

Abstract
Purpose:
The pathological hallmarks of Alzheimer’s disease (AD), amyloid, tau, and associated neurodegeneration, are present in the cortical gray matter (GM) years before symptom onset, and at significantly greater levels in carriers of the apolipoprotein E4 (APOE4) allele. Their respective biomarkers, A/T/N, have been found to correlate with aspects of brain biochemistry, measured with magnetic resonance spectroscopy (MRS), indicating a potential for MRS to augment the A/T/N framework for staging and prediction of AD. Unfortunately, the relationships between MRS and A/T/N biomarkers are unclear, largely due to a lack of studies examining them in the context of the spatial and temporal model of T/N progression. Advanced MRS acquisition and post-processing approaches have enabled us to address this knowledge gap and test the hypotheses, that glutamate-plus-glutamine (Glx) and N-acetyl-aspartate (NAA), metabolites reflecting synaptic and neuronal health, respectively, measured from regions on the Braak stage continuum, correlate with: (i) cerebrospinal fluid (CSF) p-tau181 level (T), and (ii) hippocampal volume or cortical thickness of parietal lobe GM (N). We hypothesized that these correlations will be moderated by Braak stage and APOE4 genotype.
Methods:
We conducted a retrospective imaging study of 34 cognitively unimpaired elderly individuals who received APOE4 genotyping and lumbar puncture from pre-existing prospective studies at the NYU Grossman School of Medicine between October 2014 and January 2019. Subjects returned for their imaging exam between April 2018 and February 2020. Metabolites were measured from the left hippocampus (Braak II) using a single-voxel semi-adiabatic localization by adiabatic selective refocusing sequence; and from the bilateral posterior cingulate cortex (PCC; Braak IV), bilateral precuneus (Braak V), and bilateral precentral gyrus (Braak VI) using a multi-voxel echo-planar spectroscopic imaging sequence. Pearson and Spearman correlations were used to examine the relationships between absolute levels of choline, creatine, myo-inositol, Glx, and NAA and CSF p-tau181, and between these metabolites and hippocampal volume or parietal cortical thicknesses. Covariates included age, sex, years of education, Fazekas score, and months between CSF collection and MRI exam.
Results:
There was a direct correlation between hippocampal Glx and CSF p-tau181 in APOE4 carriers (Pearson’s r = 0.76, p = 0.02), but not after adjusting for covariates. In the entire cohort, there was a direct correlation between hippocampal NAA and hippocampal volume (Spearman’s r = 0.55, p = 0.001), even after adjusting for age and Fazekas score (Spearman’s r = 0.48, p = 0.006). This relationship was observed only in APOE4 carriers (Pearson’s r = 0.66, p = 0.017), and was also retained after adjustment (Pearson’s r = 0.76, p = 0.008; metabolite-by-carrier interaction p = 0.03). There were no findings in the PCC, nor in the negative control (late Braak stage) regions of the precuneus and precentral gyrus.
Conclusions:
Our findings are in line with the spatially- and temporally-resolved Braak staging model of pathological severity in which the hippocampus is affected earlier than the PCC. The correlations, between MRS markers of synaptic and neuronal health and, respectively, T and N pathology, were found exclusively within APOE4 carriers, suggesting a connection with AD pathological change, rather than with normal aging. We therefore conclude that MRS has the potential to augment early A/T/N staging, with the hippocampus serving as a more sensitive MRS target compared to the PCC.
Keywords: Spectroscopy, Glx, CSF p-tau181, APOE genotype, Hippocampus, Cognitively unimpaired
1. Introduction
Alzheimer’s disease (AD), the most common cause of dementia in the United States, is projected to affect 7.2 million individuals aged 65 and older by the year 2025 (Alzheimer’s Association Report, 2022). While AD is biologically characterized as a continuum comprising various pathophysiological processes (De Strooper and Karran, 2016), discrete staging is still commonly used for clinical purposes. The temporal course of the disease entails an asymptomatic “preclinical” phase that progresses to symptomatic mild cognitive impairment (MCI), and eventually, to mild, moderate, and severe AD dementia (Aisen et al., 2017). Unfortunately, most individuals are diagnosed only after exhibiting objective cognitive impairment (e.g., memory loss and deficits in executive function) (Dubois et al., 2021), despite there being histologic (Price et al., 2009), structural (Petrone et al., 2019), and functional (Veitch et al., 2019; Ossenkoppele et al., 2022) imaging evidence for abnormal brain changes at least a decade before the onset of these symptoms (Holtzman et al., 2011). Many of these imaging findings are seen in normal aging, dementias, and psychiatric disorders (Steinberg et al., 2008), further complicating the possibility of a timely diagnosis (Porsteinsson et al., 2021). Therefore, the identification of novel biomarkers for predicting and monitoring disease progression are crucial for the differential identification of asymptomatic individuals at risk for progression to MCI.
There are three major pathological hallmarks of AD that can be quantified in vivo by cerebrospinal fluid (CSF), plasma, and/or imaging methodologies, yielding biomarkers of extracellular deposition of amyloid-beta (Aß) plaques (“A” for biological evidence of amyloidosis), intracellular formation of neurofibrillary tangles composed of aggregated and hyperphosphorylated tau protein (“T” for biological evidence of tauopathy), and atrophy (“N” for biological evidence of neurodegeneration) (Jack et al., 2018). Termed the A/T/N research framework (Jack et al., 2018), this biomarker-based classification system can be used to stage AD without reliance on the presentation of clinical symptoms (Jack et al., 2016). It does not take into account, however, the heterogeneous phenotypic and genotypic nature of AD (Devi and Scheltens, 2018; Birkenbihl et al., 2022) and additional biochemical and cellular dyshomeostasis (De Strooper and Karran, 2016), such as neuronal and synaptic dysfunction (Chen et al., 2019), which can pre-date or co-occur with Aß and/or tau pathology.
Proton magnetic resonance spectroscopy (1H MRS) is a non-invasive technique used to investigate neurochemical abnormalities in vivo and can augment established A/T/N biomarkers by providing insight about underlying metabolic changes in specific regions of the brain, even in the absence of structural damage and clinical features. Major MRS markers, such as choline (Cho), creatine (Cr), myo-inositol (mI), and N-acetyl-aspartate (NAA), have been found to be moderated by genetic risk for AD (Yin et al., 2015), associated with pathological severity (Graff-Radford and Kantarci, 2016); and have predicted both cognitive decline (Graff-Radford and Kantarci, 2016) and conversion to dementia (den Heijer et al., 2006). There are five MRS studies of cognitively unimpaired individuals that have examined relationships between the major MRS metabolites and A/T biomarkers (Kantarci et al., 2011; Voevodskaya et al., 2016; Nedelska et al., 2017; Kara et al., 2022; Spotorno et al., 2022): four reported correlations between higher ratios of Cho/Cr, higher ratios of mI/Cr, or lower ratios of NAA/Cr with greater Aß burden, as determined by the use of 11C-Pittsburg Compound B (PiB) positron emission tomography (PET) (Kantarci et al., 2011; Nedelska et al., 2017; Kara et al., 2022), 18F-flutametamol PET (Spotorno et al., 2022), or CSF Aß42 (Voevodskaya et al., 2016); and two reported correlations between lower ratios of NAA/Cr or higher ratios of mI/Cr and greater tau burden, as determined by 18F-flortaucipir PET (Kara et al., 2022) or 18F-RO948 PET (Spotorno et al., 2022), respectively. Together, these findings point to the potential interplay between glial (e.g., Cho, mI) and neuronal (e.g., NAA) dysfunction and neurotoxic Aß and tau aggregation, and suggests a role for MRS in detecting AD-related metabolic disruption in asymptomatic populations. We note, however, certain shared limitations. All five studies exclusively interrogated the posterior cingulate cortex (PCC) region, so it remains unknown whether relationships between metabolite levels and A/T/N biomarkers exist in other gray matter (GM) regions affected in AD, particularly in the hippocampus (the hallmark region for AD pathology, one of the earliest sites of tau accumulation (Braak and Braak, 1991; Adams et al., 2019) and atrophy (Besson et al., 2015)). Additionally, only Kara et al. (Kara et al., 2022) measured glutamate (Glu), which, along with glutamine (Gln) and their sum (Glx), has been shown to decline in MCI (Zeydan et al., 2017) and AD (Antuono et al., 2001; Fayed et al., 2011; Rupsingh et al., 2011). Importantly, Glx has been proposed as a biomarker of early AD neurodegeneration (Conway, 2020; Zeydan and Kantarci, 2021), owing to its status as a marker for the glutamate cycle between neuronal dendrites and astrocytes. All five studies (Kantarci et al., 2011; Voevodskaya et al., 2016; Nedelska et al., 2017; Kara et al., 2022; Spotorno et al., 2022) also reported findings as ratios to Cr, an approach that assumes stable Cr levels (Li et al., 2003), even though Cr levels have been shown to be altered in MCI and AD (Tumati et al., 2013). Furthermore, the statistical power of ratios may be inferior to that of absolute quantification (Foy et al., 2011), all of which can affect the interpretation of the results (Hoch et al., 2017).
