

ABSTRACT
Immunologic treatment options are uncommon in low-grade gliomas, although such therapies might be beneficial for inoperable and aggressive cases. Knowledge of the immune and stromal cells in low-grade gliomas is highly relevant for such approaches but still needs to be improved. Published gene-expression data from 400 low-grade gliomas and 193 high-grade gliomas were gathered to quantify 10 microenvironment cell populations with a deconvolution method designed explicitly for brain tumors. First, we investigated general differences in the microenvironment of low- and high-grade gliomas. Lower-grade and high-grade tumors cluster together, respectively, and show a general similarity within and distinct differences between these groups, the main difference being a higher infiltration of fibroblasts and T cells in high-grade gliomas. Among the analyzed entities, gangliogliomas and pleomorphic xanthoastrocytomas presented the highest overall immune cell infiltration. Further analyses of the low-grade gliomas presented three distinct microenvironmental signatures of immune cell infiltration, which can be divided into T-cell/dendritic/natural killer cell-, neutrophilic/B lineage/natural killer cell-, and monocytic/vascular/stromal-cell-dominated immune clusters. These clusters correlated with tumor location, age, and histological diagnosis but not with sex or progression-free survival. A survival analysis showed that the prognosis can be predicted from gene expression, clinical data, and a combination of both with a support vector machine and revealed the negative prognostic relevance of vascular markers. Overall, our work shows that low- and high-grade gliomas can be characterized and differentiated by their immune cell infiltration. Low-grade gliomas cluster into three distinct immunologic tumor microenvironments, which may be of further interest for upcoming immunotherapeutic research.
KEYWORDS: Astrocytoma, deconvolution, gene expression, low-grade glioma, tumor microenvironment
Introduction
Tumors of the central nervous system (CNS) are the second most common malignancy in children after leukemia. Gliomas constitute approximately half of all pediatric CNS tumors, of which roughly three-quarters are low-grade.1,2 Pediatric (pLGG) and adult low-grade gliomas (aLGG) differ clinically as well as molecularly. These findings were recently incorporated into the latest WHO Classification of CNS tumors with the prefix “pediatric-type” for diffuse low-grade gliomas and should also be considered in future research.3
While pLGGs, mainly when located superficially, have a comparatively good prognosis if gross total resection is achieved, pLGGs in locations such as the hypothalamus, the optic pathways, or the brain stem have a substantially worse prognosis as often only subtotal resection is feasible.2,4 This is problematic, especially in the context of concomitant adverse effects with other treatment modalities, such as chemotherapy and craniospinal irradiation therapy, the latter of which often causes long-term toxicities.5 Though modern radiotherapeutic techniques like proton beam therapy appear to show fewer toxicities, they still seem to cause reduced neurocognitive outcomes in young children.6,7 New complementary therapeutic approaches are needed to mitigate long-term effects and improve prognosis, especially in recurrent and irresectable cases.
Immune therapies have proven useful treatment modalities in several malignancies, but they are still uncommon in LGG.8 In a recent phase I clinical trial in H3K27M-mutated diffuse midline gliomas of children and young adults, CAR-T-cell therapy showed a clinical benefit in three of the four patients.9 Nonetheless, established immunotherapeutic approaches in low-grade gliomas are still generally missing. This is at least partially explicable by the incomplete knowledge of their tumor microenvironment (TME) and the significance of its composition for tumor prognosis.
The TME is essential as it not only influences a tumor’s response to conventional treatment but the TME itself is also influenced and altered by conventional treatment and immunotherapeutic approaches.10,11 Even microbial peptides seem to influence immune responses as recently shown in glioblastoma.12 All the while, stromal cells can affect tumor progression and metastasis positively and negatively.13 The microenvironment’s significance as a regulator of tumor progression is also true for CNS tumors.14 For instance, immune-related genes can be used to establish a prognostic model that correlates with the infiltration of specific immune cells, as Guo and colleagues demonstrated in medulloblastoma.15 Grabovska and colleagues were able to group pediatric CNS tumors into three broad immune clusters and found LGGs to be most associated with the one dominated by monocytes and with low CD8-T-cell infiltration.16 Wang and colleagues, who examined pediatric gliomas (PG) only, found that macrophages followed by T cells were the most common immune infiltrating cells and classified the PGs into three immune subtypes “immune hot”, “immune altered” and “immune cold”, of which the “immune hot” subtype showed the most favorable and “immune cold” the worst prognosis.17 In adult diffuse lower-grade gliomas (grade 2 and 3) Wu and colleagues identified three immune subtypes associated with distinct somatic alterations and prognoses.18
Our group previously elucidated the immunologic landscape in large-scale datasets of medulloblastoma and high-grade gliomas.19,20 Such large-scale approaches were made possible by the availability of transcriptomic data from several hundred cancer samples in various public databases. These data can be used by deconvolution methods to robustly quantify immune cell infiltration in large sample sizes retrospectively, but they are still missing for large-scale analyses specifically dedicated to pediatric low-grade glioma.19–23
Here, we gathered transcriptomic data from 10 pediatric CNS tumor datasets generally considered low-grade24 to analyze their tumor microenvironment. First, we compared these samples to high-grade gliomas, showing differences between the two subgroups. Next, we identified three immunologic clusters in LGGs, which we examined regarding their cell composition and metadata. Lastly, we analyzed the dataset for prognostically relevant genes and enriched pathways and used machine learning to predict survival.
Materials and methods
All data analyses were performed using the statistical programming language R, including the packages BiocManager, GEOquery, affy, org.HS.eg.db, AnnotationDbi, ComplexHeatmap, circlize, DESeq2, biomaRt, hgu133plus2hsentrezgcdf, hgu133ahsentrezgcdf, dendextend, factoextra, Rtsne, survival, plotrix, amap, fgsea, caret.
Datasets
Six series of microarray gene-expression data were downloaded from Gene Expression Omnibus (GEO).25–32 Further, two series of RNA-Seq gene-expression data were downloaded from the European Genome-phenome Archive (EGA).33,34 A list of all 10 datasets with additional information is provided in the supplement (Supplemental Table S1).
