 μm syringe filter and stored in suitable aliquots at −20 °C. Amplex red assay and LC-MS/MS were performed. Total cholesterol content of the LPDS was verified which was less than <5% of the level in the whole serum.
Preparation of MβCD-coated sterols
The experiment was carried out as previously described54. In brief, desired volume of ethanol-dissolved 5 mM 7-DHC, cholesterol, lanosterol, T-MAS, zymostenol and lathosterol was dried under nitrogen flow, followed by addition of same volume of 35 mM MβCD (MCE, HY-101461) prepared in serum-free DMEM. Sterol suspensions were sonicated for 5 min and fully dissolved. These MβCD-coated sterols were used immediately once prepared.
Immunoblotting
For immunoblotting, cells were washed once with ice-cold PBS and lysed in lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 5% Glycerol, and a cocktail of proteinase inhibitors) for 15 min at 4°C. The supernatant was isolated via centrifugation for 15 min at 12,000 rpm, 4°C. Proteins were resolved on SDS-PAGE gel and detected by the electrochemiluminescence (ECL) imaging system (Tanon). The following primary antibodies were used: GPX4 (1:1,000, 125066, Abcam), ACSL4 (1:3000, 22401-1-AP, Proteintech), FSP1 (1:500, sc-377120, Santa Cruz), DHODH (1:500, sc-166348, Santa Cruz), SLC7A11 (1:1000, 12691, Cell Signaling Technology), DHCR7 (1:1000, PA5-48204, Invitrogen), CYP51A1 (1:3000, 13431-1-AP, Proteintech), Flag (1:3000, 20543-1-AP, Proteintech), GAPDH (1:3000, 10494-1-AP, Proteintech), Pan-cadherin (1:1000, db4569, Diagbio), LAMP2 (1:1000, 27823-1-AP, Proteintech), HA (1:3000, 51064-2-AP, Proteintech), TOMM20 (1:1000, db50, Diagbio), Tubulin (1:1000, db3285, Diagbio), PDI (1:1000, 11245-1-AP, Proteintech), ACTIN (1:3000, db1959, Diagbio), CoxIV (1:1000, 4850T, Cell Signaling Technology), PEX19 (1:1000, 14713-1-AP, Proteintech), CD71 (1:1000, 13113S, Cell Signaling Technology), H3 (1:1000, 9715, Cell Signaling Technology), SREBP2 (1:1000, 28212-1-AP, Proteintech), Transferrin (1:500, 17435-1-AP, Proteintech), SCD1(1:1000, 28678-1-AP, Proteintech), ACSL1(1:1000, 13989-1-AP, Proteintech). The raw data covering a wide range of molecular weights in Supplementary figure 2.
FENIX assay
For sterols and compounds inhibited EggPC autoxidation.
EggPC (Avanti, 840051P) were resolved in CHCl3 to 20 mM, 3 mL of the solvent was then evaporated to yield a thin lipid film in a 100 mL vial, and placed in a vacuum oven to dry for 1 h. The film was then hydrated with 3 mL of PBS (pH 7.4) to yield a 20 mM lipid suspension. The lipid suspension was subjected to 10 freeze (dry ice, 5 min) − thaw (RT, 5 min) − sonication (RT, 5 min) cycles and extruded 20 times with a 100 nm polycarbonate membrane as previously described55. The experiment was carried out as previously described38. In brief, 1 mM liposome was mixed with 1 μM STY-BODIPY (Cayman Chemicals, 27089) and 250 µL of the mixture was added to a black 96-well polypropylene plate, sterols (7-DHC concentration gradient and other sterols in distal cholesterol biosynthesis) and compounds (AY9944 and cariprazine) were added to indicated concentration, incubated for 10 min at 37 °C and vigorous mixed for 5 min, initiated by 6 µL of 10 mM DTUN (Cayman Chemicals, 32742) in EtOH, mixed for 5 min to start autoxidation. Data was acquired by STY-BODIPY excitation at 488 nm and emission was measured at 518 nm in the BioTek Synergy H1 plate reader, Gen5 3.11 software. The data was transformed from the raw RFU values to ox-STY-BODIPY (μM) by the response factor of 3.22x104 RFU/μM. The response factor was measured for non-chain conditions by sets of uninhibited autoxidation with a dose of the STY-BODIPY (0.2, 0.4, 0.6, 0.8, 1 μM).
For 7-DHC and its analogs pre-loaded in the liposome.
The experiment was carried out as previously described20. We pre-loaded 7-DHC and other analogs into the liposome during the liposome-preparation process, leading to more efficient incorporation of sterols into the liposome membrane. In brief, ergosterol, stigmasterol, 7-DHC (Sigma, 30800) and cholesterol were dissolved to 10 mM in CHCl3, then mixed with eggPC (10 mM in CHCl3) at an indicated ratio and evaporated to yield a thin lipid film. The film was then hydrated with PBS (pH 7.4) to yield a 10 mM lipid suspension. The lipid suspension was subjected to 10 freeze (dry ice, 5 min) − thaw (RT, 5 min) − sonication (RT, 5 min) cycles and extruded 20 times with a 100 nm polycarbonate membrane. All prepared liposomes were stored at 4°C in dark and used within one week. For sterols-loaded liposomes, the experiment was carried out as follows: 250 µL of prepared 1 mM liposome and 1 μM STY-BODIPY were added to a black 96-well polypropylene plate and incubated for 10 min at 37°C and vigorous mixed for 5 min, initiated by 6 µL of 10 mM DTUN in EtOH, mixed for 5 min to start autoxidation.
Validation of carbonylated proteins in ferroptosis using m-APA with activity-based protein profiling (ABPP) assay
The experiment was carried out as previously described41. The cells were collected and counted, 8 × 106 cells were lysed by sonication in ice-cold PBS containing 0.5% TritonX-100 and centrifuged at 13, 000 x g for 30 min, and cell fragments were removed. Lysates from control or ferroptotic cells were separately labeled by 0.5 mM m-APA at pH 5.0 for 1 h at room temperature. Lysates were precipitated by adding 5 volumetric times of methanol/chloroform (v/v = 4:1) mixture and 3 volumetric times of water, and centrifuged at 10, 000 x g for 10 min. The supernatants were discarded, and the pellets were washed using cold methanol and resuspended in 0.4% (w/v) SDS/PBS. The samples were added with 1 mM CuSO4, 100 μM TBTA ligand, 100 μM biotin-(PEG)2-N3, and 1 mM TCEP for one-hour-reaction at room temperature. The samples were again precipitated by adding 5 volumetric times of methanol/chloroform (v/v = 4:1) mixture and 3 volumetric times of water, and centrifuged at 10, 000 x g for 10 min. The supernatants were discarded, and the pellets were washed using cold methanol and resuspended in 1.2% SDS/PBS, diluted to 0.2% SDS/PBS and subjected to streptavidin enrichment overnight. Samples were washed three times with 0.2% SDS/PBS, and SDS loading buffer was used to elute proteins at 95 °C for 20 min, after which SDS-page gel electrophoresis was used to detect carbonylated proteins.
Assay of 7-dehydrocholesterol reductase (DHCR7) enzymatic activity.
1 × 107 HEK293T cells overexpressing DHCR7-Flag were treated with DMSO or RSL3 for 3 h, then harvested and lysed in lysis buffer (50 mM Tris-Cl, pH 7.4, 0.5% NP-40, 150 mM NaCl, 1 mM EDTA, 10% glycerophosphate and a cocktail of proteinase inhibitors). After 30 min, the soluble fraction of the cell lysates were isolated via centrifuged at 12,000 × g for 15 min at 4 °C. For IP, the cell lysates were removed to a clean tube and incubated in M2 beads (Sigma, A2220) for 2–3 h. Flag peptide (10 μg/μL) was used to elute DHCR7-Flag from M2 beads. The concentration of DHCR7-Flag elution of DMSO treated group (DHCR7-D1st) and RSL3 treated group (DHCR7-R1st) were determined by BCA protein assay. Then, the elution of RSL3 treated group (DHCR7-R1st) was performed to activity-based protein profiling (ABPP) assay using 0.5 mM m-APA. Biotin (0.1 mM) was used to elute carbonylated DHCR7 (DHCR7-R2nd) and the concentration was measured via BCA protein assay. The in vitro enzymatic activity of DHCR7 was measured using DHCR7-D1st, DHCR7-R1st and DHCR7-R2nd (100 μg), 7-DHC (300 μM), NADPH (2 mM) with reaction buffer (20% glycerol, 0.1 M Tris-HCl, pH 7.4, 1 mM glutathione, 0.5 mM EDTA) in a final volume of 200 μL at 37 °C for 30 min; then the NADPH reduction can be calculated by measuring absorbance at 340 nm. DHCR7-D1st, DHCR7-R1st and DHCR7-R2nd enzymatic activity were detected by the reduction of NADPH.
Subcellular fraction assay
Rapid isolation of mitochondria for proteomics.
WT or DHCR7 KO HEK293T cells stably expressing 3HA-OMP25 were used for rapid isolation of mitochondria as previously described56. Briefly, 2 × 107 cells were rapidly washed twice with PBS and then gently scraped into 1 mL KPBS (136 mM KCl and 10 mM KH2PO4, pH 7.25). The cell suspension was centrifuged at 1, 000 × g for 2 min, and the cell pellet was gently resuspended in 1 mL KPBS. And then, cells were homogenized with 25 strokes of a 1 mL homogenizer. The homogenate was centrifuged at 1, 000 × g for 2 min, and the supernatant was incubated with 20 μL of prewashed anti-HA magnetic beads (Thermo, 88837) on a rotator for 15 min. Beads were quickly washed three times with 1 mL KPBS using a magnet and used for mass spectrometry-based proteomics.
Mitochondria isolation for 7-DHC quantification.
The extraction of mitochondria proteins was prepared using the commercial kit (Sigma, ER0100). Briefly, 1 x 108 WT or DHCR7 KO HEK293T cells were rinsed with ice-cold PBS. The cell suspension was centrifuged at 600 × g for 5 min, cell supernatant was removed and the cell pellet was resuspended in 3 volumes of Hypotonic Extraction Buffer. Then, cells were incubated for 20 min at 4 °C to allow the cells to swell. The cell suspension was centrifuged at 600 × g for 5 min at 4 °C, the supernatant was removed and the cell pellet was resuspended in 2 volumes of Isotonic Extraction Buffer. And then, cells were homogenized with 10 strokes of a 1 mL homogenizer. The homogenate was centrifuged at 1,000 × g for 10 min at 4 °C. The thin floating lipid layer was carefully removed, and the supernatant was transferred to another centrifuge tube and centrifuged at 12,000 × g for 15 min at 4 °C. The thin floating lipid layer was carefully removed, and the supernatant is the mitochondrial fraction. The mitochondria fractions were quantified using the BCA Protein Quantification Kit (Yeasen Biotechnology, 20201ES76). 7-DHC and cholesterol were measured by LC-MS/MS.
Plasma Membrane Isolation for 7-DHC quantification and DHCR7 localization.
The plasma membrane was separated using OrgFrontier™ plasma membrane isolation Kit (BioVision, K414-10). Briefly, 2 × 108 WT or DHCR7 KO HEK293T cells were washed once with PBS and collected by cell scraper, the cell supernatant was centrifuged at 700 × g for 5 min at 4 °C. The cell pellet was washed once with 5 mL of ice-cold PBS and centrifuged samples at 1,000 × g for 5 min at 4 °C. Resuspended the cell pellet in 1 mL ice-cold Working Buffer Solution, then homogenized with 25 strokes of a 1 mL homogenizer.
