

Summary
Tissues are organized into anatomical and functional units at different scales. New technologies for high-dimensional molecular profiling in situ have enabled the characterization of structure-function relationships in increasing molecular detail. However, it remains a challenge to consistently identify key functional units across experiments, tissues, and disease contexts, a task that demands extensive manual annotation. Here, we present spatial cellular graph partitioning (SCGP), a flexible method for the unsupervised annotation of tissue structures. We further present a reference-query extension pipeline, SCGP-Extension, that generalizes reference tissue structure labels to previously unseen samples, performing data integration and tissue structure discovery. Our experiments demonstrate reliable, robust partitioning of spatial data in a wide variety of contexts and best-in-class accuracy in identifying expertly annotated structures. Downstream analysis on SCGP-identified tissue structures reveals disease-relevant insights regarding diabetic kidney disease, skin disorder, and neoplastic diseases, underscoring its potential to drive biological insight and discovery from spatial datasets.
Keywords: spatial omics, artificial intelligence, unsupervised annotation
Graphical abstract
Highlights
-
•
SCGP is a highly flexible and efficient tool for spatial omics annotation
-
•
SCGP shows outstanding performance across 8 distinct spatial omics datasets
-
•
SCGP-Extension generalizes existing tissue structures to unseen samples
-
•
SCGP-Extension effectively addresses common data integration challenges
Motivation
Annotating tissue structures provides an important layer of biological interpretation from spatial molecular data. Uniform and consistent identification of structures across different batches, experiments, and diverse disease conditions remains a challenging task, often requiring manual intervention. The generalizability of annotations from a reference dataset to new or unseen data also remains a major challenge for methods in this arena. The current work introduces spatial cellular graph partitioning (SCGP) and its reference-query extension pipeline, SCGP-Extension, as unsupervised annotation tools that streamline and simplify this process, enhancing the consistency, reliability, and generalization of structure annotations across large datasets.
Wu et al. proposed a universal, efficient annotation tool, SCGP, for spatial omics data, which allows unsupervised recognition of multi-cellular tissue structures. They further proposed the SCGP-Extension pipeline to generalize these annotations to unseen samples, achieving robust, consistent tissue structure identification.
Introduction
All human organs exhibit characteristic cellular structures that are required for homeostasis and function. These structures are diverse in form, scale, and function and are typically composed of multiple cell types organized into spatial patterns. Disruptions to these structures usually indicate a disease process.1,2 Recent advances in in situ molecular profiling techniques, including spatial transcriptomics3,4,5,6,7 and proteomics,8,9,10,11 have allowed us to observe molecular phenotypes and cell states in their native contexts, where interactions between entities at different spatial scales—ranging from cells12 and cellular neighborhoods (CN)13,14 to tissue organization15 and patient-level characteristics16,17—can be explored. Discovering, annotating, and analyzing these functional units in consistent ways is a major goal for the field of spatial biology.
However, in practice, annotating tissue structures in a way that both incorporates complex molecular information and aligns with our histological understanding has been challenging without manual supervision. Consistency across diverse samples, experiments, or disease conditions is difficult to achieve. Moreover, existing analysis pipelines18,19 are predominantly cell centric20,21,22 or geared toward small sample numbers. Together, these issues severely limit efforts to analyze and interpret atlas-scale spatial biology datasets.
In recent studies, computational methods integrating molecular profiling with spatial information have been proposed. Some of these methods aim to improve the analysis of cell-level characteristics, such as better cell-type prediction23,24 and intercellular communication modeling.12,25 Another line of research focuses on annotating larger structures or spatial domains, exploring their interactions and disease relevance. Such annotations are performed based on clustering of cell-type composition13,26 or locally smoothed cell features,15,27 topic modeling,28 Bayesian modeling,29 optimal transport,30 graph Fourier transform,31 and graph neural networks.16,32,33 Many of these methods are unsupervised and lack the ability to generalize—that is, the resulting annotations or model cannot be extended to previously unseen samples. When new data are introduced, model retraining or refitting is necessary to annotate unseen data. Consequently, downstream analysis on structures of interest are restricted to only the training/fitting data, as consistent annotations on out-of-sample data cannot be reliably acquired. Similarly, recognizing tissue structures from unseen hematoxylin and eosin (H&E)-stained pathology images is a widely studied task often resolved in a supervised manner.34,35,36,37 However, these tools tend to be more tissue-type specific, require substantial annotated training datasets, and do not incorporate molecular information.
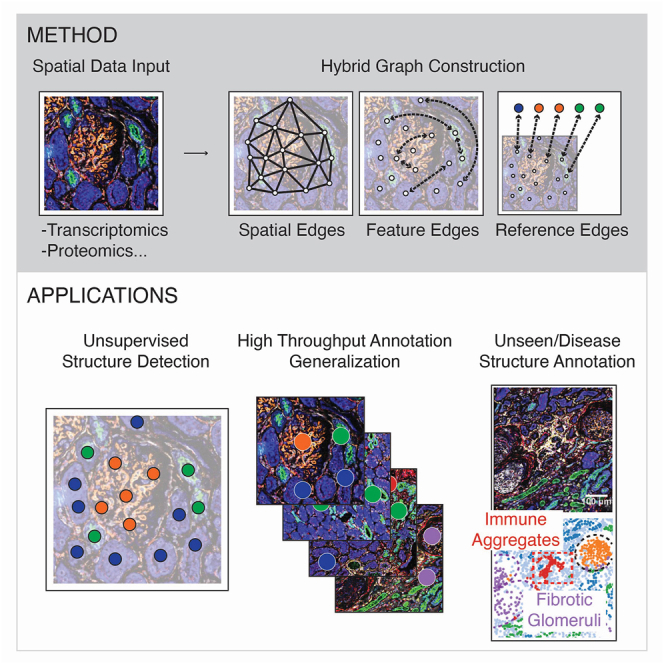
In this work, we present a universal, robust, and generalizable tissue structure segmentation tool called spatial cellular graph partitioning (SCGP). SCGP is a fast and flexible method designed to identify the anatomical and functional units in any spatial transcriptomic or proteomic dataset. We further introduce a reference-query extension pipeline, SCGP-Extension, which allows a small set of reference annotations to be generalized to previously unseen query samples. This powerful data alignment and integration method can address challenges ranging from experimental artifacts and batch effects to disease condition differences and more, greatly enhancing SCGP’s robustness and versatility. To the best of our knowledge, SCGP is the first data-type-agnostic semantic segmentation tool that is purpose built for generalization. We demonstrate its applications to eight datasets collected on different tissue types using diverse profiling techniques including CO-detection by indexing (CODEX), Visium, imaging mass cytometry (IMC), and multiplexed error-robust fluorescence in situ hybridization (MERFISH), totaling more than 2.5 million cells. The tissue structures identified by SCGP are evaluated against expert annotations, benchmarked extensively against related software tools, and applied in downstream analysis that reveals disease-relevant biological insights.
Results
Unsupervised partitioning of spatial cellular graphs
To address the task of tissue structure identification, we developed SCGP to perform community detection on specialized graph representations of tissue samples. Nodes in the graphs are small spatial units characterized by spatial coordinates and gene or protein expression at the location (STAR Methods). In the representative case of multiplexed immunofluorescence (mIF) images,38 nodes are defined on cells identified through the segmentation pipeline20 (Figure 1A). However, this concept of nodes can be further extended to accommodate broader spatial transcriptomics and proteomics data, such as spots in spatial transcriptomics sequencing measurements7 or small square patches in single-molecule fluorescence images.3 In this study, we primarily present and discuss cell- and spot-based SCGP analysis. An alternative patch-based SCGP experiment can be found in Figure S10.
Figure 1.

Workflow for SCGP and SCGP-Extension
(A) Raw mIF images of example kidney samples show multiple tissue structures.
(B) Graph representations of mIF images are constructed on nodes (white circles) representing cells or other spatial units. Spatial edges (solid lines) and feature edges (dashed arrows) are constructed to reflect spatial closeness and feature similarity.
(C) Leiden graph community detection identifies partitions representing tissue structures.
(D) The query sample shares similar structures to the reference partitions.
(E) Graph representation of the query sample is constructed with additional pseudo-nodes (colored circles in the white box) extracted from the reference partitions. Reference-query edges (brown dashed arrows) are constructed between query nodes and pseudo-nodes.
(F) Leiden graph community detection yields both existing partitions that align with reference and novel partitions that were previously unseen.
Two types of edges are constructed between nodes (Figure 1B; STAR Methods). Spatial edges are constructed between nodes based on Delaunay triangulation of node coordinates. These edges aim to capture the adjacency relationships between cells. Feature edges are constructed between nodes that share similar expression profiles.
The Leiden graph community detection algorithm39 is then applied to the graphs, yielding partitions that represent the different tissue structures (Figure 1C). The central aspect of this method is the joint contribution of two types of edges. Spatial edges guarantee the spatial continuity of the identified tissue structures, differentiating the method from cell-type clustering in that multi-cell tissue structures will appear as cohesive entities. Feature edges interrelate tissue structures of the same type even if they are spatially separated (e.g., two glomeruli from different kidney samples), ensuring the consistency of tissue structure interpretation across samples. This design resonates with a customized tool developed for identifying spatial modules in spatial enhanced resolution omics sequencing (stereo-seq),27 though SCGP used much sparser feature edges to avoid fragmentation of tissue structures (1–4 nearest neighbors; STAR Methods; Figures S3E and S3F). Ablation experiments (Figures S3A and S3B) show that both types of edges are necessary for the operation of SCGP. We first verified the effectiveness of SCGP using various types of simulated data representing diverse tissue structures (Figure S1). Results showed consistent and accurate recognition of all ground-truth structures by SCGP across all conditions.
SCGP identifies structures in kidney tissues
To examine the ability of SCGP to recognize known tissue structures, we assessed its performance on a cohort of 17 tissue sections from 12 individuals with diabetes and various stages of diabetic kidney disease (DKD).40 Tissue samples were imaged using the mIF platform CODEX9 and further annotated for four major kidney compartments: glomeruli, blood vessels, distal tubules, and proximal tubules. This cohort will be referred to as the DKD Kidney dataset (STAR Methods) in the subsequent text.