Data curation
Initially, 302 high-grade samples from two of the 10 datasets (GSE50161 and GSE16011) were included for the comparative analysis of low-grade with high-grade gliomas. Since genetic mutation data were available for these samples (Supplemental Table S2–3), we used these data to curate the samples according to the revised WHO 2021 classification that has since been published. The entirety of all the data yielded a dataset of 400 low-grade gliomas composed of 344 pilocytic astrocytomas, 36 gangliogliomas, 14 dysembryoplastic neuroepithelial tumors, and six pleomorphic xanthoastrocytomas as well as 193 additional high-grade gliomas composed of 127 glioblastomas, 22 IDH-oligodendrogliomas and 44 IDH-astrocytomas (Supplemental Table S4).
Preprocessing and batch-effect removal
The various data types required different preprocessing pipelines, which had to consider batch-correction to allow for a combined analysis of this heterogeneous data collection.
Raw microarray gene-expression data (.CEL-files) were normalized using mas5 normalization as implemented in the R-package affy. The microarray gene-expression data of one series was provided as a table already being normalized with the cubic spline algorithm implemented in the GenePattern Illumina normalizer module.25
The RNA-Seq gene-expression data were mapped with STAR using the last version of the hg19 reference genome and counted with featureCounts. The use of hg19 instead of hg38 for all RNA-Seq series was dictated by one series already having been hg19-preprocessed (.bam).34
All data were log-transformed, and data originating from the same center/study were linearly transformed for each gene such that the median of all pilocytic astrocytoma cases of the respective center/study was zero. The statistical dispersion was aligned using the same approach employing the median absolute deviation as the measure of statistical dispersion.
Sex-associated genes were removed. Analyses involving data from all platforms were restricted to 4923 available common genes between the datasets (Supplemental Table S5).
After batch-effect removal, clustering of the data for low-grade gliomas based on the 1,000 most variable genes revealed concordance of similar tumors from different datasets with respect to diagnosis and location (Supplemental Figure S1a). Further, a t-SNE based on the calculated immune cell infiltration showed no apparent batch effects (Supplemental Figure S1b).
Computation of immune and stromal signatures and purity
To infer the immune cell infiltration from gene-expression data we used published gene signatures of mRNA markers for 10 cell populations optimized by Bockmayr et al. for use in glioma20 based on published marker signatures proposed by Becht et al.21 and Danaher et al.22 (Supplemental Table S6). Infiltration scores of immune and stromal cell signatures were calculated as the average values of the corresponding marker genes available across all platforms (Supplemental Table S7). Datasets were then merged and scaled based on the mean and standard deviation for each cell signature separately. Tumor purity was quantified using PUREE35 (Supplemental Table S8).
Visualization, SVM, survival- and geneset-enrichment-analysis (GSEA)
Prior to heatmap analysis, data were trimmed at z-scores ±3. Heatmaps were then generated using the ward.D2-linkage method (ComplexHeatmap) and Spearman correlation as similarity measure. The significance of differential expression in the boxplot analysis was evaluated using the Kruskal-Wallis test and corrected for multiple testing for different metadata by applying the Benjamini-Hochberg (BH) method. The significance of the difference in the distribution of metadata between the clusters for the bar plots was evaluated using Fisher’s exact test. Survival analysis was performed using the log-rank test.
The prognostic relevance of single genes was analyzed using the proportional hazards model as implemented by the coxph function from the R-package survival. Multiple testing corrections were performed using the BH method where indicated. The regression coefficients were used for visualization and gene set enrichment analysis using the R-package fgsea and Gene Ontology sets with 100–400 genes from msigdb.36 Overlapping pathways were removed with the function collapsePathways, resulting in 12 significantly enriched pathways with a BH-corrected p-value < 0.01. Machine learning-based survival prediction were performed with a linear support vector machine (SVM) with default parameters using the packages caret, mltools, and e1071. The binary 5-year progression-free survival was used as the output variable. We trained one model based on all available clinical data (age, location, sex, histology, and resection status) and one model based on the molecular data (4923 genes). Clinical variables were one-hot encoded. The encoding of the histological diagnosis DNT was removed from the training data, as there was only a single case with available survival data. The performance was evaluated using 10-times repeated 10-fold cross-validation, such that every case was predicted 10 times to increase robustness. A case was assigned to the poor prognosis group if progression was predicted in at least 5/10 cross-validation runs. As parameter optimization was not performed, nested cross-validation was not required. An integrated prediction model was defined by combining the predictions from the clinical and the molecular SVM. Cases were assigned to the high-risk (HR) group if both models predicted tumor progression, to the intermediate-risk (IR) group if the predictions were discordant, and to the low-risk (LR) group if both models predicted no evidence of disease.
Results
Low-grade gliomas and high-grade gliomas differ in the expression of microenvironment signatures
Based on a deconvolution approach relying on reference signatures optimized for brain tumors,20 10 microenvironment signatures, eight immune and two stromal cell populations, were computed for a dataset of 593 bulk transcriptomic profiles. The dataset was compiled from 10 datasets available on GEO and EGA. Since it comprised five different platforms and two different data types, data was corrected for batch effects prior to analysis (Supplemental Figure S1). In total, 344 pilocytic astrocytomas (PA), 36 gangliogliomas (GG), 14 dysembryoplastic neuroepithelial tumors (DNT), six pleomorphic xanthoastrocytomas (PXA), 127 glioblastomas (GBM), 22 IDH-oligodendrogliomas (IDH-OD) and 44 IDH-astrocytomas (IDH-A) were included in the analysis and tumor entities were subdivided by grades.
Hierarchical clustering based on the median expression scores of all cell populations yielded a pronounced division of entities generally considered to be low grade, i.e., GG, PXA, PA, DNT, or high grade, i.e., GBM, IDH-OD, IDH-A (Figure 1(a)). Hierarchical clustering without grade subdivision yielded similar results and a similar pattern was also discernible in a t-SNE analysis based on the calculated immune cell infiltration scores (Supplemental Figure S2a,b). The most characterizing difference between the low-grade and high-grade clusters can be attributed to higher expression levels of fibroblasts and T cells in the high-grade cluster. The expression levels of myeloid dendritic cells seemed to follow a similar pattern but deviated in the case of gangliogliomas and glioblastomas (Figure 1(a,b), Supplemental Figure S2a,c).