The homogenate was transferred to a clean, pre-chilled 1.5 mL microcentrifuge tube and sonicated using two 10 s pulses, with 30 s between pulses in the ice-water bath. The cell suspension was centrifuged at 700 × g for 10 min at 4 °C. Carefully removed the thin floating lipid layer, the remaining supernatant was collected and transferred to a new pre-chilled tube. The supernatant was sonicated using two 10 s pulses, 30 s between pulses for ultracentrifuge, and mixed with 4 mL Gradient Working Solution. Then, the supernatant was placed on the bottom of a clean, ice-cold ultracentrifuge tube. Layer 10 mL of the 25% Gradient Solution carefully on top of the mixture. Then layer 2 mL of the 2.5% Gradient Solution carefully on top of the 25% solution. The mixture was ultracentrifuged at 200,000 × g for 90 min at 4 °C. After the ultracentrifuge, the plasma membrane fraction was in the visible band at the interface of the 2.5%/25% gradient solutions. The membrane fractions were quantified using the BCA Protein Quantification Kit (Yeasen Biotechnology, 20201ES76), and 40 µg of each fraction was analyzed by SDS-PAGE. 7-DHC and cholesterol were measured by LC-MS/MS.
Histology and immunohistochemistry
Tissues were collected and fixed in 4% paraformaldehyde for 24 h, dehydrated in a graded ethanol series and xylene, and then embedded in paraffin. Embedded tissues were sectioned at a thickness of 3–5 μm for periodic acid–Schiff (PAS) or immunohistochemistry (IHC) according to standard protocol. In brief, for 4-HNE staining, tissue sections were blocked using 3% H2O2 followed by antigen retrieval using 1 M sodium citrate buffer (pH 6.5), and then incubated with anti-4-HNE (1:400, ab46545, Abcam) overnight at 4 °C. After three times washing with TBST, sections were incubated with HRP-conjugated anti-Rabbit IgG (1:200, BF03008, Beijing Biodragon Immunotechnologies) for 1 h at RT. Wash three times with TBST, then DAB solution was added and the slides were counterstained with haematoxylin. The percentage of positive staining of 4-HNE was measured by ImageJ v1.52.
Metabolites measurement by mass spectrometry
Measurement of 7-DHC, cholesterol, squalene and DHCEO:
The experiment was carried out based on previously described with little modification57. Cells were trypsinized and washed once with PBS, cell pellets were stored at −80 °C. Mitochondria and plasma membrane fraction were isolated and quantified by protein concertation, stored at −80 °C. Before extraction, frozen samples were pre-equilibrated to room temperature. Added 600 μL of DCM/Methanol (v/v = 1:1) (Damas-Beta) with 50 μg/mL BHT (Sigma Aldrich, B1378) and vortexed for 1 min to ensure homogeneity, followed by sonicated for 60 s in the ice-water bath. The sample was centrifuged at 4,000 rpm for 5 min at 25 °C to pellet protein and other insoluble material. The supernatant was removed to a new tube, add another 400 μL of DCM/Methanol (v/v = 1:1) with 50 μg/mL BHT for the second extraction, and collected supernatant to the tube containing the same sample. 500 μL PBS was added to each sample and mixed gently. Samples were centrifuged at 4,000 rpm for 5 min at 25 °C. The organic (lower) layer was removed to a new tube, 500 μL DCM was added to the remaining sample for second extraction, vortexed and centrifuged, and collected organic (lower) layer to the tube containing the same sample. Samples were then dried under N2 using a 12-port drying manifold. Dried samples were redissolved in 200 μL Methanol, and placed in an ultrasonic bath for 5 min at 16 °C. 50 μL aliquot of the sample was transferred to a new autosampler vial for analysis by LC-MS/MS.
Authentic standards of 7-DHC (Acmec, D96421), cholesterol (Aladin, C104029), squalene (Selleck, S4862) and DHCEO (Kerafast, EVU139) were used for calibration. LC-MS analysis was performed using a DIONEX UltiMate 3000 UPLC system coupled to a Thermo Fisher Scientific TSQ Quantiva mass spectrometer equipped with APCI source. Chromatographic separation was carried out under gradient conditions using an Agilent poroshell 120 EC-C18 column (3.0 mm × 50 mm, 2.7 μm) for separation. The mobile phase was (A) 50% methanol containing 0.1% acetic acid and (B) methanol containing 0.1% acetic acid. The flow rate was 0.45 mL/min, and the column was maintained at 35 °C.
For 7-DHC, cholesterol and DHCEO, elution from the column was performed over 6.4 min with the following gradient: 90% B increased to 100% B in 2 min, held at 100% B for 2.5 min, then returned to 90% B in 0.2 min and equilibrated for 1.7 min.
For the measurements of squalene, the samples were run in similar LC-MS conditions as mentioned above except that the LC gradient was increased to 7.9 min.
MS instruments were operated in the positive and selective reaction monitoring (SRM) mode. The SRM transitions are follows: 7-DHC 367.4 → 159.1, cholesterol 369.4 → 161.1, DHCEO 399.3 → 381.3, squalene 411.4 → 231.
Measurement of CoQ10:
The experiment was carried out as previously described14. Cells were trypsinized and washed once with PBS, 2 × 106 cells were collected and stored at −80 °C. Cell pellets were resuspended in 200 µL ice–cold PBS, vortexed for 1 min to ensure homogeneity, followed by sonicated for 30 s in ice–water bath. Added 600 µL of pre-chilled hexane/2-propanol (v/v = 7:3) containing 0.1% BHT and vortexed for 30 s, then incubated for 20–25 min on ice to enhance extraction efficiency. Finally, samples were centrifuged for 25 min at 3,500 rpm, 4 °C. The organic layers (upper) were collected in a new tube and dried under N2 using a 12-port drying manifold. Dried samples were resuspended in 100 µL of 2-propanol/ acetonitrile /H2O (v/v/v = 55:40:5) containing 0.01% BHT. 50 μL aliquot of the sample was transferred to a new autosampler vial for analysis.
Authentic standards of CoQ10 (Sigma, C9538) were used for calibration. Chromatographic separation of extracted CoQ10 was carried out at 40 °C on Waters XSELECT CSH C18 Column, (3.0 mm X 100 mm, 2.5 µm). Mobile phase A consisted of acetonitrile/water (v/v = 7:3) and mobile phase B was 2- propanol/acetonitrile (v/v = 1:1) both containing 5 mM ammonium acetate. After injection, the gradient was held at 70% B for 0.2 min, increased to 100% B at 0.7 min, was held at 100% B for 4 min, and returned to 70% B for a 2.3 min equilibration. The total run time per sample was 7.2 min at a flow rate of 0.4 mL/min. The injection volume of standards and samples was 1 µL.
Mass spectrometric detection was performed on a Thermo Fisher Scientific TSQ Quantiva mass spectrometer, equipped with an ESI source operated in the positive and SRM mode. The SRM transitions are as follows: CoQ10 863.5 → 197.
For the above LC-MS analyses, QC sample was injected between the samples to monitor the performance and the stability of the MS platform. This QC sample was also injected at least 3 times at the beginning of the run, in order to condition the column. The data processing was performed using Thermo Xcalibur 3.0.63.
Lipidomic profiling and data analyses
The experiment was carried out as previously described58. Cells were trypsinized and washed once with PBS, for each sample, 1 × 107 cells were collected and stored at −80°C. Cell pellets were resuspended in 300 µL methanol/water (v/v = 75:25), containing of LPC 18:1(d7) as the internal standard. Then, each sample was sonicated for 30 s to ensure homogeneity and added with 750 µL MTBE, vortexed for 60 s, and gently vibrated for another 30 min. After that, the sample was added with 190 μL of nuclease-free water and vortexed for 1 min. After equilibration at room temperature for 10 min, the sample was centrifuged at 14,000 × g for 15 min. Then 400 μL aliquot of the upper lipid extract was pipetted into the new centrifuge tube and vacuum-dried. The dried sample was dissolved with 150 μL of acetonitrile/isopropanol/water (v/v/v = 65:30:5) for the instrumental analysis in the positive and negative ion mode. The lipid profiling was acquired by a Q Exactive Plus high-resolution mass spectrometer (Thermo Scientific, USA) equipped with an Ultimate 3000 UHPLC system (Thermo Scientific, USA). Parameters on the chromatographic separation in the positive ion mode were the same as those in the negative ion mode. 5 µL of the dissolved sample was injected for the lipid separation by a BEH C8 column (100 × 2.1 mm, 1.7 μm, Waters Co., USA). The column temperature was 55 °C. The lipids were eluted by the binary mobile phase A (acetonitrile/water solution, v/v = 6:4, containing 10 mM ammonium acetate) and B (isopropanol/acetonitrile solution, v/v = 9:1, containing 10 mM ammonium acetate). The flow rate was 0.3 mL/min. The elution gradient was conducted as follows: initial 32% B maintained for 1.5 min, linearly increased to 85% B from 1.5 to 12.5 min, to 97% B from 12.5 to 12.6 min, maintained at 97% B from 12.6 to 14.5 min, and then decreased to 32% B at 14.6 min, finally maintained at 32% B to 16.0 min. The temperature of the sample manager was 10 °C. The lipids eluted from the column were ionized by electrospray ionization in the mass spectrometer, and the mass signals were detected in full scan MS and -data dependent MS/MS (ddMS2) mode, with the resolution of 70,000 and 17,500, respectively. The spray voltage (kV) was +3.5 and −3.0 in the positive and negative ion mode, respectively. Other parameters were identical in the positive and negative ion mode, and set as follows: capillary temperature (°C), 300; aux gas heater temperature (°C), 350; sheath gas flow rate (arb), 45; aux gas flow rate (arb), 10; S-lens RF level, 50; mass scanning range (m/z), 150–1,500; TopN (N, the number of the fragmentation ions with the highest abundance), 10; stepped normalized collision energy (NCE), 25, 35, and 45%. Data processing was done using MS-DIAL software and statistical analysis was performed using GraphPad Prism 9. The raw data are available at https://data.mendeley.com/datasets/4prc7fy4zm/1.
Oxi-lipidomic profiling and data analyses
The experiment was carried out as previously described59. WT or DHCR7 KO HEK293T cells were treated with DMSO or 7.5 µM RSL3 for 3 h. For each sample, 2 × 106 cells were collected and washed once with ice-cold PBS, after centrifuged for 5 min at 1, 000 rpm, cell pellets were resuspended in 140 µL ice-cold nuclease-free water and vortexed for 10 s, added 100 ng PE (D16:1) and 100 ng PC (D14:1) as internal standard to each sample. Then, the Folch method was used to extract lipids, briefly, 450 µL ice-cold chloroform/methanol (v/v = 1:2) containing 0.005% BHT were added to each sample, vortexed for 1 min, then incubated for 15 min on ice to enhance extraction efficiency. Finally, samples were centrifuged for 10 min at 3, 000 x g, 4 °C. The organic layers (lower) were collected in a new tube and dried under N2 using a 12-port drying manifold. Dried samples were resuspended in 60 µL of 100% LC solvent B (methanol/isopropanol solution, v/v = 3:4, containing 5 mM Amide acetate). 50 μL aliquot of the sample was transferred to a new autosampler vial for analysis.
