

Abstract
INTRODUCTION:
Understanding the factors influencing plant pathogen spread is vital for developing disease prevention strategies. Bacterial pathogens must overcome plant immunity, compete with microbes, and resist bacteriophages to colonize plants. Host genetics affect pathogen suppression, but the role of the surrounding microbiota remains unclear. Bacteriophage (phage), viruses of bacteria, and phage-derived elements are widespread entities that selectively kill bacteria. These elements, repurposed from phage ancestors, target co-occurring bacterial strains, potentially shaping microbial communities. Despite their prevalence, their impact on plant microbiota and pathogen spread remains largely unknown.
RATIONALE:
Our previous work revealed that wild populations of Arabidopsis thaliana, unlike agricultural and clinical pathogen outbreaks, are colonized by genetically diverse Pseudomonas populations without dominant strains. Infections with Pseudomonas viridiflava comprised multiple co-occurring strains even within single plants. What prevents single pathogenic lineages from spreading? Host immune diversity likely contributes to pathogen diversity, but the plant microbiome may also play a role. Given the common occurrence of phage and phage-derived elements and their strain-specific killing activity in Pseudomonas populations, we hypothesized that differences in sensitivity to phage components could suppress specific strains.
RESULTS:
We discovered an abundant viral cluster that is conserved across pathogenic strains. This cluster does not encode an intact phage but rather encodes a tailocin, a phage-derived element that bacteria use to kill bacterial competitors. Each pathogenic Pseudomonas strain carries one of a few distinct tailocin variants, which target variable polysaccharides in the outer membrane of co-occurring pathogens. Analysis of historic herbarium samples revealed that the same tailocin and outer membrane variants have persisted in the Pseudomonas populations for at least two centuries, suggesting the continued use of a defined set of tailocin haplotypes and receptors. Our results support a model in which the tailocin and outer membranes of co-occurring Pseudomonas strains are evolving in concert in wild A. thaliana populations. The divergent tailocin and outer membrane haplotypes have been maintained across time and space.
CONCLUSION:
The presence of a limited set of tailocin haplotypes in the metapopulation could reflect a limited panel of resistance mechanisms. Our findings provide a roadmap for identifying tailocin specificities to different strains and the possibility of determining the mechanism of this specificity. Tailocin therapy, akin to phage therapy, holds promise as an alternative to traditional antibiotics. Initial studies demonstrate its efficacy in suppressing pathogens in various plant and animal models. However, as with any antimicrobial treatment, resistance may arise. Our results suggest that “tailocin cocktails” leveraging the genetic diversity of pathogens could mitigate resistance by targeting the metapopulation concurrently.
Bacteria can repurpose their own bacteriophage viruses (phage) to kill competing bacteria. Phage-derived elements are frequently strain specific in their killing activity, although there is limited evidence that this specificity drives bacterial population dynamics. Here, we identified intact phage and their derived elements in a metapopulation of wild plant–associated Pseudomonas genomes. We discovered that the most abundant viral cluster encodes a phage remnant resembling a phage tail called a tailocin, which bacteria have co-opted to kill bacterial competitors. Each pathogenic Pseudomonas strain carries one of a few distinct tailocin variants that target the variable polysaccharides in the outer membrane of co-occurring pathogenic Pseudomonas strains. Analysis of herbarium samples from the past 170 years revealed that the same tailocin and bacterial receptor variants have persisted in Pseudomonas populations. These results suggest that tailocin genetic diversity can be mined to develop targeted “tailocin cocktails” for microbial control.
Graphical Abstract
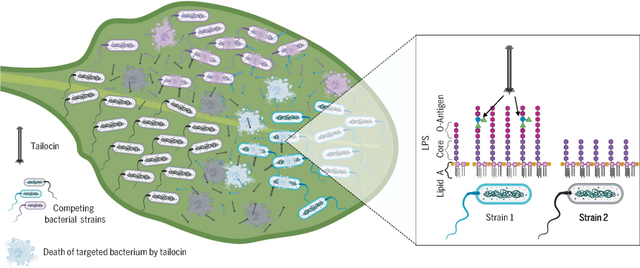
Pathogenic bacterial competitors on a leaf surface deploy tailocins as antagonistic agents. Tailocins are elements derived from bacterial viruses (bacteriophage) that have been repurposed to selectively target and kill strains of neighboring bacteria. The deployment of a diverse yet limited set of tailocins among pathogenic bacteria maintains their diversity over evolutionary timescales and shapes the coevolution of tailocins and their receptors in the outer membrane of targeted bacterial cells. [Figure created with BioRender.com]
Understanding the factors that influence the spread of pathogens is fundamental for developing disease prevention and treatment strategies. To colonize a plant, bacterial pathogens not only need to overcome the plant’s immune system, but they also must compete with surrounding microbes. Microbial disease is a frequent outcome in genetically homogeneous agricultural fields but rarer in genetically diverse wild plant populations (1). These wild plant populations are instead typically colonized at low levels by diverse pathogens, with no strain spreading widely (2). The prominent role of host genetics in suppressing pathogenic lineages is well documented (3), but how the surrounding microbiota affect disease dynamics remains largely unknown and is thus of increasing interest (4).
Bacteria have evolved diverse mechanisms of competition, and several of these mechanisms were repurposed from phage machinery (5, 6). Repurposed phage elements include injection systems such as the type VI secretion system and phage tail–like bacteriocins (tailocins) (6) in which remnants of the phage injection system have been retargeted for competition between bacteria. A common feature of many phage-related injection systems is their specificity; they can target specific strains of a bacterial species while not affecting the remainder (6). These injection systems are also widespread across bacteria (7–11). Given their prevalence, repurposed phage remnants are hypothesized to be drivers of microbiome composition in host and nonhost environments (6). To date, the impact of phage-derived elements on plant microbiota has only been observed in the laboratory under controlled conditions (12, 13). Whether these elements are a major suppressor of pathogen strains in the wild is a matter of speculation (14) [but see (15, 16)].
We previously found that wild populations of the plant Arabidopsis thaliana were colonized by a genetically diverse metapopulation of Pseudomonas bacterial pathogens (17). Our prior results suggested that these pathogen populations do not undergo the clonal expansions observed in agricultural and clinical pathogens. Infections of Pseudomonas viridiflava populations, even within a single plant, consist of several co-occurring strains with no single strain becoming dominant (2). What prevents single pathogenic lineages from spreading? Immune diversity of the host plant A. thaliana plays a role in maintaining Pseudomonas pathogen diversity; however, our previous work indicated that other members of the plant microbiome also affect the Pseudomonas composition (18–20). Because phage and their derived elements are common in Pseudomonas populations (21) and can be strain specific in their killing activity (22), we hypothesized that differences in sensitivity to phage components and their derived elements could suppress specific Pseudomonas strains in our system.
The viral cluster VC2 is strongly associated with a pathogenic clade of P. viridiflava
To determine whether phage-related elements are associated with turnover in the Pseudomonas colonizing A. thaliana, we first sought to characterize the abundant phage-homologous sequences in a wild Pseudomonas metapopulation. Viral sequences integrated within a bacterial genome can indicate a lysogeny event with a compatible phage (23). We characterized the presence and evolution of viral elements in 1524 Pseudomonas genomes all collected from A. thaliana in southwestern Germany (fig. S1) (24). More than 85% of these genomes are classified as the ATUE5 clade of P. viridiflava (Fig. 1A), an opportunistic pathogen that colonizes A. thaliana throughout Europe and the US (25, 26). We found viral sequences in 99.3% of the genomes, with an average of two viral sequences per genome (Fig. 1B). By using pairwise k-mer distances (27) and subsequent k-means clustering, we identified four viral sequence clusters (Fig. 1C).
Fig. 1. The pathogenic clade of P. viridiflava ATUE5 harbors a single highly conserved viral cluster (VC2).
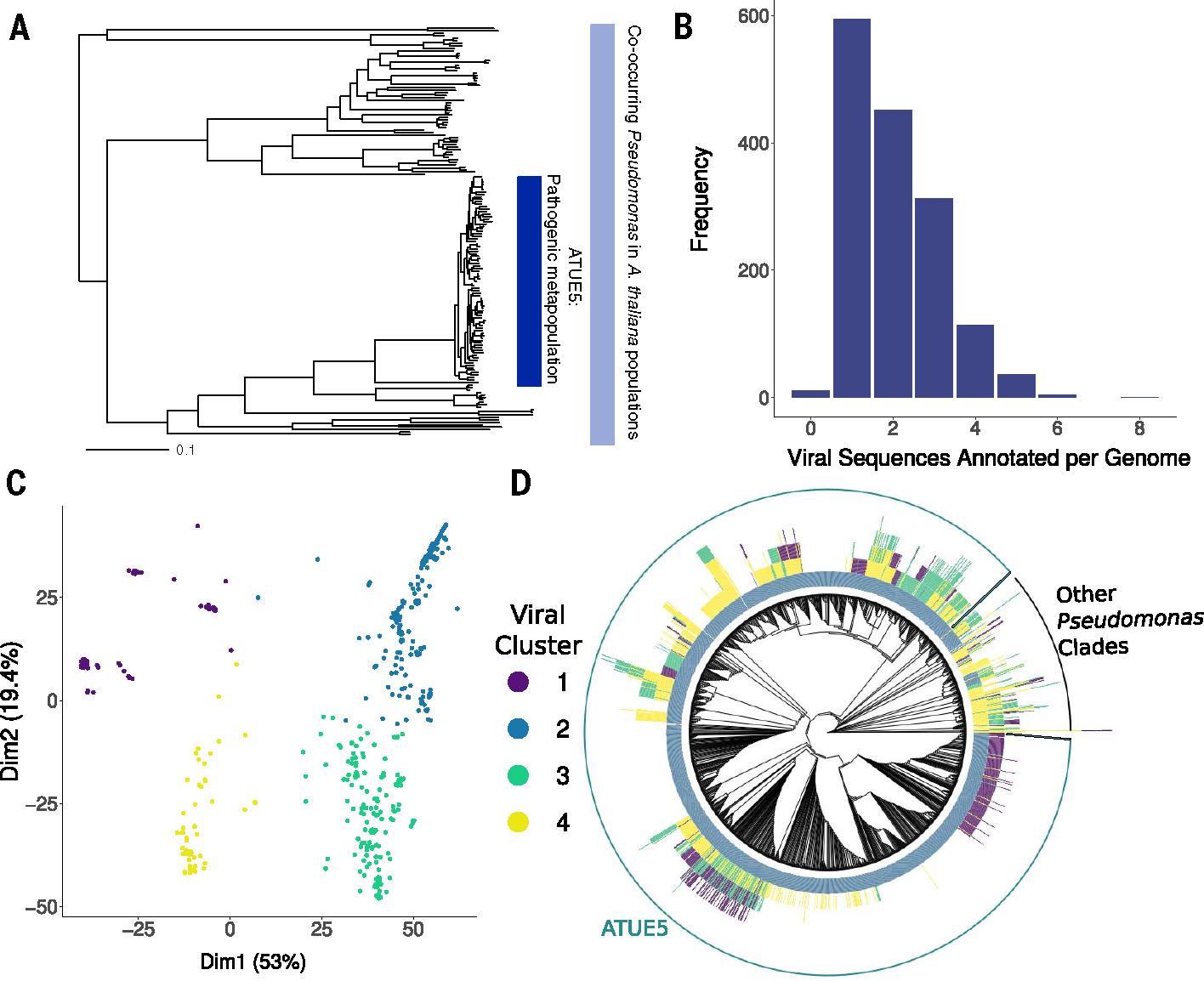
(A) Maximum likelihood concatenated core genome phylogeny of 1524 pseudomonads that co-occur in the A. thaliana phyllosphere (light blue bar). The dark blue bar indicates a pathogenic clade of P. viridiflava ATUE5 [adapted from Karasov et al. (2)]. (B) Frequency of viral elements identified in each Pseudomonas genome. Each genome harbors between one and eight viral elements. (C) Principal component analysis of viral elements based on pairwise k-mer Mash distances (27) and silhouette analysis reveals four defined clusters. Points are colored as indicated in the inset. (D) Viral elements of VC2 are present in all genomes of the pathogenic operational taxonomic unit (OTU) ATUE5. The phylogeny shows the relationships among 1524 Pseudomonas strains. Each stacked bar around the phylogeny indicates the presence of a viral element (with a maximum height of eight viral elements). The bars are colored as in (C). The outer circle shows strains that belong to ATUE5 (green) and other OTUs (black).
