

Abstract
Inefficient drug penetration hurdled by the stroma in the tumor tissue leads to a diminished therapeutic effect for drugs and a reduced infiltration level of immune cells. Herein, we constructed a PEGylated dendritic epirubicin (Epi) prodrug (Epi-P4D) to regulate the metabolism of cancer-associated fibroblasts (CAFs), thus enhancing Epi penetration into both multicellular tumor spheroids (MTSs) and tumor tissues in mouse colon cancer (CT26), mouse breast cancer (4T1) and human breast cancer (MDA-MB-231) models. Enhanced cytotoxicity against CT26 MTSs and remarkable antitumor efficacy of Epi-P4D were ascribed to reduced fibronectin, α-SMA, and collagen secretion. Besides, thinning of the tumor tissue stroma and efficient eradication of tumor cells promoted the immunogenic cell death effect for dendritic cell (DC) maturation and subsequent immune activation, including elevating the CD4+ T cell population, reducing CD4+ and CD8+ T cell hyperactivation and exhaustion, and amplifying the natural killer (NK) cell proportion and effectively activating them. As a result, this dendritic nanomedicine thinned the stroma of tumor tissues to enhance drug penetration and facilitate immune cell infiltration for elevated antitumor efficacy.
Key words: Dendritic prodrug, Enhanced penetration, Metabolic intervention, Cancer-associated fibroblasts, Immunogenic cell death, Chemotherapy, Immunotherapy, Immune microenvironment reconstitution
Graphical abstract
A dendritic polymer-based nanomedicine was constructed to disturb the amino acid metabolism of cancer-associated fibroblasts for enhancing Epi penetration, boosting tumor immune cell infiltration, and elevating its therapeutic effect.
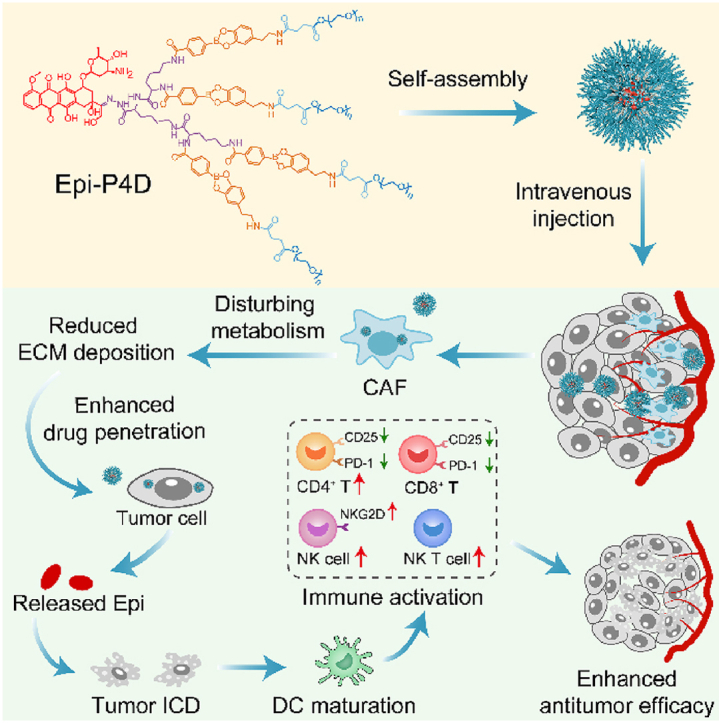
1. Introduction
Immunotherapy is one of the most promising approaches for the clinical treatment of cancer1,2, whereas insufficient stimulation of immunogenic cell death (ICD)3, inadequate infiltration of immune cells4, and ineffective modulation of the tumor immune microenvironment5,6 have severely diminished the therapeutic effect of immunotherapy. Combination of chemotherapy and immunotherapy has gained popularity as chemotherapeutic drugs in the form of nanomedicine can eradicate cancer cells to stimulate ICD7,8. However, along the delivery path of the nanomedicine to a tumor site via blood circulation9,10, poor extravasation11 and ineffective penetration of drugs in the nanomedicine12,13 are two hindering factors to achieve the expected killing effect of the nanomedicine. Consequently, unsatisfied ICD is triggered and weak/no immune response is provoked, leading to the failure of cancer treatment14.
One of the biological barriers to block the penetration of the nanomedicine is the stroma of tumor tissues15,16. Cancer-associated fibroblasts (CAFs), a key producer of the tumor extracellular matrix (ECM), play a vital role in tumor stroma deposition17,18. The accumulation of excess ECM leads to a dense stroma, which significantly impedes the delivery of nanomedicine to tumor cells and reduces the permeability of chemotherapeutic drugs19,20. Strategies of depleting CAFs have been explored to promote drug penetration into tumor tissues21, however, rapid depletion of CAFs results in profound immunosuppression and accelerated tumor progression, thus reducing the chemotherapeutic effect and the survival rate of patients.
Other strategies to enhance drug penetration include size or morphology transformation of nanomedicine at the tumor site or surface modification of nanomedicine with cell-penetrating peptides, and these strategies have been reported to promote drug penetration through the stoma barrier to some extent22,23. Nevertheless, the immunosuppressive tumor microenvironment remains unchanged for these strategies24. Alteration of the function of CAFs by reversing CAFs to normal fibroblasts has been demonstrated to improve the penetration of nanomedicine through the tumor stroma25,26. Small molecular inhibitors, such as JQ127 and losartan28, have been used to reduce ECM deposition, however, the desired therapeutic effect of these inhibitors is not achieved29. They have been combined with chemotherapeutic drugs to improve their therapeutic effect30, while the regulating action from these chemotherapeutic drugs on the ECM is still mysterious.
Herein, epirubicin (Epi) was employed as a model therapeutic agent as well as an ICD-inducing agent, and it was conjugated onto a boronate bond-linked PEGylated dendron via acid-labile hydrazone bonds. By manipulating the number of PEG molecules on a single dendron molecule, the Epi-PEGylated dendron prodrug (Epi-PD) with 4 PEG molecules was found to exhibit a uniform-sized nanostructure with superior aqueous stability. Enhanced cellular uptake of the prodrug contributed to its pro-apoptotic and antitumor effect in vitro. The prodrug disturbed the amino acid metabolism of CAFs, which helped to reduce the deposition of ECM, ultimately promoting penetration of the prodrug in multicellular tumor spheroids (MTSs) and in vivo tumor tissues. The released therapeutic drug Epi induced potent ICD, which evoked the activation and infiltration of antitumor immune cells and reduced hyperactivation and exhaustion of immunosuppressive cells, contributing to an excellent antitumor effect of the prodrug.
2. Materials and methods
2.1. Reagents
H-Lys-OMe·2HCl, Boc-Lys(Boc)-OH, 1-hydroxybenzotriazole hydrate (HOBT), and carbodiimide hydrochloride (EDC·HCl, EDCI) were purchased from GL Biochem. Ltd. (Shanghai, China). Dopamine hydrochloride, N,N′-dicyclohexylcarbodiimide (DCC), tert-butyl carbazate, N,N-diisopropylethylamine (DIEA), N-hydroxysuccinimide (NHS), 4-dimethylaminopyridine (DMAP), succinic anhydride (SA), 4-carboxybenzeneboronic acid pinacol ester, methoxypolyethylene glycol (mPEG, MW 1000 Da, mPEG1000), triethylamine (TEA), trifluoroacetic acid (TFA), and epirubicin hydrochloride (Epi·HCl) were purchased from Aladdin Reagents Company (Shanghai, China). Dimethylsulfoxide-d6 (DMSO-d6) was obtained from Sigma–Aldrich Co. (Steinheim, Germany). All other solvents were purchased from Tansoole Reagent Company (Shanghai, China).
Penicillin, streptomycin, and cell counter kit-8 (CCK-8) were purchased from MedChemExpress (MCE, USA). Hoechst 33342, DAPI, Annexin V-FITC/PI apoptosis detection kit, Matrigel matrix (Ceturegel®Matrix High Concentration, Phenol Red-Free, LDEV-Free), ATP luminescent cell viability assay kit, collagenase IV, and DNase I were purchased from YEASEN Biotechnology (Shanghai, China). Lysotracker Deep Red was purchased from Thermo Fisher Scientific (Invitrogen, USA). Roswell Park Memorial Institute (RPMI) 1640 medium, Dulbecco's modified Eagles medium (DMEM), and fetal bovine serum (FBS) were purchased from Hyclone (USA). Hyaluronidase, recombinant mouse GM-CSF, and recombinant mouse IL-4 were obtained from Absin (Shanghai, China). Anti-mouse calreticulin (CRT), primary antibody for anti-mouse high mobility group box 1 (HMGB1), and Cy5-labeled goat anti-rabbit IgG antibody were purchased from Bioss Biotechnology Co., Ltd., (Beijing, China). Purified rat anti-mouse CD16/CD32 for mouse Fc blocking, fixable viability stain 700 (FVS700), anti-CD45-FITC, anti-CD3-BV510, anti-CD4-BB700, anti-CD8-BV650, anti-CD25-PE-Cy7, anti-PD-1-BV605, and anti-CD69-BV786 antibodies were obtained from BD Pharmingen (USA). Anti-CD49b-eFluor450 antibody was obtained from eBioscience (USA). Anti-CD314-APC (NKG2D) antibody was purchased from Biolegend (USA). Enzyme-linked immunosorbent assay (ELISA) kit for granzyme B, perforin, tumor necrosis factor α (TNF-α), and interferon γ (IFN-γ) were obtained from Elabscience (Wuhan, China).