Chromatography was performed using a HILIC HPLC column (Luna 5μm, 100 Å, 50 × 2 mm, Phenomenex) at a flow rate of 0.350 mL/ min. Mass spectrometric analysis was performed in the negative ion mode using multiple-reaction monitoring (MRM) of specific precursor-product ion m/z transitions upon collision-induced dissociation. The precursor negative ions monitored were the molecular ions [M − H]− for PE, and the acetate adducts [M + CH3COO]− for PC. Identity was further verified by monitoring at the same time, using polarity switching, the positive molecular ions [M + H]+ for both PC and PE molecular species. The product ions analyzed after collision-induced decomposition, and used for data comparison, were the carboxylate anions corresponding to the non-oxidized or oxidized arachidonoyl chains. The specific precursor–product pairs monitored in negative-ion mode and used for quantification were follows: PE(16:0e_22:5(O)), 766/345; PE(18:1a_22:4(O)), 808/347; PC (18:0a_20:4(2O)), 900/335; PC(18:0a_22:4(2O)), 928/363; PE(18:0a_9’-oxo-nonanoyl), 634/171; PE(18:0a_5’-oxo-valeroyl), 578/115; PE(16:0a_22:4(O)), 782/347; PC(18:0a_HETE), 810/319. Data processing was done using LabSolutions software. The raw data are available at https://data.mendeley.com/datasets/4prc7fy4zm/1.
Procedures and characterization of synthetic compound
Synthesis of 4-((1-(Mesitylsulfonyl)piperidin-4-yl)amino)butan-2-ol (TASIN-30) was carried out as previously described42.
A mixture of piperidin-4-one (1.02 g, 7.55 mmol), 2,4,6-trimethylbenzenesulfonyl chloride (1.5 g, 6.86 mmol), and N,N-diisopropyl ethylamine (3.3 mL) in CH2Cl2 (50 mL) was stirred at room temperature overnight. The reaction solution was then poured into a saturated aqueous NaHCO3 solution (20 mL per mmol amine) and extracted with CH2Cl2 (3 × 30 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was used as crude material (1-(mesitylsulfonyl)piperidin-4-one) without further purification. A mixture of the crude 1-(mesitylsulfonyl)piperidin-4-one (1.5 g, 6.8 mmol), 4-aminobutan-2-ol (0.67 g, 7.48 mmol), AcOH (0.388 mL), Mg2SO4 (4.911 g, 40.8 mmol) and NaBH(OAc)3 (2.168 g, 10.2 mmol) in CH2Cl2 was stirred at room temperature for 36 h. The reaction was then quenched by adding saturated NaHCO3 solution at 0 °C and extracted with CH2Cl2 (3 × 30 mL). The combined organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified through flash chromatography (1:50 MeOH/CH2Cl2) to provide the title compound as a white solid (2.1 g, 87%). The analytical data were consistent with the literature report. 1H NMR (400 MHz, CDCl3) δ 6.94 (s, 2H), 3.99~3.91 (m, 1H), 3.54 (d, J =12.9 Hz, 2H), 3.03 (tt, J = 8.4, 8.4 Hz, 1H), 2.82 (ttt, J = 5.43, 5.5, 5.85 Hz, 2H), 2.74 (ddd, J = 3.76, 3.62, 3.3 Hz, 1H), 2.60 (s, 6H), 2.57~2.54 (m, 1H), 2.29 (s, 3H), 1.94 (t, J = 25.49 Hz, 2H), 1.65~1.57 (m, 1H), 1.49 ~ 1.40 (m, 1H), 1.36 – 1.25(m, 2H), 1.15 (d, J = 6.2 Hz, 3H). LC−MS (ESI) calcd for C18H30N2O3S [M + H]+ 355.4, found 355.4.
Glutathione measurement
For glutathione measurement, 2 × 105 HEK293T cells expressing sgNC, sgSC5D or sgDHCR7 were seeded into 6-well tissue culture plate. After 18 h, cells were collected by scraping and prepared for measurement of glutathione (GSH + GSSG) using the Glutathione Assay Kit (Beyotime, S0053) according to the protocol. The GSH and GSSG concentrations were calculated using the standard curve and normalized to the total protein level. Three independent biological replicates were performed.
Cellular Fe2+ measurement
For Cellular Fe2+ measurement, HEK293T cells expressing sgNC, sgSC5D or sgDHCR7 cells were seeded into 6-well tissue culture plate. After 18 h, cells were washed twice with HBSS, serum-free DMEM containing 1 μM FerroOrange probe (Dojindo, F374) was added to each well and incubated at 37 °C in the dark for 30 min, cells were washed and analyzed by flow cytometry or a multi-plate reader (TECAN, iControl software) immediately.
RNA extraction and qRT–PCR analysis
Total mRNA was isolated using TRIzol (Vazyme, R401-01), 1 μg of mRNA was used to synthesize cDNA using the HiScript III RT SuperMix for qPCR (Vazyme, R323-01). qRT-PCR was performed using ChamQ Universal SYBR qPCR Master Mix (Vazyme, Q711-02) according to standard protocol.
GPX4-F: GAGGCAAGACCGAAGTAAACTAC
GPX4-R: CCGAACTGGTTACACGGGAA
SLC7A11-F: TCTCCAAAGGAGGTTACCTGC
SLC7A11-R: AGACTCCCCTCAGTAAAGTGAC
ACSL4-F: CATCCCTGGAGCAGATACTCT
ACSL4-R: TCACTTAGGATTTCCCTGGTCC
FSP1-F: GTGAGCGGGTGAGCAATCT
FSP1-R: CTTGATGCCGGTGCAGAGAA
TF-F: GGATACACCAGAGGCAGGTGA
TF-R: TCAGATTGTCCCAGGTGAGGT
SCD1-F: ACCACAAACCCTGCTGAACA
SCD1-R: TGCCCTAGGCTGTAGGGAAT
ACSL1-F: CGCGGCCAACAAGAAGATG
ACSL1-R: CGACGAGTAGGATGAGACCG
ACTB-F: ACACTGTGCCCATCTACGAG
ACTB-R: TCAACGTCACACTTCATGATG
Animal studies
All mice were maintained under pathogen-free conditions. Animal handling and experimental procedures were approved by the Institutional Animal Care and Use Committee at Tongji University. For IRI animal model, male 8-week-old C57BL/6 were purchased from Charles River. For tumour xenograft and metastasis model, male 6-week-old C57BL/6 and 5-week-old athymic nude mice were purchased from Slaccas and kept on normal chow and fed ad libitum, 12 h light/dark cycle.
B16F10 metastasis model.
To test the effect of 7-DHC on the ability of melanoma cells to survive and form tumours after intravenous injection, mouse melanoma cells B16F10 were seeded into 10 cm dish. After 12 h, cells were pretreated with vehicle (EtOH) and 7-DHC (25 μM in EtOH) for 24 h, cells were then trypsinized and washed with PBS. For intravenous injection, 3 × 105 cells in 100 μL of PBS were injected into mice via tail vein. Mice were sacrificed after 12–16 days post-injection for analyses of their tumour burden. For the knockout cell line, B16F10 sgNC and sgDhcr7 were harvested and injected intravenously in the same way and sacrificed 16 days post-injection. For the TASIN-30 pretreat experiment, B16F10-sgNC and sgDhcr7 cells were seeded into 10 cm dish. After 12 h, cells were pretreated with vehicle and TASIN-30 (10 μM) for 24 h, 3 × 105 cells in 100 μL of PBS were intravenously injected and sacrificed after 16 days post-injection.
Subcutaneous tumour model and treatment.
For the B16F10 subcutaneous tumour model,sgNC and sgDhcr7 cells were harvested by trypsinization and washed once with cold PBS, followed by resuspension in cold serum-free DMEM and counted, 6 × 105 cells in 100 μL of serum-free DMEM were subcutaneously injected on the right flank of C57BL/6 mice. When the tumour volume reached 50–100 mm3 (length × width2 × 1/2), it was measured every 2 days until the endpoint.
For the SU-DHL-8 tumour xenograft model, cells were harvested and resuspended with cold serum-free 1640 medium and counted, followed by mixing Matrigel (CORNING, 356231) at a ratio of 2:1, 4 × 106 cells per 150 μL were subcutaneously injected on the right flank of nude mice. When the tumour volume reached 50 mm3 (length × width2 × 1/2), mice were assigned randomly into different treatment groups. TASIN-30 was intraperitoneally injected daily at a dose of 40 mg/kg (5% DMSO + 30% PEG300 + 5% Tween80 + 60% H2O). Liproxstatin-1 was intraperitoneally injected at a dose of 10 mg/kg (5% DMSO + 30% PEG300 + 5% Tween80 + 60% H2O) every 2 days. The tumour volume was measured every 2 days until the endpoint. The maximal tumour volume is 2000 mm3 and this limit was not exceeded in any of the experiments.
Induction of kidney ischemia-reperfusion injury.
Vehicle, AY9944 (25 mg/kg) or dose of cariprazine was intraperitoneally injected daily for 4 times before surgery. Kidney ischemia-reperfusion was carried out as previously described5,60. Mice were anesthetized and placed on a heating system calibrated to 37 °C to keep body temperature. A 1.5 cm abdominal incision was performed, used a blunt retractor to open the chest, and gently removed the gut to expose both kidneys and blood vessels. The blood vessel was clamped with microaneurysm clamps for 30 min and gently put the gut back in the abdominal cavity, 300 μL of pre-warmed PBS was administered intraperitoneally, covered with the gauze pieces, and placed in a warm box. After 30 min, the clamps were removed, sutured the wound after confirming that the reperfusion is successful, and placed mice in a warm box to recover. Sham-operated mice were performed identical surgical procedures, except that microaneurysm clamps were not used. Mice were sacrificed after ischemia-reperfusion for 24 h, blood samples were taken for the assessment of renal function and 7-DHC measurement. Renal function are tested using the Urea Assay Kit (Nanjing Jiancheng Bioengineering Institute, C013-2-1) and Creatinine (Cr) Assay kit (sarcosine oxidase) (Nanjing Jiancheng Bioengineering Institute, C011-2-1). Kidneys were harvested for 7-DHC measurement, and fixed in 4% PFA for periodic acid-Schiff (PAS) staining and 4-HNE immunohistochemical staining.
The severity of injury was based on the following scoring system on a scale of 0 – 5: 0= ≤ 5% area injured; 1 = 6% - 20% of area injured; 3 = 41% - 60% of area injured; 4 = 61% - 80% of area injured; 5 > 80%. The percentage of positive staining of 4-HNE was measured by ImageJ v1.52.
Statistical analysis
GraphPad Prism 9 software was used for statistical analysis. All experiments (except those described otherwise in the legend) were performed independently at least twice with similar outcome. The number of independent experiments is reported in the figure legend. Data were shown as mean ± s.d. for all cell culture experiments and mean ± s.e.m. for all animal experiments. Statistical significance was evaluated using the unpaired two-tailed t-test for comparison of two mean values. One-way ANOVA for comparison of multiple mean values and two-way ANOVA for comparison of multiple mean values under different conditions. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns, not significant. P < 0.05 was considered statistically significant and the exact p values were summarized in the Supplementary Table 4. No statistical methods were used to predetermine sample size.