Figure 1.
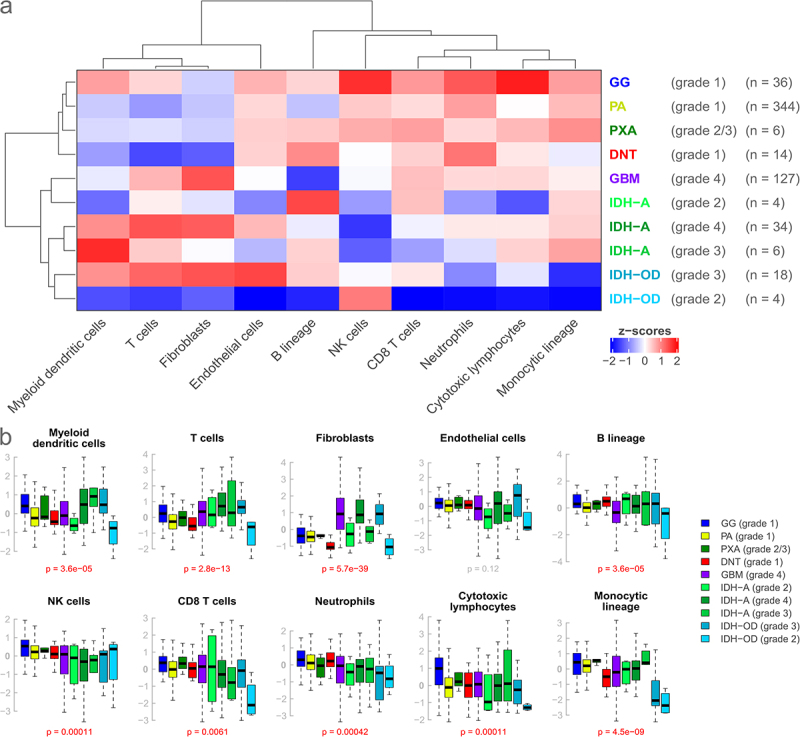
Comparison of expression scores for 10 microenvironment cell populations in seven brain tumor entities subdivided by grade. (a): Heatmap based on the median expression score of every cell population for every entity. Entities generally considered low-grade and high-grade cluster together, respectively, and show distinct expression profiles. (b): Corresponding boxplots. Boxes indicate the lower and upper quartiles, with the black band indicating the median. Whiskers extend to 1.5*IQR, outliers beyond that are not shown. P-values are calculated by the Kruskal-Wallis test. Low- and high-grade entities show differences for some cell populations, but overall dispersion is large.
Also, high expression of the fibroblast signature was only found in high-grade gliomas (GBM and IDH-A grade 4, IDH-OD grade 3), and only higher-grade IDH-mutated entities (IDH-A grade 3/4, IDH-OD grade 3) showed a high expression of myeloid dendritic cells (Figure 1(a,b)). Overall, gangliogliomas had the highest general expression levels of all the entities.
In addition to those general patterns, other observations were made, such as an elevated expression of cytotoxic lymphocytes and natural killer (NK) cells in gangliogliomas, a decreased expression of the monocytic lineage and neutrophils in IDH-OD as well as a decreased expression of natural killer cells in IDH-A (Figure 1(a,b), Supplemental Figure S2a,c). Overall, the observed dispersion of expression levels was large, and the distributions of immune and stromal signatures partially overlapped between the different groups. However, expression scores for all signatures, except for endothelial cells, were significantly different between entities or entity-grade pairings (all p < 0.0061, Figure 1(b), Supplemental Figure S2c). A correlation analysis revealed a negative association between tumor purity and immune cell signatures and highlights that the differences between immune clusters are transcriptomic variations and not solely explained by the overall immune cell abundance (Supplemental Figure S5).
Division of low-grade gliomas into immunologic clusters by microenvironment expression scores
Next, we analyzed the expression of the microenvironmental signatures for the low-grade gliomas only. Unsupervised hierarchical clustering resulted in three clusters with distinct microenvironmental expression patterns for the 400 analyzed samples. The dominant cell populations were T cells, dendritic and natural killer cells in Cluster 1, neutrophilic, B lineage and natural killer cells in Cluster 2, and monocytic, vascular, and stromal cells in Cluster 3 (Figure 2(a,b), Supplemental Figure S3a). The described cluster characteristics were strongest for Cluster 1, with the expression of T-cell population signatures, followed by Cluster 3, with the expression of the monocytic lineage and endothelial cells. In general, Cluster 1 showed the highest overall microenvironmental expression.
Figure 2.
