

Abstract
Genome-wide studies have demonstrated regulatory roles for diverse non-coding elements, but their precise and interrelated functions have often remained enigmatic. Addressing the need for mechanistic insight, we studied their roles in expression of Lhb which encodes the pituitary gonadotropic hormone that controls reproduction. We identified a bi-directional enhancer in gonadotrope-specific open chromatin, whose functional eRNA (eRNA2) supports permissive chromatin at the Lhb locus. The central untranscribed region of the enhancer contains an iMotif (iM), and is bound by Hmgb2 which stabilizes the iM and directs transcription specifically towards the functional eRNA2. A distinct downstream lncRNA, associated with an inducible G-quadruplex (G4) and iM, also facilitates Lhb expression, following its splicing in situ. GnRH activates Lhb transcription and increased levels of all three RNAs, eRNA2 showing the highest response, while estradiol, which inhibits Lhb, repressed levels of eRNA2 and the lncRNA. The levels of these regulatory RNAs and Lhb mRNA correlate highly in female mice, though strikingly not in males, suggesting a female-specific function. Our findings, which shed new light on the workings of non-coding elements and non-canonical DNA structures, reveal novel mechanisms regulating transcription which have implications not only in the central control of reproduction but also for other inducible genes.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00018-024-05398-7.
Keywords: Biological sciences, Luteinizing hormone, Steroid, Epigenetic, Chromatin conformation, Histone
Introduction
Transcriptional regulation involves multiple regions of the non-coding genome. Distal enhancers and super-enhancers direct transcription of protein-coding genes, often involving non-coding enhancer RNAs (eRNAs) which can recruit regulatory proteins and/or act to determine the chromatin landscape and contact with the target gene promoter [1–5]. Other classes of long non-coding RNAs (lncRNAs) have diverse functions in mediating gene expression through various transcriptional and post-transcriptional mechanisms which are beginning to emerge, but have often proven difficult to elucidate [6–9]. The number of enhancers and lncRNAs annotated in the human genome far outweighs that of the protein-coding genes, indicating that multiple elements likely regulate a single gene [10]. However for very few has a function towards the target gene been demonstrated, and the mechanisms of action and inter-relationships between such elements remain poorly explained [10].
Non-canonical DNA structures have also gained much recent attention, particularly in the context of cell-specific gene-expression and differentiation, and their roles in disease [11–18]. G-quadruplexes (G4s) comprise guanine stretches on a single DNA strand that fold into secondary structures via Hoogsteen hydrogen base-pairing. Enriched at regulatory regions of the genome, these structures recruit various proteins, including epigenetic modifiers and transcription factors (TFs), and those at promoters are associated with high levels of transcription [13, 19–25]. IMotifs (iMs) can form on the C-rich complementary strand, through interaction of hemi-protonated cytosine base pairing [26, 27]. IMs have been studied less than G4s, are sensitive to ambient conditions, particularly pH, and were only recently shown to form in cells [21, 26–28]. Enrichment of these structures at gene regulatory regions hints strongly at their function, but only some of these structures are found where predicted, their formation and stability depending on both the underlying and flanking DNA sequence, bound proteins and chromatin environment [27, 29, 30]. All of this points to the roles of these structures being highly cell- and genomic-context dependent, and some likely responding to specific signals. Moreover, evidence suggests that the G4 and iM structures cannot form on both strands at the same time, such that their interdependency might also provide a regulatory switch [31–33].
Much of the work identifying these potential regulatory elements and their association with transcriptional states and networks has resulted from genome-wide studies. However cataloging these elements through common observational criteria is problematic because of their inherently heterogenous characteristics, and there is a clear need to define them based on their biological activity [10]. Currently there is a paucity of reports from in-depth studies on specific loci which explain the function of such non-coding regulatory elements in their appropriate physiological context. Here we address this knowledge gap by examining a gene whose transcription is induced and repressed by known signals in the reproductive endocrine system. The gene, Lhb, encodes one of the subunits of the luteinizing hormone (LH) that controls mammalian reproductive function. Expressed exclusively in the pituitary gland gonadotropes, Lhb is regulated by the stimulatory gonadotropin releasing hormone (GnRH), and (mostly negative) feedback by gonadal steroids, which together are responsible for the undulating hormone levels during the female estrous or menstrual cycle.
We hypothesized that regulation of Lhb transcription is mediated via various distal DNA elements. Building on snATAC-seq data showing gonadotrope-specific open chromatin upstream of the Lhb gene, we identified a distal bi-directional enhancer which contains an iM, and investigated its activity and regulation, as well as those of a distinct lncRNA, associated with inducible structures. We show that this adjacent lncRNA is chromatin-enriched and functions locally, though its homolog was recently reported to encode a bovine sperm protein, suggesting flexible usage of this DNA in different reproductive tissues. Moreover, in the murine gonadotropes, its role appears female-specific. Our insights on the novel regulation and roles of these diverse non-coding elements, in the context of the complex reproductive endocrine system, have implications particularly for other tightly regulated genes.
Materials and methods
Tissues, cells, treatments and transfections
The LβT2 and αT3-1 cell lines (gifts from P Mellon, University of California, San Diego) were derived from murine gonadotropes at different stages of development; both express Lhb, but αT3-1 cells represent an earlier stage and exhibit more consistent responses to steroid hormones. Both cell lines were cultured as described [34] in minimum essential medium (MEM for αT3-1) supplemented with 1% non-essential amino acid and 1% HEPES pH 7.3, or high glucose Dulbecco’s modified Eagle’s medium (DMEM for LβT2), containing 1% sodium pyruvate,1% sodium bicarbonate, 1% penicillin-streptomycin, and 10% fetal calf serum (FCS) unless stated otherwise (all Biological Industries, Bet Ha’Emek, Israel). For some experiments (as noted), serum starvation was used for cell synchronization, and for steroid treatments the cells were incubated in charcoal-stripped FCS (Biological Industries). Alternatively, LβT2 cells were cultured in media as above with phosphate buffer (25 mM NaHPO4 for pH 7.2, or NaH2PO4 for pH 6.3).
Cells were treated with vehicle alone (non-treated controls), GnRH (100 nM), PMA (0.1 µM), forskolin (10 µM), trichostatin A (TSA, 100 ng/ml), estradiol (E2; 10 nM) or dihydrotestosterone (DHT: 10 nM, all Sigma). Primary gonadotrope-derived cell cultures have been reported [35]. Pituitary cells were harvested from male and female (at estrous) mice, sham-operated controls, ovariectomized or castrated mice, all as described previously [36], in accordance with IACUC guidelines and after protocol approval.
Stable transfections were carried out in LβT2 cells at 60% confluency, using PolyJet reagent (SignaGen) according to the manufacturer’s instructions, and fresh medium replaced after 24 h, followed by selection using G418 (400 µg/ml) or puromycin (0.5 µg/ml, both Sigma). The dCas9-VP64 was transfected using lentivirus, with 2nd generation plasmid (PAX2, MD2.G; Addgene) and HEK293 cells. Concentrated viruses were used to infect LβT2 cells, followed by puromycin selection as before. These stably transfected dCas9- KRAB and VP64 cells were then stably transfected with linearized gRNA4 (below) and selected using G418.
Plasmid constructs
The dCas9-KRAB (#50919) and dCas9-VP64 (#50918) plasmids were from Addgene. The gRNA oligos (Table S1) were designed (http://crispr.dbcls.jp), cloned into Cas9 sgRNA vector (Addgene #68463), and verified by sequencing before being linearized with HindIII and SalI and inserted into pEGFP-N1 (Clontech). The stable knockdowns were as described [1] with linearized pSR-GFP/neo plasmid (OligoEngine) expressing the shRNA sequences (Table S1). The lncRNA plasmids (CMV-Lhb -2575 to -582, CMV-Lhb -2575 to +1175, CMV-Lhb -2575 to +1175) utilized EGFp-N1 as a backbone which also served as the control. The inserts were PCR-amplified from DNA, at the locations stated, except for the U1 mutated form (Table S2), which was synthesized using Gblocks (IDT Israel). The sequence encompassing the lncRNA and genomic Lhb (-2575 to +1175) was also inserted into PGEM-Teasy (Promega) without an additional promoter. All references to location upstream of the Lhb TSS refer to the isoform NM_008497.2 and used the mm10 genome assembly.
RNA extraction and real-time quantitative PCR
Total RNA was extracted using TRIzol (Ambion), treated with DNase and purified using R1014 RNA clean & concentrator-5 kit (Zymo Research). The cDNA was synthesized using qScript cDNA Synthesis Kit (Quanta Biosciences), or High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) according to manufacturer’s instructions. Real-time quantitative PCR (qPCR) was carried out using the PerfeCTa SYBR Green FastMix (Quanta Biosciences) or SYBR green blue mix Hi-ROX (PCR Biosystems), using primers listed in Table S3. Amplicon levels were quantified using standard curves and normalized to Rplp0 or Gapdh housekeeping genes. Isolation of cytosolic, nuclear or chromatin-associated RNA was as previously reported [35].
Chromatin immunoprecipitation (ChIP)
ChIP experiments for H3K4me1 (Ab8895), H3K4me3 (Ab1012), H3K9me3 (Ab8898), H3 (Ab1791), Hmgb2 (Ab6728), IgG control (Ab6721; all from Abcam) were carried out in formaldehyde cross-linked LβT2 cells, as previously, after extensive sonication [1]. Real-time qPCR (primers in Table S3) amplified specific regions in the IP and input samples, for normalization.
ChIP for iM was carried out in (1 × 107) LβT2 cells at 80% confluency without crosslinking, sonicated using 10 × 15 s on-, 10 s off- pulses. Cells were rinsed in the culture medium without FCS before harvest in the same medium. Cells were lysed with 750 µl lysis buffer (50 mM Hepes·KOH [pH 7.5 or pH 6.5], 140 mM NaCl, 1 mM EDTA [pH 8], 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, protease inhibitors). Cell debris was pelleted (10,000 g for 5 min, 4 °C) and 50 µl of supernatant was taken as input. The remaining supernatant was immunoprecipitated using 5 µg of iMab scFv FLAG tag [26] (a gift from Daniel Christ, Garvan Institute of Medical Research, Australia), or IgG as control, and Protein L magnetic beads (10 µl; 88849 Thermo Scientific) incubated overnight on a roller at 4 °C. After three washes with the same lysis buffer (pH 7.5 or 6.5), samples were eluted (1% SDS, 100 mM NaHCO3) and incubated at 65 °C for 1 h with 2 µl RNase A (0.5 mg/mL), then purified using PCR purification kit (28106; Qiagen) for analysis as above.