Extended Data
Extended Data Fig. 1|. Identification of distal CB pathway including CYP51A1, MSMO1, EBP and SC5D as ferroptosis suppressors.
a, Integrative analysis of pooled CRISPR genetic screening using MAGeCK and identification of top candidate genes that were assigned with values generated from negative selection (anti-ferroptotic) by modified robust ranking aggregation (α-RRA) analysis. b, The normalized read counts of sgRNAs by CYP51A1, MSMO1, EBP and SC5D, generated by the MAGeCK-test module. n=5 sgRNAs per gene. c, Schema of the cholesterol pathway. Top screen hits are shown as red (pro-ferroptotic) or blue (anti-ferroptotic). d, The protein-protein interaction network was constructed using the STRING database to identify the top screening hits. e, GO analysis (conducted by https://string-db.org) evaluates global functional category enrichment of gene lists and indicates pathway-level pattern enrichment of cholesterol synthesis from the 50 top scoring genes. FDR are p-values corrected for multiple testing using the Benjamini–Hochberg procedure. f, CTRP analysis (conducted with https://portals.broadinstitute.org/ctrp.v2.1/) shows that the expression levels of CYP51A1, MSMO1, EBP and SC5D, positively correlate with their resistance to ferroptosis induced by the compounds (i.e. RSL3, ML162, ML210 and Erastin) in cancer cells. Plotted values are z-scored Pearson’s correlation coefficients with minima and maxima of the distributions; line, median; box, 25th–75th percentile; whiskers, expansion of outlier compounds (red dots) according to the interquartile multiplier (as shown in right).
Extended Data Fig. 2|. The distal cholesterol biosynthesis genes regulates ferroptosis.
a, Genotyping of subclones generated from HEK293T cells expressing sgRNAs targeting MSMO1, SC5D and EBP by Sanger sequencing. b, The protein level of CYP51A1 in CYP51A1 KO HEK293T cells was analyzed by Immunoblotting. c, Cell viability of WT and CYP51A1, MSMO1, EBP, SC5D KO HEK293T cells treated with ML210 for 8–10 h. d, e, Cell viability of WT and CYP51A1, MSMO1, EBP, SC5D KO HEK293T cells treated with RSL3 for 6–8 h following pretreatment of DMSO, Fer-1 (1 μM), DFO (20 μM) and Idebenone (10 μM) (d) or Fer-1 (1 μM), Z-VAD-FMK (20 μM) and Nec-1s (20 μM) (e). f, The protein level of SC5D-FLAG in SC5D KO HEK293T cells expressing vector and SC5D-FLAG was analyzed by Immunoblotting. g, The protein level of ACSL4 in SC5D KO HEK293T cells expressing sgRNAs targeting ACSL4 was analyzed by Immunoblotting. h, The normalized read counts of sgRNAs by DHCR7, generated by the MAGeCK-test module. n=5 sgRNAs. i, Integrative analysis of pooled CRISPR genetic screening using MAGeCK and identification of top candidate genes that were assigned with values generated from positive selection (pro-ferroptotic) by modified robust ranking aggregation (α-RRA) analysis. j, The protein level of DHCR7 in WT and DHCR7 KO HEK293T cells was detected by Immunoblotting. k, Cell viability of WT and DHCR7 KO HEK293T cells treated with ML210 for 8–10 h. l, The protein level of DHCR7-FLAG in DHCR7 KO HEK293T cells expressing vector and DHCR7-FLAG analyzed by Immunoblotting. For b-g, j-l, data are representative of three independent experiments. Data are mean ± s.d. of n=3 biological replicates.
Extended Data Fig. 3|. Distal CB pathway regulates ferroptosis independent of cholesterol and known ferroptosis defense system.
a, Levels of cholesterol in WT and CYP51A1, MSMO1, EBP, SC5D, DHCR7 KO HEK293T cells. b, Relative concentration of cholesterol in FBS and LPDS measured by Amplex® Red Cholesterol Assay Kit. c, The concentrations of cholesterol in FBS and LPDS detected by LC-MS/MS. d, e, Cell viability in HEK293T (d) and HT1080 (e) cells treated with RSL3 for 6–8 h after pretreatment with or without cholesterol (25 μM) for 24 h in FBS- or LPDS-supplemented medium. n=2 biological replicates. f, g, The protein levels of GPX4, FSP1, DHODH, ACSL4 and SLC7A11 in WT SC5D KO (f) and DHCR7 KO (g) HEK293T cells detected by immunoblotting. h, i, GSH/GSSG level measurement in WT, SC5D KO (c) and DHCR7 KO (d) HEK293T cells. j, k, FACS analysis of Fe2+ using FerroOrange probe by measuring MFI values in WT, SC5D KO (j) and DHCR7 KO (k) HEK293T cells. l, m, Levels of squalene (l) and CoQ10 (m) in WT, SC5D KO and DHCR7 KO HEK293T cells. Data are representative of two (a-c, l, m) and three (d-k) independent experiments. Data are mean ± s.d. of n=3 biological replicates (a-c, h-m). Statistical analysis was performed using one-way ANOVA (a, h-m) or unpaired two-tailed t-tests (b and c); ****P < 0.0001; ns, not significant.
Extended Data Fig. 4|. 7-DHC suppresses ferroptosis.
a, The protein level of DHCR7 in SC5D KO HEK293T cells expressing sgRNAs targeting DHCR7 detected by Immunoblotting. b, g, The concentration of 7-DHC in WT and DHCR7 KO HEK293T cells after treatment of indicated concentrations of AY9944 (b) or cariprazine (g) for 48 h. c, h, Cell viability of WT, DHCR7 KO and SC5D KO HEK293T cells treated with RSL3 for 8 h after pretreatment of AY9944 (100 nM) (c) or cariprazine (100 nM) (h) for 48 h. d, Schema of cholesterol biosynthesis pathway. e, i, Cell viability of U937 and SNU-1 cells treated with RSL3 for 18 h after pretreatment of AY9944 (100 nM) (e) or cariprazine (100 nM) (i) for 48 h. f, j, Representative autoxidation inhibited by indicated concentration of AY9944 (f) and cariprazine (j). k, Levels of 7-DHC in HEK293T cells after treatment of indicated dose of ethanol-dissolved 7-DHC or MβCD-coated 7-DHC for 24 h. l, Cell viability of HEK293T cells treated with RSL3 for 8 h after pretreatment of ethanol-dissolved 7-DHC(25 μM) or MβCD-coated 7-DHC (25 μM) for 24 h. m, Cell viability of HEK293T cells treated with RSL3 for 8 h after pretreatment of MβCD-coated sterols (25 μM) for 24 h. Data are representative of at least two (a-c and e-m) independent experiments. Data are mean ± s.d. of n=3 biological replicates (b, c, e, g-I and k-m). Statistical analysis was performed using two-way ANOVA (b, g and k); *P < 0.05, ****P < 0.0001, ns, not significant.
Extended Data Fig. 5|. 7-DHC is a general suppressor of ferroptosis.
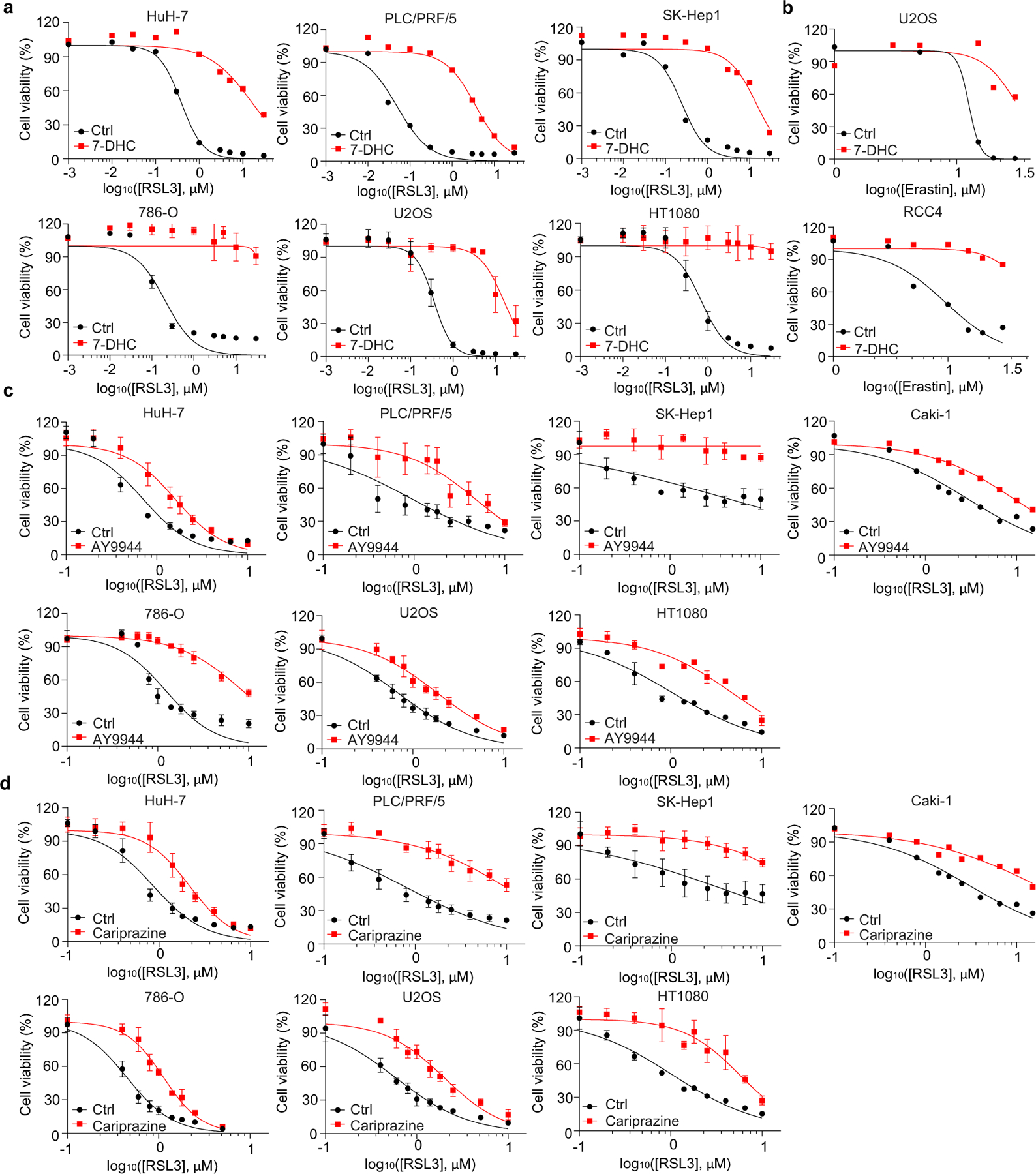
a, Cell viability of various cancer cell lines treated with indicated concentrations of RSL3 for 6–8 h after pretreatment of 7-DHC (25 μM) for 24 h. For HuH-7, PLC/PRF/5 and SK-Hep1, n=2 biological replicates and for 786-O, U2OS and HT1080, n=3 biological replicates. b, Cell viability of U2OS and RCC4 treated with indicated concentrations of Erastin for 16 h after pretreatment of 7-DHC (25 μM) for 24 h. n=2 biological replicates. c, d, Cell viability of various human cancer cell lines (human hepatocellular carcinoma cell lines HuH-7, PLC/PRF/5 and SK-Hep1; human kidney cancer cell lines Caki-1; human kidney cancer cell line 786-O; human osteosarcoma cell line U2OS; human fibrosarcoma cell line HT1080) treated with indicated concentrations of RSL3 for 8–12 h after pretreatment of AY9944 (100 nM) (c) or cariprazine (100 nM) (d) for 48 h in LPDS-supplemented medium. For Caki1, n=2 biological replicates and for the other cell lines, n=3 biological replicates. For a-d, data are representative of at least two independent experiments. Data are shown as the mean ± s.d. when n=3 biological replicates (a, c, d).