2.2. Preparation of the nanoassembly
The nanoassembly was prepared via an ultrasonic method. Briefly, 1.0 mg of Epi-conjugated PEGylated dendron (Epi-PD) dissolved in 20 μL of DMSO was dropped into 1 mL of deionized water and the mixture was subjected to ultrasound treatment for 30 min.
2.3. Epi loading amount
The Epi loading amount in Epi-PD was measured via a UV–Vis spectrometer. Briefly, Epi-PD with 1, 2, and 4 PEG chains in each dendron molecule were named Epi-P1D, Epi-P2D, and Epi-P4D, respectively, and they were weighed and dissolved in DMSO. The amount of Epi in Epi-PD was calculated from the standard curve by plotting the absorbance at 480 nm vs. the Epi·HCl concentration in DMSO.
2.4. Size distribution and morphology observation
The size distribution of the nanoassemblies at a concentration of 100 μg/mL was characterized on a Brookhaven BIC multiangle light scattering instrument at 25 °C. Each measurement was carried out in triplicates.
To observe the morphology of the nanoassembly, a field emission transmission electron microscope was used. A drop of the solution of the nanoassembly was spread onto a copper grid and dried in air at room temperature without phosphotungstic acid staining before analysis.
2.5. Cell culture
CT26 (murine colon cancer cell line) cells and 4T1 (murine mammary carcinoma cell line) cells were cultured in the RPMI 1640 medium supplemented with 10% FBS and antibiotics (100 U/mL of penicillin and 100 U/mL of streptomycin) in a humidified atmosphere containing 5% CO2 at 37 °C. MDA-MB-231 (human breast cancer cell line) cells and NIH/3T3 cells were cultured in the DMEM medium supplemented with 10% FBS and antibiotics (100 U/mL of penicillin and 100 U/mL of streptomycin).
2.6. Quantification of internalization of Epi-PD
CT26 cells at a logarithmic growth phase were seeded into a 6-well plate and incubated with Epi-P1D, Epi-P2D, and Epi-P4D (an Epi concentration of 5 μg/mL) for 3 h. Then the cells were collected and washed with cold phosphate-buffered saline (PBS) twice. The fluorescence intensity of the cells was monitored by flow cytometry and the cells without any treatment were used as a control.
2.7. Cellular internalization and intracellular trafficking
CT26 cells were seeded into a 35 mm glass-bottom dish and grew for 24 h. Epi-P1D, Epi-P2D, and Epi-P4D at an Epi concentration of 1 μg/mL were diluted with the cell culture medium and incubated with CT26 cells for different durations. The lysosomes and nuclei were stained with Lysotracker Deep Red and Hoechst 33342. These cells were washed twice with PBS and observed under a confocal laser scanning microscope (CLSM).
2.8. Penetration into multicellular tumor spheroids
Multicellular tumor spheroids (MTSs) were constructed to mimic tumor tissue and they were used to assess the penetration capacity of Epi·HCl or Epi-PD. To construct the 3D MTSs, CT26 or 4T1 cells were seeded into an ultra-low attachment 12-well plate (2 × 104 cells per well) in 2 mL culture medium for one week to grow into MTSs with a diameter of 200 μm31,32. The MDA-MB-231 MTSs were prepared via a hanging drop method33. Briefly, MDA-MB-231 cell suspensions were diluted in the DMEM medium containing 8% of the Matrigel matrix at a density of 5 × 104 cells per mL. 10 μL of the cell suspensions were dropped onto the lids of 96-well plates. After 48 h, the formed spheroids were transferred to an ultra-low attachment 12-well plate and incubated for another 5 days to obtain MDA-MB-231 MTSs.
To compare the penetration depth of Epi-PD into MTSs, MTSs were cultured with Epi·HCl, Epi-P1D, Epi-P2D, and Epi-P4D (an Epi concentration of 10 μg/mL) for 4 h. The spheroids were collected and washed with PBS twice. After they were resuspended in 200 μL of PBS in glass-bottom dishes, the MTSs were imaged with a CLSM at an interval of 10 μm.
2.9. Cytotoxicity assay
CT26 cells were seeded into a 96-well plate (5 × 103 cells per well) and the cells were cultured with Epi·HCl, Epi-P1D, Epi-P2D, and Epi-P4D at various Epi concentrations for 24 h. The medium was discarded and the cells were gently washed with PBS twice. The cell viability was quantified with the CCK-8 kit by reading the absorbance at 450 nm of each well using a microplate reader. The relative cell viability was calculated as in Eq. (1):
| Relative cell viability (%) = (ODsample − ODbackground)/(ODcontrol − ODbackground) × 100 | (1) |
2.10. Cell cycle distribution
CT26 cells were seeded into a 6-well plate and incubated with Epi·HCl, Epi-P1D, Epi-P2D, and Epi-P4D at an Epi concentration of 0.3 μg/mL or 1 μg/mL for 24 h. After they were collected and washed with cold PBS, the cells were fixed with cold 70% ethanol at 4 °C overnight. Then the cells were washed with cold PBS and stained with DAPI (2.5 μg/mL) at 37 °C in the dark for 30 min. These cells were analyzed via a flow cytometer.
2.11. Apoptosis assay
CT26 cells were seeded into a 6-well plate and grew for 24 h. The cells were cultured with Epi·HCl, Epi-P1D, Epi-P2D, and Epi-P4D at an Epi concentration of 0.3 μg/mL for 24 h, and stained with DAPI and Annexin V-FITC according to the manufacturer's protocol. These cells were analyzed by a flow cytometer.
2.12. Live/dead cell staining of CT26 cells and CT26 MTSs
CT26 cells and CT26 MTSs were cultured with Epi·HCl, or Epi-PD at an Epi concentration of 0.3 or 2 μg/mL for 24 h. The cells and the MTSs were stained with Calcein-AM (2 μmol/L) and PI (5 μg/mL) at 37 °C in the dark for 15 min. After they were washed with PBS twice, the obtained cells and MTSs were imaged with a CLSM.
2.13. Induction of ICD
The exposure of CRT, release of HMGB1, and secretion of ATP in CT26 cells were studied in vitro. CT26 cells were treated with Epi·HCl, Epi-P1D, Epi-P2D, or Epi-P4D at an equivalent Epi concentration of 1.5 μg/mL to assess the surface CRT exposure and HMGB1 release. Briefly, CT26 cells were seeded into 35 mm glass-bottom dishes and grew for 24 h. Epi·HCl, Epi-P1D, Epi-P2D, or Epi-P4D were added and incubated for another 24 h. For CRT detection, the treated CT26 cells were washed with cold PBS twice and fixed with 4% paraformaldehyde for 10 min. After blocking with 5% bovine serum albumin (BSA) in PBS for 30 min, the cells were incubated with a CRT primary antibody overnight at 4 °C. These cells were washed with cold PBS 3 times, and then incubated with a Cy5-labelled secondary antibody for 45 min and washed 3 times with PBS. After staining with DAPI, the cells were imaged with a CLSM. For HMGB1 detection, the fixed cells were permeated with 0.1% Triton X-100 for 10 min and underwent the same procedure as CRT staining. To detect ATP release, both ATP in the supernatant of the treated cells and within the treated cells were measured following the manufacturer's protocol.
2.14. In vitro DC maturation and cytokine secretion
Bone marrow-derived dendritic cells (BMDCs) were used to confirm the stimulation of DC maturation in vitro by Epi-PD. BMDCs were isolated from a 4-week-old male BALB/c mouse34. Briefly, the femur and tibiae were removed and the surrounding muscles were detached. The bone marrow was flushed out with cold PBS and filtrated through a 70 μm nylon cell strainer to remove debris. The red blood cells were lysed with a red blood cell lysis buffer. The obtained cells were resuspended into the RPMI 1640 medium containing GM-CSF (20 ng/mL) and IL-4 (10 ng/mL) and cultured in a 37 °C incubator with 5% CO2. On Day 3, an equal volume of the culture medium with GM-CSF (20 ng/mL) and IL-4 (10 ng/mL) was added gently into the cells. One half of the spent medium was removed and the same volume of fresh medium was replenished gently on Day 6. The cells were harvested on Day 8 and incubated with the supernatant of the CT26 cells cultured with Epi·HCl or Epi-PD for 24 h. The BMDCs were stained with FVS700, anti-CD11c, anti-MHC II, anti-CD80, and anti-CD86 antibodies and analyzed with a flow cytometer. The cytokines secreted by BMDCs within the supernatant were collected and detected with ELISA kits following the manufacturer's instructions.
2.15. Tumor models
All animal experiments were carried out in accordance with the guidelines of the animal ethics committee of Sichuan University (2018154A). Male BALB/c mice of 6-week-old were subcutaneously inoculated on the right flank with 5 × 105 CT26 cells resuspended in 50 μL of PBS. Female BALB/c mice and female BALB/c nude mice of 6-week-old were subcutaneously inoculated on the right flank with 5 × 105 4T1 cells in 50 μL of PBS or 8 × 106 MDA-MB-231 cells resuspended in 100 μL of PBS, respectively. When the tumor grew to 100 mm3, the mice were ready for ex vivo fluorescent imaging and tumor therapy. When the tumor length reached 1.5 cm or the body weight loss was more than 20%, the mouse was considered dead.
