

Abstract
Lamellar ichthyosis (LI) is a chronic disease, mostly caused by mutations in the TGM1 gene, marked by impaired skin barrier formation. No definitive therapies are available, and current treatments aim at symptomatic relief. LI mouse models often fail to faithfully replicate the clinical and histopathological features of human skin conditions. To develop advanced therapeutic approaches, such as combined ex vivo cell and gene therapy, we established a human cellular model of LI by efficient CRISPR-Cas9-mediated gene ablation of the TGM1 gene in human primary clonogenic keratinocytes. Gene-edited cells showed complete absence of transglutaminase 1 (TG1) expression and recapitulated a hyperkeratotic phenotype with most of the molecular hallmarks of LI in vitro. Using a self-inactivating γ-retroviral (SINγ-RV) vector expressing transgenic TGM1 under the control of its own promoter, we tested an ex vivo gene therapy approach and validate the model of LI as a platform for pre-clinical evaluation studies. Gene-corrected TGM1-null keratinocytes displayed proper TG1 expression, enzymatic activity, and cornified envelope formation and, hence, restored proper epidermal architecture. Single-cell multiomics analysis demonstrated proviral integrations in holoclone-forming epidermal stem cells, which are crucial for epidermal regeneration. This study serves as a proof of concept for assessing the potential of this therapeutic approach in treating TGM1-dependent LI.
Keywords: lamellar ichthyosis, gene therapy, TGM1, CRISPR-Cas9, primary human keratinocytes, 3D skin equivalent, regenerative medicine, multiomics analysis
Graphical abstract
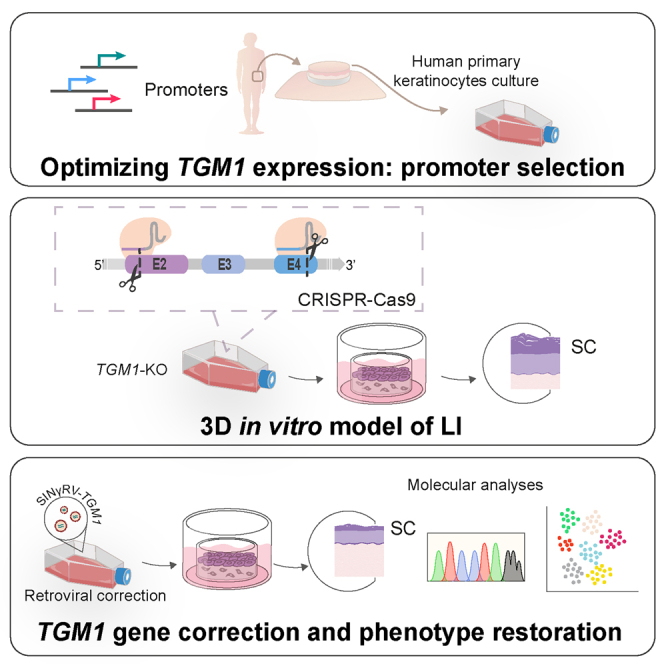
A human cellular model of lamellar ichthyosis has been established by CRISPR-Cas9-mediated TGM1 ablation in primary keratinocytes, recapitulating molecular disease hallmarks. Employing this model, they demonstrated an ex vivo gene therapy approach that corrected epidermal stem cells, providing a proof of concept for future therapeutic application.
Introduction
Lamellar ichthyosis (LI) is one of the most common forms of autosomal recessive congenital ichthyoses (ARCIs), a heterogeneous group of keratinization disorders mainly characterized by abnormal skin scaling over the whole body.1,2 Over 30% of LI is caused by mutations in TGM1,3,4 the gene encoding transglutaminase 1 (TG1), a 90-kDa, membrane-bound, calcium-dependent enzyme that catalyzes the formation of Nε-(γ-glutamyl)lysine bonds between (and within) proteins forming the cross-linked cornified cell envelopes (CEs) of the epidermis.5,6 Loss of TG1 function results in an impaired epidermal barrier, leading to transepidermal water loss and severe, often life-threatening, dehydration, especially during the first weeks of life.4,7
LI is characterized by severe skin dryness, erythema, hyperkeratosis, and massive scaling, highly impairing patients’ quality of life.8,9 Affected individuals are often born with a “collodion membrane” that sheds in the first week of life, leaving patients with a disfiguring ichthyosis, characterized by coarse and dark-brown scales.7,10 LI is commonly associated with ectropion and eclabium (reversal of the eyelids and lips because of tension from the collodion membrane) and scarring alopecia. Flexural surfaces and palms and soles are frequently involved.8,11 Depending on TGM1 mutations, LI can range from very mild to severe forms,12,13 with variable degrees of scaling, hyperkeratosis, and erythema, all of which require different approaches and even personalized therapies.
Currently, treatments for most ARCIs are not tailored, and patients lack definitive therapeutic options.14,15 Enzyme replacement therapy (ERT) with nanoparticles and liposomes or non-integrating herpes simplex virus aim at skin-topical delivery of human recombinant TG1.16,17,18 Non-viral gene transfer techniques have been also exploited for TGM1 gene delivery in organotypic cultures from LI patients’ keratinocytes with the adenovirus-enhanced, transferrin-receptor-mediated transfection (AVET) system.19 However, AVET transfection did not show the expected efficiency in primary keratinocytes, which could be due to the presence of a stratum corneum or the absence of a receptor for adenovirus entry.20 The epidermis is a highly renewing tissue, and the administered proteins or the non-integrating viral vectors have a temporary therapeutic effect, making repeated—perhaps lifelong—applications necessary. Since most ichthyoses (including TGM1-LI) are chronic life-long diseases, there is an urgent need for safe, effective, and definitive therapies. The first ex vivo gene therapy approach envisaged the delivery of the TGM1 gene into LI patient cells using an amphotropic retroviral vector or direct injection of naked DNA in a skin humanized mouse model.21,22,23 It was shown that, after TGM1-retrovirus transduction, the expression and activity of TG1 was restored in keratinocytes of LI patients in vitro.21 Although these data were relevant in showing the possibility of phenotypic correction of LI, these strategies have never reached clinical application.
Ex vivo combined cell and gene therapy has been successfully exploited in other recessively inherited genodermatoses, such as junctional and dystrophic epidermolysis bullosa.24,25,26,27 A functional copy of the defective gene can be introduced into the genome of clonogenic keratinocytes by means of replication-defective retroviral vectors. Cultured transgenic keratinocytes can then be used to prepare epidermal grafts able to permanently restore a functional epidermis.25,28 Notably, the long-term epidermal restoration strictly relies on the genetic modification of long-lived, self-renewing epidermal stem cells, detected as holoclone-forming cells.24,25
Here we report a proof-of-concept study evaluating a combined cell and gene therapy approach aimed at correcting TGM1 mutations underlying LI. A 3D TGM1-defective human cellular model was established and genetically corrected with a self-inactivating γ-retroviral (SINγ-RV) vector expressing the TGM1 cDNA under the control of its own promoter. This study confirms the valuable tool of the 3D human cellular model in replacing animal models and avoiding the need for patient biopsies and demonstrated the feasibility of a combined cell and gene therapy strategy for TGM1-LI disease.
Results
Selecting promoters driving physiological TGM1 expression in differentiated keratinocytes
Given the peculiar cross-linking, calcium-dependent enzymatic activity of TG1 and its location and function in distinct suprabasal epidermal layers,5,6,29 the selection of the appropriate promoter is a major step toward a successful gene therapy. To this end, SINγ-RV vectors expressing enhanced green fluorescent protein (EGFP) under the control of three different promoters were produced. These include a 637-bp human involucrin minimal promoter (IVLmin), a 367-bp human TGM1 minimal promoter (TGM1min), and 2.2-kb human TGM1 full-length promoter (TGM1full) (Figure 1A, top). Such promoters were initially used to transduce HaCaT cells, a spontaneously immortalized human keratinocyte cell line expressing appropriate differentiation markers, a widely used model to study the balance between keratinocyte proliferation and differentiation upon changes in calcium concentration.30,31 As detailed in materials and methods, transgenic HaCaT cells were serially diluted to isolate a homogeneous EGFP+ cell population, which was then cultured in keratinocyte serum-free medium (KSFM) in 0.03 mM calcium (Ca2+low) for at least 3 weeks and then switched to 2.8 mM calcium (Ca2+high) to induce terminal differentiation (Figure 1A, bottom). Vector copy number evaluation (VCN) of transduced HaCaT clones was also performed (Figure S5). The expression of epidermis-specific differentiation markers (K10 and TGM1) progressively increases in Ca2+high as compared to the basal level (time 0 [t0]), confirming proper differentiation stimuli also after transduction (Figure 1B). Ca2+ differentiation-dependent EGFP mRNA and protein expression were detected only with the TGM1full promoter (Figures 1C and 1D). Time-resolved promoter activity was also investigated by live-cell imaging following Ca2+high-induced terminal differentiation (Figure S1). EGFP was expressed in HaCaT cells transduced with TGM1min and IVLmin promoters independently of calcium-induced terminal differentiation. In sharp contrast, the TGM1full promoter induced EGFP expression from day 5 after switching to Ca2+high and progressively increased its activity during terminal differentiation, confirming that only the full promoter is properly regulated by differentiation stimuli.
Figure 1.