Extended Data Fig. 6|. 7-DHC protects against phospholipid peroxidation.
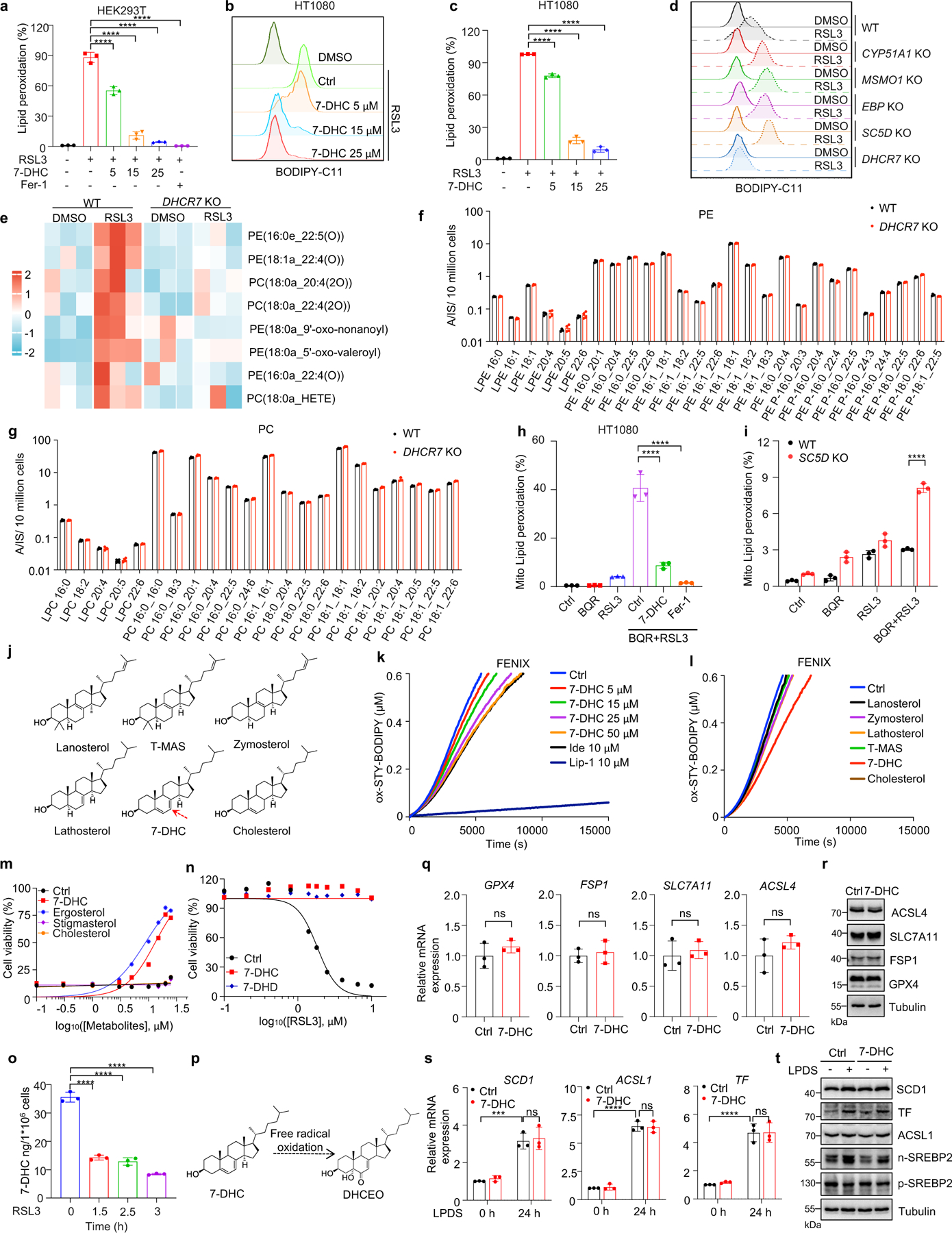
a, Lipid peroxidation assessment of HEK293T treated with RSL3 (2.5 μM) for 2 h after pretreatment of 7-DHC and Fer-1. b, c, Lipid peroxidation assessment of HT1080 treated with RSL3 (1 μM) for 1.5 h after pretreatment of 7-DHC. d, Lipid peroxidation assessment of WT and CYP51A1, MSMO1, EBP, SC5D, DHCR7 KO HEK293T treated with RSL3 (2.5 μM) for 1 h. e. Heat map showing the representation of oxidized phospholipid in WT and DHCR7 KO HEK293T treated with or without RSL3 for 4 h. Each row represents z-score-normalized intensities of the detected lipid species. a, acyl; e, plasmanyl. f, g, Lipidomic profile of phosphatidylethanolamine (PE) (f) and phosphatidylcholine (PC) (g) of WT and DHCR7 KO HEK293T. h, Mitochondrial lipid peroxidation assessment of HT1080 treated with RSL3 (0.5 μM) and BQR (200 μM) for 2 h after pretreatment of 7-DHC (25 μM) and Fer-1 for 24 h. i, Mitochondrial lipid peroxidation assessment of WT and SC5D KO HEK293T treated with RSL3 (1 μM) and BQR (200 μM). j, Chemical structures of different sterols. Red arrows point to 5, 7-diene. k, Representative autoxidation inhibited by indicated concentrations of 7-DHC. l, Representative autoxidation inhibited by 50 μM indicated sterols. m, Cell viability of HEK293T treated with RSL3 (5 μM) for 8 h after pretreatment of indicated sterols for 24 h. n=2 biological replicates. n, Cell viability of HEK293T treated with RSL3 for 8 h after pretreatment of 7-DHC (25 μM) or 7-DHD (25 μM) for 24 h. n=2 biological replicates. o, Levels of 7-DHC in HEK293T treated with RSL3 (4.5 μM) for indicated time. p, Chemical structures of 7-DHC-derived oxysterol DHCEO. q, r, The relative mRNA level(q) and protein level(r) of GPX4, FSP1, SLC7A11 and ACSL4 in HEK293T treated with 7-DHC (25 μM) for 24 h. s, The relative mRNA levels of ACSL1, TF and SCD1 in HEK293T treated with 7-DHC (25 μM) for 24 h, then cultured in LPDS-supplemented medium with 7-DHC for 24 h. t, The protein levels of ACSL1, TF, SCD1, SREBP2 (p-SREBP2) and nuclear form of SREBP2 (n-SREBP2) in HEK293T. Cells were treated with 7-DHC (25 μM) or the ethanol control (Ctrl) for 24 h, then cultured in LPDS-supplemented medium with 7-DHC for 24 h. Data are representative of two (e, k, l) or three (a-d, h, i, m-t) independent experiments, one experiment (f, g). Data are mean ± s.d. of n=3 biological replicates (a-e, h, i, o-s), n=4 biological replicates (f, g). Statistical analysis was performed using one-way ANOVA (a, c, h, o, and q) or two-way ANOVA (i and s); ***P < 0.001, ****P < 0.0001.
Extended Data Fig. 7|. Regulation of 7-DHC level during ferroptosis.
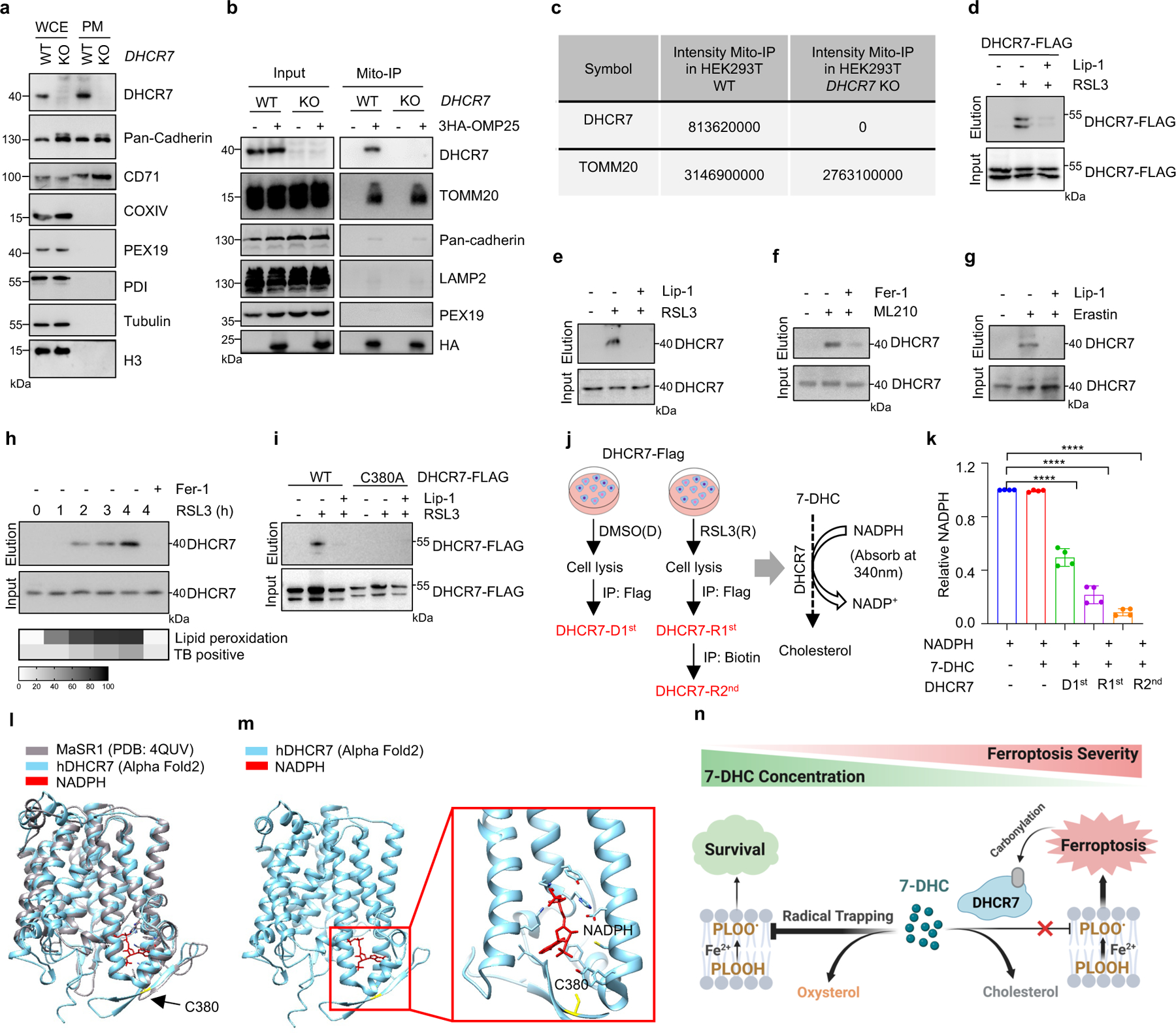
a, DHCR7 location in cell plasma membrane (PM) was detected by immunoblotting. b, Cells expressing vector or 3HA-OMP25 were performed mitochondria immunoprecipitation, DHCR7 was detected in mitochondria by immunoblotting. c, The captured mitochondria were used for LC/MS detection. Proteins identified by the mass spectrometry. d, Detection of DHCR7 carbonylation in HEK293T cells expressed DHCR7-FLAG after induced with RSL3 (6 μM) following treatment of Lip-1 (10 μM). e-g, m-APA labeled carbonylation of DHCR7 in HEK293T cells treated with RSL3 (6 μM) (e), ML210 (10 μM) (f) and in RCC4 treated with Erastin (10 μM) (g) following treatment of Lip-1 (10 μM) or Fer-1 (1 μM). h, m-APA labeled carbonylation of DHCR7 in HEK293T cells treated with RSL3 (5 μM) for indicated time. Level of lipid peroxidation and cell death (TB positive, TB: Trypan Blue) from indicated time points are depicted below in grey scale. i, m-APA selectively labeled carbonylation on C380 of HEK293T cells treated with RSL3 (4 μM). j, Schema of DHCR7 and carbonylated DHCR7 enrichment and the enzyme activity detection by NADPH-dependent reduction. DHCR7 from DMSO (DHCR7-D1st) or RSL3 (DHCR7-R1st) treatment group were enriched by Flag IP. Carbonylated DHCR7 (DHCR7-R2nd) were enriched from DHCR7-R1st by biotin IP. k, Relative DHCR7 enzyme activity detected by the reduction of NADPH. l, m, A structural comparison between the crystal structure of the delta-14 sterol reductase from M. alcaliphilum (grey) and the human DHCR7 structure predicted by AlphaFold2 (cyan), the same residues are found at the NAPDH-binding pocket (red). n, Proposed model of regulation of 7-DHC level during ferroptosis. Image created with BioRender. For a-i, data are representative of at least two independent experiments. For k, data are mean ± s.d. of n=4 biological replicates from two independent experiments. Statistical analysis was performed using one-way ANOVA (k), ***P < 0.001.