This study’s design addresses the aforementioned limitations to enable testing of the overarching hypothesis that, in cognitively unimpaired elderly, Glx and NAA levels within PET-based Braak stage-defined GM regions (Therriault et al., 2022) would correlate with T and N markers. Our approach uses two MRS acquisition schemes: a single-voxel semi-adiabatic localization by adiabatic selective refocusing (sLASER) sequence, a technique we recently adapted for the detection of Glx and mI in the hippocampus (Gajdosik et al., 2021); and MR spectroscopic imaging (MRSI) for the spatial mapping of metabolite distributions within various GM regions in the parietal lobe. The hypotheses, motivated by the reviewed studies and the well-known dependency of T and N pathology on Braak-stage (Therriault et al., 2022; Biel et al., 2021), were as follows. Lower Glx and NAA in the hippocampus and PCC would correlate with higher CSF p-tau181 (T) and, respectively, with smaller hippocampal volume (N) and PCC cortical thickness (N); and these correlations would be stronger in the hippocampus (Braak II) than in the PCC (Braak IV). No correlations were expected in the precuneus (Braak V) and precentral gyrus (Braak VI). All correlations were expected to be moderated by apolipoprotein E (APOE) genotype, as the E4 allele (APOE4) is not only associated with greater amounts of Aß burden (Baek et al., 2020), but also confers the strongest genetic risk for AD (Yamazaki et al., 2019). Overall, this study has a replication component (i.e., tests whether previous PCC findings hold), tests a novel hypothesis regarding Glx, and investigates whether the hippocampus has the potential to serve as a more robust MRS biomarker region than the PCC in prodromal AD. To increase the level of confidence in the results, the parietal GM regions affected at later Braak stages served as negative controls.
2. Materials and methods
2.1. Study population
Thirty-four cognitively unimpaired elderly subjects were recruited from the general population through pre-existing prospective studies at the NYU Grossman School of Medicine that are supported by the National Institute on Aging within the National Institutes of Health. Subjects met the following inclusion criteria: (i) male and female individuals between 50 – 95 years of age, with evidence of “normal cognition”, defined as scores of (ii) 0 on the Clinical Dementia Rating (CDR), (iii) ≥27 on the Mini-Mental State Examination (MMSE), (iv) ≤2 on the Global Deterioration Scale (GDS), and (v) ≤2 on the Brief Cognitive Rating Scale (BCRS), all of which are standard clinical screening instruments used for detecting and staging cognitive impairment (McDougall, 1990). Subjects were considered ineligible if they had a history of any neurologic, psychiatric, or hematologic conditions, or any contraindications to lumbar puncture for CSF collection, or MR imaging (MRI). Only lumbar punctures performed within 5 years of the MRI exam (e.g., before or after) were included in the analysis. For subjects who received multiple lumbar punctures within this time frame, the sample closest in time to their MRI exam was used. This study was approved by the Institutional Review Board and conducted at an outpatient MRI facility between 2018 and 2020. Written informed consent was obtained from all subjects.
2.2. Imaging acquisition, processing, and analysis
2.2.1. MRI data acquisition
All imaging was performed at 3 Tesla on a Siemens MAGNETOM Prisma (software version syngo MR E11) scanner using a 20-channel transmit-receive head coil (Siemens Healthcare, Erlangen, Germany). The protocol included the following clinically-used sequences: (i) 3D T1-weighted magnetization prepared rapid gradient echo (MPRAGE): repetition time (TR) = 2400 ms, TE = 2.2 ms, inversion time (TI) = 1060 ms, voxel size = 0.8 × 0.8 × 0.8 mm3, field-of-view (FOV) = 256 × 240 × 208 mm3, 6:38 min; and (ii) 2D T2-weighted fluid-attenuated inversion recovery (FLAIR): TR = 9000 ms, TE = 81 ms, TI = 2500 ms, voxel size = 0.7 × 0.7 × 5 mm3, FOV = 220 × 220 × 30 mm3, 2:44 min.
2.2.2. Single-Voxel MRS data acquisition
MRS of the left hippocampus was conducted as described previously (Gajdosik et al., 2021). To guide MRS placement, the high-resolution MPRAGE images were re-sliced into an axial orientation parallel to the principal axis of the hippocampus, and into a coronal orientation perpendicular to that axis. A 26 × 10 × 13 mm3 (3.4 mL) voxel was positioned to ensure coverage of the hippocampus, particularly the body and tail. Although the hippocampal head was included in most placements, we prioritized avoidance of the neighboring amygdala and inferior WM (Fig. 1) to minimize signal contamination from the surrounding functionally distinct (i.e., non-hippocampal) tissue. MRS data were acquired using a sLASER prototype sequence with an optimized sinc excitation pulse and four gradient offset independent adiabatic refocusing pulses with WURST modulation for both radiofrequency field and gradient waveforms (GOIA-W) (Andronesi et al., 2010). Metabolite spectra were acquired as 2048 complex points, with a 2000 Hz spectral bandwidth and 32-step phase cycling (TR = 1500 ms, TE = 120 ms, 9:10 min). For quantification of absolute metabolite concentrations, a separate water spectrum was also acquired over the same voxel volume (TR = 10,000 ms, TE = 25 ms, 2:40 min). Shimming was done in two steps to prevent the generation of spurious echoes from inefficient water suppression, as described previously (Gajdosik et al., 2021). A water frequency linewidth of <20 Hz was the target shim for all subjects.
Fig. 1. 1H MRS placement in the left hippocampus.