Together with SCGP, we applied a diverse set of unsupervised annotation tools13,15,24,28,32 to the DKD Kidney dataset. All methods were applied to the combination of all 17 samples containing 137,654 cells, i.e., in a joint partitioning manner. Clustering/partitioning outputs on representative samples are visualized in Figures 2A and S2A, with the leftmost images illustrating the raw mIF images with key biomarkers. Due to the unsupervised nature of the output, we reordered the output clusters of each method in accordance with manually annotated compartments. In Figure 2A, the top image for each column shows the output, and the bottom image highlights the mismatches. Across samples, partitions annotated by SCGP frequently demonstrated the highest fidelity to manual annotations, and they were robust to cell segmentation noise (Figures S3C and S3D).
Figure 2.

Unsupervised annotations of the DKD Kidney samples
(A) Annotations from SCGP and other unsupervised annotation methods recognized tissue structures aligned with manually annotated compartments. Nodes representing cells are colored according to the assigned clusters/partitions in the top images, and colors not listed in the legend (e.g., cyan) refer to clusters/partitions that cannot be matched to any compartment. Mismatched nodes are highlighted in red in the bottom images.
(B) ARIs were calculated between unsupervised annotations and manual annotations. SCGP performed significantly better than all other methods (Wilcoxon signed-rank test).
(C) For each manually annotated compartment, F1 scores were calculated between manual annotations and the most overlapped cluster/partition.
(D) Signature protein biomarkers for SCGP-identified partitions match expectations of kidney tissue structures.
(E) SCGP annotations on samples with different classes of DKD show varying levels of alignment and accuracy. Diabetes mellitus (DM, with healthy kidney) and DKD classes represent the different progression stages assigned by a nephrologist following the Tervaert classification.41
See also Figure S2.
To evaluate the performance of each method quantitatively, we calculated the adjusted Rand index (ARI) between manually annotated compartments and unsupervised partitions (Figure 2B). SCGP achieved the top performance with a median ARI of 0.60, significantly outperforming all other methods (Wilcoxon signed-rank test). Since ARI downweighs smaller/rarer compartments (e.g., glomeruli, blood vessels), we calculated additional alignment and accuracy measurements, including independent F1 scores for each compartment (Figures 2C, S2B, and S2C; STAR Methods). These metrics better reflect performances on less common structures, many of which have great functional significance. We found that unsupervised discovery of tissue architecture with graphs (UTAG)15 and SCGP performed the best at recognizing glomeruli (F1 = ∼0.8), while SpiceMix24 and SpaGCN32 excelled at recognizing tubule structures. Overall, SCGP achieved the best average F1 score in discerning all manually annotated compartments.
To further validate SCGP-identified partitions, we highlighted their relative protein expression (Figures 2D and S2D). The heatmap corresponds well to our expectation, with CCR642 and Nestin43 among the top biomarkers for glomeruli and CXCR344 and MUC145 for proximal and distal tubules. Notably, by grouping performance metrics by sample disease progression,41 we found that the quality of unsupervised partitions degraded substantially with disease progression (severe DKD, class IIB/III). This indicates how normal tissue structures and functions are dysregulated in DKD and highlights the challenge of performing consistent annotations across disease states (Figure 2E).
SCGP identifies major structures of the brain
Next, we assessed SCGP’s performance on spatial transcriptomics measurements of human and mouse brains. We first employed a Visium7 dataset of human dorsolateral prefrontal cortex (DLPFC).46 In contrast to the mIF approach, the Visium platform features a grid of spatially barcoded oligonucleotide arrays that can be used for mRNA capture and library preparation. Each array (i.e., spot) may contain multiple cells. We adapted our method by treating each spot as a node and defining edges based on the grid and gene count information (STAR Methods).
DLPFC contains 12 samples annotated with 7 compartments: 6 cortical layers (L1–L6) and white matter. We directly compared our method against existing tools developed for this modality, including BayesSpace,29 SpaGCN, and SpiceMix, in recognizing manually annotated layers. Clustering/partitioning was performed independently on each sample. Results on a representative sample are demonstrated in Figure 3A, revealing a clear layer-wise pattern in the tissue structures. Among the benchmarked methods, SCGP and SpiceMix achieved the top ARI scores, exhibiting superior alignment with the ground-truth compartments marked by dashed lines. Quantitative metrics (Figures 3B and S4E) demonstrated that SCGP achieved comparable, if not superior, performance (median ARI = 0.56, median F1 = 0.65) to unsupervised annotation tools designed specifically for Visium spatial transcriptomics.
Figure 3.

Unsupervised annotations of human and mouse brain samples
(A) A representative DLPFC sample was annotated using SCGP and other unsupervised annotation methods. Note that layer 2 and layer 4 were not fully recognized. Boundaries between ground-truth layers are annotated as dashed lines.
(B) ARIs were calculated between unsupervised annotations and ground-truth layers on 12 samples, with each sample annotated independently.
(C) A mouse brain section profiled by MERFISH was annotated using SCGP, from which major brain parcels were identified. Boundaries between parcels are annotated as dashed lines.
See also Figures S4 and S5.
We subsequently tested SCGP on a MERFISH-profiled section of a whole mouse brain,47 where SCGP readily recognized major brain parcels, exhibiting an overall ARI score of 0.75 (Figure 3C). Furthermore, SCGP with higher granularity accurately identified finer structures aligned with detailed parcellation (Figure S5).
Next, we further evaluated if jointly partitioning multiple sections would improve performance. Following the experiment design by Chidester et al.,24 we applied unsupervised annotation tools to the combination of four samples in DLPFC collected from the same donor (Figures S4A–S4C). While BayesSpace, SpaGCN, and SpiceMix performed better when jointly partitioning samples, UTAG and SCGP yielded worse results. Motivated by the aforementioned challenge of partitioning different disease states as well as how joint partitioning might suffer from inter-sample variance, we explored means of generalizing consistent annotations across conditions.
SCGP extends existing tissue structures to unseen samples
In practical applications, generalizing a set of curated annotations across different experiments, conditions, or disease states is required to conduct analyses at larger scales of data and a long-standing goal of data integration. Generalization is useful for performing inference on prospective unseen data, suppressing unwanted noise or batch effects, comparing samples across disease conditions, and detecting previously unseen and unannotated disease states. Existing unsupervised annotation tools have limited or non-existent support for this functionality. Most methods (e.g., UTAG, SpaGCN) explicitly require either retraining or refitting the clustering model or the addition of separate prediction models to extend existing partitions. Therefore, to address this need, here, we present a specialized reference-query extension pipeline, SCGP-Extension.
SCGP-Extension begins with the annotation of a small group of high-quality reference samples, for example, by using SCGP. Resulting partitions are assumed to represent the ground-truth structures and referred to as reference partitions (Figure 1D). Next, we define pseudo-nodes as representative data nodes for each reference partition (STAR Methods). These pseudo-nodes are explicitly added into the SCGP graphs of unseen query samples and connected to other query nodes based on feature similarity (Figure 1E). Finally, the graph community detection step is conducted as before, with partitions of the pseudo-nodes pre-assigned. Consequently, query nodes resembling pseudo-nodes are assigned the corresponding partition, while query nodes that do not resemble any reference partitions form their own groups and assigned as newly discovered or “novel” partitions (Figure 1F). On simulated data, SCGP-Extension robustly identified both known and novel partitions (Figures S1D and S1E).
To demonstrate how SCGP-Extension improves partitioning performance, we examined its application in the DLPFC joint partitioning experiment. Here, we established reference annotations on one sample (Figure 3A) and extended them to the rest, simulating the application scenario where a small set of gold-standard annotations are acquired and generalized to the entire dataset. SCGP-Extension considerably outperformed joint clustering approaches in alignment and accuracy (Figures S4A and S4D). In addition, we evaluated approaches that directly utilized partial ground-truth annotations, consisting of both predictive modeling and SCGP-Extension approaches, where SCGP-Extension using partial labels demonstrated the best performances (Figure S4D).
SCGP-Extension can also generalize across disease conditions and help identify unique disease states. To demonstrate this, we applied SCGP-Extension to generalize primary partitions defined on healthy DKD samples to severe DKD cases. In this scenario, glomeruli exhibit elevated collagen four and decreased native marker expression (e.g., CCR6). Unsupervised SCGP, joint partitioning methods, and supervised classifiers (XGB Prediction) all struggled to correctly identify tissue characteristics of severe DKD (Figures 4A, 2E, and S2A), often mis-recognizing fibrotic glomeruli (Figure 4A, arrows). However, SCGP-Extension preserved most of the original partitions while uncovering two additional structures: the purple partition outlined fibrotic glomeruli, characterized by the depleted native biomarkers and enriched collagen expression, and the red partition, characterized by elevated CD45 and CD68 expression (Figures 4A and 4B), suggesting the infiltration of immune cells (e.g., macrophages). Compared to joint partitioning and predictive modeling, SCGP-Extension delivered more accurate results both visually and quantitatively (Figure 4C). Furthermore, it highlighted unique disease-specific partitions with ready biological interpretations, offering valuable insights into disease progression.
Figure 4.

Versatile applications of SCGP-Extension
(A) Compared to joint partitioning or predictive models, SCGP-Extension better recognized the fibrotic glomeruli (purple dashed circles) in severe DKD samples and identified a previously unseen immune aggregates partition (red boxes).
(B) Extension to severe DKD samples improved partition alignment and accuracy.
(C) Heatmap shows signature protein biomarkers for extended partitions in the severe DKD samples; note the additional fibrotic glomeruli and immune aggregates partitions.
(D) In the TR Kidney dataset, SCGP partitioned a cohort of kidney samples with heavy inflammation (top row). SCGP-Extension further extended the partitions to control samples with minimal immune responses (bottom row).
(E) In the Lung IMC dataset, SCGP-Extension recognized an anatomical structure, submucosal glands (purple), in the query samples that was not seen in reference samples.
(F) ARIs calculated between unsupervised annotations and manual annotations show that SCGP-Extension achieved significantly better alignments (Wilcoxon signed-rank test).
(G) In the UCSF Derm dataset, SCGP-Extension consistently partitioned samples from multiple experiments with different skin conditions.
See also Figures S6–S8.