Extended Data Fig. 8|. Targeting 7-DHC biosynthesis regulates cancer cell sensitivity to ferroptosis.
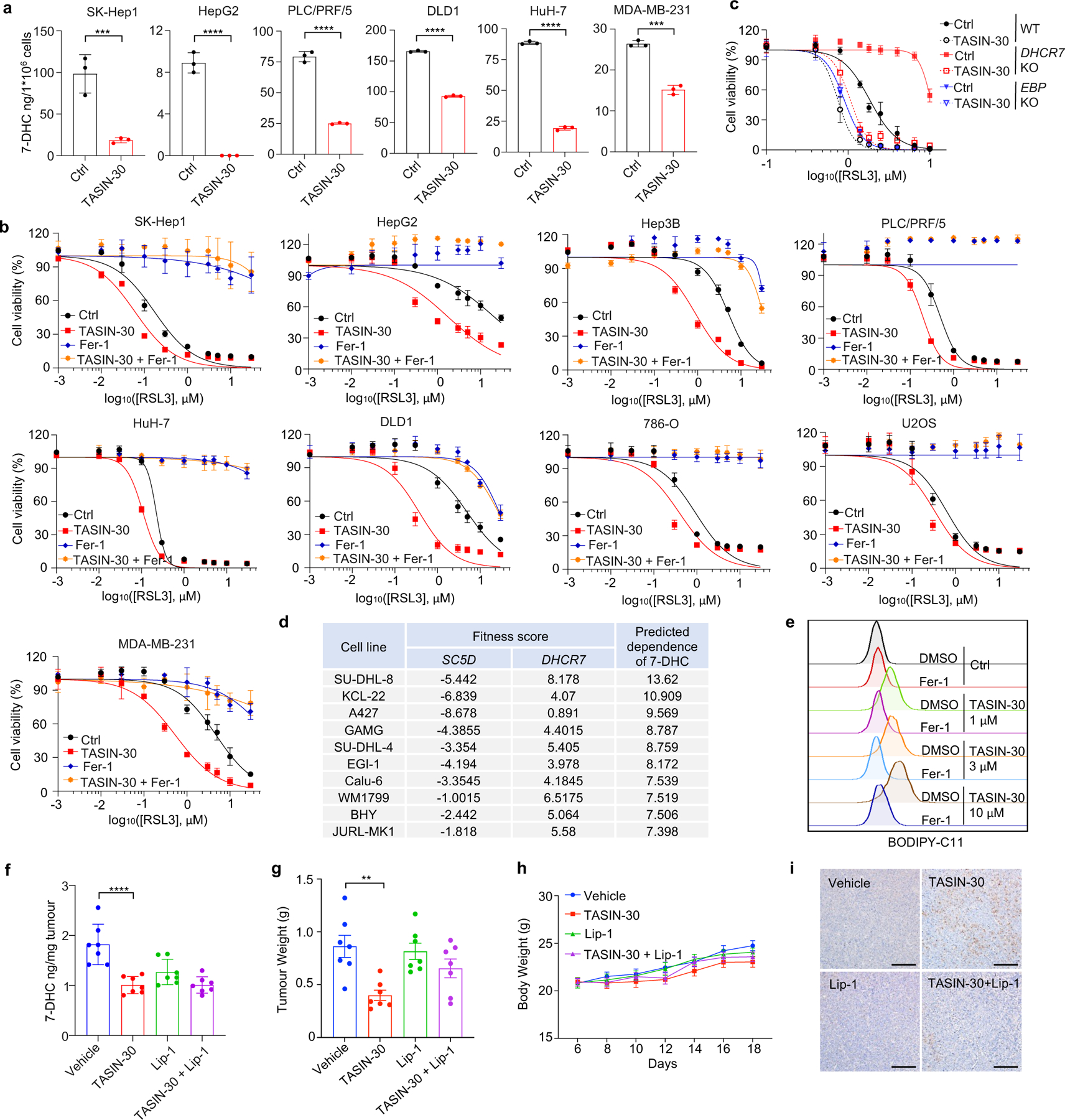
a, Levels of 7-DHC in various cancer cell lines treated with TASIN-30 (10 μM) for 8 h. b, Cell viability of various cancer cell lines treated with indicated concentrations of RSL3 for 8–10 h (SK-Hep1, HuH-7, PLC/PRF/5, U2OS and 786-O) or 18–24 h (HepG2, Hep3B, DLD1 and MDA-MB-231) following pretreatment of TASIN-30 (10 μM), Fer-1 (1 μM) or TASIN-30 (10 μM) plus Fer-1 (1 μM) for 4 h. c, Cell viability of WT, DHCR7 KO and EBP KO HEK293T cells treated with RSL3 for 8 h after pretreatment of TASIN-30 for 4 h. d, The fitness score of SC5D and DHCR7 in top 10 cell lines ranked by predicted dependence of 7-DHC. The predicted dependence of 7-DHC score is calculated by fitness score of DHCR7 minus fitness score of SC5D. e, Lipid peroxidation assessment of SU-DHL-8 cells treated with indicated concentrations of TASIN-30 for 16 h with pretreatment of Fer-1. f, Levels of 7-DHC in tumour tissue of SU-DHL-8 xenografts with indicated treatments. n=7 in each group. g, Tumour weight of SU-DHL-8 xenografts with indicated treatments over time. n=7 in each group. h, Body weight of SU-DHL-8 xenografts with indicated treatments over time. n=7 in each group. Data plotted are mean ± s.e.m. i, Representative immunohistochemical staining of 4-HNE in tumour tissue of SU-DHL-8 xenografts with indicated treatments. Representative images of 7 different samples from a single experiment are shown. Scale bar, 50 μm. For (a-c and e), data are representative of three independent experiments (b) or two independent experiments (a, c, e), and data are mean ± s.d. of n=3. For f-i, data are representative of two independent experiments (g, h) and one experiment (f, i), data plotted are mean ± s.e.m. and statistical analysis was performed using two-way ANOVA; **P < 0.01, ****P < 0.0001. Statistical analysis was performed using unpaired two-tailed t-test (a); ***P < 0.001, ****P < 0.0001.
Extended Data Fig. 9|. 7-DHC promotes metastasis.