Differentiation-related expression of EGFP in HaCaT cells transduced with SINγ-RVs
(A) Top: schematic of self-inactivating γ-retroviral vectors (SINγ-RVs) carrying the three different promoters that drive the expression of the bicistronic gene encoding EGFP and puromycin resistance. Bottom: graphic of the HaCaT differentiation protocol. (B) K10 (left) and endogenous TGM1 (ED TGM1) (right) relative mRNA expression in non-transduced (NT) and transduced (TGM1full) HaCaT cells, grown in low calcium (t0) and high calcium for 1, 4, and 6 days. GAPDH mRNA was used to normalize the RT-PCR. (C) RT-qPCR quantification of the relative mRNA expression value of EGFP. GAPDH mRNA was used to normalize the RT-PCR (∗p < 0.0001). (D) Western blot analysis of EGFP expression in low-calcium (t0) and high-calcium culture conditions for 4 or 6 days of cultivation. NT HaCaT cells 6 days after calcium addition are loaded as a reference sample. t0: time zero.
TGM1full promoter-driven expression of EGFP in primary human keratinocytes
Under appropriate culture conditions, primary human clonogenic keratinocytes give rise to colonies eventually generating cohesive epidermal sheets resembling the epidermis, which are routinely used in clinical settings for regenerative medicine.32,33 Keratinocyte confluence and initial stratification induce commitment to terminal differentiation, as demonstrated by the decrease of keratinocyte clonogenicity (Figures 2A and 2B)34 and induction of early differentiation markers, such as suprabasal keratin 10 and involucrin (Figure 2C).35
Figure 2.

The TGM1full promoter drives EGFP expression in primary differentiated human keratinocytes
(A) Schematic of the in vitro keratinocyte differentiation experimental design. (B) Representative colony-forming efficiency (CFE) of keratinocyte cultures in growing (left) (d7) and over-confluent (right) culture conditions (d14). The number of cells per dish plated in the CFE condition is indicated in brackets. Colonies were stained with Rhodamine B after 12 days of cultivation. (C) Western blot analysis of keratinocyte differentiation markers (IVL, endogenous TG1, and K10) in growing (3-day), confluent (7- to 10-day), and over-confluent (14-day) normal, healthy keratinocytes (K82). (D) Western blot analysis of keratinocyte differentiation markers (IVL, endogenous TG1, and K10) and EGFP in growing (3-day), confluent (7- to 10-day), and over-confluent (14-day) K82 keratinocytes transduced with a SINγ-RV carrying EGFP under the control of the TGM1full promoter. (E) Progressive (3- to 14-day) increase of EGFP mRNA during transgenic keratinocyte differentiation and stratification. GAPDH mRNA was used to normalize the RT-qPCR (∗p < 0.0001). (F) Representative immunofluorescence images of 7-μm-thick cryosections of 3D SEs obtained with TGM1full promoter-transduced keratinocytes, showing the expression of differentiation markers (endogenous TG1 and LEKTI) in the upper layers of the epidermis and collagen XVII and KRT14 as markers of the epidermal basal layer (n = 3, pictures are representative of what was observed in at least three independent samples or replicates). The EGFP fluorescent signal is properly restricted to the granular layer of the epidermis. White arrows indicate the presence of living fibroblasts inside the collagen matrix, expressing vimentin. A white dotted line marks the epidermal-dermal junction. DAPI (blue) stains nuclei. Scale bars, 20 μm. LEKTI, lympho-epithelial Kazal-type-related inhibitor; KRT14, cytokeratin 14; VIM, vimentin.
To investigate the ability of the TGM1full promoter to properly drive EGFP expression in primary human epidermal cells, keratinocytes were transduced with the SINγ-RV-EGFP vectors previously used on HaCaT cells. The percentage of EGFP+ colonies was approximately 60%. Transduced keratinocytes were exposed to 2 μg/mL puromycin to purify a homogeneous population of EGFP+ cells. VCN evaluation of transduced keratinocytes was also performed (Figure S5). The proper activation of the TGM1full promoter was evaluated in growing, confluent, and over-confluent transgenic primary cultures (Figures 2 and S2). As investigated previously in HaCaT cells, primary human keratinocytes also displayed differences in the expression of the reporter gene among the three different promoters following differentiation stimuli. As shown in Figures 2D and 2E, transgenic keratinocytes transduced with the TGM1full promoter showed the expected progressive, density-dependent increase of EGFP, mirroring the expression of endogenous TG1, hence confirming that only the activation of this promoter is finely controlled during epidermal differentiation. EGFP+ keratinocytes were then used to generate three-dimensional skin-equivalent (SE) cultures. To this end, transgenic keratinocytes were seeded onto a collagen matrix containing living fibroblasts and cultured for 1 week in submerged culture and then for 3 weeks in an air-liquid interface, allowing them to stratify and fully differentiate. Cultures were fixed and sectioned to examine morphology, differentiation markers, as well as EGFP protein expression in a biomimetic and physiological environment. A fully developed epidermis was obtained (Figure 2F). Differentiation markers, such as TG1 and LEKTI, were correctly localized and expressed in the upper layers of differentiated epidermis, while KRT14 and collagen XVII were correctly restricted to the epidermal basal layer (Figure 2F). EGFP fluorescence, visualized directly in sections, reflects the accumulation of EGFP signal only in the differentiated compartment when keratinocytes are transduced with both the TGM1full and IVLmin promoters (Figures 2F, bottom right, and S2C). Moreover, fibroblasts contained within the 3D SE clearly expressed vimentin, which indicates their viability, crucial for the maintenance of the 3D SE model.
These data demonstrate that retroviral vectors containing TGM1full promoter can drive the expression of a reporter gene to the upper differentiated layers of the epidermis.
Generation of the ΔTGM1 human cellular model
To create a reliable cellular model able to recapitulate the LI phenotype, TGM1 was knocked out in primary keratinocyte cultures obtained from healthy donors using the CRISPR-Cas9 system. Three different CRISPR guide RNAs (gRNAs) were designed to target both TGM1 alleles, exploiting the presence of a SNP in a wild-type strain (K81) (Figure 3A). gRNA1 and gRNA2 were designed to target exon 2 (E2) of the TGM1 gene; these gRNAs differ for 1 nt close to the protospacer adjacent motif (PAM) sequence, which makes them allele specific. gRNA3 was designed to target exon 4 (E4). E2 and E4 were chosen for their crucial role in determining the protein function. Indeed, E2 encodes for a unique TG1 N-terminal sequence that is required for membrane anchoring, while E4 encodes for a part of the catalytic core domain of the enzyme.29,36
Figure 3.

Generation of the ΔTGM1 human cellular model
(A) TGM1 KO strategy design. gRNA1 and gRNA2 target E2, while gRNA3 targets E4 of the TGM1 gene. gRNA1 and gRNA2 differ in 1 nt (underlined), allowing targeting of both alleles, exploiting the presence of a SNP in a wild-type strain (K81). gRNA sequences are enclosed in colored boxes, with the protospacer adjacent motif (PAM) shown in bold. (B) PCR analysis on both edited exons performed on genomic DNA from wild-type keratinocytes (K81) and ΔTGM1-nucleofected keratinocytes. Note the intensity reduction of the bands at 608 bp and 780 bp in ΔTGM1 nucleofected keratinocytes. (C and D) RT-qPCR (C) and western blot analysis (D) show ablation of both TGM1 mRNA (C, orange bars) and protein (D) in ΔTGM1 keratinocytes as compared to wild-type cells. (E) Relative TG1 enzymatic activity in growing (3-day) and over-confluent (14-day) wild-type and ΔTGM1 keratinocytes. n = 3 replicates, data are presented as mean ± SD (∗p < 0.0001).
Human primary keratinocytes were nucleofected with ribonucleoprotein (RNP) complexes containing the three different gRNA-Cas9 pairs. In both exons, the corresponding edited sequences were assessed by PCR amplification of fragments spanning the target sites. Specific primers, designed on target sites, clearly showed virtually complete TGM1 ablation (Figure 3B). Moreover, PCR amplification performed with primers spanning both the Cas9 target sites demonstrated that the excision of the entire genomic region (1.7 kb) between E2 and E4 is a high-frequency event (Figure S3A). Validation of efficient TGM1 ablation was obtained by TIDE analysis through Sanger sequencing of E2 and E4, respectively, as compared to wild-type sequences. The overall editing efficiency, reaching 100%, is indicated by the absence of non-edited sequences (point 0 of the graphs) and the presence of a significant amount of insertions or deletions abolishing the open reading frame and expression of TGM1 (Figure S3B).
TGM1 mRNA and TG1 protein expression, as well as TG1 enzymatic activity, tested both in growing (3-day) and high-density, differentiating (14-day) cultures, were undetectable in ΔTGM1 keratinocytes as compared to wild-type keratinocytes (Figures 3C–3E). The remarkable efficacy of TGM1 ablation achieved in bulk keratinocyte culture avoided the selection of specific clones of the keratinocytes treated with gRNA-Cas9 pairs.
Overall, these results demonstrate the feasibility and the high efficiency of TGM1 knockout in human keratinocytes using the CRISPR-Cas9 system.
ΔTGM1 model for proof-of-concept therapy studies
ΔTGM1 epidermal cells were transduced with a SINγ-RV vector containing TGM1 cDNA under the control of the TGM1full promoter (SINγ-RV-TGM1) (Figure S4A). Transgenic keratinocyte colonies clearly demonstrated the correct expression of exogenous TG1, closely resembling TG1 expression in normal keratinocyte colonies (Figure 4A).