Phylogenetic associations between Pseudomonas clades and viral sequence clusters were immediately apparent (Fig. 1D) (2), with viral cluster 2 (VC2) found in all ATUE5 pathogenic strains, but they were less frequent (29%) (Fig. 1D) in nonpathogenic isolates outside of the ATUE5 clade. Furthermore, the gene composition and synteny of VC2 is conserved across ATUE5 strains but has divergent sequences outside the closest relatives to ATUE5 (Fig. 2B). The VC2 sequences in ATUE5 consist of 24 genes that are colinearly integrated in all genomes at the same location: between the trpE and trpG bacterial genes (28, 29). These VC2 sequences are predicted to encode the structural components typical of a prophage, including base plates, tail spikes, tubes, sheaths, tail fibers, putative assembly chaperones, and transcriptional regulation proteins.
Fig. 2. VC2 encodes a structurally functional tailocin.
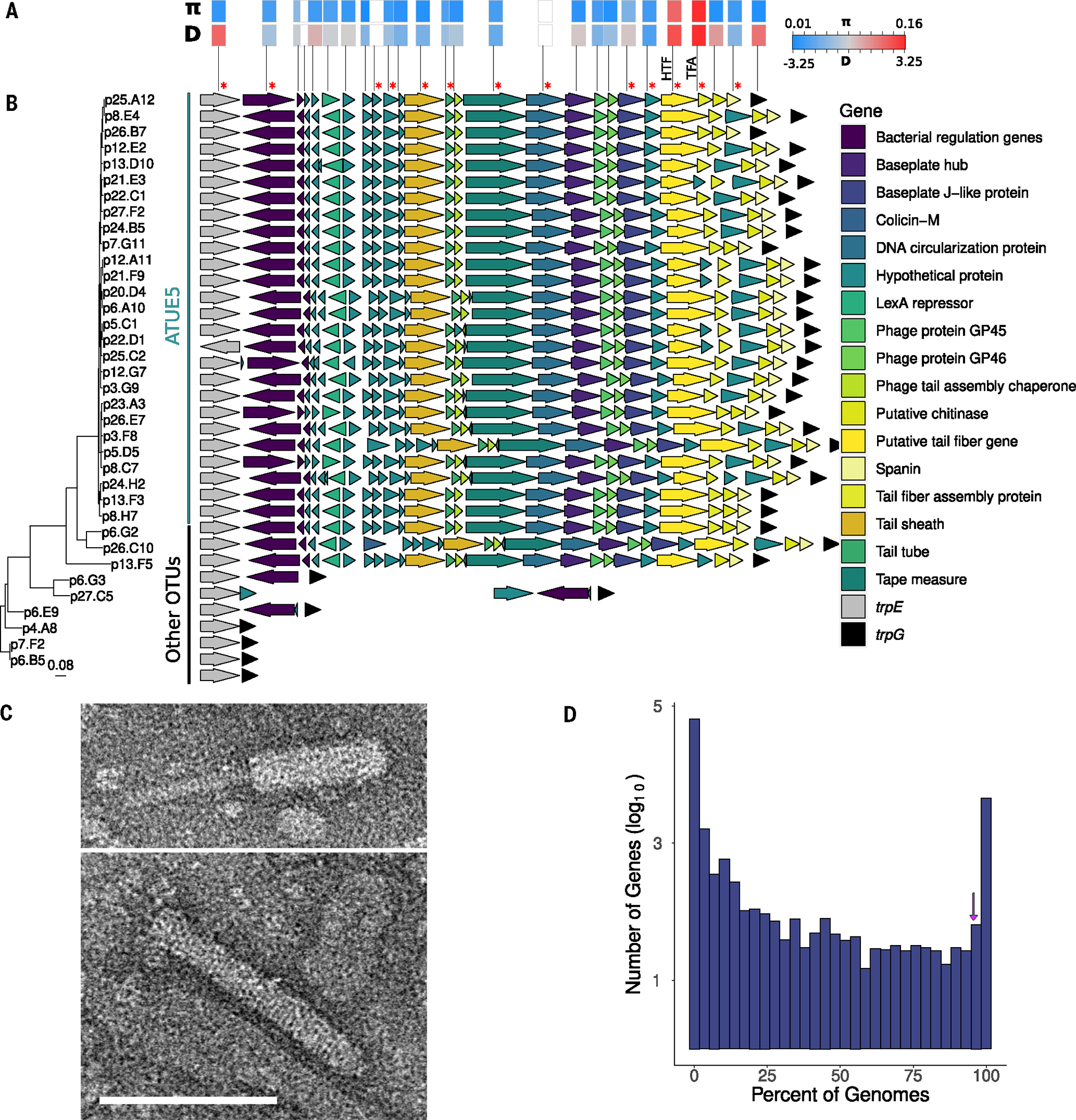
(A) Within-lineage measures of nucleotide diversity measured as pairwise nucleotide differences (π) and the site frequency spectrum measured as Tajima’s D (D) for each tailocin gene. White boxes indicate genes (statistics are excluded). Red asterisks indicate proteins found in proteomics analysis. (B) Genes of VC2 viral elements are syntenic in genomes of ATUE5. The arrows represent tailocin genes and surrounding bacterial genes (gene names are color coded in the inset). Each row is organized according to its phylogenetic placement. The phylogeny includes 36 Pseudomonas representative strains. The vertical lines indicate strains that belong to ATUE5 (green) and other OTUs (black). (C) TEM image demonstrates the presence of an assembled tailocin, showing the induced and partially purified tailocin from one representative ATUE5 strain (p25.A12) in both its contracted (top) and (bottom) uncontracted forms. Scale bar indicates 100 nm and applies to both micrographs. (D) The tailocin is part of the ATUE5 core genome. The histogram shows the frequency of each of the pangenome genes within 1399 ATUE5 genomes. The 11 most conserved tailocins genes are present in >90% of the ATUE5 genomes (marked with a purple arrow in the histogram).
VC2 encodes a structurally functional tailocin
Analysis of the VC2 gene content revealed that this cluster lacks the components necessary for phage replication and encapsulation, such as capsid, terminase, integrase, and recombinase proteins, suggesting that the VC2 genomic region does not encode a fully functional phage (30, 31). Although the structure of VC2 was not consistent with a functional phage, it did resemble previously described phage tail–like bacteriocins derived from phage, which are referred to as tailocins (32). Tailocins are repurposed phages that bacteria use to kill co-occurring bacterial strains (28) using the targeting machinery of the phage ancestor (33).
To test whether the tailocin genomic island produces the predicted tailocin protein complex, we induced tailocin production in a representative strain of ATUE5 (p25.A12) using treatment with mitomycin C (MMC) (34), partially purified the lysate, and then performed transmission electron microscopy (TEM). TEM images (Fig. 2C and fig. S2) revealed one rod-like tailocin in the MMC-induced lysates of p25.A12 existing in both the extended (sheath-uncontracted) and contracted (sheath-contracted and tube-ejected) forms (Fig. 2C). The rod-like structures suggest that this tailocin is an R-type contractile tailocin (35–37). Image comparison of the two tailocin forms indicated that the tailocins differ in average length: The contracted tailocins were on average 130 nm, whereas the extended tailocins were on average 144 nm in length. No other intact phage or bacteriocin-like structures were observed in the TEM images, only apparent sheath assemblies without tubes (fig. S2).
To validate that the observed tailocin in the TEM image corresponded to our predicted tailocin from VC2, we performed untargeted data dependent liquid chromatography–tandem mass spectrometry (LC-MS/MS) proteomics analysis (28, 38). Proteomics data confirmed the presence of tailocin proteins annotated in VC2 (Fig. 2B and table S1). No tailocin proteins were found in the sample that was not induced with MMC, and no other viral proteins or proteinaceous toxins such as S-bacteriocins were detected in the tailocin lysates. These results suggest that the highly conserved ATUE5 viral sequence is an active and inducible R-type tailocin.
Co-occurring Pseudomonas genomes encode highly diverse tailocin variants
The phylogenetic distribution of the VC2 cluster (Figs. 1 and 2B) suggested that the VC2 genomic island is well conserved across pathogenic ATUE5 strains. Across the 1524 Pseudomonas genomes analyzed (Fig. 2D), the full complement of the 27 tailocin genes was found in >95% of ATUE5 genomes, suggesting that the tailocin is in the core genome of ATUE5 strains along with essential bacterial genes (39). The conservation of the tailocin protein complex across all ATUE5 genomes, its conserved content, and its consistent location between trpE/trpG (32) suggests a single integration of the VC2 tailocin in the common ancestor of ATUE5 that is likely important for fitness in its natural ecological context.
In addition to the conservation of its gene content and collinearity, we found that the VC2 cluster harbors average levels of intralineage nucleotide diversity comparable with bona fide Pseudomonas core genes (Fig. 2A and fig. S3A), indicating that the VC2 cluster has likely been evolving under similar levels of selective constraint. However, by calculating nucleotide diversity for each of the VC2 genes individually, we identified greater levels of nucleotide diversity in two tailocin genes, the hypothetical tail fiber (HTF) and the tail fiber assembly (TFA) genes. Moreover, these two genes were also outliers for positive values of Tajima’s D (Fig. 2A and fig. S3B), a measure of the skewness of the site frequency spectrum (SFS), which in this case might indicate gene genealogies with deep divergence times and the likely existence of distinct haplotypes. It has been previously shown that the HTF and TFA genes together are key determinants of bacterial host range (40), which could explain their high level of diversity and the likely presence of multiple haplotypes.
We formally ascertained 40 and 25 haplotypes for HTF and TFA, respectively. In both cases, the five most common haplotypes accounted for ~80% of the Pseudomonas strains. The HTF haplotypes exhibited polymorphism in terms of their sequence length, with most falling into four distinct length categories (Fig. 3A). The HTF and TFA haplotypes are significantly associated (fig. S4, Fisher’s exact test, P = 0.0005). The high positive values of Tajimas’s D suggest that the divergent HTF and TFA haplotypes might have been maintained in the ATUE5 clade for long evolutionary times. Our population genomic analyses suggested that although the presence of the tailocin is conserved in pathogenic ATUE5 populations, the different ATUE5 strains encode one of a few divergent tailocin variants, with possible differences in binding and killing activity between the variants.
Fig. 3. Tailocins target closely related pathogens.
