

Abstract
Huntington’s disease (HD) is a neurodegenerative genetic disorder caused by an expansion in the CAG repeat tract of the huntingtin (HTT) gene resulting in behavioural, cognitive, and motor defects. Current knowledge of disease pathogenesis remains incomplete, and no disease course-modifying interventions are in clinical use. We have previously reported the development and characterisation of the OVT73 transgenic sheep model of HD. The 73 polyglutamine repeat is somatically stable and therefore likely captures a prodromal phase of the disease with an absence of motor symptomatology even at 5-years of age and no detectable striatal cell loss. To better understand the disease-initiating events we have undertaken a single nuclei transcriptome study of the striatum of an extensively studied cohort of 5-year-old OVT73 HD sheep and age matched wild-type controls. We have identified transcriptional upregulation of genes encoding N-methyl-D-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and kainate receptors in medium spiny neurons, the cell type preferentially lost early in HD. Further, we observed an upregulation of astrocytic glutamate uptake transporters and medium spiny neuron GABAA receptors, which may maintain glutamate homeostasis. Taken together, these observations support the glutamate excitotoxicity hypothesis as an early neurodegeneration cascade-initiating process but the threshold of toxicity may be regulated by several protective mechanisms. Addressing this biochemical defect early may prevent neuronal loss and avoid the more complex secondary consequences precipitated by cell death.
Keywords: Huntington's disease, single nuclei RNA-seq, prodromal, glutamate excitotoxicity
Introduction
Huntington’s disease (HD) is a debilitating neurodegenerative genetic disorder caused by an expanded polyglutamine-encoding CAG repeat in the huntingtin gene (HTT) [1]. Individuals with 40 or more CAG codons develop the condition with near complete penetrance [2]. Widespread loss of medium spiny neurons (MSN) in the caudate nucleus and putamen (striatum) contributes to the presenting symptoms that encompass motor, cognitive and behavioural abnormalities. Despite characterisation of the HD defect and advancements in our understanding of disease pathogenesis, no proposed disease-modifying interventions have proved successful in HD clinical trials [3].
Glutamate excitotoxicity as an initiator of neuronal death has been a longstanding hypothesis and research focus in neurodegenerative disorders [4–7]. In HD, it has been proposed that the loss of the MSNs of the striatum is due to elevated synaptic glutamate levels from reduced glutamate clearance and increased glutamate release. This excess synaptic glutamate has been proposed to cause an overactivation of ionotropic glutamate receptors including N-methyl-d-aspartate (NMDA) receptors, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, and kainate receptors, leading to an influx of calcium ions, chronic membrane depolarisation, oxidative stress, and activation of cell death pathways [4–8]. Evidence for acute ionotropic glutamate receptor-mediated excitotoxicity was shown through the experimental striatal injection of glutamate receptor agonists (glutamic acid, kainic acid, quinolinic acid) in rodents and non-human primates resulting in MSN degeneration and motor dysfunction [9–15]. Further, studies investigating several HD mouse models provided evidence for an association between excessive NMDA receptor signalling and striatal degeneration [16–18].
Our group has previously generated a transgenic ovine model of HD (named OVT73) that expresses the full-length human huntingtin (HTT) cDNA with a pure CAG repeat length of 69 codons along with a short CAA CAG tract, resulting in a polyglutamine tract of 73 units. The HTT cDNA is under the control of the 1.1 kb segment of the immediate upstream human HTT genomic sequence [19]. The transcript expression level from the transgene in the OVT73 animals is estimated to be about ~10% to 20% of a single normal allele equivalent seen in HdhQ80, YAC128 and rat BACHD HD models [20]. The germline transmission of the 73-unit glutamine coding repeat was stable over three generations [20] and somatic instability was observed to be minimal [21]. The moderate expression of the transgene combined with a somatically stable polyglutamine coding repeat might position this model for the investigation of the initiating stages of HD (prodromal phase). Animals at 6 years of age do not exhibit any striatal cell loss and there are animals that are over 10 years of age not showing any overt symptoms [20, 22–25]. Several molecular and behavioural changes comparable to early phase HD were observed in the OVT73 line, including the formation of intracellular HTT aggregates [20], circadian rhythm abnormalities [24, 25] and changes in brain, plasma and liver metabolites [22, 23, 26, 27]. We have previously reported increased urea levels in the OVT73 striatum [27] also found on post-mortem throughout HD patients brains including the striatum. The human urea results do not appear to be an end stage cell loss associated phenomena as it is also found in HD post-mortem brains with low grade neuropathology and lower levels of cell loss (Vonsattel grade 0/1) [28]. Increased urea and ammonia levels are known to cause neurological impairment and have been suggested to exacerbate neuronal excitotoxicity [29–33].
To gain a better understanding of the disease mechanism in a prodromal HD model, we have undertaken RNA-seq of single nuclei from the OVT73 striatum. The tissue utilised for this study was obtained immediately adjacent to that used for our previously reported bulk RNA-seq analysis [27], taken from an extensively investigated cohort of 5-year-old animals [20, 22–25]. Multidimensional data from these animals are also publicly available in the form of a queryable database [34]. Surprisingly, we have detected transcriptomic signatures for a process that indicates the upregulation of glutamate signalling occurring in the MSNs of the OVT73 striatum. Since our HD sheep do not display striatal cell loss or overt motor symptoms, we propose that the response to glutamate in these animals is at a “steady state” but may be the prelude to excitotoxicity mediated neurodegeneration. These results suggest that there is a treatment window when the biochemical dysfunction could be addressed before the cascade of cell death. These observations position the OVT73 HD sheep model as a valuable resource testing of therapeutics in the prodromal phase and not at a disease stage confounded by the cell death cascade.
Results
Upregulation of genes involved in synaptic transmission in OVT73 medium spiny neurons
Following quality control, a total of 28 234 nuclei were recovered from the striatum of 6 OVT73 sheep and 6 age matched wild-type controls. A total of 13 cell types were identified including 12 507 oligodendrocytes (expressing marker genes MOG and PLP1), 6093 MSNs (RGS9, PDE10A), 3024 oligodendrocyte precursor cells (PCDH15, PDGFRA), 2601 microglia (CSF1R, CX3CR1), 2181 astrocytes (AQP4, GFAP), 1068 neuroblasts (DCX¸ ADARB2), 683 interneurons (ELAVL2, CLSTN2) and 77 endothelial cells (FLT, MECOM). Interneurons were subcategorised based on markers defined in Munoz-Manchado et al., [35] into 526 PV/Th interneurons (HS3ST2, PTHLH), 107 SST/NPY interneurons (SST, NPY) and 50 cholinergic interneurons (CHAT). MSNs were subcategorised according to cell markers defined in Saunders et al., [36] into 2776 D1 MSNs (TAC1, EBF1), 2998 D2 MSNs (DRD2, PENK) and 319 eccentric MSNs (OTOF, FOXP2). Full cell marker lists are provided in Supplementary File 2, and cell type distributions shown in Fig. 1C. Across all cell types, we observed a significant reduction in the proportion of oligodendrocytes in the OVT73 derived tissue compared with control cases (ANOVA, P = 1.24×10−5).
Figure 1.
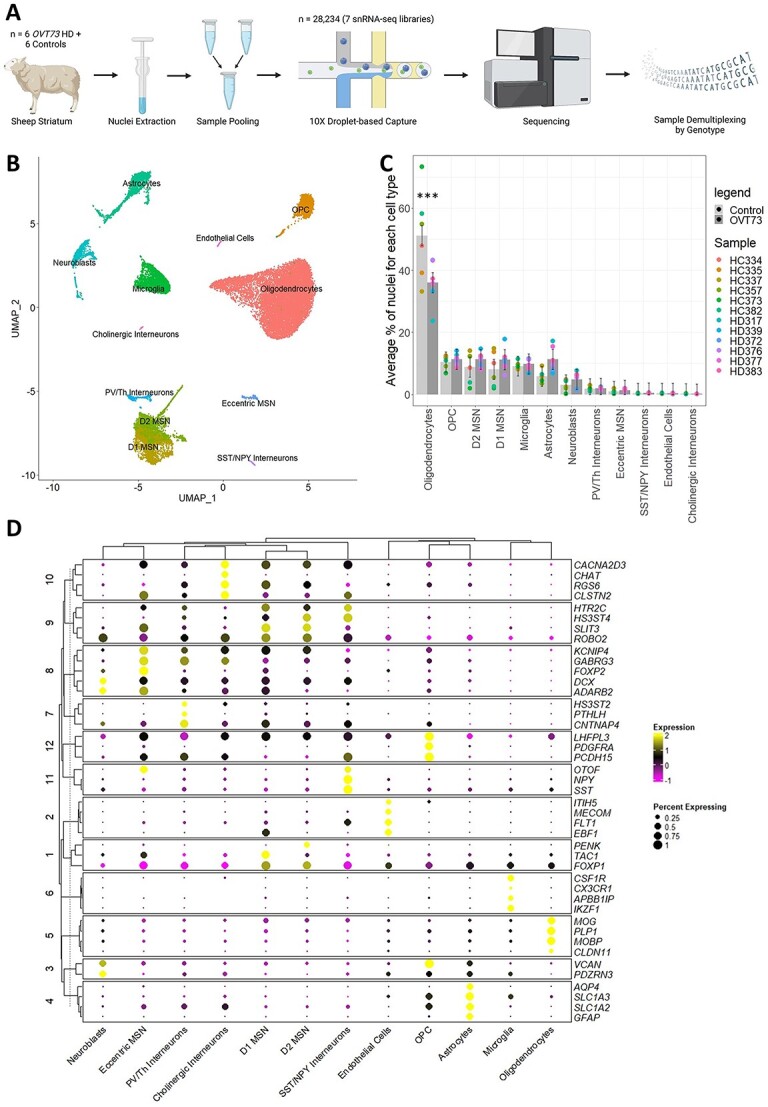
Single nuclei RNA-seq of the OVT73 sheep striatum. (A) Experimental design. Nuclei were extracted from the striatum of 12 sheep (6 OVT73 and 6 controls) and pooled to form 7 nuclei suspensions. Single nuclei RNA libraries were generated from the sample multiplexed suspensions and sequenced. Reads were demultiplexed based on the natural genetic variation between pooled samples. Panel image created with BioRender.com. (B) UMAP visualisation of clusters identified and annotated by cell marker gene expression. (C) Proportions of identified cell types across the 12 animals. A significant decrease in oligodendrocytes between OVT73 and control cases was observed (***ANOVA, P < 0.001) (D) dot plot of selected cell-type enriched markers for annotated cell types in the sheep striatum.