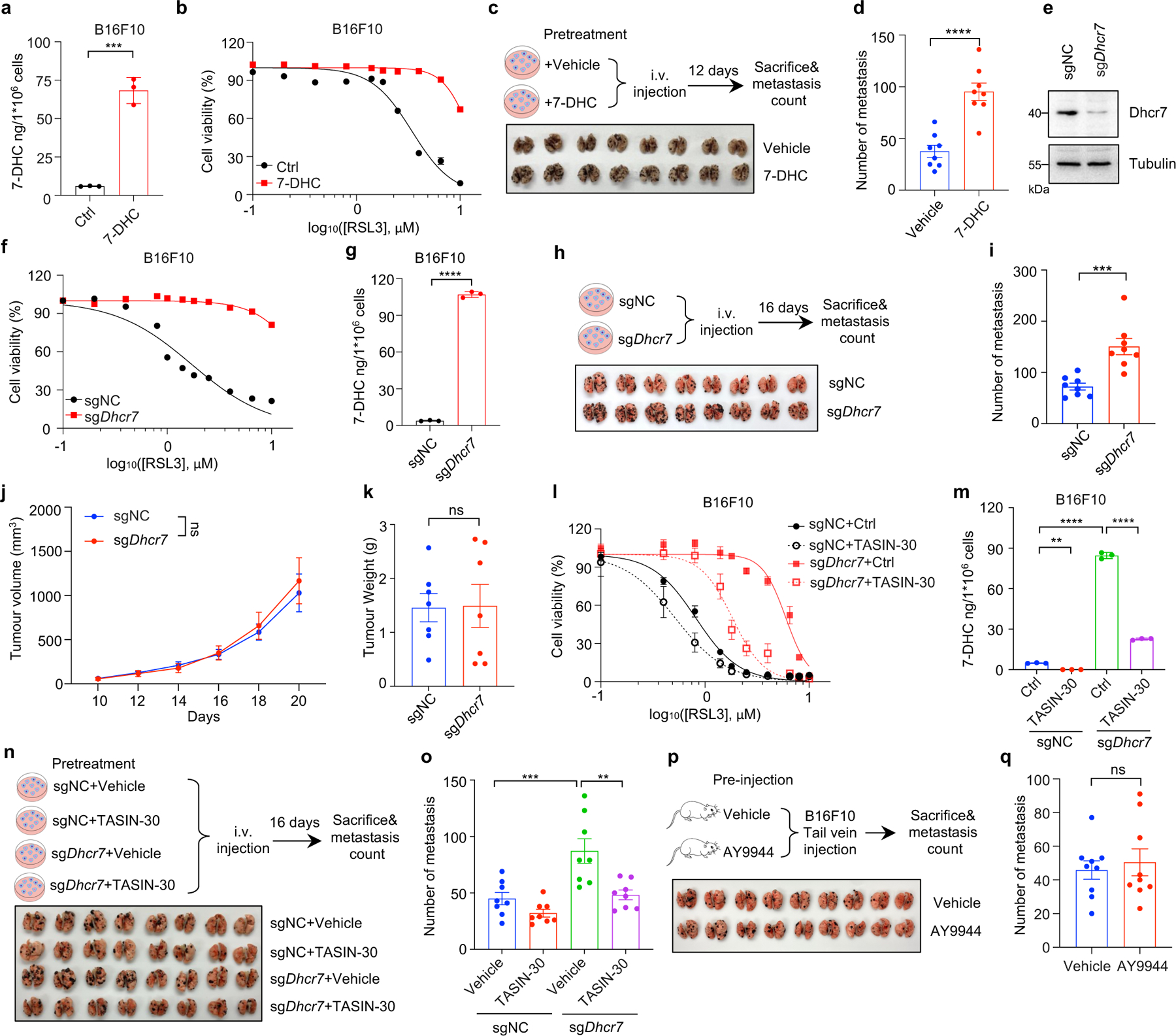
a, Levels of 7-DHC in B16F10 treated with 7-DHC (25 μM) for 24 h. b, Cell viability of B16F10 with treatment of RSL3 for 6–8 h following pretreatment of 7-DHC (25 μM) for 24 h. n=2 biological replicates. c, d, Image (c) and quantification (d) of B16F10 metastatic tumours in lungs with cells pretreated 7-DHC (25 μM) for 24 h. n=8 in each group. e, The protein level of Dhcr7 in sgNC and sgDhcr7 B16F10. f, Cell viability of sgNC and sgDhcr7 B16F10 treated with RSL3 for 6–8 h. n=2 biological replicates. g, Levels of 7-DHC in sgNC and sgDhcr7 B16F10. h, i, Image (h) and quantification (i) of sgNC and sgDhcr7 B16F10 metastatic tumours. n=8 in each group. j, k, Tumour volume (j) and tumour weight (k) of sgNC and sgDhcr7 B16F10 subcutaneous tumour. n=7 in each group,. l, Cell viability of sgNC and sgDhcr7 B16F10 treated with RSL3 for 10–12 h following pretreatment of TASIN-30 (10 μM) for 24 h. m, Levels of 7-DHC in sgNC and sgDhcr7 B16F10 treated with TASIN-30 (10 μM) for 24 h. n, o, Image (n) and quantification (o) of metastatic tumours of sgNC and sgDhcr7 B16F10 pretreated TASIN-30 (10 μM) for 24h. n=8 in each group. p, q, Image (p) and quantification (q) of metastatic tumours of B16F10 in mice pre-injected with vehicle or AY9944 (25 mg/kg) via i.p. once daily for four times prior to i.v. of B16F10. n=9 in each group. For a-q, data are representative of two independent experiments. Data are mean ± s.d. of n=3 biological replicates (a, g, i, m) for cell experiments and mean ± s.e.m for animal experiments, statistical analysis was performed using unpaired two-tailed t-test (a, d, g, i, k and q) or two-way ANOVA (m and o); **P < 0.01, ***P < 0.001, ****P < 0.0001, ns, not significant.
Extended Data Fig. 10 |. Cariprazine protects IRI in vivo.
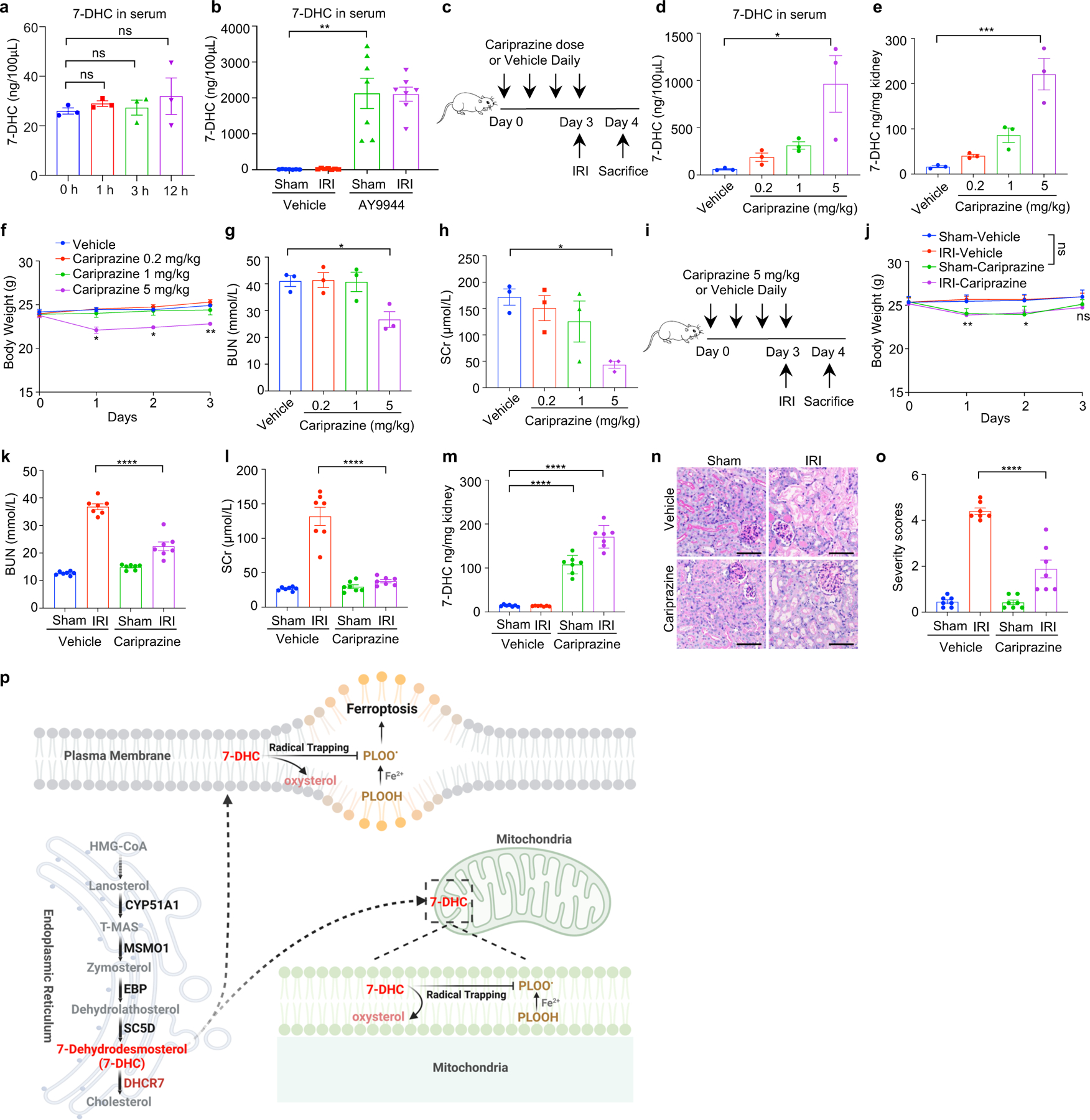
a, Levels of 7-DHC in serum of mice intraperitoneal injected of 7-DHC after indicated time. n=3 in each group. b, Levels of 7-DHC in serum of mice kidney IRI model with pretreatment of AY9944. n=7 in each group. c, Workflow of mice kidney IRI model with pretreatment of indicated dose of cariprazine via intraperitoneal injection once daily for four times before IRI. d, e, Levels of 7-DHC in serum (d) and kidneys (e) of mice kidney IRI model with pretreatment of indicated dose of cariprazine. f, Body weight of IRI model mice with indicated treatments over time. g, h, The concentration of BUN (g) and SCr (h) of mice IRI model. i, Workflow of IRI model with pretreatment of cariprazine at the dose of 5mg/kg via intraperitoneal injection once daily for four times before IRI. j, Body weight of IRI model mice with indicated treatment over time. k, l, The concentration of BUN (k) and SCr (l) of mice kidney IRI model with indicated treatment over time. m, Levels of 7-DHC in kidneys of mice IRI model with pretreatment of cariprazine. n, o, Representative PAS staining (n) and quantification of renal damage (o) from mice IRI model. Scale bar, 100 μm. For d-h, n=3 in each group, and for i-o, n=7 in each group. For (a and d-o), data plotted are mean ± s.e.m. from one experiment. For b, data are representative of two independent experiments and data plotted are mean ± s.e.m. Statistical analysis was performed using one-way ANOVA (a, d-h) and two-way ANOVA (b, j-m, o), *P < 0.05, **P < 0.01 ***P < 0.001, ****P < 0.0001, ns, not significant. p, Working model of 7-DHC. 7-dehydrocholesterol (7-DHC) protects cells from lipid peroxidation on both plasma membrane and mitochondria via diverting the peroxidation pathway from phospholipids, thus mitigating ferroptosis. PLOOH, phospholipid hydroperoxide; PLOO·, phospholipid hydroperoxyl radical. Image created with BioRender.
Supplementary Material
Acknowledgments
We thank Dr. Jose Pedro Friedmann Angeli and Dr. Cong Jiang for constructive suggestions to our manuscript. We thank Dr. Chu Wang for providing guidance to the activity-based protein profiling (ABPP) assay using m-APA. We thank Dr. Zhenwei Zhang for the structure analysis. We appreciate Dr. Yongyong Li’s help for providing platform of liposome production used in FENIX assay. We thank Dr. Hongyan Wang for providing guidance to 7-DHC and cholesterol measurement. We also thank Dr. Andong Qiu, and Wei Zhai for sharing information of kidney Ischemia-Reperfusion Injury model. We thank Dr. Hui Li for technical help. This study was supported by the National Key Research and Development Program of China (2022YFC3401500, 2021YFA1302200, 2020YFA0803201), the Grants from the National Natural Science Foundation of China (31830053, 31920103007, 82341028, 82122056, 82073155, 82073153, 32030053, 22207084, 32150710522), the Shanghai Sailing Program (22YF1433500), Shanghai Municipal Health Commission (20204Y0033), the Fundamental Research Funds for the Central Universities (22120220616, PA2023000692), the fund from City University of Hong Kong (9380154, 7006046), Shanghai Rising-Star Program (23QA1407500).
Footnotes
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary Information
This file contains Supplementary Figs. 1 and 2. Supplementary Fig. 1: An example of the gating strategy for the lipid peroxidation and mitochondrial lipid peroxidation assays. Supplementary Fig. 2: Raw images of immunoblotting. The CRISPR screen data, sgRNA sequence, IC50 summary, and p-value summary are provided in Supplementary Table.
Competing interests X.J. is an inventor on patents related to autophagy and cell death and holds equity of and consults for Exarta Therapeutics and Lime Therapeutics. The other authors declare no competing interests.
Data availability
Oxi-lipidomic and lipidomic data are available at https://data.mendeley.com/datasets/4prc7fy4zm/1. All other data are available in the article and its Supplementary Information. The CRISPR screen data are provided in Supplementary Table. Gel source images are shown in Supplementary Fig. 2. STRING and GO analysis were conducted with the STRING database at https://string-db.org. CTRP data analysis were mined from https://portals.broadinstitute.org/ctrp.v2.1/. Fitness scores data were mined from https://score.depmap.sanger.ac.uk/. Source data are provided in this paper.