2.16. Ex vivo fluorescence imaging
To map the biodistribution of Epi-P1D, Epi-P2D, and Epi-P4D in the mice, the tumor-bearing mice were intravenously injected with Epi·HCl, Epi-P1D, Epi-P2D, or Epi-P4D at an Epi dose of 8 mg/kg. At a predetermined time point, the mice were sacrificed and the major organs and tumor tissues were harvested for ex vivo imaging with 480 nm excitation and 600 nm emission. The tumor tissues were also harvested for frozen sectioning and stained with DAPI and anti-CD31 antibody to detect the distribution of Epi via CLSM.
2.17. Antitumor therapy
The antitumor effect of Epi-PD was assessed on the CT26 tumor-bearing BALB/c mice. When the tumor volume reached 100 mm3, the mice were randomly divided into 5 groups (n = 5) and intravenously administrated with saline, Epi·HCl, Epi-P1D, Epi-P2D, or Epi-P4D every three days 4 times at an Epi dosage of 5 mg/kg. The tumor volume and the mice body weight were recorded for 21 days every three days. The tumor volume was calculated from Eq. (2):
| V = (Length × Width2)/2 | (2) |
The tumors were harvested and weighed at the end of the treatment.
2.18. Histological analysis
The major organs and tumors were harvested for hematoxylin-eosin staining (H&E), CD31, ki67, fibronectin, α-SMA, masson for collagen, and terminal-deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) staining following the manufacturer's protocols.
2.19. In vivo assessment of tumor-infiltrating immune cells
When the tumor volume of the CT26 tumor-bearing mice reached 100 mm3, the mice were randomly divided into 5 groups (n = 3) and they were intravenously administrated with saline, Epi·HCl, Epi-P1D, Epi-P2D or Epi-P4D every three days 4 times (an Epi dosage of 5 mg/kg). On Day 2 after the last treatment, the tumor tissue in each treatment group was separated, cut into pieces, and digested with hyaluronidase (1 mg/mL), collagenase IV (1 mg/mL), and DNase I (10 μg/mL) at 37 °C for 30 min. After the tissue was ground and filtrated through a 70 μm nylon cell strainer, a single cell suspension of the tumor tissue was obtained. After they were stained with FVS 700, blocked the Fc receptor, and stained with antibodies, the stained cells were collected for flow cytometry analysis.
2.20. Statistical analysis
All data were presented as mean ± SD (n ≥ 3). Statistical analysis was performed with a one-way analysis of variance (ANOVA). Statistical significance was defined as ns, not significant, ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001.
3. Results and discussion
A dual-functionalized dendron was decorated with the hydrazine hydrate for conjugating epirubicin hydrochloride (Epi·HCl) via a pH-sensitive hydrazone linker and phenylboronic acid (PBA) for conjugating polyethylene glycol (PEG) to obtain an Epi-conjugated PEG-decorated dendron (Epi-PD) (Supporting Information Figs. S1–S5). The PEGylated dendrons with different generations were used as the polymeric backbone. Three Epi-PDs with 1, 2, and 4 PEG chains in each dendron molecule were prepared, and they were named Epi-P1D, Epi-P2D, and Epi-P4D, respectively. The intermediate products were characterized with 1H NMR and 13C NMR (Supporting Information Figs. S6‒S11). As illustrated in 1H NMR spectra of Epi-PD (Supporting Information Fig. S12), the prodrug displayed a characteristic peak for aromatic protons in Epi at around 7.7 ppm and a characteristic peak of protons in PEG at around 3.5 ppm. The Epi load content was calculated to be 27.61%, 17.48%, and 6.29% for Epi-P1D, Epi-P2D, and Epi-P4D, respectively, from a standard curve via a UV–Vis spectrometer (Supporting Information Fig. S13 and Table S1).
The amphiphilic structure of Epi-PD endowed them with a self-assembly property. To examine the self-assembly process of Epi-PD, dissipative particle dynamics (DPD) simulation was employed. The chemical structures and molecular models of Epi-P1D, Epi-P2D, and Epi-P4D were shown in Fig. 1A, and dynamic formation of the nanoassembly in water was illustrated in Fig. 1B and Supporting Information Figs. S14 and S15. Epi-P1D, Epi-P2D, and Epi-P4D underwent a similar assembly procedure. The formation of small fragmentary aggregates was initially driven by hydrophobic interaction, and these small aggregates gradually grew into large irregular ones. Stable nanoparticles with a relatively regular nanostructure were finally obtained. Different ratios of the hydrophilic PEG moiety to the hydrophobic Epi moiety in Epi-PD resulted in different sizes of the final stable nanostructure.
Figure 1.

Chemical structures and characterizations of Epi-PD. (A) Molecular structures and Coarse-Grained models of Epi-P1D, Epi-P2D, and Epi-P4D. (B) The self-assembly processes of nanomedicine formation from Epi-P1D, Epi-P2D, and Epi-P4D. (C) Transmission electron microscopy (TEM) images and (D) hydrodynamic size distributions of Epi-P1D, Epi-P2D, and Epi-P4D. (E) Fluorescence emission spectra of Epi·HCl, Epi-P1D, Epi-P2D, and Epi-P4D in H2O at an equivalent Epi concentration excited at 480 nm. (F) Size variations of Epi-P1D, Epi-P2D, and Epi-P4D in H2O for up to 42 weeks (data are mean ± SD, n = 3). In vitro Epi release profiles from (G) Epi-P1D, (H) Epi-P2D, and (I) Epi-P4D in phosphate-buffered saline (PBS, pH 7.4, 6.5, and 5.0) within 72 h (data are mean ± SD, n = 3).
The morphology of Epi-PD was observed under transmission electron microscopy (TEM). In agreement with the simulation results, Epi-P4D self-assembled into a more uniform spherical nanostructure with a smaller size compared to Epi-P1D and Epi-P2D (Fig. 1C). The size distribution of Epi-PD in water was analyzed via dynamic light scattering (DLS). The self-assembled Epi-PD nanoassembly had a similar negative zeta potential of about −13 to −20 mV (Supporting Information Table S2). Epi-P1D displayed an irregular nanostructure with a hydrodynamic diameter of 200.0 ± 12.3 nm, Epi-P2D exhibited a relatively large hydrodynamic diameter of 224.9 ± 7.5 nm with a rough surface, while Epi-P4D presented a uniform spherical nanostructure with the smallest hydrodynamic diameter of 150.0 ± 1.9 nm (Fig. 1D). After Epi-PD was dissolved in H2O, Epi fluorescence signal could be detected, but the fluorescent intensity decreased significantly compared with Epi·HCl. The reduction in the fluorescence intensity was in the order of Epi-P2D, Epi-P1D, and Epi-P4D (Fig. 1E), indicating that hydrophobic interaction was the driving force for the self-assembly process to yield Epi-PD nanoparticles. Besides, the stability of Epi-PD in water was investigated by measuring the size and polydispersity index (PDI) variations. Both Epi-P1D and Epi-P2D exhibited significant size variations and the maximum size reached about 600 nm and the maximum PDI variation was 0.7 (Fig. 1F and Supporting Information Fig. S16). On the contrary, Epi-P4D maintained a size variation of less than 10 nm for up to 42 weeks (Fig. 1F and Supporting Information Fig. S17), indicating its superior aqueous stability. Such colloid stability of Epi-P4D would help prolonging its blood circulation time after intravenous injection35.
Epi was conjugated onto the functionalized dendron with different PEG chains via a pH-responsive hydrazone bond and the in vitro Epi release behavior from Epi-P1D, Epi-P2D, and Epi-P4D was investigated in PBS of pH 7.4, 6.5, and 5.0. The release of Epi from Epi-P1D, Epi-P2D, and Epi-P4D shared a similar release profile. Under a normal physiological environment (pH 7.4) and a tumor tissue-mimicking microenvironment (pH 6.5)36, no more than 10% and 25% Epi was released from Epi-P1D, Epi-P2D, and Epi-P4D within 72 h, respectively. However, at an acidic condition (pH 5.0) within tumor cells, the accumulative release of Epi from Epi-P1D and Epi-P2D exceeded 70% within 24 h, while Epi-P4D displayed a much faster Epi release rate with more than 80% drug released (Fig. 1G–I). These results manifested the successful construction of Epi-conjugated dendritic nanomedicines with pH-responsive Epi release behavior.
To examine the effect of the nanostructure of the Epi-PD nanoassembly on its cellular uptake and intracellular distribution, CT26 cells from a mice colorectal cancer cell line were used as a model for Epi-PD nanoassemblies. Epi-P4D was demonstrated to have the highest internalization level by CT26 cells, evidenced with a much stronger mean fluorescence intensity (MFI) compared with that of Epi-P1D and Epi-P2D (Fig. 2A and B). CLSM was used to observe the intracellular distribution of Epi-PD. Epi-P1D displayed moderate cellular internalization and its red fluorescence was overlapped with green fluorescence from lysosomes within 6 h. Epi-P2D was trapped in lysosomes within 3 h and it slowly escaped from lysosomes after 6 h incubation. In comparison, Epi-P4D showed the brightest fluorescence signal, and its red fluorescence overlapped with the green fluorescence of lysosomes within 1 h and it escaped from lysosomes within 6 h (Fig. 2C). The acidic condition within lysosomes would help to disrupt the hydrazone bond within Epi-PD for disassembly of the nanostructure and release Epi to kill cancer cells.