To investigate differential gene regulation in the OVT73 HD striatum, differential expression analyses were conducted between OVT73 and controls for each cell type separately. The ratio of differentially expressed genes (DEGs) identified to the median number of expressed genes per nucleus was 1.75 in oligodendrocytes (1753 DEGs/1002 median number of expressed genes of oligodendrocyte nuclei), 0.41 in OPCs (742/1812), 0.98 in D2 MSNs (4444/4516), 0.47 in D1 MSNs (2262/4850), 0.01 in eccentric MSNs (45/4337), 0.21 in microglia (217/1049), 0.05 in neuroblasts (47/895), 0.18 in astrocytes (303/1687) and 0.01 in PV/Th interneurons (33/4398) (Fig. 2B). No DEGs were detected in endothelial cells, cholinergic interneurons, and SST/NPY interneurons, likely due to their small population sizes. The statistical power to detect differences in gene expression is dependent on the number of cells, consistent with the highest proportion of DEGs being observed in oligodendrocytes, with the highest number of cells. When comparing the proportion of DEG’s in D1 MSNs, D2 MSNs, OPC, microglia, astrocytes and neuroblasts all represented in the data set by similar nuclei numbers, a greater proportion of DEGs was observed in D2 MSNs. The full list of DEGs is available in Supplementary File 3 and volcano plots for individual cell type DEG analysis are available in Supplementary Fig. 5.
Figure 2.
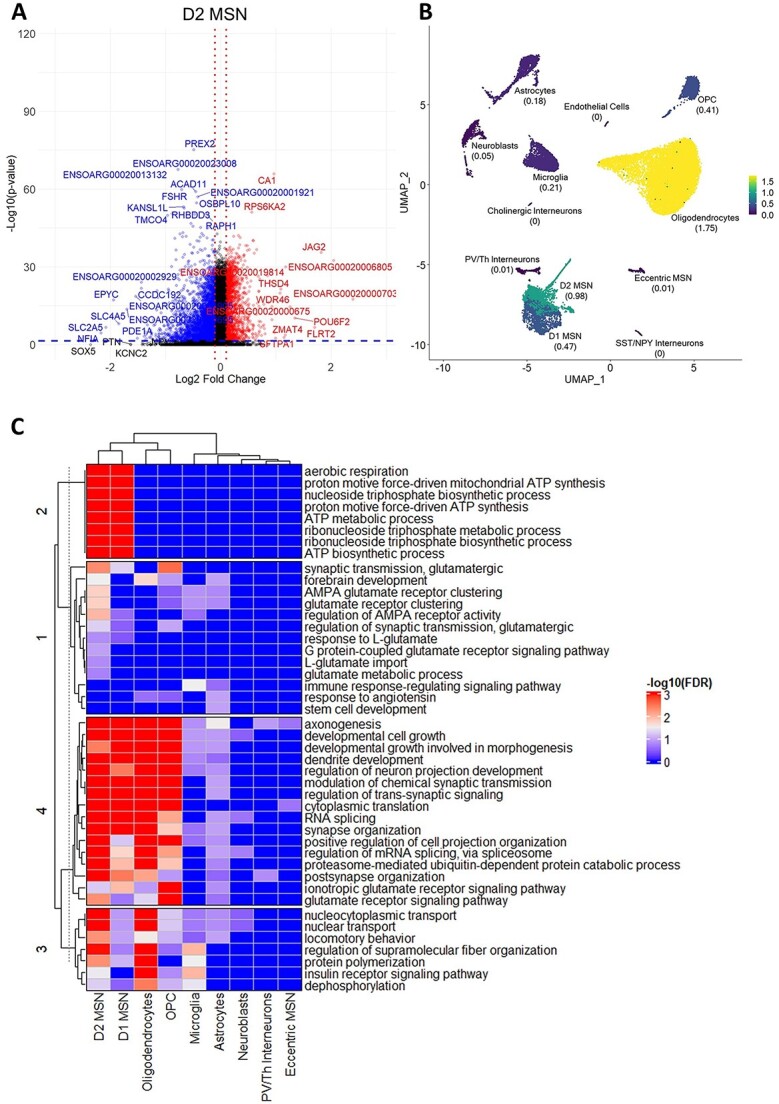
Differentially expressed genes in OVT73 versus control sheep striatum. (A) Volcano plot of differentially expressed genes (DEGs) identified in D2 medium spiny neurons (OVT73 D2 MSN vs control D2 MSN). Horizontal blue line shown at P = 0.05, vertical red lines shown at log2 fold change of −0.1 and 0.1. (B) Proportion of DEGs over the median number of expressed genes in each cell type. (C) Heatmap of most significant gene ontology terms (ordered by lowest FDR adjusted p-values) in cell types.
The top enriched Gene Ontology (GO) terms (ordered by FDR adjusted p-values) associated with DEGs included synapse assembly and organisation, GABAergic and glutamatergic synaptic transmission, axonogenesis, neuronal projection development, aerobic respiration and proton motive force-driven mitochondrial ATP synthesis (Fig. 2C). Differential expression in D1 and D2 MSNs were the most significant contributors to the enrichment of the above GO terms. The full list of enriched GO terms is available in Supplementary File 4.
Co-expressed gene modules involved in synaptic transmission show increased activity in OVT73 medium spiny neurons and astrocytes
To identify and investigate sets of genes that were co-expressed in the OVT73 versus control cell types, we performed co-expression module analysis with MEGENA [37]. We identified several gene modules involved in synaptic transmission that exhibited higher module activity in OVT73 MSNs and OVT73 astrocytes compared to controls. A module centred around DLGAP2, GRIN2B, CACNA1C, CACNA1B, CNTNAP5, SYT1, KCTD16, and GRIN2A genes (Module 2, M2) showed higher module activity in OVT73 D1 MSNs compared to control D1 MSNs (differential module activity of 0.054; P < 0.0005, randomised permutation test with 2000 permutations) and OVT73 D2 MSNs compared to control D2 MSNs (differential module activity of 0.06, P < 0.0005) (Fig. 3B, Supplementary Fig. 7 and Supplementary Fig. 8). The most significant GO terms for module genes included synapse organisation, synapse assembly, chemical synaptic transmission, dicarboxylic acid catabolic process and glutamate metabolic process (Supplementary Fig. 6 and Supplementary File 6). Furthermore, this module was enriched for D1 MSNs DEGs (gene set over-representation analysis, FDR = 0.022) and D2 MSNs DEGs (FDR = 0.0013). Additionally, a module centred around SLC1A2, SLC7A11, GLI3, IRAG1, GFAP and SLC39A12 genes (Module 11, M11) was enriched in OVT73 astrocytes compared to control astrocytes (differential module activity of 0.092, P < 0.0005). Most significant GO terms for modules genes included L-glutamate transmembrane transport and amino acid transport. The module showed an enrichment of astrocyte DEGs (FDR = 2.08×10−5). Full lists of genes in gene modules are available in Supplementary File 5. The full list of enriched GO terms for modules is available in Supplementary File 6.
Figure 3.

Co-expression gene modules. (A) Co-expression modules generated using the multiscale embedded gene Co-expression network analysis (MEGENA). A total of 12 modules were identified with the structure outlined in the top right. module hub genes are labelled with connected genes represented as dots. Genes differentially expressed between OVT73 and controls are coloured in blue. Genes with evidence to support an interaction with HTT as curated by the HDinHD database [38] were coloured in red. genes that were both differentially expressed and a known HTT interactor were coloured in green. (B) Co-expression module activity in OVT73 and control cell types. Module activity in cell types were determined by computing the module eigengene (first principal component) using normalised expression values of module genes. Eigengene values are shown with p-values of the correlation shown in parentheses in each square. Higher eigengene values indicate higher gene expression of module genes within the cell type.
Cell–cell signalling is elevated in OVT73 animals
The differential expression analyses and co-expression analyses both indicated that synaptic signalling and transmission may be increased in the OVT73 animals. This led us to examine the cell–cell communication networks using CellChat [39]. Cell–cell communication analysis also supported increased synaptic signalling in neuronal and glial cells of the OVT73 striatum. Comparison of ligand-receptor pair expression from OVT73 and control revealed a higher total number of ligand-receptor interactions (4030 and 3509 detected ligand-receptor pairs for OVT73 and control respectively) and a higher sum of all communication probabilities (200 and 130 for OVT73 and control respectively) across all cell types in the OVT73 samples. The cell types that showed the greatest increase in the number of incoming (receptor to ligand) and outgoing (ligand to receptor) ligand-receptor interactions included D2 MSNs and astrocytes. The communication probability was greater in the OVT73 dataset for all neuronal cells (D1, D2, eccentric MSNs and PV/Th, NPY/SST, cholinergic interneurons), OPCs and astrocytes indicating more cell–cell crosstalk in the OVT73 cell state (Fig. 4A and B, thicker lines indicate higher number of ligand receptor interactions or higher communication probability in OVT73). In contrast, no differences in cell–cell signalling were detected in microglia from OVT73 compared to controls (Supplementary Fig. 9).
