

Abstract
Multifaceted and divergent manifestations across tissues and cell types have curtailed advances in deciphering the cellular events that accompany advanced age and contribute to morbidities and mortalities. Increase in human lifespan during the past century has heightened awareness of the need to prevent age-associated frailty of neuronal and sensory systems to allow a healthy and productive life. In this review, we discuss molecular and physiological attributes of aging of the retina, with a goal of understanding age-related impairment of visual function. We highlight the epigenome–metabolism nexus and proteostasis as key contributors to retinal aging and discuss lifestyle changes as potential modulators of retinal function. Finally, we deliberate promising intervention strategies for promoting healthy aging of the retina for improved vision.
“Aging is the progressive accumulation of changes with time that are associated with or responsible for the ever-increasing susceptibility to disease and death which accompanies advancing age.”
—Harman (1981, p. 7124)
Keywords: vision health, epigenome, metabolism, mitochondria, age-related macular degeneration, diet, environment
1. INTRODUCTION
1.1. Conceptual Framework of Aging
Aging is a time-dependent multifactorial process associated with progressive deterioration of biological systems, leading to impaired physiological function and increased risk of disease and death. This general decline of functional capabilities through an organismal life is fairly conserved among species (Lopez-Otin et al. 2013). Aging embodies complex phenotypes because of a plethora of underlying facets, including tremendous person-to-person variability. Many theories have been proposed in an attempt to explain the molecular underpinnings of aging but none fully comprehend the cellular and system-level changes that accompany advanced age (Johnson et al. 2019). Three evolutionary concepts of aging have been formulated: (a) The theory of mutation accumulation proposes that organisms accrue harmful germline mutations, which cannot be eliminated by selective pressure because of their expression after the reproductive period; (b) the antagonistic pleiotropy theory suggests that mutations advantageous to early fitness become detrimental at an advanced age, for example, during cellular senescence; and (c) the disposable soma theory of aging proposes evolutionary tradeoffs between growth or reproduction and repair mechanisms. The latter is consistent with the accumulation of cellular damage over time and is widely considered to be the general cause of aging, influenced by genetic, environmental, and stochastic processes.
Longevity, as determined by age in years at death, has always been celebrated, and humans have eternally strived for a long life. A major difference between longevity and healthy aging studies is that the former focus on lifespan, which includes a significant genetic component, whereas the latter emphasize health span (Labat-Robert & Robert 2015). The human lifespan has enhanced markedly over the past century; however, living longer has also been associated with increased prevalence of age-related diseases, such as Alzheimer’s disease and age-related macular degeneration (AMD), commonly impacting nervous and sensory systems.
The mechanisms that determine how we age are not yet fully understood; nonetheless, contemporary understanding points to complex and tissue-specific age-associated alterations involving an array of biological processes, classified as hallmarks of aging. These hallmarks, mostly studied in mammals, encompass genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, and altered intracellular communication (Lopez-Otin et al. 2013). Whether these changes reflect necessary adaptations or unwanted turbulences is far from clear. Current appreciation of molecular pathways and environmental cues that impact aging-associated cell and tissue responses have enabled new research to focus on delaying, preventing, or reversing at least a few phenotypes of aging.
1.2. Genetics of Aging and Longevity
Aging is a universal process affecting phylogenetically diverse organisms from Saccharomyces cerevisiae and Caenorhabditis elegans to mammals including humans. The differences in lifespan among species suggest continuing evolution of the aging process. Studies in model systems, especially C. elegans and S. cerevisiae, have shown that lifespan can be changed by manipulations even in a single gene, although at the expense of fitness. Discoveries of aging-associated cellular pathways and molecules [such as sirtuins, insulin and insulin-like growth factor (IGF) signaling, mitochondrial alteration, and caloric restriction] in model organisms have provided key insights into aging in humans.
1.2.1. Studies in Caenorhabditis elegans and Saccharomyces cerevisiae.
Experiments in model organisms have underscored the importance of single genes in the regulation of lifespan. Early studies in C. elegans identified five mutants that alter lifespan (Klass 1983), mapping to a single genetic locus: age-1, which encodes a phosphatidylinositol-3 kinase (Friedman & Johnson 1988). Identification of age-1 propelled aging research by implying a genetic basis of lifespan and the possibility of modifying it. Other longevity genes were subsequently identified; these include daf-2 and daf-16 (dauer abnormal formation protein), which encode an insulin-like receptor and a FOXO-like transcription factor, respectively (Zecic & Braeckman 2020). Age-1, daf-2, and daf-16 are orthologs of mammalian genes involved in the insulin and IGF signaling pathways, suggesting their conserved role in aging. Similarly, studies in S. cerevisiae were instrumental in elucidating replicative aging (the number of cell divisions that one cell can complete) and chronological aging (the time over which a yeast cell can remain viable in a nutrient-deprived state) (Longo & Fabrizio 2012). NAD+-dependent deacetylase silent information regulator 2–related enzyme 1 (sirtuin1; SIRT1) was discovered in S. cerevisiae and shown to extend lifespan (Kaeberlein et al. 1999). Importantly, yeast studies revealed that lifespan extension is not necessarily accompanied by improved fitness, as long-lived mutants could be outcompeted by wild-type progenitors (Delaney et al. 2011). Basic investigations in S. cerevisiae and C. elegans have therefore uncovered key genes that contribute to aging and unraveled fundamental pathways associated with sirtuins and insulin signaling. We refer readers to interesting reviews summarizing mechanisms of aging in these organisms (Sampaio-Marques et al. 2019, Tissenbaum 2015).
1.2.2. Mammalian genetic studies.
Genes and genetic variations can influence lifespan in organisms, with heritability accounting for 10–15% of human longevity (Singh et al. 2019). Variants in genes including APOE, LDLR, ATXN2, and CDKN2B-AS1 have been identified through the associations of offspring health status and survival with parental lifespans, whereas others such as those in FOXO3 are correlated with extreme longevity, although none of these genes seem to be essential or sufficient to achieve longevity (Melzer et al. 2020). Importantly, mechanisms involved in healthy aging would likely be tissue dependent. For example, transcriptome analysis and genome-wide association studies (GWAS) have identified selective impact of genetic variants in TMEM106B and progranulin in the frontal cerebral cortex at an advanced age (Rhinn & Abeliovich 2017). Nonetheless, identification of variants commonly associated with many age-related disorders indicates that similar age-linked molecular mechanisms may drive the onset and/or severity of disease. Recently, aging-associated traits have been linked at 22 genetic loci that impact lifespan, as well as common disorders including cardiovascular disease, cancers, endometriosis, and glaucoma (Melzer et al. 2020). Genetic studies have also provided insights into the importance of genome integrity and maintenance in aging and longevity. Mutations in genes that affect DNA repair and DNA maintenance can cause progerias, characterized by accelerated aging (Burla et al. 2018). A strong association has been detected between accumulation of somatic DNA mutations and aging phenotypes, a topic that is expanded on further below.
Aging research has come a long way, from identification of genes that extend longevity in worms and yeast to formulation of drugs and interventions that benefit human health and lifespan. In this review, we discuss how aging impacts the retina, with an emphasis on age-related molecular events that contribute to decline in visual function. In addition, given that advanced age is a major risk factor for common blinding eye diseases, we ponder whether lifestyle habits including dietary changes can ameliorate retinal aging and consequently the clinical impact of age-related diseases such as AMD.
2. AGING OF THE RETINA
Age-related loss of sensory functions is slow and progressive, likely representing a general decline of neuronal activity. Vision is arguably the most dominant sense in human beings. Impairment of vision with advancing age can lead to lower quality of life in the elderly, impacting negatively on daily activities, social interactions, and independence. The visual system consists of the eye, optic nerve, optic chiasm, optic tract, lateral geniculate nucleus, and visual cortex (Schiller & Tehovnik 2015). The visual process is initiated in the retina, a light-sensitive tissue lining the inner surface of the eye, which converts photons into electrical signals and transmits the information to the brain for object recognition.
The mammalian retina is composed of the neural retina and the retinal pigment epithelium (RPE). The neural retina is a laminated structure consisting of six major types of neurons, organized in three nuclear layers that encompass neuronal cell bodies (or somas) and two plexiform layers of synapses [Masland 2001, Ramón y Cajal 1892 (1972)]. Rods and three types of cone photoreceptors [short (S)-, medium (M)-, and long (L)-wavelength cones] in the outer nuclear layer contain distinct visual pigments and initiate the phototransduction cascade by capturing photons and converting them to electrical signals. These light-induced neuronal responses are further codified by the inner retina neurons and sent to the brain via the optic nerve, which originates from axons of the retinal ganglion cells (RGCs) (Hoon et al. 2014). Cones are adapted to photopic conditions and permit the perception of color. In contrast, rods are highly sensitive to dim light, thereby allowing night vision. Rod photoreceptors dominate the retina, making up almost 70% of total cells in most mammals (including humans), whereas the three types of cones together constitute approximately 5% of the cells in the human retina. Unlike those of a majority of mammals, the human (and primate) retinas display a unique region located at the center, called the macula. This distinctive area is specialized for high-resolution vision and includes the fovea and foveola, which are made up of only cone (and no rod) photoreceptors. Cone abundance is higher in the macula and decreases toward the peripheral retina. The photoreceptors form a functional unit with the RPE, a monolayer of epithelial cells that carry out multiple essential functions (Strauss 2005).
Aging can lead to tissue- and even cell type–specific structural and functional aberrations. In the retina, cellular and molecular changes associated with advanced age can be categorized as primary, antagonistic or adaptive, and integrative signatures (Figure 1). Primary signatures can trigger time-dependent damage accumulation and include loss of genome integrity and stability, epigenetic modifications, alterations of post-transcriptional regulatory mechanisms, and abnormal proteostasis. Antagonistic or adaptive responses are comprised of changes in metabolic pathways, local inflammation, and cellular senescence. The inability to repair or adapt to primary signatures can lead to disruptions in cellular homeostasis and emergence of integrative signatures including impaired intercellular communication. Stem cell exhaustion is considered to be an integrative hallmark and a major driver of aging in multiple tissues. However, stem cell activity in the adult human retina is negligible, if it exists at all, and thus evidence pointing toward the capacity for retinal regeneration is scarce.
Figure 1.
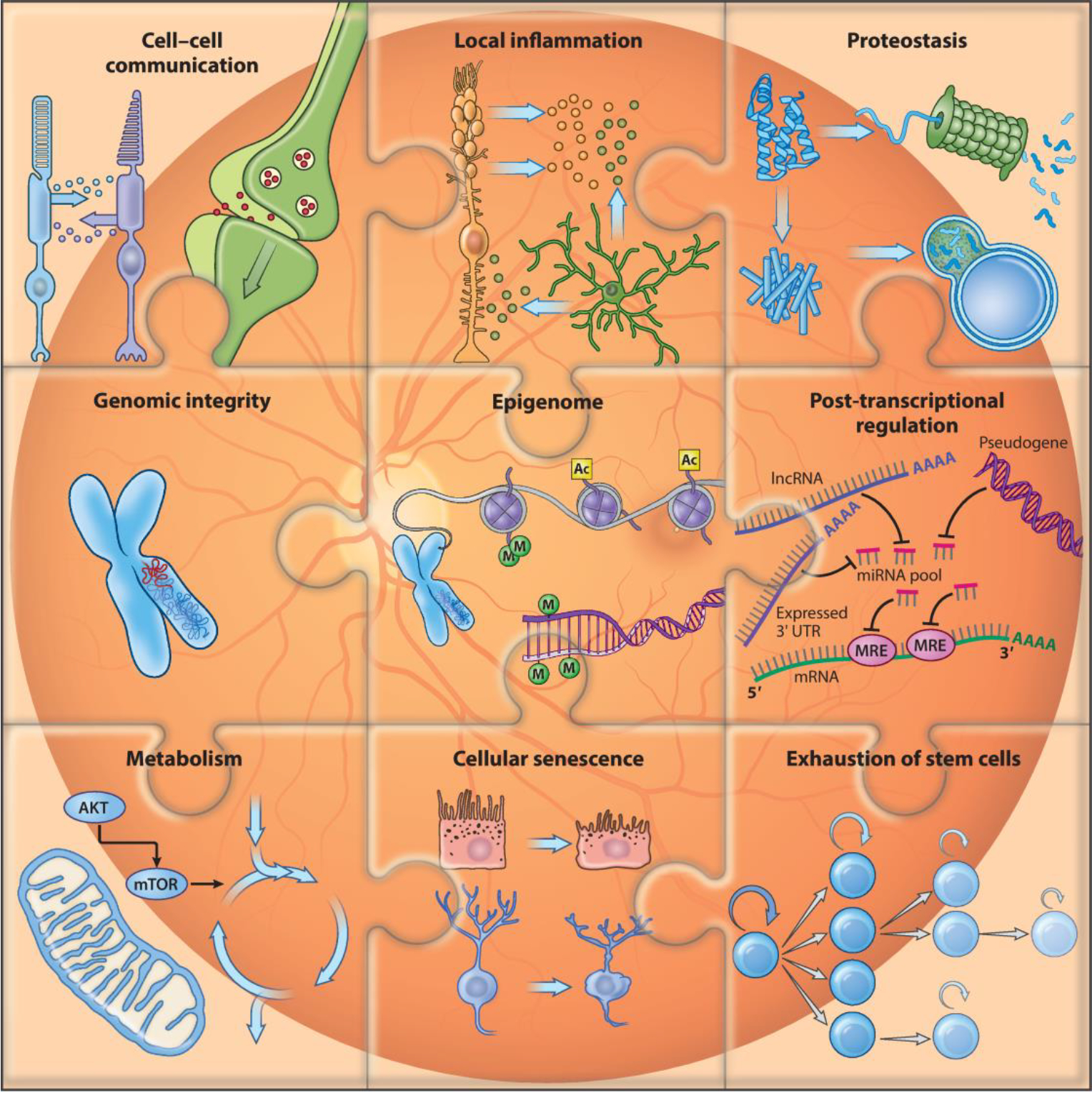
Signatures of aging in the retina. A schematic of molecular and cellular signatures of retinal aging is shown. Stem cell exhaustion and post-transcriptional regulation have been observed in other organisms but not yet established as signatures of retinal aging.
2.1. Functional Changes
Vision deteriorates during the natural course of aging (Figure 2a). Psychophysical and physiological studies in humans have demonstrated several common problems in older adults; these include lower visual acuity, impaired dark adaptation and reduction in contrast sensitivity (especially under low luminance levels), and altered sensitivity to motion and color perception (Owsley 2011). The degree of vision loss varies among elderly individuals; however, even a less severe visual deficit can impair one’s abilities to function optimally. Age-related loss of vision is attributed in part to changes in the optics of the eye, mainly due to lens opacification, optical aberrations, and loss of pupillary reactivity, which alter the light propagation to the retina (Berrio et al. 2010, Petrash 2013). Significant decline in visual function in the elderly is the result of dysfunction and/or death of retinal cells (Aggarwal et al. 2007, Curcio & Drucker 1993, Curcio et al. 1993, Gao & Hollyfield 1992). Rod photoreceptors are especially vulnerable to aging, resulting in a more prominent decline of scotopic than of photopic vision (Jackson & Owsley 2000). Older adults frequently cite vision problems at night and under dim light conditions. In standard full-field electroretinograms, which measure global retinal function, older individuals commonly exhibit a significant increase in implicit times and a reduction in the amplitudes of a- and b-waves generated by responses of photoreceptors and inner retinal cells (mainly bipolar and Müller cells), respectively (Bonnel et al. 2003). Multifocal electroretinograms that analyze local responses from different regions of the retina have revealed the greatest loss of amplitude density in the central 10° diameter of vision in the elderly (Fortune & Johnson 2002, Gerth et al. 2002, Jackson et al. 2002). Moreover, the amplitude of multifocal oscillatory potentials that are thought to reflect rod–cone interactions decreases linearly and uniformly over the retina with age, whereas their latency increases over time (Kurtenbach & Weiss 2002). At this stage, quantitative conclusions are difficult from such studies because of differences in subject age, confounding changes in pupil size and lens density, and other individual-specific variabilities.