Figure 4.

SINγ-RV-TGM1 rescues TG1 expression and activity in the ΔTGM1 human cellular model
(A) Representative images of TG1 immunofluorescence analysis of wild-type, ΔTGM1, and SINγ-RV-TGM1-transduced keratinocytes after 7-days cultivation. DAPI (blue) stains nuclei. Scale bars, 50 μm. (B) Optical micrographs of the isolated cornified cell envelopes in wild-type and SINγ-RV-TGM1-transduced keratinocytes. Note that typical envelopes were not detected in ΔTGM1 keratinocytes, and only a few cell fragments were observed. Scale bar, 20 μm. (C) Immunostaining detection of TG1 protein (top, green) and TG1 in situ enzymatic activity (bottom, red) in cryosections of collagen-based 3D SEs from wild-type, ΔTGM1, or SINγ-RV-TGM1-transduced keratinocytes. DAPI (blue) stains nuclei. A white dotted line marks the epidermal-dermal junction. Scale bars, 50 μm (n = 3, pictures are representative of what was observed in at least three independent samples or replicates).
In order to determine exogeneous TG1 in vitro enzymatic function, its cross-linking activity was investigated in stratified keratinocytes cultures. During epidermal keratinization, TG1 catalyzes the formation of cross-linked cornified cell envelopes (CEs), which are chemically resistant to ionic detergent and reducing agent, as described previously.37 Since the cross-linking process is strictly related to the presence of TG1 in terminally differentiated keratinocytes, the absence of a functional protein should result in failure to form CEs. Indeed, no CEs were detected in ΔTGM1 keratinocytes, while typical polyhedral CEs were isolated both from normal and SINγ-RV-TGM1-transduced keratinocytes (Figure 4B).
The ability to faithfully recapitulate the LI phenotype of the ΔTGM1 keratinocytes was assessed in 3D SE models. SE models were prepared using normal, ΔTGM1, or SINγ-RV-TGM1-transduced (transgenic) keratinocytes. As shown in Figure 4C, top, TG1 protein expression in both normal and transgenic keratinocytes was correctly restricted to the granular layer with the typical pericellular distribution. Furthermore, the correct expression of exogenous TG1 in transgenic keratinocytes was associated with full restoration of its enzymatic activity (Figure 4C, bottom; red fluorescence indicates the in situ evaluation of TG1 enzymatic activity). Of note, ΔTGM1 SE revealed an epidermis characterized by a thickened stratum corneum (SC) phenocopying the human ichthyosis phenotype.11,38,39 In contrast, the transgenic SE produced a normal SC virtually indistinguishable from that observed in SE models generated by wild-type keratinocytes (Figure 5).
Figure 5.

ΔTGM1 3D SEs resembling LI skin hallmarks and architecture
Histological analysis (H&E) and immunostaining of TG1 substrates (involucrin, LEKTI, and loricrin) and keratin 10 on 3D SEs from wild-type, ΔTGM1, or SINγ-RV-TGM1-transduced keratinocytes. DAPI (blue) stains nuclei. A dotted line marks the epidermal-dermal junction. Scale bars, 20 μm.
The presence and spatial distribution of the TG1 substrates involucrin, LEKTI, and loricrin were also assessed (Figure 5). TG1 substrates showed a diffuse distribution in the ΔTGM1 SE model with abnormal cytoplasmic staining, indicating the lack of protein cross-linking in the upper layers of the epidermis, as typically occurs in LI.40,41,42 In contrast, in the transgenic SE, each TG1 substrates is correctly localized at the cellular periphery of the upper differentiated layers of the epidermis, resembling the normal skin. The expression of K10, localized in the suprabasal cell layers, remained unchanged in all conditions.
Finally, gene-corrected TGM1-null keratinocytes examined at increased cell densities confirmed that exogenous TG1 protein expression is finely controlled during cell differentiation (Figure S4B), as proven by the expression of the involucrin differentiation marker.
Altogether, these findings demonstrated the restoration of TG1 expression and function using an ex vivo gene therapy approach mediated by a SINγ-RV-TGM1 vector in primary ΔTGM1 keratinocytes, further corroborating the valuable established tool for testing novel therapies for LI.
Multiomics analysis of SINγ-RV-TGM1-transduced keratinocytes
In view of a potential future clinical application of TGM1-transduced keratinocytes, it must be considered that long-term epidermal restoration strictly relies on the genetic modification of long-lived, self-renewing epidermal stem cells, detected as holoclone-forming cells.24,25 Since basal, clonogenic human keratinocytes, including stem cells, do not express TGM1 mRNA, an assessment of exogenous TGM1 expression cannot be performed solely by single-cell RNA sequencing (scRNA-seq) analysis. To this end, a single-keratinocyte multiomics analysis was conducted on SINγ-RV-TGM1-transduced cells. This approach allowed for the simultaneous evaluation of provirus integration and transcriptome profiling within a single cell via scATAC-seq and scRNA-seq, respectively.
As shown in Figure 6A, transgenic keratinocytes contain the previously defined five keratinocyte populations,43,44 three of which are clonogenic (holoclone, meroclone, and paraclone), while the other two identify terminally differentiated cells (TD1 and TD2). The holoclone population, formed by epidermal stem cells, is defined by the transcriptomic expression of a “holoclone signature”43 (Figure S6A).
Figure 6.

Single-cell multiomics analysis reveals TGM1-transduced keratinocyte stem cells
(A) UMAP of scRNA-seq profiles in SINγ-RV-TGM1-transduced keratinocytes. Keratinocytes were classified in five clonogenic and differentiated populations through label transfer and mapping to a previously reported reference of human keratinocytes.43,44 Dots represent individual cells, and the colors indicate the cell populations (holoclone [H] in orange, meroclone [M] in light blue, paraclone [P] in gray, TD1 in light brown, and TD2 in brown). Feeder layer-derived fibroblasts and low-quality keratinocyte clusters (F1 and 6,7) are shown in light gray. The percentages of cells in each population are shown on the right. (B) UMAP showing SINγ-RV-TGM1-positive cells containing at least one provirus fragment (red). The color scale indicates the number of provirus fragments retrieved in each cell. Negative cells are shown in light gray. The percentages of positive cells in each population are shown on the right.
We were able to detect TGM1-proviral integration within individual cells by mapping scATAC-seq data of TGM1-transduced cells to a custom human reference genome that includes the sequence of the SINγ-RV-TGM1 provirus. About 30% of total cells were found to be positive for at least one exogenous TGM1 fragment (Figures 6B and S6B), with the majority of them containing a unique proviral integration (Figures S6C and S5). Notably, a uniform distribution of TGM1 proviral integration is found across all five keratinocytes populations (Figures 6B and S6B) without any clonal selection, demonstrating that the genetically corrected holoclones maintained their ability to self-renew in vitro and produce progenitors that replenish terminally differentiated keratinocytes.
These data clearly show that the proposed therapeutical strategy is able to preserve and genetically correct the population of epidermal stem cells, crucial for a possible ex vivo gene therapy approach aimed at full epidermal restoration.
Discussion
TGM1-deficient LI is a rare cornification disorder associated with severe clinical complications that highly decrease the patient’s quality of life.2,9,15 There is no cure for LI, and the existing treatments only aim at relieving the symptoms.45,46,47 Thus, there is a significant unmet need for therapeutic strategies aimed at correcting the TGM1 deficiency underlying LI.48,49
Currently, the most innovative therapeutical approaches (still not commercially available), are based on ERT16,17 or in vivo gene therapy18 (phase II clinical trial, KB105; ClinicalTrials.gov: NCT05735158). Although these therapeutical strategies are quite promising and technically feasible, they have a common drawback; namely, they could likely have only a temporary beneficial effect. Recently, an adenine base editing system has been proposed for the correction of a TGM1 pathogenic mutation in human embryos.50 Even though the goal of permanently correcting point mutations is admirable, it is important to consider the effectiveness of the process and to evaluate any possible off-target RNA occurrences. Additionally, the primary challenges associated with its clinical application include the high cost of precision medicine and the application of this technology to human embryos. This latter aspect is the subject of constant debate.51
While γ-RV vectors, derived from the Moloney murine leukemia virus, are widely used in clinical trials for keratinocyte-based ex vivo gene therapy,52,53 they have the potential risk of insertional mutagenesis. To address this safety concern, SIN vectors lacking viral enhancers/promoters in their 3′ long terminal repeat have been developed.54,55,56 These vectors mitigate the risk of insertional mutagenesis and provide the opportunity to utilize an internal, often tissue-specific promoter to elicit controlled and physiological expression of the target gene.54,55,56
Combined ex vivo cell and gene therapy by means of cultured, autologous, transgenic keratinocytes can permanently restore a functional epidermis, as already shown with other devastating skin genetic diseases.24,25,26,27 In fact, combined ex vivo cell and gene therapy of generalized intermediate LAMB3-junctional epidermolysis bullosa (JEB) has proven to be life saving and able to permanently regenerate a fully functional epidermis in three JEB patients (up to 18 years of follow-up).25,28,57,58,59 But JEB is characterized by massive blisters and erosions (hence, by open wounds), allowing easier preparation of the dermal wound bed required for transplantation of the transgenic grafts. LI patients face an additional challenge, as their thickened SC must be surgically removed to expose the dermal layer. As a result, ex vivo gene therapy for LI patients becomes more invasive as compared to that for JEB patients. While it may initially seem daunting, ex vivo gene therapy treatments may improve the overall quality of life for LI patients affected by specific LI forms. For instance, localized TGM1-dependent bathing suit ichthyosis,7,60 which is characterized by a distribution of lesions especially on the trunk, could be addressed first, providing an important proof of principle for the treatment of LI lesions.
