

SUMMARY
CREB-regulated transcription co-activator (CRTC) is activated by Calcineurin (CaN) to regulate gluconeogenic genes. CaN also has roles in cardiac hypertrophy. Here, we explore a cardiac-autonomous role for CRTC in cardiac hypertrophy. In Drosophila, CRTC mutants exhibit severe cardiac restriction, myofibrillar disorganization, fibrosis, and tachycardia. Cardiac-specific CRTC knockdown (KD) phenocopies mutants, and cardiac overexpression causes hypertrophy. CaN-induced hypertrophy in Drosophila is reduced in CRTC mutants, suggesting that CRTC mediates the effects. RNA sequencing (RNA-seq) of CRTC-KD and -overexpressing hearts reveals contraregulation of metabolic genes. Genes with conserved CREB sites include the fly ortholog of Sarcalumenin, a Ca2+-binding protein. Cardiac manipulation of this gene recapitulates the CRTC-KD and -overexpression phenotypes. CRTC KD in zebrafish also causes cardiac restriction, and CRTC KD in human induced cardiomyocytes causes a reduction in Srl expression and increased action potential duration. Our data from three model systems suggest that CaN-CRTC-Sarcalumenin signaling represents an alternate, conserved pathway underlying cardiac function and hypertrophy.
In brief
Metabolic disorders such as obesity, insulin resistance, and diabetes increase the risk of heart disease, especially cardiac hypertrophy. In this paper, Dondi et al. present evidence that CRTC, a nutrient sensor in the liver, also plays a conserved and cardiac-autonomous role in maintaining heart structure and function.
Graphical Abstract
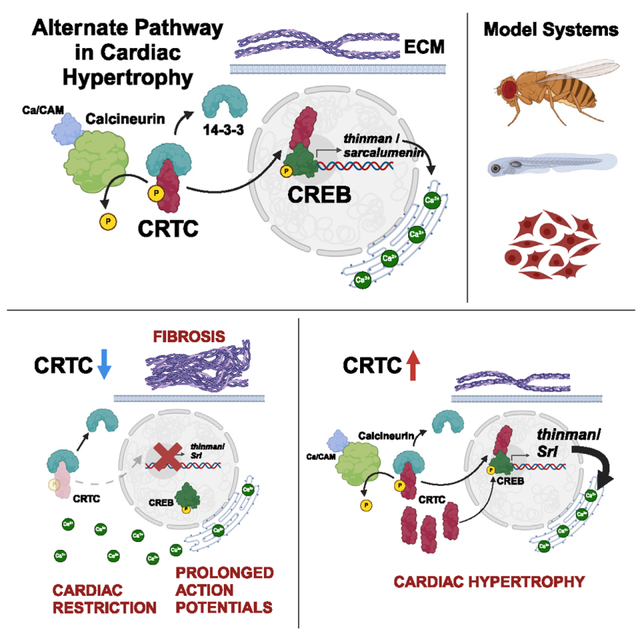
INTRODUCTION
Despite decades of research, heart disease (HD) remains the leading cause of death in the industrialized world. An increasing number of HDs are attributed to disorders of metabolism such as obesity, insulin resistance, and diabetes.1–4 Left ventricular hypertrophy has been diagnosed in as many as 50% of type 2 diabetes patients and is predictive of more adverse cardiovascular events.5 Calcineurin (CaN) has been established as part of the canonical signaling pathway underlying mammalian cardiac hypertrophy through its activation of downstream transcriptional regulators, such as Mef2 and NFAT.6–8 CaN, a Ca2+/Calmodulin-dependent phosphatase, also plays important roles in insulin-sensitive tissues, such as the liver, where it dephosphorylates and activates CRTC, a CREB (cAMP-responsive element binding protein)-regulated transcription co-activator9 (previously TORC). Under basal conditions, CRTC is phosphorylated by SIK2, a salt-inducible kinase, resulting in increased association with 14–3-3 proteins and sequestration in the cytoplasm.10 During fasting, glucagon signaling leads to activation of cAMP-dependent protein kinase A (PKA), which phosphorylates and inhibits SIK2 while also activating CREB. Simultaneous activation of CRTC (by Ca2+-dependent CaN) and CREB (by cAMP-dependent PKA) acts as a “coincidence detector” to increase transcription of target genes involved in gluconeogenesis.9
In mammals, there are three forms of CRTC, and while much is known about the role of CRTC in liver and neurons, there is little evidence to support a role for CRTC in cardiac function. A single study in mice linked systemic knockout (KO) of CRTC1 to cardiac hypertrophy, but this phenotype was likely secondary to effects on neuronal function and activation of β-adrenergic receptors.11 In addition, CRTC1 is primarily expressed in nervous tissue,12,13 whereas CRTC2 and CRTC3 are more ubiquitously expressed.14 Here, we provide evidence that CRTC functions autonomously in the heart. Using the fly model, we show a cardiac-specific effect of CRTC on cardiac structure and fibrosis that worsens with age. In the zebrafish model, CRTC3 knockdown (KD) led to defects in heart structure and function similar to those in flies. We also observed functional defects in response to KD of CRTC2 and CRTC3 in human induced cardiomyocytes (CMs). Our analysis of changes in gene expression in the fly heart in response to cardiac-specific CRTC KD and overexpression (OE) indicate that Sarcalumenin (SRL) is a downstream effector of CRTC in the heart. Overall, our data suggest that CRTC provides an alternative pathway to NFAT in mediating cardiac hypertrophy, in part by regulating the expression of Sarcalumenin (Srl) in cardiac muscle.
RESULTS
CRTC mutant flies exhibit cardiac dysfunction
Adult Drosophila with systemic CRTC KO are sensitive to starvation and oxidative stress and are very lean.15 Neuronal OE of CRTC only partially rescues these phenotypes. We initially used a cardiac pacing assay to examine the effects of stress on heart failure rates in CRTC-KO mutants and wild-type flies (w1118, the genetic background of the CRTC mutant line15). Male and female flies were paced with external electrodes (described previously16,17), and hearts were scored for rhythmic beating at 60 and 120 s post-pacing. At 60 s, approximately 40% of control hearts beat regularly compared to less than 20% of CRTC mutant hearts. By 120 s post-pacing, nearly 80% of hearts in the controls had recovered rhythmic function compared to 38% of CRTC-KO hearts (Figure S1A). Thus, CRTC mutants exhibited significantly compromised in vivo heart function.
We confirmed cardiac expression of CRTC with qPCR analysis of isolated hearts revealing robust expression in wild-type control flies and none in CRTC mutant hearts (Figure S1B). In addition, we used in situ hybridization chain reaction (HCR) to localize CRTC expression within the heart structure. HCR staining revealed the expected staining in the myocardial nuclei in controls (Figure S1C) as well as in the associated alary muscle nuclei (Figure 1C). Expression was nearly absent in myocardial cells and alary muscles in CRTC-null mutants (Figure S1D).
Figure 1. CRTC KO causes cardiac dysfunction and fibrosis that are mimicked by CREB KO.
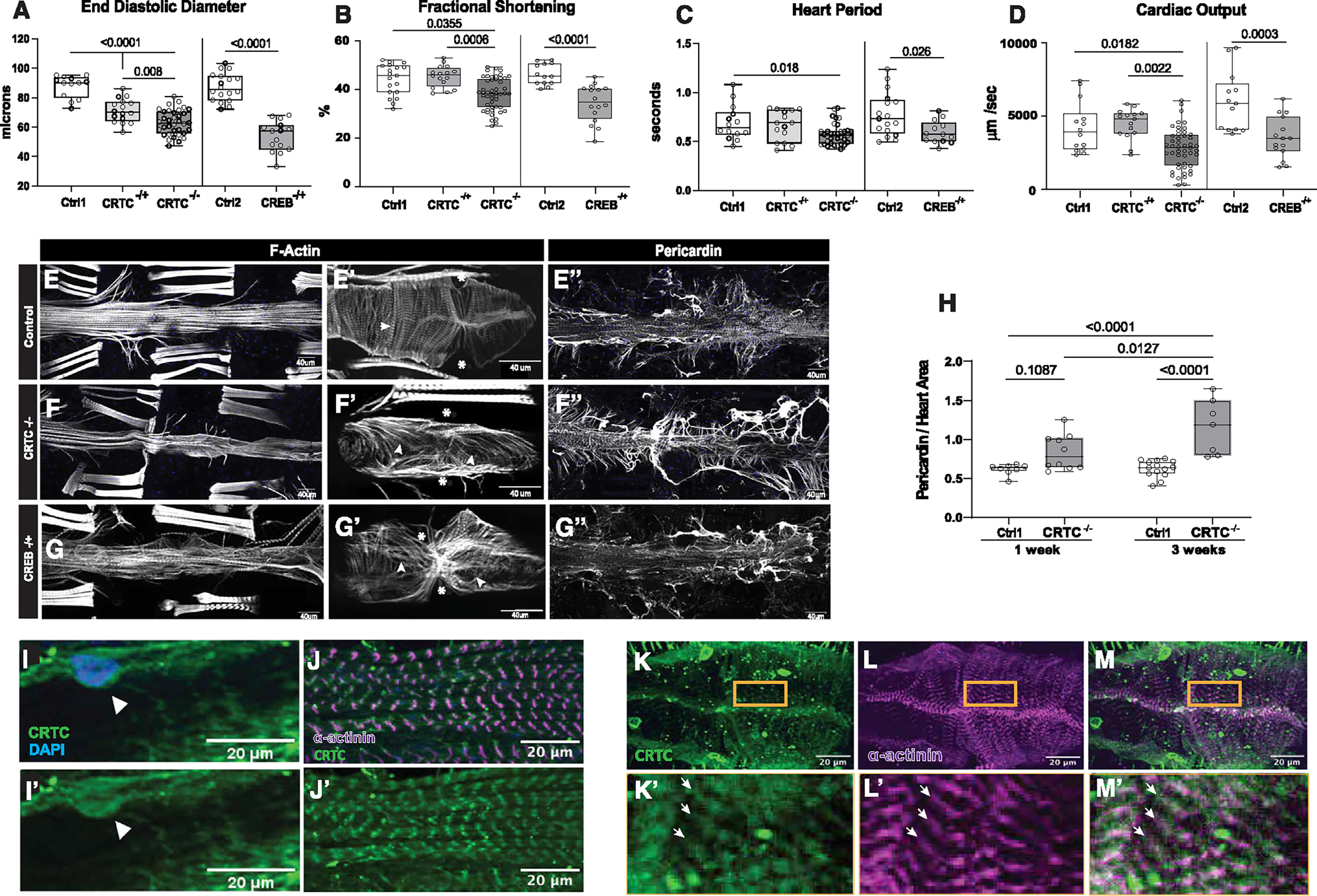
(A) (Left) KO of CRTC significantly reduced end diastolic diameter (EDD) in hearts from both heterozygous (−/+) and homozygous (−/−) mutants compared to genetic background controls (w1118). (Right) Similar cardiac restriction was observed in hearts from CREB−/+ heterozygotes.
(B) (Left) Fractional shortening was reduced in hearts from CRTC−/− mutants and (right) in CREB−/+ heterozygotes.
(C) (Left) Heart period (one contraction cycle) was reduced in CRTC-KO flies, but not in CRTC heterozygotes. (Right) CREB−/+ heterozygote flies also showed significant reductions in HP.
(D) (Left) Cardiac output was significantly reduced in CRTC−/− mutants and (right) in CREB−/+ heterozygotes.
(For A–D, flies were 1 week of age; p values by one-way ANOVA for CRTC mutants and two-tailed, unpaired t tests for CREB mutants; plots show all data points, max, min, median, and p values.)
(E) F-actin staining with phalloidin reveals the cardiac tube and (E′) tightly packed circumferential fibers (arrowhead) in an optical section from a single chamber in a control. (Asterisks denote position of ostia, anterior is left.) (E″) Control stained for collagen IV (pericardin) reveals the extensive extracellular matrix that surrounds the heart.
(F) F-actin staining in a CRTC mutant exhibiting cardiac restriction and (F′) disorganized myofibrils (arrowheads); ostia are also malformed (asterisks). (F″) The collagen matrix in CRTC−/− mutants is significantly expanded, especially in the posterior region (right).
(G) Cardiac chamber from a CREB heterozygote mutant showing cardiac restriction and (G′) disorganized myofibrils with gaps (arrowheads) and malformed ostia (asterisks). (G″) Collagen network in the CREB−/+ heterozygotes is expanded, especially in the posterior region (right).
(For E–G″, scale bars represent 40 μm).
(H) Quantification of collagen area normalized to cardiac actin area is increased in CRTC mutants compared to controls at 1 week (p values by two-tailed, unpaired t tests).
(I and I′) Immunohistochemistry shows nuclear CRTC localization in cardiomyocytes (arrowheads, CRTC staining is green, nuclear staining is blue).
(J and J′) Co-staining of non-myocardial ventral longitudinal fibers with CRTC and α-actinin antibodies shows CRTC (green) strongly associated with α-actinin in Z bands (magenta).
(K–M) Optical sections through one chamber of the heart stained for CRTC (K, green), α-actinin (L, magenta), and (M) merged image. (K′-M′) Higher magnification of regions in yellow rectangles. Arrows show location of anti-CRTC staining (K′), α-actinin staining (L′), and co-localization (M′, white).
(For I–M′, scale bars represent 20 μm).
To directly examine heart function, we used denervated, semi-intact fly heart preparations.18 Flies were collected upon eclosion, aged to 1 week, dissected to remove the central nervous system, and assayed for heart function by high-speed video imaging.19,20 Hearts from female CRTC mutants were significantly thinner compared to controls, during both diastole (Figure 1A, left) and systole (Figure S1G, left), associated with a significant reduction in contractility (measured by fractional shortening, Figure 1B, left). Hearts from female CRTC mutants also had significant reductions in stroke volume (Figure S1H, left) and heart period (length of one contraction cycle, Figure 1C, left). However, despite the increased heart rate (1/heart period), cardiac output was still reduced (Figure 1D, left; see also Video S1). Male CRTC mutant flies showed similar reductions in end diastolic diameter (EDD), end systolic diameter (ESD), and fractional shortening compared with their female counterparts (Figures S1I–S1K). Although there was no significant change in the heart period in males compared to controls (Figure S1L), they still showed significantly decreased cardiac output (Figure S1M).
The effects of CRTC in the liver have been shown to be mediated by interactions with CREB.9,21,22 Because homozygous CREB mutations are lethal, we examined heart function in female CREBbD40015 heterozygotes (hereafter CREBb−/+). We observed similar reductions in heart size, fractional shortening, heart period, and cardiac output in CREBb−/+ flies (Figures 1A–1D, right, and Figures S1G and S1H, right). These results suggested that CRTC loss of function caused the observed heart dysfunction and that CRTC acts by modulating CREB function, as previously reported15 (Data S1 contains all the raw data for each text and supplemental figure).
CRTC KO causes myofibrillar disorganization and fibrosis
We assessed the effects of CRTC loss of function on cardiac morphology. Following heart functional assessments, hearts were relaxed by the addition of EGTA, fixed, and stained for both F-actin and the cardiac extracellular matrix protein pericardin (collagen IV homolog). The linear Drosophila heart tube consists of a single layer of myocardial cells and is separated into four chambers by internal valves (Figures 1E, 1F, and 1G). The myofibrils within the myocardial cells are normally arranged circumferentially (Figures 1E′ and S2A–S2C; note vertical arrangement of myofibrils), allowing the heart tube to contract and eject hemolymph. In CRTC-mutant and -KD hearts, myofibrils exhibit significant disorganization with many gaps between the myofibrils (Figures 1F′, S2E, and S2F; note the more horizontally oriented myofibrils). Myocardial cells in hearts from CREBb−/+ flies also showed disorganized myofibrils with non-circumferential orientations and gaps (Figures 1G′ and S2G). We used the “Directionality” plugin in ImageJ to quantify the extent of myofibrillar disorganization in regions of interest (ROIs) made within individual myocardial cells (Figure S2; STAR Methods) and found a significant rearrangement from vertical to more horizontally organized myofibrils in the CRTC and CREB mutant files compared to controls (Figures S2E–S2H). In addition, immunohistochemical staining for pericardin (collagen IV) showed that the network that normally surrounds the heart tube (Figure 1E″) was significantly increased for both CRTC mutants and CREBb−/+ flies compared to controls and was especially enhanced in the posterior region of the heart (compare Figure 1E″ with 1F″ and 1G″). We used the Weka segmentation tool in ImageJ to quantify the collagen deposition from z stack images and showed an age-dependent increase in collagen deposition in CRTC mutants (Figure 1H).
Fly hearts were also stained with CRTC antibodies23 to localize CRTC protein. As expected, we observed prominent CRTC staining in myocardial cell nuclei (Figures 1I and 1I′). In addition, we saw discrete bands of CRTC staining associated with bands of α-actinin staining in both the overlying, non-cardiac longitudinal fibers (Figures 1J and J′) and the myocardial cells (Figure 1K–1M), suggesting that CRTC is localized to Z bands in muscle. To confirm specificity, we also examined CRTC-null mutants and observed a near-complete absence of staining (Figure S1N). We also examined hearts from a fly line with FLAG-tagged CRTC. Hearts were probed with anti-FLAG antibody, and we observed similar staining patterns compared with the CRTC antibody (Figure S1O).