We next assessed the performance of SCGP in another healthy versus disease comparison. Here, kidney tissue samples from patients who experienced transplant rejection were partitioned (TR Kidney dataset, STAR Methods), characterized by heavily deformed native kidney structures, substantial inflammation, and immune cell infiltration. Although the measured protein markers and disease context were different, our analysis derived the same set of tissue structures as before (Figure 4D). Extending partitions from reference samples with heavy inflammation to query samples with minimal immune activities produced consistent structures, which enabled direct comparisons between conditions (Figures 4D, S6A, S6B, and S6G). Notably, SCGP-Extension effectively partitioned a region containing background signal artifacts, demonstrating effective handling of data issues that would typically require manual intervention to find and fix (Figures S7A–S7D).
To show that SCGP-Extension can identify novel anatomical structures in query samples, we evaluated unsupervised annotation methods on an IMC dataset collected on healthy lung specimens (Lung IMC dataset, STAR Methods), which were manually annotated for anatomical structures. SCGP achieved remarkable alignment with ground truth, but tissue structures across samples were disintegrated and assigned to different partitions (Figure S8A). To address this issue, we conducted primary SCGP on a small subset of samples and extended partitions to the remaining samples. Notably, SCGP-Extension identified two anatomical structures (submucosal glands and cartilage) that were absent in the reference while preserving all known structures (Figure 4E). SCGP-Extension also achieved significantly better alignment scores (median ARI = 0.564; Figures 4F, S8B, and S8C) than all other benchmarked methods (Wilcoxon signed-rank test).
SCGP-Extension can also help mitigate batch effects between experiments. On a cohort of skin samples collected from four separate experiments (UCSF Derm dataset, STAR Methods), we employed unsupervised annotation methods to define tissue structures. The UCSF Derm dataset comprised samples of different skin conditions sharing similar anatomical structures (Table S1). Both UTAG and SCGP failed to link tissue structures from different samples (Figures S7E and S7F). We then attempted SCGP-Extension by defining reference partitions on samples from one experiment (Figure S6C) and extending them to the rest (Figures S6D–S6F). Regardless of biases in the experiments and differences in disease conditions, SCGP-Extension successfully recognized consistent partitions across samples reflecting anatomical structures (Figures 4G and S7H).
SCGP partitions assist downstream analysis of disease states
Partitions acquired by unsupervised annotation using SCGP reflect the anatomical and functional structures of the subject tissues. In this section, we demonstrate how these tissue structures can facilitate the analysis of disease states and enable the discovery of key insights regarding disease-relevant partitions.
In the DKD Kidney experiments, samples were collected from individuals with different DKD classes. Based on the partitioning of these samples, we were interested in exploring correlations between tissue structures and disease progression. Figure 5A illustrates three representative samples of different DKD classes along with their tissue structures annotated by SCGP and SCGP-Extension. Clear visual differences between samples can be observed: tubules and glomeruli were denser in the healthy sample, while these structures gradually deformed over the course of DKD, accompanied by fibrosis and infiltration of immune cells. These changes were also reflected in the tissue structures: a significant increase (p < 0.001, Jonckheere-Terpstra test48) in the area proportion of the basement membrane partition was observed across samples (Figure 5B), reflecting the degradation of normal kidney structures. Immune aggregate (red) and fibrotic glomerular (purple) partitions were identified in the severe DKD sample with SCGP-Extension, which were not present in healthy and mild DKD samples.
Figure 5.

Downstream analysis of disease states with SCGP-identified partitions
(A) Three representative samples of different DKD classes are illustrated; note the fibrosis of glomeruli and increase in the area of the basement membrane partition (light blue-colored nodes). Glomeruli are segmented by dashed circles.
(B) Boxplot shows that the area proportion of the basement membrane partition significantly increases (p < 0.001, Jonckheere-Terpstra test) in DKD samples. Each dot represents a tissue sample.
(C) Expression of native proteins (CCR6) significantly decreases (p < 0.001, Jonckheere-Terpstra test) in glomeruli of patients with DKD. Each dot represents an individual glomerulus, and each box summarizes glomeruli from one patient. Note the heterogeneity observed within a single tissue sample (A, bottom row) and between patients from the same DKD class.
(D) Two samples from the UCSF Derm dataset with different skin conditions are illustrated.
(E) Area proportion of the immune aggregate partition significantly increases in atopic dermatitis samples (p < 0.001, two-sided two-sample t test).
(F) Epidermal layers show significant thickening in atopic dermatitis samples (p < 0.001, two-sided two-sample t test).
Furthermore, to characterize how DKD affects glomerular functions, we assessed the protein biomarker expression of individual glomerulus (dashed circles in Figure 5A), annotated by deriving connected components of the SCGP glomeruli partitions. The results suggested that glomeruli undergo significant loss of native proteins (CCR6) throughout the disease progression (p < 0.001, Jonckheere-Terpstra test; Figure 5C), with high intra-sample and inter-sample heterogeneity.
We next applied the partition-based analysis to the UCSF Derm dataset comprising normal samples and atopic dermatitis samples. Representative samples visualized in Figure 5D show notable differences in terms of epidermal thicknesses and immune cell densities. We verified these visual signatures using SCGP partitions: the area proportion of the immune aggregate partition demonstrated significant increases in atopic dermatitis samples (p < 0.001, two-sided two-sample t test; Figure 5E). Thicknesses of the epidermal layers were characterized using the contours of the corresponding partition, which showed significant increases as well (p < 0.001, two-sided two-sample t test; Figure 5F). Patients with atopic dermatitis also exhibit a much more heterogeneous distribution of epidermal thickness.49 Thus, SCGP empowers rapid quantitative insights from spatial molecular data.
Atlas-scale structural annotation of TMEs
Lastly, we assessed the applicability of SCGP in identifying structures in neoplastic diseases, which are typically characterized by the complete disruption of normal cellular architectures and uncontrolled growth of abnormal cells. We focused our analysis on tumor microenvironments (TMEs),50 highly structured and complex systems containing cancer cells and a variety of non-malignant cell types, including immune cells and stromal cells.
Identifications of major structures in TMEs, such as tumor and stromal regions, are crucial tasks that facilitate further investigation of critical topics including tumoral heterogeneity, dynamics of tumor-immune cell interactions, and therapeutic targeting, among others. Extensive studies have been proposed to perform tumor classification and segmentation on histopathology images using various deep learning methods,36,51,52,53 most of which adopt supervised learning strategies. Spatial omics, on the contrary, facilitate the detection of these structures in TMEs in a fully unsupervised manner.
We benchmarked SCGP against other annotation tools to a cohort of head and neck tumor samples (University of Pittsburgh Medical Center-Head and Neck Cancer [UPMC-HNC]; STAR Methods) containing 36 tumor cores and 175,000 cells. As illustrated in Figures 6A and S9A, a distinct separation between tumor and stromal regions was observed after partitioning at a coarse resolution. Quantitative metrics showed a median ARI exceeding 0.8 and an F1 score of 0.94 from SCGP (Figure 6B), which not only significantly outperformed competing unsupervised methods but also matched the reported performance of H&E-based supervised segmentation tools. These outcomes are further verified in another cancer study (Figures S9B and S9C).
Figure 6.

Annotation of major structures in TMEs by SCGP
(A) Coarse partitioning identified tumor/stroma regions in TMEs.
(B) ARIs and F1 scores were calculated between unsupervised annotations and manual tumor/stroma annotations. SCGP achieved significantly better performances than other methods (Wilcoxon signed-rank test).
(C) Fine-grained partitions detected additional tissue structures in TMEs.
(D) Heatmaps show distinct cell-type enrichment and protein expression profiles of different tissue structures identified by SCGP.
See also Figure S9.
We also performed more granular partitioning of TMEs. By deploying our SCGP-Extension pipeline, we acquired consistent annotations of TMEs at a higher granularity (Figure 6C). In addition to the tumor/stroma separation, several novel partitions with distinct biomarker profiles and cell-type compositions were identified (Figure 6D). For example, an immune-rich partition was isolated from stroma regions, which exhibited much denser immune cell populations including B cells, T cells, and macrophages. Similarly, a unique CD15/granulocyte-rich partition was detected in the tumor regions of multiple samples, which showed distinct morphology and biomarker enrichment. Such results highlight the power of tissue structure annotations, from which distinct spatial organization of different cell types can be discovered and examined.
Discussion
In this work, we present SCGP, an unsupervised annotation tool for spatial transcriptomics and proteomics measurements. SCGP embeds spatial information and molecular features into graph representations and performs Leiden graph community detection to identify partitions corresponding to anatomical and functional structures. The reference-query extension pipeline, SCGP-Extension, robustly generalizes existing tissue structures to unseen samples and recognizes unique disease states and anatomical structures. Our experiments demonstrate the power of SCGP and SCGP-Extension in identifying tissue structures in various data cohorts and show how tissue structures assist downstream biomedical research and discoveries.
We compared SCGP against other unsupervised annotation tools that similarly utilize both spatial information and molecular profiling output. Methods such as CN, UTAG, and Spatial-LDA define different concepts of neighborhoods, usually based on distance thresholding, and annotate them through unsupervised clustering. These methods tend to be inflexible to structures of different spatial scales. Another class of methods also uses graph representations of tissue samples to model cellular organization. Computational tools including latent variable modeling and graph neural networks are applied on these graphs to annotate nodes (cells) according to their tissue context. However, spatially disconnected samples are difficult to annotate with these tools. SCGP and SCGP-Extension follow a graph representation approach and enhance it with nearest-neighbor feature edges to weave multiple samples into one cohesive graph.
Moreover, SCGP-Extension is the first method that addresses the long-standing need of generalizing structures to previously unseen samples. SCGP-Extension resembles supervised learning tools in that models apply knowledge learned from the reference samples (i.e., training data) to unseen query samples (i.e., test data). Simultaneously, SCGP-Extension can isolate previously unseen tissue structures during inference. In practice, we have demonstrated that SCGP-Extension can help overcome common challenges including experimental artifacts, batch effects, and different disease conditions. More importantly, it can help uncover unique disease states and anatomical structures.
SCGP and SCGP-Extension offer outstanding running time and memory consumption performances compared to many unsupervised annotation tools, some of which require extensive parameter estimations or optimizations (STAR Methods; Table S2). In practice, this advantage allows for applications to much larger datasets and facilitates model parameter tuning to obtain optimal partitions.
