


Abstract
We have developed a new strategy for producing subtracted cDNA libraries that is optimized for connective and epithelial tissues, where a few exceptionally abundant (super-prevalent) RNA species account for a large fraction of the total mRNA mass. Our method consists of a two-step subtraction of the most abundant mRNAs: the first step involves a novel use of oligo-directed RNase H digestion to lower the concentration of tissue-specific, super-prevalent RNAs. In the second step, a highly specific subtraction is achieved through hybridization with probes from a 3′-end ESTs collection. By applying this technique in skeletal muscle, we have constructed subtracted cDNA libraries that are effectively enriched for genes expressed at low levels. We further report on frequent premature termination of transcription in human muscle mitochondria and discuss the importance of this phenomenon in designing subtractive approaches. The tissue-specific collections of cDNA clones generated by our method are particularly well suited for expression profiling.
INTRODUCTION
Messenger RNAs are typically divided in three abundance classes: super-prevalent, intermediate and complex (1,2); however, their distribution among different tissues shows a high degree of variability. In human, tissues like brain exhibit an extreme complexity of transcripts, while tissues composed of highly differentiated cell types contain a few super-prevalent mRNAs at exceptionally high concentration. Single genes can account for more than 10% of the total mRNA mass in connective tissues and secretory epithelia (3). Extreme cases, where most messages in the cell derive from only a single or a few genes, have been documented in other species (e.g. hen oviduct, mouse or chick reticulocytes). The massive presence of super-prevalent mRNAs in a tissue often hampers large-scale EST sequencing projects (4) and connective and epithelial tissues are actually under-represented in the dbEST database. Mitochondrial transcripts are another major problem, as they constitute a large fraction of all the mRNAs in several tissues. In those cases, the abundance of mitochondrial transcripts likely reflects a high density of mitochondria, rather than overexpression of the mitochondrial genes (5). Constructing subtracted or normalized cDNA libraries can circumvent both problems (6–11).
Skeletal muscle is the prevalent tissue of the body of vertebrates and the characterization of its gene expression profile has important implications in physiology, medicine and zootechnics. Large amounts of expression data have been produced in human through systematic sequencing of cDNA libraries (12,13) and the serial analysis of gene expression (SAGE) method (5), giving generally consistent results. Not surprisingly, the most expressed genes are involved in three essential functions in muscle: contraction, energy metabolism and protein synthesis. Particularly abundant are mRNAs encoding for myofibrillar and mitochondrial proteins. Remarkably, the prevalence of a few mRNAs in differentiated muscles is the terminal effect of a dramatic change in gene expression occurring during myogenesis, as pointed out by early mRNA–cDNA hybridization experiments in chick (14). Skeletal muscle is a complex tissue, composed of a large variety of fiber types. At the molecular level, multiple isoforms of myofibrillar proteins contribute to the observed fiber variability (15–17). Recent expression profiling studies showed that understanding the processes leading to the distinct features of normal or pathological muscles is only at an initial stage (18).
Our group has been engaged for several years on an extended study of gene expression in human skeletal muscle using the expressed sequence tag (EST) approach (19,20). Through systematic sequencing of 3′-end ESTs we have built a catalog of muscle transcripts, which currently contains more than 4600 entries (http://muscle.cribi.unipd.it). We utilized special cDNA libraries optimized for ESTs production, in which short cDNA inserts (350–600 bp) containing the 3′-terminal sequences of the polyadenylated RNAs were directionally cloned in plasmid vectors (13). Taking advantage of our collection of 3′-end ESTs, we have developed a novel two-step subtractive strategy that allows the construction of cDNA libraries effectively enriched for genes expressed at low levels.
MATERIALS AND METHODS
Plasmid vectors, recombinant clones and DNA oligonucleotides
The plasmid vectors utilized in this study were pcDNA II (Invitrogen) and p0PD. The latter is a positive selection cloning vector carrying the ccdB cytotoxicity gene of Escherichia coli (21), derived from pCR-Blunt II-TOPO (Invitrogen). Both plasmids are identical with respect to multiple cloning sites.
The HM1 and HM3 cDNA libraries were constructed in BstXI+NotI-cut pcDNA II, while the cDNA inserts of the subtracted HM3/RH and HM3/S3 libraries were cloned in EcoRI+NotI-cut p0PD. Sixteen different overlapping fragments were amplified by PCR from human mitochondrial DNA using specific primers (see below) and subsequently cloned in EcoRV-cut p0PD. Mitochondrial DNA was isolated from human erythroleukemic K562 cells by alkaline extraction (22).
Synthetic DNA oligonucleotides were purchased from MWG Biotech. The complete list of oligonucleotides used in this work is reported in Supplementary Material. The list includes oligonucleotides required for cloning the human mitochondrial genomic clones, for oligo-directed RNase H digestion and for semi-quantitative PCR amplification. Vector-specific primers were: primer A, 5′-TCCGGCTCGTATGTTGTGTGGAAT-3′; primer B, 5′-GTTGTAAAACGACGGCCAGTGAATTG-3′; PC2R, 5′-CTCGGATCCACTAGTAACG-3′, sense, 5′-GCCGCCAGTGTGGTGGAATTC-3′; oligo-dT-NotI, 5′-AACCCGGCTCGAGCGGCCGCTTTTTTTTTTTTTTTTTT(AGC)-3′.
Catalog of the most expressed genes in skeletal muscle
Two independent cDNA libraries, named HM1 and HM3, were constructed from the same human pectoral muscle sample following the procedure described in detail by Lanfranchi et al. (13). Briefly, the first strand cDNA was primed with a specially designed, biotinylated oligo-dT-NotI primer. After completion of the second strand, the double-stranded cDNA was sonicated and size fractionated on agarose gels. The 3′-end-specific fragments were selected on streptavidin-coated magnetic beads, ligated to non-palindromic BstXI adapters (Invitrogen), NotI digested and directionally cloned into BstXI+NotI-cut pcDNA II vector. The length of size selected cDNA fragments was slightly different in the two libraries: 250–400 bp for HM1 and 350–550 bp for HM3. Neither library was subtracted or normalized. However, after random sequencing of the first 1000 cDNA clones, the ESTs of the 8–10 most abundant mRNAs were systematically excluded from DNA sequencing as a result of either a filter hybridization or a semi-multiplexed interference PCR pre-screening test (13,23). The relative abundance of mRNA species has been estimated after analysis of ∼37 000 independent cDNA clones from the two libraries: 22 855 high quality sequences were obtained, while the number of clones identified by pre-screening tests was calculated as described (13). Some factors in the methods used for generating the cDNA libraries and managing the data may have influenced the final results. For instance, varying the size of selected cDNAs after sonication led to different representation of some short mRNA species, while the clustering parameters are critical for the attribution of a given EST to a particular transcript when multigene families or multiple isoforms are concerned. UniGene library ID numbers of the HM1 and HM3 cDNA libraries are 203 and 500, respectively.