Reference
- 1.Dixon SJ et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072, doi: 10.1016/j.cell.2012.03.042 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Viswanathan VS et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457, doi: 10.1038/nature23007 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hangauer MJ et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250, doi: 10.1038/nature24297 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen L, Hambright WS, Na R & Ran Q Ablation of the Ferroptosis Inhibitor Glutathione Peroxidase 4 in Neurons Results in Rapid Motor Neuron Degeneration and Paralysis. J Biol Chem 290, 28097–28106, doi: 10.1074/jbc.M115.680090 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Linkermann A et al. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci U S A 111, 16836–16841, doi: 10.1073/pnas.1415518111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tonnus W et al. Dysfunction of the key ferroptosis-surveilling systems hypersensitizes mice to tubular necrosis during acute kidney injury. Nat Commun 12, 4402, doi: 10.1038/s41467-021-24712-6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kagan VE et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 13, 81–90, doi: 10.1038/nchembio.2238 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anandhan A et al. NRF2 controls iron homeostasis and ferroptosis through HERC2 and VAMP8. Sci Adv 9, eade9585, doi: 10.1126/sciadv.ade9585 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Agmon E, Solon J, Bassereau P & Stockwell BR Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci Rep 8, 5155, doi: 10.1038/s41598-018-23408-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang WS et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331, doi: 10.1016/j.cell.2013.12.010 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doll S et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698, doi: 10.1038/s41586-019-1707-0 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Bersuker K et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692, doi: 10.1038/s41586-019-1705-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mao C et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 593, 586–590, doi: 10.1038/s41586-021-03539-7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kraft VAN et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent Sci 6, 41–53, doi: 10.1021/acscentsci.9b01063 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soula M et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol 16, 1351–1360, doi: 10.1038/s41589-020-0613-y (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liang D et al. Ferroptosis surveillance independent of GPX4 and differentially regulated by sex hormones. Cell 186, 2748–2764 e2722, doi: 10.1016/j.cell.2023.05.003 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muller C, Junker J, Bracher F & Giera M A gas chromatography-mass spectrometry-based whole-cell screening assay for target identification in distal cholesterol biosynthesis. Nat Protoc 14, 2546–2570, doi: 10.1038/s41596-019-0193-z (2019). [DOI] [PubMed] [Google Scholar]
- 18.Yin H, Xu L & Porter NA Free radical lipid peroxidation: mechanisms and analysis. Chem Rev 111, 5944–5972, doi: 10.1021/cr200084z (2011). [DOI] [PubMed] [Google Scholar]
- 19.Porter NA, Xu L & Pratt DA Reactive Sterol Electrophiles: Mechanisms of Formation and Reactions with Proteins and Amino Acid Nucleophiles. Chemistry (Basel) 2, 390–417, doi: 10.3390/chemistry2020025 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu L, Davis TA & Porter NA Rate constants for peroxidation of polyunsaturated fatty acids and sterols in solution and in liposomes. J Am Chem Soc 131, 13037–13044, doi: 10.1021/ja9029076 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu L et al. DHCEO accumulation is a critical mediator of pathophysiology in a Smith-Lemli-Opitz syndrome model. Neurobiol Dis 45, 923–929, doi: 10.1016/j.nbd.2011.12.011 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pfeffer BA, Xu L, Porter NA, Rao SR & Fliesler SJ Differential cytotoxic effects of 7-dehydrocholesterol-derived oxysterols on cultured retina-derived cells: Dependence on sterol structure, cell type, and density. Exp Eye Res 145, 297–316, doi: 10.1016/j.exer.2016.01.016 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Porter FD Smith-Lemli-Opitz syndrome: pathogenesis, diagnosis and management. Eur J Hum Genet 16, 535–541, doi: 10.1038/ejhg.2008.10 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Schmitz R et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 490, 116–120, doi: 10.1038/nature11378 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park RJ et al. A genome-wide CRISPR screen identifies a restricted set of HIV host dependency factors. Nat Genet 49, 193–203, doi: 10.1038/ng.3741 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dixon SJ et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 3, e02523, doi: 10.7554/eLife.02523 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Szklarczyk D et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 47, D607–D613, doi: 10.1093/nar/gky1131 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rees MG et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat Chem Biol 12, 109–116, doi: 10.1038/nchembio.1986 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doll S et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 13, 91–98, doi: 10.1038/nchembio.2239 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stockwell BR et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 171, 273–285, doi: 10.1016/j.cell.2017.09.021 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun X et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63, 173–184, doi: 10.1002/hep.28251 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang WS et al. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A 113, E4966–4975, doi: 10.1073/pnas.1603244113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Genaro-Mattos TC et al. Dichlorophenyl piperazines, including a recently-approved atypical antipsychotic, are potent inhibitors of DHCR7, the last enzyme in cholesterol biosynthesis. Toxicol Appl Pharmacol 349, 21–28, doi: 10.1016/j.taap.2018.04.029 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garcia-Bermudez J et al. Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death. Nature 567, 118–122, doi: 10.1038/s41586-019-0945-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao M et al. Role of Mitochondria in Ferroptosis. Mol Cell 73, 354–363 e353, doi: 10.1016/j.molcel.2018.10.042 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gaschler MM et al. Determination of the Subcellular Localization and Mechanism of Action of Ferrostatins in Suppressing Ferroptosis. ACS Chem Biol 13, 1013–1020, doi: 10.1021/acschembio.8b00199 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lamberson CR et al. Propagation rate constants for the peroxidation of sterols on the biosynthetic pathway to cholesterol. Chem Phys Lipids 207, 51–58, doi: 10.1016/j.chemphyslip.2017.01.006 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shah R, Farmer LA, Zilka O, Van Kessel ATM & Pratt DA Beyond DPPH: Use of Fluorescence-Enabled Inhibited Autoxidation to Predict Oxidative Cell Death Rescue. Cell Chem Biol 26, 1594–1607 e1597, doi: 10.1016/j.chembiol.2019.09.007 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Hong X et al. The Lipogenic Regulator SREBP2 Induces Transferrin in Circulating Melanoma Cells and Suppresses Ferroptosis. Cancer Discov 11, 678–695, doi: 10.1158/2159-8290.CD-19-1500 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stockwell BR Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 185, 2401–2421, doi: 10.1016/j.cell.2022.06.003 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen Y et al. Quantitative Profiling of Protein Carbonylations in Ferroptosis by an Aniline-Derived Probe. J Am Chem Soc 140, 4712–4720, doi: 10.1021/jacs.8b01462 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Theodoropoulos PC et al. A Medicinal Chemistry-Driven Approach Identified the Sterol Isomerase EBP as the Molecular Target of TASIN Colorectal Cancer Toxins. J Am Chem Soc 142, 6128–6138, doi: 10.1021/jacs.9b13407 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dwane L et al. Project Score database: a resource for investigating cancer cell dependencies and prioritizing therapeutic targets. Nucleic Acids Res 49, D1365–D1372, doi: 10.1093/nar/gkaa882 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ubellacker JM et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature 585, 113–118, doi: 10.1038/s41586-020-2623-z (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rodgers MA, Saghatelian A & Yang PL Identification of an overabundant cholesterol precursor in hepatitis B virus replicating cells by untargeted lipid metabolite profiling. J Am Chem Soc 131, 5030–5031, doi: 10.1021/ja809949r (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xiao J et al. Targeting 7-Dehydrocholesterol Reductase Integrates Cholesterol Metabolism and IRF3 Activation to Eliminate Infection. Immunity 52, 109–122 e106, doi: 10.1016/j.immuni.2019.11.015 (2020). [DOI] [PubMed] [Google Scholar]
- 47.Xu L, Korade Z & Porter NA Oxysterols from free radical chain oxidation of 7-dehydrocholesterol: product and mechanistic studies. J Am Chem Soc 132, 2222–2232, doi: 10.1021/ja9080265 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yasunori YAOITA KA, Hiroyuki OHNUMA, Katsuyuki FURUMURA, & Akihiro MASAKI. Toshihiko MATSUKI, a. M. K. Sterol Constituents from Five Edible Mushrooms. Chem. Pharm. Bull 46, 944–950 (1998). [Google Scholar]
- 49.Kaneski CR, Hanover JA & Schueler Hoffman UH Generation of an in vitro model for peripheral neuropathy in Fabry disease using CRISPR-Cas9 in the nociceptive dorsal root ganglion cell line 50B11. Mol Genet Metab Rep 31, 100871, doi: 10.1016/j.ymgmr.2022.100871 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li W et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15, 554, doi: 10.1186/s13059-014-0554-4 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kanarek N et al. Histidine catabolism is a major determinant of methotrexate sensitivity. Nature 559, 632–636, doi: 10.1038/s41586-018-0316-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li J et al. Hiplot: a comprehensive and easy-to-use web service for boosting publication-ready biomedical data visualization. Brief Bioinform 23, doi: 10.1093/bib/bbac261 (2022). [DOI] [PubMed] [Google Scholar]
- 53.Luu W, Gelissen IC & Brown AJ Manipulating Cholesterol Status Within Cells. Methods Mol Biol 1583, 41–52, doi: 10.1007/978-1-4939-6875-6_4 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Anderson RH et al. Sterols lower energetic barriers of membrane bending and fission necessary for efficient clathrin-mediated endocytosis. Cell reports 38, 110371, doi: 10.1016/j.celrep.2022.110371 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li B et al. Besting vitamin E: sidechain substitution is key to the reactivity of naphthyridinol antioxidants in lipid bilayers. J Am Chem Soc 135, 1394–1405, doi: 10.1021/ja309153x (2013). [DOI] [PubMed] [Google Scholar]
- 56.Chen WW, Freinkman E, Wang T, Birsoy K & Sabatini DM Absolute Quantification of Matrix Metabolites Reveals the Dynamics of Mitochondrial Metabolism. Cell 166, 1324–1337 e1311, doi: 10.1016/j.cell.2016.07.040 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McDonald JG, Smith DD, Stiles AR & Russell DW A comprehensive method for extraction and quantitative analysis of sterols and secosteroids from human plasma. J Lipid Res 53, 1399–1409, doi: 10.1194/jlr.D022285 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huang H et al. Plasma Lipidomics Identifies Unique Lipid Signatures and Potential Biomarkers for Patients With Aortic Dissection. Front Cardiovasc Med 8, 757022, doi: 10.3389/fcvm.2021.757022 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yin H et al. Identification of intact oxidation products of glycerophospholipids in vitro and in vivo using negative ion electrospray iontrap mass spectrometry. J Mass Spectrom 44, 672–680, doi: 10.1002/jms.1542 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Van Coillie S et al. Targeting ferroptosis protects against experimental (multi)organ dysfunction and death. Nat Commun 13, 1046, doi: 10.1038/s41467-022-28718-6 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Oxi-lipidomic and lipidomic data are available at https://data.mendeley.com/datasets/4prc7fy4zm/1. All other data are available in the article and its Supplementary Information. The CRISPR screen data are provided in Supplementary Table. Gel source images are shown in Supplementary Fig. 2. STRING and GO analysis were conducted with the STRING database at https://string-db.org. CTRP data analysis were mined from https://portals.broadinstitute.org/ctrp.v2.1/. Fitness scores data were mined from https://score.depmap.sanger.ac.uk/. Source data are provided in this paper.