SCGP and SCGP-Extension are validated on both simulated and real datasets to model various spatial proteomics and transcriptomics measurements effectively, capturing tissue structures of diverse shapes and sizes in systems with varying levels of complexities. Minimal parameter adjustments (STAR Methods) are required. The two major parameters for SCGP are the density of feature edges and the granularity of partitioning (Figures S3E–S3G). One additional parameter, the extent of extension, is introduced in SCGP-Extension to control the balance between generalizing existing partitions and exploring new partitions. In examples involving different disease conditions, certain tissue structures may experience changes in their expression profiles (e.g., fibrosis, immune infiltration), and the decision of whether to integrate or separate these structures will depend on the specific downstream applications.
Looking ahead, SCGP opens new opportunities for analyzing and understanding spatially resolved molecular profiling data. By inserting a middle layer between cell-level annotations and sample-level characteristics, it facilitates analysis tailored to specific structures of interest at scale.
Limitations of the study
We acknowledge the presence of certain caveats in SCGP and SCGP-Extension, which hinder their applications in specific scenarios. One major shortcoming is that SCGP appears to be less suitable for identifying thinly layered structures. Due to the design of the hybrid graph, spatial edges are isotropic for the purpose of community detection. Thin-layer structures exhibiting much denser spatial connections in their normal directions than tangential directions are harder to detect (Figures S1A and S4A). In the DLPFC study, alternative methods (e.g., BayesSpace, SpiceMix) can identify thinner structures under more granular settings, while SCGP recognizes structures in the orthogonal direction. Refining the spatial edges to reflect the anisotropy of tissues would be a direction to improve the performance of SCGP.
Another limitation is that the outputs of SCGP and SCGP-Extension are hard class assignments. This lack of flexibility may limit applications in scenarios where probabilistic or mixture outputs are preferred. One way to accommodate this limitation is to collect ensembled outputs from running the algorithm using different random seeds.
Case studies presented in this work used solely biomarker expression to define tissue structures. This can be further improved by integrating multi-modal data. For instance, inclusion of morphology information (e.g., through embeddings of H&E staining) would greatly enrich the feature space and allow SCGP to account for cellular- and tissue-level morphological differences.
In addition, the concept of reference-based annotation can be further extrapolated to prior-based annotation. Specifically, instead of fully unsupervised partitioning, incorporating prior knowledge of the expected tissue structures (e.g., signature biomarkers, sizes, and shapes) could potentially yield better-aligned results and will be a direction for future improvements. Lastly, we acknowledge that evaluations in this study were not exhaustive of all possible methods and datasets. We selected representative methods spanning diverse approaches to establish the benchmark for SCGP. Many recent methods54,55,56,57 were not included due to time constraints despite their competitive performances. Moreover, quantitative performances (i.e., ARI, F1 scores) were calculated based on manual annotations, which are subjective and may contain intrinsic biases. To mitigate dataset-specific biases, we performed comparisons across diverse datasets from different institutions, though realistic performances will still vary based on the specific tissue/disease environments.
STAR★Methods
Key resources table
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Alexandro E. Trevino (alex@enablemedicine.com).
Materials availability
This study did not generate new unique materials or reagents.
Data and code availability
-
•
External spatial proteomics and transcriptomics datasets used in this study (Human DLPFC Visium dataset, adult mouse brain MERFISH dataset, Lung IMC dataset, UPMC head-and-neck cancer CODEX dataset) are publicly available as of the date of publication. Processed CODEX datasets (DKD kidney CODEX dataset, UCSF derm CODEX dataset, TR kidney CODEX dataset, Stanford pancreatic cancer CODEX dataset) have been deposited at zenodo. All accession numbers are listed in the key resources table. Raw data and images will be accessible through links posted at: https://gitlab.com/enable-medicine-public/scgp. They will also be available from lead contact (A.E.T.) upon request.
-
•
SCGP was implemented based on Annotated data61 (https://anndata.readthedocs.io/en/latest/) and emObject62 (https://docs.enablemedicine.com/emobject/). All source codes are publicly available at https://gitlab.com/enable-medicine-public/scgp. An archival DOI is listed in the key resources table.
-
•
Any additional information required to re-analyze the data reported in this paper will be available from lead contact (A.E.T.) upon request.
Experimental model and study participant details
Eight spatial proteomics and transcriptomics datasets collected from diverse tissue types were examined in this work. An overview of the statistics and primary phenotypes of these datasets can be found in Table S1. For external datasets, please refer to the source publications for full details. For datasets introduced in this study:
TR Kidney63: Kidney samples were obtained from patients who underwent allograft nephrectomy at Stanford University Medical Center under Institutional Review Board-approved protocols. One TMA was constructed using 2 mm cores of cortical tissue, then stained and acquired on the PhenoCycler Fusion using a 51-plex biomarker panel, from which 5 normal and 17 transplant rejection kidney regions were used in this study. Further clinical or patient demographic data related to these samples was not selected or considered in this study.
Stanford-PC: De-identified tissue samples were obtained from the surgical pathology archives of Stanford Medical Center under Institutional Review Board-approved protocols. Tumor cores (1 mm diameter) were sampled and profiled with CODEX using immune-cell focused biomarker panels. One coverslip containing 148 cores was analyzed in this study. Clinical or patient demographic data related to these samples was not selected or considered in this study. All cores were manually annotated for tumor regions based on epithelial protein markers.
Method details
Datasets
This section briefly describes phenotypes and properties of each dataset analyzed in this study.
DKD Kidney40: Kidney samples were obtained from patients with diabetes and healthy kidneys (DM, five individuals), DKD classes IIA, and IIB (two individuals per class), IIA-B intermediate (two individuals), and III (one individual). Twenty-three cores, each 0.5 mm in diameter, were sampled from twelve tissue blocks and assembled into a tissue microarray (TMA). The TMA block was further sectioned into 5 μm slices. One TMA section was imaged and characterized using the CO-Detection by indexing (CODEX) platform. After excluding medulla samples and quality control, a total of 17 cortical section samples across various DKD classes were acquired. Each sample was imaged for 21 protein biomarkers (see columns in Figure S1B).
DLPFC46: Spatial gene expression in human postmortem dorsolateral prefrontal cortex tissue sections was profiled using two pairs of “spatial replicates” from three independent neurotypical adult donors on the Visium platform, each pair comprising two directly adjacent, 10 μm serial tissue sections, with the second pair located 300 μm posterior to the first. In total 12 samples are collected and examined. We downloaded the filtered count matrices for all 12 samples from the spatialLIBD project.64 In the independent partitioning experiment (Figure 3), we filtered the count matrices to exclude spike-in genes, mitochondrial genes, and genes that have nonzero expression in fewer than three spots. Expression matrices were normalized to have the same total counts per spot (median of all pre-normalize spots), log-transformed and reduced to the top 50 principal components. In the joint partitioning experiment (Figure S2), we followed the preprocessing steps outlined in SpiceMix24: Genes having nonzero expression in less than 10% of spots were removed. Expression matrices were normalized to have total counts of 10,000 per spot and log-transformed. We further reduced the expression to the top principal components.
Adult Mouse Brain (ABCA-1)47: This dataset comprises in total 147 coronal sections of a single adult mouse brain, profiled with a 1122 gene panel using MERFISH. Cells were annotated for cell types and mapped to the Allen mouse common coordinate framework.65 We downloaded preprocessed biomarker expression matrices, cell types, and CCF coordinates from the source publication. Two sections (ABCA-1.071, ABCA-1.079) were used to assess SCGP and SCGP-Extension. In the experiments, the 300 most variable genes of the reference section (ABCA-1.079) were used as features.
TR Kidney63: Kidney samples were obtained from patients who underwent allograft nephrectomy. Briefly, a TMA was constructed using 2 mm cores of cortical tissue. The TMA comprised 7 samples of normal, peritumoral renal cortex (from patients undergoing native nephrectomy for tumor removal), and 43 samples of cortex from patients undergoing allograft nephrectomies. After sectioning of the tissue block, only 41 cores from allograft nephrectomies remained. An FFPE embedded tissue microarray core of human kidney samples were stained and acquired on the PhenoCycler Fusion using a 51-plex biomarker panel (Figure S3C) by Enable Medicine.
Lung IMC15,66: Lung samples were acquired from three healthy donor lung specimens. In total 26 samples were imaged using IMC with 28 biomarkers (see columns in Figure S4D). Tissue samples were collected with a particular focus on airways extending from proximal bronchi and succeeding divisions to terminal and respiratory bronchioles. Each image was manually annotated with organ-specific microanatomical domains: airways, connective tissue, submucosal glands, vessels, cartilages, and alveolar space. These manually annotated domains were used as labels for unsupervised annotations. Additionally, cells in these samples were phenotyped into seven broad clusters of cell identity: CD8 T cells, macrophages, mast cells, smooth muscle cells, endothelial cells, epithelial cells, and connective tissue cells. Cell type information was used in CN and Spatial-LDA, other methods only used biomarker expression data. We downloaded preprocessed biomarker expression matrices and domain/cell type annotations from the source publication.59
UCSF Derm60: Tissue samples were acquired as 2–4 mm punch or shave skin biopsy specimens from healthy control and atopic dermatitis patients. In total, 44 skin samples were obtained and stained in 4 experiments with varying biomarker panels. See Table S1 for a detailed breakdown of experiments and patient skin disorders. 35 shared protein biomarkers were used in the unsupervised annotation analysis with UTAG and SCGP.
Stanford-PC and UPMC-HNC16: These two datasets were derived from studies of pancreatic cancer and head-and-neck cancer. Tumor cores (1 mm diameter) were sampled and profiled with CODEX using immune-cell focused biomarker panels. Figure 6D shows some of the representative protein biomarkers in the panel. We adapted one coverslip from each study to assess SCGP and SCGP-Extension. Stanford-PC contained 148 cores in total, which were further divided into two subsets: a higher-quality reference set (63 cores) and a lower-quality query set (85 cores). UPMC-HNC contained 36 cores. Clinical data related to these samples were not used in this study. All cores were manually annotated for tumor regions by an expert, and other foreground tissues were treated as non-tumor/stroma regions.