Systematic sequencing of cDNA clones
The cDNA libraries were plated as liquid cultures in 384-well microtiter plates at a density of 20–30 colonies per 100 wells. After growth, the wells containing colonies were re-arrayed and processed for both colony stocking and PCR amplification with universal primers (A and B), without any further growth. Double-stranded PCR templates were sequenced by the ABI PRISM BigDye Terminator chemistry (Applied Biosystems), using the nested sequencing primer PC2R, located 21 bases upstream of the first nucleotide of the cDNA inserts. Sequencing gels were run in a 96-capillary ABI3700 DNA sequencer.
Computer management of the data
The base-calling software PHRED (24) read DNA sequencer trace data and sequence portions of poor quality were trimmed. Mitochondrial, repeated and vector sequences were identified using an adapted version of Repeatmasker (A.F.A.Smit and P.Green at http://ftp.genome.washington.edu/RM/RepeatMasker.html). Terminal repeats and vector sequences were removed, while internal repeats were masked. Only ESTs whose sequences exceeded 50 bp after trimming and masking were considered for further analyses. These ESTs were assembled by similarity into groups (25) and the obtained consensus sequences were searched (26) against public databases (GenBank release 126.0). Finally, each transcript was recorded and annotated in a dedicated database (Muscle TRAIT, where TRAIT stands for transcript integrated table). The database is publicly released and available for consultation at our web site (http://muscle.cribi.unipd.it).
Oligo-directed RNase H digestion
About 100 000 bacterial clones of the HM3 library were plated on solid medium, pooled by scraping after overnight growth and their plasmid DNAs isolated. RNA molecules were synthesized with T7 RNA polymerase by run-off in vitro transcription from HindIII-cut plasmid using the AmpliScribe T7 High Yield Transcription Kit (Epicentre Technologies). Approximately 5 µg of DNase I-treated synthetic RNA was incubated with a molar excess of 16 DNA oligonucleotides (10 pmol each) in the presence of 5 U Hybridase (Epicentre Technologies). A thermostable RNase H was chosen in order to minimize the background due to non-specific hybridization. The reaction proceeded for 20 min at 58°C in 50 µl of buffer containing 20 mM Tris–HCl pH 7.5, 20 mM KCl, 10 mM MgCl2, 0.1 mM EDTA and 0.1 mM DTT. The following DNA oligonucleotides were included: 16S, ACTA1, ATP6, CKM, COX2, COX3, GAPD, HBA1, MYH7, MYLC2, MYL2, NADH2, NADH3, TNNI1, TNNT1 and TPM2. Undigested RNAs were eluted from a denaturing 5% polyacrylamide gel and reverse transcription was carried out with a vector-specific primer on 1 µg of gel-purified RNA template. A high fidelity PCR step (2–10 cycles) was introduced at this point, using the PfuTurbo DNA polymerase (Stratagene) in the presence of the first strand primer PC2R and anchored oligo-dT-NotI (AGC). PCR products were digested with EcoRI and NotI, purified from 1.5% agarose gels and directionally cloned into plasmid p0PD. A more detailed report on this technique will be published elsewhere (P.Laveder et al., manuscript in preparation).
Filter hybridization
DNA inserts obtained by PCR amplification were spotted on nylon filters (Hybond N+; Amersham Pharmacia Biotech) using a Biomek 2000 Workstation (Beckman Coultard) equipped with a 96-nail tool. The detailed list of DNA probes, i.e. immobilized nucleic acids, is reported in Supplementary Material. Plasmid DNA was isolated from each cDNA library to be tested (pools of ∼100 000 clones grown on solid medium), NotI digested and RNA sense molecules were produced with SP6 RNA polymerase by run-off in vitro transcription using the AmpliScribe SP6 High Yield Transcription Kit (Epicentre Technologies). Synthesis of radioactively labeled complex cDNA targets by reverse transcription of 0.5 µg RNA in the presence of dideoxynucleotides and hybridization were carried out as described (27). Filters were washed twice at room temperature in 2× SSC, 0.1% SDS (15 min each), once for 15 min at 65°C in 1× SSC, 0.1% SDS and twice for 10 min at 65°C in 0.1× SSC, 0.1% SDS. Hybridization signals were analyzed in a Cyclone Storage Phosphor System (Packard), using the OptiQuant software supplied. The intensity values were first determined in four independent spots for each clone, then the mean ± SD was calculated and, finally, the values were normalized with respect to the maximum intensity recorded on each filter.
Semi-quantitative PCR amplification
The DNA template was isolated from a purchased skeletal muscle cDNA library (Clontech) constructed in plasmid vector pGAD10. PCR amplification was carried out in a final volume of 50 µl in the presence of 15 ng DNA template, 200 µM each dNTP, 0.2 µM each primer and 2.5 U AmpliTaq Gold enzyme (Applied Biosystems), according to the supplier’s instructions. Reaction mixtures were incubated for 10 min at 95°C (‘hot start’) in a GeneAmp PCR System 9600 (Applied Biosystems), then the following cycle was repeated 18, 21, 24, 27, 30, 33, 36 or 39 times: 20 s at 95°C, 30 s at 62°C and 40 s at 72°C. Reaction products were analyzed on 1.5% agarose gels, by loading one-tenth of the volume.