Figure 2.
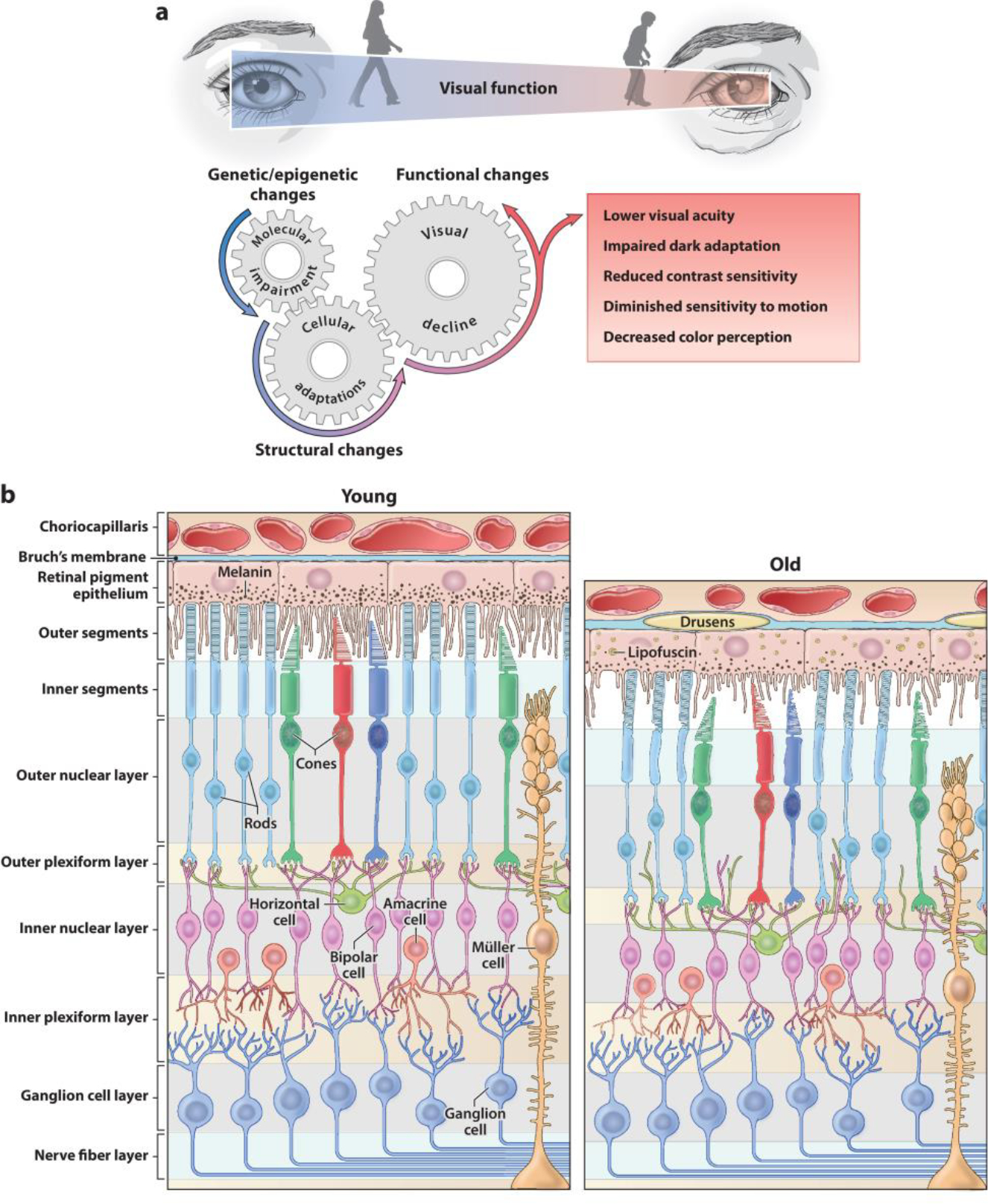
Functional and structural changes in the aging retina. (a) Visual impairment is prevalent in the elderly. Molecular alterations accumulated over time trigger cellular responses that together determine the structural and functional retinal aging phenotype. (b) Representation of retinal vertical sections showing characteristic morphological features of the young adult retina (left) and anatomical modifications in the aged retina (right).
2.2. Structural Changes
Cellular and laminar structures of the retina are largely maintained in the elderly. However, both the RPE and neural retina display distinct age-related quantitative alterations (Figure 2b). Common features of RPE aging include loss of melanin granules, accumulation of lipofuscin, formation of drusen, thickening of Bruch’s membrane, microvilli atrophy, and disorganization of the basal infoldings (Bonilha 2008, Boulton & Dayhaw-Barker 2001, Gao & Hollyfield 1992). Severity of these aforementioned changes varies among individuals and can result in RPE metabolic dysfunction contributing to age-related vision loss. Rod photoreceptors seem to be more vulnerable to the aging process than cones (Curcio et al. 1993, Gao & Hollyfield 1992). Major changes in the rod population of the peripheral retina occur between the second and fourth decades of human life (average loss of 970 cells/mm2/year), accounting for almost half of the total rod loss in aging (Gao & Hollyfield 1992). Rod loss rate slows down to 570–330 cells/mm2/year thereafter. In the central retina, rod density decreases by 30% over a lifetime, whereas cone number in the macula remains unaltered (Curcio et al. 1993).
Morphological changes are also detected in second-order neurons of the aging human retina. Dendrites of horizontal neurons, rod bipolar cells, and ON-cone bipolar cells undergo sprouting into the outer nuclear layer in aged retinas (Eliasieh et al. 2007). Aberrant processes in bipolar cells exhibit a markedly spatial gradient, increasing from the center to the periphery of the retina. The number of tyrosine hydroxylase-immunoreactive amacrine cells is reduced by half in the older (>65 years) human retina (Roufail & Rees 1997). However, this change could reflect a loss of immunoreactivity without cell loss.
RGC density is reduced by 25% near the fovea and in the nasal retina of older humans (Curcio & Drucker 1993). Irrespective of the species, the number of RGC axons is reduced by as much as 40% over a lifetime (Calkins 2013), influencing the transmission of information from the eye to the brain. A decrease of overall macular thickness and volume with age may be attributed to thinning of the retinal nerve fiber layer and inner retinal layers (Parikh et al. 2007, Subhi et al. 2016). The number of melanopsin-expressing RGCs decreases by 30% in the elderly (>70 years) compared to younger subjects (30–50 years), and the dendritic area of the remaining cells is reduced (Esquiva et al. 2017), likely explaining sleep disorders in older-age persons given the fundamental role of these cells in modulating circadian rhythms (Do 2019). Astrocytes in aged retinas from humans and rodents show hypertrophy, higher levels of glial fibrillary acidic protein, and distinctive morphological changes (Mansour et al. 2008, Ramirez et al. 2001). Microglia ramifications in the mouse retina lose motility, branching complexity, and length during aging (Damani et al. 2011, Ma & Wong 2016), resulting in reduced surveillance territory and phagocytic activity with accumulation of neurotoxic debris. Taken together, these results indicate that structural modifications of the neural retina and RPE likely contribute to age-related visual deficits by altering the elaboration and transmission of visual information from the eye to the brain.
3. GENOME AND EPIGENOME
Genomic and epigenomic alterations are widely observed during aging (Booth & Brunet 2016, Hoeijmakers 2009). Epigenome changes, in particular, are strongly linked to age-related disorders and offer a suitable therapeutic target due to their reversibility and interaction with the environment. In this section, we summarize changes in genome, transcriptome, and epigenome landscapes of the aging retina.
3.1. Genome Instability
Defects in genome maintenance and damage-response pathways are widely linked to the aging process. Accordingly, accumulation of DNA damage and activation of transposable elements have been observed in tissues from elderly individuals. In this section, we discuss the role of DNA repair and transposable elements in aging and highlight the studies performed in the retina.
3.1.1. DNA repair.
The accumulation of DNA lesions with age has been widely recognized as a hallmark of aging in various organisms (Hoeijmakers 2009). Upregulation of DNA repair pathways is associated with increased longevity in rodents (MacRae et al. 2015), whereas alterations in DNA repair are linked to premature aging, neurological disorders (Madabhushi et al. 2014), and age-related cognitive impairment (Lodato et al. 2018). DNA repair includes nucleotide excision repair (NER), base excision repair (BER), homologous recombination (HR), and nonhomologous end joining (NHEJ) (Dinant et al. 2008). NER participates in the repair of photoproducts and lesions located in transcribed regions. DNA double-strand breaks are primarily repaired by HR and NHEJ (Gillet & Scharer 2006). However, DNA damage responses can be tissue and species specific, making it difficult to use model systems to study the role of DNA damage in aging. Imbalances in BER pathways have been linked to retinal degeneration in mice (Meira et al. 2009). Adult mouse rods are reportedly deficient in ataxia-telangiectasia mutated kinase, which promotes double-strand break repair, and are unable to repair heterochromatin lesions (Frohns et al. 2014) or form 53bp1 DNA damage foci, yet human explants of retina show efficient double-strand repair and 53BP1 foci after induced DNA damage (Frohns et al. 2020). Importantly, polymorphisms in DNA repair genes have been associated with AMD; these include RAD51B, which is involved in DNA double-strand breaks (Chu et al. 2014), and ERCC2 (XPD), a helicase that participates in NER (Gorgun et al. 2010). However, the extent of DNA damage with normal aging in the retina is unknown. Mitochondrial DNA damage is shown to influence aging and age-related disease in the retina and is summarized in Section 4.2.
3.1.2. Transposable elements.
Transposable elements (TEs) are mostly silenced via epigenetic mechanisms such as histone modifications and DNA methylation (DNAm). Loss of TE silencing can influence the aging process (De Cecco et al. 2019) and contribute to neurodegenerative disorders (Jonsson et al. 2020). The function of TEs in the aging retina is poorly understood. Notably, Alu elements have been implicated in geographic atrophy, a form of advanced AMD (Kim et al. 2014). As new technologies such as long-read sequencing and bioinformatic pipelines emerge, investigations of TEs and repetitive sequences in healthy retinal aging and disease conditions will become feasible from human retina data sets, as shown for other tissues (LaRocca et al. 2020).
The retina likely evolved molecular adaptations to counteract the excessive DNA damage and TE activation that result from a high metabolism and UV exposure. DNA repair pathways seem to be altered in age-related diseases. Thus, elucidation of relevant molecules and DNA repair pathways in the human retina and their dynamics with age may offer targets to counteract loss of genomic integrity and delay vision loss.
3.2. Transcriptome
Loss of global transcriptional integrity is a prominent signature of aging. Common alterations include downregulation of genes encoding mitochondrial proteins and protein synthesis and dysregulation of genes involved in the immune system, responses to stress, and DNA damage (Frenk & Houseley 2018). Initial experiments using slide microarray analysis showed transcriptional changes in 24 genes related to energy metabolism, stress response, cell growth, and neuronal signaling by comparing young and elderly human retinas (Yoshida et al. 2002). A follow-up study, also using microarray analysis, compared young and elderly macula and peripheral retina (Cai et al. 2012). In the older macula, 85 downregulated genes were involved in cell metabolism, cell regulation, and development, whereas 55 upregulated genes were associated with cell proliferation, survival, and differentiation. In the peripheral retina, 52 downregulated and 34 upregulated genes were involved in a variety of biological functions, including an epigenetic regulator EZH1. However, whole-retina gene profiles can miss cell type–specific changes across the human lifespan. Transcriptome profiling of purified mouse rod photoreceptors (Corso-Diaz et al. 2020, Parapuram et al. 2010) at different time points of aging uncovered expression changes in immune-related genes, neuronal signaling, and aging pathways. Notably, changes in neuronal signaling and DNA damage response pathways appear to be conserved in aging Drosophila photoreceptors (Hall et al. 2017). Aging RPE cells also show changes primarily in immune response and inflammation (Chen et al. 2010, Ida et al. 2003). Importantly, single-cell RNA-seq from aging mouse retina has provided expression signatures of complement pathway genes in distinct cell types, pointing to their role in the progression of age-related diseases (Pauly et al. 2019). The expression of complement genes C1s, Cfb, and Cfi increased, whereas Cfh expression decreased in mouse retinal cells at 24 weeks of age. Thus, many gene expression signatures appear to be similar between retinal cells and other tissues. Single-cell transcriptional dynamics at distinct stages of aging would be valuable to identify primary cell type–specific alterations as potential targets for intervention.
3.3. Epigenome
The genomic DNA is packed into nucleosomes, which protects DNA from nucleases and allows for the regulation of DNA repair, transcription, and chromatin remodeling (Bannister & Kouzarides 2011). Chromatin (DNA and histones) undergoes chemical or epigenetic modifications that control gene expression and integrate stimuli from the environment into the genome (Sen et al. 2016). Several aspects of chromatin are altered with aging, including histone levels and modifications, nucleosome composition and positioning, and high-order three-dimensional (3D) conformation (Criscione et al. 2016, Sen et al. 2016). However, the relationship between the epigenome and transcriptional regulation during mammalian aging is still poorly understood.
3.3.1. Chromatin structure.
Chromatin 3D conformation and accessibility patterns have been used to identify key regulatory elements in the mouse retinal genome (Norrie et al. 2019). However, the dynamics of chromatin with age in different retinal cell types is unknown. The work of Corso-Diaz et al. (2020) on rod DNAm showed that certain chromosomal regions engaged in long-range looping are more susceptible to age-related epigenetic changes, suggesting alterations in DNA contacts with age.
A widespread reduction of chromatin accessibility has been detected in the neural retina and RPE of individuals with AMD and in iPSC-derived RPE cells after treatment with cigarette smoke (Wang et al. 2018). These changes may be mediated in part by HDAC11, as its overexpression in RPE cells grown in vitro induced a widespread decrease of chromatin accessibility and concomitant reduction of H3K27ac. Similarly, Abca4/Rdh8 double-knockout mice (a photosensitive mouse model of stress-induced photoreceptor degeneration) show a global decrease in accessibility of open chromatin regions in both the neural retina and RPE upon induction of photoreceptor degeneration by bright light stress (Luu et al. 2020). Notably, pharmacological inhibition of the histone-modifying enzymes HDAC11 and suppressor of variegation 3–9 homolog 2 (SUV39H2) can ameliorate light damage, suggesting that therapy aimed at epigenome modification is feasible. Changes in chromatin accessibility of retinal cells during photoreceptor stress, age-related disease, or environmental stress indicate that chromatin alterations are a common consequence of injury. However, it is unknown whether these changes are protective or drivers of neurodegenerative processes and whether aging-associated changes in retinal cell composition are responsible for observed chromatin alterations.
3.3.2. Histone modifications.
Profiles of histone modifications associated with gene activation or repression have been described during retinal development (Aldiri et al. 2017). Several retinal transcription factors including PAX6, NRL, CRX, and NR2E3 are shown to interact with histone-modifying enzymes to drive specific developmental programs (Corso-Diaz et al. 2018). The expression of histone deacetylase SIRT1 appears to be reduced with aging in the rat (Lamoke et al. 2015) and human (Peng et al. 2010) retina. Interestingly, resveratrol, a sirtuin activator, can increase SIRT1 expression and improve electroretinogram signals in the rat retina and the viability of stem-like cells in cultures obtained from old rats (Peng et al. 2010, Zeng & Yang 2015). Alterations in histone modifications have also been observed at genes involved in mitochondrial homeostasis and superoxide metabolism in models of diabetic retinopathy (Kowluru & Mishra 2015). Thus, targeting epigenetic enzymes can potentially ameliorate age-related functional decline in the retina and even in age-related retinal disease. Indeed, several inhibitors of methyltransferases, histone acetylases, and deacetylases have been proposed for therapeutic intervention to treat AMD (Gemenetzi & Lotery 2020).