CRTC mutants show normal embryonic heart development
To test whether the effects we observed in adults originated as congenital defects, we examined embryonic specification of cardioblasts in stage 17 embryos. At this stage the cardioblasts are fully specified and are arranged as parallel rows of cells along the dorsal midline (Figures S3A–S3C). All cardioblasts express Neuromancer-1/H15 (Nmr1/H15) and, in a subset that will form the ostia (inflow tracts), Seven-up (Svp), whereas the closely associated pericardial cells selectively express Zinc finger homeodomain 1 (zfh1). Both wild-type and mutant embryos stained for these markers exhibited similar staining patterns for all cell types and similar cell numbers/sizes (Figure S3, compare S3A–S3C with S3D–S3F), suggesting that embryonic heart specification and development occur normally in mutants.
CRTC acts cardiac autonomously to affect heart function
To address whether CRTC affects the heart cell autonomously or systemically, we used a Gal4/upstream activating sequence (UAS)-mediated RNAi approach to tissue-specifically knock down target genes. We used the myocardial cell-specific driver tinCΔ4-Gal424 crossed to UAS-CRTC RNAi lines and assayed heart function of adult female progeny at 3 weeks of age. CM-specific KD of CRTC caused a significant reduction in heart diameter (Figures 2A and S4A, Video S1) and reduced stroke volume (Figure S4B). As for CRTC mutants, the reduced heart period (increased heart rate) in CRTC-KD hearts (Figure 2C) did not compensate for the reduced volume, as cardiac output was significantly reduced (Figure 2D). In contrast, cardiac-specific CRTC OE significantly increased diastolic and systolic diameters (Figures 2A and S4A, Video S2) and decreased fractional shortening (Figure 2B). Heart period was significantly increased (decreased heart rate) (Figure 2C); nevertheless, cardiac output significantly increased due to the larger heart size (Figure 2D).
Figure 2. The effects of CRTC are cardiac autonomous.
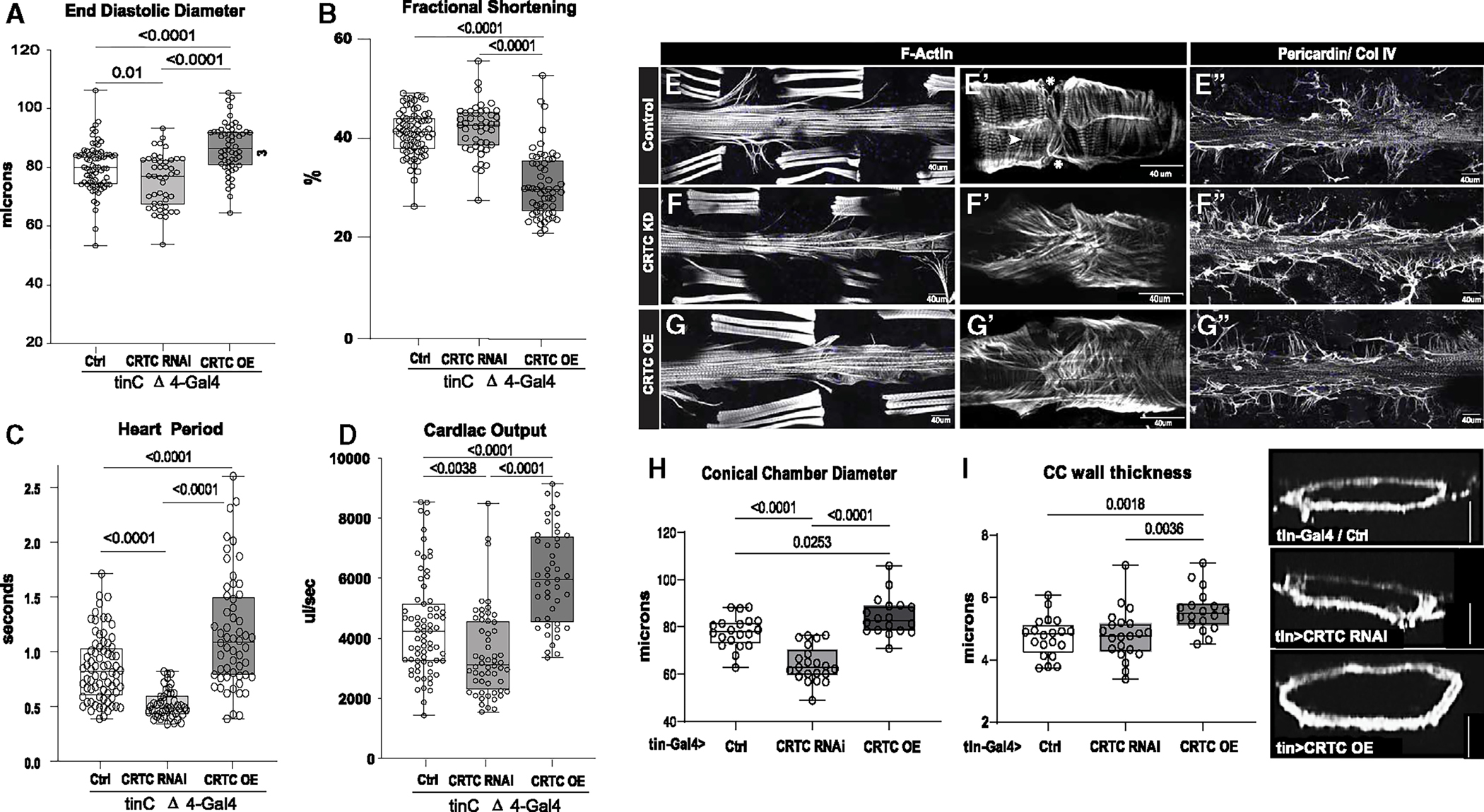
(A) Cardiac-specific CRTC KD with tinCΔ4-Gal4 significantly reduced EDD, while cardiac CRTC OE increased EDD, compared to controls (tinCΔ4-Ga4/+).
(B) Fractional shortening was significantly reduced by cardiac CRTC OE.
(C) Heart period was significantly reduced in CRTC-KD hearts and increased in hearts with CRTC OE.
(D) Cardiac output was reduced in CRTC-KD hearts and increased in hearts with CRTC OE. All flies were 3 weeks of age; plots show all data points, max, min, median, and p values; significance by one-way ANOVA with Tukey’s multiple comparisons post hoc test.
(E–G) (E) F-actin staining shows sarcomeric structure in cardiac tubes and adjacent body wall muscles. (E′) Optical section through one cardiac chamber showing the tightly packed, circumferential fibers (arrowhead) in a control heart. (Asterisk denotes position of the ostia, anterior is left in all pictures.) (F) F-actin staining of a heart with cardiac CRTC KD, exhibiting cardiac restriction and (F′) disorganized myofibrils with malformed ostia. (G) Cardiac CRTC-OE heart showing cardiac dilation and (G′) some disorganized myofibrils and gaps and malformed ostia. (E″) Wild-type control stained for collagen IV (pericardin) reveals the extensive extracellular matrix. (F″) The collagen matrix in CRTC KD is expanded, especially in the posterior heart (right), but (G″) not with CRTC OE.
(For E–G″, scale bars represent 40 μm).
(H) Diameters from optical sections of dystroglycan-stained hearts. Cardiac conical chamber (CC) diameters were significantly reduced with cardiac CRTC KD and increased with cardiac CRTC OE.
(I) Cardiomyocyte thickness, measured from optical sections of dystroglycan-stained hearts (to the right, scale bar represents 50 μm), was increased with cardiac CRTC OE compared to control and cardiac CRTC-KD hearts.
To further substantiate that the effects of CRTC were CM autonomous, we examined the effects of CRTC KD in nephrocyte-like pericardial cells, which have previously been reported to exert effects on the heart.25 CRTC KD with the Dot-Gal4 (pericardial cell) driver caused phenotypes opposite to CRTC myocardial cell KD, and OE had no effect (Figures S4D–S4F). KD with a second pericardial cell-specific driver, SNS (sticks and stones)-Gal4, had no effect on cardiac function (Data S1). Since CRTC is highly expressed in vertebrate neurons, we also modulated CRTC expression specifically in the nervous system using elav-Gal4,26 but again observed no significant effect on cardiac function (Figures S4G–S4I). CRTC also plays important roles in liver cells by mediating the effects of glucagon signaling. The fly fat body is thought to perform functions similar to those of liver cells in humans.27 However, modulating CRTC expression with fat-body-specific drivers lsp-Gal428 (Figures S4J–S4L) or CG-Gal4 (Data S1) had no significant effect on heart function. Thus, CRTC appears to play a cardiac-autonomous role in maintaining adult cardiac structure and function in the fly (Data S1).
Cardiac-specific manipulation of CRTC causes cardiac remodeling, fibrosis, and hypertrophy
To examine the role of CRTC in cardiac maintenance, fly hearts were stained for F-actin and anti-pericardin antibody to label the collagen IV network surrounding the heart. We again found cardiac restriction following CRTC KD and enlargement in response to CRTC OE (Figures 2E–2G). Images of single heart chambers at higher magnification revealed closely packed circumferential myofibrils in controls but significant myofibrillar disorganization in CRTC-KD hearts, with some disorganization in CRTC OE (Figures 2E′–2G′, S2H, and S2L). In mutants, cardiac CRTC KD increased the collagen IV staining around the heart, while CRTC OE had no effect (Figures 2E″–2G″). Thus, CM-specific CRTC KD mimicked the effects of CRTC-null mutants.
We next determined whether the enlargement in response to CRTC OE was due to overall cardiac dilation or to an increase in the thickness of myocardial cells. To measure CM thickness, we focused on the conical chamber (CC), the largest, most anterior part of the heart tube. We labeled the inner and outer plasma membrane by staining for dystroglycan, a ubiquitous membrane-bound glycoprotein. Measurements of the outer diameters from the CC confirmed chamber restriction with cardiac CRTC KD and enlargement with CRTC OE (Figure 2H). We quantified the thickness of the myocardial cells from optical sections of dystroglycan-stained CCs. Cardiac CRTC OE significantly increased CM thickness compared to controls, while CM thickness in CRTC-KD hearts was unchanged compared to controls (Figure 2I).
CRTC effects are downstream of CaN but are independent of NFAT
In the liver, CRTC is activated by CaN-mediated dephosphorylation, allowing it to enter the nucleus and modulate DNA binding by activated CREB. CaN OE has been reported to cause cardiac hypertrophy in mouse models6 and in the fly,29 and we previously showed that cardiac-specific CaN KD of the fly homolog, Pp2B, caused cardiac restriction.30 To explore the interactions between CRTC and CaN in the fly heart, we overexpressed constitutively activated Pp2B in CRTC-mutant flies. As previously reported,29 cardiac OE of activated Pp2B/CaN caused a significant enlargement of the heart with a slight but significant increase in fractional shortening (Figures 3A and 3B). Heart period was also significantly increased (bradycardia) in response to Pp2B OE (Figure 3C), and this reduced rate likely accounts for the lack of an effect on cardiac output (Figure 3D) despite the larger heart size. Notably, these effects were similar to those seen in response to cardiac CRTC OE (Figures 2A–2D). Importantly, the hypertrophy and bradycardia induced by Pp2B/CaN OE could be reversed by reduction of CRTC expression in a dose-dependent manner (Figures 3A–3C), suggesting that CRTC mediates some of the downstream effects of CaN in the fly heart.
Figure 3. Hypertrophic effects of Calcineurin/Pp2B are rescued by reductions in CRTC.
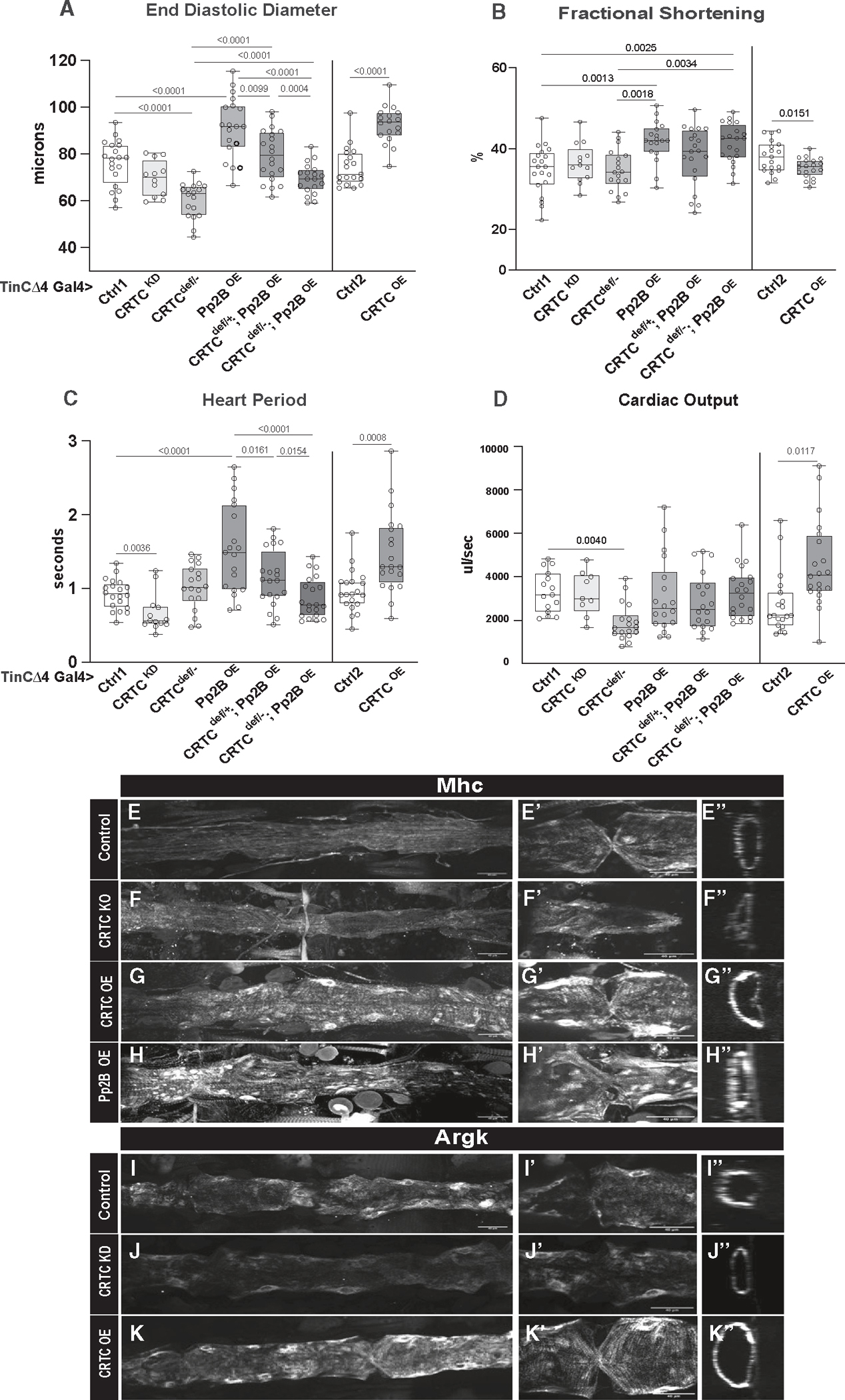
(A) (Left) EDD was reduced in hearts from cardiac-specific CRTC KD and CRTCdeficiency/− mutants compared to genetic background controls. Constitutive cardiac Pp2B OE increased EDD, which was progressively rescued in CRTCdef/+ (heterozygous) and CRTCdef/− (homozygous) mutant backgrounds. (Right) Cardiac-specific CRTC OE caused a significant increase in EDD, as for cardiac Pp2B OE.
(B) (Left) Fractional shortening increased with cardiac-specific Pp2B OE but was unaffected by reduced CRTC expression. (Right) CRTC OE caused a reduction in fractional shortening compared to genetic background controls.
(C) (Left) Cardiac-specific CRTC KD caused tachycardia (shorter heart periods), whereas Pp2B OE caused bradycardia (longer heart periods) that was progressively reversed by reducing CRTC expression. (Right) CRTC OE caused bradycardia compared to genetic background controls.
(D) (Left) Cardiac output was again reduced in response to cardiac-specific CRTC KD. (Right) Output was increased by cardiac CRTC OE.
(For A–D, plots show all data points, max, min, median, and p values; significance by one-way ANOVA with Tukey’s multiple comparisons post hoc test).
(E–H) HCR was used to examine expression of the hypertrophic marker Mhc (myosin heavy chain) in the fly heart. (E) Mhc expression in the control heart is uniformly distributed along the tube, (E′) in the myocardial cells of a single chamber, and (E″) in an optical cross section of the heart. (F) Mhc expression is slightly reduced in CRTC mutant heart, (F′) in the myocardial cells, and (F″) in an optical cross section. (G) Mhc expression is dramatically increased in the CRTC OE heart, (G′) in the thickened myocardial cells, and (G″) in a myocardial cell cross section. (H) Mhc expression is dramatically increased in the Pp2B OE heart, (H′) in the thickened myocardial cells, and (H″) in a myocardial cell cross section.
(I–K) Arginine kinase (Argk) is the fly ortholog of creatine kinase, a marker for cardiac damage. (I) Argk expression was uniformly distributed in the control heart, (I′) in myocardial cells, and (I″) in the cardiac tube cross section. (J) Argk expression was slightly reduced in the CRTC mutant heart, (J′) in the myocardial cells, and (J″) in the cardiac tube cross section. (K) Argk expression was dramatically increased in the CRTC-OE heart, (K′) in the myocardial cells, and (K″) in the cross section.