Figure 2.

Internalization and intracellular distribution of Epi-PD. (A) The internalization efficiency and (B) the mean fluorescence intensity (MFI) of Epi-PD via flow cytometry (data are mean ± SD, n = 3). ∗∗P < 0.01, and ∗∗∗P < 0.001 compared with the CT26 cells incubated with Epi-P4D. (C) Intracellular distribution of Epi-PD in CT26 cells. Nuclei and lysosome were stained with Hoechst 33342 (blue) and lysotracker (green), and Epi was shown in red.
Variations in the size and morphology of Epi-PD might affect its penetration into tumor tissues and its therapeutic effect. CT26 MTSs with a diameter of about 200 μm were established to mimic in vivo tumor tissues. After incubation with Epi·HCl or Epi-PD at an Epi concentration of 10 μg/mL for 4 h, red fluorescence signal within CT26 MTSs was scanned at an interval of 10 μm from top to bottom via CLSM. CT26 MTSs after treatment with Epi·HCl displayed a dim fluorescence signal at a depth of 60 μm and a barely detectable fluorescence signal at a depth of 90 μm, indicating poor penetration of Epi·HCl. In contrast, CT26 MTSs treated with Epi-P4D exhibited the strongest fluorescence signal at a depth of 60 μm and the fluorescence signal was distinctly detectable at a depth of 60 μm or 90 μm (Fig. 3A), demonstrating the strongest penetration ability of Epi-P4D.
Figure 3.

In vitro and in vivo penetration of Epi-PD. (A) CLSM images of CT26 multicellular tumor spheroids (MTSs) at different depths after incubation with Epi·HCl or Epi-PD (left) at an Epi concentration of 10 μg/mL for 4 h. Reconstructed 3D view of the fluorescence signal intensity (middle) and the fluorescence signal intensity along the white arrow at a depth of 60 μm (right). (B) Temporal changes in the Epi concentration in the blood of Epi·HCl, Epi-P1D, Epi-P2D, or Epi-P4D at an Epi dose of 5 mg/kg after a single intravenous administration (Data are mean ± SD, n = 4). (C) Ex vivo fluorescence images of major organs and tumor tissues after intravenous injection of Epi·HCl or Epi-PD (an Epi dose of 8 mg/kg) into the CT26 tumor-bearing mice at predetermined time points. (D) CLSM images of cryosections of CT26 tumors at 48 h post-injection. The blood vessels were stained with CD31 (green) and the nuclei were stained with DAPI (blue). The Epi signal was red.
Before examining the in vivo tumor tissue penetration of Epi-PD, the pharmacokinetics of Epi·HCl or Epi-PD were studied. The blood concentration of Epi·HCl decreased rapidly within 3 h after intravenous administration and its half-life time was about 1.11 h. Formulation of Epi in Epi-P1D, Epi-P2D, and Epi-P4D significantly prolonged the half-life time of Epi, which was 3.55-, 4.76-, and 6.05- fold of that of Epi·HCl, respectively (Fig. 3B and Supporting Information Table S3). A CT26 tumor model was established for the evaluation of tumor penetration of Epi-PD. After a single intravenous administration of Epi·HCl or Epi-PD intravenously, major organs and the tumor tissue were harvested for ex vivo imaging at a predetermined time point. Epi·HCl was rapidly eliminated and no accumulation of Epi·HCl was observed at the tumor site, on the contrary, Epi-PD exhibited distinct tumor accumulation. Among Epi-PD, Epi-P4D displayed the highest level of enrichment with the brightest fluorescence signal at the tumor tissue at 48 h (Fig. 3C and Supporting Information Fig. S18). After 48 h administration, the tumor tissues were sliced into frozen sections and blood vessels were stained to map in vivo drug distribution. The fluorescence signal was barely observed in the groups administrated with saline or Epi·HCl. In contrast, homogeneous red fluorescence signal extravasation from the blood vessels was detected after Epi-P4D administration, demonstrating it could be highly enriched in the tumor tissue, which could be credited to efficient extravasation from the blood vessel and deep drug penetration through the tumor tissue (Fig. 3D).
In addition, 4T1 murine mammary carcinoma cells and MDA-MB-231 human breast cancer cells were also applied to verify the penetration capacity of Epi-PD. 4T1 MTSs were incubated with Epi·HCl or Epi-PD for 4 h. The strongest fluorescence signal was seen within the MTSs treated with Epi-P4D, while dim fluorescence signal was confined only to the peripheral of the MTSs treated with Epi·HCl at a depth of 70 μm (Supporting Information Fig. S19). The ex vivo distribution of Epi-PD within the 4T1 tumor-bearing mice at 24 and 48 h after intravenous injection was also analyzed. The strongest fluorescence signal was found in the tumor tissue treated with Epi-P4D both at 24 and 48 h post-injection compared with other groups (Supporting Information Figs. S20 and S21) although there was no significant difference in the fluorescence intensity in the tumor tissue after treatment with Epi·HCl, Epi-P1D, and Epi-P2D. Interestingly, the ex vivo distribution pattern of Epi·HCl and Epi-PD in major organs including the heart, liver, spleen, lung, and kidney were quite similar. Fluorescence images of the cryosections of the tumor tissue revealed the greatest enrichment of Epi at the tumor section after treatment with Epi-P4D at 24 or 48 h post-injection compared with other groups (Supporting Information Fig. S22). Similar results were observed with MTSs and tumor tissues from the MDA-MB-231 cell line (Supporting Information Fig. S23‒S25). These results manifested that Epi-P4D could efficiently accumulate in the tumor tissue and deeply penetrate into MTSs from a variety of tumor cell spheroids or solid tumor tissues in tumor-bearing mice models.
To probe the mechanism underlying enhanced penetration of Epi-PD, we hypothesized that the cell matrix may become less dense after Epi-PD treatment. In the tumor microenvironment (TME), CAFs act as a key contributor to build a physical barrier for impeding the penetration of nanomedicine to protect tumor cells37,38. The CAFs could also impact tumor cell growth through their metabolite exchange with tumor cells39. In addition, CAFs may suppress the function of immune cells by hydrolyzing essential amino acids40 and promoting the expression of programmed cell death protein (PD-1) on immune cells41 to sustain an immunosuppressive microenvironment. Thus, targeted metabolomic analysis was performed to investigate the effect of Epi-P4D treatment on the CAF metabolism at a cellular metabolic level. CAFs are programmed from NIH/3T3 cells in the TME42. Compared with NIH/3T3 cells, the majority of amino acids were downregulated, while glutamine, alanine, proline, and creatine were upregulated in the CAFs (Supporting Information Fig. S26‒S28), demonstrating a significant change in amino acid metabolic pathways after fibroblasts were activated in the TME. The cytotoxicity of Epi-P4D on CAFs was analyzed, and Epi-P4D at an Epi concentration of 0.3 μg/mL barely inhibited CAF growth, and this concentration was chosen for the following experiments (Supporting Information Fig. S29). After treating CAFs with Epi-P4D at an Epi concentration of 0.3 μg/mL for 24 h, the amino acid metabolism was severely disturbed compared with CAFs without any treatment. The majority of amino acids were downregulated (Fig. 4A–C). Among these down-regulated amino acids, the concentrations of proline, alanine, and creatine were reduced to a similar level of that in the NIH/3T3 cells (Fig. 4C–F), suggesting that CAFs in the TME may be reprogrammed back to NIH/3T3 cells after Epi-P4D treatment and their contribution to the ECM could be significantly diminished. It was noted that the energy metabolism of CAFs was not impacted after Epi-P4D treatment (Fig. 4G).
Figure 4.

Disturbance of the CAF metabolism by Epi-P4D. (A) Principal component analysis (PCA) of the amino acid metabolism between the Epi-P4D-treated CAFs and CAFs without any treatment as a control. (B) Volcano plot and (C) heatmap of differential amino acids in the Epi-P4D treated CAFs compared to the CAFs without any treatment. Controls 1–5 and Epi-P4Ds 1–5 represent five replicates of the CAFs without any treatment and the Epi-P4D-treated CAFs, respectively. CAFs were treated with Epi-P4D at an Epi concentration of 0.3 μg/mL for 24 h. Concentrations of (D) proline, (E) alanine, and (F) creatine in the Epi-P4D-treated CAFs compared to CAFs without any treatment (Data are mean ± SD, n = 5). ∗∗P < 0.01, and ∗∗∗P < 0.001. (G) PCA of the energy metabolism between Epi-P4D-treated CAFs and CAFs without any treatment. (H) The proposed mechanism of disturbing the amino acid metabolism of CAFs by Epi-P4D for enhancing penetration of the nanomedicine and boosting its antitumor effect.