3.3.3. DNA methylation.
Early studies on DNAm suggested a global depletion of DNAm as we age (Unnikrishnan et al. 2018). However, methylation has been shown to change bidirectionally with aging (Maegawa et al. 2010). Importantly, variations in CpG methylation with age have permitted the development of epigenetic clocks that identify a set of CpGs strongly correlated with chronological age. Epigenetic age acceleration can be determined by contrasting DNAm age with chronological age, is predictive of mortality and longevity, and is linked to many human diseases (Bell et al. 2019).
DNAm profiles have been generated from developing and adult mouse and human retinas (Aldiri et al. 2017, Hoshino et al. 2019). The epigenetic age of the fetal retina and stem cell–derived retinal organoids is shown to be highly correlated with chronological age but accelerated in individuals with Down syndrome (Hoshino et al. 2019). However, to date, few studies have addressed the DNAm changes that occur during physiological aging in the retina. Increased DNAm at the promoter of the long-chain fatty acid elongase Elovl2 has been associated with its upregulation during aging and functional decline of the mouse retina (Chen et al. 2020). Notably, changes in methylation during normal aging are usually small and occur at cell type–specific regulatory regions (Cole et al. 2017). To get insights into cell-specific DNAm changes, Corso-Diaz et al. (2020) generated whole-genome DNAm profiles with nucleotide resolution of isolated photoreceptors at different states of mouse aging. DNAm changes are enriched at regulatory regions and correlated with alterations in known aging pathways such as energy metabolism and mitochondrial respiration (Corso-Diaz et al. 2020), suggesting crosstalk between epigenome and metabolic pathways. Similarly, Müller glial cells show age-related subtle DNAm changes at intergenic regions (Lin et al. 2019). DNA methylation patterns and age-related functional decline of mouse ganglion cells can be restored through ectopic expression of Oct4 (Pou5f1), Sox2, and Klf4 genes (OSK) (Lu et al. 2020). The presence of these factors can promote axon regeneration after injury and reverse vision loss in aging mice and in a mouse model of glaucoma. This reversal requires the DNA demethylases TET1 and TET2, presumably acting on CpGs in genes related to synaptic and neuronal processes and bound by the polycomb repressive complex 2 (PRC2). However, a direct effect of DNA methylation on gene expression has not been addressed, and simply changing the levels of TET enzymes is not sufficient to achieve restoration. OSK factors can mediate many cellular changes that need to be further explored.
DNAm studies on age-related diseases such as AMD have interrogated only a few genomic sites. Hypermethylation at the promoter of glutathione S-transferase 1 (GSTM1) has been detected in the RPE of individuals with AMD (Hunter et al. 2012). Interestingly, hypermethylation of Gstm2, Gstm5, and Gstm6 promoters is also observed in aging mouse rods (Corso-Diaz et al. 2020), suggesting crosstalk of methylation with oxidative stress pathways. Hypomethylation of Tenascin X (TNXB) and the KI proto-oncogene (SKI), as well as hypermethylation of the general transcription factor IIH subunit H4 (GTF2H4), is also observed in the RPE of individuals with AMD (Porter et al. 2019). DNA methylation levels in peripheral blood leukocytes of individuals with AMD demonstrated hypomethylation at the IL17RC promoter (Wei et al. 2012), a finding that was later challenged (Oliver et al. 2013). Hypomethylation near the ARMS2 locus in the blood of neovascular AMD patients has also been reported (Oliver et al. 2015). Incontrast, global DNAm levels measured by liquid chromatography are shown to be increased in the blood cells of diabetic retinopathy patients compared to controls (Maghbooli et al. 2015). Despite current evidence of DNAm alterations in aging, a comprehensive profile of DNAm with nucleotide resolution in different retinal cell types during normal human aging and disease is still needed, in conjunction with directed epigenomic editing experiments, to identify specific targets for epigenetic therapy.
Importantly, longer genes tend to be associated with neuronal functions and rely on gene body methylation for regulation (Gabel et al. 2015). Re-analysis of Corso-Diaz et al.’s (2020) DNAm profiles of aging rods reveals more frequent age-related DNAm changes in longer gene bodies with correspondingly more exons affecting the genes involved in aging and cellular communication (Figure 3). Neuronal genes are more susceptible to DNA damage and require epigenome editing more frequently (Wei et al. 2016). It is thus possible that the neuronal gene structure itself makes some genes more susceptible to epigenetic alterations with age. This hypothesis is consistent with observed age-related alterations in long genes associated with synaptic function in Drosophila photoreceptors (Hall et al. 2017) and with transcriptional and DNA methylation changes in mouse synaptic genes including Grm1 and Syt14 (Corso-Diaz et al. 2020). Further studies should take into account the relationship between genomic features and age-related loss of chromatin integrity.
Figure 3.
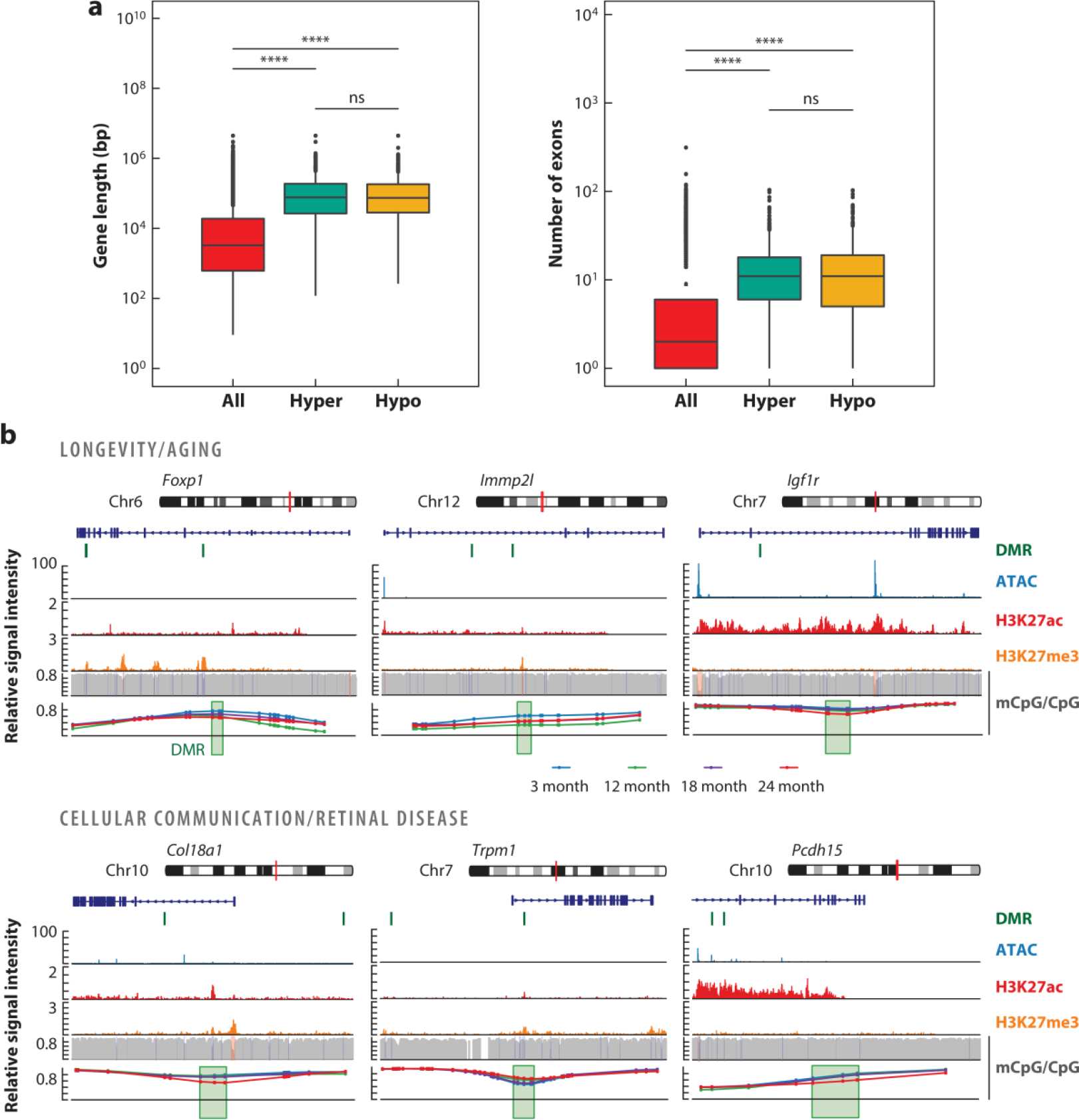
Differential methylation at gene bodies of long genes. Re-analysis of published (Corso-Diaz et al. 2020) differentially methylated regions (DMRs) identified in purified mouse rod photoreceptors at 24 months old compared to at 3 months old is shown. (a) Gene length expressed in base pairs (bp) and exon number of all genes and genes containing DMRs that are hyper- or hypo-methylated with age. ***indicates p < 2 × 10−16; ns indicates not significant. (b) Genome browser view of DMRs at genes involved in aging and longevity (Foxp1, Immp2l, and Igf1r) and cellular communication (Col18a1, Trpm1, and Pcdh15). The red vertical line on the chromosome pictogram indicates the location of the loci presented below. The location of DMRs on or in the vicinity of associated genes is shown by a green vertical line. Signal levels for open chromatin (ATAC-seq), histone modifications associated with active (H3K27ac) and repressed (H3K27me3) regions, and CpG methylation are shown for the three-month time point across the respective genes and their surroundings. The bottom panel represents a zoomed-in view of DMRs (green box) showing methylation levels of each CpG for rods from the 3-month-old (blue), 12-month-old (green), 18-month-old (purple), and 24-month-old (red) retina.
Age-associated alterations in epigenome-mediated gene regulation can act synergistically with aging hallmarks, leading to progressive accumulation of damage. Future studies focusing on individual cells may elucidate cell-intrinsic genetic networks that impact retinal homeostasis with advancing age. The interactions of environmental factors (such as diet and smoking) with the epigenome will likely offer opportunities to impede the progression of aging.
4. METABOLIC DYSREGULATION
The retina is among the most metabolically active tissues in mammals, with photoreceptors requiring large amounts of ATP for phototransduction as well as for maintaining a depolarized state in the absence of light (Linton et al. 2010, Okawa et al. 2008). Additionally, photoreceptors need steady protein synthesis and biomolecular supply for renewal of the light-sensitive ciliary outer segments (Chinchore et al. 2017). However, a major side effect of high metabolic activity is generation of metabolic stresses that often upsurge with aging (Wellen & Thompson 2010). In this section, we discuss metabolic dysfunction as a prominent signature of retinal aging in the context of the epigenome–metabolism nexus (Corso-Diaz et al. 2020) (Figure 4).
Figure 4.
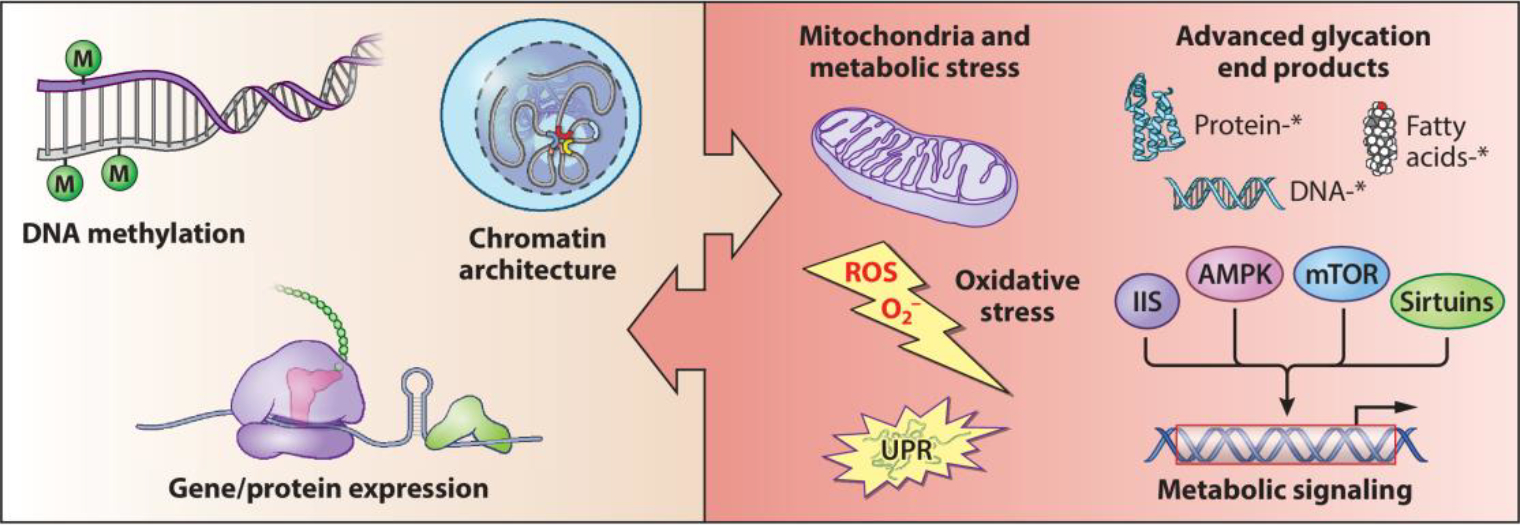
The epigenome–metabolism nexus in retinal aging. Among the signatures of retinal aging, epigenome–metabolism crosstalk is now emerging as a key determinant of age-related molecular and functional changes. Traditionally, genomic and epigenomic alterations have been considered primary drivers of aging. However, aging-related metabolic dysregulation and the possibility of epigenome moderation through metabolites are suggestive of a previously unappreciated molecular nexus. Asterisks indicate advanced glycation end product–modified biomolecules.
4.1. Dysregulated Metabolic Signaling
Efficient metabolic signaling is essential for maintaining nutrient flux, which is in turn critical for photoreceptor homeostasis. The key components involved in sensing nutrient abundance or scarcity during aging include insulin and IGF signaling; mammalian target of rapamycin (mTOR); adenosine monophosphate-activated protein kinase (AMPK); and sirtuins (Lopez-Otin et al. 2013), which also appear to contribute to the aging-associated physiological response in the retina. Loss of IGF1 leads to significant retinal dysfunction in 1-year-old mice (Rodriguez-de la Rosa et al. 2012). IGF1 is also suggested to modulate inflammation preceding retinal pathologies, including AMD (Arroba et al. 2018). The gene for insulin receptor substrate 1 (Irs1) is identified among the epigenomic targets in aging rod photoreceptors (Corso-Diaz et al. 2020). Modulation of mTOR complexes determines photoreceptor survival (Punzo et al. 2009), where mTORC1 and mTORC2 play distinct roles. Interestingly, mTORC1 activity is enhanced in the aging retina, and its stimulation can emulate photoreceptor aging and degeneration (Cheng et al. 2020). Increased mTORC1 is also shown to inhibit photoreceptor outer segment recycling in aged RPE cells (Yu et al. 2014).
AMPK and sirtuins play protective roles in aging (Lopez-Otin et al. 2013). AMPK senses energy deficit and activates several metabolic pathways including mitochondrial biogenesis and energy production (Herzig & Shaw 2018). In the retina, AMPK and liver kinase B1 (LKB1/STK11) regulate synaptic remodeling at advanced age (Samuel et al. 2014). AMPK and mTOR act antagonistically, and AMPK activity is reported to be weakened as a consequence of increased mTOR function in RPE from AMD donor eyes (Zhang et al. 2020). Sirtuins are deacetylases that sense nicotinamide adenine dinucleotide (NAD+) levels and initiate transcriptional changes triggering mitochondrial biogenesis and energy production. Though most sirtuins are expressed in the mammalian retina, SIRT1 is suggested to play a protective role against damage from oxidative stress in ocular aging (Mimura et al. 2013). Aging-associated decline in cellular NAD+ levels is observed in multiple ocular cell types (Jadeja et al. 2020), and subsequent reductions in SIRT1 may limit its protective benefits. However, additional studies are needed to clarify these relationships in the aging retina.