Preprocessing
DLPFC, Lung IMC, and ABCA-1 datasets were downloaded and normalized as specified above.
For the CODEX datasets, we followed the preprocessing pipeline established in the prior work by Wu et al.16 Briefly, a neural network-based cell segmentation tool DeepCell20 was applied to DAPI images to identify nuclei, which were further dilated to obtain whole-cell segmentation.
Next, the biomarker expression for biomarker in cell was computed following the strategy below67:
-
(1)
For channel , mean pixel intensity within the cell segmentation mask of cell was calculated and denoted as . The set of expression values for all cells in the same sample was denoted as .
-
(2)
Normalized expression value for channel was calculated using quantile normalization and arcsinh transformation:
in which represents the 20-th quantile of and is the inverse hyperbolic sine function. The set of all normalized expression values was denoted as .
-
(1)
Z score of normalized expression value was calculated:
It should be noted that SCGP does not require any cell clustering or classification inputs, it infers partitions using biomarker expression of cells.
Spatial cellular graph partitioning (SCGP)
SCGP is an unsupervised annotation tool that recognizes tissue structures by partitioning graphs constructed based on the spatial organization of cells (or other units) in the subject tissue sample(s). The SCGP pipeline comprises the following steps.
Nodes
Nodes represent small spatial regions in the tissue, and are indivisible units throughout the partitioning process. In this study, we employed three different strategies for defining nodes.
-
(1)
Cells: Nodes are defined based on individual cells, which are identified via the cell segmentation preprocessing step specified above. Biomarker expression values are calculated and normalized accordingly, and these values are set as the node features.
-
(2)
(Visium) Spots: Nodes are defined based on the barcoded spots used in the Visium platform, each measuring gene expression in a circular area 55 μm in diameter. The normalized gene expression values or top principal components are set as node features.
-
(3)
Patches: In the patch-based SCGP experiment (Figure S10), nodes are defined based on small square patches with 12 μm side lengths on the mIF images, sampled using a sliding window mechanism (stride equals patch side length). Only patches within the spatial range of tissue are retained (>85% overlap), and the average fluorescence intensities for each biomarker channel are used as node features.
Spatial edges
Spatial edges are constructed between spatially adjacent nodes to embed the spatial closeness relationship into the graph.
-
(1)
Cells: A Delaunay triangulation is conducted on centroid coordinates of all cells from the same sample. Node pairs that share edges in the triangulation output are connected, excluding any edge exceeding 35 μm in length.
-
(2)
(Visium) Spots: Nodes (i.e., spots) are spatially arranged in a close-packing manner. Each node is connected to its six closest neighboring nodes.
-
(3)
Patches: Nodes (i.e., patches) are spatially arranged in a regular 2D grid. Each node is connected to its four immediately adjacent nodes.
Feature edges
Feature edges are constructed between nodes with similar biomarker expression profiles. For node , its nearest neighboring nodes in the expression space are identified based on Euclidean distances between node features (e.g., z-scored protein expression values, principal components of the gene expression). We used the nearest neighbor descent68 approximate queries implemented in PyNNDescent for better computational efficiency.
In practice, is a hyperparameter that controls the balance between spatial coherence and expression consistency within partitions. We typically set it to an integer between 1 and 4 so that feature edges account for about 25%–50% of all edges (Figures S3E and S3F). In general, less feature edges will generate spatially smoother partitions, though minor changes in do not significantly change partition outcomes in our experiments. However, a larger (>5) will lead to spatially fragmented partitions, which resemble the results of direct Leiden clustering on feature vectors. This design choice of sparse feature edges differentiates SCGP from other Leiden-based unsupervised annotation tools.15,27
Graph community detection
Nodes, spatial edges, and feature edges define the spatial cellular graph input for SCGP. We used the Leiden algorithm39 to detect graph communities.
We adapted the python implementation in leidenalg, and we used the Constant Potts Model69 as the quality function for community detection (leidenalg.CPMVertexPartition). Additional arguments include.
-
(1)
Edge weights: For each edge, its weight is defined as the inverse of Euclidean distance between the node features of the two nodes it connects. We further normalized all edge weights by their median value.
-
(2)
Resolution parameter (γ): γ controls the density of the output communities.
Note that the resolution parameter γ is the second major hyperparameter of SCGP, regulating the granularity of the output partitions. We empirically tuned γ to generate the desired amount of partitions (e.g., 2 for TME samples, 10+ for whole brain samples). This process was informed by the understanding and expectations of the subject tissues (Figure S3G).
Note that variances observed among nodes may originate from their underlying tissue structures or sample-specific biases (i.e., batch effect). Given the unsupervised nature of SCGP, scenarios in which biases overwhelm the biological differences between structures can lead to a situation where increasing γ will cause structures from different samples to disintegrate and thus be assigned to distinct partitions. This caveat is addressed in practice by curating a well-integrated reference set for SCGP followed by applying SCGP-Extension.
Post-processing
Upon acquiring the initial partition outcomes, optional post-processing steps can be executed to refine the results.
-
(1)
Size Filtering: Partitions accounting for less than 0.2% of all nodes are discarded.
-
(2)
Spatial smoothing: For any node who holds a different partition assignment from its spatial neighbors, we reassigned it to the partition held by the majority of its spatial neighbors (>50%) if applicable.
SCGP-extension
SCGP-Extension extends a given set of reference partitions to unseen query samples. Query samples are processed in the same manner as specified in the SCGP pipeline for graph construction. Reference partitions are defined on the reference nodes, which should be in the same format as nodes in the query samples. These partitions are usually generated through a primary SCGP run on the reference samples. It is worth noting that technically any form of discrete labels can be employed as reference partitions. Figures S4B and S4C demonstrates experiments that extend ground truth labels or noisy labels defined by arbitrary annotation tools.
Pseudo-nodes
SCGP-Extension introduces pseudo-nodes as guidance for unseen query sample partitioning. Based on the reference nodes and their reference partitions, pseudo-nodes can be created for each partition via two strategies.
-
(1)
Selection: A median node feature vector is calculated using all reference nodes affiliated with the partition. The Euclidean distance between each node’s feature vector and the median vector is calculated. Pseudo-nodes are sampled from all reference nodes based on their distances to the median vector.
-
(2)
Random sampling: The mean and covariance of the node feature vector are calculated using all reference nodes affiliated with the partition. Pseudo-nodes are generated by sampling multivariate normal random variables based on the mean and covariance.
In practice, we typically generate 100 pseudo-nodes for each partition, but the size can be adjusted based on the number of nodes in the reference and query samples. The two strategies tend to yield comparable results, we empirically prefer the selection strategy.
Prior to their integration into the query graph, dense feature edges are added to the pseudo-nodes – 20 nearest neighbors for each pseudo-node if in total 100 nodes per partition are used. This is to guarantee well-structured communities within the pseudo-nodes.
Reference-query edges
Pseudo-nodes are integrated into the query graph via additional reference-query edges. For each node in the query graph, its nearest neighbors in the pseudo-nodes based on Euclidean distances between node features are identified. An additional ratio parameter () is included to regulate the strengths of reference-query edges. Given a total of nodes in the query graph, the initial nearest neighbor search will yield reference-query edges. These edges are sorted by edge weights (i.e., Euclidean distances), with only the top edges with the highest weights or closest distances retained.
The completed query graph comprises spatial edges, intra-sample feature edges (-nearest neighbors within the query nodes), and reference-query feature edges (-downsampled -nearest neighbors between query nodes and pseudo-nodes). and are two additional hyperparameters in SCGP-Extension, which control the degree of matching between query nodes and reference partitions. In practice we adjusted , and so that the total number of feature edges matches the number of spatial edges, with a comparable number of intra-sample feature edges and reference-query feature edges. is typically set to a value between 0.1 and 0.5 based on expectations of matching level between query and reference samples. By default we set , , and .
Graph community detection with fixed membership assignment
The same community detection strategy is used to identify partitions in the query sample. Additional arguments including edge weights and resolution parameter are specified in the same manner as specified in the SCGP pipeline.
Notably, as pseudo-nodes are created for reference partitions, their assignments are predetermined and fixed throughout the partition optimization process70 using the is_membership_fixed argument of the leidenalg.Optimiser().optimise_partition method. As a result, query nodes that are similar to any of the existing reference partitions will be assigned to the corresponding group, while nodes distinct from the reference will be assigned to novel partitions.
Predictive modeling for partition extension
An alternative approach to extend existing partitions to unseen samples is through constructing a predictive model and applying it for inference. This is demonstrated in two experiments in Figures 4A and S4B. In these experiments, we used manual annotations of reference samples to train gradient boosted tree classifiers, which were subsequently applied to unseen query samples.
The training dataset was constructed using the reference nodes and used manual annotations as labels. The inputs contained reference node features, as well as 1-hop aggregated node features, which were computed by averaging features from the center nodes and their immediate spatial neighbors (defined by distance thresholding). This augmentation was inspired by the UTAG and SpaGCN methods and allowed the model to have larger fields of view. The test inputs for the query nodes were formulated similarly, and the trained models were employed to infer their cluster/partition assignments.
We evaluated a range of common machine learning methods including logistic regression, linear SVR, k-nearest neighbor classifier, and random forest. Gradient boosted trees implemented via XGBoost71 yielded the best performance.
Evaluation metrics
On datasets with ground truth annotations, we assessed the performances of SCGP, SCGP-Extension and other unsupervised annotation tools using the following metrics.
-
(1)
Adjusted Rand Index72 (ARI) and Rand Index (RI): ARI and RI are measures that evaluate the similarity between two data clusterings, in which ARI also takes into account the probabilities of random agreement between two clusterings. ARI ranges from −1 to 1, where 1 indicates perfect agreement and 0 indicates a random agreement. We adapted the implementation in scikit-learn58: sklearn.metrics.adjusted_rand_score. RI ranges from 0 to 1, where 1 indicates perfect agreement. RI is only used in the Lung IMC dataset to reproduce metrics reported in its source publication. We adapted the implementation in scikit-learn: sklearn.metrics.rand_score.