Figure 4.

CellChat cell–cell communication networks inferred from expression of ligand-receptor pairs. Circle plot showcasing the (A) differential number of ligand-receptor interactions and (B) differential communication probability between OVT73 and controls for any two cell types. Red arrows indicate increased number/communication probability in OVT73, blue arrows indicate decreased number/communication probability in OVT73. Thickness of line indicates greater number/communication probability. The total number of ligand-receptor interactions and total interaction strength for OVT73 and controls is also shown as bar graphs on the top right. (C) Differential information flow between OVT73 and control cell types for outgoing (ligand to receptor) and incoming (receptor to ligand) signalling pathways. The information flow for a given signalling pathway is defined as the sum of communication probabilities of all ligand receptor pairs in the pathway. A positive value indicates more communication in OVT73.
Examination of signalling pathways showed greater information flow (defined as the sum of communication probabilities of all ligand receptor pairs in the signalling pathway) in neurexin (NRXN), neuregulin (NRG), protein tyrosine phosphatase receptor M (PTPRM), contactin-1 (CNTN), neuronal growth regulator (NEGR), laminin, cell adhesion molecule (CADM), neural cell adhesion molecule (NCAM), ephrin type receptor A (EPHA) and pleiotrophin (PTN) pathways in OVT73 D2 MSNs. Interestingly, the greatest increase in communication was observed in myelin protein zero (MPZ), collagen, bone morphogenetic protein (BMP), and fibroblast growth factor (FGF) signalling pathways (Fig. 4C, Supplementary Fig. 10). Individual ligand receptor pairs that showed the greatest increase in communication probability between OVT73 MSNs and control included NEGR1-NEGR1, NRXN1-NLGN1, CADM1-CADM1, NRXN3-NLGN1, CDH2–CDH2, NRG1-ERBB4 and NRG3-ERBB4. These ligand-receptor pairs have been implicated in various neurodevelopmental processes including myelination [40–43], glutamatergic and GABAergic synapse development [44, 45], neurite outgrowth and general nervous system development [46–53]. Individual ligand receptor pair interactions for astrocytes and MSNs (D1, D2) are shown in Supplementary Fig. 11. Communication probabilities between all ligand receptor pairs across all cell types is available in Supplementary File 7.
Upregulation of glutamate signalling is attenuated by protective mechanisms
The glutamate excitotoxicity hypothesis proposes neuronal stress and eventual initiation of cell death pathways arising from elevated levels of synaptic glutamate and excessive signalling through ionotropic glutamate receptors [4–7]. We have identified transcriptional evidence in support of elevated glutamate signalling in the OVT73 striatum through a widespread transcriptional upregulation of ionotropic glutamate receptors (NMDA, AMPA, kainate and delta receptors) (Fig. 5A). We also observed an upregulation of transcription of the glutamine to glutamate conversion enzyme glutaminase (GLS) in OVT73 D1 and D2 MSNs and OVT73 astrocytes. Upregulation of GLS suggests increased production of glutamate which may be released into the synaptic space, bind glutamate receptors, and trigger a feedback loop to upregulate glutamate receptor expression. Elevated glutamate signalling is commonly observed with oxidative stress signatures represented by reduced activity of the oxidative phosphorylation complexes [54–56]. In keeping with this we have observed a downregulation of several genes encoding oxidative phosphorylation complexes including complex I, II, III, IV and V in OVT73 D1 and D2 MSNs.
Figure 5.

Heatmap of log2 fold change of selected genes between OVT73 and control cell types implicated in glutamate signalling. Differential gene transcriptional regulation of (A) glutamate receptors (NMDAR, AMPAR, Kainate, delta receptors), (B) GABAA receptors and glutamate to GABA conversion genes, (C) glutamate uptake transporters and glutamine-glutamate cycle genes and (D) genes encoding oxidative phosphorylation complexes.
In addition, we observed transcriptional upregulation of several genes likely due to a compensatory response to elevated glutamate levels. The transcription of genes encoding glutamate uptake transporters GLT (SLC1A2) and GLAST (SLC1A3) were upregulated in OVT73 astrocytes suggesting a response to remove excess synaptic glutamate (Fig. 5C). We also describe a transcriptional upregulation of genes encoding the gamma-aminobutyric acid A receptor subunits (GABAA) including the α, β and γ subunits in OVT73 D1 and D2 MSNs compared to controls. Additionally, an upregulation of genes encoding GABA transporter GAT4 (SLC6A11) and genes encoding the glutamate to GABA conversion enzymes, glutamate decarboxylase (GAD1 and GAD2) was also observed in OVT73 D2 MSNs (Fig. 5B). Taken together, these observations suggested increased GABAA receptor signalling is likely a result of increased glutamate conversion to GABA.
Transcription analysis of gene regulatory networks indicate reduced CREB regulon activity in the OVT73 striatum
We assessed the transcription factor regulation of highly variable genes in OVT73 through construction of gene regulatory networks using SCENIC. We focused on the regulon activity of the cAMP-responsive element-binding protein (CREB) family of transcription factors that has been shown to be activated following excessive glutamate signalling [57, 58]. When examining differential regulon activity between OVT73 and controls, we observed a reduction in regulon activity for CREB1 in astrocytes (differential module activity of −0.952, P = 0.0005, randomised permutation test with 2000 permutations), D1 MSNs (−1.43, P < 0.0005) and D2 MSNs (−1.367, P < 0.0005) (Fig. 6). ATF2 regulon activity was reduced between OVT73 and controls for D2 MSN (−0.755, P < 0.0005). ATF4 regulon activity was reduced between OVT73 and controls for astrocytes (−0.162, P = 0.001), D1 MSNs (−0.087, P = 0.006) and D2 MSNs (−0.448, P < 0.0005). ATF7 regulon activity was reduced between OVT73 and controls for D2 MSNs (−0.265, P < 0.0005) (Fig. 6B, Supplementary Fig. 14). The greatest sum of difference in CREB regulon activity between OVT73 and controls (sum of CREB1, ATF2, ATF4, ATF7 regulon activity in OVT73 subtracted by the sum of CREB1, ATF2, ATF4, ATF7 regulon activity in control) was observed in the D2 MSNs. Interestingly, CREB regulon activity was reduced to a lesser degree in D1 MSNs compared to D2 MSNs. Regulon activity of all identified regulons is available in Supplementary Fig. 12 and Supplementary Fig. 13. Gene members of each regulon are available in Supplementary File 8.
Figure 6.

Gene regulatory networks show reduced CREB regulon activity in OVT73. (A) CREB-related transcription factor regulated gene modules (regulons) network. The associated transcription factor regulon is shown as triangles with gene members of the regulon connected as dots. The two panels on the right show the log2 fold change of differentially expressed gene members of the regulon between OVT73 and controls in D1 MSNs and D2 MSNs. (B) Differential regulon activity was computed by subtraction of regulon activity between OVT73 and controls. A randomised permutation test with 2000 permutations was performed to determine significant differential regulon activity between OVT73 and control cell types. P-values of the randomised permutation test are shown in the parentheses.
Discussion
Revealing the HD pathogenic mechanisms prior to striatal cell loss and motor and cognitive deficits is likely to reveal disease modifying pharmaceutical targets. Our group has generated and characterised a transgenic sheep model of HD (named OVT73) that displays no striatal cell loss, or overt symptoms but exhibits many of the molecular changes of prodromal HD [20, 22–26]. To gain further insight into cellular changes in a prodromal HD model we undertook a single nuclei transcriptomic study in striatal tissue from 5-year-old OVT73 animals. Differential gene expression analysis, gene co-expression network analysis and cell–cell signalling analysis indicate alterations in glutamatergic and GABAergic synapses in striatal MSN’s, the most vulnerable cells in HD.
With respect to the glutamatergic synapse, we observed transcriptional upregulation of genes encoding ionotropic glutamate receptors including NMDA, AMPA and kainate receptors in both the D1 and D2 type OVT73 MSNs. Co-expression analysis also revealed increased transcriptional activity of the gene modules containing the aforementioned glutamate receptor genes in the same cell types. Moreover, cell–cell signalling inferred from ligand-receptor co-expression revealed a disease state increase in signalling of pathways including neurexin, neuroligin, neuregulin, neural cell adhesion molecule and ephrin. These pathways have been implicated in neurite outgrowth, glutamatergic synapse formation and upregulation result in an increase in ionotropic glutamate receptor abundance [44, 59–69]. Previous studies of striatal MSNs from post-mortem human and HD mouse models found positive correlations between ionotropic glutamate receptor mRNA levels and receptor protein abundance. Further, an upregulation of receptor signalling inferred from ligand binding studies and changes in receptor currents were also identified [70–74]. Therefore, it is likely that the observed upregulation of ionotropic glutamate receptor gene transcription in OVT73 medium spiny neurons indicates increased glutamate signalling.