4.2. Mitochondrial Damage and Compromised Energy Metabolism
Mitochondrial damage and declining energy production are the bases of several theories of aging (Balaban et al. 2005). The retina is particularly vulnerable to mitochondrial dysfunction owing to its critical role in homeostasis (Schrier & Falk 2011). Aging is concurrent with weakening of protective mechanisms such as mitophagy by sirtuins and mitochondrial DNA (mtDNA) repair by poly (ADP-ribose) polymerase 1 (PARP1), mutY homolog (MYH), and endonuclease III homolog 1 (NTH1) (Wang et al. 2010). Furthermore, mitochondria in retinal cells undergo structural changes with advancing age (Kam & Jeffery 2015). In addition to there being fewer mitochondria in aging photoreceptors, the mitochondrial network is less elongated, with reduced branching, suggestive of declining fidelity (Kam et al. 2019). Structural analysis has uncovered loss of cristae and substantial decrease in complex I at older age, indicative of a possible discourse for deregulated ATP production (Nag & Wadhwa 2012, 2016). Moreover, the mtDNA is more susceptible than the nuclear DNA to damages from reactive oxygen species (ROS) primarily generated from mitochondrial metabolism (Balaban et al. 2005, Wang et al. 2010). Aging-related ROS have been linked to specific mtDNA damage, including the 4,977-bp deletion common in aging retinal cells, and mtDNA damage is enhanced and widespread in age-related retinal diseases (Barreau et al. 1996, Karunadharma et al. 2010). Aging has been further linked with mitochondrial gene regulation and metabolite differences (Corso-Diaz et al. 2020, Wang et al. 2018). Oxidative phosphorylation is compromised in aging photoreceptors, with a detectable shift toward fatty acids as substrates for energy production (Corso-Diaz et al. 2020). Additionally, age-related decline of nicotinamide phosphoribosyltransferase (NAMPT) activity and reduced NAD+ levels (Jadeja et al. 2020) can also impact energy homeostasis in photoreceptors. The significance of mitochondria in retinal aging is apparent, and it presents an encouraging target for intervention and therapy.
4.3. Metabolic Stress
Cellular aging is marked by accumulation of metabolic byproducts due to a progressive decline in homeostatic regulation. Metabolic byproducts can be toxic or catalyze undesired alterations of biomolecules, which when unchecked can lead to damaging consequences. Cellular stresses in aging retina are well characterized and have been linked to functional decline and frailty.
4.3.1. Reactive oxygen species and oxidative stress.
ROS are unstable oxygen-containing molecules that can cause unwanted modification(s) of cellular biomolecules. A major source of intracellular ROS is the mitochondrial electron transport chain, where electron leakage and subsequent reaction with molecular oxygen generates free oxygen radicals (Balaban et al. 2005) that are rapidly eliminated by enzymes (superoxide dismutases, catalase) or nonenzymatic antioxidants (glutathione, carotenoids) (Tan et al. 2018). However, age-dependent attenuation of antioxidant processes and enhanced ROS production because of decline in mitochondrial integrity can lead to progressive cellular damage (Balaban et al. 2005, Maeda et al. 2005). Despite an efficient antioxidant defense system, the aging retina, especially photoreceptors and the RPE, is extremely susceptible to ROS damage because of high requirements for metabolic and mitochondrial activities (Domènech & Marfany 2020). Of particular significance is oxidation of the long-chain poly unsaturated fatty acids (PUFAs) that are prevalent in photoreceptor membranes. Photoreceptor outer segment recycling also contributes to ROS accumulation in the aging RPE, and the resulting oxidative stress is concomitant with the decrease in phagocytosis potential (Datta et al. 2017). Oxidative stress can impair mitophagy and, together with other aging-associated alterations, including in the epigenome, can have broader effects on health of the aging retina. Furthermore, unchecked ROS can instigate secondary stresses, including excessive advanced glycation end products (AGEs). Oxidative stress is linked to retinal pathologies, and ROS have been further implicated in mitochondrial DNA aberrations of AMD (Kaarniranta et al. 2020). Therefore, managing the balance of ROS and antioxidants is essential for retinal functioning at advanced age.
4.3.2. Accumulation of advanced glycation end products.
AGE are formed from chemical modifications of proteins, lipids, and DNA, with reactive dicarbonyl intermediates arising from metabolic pathways including glycolysis. Modifications of long-lived proteins prevent degradation and activate proinflammatory signaling, leading to harmful outcomes that correlate with progressing age. AGE can exert wider impact through the receptor for AGE (RAGE) and galectin-3, which are broadly expressed in retinal cells (Uehara et al. 2001). Deglycases including DJ1/PARK7 efficiently maintain homeostasis (Chaudhuri et al. 2018), yet weakening of detoxifying mechanisms can result in AGE accumulation. Owing to metabolic diversity among ocular tissues, aging-related increases in AGE can have divergent effect on the neural retina and RPE, as well as on the lens, cornea, and choroid (Nagaraj et al. 2012, Raghavan et al. 2016). A glucose- and lipid-rich microenvironment predisposes the retina to AGE buildup and advanced lipoxidation end products. Notably, the retinas of older individuals exhibit elevated AGE and RAGE, especially in the inner layers, RPE, and Bruch’s membrane (Tian et al. 2005). AGE accumulation and RAGE activation are well established in the AMD retina, and glycation moieties with attached proteins have been detected in AMD drusen (Crabb et al. 2002). Additionally, AGE can inhibit the neurotrophic effect of pigment epithelium–derived factor (PEDF) and promote neovascularization by activating VEGF (Lu et al. 1998), which may cause or aggravate AMD.
5. PROTEOSTASIS
Proteostasis refers to an integrated network of molecular chaperones, proteolytic systems, and pathways that maintain a stable functional proteome in the cell by controlling protein synthesis, folding, transport, and removal of misfolded proteins by autophagy or proteasome system (Hipp et al. 2019). Proteostasis declines with advanced age because of changes in protein expression and degradation of misfolded or damaged proteins (Anisimova et al. 2018). In the retina, the key players in proteostasis maintenance include chaperones, the ubiquitin-proteasome system (UPS) and autophagy, which determine whether an unfolded or misfolded protein can be restored to a stable conformation or should be eliminated through proteolysis (Labbadia & Morimoto 2015). The unfolded protein response (UPR) and heat-shock response are equally essential stress response pathways to dynamically control protein homeostasis.
A comprehensive analysis of the retinal proteome during distinct stages of aging would be valuable for gaining insights into aberrations in proteostasis. A detailed analysis of foveomacular, juxtamacular, and peripheral regions of donor human retinas identified 697 proteins that are differentially expressed in distinct parts (Velez et al. 2018); however, such an analysis is not yet available to distinguish aging-associated alterations. Notably, comparative proteomic analysis of young and aged retinas of marmosets and rats has revealed alterations of four proteins (PARK7/DJ1, Stathmin, Peroxiredoxin, and β-synuclein) during retinal aging (Bohm et al. 2013). In humans, quantitative proteomic studies have been performed on normal and AMD donor retinas, revealing differential expression of complement proteins, TlMP3, and vitronectin (Hollyfield et al. 2003). Additional studies are needed to determine the disruptions in proteostasis and to identify biomarkers associated with human retinal aging.
5.1. The Unfolded Protein Response
The UPR constitutes an intracellular signaling pathway activated by cells in response to endoplasmic reticulum (ER) stress that results from accumulation of unfolded proteins. The UPR tracks protein folding levels in the ER and adjusts the folding capacity to match the synthesis load, thus providing a successful balance for protein homeostasis (Wang & Kaufman 2016). The UPR is comprised of three sensors that are activated upon stress: activating transcriptional factor 6 (ATF6), protein kinase RNA-like ER kinase (PERK), and inositol-requiring kinase 1 (IRE1). In aged rat retinas, the UPR-PERK arm, phosphorylated (p)eIF2a, ATF4, and GADD34 are significantly altered with upregulation of cleaved pATF6 and CHOP proteins, indicating the activation of ATF6 (Lenox et al. 2015). Older mouse retinas also exhibit significantly reduced levels of X-box binding protein 1 (XBP1), a primary effector of the IRE1 pathway, suggesting compromised activation of the ER stress response (McLaughlin et al. 2018). Low levels of XBP1 are observed in the RPE from old mice, as well (Zhong et al. 2012). Thus, aged retinas are at risk because of UPR activation.
5.2. The Ubiquitin Proteasome System
The UPS is involved in cellular quality control by degrading unassembled, misfolded, or damaged proteins that can form toxic aggregates. The proteasome is responsible for removal of oxidized proteins, and this function declines in aging, suggesting that aged tissue is at greater risk of oxidative damage (Campello et al. 2013). Aging- or stress-related damage of the UPS in the retina results in accumulation of abnormal or damaged proteins, contributing to drusen formation and accumulation (Louie et al. 2002). Retinal homogenates from old rats showed that chymotrypsin-like activity of the proteasome decreases with aging (Kapphahn et al. 2007, Louie et al. 2002). Accumulation of oxidized retinal proteins occurs during aging, leading to disruptions in proteosome function.
Aging involves gradual accumulation of protein aggregates that contribute to neuronal dysfunction and senescence. A study on human retinas obtained from patients aged 49 to 87 years identified tau aggregates within the cytoplasm of several photoreceptor cells and showed a positive correlation with age (Leger et al. 2011). The percentage of aged patients displaying α-synuclein and ubiquitin intracytoplasmic inclusions is significantly higher than that of younger individuals and suggests that protein aggregation in the retina increases with advanced age. Aggregation of lipofuscin in RPE cells is considered a characteristic feature of aging. During aging, human RPE cells show a decrease in the activity of 20S proteasomes (Li et al. 2008).
5.3. Autophagy
Autophagy is an intracellular degradative pathway that plays a critical role in the removal of damaged cell components to maintain proteostasis. In mammalian cells, three main autophagic pathways are involved: macroautophagy, microautophagy, and chaperone-mediated autophagy. The autophagic activity decreases with age, contributing to the accumulation of damaged macromolecules and organelles (Martinez-Lopez et al. 2015). Various proteins involved in autophagy include autophagy-related proteins (ATGs), mTOR, the serine/threonine kinase (ULK1), FIP-200, p62 (SQSTM1), and microtubule-associated protein light chain 3 (LC3). Autophagy plays a dual role in the retina: Normal autophagy helps retinal cells to protect themselves against harmful stress, and alterations in autophagy result in retinal degeneration (Hargrove-Grimes et al. 2020, Lin & Xu 2019). During aging in the retina, autophagy plays an important role in protecting the cells by removing damaged components that would otherwise cause accumulation and promote the formation of cytotoxic molecules in situ under oxidative stress (Ferrington et al. 2016). Unbalanced autophagy causes accumulation of intracellular debris, inflammasome activation, and cell death (Frost et al. 2014). In aged mouse retina, complement system deficiency results in cellular clearance of proteins through autophagy mechanisms (Roginska et al. 2017). The macroautophagy pathway in the retinas of aged mice contributes to age-associated reduction in visual function, lipofuscin accumulation, and neurodegeneration in multiple retinal layers (Rodriguez-Muela et al. 2013). Retinas of 18-month-old senescence-accelerated OXYS rats showed significant changes in the expression of autophagy proteins, including LC3, Atg7, and Atg12–Atg5 (Kozhevnikova et al. 2018). Similarly, dysregulated autophagy in retina tissues was observed in 18-month-old mice with significant loss of RGCs and presence of autophagic structures in the degenerating axons (Nettesheim et al. 2020).
Autophagy is a critical regulator of homeostasis in the RPE and plays an essential function in protection against oxidative stress (Ferrington et al. 2016). Multiple studies on aged mouse RPEs and healthy human donors showed evidence for elevated autophagy and increased content of autophagy proteins and autophagic vesicles (Mitter et al. 2014; Wang et al. 2009a, b). Aggregates of lipofuscin-like material and inefficient lysosomal clearance were observed in the RPEs of 1-year-old rats with a mutation in the Cryba1 gene (which participates in lysosomal-mediated clearance) (Sinha et al. 2016). In aged mouse RPEs, overexpression of SIRT6 (a nuclear protein that is also known to regulate autophagy) and autophagic markers was found with accumulation of subretinal deposition of amyloid-β (Feng et al. 2018). Deletion of the Rb1cc1 gene (essential for induction of autophagy) in aged mice caused multiple autophagy defects within the RPE, including the accumulation of autophagy-targeted precursors and increased numbers of mitochondria (Yao et al. 2015). These observations are consistent with autophagy playing a critical role in the aging of the retina and suggest that perturbation of this pathway can lead to age-related degeneration.
6. IMMUNE RESPONSE AND SENESCENCE
Aging is accompanied by a gradual deterioration of the immune system (immunosenescence), with consequences such as decreased response to new antigens, impaired ability to fight infection, increased autoimmunity, and persistent low-grade inflammation (known as inflammaging) (Goronzy & Weyand 2013). Oxidative stress, cellular instability, nutrition, and gut microbiota contribute to the inflammatory response during aging (Franceschi et al. 2018). Inflammaging is considered a major risk factor for morbidity and mortality in the aging population, and it is counteracted by a simultaneous increase in anti-inflammatory factors (anti-inflammaging). Notably, secretion of proinflammatory cytokines can also be accompanied by expression of matrix metalloproteases and growth factors that together constitute the senescence-associated secretory phenotype (SASP) characteristic of senescent cells (Tchkonia et al. 2013). These cells accumulate with age and display irreversible replicative arrest, tumor suppressor activation, chromatin changes, apoptosis resistance, and increased protein synthesis (Tchkonia et al. 2013). The SASP is also evident in postmitotic cells, and some of its components induce the recruitment of the immune system, stimulate cellular reprogramming, and promote angiogenesis (Sapieha & Mallette 2018).
The neural retina is an immune-privileged tissue protected from immune cells by several layers, the first being the blood–retina barrier (BRB) maintained by the neurovascular unit consisting of glial cells, pericytes, vascular endothelial cells, and neurons. The second layer of protection is through induction of retinal cell tolerance and suppression of the inflammatory response. Immune suppression in the retina is achieved by various immunoregulatory proteins such as CX3CL1 and CD47 (macrophages and microglia activation), as well as CFH and CD55 (complement activation) (Chen et al. 2019). The structural and functional integrity of the BRB is affected in the elderly. The neovascular unit shows loss of endothelial cells, senescence of neurons, altered structure of desmin filaments of pericytes, and thickening of the basement membrane, resulting in microglia and complement activation. Indeed, alterations in genes involved in the immune response were observed in old mouse retinas, suggesting a low-grade proinflammatory state (Chen et al. 2010). These changes include overexpression of Calcr (encoding the calcitonin receptor, a biomarker for inflammation) and other genes related to complement activation, myeloid cell differentiation, endocytosis, phagocytosis, and chemotaxis. The aging retina also exhibits an increase in the number and density of microglia (Chen et al. 2010). Therefore, elucidating pathways involved in microglia activation is of interest.
The RPE plays an important role in immune regulation, as it segregates the neural retina from choroidal circulation and inhibits immune activation. The aging RPE becomes immunologically active and upregulates genes involved in leukocyte extravasation signaling, the complement system, natural killer (NK) cells, IL-10 IL-2, chemokines, and B and T cell signaling (Chen et al. 2008). Upregulation of anti-inflammatory responses in RPE suggests that a homeostatic balance of both pro- and anti-inflammatory responses is crucial for normal aging. Mild RPE damage during aging can also lead to accumulation of phagocytes in the subretinal space (Xu et al. 2008). However, the mechanisms underlying infiltration of macrophages, phagocytes, and leukocytes remain elusive.