CAFs conduct biosynthesis of structural proteins in the ECM such as collagen and fibronectin, which often contain rich amino acid residues43. Collagen is composed of Gly–X–Y motifs, and X and Y are usually proline and hydroxyproline44. Ornithine, the hydrolysate of arginine, is a raw material for the synthesis of proline with the least energy cost45. Down-regulation of glycine, proline, hydroxyproline, and ornithine in the CAFs after Epi-P4D treatment would result in a decrease in the deposition of the ECM, which could promote the penetration of the nanomedicine (Fig. 4D and Supporting Information Fig. S30). Alanine, a non-essential amino acid derived from CAFs, serves as an alternative carbon source for tumor cells to reduce their reliance on glucose and serum nutrients46. Deprivation of alanine could lead to the interruption of this energy source to support the mitochondrial metabolism in the CAFs (Fig. 4E). In addition, a reduction in creatine could help impairing the polarization of macrophages to the M2-like phenotype47, which would relieve an immunosuppression microenvironment (Fig. 4F). Thus, the intervention of CAFs by Epi-P4D could decrease the deposition of structural proteins for the ECM, reduce the energy supply to cancer cells and relieve immunosuppression, thus enhancing nanomedicine penetration and promoting its antitumor effect (Fig. 4H).
The in vitro antitumor effect of three Epi-PD compounds was examined by assessing their cytotoxicity on CT26 cells. Both Epi·HCl and Epi-PD displayed their cytotoxic effects in a dosage-dependent manner. Epi·HCl had the smallest IC50 value, while Epi-P4D exhibited a comparable cytotoxic effect as Epi·HCl (Fig. 5A). Epi·HCl, an anthracycline, destroys cancer cells by binding to the DNA in the nucleus and arresting the cell cycle at the G2/M phase48. In the control group, a large population of tumor cells stayed at the G0/G1 phase. The fraction of CT26 cells at the G2/M phase significantly increased from 21% to 43% after treatment of CT26 cells with Epi-P2D at an Epi concentration of 0.3 μg/mL, while this fraction was elevated to over 60% after Epi-P1D and Epi-P4D treatment at the same Epi concentration. Treatment with Epi·HCl at an Epi concentration of 0.3 μg/mL led to around 76% of CT26 cells at the G2/M phase (Fig. 5B and C). The proportion of CT26 cells at the G2/M phase was elevated after treatment with Epi·HCl and three Epi-PD compounds at an Epi concentration of 1 μg/mL, compared to that treated at an Epi concentration of 0.3 μg/mL (Supporting Information Fig. S31).
Figure 5.

In vitro antitumor efficacy of Epi-PD against CT26 cells. (A) Cytotoxicity of CT26 cells after incubation with Epi·HCl or Epi-PD for 24 h (Data are mean ± SD, n = 5). (B) Cell cycle distribution and (C) quantification of CT26 cell populations at different cell cycle phases after incubation with Epi·HCl or Epi-PD (an Epi concentration of 0.3 μg/mL) for 24 h. (D) Apoptosis analysis of CT26 cells via flow cytometry. (E) Calcein-AM/PI staining of CT26 MTSs after different treatments.
Cell apoptosis was further examined by Annexin V-FITC/DAPI staining. Epi·HCl treatment led to 13.1 ± 0.6% of cells with late apoptosis and 15.5 ± 0.6% with early apoptosis. Compared with Epi-P1D or Epi-P2D, 10.0 ± 1.3% of cells with late apoptosis and 15.4 ± 3.4% with early apoptosis were found after treatment with Epi-P4D. The difference in the percentage of tumor cells with late apoptosis after treatment with Epi-P4D compared to that after treatment with Epi·HCl might be explained by the gradual release of Epi from Epi-P4D in the tumor cells after its internalization, but rapid diffusion of Epi·HCl from an extracellular environment into the nuclei, thus, more tumor cells after Epi-P4D treatment entered early apoptosis but less entered late apoptosis (Fig. 5D and Supporting Information Fig. S32). To visualize the antitumor effect on CT26 cells or MTSs, CT26 cells and MTSs were stained with Calcein-AM for live cells and PI for dead cells after they were treated with Epi·HCl or Epi-PD for 24 h at an Epi concentration of 0.3 and 2 μg/mL, respectively. A distinct red signal, which was generated from dead cells, was observed in CT26 cells after treatment with Epi-P4D (Supporting Information Fig. S33). A small fraction of dead cells were observed in the Epi·HCl-treated MTSs, which might be ascribed to poor penetration of Epi·HCl. An increase in dead cells was found in both Epi-P1D and Epi-P2D-treated MTSs owing to an improvement in their penetration compared to Epi·HCl. More encouragingly, the highest number of dead cells was observed in the MTSs treated with Epi-P4D. These dead cells were distributed from the periphery to the inner core of the MTSs, manifesting superior penetration and potent therapeutic efficacy of Epi-P4D (Fig. 5E).
The antitumor drug, Epi, has been reported to induce ICD of cancer cells by the release of damage-associated molecular patterns (DAMPs), such as calreticulin (CRT), high mobility group box 1 (HMGB1) and adenosine triphosphate (ATP), to activate the systemic immunity. Dendritic cells (DCs), one of the most common antigen-presenting cells49, were chosen to assess the potency of Epi-PD in stimulating immature DCs (imDCs) into mature DCs (mDCs) (Fig. 6A). Treatment with Epi·HCl or Epi-PD led to evident surface exposure of CRT (Fig. 6B) and pronounced release of HMGB1 from the nuclei (Fig. 6C). In addition, a much smaller intracellular ATP concentration was seen in the cells treated with Epi·HCl and Epi-P4D compared with that in other groups (Fig. 6D), suggesting that more intracellular ATP was released into the supernatant. The greatest level of ATP release was found in the cells treated with Epi-P4D (Fig. 6E). After successfully inducing ICD of CT26 cells by Epi-PD, DCs could be stimulated to become mature. The percentage of matured BMDCs (CD80+CD86+ cells within CD11c+MHC II+ cells) was investigated via flow cytometry. 21.6 ± 1.0% of mDCs were obtained after stimulation by the supernatant of Epi·HCl-treated CT26 cells, and 18.2 ± 1.0% by the supernatant of the Epi-P4D-treated cells. The stimulation of imDCs was less effective by the supernatant of cells treated with Epi-P1D or Epi-P2D (Fig. 6F and G). In addition, the stimulation of BMDC maturation by Epi-P4D led to significant upregulation of secreted cytokines, including TNF-α and IFN-γ (Supporting Information Fig. S34). These results demonstrated that Epi-P4D could serve as a powerful ICD-inducing agent to evoke robust systemic immunity and enhance its antitumor potency.
Figure 6.

In vitro ICD effect and DC maturation stimulation. (A) Schematic illustration of the ICD effect induced by CT26 cells treated with different Epi formulations to stimulate mouse bone marrow DC (BMDC) maturation. Representative CLSM images of (B) surface calreticulin exposure and (C) release of high mobility group box 1 (HMGB1) in CT26 cells after culturing with Epi·HCl or Epi-PD for 24 h. (D) Intracellular and (E) extracellular adenosine triphosphate (ATP) concentrations in CT26 cells after the same treatment as (B) (Data are mean ± SD, n = 4). (F) Flow cytometry analysis and (G) statistical data of mature BMDCs (CD80+CD86+ cells in CD11c+MHC II+ cells) in vitro (Data are mean ± SD, n = 3). ns, not significant, ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 compared with the Epi-P4D-treated group.
The antitumor effect of Epi-PD on CT26 MTSs and CT26 tumor-bearing mice was assessed. CT26 MTSs were cultured with Epi·HCl or Epi-PD with an equivalent Epi dosage 3 times every other day. As shown in Fig. 7A, CT26 MTSs without any treatment rapidly grew during the 15-day treatment. Treatment with Epi·HCl, Epi-P1D, or Epi-P2D at 2.5 μg/mL distinctly hindered the growth of MTSs, and these MTSs appeared to have a very similar size on Days 1 and 15. Epi-P4D treatment significantly inhibited the growth of MTSs with distinct size shrinkage. The volume of MTSs in the control group had a 4.5-time increase compared to its initial volume. In contrast, Epi-P4D treatment resulted in a reduced volume of MTSs, which was 50% of the initial volume (Fig. 7B). Suppression of tumor growth could also be observed in the MTSs treated with Epi-P4D after reducing the Epi concentration down to 1 μg/mL, while slightly growth of MTSs was observed after Epi·HCl, Epi-P1D, or Epi-P2D treatment at such a low Epi concentration (Supporting Information Fig. S35). These results were in line with the penetration performance of Epi-P4D, suggesting the deepest penetration could be correlated with the best anti-MTS growth effect.
Figure 7.

Shrinkage of MTSs and in vivo antitumor effect. (A) Images of CT26 MTSs on Days 1, 5, 9, 13, and 15 after different treatments at an Epi concentration of 2.5 μg/mL every other day 3 times. (B) Temporal changes in the volume of MTSs in different groups (Data are mean ± SD, n = 5). (C) Changes in the individual tumor volume of the mice after intravenous administration of different Epi formulations at an Epi dose of 5 mg/kg every 3 days 4 times. Mice administrated with saline were used as a control. (D) Tumor growth inhibition (TGI) profile of CT26 tumor-bearing mice (Data are mean ± SD, n = 5). (E) Changes in the body weight of the CT26 tumor-bearing mice during the treatment course (Data are mean ± SD, n = 5). ns, not significant, ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 compared with the Epi-P4D-treated group. (F) Hematoxylin-eosin (H&E) staining and immunohistochemistry images of fibronectin (brown) and α-SMA (brown) in the tumor tissue.