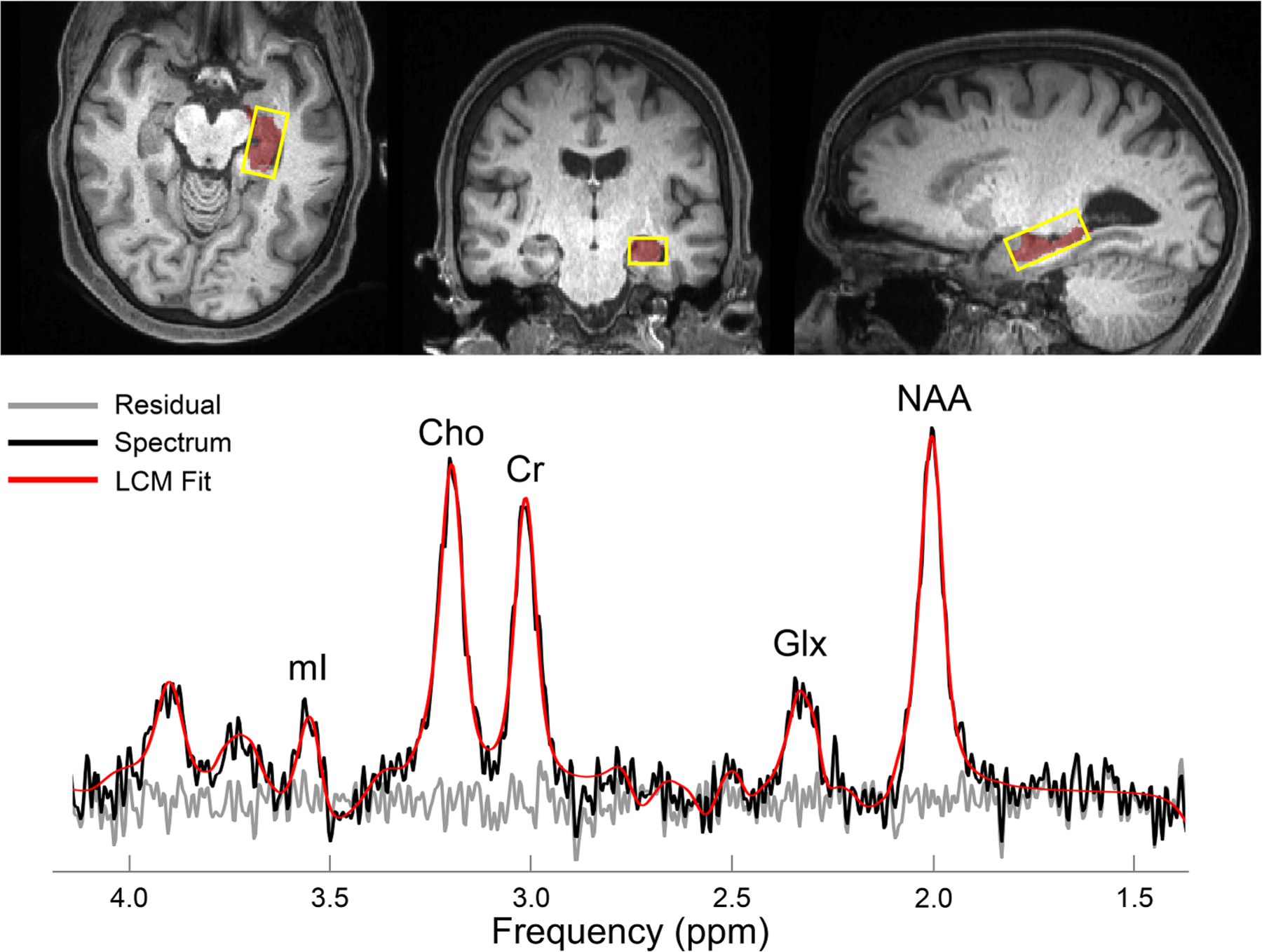
Top: Reconstructed axial (left), coronal (middle) and sagittal (right) T1-weighted MPRAGE images from a single subject. The 26×10×13 mm3 (left-right x anterior-posterior x inferior-superior) volume-of-interest is superimposed (yellow), along with the left hippocampus (red). Bottom: A spectrum (black) acquired from the same subject, along with its fitted function (red) and residual signal (gray). Quantified metabolites are labeled by their peaks.
Abbreviations: Choline = Cho; Creatine = Cr; Glutamate-plus-glutamine = Glx; myo-inositol = mI; N-acetyl-aspartate = NAA.
2.2.3. Whole-Brain MRSI data acquisition
To guide MRSI placement, the high-resolution MPRAGE images were re-sliced into an axial orientation parallel to the anterior commissure – posterior commissure (AC-PC) axis with a 1 mm slice resolution. MRSI was conducted using a 280 × 280 × 100 mm3 excitation slab that was manually positioned to ensure coverage of the brain, in its entirety from the base of the occipital lobe through the base of the skull, as done in an unrelated study (Chen et al., 2023) (Fig. 2). An outer volume saturation band was placed over the sinuses and eyeballs to reduce magnetic field inhomogeneity effects. Metabolite spectra were acquired using a validated (Ding et al., 2015) echo-planar spectroscopic imaging (EPSI) prototype sequence with lipid inversion nulling (TR = 1710 ms, TE = 17.6 ms, flip angle = 73°, TI = 198 ms, FOV = 280 × 280 × 135 mm3, matrix = 50 × 50 × 18, nominal voxel volume = 0.235 cm3, 16:49 min). Internal water spectra were acquired using an interleaved, gradient-echo acquisition with the same spatial resolution as that of the metabolite acquisition (TR = 591 ms, TE = 6.3 ms, flip angle = 20°) (Barker et al., 1993). Automatic shimming, followed by a manual adjustment of first-order shims to a water frequency linewidth of <27 Hz was the target for all subjects.
Fig. 2. 1H MRSI of the whole brain.
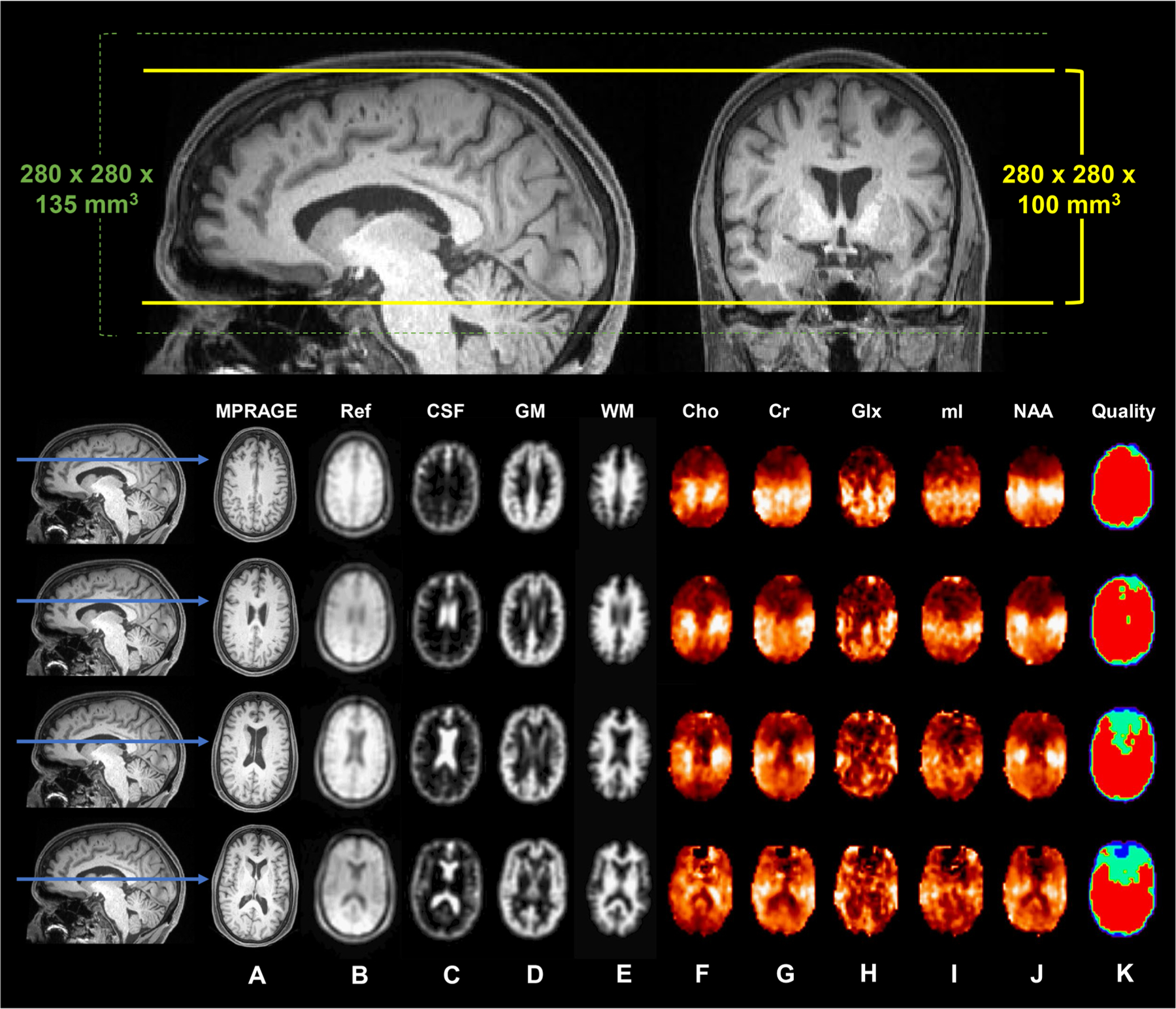
Top: Reconstructed sagittal and coronal T1-weighted MPRAGE images from a single subject. The 280 × 280 × 135 mm3 (left-right x anterior-posterior x inferior-superior) field-of-view is superimposed (dotted green lines), along with the 280 × 280 × 100 mm3 excitation slab (solid yellow lines). Bottom: Four reconstructed axial T1-weighted MPRAGE images (A) and their indicated positions along the sagittal plane (blue arrows). Shown in (B) is the water reference image, used for image registration, B0- and eddy-current correction, and signal normalization; (C-E) are the segmented tissue maps of cerebrospinal fluid (CSF), gray matter (GM), and white matter (WM), down-sampled to the MRSI resolution; (F-J) are the metabolite maps of choline (Cho), creatine (Cr), glutamate-plus-glutamine (Glx), myo-inositol (mI), and N-acetyl-aspartate (NAA); and (K) is the quality map, in which red indicates voxels that fulfilled all quality criteria, including metabolite linewidth <12 Hz and signal <3 standard deviations from the mean.