Senescence in human RPE cells can be induced by oxidative stress (Zhu et al. 2009), reduced levels of NAD+ (Jadeja et al. 2018), and amyloid β-peptide (Liu et al. 2015). Senescence has also been identified in retinal neurons and blood vessels and is associated with microaneurysms (Lopez-Luppo et al. 2017). Intriguingly, a primary open-angle glaucoma risk variant of SIX6 induces senescence of mouse RGCs, suggesting a role of cellular senescence in glaucoma pathogenesis (Skowronska-Krawczyk et al. 2015). Moreover, removal of senescent cells after experimental ocular hypertension protects other RGCs from senescence and apoptosis (Rocha et al. 2020). Intriguingly, in mouse models of ischemic retinopathy, retinal neurons exhibit lower rates of apoptosis and secrete SASP-associated cytokines, which are also present in the vitreous humor of patients with proliferative diabetic retinopathy (Oubaha et al. 2016). Overall, these studies highlight the importance of senescence in postmitotic cells and the potential contribution of this aging phenotype to disease.
In summary, inflammation, cellular senescence, and impairment of the BRB are aging mechanisms that together can contribute to retinal disease. We suggest that immunoregulatory and SASP factors should be investigated in all retinal cell populations at different stages of aging due to the inherent heterogeneity of senescent cells (Hernandez-Segura et al. 2017) and variability of inflammatory responses. Identifying the pathways that trigger senescence in neurons and determining whether they are similar to the ones driving senescence of mitotic cells are exciting new avenues for research.
7. AGE-RELATED RETINAL DISEASES
Complex interplays among genetic, environmental, and lifestyle factors contribute to human disease susceptibility in discrete tissues, including the retina. Age-related dysfunction and dysregulation of homeostatic processes in the retina can lead to vision impairment in disorders such as glaucoma, diabetic retinopathy, and AMD. Glaucoma is characterized by progressive optical atrophy due to the death of RGCs, whereas diabetic retinopathy demonstrates marked glial, neuronal, and microvascular abnormalities that progressively disrupt retinal function. In this section, we focus on AMD, for which advanced age is arguably the most important risk factor (Fritsche et al. 2014, Swaroop et al. 2009).
AMD patients display loss of central vision with a range of clinical phenotypes including lipid-rich extracellular deposits, localized inflammation, and subsequent neurodegeneration starting in the macular region (Swaroop et al. 2009). Advanced age and family history are major risk factors, while smoking and nutrition significantly contribute to disease progression. The late stage of the disease can result in choroidal neovascularization or geographic atrophy, accompanied by loss of photoreceptors and RPE abnormalities. Anatomic alterations observed in the healthy aging retina, such as drusen growth, accumulation of lipofuscin, and changes in Bruch’s membrane, precede the development of AMD pathology (Fritsche et al. 2014). Healthy retina develops a modest immune response as a function of age, whereas para-inflammation in AMD turns into a chronic inflammatory immune response with activation of microglia and macrophages. Similarly, normal aging is accompanied by mtDNA damage, oxidative stress, and an aberrant epigenome that could contribute to the onset of AMD when co-occurring with susceptibility factors such as genetic polymorphisms, smoking, a high-fat diet, and stress.
GWAS have identified 52 genetic variants at 34 AMD-associated loci (Fritsche et al. 2016). These variants can influence gene expression quantitatively and are called expression quantitative trait loci (eQTLs). The integration of eQTLs with AMD GWAS and transcriptome-wide association studies (TWAS) has suggested candidate target genes at six loci determined by AMD GWAS and regulation of several potential associated genes (Ratnapriya et al. 2019). Given that epigenetic alterations are commonly detected with age, identification of variants that regulate the epigenome, including DNA methylation QTLs (mQTLs), will further augment our genetic understanding of AMD pathology. The precise contribution of aging to AMD is difficult to assess at this stage, since studies of normal human retinal aging are lacking. Further investigations are required for identification and integration of age-specific eQTLs and mQTLs to AMD GWAS and to decipher the interactions among age, genetics, and environment in clinical progression from early to advanced AMD.
8. ENVIRONMENT AND LIFESTYLE
Retinal homeostasis depends on the balance of in vivo stress generation (byproducts of phototransduction and high metabolic activity) and adaptive processes countering it. Environment and lifestyle variables such as light exposure, tobacco smoking, and specific dietary habits can aggravate retinal stress through induction of several cellular aberrations including oxidative stress (Figure 5). Healthy aging is acknowledged to cause decline of adaptive mechanisms, and therefore, environmental factors can exacerbate the aging process or increase risk of age-related retinal pathologies. Light directly projects onto the retina, making it an important extrinsic factor affecting eye health and age-related vision disorders (Ozawa 2020). Artificially generated blue light from common electronic and lighting devices are increasingly being recognized to be hazardous as well. Exposure to unsafe light wavelengths causes oxidative damage and inflammation in retinal cells, often involving the mitochondrial machinery (Tao et al. 2019). Light-induced damage in the retina can accumulate over the years and poses additional risk by shifting our natural circadian rhythm. Furthermore, cigarette smoking is an important lifestyle factor with validated association with retinal health. Observational studies have found that smoking exacerbates age-dependent cognitive decline, while also identifying it as a risk factor for AMD (Durazzo et al. 2014, Seddon et al. 1996). Smoking generates oxidative stress in the retina, alters the extracellular matrix, and promotes neovascularization, potentially contributing to AMD (Velilla et al. 2013).
Figure 5.
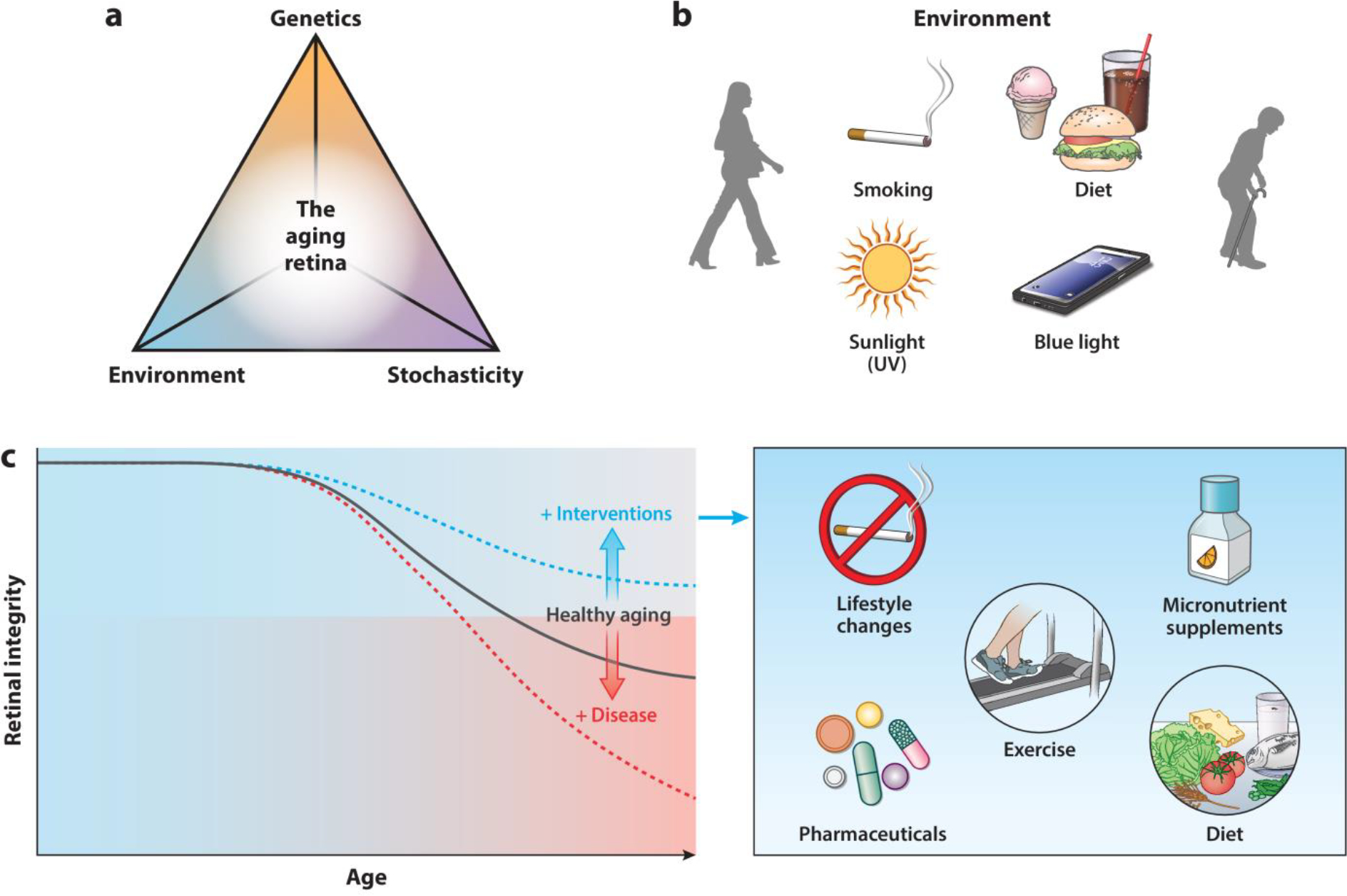
Principles of retinal aging and potential strategies to alleviate its impact. (a) Aging of the retina is influenced by genetic, environmental, and stochastic events. (b) The retina is subjected to constant environmental stressors, including UV and blue light, and lifestyle variables, such as diet and smoking, that contribute to loss of functional and cellular integrity with age. (c) Age-dependent shifts in retinal integrity. Genetic variants, comorbidities, and environmental risk factors can exacerbate retinal aging, whereas specific cellular and molecular alterations can be ameliorated by pharmaceutical compounds or modifications in lifestyle. These interventions could prevent or delay functional decline and emergence of disease.
A nutritionally balanced diet improves health; however, unhealthy eating habits can have severe outcomes in the aging retina. Unhealthy diets increase the risk of comorbidities like diabetes and hypertension, further predisposing elderly individuals to retinal neurodegenerative diseases. Epidemiological studies have identified dietary glycemic index as a risk factor for AMD (Chiu et al. 2007). In aging mice, a hyperglycemic diet has been shown to induce retinal inflammation, AMD-like features, and AGE accumulation (Rowan et al. 2017, Uchiki et al. 2012). Additionally, a hyperglycemic diet was also linked to retinal cell death and alterations in the gut–retina axis (Rowan et al. 2017). On similar lines, a high-fat diet is associated with neuroinflammatory outcomes and causes retinopathies at older age (Asare-Bediako et al. 2020, Cai 2013). To conclude, lifestyle choices and environmental factors are significant determinants of ocular aging, and their targeted modulation could be a candidate for antiaging interventions.
9. INTERVENTIONS TO SLOW RETINAL AGING
With increasing human lifespan, there is a need for interventions that preserve retinal function at an advanced age. Early antiaging therapies like caloric restriction (CR) were serendipitous findings; however, our knowledge of molecular signatures provides for directed research into interventions for healthier eyesight. Of special interest is the epigenome–metabolism nexus, which can be targeted to mitigate the effects of retinal aging (Figure 4). In this section, we discuss potential interventions, with a focus on lifestyle strategies as nonpharmaceutical alternatives, for countering vision aging.
9.1. Lifestyle Behaviors
Retinal protective mechanisms or damaging aggressors can be modulated through lifestyle changes such as exercise and avoiding stressors (e.g., smoking). Regular exercise allows calorie and glycemic control and activates mechanisms involving metabolic signaling and mitochondria to facilitate cellular health (Hawley et al. 2014). Exercise reportedly attenuates photoreceptor cell death through a BDNF-dependent mechanism and by suppressing choroidal neovascularization (Chrysostomou et al. 2016, Makin et al. 2020). Tobacco smoking is the best-characterized lifestyle habit linked to oxidative stress, complement activation, and AMD (Rohrer et al. 2019, Seddon et al. 2020).
9.2. Diet
CR during aging is reportedly beneficial in vertebrate models and humans (Mattison et al. 2017). CR and related strategies (such as intermittent fasting and time-restricted eating) suppress metabolism, oxidative stress, and inflammation (Flanagan et al. 2020) and retard retinal changes during aging (Li et al. 2003). Diet patterns can alter progression of chronic ailments like diabetes and hypertension, which could have retinal consequences. Notably, the Mediterranean diet is associated with reduced risk of progression to advanced AMD (Keenan et al. 2020). The keto diet is shown to improve retinal viability through activating AMPK and mitochondrial biogenesis in a glaucoma model (Harun-Or-Rashid et al. 2018), suggesting its potential in modifying aging hallmarks. However, more comprehensive mechanistic studies are desirable to establish the impact of these diets on retinal aging phenotypes.
9.3. Micronutrient Supplementation
Vitamins and micronutrients ensure retinal homeostasis by acting as cofactors to cellular processes or through antioxidant properties. Micronutrients as supplements can interact with and modify key aging hallmarks, especially the epigenome–metabolism nexus, a key driver of retinal aging. Vitamins B6, B9, and C can alter the DNA methylome and gene regulatory outcomes through the DNMT1 and TET enzymes (Crider et al. 2012, Young et al. 2015, Zhong et al. 2017). Zinc, NAD+, lutein, and zeaxanthin may act as cofactors or antioxidants for cellular reactions that scavenge free radicals from mitochondrial metabolism or improve mitochondrial health (Arend et al. 2015,Chae et al. 2018,Gilbert et al. 2019). Vitamin D3 supplementation combined with exposure to 670-nm light reduces inflammation in the aging retina (Lee et al. 2012). Furthermore, dietary supplementation of PUFAs can improve retinal health (Querques et al. 2011). PUFA-derived elovanoids are shown to suppress oligomeric β-amyloid-induced senescence gene expression changes in the mouse RPE, highlighting PUFA’s antiaging potential (Do 2019). Predictably, large longitudinal studies and clinical trials have obtained promising results related to reduction of age-related retinal diseases by micronutrient supplementation (Age-Relat. Eye Dis. Study Res. Group 2001, AREDS2 Res. Group et al. 2012, Moeller et al. 2006), indicating the benefits of micronutrients in healthy aging of the retina.
9.4. Pharmaceuticals
Resveratrol, rapamycin, and metformin are among the drugs that can modify aging-associated metabolic processes and impact AMD and glaucoma progression (Brown et al. 2019, Osborne et al. 2014). These pharmaceuticals modulate metabolic signaling by targeting mTOR activity. Rapamycin directly represses mTOR, whereas metformin activates AMPK, a negative regulator of mTOR. Metformin also improves mitochondrial health and regulates oxidative phosphorylation. Furthermore, CR mimetics (CRMs) induce responses comparable to CR, and CRMs such as resveratrol and spermidine have been reported to promote retinal health. Resveratrol increases mitochondrial fidelity, upregulates AMPK and SIRT1 pathways, suppresses mTOR activity, and provides antiaging effects (Wang et al. 2019). Spermidine activates autophagy, inhibits proinflammatory factors, and is an antioxidant. Although spermidine remains to be tested in the context of retinal aging, its neuroprotective roles have been documented in RGCs (Noro et al. 2015). Additionally, molecules that suppress AGE pathology are also useful and can be broadly classified as AGE inhibitors or crosslink breakers. AGE inhibition can be achieved through aminoguanidine (Hammes et al. 1991) or by interrupting AGE–RAGE signaling. Using crosslink breakers is another AGE-detoxification method that cleaves the covalent association between AGE-protein derivatives and opens them up for clearance (Wolffenbuttel et al. 1998). Regulation of senescence by senolytics can improve retinal health, and they are now being tested for age-related disorders (van Deursen 2019). One such compound, UBX1967, a Bcl-2 family inhibitor, is entering clinical trials to treat neovascular AMD, proliferative diabetic retinopathy, and diabetic macular edema.