The in vivo antitumor effect was investigated on the CT26 tumor-bearing mice that received saline, Epi·HCl, Epi-P1D, Epi-P2D, or Epi-P4D at an intravenous dosage of 5 mg Epi/kg 4 times every 3 days. Epi-P1D and Epi-P2D slightly curbed tumor growth. Compared to a tumor volume of 1500 mm3 in the saline-treated mice, the tumor volume reached about 300 mm3 after treatment with Epi-P4D. The tumor volume was even smaller than that of the Epi·HCl-treated mice (Fig. 7C and Supporting Information Figs. S36 and S37). When the mice were sacrificed on Day 21 after the first treatment, the major organs and tumor tissues were harvested for pathological and immunohistochemical analysis. The harvested tumors were weighed and the tumor growth inhibition (TGI) after Epi-P4D treatment was up to 80%, which was significantly higher than that in other groups (Fig. 7D). Nevertheless, more than 15% body weight loss occurred after administration of Epi·HCl that was not regained at the end of the experiment, manifesting serious side effects of Epi·HCl (Fig. 7E).
Fibronectin, α-SMA, and collagen have been reported to be the main components of the stroma in tumor tissues that hinder the penetration of nanomedicine. The influence of Epi-PD treatment on the stroma components was assessed. A pronounced reduction in fibronectin (brown spot), α-SMA (brown spot), and collagen (blue) was noticed after administration of Epi-PD, and there was the smallest amount of these proteins after Epi-P4D treatment (Fig. 7F and Supporting Information Figs. S38 and S39). Additionally, Epi-P4D treatment led to an increase in abnormal cells, a remarkable reduction in the levels of proliferation and angiogenesis, and an increased level of tumor cell apoptosis (Fig. 7F and Supporting Information Figs. S40 and S41), indicating a potent antitumor effect of Epi-P4D. Encouragingly, no noticeable pathological changes were found in the heart, liver, spleen, lung, and kidney after treatment with Epi-PD (Supporting Information Fig. S42). The expression of the fibronectin, α-SMA, and collagen in 4T1 tumor tissues was also analyzed. Similarly, an obvious reduction of structural proteins could be observed after Epi-P4D treatment (Supporting Information Figs. S43 and S44).
The excellent antitumor effect of Epi-PD might be credited to a change in the tumor immune microenvironment in the tumor tissue. The population of infiltrated immune cells at the tumor site was analyzed by flow cytometry and the gating strategies were shown in Supporting Information Fig. S45. The percentage of the infiltrated natural killer (NK) T cells (CD45+CD3+CD49+ cells) was elevated from 4.17 ± 1.61% in the saline-injected group to 6.73 ± 0.71% in the group after Epi-P4D treatment (Fig. 8A and B). The populations of NK T cells including activated (CD69+)50,51, hyperactivated (CD25+)52, and exhausted (PD-1+)53 were analyzed and no significant difference was detected in these NK T cell populations before or after treatments (Supporting Information Figs. S46–S48). The populations of NK cells (CD45+CD3−CD49b+ cells) in the Epi-PD-treated groups increased at varying degrees. Compared to 12.43 ± 2.20% of NK cells in the control group, the highest NK cell percentage of 18.20 ± 1.91% was achieved in the Epi-P4D-treated group. It was noted that Epi·HCl treatment led to a decrease in the NK cell portion down to 8.75 ± 2.70% (Fig. 8A and C). The expression of natural killer group 2 member D (NKG2D) has been demonstrated to activate NK cell immunity for antitumor activity54. The percentage of NKG2D+ NK cells after Epi-P4D treatment reached up to 48.10 ± 1.13%, which was significantly higher than that in other groups, indicating that about half of the NK cells were in an activated status (Fig. 8D and E). Exhausted NK (PD-1+ NK) cells in all treatment groups were at a low level (less than 10%) and no significant difference was found in the exhausted NK cells in these treated mice except those treated with Epi-P1D (Supporting Information Fig. S49).
Figure 8.

Modulation of the tumor immune microenvironment. (A) Flow cytometry diagrams and (B) quantitative results of natural killer (NK) T cells (CD45+CD3+CD49b+) and (C) NK cells (CD45+CD3−CD49b+) in the tumors of the CT26 tumor-bearing mice after treatment with Epi·HCl, Epi-P1D, Epi-P2D, or Epi-P4D (Data are mean ± SD, n = 3). (D) Flow cytometry analysis and (E) statistical results of activated (NKG2D+) NK cells (Data are mean ± SD, n = 3). (F) Flow cytometry analysis of CD4+ T cells (CD45+CD3+CD4+) and CD8+ T cells (CD45+CD3+CD8+) in the tumors after different treatments. (G) Illustration of changes in the populations of immune cells in the tumor after treatment with Epi-P4D. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 compared with the Epi-P4D-treated group.
We further analyzed the populations of cytotoxic T lymphocytes (CD8+ T cells) and helper T cells (CD4+ T cells). A comparable level of the CD8+ T cell portion was observed in the mice after saline or Epi-PD treatment, while Epi·HCl treatment led to a decrease in the population of CD8+ T cells (Fig. 8F and Supporting Information Fig. S50). Activated CD8+CD69+ T cells at a high portion of about 75% were seen in the groups treated with saline and Epi·HCl, while 60% in the group after Epi-PD treatment (Supporting Information Fig. S51). Besides, hyperactivated CD8+ T cells (CD8+CD25+ T cells) in the saline-treated group were notably higher than those in other groups (Supporting Information Fig. S52). Since a high level of PD-1 expression is positively correlated with a great level of T cell exhaustion, the PD-1 expression level of CD8+ T cells was assessed. About 88% of CD8+ T cells were exhausted after treating with saline or Epi·HCl, on the contrary, a decrease in the population of CD8+PD-1+ T cells was seen in the mice after treatment with Epi-PD (Supporting Information Fig. S53).
In addition, a relatively higher proportion of CD4+ T cells, up to 26%, was found in the mice after treatment with Epi-P4D, compared to that in saline, Epi·HCl and Epi-P1D groups (Fig. 8F and Supporting Information Fig. S50). The proportion of activated CD4+ T cells (CD4+CD69+ T cells) in the Epi-P4D-treated group was remarkably higher than that in the groups treated with Epi-P1D and Epi-P2D (Supporting Information Fig. S54). Besides, the populations of hyperactivated CD4+CD25+ T cells and dysregulated CD4+PD-1+ T cells after Epi-P4D treatment were lower than those in the groups administrated with Epi·HCl or Epi-P1D (Supporting Information Figs. S55 and S56). The influence of different treatments on cytokine secretion was also analyzed. The levels of granzyme B and perforin in the serum and tumor tissues were significantly upregulated after Epi-P4D treatment, which might be attributed to the increased population of NK cells55. Furthermore, the levels of TNF-α and IFN-γ after Epi-P4D treatment were also upregulated significantly compared with the other four groups (Supporting Information Figs. S57 and S58). The number of immune cells in the blood, including white blood cells (WBCs), neutrophils, lymphocytes, and monocytes, were counted through blood routine analysis. The number of WBCs and neutrophils in the blood did not have a significant change before or after Epi·HCl or Epi-PD treatment, while the number of lymphocytes decreased after Epi·HCl or Epi-P1D treatment. It was noted that the number of monocytes after Epi·HCl treatment increased significantly compared to that in the Epi-PD-treated groups (Supporting Information Fig. S59). The decrease in the number of lymphocytes and the increase in the number of monocytes after Epi·HCl treatment compared with Epi-P4D treatment might be attributed to the side effect of myelosuppressive effect caused by the poor tumor accumulation of Epi·HCl. These results demonstrated that Epi-P4D treatment provoked strong immune surveillance to achieve an enhanced antitumor effect by promoting the population of NK cells and activating them, reducing hyperactivated CD4+CD25+/CD8+CD25+ T cells and dysregulated CD4+PD-1+/CD8+PD-1+ T cells (Fig. 8G), and boosting the secretion of cytokines.
4. Conclusions
We constructed an Epi-conjugated PEGylated dendron (Epi-PD) by manipulating the number of PEG chains in each dendron molecule. Epi-PD with 4 PEG chains (Epi-P4D) exhibited a uniform-sized nanostructure, great aqueous stability for up to 42 weeks, and an enhanced level of cellular internalization. Disturbance of the amino acid metabolism of CAFs by treatment with Epi-P4D led to a reduced level of ECM deposition, thus achieving superior accumulation and remarkable penetration of Epi-P4D in MTSs and tumor tissues from CT26, 4T1, and MDA-MB-231 cell lines. As a result, a potent antitumor effect with a tumor growth inhibition of about 80% was obtained after Epi-P4D treatment, encouragingly, the systemic toxic effect of Epi was reduced after the Epi nanomedicine, Epi-P4D, was administrated. Furthermore, the infiltration of antitumor immune cells, including NK cells, NK T cells, and CD4+ T cells, was elevated. Among these immune cells, NK cell activation was boosted, and CD4+ and CD8+ T cell hyperactivation and exhaustion were mitigated, which enhanced the antitumor effect of Epi-P4D. Therefore, the dendritic nanomedicine from a facile fabrication method could enhance drug penetration, promote immune cell infiltration, and activate the function of immune cells by disturbing the amino acid metabolism of CAFs.