ChIP for G4 was using the BG4 antibody scFv fragment (Absolute Antibodies ab00174-30.125 lot: T2345840), or IgG as control, essentially as custom [37]. Serum-starved LβT2 cells (1 × 107) at 80% confluency were formaldehyde cross-linked (1% final concentration in ice cold PBS, for 10 min with rotation), quenched with glycine (125 mM final, 5 min rotation), rinsed and collected into cold PBS, before storing the pellet at -80 °C. The pellet was resuspended in hypotonic buffer (25 mM HEPES pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 1 mM DTT, 0.1% NP-40) with PMSF and protease inhibitor cocktail, and incubated for 10 min on ice. After centrifugation, the pellet was resuspended in 1 ml of 50 mM Tris, 10 mM EDTA, 0.5% SDS with PMSF and protease inhibitor cocktail, and kept on ice for 10 min, before transferring to Covaris millitubes (1 ml), for sonication (“high cell” parameters: peak incident power 140 W, duty factor 5%, cycles per burst 200, 6 ˚C) for 25 min. After centrifugation (5 min, 13,000 rpm), the supernatant was transferred to a new tube. To 25 µl of the supernatant, 100 µl elution buffer (50 mM Tris pH 8, 10 mM EDTA, 1% SDS) with 2 µl RNAseA (0.5 µg/µl) was added, and incubated at 65 °C for 4 h. After cleanup (Qiagen #28106 PCR purification kit), concentration was checked using Qubit B.R., and fragmentation by Tape Station. The remainder was cleared by centrifugation (5 min at 13,000 rpm) and divided into 100 µl aliquots, kept at -80 °C. This typically resulted in 100–500 bp fragments (peak 215 bp), at concentration of 100 ng/µl. After thawing, Triton X (1% final) was added to each aliquot, and incubated for 10 min at RT. Blocking buffer (900 µl of 25 mM HEPES pH 7.5, 10.5 mM NaCl, 110 mM KCl, 1 mM MgCl2, 1% wt/vol BSA; with 10 µl PMSF, and 0.5 µl RNAseA [24 mg/ml]) was added and incubated for 30 min at 37 ˚C, before storing on ice. From this, 50 µl was removed (for “input”), to which 50 µl 2x elution buffer was added, and samples de-cross-linked for 4 h at 65 ˚C. Following cleanup (Qiagen PCR purification kit), DNA was eluted and diluted in DDW for qPCR. For the remainder of the sample, 3 µg of antibody (1.5 µl of IgG [ab6721 Abcam goat anti rabbit HRP 2 µg/µl] as control; or 3 µl of BG4 (1 µg/µl) was added for 1 h at RT on a roller. FLAG-agarose beads (Sigma A2220; 50 µl) were pre-incubated in 100 µl blocking buffer with oligo (CCCAAATCTCCAATCTTCTTAAATAC; 75 ng/µl beads) on a roller for 30 min at RT, and then rinsed in the same buffer. These beads were added to the samples and kept ON on a roller at 4 °C. The following day, they were rinsed (x5) with wash buffer (10 mM TRIS pH 7.4, 100 mM KCl, 0.1% Tween 20), and eluted in 150 µl elution buffer (15 min at 65 °C). After centrifugation, the supernatant underwent de-cross-linking (4 h at 65 °C), and clean-up as above, before analysis by qPCR. Positive and negative control regions were picked based on published data (GSM6634325, GSM6634326) [38], combined with G4Hunter [39] and pqsfinder [40] predictions, and known expression of the positive control gene in gonadotrope cells.
Circular dichroism
The putative iM and G4-forming oligonucleotides (Table S4) were prepared at 5 µM in 50 mM sodium-cacodylate buffer (pH 6.5, 0.1 M sodium acetate, 1.4 M solution; Sigma) and pH adjusted with 10 M NaOH or 17.4 M acetic acid. The oligonucleotides (500 µl) were allowed to fold for 20 min in a boiling water bath and cooled down slowly overnight. The circular dichroism (CD) spectra were measured on Applied Photo-physics PiStar circular dichroism spectropolarimeter (Leatherhead, Surrey, UK), using a 1 mm path length quartz cuvette at 25 oC, over 230–350 nm wavelengths. Three measurements were recorded, averaged and baseline corrected for signal contribution from buffers, analysed by the Pro-Data viewer software. These averages are shown in the plots which are representative of experiments that were repeated on various occasions, after smoothing using the same software, based on the Savitzky–Golay filter, with default settings.
Binding assays
Fluorescence binding experiments were carried out using a similar approach as in published studies [41], with a PC1 photon counting spectrofluorometer (ISS, Champaign, IL) with Quantum Northwest TC125 temperature controller system set at 25 oC. All equilibrium-binding reactions were performed in a 20 µl Precision cell fluorescence cuvette (Farmingdale, NY, USA). The DNA oligonucleotides (CCCCACCCCACCCCACCCCCACCCCCACCTTTCC: 100 µM) were allowed to fold in 10 mM potassium phosphate buffer (K2HPO3/KH2PO4) with 70 mM KCl (pH 6 or pH 7) at 90 oC for 5 min, slowly cooled O.N and were kept at 4oC until use. Samples for the experiments were prepared in the same potassium phosphate buffer in a 0–20 µM concentration range of the DNA, with or without constant concentration of 1 µM His-tagged HMGB2 (Abcam; Ab109962). Buffer of the dissolved HMGB2 protein (made as in manufacturer’s instructions: 0.0154% DTT, 0.316% Tris HCl pH 8, 30% glycerol, 0.058% sodium chloride) was added to samples without HMGB2 in the same ratio, to account for background from the buffer. After excitation at 280 nm, emission spectra were recorded between 305 and 500 nm with 2 mM slit width. The fraction of bound protein (α) was calculated at 331 nm using the following equation:
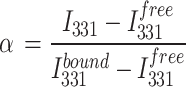 |
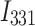 is the fluorescence intensity at 331 nm at each DNA concentration that was examined,
is the fluorescence intensity at 331 nm at each DNA concentration that was examined, 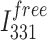 is the fluorescence intensity of the free protein and
is the fluorescence intensity of the free protein and 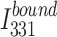 is the fluorescence intensity at saturating protein concentrations. The
is the fluorescence intensity at saturating protein concentrations. The 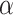 was plotted against DNA concentration and curve fitting was done using nonlinear regression to a theoretical binding model described [42] (equation to fit the data is shown in Fig S6).
was plotted against DNA concentration and curve fitting was done using nonlinear regression to a theoretical binding model described [42] (equation to fit the data is shown in Fig S6).
Chromatin conformation capture assay (3 C) and UMI-4 C
Chromatin conformation capture (3 C) assay was carried out in αT3-1 cells as previously [1]. PCR was performed using 1 µl of the library per 50 µl reaction. The primers comprised nested forward primers targeting + 344 and + 379 from the Lhb TSS, and sets of primers targeting various upstream sites as shown and detailed in Table S3. The first round of PCR was at 55 °C for 35 cycles, and the second round at 55 °C for 25 cycles. All amplicons were verified by sequencing.
UMI-4 C [43] was performed using 1 × 106 LβT2 cells, with or without 2 h exposure to GnRH (100 nM), and initial libraries prepared essentially as for the 3 C. This 3 C DNA was re-suspended in TE and sonicated to 300–400 bp (Covaris S2 with default parameter of Intensity (4), duty cycle (10%), cycles per burst (200), and time (80 s)). The fragmented DNA was end-repaired and dA-tailed (NEBNext® Ultra™ II End Repair/dA-Tailing Module; NEB #E7546), and 5′-end dephosphorylated with calf intestinal alkaline phosphatase (New England Biolabs, M0525) for 1 h at 37 °C. The DNA was purified with 1x Ampure XP beads, eluted in 60 µl of water, and then ligated with the Illumina adapter using the NEBNext Ultra II Ligation Module, with 2.5 µl NEBNext Illumina adapter (final 0.4 µM), 30 µl NEBNext Ultra II Ligation Master Mix, and 1 µl of Ligation Enhancer, at 20 °C for 15 min, followed by treatment with 3 µl of USER Enzyme (NEB) at 37 °C for 15 min. To release the unligated strand of the adapter, the DNA was denatured at 95 °C for 2 min and cleaned with 1x AmpureXP beads. DNA concentration was measured using Qubit ssDNA Assay kit.
Nested PCR was performed, initially for the upstream PCR, 20 cycles with the upstream (US) primer (in 50 µl: 200 ng DNA, NEBNext Q5 Hot Start HiFi PCR Master Mix Kit ([EB. #M0543S], 0.4 µM of US viewpoint primer and 0.4 µM Illumina universal reverse primer: 98 °C for 30s, 20 cycles of 98 °C for 10s, 65 °C for 10s, and 72 °C for 60s, final extension at 72 °C for 5 min). The reactions were pooled and cleaned with 1x Ampure XP beads before the nested downstream PCR (with NEBNext® Q5® Hot Start HiFi PCR Master Mix, 0.4 µM of downstream (DS) viewpoint primer and 0.4 µM Illumina universal reverse primer), and amplified for 15 cycles similarly.
The final libraries were purified and quantified using the Qubit dsDNA HS assay kit (Q32854) as before, examined by Bioanalyzer (Applied Biosystems), and diluted to 10 nM for 2 × 150 bp paired-end sequencing on MGI G400 platform (Syntezza Bioscence). The resulting FASTQ files were processed using the umi4cPackage pipeline (https://github.com/tanaylab/umi4cpackage) [43] to generate interaction tracks representing UMI counts per genomic restriction fragment. The viewpoint-specific interaction profiles were generated and normalized to the total UMI count within the profile, to generate the comparative plot for GnRH-treated and non-treated cells.
Characterization of eRNAs and lncRNA
Chromatin-enriched RNA was extracted using TRIzol (Ambion), treated with DNase and purified with Zymo RNA cleanup kit. The RNA was reverse transcribed (Superscript II, Invitrogen), using adaptor stem-loop Nx7, AGATCGGAAGAGAGACGTGTGCTCTTCCGATCTNNNNNNN. For the 3’ ends, and determining the orientation, PCR was performed with the adaptor AGACGTGTGCTCTTCCGATCT over two rounds of PCR with primers Lhb (-2802 F) then Lhb (-2655 F) for eRNA1, and Lhb (-4796R) then Lhb (-4856R) for eRNA2. The products were cloned and sequenced. For the 5’ ends, PCR was performed using various sets of primers with DNA as control for the reactions.