10. CONCLUDING REMARKS
Aging is a universal process affecting all aspects of the biological, social, and psychological spheres of an individual’s life and is accompanied by gradual decline in physiological function over time. Variability observed in human aging is a reflection of the interplay among genetic, environmental, and stochastic elements (Figure 5a), and thus, no two humans age identically. Stochasticity is exemplified by the NASA Twins Study (Garrett-Bakelman et al. 2019). Although aging per se is not a disease, physiological changes with advancing age make us more susceptible to common aging-associated disorders. In general, aging exacerbates clinical manifestations of many diseases, but the phenotypes are especially severe in those involving the neuronal system. The key role of vision in multiple higher-order functions associated with the central nervous system and life-long exposure to environmental insults (including light and oxidative stress) make the retina an attractive tissue to investigate complex phenomena of aging. Progressive deterioration in retinal anatomy and function is apparent with advanced age but likely varies depending on environmental exposure. A thorough understanding of molecular and metabolic signatures at different levels of the biological hierarchy in aging is crucial to developing interventions for healthy vision and to delaying or preventing late-onset retinal neurodegeneration (Figure 5b,c).
We propose that an excess of DNA damage, combined with aberrant DNA repair and/or epigenome editing, especially of genes involved in neuronal communication, mediates at least a part of the age-related functional decline of the retina. Neurons are postmitotic cells and cannot utilize cell division to repair molecular damage. Genes encoding neuronal proteins are usually longer and more susceptible to age-related DNA damage and methylation changes than most other genes. Furthermore, retinal neurons are challenged by constant UV exposure and high metabolic activity. The crosstalk that exists between the nucleus and the mitochondria probably exacerbates this cellular damage with aging. In this context, excess nuclear DNA damage might cause distress signals to be sent to the mitochondria, while other stressors, such as protein aggregates, further induce mitochondrial stress responses that, in turn, signal back to the nucleus. Impairments can be more or less pronounced based on individual-specific genetic variations and/or comorbidities. In this scenario, one can reasonably speculate that the imbalance between the cumulative cellular damage and repair mechanisms, together with the synergy among deregulated signaling pathways, likely results in vision impairment during aging. Simultaneous DNA and protein damage may induce cellular adaptations that ultimately lead to integrative responses and establish a new normal homeostasis, creating a different cellular order to counter aging. These changes could potentially make cells more vulnerable to extracellular and intrinsic stresses and expedite disease pathology. Several outstanding questions remain: What are the early molecular mechanisms that lead to vision decline with age? Is there a molecular clock of aging? Is there a threshold capability for repairing the accumulated cellular damage? Are the molecular changes observed during aging intrinsic or an outcome of adaptive responses? Are these changes protective, permissive, or causative of age-related functional decline?
Aging research is challenging. In addition to the inherent biological complexity, practical limitations exist to designing and conducting long-term experiments. Enrolling elderly volunteers represents a major challenge due to ethical issues, environmental and social factors, genetic variations, and differences in lifespan. Animal models have proven useful but do not always mimic the biology of human aging. Additionally, there are logistic challenges related to the duration of and resources required to perform aging experiments. We believe that aging research would greatly benefit from recent advances in high-throughput technologies. A systems biology approach of integrating DNA damage, genomics, transcriptomics, epigenomics, and metabolomics would help delineate the complexity and interconnections of the chronological events that alter cellular homeostasis. Furthermore, genome-wide epigenetic, epitranscriptomic, and protein expression studies with human retinal samples can help us understand the effect of age on genetic regulation and identification of age-specific QTLs. Isolating the contribution of healthy aging to diseases like AMD could provide a better path for innovative therapies. Interventions aimed at reversing epigenomic changes (e.g., pharmacological reprogramming) and preventive approaches (such as lifestyle modifications including the Mediterranean diet, intermittent fasting, and CR) can effectively increase the retinal health span. We hope that, eventually, personalized medicine that integrates genetic variations with specific aging hallmarks and environmental factors will permit optimization of interventions for long-term preservation of retinal function.
ACKNOWLEDGMENTS
We sincerely thank Drs. Nicolás Cuenca, Isabel Pinilla, and Emily Chew and members of the Neurobiology, Neurodegeneration and Repair Laboratory for fruitful and stimulating discussions. We apologize to those authors whose articles are not cited due to space limitations. The figures were created by Alan Hoofring at the National Institutes of Health Medical Arts department. This research was supported by the Intramural Research Program of the National Eye Institute (ZIAEY000450 and ZIAEY000546 to A.S.).
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- Age-Relat. Eye Dis. Study Res. Group. 2001. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch. Ophthalmol. 119:1417–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal P, Nag TC, Wadhwa S. 2007. Age-related decrease in rod bipolar cell density of the human retina: an immunohistochemical study. J. Biosci. 32:293–98 [DOI] [PubMed] [Google Scholar]
- Aldiri I, Xu B, Wang L, Chen X, Hiler D, et al. 2017. The dynamic epigenetic landscape of the retina during development, reprogramming, and tumorigenesis. Neuron 94:550–68.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anisimova AS, Alexandrov AI, Makarova NE, Gladyshev VN, Dmitriev SE.2018.Protein synthesis and quality control in aging. Aging 10:4269–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- AREDS2 Res. Group, Chew EY, Clemons T, SanGiovanni JP, Danis R, et al. 2012. The Age-Related Eye Disease Study 2 (AREDS2): study design and baseline characteristics (AREDS2 report number 1). Ophthalmology 119:2282–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arend N, Wertheimer C, Laubichler P, Wolf A, Kampik A, Kernt M. 2015. Idebenone prevents oxidative stress, cell death and senescence of retinal pigment epithelium cells by stabilizing BAX/Bcl-2 ratio. Ophthalmologica 234:73–82 [DOI] [PubMed] [Google Scholar]
- Arroba AI, Campos-Caro A, Aguilar-Diosdado M, Valverde AM. 2018. IGF-1, inflammation and retinal degeneration: a close network. Front. Aging Neurosci. 10:203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asare-Bediako B, Noothi SK, Li Calzi S, Athmanathan B, Vieira CP, et al. 2020. Characterizing the retinal phenotype in the high-fat diet and Western diet mouse models of prediabetes. Cells 9:464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T. 2005. Mitochondria, oxidants, and aging. Cell 120:483–95 [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T. 2011. Regulation of chromatin by histone modifications. Cell Res. 21:381–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreau E, Brossas JY, Courtois Y, Treton JA. 1996. Accumulation of mitochondrial DNA deletions in human retina during aging. Investig. Ophthalmol. Vis. Sci. 37:384–91 [PubMed] [Google Scholar]
- Bell CG, Lowe R, Adams PD, Baccarelli AA, Beck S, et al. 2019. DNA methylation aging clocks: challenges and recommendations. Genome Biol. 20:249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrio E, Tabernero J, Artal P. 2010. Optical aberrations and alignment of the eye with age. J. Vis. 10:34. [DOI] [PubMed] [Google Scholar]
- Bohm MR, Mertsch S, Konig S, Spieker T, Thanos S. 2013. Macula-less rat and macula-bearing monkey retinas exhibit common lifelong proteomic changes. Neurobiol. Aging 34:2659–75 [DOI] [PubMed] [Google Scholar]
- Bonilha VL. 2008. Age and disease-related structural changes in the retinal pigment epithelium. Clin. Ophthalmol. 2:413–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnel S, Mohand-Said S, Sahel JA. 2003. The aging of the retina. Exp. Gerontol. 38:825–31 [DOI] [PubMed] [Google Scholar]
- Booth LN, Brunet A. 2016. The aging epigenome. Mol. Cell 62:728–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulton M, Dayhaw-Barker P. 2001. The role of the retinal pigment epithelium: topographical variation and ageing changes. Eye 15:384–89 [DOI] [PubMed] [Google Scholar]
- Brown EE, Ball JD, Chen Z, Khurshid GS, Prosperi M, Ash JD. 2019. The common antidiabetic drug metformin reduces odds of developing age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 60:1470–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burla R, La Torre M, Merigliano C, Verni F, Saggio I. 2018. Genomic instability and DNA replication defects in progeroid syndromes. Nucleus 9:368–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D 2013.Neuroinflammation and neurodegeneration in overnutrition-induced diseases. Trends Endocrinol. Metab. 24:40–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Fields MA, Hoshino R, Priore LV. 2012. Effects of aging and anatomic location on gene expression in human retina. Front. Aging Neurosci. 4:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkins DJ. 2013. Age-related changes in the visual pathways: blame it on the axon. Investig. Ophthalmol. Vis. Sci 54:ORSF37–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campello L, Esteve-Rudd J, Cuenca N, Martin-Nieto J. 2013. The ubiquitin-proteasome system in retinal health and disease. Mol. Neurobiol. 47:790–810 [DOI] [PubMed] [Google Scholar]
- Chae SY, Park SY, Park G. 2018. Lutein protects human retinal pigment epithelial cells from oxidative stress-induced cellular senescence. Mol. Med. Rep. 18:5182–90 [DOI] [PubMed] [Google Scholar]
- Chaudhuri J, Bains Y, Guha S, Kahn A, Hall D, et al. 2018. The role of advanced glycation end products in aging and metabolic diseases: bridging association and causality. Cell Metab. 28:337–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Chao DL, Rocha L, Kolar M, Nguyen Huu VA, et al. 2020. The lipid elongation enzyme ELOVL2 is a molecular regulator of aging in the retina. Aging Cell 19:e13100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Liu B, Lukas TJ, Neufeld AH. 2008. The aged retinal pigment epithelium/choroid: a potential substratum for the pathogenesis of age-related macular degeneration. PLOS ONE 3:e2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Luo C, Zhao J, Devarajan G, Xu H. 2019. Immune regulation in the aging retina. Prog. Retin. Eye Res. 69:159–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Muckersie E, Forrester JV, Xu H. 2010. Immune activation in retinal aging: a gene expression study. Investig. Ophthalmol. Vis. Sci. 51:5888–96 [DOI] [PubMed] [Google Scholar]
- Cheng SY, Cipi J, Ma S, Hafler BP, Kanadia RN, et al. 2020. Altered photoreceptor metabolism in mouse causes late stage age-related macular degeneration-like pathologies. PNAS 117:13094–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinchore Y, Begaj T, Wu D, Drokhlyansky E, Cepko CL. 2017. Glycolytic reliance promotes anabolism in photoreceptors. eLife 6:e25946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu CJ, Milton RC, Gensler G, Taylor A. 2007. Association between dietary glycemic index and age-related macular degeneration in nondiabetic participants in the Age-Related Eye Disease Study. Am. J. Clin. Nutr. 86:180–88 [DOI] [PubMed] [Google Scholar]
- Chrysostomou V, Galic S, van Wijngaarden P, Trounce IA, Steinberg GR, Crowston JG. 2016. Exercise reverses age-related vulnerability of the retina to injury by preventing complement-mediated synapse elimination via a BDNF-dependent pathway. Aging Cell 15:1082–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu XK, Meyerle CB, Liang X, Chew EY, Chan CC, Tuo J.2014.In-depth analyses unveil the association and possible functional involvement of novel RAD51B polymorphisms in age-related macular degeneration. Age 36:9627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JJ, Robertson NA, Rather MI, Thomson JP, McBryan T, et al. 2017. Diverse interventions that extend mouse lifespan suppress shared age-associated epigenetic changes at critical gene regulatory regions. Genome Biol. 18:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corso-Diaz X, Gentry J, Rebernick R, Jaeger C, Brooks MJ, et al. 2020.Genome-wideprofilingidentifiesDNA methylation signatures of aging in rod photoreceptors associated with alterations in energy metabolism. Cell Rep. 31:107525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corso-Diaz X, Jaeger C, Chaitankar V, Swaroop A. 2018. Epigenetic control of gene regulation during development and disease: a view from the retina. Prog. Retin. Eye Res. 65:1–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabb JW, Miyagi M, Gu X, Shadrach K, West KA, et al. 2002. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. PNAS 99:14682–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crider KS, Yang TP, Berry RJ, Bailey LB. 2012. Folate and DNA methylation: a review of molecular mechanisms and the evidence for folate’s role. Adv. Nutr. 3:21–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criscione SW, Teo YV, Neretti N. 2016. The chromatin landscape of cellular senescence. Trends Genet. 32:751–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curcio CA, Drucker DN.1993.Retinal ganglion cells in Alzheimer’s disease and aging.Ann.Neurol.33:248–57 [DOI] [PubMed] [Google Scholar]
- Curcio CA, Millican CL, Allen KA, Kalina RE. 1993. Aging of the human photoreceptor mosaic: evidence for selective vulnerability of rods in central retina. Investig. Ophthalmol. Vis. Sci. 34:3278–96 [PubMed] [Google Scholar]
- Damani MR, Zhao L, Fontainhas AM, Amaral J, Fariss RN, Wong WT. 2011. Age-related alterations in the dynamic behavior of microglia. Aging Cell 10:263–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S, Cano M, Ebrahimi K, Wang L, Handa JT. 2017. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog. Retin. Eye Res. 60:201–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cecco M, Ito T, Petrashen AP, Elias AE, Skvir NJ, et al. 2019. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 566:73–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney JR, Murakami CJ, Olsen B, Kennedy BK, Kaeberlein M. 2011. Quantitative evidence for early life fitness defects from 32 longevity-associated alleles in yeast. Cell Cycle 10:156–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinant C, Houtsmuller AB, Vermeulen W. 2008. Chromatin structure and DNA damage repair. Epigenetics Chromatin 1:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do MTH.2019.Melanopsin and the intrinsically photosensitive retinal ganglion cells: biophysics to behavior. Neuron 104:205–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domènech EB, Marfany G. 2020. The relevance of oxidative stress in the pathogenesis and therapy of retinal dystrophies. Antioxidants 9:347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durazzo TC, Mattsson N, Weiner MW, Alzheimer’s Dis. Neuroimaging Initat. 2014. Smoking and increased Alzheimer’s disease risk: a review of potential mechanisms. Alzheimers Dement. 10:S122–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliasieh K, Liets LC, Chalupa LM. 2007. Cellular reorganization in the human retina during normal aging. Investig. Ophthalmol. Vis. Sci. 48:2824–30 [DOI] [PubMed] [Google Scholar]
- Esquiva G, Lax P, Perez-Santonja JJ, Garcia-Fernandez JM, Cuenca N. 2017. Loss of melanopsin-expressing ganglion cell subtypes and dendritic degeneration in the aging human retina. Front. Aging Neurosci. 9:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Liang J, Zhai Y, Sun J, Wang J, et al. 2018. Autophagy activated by SIRT6 regulates Aβ induced inflammatory response in RPEs. Biochem. Biophys. Res. Commun. 496:1148–54 [DOI] [PubMed] [Google Scholar]