Author contributions
Yunkun Li and Kui Luo designed the research. Yunkun Li carried out the experiment, and data analysis, and wrote the manuscript. Xiaoding Shen, Haitao Ding, Yuxin Zhang, and Dayi Pan participated part of the cellular experiments and data collection. Haitao Ding, Yuxin Zhang, Liping Su, Yahui Wu, Zaixiang Fang, and Jie Zhou participated part of the animal experiments and data collection. Qiyong Gong and Kui Luo supervised the work. Kui Luo revised the manuscript. All the authors have read and approved the final manuscript.
Conflicts of interest
The authors have no conflicts of interest to declare.
Acknowledgments
This work was supported by the National Science and Technology Major Project of China (2023YFB3810004), the National Natural Science Foundation of China (32271445, 52103175, 32271382, 32101145), the National Key Research and Development Program of China (2022YFC2009900), Department of Science and Technology of Sichuan Province (24NSFJQ0167, China), 1·3·5 Project for Disciplines of Excellence, West China Hospital, Sichuan University (ZYJC21013, China), Post-Doctor Research Project, West China Hospital, Sichuan University (2021HXBH055, 2021HXBH057, China) and China Postdoctoral Science Foundation (2021M692314). The authors would like to thank the Analytical and Testing Center of Sichuan University for morphology characterization and NMR spectroscopy by Prof. Shanling Wang and Prof. Pengchi Deng. The authors thank Zhiqian Li, Yaping Wu, Lei Wu, Qiuxiao Shi, Lin Bai, Xiaoting Chen, Yang Yang, Yan Wang, Dan Li, Cong Li, Jian Yang, Qiaorong Huang, Xue Li, Wentong Meng, Li Li, and Fei Chen (Histology and Imaging Platform, Experimental Animal Center of West China Hospital, Research Core Facility, Laboratory of Stem Cell Biology, Institute of Clinical Pathology, West China Hospital, Sichuan University) for sample analysis, imaging, histological staining, cellular experiment and animal experiment.
Footnotes
Peer review under the responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting information to this article can be found online at https://doi.org/10.1016/j.apsb.2024.03.010.
Appendix A. Supporting information
The following is the Supporting information to this article.
References
- 1.Dagher O.K., Schwab R.D., Brookens S.K., Posey A.D. Advances in cancer immunotherapies. Cell. 2023;186:1814–1814e1. doi: 10.1016/j.cell.2023.02.039. [DOI] [PubMed] [Google Scholar]
- 2.Yang K.L., Halima A., Chan T.A. Antigen presentation in cancer-mechanisms and clinical implications for immunotherapy. Nat Rev Clin Oncol. 2023;20:604–623. doi: 10.1038/s41571-023-00789-4. [DOI] [PubMed] [Google Scholar]
- 3.Li H.N., Luo Q., Zhang H., Ma X.L., Gu Z.W., Gong Q.Y., et al. Nanomedicine embraces cancer radio-immunotherapy: mechanism, design, recent advances, and clinical translation. Chem Soc Rev. 2023;52:47–96. doi: 10.1039/d2cs00437b. [DOI] [PubMed] [Google Scholar]
- 4.Zhang X.K., Yang Y.Z., Li J.X., Xie X.R., Hao Y.Q., Zhu Y.C., et al. Engineered oxygen factories synergize with STING agonist to remodel tumor microenvironment for cancer immunotherapy. Adv Funct Mater. 2023;33 [Google Scholar]
- 5.Li Y.K., Wu Y.H., Fang Z.X., Zhang Y.X., Ding H.T., Ren L., et al. Dendritic nanomedicine with boronate bonds for augmented chemo-immunotherapy via synergistic modulation of tumor immune microenvironment. Adv Mater. 2024;36 doi: 10.1002/adma.202307263. [DOI] [PubMed] [Google Scholar]
- 6.Li M.O., Wolf N., Raulet D.H., Akkari L., Pittet M.J., Rodriguez P.C., et al. Innate immune cells in the tumor microenvironment. Cancer Cell. 2021;39:725–729. doi: 10.1016/j.ccell.2021.05.016. [DOI] [PubMed] [Google Scholar]
- 7.Yap T.A., Parkes E.E., Peng W.Y., Moyers J.T., Curran M.A., Tawbi H.A. Development of immunotherapy combination strategies in cancer. Cancer Discov. 2021;11:1368–1397. doi: 10.1158/2159-8290.CD-20-1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nam J., Son S., Park K.S., Zou W.P., Shea L.D., Moon J.J. Cancer nanomedicine for combination cancer immunotherapy. Nat Rev Mater. 2019;4:398–414. [Google Scholar]
- 9.Li H.N., Sun J.Y., Zhu H.Y., Wu H.X., Zhang H., Gu Z.W., et al. Recent advances in development of dendritic polymer-based nanomedicines for cancer diagnosis. WIRES Nanomed Nanobi. 2021;13 doi: 10.1002/wnan.1670. [DOI] [PubMed] [Google Scholar]
- 10.Li H.N., Feng Y., Luo Q., Li Z.Q., Li X., Gan H.T., et al. Stimuli-activatable nanomedicine meets cancer theranostics. Theranostics. 2023;13:5386–5417. doi: 10.7150/thno.87854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou Q., Li J.J., Xiang J.J., Shao S.Q., Zhou Z.X., Tang J.B., et al. Transcytosis-enabled active extravasation of tumor nanomedicine. Adv Drug Delivery Rev. 2022;189 doi: 10.1016/j.addr.2022.114480. [DOI] [PubMed] [Google Scholar]
- 12.Tang D.S., Zhou H.L., Cui M.H., Liang G.H., Zhang H.C., Xiao H.H. NIR-II light accelerated prodrug reduction of Pt(IV)-incorporating pseudo semiconducting polymers for robust degradation and maximized photothermal/chemo-immunotherapy. Adv Mater. 2023;35 doi: 10.1002/adma.202300048. [DOI] [PubMed] [Google Scholar]
- 13.Kim J., Lee S., Kim Y., Choi M., Lee I., Kim E., et al. In situ self-assembly for cancer therapy and imaging. Nat Rev Mater. 2023;8:710–725. [Google Scholar]
- 14.Sen S., Won M., Levine M.S., Noh Y., Sedgwick A.C., Kim J.S., et al. Metal-based anticancer agents as immunogenic cell death inducers: the past, present, and future. Chem Soc Rev. 2022;51:1212–1233. doi: 10.1039/d1cs00417d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han H., Xing L., Chen B.T., Liu Y., Zhou T.J., Wang Y., et al. Progress on the pathological tissue microenvironment barrier-modulated nanomedicine. Adv Drug Delivery Rev. 2023;200 doi: 10.1016/j.addr.2023.115051. [DOI] [PubMed] [Google Scholar]
- 16.Li X., Gao Y., Li H.L., Majoral J.P., Shi X.Y., Pich A. Smart and bioinspired systems for overcoming biological barriers and enhancing disease theranostics. Prog Mater Sci. 2023;140 [Google Scholar]
- 17.Rimal R., Desai P., Daware R., Hosseinnejad A., Prakash J., Lammers T., et al. Cancer-associated fibroblasts: Origin, function, imaging, and therapeutic targeting. Adv Drug Delivery Rev. 2022;189 doi: 10.1016/j.addr.2022.114504. [DOI] [PubMed] [Google Scholar]
- 18.Caligiuri G., Tuveson D.A. Activated fibroblasts in cancer: Perspectives and challenges. Cancer Cell. 2023;41:434–449. doi: 10.1016/j.ccell.2023.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murthy T., Podojil J., Kwak T., Vonteddu P., Elhofy A., Boyne M., et al. Biodegradable nanoparticles inhibit tumor growth by altering tumor-associated macrophages and cancer-associated fibroblasts. J Clin Oncol. 2022;40 doi: 10.7150/jca.69338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saw P.E., Chen J.N., Song E.W. Targeting CAFs to overcome anticancer therapeutic resistance. Trends Cancer. 2022;8:527–555. doi: 10.1016/j.trecan.2022.03.001. [DOI] [PubMed] [Google Scholar]
- 21.Zhang H., Yue X.H., Chen Z., Liu C., Wu W.T., Zhang N., et al. Define cancer-associated fibroblasts (CAFs) in the tumor microenvironment: New opportunities in cancer immunotherapy and advances in clinical trials. Mol Cancer. 2023;22:159. doi: 10.1186/s12943-023-01860-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cong Z.Q., Zhang L., Ma S.Q., Lam K.S., Yang F.F., Liao Y.H. Size-transformable hyaluronan stacked self-assembling peptide nanoparticles for improved transcellular tumor penetration and photo-chemo combination therapy. ACS Nano. 2020;14:1958–1970. doi: 10.1021/acsnano.9b08434. [DOI] [PubMed] [Google Scholar]
- 23.Xu F.N., Huang X.H., Wang Y., Zhou S.B. A size-changeable collagenase-modified nanoscavenger for increasing penetration and retention of nanomedicine in deep tumor tissue. Adv Mater. 2020;32 doi: 10.1002/adma.201906745. [DOI] [PubMed] [Google Scholar]