MRS and MRSI data acquisition parameters are summarized in Supplementary Table 1 – Sections 1 and 2. In Sections 2.2.5 and 2.2.6, we provide a detailed overview of MRS and MRSI data processing parameters, respectively, which are summarized in Supplementary Table 1 – Sections 3 and 4.
2.2.4. MRI data processing and analysis
An experienced neuroradiologist utilized the FLAIR images to grade the degree of hyperintense white matter (WM) signal abnormalities observed in each subject, following the frequently used Fazekas rating scale (Fazekas et al., 1987), which ranges from 0 (none or a single punctate lesion) to 3 (large, confluent lesions) (Vernooij and Smits, 2012). Separate Fazekas scores were provided for the frontal and posterior regions of the brain, and then averaged to yield a single rating for global WM lesion load.
Each subject’s axial MPRAGE was segmented into masks of CSF, GM, and WM using SPM12 (Ashburner and Friston, 2005) to correct for partial volume effects within the MRS voxel (Section 2.2.5), and using the Metabolite Imaging and Data Analyses System (MIDAS) software package (Maudsley et al., 2009; Sabati et al., 2015; Maudsley et al., 2006) to correct for partial volume effects within the MRSI voxels (Section 2.2.6); and masks of the left hippocampus, bilateral PCC, bilateral precuneus, and bilateral precentral gyrus, using the recon-all standard processing pipeline from FreeSurfer (version 6.0.0) (Fischl, 2012) (Fig. 3), to extract regional volume or cortical thickness measures, and to assess regional distributions of cerebral metabolites (Section 2.2.6). All segmentations were visually inspected for errors.
Fig. 3. Segmented regions-of-interest (ROIs) and their fitted spectra.
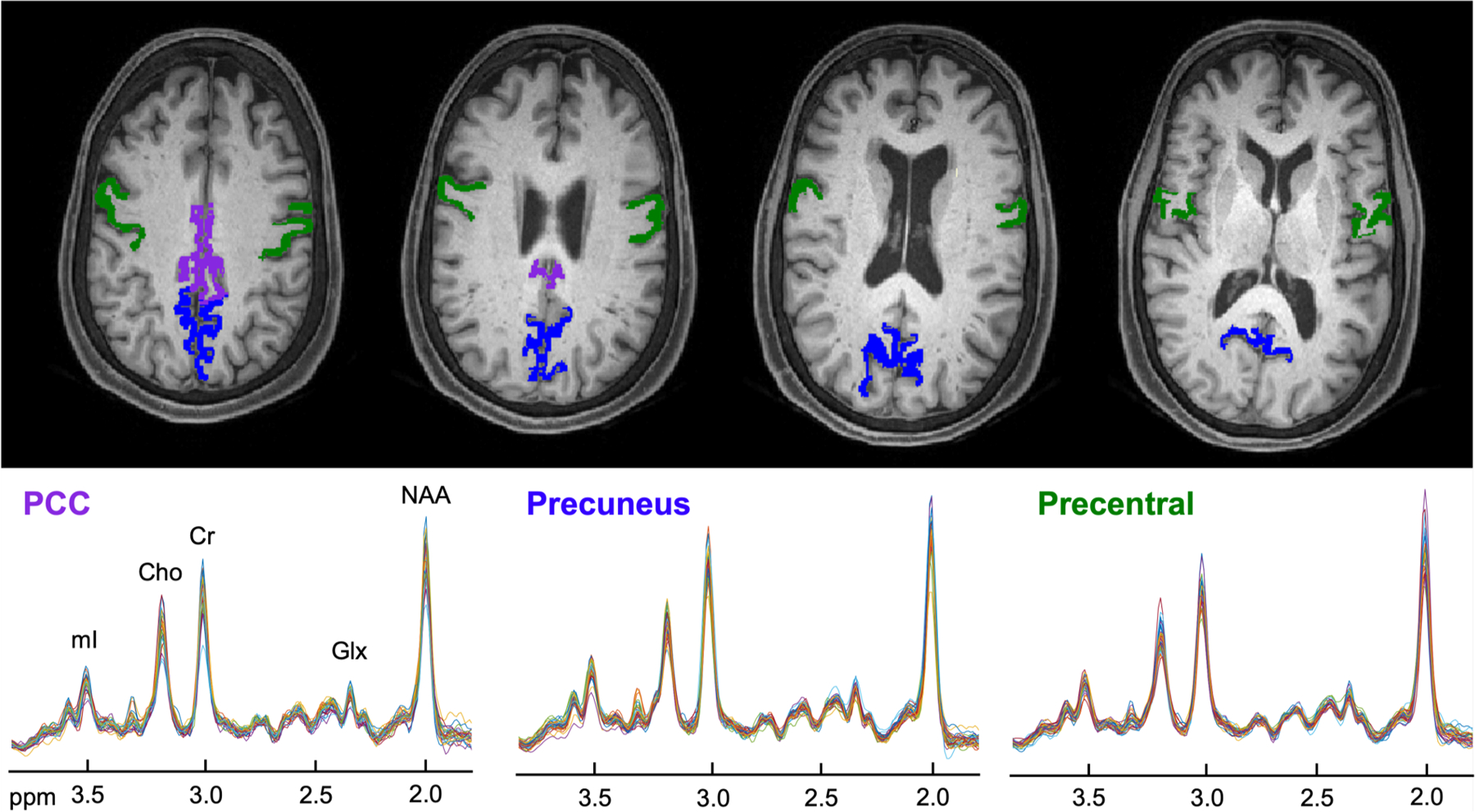
Top: Reconstructed axial T1-weighted MPRAGE images from a single subject (as shown in Figs. 1 and 2) overlaid with the selected atlas-defined cortical gray matter (GM) ROIs, obtained from FreeSurfer’s automated segmentation pipeline (Fischl, Neuroimage, 2012). Bottom: For each region, individual spectra from all subjects are overlaid on the same frequency and intensity scales. Note that 27 subjects contributed metabolite data from the posterior cingulate cortex (PCC, left), 31 from the precuneus (middle), and 32 from the precentral gyrus (right).
Abbreviations: Choline = Cho; Creatine = Cr; Glutamate-plus-glutamine = Glx; myo-inositol = mI; N-acetyl-aspartate = NAA; Posterior cingulate cortex = PCC.
Left hippocampal and estimated intracranial (eTIV) volumes were obtained from FreeSurfer’s output aseg.stats files. Each subject’s left hippocampal volume was normalized to their eTIV, one of the most common registration-based intracranial volume estimation methods (Nerland et al., 2022), to correct for differences in head size (Voevodskaya et al., 2014).
PCC, precuneal, and precentral cortical thicknesses were obtained from FreeSurfer’s output lh.aparc.stats and rh.aparc.stats files, respectively, and then averaged between hemispheres without normalization as recommended (Westman et al., 2013; Seiger et al., 2018), as these parameters are derived from a surface mesh-based algorithm and not a volumetric one (Tustison et al., 2014).