The limited availability of patients with rare diseases and ethical concerns related to the need for tissue samples often hamper studies aimed at the development of this type of advanced therapies. Although LI murine models have been generated, TGM1−/− mice exhibited an early lethality and poorly recapitulated the human phenotype.61,62 Our studies were conducted using an in vitro human LI model (i) allowing the cultivation of a fully differentiated human epidermis (ii) able to recapitulate the LI phenotype starting from keratinocytes obtained from surgical waste skin samples and (iii) allowing in-depth studies on molecular, biochemical, and functional restoration of TG1. More importantly, we provide evidence that long-lived self-renewing stem cells have been targeted and genetically corrected, thus maintaining the ability to permanently regenerate a functional epidermis. Such an experimental approach can not only avoid or limit animal experiments but, in the case of human skin diseases, also minimize the significant differences that exist between human and murine skin architecture and physiology. In fact, disease-resembling SEs models using the knockout (KO) strategy have been produced for other types of genodermatoses. For example, an effective cellular model of Netherton syndrome was developed by disrupting SPINK5 through efficient CRISPR-Cas9-mediated gene ablation in human primary keratinocytes.63 Both studies aim to develop surrogate models using human primary cells to investigate genodermatosis diseases that efficiently replicate the phenotype and molecular hallmarks of Netherton syndrome and TGM1-LI, respectively.
However, to achieve clinical application, two additional steps are required: (1) the development of a clinical-grade stable packaging cell line, which will enable high-titer, large-scale production of SINγ-RVs, an essential step in transitioning from bench to bedside, and (2) the validation of these findings using LI patient-derived cells from multiple donors with diverse genetic backgrounds.
Taken together, these findings support the development of a human model for LI and pave the way for the prospective implementation of combined cell and gene therapy targeting at least specific forms of TGM1-dependent LI.
Materials and methods
Human tissues
All human tissues were collected after obtaining informed consent and in compliance with Italian regulations (Comitato Etico dell’Area Vasta Emilia Nord, number 178/09 for healthy donor skin samples).
Primary human cell cultures
Human skin samples were obtained as anonymized surgical waste, typically from abdominoplasty or mammoplasty. Briefly, skin biopsies were minced and trypsinized (0.05% trypsin/0.01% EDTA) at 37°C for 4 h. Cells were collected every 30 min, plated (2.5–3 × 104/cm2) on lethally irradiated 3T3-J2 cells (2.4 × 104/cm2), and cultured in 5% CO2 and a humidified atmosphere in keratinocyte growth medium (KGM): DMEM and Ham’s F12 medium (3:1 mixture) containing fetal bovine serum (FBS) (10%), insulin (5 μg/mL), adenine (0.18 mM), hydrocortisone (0.4 μg/mL), cholera toxin (0.1 nM), triiodothyronine (liothyronine sodium) (2 nM), EGF (10 ng/mL), glutamine (4 mM), and penicillin-streptomycin (50 IU/mL).64 Subconfluent primary cultures were serially propagated as described previously.43,65
3T3-J2 cell line
Mouse 3T3-J2 cells were a gift from Prof. Howard Green (Harvard Medical School, Boston, MA, USA). Fibroblasts were cultivated in DMEM supplemented with 10% gamma-irradiated donor adult bovine serum, penicillin-streptomycin (50 IU/mL), and glutamine (4 mM).
HaCaT cell line
HaCaT cells, a spontaneously immortalized human keratinocyte line, were cultured in 5% CO2 at 37°C in three different culture media as needed. Standard culture medium contained DMEM supplemented with 10% heat-inactivated FBS, glutamine (2 mM), and penicillin-streptomycin (100 IU/mL). Low-calcium medium contained KSFM (17005042, Thermo Fisher Scientific) supplemented with 0.03 mM calcium chloride, 25 μg/mL bovine pituitary extract (BPE; Thermo Fisher Scientific), 0.2 ng/mL of recombinant EGF (Thermo Fisher Scientific), and penicillin-streptomycin (100 IU/mL). High-calcium medium contained KSFM supplemented with 2.8 mM of calcium chloride, 25 μg/mL BPE, 0.2 ng/mL of recombinant EGF (Thermo Fisher Scientific), and penicillin-streptomycin (100 IU/mL).
Plasmid constructs and retrovirus production
Genomic DNA from human primary keratinocytes (K81) was used as template to generate (1i) TGM1min, (2) IVLmin, and (3) TGM1full by PCR using high-fidelity Taq polymerase (LA Taq DNA Polymerase, Takara). Specific primers were used to amplify the distal region (−1.6 kb/−1.4 kb) (containing the AP1 and Sp1 binding sites) and the proximal region (−90 bp/+67 bp) of the wild-type human TGM1 promoter,66 the distal region (−2.473 kb/−2.036 kb) and the proximal region (−221 bp/+6 bp) of the wild-type human involucrin promoter,67 and the full-length (2.2 kb) human TGM1 promoter. A list of PCR primers is shown in Table S1. The PCR fragments were verified by Sanger sequencing and subsequently cloned into SINγ-RV HSC1-GiP, a gift from James Ellis (Addgene plasmid 58254), deprived of the internal human EF1alpha promoter. The obtained constructs carry one of the three different promoters guiding the expression of a bicistronic cassette EGFP ires Puromycin. The HSC1-GiP vector was used as control. A retroviral vector expressing the full-length 2.9-kb TGM1 cDNA (GeneArt, Invitrogen) was constructed by cloning the TGM1 cDNA under the control of the TGM1 full promoter into the HSC1-GiP backbone.
Phoenix-AMPHO cells were cultured in DMEM supplemented with 10% heat-inactivated FBS, 1% glutamine, and 1% penicillin-streptomycin antibiotics (Thermo Fisher Scientific). High-quality DNA of each transfer vector was used for the transient transfection of Phoenix-AMPHO cells with branched high-performance polyethylenimine PEI (Sigma), for retroviral production. Infectious retroviruses were harvested 48 h post transfection, filtered through 0.45-μm pore cellulose acetate filters, and concentrated by ultracentrifugation. Primary keratinocytes or HaCaT cells were infected with the retroviral stock at various multiplicities of infection (MOIs) with Vectofusin-1 (Miltenyi Biotec) or RetroNectin (Takara) addition.
Generation of genetically modified HaCaT cell cultures
HaCaT cells were transduced with SINγ-RV-EGFP supernatants at MOI 5 with RetroNectin recombinant human fibronectin fragment solution (Takara; 20 μg/mL in DPBS [Gibco]) at 10 μg/cm2. Each condition of SINγ-RV-EGFP-transduced HaCaT cells was serially diluted (1 cell/well) for positive clone isolation. After 7 days of cultivation, fluorescent positive clones were selected using an inverted florescence microscope. EGFP+ clones were picked and expanded in serial passages. Transduced HaCaT cells were cultured in low-calcium conditions (0.03 mM Ca2+) for at least 3 weeks in order to induce a basal-like state that is characteristic of undifferentiated cells. HaCaT cells were then cultured in high-calcium conditions (2.8 mM Ca2+) to induce differentiation. Samples were harvested at t0 (low Ca2+) and 8 h, 24 h, 48 h, 4 days, and 6 days, after calcium addition.
Live-cell imaging of genetically modified HaCaT cell cultures
EGFP fluorescent emission of transduced HaCaT cells under differentiation conditions was evaluated with a Cell Observer microscope (Axio Observer Z1, 10× objective). Fluorescence and bright-field micrographs of the samples were acquired every 24 h for 6 days. Non-transduced HaCaT cells were used as a negative control.
Generation of genetically modified human primary keratinocytes
Sub-confluent keratinocytes mass cultures (K82) were treated with 0.05% trypsin and 0.01% EDTA for 15–20 min at 37°C and plated (3 × 104 cells/cm2) onto lethally irradiated 3T3-J2 cells (3 × 104 cells/cm2) in KGM without FBS or penicillin-streptomycin. SINγ-RV-EGFP viral supernatants were added separately at MOI 1.5 with Vectofusin-1 (Miltenyi Biotec). After 3 days of cultivation, cells were collected and cultured in KGM on a regular 3T3-J2 feeder layer. Keratinocytes were treated with 2 μg/mL of puromycin (Sigma) for 72 h, enabling the selection of cells. The selected sub-confluent transduced primary keratinocyte cultures were trypsinized and plated in over-confluent conditions to induce differentiation. Samples were feeder depleted and harvested at 3, 7, 10, and 14 days of culture.
3D SE culture preparation
3T3-J2 -populated collagen gels (final concentration of 5 mg/mL) were prepared as reported previously68 and seeded with primary human keratinocytes (1 × 105 cells/scaffold) onto lethally irradiated 3T3-J2 cells (5 × 104 cells/scaffold) in KGM. After 6 days in submerged culture, the medium was removed, and the collagen gels were gently moved in Millicell cell culture (Merck). The 3D SE cultures were further cultured for 20–24 days in the air-liquid interface (ALI) condition to induce epidermal differentiation.