Overactivation of ionotropic glutamate receptors has been shown to lead to an influx of calcium ions promoting excitotoxic stress, mitochondrial dysfunction, and eventual initiation of cell death [8, 75]. Mitochondrial dysfunction and oxidative stress induced by excitotoxic stress is evident by a decrease in the activity of oxidative phosphorylation (OXPHOS) complexes [76–78]. In support of this, a recent single nuclei RNA-seq study of HD patient striatum and HD mouse model tissue also reports transcriptional downregulation of these OXPHOS complexes in the MSNs [79]. Similarly, in the OVT73 sheep striatum, we have detected downregulation of transcription from genes encoding OXPHOS complexes including complex I, II, III and IV and V in the OVT73 D1 and D2 MSNs. This downregulation is occurring in the absence of cell loss in the sheep indicating there must be a compensatory mechanism.
Previous studies have also shown that excess influx of calcium ions due to overactivated ionotropic glutamate receptors can also trigger neuroprotective pathways including phosphorylation of the transcription factor CREB and activation by CREB binding protein [57]. Interestingly, we have observed a downregulation of genes encoding CREB transcription factors (CREB1, ATF2, ATF4 and ATF7) and a reduction of CREB regulon activity inferred from gene regulatory networks in OVT73 D1 and D2 MSNs, astrocytes, microglia, neuroblasts and oligodendrocytes. This would suggest that the CREB transcriptional machinery may be dysregulated in OVT73. Studies in HD mouse models that have shown that CREB phosphorylation and activation is lost prior to cell death [58, 80–82]. In keeping with the earliest stage selective vulnerability of D2 MSN, the greatest reduction in CREB regulon activity was observed in the OVT73 D2 MSNs. These results provide further evidence for CREB transcriptional dysregulation in HD and importantly that this precedes the cell death cascade.
In HD, the mutant CAG tract has been shown to expand somatically throughout disease course particularly in the striatum [83–87]. It has been proposed that further repeat expansion in individual cells leads to increased excitotoxic stress in those cells and once a cell specific repeat threshold has been exceeded, cell death processes are activated [88]. The 73-unit polyglutamine repeat coding tract in the OVT73 line is somatically stable which may explain why there is an absence of striatal neuronal loss. It is possible that this allows us to observe in this model a steady state of compensatory mechanisms activated to prevent damage due to excitotoxic stress. A core mechanism for the clearance of synaptic glutamate is through uptake into astrocytic glutamate transporters (SLC1A2, SLC1A3) and degradation via the glutamine-glutamate cycle [89, 90]. A reduction in astrocytic glutamate transporters at both RNA and protein level have been reported in HD patients and HD mouse models [74, 91, 92]. Conversely, we have observed an increase in transcription of SLC1A2 and SLC1A3 in OVT73 astrocytes. Given the prodromal nature of the OVT73 model, we postulate that we are observing a disease timepoint where these compensatory mechanisms remain operational and have not been overwhelmed as per the later stages of the disease. Our sheep dataset exclusively only comprises of transcription, however it has been shown in HD mouse models and HD post mortem tissue that SLC1A2 and SLC1A3 mRNA levels were positively correlated with uptake of synaptic glutamate [74, 93, 94]. It is therefore possible that the upregulation of SLC1A2 and SLC1A3 in the OVT73 striatum represent a compensatory response to remove excess synaptic glutamate.
The loss of GABAA receptor mediated neuronal inhibition is considered a key feature of neuronal dysfunction in HD [95, 96]. We have found in the OVT73 model striatum transcriptional upregulation of genes encoding GABAA receptor subunits α, β, and γ specifically in D1 and D2 MSNs. Other studies have revealed that mRNA levels of GABAA receptors in striatal MSNs (human cases and HD mouse models) correlate with receptor protein abundance and receptor signalling inferred through receptor binding studies and changes in receptor currents [95, 96]. Increased striatal GABAA receptor activity have been postulated to have a neuroprotective effect against excitotoxicity [97, 98] and therefore increased GABAA signalling in OVT73 may be another mechanism that mitigates glutamate induced stress providing neuronal protection in the OVT73 striatum.
A limitation of our study is the absence of functional or other analyses to confirm the transcriptional differences identified in the present data. Unfortunately, we did not have further tissue from these animals for further analysis. Future functional characterisation in other cohorts will be critical, not only to confirm results summarised in this study, but also for comprehensive characterisation of the overall OVT73 sheep model.
The transcriptional alterations are encouraging evidence for an active cell stress response to excess synaptic glutamate. This protective response has prevented or delayed the excitotoxic process in the OVT73 striatum as there is no cell loss. These observations also suggest that glutamate induced stress may be present long before the initiation of the cell death cascade. Our single nuclei RNA-seq observations corroborates reports of HD mouse models that showcase excitotoxicity begins early on in disease phase before the onset of symptoms [99–101]. Current experimental HD therapeutics targeting glutamate mediated excitotoxicity have included NMDA receptor antagonists (Memantine, Amantadine) that reduce the overactivation of NMDA receptors which have been utilised in clinical trials to limited success [102–105]. One potential explanation for the lack of efficacy of these drugs is that the therapeutic agents were administrated too late in disease progression where the consequences of cell death (neuroinflammatory cytokines, proinflammatory markers and reactive oxygen species) takes over the momentum [106]. In support of this proposition striatal neurons of HD mouse models have shown to gradually become resistant to excitotoxicity modifying agents following disease progression [107–110]. The findings of our single nuclei RNA-seq study indicate that striatal cells can maintain steady state disease resilience and early intervention for instance glutamate depletion may prevent the cell death cascade and lead to better clinical outcomes.
Methods and materials
Ovine samples
The cohort maintenance and tissue sampling has been reported in previous publications [22, 27]. Briefly, animals were maintained in a certified, purpose-made research facility at the South Australian Research and Development Institute (SARDI). Animals were kept in large paddocks as a mixture of wild-type controls and transgenic animals and fed ad libitum. Post-mortem striatal samples were taken from six 5-year-old OVT73 (3 females, 3 males) HD sheep as previously described [22, 27] and six age-matched wild-type controls (2 females, 4 males). Tissue collection was performed in accordance with the SARDI/PIRSA Animal Ethics Committee (approval no. 19/02 and 05/12). All experiments performed adhered to the recommendations in the ARRIVE guidelines [111]. In brief, a lethal dose of pentobarbitone sodium solution (Lethabarb, 1 ml/2 kg body weight) was administrated intravenously, the brains were removed from the skulls, dissected into the 5 distinct blocks (Fig. 1) and snap frozen initially on dry ice and then in liquid nitrogen. Samples were wrapped in tinfoil and stored at −80°C until further use.
Nuclei isolation for single nuclei RNA-seq
Nuclei were extracted from frozen anterior ventral-medial striatal (Supplementary Fig. 1) tissue utilising an adapted protocol from Krishnaswami et al., 2016 [112]. Briefly, approximately 50–100 mg of brain tissue was transferred to a dounce homogenizer containing 1 ml homogenization buffer—HB (250 mM sucrose, 25 mM KCl, 5 mM MgCl2, 10 mM Tris buffer pH 8.0, 1 μM DTT, 1X protease inhibitor (Sigma), 0.4 U/μl RNaseIn (ThermoFisher Scientific), 0.2 U/μl SuperaseIn (ThermoFisher Scientific), 0.1% Triton X-100). Tissue was homogenised using 5 strokes with the loose dounce pestle A, followed by 10–15 strokes of tight dounce pestle B. The homogenate was filtered through a 40 μm strainer into 5 ml Eppendorf tubes and centrifuged at 1000 rcf (4°C) for 8 min. The supernatant was removed, and the pellet was resuspended in 250 μl of HB. A 50%–29% iodixanol gradient (OptiPrep™ Density Gradient Medium, Sigma) was prepared to allow removal of the myelin layer. 250 μl of 50% iodixanol was added to the nuclei-HB mixture and slowly layered on top of 500 μl of 29% iodixanol in a new Eppendorf tube. The resultant gradient was centrifuged at 13 000 rcf (4°C) for 40 min. The supernatant and myelin were removed, and the purified nuclei pellet was resuspended in a solution containing 1 ml PBS, 1% BSA and 0.2 U/μl RNAse inhibitor. 5 μl of resuspended nuclei was stained with 5 μl trypan blue and the quality and number of nuclei was assessed using the Countess II FL Automated Cell Counter (ThermoFisher Scientific). To reduce the cost per library, nuclei suspensions from different samples were pooled at equal concentrations in groups of 2 (Supplementary Fig. 2) prior to library preparation and demultiplexed as described below.
Library preparation and single nuclei RNA sequencing
The droplet-based Chromium methodology from 10X Genomics was utilised for the generation of single nuclei libraries. Libraries were prepared according to the Chromium Next GEM Single Cell 3’ Reagent Kits v3.1 as per manufacturer’s instructions. The single nuclei RNA-seq libraries were sequenced on the HiSeq XTen platform. Alignment of reads was performed using the CellRanger v7.0.0 pipeline with STAR v2.7.2a to the sheep Oar_rambouillet_v1.0 reference genome and annotation (Ensembl release 107). Summary statistics for single nuclei RNA-seq libraries are shown in Supplementary Fig. 3.
Sample demultiplexing
Demultiplexing of pooled nuclei associated barcodes in the CellRanger computed alignments utilized the genetic variation between individual samples. In brief, regions of the transcriptome with high read coverage (> 50) were identified using the featureCounts function from the Subread package [113, 114]. These regions were used as input into Freebayes variant caller to find genomic variants for each barcode. A filtering step was subsequently applied using BCFtools [115] to remove low confidence variant calls (QUAL score < 30). Barcodes were assigned to sample ID by genotype at variant loci with the scSplit algorithm [116]. scSplit employs a hidden state model to assign nuclei associated barcodes from the pooled sample to respective groups based on an expectation-maximisation framework [116]. scSplit input parameters included an expected number of mixed samples of 2 and an estimated doublet percentage of 4% (scSplit -n 2 -d 0.04). scSplit demultiplexing outcome of barcodes is available in Supplementary File 1. The filtered unique molecular identifiers (UMI) feature-barcode matrices generated by CellRanger were split according to the demultiplexed barcodes.