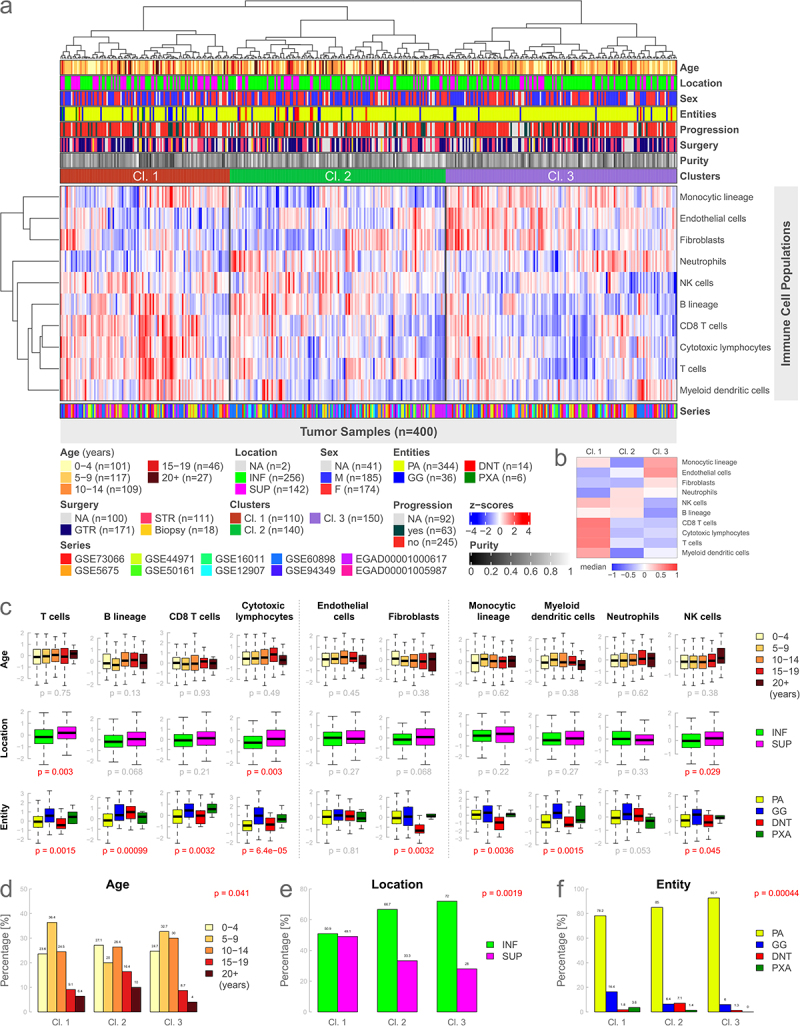
Immunologic clusters and immune cell signatures analyzed by age, location, and entity. (a): Heatmap based on the gene expression of 10 microenvironment cell populations using unsupervised hierarchical clustering. Three distinct immune clusters can be identified. (b): Visual representation of medians of each cluster for every cell population of Figure 2a. (c): Boxplots illustrating gene expression of individual immune cell signatures for different types of metadata. Data is presented analogously to Figure 1b. (d-f): Analysis of the difference in the distribution of different metadata between the clusters using Fisher’s exact test. INF, infratentorial; SUP, supratentorial; NA, not available.
We further analyzed the differences in the expression of the individual immune cell signatures in relationship to different metadata such as age, location, sex, entity, tumor progression, the extent of surgery, and the newly introduced clusters. Immune-cell-specific gene expression did not significantly differ for age, sex, or tumor progression (Supplemental Figure S3a). Concerning tumor location, supratentorial cases had a significantly higher expression of cytotoxic lymphocytes (p = 0.003), T cells (p = 0.003), and natural killer cells (p = 0.029) (Figure 2(c)). The remaining cell populations showed no significant difference for tumor location. Expression scores between the different low-grade entities were significant for all immune cell signatures except for endothelial cells and neutrophils. The differences in gene expression between the immune clusters were highly significant for all 10 cell populations (all p < 2e-06) (Supplemental Figure S3a).
Patient age groups, tumor location, and entity significantly differed between the defined immune cell clusters (Figure 2(d-f), all p < 0.04). Cluster 2 was enriched for older patients as well as DNTs. Cluster 1 was associated with comparatively more supratentorial samples and GGs (Figure 2(e,f)). Distributional differences between immune clusters were not significant for sex, tumor progression, and type of surgery (Supplemental Figure S3b-d).
Furthermore, we performed a survival analysis for progression-free survival, which did not significantly differ between the clusters (p = 0.1, Supplemental Figure S3e). Survival analysis for age, location, sex, and entity was not significant, solely the type of surgery showed a highly significant (p = 0.00016) difference in progression-free survival in favor of tumors treated with a gross total resection (GTR) compared to a subtotal resection (STR) (Supplemental Figure S3f-j). Data for progression-free survival were available for 43.8% of the low-grade glioma samples (Supplemental Figure S4).
Gene expression-based survival analysis
To get a more detailed insight into the prognostic relevance of the available gene expression data (5-year progression-free survival), we analyzed each of the 4923 genes included in all 10 compiled datasets separately using Cox proportional hazards modeling. From these genes, 163 were associated with worse survival, and 130 were associated with better survival (all p < 0.05, Figure 3(a)). However, only the endothelial marker VE-cadherin (CDH5) was significantly associated with poor outcomes after correction for multiple testing (p-BH = 0.027).
Figure 3.
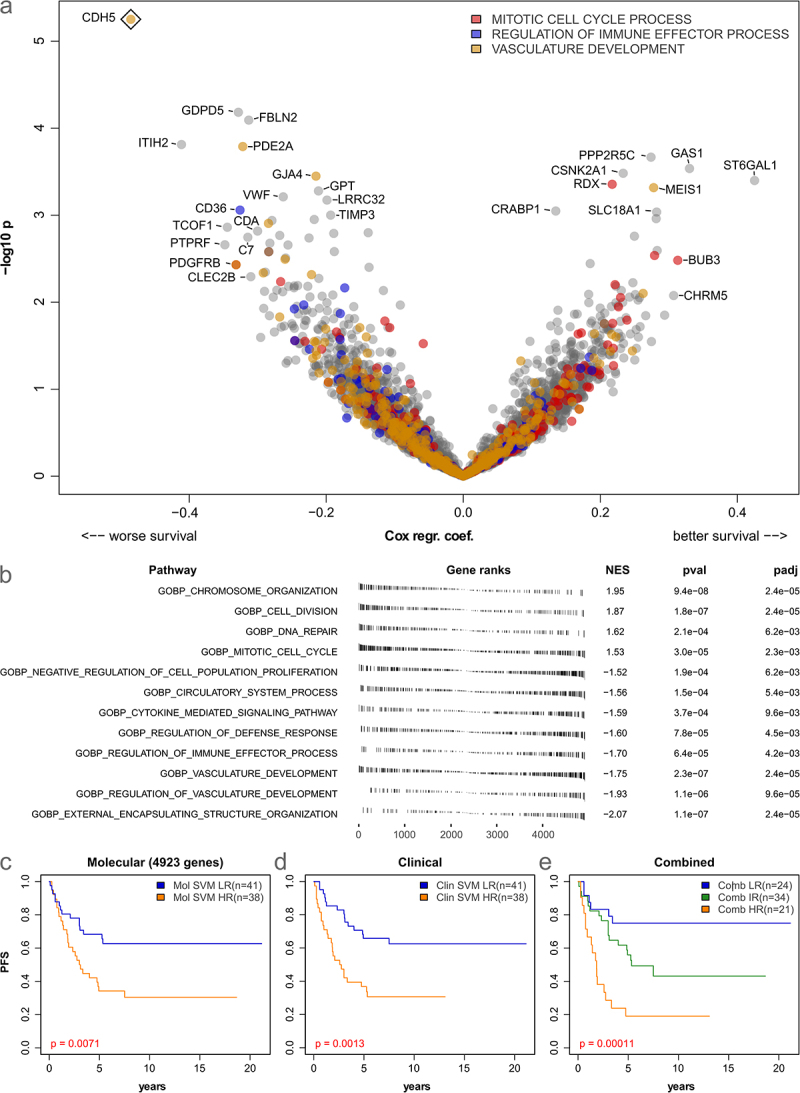
Prognostic relevance of gene expression and immuno-stromal markers in low-grade glioma. (a): Volcano plot showing log hazard ratios and p-values from Cox-regression for 5-year progression-free survival (PFS) based on the 4923 genes present in all datasets. Three significantly enriched gene expression signatures are color-coded. A rhombus indicates significance after multiple testing correction (FDR 5%). (b): results of gene set enrichment analysis of prognostically relevant genes (5-year PFS). (c-e): Kaplan-Meier curves showing results of SVM-based prediction of 5-year PFS based on 4923 genes (c) clinical data (age, location, sex, histology, and resection status, (d) and a combined model (e) in 79 patients.