(For E–K″, all images were taken with the same exposure settings; scale bars represent 40 μm).
In vertebrates, the effects of CaN OE on cardiac hypertrophy were reported to be mediated by the transcription factor NFAT.31 We used the tinCΔ4-Gal4 driver to heart-specifically knock down NFAT; we also tested for interactions in a CRTC-heterozygous “sensitized” background. Cardiac KD of NFAT alone had no effect on heart size, and there was no genetic interaction between CRTC+/− and NFAT (Figures S5A and S5B), suggesting that NFAT does not affect heart structure and function in flies, nor does it act through CRTC or vice versa in the fly heart.
Cardiac CRTC OE causes increased expression of hypertrophy markers
Altered expression and function of myosin heavy chain (Mhc) have been associated with cardiac hypertrophy.32 We used HCR to quantify cardiac expression of Mhc and observed ubiquitous, diffuse expression of Mhc within myocardial cells in both control and CRTC-KO hearts (Figures 3E and 3F″). Cardiac CRTC OE showed increased Mhc expression throughout the myocardial tissue (Figures 3G and 3G″). Similar results were observed in hearts with cardiac Pp2B/CaN OE (Figures 3H and 3H″). We also examined expression of arginine kinase (Argk), the fly ortholog of creatine kinase, which can be a marker for cardiac damage in humans,33 and isoform switching has been observed in hypertrophic hearts.34 We observed ubiquitous Argk expression within myocardial cells in controls (Figures 3I and 3I″), decreased expression in CRTC-KD hearts (Figures 3J and 3J″), but increased expression in CRTC-OE hearts (Figures 3K and 3K″). These results support the conclusion that CRTC functions cardiac autonomously and that CRTC plays a role in cardiac hypertrophy.
CRTC affects somatic muscle function
Climbing assays were performed with CRTC systemic mutant and CRTC cardiac-specific KD female and male flies at 1 and 3 weeks of age. We assessed the number of flies that were able to climb more than 2 cm in 10 s. We observed that less than 30% of CRTC systemic mutant flies were able to cross the 2 cm threshold, whereas almost 80% of control flies climbed above the set threshold in 10 s (Figures S5C and S5D). Notably, climbing performance in cardiac-specific CRTC-KD flies was not significantly different from that of controls (Figures S5E and S5F), suggesting that it was the loss of CRTC in somatic muscles that impaired climbing ability in mutants.
CRTC is highly expressed in the zebrafish heart, and KD causes cardiac restriction
We used zebrafish to examine the role of CRTC in a vertebrate model. We used qPCR to quantify cardiac CRTC1,2, and 3 expression in hearts isolated from adult fish (10 months of age, eight hearts per sample). qPCR analysis showed that all three forms of CRTC were expressed in the heart, but CRTC3 was by far the most prominent form (Figure S5G). We co-stained larval zebrafish hearts with antibodies against CRTC and MF20 (muscle myosin). The CRTC antibody was specific for a region in human CRTC that is highly conserved across all three paralogs and conserved in CRTC 2 and 3 in fish. Similar to flies, CRTC localized to nuclei and also exhibited a striated pattern, localizing to regions between M bands, i.e., where Z bands are located (Figures 4A′ and 4A‴). To assess the cardiac role of CRTC, we injected morpholinos (MO) targeting CRTC3 (Figure S5H) into fertilized embryos at the one-cell stage and analyzed heart function at 72 h post-fertilization (hpf), a stage when the fish are still transparent and the heart can be readily visualized. To confirm CRTC3 morpholino KD, we stained larval zebrafish hearts with anti-CRTC antibodies and observed significant reductions in nuclear staining as well as a loss of the banded staining pattern associated with sarcomeres (Figures 4B and 4B‴). CRTC3 KD did not noticeably affect the overall morphology of the fish35; cardiac looping, tail musculature, and fin development were unperturbed (Figures 4C and 4D). Analysis of heart function showed that the primary effect of CRTC3 KD was a small but significant reduction in the diastolic and systolic surface areas of hearts in KD fish compared to controls (Figures 4E and 4F; Video S3). This cardiac restriction occurred in both atria and ventricles but was most prominent in the ventricles and paralleled the cardiac restriction seen with CRTC KO and KD in the fly heart (Figures 1 and 2).
Figure 4. CRTC affects cardiac structure and function in zebrafish.
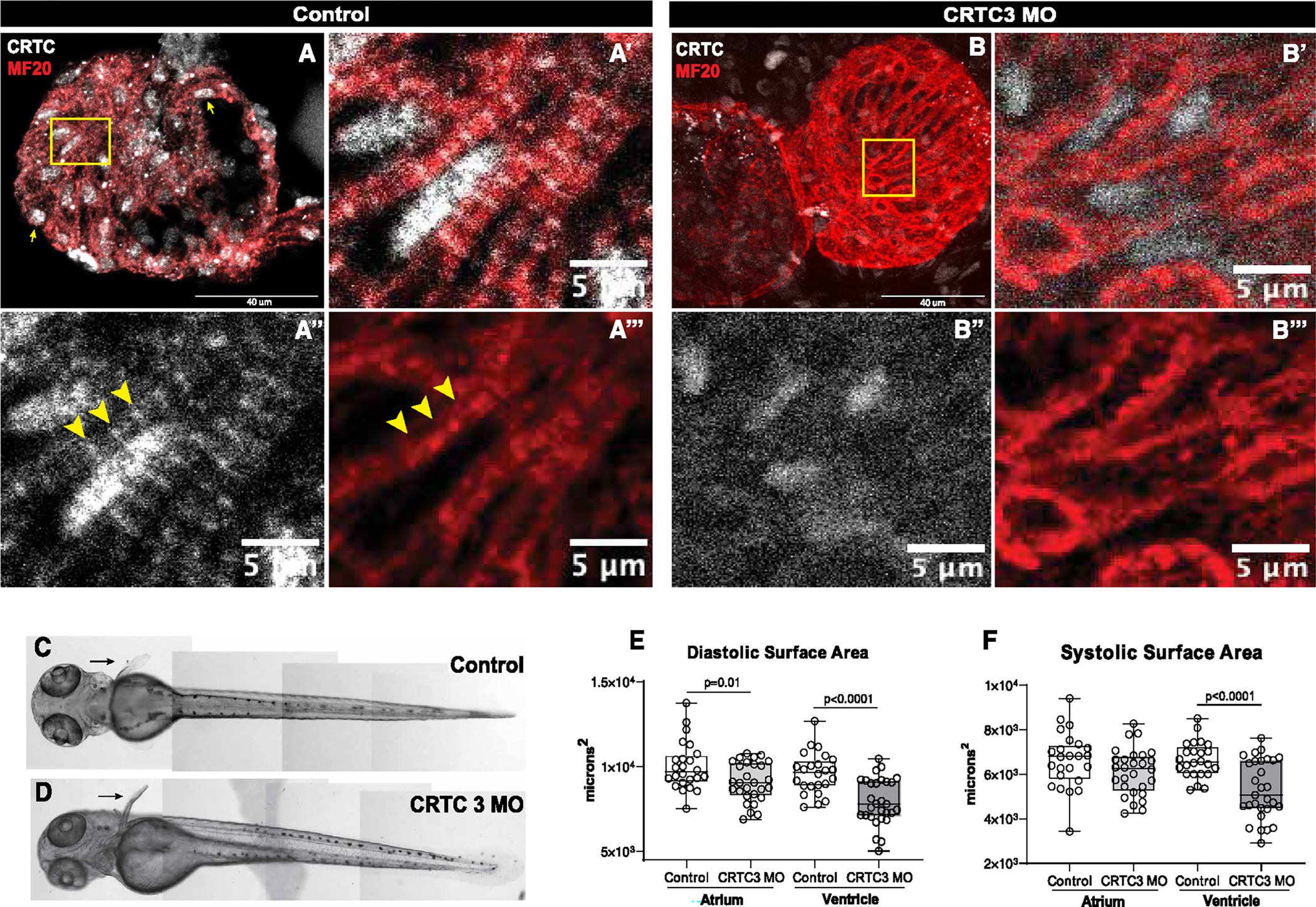
(A) Hearts from 72 hpf zebrafish were stained for myosin (MF20, red) and CRTC (white). Scale bar, 40 μm. (A′) Magnified inset from (A) showing significant nuclear localization of CRTC. (A″) CRTC also showed a distinct banded structure in cardiomyocytes (arrowheads; scale bar, 5 μm). (A‴) Myosin staining showing cardiac myofibrils. The position of CRTC staining in A″ is shown by arrowheads and appears to localize between bands of myosin staining. Scale bar, 5 μm.
(B) CRTC3 morpholino-KD hearts stained for myosin (MF20, red) and CRTC (white; note that all images in the CRTC channel were significantly overexposed). Scale bar, 40 μm. (B′) Magnified inset from (B) showing some residual CRTC in nuclei. (B″) CRTC staining in the morpholino heart was reduced and diffuse and did not show any structure pattern. Scale bar, 5 μm. (B‴) Myosin staining showing cardiac myofibrils. Scale bar, 5 μm.
(C and D) Images of (C) control (DMSO injected) and (D) CRTC-KD fish at 72 hpf show normal development of body-wall muscle and fins (arrows).
(E) End diastolic surface area of hearts in 72 hpf fish was significantly reduced in atria and especially in the ventricles, following MO KD of CRTC3.
(D) End systolic surface area in ventricles was also significantly reduced. (For D and E, plots show all data points, max, min, median, and p values; significance by two-way unpaired t test).
RNA-sequencing analysis of CRTC-KD and -OE hearts reveals concerted regulation of cardiac metabolic pathways
To examine the effects of CRTC on cardiac gene expression, we performed whole-transcriptome analysis of isolated fly hearts (GEO: GSE271481). Hearts from CM-specific (tinCΔ4-Gal4) CRTC-KD flies exhibited upregulation of 357 and downregulation of 186 transcripts, whereas cardiac CRTC OE resulted in the upregulation of 426 and downregulation of 390 transcripts relative to their respective control flies (fold change ±1.25 log2 fold; adj. q < 0.05) (Figures S6A and S6B). Comparative analysis revealed that a total of 140 genes were contraregulated between CRTC-KD and -OE flies, suggesting CRTC-dependent regulation of specific transcriptional programs (Figure 5A; Data S2). Gene Ontology (GO) analysis showed that loss of CRTC enhanced, while gain of CRTC suppressed, pathways involved in oxidative-phosphorylation, innate immune response, glycolysis/gluconeogenesis, amino acid biosynthesis, and fatty acid elongation. Conversely, loss of CRTC suppressed, and gain of CRTC enhanced, expression of fatty acid degradative genes (Figure 5B; Data S3).
Figure 5. Cardiac CRTC KD and OE concertedly regulate metabolism in the heart.
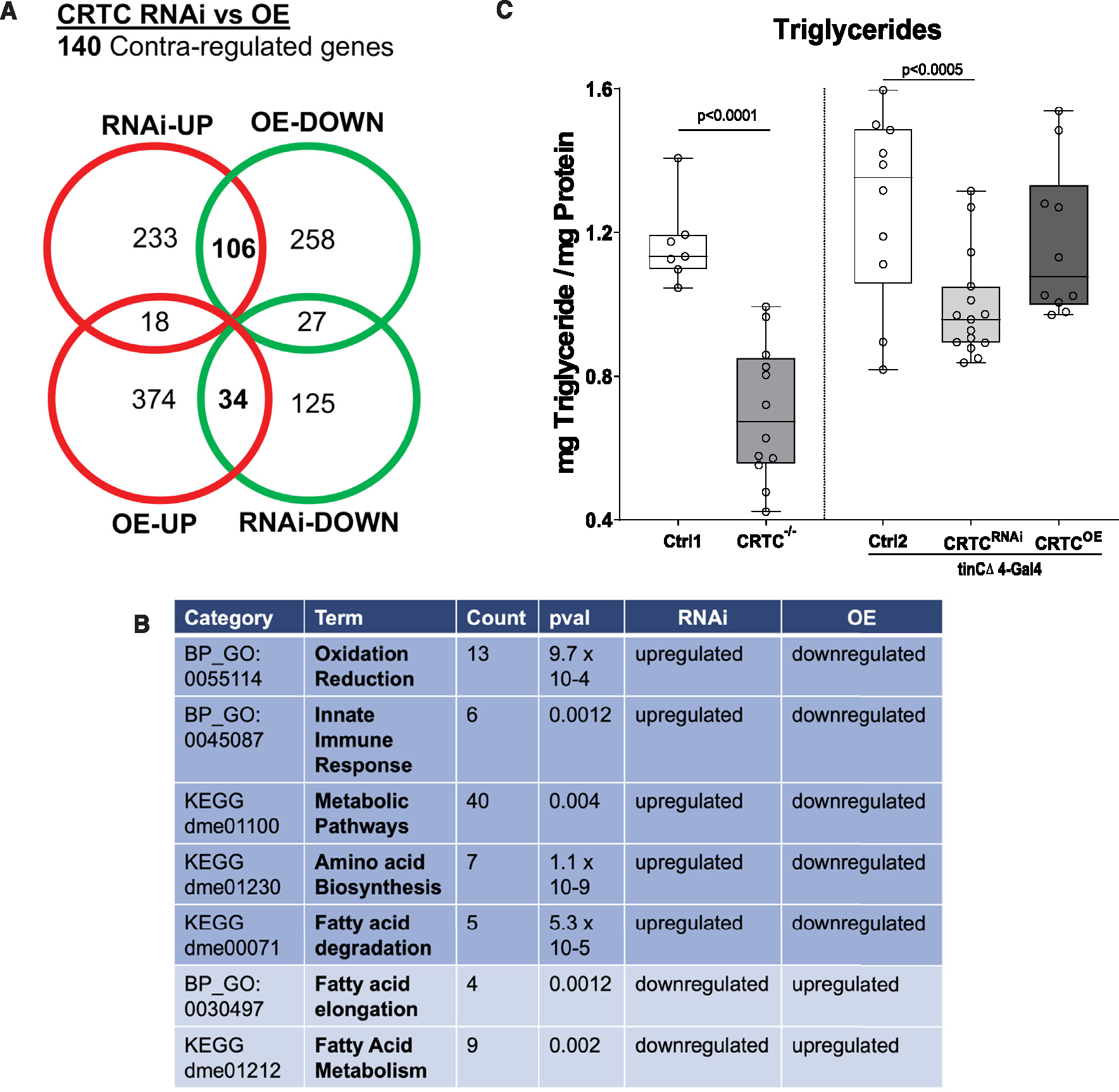
(A) Venn diagram showing up- and downregulated genes in response to cardiac-specific KD or OE of CRTC. Bolded numbers in the center represent genes that are concertedly regulated by CRTC; 106 are DOWN with CRTC OE and UP with CRTC KD, 34 are UP with CRTC OE and DOWN with CRTC KD.
(B) Significantly affected GO categories for the 140 concertedly regulated genes are primarily pathways involved in metabolic regulation. (“Count” indicates the number of genes affected in the GO category).
(C) Whole-body triglyceride levels were significantly reduced in CRTC systemic mutant flies (left) and were also significantly reduced in cardiac-specific CRTC-KD flies (right). Ctrl1 flies were w1118 and Crtl2 flies were tinCD4-Ga4/+. (Plots show all data points, max, min, median, and p values; significance by unpaired t test [left] and one-way ANOVA with Tukey’s multiple comparisons post hoc test [right]).
These results are consistent with previous observations that CRTC-null fly mutants exhibited dramatically reduced lipid stores, a reduction that was only partially restored with neuronal OE of CRTC.15 Because our RNA-sequencing (RNA-seq) results suggest that CRTC regulates metabolism in myocardial cells and because the heart is an energetically demanding organ, we wondered if cardiac CRTC levels also played a role in systemic lipid storage. We analyzed whole-body triacylglyceride (TAG) content in CRTC-null mutants as well as with cardiac-specific modulation of CRTC expression. As previously reported, we found that CRTC mutants exhibited a dramatic reduction in systemic triglyceride levels (Figure 5C, left). Remarkably, cardiac-specific KD of CRTC also resulted in a significant decrease in whole-body fat levels (Figure 5C, right). Taken together, these data suggest that CRTC may act as a metabolic switch in the heart to regulate lipid, carbohydrate, and protein metabolism and that cardiac CRTC KD has a systemic effect on organismal metabolism.
RNA-seq also showed upregulation of a number of hypertrophy marker genes, including Mhc and Argk (Figure S6C), in response to CRTC OE, consistent with our HCR results (Figures 3E–3K″). Genes for other hypertrophy marker genes, including actin, myosin light chain, and troponin, were also significantly upregulated by CRTC OE, further supporting a role for CRTC in cardiac hypertrophy.