2.2.5. MRS data processing and analysis
Raw MRS data were first coil combined, signal averaged over one phase cycle, and corrected for eddy current artifacts using the unsuppressed internal water reference signal. Spectra were then frequency- and phase-corrected over 1.0 to 4.2 ppm using MATLAB and FID-A toolkit, as described previously (Gajdosik et al., 2021). A basis set containing eight metabolites (mI, scyllo-inositol, Cho, Cr, Glu, Gln, NAA, and lactate) was produced in MARSS (Landheer et al., 2021), and linear combination model fitting was performed in INSPECTOR (Gajdosik et al., 2021). The fitted metabolite and water signals were then scaled by the fractions of CSF, GM, and WM contained within the single voxel, as well as their relative literature T1 and T2 relaxation times (Supplementary Table 2) to correct for partial volume and relaxation effects, respectively. Corrected metabolite signals were then normalized to the corrected water signal to yield absolute metabolite concentrations in millimolar (mM) units (additional details are available in Supplementary Section 1 [Part A]).
All spectra were visually inspected to detect spurious artifacts or lipid contamination, as recommended by Near et al. (Near et al., 2021). Signal-to-noise ratio (SNR) was calculated as the ratio of the metabolite peak amplitude to the standard deviation of the noise signal estimated between 10 and 12 ppm. A metabolite spectrum, averaged across the 33 subjects who contributed adequate data from the left hippocampus, is shown in Fig. 4.
Fig. 4. Averaged fitted 1H spectrum from the left hippocampus.
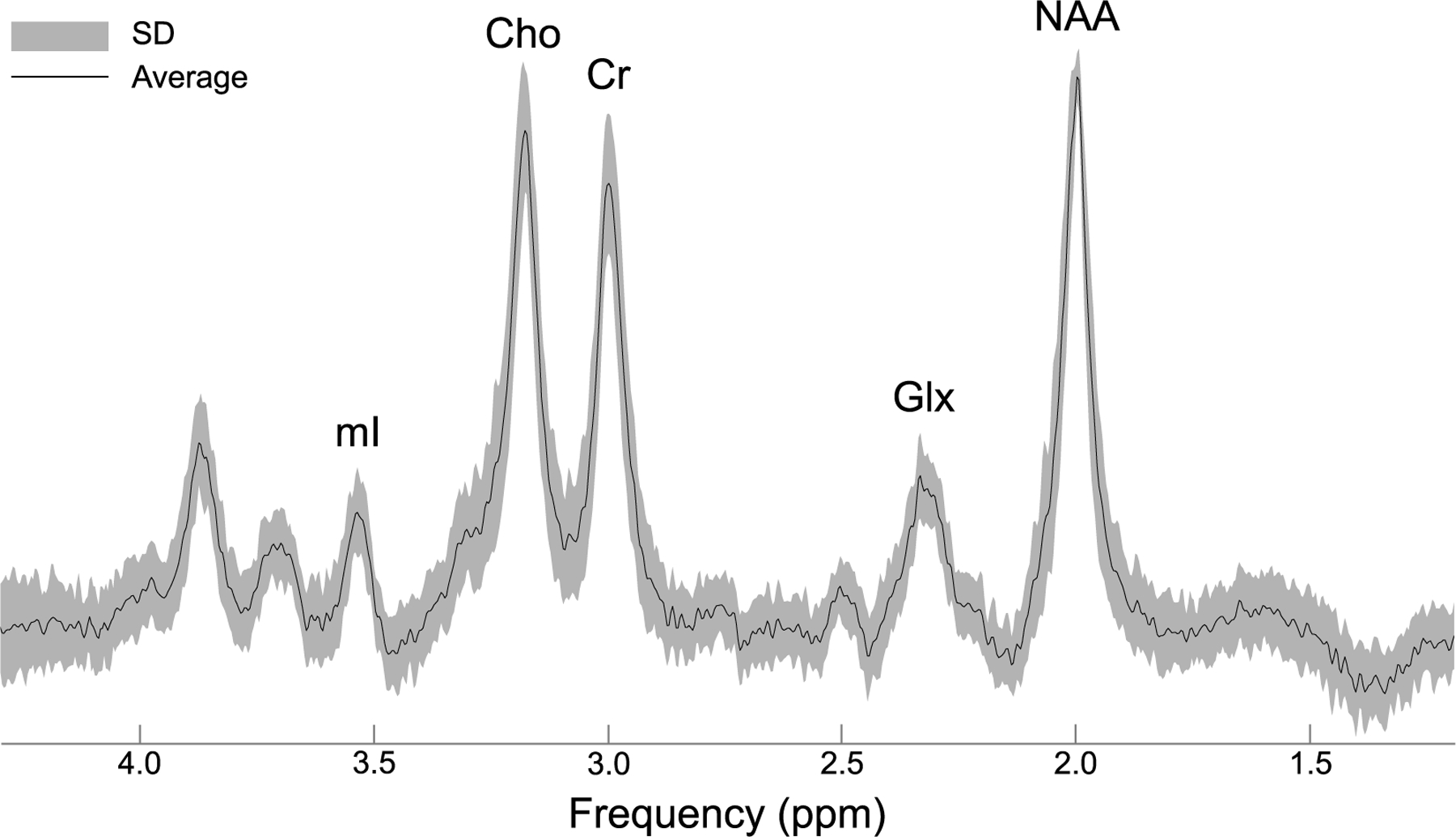
An averaged spectrum (black) from 33 subjects who provided adequate data from the left hippocampus, along with its standard deviation (SD; gray). Quantified metabolites are labeled by their peaks choline (Cho), creatine (Cr), glutamate-plus-glutamine (Glx), myo-inositol (mI), and N-acetyl-aspartate (NAA).
2.2.6. MRSI data processing and analysis
Raw MRSI data were processed entirely with MIDAS (Maudsley et al., 2009; Sabati et al., 2015; Maudsley et al., 2006), which included image reconstruction and parametric spectral analysis for determination of volumetric spectroscopic images (“maps”) of five metabolites (Cho, Cr, Glx, mI, and NAA), as described previously (Soher et al., 1998). Analogous to what was done for MRS data processing (Section 2.2.5), metabolite maps were corrected for local B0-, frequency-, and phase-shifts, eddy currents, and lineshape distortions at each voxel location using the internal water reference map, and reconstructed to 64×64×32 points with a voxel volume of approximately 0.08 mL. In parallel, the high-resolution axial MPRAGE image was segmented into CSF, GM, and WM and convolved with the MRSI point-spread function to yield tissue maps interpolated to the same spectroscopic image resolution. Within voxels, fitted metabolite and internal water spectra were scaled by fixed tissue water fractions (CSF = 0.98, GM = 0.726, WM = 0.634) (Neeb et al., 2006) and tissue water T1 relaxation values (CSF = 4300 ms, GM = 1350 ms, WM = 840 ms) (Wansapura et al., 1999; Lu et al., 2005) derived from literature (Supplementary Table 2) to reduce local signal intensity variations due to possible tissue misclassification and signal losses due to significant CSF partial volume contribution (additional details are available in Supplementary Section 1 [Part B]). Corrected metabolite signals were then normalized to the corrected internal water signal, yielding relative metabolite levels in institutional units (i.u.).
A visual inspection of metabolite maps and spectral patterns was performed for identification of contamination artifacts, and a set of inclusion criteria was applied to all data: (i) voxels with spectral linewidth <12 Hz, defined as a single Lorentzian-Gaussian lineshape fitted to all metabolite peaks (Ebel et al., 2007), and (ii) voxels with values <3 standard deviations from the mean parameter value over a 3 × 3 voxel neighborhood (Kreis, 2004; Maudsley et al., 2010). SNR was calculated as the ratio of the metabolite peak amplitude to the standard deviation of the noise signal estimated between 0 and 1.0 ppm, as done in other studies (Goryawala et al., 2016; Goryawala et al., 2018).
FreeSurfer-derived masks of the PCC, precuneus, and precentral gyrus (Section 2.2.4) were then mapped to the MRSI data using the Map Integrated Spectrum (MINT) module in MIDAS, which performed spectral averaging to yield a single fitted spectrum per region for each subject (Goryawala et al., 2016; Zhang et al., 2018) (Fig. 3). An additional set of inclusion criteria was applied to the regional data: (i) voxels with >95 % mask content, (ii) voxels with a CSF fraction ≤5 %, (iii) Cramér-Rao lower bound (CRLB) ≤15 % for Cho, Cr, and NAA, and (iv) regions containing ≥10 voxels following filters (i) to (iii).