For the lncRNA, total RNA was extracted using TRIzol, DNAse-treated and purified as above, and poly(A) tailed (NEB M0276) and reverse transcribed (Quanta Flex 95049), using adaptor dT GACTCGAGTCGACATCGATTTTTTTTTTTTTTTTT. For the 3’ ends and determining the orientation, PCR was performed with the adaptor GACTCGAGTCGACATCGAT over two rounds of PCR with primers Lhb (-2575 F) then Lhb (-1966 F) for forward orientation, and for reverse orientation: Lhb (-582R) then Lhb (-1117R), or Lhb (-1803R) and then Lhb (-2061R). For the 5’ ends, PCR utilized various sets of primers with DNA as control.
Protein pulldown and mass spectrometry
Protein pull-down was according to a published protocol [44]. For each stable clone (expressing one of two different gRNAs and FLAG-dCas9 from the same plasmid, or FLAG-dCas9 alone as control: Table S5), 2 × 107 cells were cross linked with 1% formaldehyde for 10 min at RT. Glycine was added (final 0.125 M) and cells incubated for 5 min at RT, before two rinses with PBS. They were then collected into PBS, centrifuged and suspended in 10 ml CLB (10 mM Tris pH 8, 1 mM EDTA, 0.5% IGEPAL CA-630, 1x protease inhibitors), incubated for 10 min on ice and centrifuged (930 g at 4 ˚C for 8 min). The pellet was resuspended in 10 ml NLB (10 mM Tris pH 8, 1 mM EDTA, 0.5 M NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.5% lauroyl sarcosine, 1x protease inhibitor), incubated on ice for 10 min, with vortex at max speed for 2–3 s every 2–3 min, and then centrifuged (930 g at 4 ˚C for 8 min). The pellet was resuspended in 10 ml PBS and centrifuged as before.
The pelleted fraction was suspended in 0.8 ml MLB3 (10 mM Tris pH 8, 1 mM EDTA, 0.5 mM EGTA, 150 mM NaCl, 0.1% sodium deoxycholate, 0.1% SDS, 1x protease inhibitor) and sonicated for a total of 1 min and 15 sec (15 s on, 10 s off), and centrifuged (16,000 g, at 4 ˚C for 10 min). The supernatant was collected for pulldown: Triton X-100 was added (1% final), 10 µg anti-FLAG (Sigma F1804) and 40 µl protein G magnetic beads (Invitrogen 10004D) for overnight incubation on a roller at 4 ˚C. The next day, a series of washes was performed and in each step the beads were concentrated on a magnetic stand. Each wash was incubated for 5 min, at 4 ˚C on the roller. The washes comprised: 2x low salt (20 mM Tris pH 8, 2 mM EDTA, 150 mM NaCl, 1% Triton X-100, 0.1%SDS); 2x high salt (20 mM Tris pH 8, 2 mM EDTA, 500 mM NaCl, 1% Triton X-100, 0.1% SDS); LiCl wash (10 mM Tris pH 8, 1 mM EDTA, 0.25 M LiCl, 0.5% IGEPAL CA-630, 0.5% sodium deoxycholate); TBS-IGEPAL (50 mM Tris pH 8, 150 mM NaCl, 0.1% IGEPAL CA-630).
For elution, the beads were suspended in 200 µl elution buffer (50 mM Tris pH 8, 150 mM NaCl, 0.1% IGEPAL CA-630, 500 µg/ml 3xFLAG peptide [F4799 Sigma]), incubated at 37 ˚C for 20 min. The beads were separated and the supernatant collected for precipitation: 200 µl supernatant, 500 µl isopropanol, 25 µl of 3 M sodium acetate (pH 5.2) and 5 µl glycogen (20 mg/ml), and incubation overnight at -20 ˚C. The tube was centrifuged (16,000 g, at 4 ˚C for 30 min), washed with 70% ethanol, and the pellet suspended in 40 µl 2x Laemmli sample buffer. To reverse cross linking, the tube was incubated for 10 min at 95 ˚C. The sample was run on a premade 4–20% gradient protein gel (#4561094 BioRad) for 1 cm, stained with Imperial protein stain (#24615 Thermo), cut and analyzed at the Smoler Proteomics Center (Lorry I. Lokey Interdisciplinary Center for Life Sciences & Engineering, Technion). Following trypsin digestion, the samples were analyzed by LC-MS/MS on HF (Thermo), and proteins identified with Sequest (Thermo) and Mascot (Matrix science) search algorithms and Discoverer software (Thermo), using the Mus musculus proteome database in Uniprot, and a decoy database, to determine the false discovery rate. The identified peptides were filtered with high confidence, top rank, mass accuracy, and high confidence peptides passed the 1% false discovery rate threshold. Semi quantitation was performed by calculating the peak area of each peptide; the area of the protein is the average of the three most intense peptides from each protein.
Statistical analysis
Data are from multiple biological repeats (n-value) which were assayed individually. F-test determined similarity of variance and data analyzed accordingly by Student’s t-test (two-tailed), or ANOVA followed by Tukey HSD or Bonferroni t-test for multiple comparisons to establish significance, defined as P < 0.05. Graphs show mean ± SEM, and for box plots, whiskers represent minimum and maximum values, boxes are 25–75% data ranges, and horizontal lines within boxes indicate the median values. Pearson’s analysis of correlation was performed in R package. For the 4 C-UMI, comparison of contact intensities at specific genomic intervals and P-value calculation (χ2 test) were conducted using the P4cIntervalMean function (umi4cPackage), default parameters.
Results
Identification of a putative upstream regulatory element for the Lhb gene
The chromatin encompassing the Lhb gene in the murine pituitary is open only in gonadotrope cells, and this extends upstream to the neighboring Ntf5 gene (Fig. 1A; data from [45]). Predicting that this region might contain a transcriptional enhancer, we looked for enrichment of the enhancer-characteristic histone modification, H3K4 monomethylation (H3K4me1), in a gonadotrope-derived cell line (LβT2). A peak of H3K4me1 was evident around -3200 bp upstream of the Lhb TSS (Fig. 1B). H3K4 trimethylation (H3K4me3), which marks active protein-coding genes, was not evident other than a small but statistically significant signal around -2802 to -2633 bp (Fig. 1C). The ability of the H3K4me1-enriched region to regulate Lhb transcription was established by gRNA-mediated targeting of dCas9-VP64 or dCas9-KRAB bp to the locus (~-3550 bp), which resulted in 4-fold activation or 80% repression, respectively, of Lhb mRNA levels compared to the controls that lacked the targeting gRNAs (Fig. 1D).
Fig. 1.

Identification of an upstream regulatory element for the Lhb gene. (A) Open chromatin specific to the gonadotropes (boxed in red) is shown in ATAC-seq data, from Single Nucleus Pituitary Atlas (snpituitaryatlas.princeton.edu)[45]. (B-C) ChIP was performed in LβT2 cells for (B) H3K4me1 and (C) H3K4me3. IP/input levels, at this region upstream of Lhb, shown relative to total H3 levels at the same locations, and as H3K4me3/input with IgG controls (-Ab). Mean±SEM, n=3. (D) The mRNA levels of Lhb (mean±SEM, n=3) in dCas9-KRAB/VP64 expressing cells with or without (control) gRNA targeted to -3550 bp; shown relative to levels in control cells. Student’s t-test compared mean values with controls: *: P<0.05; **: P<0.01. (E) Chromatin conformation capture (3 C) was performed in αT3-1 cells after Dpn2 restriction (sites in red), and analyzed by PCR using nested forward primers (+344 and +379) and those targeting the various upstream regions noted; amplicons (Fig S1) were confirmed by sequencing. Horizontal blue bars represent regions found in proximity with the Lhb TSS. (F) UMI-4C normalized counts across the Lhb gene locus are shown for untreated (grey line) and GnRH-treated (blue line) LβT2 cells. The Lhb promoter viewpoint is demarked by dashed green lines, shaded areas represent the eRNAs (pale blue) with central untranscribed region (shaded purple, to which 200 bp was added on either side to reach minimum size for the analysis), and the lncRNA (shaded beige). Locations of the Lhb and Ntf5 genes are noted above the schematic by a black bar. Comparison of counts at 1 kbp genomic intervals and their P-values were calculated (umi4cPackage) upstream from the Lhb gene viewpoint, and the P-values are shown in the bar chart, as -log10 values, both panels aligned with the snATAC-seq in gonadotropes (as in A)
To determine whether this locus interacts physically with the 5’ end of the Lhb gene, we carried out chromatin conformation capture (3 C) in αT3-1 gonadotrope cells. This technique identified a number of specifically-queried upstream sites to be in close proximity with the Lhb TSS (Fig. 1E, S1). We performed also unique molecular identifier (UMI)-4 C [43], in untreated and GnRH-treated LβT2 cells. Although this technique is less suited for identifying interacting sites so close to the viewpoint (i.e. the Lhb TSS), it takes an unbiased approach to search for interacting sites, enabled quantification of the contacts and revealed two sites of significantly increased interaction after GnRH treatment (Fig. 1F).
The enhancer is transcribed bi-directionally to two eRNAs, and a distinct spliced lncRNA
To understand the mechanisms through which this regulatory region might function, we looked for RNAs transcribed from the locus. PCR on DNase-treated, reverse-transcribed RNA from the gonadotrope cell line, amplified transcripts in the -5000 to -2152 bp region (relative to the Lhb TSS), though not between -3682 to -3460 bp (control amplifications were from DNA templates). We mapped the 5’ and 3’ ends of these RNAs and their directionality to reveal two unspliced enhancer (e)RNAs that are transcribed bidirectionally. The more upstream (i.e. further from Lhb) eRNA2, starts at -3682 bp and ends at -5046. This eRNA2 is transcribed from one of the regions whose interaction with Lhb was seen to increase after GnRH treatment (Fig. 1F), and overlaps with the 3’ end of Ntf5. The other, eRNA1, is transcribed between -3460 and -2151 bp, in the direction of Lhb (Fig. 2, S2A-D). Transcription of these RNAs was confirmed in primary gonadotrope cells, and they were both found to lack polyA tails and be enriched specifically at the chromatin (Fig S2E).