Quality control and cell clustering
The filtered unique molecular identifiers (UMI) feature-barcode matrices were processed with ICARUS software [117, 118] developed by our group which utilizes the Seurat v4.0 R package [119]. A quality control filter was applied to remove low quality nuclei with gene counts less than 200 or more than 7500. Additionally, nuclei with mitochondrial reads (> 5%) were removed. From the 12 samples, a total of 28 234 high quality nuclei were recovered with an average of 1357 median genes per nucleus (range: 1292–2607), an average of 2672 median unique molecular identifiers (UMIs) per nucleus (range: 1569–3877) and an average percentage of transcripts originating from mitochondrial genes of 1.04% (range: 0.5%–2.02%) (Supplementary Fig. 4).
The read counts were normalised and scaled using the NormalizeData function in Seurat with parameters normalisation.method = LogNormalize and scale.factor = 10 000. Dimensionality reduction was performed on normalised read counts using a set of 3000 top variable genes identified through the FindVariableFeatures function in Seurat. All sheep datasets (6 OVT73 and 6 Control) were integrated using harmony [120] and cell clustering was performed with the first 30 dimensions, k-nearest neighbour value of 20 and the Louvain community detection algorithm. Cell type annotation was performed by comparison of cluster marker genes identified using the FindAllMarkers function in Seurat with parameters min.pct = 0.25 and logfc.threshold = 0.25 (Supplementary File 2 for list of cell type marker genes) against known striatal cell type markers identified in published single cell RNA-seq datasets [36, 79, 121]. Medium spiny neurons were classified into 3 sub-categories (D1, D2 and Eccentric) using cell markers described in the DropViz atlas [36].
Differential expression analyses and gene ontology enrichment
Differential expression analyses were conducted for each cell type separately between OVT73 cases and controls. The mixed model-based MAST statistical test [122] was performed to identify differentially expressed genes (DEGs). Genes with a log2 fold change of > 0.1, p-value adjusted for false discovery rate (FDR < 0.05) and expressed in at least 10% of nuclei from either condition (OVT73 or control) were considered to be differentially expressed. A Gene Ontology (GO) over-representation analysis was performed on differentially expressed genes using the enrichGO function in the R package ClusterProfiler [123], which identifies enriched GO terms using Fisher’s exact test. A list of DEGs was used as input into enrichGO. GO terms were extracted from the NCBI annotation of Ovis aries, retrieved with record AH107722 with the R package AnnotationHub [124].
Gene co-expression analysis
Co-expression analysis was performed for the 3000 most variable genes (identified by the FindVariableFeatures function in Seurat) across all OVT73 and control samples. MEGENA (v1.5) was used to extract significant gene interactions and construct co-expression gene modules [37]. Default parameters detailed in the MEGENA vignette were used, including the use of Pearson’s correlations, FDR threshold of 0.05, module and hub significance P-values of 0.05 and a minimum module size of 10 genes. Modules were visualised graphically using Cystoscape v3.9.1 [125]. Module activity in cell types were determined by computing the module eigengene (first principal component) using normalised expression values of module genes. Higher eigengene values indicate higher gene expression of module genes within the cell type. The WGCNA package was used to compute module eigengenes with the moduleEigengenes function. Differential module activity between OVT73 and control cell types was computed as a subtraction of module eigengenes of the two conditions. A randomised permutation test was used with 2000 permutations to identify significant differential module activity between OVT73 and control cell types.
Additionally, Gene Ontology analysis of module genes was performed using the moduleGO function in the DGCA R package with a p-value threshold of 0.05. Gene sets in each module were examined for its enrichment of OVT73 versus control differentially expressed genes. Gene set enrichment analysis was performed using the enricher function in the clusterProfiler v4.6.0 R package [123]. A minimum and maximum gene set size of 10 and 500 was used with adjusted p-value FDR threshold of 0.05.
Gene regulatory network analysis
Examination of gene regulatory networks for cell types across OVT73 and control samples was conducted using R SCENIC v1.2.0 [126]. SCENIC performs cis-regulatory transcription factor binding motif analysis on a set of co-expressed transcription factors and genes. Transcription factor genes were defined from the curated list by Lambert et al., [127]. Given, a public sheep transcription factor motif database was not available, a custom transcription factor motif database for sheep was made for this study. The motif database was generated as follows, the 500 bp sequence upstream and 100 bp sequence downstream of the transcriptional start site (TSS) for each gene was extracted from the oar_rambouillet v1.0 reference genome (ensemble version 107). Additionally, the 10 kb sequence upstream and downstream of the TSS were also extracted. The files are available on zenodo (https://doi.org/10.5281/zenodo.8057929) under oar_rambouillet_v1.0_500bp_up_100bp_down.fa and oar_rambouillet_v1.0_10kbp_up_10kbp_down.fa. These regions were assessed for transcription factor binding motifs based on the 2022 SCENIC+ motif collection (https://resources.aertslab.org/cistarget/motif2tf/) using the create_cistarget_motif_databases.py function of the create_cisTarget_databases package (https://github.com/aertslab/create_cisTarget_databases). Code used to generate database is shared under https://doi.org/10.5281/zenodo.8057929.
The 3000 most variable genes (identified by the FindVariableFeatures function in Seurat) across all OVT73 and control samples were used as inputs into SCENIC. Co-expression modules were constructed using GENIE3 [126, 128] and transcription factor motifs were scored using RcisTarget v1.18.2. Transcription factor-regulated gene modules (regulons) with 10 or more genes were considered. The activity of each regulon in OVT73 and control cell types (regulon activity) were scored using the AUCell algorithm which computes the enrichment of regulons as an area under the recovery curve across a ranking of all genes in a cell based on their normalised expression values. A high regulon activity indicates genes within the regulon are positively regulated by the transcription factor. Regulons were visualised graphically using Cystoscape v3.9.1 [125]. Differential regulon activity between OVT73 and control cell types was computed as a subtraction of regulon activity of the two conditions. A randomised permutation test was used with 2000 permutations to identify significant differential regulon activity between OVT73 and control cell types. Additionally, a regulon specificity score (RSS) was determined which employs the Jensen-Shannon divergence metric to assess cell-type specificity of regulon activity [126]. Regulons in cell types with a specificity score of 1 indicates exclusive expression of the regulon in that one cell type, while a specificity score of 0 indicates the regulon is evenly expressed across all cell types.
Cell–cell communication analysis
The inference of cell signalling crosstalk through the transcription levels of ligand receptor pairs was examined using CellChat. CellChat incorporates a comprehensive database of ligand receptor interactions, soluble agonists/antagonists and stimulatory/inhibitory membrane bound co-receptors to infer cell–cell communications from single cell RNA-seq data based on mass action models, social network analysis tools and pattern recognition methods [39]. In brief, intercellular communications were inferred through 3 steps; (1) identification of differentially expressed ligands and receptors genes for each cell type using the Wilcoxon rank sum test with a p-value threshold of 0.05. (2) A communication probability (interaction strength) is computed by modelling ligand-receptor interactions using the law of mass action on average expression values of a ligand in one cell group and a receptor of another cell group. This calculation accounts for the number of cells within each cell group. (3) Significant interactions are identified using a permutation test that randomly permutes cell group labels and recomputes the interaction probability.
A comparison of cell–cell communication in OVT73 versus control cell types was performed using default parameters detailed in the CellChat v1.6.1 vignettes. Ligand-receptor interactions were extracted from the human CellChat database. Interactions involving 50 or less nuclei were filtered out using the filterCommunication function.
Supplementary Material
Acknowledgements
The authors wish to thank the incredible on-farm SARDI team for all-animal management. This work was supported by the CHDI Foundation, Freemasons New Zealand and the New Zealand–China Non-Communicable Diseases Research Collaboration Centre (NCD CRCC).
Contributor Information
Andrew Jiang, Applied Translational Genetics Group, Centre for Brain Research, School of Biological Sciences, The University of Auckland, 3 Symonds Street, Auckland 1010, New Zealand.
Linya You, Department of Human Anatomy & Histoembryology, School of Basic Medical Sciences, Fudan University, 131 Dong'an Road, Shanghai 200032, China; Key Laboratory of Medical Imaging Computing and Computer Assisted Intervention of Shanghai, 130 Dong'an Road, Shanghai 200032, China.
Renee R Handley, Applied Translational Genetics Group, Centre for Brain Research, School of Biological Sciences, The University of Auckland, 3 Symonds Street, Auckland 1010, New Zealand.
Victoria Hawkins, Applied Translational Genetics Group, Centre for Brain Research, School of Biological Sciences, The University of Auckland, 3 Symonds Street, Auckland 1010, New Zealand.
Suzanne J Reid, Applied Translational Genetics Group, Centre for Brain Research, School of Biological Sciences, The University of Auckland, 3 Symonds Street, Auckland 1010, New Zealand.
Jessie C Jacobsen, Applied Translational Genetics Group, Centre for Brain Research, School of Biological Sciences, The University of Auckland, 3 Symonds Street, Auckland 1010, New Zealand.
Stefano Patassini, Applied Translational Genetics Group, Centre for Brain Research, School of Biological Sciences, The University of Auckland, 3 Symonds Street, Auckland 1010, New Zealand.