RNA extraction, real-time qPCR, and droplet digital PCR
Total RNA was isolated from cultured cells using the PureLink RNA Mini Kit (Thermo Fisher Scientific). cDNA was generated using the SuperScript VILO cDNA Synthesis Kit (Thermo Fisher Scientific). Real-time qPCR analyses were carried out on triplicate samples of retrotranscribed cDNA with TaqMan Universal PCR Master Mix (Thermo Fisher Scientific) on a 7900H Real-Time PCR System (Thermo Fisher Scientific).69 Expression levels are given relative to human GAPDH. Data were analyzed using the 2−ΔCt quantification and visualized with Prism 8. Droplet digital PCR (ddPCR) was carried out for VCN evaluation on genomic DNA isolated from cultured cells using the QIAamp DNA Mini Kit (QIAGEN, 51304). Droplets were generated using the QX200 Droplet Generator (Bio-Rad) according to the manufacturer’s instructions. PCR amplifications were performed in a thermal cycler and analyzed using a QX200 Droplet Reader (Bio-Rad). VCN was absolutely quantified considering an internal reference gene (VCN = 2). Data were analyzed using QuantaSoft Analysis Pro software and visualized with Prism 8. For both real-time qPCR and ddPCR, a list of TaqMan probes (Thermo Fisher Scientific) is provided in Table S2.
Western blotting
Samples of human primary keratinocytes destined for western blot analysis were depleted of feeder layer in 20 mM cold PBS/EDTA. Cells were lysed in 1× RIPA buffer (Sigma-Aldrich) supplemented with phosphatase and protease inhibitor cocktail (Thermo Fisher Scientific). BCA kits (Pierce) were used to quantify the total protein amount. Equivalent quantities of RIPA-solubilized proteins were resolved by 4%–12% NuPAGE BisTris gels or 10% NuPage Tris acetate gels (Thermo Fisher Scientific) at 100 V for 2 h and transferred to 100 V at 4°C for 2 h on a nitrocellulose membrane (Millipore). Membranes were treated with EveryBlot blocking buffer (Bio-Rad) for 5 min with agitation. Primary antibodies were diluted in blocking buffer as indicated in Table S3 and added overnight at 4°C to the membranes. Secondary antibodies were diluted in blocking buffer as indicated in Table S3 and added to the corresponding membranes for 1 h at room temperature. Signal was visualized with Clarity Western ECL Substrate (Bio-Rad) using ChemiDoc (Bio-Rad). Bio-Rad’s Image Lab software 6.0 was used for band densitometry. A gray background on the images was homogeneously added for graphical purposes.
Immunofluorescence and H&E staining
For immunofluorescence analyses of in vitro-cultured keratinocytes, cells were plated at 1,000/well onto glass coverslips and grown as described previously. Cells were fixed with 3% paraformaldehyde (PFA) for 10 min at room temperature, carefully washed with 1× PBS, and permeabilized in 0.3% PBS/Triton for 15 min. Blocking solution (5% FBS and 2% BSA in 0.1% PBS/Triton) was added for 1 h at room temperature. Primary antibodies were diluted in blocking solution as described in Table S3 and added to the samples overnight at 4°C. Secondary antibodies were diluted in blocking solution as described in Table S3 and added to the samples for 1 h at room temperature. Cell nuclei were stained with DAPI. Dako mounting medium was used to mount coverslips. A Carl Zeiss confocal microscope (LSM510meta) with a Carl Zeiss EC Plan-Neofluar 40×/1.3 oil immersion objective was used to visualize fluorescent signals.
For 3D human SE immunofluorescence, the 3D cultures were dehydrated in a sucrose gradient (0.9 M and 2 M) for 30 min at room temperature, embedded in Killik-OCT Cryostat Embedding Medium (Bio-Optica), and frozen. 7-μm sections of embedded SE were obtained with a histological cryomicrotome (Leica CM1850 UV). Immunofluorescence was performed on 7-μm cryosections. In brief, sections were fixed in 3% PFA or methanol, permeabilized for 15 min with 0.3% PBS/Triton, and blocked for 30 min at 37°C with blocking solution (5% BSA in PBS). The primary antibody, diluted in blocking solution, was added to skin sections for 2 h at 37°C. Sections were washed three times in wash solution (0.1% PBS/Triton) and incubated with secondary antibody diluted in blocking solution for 1 h at room temperature. Cell nuclei were stained with DAPI for 3 min at room temperature. Glasses were then mounted with Dako mounting medium, and fluorescent signals were monitored under a Carl Zeiss confocal microscope LSM900 with a Carl Zeiss EC Plan-Neofluar 40×/1.3 oil immersion objective. The antibodies used are described in Table S3.
H&E staining was performed on 7-μm cryosections of 3D SEs (Harris hematoxylin for 1 min, running tap water for 1 min, eosin Y 50% in ethanol for 30 s, 95% ethanol for 1 min, 100% ethanol for 1 min, and two rinses in fresh 100% ethanol for 1 min each). Color bright-field images were taken with a Carl Zeiss microscope (Axio Imager M2) with an EC Plan-Neofluar 40×/0.75 M27 air objective.
Colony-forming efficiency assay
Colony-forming efficiency was calculated at each passage from the indicator dish stained with rhodamine B after 12 days of cultivation. The percentages of clonogenic cells correspond to the number of colonies on the total amount of plated cells; the percentage of abortive colonies was calculated as the number of abortive colonies among the total number of colonies.
Generation of the ΔTGM1 human cellular model
gRNA CRISPR guides (Invitrogen) were designed to target TGM1 E2 (gRNA1, 5′-AAATGCGGCAGATGACGACT-3′ and gRNA2, 5′-AAATGCGGCAGATGACGACG-3′), which differ in 1 nt (underlined), allowing targeting of both TGM1 alleles, exploiting the presence of a SNP in a wild-type strain [K81], and TGM1 E4 (gRNA3, 5′-GGATGTAGATCTCATTGCGG-3′). gRNAs were checked for the minimal off-target activity predicted by an online tool (http://www.rgenome.net/cas-offinder/). gRNAs were mixed with the Cas9 protein (Alt-R S.p. Cas9 Nuclease V3, Integrated DNA Technologies [IDT], 1081058) in a 0.3:0.3:0.3:1 ratio and incubated at room temperature for 20 min, forming the RNP complex. 5 × 105 primary human keratinocytes (K81 strain) were resuspended in a final volume of 100 μL of primary cell nucleofection solution (P3 Primary Cell 4D-Nucleofector Kit, Lonza), mixed with the RNP complex solution and 4 mM Cas9 electroporation enhancer (IDT), and nucleofected using a 4D-Nucleofector (Lonza).70
Genomic analysis by sequence decomposition (TIDE) of the ΔTGM1 human cellular model
Genomic DNA isolated from ΔTGM1 and non-treated keratinocytes was used for the screening of the TGM1 sequence through PCR amplification. Two couples of primers (11-12 and 13-14; Table S2) were used to investigate the genomic sequence flanking both edited exons (e.g., E2 and E4, respectively). Moreover, PCR fragments spanning the Cas9 target sites were generated to evaluate the excision of the entire genomic region of above 1.7 kb between E2 and E4 with 11–14 primers (Table S2). PCR products were analyzed in 1% agarose gel. The molecular weight markers were the 100-bp DNA Ladder (Invitrogen) and 1-kb Plus DNA Ladder (Invitrogen). PCR amplicons were subjected to conventional Sanger sequencing. The resulting sequence trace files were uploaded on the TIDE web tool with the gRNA sequence as input. Obtained data were visualized with Prism 8.
Isolation of cross-linked cornified cell envelopes
For envelope formation, human primary keratinocytes were seeded onto a Millicell cell culture insert of 12-mm diameter with 0.4-μm pore size (Merck) for 5 days in submerged culture and further cultured for 20–24 days in the ALI condition to induce epidermal differentiation. Thereafter, the insert was carefully removed with tweezers and submerged in a buffer solution consisting of 1 M Tris-HCl (pH 8.0), 2% 2-mercaptoethanol, and 2% SDS.37 The suspension was heated to 100°C for 10 min and vortex mixed for 10 min. Envelope suspension was assessed by phase-contrast microscopy.
TG1 in situ enzymatic activity assay
TG1 in situ activity was performed on unfixed cryosections (7 μm thick) as described previously.71 The 3D SE cryosections (from normal, ΔTGM1, or SINγ-RV-TGM1-transduced keratinocytes) were air dried for 10 min at room temperature and then blocked with 1% BSA in PBS for 30 min at room temperature. Sections were incubated in 100 mmol/L Tris-HCl (pH 7.4), 5 mmol/L CaCl2, and 0.1 mmol/L Alexa Fluor 594 Cadaverine (A30678, Thermo Fisher Scientific) for 90 min at room temperature. Cell nuclei were stained with DAPI. Glasses were mounted with Dako mounting medium, and fluorescent images were obtained using a Carl Zeiss confocal microscope (LSM900) with a Carl Zeiss EC Plan-Neofluar 20×/0.50 M27 air objective.
TG1 in vitro enzymatic activity assay
The TG1 in vitro activity assay was performed with 50 μg of protein extracts obtained from normal or ΔTGM1 keratinocytes in growing (3-day) and over-confluent (14-day) conditions using the Transglutaminase Assay Kit (CS1070, Merck) according to the manufacturer’s instructions. Briefly, 50 μg of protein extracts was added to each well, and 2 milliunits/mL of transglutaminase enzyme from guinea pig liver was used as a positive control. 50 μL of assay mixture was added to each well, and the plate was incubated for 30 min at room temperature. Streptavidin-peroxidase and TMB substrate were added to each well, and the absorption was read at 450 nm with a plate-reading spectrophotometer (Glomax, Promega).