Specific self-subtracted cDNA library
Preparation of the cDNA tracer. Plasmid DNA was isolated from ∼1 000 000 bacterial clones of the HM3/RH library after overnight growth on solid medium. The cDNA inserts were excised by HindIII+NotI digestion and then purified from agarose gels. To generate single-stranded tracer, 250 ng cDNAs template were added to a 100 µl reaction mixture containing 0.5 µM sense primer, 250 µM each dNTP, 5 U PfuTurbo enzyme (Stratagene) and linear amplification was carried out according to the supplier’s instructions. Cycling conditions were: 20 s at 95°C, 40 s at 55°C and 90 s at 72°C, repeated 40 times. The sense primer spans the 21 bases of the vector closest to the 5′-end of the cDNA inserts. Single-stranded cDNAs were resolved from double-stranded DNA template through gel electrophoresis in 1.5% low melting point agarose and subsequently purified. The concentration of nucleic acids was determined by measuring optical density at 260 nm.
Preparation of the RNA driver. Single bacterial clones were inoculated into 1 ml aliquots of SOB medium plus the appropriate antibiotic and grown in sterile 96-deep-well blocks (Beckman Coulter) at 37°C for 20–24 h. Plasmid DNA was isolated by alkaline lysis either from grouped cultures or individually from single clones using MultiScreen filtration plates (Millipore). DNA templates from clones of our collection of 3′-end ESTs were digested with EcoRI. To compensate for the presence of EcoRI restriction sites in some mitochondrial genomic clones, such DNA templates were digested separately with both EcoRI and PstI; in the latter case 3′-protruding ends were converted to blunt ends with T4 DNA polymerase. Antisense, biotinylated RNA molecules were obtained by run-off in vitro transcription in the presence of biotin-16-UTP (Roche). DNA templates (3–5 µg) were incubated for 2 h at 37°C in 100 µl of reaction medium containing 40 mM Tris–HCl pH 7.5, 10 mM NaCl, 6 mM MgCl2, 2 mM spermidine, 1 mM each ATP, GTP and CTP, 0.65 mM UTP, 0.35 mM biotin-16-UTP, 10 mM DTT and 200 U T7 RNA polymerase (Epicentre Technologies). RNA was treated with DNase I (RNase-free; Epicentre Technologies), phenol extracted and subjected to two rounds of ethanol precipitation in the presence of 2.5 M ammonium acetate.
Hybridization. Blocking oligonucleotides were not necessary during hybridization, since the cDNAs tracer had no vector sequences 3′ to the cDNA inserts and the antisense RNA transcripts did not carry sequences of the sense primer at their 3′-end. Furthermore, the anchored oligo-dT-NotI used in the first subtractive step shortened the poly(A) tract of cDNAs to only 18 nt. Approximately 1 µg cDNA tracer and 25 µg RNA driver were ethanol precipitated in a 1.5 ml silanized tube, resuspended in deionized formamide, heated at 80°C for 3 min and incubated overnight (at least 12 h) at 42°C in 25 µl of buffer containing 0.5 M NaCl, 10 mM Tris–HCl pH 7.5, 5 mM EDTA and 50% formamide. Due to low sequence complexity of the molecules involved in the reaction, R0t values < 1 (s mol/l) were assumed to give complete RNA–cDNA reassociation. After hybridization, the sample was precipitated with 3 vol of absolute ethanol, briefly washed with 70% ethanol and the pellet redissolved in 25 µl of water.
Cloning. Meanwhile, 750 µl of streptavidin-coated magnetic beads (10 mg/ml; Dynal) were washed twice with 1.5 ml of binding buffer (BWT, 1 M NaCl, 10 mM Tris–HCl pH 7.5, 5 mM EDTA and 0.1% Tween 20), resuspended in the original volume and preincubated for 15–20 min at room temperature with 150 µg tRNA to saturate all possible non-specific binding sites. After three washes with 5 ml of BWT, 750 µl of saturated beads in BWT were added to the hybridized sample, mixed gently and incubated for 20 min at room temperature under low speed rolling. The beads were immobilized by inserting the tube into a magnetic stand and the supernatant transferred to a new silanized tube. This step was repeated twice. Unbound nucleic acids were precipitated with 2 vol of absolute ethanol and RNA molecules hydrolyzed by alkaline treatment (30 min at 55°C in 0.1 N NaOH). After ethanol precipitation, single-stranded cDNAs were purified from 1.5% low melting point agarose gel and used as a template for high fidelity PCR (PfuTurbo DNA polymerase; Stratagene) with the sense and oligo-dT-NotI (AGC) primers. Products amplified after 2, 4, 6, 8 and 10 PCR cycles were analyzed on agarose gels and the sample giving the weakest detectable signal after ethidium bromide staining was chosen for the subsequent steps. Gel-purified PCR products were digested with EcoRI and NotI and directionally cloned into plasmid p0PD.
The ESTs of the HM3/S3 cDNA library analyzed in this work have been submitted to the EMBL database (accession nos AJ345961–AJ346853).
RESULTS
Subtractive strategy
Tables S1–S3 are presented as Supplementary Material and are also available at http://muscle.cribi.unipd.it/S3_libraries. Tables S1 and S2 summarize data on the nuclear and mitochondrial encoded most expressed genes in human skeletal muscle, respectively. The expression profile of skeletal muscle shows the following traits. First, some mRNAs are extremely abundant (herein referred to as ‘super-prevalent’). Second, mitochondrial mRNAs account for 20–25% of the polyadenylated RNA, much more than normally found in other tissues (4). Third, a large fraction of the mRNA mass derives from a relatively small group of genes. We estimated that the 100 most expressed nuclear genes account for ∼35% of the mRNA population in skeletal muscle. In good agreement with our data, Kawamoto et al. (3) recently calculated that ∼500 genes account for 50% of the mRNA mass in connective and epithelial tissues. Based on these data, subtractive approaches seem particularly attractive in muscle, because by constructing probes directed against a small population of genes it should be possible to remove more than half of the total mRNA mass.
We took advantage of our collection of 3′-end ESTs to prepare a battery of probes specific for the 96 most abundant nuclear encoded transcripts. A different approach was chosen for probes directed against the genes encoded by the H strand of the human mitochondrial genome, because of the peculiar characteristics of the mitochondrial transcripts (see below). The efficiency of subtraction was monitored by filter hybridization experiments: 50 probes specific for the most expressed nuclear genes and 16 mitochondrial genomic clones were arrayed on nylon filters and hybridized with radioactive targets derived from the cDNA libraries to be tested. Figure 1A shows the results obtained with a 3′-end cDNA library named HM3, which was not subtracted or normalized. In general, we observed a good correlation between signal intensity and abundance of the corresponding cDNA in the library (Table S3), although these experiments gave only semi-quantitative results.