Srl/thinman is a candidate downstream effector of CRTC
To identify potential direct targets of CREB/CRTC, we focused on the 140 contraregulated candidate genes from our RNA-seq data. We first determined which Drosophila genes contained a CREBa or CREBb binding site around the transcriptional start site (−2 kb upstream/+1 kb downstream). We used binding-site motifs from FlyFactorSurvey,36 and genes were chosen only if the binding sites were also found to be conserved between two divergent fly species, Drosophila melanogaster and Drosophila persimilis (Figure S7A). This approach yielded 915 unique genes (309 with CREBb sites, 714 with CREBa sites, 108 with both; Figure S7B). We determined which of our 140 candidate genes were among those with CREB binding sites and tested if there was enrichment in our candidate pool. Indeed, genes with CREB binding sites in the dysregulated set of genes were more abundant (12%) compared to all genes (9.2%) or dysregulated genes with no CREB binding sites (8.9%) (hypergeometric test; p < 0.05, Figure S7C). The 140 contraregulated candidate genes from our RNA-seq data (Data S2) were subsequently filtered for genes that were expressed in myocardial cells as determined by Fly Cell Atlas single-cell sequencing data.37 We identified 15 genes that met all these criteria (Figure S7D). Among them, CG9297 was upregulated upon CRTC OE and downregulated with CRTC KD; it also had the strongest cardiac expression of genes with human orthologs and had the consensus CREB binding site sequence TATGACGTGGCT (half-site). CG9297 is orthologous to the human gene Srl, a Ca2+ binding protein that buffers and transports Ca2+ within the sarcoplasmic reticulum (SR) in skeletal and myocardial cells.38
We performed HCR on exposed fly hearts and found that CG9297 mRNA is expressed throughout the Drosophila cardiac tube. Expression of CG9297 was significantly reduced in hearts from CRTC mutants as well as in hearts with cardiac-specific CRTC KD (Figure 6A). Conversely, hearts with cardiac-specific CRTC OE showed increased CG9297 mRNA expression (Figure 6A). These results are consistent with our RNA-seq data and suggest that CRTC regulates CG9297 expression.
Figure 6. CG9297/thinman is regulated by CRTC in the heart.
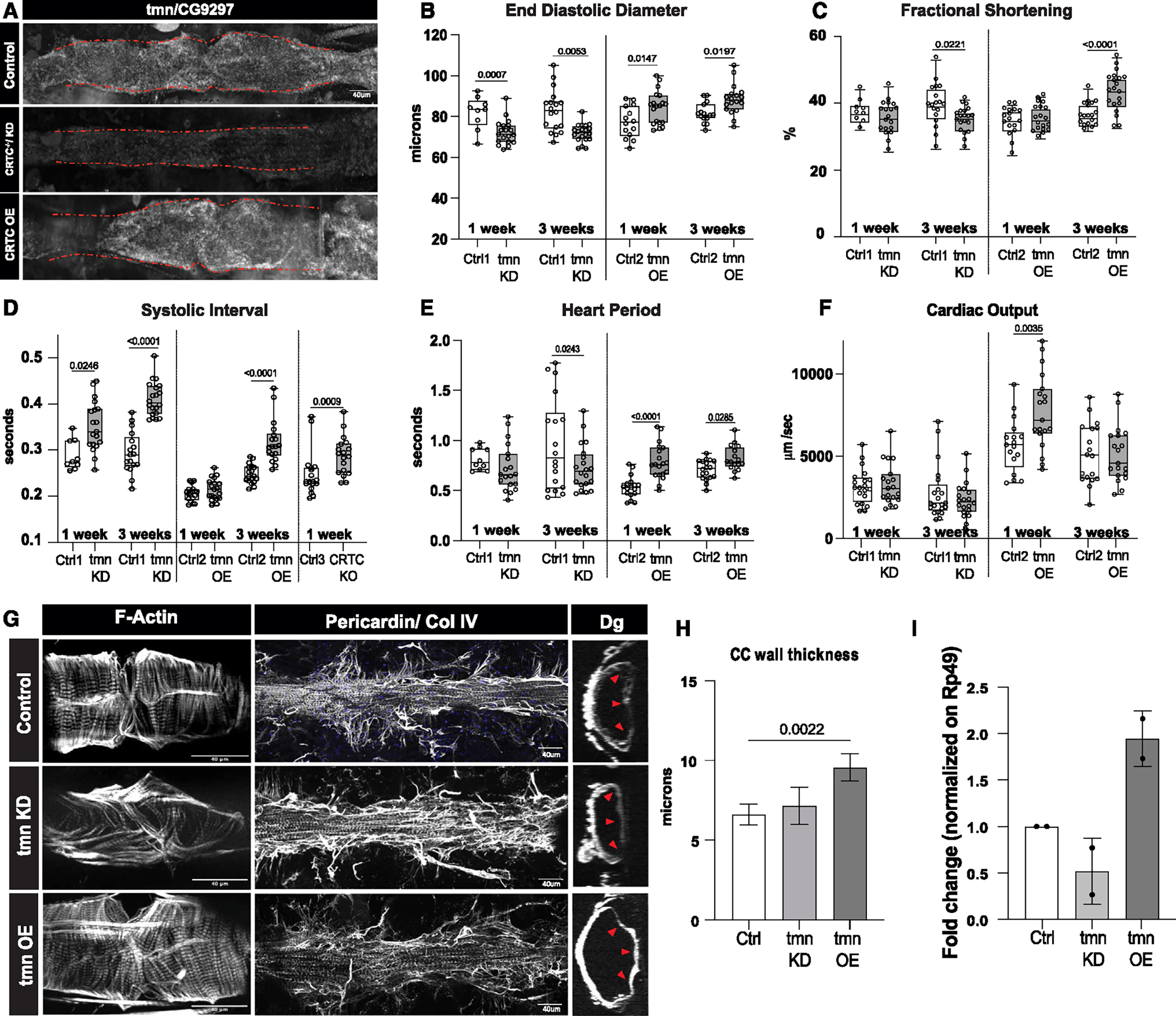
(A) HCR in Drosophila hearts shows that tmn (CG9297)/Srl is ubiquitously expressed in control hearts (tinCΔ4-Ga4/+); expression was significantly reduced in the CRTC mutant and KD hearts, while expression was increased in CRTC-OE hearts. Scale bar, 40 μm
(B) (Left) EDD was significantly reduced in response to cardiac tmn/Srl KD at both 1 and 3 week of age compared to controls. (Right) EDD was significantly increased in response to cardiac tmn/Srl OE at both 1 and 3 week of age compared to controls. (For B–F, Crtl1 flies were tinCΔ4-Ga4/+ [GD Cntrl], Ctrl2 flies were tinCΔ4-Ga4/w1118, and Ctrl3 flies were w1118).
(C) (Left) Fractional shortening was reduced by cardiac tmn/Srl KD at 3 week of age compared to controls (+). (Right) Fractional shortening increased in response to cardiac tmn/SRL OE at 3 week of age compared to controls.
(D) (Left) Systolic intervals are increased in response to cardiac tmn/Srl KD at both 1 and 3 week of age compared to wild-type controls (+). (Middle) Systolic intervals are increased in response to cardiac tmn/SRL OE at 3 week of age compared to controls. (Right) Systolic intervals in fly hearts are increased in response to cardiac-specific KO of CRTC at 1 week of age compared to controls.
(E) (Left) Heart period was decreased in response to cardiac tmn/Srl KD at 3 week of age compared to controls (+). (Right) Heart period was also significantly increased in response to cardiac tmn/Srl OE at both 1 and 3 week of age compared to controls.
(F) (Left) Cardiac output was not significantly affected by cardiac tmn/Srl KD. (Right) Cardiac output was significantly increased in response to cardiac tmn/Srl OE at 1 week of age. (Plots show all data points, max, min, median, and p values; significance by one-way ANOVA with Tukey’s multiple comparisons post hoc test.)
(G) Compared to the closely packed myofibrils in controls (top), F-actin staining shows disorganized myofibrils in tmn/Srl-KD hearts (middle), but relatively normal circumferential myofibrillar arrangement in tmn/Srl-OE hearts (bottom). Anti-pericardin (collagen IV) staining shows a more extensive collagen network surrounding the tmn/Srl-KD heart compared to control. Dystroglycan staining followed by optical sectioning showed an increased thickness in tmn/Srl-OE hearts, especially noticeable at arrowheads. (Dorsal is right, ventral is left.) (For G–I, control flies were tinCΔ4-Ga4/+). Scale bars, 40 μm.
(H) Myocardial cell thickness, made at three points along the heart tube (red arrowheads in G), was significantly increased by cardiac-specific tmn/Srl-OE. (Significance by one-way ANOVA and Tukey’s multiple comparisons post hoc test; p value shown).
(I) RT-qPCR quantification of tmn expression in isolated hearts from genetic background controls (+) and cardiac tmn-KD and -OE hearts. (Normalized to ribosomal protein 49, two biological replicates in triplicate).
Further, CM-specific KD of CG9297 caused cardiac restriction and myofibrillar disarray in both 1- and 3-week-old flies (Figures 6B, 6G, S2J, and S2J′), similar to the effects of CRTC KD and KO (Figures 1 and 2) and CRTC KD in fish (Figure 4, Videos S1 and S3). Due to this cardiac restriction phenotype we propose to rename CG9297 as thinman (tmn). We generated a UAS tmn-OE fly line and found that cardiac OE of tmn caused increased EDD, ESD, fractional shortening, heart period, and cardiac output (Figures 6B–6F, right), similar to cardiac CRTC OE (Figures 2A, 2C, and 2D; Video S2), suggesting that tmn/Srl also plays a role in cardiac function and structure. In addition, we observed a fibrotic phenotype (Figure 6G) similar to CRTC-KO and -KD hearts (Figures 1E″–1H″ and 2E″–2G″). Further functional analysis of flies with cardiac tmn KD showed a progressive, age-dependent prolongation of systolic intervals (SI) in response to cardiac tmn KD (Figure 6D, left), reminiscent of the longer SIs observed in CRTC KO (Figure 6D, right). We previously showed that the length of SIs correlates with intracellularly recorded action potentials (APs) and can be used as a surrogate for AP duration (APD) in this single-cell-layer heart tube.39 Reductions in tmn/Srl expression would be expected to decrease SR Ca2+ buffering capacity and increase cytosolic Ca2+ levels40 (see model in Figure 7D), consistent with the prolonged APD/SI.
Figure 7. CRTC affects cardiac function in hiPSC-cardiomyocytes.
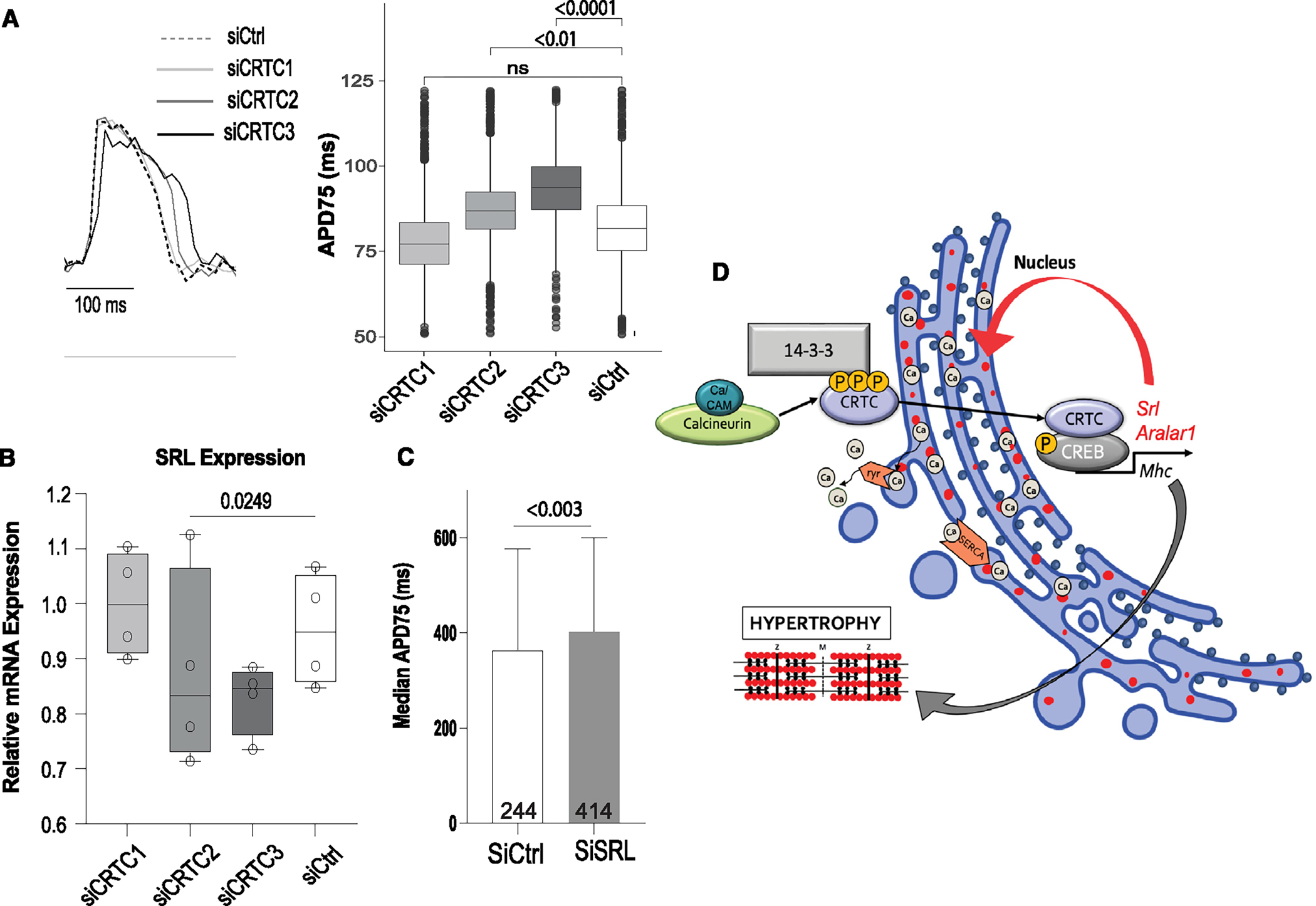
(A) Representative voltage traces from control siRNA- and siCRTC-transfected cardiomyocytes. APs were recorded optically from individual cardiomyocytes. APD at 75% of repolarization (APD75) was significantly increased with siRNA-mediated KD of CRTC2 and CRTC3. (Plots show max, min, median, and p values; siCtrl n = 1,338, siCRTC1 n = 1,459, siCRTC2 n = 1,427, siCRTC3 n = 1,326; significance by one-way ANOVA with Tukey’s multiple comparisons post hoc test).
(B) RT-qPCR quantification showing reduction in SRL expression in iPSC-CMs after siRNA CRTC KD. (Significance by one-way ANOVA with Tukey’s multiple comparisons post hoc test).
(C) siRNA KD of SRL prolongs APD75 in hiPSC-CMs (unpaired t test).
(D) Model of proposed CaN-CRTC-SRL signaling pathway.
CRTC KD affects APD in hiPSC-CMs
We previously established a cardiac role in the fly for the voltage-dependent K+ channel encoded by seizure (sei, the fly homolog of hERG). As observed for both CRTC mutants and tmn-KD flies, hearts from sei mutants were constricted with disorganized myofibrils, and they exhibited significantly prolonged APDs and SIs, suggesting links between myocardial cell maintenance and ion-channel dysfunction.41 Notably, sei mutants also showed significant reductions in cardiac CRTC expression.41 To explore a role for CRTC in channel function, we turned to human CMs obtained from induced pluripotent stem cells (hiPSC-CMs) to test whether electrical activity could be altered by CRTC KD in beating induced CMs. hiPSC-CMs were differentiated, and the cells were dissociated (day 25), replated, and transfected with small interfering RNAs (siRNAs) against all three paralogs of CRTC. Differentiated hiPSC-CMs expressed all three CRTC paralogs (Figure S8A), and siRNA KD significantly reduced expression (Figures S8B–S8D). Three days post-transfection, cells were loaded with a voltage-sensitive dye, imaged, and processed to obtain single-cell voltage traces for all cells in each well. Representative traces of median APs from cells with KD of CRTC1, 2, and 3 compared to controls are shown in Figure 7A. APD was quantified from individual cells at 50%, 75%, and 90% repolarization. KD of CRTC2 and CRTC3 significantly increased APD, whereas KD of CRTC1, the primary form expressed in neurons, did not affect APD duration relative to controls (Figures 7A and S8F–S8H). It should be noted that under these in vitro conditions we did not see any differences in CM size in response to CRTC KD (Figure S8I).
Given our results in the fly heart, we quantified human Srl expression in hiPSC-CMs in response to CRTC KD and observed a significant reduction in the expression of SRL with CRTC2 KD (Figure 7B). Finally, Srl KD in hiPSC-CMs also caused significant increases in APD75 (Figure 7C). Together, these data suggest that CRTC2 and/or CRTC3 has a specific role in vertebrate myocardial cell function that is distinct from that of CRTC1. Reduced expression of Srl would be expected to increase cytosolic Ca2+ levels, consistent with our observed increases in CM APD in response to KD of CRTC or Srl in CMs (Figures 7A and 7C) and with the increased SI in response to cardiac CRTC or Srl KD in the fly heart (Figure 6D).