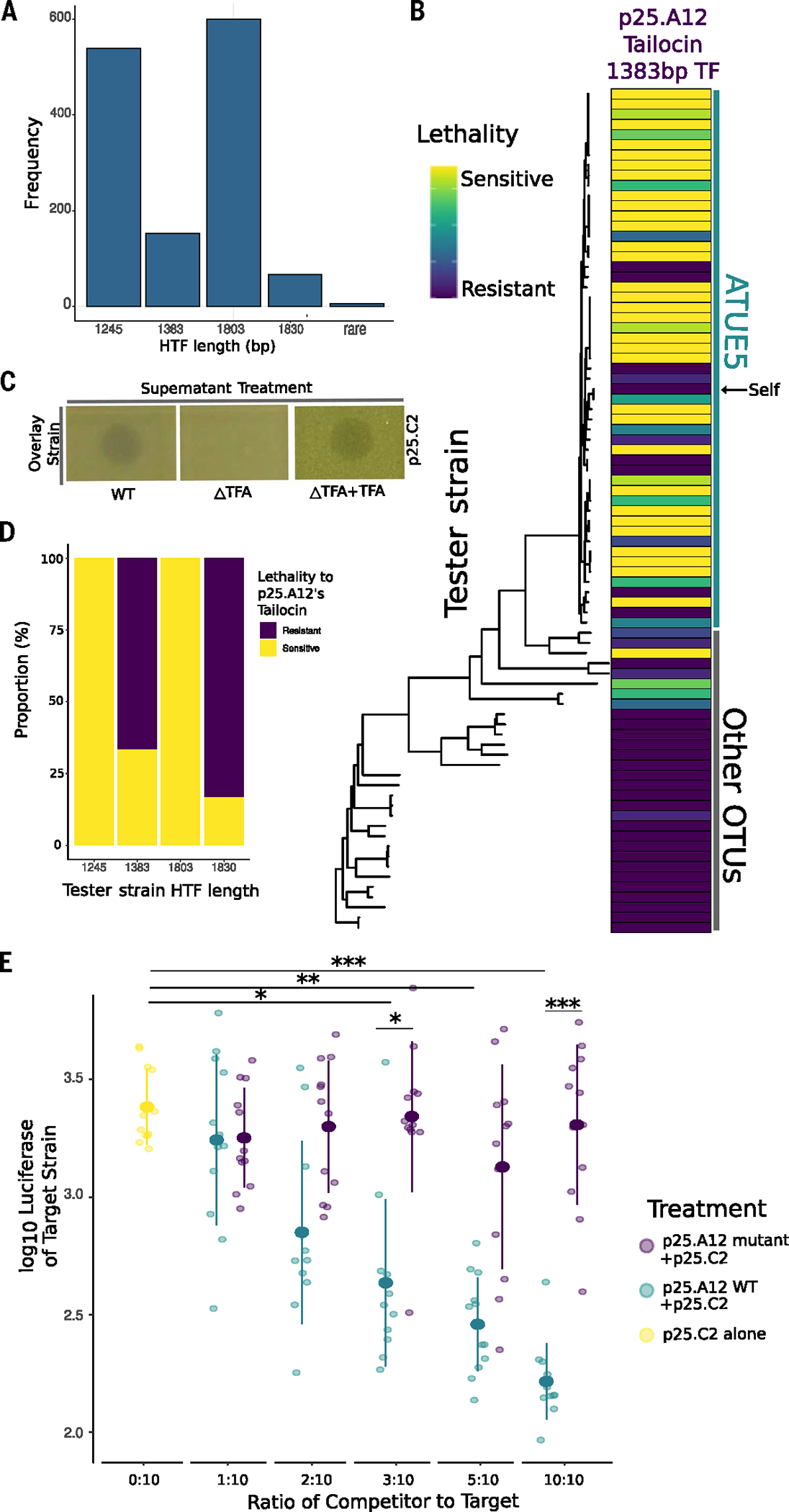
(A) Frequency of HTF nucleotide sequence lengths. There are four highly conserved lengths within the Pseudomonas populations. (B) Tailocins are preferentially used for intralineage killing. Soft agar cultures of the Pseudomonas strains (rows) were challenged with viral particles extracted from cultures of one strain, p25.A12 (from the 1383-bp hypothetical tail fiber length haplotype, column), in three technical and three biological replicates. The phylogeny includes 83 Pseudomonas representative strains and is displayed according to phylogenetic placement. Vertical lines indicate strains that belong to ATUE5 (green) and other OTUs (gray). Interactions with the strain’s own tailocin are indicated by the black arrow pointing to self. For each replicate, a strain was given a score of 3 for clear zone of inhibition, a 2 for semiclear, a 1 for opaque, or a 0 for no killing, and then added together after all three replicates. (C) Knockout of the tail fiber assembly gene disrupts tailocin bactericidal activity. Killing activity is indicated by a clearing in the lawn of the overlay strain. Complementation of the gene on an overexpression plasmid in the knockout strain restores the killing phenotype. (D) The proportion of tester strains sensitive or resistant to p25.A12’s tailocin, a 1383-bp hypothetical tail fiber, significantly correlated with the tester strain’s hypothetical tail fiber length haplotype (Fisher’s exact test, P = 10–8). (E) In planta coinfections of p25.A12 (competitor) and p25.C2 (target, known to be sensitive to p25. A12). Different ratios of competitor and constant amounts of target strain were used. p25.C2 was grown alone as the control. ANOVA test P values are shown. ***P < 0.001, **P < 0.01, or *P < 0.05.
Different tailocin variants kill closely related pathogenic Pseudomonas
We hypothesized that like other tailocins, the tailocin variants encoded by the Pseudomonas strains are used for interference competition against closely related competitor strains (28, 41). Our system provides an opportunity to characterize the killing activity of tailocins produced by co-occurring microbes in the same wild plant populations. The relatedness of these microbes spans the spectrum from closely related (ATUE5, <1% genome-wide sequence divergence) to distant commensals (other ATUEs, >20% genome-wide sequence divergence).
We hypothesized that the ATUE5 tailocin is used for interference competition against (i) other pathogenic pseudomonads that also produce a tailocin, (ii) commensal pseudomonads that mostly do not produce the VC2 tailocin, or (iii) other bacterial strains in the phyllosphere community. To test this, the tailocins from three strains and two representative HTF length haplotypes were induced and then partially purified, and then concentrated tailocin lysates were applied to 55 ATUE5 strains, 28 other ATUE strains, and >50 other bacterial strains isolated from the phyllosphere (taxonomy unknown). We found that, as expected, ATUE5 strains were resistant to their own produced tailocin (28). The killing assay also revealed that non-ATUE5 Pseudomonas strains were sensitive in only 6% of ATUE5-derived tailocin treatments, whereas 40% of ATUE5 treatments with ATUE5-derived tailocins exhibited sensitivity (Fig. 3B and fig. S6). To test whether other non-Pseudomonas community members were sensitive to the tailocin treatments, we tested 50 colonies (taxonomy and diversity unknown) from the surrounding phyllosphere and found that none was sensitive to tailocin treatment. An agriculturally relevant Pseudomonas strain, DC3000, was also tested and found to be sensitive to one of the tested tailocins (p25.A12). The different tailocin variants appear to exhibit partial killing specificity to different subsets of other ATUE5 pathogenic Pseudomonas, only 6% of the other Pseudomonas commensals, and not to other phyllosphere strains except for the above-mentioned DC3000.
To verify that the tailocin was responsible for the observed killing activity, we generated a TFA-deficient mutant. Tailocins collected from the mutant (p25.A12ΔTFA) lost killing activity against all strains tested (Fig. 3C and fig. S5), and the killing phenotype could be restored with an overexpression plasmid containing the TFA gene. These results suggest that tailocin was necessary for the observed killing activity.
To determine whether the TFA or HTF haplotypes are associated with killing activity, we analyzed whether the tailocin-killing spectrum correlates with a bacterial tester strain’s TFA or HTF haplotype. Rather than a correlation, we found an array of different killing spectra, with the tail TFA and/or HTF haplotypes tested all displaying broad killing against other ATUE5 strains (Fig. 3B and fig. S6). Although we found there to be no significant associations between killing spectra and TFA or HTF haplotype, we did identify a significant association between the length of the HTF and its killing spectrum (Fig. 3, A and D, Fisher’s exact test, P = 10−8). This suggests that the tail fibers in the characterized tail fiber sequence length haplotypes could be important in binding to target cells, and that the tail fiber haplotypes are likely associated with an outer membrane receptor gene.
We next aimed to determine whether tailocin killing is observed in the plant host. We performed competition assays in planta. Strain p25.12’s tailocin was able to kill a particularly pathogenic strain, p25.C2, in all three killing assays (Fig. 3B, third row). To test whether the target strains would also be killed by the strain p25.A12, we grew strains together using a variant of p25.C2 tagged with luciferase (42) to measure its relative abundance in three replicates. As a control, we also competed p25.C2 with p25.A12ΔTFA. Five ratios of p25.A12 and p25.C2 were used to determine whether the concentration of the tailocin strain affected killing while controlling the abundance of the target strain. We found that p25.A12 significantly reduced the growth of p25.C2 in the three highest ratios tested (Fig. 3E). The 10:10 and 3:10 ratios significantly differed from the p25.A12ΔTFA competitive assay, suggesting that having a functional tailocin suppresses the growth of p25.C2 and that p25.A12 is no longer able to significantly reduce the growth of p25.C2 when lacking the tailocin. The 5:10 ratio did not significantly differ from the p25.A12ΔTFA competitive assay, perhaps due to technical error. We found that p25.C2 grown with p25.A12ΔTFA was never significantly different from p25.C2 grown alone, suggesting that tailocin is an effective competitive mechanism in the plant host. Additionally, the mutant and wild-type p25.A12 were shown to have no growth differences in planta (fig. S7). These results were replicated in competition assays done in test tubes (fig. S8); however, in these trials, p25.A12ΔTFA competing with p25. C2 significantly differed from the control for the highest ratio. This result could indicate that p25.A12 uses another competitive mechanism in the absence of tailocin, which proves ineffective at lower starting abundances. These results suggest that tailocin killing is important in killing closely related competitors in planta.
The tail fiber is coevolving with its O-antigen receptor in natural populations
The differences that we observed between strains in their susceptibility to tailocin (Fig. 3B) indicated that these strains may encode an unidentified receptor that has different variants. To identify the receptor, we conducted a transposon mutagenesis screen (43) in which we screened a mutant library of a susceptible strain (p25.C2) for insertions that render the strain resistant to the p25.A12 tailocin (Fig. 4A). Insertions (putative knockouts) in 70 genes were significantly associated with resistance to the tailocin. Six of these genes lie within an O-antigen biosynthesis gene cluster, a locus previously implicated in resistance to phage and tailocin tail fibers (44, 45). These results suggested that differences between strains in their O-antigen may explain differences in tailocin susceptibility. Comparative deoxycholic acid (DOC)–PAGE analysis of the lipopolysaccharide (LPS) isolated from two resistant and two susceptible strains showed that the resistant strains lacked the high-molecular-weight polysaccharide chain that was present in the susceptible strains. Subsequent gas chromatography–MS (GC-MS) (Fig. 4C) revealed that the susceptible strains had relatively high amounts of rhamnose compared with the resistant strains (table S2). Our finding of the higher content of rhamnose, along with the expanded size of the LPS in tailocin-resistant strains, agrees with previous studies suggesting that the affected gene locus might be involved in the biosynthesis of the O-antigen in Pseudomonas (46–48). Here, we propose that the O-antigen is likely a receptor for the focal tailocin.
Fig. 4. The O-antigen is important for tailocin lethality and is coevolving with the tailocin HTFP.