Figure 1.
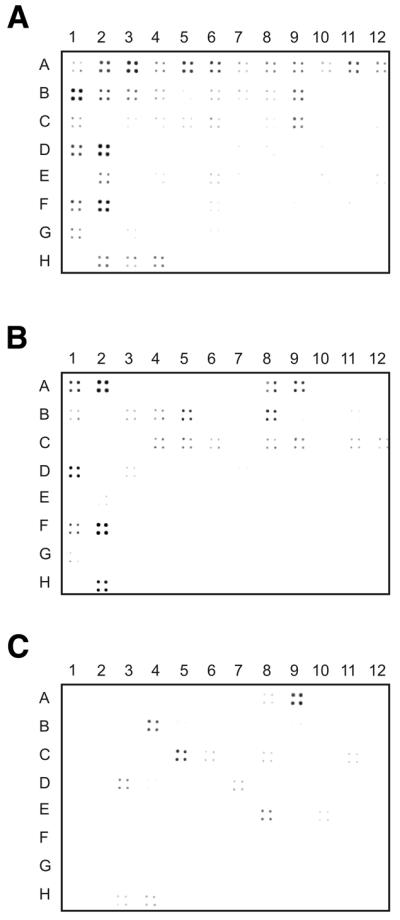
Filter hybridization results. DNA probes obtained by PCR amplification were arrayed on nylon filters and hybridized with radioactive targets derived from 3′-end-specific cDNA libraries at different stages of the subtractive process. Sixteen overlapping fragments of the human mitochondrial genome were placed in columns 1–2 (rows A–H). Other probes representing either genes expressed at different levels in human skeletal muscle or simple controls were arrayed as follows: rows A–E (columns 3–12), the fifty most abundant muscle transcripts; rows F–G (columns 3–12), muscle transcripts of low abundance; row H (columns 3–12), controls. The detailed list of DNA probes, controls included, is available as Supplementary Material. Hybridization signals, quantified as described (Materials and Methods), are reported in Table S3. The analysis of mitochondrial clones is not straightforward, since they often overlap different genes. (A) HM3 library, not subtracted or normalized. The highest hybridization signals are found in the correspondence of super-prevalent mRNAs, which include several mitochondrial encoded species. (B) HM3/RH library produced after the first subtractive step. Note the lower intensity observed in the super-prevalent mRNAs digested with RNase H (boxed values in Table S3). The signal visible in some mitochondrial clones (e.g. A2) is likely due to ‘truncated’ RNAs that escaped digestion. (C) Subtracted HM3/S3 library. It is apparent that all mitochondrial RNAs were efficiently removed. The high hybridization signals recorded were the first clue that some probes could have not worked properly during the subtractive process.
In preliminary subtractive experiments we tested all the probes in a single hybridization reaction with unsatisfactory results. We then observed that the relative abundance of the target RNA species varied >100-fold between super-prevalent mRNAs (e.g. α-actin = 8.5%) and other intermediate but still abundant mRNAs (the hundredth transcript is 0.08%). We reasoned that it would be difficult to find an optimal concentration of driver for both target species and thus we eventually followed a two-step strategy. The HM3 library went through to a first subtractive step, as described in detail in Materials and Methods.
First step: oligo-directed RNase H digestion
The first subtraction was limited to only 15 mRNAs belonging to the super-prevalent class. In order to drastically lower the concentration of these mRNAs, we made use of the enzyme RNase H, which specifically degrades the RNA moiety in a DNA:RNA hybrid, without affecting DNA or unhybridized RNA (28). The strategy is outlined in Figure 2. A set of DNA oligonucleotides complementary to the 3′-untranslated region (3′-UTR) of the RNA targets drives digestion only of the super-prevalent mRNAs. Importantly, in vitro transcription of the HM3 library produces RNAs of roughly uniform length (400–600 nt). Specific digestion is obtained by incubating RNAs and DNA oligonucleotides in the presence of thermostable RNase H. Undigested RNAs are then purified from polyacrylamide gels, reverse transcribed and cloned in plasmid vectors. The cDNA library obtained after the first round of subtraction was named HM3/RH. Figure 1B clearly shows a substantial reduction of all target cDNAs in the HM3/RH library.
Figure 2.

Outline of the first subtractive step. Synthetic RNAs were produced by run-off in vitro transcription from a 3′-end-specific cDNA library (HM3). The resulting RNA molecules are nearly uniform in length (400–600 nt) and their composition reflects the relative abundance of each cDNA in the starting library. Super-prevalent RNAs are shown as black wavy lines. We designed DNA oligonucleotides complementary to the 3′-UTR of the super-prevalent mRNAs, then RNAs and DNA oligonucleotides were incubated in the presence of RNase H, which specifically degrades the RNA moiety in a DNA:RNA hybrid. The undigested RNAs were purified from preparative gels, reverse transcribed and cloned in plasmid vectors.
Mitochondrial transcripts in human skeletal muscle
While building the catalog of muscle transcripts we verified whether the ESTs of our 3′-end cDNA libraries really matched the 3′-portion of the mRNAs. This was true for the large majority of nuclear transcripts examined. We noted, however, that the 3′ polyadenylated ends of mitochondrial transcripts often occurred at unexpected locations (13). Such a strange phenomenon is illustrated in Figure 3, where a sample of 100 3′-end ESTs representative of all genes encoded by the H strand in the human mitochondrion has been aligned with the corresponding full-length RNAs. It is conceivable that the selected ESTs represent authentic RNA terminations, because most of them have long poly(A) stretches at their 3′-end. From the alignment it is evident that mitochondrial RNAs are often prematurely terminated and that mitochondrial genes are affected to a different extent by this phenomenon. We explain the frequent presence of ‘truncated’ mRNAs in mitochondria by the assumption that poly(A) tracts might be added to every free 3′-end of a terminated mRNA once it becomes available to the polyadenylation machinery.
Figure 3.