Encapsulation with the 10X Genomics chromium system and bioinformatics analysis on single-cell multiome data (ATAC and gene expression)
Fully confluent keratinocytes were detached, and cells were accurately resuspended to obtain a single-cell suspension in PBS containing 0.04% BSA. About 8 × 104 cells of SINγ-RV-TGM1-transduced keratinocyte samples were subjected to nucleus isolation and DNA fragmentation according to the manufacturer’s instructions (CG000338 Rev F, 10X Genomics). About 15,000 nuclei were loaded into the Chromium Next GEM Chip J using the Single Cell Multiome ATAC + Gene Expression Kit (10× Genomics). The sample was pre-amplified and half-divided to generate the Illumina sequencing ATAC and GEX libraries. Sequencing was performed on the NovaSeq6000 SP platform following the 10× Genomics instructions for read generation (150PE), reaching, on average, 67,000 reads per cell for the GEX library and 213,000 reads per cell for the ATAC library.
For the bioinformatics analysis, the Cell Ranger ARC pipeline (v.2.0.2) was used to generate FASTQ files, to align reads to the reference transcriptome (GRCh38) for the GEX library or to the reference genome (GRCh38) plus the exogenous SINγ-RV-TGM1 (provirus sequence) for the ATAC library, and to calculate unique molecular identifier counts from the mapped reads. The feature-barcode matrix was imported in R (v.4.2.2) and analyzed using the Seurat72 (v.4.3.0), Signac73 (v.1.9.0), and popsicleR74 (v.0.2.1) R packages. Low-quality cells were identified as outliers for both GEX and ATAC quality metrics and subsequently discarded. Doublets were identified and discarded using scDblFinder75 (v.1.8.0). Cell cycle scores were assigned to each remaining cell and regressed out before dimensionality reduction with PCA on GEX data. Then, cells were clustered at low resolution to discriminate major cell populations. Cells were classified into holoclones, meroclones, paraclones, and terminally differentiated cells using an annotated scRNA-seq dataset of human keratinocytes43,44 as a reference and the FindTransferAnchors, TransferData, and MapQuery functions in Seurat with default parameters. Clusters representing fibroblasts were identified using the expression level of VIM and COL1A1. We assessed the quality of the assigned labels monitoring the expression of known markers. Finally, to identify transduced keratinocytes, we recovered, for each cell, the number of fragments mapped to the provirus sequence from the “ATAC Per Fragment Information” file generated by Cell Ranger ARC. Expression data are available in the Gene Expression Omnibus (GEO: GSE268321).
Statistical analyses
GraphPad Prism v.8 was used for statistical analyses in the main and supplemental figures. RT-qPCR data are presented as the mean ± SD of at least three independent experiments (n = 3). Significance was calculated from ΔCt values with ANOVA. A value of p < 0.0001 was considered statistically significant.
Replications
All experiments were replicated a number of times depending on the experiment performed. For qualitative data, such as those obtained from immunofluorescence, images are representative of what was observed in at least three independent samples or replicates.
Data and code availability
All data generated or analyzed during this study are available from the corresponding author upon reasonable request.
Acknowledgments
This project was supported by the European Union “National Center for Gene Therapy and Drugs based on RNA Technology” NextGenerationEU (PNRR) (mission 4, component 2, investment 1.4, CN 00000041, CUP E93C22001080001), European Research Council (ERC) advanced grant HOLO-GT (101019289), and Telethon (grant GGP20088). We thank Le Ali di Camilla for providing assistance to patients.
Author contributions
L.S. performed experiments, analyzed data, assembled all input data, and edited the manuscript. G.M. and E.E. contributed to the multiome experiment. O.R. and M.F. performed multiome bioinformatics analyses. L.D.R. and M.D.L. coordinated the study, defined strategic procedures, performed the experiments, and revised the manuscript.
Declaration of interests
M.D.L. is a consultant for J-TEC-Japan Tissue Engineering, Ltd.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2024.101311.
Contributor Information
Laura De Rosa, Email: laura.derosa@unimore.it.
Michele De Luca, Email: michele.deluca@unimore.it.
Supplemental information
References
- 1.Elias P.M., Williams M.L., Crumrine D., Schmuth M. Focuses solely on generalized, inherited (Mendelian) disorders of cornification (DOC or MeDOC). Introduction. Curr. Probl. Dermatol. 2010;39:1–29. doi: 10.1159/000321082. [DOI] [PubMed] [Google Scholar]
- 2.Oji V., Tadini G., Akiyama M., Blanchet Bardon C., Bodemer C., Bourrat E., Coudiere P., DiGiovanna J.J., Elias P., Fischer J., et al. Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Soreze 2009. J. Am. Acad. Dermatol. 2010;63:607–641. doi: 10.1016/j.jaad.2009.11.020. [DOI] [PubMed] [Google Scholar]
- 3.Fischer J., Bourrat E. Genetics of Inherited Ichthyoses and Related Diseases. Acta Derm. Venereol. 2020;100:adv00096. doi: 10.2340/00015555-3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vahlquist A., Fischer J., Törmä H. Inherited nonsyndromic ichthyoses: an update on pathophysiology, diagnosis and treatment. Am. J. Clin. Dermatol. 2018;19:51–66. doi: 10.1007/s40257-017-0313-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abernethy J.L., Hill R.L., Goldsmith L.A. epsilon-(gamma-Glutamyl) lysine cross-links in human stratum corneum. J. Biol. Chem. 1977;252:1837–1839. [PubMed] [Google Scholar]
- 6.Rice R.H., Green H. The cornified envelope of terminally differentiated human epidermal keratinocytes consists of cross-linked protein. Cell. 1977;11:417–422. doi: 10.1016/0092-8674(77)90059-9. [DOI] [PubMed] [Google Scholar]
- 7.Takeichi T., Akiyama M. Inherited ichthyosis: Non-syndromic forms. J. Dermatol. 2016;43:242–251. doi: 10.1111/1346-8138.13243. [DOI] [PubMed] [Google Scholar]
- 8.Fleckman P., DiGiovanna J.J. In: Fitzpatrick's Dermatology in General Medicine, 8e. Goldsmith L.A., Katz S.I., Gilchrest B.A., Paller A.S., Leffell D.J., Wolff K., editors. The McGraw-Hill Companies; 2012. Chapter 49. The Ichthyoses. [Google Scholar]
- 9.Schmuth M., Martinz V., Janecke A.R., Fauth C., Schossig A., Zschocke J., Gruber R. Inherited ichthyoses/generalized Mendelian disorders of cornification. Eur. J. Hum. Genet. 2013;21:123–133. doi: 10.1038/ejhg.2012.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmuth M., Gruber R., Elias P.M., Williams M.L. Ichthyosis update: towards a function-driven model of pathogenesis of the disorders of cornification and the role of corneocyte proteins in these disorders. Adv. Dermatol. 2007;23:231–256. doi: 10.1016/j.yadr.2007.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arkin L., Lee L.W., Rubin A.I., Yan A.C. In: Lever’s Histopathology of the Skin. 11th Edition. Elder D.E., editor. 2015. Congenital Diseases (Genodermatoses) [Google Scholar]
- 12.Farasat S., Wei M.-H., Herman M., Liewehr D.J., Steinberg S.M., Bale S.J., Fleckman P., Toro J.R. Novel transglutaminase-1 mutations and genotype–phenotype investigations of 104 patients with autosomal recessive congenital ichthyosis in the USA. J. Med. Genet. 2009;46:103–111. doi: 10.1136/jmg.2008.060905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simpson J.K., Martinez-Queipo M., Onoufriadis A., Tso S., Glass E., Liu L., Higashino T., Scott W., Tierney C., Simpson M.A., et al. Genotype-phenotype correlation in a large English cohort of patients with autosomal recessive ichthyosis. Br. J. Dermatol. 2020;182:729–737. doi: 10.1111/bjd.18211. [DOI] [PubMed] [Google Scholar]
- 14.Chulpanova D.S., Shaimardanova A.A., Ponomarev A.S., Elsheikh S., Rizvanov A.A., Solovyeva V.V. Current Strategies for the Gene Therapy of Autosomal Recessive Congenital Ichthyosis and Other Types of Inherited Ichthyosis. Int. J. Mol. Sci. 2022;23:2506. doi: 10.3390/ijms23052506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gutiérrez-Cerrajero C., Sprecher E., Paller A.S., Akiyama M., Mazereeuw-Hautier J., Hernández-Martín A., González-Sarmiento R. Ichthyosis. Nat. Rev. Dis. Primers. 2023;9:2. doi: 10.1038/s41572-022-00412-3. [DOI] [PubMed] [Google Scholar]