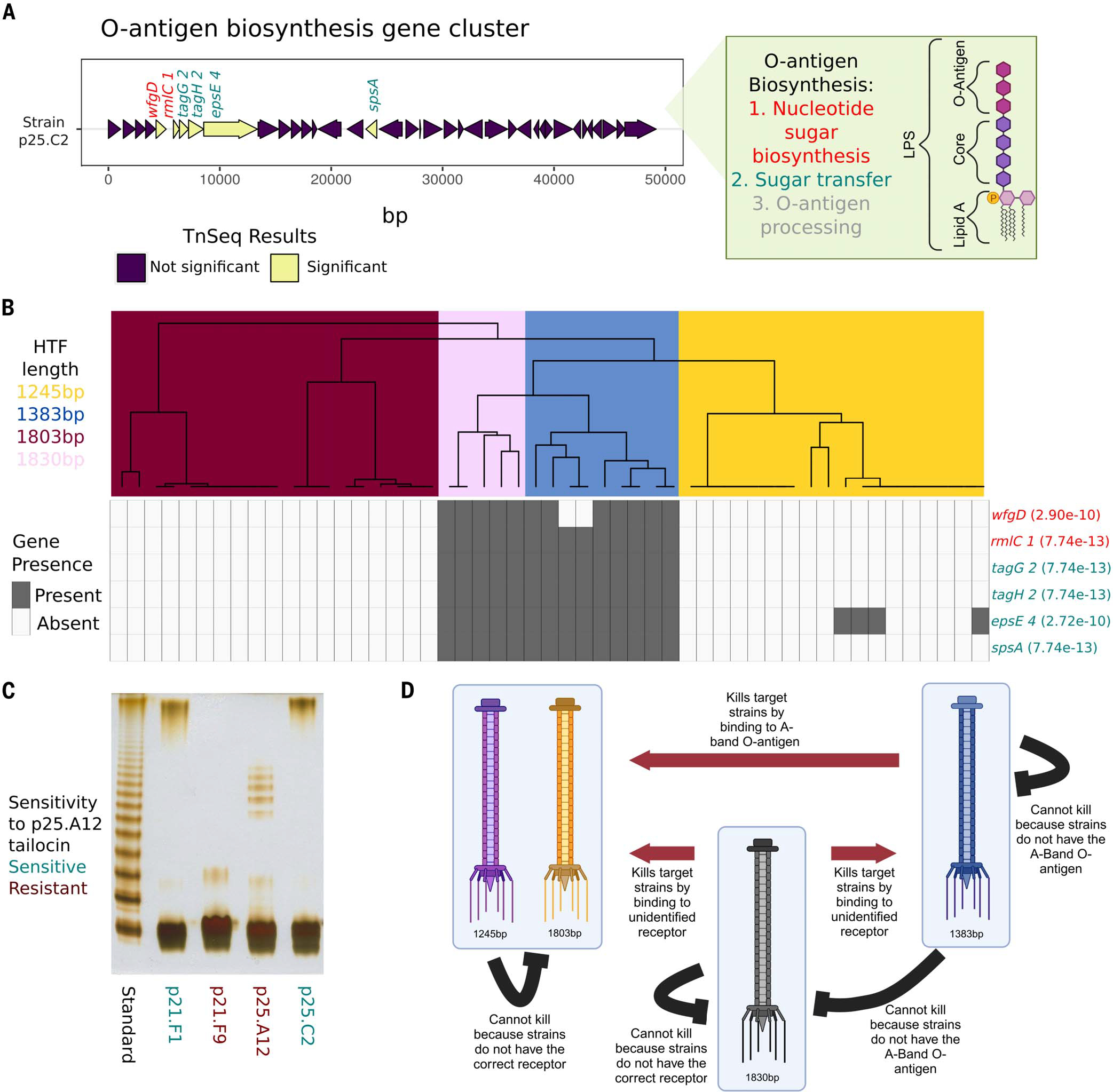
(A) Gene plot of the previously characterized O-antigen gene cluster found in the ATUE5 strains. Six of the 70 significant TnSeq genes in this study were found in this gene cluster and are shown in yellow. Gene names are colored by the step in the O-antigen biosynthesis pathway. (B) The top dendrogram is from hierarchical clustering of the PanKmer (92) output for the hypothetical tail fiber gene and colored by length. Rows represent the six significant O-antigen genes. Gray corresponds to gene presence and white to gene absence. Fisher’s exact test P values between genes and length-based clusters are shown in parentheses. (C) DOC-PAGE profile of silver-stained LPS (1 μg each lane) isolated from a subset of ATUE5 strains. The standard is of Salmonella enterica Ser. typhimurium (S-type LPS). Gray boxes indicate the high-molecular-weight O-chain. (D) Our working model of tailocin killing and lethality in P. viridiflava isolates. Red arrows indicate known significant patterns of killing. Black lines show known significant patterns of nonkilling. [Figure created with BioRender.com].
Given that strains are resistant to their own tailocins (33), we posited that the tailocin and receptor loci should coevolve. To test this, we measured congruence between tail fiber and LPS haplotypes (Fig. 4B) and found that the tail fiber and LPS haplotypes statistically co-varied (Fisher’s exact test, P < 2.72 × 10–10 for every association between presence or absence of the gene and the length-based clusters; Fig. 4B). Knowing that the tail fiber haplotype allows for prediction of the LPS haplotype. These experiments support a model in which the tail fiber and LPS of co-occurring Pseudomonas strains are evolving in concert in wild A. thaliana populations (Fig. 4D).
The divergent tailocin and LPS haplotypes have been maintained across time and space
To ascertain the time scales of the emergence of the tailocin and different tailocin variants, we sought to retrieve Pseudomonas genomes from historical A. thaliana herbarium specimens collected over the past two centuries. Only historical Pseudomonas genomes can reveal whether extant populations are the result of population continuity or population replacement. We used state-of-the-art ancient DNA retrieval and sequencing techniques (49) to sequence leaves of 35 A. thaliana herbarium specimens using shotgun genomics (table S3). We mapped the herbarium-derived reads to the ATUE5 reference genome strain to identify Pseudomonas reads and found seven samples with >60% of the genome covered by at least one sequencing read (mean genome coverage 0.97 to 8.7x) (fig. S9 and table S3). The Pseudomonas-derived reads showed patterns of DNA damage and fragmentation typical of ancient DNA (50), which authenticated their historical origin (fig. S10). Our analysis revealed that herbarium specimens preserve the genomes of highly abundant bacterial genera of the A. thaliana phyllosphere.
To place the historical Pseudomonas genomes in the context of extant genetic diversity, we first used patterns of homozygosity support to identify herbarium specimens most likely to be colonized by a single dominating Pseudomonas strain (fig. S11).
This analysis allowed us to select three historical samples (mean genome depth of 8.2 to 9.6x), which we combined with 65 present-day Pseudomonas genomes to build a phylogenetic tree using whole-genome single-nucleotide polymorphisms (SNPs). The three historical samples were placed within the present-day ATUE5 diversity (Fig. 5), showing the genetic continuity of this clade for at least the past 177 years. Moreover, the mappings of the historical samples against the ATUE5 reference genome identified the presence of the tailocin in all three historical bacterial genomes (Fig. 5A). To ascertain the tailocin variants encoded in the historical genomes and to assess whether the gene colinearity and the insertion location in the bacterial genome are conserved in the historical genomes, we performed de novo assembly of historical Pseudomonas-derived reads. We obtained an average of nine contigs per sample (average sequence length of ~3.4 kb) that were sufficient to show that both the insertion place of the tailocin and the colinearity of its genes are conserved in the historical genomes (Fig. 5A). Moreover, we ascertained that each historical genome carries a different tailocin variant and that all historic variants are segregating in present-day populations (Fig. 5B and fig. S12A). The polyphyletic distribution of the HTF and TFA gene haplotypes (Fig. 5B and fig. S12A) is likely the result of recombination at this locus (40).
Fig. 5. The same length haplotypes of HTF are present in contemporary and century-old historical samples, and the HTF haplotype diversity is maintained at the leaf scale and broader geographical scales.
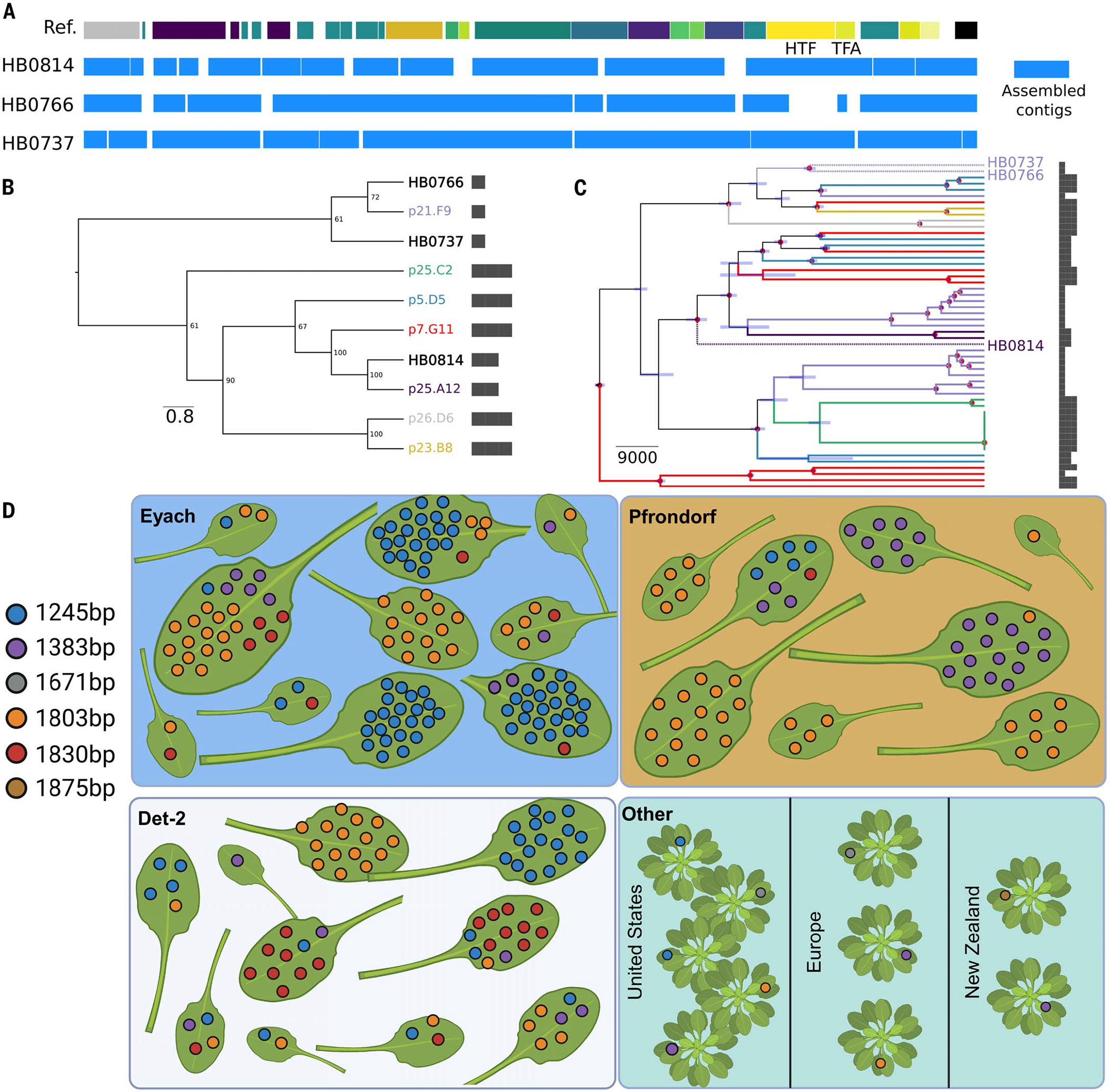
(A) De novo assembled contigs spanning the tailocin genomic region for the historical samples. The reference track on the top represents the genes present in the ATUE5 reference genome. The blue boxes depict the de novo assembled contigs. (B) Maximum-likelihood neighbor-joining tree of the HTF gene translated sequence. Historical samples (HB) are placed in the context of the most common haplotypes. HTF lengths are shown with bars. (C) Bayesian tip-date calibrated phylogeny representing the evolutionary relationship between historical and modern Pseudomonas strains. The tips and branches are colored based on the HTF gene haplotype. Only historical sample labels are shown with stars. The node bars represent the 95% highest posterior density intervals of the estimated time, and the nodes marked with red dots represent those with a posterior probability of 1. HTF lengths are shown with bars. (D) A subset of eight to 10 plants from the three German populations (blue, orange, and white panels) and 10 P. syringae isolates collected from plants globally, which were downloaded from NCBI (green). Each leaf is from a single plant. Colored dots indicate a single bacterial isolate found and its corresponding HTF length. Plants in the bottom right panel indicate those not found within the same population. [Figure created with BioRender.com]
We used our phylogenetic reconstruction to determine that the tail fiber assembly gene haplotypes of the historical genomes are closely related to different modern haplotypes (Fig. 5C and fig. S12B), suggesting that the same haplotypes have circulated in the Pseudomonas metapopulation for the past 177 years.
The observed persistence of HTF length haplotypes within the bacterial metapopulation may result from a potential artifact introduced by population structure at the plant, plant patch, or site level. At these levels, specific haplotypes could reach fixation, creating the appearance of the maintenance of multiple haplotypes when analyzing all populations collectively. To rule out this possibility, we looked at the co-occurrence of the HTF length haplotypes at the level of single leaves, all collected within a single site in the German metapopulation, as well as at broader geographic locations using 10 P. syringae genomes from the National Center for Biotechnology Information (NCBI) (five isolates collected from the US, three from Europe, and two from New Zealand). We found that HTF lengths co-occurred in single leaves and in all three German populations (Fig. 5D). Additionally, we found that the 1245-, 1383-, and 1803-bp HTF lengths in isolates from other countries, in addition to two other lengths. These results suggest that HTF lengths are conserved and maintained at the microscale and over great geographic distances.