Next, gene set enrichment analysis was performed to identify functional sets associated with survival. Overall, after filtering and removal of overlapping categories (see Methods), 12 gene sets were significantly associated with the outcome (all p-BH < 0.006, Figure 3(b)). Interestingly, the GO categories “chromosome organization”, “DNA repair”, and “mitotic cell cycle” were associated with better outcomes (Figure 3(b)). Further, the immune and stromal/endothelial categories “regulation of defense response”, “cytokine mediated signaling pathway”, “regulation of immune effector process”, and “vascular development” were associated with worse outcome. Six of the 12 enriched categories were linked to cell replication and DNA repair, while four were associated with the tumor microenvironment.
As the previously defined immune clusters showed no clear prognostic relevance, we finally investigated if 5-year progression-free survival could instead be directly predicted from gene expression and clinical data using machine learning. To this end, a linear support vector machine (SVM) was used and evaluated using 10-fold cross-validation. The SVM trained on expression data was able to divide the cohort into two groups (low-risk = LR and high-risk = HR) with significantly differing PFS (p = 0.0071, Figure 3(c)). Similarly, training an SVM on clinical data identified two groups with significantly differing PFS (p = 0.0013, Figure 3(d)). Finally, a combined model based on molecular and clinical data allowed to further improve predictions into three prognostic groups (p = 0.00011, Figure 3(e)), highlighting the relevance of both molecular and clinical data for the prognostic stratification of pLGG.
Discussion
Pediatric low-grade gliomas have a relatively good prognosis if the tumor is resected in its entirety. Still, the patient’s prognosis drastically declines if this is not achieved or not possible because of tumor location.2,4 Other used treatment modalities, such as craniospinal radiation therapy, are concomitant with adverse effects and long-term toxicities.5–7 In the last decade, immunotherapies have been successfully implemented in the treatment of various malignancies8,9 but are generally absent in low-grade gliomas. Knowledge of the TME is pivotal for the development, implementation, and improvement of the effectiveness of immune therapies. Previous studies on pediatric CNS tumors, pediatric gliomas, and adult LGGs showed great inter- and intratumor heterogeneity in immune cell infiltration,16–18 so it can be hypothesized that this also applies to pediatric low-grade gliomas.
Our study analyzes a cohort compiled from 10 published datasets analyzed on five platforms. Although this resulted in a reduced set of 4923 genes and different steps of data preprocessing as well as batch correction were necessary, we opted for this approach to get a comprehensive overview of the immuno-stromal landscape in pediatric low-grade glioma (pLGG) in a large cohort. The analysis includes 400 pLGG samples, which adequately addresses intertumoral heterogeneity. It is currently the most extensive analysis addressing the immune microenvironment in pLGG. The deconvolution approach relies on transcriptomic data, which are available for large sample sizes while at the same time offering a sufficient resolution for a precise characterization of the tumor microenvironment.
Higher resolution techniques like single-cell RNA-Seq spatial transcriptomics, or flow cytometry using fresh material can better characterize the immune microenvironment for selected tumors.37 Our approach results in a less detailed characterization of the TME, but it can be applied to much larger cohorts, and it complements further analyses relying on high-resolution methods.
Besides transcriptomic data, bulk DNA methylation data, which are also available for large cohorts of pLGG, can also be used to characterize the tumor microenvironment using deconvolution methods.16,38 However, although widely available, this data type is less suitable for deconvolution analyses in samples with a relatively low number of tumor-infiltrating immune cells like pediatric glioma.38 Tumor-infiltrating immune cells can, in principle, also be detected from standard histopathological images using machine-learning techniques.39 However, this approach is only of limited utility in pediatric brain tumors as morphological characterization and differentiation of small numbers of infiltrating immune cells is very challenging on H&E-stained slides and as no large series of immunohistological data are available.