Fig. 2.

The enhancer is transcribed bi-directionally to two eRNAs and a distinct spliced lncRNA. The region upstream of Lhb, modified from the mm10 UCSC genome browser, showing locations of two bidirectional eRNAs, the annotated AC151602.1 and Lhb gene. Start and stop sites of the eRNAs are noted in red, and for the gonadotrope lncRNA, which differs slightly from the annotated transcript, these are noted in purple: the lncRNA termination site is not discrete but it was not detected after -643 (see also Figs S2, S3). All distances are shown relative to Lhb NM_008497 TSS
Although eRNA1 is not spliced (Fig S2), it is transcribed from a region partly overlapping an annotated spliced RNA, AC151602.1 (Fig. 2). We detected this distinct spliced transcript, starting just upstream from the annotated sequence (-2575 bp from the Lhb TSS), in both primary pituitary cells and the LβT2 gonadotrope cell line (S3A-D). This sequence is annotated as a polycomb-associated lncRNA [46], has been reported in the other tissues of the HPG axis, including the hypothalamus [47], and isoforms detected with varying 3’ ends (Fig S3A). Moreover, in both testes and ovaries it was found to form part of a long Lhb mRNA variant [48, 49]. However, in primary pituitary cells and the gonadotrope cell line, this lncRNA terminates before the end of the last annotated exon 5 (Fig S3C), and we did not detect any transcript spanning the lncRNA and Lhb mRNA (Fig S3D). Although the transcript does contain a possible ORF, the RNA lacked a polyA tail (Fig S3E), suggesting that any function is mediated by the RNA, or perhaps through the process of its transcription near to the Lhb gene.
The central untranscribed region of the enhancer can form G4 and iM DNA structures
The central untranscribed region of this enhancer (between -3682 to -3460 bp) contains a sequence (GGGGGTGGGGGTGGGGTGGGGTGGGG) that is strongly predicted to form a G4 (Table S6). To verify that this sequence folds into a G4, and the complementary sequence an iM, we performed circular dichroism (CD) using oligonucleotides, similar sequences with mutations that are not predicted to give rise to these structures, and the previously reported iM and G4 sequences of human C-MYC [32, 50] as positive controls. The CD spectra for the G-rich strand had a maximum positive peak around 260 nm which is characteristic for the parallel form of the G4 [51], and was similar but stronger than those of the C-MYC G4 (Fig. 3A, S4). The CD spectra for the complimentary C-rich sequence peaked around 286 nm, with a negative peak around 260 nm, characteristic for iM structures, and similar to the spectra of the C-MYC iM (Fig. 3B, S4). Both these C-rich sequences emitted similar spectra at pH 6.19 and 6.5, while at pH 7.26 they resembled those of the mutant sequence, contrasting with the G4 which was insensitive to the change in pH (Fig. 3A, B).
Fig. 3.

The central region of the Lhb enhancer forms a stable iM structure in gonadotropes. Circular dichroism was performed with ssDNA from the central untranscribed region of the Lhb enhancer and previously reported human c-MYC or mutated sequences as controls. (A) The G-rich sequence spectra are characteristic of the G4, regardless of pH, and (B) the complementary C-rich sequence spectra characteristic of the iM only at the lower pHs (see also Fig S4). (C) IGV view of the locus showing published data from BG4 ChIP-seq or CUT&RUN experiments in mouse skin [52] and mESCs [38] respectively, with or without PDS treatment; site of the G/C rich sequence at the central untranscribed region of the enhancer is marked with an arrowhead. (D) BG4 ChIP at the enhancer, lncRNA promoter and 5’ end in LβT2 cells, with or without GnRH treatment (2 h, 100 nM). Levels (mean ± SEM, n = 3) are shown as IP/input; ANOVA followed by Bonferroni t-test compared all levels in non-treated cells, and those sharing the same letter are not significantly different (P > 0.05); asterisks mark significantly higher levels following GnRH treatment. The GnRH-responsive gene, Cga, is positive control. (E, F) ChIP for iM in LβT2 cells using iMab antibody. Levels (mean ± SEM, n = 3) are shown as IP/input; ###: P < 0.0001 compared to all other groups in ANOVA followed by Tukey HSD. (F) As part of the same experiment (controls are as in E), some of the cells were treated with GnRH (2 h, 100 nM) or TSA (24 h, 100 ng/ml). Student’s t-test compared levels in each treatment group with those in untreated cells at the same region, *: P < 0.05; **: P < 0.01
Structures are found at several sites in this region in gonadotropes
We hypothesized that one or both of these structures might play a role in regulating the enhancer activity, and next looked for evidence that they are formed in the cellular environment. Published data from mouse G4-seq in skin cells [52] and CUT&RUN in embryonic stem cells (mESCs) [38] both show a G4 at this site of the Lhb enhancer (in skin cells shown specifically on the G-rich reverse strand: Fig. 3C). Additional signals were evident at several sites along the lncRNA-coding region where G4s are also predicted (Table S6), and these were increased following treatment with the G4-stabilizing agent, pyridostatin (PDS; Fig. 3C). To examine their presence in gonadotropes, we performed ChIP using the same BG4 antibody (specific to G4s [53]) in serum-starved LβT2 cells, some of which were treated with GnRH, and examined these specific sites by qPCR. A weak G4 signal was seen at the enhancer site which was not affected by GnRH (Fig. 3D). However stronger signals were evident at the lncRNA promoter and 5’end, especially after GnRH treatment; GnRH also increased the positive control signal located in the GnRH-responsive gene Cga (Fig. 3D).
Given that iMs might form at some of these sites, we next performed ChIP with the iMab antibody [26]. In untreated cells, the iM signals were similar across the region, except at the center of the enhancer where there was a clear peak (Fig. 3E). We also examined cells treated with GnRH, or the histone deacetylase (HDAC) inhibitor, trichostatin A (TSA) which increases global acetylation. GnRH-stimulated HDAC nuclear export increases Lhb expression [35, 54, 55], and HDAC inhibition reduces intracellular pH [56], such that either treatment might be expected to facilitate iM formation. In fact, the enhancer iM was not affected significantly by either treatment, although the TSA increased the iM signal at the lncRNA promoter by more than three-fold (Fig. 3F), and elevated levels of these non-coding RNAs in addition to Lhb mRNA (Fig S5).
Hmgb2 binding the enhancer iM directs transcription specifically to eRNA2
The above results pointed to the presence of a stable iM at the central untranscribed region of the enhancer. We considered that any function in regulating enhancer activity might be via protein recruitment, and looked to identify the binding proteins by precipitation of the cross-linked region, and analysis by mass spectrometry. Using a stringent cut-off, seven proteins (Table S7) were precipitated independently using two probes to distinct sites in the locus, and not precipitated in control cells which lacked the targeting gRNAs. Of these proteins, most notable was Hmgb2, which is known to bind single-stranded or structured DNA [57, 58], and was found previously enriched at G4-forming regions, though it did not bind the G4 in vitro [59].
To verify the Hmgb2 binding at this site, we performed ChIP in LβT2 cells. Hmgb2 was enriched at the center of the enhancer, and also at the 5’ end of the lncRNA and Lhb first intron (Fig. 4A). Although iMs were not detected at the latter loci (Fig. 3E), Hmgb2 binds structured and unstructured DNA frequently and though various mechanisms [60, 61]. We thus looked to examine further the connection between Hmgb2 and the enhancer iM. We performed Hmgb2 ChIP in cells cultured in a different buffer, at pH 7.2, or pH 6.3 where iMs are more stable (Fig. 3B [26]). Hmgb2 binding at the enhancer was nearly four-fold higher in cells cultured at the lower pH and much higher at this site than at the other locations examined where the Hmgb2 binding also appeared less stable in this different media (Fig. 4B).
These findings strongly suggested that Hmgb2 binds the enhancer iM, which we sought to confirm with fluorescence emission binding assays. In the presence of HMGB2, we detected a decrease in fluorescence as a function of increasing concentration of the single-stranded enhancer iM DNA (Fig. 4C). Under steady-state equilibrium conditions, hyperbolic dependence is observed (Fig S6), confirming the direct binding of HMGB2, with estimated binding affinity (Kd 12.86 × 10− 7) akin to that of HMGB1 binding G4 DNA in similar studies [41, 62]. Unlike the iM sequence, the Lhb complimentary G4 sequence showed very high levels of autofluorescence (Fig S7A-D), so is not suited to this kind of analysis, though HMGB2 was previously reported not to bind the G4 structure [59].
To establish the role of Hmgb2 in this context, we knocked down Hmgb2 expression, which reduced its mRNA levels by around 60% (Fig. 4D), and the enhancer iM signal in these cells dropped by ~ 40% (Fig. 4E). Strikingly, although Lhb mRNA, lncRNA and eRNA2 levels were all reduced quite dramatically in these cells, the level of eRNA1 increased significantly (Fig. 4D). Taken together, these results suggest that Hmgb2 binds and stabilizes the enhancer iM to promote transcription specifically in the direction of eRNA2.
Fig. 4.

Hmgb2 binding the enhancer iM directs transcription specifically to eRNA2. (A, B) ChIP for Hmgb2 was carried out in LβT2 cells. In (A) cells were cultured in standard media; and IP/input levels presented as mean±SEM, n = 3; ANOVA followed by Bonferroni t-test compared all means, and those that are similar (P>0.05) share the same letter. In (B) the cells were treated for 2.5 h with phosphate buffer at pH 7.2 or pH 6.3; t-test compared means at each locus in the different conditions, and annotations are as before; n = 4. (C) Fluorescence emission spectra of HMGB2 (1 µM) at 280 nm in the presence of Lhb enhancer iM DNA (0-18 µM; see also Fig S6, S7). (D,E) Hmgb2 was knocked down with shRNA, and (D) Hmgb2 and Lhb mRNA, lncRNA and eRNA levels measured in these (n = 4) and control WT cells (n = 3); whiskers show minimum and maximum values relative to mean levels in WT control cells, boxes 25–75% ranges, and internal horizontal lines indicate median; Student’s t-test compared values with those in WT controls, annotated as before; ***: P < 0.001 (E) IMab ChIP was carried out in these Hmgb2 KD cells (part of the same experiment as shown in Fig 3E), and data presented as before, with Znrf2 as control; n = 3
The enhancer and eRNA2 regulate the chromatin landscape
We next examined whether eRNA2 plays a role in regulating Lhb expression, and found that its knockdown (KD) by ~ 60% resulted in a 50% drop in Lhb mRNA levels, while eRNA1 was not affected (Fig. 5A). A similar drop was seen using a different sequence targeting eRNA2, while targeting eRNA1 did not have the same effect and even increased Lhb mRNA levels (Fig S8). ChIP revealed that the eRNA2 KD affected the chromatin landscape at the Lhb gene locus, leading to an elevation in H3 levels at sites normally depleted of nucleosomes [63] in the proximal and distal promoter, and start of the first intron (Fig. 5B). The repressive histone modification, H3K9me3, also increased across this entire region (Fig. 5C).