DISCUSSION
Obesity, diabetes, and metabolic syndrome are prominent risk factors thought to contribute to cardiomyopathies. CRTC is known to be a key regulator of metabolism in mammalian hepatocytes and neurons, but a cardiac-autonomous, regulatory role for CRTC has yet to be identified. Our data here show that hearts from CRTC mutant flies exhibit significant cardiac restriction, myofibrillar disorganization, and fibrosis (Figures 1, 2, S1, S2, and S4). Cardiac KD of either the CRTC co-modulator CREB or the CRTC activator CaN30/Pp2B recapitulates the CRTC mutant phenotype (Figures 1, 2, and 3). Finally, the cardiac-specific effects of CRTC appear to regulate the maintenance of adult myocardial cells, as heart development was seemingly unaffected in the CRTC systemic mutants (Figure S3).
Based on our data from zebrafish, a cardiac role for CRTC is likely conserved: all three paralogs of CRTC were detected in isolated hearts from adult fish, but CRTC3 had by far the highest expression levels (Figure S5G), and KD of CRTC3 caused significant cardiac restriction, similar to the fly cardiac phenotype (Figures 4E and 4F). In addition, all three CRTC genes were also expressed in 25 day iPSC-CMs, but CRTC1 was expressed at very low levels compared to CRTC2 and 3 (Figure S8A). Consistent with this, siRNA KD of CRTC2 or 3 in hiPSC-CMs caused significant AP broadening, while KD of the predominantly neuronal form, CRTC1, had no effect (Figures 7A and S8F–S8H). Also consistent with these results is a report that systemic KO of CRTC1 had no effect on cultured mouse CMs.11 On the other hand, this same study documented increased ventricular CRTC1 levels in patients with aortic stenosis and hypertrophic cardiomyopathy; however, the authors did not analyze the CRTC2 or CRTC3 paralogs, which are the predominant cardiac genes.11
One way that CRTC may regulate myocardial cell function is by controlling metabolic pathways in the heart, as it does in mammalian liver.9 CRTC-null mutants, as previously reported,23 or cardiac KD of CRTC (this report) exhibited lean phenotypes, and surprisingly, flies with cardiac-specific CRTC KD had significantly reduced whole-body TAG levels (Figure 5C). This may account for the observation that neuronal CRTC OE did not completely rescue TAG levels in previous studies.15 Comparisons of RNA-seq data between hearts with cardiac CRTC KD and OE suggested that CRTC acts as a metabolic switch between fatty acid degradation and synthesis in the heart. The fly data therefore suggest that cardiac metabolism impacts systemic fat content. A similar situation was observed in mice and flies, where cardiac KD of a subunit of the mediator complex (Med13/Skd) makes the animals prone to fat accumulation under a high-fat diet (HFD), whereas cardiac OE was protective.42,43 It will be interesting to see if the systemic leanness of cardiac KD flies also depends on Med13/Skd function.
CRTC has been implicated in skeletal muscle hypertrophy,44 and our data from fly climbing assays also suggest a separate role for CRTC in somatic muscle function. In a previous study we reported no effect on climbing in CRTC mutants, but only very young flies were tested.23 We tested older flies (1 and 3 weeks) and found that climbing was impaired in systemic CRTC mutants and that performance in both wild-type and mutant flies declined in an age-dependent manner. Interestingly, flies with cardiac-specific CRTC KD performed the same as or slightly better than controls (Figures S5C–S5F), suggesting that somatic muscle function was compromised in the systemic CRTC mutants but not in the cardiac-KD flies. Consistent with a downstream role in CRTC signaling, tmn/Srl was previously identified in a screen for genes that are important in muscle function45 and has been shown to be highly expressed in muscle37 and in the heart.46
OE of CaN has been shown to induce cardiac hypertrophy in mice and flies,6–8,29,31 and we again observed hypertrophy with OE of CaN/Pp2B in the fly (Figure 3A). This hypertrophy was partially reversed in CRTC heterozygotes and was further reduced in CRTC-homozygous null mutants (Figure 3A). These data all point to a cardiac-autonomous role for CRTC acting downstream of CaN in the fly heart. Further, the lack of a robust effect of NFAT KD on fly heart function (Figures S5A and S5B) suggests that CRTC signaling may mediate an alternate hypertrophic signaling pathway. CRTC has been shown to modulate the activity of other transcription factors, such as AP1 and ATF6, as well as having non-canonical roles, for example, preventing SREBP activation by sequestering Sec31 in the cytosol and possibly even as a secreted signal.21 Thus, CRTC may play roles in cardiac function beyond the regulation of gene expression.
We identified several genes that were contraregulated in response to CRTC KD and OE that are expressed in myocardial cells and have upstream CREBb binding sites conserved between distant Drosophila species. One of these genes, tmn, orthologous to vertebrate Srl, had cardiac-specific effects that paralleled those seen with CRTC KD and KO (Figures 1, 2, and 6). SRL was first purified and characterized in 198047 and remains an underinvestigated Ca2+-binding protein that is abundant within the SR. SRL regulates Ca2+ reuptake by interacting with cardiac sarco(endo)plasmic reticulum Ca2+-ATPase 2a (SERCA2a).38,40 It has been reported to have a role in maintaining cardiac function during endurance exercise training and maintaining Ca2+ handling function of the SR in skeletal and cardiac muscle. One of SRL’s predicted partners, Atp2a1, has been proposed to interfere with hypoxia preconditioning and survival.44 Systemic ablation of Srl caused enhanced resistance to muscle fatigue by compensatory changes in Ca2+-regulatory proteins that affect store-operated Ca2+ entry.48 Interestingly, human gene network analysis predicted interactions between Srl and YWHAE, which encodes 14–3-3 epsilon, the protein that sequesters and inactivates cytoplasmic CRTC.10 In C. elegans, col18 (Srl ortholog) was shown to be 253 upregulated in glucose-fed animals,49 also suggesting a connection to CRTC signaling. In addition, our data suggest that abnormal Ca2+ handling may play a key role in muscular dystrophy, where drastic reductions in SRL in Dp427 (dystrophin of 427 kDa)-deficient fibers have been documented.50 In Drosophila, tmn is expressed in the developing and mature muscle in the embryo51 and adult muscle tissue,37 and mesodermal KD of tmn caused larval lethality,45 indicating a critical role for tmn in muscle function. It is thus possible that our observation of impaired climbing capacity of CRTC-null flies could, in fact, be mediated by impaired muscle function due to reduction of tmn.
Our data, showing significant increases in the APD in response to KD of CRTC2 and CRTC3 in hiPSC-CMs (Figures 7 and S8), and significant increases in SI upon tmn/Srl or CRTC KD in flies (Figure 6D), are all consistent with a Ca2+-overload hypothesis and with our previously published data showing prolonged AP and reduced CRTC expression in sei/hERG mutants (0.19-fold, adj p = 0.00341). Significantly, a reexamination of cardiac expression data from sei/hERG mutant hearts also showed reduced tmn/srl expression (fold change −0.82879 log2; p = 0.04541). Concerted regulation by CRTC of the Ca2+-binding protein Aralar (Figure S7D) further supports a role for CRTC in Ca2+ homeostasis. Together, our data suggest a model whereby loss of CRTC causes reductions in Srl/tmn expression, cytosolic Ca2+ overload (Figure 7D), and, ultimately, mechanical and structural abnormalities with impaired contractility. Our consistent results across three cardiac model systems suggest that CRTC signaling likely plays a conserved and cardiac-autonomous role in maintaining heart structure and function. Thus, CaN-CRTC-SRL may represent a parallel pathway to CaN-NFAT signaling in mediating cardiac hypertrophy and may also play more general roles in muscle cell maintenance.
Limitations of the study
Using tissue-specific drivers, we provide evidence in Drosophila of a cardiac-autonomous role for CRTC in adult flies, but we cannot rule out that these cardiac phenotypes were not also due to contributions during cardiac development. Despite the normal heart and tail muscle development in zebrafish, we cannot rule out a role in heart development, because the CRTC F0 CRISPR KO was systemic. We took advantage of the flies’ short lifespan to examine effects on aging but did not investigate such effects in fish because their lifespan is much longer than that of flies. A limitation of iPSC-induced CM experiments is that it is an in vitro situation, and they are not fully mature, but they have the advantage that they are human and thus indicate potential human relevance. Our RNA-seq analysis suggests additional CRTC downstream candidates that potentially contribute to the observed phenotypes upon CRTC manipulation, which we did not test. However, their characterization provides candidates for future studies.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Karen Ocorr (kocorr@sbpdiscovery.org).
Materials availability
Newly generated materials from this study are available upon reasonable request.
Data and code availability
All data supporting the findings of this study are available from the corresponding author upon reasonable request.
The R code used in this study is available from Github at https://github.com/gvogler/Dondi_2024. Semi-automated Optical Heartbeat Analysis software is available free of change at www.SOHAsoftware.com
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
RNA-Seq dataset was deposited in GEO-Gene Expression Omnibus. GEO: GSE271481
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Study approvals
All animal procedures were approved by the Institutional Animal Care and Use Committees (IACUC) at the University of California Irvine and the Tibor Rubin VA Long Beach Medical Center (AUF22–073). All experimental protocols were conducted following the NIH Guide for the Care and Use of Laboratory Animals, the recommendations of the American Veterinary Medical Association Panel on Euthanasia. Veterinary services are available at Sanford Burnham Prebys Medical Discovery Institute and provided as needed or in emergencies.
Vertebrate animal care and husbandry
We use the zebrafish (Danio Rerio) as vertebrate cardiac model to verify interactions among genes and ion channels identified in the fly. Our studies use breeding male and female fish aged 3 months to 1 year that that are housed in an electronically and manually monitored aquatic system. Our studies used transgenic zebrafish by using well established morpholino microinjections into the one-cell stage of zebrafish embryo. Our analysis of the cardiac system during embryonic stages will be carried out by high-speed optical recording of intact zebrafish larva, a non-invasive procedure. Heart function parameters was quantified from videos using our established Semiautomatic Optical Heartbeat Analysis system (Fink et al, 2009). Within the first seven days of life, zebrafish cannot perceive pain. However, spontaneous muscle twitches occur making it difficult to score phenotypes. All fish examined is this study were 72 hours post-fertilization or less.
METHOD DETAILS
Generation and maintenance of fly and fish stocks
Drosophila stocks were reared at 25°C on a standard laboratory diet consisting of yeast, corn starch, and molasses. All experiments were conducted on one or three-week old adult females. The CRTC mutant (CRTC, 25–3 line) and UAS-CRTC (ii)-OE (overexpression) lines were a gift from Dr. Marc Montminy15. The CREB mutant (yCREBb[Δ400]/FM7c) was a gift from Dr. John Thomas (Salk Institute). Transgenic UAS-RNAi fly lines were obtained from the Vienna Drosophila Stock Center; the Bloomington Drosophila Stock Center; and the Kyoto Stock Center. The following Gal4 drivers were used: (cardiac) tinCΔ4-Gal4 24 and Hand4.2-Gal454; (pericardial cell) Dot-Gal4 (#BL690356) and sns-Gal4 (#BL7616057), (neurons) elav-Gal4 (#BL876558), (fat body) Cg-Gal4 (#BL6514759) and Lsp2-Gal4 (#BL635727,52), and (muscle) Mef2-Gal4 (#BL50742); all #BL are from the Bloomington Stock Center, Bloomington, IN, USA. The following UAS-RNAi lines were used: CRTC (#V100974), Pp2B (#V103144), CRTC-Flag (#V318324), Nfat (#V107032, all #V from Vienna Stock Center, Biocenter 1, 1030 Wien, At.), CG9297 (#V43222). The following UAS-OE lines were used: constitutively active Pp2B-14B (#DGRC116254, Drosophila Genomic Resource Center, 1001 E. 3rd St. Bloomington, IN). A transgenic fly line was generated for the overexpression of CG9297 with UAS/Gal4. A CG9297 cDNA clone RE20510 (in pFLC-I) was obtained from DGRC and verified with insert PCR and sequencing of the amplicon (PCR primers: 5p: ATGGG GCGGCTAACCATTTG, 3p: TCAACT CGTCCTCTTAGGCT; primer-free sequencing by Plasmidsaurus/Primordium Labs). The confirmed clone was digested with XbaI to release the cDNA insert, followed by gel-purification. In parallel, pUASTattB (EF362409) plasmid was digested with EcoRI. The released inserts (2859bp) and cut pUASTattB vector (8489bp) were then ligated using the Gibson Assembly Cloning Kit following the manufacturer instructions (New England bioLabs Inc. #E5510S). The final construct was cloned and confirmed by sequencing. Transgenic lines were created by PhiC31-mediated integration into attP5 landing site (BDSC #34765) using the services of BestGene Inc. Successful integration indicated by red-eye color was confirmed by sequencing, RT-qPCR and HCR in situ following overexpression by tinCΔ4-Gal4. The following lines served as controls: w1118 (#BL3605), KK (#V60100), GD (#V66000), y[1]w[67c23] (#BL6599). Homozygous CREBb mutant females were sterile and difficult to produce, thus, mutant males were also included in the experiments involving CREBb systemic mutants. (See Data File 1 – Excel file 1 for a complete list of fly strains, crosses, and functional data).
Zebrafish stocks (Oregon AB wild-type) were maintained under standard laboratory conditions at 28.5°C. Gene expression was manipulated using standard microinjection of morpholino (MO) antisense oligonucleotides. Subsequently, zebrafish were raised to 72 hours postfertilization (hpf); embryos were staged according to Kimmel et al 35. All zebrafish experiments were performed in accordance with the protocols approved by SBP IACUC. For gene KD we used a previously characterized morpholino against CRTC3 (MO sequence: TCCTAATTTGGCTGAGCTTACCCTT, Gene Tools, LLC.) 44
Drosophila and zebrafish heart physiology
Drosophila:
Electrical pacing assays were carried as previously described17. Intact adult flies were anesthetized with FlyNap (Carolina Biological Supply Co, Burlington, NC) placed between two rows of conductive gel overlying two electrodes and paced by applying a 40 V, 6 Hz square wave for 30 seconds. Abdominally located hearts were observed under a dissecting microscope; heart failure rate was defined as the percentage of hearts that did not beat or were visibly fibrillating 2 minutes after the end of the pacing regime.
Semi-intact preparations of the fly hearts were made as described previously18 Briefly, flies were dissected in oxygenated artificial hemolymph (AHL) to expose the linear tube-like heart; excess fat was suctioned off with a micropipette. Following 15–20 minutes of recovery in fresh oxygenated AHL, 30-second high-speed movies (>140 fps) of contracting hearts were captured using via a Hamamatsu digital camera (EMCCD-C9300) on an Olympus BX61WI microscope with a 10x immersion objective. These movies were analyzed with the SOHA software (sohasoftware.com)18,19. The following key heart function parameters were measured: Diastolic Interval (DI), Systolic Interval (SI), Heart Period (HP, one contraction cycle), Diastolic Diameter (DD), Systolic Diameter (SD), and contractility (measured as fractional shortening). Stroke Volume (SV) was estimated based on the assumption of a cylindrical heart tube using SV=(r2) *h, where h=1 and “r” is the tube radius and derived from the DD and SD measurements, i.e. SV =(1/2) DD)2 – (1/2SD)2. Cardiac output (CO) was estimated as SV x HR.
Zebrafish:
In-depth quantitative analyses of zebrafish cardiac function and conduction dynamics was performed as previously described (16). Larval zebrafish at 72 hpf were immobilized in a small volume of low melt agarose (1.5–2%) and submerged in conditioned water. Hearts were imaged in vivo at room temperature (20–21°C) with direct immersion optics and a digital high-speed camera (Orca Flash, Hamamatsu Photonics). High-speed movies (~150 fps) were analyzed using SOHA )60,61 to quantify heart period (R-R interval) and chamber size.
Immunohistochemistry and optical sectioning
To assess morphological differences, immunohistochemistry was performed as described previously62. Briefly, hearts were dissected, arrested/relaxed with 10mM EGTA and fixed for 20 min with 4% paraformaldehyde. The trimmed hearts were transferred to a 96-well plate (a maximum of ten hearts per well) and washed with PBSTx (phosphate-buffered saline, 0.1% Triton X-100). Dissected abdomens were incubated with primary antibodies diluted in PBTx for 2 hours at room temperature or overnight at 4°C and then washed in PBTx and incubated in secondary antibodies diluted in PBTx. Following 3 additional washes, hearts were mounted onto glass slides in Prolong Diamond anti-fade mounting medium with DAPI (ThermoFisher Scientific, Waltham, MA, USA). Z-stack images were obtained with a Zeiss ApoTome.2 and Zeiss Imager.Z1 Microscope system (Carl Zeiss, White Plains, NY, USA), at 10x and 25x magnification. Primary Antibodies used were: pericardin antibody (1:100, EC11, DSHB) to stain for the collagen-IV like extracellular matrix protein; α-actinin at 1:100 (gift from J. Saide). Fly anti-CRTC antibody at 1:100 was previously generated using a synthetic peptide covering amino acids 103–118 of fly CRTC and validated (see M. Montminy; 23). The vertebrate CRTC antibody is commercially available and was generated against a peptide covering a sequence that is highly conserved among all three human paralogs and is also conserved in CRTC2 and CRTC3 in fish; it was used at 1:100 (MRC PPU Reagents and Services), anti-flag antibody (Sigma F3165), anti-H15/Nmr1 at 1:2000 (gift from J. Skeath 63, anti-Dystroglycan at 1:1000 (gift from A. Wodarz (44), Zfh1 at 1:1000 (45), anti-DMef2 at 1:2000 (gift from E. Olson 64, anti-Svp at 1:200 (gift from R. Cripps65). Secondary antibodies used were Alexa Fluor 488 donkey anti-sheep at 1:500 (Invitrogen, Carlsbad, CA, USA), Alexa Fluor 488 goat anti-mouse at 1:500 (Invitrogen, Carlsbad, CA, USA), Alexa Fluor 647 donkey anti-rabbit at 1:500 (Invitrogen, Carlsbad, CA, USA), Cy5 goat anti-guinea pig at 1:500 (Abcam, Cambridge, MA, USA). Alexa Fluor 594 phalloidin (Invitrogen, Carlsbad, CA, USA) was used to stain for F-actin or filamentous actin.