The deconvolution analysis relied on a method specifically developed for use with brain tumors, which reduces the risk of nonspecific signatures.19,20 These signatures were optimized based on marker signatures proposed by Becht et al.21 and Danaher et al.22 In contrast to methods based on support vector regression like CIBERSORT,40 this methodologically more straightforward approach is more stable in cases with overall low immune infiltration.38,41
Our analysis of the immune and stromal microenvironment of pediatric low-grade gliomas revealed three different clusters of immune cell infiltration that can be described as T-cell/dendritic/natural killer cell- (Cl. 1), neutrophilic/B lineage/natural killer cell- (Cl. 2), and monocytic/vascular/stromal-cell-dominated (Cl. 3). Overall, microenvironmental expression was highest and most pronounced in Cl. 1 and the immunologic clusters correlated with location, age, and histological diagnosis. Our data also showed that HGGs and pediatric LGGs differ in their immune and stromal microenvironment, especially with T cells and fibroblasts being more present in HGGs. A relatively high presence of fibroblasts correlated with higher tumor grades (grade 3 and 4), which is in concordance with the findings by Chen et al.42 Differences in progression-free survival were not significant between the defined immune clusters, but we showed that 5-year PFS can be predicted from the molecular and clinical data with machine learning. While this underlines the prognostic relevance of the studied data, large datasets analyzed with high-resolution molecular profiling techniques are likely necessary to identify the specific role of the different immune cell populations. More specifically, we show that the vascular marker VE-Cadherin (CDH5) was associated with poor prognosis in our cohort. The expression of CDH5 was previously correlated with tumor grade in glioma, and an association with neovasculogenesis has been postulated.43 This could indicate that overexpression of CDH5 is associated with higher aggressiveness, which is usually present in higher-grade glioma.
Previously published work on immune infiltration in smaller series of pLGG reported mixed results. Using multicolor FACS and gene expression analysis (including some samples re-analyzed here), Griesinger et al. found a higher immune cell infiltration, especially for myeloid cells, in PA than in GBM, which we could not fully replicate.28 While some cell lines of myeloid lineage tended to be slightly more prevalent in PA than in GBM on average (monocytic lineage and neutrophils), others did not (myeloid dendritic cells). Robinson et al. examined T-cell infiltration in a small sample size of pediatric glioma, implementing multiplex immunofluorescence immunohistochemistry, machine learning, and single-cell mass cytometry. They found T-cell infiltration higher in LGGs than HGGs.44 In our analysis, T-cell infiltration was higher in HGGs than in LGGs, except for GG, although the distribution partially overlapped. Using immunohistochemical markers, Yang and colleagues found the (CD8) T-cell infiltration higher in GBM than in PA.45 Among LGGs, in concordance with Robinson et al. T cells, CD8 T cells, and cytotoxic lymphocytes were indeed more prevalent in GGs and PXAs in our study than in PAs and, in our case, DNTs.44 Taken together, these results underline the great heterogeneity of immune infiltration in pLGG, which is also suggested by a recent pan-cancer analysis of immunogenomics in pediatric brain tumors.46
In summary, our analysis emphasizes the heterogeneity of the immune and stromal microenvironment in pLGG and identifies three main immuno-stromal patterns. Vascular markers were associated with worse overall survival. The differences in immune cell infiltration in pediatric low-grade gliomas should be considered in upcoming immunotherapeutic research.
Supplementary Material
Acknowledgments
MB and US were supported by Fördergemeinschaft Kinderkrebs-Zentrum Hamburg.
Funding Statement
The work was supported by the Fördergemeinschaft Kinderkrebs-Zentrum Hamburg and the Open Access Publication Fund of UKE - Universitätsklinikum Hamburg-Eppendorf.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Authors’ contribution (CRediT)
Conceptualization: MK, MB
Methodology: MK, US, MB
Formal Analysis: MK, MS, MB
Investigation: all authors
Resources: US, MB
Data Curation: MK
Writing – Original Draft: MK
Writing – Review & Editing: all authors
Visualization: MK, MB
Supervision: US, MB
Data availability statement
The raw gene expression and RNA-Seq data analyzed in this study are available from Gene Expression Omnibus (GEO) or the European Genome-phenome Archive (EGA). Immune and stromal signatures and clinical metadata are available in the Supplementary material. The code is available from the corresponding author upon reasonable request.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2024.2386789
References
- 1.Erdmann F, Grabow KP, Spix DC.. German childhood cancer registry - annual report 2019 (1980-2018): institute of medical biostatistics, epidemiology and informatics (IMBEI) at the University Medical Center of the Johannes Gutenberg University Mainz; 2020. [Google Scholar]
- 2.Qaddoumi I, Sultan I, Gajjar A. Outcome and prognostic features in pediatric gliomas: a review of 6212 cases from the surveillance, epidemiology, and end results database. Cancer. 2009;115(24):5761–10. doi: 10.1002/cncr.24663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, Hawkins C, Ng HK, Pfister SM, Reifenberger G, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro-Oncology. 2021;23(8):1231–1251. doi: 10.1093/neuonc/noab106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stokland T, Liu J-F, Ironside JW, Ellison DW, Taylor R, Robinson KJ, Picton SV, Walker DA. A multivariate analysis of factors determining tumor progression in childhood low-grade glioma: a population-based cohort study (CCLG CNS9702). Neuro-oncology. 2010;12:1257–1268. doi: 10.1093/neuonc/noq092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benesch M, Lackner H, Sovinz P, Suppan E, Schwinger W, Eder H-G, Dornbusch HJ, Moser A, Triebl-Roth K, Urban C. Late sequela after treatment of childhood low-grade gliomas: a retrospective analysis of 69 long-term survivors treated between 1983 and 2003. J Neuro-Oncol. 2006;78(2):199–205. doi: 10.1007/s11060-005-9091-z. [DOI] [PubMed] [Google Scholar]