Fig. 5.

The eRNA2 promotes open chromatin. (A) LβT2 cells were stably transfected with shRNA targeting eRNA2 (sheRNA2). Levels of normalized RNA are shown compared to those in WT cells, presented and analysed as before; n = 4 (see also Fig S8). (B-E) ChIP for (B, D) H3 and (C, E) H3K9me3 was performed in (B, C) WT or sheRNA2 cells, or (D, E) dCas9-KRAB cells without (control) or with the gRNA (as in Fig. 1D). The IP levels are shown relative to input and normalized to the positive control, Atoh. Mean ± SEM, n = 3; statistical analysis as before
The ability of the enhancer to affect the chromatin at the proximal promoter was also seen following the targeting of dCas9-KRAB to the enhancer (as in Fig. 1D), which increased H3 levels at the proximal promoter and at two sites in the enhancer (Fig. 5D). Asides from an increase at the distal regions of the enhancer, H3K9me3 was also elevated at the Lhb TSS where its levels reached more than double those in control cells which lacked the site-specific targeting gRNAs (Fig. 5E). Thus, the enhancer, and specifically eRNA2, play a role in facilitating open chromatin at the Lhb gene.
Levels of the eRNAs and lncRNA are hormonally-regulated and correlate with Lhb mRNA in female but not male mice
We hypothesized that if eRNA2 exerts a regulatory effect on Lhb, their transcription should be coordinated. We thus examined the levels of these RNAs in the LβT2 gonadotropes following exposure to treatments known to affect Lhb expression. GnRH, given in different pulse regimes, elevated the levels of both eRNAs and the lncRNA, with eRNA2 consistently responding the most (Fig. 6A). The phorbol ester, PMA, which activates the protein kinase C pathway that mediates GnRH signaling [64], similarly increased the levels of both eRNAs, eRNA2 more markedly, while forskolin (activates protein kinase A, a minor GnRH-activated pathway) had less of an effect (Fig. 6B). We then exposed αT3-1 gonadotrope cells to estradiol (E2) or the androgen, dihydrotestosterone (DHT). The E2 repressed Lhb mRNA, eRNA2 and lncRNA levels significantly without affecting eRNA1, while DHT had no effect on any of these RNAs (Fig. 6C).
Fig. 6.

Levels of the eRNAs and lncRNA are differentially responsive to hormonal treatments and correlate with Lhb mRNA levels in female but not male mice. Lhb mRNA, eRNA and lncRNA levels in LβT2 cells after (A) GnRH (100 nM; 5 min pulses every 30’ or 2 h, or constant, for 8 h), n = 3; (B) PMA or forskolin (0.1 µM or 10 µM, both 4 h), n = 3; (C) estradiol (E2) or DHT (both 10 nM, 24 h), and Greb1 as control, n = 6–9. Normalized levels of the RNAs are shown relative to those in NT cells. Presentation and analysis as before. (D-F) RNA levels were measured in pituitaries of female and male mice of varying ages. (D) Mean levels in young (22 d) or adult (45 d) mice are shown in arbitrary units, mean ± SEM, n = 6–8. (E, F) Levels of the non-coding RNAs in mice (aged 16–61 d) are shown relative to levels of Lhb mRNA in the same samples; analyzed with Pearson’s correlation (see also Fig S9). (G) Female and (H) male mice, after sham-operation, (G) ovariectomy or (H) castration. Box plots and analysis are as before, n = 4. The correlations with Lhb in individual samples are shown for females (top lncRNA; middle eRNAs) and for males (bottom), analyzed as before
To determine the relevance of these hormonal effects seen in culture, to levels of the RNAs in vivo, we measured them in mice pituitaries. The mean levels prior to puberty (22 d) were similar between the sexes for all these RNAs, but in adults (45 d) they were significantly higher in males (Fig. 6D). In female mice across the lifespan (aged 16–61 d), levels of Lhb mRNA correlated best with those of the lncRNA, and significantly with both eRNAs (Fig. 6E, S9A). Strikingly, however, in pituitaries of male mice, none of these RNAs correlated positively with levels of Lhb mRNA, the lncRNA levels even showing a negative trend (Fig. 6F, S9B).
Subsequently, we removed the gonadal steroid feedback through ovariectomy or castration, reducing their negative regulation of the gonadotropes directly and via the hypothalamus. In ovariectomized females, the mean Lhb mRNA levels were nearly 4-fold those in sham-operated controls after 2 w, and eRNA2 levels also increased significantly, though those of eRNA1 were unchanged (Fig. 6G). The lncRNA levels were variable in these mice pituitaries, but their positive correlation with those of Lhb mRNA in the individual samples was very clear (R = 0.91), and those of eRNA2 with Lhb even more so (R = 0.97: Fig. 6G). In males, castration led similarly to an increase in Lhb mRNA and eRNA2 levels, albeit less than in the females, and levels of eRNA1 and the lncRNA were unaltered; in the individual samples, only eRNA2 levels were correlated with those of Lhb mRNA (R = 0.83, P = 0.011; Fig. 6H).
Splicing of the lncRNA in cis facilitates Lhb expression
The findings above suggested that the lncRNA might have a role in Lhb transcription, distinct from that of eRNA2. To determine whether this lncRNA affects Lhb expression directly, we over-expressed it in trans. Despite an increase in the levels of the spliced lncRNA by more than 400-fold, there was no change in Lhb mRNA levels (Fig. 7A). Considering that the lncRNA might affect Lhb only when transcribed in cis, we included the entire genomic region spanning the lncRNA- and the Lhb-encoding sequences, with or without the CMV promoter. In these cells, the spliced and edited Lhb mRNA levels reached ~ 23-fold those in controls, and the edited lncRNA levels increased ~ 800-fold (Fig. 7A).
Fig. 7.

Splicing of the lncRNA in cis is required for Lhb expression. (A) The lncRNA genomic fragment was inserted into a plasmid alone (CMV lncRNA) or upstream of the genomic fragment encompassing Lhb, without (lncRNA-Lhb) or with a CMV promoter (CMV lncRNA-Lhb). Levels of spliced lncRNA (on left) or spliced Lhb mRNA (on right) were measured; box plots and ANOVA with Bonferroni t-test as before; n = 3. (B) The same CMV-lncRNA-Lhb constructs, with or without mutations in the 6 U1 sites were transfected and levels of spliced lncRNA or Lhb mRNA were measured and presented similarly, with comparisons between the two groups by Student’s t-test; GFP-transfections served as control; n = 3
These results indicate that transcription or processing in situ of the lncRNA, rather than the resulting lncRNA itself, facilitates Lhb expression. To investigate this further, we transfected the same CMV-driven lncRNA-Lhb genomic DNA plasmid, intact as before, or after mutation of six U1 splice sites in the lncRNA. These U1 mutations reduced the amount of edited lncRNA to below 15% that in cells with the intact sequence, and levels of Lhb mRNA were reduced by more than half (Fig. 7B). Together, this reveals a role for splicing of the lncRNA in cis, which appears to play a role in regulating Lhb expression in female mice.
Discussion
We report here an upstream element that modulates Lhb expression through a novel mechanism involving an iM-containing bi-directional enhancer whose transcription is directed to the functional eRNA2 by Hmgb2. The enhancer activity, through this eRNA2, facilitates open chromatin at the Lhb gene, complemented by the splicing in situ of an adjacent but distinct lncRNA (Fig. 8). Activation and repression of this composite element by the reproductive regulatory hormones endows it with a role indicated to be female-specific, in accordance with the dynamic hormonal changes during the estrous cycle.
Fig. 8.

Working model of the organization of the Lhb enhancer locus and roles of eRNA2 and the lncRNA in Lhb expression. The Lhb enhancer locus comprises a bi-directional trancribed region with a stable iM at its center, bound by Hmgb2 which directs trancription to the more distal and functional eRNA2. This distal region is in physical contact with the Lhb gene, and eRNA2 promotes its open chromatin. Further downstream, the lncRNA is transcribed from a region with inducible iM/G4 structures at its promoter and/or first intron; the locus is also enriched with Hmgb2, all with likely roles in regulating lncRNA trancription through mechanims that are less clear. The splicing of this lncRNA in situ is required for optimal Lhb expression
The function of this eRNA2 is reminiscent of an eRNA that we characterized previously to be transcribed upstream of the Cga gene, and also plays a role in maintaining open chromatin at the target locus [1, 63]. As at the Cga enhancer, we saw no evidence for a function of the second eRNA, eRNA1. In fact, the Lhb enhancer eRNA1 is encoded from a sequence that overlaps with that encoding the lncRNA which is both detected at higher levels and regulated differently. This lncRNA has a unique role requiring its splicing and/or recruitment of the spliceosome to the locus for optimal Lhb expression, perhaps facilitating co-transcriptional Lhb splicing that might be particularly important during times of elevated Lhb expression. Like other lncRNAs [65, 66], this lncRNA is not spliced optimally, and similar activity towards a target gene through localized splicing has been reported in other contexts [7, 67]. Further studies in vivo will be needed to elucidate its role specifically during the estrous cycle. Remarkably, however, this lncRNA was recently reported to be expressed as a structural protein in the bovine sperm tail [68]. We found no evidence of a protein being produced from this sequence in the gonadotrope, supported by the fact that the lncRNA lacked a polyA tail, suggesting that flexible usage of the sequence has evolved to facilitate reproduction in very different ways along the reproductive axis. Notably, GnRH and its receptor are expressed in male germ cells, it is thus conceivable that GnRH might play a similar role in regulating expression of this newly-reported protein in sperm cells.