2.3. CSF collection and analysis
CSF samples were collected by a radiologist using fluoroscopically guided lumbar puncture performed at the L3/4 interspace with the subject in a prone position. Sterile techniques were used. Samples were processed in accordance with standard protocols (de Leon et al., 2018). Briefly, samples were collected in 10 mL sterile Starstedt polypropylene tubes and centrifuged within 2 h at 2000 g for 10 min at 4°C. After centrifuging, 0.25 mL of CSF were stored in 1 mL polypropylene tubes at −80°C until assayed. In this study, 510 μl from each subject were analyzed as a single batch with a p-tau181 kit using Lumipulse Fujirebio (Leitão et al., 2019).
2.4. APOE genotyping
APOE genotyping was performed by LGC genomics. Each of the two copies of the APOE gene has three allelic variants (E2, E3, and E4). Subjects were stratified into groups based on carriership of at least one E4 allele: APOE4 carriers were classified as those with either E3/E4 or E4/E4 genotypes, whereas APOE4 non-carriers were classified as those with either E2/E2, E2/E3, or E3/E3 genotypes.
2.5. Statistical analyses
The associations between each outcome (CSF p-tau181 level, left hippocampal volume, and parietal cortical thicknesses) and the level of each metabolite (Cho, Cr, Glx, mI, NAA) measured from each brain region (left hippocampus; and bilateral PCC, precuneus, and precentral gyrus) were assessed using Pearson and Spearman correlations. Direct and partial correlations were computed, the latter adjusting for selected covariates based on known demographic and genetic risk factors (age, sex, race, ethnicity) (Silva et al., 2019), acquired protective factors (years of education) (Silva et al., 2019), vascular risk factors (Fazekas score) (Vernooij and Smits, 2012; Damulina et al., 2019; Kynast et al., 2018), and time-varying covariates (months from CSF collection to MRI) (Zhang et al., 2018). Relevant covariates for each outcome were identified using the least absolute shrinkage and selection operator (LASSO). For the prediction of CSF p-tau181, the covariates allowed to compete for inclusion in the model consisted of age, years of education, Fazekas score, and months from CSF collection to MRI as numeric factors, and sex, race, and ethnicity as binary factors. For the prediction of left hippocampal volume and parietal cortical thicknesses, the covariates allowed to compete for inclusion in the model comprised all of the above mentioned factors, except months from CSF collection to MRI. Partial correlations adjusted for a consistent set of covariates, including age, sex, years of education, Fazekas score, and months from CSF collection to MRI, were also performed.
Using data from the left hippocampus and PCC, mixed model regression was used to test whether correlations between fluid or imaging outcomes and metabolite levels were stronger in the left hippocampus than in the PCC. Metabolite levels and outcomes were standardized to have zero mean and unit variance in the whole sample so that the regression coefficient for an outcome as a predictor of a regional metabolite corresponds to the Pearson correlation between these variables. The model to predict each metabolite as dependent variable included the outcome as a numeric factor, brain region (left hippocampus, PCC) as a binary factor and the term representing the outcome-by-region interaction. An anonymized subject identification number was incorporated into the analysis as a random classification factor in order to account for the lack of statistical independence between metabolite levels in the two regions of the same subject.
Linear regression was used to test whether APOE genotype moderated the association of each regional metabolite with each outcome. Since each subject contributed at most one observation to each analysis, there was no need to account for intra-subject correlations, and subject-level outcomes could be treated as dependent variables. The model to predict each outcome as a dependent variable included the metabolite as a numeric factor, APOE4 carrier status as a binary factor, and the term representing the metabolite-by-carrier interaction.
For all continuous demographic and clinical variables (age, years of education, Fazekas score, months between CSF collection and MRI exam) and outcomes (CSF p-tau181, left hippocampal volume, eTIV, and PCC, precuneus, and precentral cortical thicknesses), normality was assessed with the Shapiro-Wilk test, and homoscedasticity was assessed with Levene’s test. If data were normally distributed and exhibited homogeneity of variance, Student’s t tests were used to compare APOE4 carriers to non-carriers. Otherwise, Mann-Whitney U tests were performed. For categorical demographic variables (sex, race, ethnicity), Chi-Squared tests were performed. Modified z-scores (Iglewicz and Hoaglin, 1993) were calculated for structural outcomes (normalized left hippocampal volume, and PCC, precuneus, and precentral cortical thicknesses) to detect the presence of potential outliers (i.e., individuals with substantial atrophy), defined as a z-score greater than 3.5 or less than 3.5.
Statistical tests were conducted at the two-sided 5 % significance level using SAS 9.4 (SAS Institute, Cary, NC) or R Statistical Software version 4.3.1 (R Core Team 2023).
3. Results
3.1. Study population
Demographic and clinical characteristics and selected outcomes are compiled in Table 1. Of the 34 subjects, on average 69 ± 9 years old (mean ± standard deviation; range = 56 – 89 years) with 17 ± 2 years of education (range = 14 – 21 years), 22 (65 %) were female, 8 (24 %) identified as non-White or Hispanic/other, and 12 (35 %) were APOE4 carriers. Moderate-to-severe WM hyperintensities were seen in 16 (47 %) subjects, as indicated by their average Fazekas score of 2 or greater. A valid CSF p-tau181 sample was obtained from 26 (76 %) subjects within 20 ± 14 months (range 0 – 52 months) of their MRI exam.
Table 1.
Summary of subject demographics, clinical characteristics, and selected outcomes. Data are expressed as mean (and standard deviation) or number of subjects (and percentage), as appropriate. Subjects were stratified into two groups based on their APOE genotype. Carriers were defined as having at least one E4 allele, whereas non-carriers were defined has having no E4 allele. APOE4 carriers and non-carriers were then tested for differences between groups, for each parameter. Note that no subject exhibited a genotype of E2/E2, which has been associated with reduced cognitive decline and thus thought to exert a protective effect against neurodegeneration (Sweigart et al., 2021).
Abbreviations: Apolipoprotein E = APOE; Cerebrospinal fluid = CSF; Posterior cingulate cortex = PCC; Standard deviation = SD.
| Parameter | Cohort (N = 34) | Carriers (N = 12) | Non-Carriers (N = 22) | P Value‡ | |
|---|---|---|---|---|---|
| Age | 69 (9) | 67 (8) | 70 (9) | 0.43 | |
| Sex | Female | 22 (65 %) | 8 (67 %) | 14 (70%) | 1.00 |
| Male | 12 (35 %) | 4 (33 %) | 8 (36 %) | ||
| Years of education | 17 (2) | 18 (2) | 17 (2) | 0.15 | |
| Race / Ethnicity | White | 26 (76 %) | 9 (75 %) | 17 (77 %) | 1.00 |
| Hispanic / Other | 8 (24 %) | 3 (25 %) | 5 (23 %) | ||
| Average Fazekas score | 0 | 7 (21 %) | 3 (25 %) | 4 (18 %) | 0.29 |
| 1 | 11 (32 %) | 4 (33 %) | 7 (32 %) | ||
| 2 | 10 (29 %) | 5 (42 %) | 5 (23 %) | ||
| 3 | 6 (18 %) | – | 6 (27 %) | ||
| APOE genotype | E2/E3 | 3 (9 %) | – | 3 (14 %) | – |
| E3/E3 | 19 (56 %) | – | 19 (86 %) | – | |
| E3/E4 | 9 (26 %) | 9 (75 %) | – | – | |
| E4/E4 | 3 (9 %) | 3 (25 %) | – | – | |
| CSF p-tau181, pg/mL† | 33 (15) | 40 (19) | 31 (12) | 0.21 | |
| Months between CSF collection and MRI exam † | 24 (27) | 10 (12) | 24 (13) | 0.01** | |
| Estimated total intracranial volume, x106 mm3 | 1.59 (0.15) | 1.61 (0.13) | 1.60 (0.16) | 0.72 | |
| Left hippocampal volume, x103 mm3 | 3.80 (0.44) | 3.86 (0.38) | 3.77 (0.48) | 0.99 | |
| PCC cortical thickness, mm | 2.50 (0.16) | 2.52 (0.14) | 2.49 (0.17) | 0.57 | |
| Precuneus cortical thickness, mm | 2.37 (0.09) | 2.38 (0.10) | 2.36 (0.09) | 0.74 | |
| Precentral cortical thickness, mm | 2.56 (0.16) | 2.59 (0.11) | 2.55 (0.18) | 0.40 | |
Values are given as mean (standard deviation) or count (percent).