To quantify the heart associated collagen IV (pericardin) hearts were stained with anti-pericardin and phalloidin for F-actin and Z stack images were obtained at a constant exposure. The area of pericardin staining around the heart was quantified using the Weka Segmentation tool in Image J66. F-actin staining was used to define the total heart area underlying the pericardin staining. The outer edges of the heart tubes were traced, and the area was quantified. Pericardin area was then normalized to the area of the F-actin-stained heart tube.
Optical sectioning for CM thickness measurements was done on both sides of the conical chamber ostia (anterior-most heart chamber). Optical sections were converted to binary images and the heart walls were identified using the Weka segmentation tool in ImageJ. Heart wall measurements were made at 4 points (2 dorsal and 2 ventral) for each of the 2 images per chamber and averaged. Care was taken to avoid measuring at locations containing nuclei, which expand the CM membranes.
Zebrafish hearts were fixed in 4% paraformaldehyde in PBS for 20 min and whole-mount immunofluorescence was performed as previously described 67,68, using primary monoclonal antibodies against sarcomeric myosin heavy chain (MF20). MF20 was obtained from the Developmental Studies Hybridoma Bank maintained by the Department of Biological Sciences, University of Iowa. Secondary antibody, either donkey anti-chicken AlexaFluor488 (Jackson ImmunoResearch, 1:200) or Donkey anti-Rabbit AlexaFluor594 (Invitrogen, 1:200), was used in 1:200 dilution. Fluorescence images were acquired using an LSM 510 confocal microscope (Zeiss, Germany) with a 40X water objective.
Myofibrillar disorganization assay
To quantify myofibrillar disorganization in cardiomyocytes we used the Directionality” plugin in FIJI. This plugin is used to infer the preferred orientation of structures present in input images. It quantifies structures and their orientation and outputs the number of structures oriented in different directions. Images with completely isotropic content will give a flat histogram, whereas images in which there is a preferred orientation will give a histogram with a peak at that orientation. Using Zstacks of F-actin stained, horizontally oriented heart tubes, we defined ROIs within individual myocardial cells (Figures S2A and S2B). Averaged output from these analyses is graphed in Figure S2H. This plugin also fits the highest peak by a Gaussian function and reports the overall ‘Direction (°)’ as the center of the gaussian and the “Goodness” of fit. For a number of controls this plugin mis-identified myofibrils and tried to connect adjacent F actin-stained regions (blue and green lines in Figure S2C’), also resulting in low “goodness” scores. To permit correct myofibril identification in these cases, it was necessary to “fill in” the dark regions containing the unstained myosin bands (Figure S2C″). This permitted a more accurate identification and better fit of the vertically oriented myofibrils (compare C′ and C‴ in Figure S2). Hearts with “Goodness” values of < 0.70 were rejected.
Climbing assay
The negative geotaxis assays are modified from the RING (Rapid Iterative Negative Geotaxis) assays described in 69. Flies were sorted on CO2, by sex, into groups of twenty or less. After one hour of recovery time, they were transferred into polystyrene vials, marked with 1cm intervals. To induce the geotaxis response, each vial was then placed against a white background and tapped down firmly until all the flies fell to the bottom of the vial. Climbing ability/Negative geotaxis response was filmed with a digital camera over a ten second interval for each trial. Assays were performed three times for each set of flies with one minute recovery time in between each trial. For the analysis, video to jpeg conversion software was used to create still images at one second intervals for the 10 sec trial. The flies in each image were counted based on the 1 cm intervals marked on the vial. Flies that were found to be directly on a centimeter mark were counted in the higher distance bracket. Number of flies crossing the height of 2 and 10 cm were recorded. Measurements from all three trials were averaged.
Triacylglyceride (TAG) assay
Triacylglyerides were measured using the TAG assay as described previously 70. Extracts obtained from three-week old intact female flies were used in a colorimetric assay whereby triacylglycerides are enzymatically cleaved into free fatty acids and glycerol, glycerol 3-phosphate and finally H2O2 which reacts with 4-aminoantipyrine (4-AAP) and 3,5-dichloro-2-hydroxybenzene sulfonate (3,5 DHBS) to produce a red colored product that was measured using a 96-well spectrophotometer. The level of TAGs in the samples was based on a standard curve generated using TAG standards (Stanbio Life Sciences) of known concentrations (0.0625 – 2 μg/μL). TAG content was normalized to total protein determined using a standard Bradford Protein assay (Bio-Rad Laboratories, Hercules, CA, USA).
DNA extractions
DNA for PCR was extracted from whole flies using a standard organic extraction protocol. Materials included homogenizer tubes with polyacetal pestle (#K7496250030), Cell Lysis Solution (Qiagen #158906), RNase A Solution (Qiagen #158922), Protein Precipitation Solution (Qiagen #158910), DNA Hydration Solution (Qiagen #158914, QIAGEN, Germantown, MD, USA), 100% alcohol (Decon Laboratories, Inc.), and Isopropanol (ThermoFisher Scientific, Waltham, MA, USA). The final samples were assessed for quantity and quality using a NanoDrop spectrophotometer.
RNA extractions
RNA was extracted from the hearts of either one-week old or three-week old female flies using the miRNAeasy Mini Kit (QIAGEN, Germantown, MD, USA) as per manufacturer’s protocol. The final samples were assessed for quantity and quality using a NanoDrop spectrophotometer.
RNA extraction, cDNA synthesis and quantitative PCR (Q-PCR)
Total RNA was isolated from fly hearts (15 per sample) with a RNeasy Isolation kit (QIAGEN, Germantown, MD, USA) per manufacturer’s protocol. Total RNA (25–30 ng) was quantified and checked for purity using a Nanodrop spectrophotometer. RNA was subsequently reverse-transcribed using a QuantiTect Reverse Transcription Kit (QIAGEN, Germantown, MD, USA), per manufacturer’s protocol. Perl Primer software (IDT) was used to design primers with optimal annealing temperatures and primer efficiency. The FastStart Essential DNA Green Master kit (Sigma-Aldrich Corp. St. Louis, MO, USA) was used to carry out qPCR in a Roche LightCycler® 96. RPL32 and GAPDH, and actin were used as reference genes.
Total RNA was extracted from zebrafish adult hearts using TRIzol (Invitrogen) and stabilized in RNA later (Thermo Fisher) and processed according to the RNeasy Micro Kit (Qiagen). Eight hearts were pooled together. RNA (1 μg) was reverse transcribed to cDNA with SuperScript reverse transcriptase (Invitrogen) using random hexamers. β-actin (actb2) and GADPH were used to normalize gene expression in the RT-qPCR experiments. At least two or three independent biological replicates were performed.
In situ hybridization
Gene expression in adult hearts was assessed for Gal4 using RNAscope (ACDbio, Newark, USA), and for CRTC and CG9297 using HCR (Molecular Instruments). Flies were dissected in oxygenated artificial hemolymph (AHL) to expose the linear tube-like heart; excess fat was suctioned off with a micropipette, and hearts were fixed for 20 minutes in 4% paraformaldehyde. Hybridization and fluorescent labeling were performed according to manufacturers’ protocols.
RNA sequencing and analysis
Hearts from 15 female flies were dissected as described previously, removed with fine forceps and pooled as a single sample. RNA was extracted using the Qiagen RNAeasy mini kit (QIAGEN, Germantown, MD, USA). Six replicate samples were obtained from 1-week old wildtype (w1118) and CRTC−/− knockout flies. Six to seven replicate samples were obtained from 3-week-old TincΔ4-Gal4>UAS-CRTC-RNAi, TincΔ4-Gal4>UAS-CRTC-OE, and TincΔ4-Gal4>KK-control flies. PolyA RNA was isolated using the NEBNext® Poly(A) mRNA Magnetic Isolation Module and barcoded libraries were made using the NEBNext® Ultra II™ Directional RNA Library Prep Kit for Illumina® (NEB, Ipswich, MA, USA). Libraries were pooled and paired-end sequenced (2X75) on the Illumina NextSeq 500 using the High output V2.5 kit (Illumina Inc., San Diego, CA, USA). Average read count for w1118 and CRTC mutant transcripts was approximately 15.3 million reads per sample, as 26 samples were shared on a single flow cell with maximum read count of 400 million reads. Average read count for CRTC-RNAi, CRTC-OE, and ControlKK was 19 million reads per sample, as 21 samples were shared on a single flow cell with maximum read count of 400 million reads. Read data were processed in BaseSpace (basespace.illumina.com). Reads were aligned to Drosophila melanogaster genome (Dm6) using STAR aligner (https://code.google.com/p/rna-star/) with default settings. Differential transcript expression was determined using the Cufflinks Cuffdiff package (https://github.com/cole-trapnell-lab/cufflinks).
iPSC-CM assay
Generation of hiPSC-CMs
Id1 overexpressing hiPSCs71,72 were dissociated with 0.5 mM EDTA (ThermoFisher Scientific, Waltham, MA, USA) in PBS without CaCl2 and MgCl2 (Corning) for 7 min at room temperature. hiPSC were resuspended in mTeSR-1 media (StemCell Technologies, Cambridge, MA 02142) supplemented with 2 μM Thiazovivin (StemCell Technologies, Cambridge, MA 02142) and plated in a Matrigel-coated 12-well plate at a density of 3 × 105 cells per well. After 24 hours after passage, cells were fed daily with mTeSR-1 media (without Thiazovivin) for 3–5 days until they reached ≥ 90% confluence to begin differentiation. hiPSC-CMs were differentiated as previously described (40, 41). At day 0, cells were treated with 6 μM CHIR99021 (Selleck Chemicals, Houston, TX, USA) in S12 media for 48 hours. At day 2, cells were treated with 2 μM Wnt-C59 (Selleck Chemicals, Houston, TX, USA), an inhibitor of WNT pathway, in S12 media. 48 hours later (at day 4), S12 media is fully changed. At day 5, cells were dissociated with TrypLE Express (ThermoFisher Scientific, Waltham, MA, USA) for 2 min and blocked with RPMI supplemented with 10% FBS (Omega Scientific, Tarzana, CA, USA). Cells were resuspended in S12 media supplemented with 4 mg/L Recombinant Human Insulin (ThermoFisher Scientific, Waltham, MA, USA) (S12+ media) and 2 μM Thiazovivin and plated onto a Matrigel-coated 12-well plate at a density of 9 × 105 cells per well. S12+ media was changed at day 8 and replaced at day 10 with RPMI (ThermoFisher Scientific, Waltham, MA, USA) media supplemented with 213 μg/μL L-ascorbic acid (Sigma-Aldrich, St. Louis, MO, USA), 500 mg/L BSA-FV (Gibco), 0.5 mM L-carnitine (Sigma-Aldrich, St. Louis, MO, USA) and 8 g/L AlbuMAX Lipid-Rich BSA (CM media, ThermoFisher Scientific, Waltham, MA, USA). Typically, hiPSC-CMs start to beat around day 9–10. At day 15, cells were purified with lactate media (RPMI without glucose, 213 μg/μL L-ascorbic acid, 500 mg/L BSA-FV and 8 mM Sodium-DL-Lactate (Sigma-Aldrich, St. Louis, MO, USA), for 4 days. At day 19, media was replaced with CM media.
Voltage assay in hiPSC-CMs
Voltage assay is performed using labeling protocol described in71. Briefly, hiPSC-CMs at day 25 of differentiation were dissociated with TrypLE Select 10X for up to 10 min and action of TrypLE was neutralized with RPMI supplemented with 10% FBS. Cells were resuspended in RPMI with 2% KOSR (Gibco) and 2% B27 50X with vitamin A (Life Technologies, Carlsbad, CA, USA) supplemented with 2 μM Thiazovivin and plated at a density of 6,000 cells per well in a Matrigel-coated 384-well plate. hiPSC-CMs were then transfected with siRNAs directed against each CRTC complex components (ON-TARGETplus SMART pool, siCRTC1: L-014026–01-0005, siCRTC2: L-018947–00-0005, siCRTC3: L-014210–01-0005) using lipofectamine RNAi Max (ThermoFisher Scientific, Waltham, MA, USA). Each siRNA was tested individually and in combination in 8-plicates. Three days post-transfection, cells were first washed with pre-warmed Tyrode’s solution (Sigma-Aldrich, St. Louis, MO, USA) by removing 50 μL of media and adding 50 μL of Tyrode’s solution 5 times using a 16-channel pipette. After the fifth wash, 50 μL of 2x dye solution consisting in voltage sensitive dye Vf2.1 Cl (Fluovolt, 1:2000, ThermoFisher Scientific, Waltham, MA, USA) diluted in Tyrode’s solution supplemented with 1 μL of 10% Pluronic F127 (diluted in water, ThermoFisher Scientific, Waltham, MA, USA) and 20 μg/mL Hoescht 33258 (diluted in water, ThermoFisher Scientific, Waltham, MA, USA) was added to each well. The plate was placed back in the 37°C 5% CO2 incubator for 45 min. After incubation time, cells were washed 4 times with fresh pre-warmed Tyrode’s solution using the same method described above. hiPSC-CMs were then automatically imaged with ImageXpress Micro XLS microscope at an acquisition frequency of 100 Hz for a duration of 5 sec with excitation wavelength of 485/20 nm and emission filter 525/30 nm. A single image of Hoescht was acquired before the time series. Fluorescence over time quantification and trace analysis were automatically quantified using custom software packages developed by Molecular Devices and Colas lab. Two independent experiments were performed. KD efficiency was confirmed 2 days post-siRNA-transfection by RT-qPCR.
RNA extraction and qPCR assay with hiPSC-CMs
The hiPSC-CMs were dissociated at day 25 of differentiation with TrypLE Select 10X for up to 10 min and action of TrypLE was neutralized with RPMI supplemented with 10% FBS. Cells were resuspended in RPMI (ThermoFisher Scientific, Waltham, MA, USA) media supplemented with 213 μg/μL L-ascorbic acid (Sigma-Aldrich, St. Louis, MO, USA), 500 mg/L BSA-FV (Gibco), 0.5 mM L-carnitine (Sigma-Aldrich, St. Louis, MO, USA) and 8 g/L AlbuMAX Lipid-Rich BSA (CM media, ThermoFisher Scientific, Waltham, MA, USA) supplemented with 2 μM Thiazovivin and plated at a density of 6,000 cells per well in a Matrigel-coated 384-well plate. hiPSC-CMs were then transfected with siRNAs directed against each CRTC complex components (ON-TARGETplus SMART pool, siCRTC1: L-014026–01-0005, siCRTC2: L-018947–00-0005, siCRTC3: L-014210–01-0005) using lipofectamine RNAi Max (ThermoFisher Scientific, Waltham, MA, USA). Each siRNA was tested individually and in combination in 4-plicates. Three days post-transfection, total RNA was extracted using Zymo Research Quick-RNA MircoPrep Kit (Zymo Research, R1051) following the manufacturers’ recommendations. RNA concentration was measured by Nanodrop (Thermo Scientific). Aliquots of 1 μg of RNA were reverse transcribed using a QuantiTect Reverse Transcription kit (Qiagen, 205314), and qPCR was performed with iTaq SYBR Green (Life Technologies) using a 7900HT Fast Real-Time PCR system (Applied Biosystems). Gene expression was normalized to that of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) for human iPSC-derived cardiomyocyte samples using the 2–ΔΔCt method. Human primer sequences for qRT-PCR were obtained from Harvard Primer Bank.
In silico TFBS analysis in R
Binding site motifs for CrebB-17A and CrebA were imported from MotifDB72 and mapped in aligned genome assemblies of D. melanogaster/persimilis and D.melanogaster/yakuba. This identified all sites that were conserved between those genome pairs. Next, this was further narrowed down to those sites that localize within a −2000bp/+1000bp window of annotated transcriptional start sites (dm6 assembly). The R script for this analysis is available at https://github.com/gvogler/Dondi_2024.
QUANTIFICATION AND STATISTICAL ANALYSIS
Fly and fish cardiac function data was analyzed using the D’Agostino and Pearson omnibus normality test for Gaussian distribution. For normally distributed data, statistical significance was determined using a 1-way ANOVA for simple comparisons between more than two groups and 2-way ANOVA (for multiple manipulations) followed by multiple comparisons post-hoc tests as indicated in Figure legends. Data sets that did not show a normal distribution were analyzed using a nonparametric 2-tailed unpaired t-test, Wilcoxcon Rank Sum test, or Kruskal-Wallis test followed by Dunn multiple comparisons post-hoc tests.
For hiPSC-ACM data we used the nonparametric Kolmogorov-Smirnov test to compare the differences in the cumulative distributions of APD data. Population distribution of control and siRNA-treated hiPSC-CMs was generated with GraphPad Prism software (2019) using nonlinear regression. To determine statistical significance between experimental and control groups, we used two-tailed, unpaired Student’s t-test. All statistical analyses were performed using GraphPad Prism software.