Although we had hypothesized that the G4/iM structure at the enhancer might be regulated by GnRH, this iM seemed very stable, endowed presumably by the DNA sequence as well as the binding of Hmgb2. Accordingly, previous studies have found HMGB1/2 enriched at G4/iM forming sequences [59, 62], and although HMGB2 did not bind G4 structures in vitro [59], HMGB1 was reported to stabilize G4s on the KRAS promoter where it represses transcription [62]. Notably, female mice lacking Hmgb2 suffer from reduced fertility [69–71], although this protein has diverse and broad functions in genomic organization and transcriptional regulation [57, 58, 60, 72]. Interestingly, HMGB1/2 proteins can be recruited to DNA through interaction with the estrogen receptor, ESR1, and are reported to increase ESR1 binding [73–76]. However, an estrogen response element (ERE) is not present at the enhancer iM site which is enriched with Hmgb2, and ESR1 was not found to bind the locus in other contexts (ChIP-Atlas.org). Moreover, the similar impact of Hmgb2 KD, and E2 treatment on the RNA levels suggests that these regulatory mechanisms are not coupled, unless, perhaps, E2 induced ESR1 inhibits the actions of HMGB2 in this context.
The stability of the enhancer iM contrasted with the structures detected at the 5’ end of the lncRNA that were induced by GnRH and TSA, treatments which also up-regulated lncRNA expression, suggesting possible mediating roles, although either treatment might facilitate transcription via elevating histone acetylation, which is increased globally by TSA and at specific genomic sites in gonadotropes after GnRH treatment [54, 77, 78]. Still, GnRH was reported to cause a biphasic change in gonadotrope pH [79], and an increase in pH in fish somatotropes which it also activates [80, 81]. These GnRH-induced changes in pH were short-lived, and G4 structures are not affected directly by pH, though it is possible that a transient increase in intracellular pH might favor the formation of G4s opposite unstable (and difficult to detect) iMs which are highly sensitive [26], and considered mutually exclusive at the same site [31, 32]. However, both structures are affected also by negative torsional stress behind transcribing RNAPII [82] and DNA binding proteins, and SP1-induced negative super helicity at the CMYC promoter was proposed to cause a switch between a repressive G4 and activating iM [32]. Various studies have reported that G4s repress transcription [32, 50], yet recent findings suggested that, at promoters, both iMs and G4s are largely associated with activating transcription [13, 21], G4s at genes with higher rates of transcription, as compared to lower rates at iM-associated genes [21]. GC-rich regions harbor binding sites for various TFs, and SP1 and AP-1 were seen to cooperate with G4s to direct cell-specific transcription [13]. In response to GnRH, the TF Egr1 binds the Lhb promoter at several sites, including CACCCCCAC [83, 84], which is also found in the sequence forming the enhancer iM, and similar sequences are located at the lncRNA TSS, suggesting another possible mechanism of GnRH-induced modification of these structures. Single-cell and single-molecule studies will be required to understand how exactly these less stable and dynamic structures are regulated, and their precise roles in expression of this lncRNA.
The striking sex-differences in activity of this enhancer element, pointing to a female-specific role in the control of Lhb, together the differential responses to gonadal steroids, suggest regulation arising from the intricate hormonal dynamics across the estrous cycle. Although GnRH was seen to stimulate eRNA2 and lncRNA expression, high levels of E2 would act directly on the gonadotrope to restrain their levels, providing tighter control on their activity as compared to males. In males, the gonadal negative feedback on GnRH occurs after testosterone aromatization to E2 in the brain; removal of this inhibition by castration would allow GnRH to up-regulate levels of eRNA2 which was the most responsive of these RNAs. However, aromatase is not expressed in pituitary gonadotropes. Thus, the effects of castration on the gonadotrope would not include removal of E2-mediated repression, explaining perhaps the different response to gonadectomy and also the higher levels in the intact adult males. The effects of E2 on this locus in females might be mediated via a site at the 3’ end of the lncRNA which, though lacking a consensus binding motif, was found to bind ESR1 in other contexts (in Re-Map [85]), and ESR1 is likely recruited also to the promoter via interactions with other transcription factors [86]. Additional work will be required to ascertain the molecular mechanisms through which E2 regulates the enhancer and lncRNA, and whether connected to the role of Hmgb2.
In conclusion, our study has revealed novel mechanisms regulating transcription via a composite enhancer which, while illustrated through study of Lhb, has wider implications particularly for other inducible genes. Further, although enhancers have traditionally been considered “location-independent” our study stresses the crucial importance of the genomic context for some of these elements, which needs to be considered in the study of their regulation and function. This is particularly relevant when looking at the therapeutic potential of targeting such elements, non-canonical DNA structures or lncRNAs, which holds much promise [14, 87–89], yet requires a thorough understanding of the mechanisms involved.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We thank Pamela Mellon for the gonadotrope cell lines, Daniel Christ for the iMab antibody, and Frederique Ruf-Zamojski for providing the snATAC-seq data files. Thanks also to Silvia Galli for advice on the G4-ChIP.
Author contributions
Conceptual design of the project was by Tal Refael, Lilach Pnueli and Philippa Melamed. Experiments were performed by Tal Refael and Lilach Pnueli, with help from Maya Sudman, Gil Golan, Sujay Naik and Arnon Henn. Funding was secured by Tal Refael, Philippa Melamed, Sujay Naik, Ella Preger-Ben Noon and Ariel Kaplan. The manuscript draft was written by Tal Refael and Philippa Melamed; all authors commented and provided suggestions, read and approved the final manuscript.
Funding
This study was supported by funding from the Israel Science Foundation (1850/17, 334/23 to PM, 712/18 to AH and 927/20 to AK), the United States-Israel Binational Science Foundation (BSF 2023086 to PM) and the Ministry of Science and Technology through a Levi Eshkol Doctoral Fellowship (TR). Additional funding was provided from the Russell Barrie Nanotechnology NEVET program (PM and AK), and Gellman-Lasser Fund (TR and SN).
Data availability
The data supporting the findings of this study are found within the article and its Supplementary Data. The mass-spectrometry data have been deposited into the PRIDE proteomics identification database.
Declarations
Ethical approval
The mice were held and handled humanely, following IACUC gudleines. All experimental protocols were according to the approvals which were granted by the Technion Ethics Committee.
Consent to publish
Not applicable.
Conflict of interest
There are no competing interests to declare.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Pnueli L, Rudnizky S, Yosefzon Y, Melamed P (2015) RNA transcribed from a distal enhancer is required for activating the chromatin at the promoter of the gonadotropin α-subunit gene. Proc Natl Acad Sci U S A 112:4369–4374. 10.1073/pnas.1414841112 10.1073/pnas.1414841112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Melamed P, Yosefzon Y, Rudnizky S, Pnueli L (2016) Transcriptional enhancers: transcription, function and flexibility. Transcription 7:26–31. 10.1080/21541264.2015.1128517 10.1080/21541264.2015.1128517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sabari BR, Dall’Agnese A, Boija A et al (2018) Coactivator condensation at super-enhancers links phase separation and gene control. Sci (80-) 361:eaar3958. 10.1126/science.aar3958 10.1126/science.aar3958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lam M, Li W, Rosenfeld M, Glass C (2014) Enhancer RNAs and regulated transcriptional programs. Trends Biochem Sci 39:170–182. 10.1016/J.TIBS.2014.02.007 10.1016/J.TIBS.2014.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Natoli G, Andrau JC (2012) Noncoding transcription at enhancers: General principles and functional models. Annu Rev Genet 46:1–19 10.1146/annurev-genet-110711-155459 [DOI] [PubMed] [Google Scholar]
- 6.Mattick JS, Amaral PP, Carninci P et al (2023) Long non-coding RNAs: definitions, functions, challenges and recommendations. Nat Rev Mol Cell Biol 24:430–447. 10.1038/s41580-022-00566-8 10.1038/s41580-022-00566-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gil N, Ulitsky I (2020) Regulation of gene expression by cis-acting long non-coding RNAs. Nat Rev Genet 21:102–117 10.1038/s41576-019-0184-5 [DOI] [PubMed] [Google Scholar]
- 8.Gil N, Perry RBT, Mukamel Z et al (2023) Complex regulation of eomes levels mediated through distinct functional features of the Meteor long non-coding RNA locus. Cell Rep 42. 10.1016/j.celrep.2023.112569 [DOI] [PMC free article] [PubMed]
- 9.Unfried JP, Ulitsky I (2022) Substoichiometric action of long noncoding RNAs. Nat Cell Biol 24:608–615 10.1038/s41556-022-00911-1 [DOI] [PubMed] [Google Scholar]