-
(2)
V Measure: V Measure is the harmonic mean of homogeneity and completeness, two intrinsic metrics used to evaluate the quality of clustering. Homogeneity measures if all points within one unsupervised cluster are members of a single label class, while completeness verifies that all members of a given label class are assigned to the same cluster. All three metrics range from 0 to 1, where higher values indicate better clustering performance. We adapted the implementation in scikit-learn: sklearn.metrics.homogeneity_completeness_v_measure. Homogeneity score is used in the Lung IMC dataset to reproduce metrics reported in its source publication.
-
(3)F1 Score: F1 is an accuracy measure calculated as the harmonic mean of precision and recall. For each label compartment, the F1 score is calculated through the following process:
-
○Labels: 1s are assigned to all nodes affiliated with the target label compartment, 0s are assigned to the rest.
-
○Predictions: For a given partition, predictions are calculated using the indicator function of whether a node is assigned to that specific partition. A series of predictions will be derived for all partitions identified by the unsupervised annotation tool.
-
○Metrics: Multiple F1s are calculated based on the labels and the series of predictions. The highest F1 score corresponds to the partition that has the best match with the target label compartment, and this score is taken as the final score.
-
○
To compare performances of different methods, we used the following statistical test.
-
(1)
Wilcoxon signed-rank test: this test is employed to compare paired metrics (ARI, F1) calculated between ground truths and partitions defined by unsupervised annotation methods, assessing the significance of differences between groups of metrics.
In the analysis of disease states, we used the following statistical tests.
-
(1)
Two-sample t-test: this test is employed to determine if there is a significant difference between the means of two independent groups. It is used to compare characteristics of samples with different disease conditions.
-
(2)
Jonckheere-Terpstra test: this test is employed to assess ordered differences among multiple independent groups. It is used to determine if certain characteristics of tissue structures have ordered changes with respect to disease stages.
Running time and memory usage
We profiled running time and peak memory usage for all unsupervised annotation methods using two major tasks.
-
(1)
Joint clustering/partitioning of 17 samples from the DKD kidney dataset, containing 137,654 cells;
-
(2)
Joint clustering/partitioning of 4 samples from the same specimen (Br8100) from the DLPFC Visium dataset, containing 14,364 spots.
The profiling is performed on an amazon cloud service ec2 instance (r6i.16xlarge or g4dn.16xlarge if GPU is required). See Table S2 for full results.
CN, UTAG, SCGP, and SCGP-Extension had the shortest running times. KMeans-based methods including CN required minimum memory usage, while memory consumption of Leiden-based methods relied on the number of nearest neighbors in the graph construction, in which SCGP and SCGP-Extension were less demanding.
Hyperparameter optimizations
For unsupervised annotation methods benchmarked in this work, we searched the hyperparameter space for each method and chose the best results. Below we briefly described the pipelines for these methods, please refer to their source publications and code bases for full details. Note that parameters that are optimized during the hyperparameter search are marked as “searched”.
-
(1)KMeans.
-
○Input: Biomarker expression vectors of cells/spots;
-
○Clustering: KMeans algorithm (, searched), implemented by scikit-learn.58
-
○
-
(2)Leiden.39
-
○Input: -nearest neighbor graph (, searched) constructed using the biomarker expression vectors of cells/spots;
-
○Clustering: Leiden algorithm, implemented by leidenalg (https://github.com/vtraag/leidenalg). Resolution parameter is searched.
-
○
-
(3)Cellular Neighborhood (CN).13,26
-
○Input: Cell types are first identified through leiden clustering. For each cell, a composition (frequency of cell types) vector is calculated based on a window of 10–20 nearest neighboring cells (searched) as measured by Euclidean distance between X/Y coordinates.
-
○Clustering: KMeans algorithm (, searched), implemented by scikit-learn.
-
○
-
(4)UTAG.15
-
○Developed and validated on spatial proteomics (mIF) data: IMC, CyCIF.
-
○Input: For each cell/spot, an average biomarker expression vector is calculated over all neighboring cells/spots and the center cell/spot within a window surrounding the center cell/spot thresholded by Euclidean distance (10–20 μm, searched), referred to as the spatially aggregated feature matrix. -nearest neighbor graph (, searched) is then constructed using the aggregated expression vectors.
-
○Clustering: Leiden algorithm, implemented by leidenalg. Resolution parameter is searched.
-
○Results on the LungIMC dataset are directly downloaded from https://zenodo.org/records/6376767.
-
○
-
(5)Spatial-LDA.28
-
○Developed and validated on spatial proteomics data: CODEX, MIBI-TOF.
-
○Input: Cell types are first identified through leiden clustering. For each cell, its local environment is encoded as the count of cell types (bag-of-cell) within a window surrounding the center cell thresholded by Euclidean distance (10–30 μm, searched). Spatial prior (i.e., adjacency between cells) is first constructed by computing the Voronoi partitioning of cell positions, in which pairs of cells that share a facet in the Voronoi partitioning are connected, then reduced to a minimum spanning tree based on the edges.
-
○Clustering: Latent Dirichlet Allocation with spatial prior, implemented in https://github.com/calico/spatial_lda. Major parameters (i.e., difference penalty) are searched.
-
○
-
(6)BayesSpace.29
-
○Developed and validated on spatial transcriptomics data: Visium.
-
○Input: Top principal components of the log transformed and normalized gene expression counts.
-
○Clustering: Spots are modeled using a fully Bayesian model with a Markov random field prior, specified by the Potts model. Model parameters are estimated using a Markov chain Monte Carlo method. We adapted codes (in R) from https://edward130603.github.io/BayesSpace/articles/BayesSpace.html. Major parameters (i.e., number of clusters) are searched.
-
○Only used on the DLPFC data.
-
○
-
(7)SpaGCN.32
-
○Developed and validated on spatial transcriptomics data: Visium, SlideSeqV2, MERFISH, STARmap.
-
○Input: Biomarker expression vectors of cells/spots are reduced to their top 20 principal components and then constructed into a weighted undirected graph, in which edges are weighted by Euclidean distances (histology information is not included in this study).
-
○Clustering: A one-layer graph convolutional network on the input graph generates initial embeddings for cells/spots, which are clustered using the Louvain algorithm (resolution searched). Network parameters and cluster centroids are optimized by minimizing a soft assignment-based loss function using stochastic gradient descent with momentum until convergence. We adapted codes from https://github.com/jianhuupenn/SpaGCN.
-
○
-
(8)SpiceMix.24
-
○Developed and validated on spatial transcriptomics data: Visium, seqFISH+, STARmap.
-
○Input: a Hidden Markov Random Field model is constructed based on the graphical model, which treats cells/spots as nodes and connects spatially adjacent pairs (through Delaunay triangulation) with edges.
-
○Clustering: Cells/spots are first clustered using the Louvain algorithm to initialize estimates of hidden states and model parameters, which are further iteratively optimized via coordinate ascent. A total of 6–15 metagenes (searched) are used. We adapted codes from https://github.com/ma-compbio/SpiceMix.
-
○
Quantification and statistical analysis
Details of statistical tests are described in the figure legends and in STAR Methods (see the subsection “evaluation metrics”).
Acknowledgments
J.Z. is supported by NSF CAREER 1942926 and a Chan-Zuckerberg Biohub Investigator Award. We would like to extend our sincere gratitude to My Thoi, Erica Bauer, Haley Hauser, Ryan Preska, Blaize D'Angio, David Han, and Nathan Le from the Enable Medicine lab team for their invaluable contributions to this research.
Author contributions
Conceptualization, Z.W. and A.E.T.; methodology, Z.W.; software, Z.W., E.A.G.B., and B.C.; validation, A.K., E.W., and M.K.R.; investigation, Z.W.; resources, N.A.B., V.C., R.J.C., J.B.C., and M.A.; data curation, A.K. and M.M.; writing – original draft, Z.W. and A.E.T.; writing – review & editing, Z.W. and A.E.T.; supervision, J.Z., A.T.M., and A.E.T.
Declaration of interests
Z.W., A.K., M.M., E.A.G.B., B.C., M.K.R., A.T.M., and A.E.T. are affiliated with Enable Medicine as employees and/or shareholders. J.Z. is a member of Enable Medicine’s scientific advisory board.
Declaration of generative AI and AI-assisted technologies in the writing process
During the preparation of this work, the authors used GPT-4 in order to polish the writing of the manuscript. After using this tool/service, the authors reviewed and edited the content as needed and took full responsibility for the content of the publication.
Published: August 9, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2024.100838.
Contributor Information
Zhenqin Wu, Email: zhenqin@enablemedicine.com.
James Zou, Email: jamesz@stanford.edu.
Aaron T. Mayer, Email: aaron@enablemedicine.com.
Alexandro E. Trevino, Email: alex@enablemedicine.com.