- Ferrington DA, Sinha D, Kaarniranta K. 2016. Defects in retinal pigment epithelial cell proteolysis and the pathology associated with age-related macular degeneration. Prog. Retin. Eye Res. 51:69–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan EW, Most J, Mey JT, Redman LM. 2020. Calorie restriction and aging in humans. Annu. Rev. Nutr. 40:105–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortune B, Johnson CA. 2002. Decline of photopic multifocal electroretinogram responses with age is due primarily to preretinal optical factors. J. Opt. Soc. Am. A 19:173–84 [DOI] [PubMed] [Google Scholar]
- Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. 2018. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 14:576–90 [DOI] [PubMed] [Google Scholar]
- Frenk S, Houseley J. 2018. Gene expression hallmarks of cellular ageing. Biogerontology 19:547–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman DB, Johnson TE. 1988. A mutation in the age-1 gene in Caenorhabditis elegans lengthens life and reduces hermaphrodite fertility. Genetics 118:75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsche LG, Fariss RN, Stambolian D, Abecasis GR, Curcio CA, Swaroop A. 2014. Age-related macular degeneration: genetics and biology coming together. Annu. Rev. Genom. Hum. Genet. 15:151–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsche LG, Igl W, Bailey JN, Grassmann F, Sengupta S, et al. 2016. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 48:134–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohns A, Frohns F, Naumann SC, Layer PG, Lobrich M. 2014. Inefficient double-strand break repair in murine rod photoreceptors with inverted heterochromatin organization. Curr. Biol. 24:1080–90 [DOI] [PubMed] [Google Scholar]
- Frohns F, Frohns A, Kramer J, Meurer K, Rohrer-Bley C, et al. 2020. Differences in the response to DNA double-strand breaks between rod photoreceptors of rodents, pigs, and humans. Cells 9:947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost LS, Mitchell CH, Boesze-Battaglia K. 2014. Autophagy in the eye: implications for ocular cell health. Exp. Eye Res. 124:56–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabel HW, Kinde B, Stroud H, Gilbert CS, Harmin DA, et al. 2015. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 522:89–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H, Hollyfield JG. 1992. Aging of the human retina: differential loss of neurons and retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 33:1–17 [PubMed] [Google Scholar]
- Garrett-Bakelman FE, Darshi M, Green SJ, Gur RC, Lin L, et al. 2019. The NASA Twins Study: a multidimensional analysis of a year-long human spaceflight. Science 364:eaau8650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemenetzi M, Lotery AJ. 2020. Epigenetics in age-related macular degeneration: new discoveries and future perspectives. Cell Mol. Life Sci. 77:807–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerth C, Garcia SM, Ma L, Keltner JL, Werner JS. 2002. Multifocal electroretinogram: age-related changes for different luminance levels. Graefes Arch. Clin. Exp. Ophthalmol. 240:202–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert R, Peto T, Lengyel I, Emri E. 2019. Zinc nutrition and inflammation in the aging retina. Mol. Nutr. Food Res. 63:e1801049. [DOI] [PubMed] [Google Scholar]
- Gillet LC, Scharer OD.2006.Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem. Rev. 106:253–76 [DOI] [PubMed] [Google Scholar]
- Gorgun E, Guven M, Unal M, Batar B, Guven GS, et al. 2010. Polymorphisms of the DNA repair genes XPD and XRCC1 and the risk of age-related macular degeneration. Investig. Ophthalmol. Vis. Sci 51:4732–37 [DOI] [PubMed] [Google Scholar]
- Goronzy JJ, Weyand CM. 2013. Understanding immunosenescence to improve responses to vaccines. Nat. Immunol. 14:428–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall H, Medina P, Cooper DA, Escobedo SE, Rounds J, et al. 2017. Transcriptome profiling of aging Drosophila photoreceptors reveals gene expression trends that correlate with visual senescence. BMC Genom. 18:894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes HP, Martin S, Federlin K, Geisen K, Brownlee M. 1991. Aminoguanidine treatment inhibits the development of experimental diabetic retinopathy. PNAS 88:11555–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargrove-Grimes P, Mondal AK, Gumerson J, Nellissery J, Aponte A, et al. 2020. Loss of endocytosis-associated RABGEF1 causes aberrant morphogenesis and altered autophagy in photoreceptors leading to retinal degeneration. PLOS Genet. 16:e1009259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman D 1981. The aging process. PNAS 78:7124–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harun-Or-Rashid M, Pappenhagen N, Palmer PG, Smith MA, Gevorgyan V, et al. 2018.Structural and functional rescue of chronic metabolically stressed optic nerves through respiration. J. Neurosci. 38:5122–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley JA, Hargreaves M, Joyner MJ, Zierath JR. 2014. Integrative biology of exercise. Cell 159:738–49 [DOI] [PubMed] [Google Scholar]
- Hernandez-Segura A, de Jong TV, Melov S, Guryev V, Campisi J, Demaria M. 2017. Unmasking transcriptional heterogeneity in senescent cells. Curr. Biol. 27:2652–60.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S, Shaw RJ. 2018. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 19:121–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hipp MS, Kasturi P, Hartl FU. 2019. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 20:421–35 [DOI] [PubMed] [Google Scholar]
- Hoeijmakers JH. 2009. DNA damage, aging, and cancer. N. Engl. J. Med. 361:1475–85 [DOI] [PubMed] [Google Scholar]
- Hollyfield JG, Salomon RG, Crabb JW. 2003. Proteomic approaches to understanding age-related macular degeneration. Adv. Exp. Med. Biol. 533:83–89 [DOI] [PubMed] [Google Scholar]
- Hoon M, Okawa H, Della Santina L, Wong RO. 2014. Functional architecture of the retina: development and disease. Prog. Retin. Eye Res. 42:44–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino A, Horvath S, Sridhar A, Chitsazan A, Reh TA. 2019. Synchrony and asynchrony between an epigenetic clock and developmental timing. Sci. Rep. 9:3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter A, Spechler PA, Cwanger A, Song Y, Zhang Z, et al. 2012. DNA methylation is associated with altered gene expression in AMD. Investig. Ophthalmol. Vis. Sci. 53:2089–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ida H, Boylan SA, Weigel AL, Hjelmeland LM. 2003. Age-related changes in the transcriptional profile of mouse RPE/choroid. Physiol. Genom. 15:258–62 [DOI] [PubMed] [Google Scholar]
- Jackson GR, Ortega J, Girkin C, Rosenstiel CE, Owsley C. 2002. Aging-related changes in the multifocal electroretinogram. J. Opt. Soc. Am. A 19:185–89 [DOI] [PubMed] [Google Scholar]
- Jackson GR, Owsley C. 2000. Scotopic sensitivity during adulthood. Vis. Res. 40:2467–73 [DOI] [PubMed] [Google Scholar]
- Jadeja RN, Powell FL, Jones MA, Fuller J, Joseph E, et al. 2018. Loss of NAMPT in aging retinal pigment epithelium reduces NAD+ availability and promotes cellular senescence. Aging 10:1306–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadeja RN, Thounaojam MC, Bartoli M, Martin PM. 2020. Implications of NAD+ metabolism in the aging retina and retinal degeneration. Oxid. Med. Cell Longev. 2020:2692794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AA, Shokhirev MN, Shoshitaishvili B. 2019. Revamping the evolutionary theories of aging. Ageing Res. Rev. 55:100947. [DOI] [PubMed] [Google Scholar]
- Jonsson ME, Garza R, Johansson PA, Jakobsson J. 2020. Transposable elements: a common feature of neurodevelopmental and neurodegenerative disorders. Trends Genet. 36:610–23 [DOI] [PubMed] [Google Scholar]
- Kaarniranta K, Uusitalo H, Blasiak J, Felszeghy S, Kannan R, et al. 2020. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog. Retin. Eye Res. 79:100858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M, McVey M, Guarente L. 1999. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 13:2570–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kam JH, Jeffery G.2015.To unite or divide: mitochondrial dynamics in the murine outer retina that preceded age related photoreceptor loss. Oncotarget 6:26690–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kam JH, Weinrich TW, Shinhmar H, Powner MB, Roberts NW, et al. 2019. Fundamental differences in patterns of retinal ageing between primates and mice. Sci. Rep. 9:12574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapphahn RJ, Bigelow EJ, Ferrington DA. 2007. Age-dependent inhibition of proteasome chymotrypsin-like activity in the retina. Exp. Eye Res. 84:646–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunadharma PP, Nordgaard CL, Olsen TW, Ferrington DA. 2010. Mitochondrial DNA damage as a potential mechanism for age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 51:5470–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan TD, Agron E, Mares J, Clemons TE, van Asten F, et al. 2020. Adherence to the Mediterranean diet and progression to late age-related macular degeneration in the Age-Related Eye Disease Studies 1 and 2. Ophthalmology 127:1515–28 [DOI] [PubMed] [Google Scholar]
- Kim Y, Tarallo V, Kerur N, Yasuma T, Gelfand BD, et al. 2014. DICER1/Alu RNA dysmetabolism induces Caspase-8-mediated cell death in age-related macular degeneration. PNAS 111:16082–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klass MR. 1983. A method for the isolation of longevity mutants in the nematode Caenorhabditis elegans and initial results. Mech. Ageing Dev. 22:279–86 [DOI] [PubMed] [Google Scholar]
- Kowluru RA, Mishra M. 2015. Contribution of epigenetics in diabetic retinopathy. Sci. China Life Sci. 58:556–63 [DOI] [PubMed] [Google Scholar]
- Kozhevnikova OS, Telegina DV, Devyatkin VA, Kolosova NG. 2018. Involvement of the autophagic pathway in the progression of AMD-like retinopathy in senescence-accelerated OXYS rats. Biogerontology 19:223–35 [DOI] [PubMed] [Google Scholar]
- Kurtenbach A, Weiss M.2002.Effect of aging on multifocal oscillatory potentials. J. Opt. Soc. Am. A 19:190–96 [DOI] [PubMed] [Google Scholar]
- Labat-Robert J, Robert L. 2015. Longevity and aging: mechanisms and perspectives. Pathol. Biol. 63:272–76 [DOI] [PubMed] [Google Scholar]
- Labbadia J, Morimoto RI. 2015. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 84:435–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamoke F, Shaw S, Yuan J, Ananth S, Duncan M, et al. 2015.Increased oxidative and nitrative stress accelerates aging of the retinal vasculature in the diabetic retina. PLOS ONE 10:e0139664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRocca TJ, Cavalier AN, Wahl D. 2020. Repetitive elements as a transcriptomic marker of aging: evidence in multiple datasets and models. Aging Cell 19:e13167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee V, Rekhi E, Hoh Kam J, Jeffery G. 2012. Vitamin D rejuvenates aging eyes by reducing inflammation, clearing amyloid beta and improving visual function. Neurobiol. Aging 33:2382–89 [DOI] [PubMed] [Google Scholar]
- Leger F, Fernagut PO, Canron MH, Leoni S, Vital C, et al. 2011. Protein aggregation in the aging retina. J. Neuropathol. Exp. Neurol. 70:63–68 [DOI] [PubMed] [Google Scholar]
- Lenox AR, Bhootada Y, Gorbatyuk O, Fullard R, Gorbatyuk M. 2015. Unfolded protein response is activated in aged retinas. Neurosci. Lett. 609:30–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Sun F, Wang K. 2003. Caloric restriction retards age-related changes in rat retina. Biochem. Biophys. Res. Commun. 309:457–63 [DOI] [PubMed] [Google Scholar]
- Li Y, Wang YS, Shen XF, Hui YN, Han J, et al. 2008. Alterations of activity and intracellular distribution of the 20S proteasome in ageing retinal pigment epithelial cells. Exp. Gerontol. 43:1114–22 [DOI] [PubMed] [Google Scholar]
- Lin S, Guo J, Chen S. 2019. Transcriptome and DNA methylome signatures associated with retinal Müller glia development, injury response, and aging. Investig. Ophthalmol. Vis. Sci 60:4436–50 [DOI] [PubMed] [Google Scholar]
- Lin W, Xu G. 2019. Autophagy: a role in the apoptosis, survival, inflammation, and development of the retina. Ophthalmic Res. 61:65–72 [DOI] [PubMed] [Google Scholar]
- Linton JD, Holzhausen LC, Babai N, Song H, Miyagishima KJ, et al. 2010. Flow of energy in the outer retina in darkness and in light. PNAS 107:8599–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Cao L, Yang S, Xu L, Liu P, et al. 2015. Subretinal injection of amyloid-beta peptide accelerates RPE cell senescence and retinal degeneration. Int. J. Mol. Med. 35:169–76 [DOI] [PubMed] [Google Scholar]
- Lodato MA, Rodin RE, Bohrson CL, Coulter ME, Barton AR, et al. 2018. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science 359:555–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo VD, Fabrizio P. 2012. Chronological aging in Saccharomyces cerevisiae. Subcell. Biochem. 57:101–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Luppo M, Catita J, Ramos D, Navarro M, Carretero A, et al. 2017. Cellular senescence is associated with human retinal microaneurysm formation during aging. Investig. Ophthalmol. Vis. Sci. 58:2832–42 [DOI] [PubMed] [Google Scholar]
- Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G.2013.The hallmarks of aging. Cell 153:1194–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louie JL, Kapphahn RJ, Ferrington DA. 2002. Proteasome function and protein oxidation in the aged retina. Exp. Eye Res. 75:271–84 [PubMed] [Google Scholar]
- Lu M, Kuroki M, Amano S, Tolentino M, Keough K, et al. 1998. Advanced glycation end products increase retinal vascular endothelial growth factor expression. J. Clin. Investig. 101:1219–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Brommer B, Tian X, Krishnan A, Meer M, et al. 2020. Reprogramming to recover youthful epigenetic information and restore vision. Nature 588:124–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luu J, Kallestad L, Hoang T, Lewandowski D, Dong Z, et al. 2020. Epigenetic hallmarks of age-related macular degeneration are recapitulated in a photosensitive mouse model. Hum. Mol. Genet. 29:2611–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Wong WT. 2016. Aging changes in retinal microglia and their relevance to age-related retinal disease. Adv. Exp. Med. Biol. 854:73–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacRae SL, Croken MM, Calder RB, Aliper A, Milholland B, et al. 2015. DNA repair in species with extreme lifespan differences. Aging 7:1171–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madabhushi R, Pan L, Tsai LH. 2014. DNA damage and its links to neurodegeneration. Neuron 83:266–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda A, Crabb JW, Palczewski K. 2005. Microsomal glutathione S-transferase 1 in the retinal pigment epithelium: protection against oxidative stress and a potential role in aging. Biochemistry 44:480–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maegawa S, Hinkal G, Kim HS, Shen L, Zhang L, et al. 2010.Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res. 20:332–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maghbooli Z, Hossein-nezhad A, Larijani B, Amini M, Keshtkar A. 2015. Global DNA methylation as a possible biomarker for diabetic retinopathy. Diabetes Metab. Res. Rev. 31:183–89 [DOI] [PubMed] [Google Scholar]
- Makin RD, Argyle D, Hirahara S, Nagasaka Y, Zhang M, et al. 2020. Voluntary exercise suppresses choroidal neovascularization in mice. Investig. Ophthalmol. Vis. Sci. 61:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour H, Chamberlain CG, Weible MW 2nd, Hughes S, Chu Y, Chan-Ling T. 2008. Aging-related changes in astrocytes in the rat retina: imbalance between cell proliferation and cell death reduces astrocyte availability. Aging Cell 7:526–40 [DOI] [PubMed] [Google Scholar]
- Martinez-Lopez N, Athonvarangkul D, Singh R. 2015. Autophagy and aging. Adv. Exp. Med. Biol. 847:73–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masland RH. 2001. The fundamental plan of the retina. Nat. Neurosci. 4:877–86 [DOI] [PubMed] [Google Scholar]
- Mattison JA, Colman RJ, Beasley TM, Allison DB, Kemnitz JW, et al. 2017. Caloric restriction improves health and survival of rhesus monkeys. Nat. Commun. 8:14063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin T, Falkowski M, Park JW, Keegan S, Elliott M, et al. 2018. Loss of XBP1 accelerates age-related decline in retinal function and neurodegeneration. Mol. Neurodegener. 13:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meira LB, Moroski-Erkul CA, Green SL, Calvo JA, Bronson RT, et al. 2009.Aag-initiated base excision repair drives alkylation-induced retinal degeneration in mice. PNAS 106:888–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melzer D, Pilling LC, Ferrucci L. 2020. The genetics of human ageing. Nat. Rev. Genet. 21:88–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimura T, Kaji Y, Noma H, Funatsu H, Okamoto S. 2013. The role of SIRT1 in ocular aging. Exp. Eye Res. 116:17–26 [DOI] [PubMed] [Google Scholar]