- 10.Gasperini M, Tome JM, Shendure J (2020) Towards a comprehensive catalogue of validated and target-linked human enhancers. Nat Rev Genet 21:292–310. 10.1038/s41576-019-0209-0 10.1038/s41576-019-0209-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Varshney D, Spiegel J, Zyner K et al (2020) The regulation and functions of DNA and RNA G-quadruplexes. Nat Rev Mol Cell Biol 21:459–474 10.1038/s41580-020-0236-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spiegel J, Adhikari S, Balasubramanian S (2020) The structure and function of DNA G-Quadruplexes. Trends Chem 2:123–136 10.1016/j.trechm.2019.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lago S, Nadai M, Cernilogar FM et al (2021) Promoter G-quadruplexes and transcription factors cooperate to shape the cell type-specific transcriptome. Nat Commun 12. 10.1038/s41467-021-24198-2 [DOI] [PMC free article] [PubMed]
- 14.Hänsel-Hertsch R, Di Antonio M, Balasubramanian S (2017) DNA G-quadruplexes in the human genome: detection, functions and therapeutic potential. Nat Rev Mol Cell Biol 18:279–284. 10.1038/nrm.2017.3 10.1038/nrm.2017.3 [DOI] [PubMed] [Google Scholar]
- 15.Carvalho J, Mergny JL, Salgado GF et al (2020) G-quadruplex, friend or foe: the role of the G-quartet in anticancer strategies. Trends Mol Med 26:848–861 10.1016/j.molmed.2020.05.002 [DOI] [PubMed] [Google Scholar]
- 16.Robinson J, Raguseo F, Nuccio SP et al (2021) DNA G-quadruplex structures: more than simple roadblocks to transcription? Nucleic Acids Res 49:8419–8431. 10.1093/nar/gkab609 10.1093/nar/gkab609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maizels N (2015) G4-associated human diseases. EMBO Rep 16:910–922. 10.15252/embr.201540607 10.15252/embr.201540607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duardo RC, Guerra F, Pepe S, Capranico G (2023) Non-B DNA structures as a booster of genome instability. Biochimie 214:176–192 10.1016/j.biochi.2023.07.002 [DOI] [PubMed] [Google Scholar]
- 19.Spiegel J, Cuesta SM, Adhikari S et al (2021) G-quadruplexes are transcription factor binding hubs in human chromatin. Genome Biol. 10.1186/s13059-021-02324-z. 22: 10.1186/s13059-021-02324-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mao SQ, Ghanbarian AT, Spiegel J et al (2018) DNA G-quadruplex structures mold the DNA methylome. Nat Struct Mol Biol 25:951–957. 10.1038/s41594-018-0131-8 10.1038/s41594-018-0131-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zanin I, Ruggiero E, Nicoletto G et al (2023) Genome-wide mapping of i-motifs reveals their association with transcription regulation in live human cells. Nucleic Acids Res 51:8309–8321. 10.1093/nar/gkad626 10.1093/nar/gkad626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hänsel-Hertsch R, Beraldi D, Lensing SV et al (2016) G-quadruplex structures mark human regulatory chromatin. Nat Genet 48:1267–1272. 10.1038/ng.3662 10.1038/ng.3662 [DOI] [PubMed] [Google Scholar]
- 23.Shen J, Varshney D, Simeone A et al (2021) Promoter G-quadruplex folding precedes transcription and is controlled by chromatin. Genome Biol 22. 10.1186/s13059-021-02346-7 [DOI] [PMC free article] [PubMed]
- 24.Williams JD, Houserova D, Johnson BR et al (2020) Characterization of long G4-rich enhancer-associated genomic regions engaging in a novel loop:loop G4 kissing interaction. Nucleic Acids Res 48:5907–5925. 10.1093/NAR/GKAA357 10.1093/NAR/GKAA357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liano D, Monti L, Chowdhury S et al (2022) Long-range DNA interactions: inter-molecular G-quadruplexes and their potential biological relevance. Chem Commun 204. 10.1039/d2cc04872h [DOI] [PMC free article] [PubMed]
- 26.Zeraati M, Langley DB, Schofield P et al (2018) I-motif DNA structures are formed in the nuclei of human cells. Nat Chem 10:631–637. 10.1038/s41557-018-0046-3 10.1038/s41557-018-0046-3 [DOI] [PubMed] [Google Scholar]
- 27.Assi HA, Garavís M, González C, Damha MJ (2018) I-motif DNA: structural features and significance to cell biology. Nucleic Acids Res 46:8038–8056 10.1093/nar/gky735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abdelhamid MAS, Gates AJ, Waller ZAE (2019) Destabilization of i-Motif DNA at Neutral pH by G-Quadruplex ligands. Biochemistry 58:245–249. 10.1021/acs.biochem.8b00968 10.1021/acs.biochem.8b00968 [DOI] [PubMed] [Google Scholar]
- 29.Wright EP, Abdelhamid MAS, Ehiabor MO et al (2020) Epigenetic modification of cytosines fine tunes the stability of i-motif DNA. Nucleic Acids Res 48:55–62. 10.1093/nar/gkz1082 10.1093/nar/gkz1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rajendran A, Nakano SI, Sugimoto N (2010) Molecular crowding of the cosolutes induces an intramolecular i-motif structure of triplet repeat DNA oligomers at neutral pH. Chem Commun (Camb) 46:1299–1301. 10.1039/B922050J 10.1039/B922050J [DOI] [PubMed] [Google Scholar]
- 31.King JJ, Irving KL, Evans CW et al (2020) DNA G-Quadruplex and i-Motif structure formation is interdependent in human cells. J Am Chem Soc 142:20600–20604 10.1021/jacs.0c11708 [DOI] [PubMed] [Google Scholar]
- 32.Sutherland C, Cui Y, Mao H, Hurley L (2016) A Mechanosensor mechanism controls the G-Quadruplex/i-Motif molecular switch in the MYC promoter NHE III 1. J Am Chem Soc 138:14138–14151. 10.1021/JACS.6B09196 10.1021/JACS.6B09196 [DOI] [PubMed] [Google Scholar]
- 33.Dhakal S, Yu Z, Konik R et al (2012) G-quadruplex and i-motif are mutually exclusive in ILPR double-stranded DNA. Biophys J 102:2575–2584. 10.1016/j.bpj.2012.04.024 10.1016/j.bpj.2012.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luo Z, Wijeweera A, Oh Y et al (2010) Pin1 facilitates the phosphorylation-dependent ubiquitination of SF-1 to regulate Gonadotropin β-Subunit gene transcription. Mol Cell Biol 30:745–763. 10.1128/mcb.00807-09 10.1128/mcb.00807-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pnueli L, Shalev D, Refael R et al (2021) Proliferating primary pituitary cells as a model for studying regulation of gonadotrope chromatin and gene expression. Mol Cell Endocrinol 533:111349. 10.1016/J.MCE.2021.111349 10.1016/J.MCE.2021.111349 [DOI] [PubMed] [Google Scholar]
- 36.Yosefzon Y, David C, Tsukerman A et al (2017) An epigenetic switch repressing Tet1 in gonadotropes activates the reproductive axis. Proc Natl Acad Sci U S A 114:10131–10136. 10.1073/pnas.1704393114 10.1073/pnas.1704393114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Galli S, Flint G, Růžičková L, Di Antonio M (2024) Genome-wide mapping of G-quadruplex DNA: a step-by-step guide to select the most effective method. RSC Chem Biol 5:426–438 10.1039/D4CB00023D [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wulfridge P, Yan Q, Rell N et al (2023) G-quadruplexes associated with R-loops promote CTCF binding. Mol Cell 83:3064–3079e5. 10.1016/j.molcel.2023.07.009 10.1016/j.molcel.2023.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brázda V, Kolomazník J, Lýsek J et al (2019) G4Hunter web application: a web server for G-quadruplex prediction. Bioinformatics 35:3493–3495. 10.1093/bioinformatics/btz087 10.1093/bioinformatics/btz087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hon J, Martínek T, Zendulka J, Lexa M (2017) Pqsfinder: an exhaustive and imperfection-tolerant search tool for potential quadruplex-forming sequences in R. Bioinformatics 33:3373–3379. 10.1093/bioinformatics/btx413 10.1093/bioinformatics/btx413 [DOI] [PubMed] [Google Scholar]
- 41.Amato J, Cerofolini L, Brancaccio D et al (2019) Insights into telomeric G-quadruplex DNA recognition by HMGB1 protein. Nucleic Acids Res 47:9950–9966. 10.1093/nar/gkz727 10.1093/nar/gkz727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Giancola C, Pagano B (2012) Energetics of Ligand Binding to G-Quadruplexes. In: Topics in Current Chemistry. Top Curr Chem, pp 211–242 [DOI] [PubMed]
- 43.Schwartzman O, Mukamel Z, Oded-Elkayam N et al (2016) UMI-4 C for quantitative and targeted chromosomal contact profiling. Nat Methods 13:685–691. 10.1038/NMETH.3922 10.1038/NMETH.3922 [DOI] [PubMed] [Google Scholar]
- 44.Fujita T, Fujii H (2013) Efficient isolation of specific genomic regions and identification of associated proteins by engineered DNA-binding molecule-mediated chromatin immunoprecipitation (enChIP) using CRISPR. Biochem Biophys Res Commun 439:132–136. 10.1016/J.BBRC.2013.08.013 10.1016/J.BBRC.2013.08.013 [DOI] [PubMed] [Google Scholar]
- 45.Ruf-Zamojski F, Zhang Z, Zamojski M et al (2021) Single nucleus multi-omics regulatory landscape of the murine pituitary. Nat Commun 12:2677. 10.1038/s41467-021-22859-w 10.1038/s41467-021-22859-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao J, Ohsumi TK, Kung JT et al (2010) Genome-wide identification of polycomb-Associated RNAs by RIP-seq. Mol Cell 40:939–953. 10.1016/j.molcel.2010.12.011 10.1016/j.molcel.2010.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carninci P, Shibata Y, Hayatsu N et al (2000) Normalization and subtraction of cap-trapper-selected cDNAs to prepare full-length cDNA libraries for rapid discovery of new genes. Genome Res 10:1617–1630. 10.1101/GR.145100 10.1101/GR.145100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schirman-Hildesheim TD, Gershon E, Litichever N et al (2008) Local production of the gonadotropic hormones in the rat ovary. Mol Cell Endocrinol 282:32–38. 10.1016/j.mce.2007.11.014 10.1016/j.mce.2007.11.014 [DOI] [PubMed] [Google Scholar]
- 49.Zhang FP, Rannikko A, Huhtaniemi I (1995) Isolation and characterization of testis-specific cDNAs for luteinizing hormone β-subunit in the rat. Biochem Biophys Res Commun 210:858–865. 10.1006/bbrc.1995.1737 10.1006/bbrc.1995.1737 [DOI] [PubMed] [Google Scholar]
- 50.Siddiqui-Jain A, Grand C, Bearss D, Hurley L (2002) Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc Natl Acad Sci U S A 99:11593–11598. 10.1073/PNAS.182256799 10.1073/PNAS.182256799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vorlíčková M, Kejnovská I, Bednářová K et al (2012) Circular dichroism spectroscopy of DNA: from duplexes to quadruplexes. In: Chirality, Chirality, pp 691–698 [DOI] [PubMed] [Google Scholar]