Supplemental information
References
- 1.Nelson C.M., Bissell M.J. Of extracellular matrix, scaffolds, and signaling: tissue architecture regulates development, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol. 2006;22:287–309. doi: 10.1146/annurev.cellbio.22.010305.104315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rao A., Barkley D., França G.S., Yanai I. Exploring tissue architecture using spatial transcriptomics. Nature. 2021;596:211–220. doi: 10.1038/s41586-021-03634-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen K.H., Boettiger A.N., Moffitt J.R., Wang S., Zhuang X. RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science. 2015;348 doi: 10.1126/science.aaa6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eng C.-H.L., Lawson M., Zhu Q., Dries R., Koulena N., Takei Y., Yun J., Cronin C., Karp C., Yuan G.-C., Cai L. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature. 2019;568:235–239. doi: 10.1038/s41586-019-1049-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rodriques S.G., Stickels R.R., Goeva A., Martin C.A., Murray E., Vanderburg C.R., Welch J., Chen L.M., Chen F., Macosko E.Z. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science. 2019;363:1463–1467. doi: 10.1126/science.aaw1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang X., Allen W.E., Wright M.A., Sylwestrak E.L., Samusik N., Vesuna S., Evans K., Liu C., Ramakrishnan C., Liu J., et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science. 2018;361 doi: 10.1126/science.aat5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ståhl P.L., Salmén F., Vickovic S., Lundmark A., Navarro J.F., Magnusson J., Giacomello S., Asp M., Westholm J.O., Huss M., et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 2016;353:78–82. doi: 10.1126/science.aaf2403. [DOI] [PubMed] [Google Scholar]
- 8.Giesen C., Wang H.A.O., Schapiro D., Zivanovic N., Jacobs A., Hattendorf B., Schüffler P.J., Grolimund D., Buhmann J.M., Brandt S., et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods. 2014;11:417–422. doi: 10.1038/nmeth.2869. [DOI] [PubMed] [Google Scholar]
- 9.Goltsev Y., Samusik N., Kennedy-Darling J., Bhate S., Hale M., Vazquez G., Black S., Nolan G.P. Deep Profiling of Mouse Splenic Architecture with CODEX Multiplexed Imaging. Cell. 2018;174:968–981.e15. doi: 10.1016/j.cell.2018.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Angelo M., Bendall S.C., Finck R., Hale M.B., Hitzman C., Borowsky A.D., Levenson R.M., Lowe J.B., Liu S.D., Zhao S., et al. Multiplexed ion beam imaging of human breast tumors. Nat. Med. 2014;20:436–442. doi: 10.1038/nm.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin J.-R., Fallahi-Sichani M., Chen J.-Y., Sorger P.K. Cyclic Immunofluorescence (CycIF), A Highly Multiplexed Method for Single-cell Imaging. Curr. Protoc. Chem. Biol. 2016;8:251–264. doi: 10.1002/cpch.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fischer D.S., Schaar A.C., Theis F.J. Modeling intercellular communication in tissues using spatial graphs of cells. Nat. Biotechnol. 2023;41:332–336. doi: 10.1038/s41587-022-01467-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schürch C.M., Bhate S.S., Barlow G.L., Phillips D.J., Noti L., Zlobec I., Chu P., Black S., Demeter J., McIlwain D.R., et al. Coordinated Cellular Neighborhoods Orchestrate Antitumoral Immunity at the Colorectal Cancer Invasive Front. Cell. 2020;183:838. doi: 10.1016/j.cell.2020.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jackson H.W., Fischer J.R., Zanotelli V.R.T., Ali H.R., Mechera R., Soysal S.D., Moch H., Muenst S., Varga Z., Weber W.P., Bodenmiller B. The single-cell pathology landscape of breast cancer. Nature. 2020;578:615–620. doi: 10.1038/s41586-019-1876-x. [DOI] [PubMed] [Google Scholar]
- 15.Kim J., Rustam S., Mosquera J.M., Randell S.H., Shaykhiev R., Rendeiro A.F., Elemento O. Unsupervised discovery of tissue architecture in multiplexed imaging. Nat. Methods. 2022;19:1653–1661. doi: 10.1038/s41592-022-01657-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu Z., Trevino A.E., Wu E., Swanson K., Kim H.J., D’Angio H.B., Preska R., Charville G.W., Dalerba P.D., Egloff A.M., et al. Graph deep learning for the characterization of tumour microenvironments from spatial protein profiles in tissue specimens. Nat. Biomed. Eng. 2022;6:1435–1448. doi: 10.1038/s41551-022-00951-w. [DOI] [PubMed] [Google Scholar]
- 17.Fischer D.S., Ali M., Richter S., Ertürk A., Theis F. Graph neural networks learn emergent tissue properties from spatial molecular profiles. bioRxiv. 2022 doi: 10.1101/2022.12.08.519537. Preprint at. [DOI] [Google Scholar]
- 18.Palla G., Spitzer H., Klein M., Fischer D., Schaar A.C., Kuemmerle L.B., Rybakov S., Ibarra I.L., Holmberg O., Virshup I., et al. Squidpy: a scalable framework for spatial omics analysis. Nat. Methods. 2022;19:171–178. doi: 10.1038/s41592-021-01358-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greenwald N.F., Miller G., Moen E., Kong A., Kagel A., Dougherty T., Fullaway C.C., McIntosh B.J., Leow K.X., Schwartz M.S., et al. Whole-cell segmentation of tissue images with human-level performance using large-scale data annotation and deep learning. Nat. Biotechnol. 2022;40:555–565. doi: 10.1038/s41587-021-01094-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Becht E., McInnes L., Healy J., Dutertre C.-A., Kwok I.W.H., Ng L.G., Ginhoux F., Newell E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2018;37:38–44. doi: 10.1038/nbt.4314. [DOI] [PubMed] [Google Scholar]
- 22.Kiselev V.Y., Andrews T.S., Hemberg M. Challenges in unsupervised clustering of single-cell RNA-seq data. Nat. Rev. Genet. 2019;20:273–282. doi: 10.1038/s41576-018-0088-9. [DOI] [PubMed] [Google Scholar]
- 23.Brbić M., Cao K., Hickey J.W., Tan Y., Snyder M.P., Nolan G.P., Leskovec J. Annotation of spatially resolved single-cell data with STELLAR. Nat. Methods. 2022;19:1411–1418. doi: 10.1038/s41592-022-01651-8. [DOI] [PubMed] [Google Scholar]
- 24.Chidester B., Zhou T., Alam S., Ma J. SPICEMIX enables integrative single-cell spatial modeling of cell identity. Nat. Genet. 2023;55:78–88. doi: 10.1038/s41588-022-01256-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cang Z., Zhao Y., Almet A.A., Stabell A., Ramos R., Plikus M.V., Atwood S.X., Nie Q. Screening cell–cell communication in spatial transcriptomics via collective optimal transport. Nat. Methods. 2023;20:218–228. doi: 10.1038/s41592-022-01728-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhate S.S., Barlow G.L., Schürch C.M., Nolan G.P. Tissue schematics map the specialization of immune tissue motifs and their appropriation by tumors. Cell. 2022;13:109–130.e6. doi: 10.1016/j.cels.2021.09.012. [DOI] [PubMed] [Google Scholar]
- 27.Chen A., Liao S., Cheng M., Ma K., Wu L., Lai Y., Qiu X., Yang J., Xu J., Hao S., et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell. 2022;185:1777–1792.e21. doi: 10.1016/j.cell.2022.04.003. [DOI] [PubMed] [Google Scholar]
- 28.Chen Z., Soifer I., Hilton H., Keren L., Jojic V. Modeling Multiplexed Images with Reveals Novel Tissue Microenvironments. J. Comput. Biol. 2020;27:1204–1218. doi: 10.1089/cmb.2019.0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao E., Stone M.R., Ren X., Guenthoer J., Smythe K.S., Pulliam T., Williams S.R., Uytingco C.R., Taylor S.E.B., Nghiem P., et al. Spatial transcriptomics at subspot resolution with BayesSpace. Nat. Biotechnol. 2021;39:1375–1384. doi: 10.1038/s41587-021-00935-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mani S., Haviv D., Kunes R., Pe’er D. NeurIPS 2022 Workshop on Learning Meaningful Representations of Life. 2023. SPOT: Spatial Optimal Transport for Analyzing Cellular Microenvironments. [Google Scholar]
- 31.Chang Y., Liu J., Ma A., Jiang S., Krull J., Yeo Y.Y., Liu Y., Rodig S.J., Barouch D.H., Fan R., et al. Spatial omics representation and functional tissue module inference using graph Fourier transform. bioRxiv. 2023 doi: 10.1101/2022.12.10.519929. Preprint at. [DOI] [Google Scholar]
- 32.Hu J., Li X., Coleman K., Schroeder A., Ma N., Irwin D.J., Lee E.B., Shinohara R.T., Li M. SpaGCN: Integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network. Nat. Methods. 2021;18:1342–1351. doi: 10.1038/s41592-021-01255-8. [DOI] [PubMed] [Google Scholar]
- 33.Hu Y., Rong J., Xie R., Xu Y., Peng J., Gao L., Tan K. Learning predictive models of tissue cellular neighborhoods from cell phenotypes with graph pooling. bioRxiv. 2022 doi: 10.1101/2022.11.06.515344. Preprint at. [DOI] [Google Scholar]
- 34.Hermsen M., de Bel T., den Boer M., Steenbergen E.J., Kers J., Florquin S., Roelofs J.J.T.H., Stegall M.D., Alexander M.P., Smith B.H., et al. Deep Learning-Based Histopathologic Assessment of Kidney Tissue. J. Am. Soc. Nephrol. 2019;30:1968–1979. doi: 10.1681/ASN.2019020144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.BenTaieb A., Hamarneh G. Medical Image Computing and Computer-Assisted Intervention – MICCAI 2016. 2016. Topology Aware Fully Convolutional Networks for Histology Gland Segmentation; pp. 460–468. [DOI] [Google Scholar]
- 36.Xu J., Luo X., Wang G., Gilmore H., Madabhushi A. A Deep Convolutional Neural Network for segmenting and classifying epithelial and stromal regions in histopathological images. Neurocomputing. 2016;191:214–223. doi: 10.1016/j.neucom.2016.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xie Y., Zhang Z., Sapkota M., Yang L. Spatial Clockwork Recurrent Neural Network for Muscle Perimysium Segmentation. Med. Image Comput. Comput. Assist. Interv. 2016;9901:185–193. doi: 10.1007/978-3-319-46723-8_22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tan W.C.C., Nerurkar S.N., Cai H.Y., Ng H.H.M., Wu D., Wee Y.T.F., Lim J.C.T., Yeong J., Lim T.K.H. Overview of multiplex immunohistochemistry/immunofluorescence techniques in the era of cancer immunotherapy. Cancer Commun. 2020;40:135–153. doi: 10.1002/cac2.12023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Traag V.A., Waltman L., van Eck N.J. From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep. 2019;9:5233. doi: 10.1038/s41598-019-41695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kondo A., McGrady M., Nallapothula D., Ali H., Trevino A.E., Lam A., Preska R., D’Angio H.B., Wu Z., Lopez L., et al. Spatial proteomics of human diabetic kidney disease, from health to class III. bioRxiv. 2023 doi: 10.1101/2023.04.12.534028. Preprint at. [DOI] [PubMed] [Google Scholar]