Discussion
We discovered that a tailocin, or repurposed phage, is conserved as part of the core genome of metapopulations of the pathogen P. viridiflava. The sequence conservation of its genes (other than TFA and HTF) indicates that the tailocin is likely essential for survival in P. viridiflava pathogen metapopulations. This finding is consistent with previous work suggesting that antimicrobial nanomachines such as the type VI secretion system are important for survival in the plant environment (51). Despite the conservation of the entire tailocin gene cluster, multiple allelic variants of the tailocin have been maintained in these populations. We found that the allelic variants target different strains of related pathogenic P. viridiflava that differ in the LPS-enriched outer leaflet of the outer membrane (28). Similar to the coevolution of toxin-antitoxin systems, the tail fiber and assembly proteins of an ATUE5 strain are coevolving with the LPS membrane composition (52). The strong evolutionary conservation and correlation between loci suggests that inter-Pseudomonas strain competition through the tailocin is a strong and persistent selective pressure on Pseudomonas pathogens.
Second, our results showed that the same tail fiber and/or LPS variants have been maintained in the Pseudomonas populations for the past 200 years. Bacteria are capable of rapid adaptation (53). The fact that a defined set of variants was maintained in these populations indicates selective constraints on the evolution of this system, consistent with what has been found in viral evolution (54). The presence of a limited set of tail fiber haplotypes in the metapopulation could reflect a limited panel of resistance mechanisms.
Our findings provide a roadmap for identifying tail fiber specificities to different strains and introduce the possibility of determining the mechanism of this specificity. Tailocin therapy, like phage therapy, has been proposed as a possible alternative to small-molecule antibiotics (12, 13, 55). Proof-of-concept studies have shown that tailocins can be used to suppress specific pathogens in animal (55) and plant (12) models. With any antimicrobial treatment, there is a risk that the target bacterium will evolve resistance. Our findings indicate that, considering the probable constrained array of resistance mechanisms, the tailocin genetic diversity of a pathogen metapopulation can be mined to discover “tailocin cocktails.” These cocktails have the potential to target the metapopulation in parallel, thereby reducing the likelihood of resistance evolution. In the future, the mining and characterization of the tailocin repertoire from diverse wild Pseudomonas populations should unveil possible combinations between the tail fiber and LPS composition, as well as the time scales in natural settings over which tailocin resistance emerges.
Materials and Methods
Bacterial strains, plasmids, and growth conditions
The strains used in this study are detailed in table S2. Bacteria were grown on nutrient lysogeny broth (LB) agar and in nutrient LB medium at 28°C for Pseudomonas strains and at 37°C for Escherichia coli strains. All liquid cultures were incubated with shaking at 180 rpm. Growth media were supplemented with kanamycin (50 mg/ml, Km50), nitrofurantoin (100 mg/ml, NFN100), or tetracycline (20 mg/ml, TET20). All strains were stored at −80°C in 20% glycerol [v/v]. For plasmid preparation, E. coli transformants were cultured in LB. TET20 was used to select E. coli transformants. LB supplemented with sucrose, and nitrofurantoin (10 g/L tryptone, 5 g/L yeast extract, 10% w/v filtered sucrose, 15 g/L agar, NFN100) plates were used in the selection of the second P. syringae homologous recombination event.
Viral identification and clustering
VIBRANT v1.2.1 (24) was run on the 1524 assembled Pseudomonas genomes and 10 NCBI genomes to identify viral sequences. To group the 3104 annotated viral sequences from VIBRANT into genetically similar clusters, the pairwise mutation distance was calculated for all 3104 viral sequences with Mash v2.2.2 (27), a MinHash-based technique that reduces sequences to compressed k-mer representations (sketches). Mash was run ignoring single-copy k-mers that are more likely the result of sequencing errors, and a sketch size of 10,000, which corresponds to the number of nonredundant MinHash values that are kept for the pairwise distance calculation. This resulted in a 3104 × 3104 pairwise mutation distance matrix. Next, the viral sequences were clustered from the pairwise mutation distance matrix with principal component analysis and k-means clustering (56, 57). Silhouette analysis (58) was performed to determine the optimal number of clusters for the data.
Annotation of the VC2 gene cluster
A highly conserved viral sequence, the VC2 gene cluster, was identified in assembled genomes using VIBRANT v1.2.1, and groups of orthologous genes (orthogroups) were identified using panX (59). Insertion sites for VC2 gene clusters were identified by searching for orthologs of the known viral sequence flanking genes mutS, cinA, trpE, and trpG. VC2 was always found between trpE and trpG. Gene function predictions were determined by orthologous viral genes previously characterized in VIBRANT and BLAST. Tailocin gene clusters were visualized with the R package “gggenes.”
Viral particle extraction and partial purification
Overnight cultures were back-diluted into 50 ml of fresh LB to extract and isolate tailocins from the various Pseudomonas strains. When cultures reached anexponential growth phase [an optical density at 600 nm (OD600) of 0.4 to 0.6), 5 μg/ml MMC (Selleck, catalog no. S8146) was added to induce the bacterial SOS (Save-Our-Soul) response and tailocin induction. The cultures were then incubated at 28°C for a minimum of 18 hours and then centrifuged at 4°C for 1 hour at 1400 × g to pellet cell debris. The supernatants were sterilized by filtration with 0.2-μm cellulose acetate filters. To precipitate viral particles, 40% (w/v) of ammonium sulfate was slowly added to the filter-sterilized tailocin lysate while stirring on ice. The lysate was left stirring on ice for at least 18 hours. After 18 hours at 4°C, the ammonium sulfate was pelleted by centrifugation at 2090 × g for 2 hours at 4°C. The supernatant was discarded and the pellet was resuspended in 500 μl of cold P-buffer (100 mM NaCl, 8 mM MgSO4, 50 mM Tris-HCl, pH 7.5) and stored at 4°C. Tailocin lysates were used fresh or for up to 2 weeks.
Visualization of viral particles using TEM
One negatively stained specimen (from strain p25.A12) was prepared and imaged in the following manner. First, 3.5 μl of sample was placed on a carbon-coated, TEM grid that had been glow discharged to make it more hydrophilic. After ~1 min, filter paper was used to remove excess solution from the grid by touching the side of the grid. This was followed by two steps of brief (~1 to 2 s) washing in P-buffer and blotting with filter paper in the same manner. Next, this process was repeated twice but with the negative stain solution (1% ammonium molybdate). Finally, the grid was incubated for 15 to 20 s in a droplet of 1% ammonium molybdate, blotted again, and allowed to air dry. Negatively stained specimens were viewed on Tecnai 12 (ThermoFisher Scientific, Waltham, MA) and JEM-1400 (JEOL, Tokyo, Japan) transmission electron microscopes operated at 120 kV. Images were recorded on an UltraScan and Orius cameras (Gatan, Pleasanton, CA) respectively.
MS characterization of tailocin proteins
Tailocins partially purified as described above were treated with or without MMC and resuspended in 50 mM triethylammonium bicarbonate and 5% SDS (1× S-TRAP buffer) and frozen at −80°C until digestion. Total protein content was quantified using the Pierce BCA Protein Assay Kit (ThermoFisher Scientific), and 10 μg of protein was diluted into 25 μl of 1× S-TRAP buffer. The protein was reduced with 20 mM dithiothreitol at 37°C for 30 min and alkylated with 40 mM iodoacetamide at room temperature for 45 min in the dark. Samples were acidified with phosphoric acid and digested using micro-S-TRAP columns (Protifi, Fairport, NY) with 0.5 μg of trypsin/Lys C per sample according to the manufacturer’s instructions. The peptides were dried to completion, resuspended in 300 μl of 0.1% trifluoroacetic acid, and desalted using Pierce Peptide Desalting Spin Columns (ThermoFisher Scientific) according to the manufacturer’s instructions. The peptides were resuspended in 40 μl of 0.1% formic acid for LC-MS/MS analysis.
Reversed-phase nano-LC-MS/MS was performed on an Dionex UltiMate 3000 RSLCnano system coupled to a ThermoFisher Scientific Q Exactive-HF orbitrap mass spectrometer equipped with a nanoelectrospray source. One microgram of each sample was first trapped on a 2-cm Acclaim PepMap-100 column (ThermoFisher Scientific) with 5% acetonitrile at 5 μl/min. Then at 5 min, the sample was injected onto the LC reverse-phase Acclaim PepMap 100 C18 2.0 μm nanocolumn (ThermoFisher Scientific). A 500-mm-long, 0.075-mm inner-diameter nanocolumn heated to 35°C was used for chromatographic separation. The peptides were eluted with a gradient of reversed-phase buffers (buffer A: 0.1% formic acid in water; buffer B: 0.1% formic acid in 100% acetonitrile) at a flow rate of 0.2 μl/min. The LC run lasted for 85 min with a starting concentration of 5% buffer B, increasing to 28% buffer B over 75 min, up to 40% buffer B over 10 min, and held at 90% buffer B for 10 min. The column was allowed to equilibrate at 5% buffer B for 20 min before starting the next data acquisition. The Q Exactive-HF mass spectrometer was operated in data-dependent acquisition MS/MS analysis mode selecting the top 20 most abundant precursor ions between 375 and 1650 m/z at 60,000 resolution for fragmentation at 15,000 resolution.
The raw data were analyzed using Proteome Discoverer 3.0 software with the SEQUEST algorithm against the uniprot_ref_pseudomonas_viridiflava database (1–18-2023 version with 4389 proteins) or the p25.A12 database. An allowance was made for two missed cleavages after trypsin and Lys C digestion. No fixed modifications were considered. The variable modifications of methionine oxidation and cysteine carbamidomethylation were considered with a mass tolerance of 15 ppm for precursor ions and a mass tolerance of 0.02 Da for fragment ions. The results were filtered with a false discovery rate of 0.01. A minimum of one unique peptide was reported for all proteins identified.
Allelic diversity and nucleotide sequence data analysis
Orthogroups that were previously identified using panX were included in the analysis. The nucleotide and peptide sequences were aligned with Clustal Omega (60), and then a codon-based nucleic acid alignment was generated with pal2nal (61). This step is critical to ensure that the nucleic acid alignment is aligned codon by codon to determine whether substitutions result in a synonymous or nonsynonymous amino acid change. The output aligned file was saved in fasta format and used as an input file for population summary statistic calculations in R using the package PopGenome v2.7.5 (62). The level of sequence diversity among ortho groups was compared using the summary statistics π (average pairwise difference or average number of nucleotide diversity per site) (63) and θ (population mutation parameter or number of segregating sites) (64), whereas Tajima’sD (65) was used as a proxy for the SFS, and thus to determine the demographic and selective forces shaping the SFS of tailocin genes.
Ascertainment of tailocin haplotypes
To determine how many haplotypes of tail fiber assembly and hypothetical tail fiber genes there are in the pathogenic strains in our study, nucleotide and peptide alignments from Clustal Omega (60) were analyzed using theR package pegas (66). The haplotype function was used to determine how many unique haplotypes are in the ATUE5 strains. The most common haplotypes were used for downstream analyses.
Mapping of reads from historical samples to ATUE5
A set of 35 herbarium A. thaliana samples collected between 1817 and 1957 from southern Germany (table S3) (67) were screened for Pseudomonas presence. Adapter sequences from the raw reads were trimmed and merged using Adapterremoval V.2 (68) and subsequently mapped to the Pseudomonas p25. A12 assembly using bwa aln v.0.7.17 (69) with seed deactivation to allow the alignment of of substitutions present at the termini of the reads, for example, those arising from DNA deamination in historic material (49). A total of seven samples for which the Pseudomonas reference genome was covered in at least 60% were kept (table S2). To authenticate the historic origin of the bacterial reads, the proportion of C-to-T (or G-to-A) substitutions, as well as the fragment size distribution of the reads, were computed using mapDamage v.2.2.1 (70) (fig. S8). Patterns of homozygosity in the mapped reads were analyzed and only samples that were likely dominated by single Pseudomonas lineages were retained (fig. S10). As a result, only the samples HB0737, HB0766, and HB0814 were kept for downstream analyses.