Skye R Rudiger, Molecular Biology and Reproductive Technology Laboratories, South Australian Research and Development Institute, 129 Holland Road, Adelaide, SA 5350, Australia.
Clive J Mclaughlan, Molecular Biology and Reproductive Technology Laboratories, South Australian Research and Development Institute, 129 Holland Road, Adelaide, SA 5350, Australia.
Jennifer M Kelly, Molecular Biology and Reproductive Technology Laboratories, South Australian Research and Development Institute, 129 Holland Road, Adelaide, SA 5350, Australia.
Paul J Verma, Aquatic and Livestock Sciences, South Australian Research and Development Institute, 129 Holland Road, Adelaide, SA 5350, Australia.
C Simon Bawden, Molecular Biology and Reproductive Technology Laboratories, South Australian Research and Development Institute, 129 Holland Road, Adelaide, SA 5350, Australia.
James F Gusella, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, 185 Cambridge Street, Boston, MA 02114, United States; Department of Genetics, Harvard Medical School, 25 Shattuck Street, Boston, MA 02115, United States.
Marcy E MacDonald, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, 185 Cambridge Street, Boston, MA 02114, United States; Department of Neurology, Harvard Medical School, 25 Shattuck Street, Boston, MA 02115, United States.
Henry J Waldvogel, Department of Anatomy and Medical Imaging, Centre for Brain Research, Faculty of Medical and Health Science, The University of Auckland, 85 Park Road, Auckland 1023, New Zealand.
Richard L M Faull, Department of Anatomy and Medical Imaging, Centre for Brain Research, Faculty of Medical and Health Science, The University of Auckland, 85 Park Road, Auckland 1023, New Zealand.
Klaus Lehnert, Applied Translational Genetics Group, Centre for Brain Research, School of Biological Sciences, The University of Auckland, 3 Symonds Street, Auckland 1010, New Zealand.
Russell G Snell, Applied Translational Genetics Group, Centre for Brain Research, School of Biological Sciences, The University of Auckland, 3 Symonds Street, Auckland 1010, New Zealand.
Conflict of interest statement
J.F.G. was a founder and scientific advisory board member with a financial interest in Triplet Therapeutics Inc. His NIH-funded project is using genetic and genomic approaches to uncover other genes that significantly influence when diagnosable symptoms emerge and how rapidly they worsen in Huntington’s disease. The company was developing new therapeutic approaches to address triplet repeat disorders such Huntington’s disease, myotonic dystrophy and spinocerebellar ataxias. His interests were reviewed and are managed by Massachusetts General Hospital and Mass General Brigham in accordance with their conflict of interest policies. J.F.G. has also been a consultant for Wave Life Sciences USA Inc., Biogen Inc. and Pfizer Inc.
All other authors have declared that they have no competing interests.
Funding
This work was funded by the New Zealand Ministry of Business Innovation and Employment funding for New Zealand–China Non-Communicable Diseases Research [UOOX1601]. Science Innovation 2030—Brain Science and Brain-Inspired Intelligence Technology Major Project (#2021ZD0201100 Task 4 #2021ZD0201104) from the Ministry of Science and Technology (MOST), China.
Data availability
The data have been deposited in NCBI’s Gene Expression Omnibus (GEO) [129] and are accessible through accession number GSE229839 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc= GSE229839).
The code used in this study are deposited on zenodo available at https://doi.org/10.5281/zenodo.8057929.
References
- 1. The Huntington's Disease Collaborative Research Group . A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's disease collaborative research group. Cell 1993;72:971–983. [DOI] [PubMed] [Google Scholar]
- 2. Snell RG, MacMillan JC, Cheadle JP. et al. Relationship between trinucleotide repeat expansion and phenotypic variation in Huntington's disease. Nat Genet 1993;4:393–397. [DOI] [PubMed] [Google Scholar]
- 3. Jiang A, Handley RR, Lehnert K. et al. From pathogenesis to therapeutics: a review of 150 years of Huntington's disease research. Int J Mol Sci 2023;24:13021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Doble A. The role of excitotoxicity in neurodegenerative disease: implications for therapy. Pharmacol Ther 1999;81:163–221. [DOI] [PubMed] [Google Scholar]
- 5. Salinska E, Danysz W, Lazarewicz JW. The role of excitotoxicity in neurodegeneration. Folia Neuropathol 2005;43:322–339. [PubMed] [Google Scholar]
- 6. Dong X-X, Wang Y, Qin Z-H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol Sin 2009;30:379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mehta A, Prabhakar M, Kumar P. et al. Excitotoxicity: bridge to various triggers in neurodegenerative disorders. Eur J Pharmacol 2013;698:6–18. [DOI] [PubMed] [Google Scholar]
- 8. Sepers MD, Raymond LA. Mechanisms of synaptic dysfunction and excitotoxicity in Huntington's disease. Drug Discov Today 2014;19:990–996. [DOI] [PubMed] [Google Scholar]
- 9. Beal MF, Kowall NW, Ellison DW. et al. Replication of the neurochemical characteristics of Huntington's disease by quinolinic acid. Nature 1986;321:168–171. [DOI] [PubMed] [Google Scholar]
- 10. Bordelon YM, Chesselet M-F, Nelson D. et al. Energetic dysfunction in quinolinic acid-lesioned rat striatum. J Neurochem 1997;69:1629–1639. [DOI] [PubMed] [Google Scholar]
- 11. Emerich DF, Thanos CG, Goddard M. et al. Extensive neuroprotection by choroid plexus transplants in excitotoxin lesioned monkeys. Neurobiol Dis 2006;23:471–480. [DOI] [PubMed] [Google Scholar]
- 12. Foster AC, Miller LP, Oldendorf WH. et al. Studies on the disposition of quinolinic acid after intracerebral or systemic administration in the rat. Exp Neurol 1984;84:428–440. [DOI] [PubMed] [Google Scholar]
- 13. Beal MF, Ferrante RJ, Swartz KJ. et al. Chronic quinolinic acid lesions in rats closely resemble Huntington's disease. J Neurosci 1991;11:1649–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Coyle JT, Schwarcz R. Lesion of striatal neurones with kainic acid provides a model for Huntington's chorea. Nature 1976;263:244–246. [DOI] [PubMed] [Google Scholar]
- 15. McGeer EG, McGeer PL. Duplication of biochemical changes of Huntington's chorea by intrastriatal injections of glutamic and kainic acids. Nature 1976;263:517–519. [DOI] [PubMed] [Google Scholar]
- 16. Milnerwood AJ, Gladding CM, Pouladi MA. et al. Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington's disease mice. Neuron 2010;65:178–190. [DOI] [PubMed] [Google Scholar]
- 17. Joshi PR, Wu NP, Andre VM. et al. Age-dependent alterations of corticostriatal activity in the YAC128 mouse model of Huntington disease. J Neurosci 2009;29:2414–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Heng MY, Detloff PJ, Wang PL. et al. In vivo evidence for NMDA receptor-mediated excitotoxicity in a murine genetic model of Huntington disease. J Neurosci 2009;29:3200–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jacobsen JC, Bawden CS, Rudiger SR. et al. An ovine transgenic Huntington's disease model. Hum Mol Genet 2010;19:1873–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huntington's Disease Sheep Collaborative Research, G, Reid SJ, Patassini S. et al. Further molecular characterisation of the OVT73 transgenic sheep model of Huntington's disease identifies cortical aggregates. J Huntingtons Dis 2013;2:279–295. [DOI] [PubMed] [Google Scholar]
- 21. Handley RR, Reid SJ, Burch Z. et al. Somatic CAG repeat stability in a transgenic sheep model of Huntington's disease. J Huntingtons Dis 2024;13:33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Handley RR, Reid SJ, Patassini S. et al. Metabolic disruption identified in the Huntington's disease transgenic sheep model. Sci Rep 2016;6:20681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Skene DJ, Middleton B, Fraser CK. et al. Metabolic profiling of presymptomatic Huntington’s disease sheep reveals novel biomarkers. Sci Rep 2017;7:43030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Morton AJ, Rudiger SR, Wood NI. et al. Early and progressive circadian abnormalities in Huntington's disease sheep are unmasked by social environment. Hum Mol Genet 2014;23:3375–3383. [DOI] [PubMed] [Google Scholar]
- 25. Vas S, Nicol AU, Kalmar L. et al. Abnormal patterns of sleep and EEG power distribution during non-rapid eye movement sleep in the sheep model of Huntington's disease. Neurobiol Dis 2021;155:105367. [DOI] [PubMed] [Google Scholar]
- 26. Morton AJ, Middleton B, Rudiger S. et al. Increased plasma melatonin in presymptomatic Huntington disease sheep (Ovis aries): compensatory neuroprotection in a neurodegenerative disease? J Pineal Res 2020;68:e12624. [DOI] [PubMed] [Google Scholar]
- 27. Handley RR, Reid SJ, Brauning R. et al. Brain urea increase is an early Huntington's disease pathogenic event observed in a prodromal transgenic sheep model and HD cases. Proc Natl Acad Sci USA 2017;114:E11293–E11302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Patassini S, Begley P, Reid SJ. et al. Identification of elevated urea as a severe, ubiquitous metabolic defect in the brain of patients with Huntington's disease. Biochem Biophys Res Commun 2015;468:161–166. [DOI] [PubMed] [Google Scholar]
- 29. De Deyn PP, D'Hooge R, Van Bogaert P-P. et al. Endogenous guanidino compounds as uremic neurotoxins. Kidney Int 2001;59:S77–S83. [DOI] [PubMed] [Google Scholar]
- 30. Gropman AL, Summar M, Leonard JV. Neurological implications of urea cycle disorders. J Inherit Metab Dis 2007;30:865–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Robinson MB, Hopkins K, Batshaw ML. et al. Evidence of excitotoxicity in the brain of the ornithine carbamoyltransferase deficient sparse fur mouse. Brain Res Dev Brain Res 1995;90:35–44. [DOI] [PubMed] [Google Scholar]