All parameters shown are Mean ± Std. Dev., every animal used are represented as individual data points in the figures, p values are shown in all the graphs (we used p<0.05 as significant). Ages and sex of animals used are indicated in the Figure Legend and/or on the graphs.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Mouse α-pericardin | DSHB | RRID: AB_528431 |
| Rat α-actinin | Gift from J.Saide | N/A |
| Rabbit α-CRTC | Gift from M.Montminy | N/A |
| Sheep α-CRTC (Vertebrate) | MRC PPU Reagents and Services | #S279D |
| Mouse α-FLAG | Sigma-Aldrich | RRID: AB_259529 |
| α-H15/Nmr1 | Gift from J. Skeath | N/A |
| Mouse α-dystroglican | Gift from A.Wodarz | N/A |
| α- Zfh1 | Gift from E.Olson | N/A |
| α- DMef2 | Gift from E.Olson | N/A |
| α-Svp | Gift from R.Cripps | N/A |
| Alexa Fluor 488 donkey anti-sheep | Invitrogen | #A-11015; RRID: AB_2534082 |
| Alexa Fluor 488 goat anti-mouse | Invitrogen | #A-11001; RRID: AB_2534069 |
| Alexa Fluor 647 donkey anti-rabbit | Invitrogen | #A-31573; RRID: AB_2536183 |
| Alexa Fluor 594 phalloidin | Invitrogen | #A-12381; RRID: AB_2315633 |
| Alexa Fluor 647 goat anti-guinea pig | Invitrogen | #A-21450; RRID: AB_2535867 |
| Alexa Fluor 594 donkey anti-rabbit | Invitrogen | #A-21207; RRID: AB_141637 |
| MF20 (Myosin) | Developmental Studies Hybridoma Bank | Antibody Registry ID: AB_2147781 |
| AlexaFluor488 donkey anti-chicken | Invitrogen | #A-78948; RRID: AB_2921070 |
| AlexaFluor488 donkey anti-rabbit | Invitrogen | #A-21206; RRID: AB_2535792 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Trizol | Invitrogen | #15596026 |
| mTeSR-1 media | StemCell Technologies | #85850 |
| Thiazovivin | StemCell Technologies | #100–0247 |
| CHIR99021 (GSK-3β and GSK-3α inhibitor) | Selleck Chemicals | #1263 |
| Wnt-C59 (WNT pathway inhibitor) | Selleck Chemicals | #S7037 |
| TrypLE Express | ThermoFisher Scientific | #12604013 |
| RPMI | ThermoFisher Scientific | #11875093 |
| Recombinant Human Insulin | ThermoFisher Scientific | #RP-10935 |
| L-ascorbic acid | Sigma-Aldrich | #A92902 |
| L-carnitine | Sigma-Aldrich | #8.40092 |
| AlbuMAX Lipid-Rich BSA | ThermoFisher Scientific | #11020021 |
| Sodium-DL-Lactate | Sigma-Aldrich | #041529.K2 |
| vitamin A | Life Technologies | #1257010 |
| lipofectamine RNAi Max | ThermoFisher Scientific | #13778150 |
| Tyrode’s solution | Sigma-Aldrich | #T1788 |
| voltage sensitive dye Vf2.1 Cl, Fluovolt | ThermoFisher Scientific | #F10488 |
| Pluronic F127 | ThermoFisher Scientific | #P6867 |
| CRTC in situ hybridization probe | Molecular Instrument | N/A |
| CG9297 in situ hybridization probe | Molecular Instrument | N/A |
| MHC in situ hybridization probe | Molecular Instrument | N/A |
| Argk in situ hybridization probe | Molecular Instrument | N/A |
|
| ||
| Critical commercial assays | ||
|
| ||
| Gibson Assembly Cloning Kit | New England BioLabs | #E5510S |
| miRNAeasy Mini Kit | Qiagen | #217084 |
| QuantiTect Reverse Transcription Kit | Qiagen | #205311 |
| FastStart Essential DNA Green Master kit | Sigma-Aldrich Corp. | #06402712001 |
| SuperScript™ III Reverse Transcriptase | Invitrogen | #18080093 |
| RNAscope™ ISH Kit | ACD Bio | #322786 |
| NEBNext® Poly(A) mRNA Magnetic Isolation Module | New England Biolabs | NEB#E3370 |
| NEBNext® Ultra II™ Directional RNA Library Prep Kit for Illumina® | New England Biolabs | NEB #E7765 |
| Bradford Protein assay Kit | Bio-Rad Laboratories | #5000001 |
| Zymo Research Quick-RNA MircoPrep Kit | Zymo Research | #R1051 |
| QuantiTect Reverse Transcription kit | Qiagen | #205314 |
| iTaq SYBR Green | Life Technologies | #1725120 |
|
| ||
| Deposited data | ||
|
| ||
| R script for in silico TFBS analysis | https://github.com/gvogler/Dondi_2024 | N/A |
| RNA-Seq dataset | GEO - Gene Expression Omnibus | GEO: GSE271481 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Id1 overexpressing cell line | Gift from the Wu lab | derived from Burridge PW et al., 201553 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| D. melanogaster: CRTC[25–3] | Gift from M.Montminy | N/A |
| D. melanogaster: UAS-CRTC | Gift from M.Montminy | N/A |
| D. melanogaster: y, CREBb[Δ400]/FM7c | Gift from J. Thomas | N/A |
| D. melanogaster: CRTC RNAi (P{KK106414} VIE-260B) | Vienna Drosophila Stock Center | #100974 |
| D. melanogaster: Pp2B RNAi (P{KK107714} VIE-260B) | Vienna Drosophila Stock Center | #103144 |
| D. melanogaster: CRTC-Flag- (PBac{fTRG01044.sfGFP-TVPTBF}VK00033) | Vienna Drosophila Stock Center | #318324 |
| D. melanogaster: NFAT RNAi (P{KK102385} VIE-260B) | Vienna Drosophila Stock Center | #107032 |
| D. melanogaster: CG9297 (tmn) RNAi (w1118; P{GD5649} v43222) | Vienna Drosophila Stock Center | #43222 |
| D. melanogaster: KK background control | Vienna Drosophila Stock Center | #60100 |
| D. melanogaster: GD background control | Vienna Drosophila Stock Center | #60000 |
| D. melanogaster: tinCΔ4-GaI4 | Lo, P.C., and Frasch, M., 2001 | N/A |
| D. melanogaster: Hand4.2-Gal4 | Han and Olson, 2005, Dev54 | N/A |
| D. melanogaster: Dot-Gal4 (P{Ugt36A1-GAL4.K}43A, y1 w*) | Bloomington Drosophila Stock Center | #6903 |
| D. melanogaster: sns-Gal4 (y1 w*; Mi{Trojan-GAL4.1}snsMI03001-TG4.1) | Bloomington Drosophila Stock Center | #76160 |
| D. melanogaster: elav-Gal4 (P{GAL4-elav.L}2/CyO) | Bloomington Drosophila Stock Center | #8765 |
| D. melanogaster: CG-Gal4 (y1 sc* v1 sev21; P{TRiP.HMC05950}attP40) | Bloomington Drosophila Stock Center | #65147 |
| D. melanogaster: Lsp2-Gal4 (y1 w1118; P{Lsp2-GAL4.H}3) | Bloomington Drosophila Stock Center | #6357 |
| D. melanogaster: Mef2-Gal4 (w*; P{Mef2-GAL4.247}3) | Bloomington Drosophila Stock Center | #50742 |
| D. melanogaster: CG9297 (tmn) OE | This paper | N/A |
| D. melanogaster: Pp2B-14B OE | Drosophila Genomic Resource Center | #116254 |
| D. melanogaster: y[1]w[67c23] | Bloomington Drosophila Stock Center | #6599 |
| D. melanogaster: w1118 | Bloomington Drosophila Stock Center | #3605 |
| Danio rerio: Tg(myl7:nls-EGFP) | Boston Children’s Hospital (Burns group) | Gonzalez-Rosa J.M. et al., 201855 |
|
| ||
| Oligonucleotides | ||
|
| ||
| CRTC3 (MO sequence: TCCTAATTTGGCTGAGCTTACCCTT) | Gene Tools, LLC | Manchenkov T. et al., 201544 |
| D. melanogaster RP49 qRT-PCR primers 5’ AAACGCGGTTCTGCATGAG 3’ 3’GCCACCAGTCGGATCGATAT 3’ | IDT (Integrated DNA technologies) | N/A |
| D. melanogaster CRTC qRT-PCR primers 5’ CCCCTCCCTTTGATTGCCTA 3’ 3’ TCGGAAGTGTAAGGCCCATT 3’ | IDT (Integrated DNA technologies) | N/A |
| D. melanogaster CG9297 qRT-PCR primers 5’ GGAAAAGCCAGCACCAGTTG 3’ 3’ GTCCCGCAGGATGATCTCAG 3’ | IDT (Integrated DNA technologies) | N/A |
| Danio rerio Ef1a qRT-PCR primers 5’ AGAAGGCTGCCAAGACCAAG 3’ 3’ AGAGGTTGGGAAGAACACGC 5’ | IDT (Integrated DNA technologies) | N/A |
| Danio rerio CRTC1 qRT-PCR primers 5’ AGCAGTACCAGCAACCTGAC3’ 3’ GAGTTGGCAGTTCCACCTGT 5’ | IDT (Integrated DNA technologies) | N/A |
| Danio rerio CRTC2 qRT-PCR primers 5’ CTCGGGGAAATAGGACACGG 3’ 3’ CTGGGTAAGGCAACTTGGGT 5’ | IDT (Integrated DNA technologies) | N/A |
| Danio rerio CRTC3 qRT-PCR primers 5’ CTCTCAGCTGCACTCGTCTC 3’ 3’ CTCCATTGGCAAGTTGCTGG 5’ | IDT (Integrated DNA technologies) | N/A |
| hIPSC-CMs hGAPDH qRT-PCR primers 5’ GGAGCGAGATCCCTCCAAAAT 3’ 3’ GGCTGTTGTCATACTTCTCATGG 5’ | Harvard Primer Bank | N/A |
| hIPSC-CMs hCRTC1 qRT-PCR primers 5’ TCAGTGGACAAACACGGACG 3’ 3’ CAGGGCGGAGTCAGAATTG 5’ | Harvard Primer Bank | N/A |
| hIPSC-CMs hCRTC2 qRT-PCR primers 5’ GGGGCAGTTGTTTCGACTACC 3’ 3’ GGACTGGGGTTCATCACACTT 5’ | Harvard Primer Bank | N/A |
| hIPSC-CMs hCRTC3 qRT-PCR primers 5’ GCAACTGCGCCTTACACAGTA 3’ 3’ TGAAATGACGGCTGAAACTCTG 5’ | Harvard Primer Bank | N/A |
| hIPSC-CMs hSrl qRT-PCR primers 5’ GAAGAAGCCCCATTGAGGGAC 3’ 3’ CACCGCAGAGTAGTCATCGG 5’ | Harvard Primer Bank | N/A |
| CG9297 PCR primers (cloning): 5p: ATGGG GCGGCTAACCATTTG 3p: TCAACTCGTCCTCTTAGGCT | Harvard Primer Bank | N/A |
| hIPSC siRNA targeting sequence control UGGUUUACAUGUCGACUAAUGGUUUACAUGUUGUGUGAUGGUUUACAUGUUUUCUGAUGGUUUACAUGUUUUCCUA | Dharmacon- ON TARGET plus | D-001810–10-20 |
| hIPSC siRNA targeting sequence hCRTC1 GCGGAAAUUCAGCGAGAAGGCACAGAGGCAAGGCGUACAGGACAAACACGGACGGCAGAGUUCAACAUGAUGGAGAA | Dharmacon- ON TARGET plus | L-014026–01-0005 |
| hIPSC siRNA targeting sequence hCRTC2 GGCUAAACAUGAGUGAGCAAAUUUAGUGAGAAGAUGCUCAGGGCCUAACAUCAUCAGGCAGGCAGUCUCAUUA | Dharmacon- ON TARGET plus | L-018947–00-0005 |
| hIPSC siRNA targeting sequence hCRTC3 CGUCAGAGUUUCAGCCGUC CUUCAGCAACUGCGCCUUA GAAGAGACGUUUCGAGCUG GCGACAAUUUGAUGGUAGU | Dharmacon- ON TARGET plus | L-014210–01-0005 |
| hIPSC siRNA targeting sequence hSrl AAAACAGGGUGUAGCGAAA | Dharmacon- ON TARGET plus | J-022618–19-0010 |
|
| ||
| Recombinant DNA | ||
|
| ||
| CG9297 cDNA clone in pFLC-I vector | DGRC | #RE20510 |
| pUASTattB vector | DGRC | #EF362409 |
|
| ||
| Software and algorithms | ||
|
| ||
| ImageJ | https://imagej.nih.gov/ij/ | N/A |
| SOHA | SOHAsoftware.com | N/A |
| STAR aligner | https://code.google.com/p/rna-star/ | N/A |
| Cufflinks Cuffdiff package | https://github.com/cole-trapnell-lab/cufflinks | N/A |
|
| ||
| Other | ||
|
| ||
| Sequencing | Plasmidsaurus | N/A |
| Primordium Labs | ||
Highlights.
Cardiac CRTC knockdown (KD) in flies causes restriction, remodeling, and fibrosis
CRTC3 KD significantly reduces the size of ventricles in the zebrafish model
CRTC2/3 KD in hiPSC-cardiomyocytes causes action potential broadening
Thinman, an ortholog of Sarcalumenin, is identified as a CRTC downstream effector
ACKNOWLEDGMENTS
We thank Mr. Sean Paknoosh and Dr. Xin-Xin I. Zeng for technical assistance. This work was supported by NIH R01HL149992 and NIH R01HL054732 (R.B.); NIH R01HL153645 and R01HL148827 (A.C.); NIH 5F31HL134305–02 (A.G.); NIH R01DK083834, the Leona M. and Harry B. Helmsley Charitable Trust, the Clayton Foundation for Medical Research, and the Kieckhefer Foundation (M.M.); the Sanford Burnham Prebys Medical Discovery Institute, AHA 14GRNT20490239, and NIH R01HL132241–04 (K.O.); and NIH R01DK077979 (J.B.T.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.114549.