Ascertainment of tailocin variants in historical genomes
To identify reads aligning to the whole spectrum of tailocin genetic diversity, different haplotypes of the highly polymorphic tail fiber assembly and hypothetical tail fiber genes (Fig. 3A) were added to the p25.A12 reference genome before the historical samples were mapped as described above. Reads covering the Tailocin region surrounded by the flanking genes trpE and trpG, as well as all reads covering any haplotype of the mentioned genes, were subset from the raw reads and used as input for de novo assembly using SPAdes v.3.13.0 (71). Samples HB0737, HB0766, and HB0814 yielded 10, 9, and 14 assembled contigs, respectively (table S4). To find collinearity between the de novo assemblies and the reference Tailocin region, we used minimap v.2.1 (72). Amino acid–translated sequences of the historical isolate–specific haplotypes for the tail fiber assembly protein and putative tail fiber protein were merged together with the haplotypes segregating in the observed diversity. Finally, Clustal Omega v.1.2.4 (73) was used to generate a multiple alignment and to classify the haplotypes in the historical samples.
Whole-genome phylogenetic reconstruction combining present-day and historical genomes
A set consisting of 50 ATUE5 modern and three historic Pseudomonas strains was used to create a phylogenetic reconstruction. We used the mapped reads to perform individual de novo variant callings using bcftools mpileup and bcftools call v1.11 (74) removing those reads with mapping quality values lower than 30. We then merged all individual calls using bcftools merge v.1.11 (74) and removed those samples with <60% of the called variants. We kept only biallelic SNPs and sites with a maximum missing information of 10% of the remaining samples. The number of retrieved variants were 176,528 SNPs segregating among 53 strains (50 modern and three historic).
Using the genomic SNPs, we sought to reconstruct the phylogenetic relations among the 53 strains. We used IQ-TREE V2.2.0.3 (75) in combination with ModelFinder (76), and ultrafast bootstrap UFBoot (77) to estimate a maximum-likelihood phylogenetic tree. Variant sites that are likely to arise due to putative recombination were identified using ClonalFrameML v.1.12 (78), and the output was used to mask putative recombinant positions in the original genomic SNP array. BEAST2 (79) was used to jointly estimate the phylogenetic relations among the strains and the relative time of emergence for all the tree nodes. For this purpose, we used the collection dates (table S3) and a previously estimated evolutionary rate (80) as priors. To minimize parameter estimation, we chose HYK as the evolutionary model, and to avoid demographic history assumptions, we used a coalescent extended Bayesian skyline approach (81).
Testing bacterial sensitivity to tailocins with spot test phenotypic assays
To test the sensitivity of the different bacterial strains to the tailocins, soft agar assays were performed using an adaptation of a protocol from Vacheron et al. (41). Overnight cultures (750 μl) of each strain were mixed with 25 ml of LB soft agar (0.8%), and the mixture was poured into a square plate and left to harden. Then, aliquots of 3 to 5 μl of concentrated viral particles suspension were applied to the agar, along with a control of P-buffer alone and noninduced cultures in serial dilutions. The plates were incubated overnight at 30°C. Bacterial sensitivity or resistance to the viral particles was assessed after 24 hours. When testing serially diluted tailocins, no plaques were observed, suggesting that the killing agent is nonreplicative and not a phage. When testing the uninduced tailocin control samples (not induced with mitomycin C), if lethality was observed, the interaction was deemed to be inconclusive. Spot tests were performed in three biological and three technical triplicates.
Testing bacterial sensitivity to tailocins in culture
To test the sensitivity of a known sensitive strain (p25.C2) to the mutant tailocin from 25.A12ΔTFA, p25.C2 was grown overnight and then diluted by a factor of 1:10 in fresh LB medium. Once the culture reached the growth phase, cultures were supplemented with 5% p25.A12 tailocin, 5% p25.A12ΔTFA tailocin, or a buffer control. OD600 was measured in a 96-well plate using a microplate reader (TECAN Spark).
Construction of the Pseudomonas p25.A12 tail fiber assembly gene deletion
The p25.A12ΔTFA mutant strain was cloned using the Gateway cloning system with the donor vector pDONR1K18ms (Addgene plasmid no. 72644) and the destination vector pDEST2T18ms (Addgene plasmid no. 72647). Briefly, the upstream and downstream sequences of the TFA were amplified using primers with 5′-end sequences identical to the attB sites of a suicide plasmid (table S4). The polymerase chain reaction (PCR) products were analyzed by gel electrophoresis and purified with the GeneJET gel extraction kit (ThermoFisher Scientific, catalog no. K0691). BP and LR reactions were conducted in the one-tube format using the purified attB-flanked PCR product. The resulting expression clone was transformed into competent E. coli DH5α and then introduced into recipient cells (P. viridiflava p25.A12) by conjugation. Recombinant cells (merodiploids) were selected for with the antibiotic resistance conferred by the expression clone and verified with PCR and gel electrophoresis. Positive colonies were purified by streaking onto new plates. Purified merodiploids were grown overnight without the antibiotic conferred by the expression clone to allow for another recombination event and then plated on 10% D-sucrose for sacB counterselection. Colonies were examined by PCR and verified by DNA sequencing (GeneWiz).
Construction of the Pseudomonas p25.A12 tail fiber assembly gene overexpression rescue vector
The HTF gene was cloned into pMCSG11 (82) by restriction cloning (83). This rescue vector was transformed into p25.A12ΔTFA, tailocins were induced and partially purified, and killing assays were performed to assess rescue lethality.
Plant infections of wild-type or mutant strains
To evaluate the infection outcome of p25.A12 wild type and mutant (p25.A12ΔTFA), Columbia-0 wild-type A. thaliana plants were grown under long-day conditions (16 hours light, 8 hours dark) in an AR41L3 Percival detector with 60% intensity of the SciWhite LED lights. Seedlings were grown in 24-well plates (Greiner Bio-One, catalog no. 6621665). Thirteen-day-old seedlings were used for the infections. Plants were infected with bacterial suspension of the P. viridiflava strain p25.A12, p25.A12ΔTFA, or buffer (10 mM MgSO4). Both p25.A12 strains were tagged with luciferase (42).
Bacteria were grown overnight and diluted 1:10 the day of the infection. Cells were grown for another 3 hours and then collected and resuspended in 10 mM MgSO4 to a final OD600 of 0.01. A 200-μl sample was used to spot inoculate the plants in a randomized manner. Plants were grown for 7 days and then collected. The collected plants were ground in 1 ml of 10 mM MgSO4 using the Qiagen tissue lyser II, and 200 μl of the suspension was used to measure luciferase in the TECAN using flat white plates (TECAN, catalog no. 30122300). Plants infected with MgSO4 were used as controls. In total, 24 plants were used for each strain or buffer.
In vitro competition assays
Strain p25.C2 cultures tagged with a luciferase cassette were grown together with p25.A12 wild type or the p25.A12ΔTFA strain. Five different ratios were used for mixing the two strains (10:10, 5:10, 3:10, 2:10, and 1:10) keeping the p25.C2 strain constant at a final OD600 of 0.005. Bacteria were grown overnight and then diluted 1:10 the day of the experiment. Cells were grown for another 3 hours with shaking and then collected and mixed accordingly. The co-inoculated cells were grown in a TECAN flat white plate (TECAN, catalog no. 30122300) in a final volume of 200 μl in a randomized manner. The plates were placed in the TECAN plate reader (TECAN Spark) for 21 hours at 28°C for 55 kinetic cycles. Each cycle consisted of 15 min or orbital shaking at 220 rpm, resting for 5 min, and then measuring the luciferase-tagged p25.C2 cells in each well. For the analysis, the three highest values from each sample were used, and the average luciferase value was calculated for each condition.
In planta coinfections
Columbia-0 wild-type A. thaliana plants were grown in 24-well plates (Greiner Bio-One, catalog no. 6621665) under long-day conditions (16 hours light, 8 hours dark) in an AR41L3 Percival detector with 60% intensity of the SciWhite LED lights. Thirteen-day-old seedlings were used for the infections. Plants were infected with bacterial suspension of the Pseudomonas strain p25.C2 (tagged with luciferase) and either the p25.A12 wild type or the p25.A12ΔTFA strain. The same five ratios as the in vitro assays were used, keeping the p25.C2 constant at a final OD600 of 0.005.
Bacteria were grown overnight and diluted 1:10 the day of the infection. Cells were grown for another 3 hours and then collected and resuspended in 10 mM MgSO4. Cells were then mixed according to the ratios described above, and the seedlings were submerged in 10 ml of cell suspension for 10 min in a randomized manner. Sampls of 850 μl were removed and the plants were placed back in the AR41L3 Percival detector. Three days after infection, plants were collected in 2-ml deep-well plates (Thermo Fisher Nunc, catalog no. 12–565-605) containing 1 ml of MgSO4. Plants were ground using the Qiagen tissue lyser II, and 200 μl of the suspension was used to measure luciferase in the TECAN using flat white plates (TECAN, catalog no. 30122300). Plants infected with MgSO4 or p25.C2 alone were used as controls. Each treatment was done in 12 replicates. The entire experiment was repeated three times with similar results.
Constructing the BarSeq mutant fitness library
Methods were adapted from Wetmore et al. (43). Briefly, we created the p25.C2 transposon mutant library by conjugating p25.C2 with the E. coli conjugation donor (WM3064) harboring the pHLL250 mariner transposon vector library (AMD290) (84). Equal cell numbers of mid-log-phase p25.C2 and AMD290 were conjugated for 6 hours on 0.45-μm nitrocellulose filters (Millipore) overlaid on LB agar plates containing diaminopimelic acid. The resuspended cells were plated on LB plates with 50 g/ml kanamycin to select for mutants. After 2 days, the kanamycin-resistant colonies were scraped into LB, the OD600 of the mixture was measured, and the mutant library was diluted to a starting OD600 of 0.2 in 250 ml of LB with 50 g/ml kanamycin. The diluted mutant library was grown at 28°C to a final OD600 of 1.0, glycerol was added to a final volume of 10%, and samples were stored at −80°C as freezer stocks. Cells were then collected for genomic DNA extraction.
Competitive mutant fitness assays
Assays were adapted from Carim et al. (28). Briefly, assays were performed in glass tubes. Partially purified tailocins were used as stressors at final concentrations of 0.05× of the stock preparation. P-buffer was used as a control. LB and Km50 was supplied for the mutant library as the growth medium. The tubes were incubated with shaking and mutants were harvested when the OD600 reached the mid-log phase. Cells were pelleted by centrifugation (8000 × g for 5 min) and stored at −20°C to await genomic DNA extraction. Each condition was assayed in 10 individual replicates.
BarSeq and analysis of BarSeq data
Genomic DNA was extracted and barcode PCR was performed as described in Wetmore et al. (43). DNA extractions were quantified with NanoDrop 1000 (ThermoFisher Scientific). Barcode sequence data were obtained by multiplexing on a lane of HiSeq at the University of Utah High-Throughput Genomics core. Fitness data were calculated and analyzed from these reads with the DESeq2 R package (85), and scripts can be found on our GitHub page.
LPS isolation
The cell culture was washed two times with PBS and inactivated using a solution of 1% phenol in PBS. The cell material was suspended in water and preincubated to 68°C with gentle stirring. The extraction was performed by using the corresponding volume of 90% (v/v) liquefied phenol following the standard procedure (86) and extracted for 20 min at 68°C. The sample was cooled on an ice bath and centrifuged for 20 min at 4°C, 5000 × g. The aqueous upper phase was collected, and the remaining phenol phase and cell debris were extracted two more times with water following the same protocol. The combined aqueous phases and the final phenol phase were dialyzed against several exchanges of water for 4 days 12- to 14-kDa [molecular weight cutoff (MWCO)].