Alignment of the ESTs generated from skeletal muscle 3′-end cDNA libraries along the human mitochondrial genome. One hundred ESTs for each gene encoded by the mitochondrial H strand were selected and assembled with the corresponding full-length RNA transcript (Table S2), using the program SeqMan II (DNAStar). We chose the 100 longest ESTs produced for each mitochondrial gene, after trimming the poor quality regions, but only 92 sequences were available for the NADH 5 gene. It is conceivable that the scattered ESTs identify real RNA terminations, since the cDNA libraries were constructed from poly(A)+ RNA; actually many of the selected ESTs have long poly(A) stretches at their 3′-end. From the alignment we conclude that a premature termination of transcription occurs frequently in mitochondria from skeletal muscle. The phenomenon is clearly evident for longer genes and reaches extreme proportions in a few cases. Note that the figure does not reflect the relative abundance of mitochondrial RNAs in human skeletal muscle, reported in Table S2. We also note that the most 3′-terminal ESTs selected for the NADH 5 gene identified a different 3′-end compared to that annotated in the reference sequence (Table S2). Further information on the alignments shown is available upon request.
The abnormal traits of mitochondrial mRNAs had some consequences for our subtractive approach. First, we noted that truncated mRNAs could not be removed in the first step, since oligo-directed RNase H digestion was restricted to the 3′-ends of mRNAs. More importantly, we realized that in the next subtractive hybridization the probes must cover the entire length of the genes to efficiently capture all mitochondrial RNAs. We therefore created a set of 16 overlapping fragments, spanning all the human mitochondrial genome (Materials and Methods).
Second step: subtractive hybridization
The RNase H step alone is not productive in terms of enrichment for rare mRNAs and therefore it was necessary to couple it with a very efficient subtractive hybridization, involving a much larger number of transcripts. The second step consists of classic RNA:cDNA reassociation reactions in solution (29). We took advantage of our collection of 3′-end ESTs to generate highly specific probes directed against the most abundant nuclear encoded mRNAs. It is well known that the 3′-sequences are unique for each mRNA and thus a probe restricted to this region should anneal only to its correct counterpart (6). As mentioned above, the probes directed against the mitochondrial encoded RNAs derived instead from genomic clones. The complete list of probes is available at our web site and includes the super-prevalent mRNAs already the subject of the first round of subtraction.
Sense cDNA tracer was produced from the HM3/RH library and incubated with an excess of antisense, biotinylated RNA driver produced by in vitro transcription, as described (Materials and Methods). After overnight hybridization in a high stringency buffer, the cDNA:RNA hybrids were immobilized on streptavidin-coated beads, whereas the unbound cDNA was made double stranded and cloned into plasmid vectors. The resulting cDNA library was named HM3/S3, to underline our specific self-subtractive approach.
From the hybridization results it is evident that most target mRNAs were efficiently subtracted (Fig. 1C). While all mitochondrial mRNAs were remarkably reduced, the signal intensities of some of the most abundant nuclear encoded mRNAs remained significant. This latter result is discussed below. The specificity of subtraction was tested by examining ESTs of the actin multigene family. No EST in the HM3/S3 library could be assigned to the skeletal muscle isoform of α-actin, for which specific probes were included in both subtractive steps. Instead, we could find ESTs of γ-actin and a single EST identifying the cardiac isoform of α-actin, previously undetected in our cDNA libraries. We noted that these genes are highly similar at the nucleotide level in the coding regions (∼86% identity) and only the 3′-UTRs are clearly divergent.
In order to quantify the efficiency of subtraction, we analyzed 1000 ESTs from both the parent (HM3) and the subtracted (HM3/S3) cDNA libraries. Sequencing results confirmed that all mitochondrial RNAs were efficiently removed (Table S2), while subtraction of the nuclear encoded mRNAs proved to be more problematic. In particular, four of them were clearly not subtracted (Table S3). Control experiments suggested that in vitro transcription of RNA drivers did not work with the same efficiency for all the 96 EST clones (see Discussion). Altogether, 167 ESTs in the HM3/S3 library corresponded to the gene targets of the subtraction, compared to 618 in the HM3 library. The result is even more impressive considering that 76 ESTs belong to the four transcripts for which the RNA drivers did not work properly.
Transcripts identified in the subtracted cDNA library
All ESTs carrying repetitive sequences were deliberately excluded from the pool used to generate subtractive probes. Significantly, 134 ESTs in the HM3/S3 library contained interspersed repeats, which is a figure ∼3-fold higher than in the parent library. Among the families of repeats, Alu sequences were the most frequent, as they were found in 108/134 and 30/47 cases, respectively. These ESTs were not further analyzed.
The remaining ESTs were processed through computer programs as described (Materials and Methods). Of the ESTs in the HM3/S3 library 655 were selected for further analysis and assembled in 357 distinct groups. A minimal but detectable redundancy was observed, as a consequence of the few PCR cycles introduced during the cloning step. Of these, 113 groups identified transcripts not already cataloged in the Muscle TRAIT database. To experimentally verify that these genes are really expressed at low level in muscle, as one would expect, we randomly selected 11 transcripts and measured their abundance in a skeletal muscle cDNA library by semi-quantitative PCR amplification with specific primers. The results shown in Figure 4 confirm that the corresponding amplification products originated at much later PCR cycles than those of well characterized genes expressed at high to intermediate level in striated muscle.
Figure 4.

Semi-quantitative PCR. We designed specific primers for 11 transcripts randomly selected among those isolated from the subtracted HM3/S3 library and measured the corresponding level of expression in skeletal muscle by semi-quantitative PCR amplification. The PCR reaction was stopped after the indicated number of cycles. Shown on the right is the EST ID of some selected transcripts. As controls we chose well characterized genes expressed at high or intermediate levels in striated muscle. GADP, glyceraldehyde 3-phosphate dehydrogenase, a popular standard in several cDNA expression panels; TELE, telethonin/titin cap, expressed at high level (Table S1); ANKRD2, ankyrin repeat domain 2 protein, intermediate abundance in our cDNA libraries (0.06%). For the last two genes the expression level in muscle was measured by a variety of other techniques in our laboratory (38,39).
Finally, extensive similarity searches were carried out on public databases, in order to better characterize the 113 newly recorded muscle transcripts: 83 identify known genes and 25 correspond or are similar to ESTs already present in GenBank. The low frequency of novel genes is not surprising, due to the sequencing and annotation effort promoted by the Human Genome Project. However, four different ESTs showed no similarity to any other EST and were considered unique to the HM3/S3 library; three of them did match human genomic sequences (Table 1). Other ESTs showed similarity only to a very limited number of sequences and, simply on the basis of these in silicio analyses, might potentially correspond to very rare transcripts (Table 1).