- 16.Aufenvenne K., Larcher F., Hausser I., Duarte B., Oji V., Nikolenko H., Del Rio M., Dathe M., Traupe H. Topical enzyme-replacement therapy restores transglutaminase 1 activity and corrects architecture of transglutaminase-1-deficient skin grafts. Am. J. Hum. Genet. 2013;93:620–630. doi: 10.1016/j.ajhg.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Plank R., Yealland G., Miceli E., Lima Cunha D., Graff P., Thomforde S., Gruber R., Moosbrugger-Martinz V., Eckl K., Calderón M., et al. Transglutaminase 1 replacement therapy successfully mitigates the autosomal recessive congenital ichthyosis phenotype in full-thickness skin disease equivalents. J. Invest. Dermatol. 2019;139:1191–1195. doi: 10.1016/j.jid.2018.11.002. [DOI] [PubMed] [Google Scholar]
- 18.Freedman J.C., Parry T.J., Zhang P., Majumdar A., Krishnan S., Regula L.K., O’Malley M., Coghlan S., Yogesha S.D., Ramasamy S., Agarwal P. Preclinical Evaluation of a Modified Herpes Simplex Virus Type 1 Vector Encoding Human TGM1 for the Treatment of Autosomal Recessive Congenital Ichthyosis. J. Invest. Dermatol. 2021;141:874–882.e6. doi: 10.1016/j.jid.2020.07.035. [DOI] [PubMed] [Google Scholar]
- 19.Huber M., Limat A., Wagner E., Hohl D. Efficient in vitro transfection of human keratinocytes with an adenovirus-enhanced receptor-mediated system. J. Invest. Dermatol. 2000;114:661–666. doi: 10.1046/j.1523-1747.2000.00942.x. [DOI] [PubMed] [Google Scholar]
- 20.Mustfa S.A., Maurizi E., McGrath J., Chiappini C. Nanomedicine approaches to negotiate local biobarriers for topical drug delivery. Adv. Therapeut. 2021;4:2000160. [Google Scholar]
- 21.Choate K.A., Kinsella T.M., Williams M.L., Nolan G.P., Khavari P.A. Transglutaminase 1 delivery to lamellar ichthyosis keratinocytes. Hum. Gene Ther. 1996;7:2247–2253. doi: 10.1089/hum.1996.7.18-2247. [DOI] [PubMed] [Google Scholar]
- 22.Choate K.A., Medalie D.A., Morgan J.R., Khavari P.A. Corrective gene transfer in the human skin disorder lamellar ichthyosis. Nat. Med. 1996;2:1263–1267. doi: 10.1038/nm1196-1263. [DOI] [PubMed] [Google Scholar]
- 23.Choate K.A., Khavari P.A. Direct cutaneous gene delivery in a human genetic skin disease. Hum. Gene Ther. 1997;8:1659–1665. doi: 10.1089/hum.1997.8.14-1659. [DOI] [PubMed] [Google Scholar]
- 24.De Rosa L., Enzo E., Palamenghi M., Sercia L., De Luca M. Stairways to Advanced Therapies for Epidermolysis Bullosa. Cold Spring Harbor Perspect. Biol. 2023;15:a041229. doi: 10.1101/cshperspect.a041229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirsch T., Rothoeft T., Teig N., Bauer J.W., Pellegrini G., De Rosa L., Scaglione D., Reichelt J., Klausegger A., Kneisz D., et al. Regeneration of the entire human epidermis using transgenic stem cells. Nature. 2017;551:327–332. doi: 10.1038/nature24487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eichstadt S., Barriga M., Ponakala A., Teng C., Nguyen N.T., Siprashvili Z., Nazaroff J., Gorell E.S., Chiou A.S., Taylor L., et al. Phase 1/2a clinical trial of gene-corrected autologous cell therapy for recessive dystrophic epidermolysis bullosa. JCI Insight. 2019;4:e130554. doi: 10.1172/jci.insight.130554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Siprashvili Z., Nguyen N.T., Gorell E.S., Loutit K., Khuu P., Furukawa L.K., Lorenz H.P., Leung T.H., Keene D.R., Rieger K.E., et al. Safety and Wound Outcomes Following Genetically Corrected Autologous Epidermal Grafts in Patients With Recessive Dystrophic Epidermolysis Bullosa. JAMA. 2016;316:1808–1817. doi: 10.1001/jama.2016.15588. [DOI] [PubMed] [Google Scholar]
- 28.Kueckelhaus M., Rothoeft T., De Rosa L., Yeni B., Ohmann T., Maier C., Eitner L., Metze D., Losi L., Secone Seconetti A., et al. Transgenic Epidermal Cultures for Junctional Epidermolysis Bullosa - 5-Year Outcomes. N. Engl. J. Med. 2021;385:2264–2270. doi: 10.1056/NEJMoa2108544. [DOI] [PubMed] [Google Scholar]
- 29.Lorand L., Graham R.M. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat. Rev. Mol. Cell Biol. 2003;4:140–156. doi: 10.1038/nrm1014. [DOI] [PubMed] [Google Scholar]
- 30.Deyrieux A.F., Wilson V.G. In vitro culture conditions to study keratinocyte differentiation using the HaCaT cell line. Cytotechnology. 2007;54:77–83. doi: 10.1007/s10616-007-9076-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilson V.G. Growth and differentiation of HaCaT keratinocytes. Methods Mol. Biol. 2014;1195:33–41. doi: 10.1007/7651_2013_42. [DOI] [PubMed] [Google Scholar]
- 32.Barrandon Y., Green H. Three clonal types of keratinocyte with different capacities for multiplication. Proc. Natl. Acad. Sci. USA. 1987;84:2302–2306. doi: 10.1073/pnas.84.8.2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ronfard V., Rives J.M., Neveux Y., Carsin H., Barrandon Y. Long-term regeneration of human epidermis on third degree burns transplanted with autologous cultured epithelium grown on a fibrin matrix. Transplantation. 2000;70:1588–1598. doi: 10.1097/00007890-200012150-00009. [DOI] [PubMed] [Google Scholar]
- 34.Borowiec A.-S., Delcourt P., Dewailly E., Bidaux G. Optimal differentiation of in vitro keratinocytes requires multifactorial external control. PLoS One. 2013;8:e77507. doi: 10.1371/journal.pone.0077507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poumay Y., Pittelkow M.R. Cell density and culture factors regulate keratinocyte commitment to differentiation and expression of suprabasal K1/K10 keratins. J. Invest. Dermatol. 1995;104:271–276. doi: 10.1111/1523-1747.ep12612810. [DOI] [PubMed] [Google Scholar]
- 36.Boeshans K.M., Mueser T.C., Ahvazi B. A three-dimensional model of the human transglutaminase 1: insights into the understanding of lamellar ichthyosis. J. Mol. Model. 2007;13:233–246. doi: 10.1007/s00894-006-0144-9. [DOI] [PubMed] [Google Scholar]
- 37.Jeon S., Djian P., Green H. Inability of keratinocytes lacking their specific transglutaminase to form cross-linked envelopes: absence of envelopes as a simple diagnostic test for lamellar ichthyosis. Proc. Natl. Acad. Sci. USA. 1998;95:687–690. doi: 10.1073/pnas.95.2.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chu D.H. In: Fitzpatrick's Dermatology in General Medicine, 7e. Goldsmith L.A., Wolff K., Katz S.I., Gilchrest B.A., Paller A.S., Leffell D.J., editors. The McGraw-Hill Companies; 2008. Chapter 7. Development and Structure of Skin. [Google Scholar]
- 39.Elias P.M., Williams M.L., Crumrine D., Schmuth M. Inherited disorders of corneocyte proteins. Curr. Probl. Dermatol. 2010;39:98–131. doi: 10.1159/000321086. [DOI] [PubMed] [Google Scholar]
- 40.Aufenvenne K., Rice R.H., Hausser I., Oji V., Hennies H.C., Rio M.D., Traupe H., Larcher F. Long-term faithful recapitulation of transglutaminase 1-deficient lamellar ichthyosis in a skin-humanized mouse model and insights from proteomic studies. J. Invest. Dermatol. 2012;132:1918–1921. doi: 10.1038/jid.2012.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hohl D., Huber M., Frenk E. Analysis of the cornified cell envelope in lamellar ichthyosis. Arch. Dermatol. 1993;129:618–624. [PubMed] [Google Scholar]
- 42.Wiegmann H., Valentin F., Tarinski T., Liebau E., Loser K., Traupe H., Oji V. LEKTI domains D6, D7 and D8+ 9 serve as substrates for transglutaminase 1: Implications for targeted therapy of Netherton syndrome. Br. J. Dermatol. 2019;181:999–1008. doi: 10.1111/bjd.17820. [DOI] [PubMed] [Google Scholar]
- 43.Enzo E., Secone Seconetti A., Forcato M., Tenedini E., Polito M.P., Sala I., Carulli S., Contin R., Peano C., Tagliafico E., et al. Single-keratinocyte transcriptomic analyses identify different clonal types and proliferative potential mediated by FOXM1 in human epidermal stem cells. Nat. Commun. 2021;12:2505. doi: 10.1038/s41467-021-22779-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Polito M.P., Marini G., Fabrizi A., Sercia L., Enzo E., De Luca M. Biochemical role of FOXM1-dependent histone linker H1B in human epidermal stem cells. Cell Death Dis. 2024;15:508. doi: 10.1038/s41419-024-06905-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cortés H., Del Prado-Audelo M.L., Urbán-Morlán Z., Alcalá-Alcalá S., González-Torres M., Reyes-Hernández O.D., González-Del Carmen M., Leyva-Gómez G. Pharmacological treatments for cutaneous manifestations of inherited ichthyoses. Arch. Dermatol. Res. 2020;312:237–248. doi: 10.1007/s00403-019-01994-x. [DOI] [PubMed] [Google Scholar]