The freeze-dried dialysates were resuspended in a sterile buffer (10 mg/ml to 50 mM MgCl2•6H2O and 20 mM NaOAc•3H2O), and the nucleic acids were digested with benzonase (2 μl, 18 hours at 37°C) with gentle agitation. Proteins were digested with proteinase K (50 μg/ml per ml of the LPS solution) by overnight incubation at 37°C. The buffer, nucleotides, and peptides were removed by dialysis against water (12- to 14-kDa MWCO) at 4°C, followed by ultracentrifugation at 100,000 × g for 16 hours at 4°C. The LPS pellets were suspended in water and freeze-dried, and were then ready for use.
DOC-PAGE of LPS samples
The purified LPS samples were resolved in PAGE (4% stacking gel and 18% resolving gel) in the presence of DOC buffer (87). The PAGE was visualized with a silver stain reagent kit (Bio-Rad).
Glycosyl composition analysis using the TMS method
Glycosyl composition analysis was performed by combined GC-MS of the O-trimethylsilyl (TMS) methyl glycoside derivatives produced from the sample by acidic methanolysis followed by trimethylsilylation (88). GC-MS analysis of the TMS methyl glycosides was performed on an Agilent AT 7890A GC system interfaced to a 5975B MSD using an Equity-1 (Supelco) fused silica capillary column (30 m length × 0.25 mm ID × 0.25 μm film thickness). The temperature gradient was was 80°C for 2 min, increased to 140°C at 20°C/min with a 2-min hold, increased to 200°C at 2°C/min, and then increased to 250°C at 30°C/min with 5-min hold.
Supplementary Material
ACKNOWLEDGMENTS
We thank S. Kamoun, P. Lang, G. Ofir, D. Petrov, and D. Weigel for comments that improved this manuscript and C. Krause, A. Rosenbauer, and M, Thiv from the Stuttgart State Museum of Natural History and O. Bossdorf from the Herbarium Tubingense for their introduction to and help in the herbaria and kind permission to sample specimens.
Funding:
This study was supported by startup funds from the University of Utah and by the National Institutes of Health (NIH grant R35 GM150722–01 to T.L.K.), The Leverhulme Trust (Philip Leverhulme Prize to H.A.B.), the Royal Society (grant RSWF\R1\191011 to H.A.B.), the UK Biological Sciences Research Council (BBSRC equipment grant BB/R01356X/1 to University College London), and the Max Planck Society (funding to the Department of Molecular Biology of the Max Planck Institute for Biology led by D. Weigel). T.B. was supported by the NIH Training Program in Microbial Pathogenesis (grant T32 AI055434). Proteomics mass spectrometry analysis was performed at the Mass Spectrometry and Proteomics Core Facility at the University of Utah. Transmission electron microscopy was performed at the Electron Microscopy Core Laboratory at the University of Utah. Mass spectrometry instrumentation was obtained through the University of Utah Research Instrumentation Fund. This work was supported in part by the US Department of Energy, Office of Science, Basic Energy Sciences, Chemical Sciences, Geosciences and Biosciences Division at DOE Center for Plant and Microbial Complex Carbohydrates at the CCRC (award DE-SC0015662 to P.A.) and by NIH National Glycoscience Resource-CCRC Service and Training (grant R24GM137782 to P.A.). A.D.G. was supported by the Dropkin Foundation and the University of Chicago Hutchinson fund.
Footnotes
Competing interests: The authors declare no competing interests.
License information: Copyright © 2024 the authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original US government works. https://www.science.org/about/science-licenses-journal-article-reuse. This research was funded in whole or in part by UK Biological Sciences Research Council (BBSRC equipment grant BB/R01356X/1), a cOAlition S organization. The author will make the Author Accepted Manuscript (AAM) version available under a CC BY public copyright license.
Data and materials availability:
All data and code are available in the supplementary materials and have been deposited in the following repositories. Data have been deposited to Zenodo (67) and Dryad (89). Code has been deposited to GitHub (90, 91).
REFERENCES AND NOTES
- 1.Goss EM, Timilsina S, A bacterial epidemic in wild plants. Nat. Ecol. Evol. 2, 1529–1530 (2018). doi: 10.1038/s41559-018-0692-2 [DOI] [PubMed] [Google Scholar]
- 2.Karasov TL et al. , Arabidopsis thaliana and Pseudomonas pathogens exhibit stable associations over evolutionary timescales. Cell Host Microbe 24, 168–179.e4 (2018). doi: 10.1016/j.chom.2018.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Der Biezen EA, Jones JD, Plant disease-resistance proteins and the gene-for-gene concept. Trends Biochem. Sci. 23, 454–456 (1998). doi: 10.1016/S0968-0004(98)01311-5 [DOI] [PubMed] [Google Scholar]
- 4.Ruiz-Bedoya T, Wang PW, Desveaux D, Guttman DS, Cooperative virulence via the collective action of secreted pathogen effectors. Nat. Microbiol. 8, 640–650 (2023). doi: 10.1038/s41564-023-01328-8 [DOI] [PubMed] [Google Scholar]
- 5.Leiman PG et al. , Type VI secretion apparatus and phage tail-associated protein complexes share a common evolutionary origin. Proc. Natl. Acad. Sci. U.S.A. 106, 4154–4159 (2009). doi: 10.1073/pnas.0813360106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heiman CM, Vacheron J, Keel C, Evolutionary and ecological role of extracellular contractile injection systems: From threat to weapon. Front. Microbiol. 14, 1264877 (2023). doi: 10.3389/fmicb.2023.1264877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dong TG, Ho BT, Yoder-Himes DR, Mekalanos JJ, Identification of T6SS-dependent effector and immunity proteins by Tn-seq in Vibrio cholerae. Proc. Natl. Acad. Sci. U.S.A. 110, 2623–2628 (2013). doi: 10.1073/pnas.1222783110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sarris PF, Ladoukakis ED, Panopoulos NJ, Scoulica EV, A phage tail-derived element with wide distribution among both prokaryotic domains: A comparative genomic and phylogenetic study. Genome Biol. Evol. 6, 1739–1747 (2014). doi: 10.1093/gbe/evu136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Granato ET, Meiller-Legrand TA, Foster KR, The evolution and ecology of bacterial warfare. Curr. Biol. 29, R521–R537 (2019). doi: 10.1016/j.cub.2019.04.024 [DOI] [PubMed] [Google Scholar]
- 10.Chen L et al. , Genome-wide Identification and Characterization of a Superfamily of Bacterial Extracellular Contractile Injection Systems. Cell Rep. 29, 511–521.e2 (2019). doi: 10.1016/j.celrep.2019.08.096; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geller AM et al. , The extracellular contractile injection system is enriched in environmental microbes and associates with numerous toxins. Nat. Commun. 12, 3743 (2021). doi: 10.1038/s41467-021-23777-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Príncipe A, Fernandez M, Torasso M, Godino A, Fischer S, Effectiveness of tailocins produced by Pseudomonas fluorescens SF4c in controlling the bacterial-spot disease in tomatoes caused by Xanthomonas vesicatoria. Microbiol. Res. 212–213, 94–102 (2018). doi: 10.1016/j.micres.2018.05.010; pmid: [DOI] [PubMed] [Google Scholar]
- 13.Baltrus DA et al. , Prophylactic application of tailocins prevents infection by Pseudomonas syringae. Phytopathology 112, 561–566 (2022). doi: 10.1094/PHYTO-06-21-0269-R [DOI] [PubMed] [Google Scholar]
- 14.Rasoamanana H et al. , Bacteriocin production correlates with epidemiological prevalence of phylotype I sequevar 18 Ralstonia pseudosolanacearum in Madagascar. Appl. Environ. Microbiol. 89, e0163222 (2023). doi: 10.1128/aem.01632-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dorosky RJ, Yu JM, Pierson LS 3rd, Pierson EA, Pseudomonas chlororaphis produces two distinct R-tailocins that contribute to bacterial competition in biofilms and on roots. Appl. Environ. Microbiol. 83, e00706–17 (2017). doi: 10.1128/AEM.00706-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dorosky RJ, Pierson LS 3rd, Pierson EA, Pseudomonas chlororaphis produces multiple R-tailocin particles that broaden the killing spectrum and contribute to persistence in rhizosphere communities. Appl. Environ. Microbiol. 84, e01230–18 (2018). doi: 10.1128/AEM.01230-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanski I, Metapopulation dynamics. Nature 396, 41–49 (1998). doi: 10.1038/23876 [DOI] [Google Scholar]
- 18.Karasov TL et al. , The long-term maintenance of a resistance polymorphism through diffuse interactions. Nature 512, 436–440 (2014). doi: 10.1038/nature13439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shalev O et al. , Commensal Pseudomonas strains facilitate protective response against pathogens in the host plant. Nat. Ecol. Evol. 6, 383–396 (2022). doi: 10.1038/s41559-022-01673-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lundberg DS et al. , Contrasting patterns of microbial dominance in the Arabidopsis thaliana phyllosphere. Proc. Natl. Acad. Sci. U.S.A. 119, e2211881119 (2022). doi: 10.1073/pnas.2211881119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Smet J, Hendrix H, Blasdel BG, Danis-Wlodarczyk K, Lavigne R, Pseudomonas predators: Understanding and exploiting phage-host interactions. Nat. Rev. Microbiol. 15, 517–530 (2017). doi: 10.1038/nrmicro.2017.61 [DOI] [PubMed] [Google Scholar]
- 22.Weaver SL, Zhu L, Ravishankar S, Clark M, Baltrus DA, Interspecies killing activity of Pseudomonas syringae tailocins. Microbiology 168, mic.0.001258 (2022). [DOI] [PubMed] [Google Scholar]
- 23.Silveira CB, Luque A, Rohwer F, The landscape of lysogeny across microbial community density, diversity and energetics. Environ. Microbiol. 23, 4098–4111 (2021). doi: 10.1111/1462-2920.15640 [DOI] [PubMed] [Google Scholar]
- 24.Kieft K, Zhou Z, Anantharaman K, VIBRANT: Automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome 8, 90 (2020). doi: 10.1186/s40168-020-00867-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jakob K et al. , Pseudomonas viridiflava and P. syringae—Natural pathogens of Arabidopsis thaliana. Mol. Plant Microbe Interact. 15, 1195–1203 (2002). doi: 10.1094/MPMI.2002.15.12.1195 [DOI] [PubMed] [Google Scholar]
- 26.Karasov TL et al. , Drought selection on Arabidopsis populations and their microbiomes, bioRxiv (2022) p. 2022.04.08.487684. [Google Scholar]
- 27.Ondov BD et al. , Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 17, 132 (2016). doi: 10.1186/s13059-016-0997-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carim S et al. , Systematic discovery of pseudomonad genetic factors involved in sensitivity to tailocins. ISME J. 15, 2289–2305 (2021). doi: 10.1038/s41396-021-00921-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakayama K et al. , The R-type pyocin of Pseudomonas aeruginosa is related to P2 phage, and the F-type is related to lambda phage. Mol. Microbiol. 38, 213–231 (2000). doi: 10.1046/j.1365-2958.2000.02135.x [DOI] [PubMed] [Google Scholar]
- 30.Roos WH, Ivanovska IL, Evilevitch A, Wuite GJL, Viral capsids: Mechanical characteristics, genome packaging and delivery mechanisms. Cell. Mol. Life Sci. 64, 1484–1497 (2007). doi: 10.1007/s00018-007-6451-1; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen X et al. , Functional identification of the DNA packaging terminase from Pseudomonas aeruginosa phage PaP3. Arch. Virol. 157, 2133–2141 (2012). doi: 10.1007/s00705-012-1409-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hockett KL, Renner T, Baltrus DA, Independent co-option of a tailed bacteriophage into a killing complex in Pseudomonas. mBio 6, e00452 (2015). doi: 10.1128/mBio.00452-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghequire MGK, De Mot R, The tailocin tale: Peeling off phage tails. Trends Microbiol. 23, 587–590 (2015). doi: 10.1016/j.tim.2015.07.011 [DOI] [PubMed] [Google Scholar]