Table 1. Potentially rare transcripts identified in the HM3/S3 library.
| ESTa | Identified gene | Hits dbESTb | Best hit dbESTc | Human genomed |
|---|---|---|---|---|
| HSPD50214 | AL590138 | AA523714 | ||
| HSPD50335 | Adenylosuccinate lyase gene | AA743968 | AL022238 | |
| HSPD50370 | Hypothetical gene supported by AK025812 | 1 | BI834115 | AL391665 |
| HSPD50442 | AL158817 | |||
| HSPD50489 | 2 | AI075139 | AP001491 | |
| HSPD50648 | Heparan sulfate proteoglycan 2 (HSPG2), mRNA | 3 | BG015448 | AC020565 |
| HSPD50704 | BG616650 | |||
| HSPD50728 | AC005216 | |||
| HSPD50873 | ||||
| HSPD50980 | Leucine-rich repeat-containing 2 (LRRC2), mRNA | 1 | C02806 | AC068720 |
| HSPD51059 | AL049537 |
aEST ID number.
bOnly sequences showing blast E values <10–80 were counted.
cIncluding sequences with blast E values <10–30.
dAccession number of both finished and unfinished genomic clones.
DISCUSSION
In summary, our method consists of three points. (i) Oligo-directed RNase H digestion brings the frequency of tissue-specific, super-prevalent mRNAs to acceptable levels. In principle, the same result might be obtained through hybridization capture of these mRNA species, although we did not investigate this possibility. (ii) Probes generated from overlapping genomic fragments eliminate all mRNAs transcribed from mitochondrial genes, thus removing a large fraction of the mRNA mass. A complete set of mitochondrial full-length cDNAs clones would be equally suitable for this purpose. (iii) Subtraction of the most abundant nuclear encoded RNAs is achieved by using 3′-end-specific probes, thus ensuring maximum specificity of the hybridization reactions. This feature is very important when alternatively spliced genes or multigene families are concerned, as in the case of myofibrillar proteins in muscle (15). Notably, we have shown that 3′-end probes allow discrimination between the skeletal muscle and cardiac isoforms of α-actin, despite the high similarity in the coding region of the two genes. We stress that other sets of cDNA clones might cause a loss of tissue-specific isoforms during the hybridization process. In this respect, our procedure differs from previously published methods, which also utilized run-off transcripts from mini-libraries of highly expressed genes to prepare RNA drivers (10,11). Furthermore, by following a two-step strategy we could overcome the non-specific loss of low abundance mRNAs due to pairing between long poly(dA) and poly(T) regions of unrelated sequences, which represents a serious problem in most subtractive reactions (30).
The results of semi-quantitative PCR amplification shown in Figure 4 are consistent with the assumption that the subtracted cDNA libraries constructed with our method are really enriched for genes expressed at low levels. To further support this conclusion, we analyzed the results of expression profiling studies on high density cDNA filters, which included nearly half the transcripts identified in the subtracted cDNA library. When hybridized with complex targets from skeletal muscle, the hybridization signals of the large majority of these transcripts were intermediate (23.8%) or low (61.6%), as expected (G. Lanfranchi et al., manuscript in preparation).
We envisage a few possible improvements of the method described here. (i) The starting cDNA library should preferably be constructed in phagemid vectors, from which the DNA tracer can be produced in the form of single-stranded circles. This would allow the PCR step after subtraction to be skipped, since circular plasmids can directly transform E.coli (10,31). (ii) More 3′-end probes might be included in the subtractive step. Disappointingly, we observed that not all cDNA inserts were transcribed in vitro with the same efficiency, thus causing failure of the subsequent hybridization. A similar problem has been reported and discussed by other authors (10). In our hands some clones produced apparently normal amounts of in vitro transcribed RNA if tested individually and gave little or no RNA products when placed in a complex mixture of DNA templates (not shown). Carrying out single in vitro transcription reactions for each difficult probe and then combining the appropriate quantities of RNA drivers in the hybridization reactions could circumvent the problem. (iii) All probes were checked for the absence of repeats, in order to prevent the loss of rare mRNAs carrying repetitive sequences during hybridization. We are working on appropriate strategies aimed at recovering information in the group of transcripts containing Alu repeats whose abundance increased significantly in the subtracted library. However, the retention of cDNA clones carrying repetitive sequences is a common problem of several subtractive and normalizing procedures (6–9).
We have shown that many transcripts are prematurely terminated in human muscle mitochondria and discussed the importance of this phenomenon in designing subtractive approaches. Of course, truncated mRNAs would not give rise to functional products, since most mitochondrial genes encode proteins. At the moment the significance of our observation is unclear and a more exhaustive discussion is beyond the purposes of this article. However, the phenomenon illustrated here will clearly have implications in several studies on the expression of mitochondrial genes. Truncated molecules have been documented at lower frequencies in human cDNA libraries from other tissues (10). We found a similar situation in mitochondria from mouse skeletal muscle (P.Laveder, G.Valle and G.Lanfranchi, unpublished results), suggesting that the premature termination of transcription may be widely diffused, at least in mammals.
Although all the data have been produced in a muscle system, our method would be appropriate in general for connective and epithelial tissues, since they possess similar expression profiles (3). Several probes described in this work are not strictly muscle-specific (e.g. ribosomal proteins and other housekeeping genes) and others might be selected from our collection of 3′-end ESTs. Because super-prevalent RNAs are extremely frequent in every cDNA library, it would be easy for researchers to obtain a small set of tissue-specific cDNA clones restricted to 3′ sequences, even without the preliminary production of a 3′-end-specific cDNA library. We are currently exploiting small adjustments of the method described here that should allow application of the same two-step strategy directly to mRNA preparations or traditional cDNA libraries constructed in transcription vectors.