- 32. Szerb JC, Butterworth RF. Effect of ammonium ions on synaptic transmission in the mammalian central nervous system. Prog Neurobiol 1992;39:135–153. [DOI] [PubMed] [Google Scholar]
- 33. Felipo V, Butterworth RF. Mitochondrial dysfunction in acute hyperammonemia. Neurochem Int 2002;40:487–491. [DOI] [PubMed] [Google Scholar]
- 34. Mears ER, Handley RR, Grant MJ. et al. A multi-omic Huntington's disease transgenic sheep-model database for investigating disease pathogenesis. J Huntingtons Dis 2021;10:423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Munoz-Manchado AB, Bengtsson Gonzales C, Zeisel A. et al. Diversity of interneurons in the dorsal striatum revealed by single-cell RNA sequencing and PatchSeq. Cell Rep 2018;24:2179–2190.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Saunders A, Macosko EZ, Wysoker A. et al. Molecular diversity and specializations among the cells of the adult mouse brain. Cell 2018;174:1015–1030.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Song W-M, Zhang B. Multiscale embedded gene Co-expression network analysis. PLoS Comput Biol 2015;11:e1004574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Aaronson J, Beaumont V, Blevins RA. et al. HDinHD: a rich data portal for Huntington's disease research. J Huntingtons Dis 2021;10:405–412. [DOI] [PubMed] [Google Scholar]
- 39. Jin S, Guerrero-Juarez CF, Zhang L. et al. Inference and analysis of cell-cell communication using CellChat. Nat Commun 2021;12:1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nelis E, Haites N, Van Broeckhoven C. Mutations in the peripheral myelin genes and associated genes in inherited peripheral neuropathies. Hum Mutat 1999;13:11–28. [DOI] [PubMed] [Google Scholar]
- 41. Shy ME, Jáni A, Krajewski K. et al. Phenotypic clustering in MPZ mutations. Brain 2004;127:371–384. [DOI] [PubMed] [Google Scholar]
- 42. Gregorio I, Braghetta P, Bonaldo P. et al. Collagen VI in healthy and diseased nervous system. Dis Model Mech 2018;11:dmm032946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Klimaschewski L, Claus P. Fibroblast growth factor signalling in the diseased nervous system. Mol Neurobiol 2021;58:3884–3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Craig AM, Kang Y. Neurexin-neuroligin signaling in synapse development. Curr Opin Neurobiol 2007;17:43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mo X, Liu M, Gong J. et al. PTPRM is critical for synapse formation regulated by zinc ion. Front Mol Neurosci 2022;15:822458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chatterjee M, Schild D, Teunissen CE. Contactins in the central nervous system: role in health and disease. Neural Regen Res 2019;14:206–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Luckenbill-Edds L. Laminin and the mechanism of neuronal outgrowth. Brain Res Brain Res Rev 1997;23:1–27. [DOI] [PubMed] [Google Scholar]
- 48. Jin J, Liu L, Chen W. et al. The implicated roles of cell adhesion molecule 1 (CADM1) gene and altered prefrontal neuronal activity in attention-deficit/hyperactivity disorder: a "gene-brain-behavior relationship"? Front Genet 2019;10:882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Miao H, Wang B. EphA receptor signaling—complexity and emerging themes. Semin Cell Dev Biol 2012;23:16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gamez B, Rodriguez-Carballo E, Ventura F. BMP signaling in telencephalic neural cell specification and maturation. Front Cell Neurosci 2013;7:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. González-Castillo C, Ortuño-Sahagún D, Guzmán-Brambila C. et al. Pleiotrophin as a central nervous system neuromodulator, evidences from the hippocampus. Front Cell Neurosci 2015;8:443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ditlevsen DK, Povlsen GK, Berezin V. et al. NCAM-induced intracellular signaling revisited. J Neurosci Res 2008;86:727–743. [DOI] [PubMed] [Google Scholar]
- 53. Ebnet K. Junctional adhesion molecules (JAMs): cell adhesion receptors with pleiotropic functions in cell physiology and development. Physiol Rev 2017;97:1529–1554. [DOI] [PubMed] [Google Scholar]
- 54. Bondy SC, LeBel CP. The relationship between excitotoxicity and oxidative stress in the central nervous system. Free Radic Biol Med 1993;14:633–642. [DOI] [PubMed] [Google Scholar]
- 55. Wullner U, Young AB, Penney JB. et al. 3-Nitropropionic acid toxicity in the striatum. J Neurochem 1994;63:1772–1781. [DOI] [PubMed] [Google Scholar]
- 56. Kumar A, Ratan RR. Oxidative stress and Huntington's disease: the good, the bad, and the ugly. J Huntingtons Dis 2016;5:217–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. West AE, Chen WG, Dalva MB. et al. Calcium regulation of neuronal gene expression. Proc Natl Acad Sci USA 2001;98:11024–11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Choi YS, Lee B, Cho HY. et al. CREB is a key regulator of striatal vulnerability in chemical and genetic models of Huntington's disease. Neurobiol Dis 2009;36:259–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Budreck EC, Kwon O-B, Jung JH. et al. Neuroligin-1 controls synaptic abundance of NMDA-type glutamate receptors through extracellular coupling. Proc Natl Acad Sci USA 2013;110:725–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Espinosa F, Xuan Z, Liu S. et al. Neuroligin 1 modulates striatal glutamatergic neurotransmission in a pathway and NMDAR subunit-specific manner. Front Synaptic Neurosci 2015;7:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Graf ER, Zhang X, Jin SX. et al. Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell 2004;119:1013–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sudhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature 2008;455:903–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Deng W, Luo F, Li B-M. et al. NRG1–ErbB4 signaling promotes functional recovery in a murine model of traumatic brain injury via regulation of GABA release. Exp Brain Res 2019;237:3351–3362. [DOI] [PubMed] [Google Scholar]
- 64. Li B, Woo R-S, Mei L. et al. The Neuregulin-1 receptor ErbB4 controls glutamatergic synapse maturation and plasticity. Neuron 2007;54:583–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Noll JM, Li Y, Distel TJ. et al. Neuroprotection by exogenous and endogenous Neuregulin-1 in mouse models of focal ischemic stroke. J Mol Neurosci 2019;69:333–342. [DOI] [PubMed] [Google Scholar]
- 66. Filosa A, Paixão S, Honsek SD. et al. Neuron-glia communication via EphA4/ephrin-A3 modulates LTP through glial glutamate transport. Nat Neurosci 2009;12:1285–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dalva MB, Takasu MA, Lin MZ. et al. EphB receptors interact with NMDA receptors and regulate excitatory synapse formation. Cell 2000;103:945–956. [DOI] [PubMed] [Google Scholar]
- 68. Fux CM, Krug M, Dityatev A. et al. NCAM180 and glutamate receptor subtypes in potentiated spine synapses: an immunogold electron microscopic study. Mol Cell Neurosci 2003;24:939–950. [DOI] [PubMed] [Google Scholar]
- 69. Sytnyk V, Leshchyns'ka I, Nikonenko AG. et al. NCAM promotes assembly and activity-dependent remodeling of the postsynaptic signaling complex. J Cell Biol 2006;174:1071–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cha JH, Kosinski CM, Kerner JA. et al. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human Huntington disease gene. Proc Natl Acad Sci USA 1998;95:6480–6485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cha JH, Frey AS, Alsdorf SA. et al. Altered neurotransmitter receptor expression in transgenic mouse models of Huntington's disease. Philos Trans R Soc Lond Ser B Biol Sci 1999;354:981–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Landwehrmeyer GB, Standaert DG, Testa CM. et al. NMDA receptor subunit mRNA expression by projection neurons and interneurons in rat striatum. J Neurosci 1995;15:5297–5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ali NJ, Levine MS. Changes in expression of N-methyl-D-aspartate receptor subunits occur early in the R6/2 mouse model of Huntington's disease. Dev Neurosci 2006;28:230–238. [DOI] [PubMed] [Google Scholar]
- 74. Arzberger T, Krampfl K, Leimgruber S. et al. Changes of NMDA receptor subunit (NR1, NR2B) and glutamate transporter (GLT1) mRNA expression in Huntingtonʼs disease—an In situ hybridization study. J Neuropathol Exp Neurol 1997;56:440–454. [DOI] [PubMed] [Google Scholar]
- 75. Pregi N, Belluscio LM, Berardino BG. et al. Oxidative stress-induced CREB upregulation promotes DNA damage repair prior to neuronal cell death protection. Mol Cell Biochem 2017;425:9–24. [DOI] [PubMed] [Google Scholar]
- 76. Browne SE, Bowling AC, MacGarvey U. et al. Oxidative damage and metabolic dysfunction in Huntington's disease: selective vulnerability of the basal ganglia. Ann Neurol 1997;41:646–653. [DOI] [PubMed] [Google Scholar]
- 77. Brennan WA Jr, Bird ED, Aprille JR. Regional mitochondrial respiratory activity in Huntington's disease brain. J Neurochem 1985;44:1948–1950. [DOI] [PubMed] [Google Scholar]
- 78. Gu M, Gash MT, Mann VM. et al. Mitochondrial defect in Huntington's disease caudate nucleus. Ann Neurol 1996;39:385–389. [DOI] [PubMed] [Google Scholar]
- 79. Lee H, Fenster RJ, Pineda SS. et al. Cell type-specific Transcriptomics reveals that mutant huntingtin leads to mitochondrial RNA release and neuronal innate immune activation. Neuron 2020;107:891–908.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Steffan JS, Kazantsev A, Spasic-Boskovic O. et al. The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci USA 2000;97:6763–6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Moily NS, Ormsby AR, Stojilovic A. et al. Transcriptional profiles for distinct aggregation states of mutant huntingtin exon 1 protein unmask new Huntington's disease pathways. Mol Cell Neurosci 2017;83:103–112. [DOI] [PubMed] [Google Scholar]