- 46.DiGiovanna J.J., Mauro T., Milstone L.M., Schmuth M., Toro J.R. Systemic retinoids in the management of ichthyoses and related skin types. Dermatol. Ther. 2013;26:26–38. doi: 10.1111/j.1529-8019.2012.01527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fleckman P., Newell B.D., van Steensel M.A., Yan A.C. Topical treatment of ichthyoses. Dermatol. Ther. 2013;26:16–25. doi: 10.1111/j.1529-8019.2012.01526.x. [DOI] [PubMed] [Google Scholar]
- 48.Chulpanova D.S., Shaimardanova A.A., Ponomarev A.S., Elsheikh S., Rizvanov A.A., Solovyeva V.V. Current Strategies for the Gene Therapy of Autosomal Recessive Congenital Ichthyosis and Other Types of Inherited Ichthyosis. Int. J. Mol. Sci. 2022;23:2506. doi: 10.3390/ijms23052506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Joosten M.D.W., Clabbers J.M.K., Jonca N., Mazereeuw-Hautier J., Gostyński A.H. New developments in the molecular treatment of ichthyosis: review of the literature. Orphanet J. Rare Dis. 2022;17:269. doi: 10.1186/s13023-022-02430-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dang L., Zhou X., Zhong X., Yu W., Huang S., Liu H., Chen Y., Zhang W., Yuan L., Li L., et al. Correction of the pathogenic mutation in TGM1 gene by adenine base editing in mutant embryos. Mol. Ther. 2022;30:175–183. doi: 10.1016/j.ymthe.2021.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ledford H. Why CRISPR babies are still too risky - embryo studies highlight challenges. Nature. 2023;615:568–569. doi: 10.1038/d41586-023-00756-0. [DOI] [PubMed] [Google Scholar]
- 52.Ginn S.L., Amaya A.K., Alexander I.E., Edelstein M., Abedi M.R. Gene therapy clinical trials worldwide to 2017: An update. J. Gene Med. 2018;20:e3015. doi: 10.1002/jgm.3015. [DOI] [PubMed] [Google Scholar]
- 53.Chakravarti S., Enzo E., Rocha Monteiro de Barros M., Maffezzoni M.B.R., Pellegrini G. Genetic Disorders of the Extracellular Matrix: From Cell and Gene Therapy to Future Applications in Regenerative Medicine. Annu. Rev. Genom. Hum. Genet. 2022;23:193–222. doi: 10.1146/annurev-genom-083117-021702. [DOI] [PubMed] [Google Scholar]
- 54.Cavazza A., Cocchiarella F., Bartholomae C., Schmidt M., Pincelli C., Larcher F., Mavilio F. Self-inactivating MLV vectors have a reduced genotoxic profile in human epidermal keratinocytes. Gene Ther. 2013;20:949–957. doi: 10.1038/gt.2013.18. [DOI] [PubMed] [Google Scholar]
- 55.Kraunus J., Schaumann D.H.S., Meyer J., Modlich U., Fehse B., Brandenburg G., von Laer D., Klump H., Schambach A., Bohne J., Baum C. Self-inactivating retroviral vectors with improved RNA processing. Gene Ther. 2004;11:1568–1578. doi: 10.1038/sj.gt.3302309. [DOI] [PubMed] [Google Scholar]
- 56.Yi Y., Hahm S.H., Lee K.H. Retroviral gene therapy: safety issues and possible solutions. Curr. Gene Ther. 2005;5:25–35. doi: 10.2174/1566523052997514. [DOI] [PubMed] [Google Scholar]
- 57.Bauer J.W., Koller J., Murauer E.M., De Rosa L., Enzo E., Carulli S., Bondanza S., Recchia A., Muss W., Diem A., et al. Closure of a Large Chronic Wound through Transplantation of Gene-Corrected Epidermal Stem Cells. J. Invest. Dermatol. 2017;137:778–781. doi: 10.1016/j.jid.2016.10.038. [DOI] [PubMed] [Google Scholar]
- 58.De Rosa L., Latella M.C., Secone Seconetti A., Cattelani C., Bauer J.W., Bondanza S., De Luca M. Toward Combined Cell and Gene Therapy for Genodermatoses. Cold Spring Harbor Perspect. Biol. 2020;12:a035667. doi: 10.1101/cshperspect.a035667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mavilio F., Pellegrini G., Ferrari S., Di Nunzio F., Di Iorio E., Recchia A., Maruggi G., Ferrari G., Provasi E., Bonini C., et al. Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nat. Med. 2006;12:1397–1402. doi: 10.1038/nm1504. [DOI] [PubMed] [Google Scholar]
- 60.Oji V., Traupe H. Ichthyosis. Clinical Manifestations and Practical Treatment Options. Am. J. Clin. Dermatol. 2009;10:351–364. doi: 10.2165/11311070-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 61.Kuramoto N., Takizawa T., Takizawa T., Matsuki M., Morioka H., Robinson J.M., Yamanishi K. Development of ichthyosiform skin compensates for defective permeability barrier function in mice lacking transglutaminase 1. J. Clin. Invest. 2002;109:243–250. doi: 10.1172/JCI13563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Matsuki M., Yamashita F., Ishida-Yamamoto A., Yamada K., Kinoshita C., Fushiki S., Ueda E., Morishima Y., Tabata K., Yasuno H., et al. Defective stratum corneum and early neonatal death in mice lacking the gene for transglutaminase 1 (keratinocyte transglutaminase) Proc. Natl. Acad. Sci. USA. 1998;95:1044–1049. doi: 10.1073/pnas.95.3.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gálvez V., Chacón-Solano E., Bonafont J., Mencía Á., Di W.L., Murillas R., Llames S., Vicente A., Del Rio M., Carretero M., Larcher F. Efficient CRISPR-Cas9-Mediated Gene Ablation in Human Keratinocytes to Recapitulate Genodermatoses: Modeling of Netherton Syndrome. Mol. Ther. Methods Clin. Dev. 2020;18:280–290. doi: 10.1016/j.omtm.2020.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dellambra E., Vailly J., Pellegrini G., Bondanza S., Golisano O., Macchia C., Zambruno G., Meneguzzi G., De Luca M. Corrective transduction of human epidermal stem cells in laminin-5-dependent junctional epidermolysis bullosa. Hum. Gene Ther. 1998;9:1359–1370. doi: 10.1089/hum.1998.9.9-1359. [DOI] [PubMed] [Google Scholar]
- 65.Enzo E., Cattaneo C., Consiglio F., Polito M.P., Bondanza S., De Luca M. Clonal analysis of human clonogenic keratinocytes. Methods Cell Biol. 2022;170:101–116. doi: 10.1016/bs.mcb.2022.02.009. [DOI] [PubMed] [Google Scholar]
- 66.Phillips M.A., Jessen B.A., Lu Y., Qin Q., Stevens M.E., Rice R.H. A distal region of the human TGM1 promoter is required for expression in transgenic mice and cultured keratinocytes. BMC Dermatol. 2004;4:2. doi: 10.1186/1471-5945-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ghazizadeh S., Doumeng C., Taichman L.B. Durable and stratum-specific gene expression in epidermis. Gene Ther. 2002;9:1278–1285. doi: 10.1038/sj.gt.3301800. [DOI] [PubMed] [Google Scholar]
- 68.Banerjee N.S., Chow L.T., Broker T.R. Retrovirus-mediated gene transfer to analyze HPV gene regulation and protein functions in organotypic "raft" cultures. Methods Mol. Med. 2005;119:187–202. doi: 10.1385/1-59259-982-6:187. [DOI] [PubMed] [Google Scholar]
- 69.Maurizi E., Martella D.A., Schiroli D., Merra A., Mustfa S.A., Pellegrini G., Macaluso C., Chiappini C. Nanoneedles Induce Targeted siRNA Silencing of p16 in the Human Corneal Endothelium. Adv. Sci. 2022;9:e2203257. doi: 10.1002/advs.202203257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cattaneo C., Enzo E., De Rosa L., Sercia L., Consiglio F., Forcato M., Bicciato S., Paiardini A., Basso G., Tagliafico E., et al. Allele-specific CRISPR-Cas9 editing of dominant epidermolysis bullosa simplex in human epidermal stem cells. Mol. Ther. 2024;32:372–383. doi: 10.1016/j.ymthe.2023.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Takeda M., Nomura T., Sugiyama T., Miyauchi T., Suzuki S., Fujita Y., Shimizu H. Compound heterozygous missense mutations p. Leu207Pro and p. Tyr544Cys in TGM1 cause a severe form of lamellar ichthyosis. J. Dermatol. 2018;45:1463–1467. doi: 10.1111/1346-8138.14675. [DOI] [PubMed] [Google Scholar]
- 72.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stuart T., Srivastava A., Madad S., Lareau C.A., Satija R. Single-cell chromatin state analysis with Signac. Nat. Methods. 2021;18:1333–1341. doi: 10.1038/s41592-021-01282-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Grandi F., Caroli J., Romano O., Marchionni M., Forcato M., Bicciato S. popsicleR: A R Package for Pre-processing and Quality Control Analysis of Single Cell RNA-seq Data. J. Mol. Biol. 2022;434:167560. doi: 10.1016/j.jmb.2022.167560. [DOI] [PubMed] [Google Scholar]
- 75.Germain P.L., Lun A., Garcia Meixide C., Macnair W., Robinson M.D. Doublet identification in single-cell sequencing data using scDblFinder. F1000Res. 2021;10:979. doi: 10.12688/f1000research.73600.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are available from the corresponding author upon reasonable request.