The primary application of this technology is building collections of tissue-specific cDNA clones for expression profiling, using the emerging cDNA microarrays technology (32). In this respect, we favor the construction of 3′-end-specific cDNA libraries as natural start and end products of the procedure, since they offer several advantages: (i) the short cDNA inserts can be efficiently amplified by PCR and sequenced without the need for DNA extraction, thus limiting the running costs; (ii) the 3′-UTRs are unique for each transcript and therefore the most suitable for expression studies; (iii) the complexity of the bioinformatic analyses decreases substantially, since each transcript is represented only by its 3′-end. The tissue-specific cDNA collections are unique experimental tools in those organisms where little or no genomic information is available. However, we note that EST sequencing remains a primary research tool even in the post-genome era, because the informatic prediction of genes from the genome is not yet completely reliable.
A number of existing methods might integrate our approach, in order to enrich the cDNA collections for differentially expressed genes (33). In the muscle system the subtractive hybridization step could be applied to deplete abundant RNA tracers obtained from other muscle tissues (e.g. cardiac muscle), as well as from muscles during various stages of differentiation or in particular physiological and pathological conditions. In order to maximize the extent of subtraction, specific subsets of probes could be created after sequencing a few hundred clones from the cDNA libraries of interest. Adjustable approaches are required in muscle, since fast and slow fibers differ in the relative abundance of the most expressed genes (34,35).
All subtractive approaches have the peculiarity that the relative abundance of individual mRNA species in the unhybridized fraction is not equalized at any time (6). Consequently, our method will lead to a more frequent identification of mRNAs of the intermediate class or up-regulated genes, although some biases are inevitably introduced. In contrast, the construction of normalized cDNA libraries (36) and other procedures designed for cloning of rarely expressed genes, like suppression subtractive hybridization (37), equalize the concentration of all mRNA species in a first hybridization step. It follows that any information on the relative abundance of the mRNAs in a tissue (or a mixture of tissues) is lost. Subtraction and normalization are not mutually exclusive approaches; however, great care should be taken in choosing the proper strategy before engaging in a systematic sequencing project.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
The following people were involved in systematic sequencing of the ESTs: Beniamina Pacchioni, Silvia Trevisan, Barbara Simionati, Michela D’Angelo and Rosanna Zimbello. We thank Nicola Cannata, Nicola Vitulo, Paolo Scannapieco, Rosario Dioguardi for help in computational analyses and Dr Chiara Romualdi for managing the hybridization data. This work was financed by the Fondazione Telethon, Italy (grants B41 and B57).
DDBJ/EMBL/GenBank accession nos AJ345961–AJ346853
REFERENCES
- 1.Bishop J.O., Morton,J.G., Rosbash,M. and Richardson,M. (1974) Three abundance classes in HeLa cell messenger RNA. Nature, 250, 199–204. [DOI] [PubMed] [Google Scholar]
- 2.Davidson E.H. and Britten,R.J. (1979) Regulation of gene expression: possible role of repetitive sequences. Science, 204, 1052–1059. [DOI] [PubMed] [Google Scholar]
- 3.Kawamoto S., Yoshii,J., Mizuno,K., Ito,K., Miyamoto,Y., Ohnishi,T., Matoba,R., Hori,N., Matsumoto,Y., Okumura,T. et al. (2000) BodyMap: a collection of 3′ ESTs for analysis of human gene expression information. Genome Res., 10, 1817–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adams M.D., Kerlavage,A.R., Fleischmann,R.D., Fuldner,R.A., Bult,C.J., Lee,N.H., Kirkness,E.F., Weinstock,K.G., Gocayne,J.D., White,O. et al. (1995) Initial assessment of human gene diversity and expression patterns based upon 83 million nucleotides of cDNA sequence. Nature, 377 (suppl.), 3–174. [PubMed] [Google Scholar]
- 5.Welle S., Bhat,K. and Thornton,C.A. (1999) Inventory of high-abundance mRNAs in skeletal muscle of normal men. Genome Res., 9, 506–513. [PMC free article] [PubMed] [Google Scholar]
- 6.Ko M.S.H. (1990) An equalized cDNA library by the reassociation of short double-stranded cDNAs. Nucleic Acids Res., 18, 5705–5711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patanjali S.R., Parimoo,S. and Weissman,S.M. (1991) Construction of a uniform-abundance (normalized) cDNA library. Proc. Natl Acad. Sci. USA, 88, 1943–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sasaki Y.F., Ayusawa,D. and Oishi,M. (1994) Construction of a normalized cDNA library by introduction of a semi-solid mRNA-cDNA hybridization system. Nucleic Acids Res., 22, 987–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soares M.B., Bonaldo,M.F., Jelene,P., Su,L., Lawton,L. and Efstratiadis,A. (1994) Construction and characterization of a normalized cDNA library. Proc. Natl Acad. Sci. USA, 91, 9228–9232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonaldo M.F., Lennon,G. and Soares,M.B. (1996) Normalization and subtraction: two approaches to facilitate gene discovery. Genome Res., 6, 791–806. [DOI] [PubMed] [Google Scholar]
- 11.Carninci P., Shibata,Y., Hayatsu,N., Sugahara,Y., Shibata,K., Itoh,M., Konno,H., Okazaki,Y., Muramatsu,M. and Hayashizaki,Y. (2000) Normalization and subtraction of cap-trapper-selected cDNAs to prepare full-length cDNA libraries for rapid discovery of new genes. Genome Res., 10, 1617–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Houlgatte R., Mariage-Samson,R., Duprat,S., Tessier,A., Bentolila,S., Lamy,B. and Auffray,C. (1995) The Genexpress Index: a resource for gene discovery and the genic map of the human genome. Genome Res., 5, 272–304. [DOI] [PubMed] [Google Scholar]