REFERENCES
- 1.Ashrafian H, Frenneaux MP, and Opie LH (2007). Metabolic mechanisms in heart failure. Circulation 116, 434–448. 10.1161/CIRCULATIONAHA.107.702795. [DOI] [PubMed] [Google Scholar]
- 2.Ritterhoff J, and Tian R (2023). Metabolic mechanisms in physiological and pathological cardiac hypertrophy: new paradigms and challenges. Nat. Rev. Cardiol. 20, 812–829. 10.1038/s41569-023-00887-x. [DOI] [PubMed] [Google Scholar]
- 3.Jia G, DeMarco VG, and Sowers JR (2016). Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat. Rev. Endocrinol. 12, 144–153. 10.1038/nrendo.2015.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nollet EE, Westenbrink BD, de Boer RA, Kuster DWD, and van der Velden J (2020). Unraveling the Genotype-Phenotype Relationship in Hypertrophic Cardiomyopathy: Obesity-Related Cardiac Defects as a Major Disease Modifier. J. Am. Heart Assoc. 9, e018641. 10.1161/JAHA.120.018641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mohan M, Dihoum A, Mordi IR, Choy AM, Rena G, and Lang CC (2021). Left Ventricular Hypertrophy in Diabetic Cardiomyopathy: A Target for Intervention. Front. Cardiovasc. Med. 8, 746382. 10.3389/fcvm.2021.746382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, and Olson EN (1998). A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 93, 215–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Molkentin JD (2004). Calcineurin-NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc. Res. 63, 467–475. 10.1016/j.cardiores.2004.01.021. [DOI] [PubMed] [Google Scholar]
- 8.Wilkins BJ, Dai YS, Bueno OF, Parsons SA, Xu J, Plank DM, Jones F, Kimball TR, and Molkentin JD (2004). Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ. Res. 94, 110–118. 10.1161/01.RES.0000109415.17511.18. [DOI] [PubMed] [Google Scholar]
- 9.Altarejos JY, and Montminy M (2011). CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat. Rev. Mol. Cell Biol. 12, 141–151. 10.1038/nrm3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi S, Kim W, and Chung J (2011). Drosophila salt-inducible kinase (SIK) regulates starvation resistance through cAMP-response element-binding protein (CREB)-regulated transcription coactivator (CRTC). J. Biol. Chem. 286, 2658–2664. 10.1074/jbc.C110.119222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morhenn K, Quentin T, Wichmann H, Steinmetz M, Prondzynski M, Sohren KD, Christ T, Geertz B, Schroder S, Schondube FA, et al. (2019). Mechanistic role of the CREB-regulated transcription coactivator 1 in cardiac hypertrophy. J. Mol. Cell. Cardiol. 127, 31–43. 10.1016/j.yjmcc.2018.12.001. [DOI] [PubMed] [Google Scholar]
- 12.Kovacs KA, Steullet P, Steinmann M, Do KQ, Magistretti PJ, Halfon O, and Cardinaux JR (2007). TORC1 is a calcium- and cAMP-sensitive coincidence detector involved in hippocampal long-term synaptic plasticity. Proc. Natl. Acad. Sci. USA 104, 4700–4705. 10.1073/pnas.0607524104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li S, Zhang C, Takemori H, Zhou Y, and Xiong ZQ (2009). TORC1 regulates activity-dependent CREB-target gene transcription and dendritic growth of developing cortical neurons. J. Neurosci. 29, 2334–2343. 10.1523/JNEUROSCI.2296-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Altarejos JY, Goebel N, Conkright MD, Inoue H, Xie J, Arias CM, Sawchenko PE, and Montminy M (2008). The Creb1 coactivator Crtc1 is required for energy balance and fertility. Nat. Med. 14, 1112–1117. 10.1038/nm.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang B, Goode J, Best J, Meltzer J, Schilman PE, Chen J, Garza D, Thomas JB, and Montminy M (2008). The insulin-regulated CREB coactivator TORC promotes stress resistance in Drosophila. Cell Metabol. 7, 434–444. 10.1016/j.cmet.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wessells RJ, Fitzgerald E, Cypser JR, Tatar M, and Bodmer R (2004). Insulin regulation of heart function in aging fruit flies. Nat. Genet. 36, 1275–1281. 10.1038/ng1476. [DOI] [PubMed] [Google Scholar]
- 17.Wessells RJ, and Bodmer R (2004). Screening assays for heart function mutants in Drosophila. Biotechniques 37, 58–60, 62, 64.passim. [DOI] [PubMed] [Google Scholar]
- 18.Vogler G, and Ocorr K (2009). Visualizing the beating heart in Drosophila. J. Vis. Exp. 1425, 1425. 10.3791/1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ocorr K, Reeves NL, Wessells RJ, Fink M, Chen HS, Akasaka T, Yasuda S, Metzger JM, Giles W, Posakony JW, and Bodmer R (2007). KCNQ potassium channel mutations cause cardiac arrhythmias in Drosophila that mimic the effects of aging. Proc. Natl. Acad. Sci. USA 104, 3943–3948. 10.1073/pnas.0609278104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fink M, Callol-Massot C, Chu A, Ruiz-Lozano P, Izpisua Belmonte JC, Giles W, Bodmer R, and Ocorr K (2009). A new method for detection and quantification of heartbeat parameters in Drosophila, zebrafish, and embryonic mouse hearts. Biotechniques 46, 101–113. 10.2144/000113078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mair W, Morantte I, Rodrigues AP, Manning G, Montminy M, Shaw RJ, and Dillin A (2011). Lifespan extension induced by AMPK and calcineurin is mediated by CRTC-1 and CREB. Nature 470, 404–408. 10.1038/nature09706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Conkright MD, Canettieri G, Screaton R, Guzman E, Miraglia L, Hogenesch JB, and Montminy M (2003). TORCs: transducers of regulated CREB activity. Mol. Cell 12, 413–423. 10.1016/j.molcel.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 23.Wang T, Wiater E, Zhang X, Thomas JB, and Montminy M (2021). Crtc modulates fasting programs associated with 1-C metabolism and inhibition of insulin signaling. Proc. Natl. Acad. Sci. USA 118, e2024865118. 10.1073/pnas.2024865118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lo PC, and Frasch M (2001). A role for the COUP-TF-related gene seven-up in the diversification of cardioblast identities in the dorsal vessel of Drosophila. Mech. Dev. 104, 49–60. 10.1016/s0925-4773(01)00361-6. [DOI] [PubMed] [Google Scholar]
- 25.Lim HY, Wang W, Chen J, Ocorr K, and Bodmer R (2014). ROS regulate cardiac function via a distinct paracrine mechanism. Cell Rep. 7, 35–44,. 10.1016/j.celrep.2014.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suster ML, Karunanithi S, Atwood HL, and Sokolowski MB (2004). Turning behavior in Drosophila larvae: a role for the small scribbler transcript. Gene Brain Behav. 3, 273–286. 10.1111/j.1601-183X.2004.00082.x. [DOI] [PubMed] [Google Scholar]
- 27.Lazareva AA, Roman G, Mattox W, Hardin PE, and Dauwalder B (2007). A role for the adult fat body in Drosophila male courtship behavior. PLoS Genet. 3, e16. 10.1371/journal.pgen.0030016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baker KD, and Thummel CS (2007). Diabetic larvae and obese flies-emerging studies of metabolism in Drosophila. Cell Metabol. 6, 257–266. 10.1016/j.cmet.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee TE, Yu L, Wolf MJ, and Rockman HA (2014). Galactokinase is a novel modifier of calcineurin-induced cardiomyopathy in Drosophila. Genetics 198, 591–603. 10.1534/genetics.114.166777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zarndt R, Walls SM, Ocorr K, and Bodmer R (2017). Reduced Cardiac Calcineurin Expression Mimics Long-Term Hypoxia-Induced Heart Defects in Drosophila. Circ. Cardiovasc. Genet. 10, e001706. 10.1161/CIRCGENETICS.117.001706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilkins BJ, and Molkentin JD (2004). Calcium-calcineurin signaling in the regulation of cardiac hypertrophy. Biochem. Biophys. Res. Commun. 322, 1178–1191. 10.1016/j.bbrc.2004.07.121. [DOI] [PubMed] [Google Scholar]
- 32.Carniel E, Taylor MR, Sinagra G, Di Lenarda A, Ku L, Fain PR, Boucek MM, Cavanaugh J, Miocic S, Slavov D, et al. (2005). Alpha-myosin heavy chain: a sarcomeric gene associated with dilated and hypertrophic phenotypes of cardiomyopathy. Circulation 112, 54–59. 10.1161/CIRCULATIONAHA.104.507699. [DOI] [PubMed] [Google Scholar]
- 33.Cao F, Zervou S, and Lygate CA (2018). The creatine kinase system as a therapeutic target for myocardial ischaemia-reperfusion injury. Biochem. Soc. Trans. 46, 1119–1127. 10.1042/BST20170504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schultz D, Su X, Bishop SP, Billadello J, and Dell’Italia LJ (1997). Selective induction of the creatine kinase-B gene in chronic volume overload hypertrophy is not affected by ACE-inhibitor therapy. J. Mol. Cell. Cardiol. 29, 2665–2673. 10.1006/jmcc.1997.0498. [DOI] [PubMed] [Google Scholar]
- 35.Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, and Schilling TF (1995). Stages of embryonic development of the zebrafish. Dev. Dynam. 203, 253–310. 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- 36.Zhu LJ, Christensen RG, Kazemian M, Hull CJ, Enuameh MS, Basciotta MD, Brasefield JA, Zhu C, Asriyan Y, Lapointe DS, et al. (2011). FlyFactorSurvey: a database of Drosophila transcription factor binding specificities determined using the bacterial one-hybrid system. Nucleic Acids Res. 39, D111–D117. 10.1093/nar/gkq858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H, Janssens J, De Waegeneer M, Kolluru SS, Davie K, Gardeux V, Saelens W, David FPA, Brbic M, Spanier K, et al. (2022). Fly Cell Atlas: A single-nucleus transcriptomic atlas of the adult fruit fly. Science 375, eabk2432. 10.1126/science.abk2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leberer E, Charuk JH, Green NM, and MacLennan DH (1989). Molecular cloning and expression of cDNA encoding a lumenal calcium binding glycoprotein from sarcoplasmic reticulum. Proc. Natl. Acad. Sci. USA 86, 6047–6051. 10.1073/pnas.86.16.6047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kervadec A, Kezos J, Ni H, Yu M, Marchant J, Spiering S, Kannan S, Kwon C, Andersen P, Bodmer R, et al. (2023). Multiplatform modeling of atrial fibrillation identifies phospholamban as central regulator of cardiac rhythm. Dis Model Mech 16, dmm049962. 10.1242/dmm.049962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoshida M, Minamisawa S, Shimura M, Komazaki S, Kume H, Zhang M, Matsumura K, Nishi M, Saito M, Saeki Y, et al. (2005). Impaired Ca2+ store functions in skeletal and cardiac muscle cells from sarcalumenin-deficient mice. J. Biol. Chem. 280, 3500–3506. 10.1074/jbc.M406618200. [DOI] [PubMed] [Google Scholar]
- 41.Ocorr K, Zambon A, Nudell Y, Pineda S, Diop S, Tang M, Akasaka T, and Taylor E (2017). Age-dependent electrical and morphological remodeling of the Drosophila heart caused by hERG/seizure mutations. PLoS Genet. 13, e1006786. 10.1371/journal.pgen.1006786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grueter CE, van Rooij E, Johnson BA, DeLeon SM, Sutherland LB, Qi X, Gautron L, Elmquist JK, Bassel-Duby R, and Olson EN (2012). A cardiac microRNA governs systemic energy homeostasis by regulation of MED13. Cell 149, 671–683. 10.1016/j.cell.2012.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee JH, Bassel-Duby R, and Olson EN (2014). Heart- and muscle-derived signaling system dependent on MED13 and Wingless controls obesity in Drosophila. Proc. Natl. Acad. Sci. USA 111, 9491–9496. 10.1073/pnas.1409427111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Manchenkov T, Pasillas MP, Haddad GG, and Imam FB (2015). Novel Genes Critical for Hypoxic Preconditioning in Zebrafish Are Regulators of Insulin and Glucose Metabolism. G3 (Bethesda). 5, 1107–1116. 10.1534/g3.115.018010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schnorrer F, Schonbauer C, Langer CC, Dietzl G, Novatchkova M, Schernhuber K, Fellner M, Azaryan A, Radolf M, Stark A, et al. (2010). Systematic genetic analysis of muscle morphogenesis and function in Drosophila. Nature 464, 287–291. 10.1038/nature08799. [DOI] [PubMed] [Google Scholar]
- 46.Cammarato A, Ahrens CH, Alayari NN, Qeli E, Rucker J, Reedy MC, Zmasek CM, Gucek M, Cole RN, Van Eyk JE, et al. (2011). A mighty small heart: the cardiac proteome of adult Drosophila melanogaster. PLoS One 6, e18497. 10.1371/journal.pone.0018497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Campbell KP, and MacLennan DH (1981). Purification and characterization of the 53,000-dalton glycoprotein from the sarcoplasmic reticulum. J. Biol. Chem. 256, 4626–4632. [PubMed] [Google Scholar]
- 48.Jiao Q, Bai Y, Akaike T, Takeshima H, Ishikawa Y, and Minamisawa S (2009). Sarcalumenin is essential for maintaining cardiac function during endurance exercise training. Am. J. Physiol. Heart Circ. Physiol. 297, H576–H582. 10.1152/ajpheart.00946.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garcia AM, Ladage ML, Dumesnil DR, Zaman K, Shulaev V, Azad RK, and Padilla PA (2015). Glucose induces sensitivity to oxygen deprivation and modulates insulin/IGF-1 signaling and lipid biosynthesis in Caenorhabditis elegans. Genetics 200, 167–184. 10.1534/genetics.115.174631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dowling P, Doran P, and Ohlendieck K (2004). Drastic reduction of sarcalumenin in Dp427 (dystrophin of 427 kDa)-deficient fibres indicates that abnormal calcium handling plays a key role in muscular dystrophy. Biochem. J. 379, 479–488. 10.1042/BJ20031311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weiszmann R, Hammonds AS, and Celniker SE (2009). Determination of gene expression patterns using high-throughput RNA in situ hybridization to whole-mount Drosophila embryos. Nat. Protoc. 4, 605–618. 10.1038/nprot.2009.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cherbas L, Hu X, Zhimulev I, Belyaeva E, and Cherbas P (2003). EcR isoforms in Drosophila: testing tissue-specific requirements by targeted blockade and rescue. Development 130, 271–284. 10.1242/dev.00205. [DOI] [PubMed] [Google Scholar]
- 53.Burridge PW, Holmström A, and Wu JC (2015). Chemically Defined Culture and Cardiomyocyte Differentiation of Human Pluripotent Stem Cells. Curr. Protoc. Hum. Genet. 87, 21.3.1–21.3.15. 10.1002/0471142905.hg2103s87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Han Z, and Olson EN (2005). Hand is a direct target of Tinman and GATA factors during Drosophila cardiogenesis and hematopoiesis. Dev Camb Engl. 132, 3525–3536. 10.1242/dev.01899. [DOI] [PubMed] [Google Scholar]
- 55.González-Rosa JM, Sharpe M, Field D, Soonpaa MH, Field LJ, Burns CE, and Burns CG (2018). Myocardial Polyploidization Creates a Barrier to Heart Regeneration in Zebrafish. Dev. Cell 44, 433–446.e7. 10.1016/j.devcel.2018.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kimbrell DA, Hice C, Bolduc C, Kleinhesselink K, and Beckingham K (2002). The Dorothy enhancer has Tinman binding sites and drives hopscotch-induced tumor formation. Genesis 34, 23–28. 10.1002/gene.10134. [DOI] [PubMed] [Google Scholar]
- 57.Zhang F, Zhao Y, and Han Z (2013). An in vivo functional analysis system for renal gene discovery in Drosophila pericardial nephrocytes. J. Am. Soc. Nephrol. 24, 191–197. 10.1681/ASN.2012080769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Berger C, Renner S, Luer K, and Technau GM (2007). The commonly used marker ELAV is transiently expressed in neuroblasts and glial cells in the Drosophila embryonic CNS. Dev. Dynam. 236, 3562–3568. 10.1002/dvdy.21372. [DOI] [PubMed] [Google Scholar]
- 59.Asha H, Nagy I, Kovacs G, Stetson D, Ando I, and Dearolf CR (2003). Analysis of Ras-induced overproliferation in Drosophila hemocytes. Genetics 163, 203–215. 10.1093/genetics/163.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cammarato A, Ocorr S, and Ocorr K (2015). Enhanced assessment of contractile dynamics in Drosophila hearts. Biotechniques 58, 77–80. 10.2144/000114255. [DOI] [PubMed] [Google Scholar]
- 61.Ocorr K, Fink M, Cammarato A, Bernstein S, and Bodmer R (2009). Semi-automated Optical Heartbeat Analysis of small hearts. J. Vis. Exp. 31, 1435. 10.3791/1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Alayari NN, Vogler G, Taghli-Lamallem O, Ocorr K, Bodmer R, and Cammarato A (2009). Fluorescent labeling of Drosophila heart structures. J. Vis. Exp. 32, 1423. 10.3791/1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Leal SM, Qian L, Lacin H, Bodmer R, and Skeath JB (2009). Neuromancer1 and Neuromancer2 regulate cell fate specification in the developing embryonic CNS of Drosophila melanogaster. Dev. Biol. 325, 138–150. 10.1016/j.ydbio.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lilly B, Zhao B, Ranganayakulu G, Paterson BM, Schulz RA, and Olson EN (1995). Requirement of MADS domain transcription factor D-MEF2 for muscle formation in Drosophila. Science 267, 688–693. 10.1126/science.7839146. [DOI] [PubMed] [Google Scholar]
- 65.Ryan KM, Hoshizaki DK, and Cripps RM (2005). Homeotic selector genes control the patterning of seven-up expressing cells in the Drosophila dorsal vessel. Mech. Dev. 122, 1023–1033. 10.1016/j.mod.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 66.Arganda-Carreras I, Kaynig V, Rueden C, Eliceiri KW, Schindelin J, Cardona A, and Sebastian Seung H (2017). Trainable Weka Segmentation: a machine learning tool for microscopy pixel classification. Bioinformatics 33, 2424–2426. 10.1093/bioinformatics/btx180. [DOI] [PubMed] [Google Scholar]
- 67.Alexander J, Stainier DY, and Yelon D (1998). Screening mosaic F1 females for mutations affecting zebrafish heart induction and patterning. Dev. Genet. 22, 288–299. . [DOI] [PubMed] [Google Scholar]
- 68.Zeng XX, and Yelon D (2014). Cadm4 restricts the production of cardiac outflow tract progenitor cells. Cell Rep. 7, 951–960. 10.1016/j.celrep.2014.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gargano JW, Martin I, Bhandari P, and Grotewiel MS (2005). Rapid iterative negative geotaxis (RING): a new method for assessing age-related locomotor decline in Drosophila. Exp. Gerontol. 40, 386–395. 10.1016/j.exger.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 70.Diop SB, Birse RT, and Bodmer R (2017). High Fat Diet Feeding and High Throughput Triacylglyceride Assay in Drosophila Melanogaster. J. Vis. Exp. 127, 56029. 10.3791/56029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McKeithan WL, Savchenko A, Yu MS, Cerignoli F, Bruyneel AAN, Price JH, Colas AR, Miller EW, Cashman JR, and Mercola M (2017). An Automated Platform for Assessment of Congenital and Drug-Induced Arrhythmia with hiPSC-Derived Cardiomyocytes. Front. Physiol. 8, 766. 10.3389/fphys.2017.00766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shannon P, and Richards M (2023). An Annotated Collection of Protein-DNA Binding Sequence Motifs. MptofDb Version 1.42.0. https://www.bioconductor.org/packages/release/bioc/html/MotifDb.html.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this study are available from the corresponding author upon reasonable request.
The R code used in this study is available from Github at https://github.com/gvogler/Dondi_2024. Semi-automated Optical Heartbeat Analysis software is available free of change at www.SOHAsoftware.com
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
RNA-Seq dataset was deposited in GEO-Gene Expression Omnibus. GEO: GSE271481