- Mitter SK, Song C, Qi X, Mao H, Rao H, et al. 2014. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy 10:1989–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller SM, Parekh N, Tinker L, Ritenbaugh C, Blodi B, et al. 2006. Associations between intermediate age-related macular degeneration and lutein and zeaxanthin in the Carotenoids in Age-Related Eye Disease Study (CAREDS): ancillary study of the Women’s Health Initiative. Arch. Ophthalmol. 124:1151–62 [DOI] [PubMed] [Google Scholar]
- Nag TC, Wadhwa S.2012. Ultrastructure of the human retina in aging and various pathological states. Micron 43:759–81 [DOI] [PubMed] [Google Scholar]
- Nag TC, Wadhwa S. 2016. Immunolocalisation pattern of complex I-V in ageing human retina: correlation with mitochondrial ultrastructure. Mitochondrion 31:20–32 [DOI] [PubMed] [Google Scholar]
- Nagaraj RH, Linetsky M, Stitt AW. 2012. The pathogenic role of Maillard reaction in the aging eye. Amino Acids 42:1205–20 [DOI] [PubMed] [Google Scholar]
- Nettesheim A, Dixon A, Shim MS, Coyne A, Walsh M, Liton PB. 2020. Autophagy in the aging and experimental ocular hypertensive mouse model. Investig. Ophthalmol. Vis. Sci. 61:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noro T, Namekata K, Kimura A, Guo X, Azuchi Y, et al. 2015. Spermidine promotes retinal ganglion cell survival and optic nerve regeneration in adult mice following optic nerve injury. Cell Death Dis. 6:e1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norrie JL, Lupo MS, Xu B, Al DiriI, Valentine M, et al. 2019. Nucleome dynamics during retinal development. Neuron 104:512–28.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okawa H, Sampath AP, Laughlin SB, Fain GL. 2008. ATP consumption by mammalian rod photoreceptors in darkness and in light. Curr. Biol. 18:1917–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver VF, Franchina M, Jaffe AE, Branham KE, Othman M, et al. 2013. Hypomethylation of the IL17RC promoter in peripheral blood leukocytes is not a hallmark of age-related macular degeneration. Cell Rep. 5:1527–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver VF, Jaffe AE, Song J, Wang G, Zhang P, et al. 2015. Differential DNA methylation identified in the blood and retina of AMD patients. Epigenetics 10:698–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne NN, Alvarez CN, del Olmo Aguado S. 2014. Targeting mitochondrial dysfunction as in aging and glaucoma. Drug Discov. Today 19:1613–22 [DOI] [PubMed] [Google Scholar]
- Oubaha M, Miloudi K, Dejda A, Guber V, Mawambo G, et al. 2016. Senescence-associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Sci. Transl. Med. 8:362ra144. [DOI] [PubMed] [Google Scholar]
- Owsley C 2011. Aging and vision. Vis. Res. 51:1610–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa Y 2020. Oxidative stress in the light-exposed retina and its implication in age-related macular degeneration. Redox Biol. 37:101779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parapuram SK, Cojocaru RI, Chang JR, Khanna R, Brooks M, et al. 2010. Distinct signature of altered homeostasis in aging rod photoreceptors: implications for retinal diseases. PLOS ONE 5:e13885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh RS, Parikh SR, Sekhar GC, Prabakaran S, Babu JG, Thomas R. 2007. Normal age-related decay of retinal nerve fiber layer thickness. Ophthalmology 114:921–26 [DOI] [PubMed] [Google Scholar]
- Pauly D, Agarwal D, Dana N, Schafer N, Biber J, et al. 2019. Cell-type-specific complement expression in the healthy and diseased retina. Cell Rep. 29:2835–48.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng CH, Chang YL, Kao CL, Tseng LM, Wu CC, et al. 2010. SirT1: a sensor for monitoring self-renewal and aging process in retinal stem cells. Sensors 10:6172–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrash JM. 2013. Aging and age-related diseases of the ocular lens and vitreous body. Investig. Ophthalmol. Vis. Sci 54:ORSF54–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter LF, Saptarshi N, Fang Y, Rathi S, den Hollander AI, et al. 2019. Whole-genome methylation profiling of the retinal pigment epithelium of individuals with age-related macular degeneration reveals differential methylation of the SKI, GTF2H4, and TNXB genes. Clin. Epigenetics 11:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punzo C, Kornacker K, Cepko CL. 2009. Stimulation of the insulin/mTOR pathway delays cone death in a mouse model of retinitis pigmentosa. Nat. Neurosci. 12:44–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querques G, Forte R, Souied EH. 2011. Retina and omega-3. J. Nutr. Metab. 2011:748361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavan CT, Smuda M, Smith AJO, Howell S, Smith DG, et al. 2016. AGEs in human lens capsule promote the TGFβ2-mediated EMT of lens epithelial cells: implications for age-associated fibrosis. Aging Cell 15:465–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez JM, Ramirez AI, Salazar JJ, de Hoz R, Trivino A. 2001. Changes of astrocytes in retinal ageing and age-related macular degeneration. Exp. Eye Res. 73:601–15 [DOI] [PubMed] [Google Scholar]
- Ramón y Cajal S 1892 (1972). The Structure of the Retina, transl. Thorpe SA, Glickstein M. Springfield, IL: Charles C Thomas Publ. [Google Scholar]
- Ratnapriya R, Sosina OA, Starostik MR, Kwicklis M, Kapphahn RJ, et al. 2019. Retinal transcriptome and eQTL analyses identify genes associated with age-related macular degeneration. Nat. Genet. 51:606–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhinn H, Abeliovich A. 2017. Differential aging analysis in human cerebral cortex identifies variants in TMEM106B and GRN that regulate aging phenotypes. Cell Syst. 4:404–15.e5 [DOI] [PubMed] [Google Scholar]
- Rocha LR, Nguyen Huu VA, Palomino La Torre C, Xu Q, Jabari M, et al. 2020. Early removal of senescent cells protects retinal ganglion cells loss in experimental ocular hypertension. Aging Cell 19:e13089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-de la Rosa L, Fernandez-Sanchez L, Germain F, Murillo-Cuesta S, Varela-Nieto I, et al. 2012. Age-related functional and structural retinal modifications in the Igf1−/− null mouse. Neurobiol. Dis. 46:476–85 [DOI] [PubMed] [Google Scholar]
- Rodriguez-Muela N, Koga H, Garcia-Ledo L, de la Villa P, de la Rosa EJ, et al. 2013. Balance between autophagic pathways preserves retinal homeostasis. Aging Cell 12:478–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roginska D, Kawa MP, Pius-Sadowska E, Lejkowska R, Luczkowska K, et al. 2017. Depletion of the third complement component ameliorates age-dependent oxidative stress and positively modulates autophagic activity in aged retinas in a mouse model. Oxid. Med. Cell Longev. 2017:5306790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer B, Frazer-Abel A, Leonard A, Ratnapriya R, Ward T, et al. 2019. Association of age-related macular degeneration with complement activation products, smoking, and single nucleotide polymorphisms in South Carolinians of European and African descent. Mol. Vis. 25:79–92 [PMC free article] [PubMed] [Google Scholar]
- Roufail E, Rees S.1997.Ageing has a differential effect on nitric oxide synthase-containing and catecholaminergic amacrine cells in the human and rat retina. J. Comp. Neurol. 389:329–47 [PubMed] [Google Scholar]
- Rowan S, Jiang S, Korem T, Szymanski J, Chang ML, et al. 2017.Involvement of a gut-retina axis in protection against dietary glycemia-induced age-related macular degeneration. PNAS 114:E4472–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampaio-Marques B, Burhans WC, Ludovico P. 2019. Yeast at the forefront of research on ageing and age-related diseases. Prog. Mol. Subcell. Biol. 58:217–42 [DOI] [PubMed] [Google Scholar]
- Samuel MA, Voinescu PE, Lilley BN, de Cabo R, Foretz M, et al. 2014. LKB1 and AMPK regulate synaptic remodeling in old age. Nat. Neurosci. 17:1190–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapieha P, Mallette FA. 2018. Cellular senescence in postmitotic cells: beyond growth arrest. Trends Cell Biol. 28:595–607 [DOI] [PubMed] [Google Scholar]
- Schiller PH, Tehovnik EJ. 2015. Vision and the Visual System. Oxford, UK: Oxford Univ. Press [Google Scholar]
- Schrier SA, Falk MJ. 2011. Mitochondrial disorders and the eye. Curr. Opin. Ophthalmol. 22:325–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seddon JM, Widjajahakim R, Rosner B. 2020. Rare and common genetic variants, smoking, and body mass index: progression and earlier age of developing advanced age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 61:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seddon JM, Willett WC, Speizer FE, Hankinson SE. 1996. A prospective study of cigarette smoking and age-related macular degeneration in women. JAMA 276:1141–46 [PubMed] [Google Scholar]
- Sen P, Shah PP, Nativio R, Berger SL. 2016. Epigenetic mechanisms of longevity and aging. Cell 166:822–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh PP, Demmitt BA, Nath RD, Brunet A. 2019. The genetics of aging: a vertebrate perspective. Cell 177:200–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha D, Valapala M, Shang P, Hose S, Grebe R, et al. 2016.Lysosomes: regulators of autophagy in the retinal pigmented epithelium. Exp. Eye Res. 144:46–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skowronska-Krawczyk D, Zhao L, Zhu J, Weinreb RN, Cao G, et al. 2015. P16INK4a upregulation mediated by SIX6 defines retinal ganglion cell pathogenesis in glaucoma. Mol. Cell 59:931–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss O 2005. The retinal pigment epithelium in visual function. Physiol. Rev. 85:845–81 [DOI] [PubMed] [Google Scholar]
- Subhi Y, Forshaw T, Sorensen TL. 2016. Macular thickness and volume in the elderly: a systematic review. Ageing Res. Rev. 29:42–49 [DOI] [PubMed] [Google Scholar]
- Swaroop A, Chew EY, Rickman CB, Abecasis GR. 2009. Unraveling a multifactorial late-onset disease: from genetic susceptibility to disease mechanisms for age-related macular degeneration. Annu. Rev. Genom. Hum. Genet. 10:19–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan BL, Norhaizan ME, Liew WP, Sulaiman Rahman H. 2018. Antioxidant and oxidative stress: a mutual interplay in age-related diseases. Front. Pharmacol. 9:1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao JX, Zhou WC, Zhu XG. 2019. Mitochondria as potential targets and initiators of the blue light hazard to the retina. Oxid. Med. Cell Longev. 2019:6435364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. 2013. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J. Clin. Investig. 123:966–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian J, Ishibashi K, Ishibashi K, Reiser K, Grebe R, et al. 2005. Advanced glycation endproduct-induced aging of the retinal pigment epithelium and choroid: a comprehensive transcriptional response. PNAS 102:11846–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissenbaum HA. 2015. Using C. elegans for aging research. Invertebr. Reprod. Dev. 59:59–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchiki T, Weikel KA, Jiao W, Shang F, Caceres A, et al. 2012. Glycation-altered proteolysis as a pathobiologic mechanism that links dietary glycemic index, aging, and age-related disease (in nondiabetics). Aging Cell 11:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara F, Ohba N, Ozawa M. 2001. Isolation and characterization of galectins in the mammalian retina. Investig. Ophthalmol. Vis. Sci. 42:2164–72 [PubMed] [Google Scholar]
- Unnikrishnan A, Hadad N, Masser DR, Jackson J, Freeman WM, Richardson A.2018.Revisiting the genomic hypomethylation hypothesis of aging. Ann. N. Y. Acad. Sci. 1418:69–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Deursen JM. 2019. Senolytic therapies for healthy longevity. Science 364:636–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velez G, Machlab DA, Tang PH, Sun Y, Tsang SH, et al. 2018. Proteomic analysis of the human retina reveals region-specific susceptibilities to metabolic- and oxidative stress-related diseases. PLOS ONE 13:e0193250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velilla S, Garcia-Medina JJ, Garcia-Layana A, Dolz-Marco R, Pons-Vazquez S, et al. 2013. Smoking and age-related macular degeneration: review and update. J. Ophthalmol. 2013:895147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang AL, Lukas TJ, Yuan M, Du N, Tso MO, Neufeld AH. 2009a. Autophagy and exosomes in the aged retinal pigment epithelium: possible relevance to drusen formation and age-related macular degeneration. PLOS ONE 4:e4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang AL, Lukas TJ, Yuan M, Neufeld AH. 2010. Age-related increase in mitochondrial DNA damage and loss of DNA repair capacity in the neural retina. Neurobiol. Aging 31:2002–10 [DOI] [PubMed] [Google Scholar]
- Wang J, Ohno-Matsui K, Yoshida T, Shimada N, Ichinose S, et al. 2009b. Amyloid-beta up-regulates complement factor B in retinal pigment epithelial cells through cytokines released from recruited macrophages/microglia: another mechanism of complement activation in age-related macular degeneration. J. Cell Physiol. 220:119–28 [DOI] [PubMed] [Google Scholar]
- Wang J, Zibetti C, Shang P, Sripathi SR, Zhang P, et al. 2018. ATAC-Seq analysis reveals a widespread decrease of chromatin accessibility in age-related macular degeneration. Nat. Commun. 9:1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Kaufman RJ. 2016. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 529:326–35 [DOI] [PubMed] [Google Scholar]
- Wang N, Luo Z, Jin M, Sheng W, Wang HT, et al. 2019. Exploration of age-related mitochondrial dysfunction and the anti-aging effects of resveratrol in zebrafish retina. Aging 11:3117–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L, Liu B, Tuo J, Shen D, Chen P, et al. 2012. Hypomethylation of the IL17RC promoter associates with age-related macular degeneration. Cell Rep. 2:1151–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei PC, Chang AN, Kao J, Du Z, Meyers RM, et al. 2016. Long neural genes harbor recurrent DNA break clusters in neural stem/progenitor cells. Cell 164:644–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Thompson CB. 2010. Cellular metabolic stress: considering how cells respond to nutrient excess. Mol. Cell 40:323–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolffenbuttel BH, Boulanger CM, Crijns FR, Huijberts MS, Poitevin P, et al. 1998. Breakers of advanced glycation end products restore large artery properties in experimental diabetes. PNAS 95:4630–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Chen M, Manivannan A, Lois N, Forrester JV. 2008. Age-dependent accumulation of lipofuscin in perivascular and subretinal microglia in experimental mice. Aging Cell 7:58–68 [DOI] [PubMed] [Google Scholar]
- Yao J, Jia L, Khan N, Lin C, Mitter SK, et al. 2015. Deletion of autophagy inducer RB1CC1 results in degeneration of the retinal pigment epithelium. Autophagy 11:939–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida S, Yashar BM, Hiriyanna S, Swaroop A. 2002. Microarray analysis of gene expression in the aging human retina. Investig. Ophthalmol. Vis. Sci. 43:2554–60 [PubMed] [Google Scholar]
- Young JI, Zuchner S, Wang G. 2015. Regulation of the epigenome by vitamin C. Annu. Rev. Nutr. 35:545–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B, Xu P, Zhao Z, Cai J, Sternberg P, Chen Y. 2014. Subcellular distribution and activity of mechanistic target of rapamycin in aged retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 55:8638–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecic A, Braeckman BP. 2020. DAF-16/FoxO in Caenorhabditis elegans and its role in metabolic remodeling. Cells 9:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Yang K. 2015. Sirtuin 1 participates in the process of age-related retinal degeneration. Biochem. Biophys. Res. Commun. 468:167–72 [DOI] [PubMed] [Google Scholar]
- Zhang M, Jiang N, Chu Y, Postnikova O, Varghese R, et al. 2020. Dysregulated metabolic pathways in age-related macular degeneration. Sci. Rep. 10:2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J, Karlsson O, Wang G, Li J, Guo Y, et al. 2017. B vitamins attenuate the epigenetic effects of ambient fine particles in a pilot human intervention trial. PNAS 114:3503–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y, Li J, Wang JJ, Chen C, Tran JT, et al. 2012. X-box binding protein 1 is essential for the anti-oxidant defense and cell survival in the retinal pigment epithelium. PLOS ONE 7:e38616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu D, Wu J, Spee C, Ryan SJ, Hinton DR.2009.BMP4 mediates oxidative stress-induced retinal pigment epithelial cell senescence and is overexpressed in age-related macular degeneration.J.Biol.Chem.284:9529–39 [DOI] [PMC free article] [PubMed] [Google Scholar]