- 24.Yuan S.J., Mu W.W., Liu S.J., Liu M.C., Xia Z.X., Liang S., et al. Transforming cancer-associated fibroblast barrier into drug depots to boost chemo-immunotherapy in “shooting fish in a barrel” pattern. ACS Nano. 2023;17:13611–13626. doi: 10.1021/acsnano.3c02272. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y., Yu R.R., Zhao C., Liang J.W., Zhang Y.X., Su H.C., et al. CAFs homologous biomimetic liposome bearing BET inhibitor and pirfenidone synergistically promoting antitumor efficacy in pancreatic ductal adenocarcinoma. Adv Sci. 2024;11 doi: 10.1002/advs.202305279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li X., Yong T.Y., Wei Z.H., Bie N.N., Zhang X.Q., Zhan G.T., et al. Reversing insufficient photothermal therapy-induced tumor relapse and metastasis by regulating cancer-associated fibroblasts. Nat Commun. 2022;13:2794. doi: 10.1038/s41467-022-30306-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Labrie M., Brugge J.S., Mills G.B., Zervantonakis I.K. Therapy resistance: opportunities created by adaptive responses to targeted therapies in cancer. Nat Rev Cancer. 2022;22:323–339. doi: 10.1038/s41568-022-00454-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hou L., Chen D.D., Wang R.T., Wang R.B., Zhang H.J., Zhang Z.Z., et al. Transformable honeycomb-like nanoassemblies of carbon dots for regulated multisite delivery and enhanced antitumor chemoimmunotherapy. Angew Chem Int Ed. 2021;60:6581–6592. doi: 10.1002/anie.202014397. [DOI] [PubMed] [Google Scholar]
- 29.Wu S.Y., He Y., Zhou R.Q., Chen C.L., Chen D.W., Hu H.Y. Eliciting immunogenic cell death via tumor-associated fibroblast inactivation and autophagy induction with doxorubicin and silybin loaded molecularly imprinted nanosystem. Nano Today. 2024;54 [Google Scholar]
- 30.Liu H.-J., Wang J.F., Wang M.M., Wang Y.Z., Shi S.S., Hu X.X., et al. Biomimetic nanomedicine coupled with neoadjuvant chemotherapy to suppress breast cancer metastasis via tumor microenvironment remodeling. Adv Funct Mater. 2021;31 [Google Scholar]
- 31.Ye J.Y., Hou B., Chen F.M., Zhang S.N., Xiong M.Y., Li T.L., et al. Bispecific prodrug nanoparticles circumventing multiple immune resistance mechanisms for promoting cancer immunotherapy. Acta Pharm Sin B. 2022;12:2695–2709. doi: 10.1016/j.apsb.2021.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y.K., Zhong D., Zhou C., Tu Z.X., Mao H.L., Yang J., et al. Sub-50 nm supramolecular nanohybrids with active targeting corona for image-guided solid tumor treatment and metastasis inhibition. Adv Funct Mater. 2021;31 [Google Scholar]
- 33.Dunne M., Dou Y.N., Drake D.M., Spence T., Gontijo S.M.L., Wells P.G., et al. Hyperthermia-mediated drug delivery induces biological effects at the tumor and molecular levels that improve cisplatin efficacy in triple negative breast cancer. J Control Release. 2018;282:35–45. doi: 10.1016/j.jconrel.2018.04.029. [DOI] [PubMed] [Google Scholar]
- 34.Li Q.Y., Chen C., Kong J.X., Li L., Li J.L., Huang Y. Stimuli-responsive nano vehicle enhances cancer immunotherapy by coordinating mitochondria-targeted immunogenic cell death and PD-L1 blockade. Acta Pharm Sin B. 2022;12:2533–2549. doi: 10.1016/j.apsb.2021.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng X.L., Pan D.Y., Chen X.T., Wu L., Chen M., Wang W.J., et al. Self-stabilized supramolecular assemblies constructed from PEGylated dendritic peptide conjugate for augmenting tumor retention and therapy. Adv Sci. 2021;8 doi: 10.1002/advs.202102741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu M.D., Huang L.Q., Zhang W.N., Wang X.C., Geng Y.Y., Zhang Y.H., et al. A transistor-like pH-sensitive nanodetergent for selective cancer therapy. Nat Nanotechnol. 2022;17:541–551. doi: 10.1038/s41565-022-01085-5. [DOI] [PubMed] [Google Scholar]
- 37.Zhang M.M., Gao S., Yang D.J., Fang Y., Lin X.J., Jin X.C., et al. Influencing factors and strategies of enhancing nanoparticles into tumors in vivo. Acta Pharm Sin B. 2021;11:2265–2285. doi: 10.1016/j.apsb.2021.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu Y.F., Yu X.R., Thamphiwatana S.D., Zheng Y., Pang Z.Q. Nanomedicines modulating tumor immunosuppressive cells to enhance cancer immunotherapy. Acta Pharm Sin B. 2020;10:2054–2074. doi: 10.1016/j.apsb.2020.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elia I., Haigis M.C. Metabolites and the tumour microenvironment: from cellular mechanisms to systemic metabolism. Nat Metab. 2021;3:21–32. doi: 10.1038/s42255-020-00317-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pan J.X., Lai Y., Zhang S.N., Zhang H.J., Shan Y.M., Huang L.J., et al. Self-adaptive nanoregulator to mitigate dynamic immune evasion of pancreatic cancer. Adv Mater. 2023;35 doi: 10.1002/adma.202305798. [DOI] [PubMed] [Google Scholar]
- 41.Pei L.P., Liu Y., Liu L., Gao S.C., Gao X.Y., Feng Y.D., et al. Roles of cancer-associated fibroblasts (CAFs) in anti-PD-1/PD-L1 immunotherapy for solid cancers. Mol Cancer. 2023;22:29. doi: 10.1186/s12943-023-01731-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Y.X., Zhou J., Chen X.T., Li Z.Q., Gu L., Pan D.Y., et al. Modulating tumor-stromal crosstalk via a redox-responsive nanomedicine for combination tumor therapy. J Control Release. 2023;356:525–541. doi: 10.1016/j.jconrel.2023.03.015. [DOI] [PubMed] [Google Scholar]
- 43.Mohan V., Das A., Sagi I. Emerging roles of ECM remodeling processes in cancer. Semin Cancer Biol. 2020;62:192–200. doi: 10.1016/j.semcancer.2019.09.004. [DOI] [PubMed] [Google Scholar]
- 44.Li X.J., Zhang Q., Yu S.M., Li Y. The chemistry and biology of collagen hybridization. J Am Chem Soc. 2023;145:10901–10916. doi: 10.1021/jacs.3c00713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tran D.H., Kesavan R., Rion H., Soflaee M.H., Solmonson A., Bezwada D., et al. Mitochondrial NADP+ is essential for proline biosynthesis during cell growth. Nat Metab. 2021;3:571–585. doi: 10.1038/s42255-021-00374-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bantug G.R., Hess C. The immunometabolic ecosystem in cancer. Nat Immunol. 2023;24:2008–2020. doi: 10.1038/s41590-023-01675-y. [DOI] [PubMed] [Google Scholar]
- 47.Kazak L., Cohen P. Creatine metabolism: energy homeostasis, immunity and cancer biology. Nat Rev Endocrinol. 2020;16:421–436. doi: 10.1038/s41574-020-0365-5. [DOI] [PubMed] [Google Scholar]
- 48.Fu S.W., Li G.T., Zang W.L., Zhou X.Y., Shi K.X., Zhai Y.L. Pure drug nano-assemblies: a facile carrier-free nanoplatform for efficient cancer therapy. Acta Pharm Sin B. 2022;12:92–106. doi: 10.1016/j.apsb.2021.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wculek S.K., Cueto F.J., Mujal A.M., Melero I., Krummel M.F., Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20:7–24. doi: 10.1038/s41577-019-0210-z. [DOI] [PubMed] [Google Scholar]
- 50.Liu H.Y., Luo H.X., Xue Q., Qin S., Qiu S., Liu S.B., et al. Antigen-specific T cell detection via photocatalytic proximity cell labeling (PhoXCELL) J Am Chem Soc. 2022;144:5517–5526. doi: 10.1021/jacs.2c00159. [DOI] [PubMed] [Google Scholar]
- 51.O'Donoghue G.P., Bugaj L.J., Anderson W., Daniels K.G., Rawlings D.J., Lim W.A. T cells selectively filter oscillatory signals on the minutes timescale. Proc Natl Acad Sci U S A. 2021;118 doi: 10.1073/pnas.2019285118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zheng C.X., Snow B.E., Elia A.J., Nechanitzky R., Dominguez-Brauer C., Liu S.F., et al. Tumor-specific cholinergic CD4+ T lymphocytes guide immunosurveillance of hepatocellular carcinoma. Nature Cancer. 2023;4:1437–1454. doi: 10.1038/s43018-023-00624-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu Y.R., Imrichova H., Wang H.P., Chao T., Xiao Z.T., Gao M., et al. Disturbed mitochondrial dynamics in CD8+ TILs reinforce T cell exhaustion. Nat Immunol. 2020;21:1540–1551. doi: 10.1038/s41590-020-0793-3. [DOI] [PubMed] [Google Scholar]
- 54.Cózar B., Greppi M., Carpentier S., Narni-Mancinelli E., Chiossone L., Vivier E. Tumor-infiltrating natural killer cells. Cancer Discov. 2021;11:34–44. doi: 10.1158/2159-8290.CD-20-0655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nicolai C.J., Raulet D.H. Killer cells add fire to fuel immunotherapy. Science. 2020;368:943–944. doi: 10.1126/science.abc2502. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.