- 34.Hockett KL, Baltrus DA, Use of the soft-agar overlay technique to screen for bacterially produced inhibitory compounds. J. Vis. Exp. 119, 55064 (2017). doi: 10.3791/55064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ishii SI, Nishi Y, Egami F, The fine structure of a pyocin. J. Mol. Biol. 13, 428–431 (1965). doi: 10.1016/S0022-2836(65)80107-3 [DOI] [PubMed] [Google Scholar]
- 36.Govan JR, Studies on the pyocins of Pseudomonas aeruginosa: Morphology and mode of action of contractile pyocins. J. Gen. Microbiol. 80, 1–15 (1974). doi: 10.1099/00221287-80-1-1 [DOI] [PubMed] [Google Scholar]
- 37.Higerd TB, Baechler CA, Berk RS, Morphological studies on relaxed and contracted forms of purified pyocin particles. J. Bacteriol. 98, 1378–1389 (1969). doi: 10.1128/jb.98.3.1378-1389.1969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schirmer EC, Yates JR 3rd, Gerace L, MudPIT: A powerful proteomics tool for discovery. Discov. Med. 3, 38–39 (2003). [PubMed] [Google Scholar]
- 39.Medini D, Donati C, Tettelin H, Masignani V, Rappuoli R, The microbial pan-genome. Curr. Opin. Genet. Dev. 15, 589–594 (2005). doi: 10.1016/j.gde.2005.09.006 [DOI] [PubMed] [Google Scholar]
- 40.Baltrus DA, Clark M, Smith C, Hockett KL, Localized recombination drives diversification of killing spectra for phage-derived syringacins. ISME J. 13, 237–249 (2019). doi: 10.1038/s41396-018-0261-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vacheron J, Heiman CM, Keel C, Live cell dynamics of production, explosive release and killing activity of phage tail-like weapons for Pseudomonas kin exclusion. Commun. Biol. 4, 87 (2021). doi: 10.1038/s42003-020-01581-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duque-Jaramillo A et al. , The genetic and physiological basis of Arabidopsis thaliana tolerance to Pseudomonas viridiflava. New Phytol. 240, 1961–1975 (2023). doi: 10.1111/nph.19241 [DOI] [PubMed] [Google Scholar]
- 43.Wetmore KM et al. , Rapid quantification of mutant fitness in diverse bacteria by sequencing randomly bar-coded transposons. mBio 6, e00306–e00315 (2015). doi: 10.1128/mBio.00306-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scanlan PD et al. , Coevolution with bacteriophages drives genome-wide host evolution and constrains the acquisition of abiotic-beneficial mutations. Mol. Biol. Evol. 32, 1425–1435 (2015). doi: 10.1093/molbev/msv032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jayaraman J et al. , Variation at the common polysaccharide antigen locus drives lipopolysaccharide diversity within the Pseudomonas syringae species complex. Environ. Microbiol. 22, 5356–5372 (2020). doi: 10.1111/1462-2920.15250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baltrus DA, Weaver S, Krings L, Nguyen AE, Genomic correlates of tailocin sensitivity in Pseudomonas syringae. bioRxiv 538177 [Preprint] (2023); 10.1101/2023.04.24.538177. [DOI] [Google Scholar]
- 47.King JD, Kocíncová D, Westman EL, Lam JS, Review: Lipopolysaccharide biosynthesis in Pseudomonas aeruginosa. Innate Immun. 15, 261–312 (2009). doi: 10.1177/1753425909106436 [DOI] [PubMed] [Google Scholar]
- 48.Köhler T, Donner V, van Delden C, Lipopolysaccharide as shield and receptor for R-pyocin-mediated killing in Pseudomonas aeruginosa. J. Bacteriol. 192, 1921–1928 (2010). doi: 10.1128/JB.01459-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Latorre SM, Lang PLM, Burbano HA, Gutaker RM, Isolation, library preparation, and bioinformatic analysis of historical and ancient plant DNA. Curr. Protoc. Plant Biol. 5, e20121 (2020). doi: 10.1002/cppb.20121 [DOI] [PubMed] [Google Scholar]
- 50.Weiß CL et al. , Temporal patterns of damage and decay kinetics of DNA retrieved from plant herbarium specimens. R. Soc. Open Sci. 3, 160239 (2016). doi: 10.1098/rsos.160239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bernal P, Allsopp LP, Filloux A, Llamas MA, The Pseudomonas putida T6SS is a plant warden against phytopathogens. ISME J. 11, 972–987 (2017). doi: 10.1038/ismej.2016.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rankin DJ, Turner LA, Heinemann JA, Brown SP, The coevolution of toxin and antitoxin genes drives the dynamics of bacterial addiction complexes and intragenomic conflict. Proc. Biol. Sci. 279, 3706–3715 (2012). doi: 10.1098/rspb.2012.0942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moxon ER, Rainey PB, Nowak MA, Lenski RE, Adaptive evolution of highly mutable loci in pathogenic bacteria. Curr. Biol. 4, 24–33 (1994). doi: 10.1016/S0960-9822(00)00005-1 [DOI] [PubMed] [Google Scholar]
- 54.Druelle V, Neher RA, Reversions to consensus are positively selected in HIV-1 and bias substitution rate estimates. Virus Evol. 9, veac118 (2022). doi: 10.1093/ve/veac118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gebhart D et al. , A modified R-type bacteriocin specifically targeting Clostridium difficile prevents colonization of mice without affecting gut microbiota diversity. mBio 6, e02368–14 (2015). doi: 10.1128/mBio.02368-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pearson K LIII. On lines and planes of closest fit to systems of points in space. Lond. Edinb. Dublin Philos. Mag. J. Sci. 2, 559–572 (1901). doi: 10.1080/14786440109462720 [DOI] [Google Scholar]
- 57.Lloyd S, Least squares quantization in PCM. IEEE Trans. Inf. Theory 28, 129–137 (1982). doi: 10.1109/TIT.1982.1056489 [DOI] [Google Scholar]
- 58.Rousseeuw PJ, Silhouettes: A graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 20, 53–65 (1987). doi: 10.1016/0377-0427(87)90125-7 [DOI] [Google Scholar]
- 59.Ding W, Baumdicker F, Neher RA, panX: Pan-genome analysis and exploration. Nucleic Acids Res. 46, e5 (2018). doi: 10.1093/nar/gkx977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sievers F, Higgins DG, Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 27, 135–145 (2018). doi: 10.1002/pro.3290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Suyama M, Torrents D, Bork P, PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34, W609–W612 (2006). doi: 10.1093/nar/gkl315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pfeifer B, Wittelsbürger U, Ramos-Onsins SE, Lercher MJ, PopGenome: An efficient Swiss army knife for population genomic analyses in R. Mol. Biol. Evol. 31, 1929–1936 (2014). doi: 10.1093/molbev/msu136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nei M, Li WH, Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. U.S.A. 76, 5269–5273 (1979). doi: 10.1073/pnas.76.10.5269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fu YX, Statistical properties of segregating sites. Theor. Popul. Biol. 48, 172–197 (1995). doi: 10.1006/tpbi.1995.1025 [DOI] [PubMed] [Google Scholar]
- 65.Tajima F, Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595 (1989). doi: 10.1093/genetics/123.3.585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Paradis E, pegas: An R package for population genetics with an integrated-modular approach. Bioinformatics 26, 419–420 (2010). doi: 10.1093/bioinformatics/btp696 [DOI] [PubMed] [Google Scholar]
- 67.Latorre SM, Lang PLM, Burbano HA, Historical Arabidopsis thaliana genomes from Germany, Zenodo; (2022); https://zenodo.org/record/7156189. [Google Scholar]
- 68.Schubert M, Lindgreen S, Orlando L, AdapterRemoval v2: Rapid adapter trimming, identification, and read merging. BMC Res. Notes 9, 88 (2016). doi: 10.1186/s13104-016-1900-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li H, Durbin R, Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). doi: 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jónsson H, Ginolhac A, Schubert M, Johnson PLF, Orlando L, mapDamage2.0: Fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 29, 1682–1684 (2013). doi: 10.1093/bioinformatics/btt193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Prjibelski A, Antipov D, Meleshko D, Lapidus A, Korobeynikov A, Using SPAdes De Novo Assembler. Curr. Protoc. Bioinformatics 70, e102 (2020). doi: 10.1002/cpbi.102 [DOI] [PubMed] [Google Scholar]
- 72.Li H, Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018). doi: 10.1093/bioinformatics/bty191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sievers F, Higgins DG, The Clustal Omega Multiple Alignment Package. Methods Mol. Biol. 2231, 3–16 (2021). doi: 10.1007/978-1-0716-1036-7_1 [DOI] [PubMed] [Google Scholar]
- 74.Danecek P et al. , Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021). doi: 10.1093/gigascience/giab008; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Minh BQ et al. , IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020). doi: 10.1093/molbev/msaa015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS, ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017). doi: 10.1038/nmeth.4285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS, UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522 (2018). doi: 10.1093/molbev/msx281; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Didelot X, Wilson DJ, ClonalFrameML: Efficient inference of recombination in whole bacterial genomes. PLOS Comput. Biol. 11, e1004041 (2015). doi: 10.1371/journal.pcbi.1004041; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Drummond AJ, Bouckaert RR, Bayesian Evolutionary Analysis with BEAST (Cambridge Univ. Press, 2015). [Google Scholar]
- 80.McCann EP et al. , The genotype-phenotype landscape of familial amyotrophic lateral sclerosis in Australia. Clin. Genet. 92, 259–266 (2017). doi: 10.1111/cge.12973 [DOI] [PubMed] [Google Scholar]
- 81.Drummond AJ, Rambaut A, Shapiro B, Pybus OG, Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 22, 1185–1192 (2005). doi: 10.1093/molbev/msi103 [DOI] [PubMed] [Google Scholar]
- 82.Eschenfeldt WH, Lucy S, Millard CS, Joachimiak A, Mark ID, A family of LIC vectors for high-throughput cloning and purification of proteins. Methods Mol. Biol. 498, 105–115 (2009). doi: 10.1007/978-1-59745-196-3_7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nathans D, Smith HO, Restriction endonucleases in the analysis and restructuring of dna molecules. Annu. Rev. Biochem. 44, 273–293 (1975). doi: 10.1146/annurev.bi.44.070175.001421 [DOI] [PubMed] [Google Scholar]
- 84.Adler BA et al. , The genetic basis of phage susceptibility, cross-resistance and host-range in Salmonella. Microbiology (Reading) 167, ••• (2021). doi: 10.1099/mic.0.001126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). doi: 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Westphal O, Bacterial lipopolysaccharides: Extraction with phenol-water and further applications of the procedure. Methods Carbohydr. Chem. 5, 83 (1965). [Google Scholar]
- 87.Krauss JH, Weckesser J, Mayer H, Electrophoretic analysis of lipopolysaccharides of purple nonsulfur bacteria. Int. J. Syst. Bacteriol. 38, 157–163 (1988). doi: 10.1099/00207713-38-2-157 [DOI] [Google Scholar]
- 88.York WS, Darvill AG, McNeil M, Stevenson TT, Albersheim P, “Isolation and characterization of plant cell walls and cell wall components” in Methods in Enzymology (Academic, 1986), vol. 118, pp. 3–40. [Google Scholar]
- 89.Backman T, A domesticated phage suppresses competitors in historical and modern metapopulations of pathogenic bacteria, Dryad; (2024); 10.5061/dryad.hx3ffbgn9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Latorre SM, Pseudomonas_tailocin, Zenodo; (2024); 10.5281/zenodo.10794071. [DOI] [Google Scholar]
- 91.Backman T, Ps1524_tailocin, Zenodo; (2024); 10.5281/zenodo.10729592. [DOI] [Google Scholar]
- 92.Aylward AJ, Petrus S, Mamerto A, Hartwick NT, Michael TP, PanKmer: K-mer-based and reference-free pangenome analysis. Bioinformatics 39, btad621 (2023). doi: 10.1093/bioinformatics/btad621 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data and code are available in the supplementary materials and have been deposited in the following repositories. Data have been deposited to Zenodo (67) and Dryad (89). Code has been deposited to GitHub (90, 91).