- 6.Greenberger BA, Pulsifer MB, Ebb DH, MacDonald SM, Jones RM, Butler WE, Huang MS, Marcus KJ, Oberg JA, Tarbell NJ, et al. Clinical outcomes and late endocrine, neurocognitive, and visual profiles of proton radiation for pediatric low-grade gliomas. Int J Radiat Oncol, Biol, Phys. 2014;89(5):1060–1068. doi: 10.1016/j.ijrobp.2014.04.053. [DOI] [PubMed] [Google Scholar]
- 7.Indelicato DJ, Rotondo RL, Uezono H, Sandler ES, Aldana PR, Ranalli NJ, Beier AD, Morris CG, Bradley JA. Outcomes following proton therapy for pediatric low-grade glioma. Int J Radiat Oncol, Biol, Phys. 2019;104(1):149–156. doi: 10.1016/j.ijrobp.2019.01.078. [DOI] [PubMed] [Google Scholar]
- 8.Esfahani K, Roudaia L, Buhlaiga N, Del Rincon SV, Papneja N, Miller WH. A review of cancer immunotherapy: from the past, to the present, to the future. Curr Oncol. 2020;27(12):S87–S97. doi: 10.3747/co.27.5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Majzner RG, Ramakrishna S, Yeom KW, Patel S, Chinnasamy H, Schultz LM, Richards RM, Jiang L, Barsan V, Mancusi R, et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature. 2022;603(7903):934–941. doi: 10.1038/s41586-022-04489-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirata E, Sahai E. Tumor microenvironment and differential responses to therapy. Cold Spring Harbor Perspect Med. 2017;7(7):a026781. doi: 10.1101/cshperspect.a026781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mohme M, Neidert MC. Tumor-specific T cell activation in malignant brain tumors. Front Immunol. 2020;11:205. doi: 10.3389/fimmu.2020.00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Naghavian R, Faigle W, Oldrati P, Wang J, Toussaint NC, Qiu Y, Medici G, Wacker M, Freudenmann LK, Bonté P-E, et al. Microbial peptides activate tumour-infiltrating lymphocytes in glioblastoma. Nature. 2023;617(7962):807–817. doi: 10.1038/s41586-023-06081-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quail DF, Joyce JA. The microenvironmental landscape of brain tumors. Cancer Cell. 2017;31(3):326–341. doi: 10.1016/j.ccell.2017.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo Y, Li S, Huang P, Zhang H, Yu C. Development of a prognostic model based on an immunogenomic landscape analysis of medulloblastoma. Biosci Rep. 2021;41(1). doi: 10.1042/BSR20202907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grabovska Y, Mackay A, O’Hare P, Crosier S, Finetti M, Schwalbe EC, Pickles JC, Fairchild AR, Avery A, Cockle J, et al. Pediatric pan-central nervous system tumor analysis of immune-cell infiltration identifies correlates of antitumor immunity. Nat Commun. 2020;11(1):4324. doi: 10.1038/s41467-020-18070-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Z, Guo X, Gao L, Wang Y, Guo Y, Xing B, Ma W. Classification of pediatric gliomas based on immunological profiling: implications for immunotherapy strategies. Mol Ther - Oncolytics. 2021;20:34–47. doi: 10.1016/j.omto.2020.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu F, Wang Z-L, Wang K-Y, Li G-Z, Chai R-C, Liu Y-Q, Jiang H-Y, Zhai Y, Feng Y-M, Zhao Z, et al. Classification of diffuse lower-grade glioma based on immunological profiling. Mol Oncol. 2020;14(9):2081–2095. doi: 10.1002/1878-0261.12707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bockmayr M, Mohme M, Klauschen F, Winkler B, Budczies J, Rutkowski S, Schüller U. Subgroup-specific immune and stromal microenvironment in medulloblastoma. OncoImmunology. 2018;7(9):e1462430. doi: 10.1080/2162402X.2018.1462430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bockmayr M, Klauschen F, Maire CL, Rutkowski S, Westphal M, Lamszus K, Schüller U, Mohme M. Immunologic profiling of mutational and transcriptional subgroups in pediatric and adult high-grade gliomas. Cancer Immunol Res. 2019;7(9):1401–1411. doi: 10.1158/2326-6066.CIR-18-0939. [DOI] [PubMed] [Google Scholar]
- 21.Becht E, Giraldo NA, Lacroix L, Buttard B, Elarouci N, Petitprez F, Selves J, Laurent-Puig P, Sautès-Fridman C, Fridman WH, et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016;17(1):218. doi: 10.1186/s13059-016-1070-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Danaher P, Warren S, Dennis L, D’Amico L, White A, Disis ML, Geller MA, Odunsi K, Beechem J, Fling SP. Gene expression markers of tumor infiltrating leukocytes. J For Immunother of Cancer. 2017;5(1):18. doi: 10.1186/s40425-017-0215-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Becht E, de Reyniès A, Giraldo NA, Pilati C, Buttard B, Lacroix L, Selves J, Sautès-Fridman C, Laurent-Puig P, Fridman WH. Immune and stromal classification of colorectal cancer is associated with molecular subtypes and relevant for precision immunotherapy. Clin Cancer Res. 2016;22(16):4057–4066. doi: 10.1158/1078-0432.CCR-15-2879. [DOI] [PubMed] [Google Scholar]
- 24.Bauchet L. Epidemiology of diffuse low grade gliomas. In: Duffau H. editor. Diffuse low-grade gliomas in adults. 2nd ed. Cham: Springer International Publishing; 2017. p. 13–53. [Google Scholar]
- 25.Bergthold G, Bandopadhayay P, Hoshida Y, Ramkissoon S, Ramkissoon L, Rich B, Maire CL, Paolella BR, Schumacher SE, Tabak B, et al. Expression profiles of 151 pediatric low-grade gliomas reveal molecular differences associated with location and histological subtype. Neuro-oncology. 2015;17(11):1486–1496. doi: 10.1093/neuonc/nov045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Donson AM, Apps J, Griesinger AM, Amani V, Witt DA, Anderson RCE, Niazi TN, Grant G, Souweidane M, Johnston JM, et al. Molecular analyses reveal inflammatory mediators in the solid component and cyst fluid of human Adamantinomatous Craniopharyngioma. J Neuropathol & Exp Neurol. 2017;76(9):779–788. doi: 10.1093/jnen/nlx061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gravendeel LAM, Kouwenhoven MCM, Gevaert O, de Rooi JJ, Stubbs AP, Duijm JE, Daemen A, Bleeker FE, Bralten LBC, Kloosterhof NK, et al. Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res. 2009;69(23):9065–9072. doi: 10.1158/0008-5472.CAN-09-2307. [DOI] [PubMed] [Google Scholar]