- 52.Marsico G, Chambers VS, Sahakyan AB et al (2019) Whole genome experimental maps of DNA G-quadruplexes in multiple species. Nucleic Acids Res 47:3862–3874 10.1093/nar/gkz179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Biffi G, Tannahill D, McCafferty J, Balasubramanian S (2013) Quantitative visualization of DNA G-quadruplex structures in human cells. Nat Chem 5:182–186. 10.1038/nchem.1548 10.1038/nchem.1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lim S, Luo M, Koh M et al (2007) Distinct mechanisms involving diverse histone deacetylases repress expression of the two gonadotropin beta-subunit genes in immature gonadotropes, and their actions are overcome by gonadotropin-releasing hormone. Mol Cell Biol 27:4105–4120. 10.1128/MCB.00248-07 10.1128/MCB.00248-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Melamed P (2008) Histone deacetylases and repression of the gonadotropin genes. Trends Endocrinol Metab 19:25–31. 10.1016/j.tem.2007.10.008 10.1016/j.tem.2007.10.008 [DOI] [PubMed] [Google Scholar]
- 56.McBrian M, Behbahan I, Ferrari R et al (2013) Histone acetylation regulates intracellular pH. Mol Cell 49:310–321. 10.1016/J.MOLCEL.2012.10.025 10.1016/J.MOLCEL.2012.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Murugesapillai D, McCauley M, Maher L, Williams M (2016) Single-molecule studies of high-mobility group B architectural DNA bending proteins. Biophys Rev 9:17–40. 10.1007/S12551-016-0236-4 10.1007/S12551-016-0236-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stros M (2010) HMGB proteins: interactions with DNA and chromatin. Biochim Biophys Acta 1799:101–113. 10.1016/J.BBAGRM.2009.09.008 10.1016/J.BBAGRM.2009.09.008 [DOI] [PubMed] [Google Scholar]
- 59.Zhang X, Spiegel J, Martínez Cuesta S et al (2021) Chemical profiling of DNA G-quadruplex-interacting proteins in live cells. Nat Chem 13:626. 10.1038/S41557-021-00736-9 10.1038/S41557-021-00736-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thomas J (2001) HMG1 and 2: architectural DNA-binding proteins. Biochem Soc Trans 29:395–401. 10.1042/BST0290395 10.1042/BST0290395 [DOI] [PubMed] [Google Scholar]
- 61.Voong CK, Goodrich JA, Kugel JF (2021) Interactions of hmgb proteins with the genome and the impact on disease. Biomolecules 11 [DOI] [PMC free article] [PubMed]
- 62.Amato J, Madanayake TW, Iaccarino N et al (2018) HMGB1 binds to the KRAS promoter G-quadruplex: a new player in oncogene transcriptional regulation? Chem Commun 54:9442–9445. 10.1039/c8cc03614d 10.1039/c8cc03614d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rudnizky S, Bavly A, Malik O et al (2016) H2A.Z controls the stability and mobility of nucleosomes to regulate expression of the LH genes. Nat Commun 7:12958. 10.1038/ncomms12958 10.1038/ncomms12958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Naor Z, Harris D, Shacham S (1998) Mechanism of GnRH receptor signaling: combinatorial cross-talk of CA2 + and protein kinase C. Front Neuroendocrinol 19:1–19. 10.1006/frne.1997.0162 10.1006/frne.1997.0162 [DOI] [PubMed] [Google Scholar]
- 65.Melé M, Mattioli K, Mallard W et al (2017) Chromatin environment, transcriptional regulation, and splicing distinguish lincRNAs and mRNAs. Genome Res 27:27–37. 10.1101/gr.214205.116 10.1101/gr.214205.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tilgner H, Knowles DG, Johnson R et al (2012) Deep sequencing of subcellular RNA fractions shows splicing to be predominantly co-transcriptional in the human genome but inefficient for lncRNAs. Genome Res 22:1616–1625. 10.1101/gr.134445.111 10.1101/gr.134445.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gil N, Ulitsky I (2018) Production of spliced long noncoding RNAs specifies regions with increased enhancer activity. Cell Syst 7:537–547. .e3 10.1016/j.cels.2018.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Leung MR, Zeng J, Wang X et al (2023) Structural specializations of the sperm tail. Cell 186:2880–2896. .e17 10.1016/j.cell.2023.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ronfani L, Ferraguti M, Croci L et al (2001) Reduced fertility and spermatogenesis defects in mice lacking chromosomal protein Hmgb2. Development 128:1265–1273 10.1242/dev.128.8.1265 [DOI] [PubMed] [Google Scholar]
- 70.Yamaguma Y, Sugita N, Choijookhuu N et al (2022) Crucial role of high-mobility group box 2 in mouse ovarian follicular development through estrogen receptor beta. Histochem Cell Biol 157:359–369. 10.1007/S00418-022-02074-4 10.1007/S00418-022-02074-4 [DOI] [PubMed] [Google Scholar]
- 71.Shirouzu S, Sugita N, Choijookhuu N et al (2022) Pivotal role of High-Mobility Group Box 2 in ovarian folliculogenesis and fertility. J Ovarian Res 15. 10.1186/S13048-022-01071-4 [DOI] [PMC free article] [PubMed]
- 72.Zirkel A, Nikolic M, Sofiadis K et al (2018) HMGB2 loss upon Senescence Entry disrupts genomic Organization and induces CTCF clustering across cell types. Mol Cell 70:730–744e6. 10.1016/j.molcel.2018.03.030 10.1016/j.molcel.2018.03.030 [DOI] [PubMed] [Google Scholar]
- 73.Boonyaratanakornkit V, Melvin V, Prendergast P et al (1998) High-mobility group chromatin proteins 1 and 2 functionally interact with steroid hormone receptors to enhance their DNA binding in vitro and transcriptional activity in mammalian cells. Mol Cell Biol 18:4471–4487. 10.1128/MCB.18.8.4471 10.1128/MCB.18.8.4471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Das D, Peterson RC, Scovell WM (2004) High mobility group B proteins facilitate strong estrogen receptor binding to classical and half-site estrogen response elements and relax binding selectivity. Mol Endocrinol 18:2616–2632. 10.1210/ME.2004-0125 10.1210/ME.2004-0125 [DOI] [PubMed] [Google Scholar]
- 75.Melvin VS, Harrell C, Adelman JS et al (2004) The role of the C-terminal extension (CTE) of the estrogen receptor α and β DNA binding domain in DNA binding and Interaction with HMGB. J Biol Chem 279:14763–14771. 10.1074/jbc.M313335200 10.1074/jbc.M313335200 [DOI] [PubMed] [Google Scholar]
- 76.Melvin VS, Roemer SC, Churchill MEA, Edwards DP (2002) The C-terminal extension (CTE) of the nuclear hormone receptor DNA binding domain determines interactions and functional response to the HMGB-1/-2 co-regulatory proteins. J Biol Chem 277:25115–25124. 10.1074/jbc.M110400200 10.1074/jbc.M110400200 [DOI] [PubMed] [Google Scholar]
- 77.Haj M, Wijeweera A, Rudnizky S et al (2017) Mitogen- and stress-activated protein kinase 1 is required for gonadotropin-releasing hormone-mediated activation of gonadotropin α-subunit expression. J Biol Chem 292:20720–20731. 10.1074/jbc.M117.797845 10.1074/jbc.M117.797845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bohaczuk SC, Thackray VG, Shen J et al (2021) FSHB Transcription is regulated by a Novel 5′ distal enhancer with a fertility-Associated single nucleotide polymorphism. Endocrinol (United States) 162. 10.1210/endocr/bqaa181 [DOI] [PMC free article] [PubMed]
- 79.Bouali-Benazzouz R, Mariot P, Audy MC et al (1993) Gonadotropin-releasing hormone-induced changes of intracellular pH in pituitary gonadotrophs: influence of estradiol. Endocrinology 132:855–861. 10.1210/endo.132.2.8381077 10.1210/endo.132.2.8381077 [DOI] [PubMed] [Google Scholar]
- 80.Melamed P, Rosenfeld H, Elizur A, Yaron Z (1998) Endocrine regulation of gonadotropin and growth hormone gene transcription in fish. Comp Biochem Physiol C Pharmacol Toxicol Endocrinol 119:325–338. 10.1016/S0742-8413(98)00021-8 10.1016/S0742-8413(98)00021-8 [DOI] [PubMed] [Google Scholar]
- 81.Li GL, Parks SK, Goss GG, Chang JP (2010) PKC mediates GnRH activation of a Na+/H + exchanger in goldfish somatotropes. Gen Comp Endocrinol 166:296–306. 10.1016/J.YGCEN.2009.10.002 10.1016/J.YGCEN.2009.10.002 [DOI] [PubMed] [Google Scholar]
- 82.Selvam S, Mandal S, Mao H (2017) Quantification of Chemical and Mechanical effects on the formation of the G-Quadruplex and i-Motif in duplex DNA. Biochemistry 56:4616–4625. 10.1021/acs.biochem.7b00279 10.1021/acs.biochem.7b00279 [DOI] [PubMed] [Google Scholar]
- 83.Rudnizky S, Khamis H, Malik O et al (2018) Single-molecule DNA unzipping reveals asymmetric modulation of a transcription factor by its binding site sequence and context. Nucleic Acids Res 46:1513–1524. 10.1093/nar/gkx1252 10.1093/nar/gkx1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tremblay JJ, Drouin J (1999) Egr-1 is a downstream Effector of GnRH and synergizes by Direct Interaction with Ptx1 and SF-1 to enhance luteinizing hormone β gene transcription. Mol Cell Biol 19:2567–2576. 10.1128/mcb.19.4.2567 10.1128/mcb.19.4.2567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hammal F, De Langen P, Bergon A et al (2022) ReMap 2022: a database of Human, Mouse, Drosophila and Arabidopsis regulatory regions from an integrative analysis of DNA-binding sequencing experiments. Nucleic Acids Res 50:D316–D325. 10.1093/nar/gkab996 10.1093/nar/gkab996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Luo M, Koh M, Feng J et al (2005) Cross Talk in Hormonally regulated gene transcription through induction of Estrogen Receptor Ubiquitylation. Mol Cell Biol 25:7386–7398. 10.1128/mcb.25.16.7386-7398.2005 10.1128/mcb.25.16.7386-7398.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xu H, Di Antonio M, McKinney S et al (2017) CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat Commun 8. 10.1038/ncomms14432 [DOI] [PMC free article] [PubMed]
- 88.Winkle M, El-Daly SM, Fabbri M, Calin GA (2021) Noncoding RNA therapeutics — challenges and potential solutions. Nat Rev Drug Discov 2021 208 20:629–651. 10.1038/s41573-021-00219-z 10.1038/s41573-021-00219-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li Y, Tan Z, Zhang Y et al (2021) A noncoding RNA modulator potentiates phenylalanine metabolism in mice. Sci (80-) 373:662–673. 10.1126/science.aba4991 10.1126/science.aba4991 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of this study are found within the article and its Supplementary Data. The mass-spectrometry data have been deposited into the PRIDE proteomics identification database.