- 82. Chaturvedi RK, Hennessey T, Johri A. et al. Transducer of regulated CREB-binding proteins (TORCs) transcription and function is impaired in Huntington's disease. Hum Mol Genet 2012;21:3474–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kennedy L, Evans E, Chen CM. et al. Dramatic tissue-specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Hum Mol Genet 2003;12:3359–3367. [DOI] [PubMed] [Google Scholar]
- 84. Shelbourne PF, Keller-McGandy C, Bi WL. et al. Triplet repeat mutation length gains correlate with cell-type specific vulnerability in Huntington disease brain. Hum Mol Genet 2007;16:1133–1142. [DOI] [PubMed] [Google Scholar]
- 85. Gonitel R, Moffitt H, Sathasivam K. et al. DNA instability in postmitotic neurons. Proc Natl Acad Sci USA 2008;105:3467–3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Swami M, Hendricks AE, Gillis T. et al. Somatic expansion of the Huntington's disease CAG repeat in the brain is associated with an earlier age of disease onset. Hum Mol Genet 2009;18:3039–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Loupe JM, Pinto RM, Kim KH. et al. Promotion of somatic CAG repeat expansion by Fan1 knock-out in Huntington's disease knock-in mice is blocked by Mlh1 knock-out. Hum Mol Genet 2020;29:3044–3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kacher R, Lejeune FX, Noel S. et al. Propensity for somatic expansion increases over the course of life in Huntington disease. elife 2021;10:e64674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Anderson CM, Swanson RA. Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia 2000;32:1–14. [PubMed] [Google Scholar]
- 90. Bak LK, Schousboe A, Waagepetersen HS. The glutamate/GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J Neurochem 2006;98:641–653. [DOI] [PubMed] [Google Scholar]
- 91. Lievens JC, Woodman B, Mahal A. et al. Impaired glutamate uptake in the R6 Huntington's disease transgenic mice. Neurobiol Dis 2001;8:807–821. [DOI] [PubMed] [Google Scholar]
- 92. Tong X, Ao Y, Faas GC. et al. Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington's disease model mice. Nat Neurosci 2014;17:694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Hassel B, Tessler S, Faull RL. et al. Glutamate uptake is reduced in prefrontal cortex in Huntington's disease. Neurochem Res 2008;33:232–237. [DOI] [PubMed] [Google Scholar]
- 94. Estrada-Sanchez AM, Montiel T, Segovia J. et al. Glutamate toxicity in the striatum of the R6/2 Huntington's disease transgenic mice is age-dependent and correlates with decreased levels of glutamate transporters. Neurobiol Dis 2009;34:78–86. [DOI] [PubMed] [Google Scholar]
- 95. Hsu YT, Chang YG, Chern Y. Insights into GABA(a)ergic system alteration in Huntington's disease. Open Biol 2018;8:180165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Garret M, Du Z, Chazalon M. et al. Alteration of GABAergic neurotransmission in Huntington's disease. CNS Neurosci Ther 2018;24:292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Santhakumar V, Jones RT, Mody I. Developmental regulation and neuroprotective effects of striatal tonic GABAA currents. Neuroscience 2010;167:644–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Bayon-Cordero L, Ochoa-Bueno BI, Ruiz A. et al. GABA receptor agonists protect from excitotoxic damage induced by AMPA in oligodendrocytes. Front Pharmacol 2022;13:897056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Levine MS, Klapstein GJ, Koppel A. et al. Enhanced sensitivity to N-methyl-D-aspartate receptor activation in transgenic and knockin mouse models of Huntington's disease. J Neurosci Res 1999;58:515–532. [PubMed] [Google Scholar]
- 100. Cepeda C, Ariano MA, Calvert CR. et al. NMDA receptor function in mouse models of Huntington disease. J Neurosci Res 2001;66:525–539. [DOI] [PubMed] [Google Scholar]
- 101. Starling AJ, Andre VM, Cepeda C. et al. Alterations in N-methyl-D-aspartate receptor sensitivity and magnesium blockade occur early in development in the R6/2 mouse model of Huntington's disease. J Neurosci Res 2005;82:377–386. [DOI] [PubMed] [Google Scholar]
- 102. Beister A, Kraus P, Kuhn W. et al. The N-methyl-D-aspartate antagonist memantine retards progression of Huntington's disease. J Neural Transm 2004;68:117–122. [DOI] [PubMed] [Google Scholar]
- 103. Ondo WG, Mejia NI, Hunter CB. A pilot study of the clinical efficacy and safety of memantine for Huntington's disease. Parkinsonism Relat Disord 2007;13:453–454. [DOI] [PubMed] [Google Scholar]
- 104. Lucetti C, Del Dotto P, Gambaccini G. et al. IV amantadine improves chorea in Huntington's disease: an acute randomized, controlled study. Neurology 2003;60:1995–1997. [DOI] [PubMed] [Google Scholar]
- 105. Verhagen Metman L, Morris MJ, Farmer C. et al. Huntington's disease: a randomized, controlled trial using the NMDA-antagonist amantadine. Neurology 2002;59:694–699. [DOI] [PubMed] [Google Scholar]
- 106. Galluzzi L, Vitale I, Aaronson SA. et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ 2018;25:486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Hansson O, Petersen A, Leist M. et al. Transgenic mice expressing a Huntington's disease mutation are resistant to quinolinic acid-induced striatal excitotoxicity. Proc Natl Acad Sci USA 1999;96:8727–8732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Hansson O, Castilho RF, Korhonen L. et al. Partial resistance to malonate-induced striatal cell death in transgenic mouse models of Huntington's disease is dependent on age and CAG repeat length. J Neurochem 2001;78:694–703. [DOI] [PubMed] [Google Scholar]
- 109. Hansson O, Guatteo E, Mercuri NB. et al. Resistance to NMDA toxicity correlates with appearance of nuclear inclusions, behavioural deficits and changes in calcium homeostasis in mice transgenic for exon 1 of the Huntington gene. Eur J Neurosci 2001;14:1492–1504. [DOI] [PubMed] [Google Scholar]
- 110. Strong MK, Southwell AL, Yonan JM. et al. Age-dependent resistance to Excitotoxicity in Htt CAG140 mice and the effect of strain background. J Huntingtons Dis 2012;1:221–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Percie du Sert N, Hurst V, Ahluwalia A. et al. The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. PLoS Biol 2020;18:e3000410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Krishnaswami SR, Grindberg RV, Novotny M. et al. Using single nuclei for RNA-seq to capture the transcriptome of postmortem neurons. Nat Protoc 2016;11:499–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014;30:923–930. [DOI] [PubMed] [Google Scholar]
- 114. Liao Y, Smyth GK, Shi W. The subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res 2013;41:e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Danecek P, Bonfield JK, Liddle J. et al. Twelve years of SAMtools and BCFtools. Gigascience 2021;10:giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Xu J, Falconer C, Nguyen Q. et al. Genotype-free demultiplexing of pooled single-cell RNA-seq. Genome Biol 2019;20:290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Jiang A, Lehnert K, You L. et al. ICARUS, an interactive web server for single cell RNA-seq analysis. Nucleic Acids Res 2022;50:W427–W433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Jiang A, You L, Snell RG. et al. Delineation of complex gene expression patterns in single cell RNA-seq data with ICARUS v2.0. NAR Genom Bioinform 2023;5:lqad032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Hao Y, Hao S, Andersen-Nissen E. et al. Integrated analysis of multimodal single-cell data. Cell 2021;184:3573–3587.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Korsunsky I, Millard N, Fan J. et al. Fast, sensitive and accurate integration of single-cell data with harmony. Nat Methods 2019;16:1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Malaiya S, Cortes-Gutierrez M, Herb BR. et al. Single-nucleus RNA-Seq reveals dysregulation of striatal cell identity due to Huntington's disease mutations. J Neurosci 2021;41:5534–5552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Finak G, McDavid A, Yajima M. et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol 2015;16:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Wu T, Hu E, Xu S. et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation 2021;2:100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Morgan M, Shepherd L. AnnotationHub: Client to access AnnotationHub resources. R package version 3.12.0. 2024.
- 125. Shannon P, Markiel A, Ozier O. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003;13:2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Aibar S, González-Blas CB, Moerman T. et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods 2017;14:1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Lambert SA, Jolma A, Campitelli LF. et al. The human transcription factors. Cell 2018;175:598–599. [DOI] [PubMed] [Google Scholar]
- 128. Huynh-Thu VA, Irrthum A, Wehenkel L. et al. Inferring regulatory networks from expression data using tree-based methods. PLoS One 2010;5:e12776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 2002;30:207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data have been deposited in NCBI’s Gene Expression Omnibus (GEO) [129] and are accessible through accession number GSE229839 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc= GSE229839).
The code used in this study are deposited on zenodo available at https://doi.org/10.5281/zenodo.8057929.