- 28.Griesinger AM, Birks DK, Donson AM, Amani V, Hoffman LM, Waziri A, Wang M, Handler MH, Foreman NK. Characterization of distinct immunophenotypes across pediatric brain tumor types. J Immunol. 2013;191(9):4880–4888. doi: 10.4049/jimmunol.1301966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lambert SR, Witt H, Hovestadt V, Zucknick M, Kool M, Pearson DM, Korshunov A, Ryzhova M, Ichimura K, Jabado N, et al. Differential expression and methylation of brain developmental genes define location-specific subsets of pilocytic astrocytoma. Acta Neuropathologica. 2013;126(2):291–301. doi: 10.1007/s00401-013-1124-7. [DOI] [PubMed] [Google Scholar]
- 30.Sharma MK, Mansur DB, Reifenberger G, Perry A, Leonard JR, Aldape KD, Albin MG, Emnett RJ, Loeser S, Watson MA, et al. Distinct genetic signatures among pilocytic astrocytomas relate to their brain region origin. Cancer Res. 2007;67(3):890–900. doi: 10.1158/0008-5472.CAN-06-0973. [DOI] [PubMed] [Google Scholar]
- 31.Wong K-K, Chang Y-M, Tsang YTM, Perlaky L, Su J, Adesina A, Armstrong DL, Bhattacharjee M, Dauser R, Blaney SM, et al. Expression analysis of juvenile pilocytic astrocytomas by oligonucleotide microarray reveals two potential subgroups. Cancer Res. 2005;65(1):76–84. doi: 10.1158/0008-5472.76.65.1. [DOI] [PubMed] [Google Scholar]
- 32.Zakrzewski K, Jarząb M, Pfeifer A, Oczko-Wojciechowska M, Jarząb B, Liberski PP, Zakrzewska M. Transcriptional profiles of pilocytic astrocytoma are related to their three different locations, but not to radiological tumor features. BMC Cancer. 2015;15(1):778. doi: 10.1186/s12885-015-1810-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones DTW, Hutter B, Jäger N, Korshunov A, Kool M, Warnatz H-J, Zichner T, Lambert SR, Ryzhova M, Quang DAK, et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet. 2013;45(8):927–932. doi: 10.1038/ng.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ryall S, Zapotocky M, Fukuoka K, Nobre L, Guerreiro Stucklin A, Bennett J, Siddaway R, Li C, Pajovic S, Arnoldo A, et al. Integrated molecular and clinical analysis of 1,000 pediatric low-grade gliomas. Cancer Cell. 2020;37(4):569–583.e5. doi: 10.1016/j.ccell.2020.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Revkov E, Kulshrestha T, Sung KWK, Skanderup AJ. PUREE: accurate pan-cancer tumor purity estimation from gene expression data. Commun Biol. 2023;6(1):394. doi: 10.1038/s42003-023-04764-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America; Vol. 102. 2005. p. 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gershon, R., Polevikov A, Karepov Y, Shenkar A, Ben-Horin I, Alter Regev T, Dror-Levinsky M, Lipczyc K, Gasri-Plotnitsky L, Diamant G, et al. Frequencies of 4 tumor-infiltrating lymphocytes potently predict survival in glioblastoma, an immune desert. Neuro-Oncology. 2024;26:473–487. doi: 10.1093/neuonc/noad204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Safaei S, Mohme M, Niesen J, Schüller U, Bockmayr M. Dimeimmune: robust estimation of infiltrating lymphocytes in CNS tumors from DNA methylation profiles. OncoImmunology. 2021;10(1):1932365. doi: 10.1080/2162402X.2021.1932365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klauschen F, Müller K-R, Binder A, Bockmayr M, Hägele M, Seegerer P, Wienert S, Pruneri G, de Maria S, Badve S, et al. Scoring of tumor-infiltrating lymphocytes: from visual estimation to machine learning. Semin Cancer Biol. 2018;52:151–157. doi: 10.1016/j.semcancer.2018.07.001. [DOI] [PubMed] [Google Scholar]
- 40.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12(5):453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ali HR, Chlon L, Pharoah PDP, Markowetz F, Caldas C. Patterns of immune infiltration in breast cancer and their clinical implications: a gene-expression-based retrospective study. PLOS Med. 2016;13(12):e1002194. doi: 10.1371/journal.pmed.1002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen Z, Zhuo S, He G, Tang J, Hao W, Gao W-Q, Yang K, Xu H. Prognosis and immunotherapy significances of a cancer-associated fibroblasts-related gene signature in gliomas. Front Cell Dev Biol. 2021;9:721897. doi: 10.3389/fcell.2021.721897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mao X-G, Xue X-Y, Wang L, Zhang X, Yan M, Tu Y-Y, Lin W, Jiang X-F, Ren H-G, Zhang W, et al. CDH5 is specifically activated in glioblastoma stemlike cells and contributes to vasculogenic mimicry induced by hypoxia. Neuro-Oncology. 2013;15(7):865–879. doi: 10.1093/neuonc/not029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robinson MH, Vasquez J, Kaushal A, MacDonald TJ, Velázquez Vega JE, Schniederjan M, Dhodapkar K. Subtype and grade-dependent spatial heterogeneity of T-cell infiltration in pediatric glioma. J For Immunother Of Cancer. 2020;8(2):e001066. doi: 10.1136/jitc-2020-001066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang I, Han SJ, Sughrue ME, Tihan T, Parsa AT. Immune cell infiltrate differences in pilocytic astrocytoma and glioblastoma: evidence of distinct immunological microenvironments that reflect tumor biology. J Neurosurg. 2011;115(3):505–511. doi: 10.3171/2011.4.JNS101172. [DOI] [PubMed] [Google Scholar]
- 46.Nabbi A, Beck P, Delaidelli A, Oldridge DA, Sudhaman S, Zhu K, Yang SYC, Mulder DT, Bruce JP, Paulson JN, et al. Transcriptional immunogenomic analysis reveals distinct immunological clusters in paediatric nervous system tumours. Genome Med. 2023;15(1):67. doi: 10.1186/s13073-023-01219-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw gene expression and RNA-Seq data analyzed in this study are available from Gene Expression Omnibus (GEO) or the European Genome-phenome Archive (EGA). Immune and stromal signatures and clinical metadata are available in the Supplementary material. The code is available from the corresponding author upon reasonable request.