CSF p-tau181 data were only available for 26 subjects: Carriers (n = 8), Non-Carriers (n = 18).
A Chi-Squared test was performed for categorical variables; either a Student’s t-test or a Mann-Whitney test was performed for continuous variables.
Significant difference between APOE4 carriers and non-carriers (p < 0.05).
All subjects underwent the imaging protocol and yielded adequate MPRAGE images for left hippocampal volumetry and parietal cortical morphometry. However, one subject did not complete the MRS exam, leaving 33 subjects with hippocampal metabolite data. All subjects completed the MRSI exam and yielded suitable data for processing, but some subjects’ post-processed data did not fulfill all quality criteria (Section 2.2.6) and were thus excluded from the final analyses: 27 subjects contributed metabolite data from the PCC, 31 from the precuneus, and 32 from the precentral gyrus. As per the minimum reporting standards for in vivo MRS (Lin et al., 2021), summaries of water line-width, CRLB, and SNR quality-control parameters are provided in Supplementary Table 1 – Section 2, Supplementary Table 1 – Section 4, and Supplementary Table 3, respectively.
Subject age, months between CSF collection and MRI exam, eTIV, and PCC and precentral cortical thicknesses satisfied the basic assumptions of normality and homoscedasticity for parametric analyses. A significant difference between APOE4 carriers and non-carriers was found only for the number of months between CSF collection and MRI exam (Student’s t-test, p = 0.01). No differences were found between APOE4 carriers and non-carriers, for all other continuous and categorical variables. Modified z-scores revealed no outliers for any of the structural outcomes: normalized left hippocampal volume (range = −2.4 – 1.5), PCC cortical thickness (range = −2.5 – 1.3), precuneus cortical thickness (range = −1.3 – 1.1), and precentral cortical thickness (range = −1.6 – 2.2).
3.2. Correlations with CSF p-tau181
In the analysis of all subjects, there were no associations between metabolite levels (Cho, Cr, Glx, mI, NAA) in any region and CSF p-tau181 (Supplementary Table 4).
In APOE4 carriers, however, greater concentrations of Glx measured from the left hippocampus correlated with greater levels of CSF p-tau181 (Pearson’s r = 0.76, p = 0.024) (Fig. 5, Supplementary Table 5). This finding, however, was not retained after adjusting for the potential confounding effect of years of education (partial Pearson’s r = 0.75, p = 0.054) (Supplementary Table 5), which was selected as the only covariate significantly related to CSF p-tau181 (p = 0.020) and explained 21 % of the variance in CSF p-tau181. This finding was also not retained after adjusting for the potential confounding effects of age, sex, years of education, Fazekas score, and months from CSF draw to MRI exam (partial Pearson’s r = 0.52, p = 0.652) (Supplementary Table 5).
Fig. 5. Correlations between left hippocampal metabolites and CSF p-tau181, stratified by APOE status.
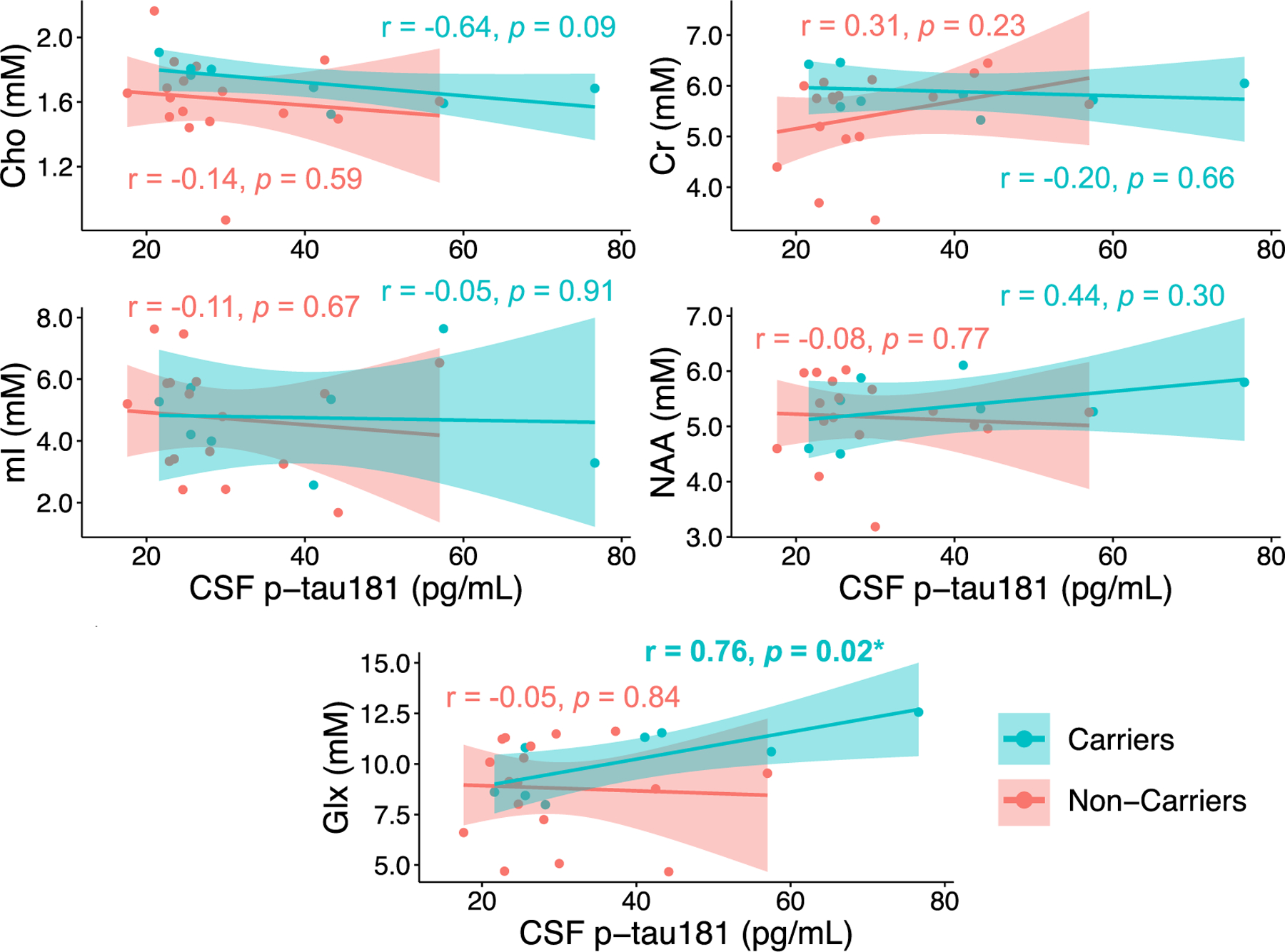
Presented are Pearson correlation coefficients (r) and p values. Only APOE4 carriers exhibited a significant association between greater millimolar (mM) concentrations of Glx measured from the left hippocampus and greater levels of CSF p-tau181 (*p < 0.05).
Abbreviations: Cerebrospinal fluid = CSF; Choline = Cho; Creatine = Cr; Glutamate-plus-glutamine = Glx; myo-inositol = mI; N-acetyl-aspartate = NAA.