- 41.Tervaert T.W.C., Mooyaart A.L., Amann K., Cohen A.H., Cook H.T., Drachenberg C.B., Ferrario F., Fogo A.B., Haas M., de Heer E., et al. Pathologic classification of diabetic nephropathy. J. Am. Soc. Nephrol. 2010;21:556–563. doi: 10.1681/ASN.2010010010. [DOI] [PubMed] [Google Scholar]
- 42.Welsh-Bacic D., Lindenmeyer M., Cohen C.D., Draganovici D., Mandelbaum J., Edenhofer I., Ziegler U., Regele H., Wüthrich R.P., Segerer S. Expression of the chemokine receptor CCR6 in human renal inflammation. Nephrol. Dial. Transplant. 2011;26:1211–1220. doi: 10.1093/ndt/gfq560. [DOI] [PubMed] [Google Scholar]
- 43.Bertelli E., Regoli M., Fonzi L., Occhini R., Mannucci S., Ermini L., Toti P. Nestin expression in adult and developing human kidney. J. Histochem. Cytochem. 2007;55:411–421. doi: 10.1369/jhc.6A7058.2007. [DOI] [PubMed] [Google Scholar]
- 44.Bek M.J., Reinhardt H.C., Fischer K.-G., Hirsch J.R., Hupfer C., Dayal E., Pavenstädt H. Up-regulation of early growth response gene-1 via the CXCR3 receptor induces reactive oxygen species and inhibits Na+/K+-ATPase activity in an immortalized human proximal tubule cell line. J. Immunol. 2003;170:931–940. doi: 10.4049/jimmunol.170.2.931. [DOI] [PubMed] [Google Scholar]
- 45.Cao Y., Karsten U., Zerban H., Bannasch P. Expression of MUC1, Thomsen-Friedenreich-related antigens, and cytokeratin 19 in human renal cell carcinomas and tubular clear cell lesions. Virchows Arch. 2000;436:119–126. doi: 10.1007/pl00008210. [DOI] [PubMed] [Google Scholar]
- 46.Maynard K.R., Collado-Torres L., Weber L.M., Uytingco C., Barry B.K., Williams S.R., Catallini J.L., Tran M.N., Besich Z., Tippani M., et al. Transcriptome-scale spatial gene expression in the human dorsolateral prefrontal cortex. Nat. Neurosci. 2021;24:425–436. doi: 10.1038/s41593-020-00787-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang M., Pan X., Jung W., Halpern A.R., Eichhorn S.W., Lei Z., Cohen L., Smith K.A., Tasic B., Yao Z., et al. Molecularly defined and spatially resolved cell atlas of the whole mouse brain. Nature. 2023;624:343–354. doi: 10.1038/s41586-023-06808-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Terpstra T.J. The asymptotic normality and consistency of kendall’s test against trend, when ties are present in one ranking. Indagat. Math. 1952;55:327–333. doi: 10.1016/S1385-7258(52)50043-X. [DOI] [Google Scholar]
- 49.Werfel T., Allam J.-P., Biedermann T., Eyerich K., Gilles S., Guttman-Yassky E., Hoetzenecker W., Knol E., Simon H.-U., Wollenberg A., et al. Cellular and molecular immunologic mechanisms in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2016;138:336–349. doi: 10.1016/j.jaci.2016.06.010. [DOI] [PubMed] [Google Scholar]
- 50.de Visser K.E., Joyce J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell. 2023;41:374–403. doi: 10.1016/j.ccell.2023.02.016. [DOI] [PubMed] [Google Scholar]
- 51.Lerousseau M., Vakalopoulou M., Classe M., Adam J., Carré A., Carré A., Estienne T., Henry T., Deutsch E., Paragios N. Weakly Supervised Multiple Instance Learning Histopathological Tumor Segmentation. Medical Image Computing and Computer Assisted Intervention – MICCAI. 2020;2020:470–479. doi: 10.1007/978-3-030-59722-1_45. [DOI] [Google Scholar]
- 52.Xu Y., Jia Z., Ai Y., Zhang F., Lai M., Chang E.I.-C. 2015 IEEE International Conference on Acoustics, Speech and Signal Processing (ICASSP) 2015. Deep convolutional activation features for large scale Brain Tumor histopathology image classification and segmentation; pp. 947–951. [DOI] [Google Scholar]
- 53.Qaiser T., Tsang Y.-W., Taniyama D., Sakamoto N., Nakane K., Epstein D., Rajpoot N. Fast and accurate tumor segmentation of histology images using persistent homology and deep convolutional features. Med. Image Anal. 2019;55:1–14. doi: 10.1016/j.media.2019.03.014. [DOI] [PubMed] [Google Scholar]
- 54.Varrone M., Tavernari D., Santamaria-Martínez A., Walsh L.A., Ciriello G. CellCharter reveals spatial cell niches associated with tissue remodeling and cell plasticity. Nat. Genet. 2024;56:74–84. doi: 10.1038/s41588-023-01588-4. [DOI] [PubMed] [Google Scholar]
- 55.Yuan Z. MENDER: fast and scalable tissue structure identification in spatial omics data. Nat. Commun. 2024;15:207. doi: 10.1038/s41467-023-44367-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shang L., Zhou X. Spatially aware dimension reduction for spatial transcriptomics. Nat. Commun. 2022;13:7203. doi: 10.1038/s41467-022-34879-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Martin P.C.N., Kim H., Lövkvist C., Hong B.-W., Won K.J. Vesalius: high-resolution in silico anatomization of spatial transcriptomic data using image analysis. Mol. Syst. Biol. 2022;18 doi: 10.15252/msb.202211080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Garreta R., Moncecchi G. Packt Publishing Ltd; 2013. Learning Scikit-Learn: Machine Learning in Python. [Google Scholar]
- 59.Kim J., Samir R., Mosquera J.M., Randell S.H., Shaykhiev R., Rendeiro A.F., Elemento O. 2022. Unsupervised discovery of tissue architecture in multiplexed imaging. Preprint at Zenodo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu D., Hailer A.A., Wang S., Yuan M., Chan J., El Kurdi A., Rahim M., Kondo A., Han D., Ali H., et al. A single-cell atlas of IL-23 inhibition in cutaneous psoriasis distinguishes clinical response. Sci. Immunol. 2024;9 doi: 10.1126/sciimmunol.adi2848. [DOI] [PubMed] [Google Scholar]
- 61.Virshup I., Rybakov S., Theis F.J., Angerer P., Wolf F.A. anndata: Annotated data. bioRxiv. 2021 doi: 10.1101/2021.12.16.473007. Preprint at. [DOI] [Google Scholar]
- 62.Baker E.A.G., Huang M.-Y., Lam A., Rahim M.K., Bieniosek M.F., Wang B., Zhang N.R., Mayer A.T., Trevino A.E. emObject: domain specific data abstraction for spatial omics. bioRxiv. 2023 doi: 10.1101/2023.06.07.543950. Preprint at. [DOI] [Google Scholar]
- 63.Zarkhin V., Kambham N., Li L., Kwok S., Hsieh S.-C., Salvatierra O., Sarwal M.M. Characterization of intra-graft B cells during renal allograft rejection. Kidney Int. 2008;74:664–673. doi: 10.1038/ki.2008.249. [DOI] [PubMed] [Google Scholar]
- 64.Pardo B., Spangler A., Weber L.M., Page S.C., Hicks S.C., Jaffe A.E., Martinowich K., Maynard K.R., Collado-Torres L. spatialLIBD: an R/Bioconductor package to visualize spatially-resolved transcriptomics data. BMC Genom. 2022;23:434–435. doi: 10.1186/s12864-022-08601-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang Q., Ding S.-L., Li Y., Royall J., Feng D., Lesnar P., Graddis N., Naeemi M., Facer B., Ho A., et al. The Allen Mouse Brain Common Coordinate Framework: A 3D Reference Atlas. Cell. 2020;181:936–953.e20. doi: 10.1016/j.cell.2020.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rustam S., Hu Y., Mahjour S.B., Randell S.H., Rendeiro A.F., Ravichandran H., Urso A., D’Ovidio F., Martinez F.J., Richmond B., et al. A unique cellular organization of human distal airways and its disarray in chronic obstructive pulmonary disease. bioRxiv. 2022 doi: 10.1101/2022.03.16.484543. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hickey J.W., Tan Y., Nolan G.P., Goltsev Y. Strategies for Accurate Cell Type Identification in CODEX Multiplexed Imaging Data. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.727626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dong W., Moses C., Li K. WWW '11: Proceedings of the 20th international conference on World wide web. Association for Computing Machinery; 2011. Efficient K-Nearest Neighbor Graph Construction for Generic Similarity Measures; pp. 577–586. [DOI] [Google Scholar]
- 69.Traag V.A., Van Dooren P., Nesterov Y. Narrow scope for resolution-limit-free community detection. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2011;84 doi: 10.1103/PhysRevE.84.016114. [DOI] [PubMed] [Google Scholar]
- 70.Zanini F., Berghuis B.A., Jones R.C., Nicolis di Robilant B., Nong R.Y., Norton J.A., Clarke M.F., Quake S.R. Northstar enables automatic classification of known and novel cell types from tumor samples. Sci. Rep. 2020;10 doi: 10.1038/s41598-020-71805-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen T., Guestrin C. KDD '16: Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. Association for Computing Machinery; 2016. XGBoost: A Scalable Tree Boosting System; pp. 785–794. [DOI] [Google Scholar]
- 72.Steinley D. Properties of the Hubert-Arabie adjusted Rand index. Psychol. Methods. 2004;9:386–396. doi: 10.1037/1082-989X.9.3.386. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
External spatial proteomics and transcriptomics datasets used in this study (Human DLPFC Visium dataset, adult mouse brain MERFISH dataset, Lung IMC dataset, UPMC head-and-neck cancer CODEX dataset) are publicly available as of the date of publication. Processed CODEX datasets (DKD kidney CODEX dataset, UCSF derm CODEX dataset, TR kidney CODEX dataset, Stanford pancreatic cancer CODEX dataset) have been deposited at zenodo. All accession numbers are listed in the key resources table. Raw data and images will be accessible through links posted at: https://gitlab.com/enable-medicine-public/scgp. They will also be available from lead contact (A.E.T.) upon request.
-
•
SCGP was implemented based on Annotated data61 (https://anndata.readthedocs.io/en/latest/) and emObject62 (https://docs.enablemedicine.com/emobject/). All source codes are publicly available at https://gitlab.com/enable-medicine-public/scgp. An archival DOI is listed in the key resources table.
-
•
Any additional information required to re-analyze the data reported in this paper will be available from lead contact (A.E.T.) upon request.