- 13.Lanfranchi G., Muraro,T., Caldara,F., Pacchioni,B., Pallavicini,A., Pandolfo,D., Toppo,S., Trevisan,S., Scarso,S. and Valle,G. (1996) Identification of 4370 expressed sequence tags from a 3′-end-specific cDNA library of human skeletal muscle by DNA sequencing and filter hybridization. Genome Res., 6, 35–42. [DOI] [PubMed] [Google Scholar]
- 14.Paterson B.M. and Bishop,J.O. (1977) Changes in the mRNA population of chick myoblasts during myogenesis in vitro. Cell, 12, 751–765. [DOI] [PubMed] [Google Scholar]
- 15.Schiaffino S. and Reggiani,C. (1996) Molecular diversity of myofibrillar proteins: gene regulation and functional significance. Physiol. Rev., 76, 371–423. [DOI] [PubMed] [Google Scholar]
- 16.Schiaffino S. and Salviati,G. (1997) Molecular diversity of myofibrillar proteins: isoforms analysis at the protein and mRNA level. Methods Cell Biol., 52, 349–369. [DOI] [PubMed] [Google Scholar]
- 17.Pette D. and Staron,R.S. (1997) Mammalian skeletal muscle fiber type transitions. Int. Rev. Cytol., 170, 143–223. [DOI] [PubMed] [Google Scholar]
- 18.Hampson R. and Hughes,S.M. (2001) Muscular expressions: profiling genes in complex tissues. Genome Biol., 2, 1033.1–1033.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adams M.D., Kelley,J.M., Gocayne,J.D., Dubnick,M., Polymeropoulos,M.H., Xiao,H., Merril,C.R., Wu,A., Olde,B., Moreno,R.F. et al. (1991) Complementary DNA sequencing: expressed sequence tags and human genome project. Science, 252, 1651–1656. [DOI] [PubMed] [Google Scholar]
- 20.Adams M.D., Dubnick,M., Kerlavage,A.R., Moreno,R., Kelley,J.M., Utterback,T.R., Nagle,J.W., Fields,C. and Venter,J.C. (1992) Sequence identification of 2,375 human brain genes. Nature, 355, 632–634. [DOI] [PubMed] [Google Scholar]
- 21.Bernard P., Gabant,P., Bahassi,E.M. and Couturier,M. (1994) Positive-selection vectors using the F plasmid ccdB killer gene. Gene, 148, 71–74. [DOI] [PubMed] [Google Scholar]
- 22.Palva T.K. and Palva,E.T. (1985) Rapid isolation of animal mitochondrial DNA by alkaline extraction. FEBS Lett., 192, 267–270. [DOI] [PubMed] [Google Scholar]
- 23.Pacchioni B., Trevisan,S., Gomirato,S., Toppo,S., Valle,G. and Lanfranchi,G. (1996) Semi-multiplex PCR technique for screening of abundant transcripts during systematic sequencing of cDNA libraries. Biotechniques, 21, 644–649. [DOI] [PubMed] [Google Scholar]
- 24.Ewing B., Hillier,L., Wendl,M.C. and Green,P. (1998) Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res., 8, 175–185. [DOI] [PubMed] [Google Scholar]
- 25.Liang F., Holt,I., Pertea,G., Karamycheva,S., Salzberg,S.L. and Quackenbush,J. (2000) An optimized protocol for analysis of EST sequences. Nucleic Acids Res., 28, 3657–3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Altschul S.F., Gish,W., Miller,W., Myers,E.W. and Lipman,D.J. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 27.Decraene C., Reguigne-Arnould,I., Auffray,C. and Pietu,G. (1999) Reverse transcription in the presence of dideoxynucleotides to increase the sensitivity of expression monitoring with cDNA arrays. Biotechniques, 27, 962–966. [DOI] [PubMed] [Google Scholar]
- 28.Donis-Keller H. (1979) Site specific enzymatic cleavage of RNA. Nucleic Acids Res., 7, 179–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Young B.D. and Anderson,M.L.M. (1985) Quantitative analysis of solution hybridization. In Hames,B.D. and Higgins,S.J. (eds), Nucleic Acid Hybridization: A Practical Approach. IRL Press, Oxford, UK, pp. 47–71.
- 30.Wang S.M., Fears,S.C., Zhang,L., Chen,J.J. and Rowley,J.D. (2000) Screening poly(dA/dT)– cDNAs for gene identification. Proc. Natl Acad. Sci. USA, 97, 4162–4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rubenstein J.L., Brice,A.E., Ciaranello,R.D., Denney,D., Porteus,M.H. and Usdin,T.B. (1990) Subtractive hybridization system using single-stranded phagemids with directional inserts. Nucleic Acids Res., 18, 4833–4842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duggan D.J., Bittner,M., Chen,Y., Meltzer,P. and Trent,J.M. (1999) Expression profiling using cDNA microarrays. Nature Genet., 21 (suppl.), 10–14. [DOI] [PubMed] [Google Scholar]
- 33.Soares M.B. (1997) Identification and cloning of differentially expressed genes. Curr. Opin. Biotechnol., 8, 542–546. [DOI] [PubMed] [Google Scholar]
- 34.Campbell W.G., Gordon,S.E., Carlson,C.J., Pattison,J.S., Hamilton,M.T. and Booth,F.W. (2001) Differential global gene expression in red and white skeletal muscle. Am. J. Physiol. Cell Physiol., 280, C763–C768. [DOI] [PubMed] [Google Scholar]
- 35.St-Amand J., Okamura,K., Matsumoto,K., Shimizu,S. and Sogawa,Y. (2001) Characterization of control and immobilized skeletal muscle: an overview from genetic engineering. FASEB J., 15, 684–692. [DOI] [PubMed] [Google Scholar]
- 36.Soares M.B and Bonaldo,M.F. (1998) Constructing and screening normalized cDNA libraries. In Birren,B., Green,E.D., Klapholz,S. Myers,R.M., Riethman,H. and Roskams,J. (eds), Genome Analysis: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, Vol. 2, pp. 49–157.
- 37.Diatchenko L., Lukyanov,S., Lau,Y.F. and Siebert,P.D. (1999) Suppression subtractive hybridization: a versatile method for identifying differentially expressed genes. Methods Enzymol., 303, 349–380. [DOI] [PubMed] [Google Scholar]
- 38.Valle G., Faulkner,G., De Antoni,A., Pacchioni,B., Pallavicini,A., Pandolfo,D., Tiso,N., Toppo,S., Trevisan,S. and Lanfranchi,G. (1997) Telethonin, a novel sarcomeric protein of heart and skeletal muscle. FEBS Lett., 415, 163–168. [DOI] [PubMed] [Google Scholar]
- 39.Pallavicini A., Kojic,S., Bean,C., Vainzof,M., Salamon,M., Ievolella,C., Bortoletto,G., Pacchioni,B., Zatz,M., Lanfranchi,G., Faulkner,G. and Valle,G. (2001) Characterization of human skeletal muscle ankrd2. Biochem. Biophys. Res. Commun., 285, 378–386. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}