3.3. Correlations with volume or cortical thickness
In the analysis of all subjects, lower levels of NAA measured from the left hippocampus correlated with smaller left hippocampal volume (Pearson’s r = 0.41, p = 0.019; Spearman’s r = 0.55, p = 0.001) (Supplementary Table 6). This finding was retained after adjusting for the potential confounding effects of age and Fazekas score (partial Spearman’s r = 0.48, p = 0.006) (Supplementary Table 6), both of which were selected as significant independent predictors of left hippocampal volume. Together, age (p = 0.038) and Fazekas score (p = 0.026) explained 31 % of the variance in left hippocampal volume. This finding was also retained after adjusting for the potential confounding effects of age, sex, years of education, Fazekas score, and months from CSF draw to MRI exam (partial Spearman’s r = 0.56, p = 0.009) (Supplementary Table 6).
In APOE4 carriers, lower levels of NAA measured from the left hippocampus correlated with smaller left hippocampal volume (Pearson’s r = 0.66, p = 0.017), even after adjusting for age and Fazekas score (partial Pearson’s r = 0.76, p = 0.008) (Fig. 6, Supplementary Table 7). A significant NAA-by-carrier interaction was also observed for the comparison of adjusted correlations involving left hippocampal NAA and left hippocampal volume (p = 0.03) (Supplementary Table 7). However, the association between left hippocampal NAA and left hippocampal volume was not retained after adjusting for the potential confounding effects of age, sex, years of education, Fazekas score, and months from CSF draw to MRI exam (partial Pearson’s r = 0.99, p = 0.092) (Supplementary Table 7).
Fig. 6. Correlations between left hippocampal metabolites and left hippocampal volume, stratified by APOE status.
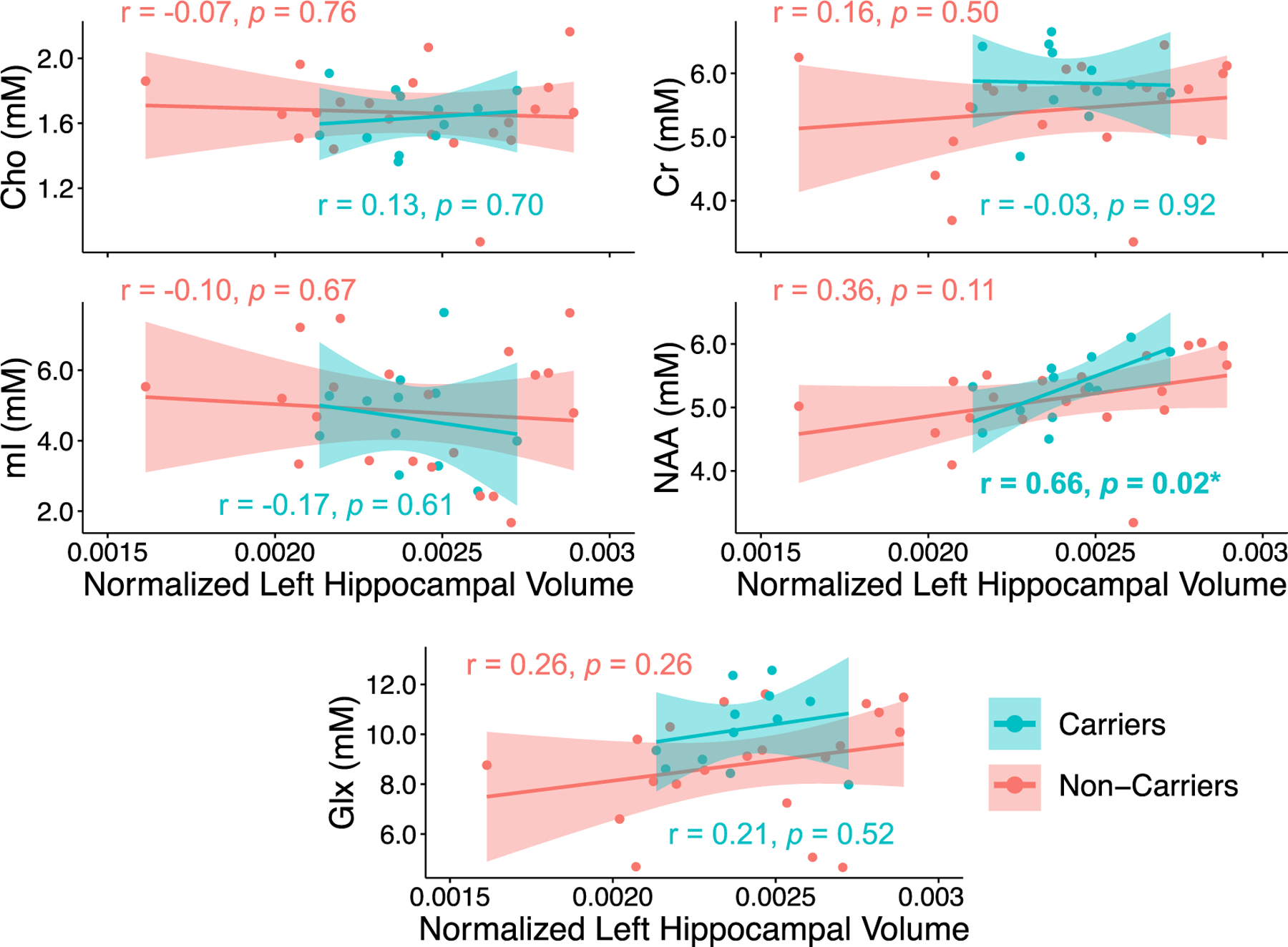
Presented are Pearson correlation coefficients (r) and p values. Only APOE4 carriers exhibited a significant association between reduced millimolar (mM) concentrations of NAA measured from the left hippocampus and reduced left hippocampal volume, after correcting for differences in head size across subjects (*p < 0.05).
Abbreviations: Choline = Cho; Creatine = Cr; Glutamate-plus-glutamine = Glx; myo-inositol = mI; N-acetyl-aspartate = NAA.
No correlations were observed between any metabolite from any parietal cortex region (i.e., PCC, precuneus, or precentral gyrus) and their corresponding cortical thicknesses, in either the whole-group (Supplementary Table 8) or the APOE4 stratified-group analysis (Supplementary Table 9). For correlations involving Glx and NAA, mixed model regression demonstrated no differences between those measured from the left hippocampus compared to those measured from the PCC (Supplementary Table 10).
4. Discussion
The current study sought to determine whether, in cognitively unimpaired individuals, there are Braak stage-dependent relationships between MRS and T/N markers in the context of a genetic risk for AD. Such relationships would indicate that specific biochemical imbalances accompany hallmark AD pathology, and motivate studies that explore the added value of MRS to the A/T/N framework on staging AD and predicting cognitive decline. The study’s novelty rests on its conceptual design of examining such correlations in regions involved in a wide range of Braak stages. This allowed us to formulate temporal-spatial hypotheses, and to have negative control regions (Section 4.1). We employed consensus-recommended, region-tailored acquisition approaches and post-processing (Section 4.2). Our findings (Section 4.3 and 4.4) have the following noteworthy aspects. First, the associations with T and N were with metabolites which reflect synaptic and neuronal health (although caution is needed in interpreting the association with T, because it did not survive adjustment for years of education). This is in line with the neuronal dysfunction and death that is known to be caused by tau and indicated by atrophy. Second, such associations were seen only in the hippocampus, which is in line with the Braak-stage-defined disease progression. And, third, these associations were related to APOE4 carriership, which is the genotype most commonly associated with risk for Aß deposition; suggesting that the associations are related to the AD trajectory of pathological change, and not to normal aging. While the study’s main limitation was the lack of an A marker, which renders our conclusions speculative (Section 4.5), we found that its results support the following notions (Section 4.6): MRS markers of compromised synaptic and neuronal health in the hippocampus are associated with, respectively, T and N pathology; and the hippocampus is an earlier MRS biomarker region than the PCC.
4.1. Background and hypotheses generation
The hippocampus, a critical unit for learning and memory (Bartsch, 2012), is one of the earliest sites of tau pathology (Braak and Braak, 1991) and volume loss (Jack et al., 1999; McRae-McKee et al., 2019; Pennanen et al., 2004) in AD. Therefore, in cognitively unimpaired elderly, it is expected that any relationships between MRS and T/N markers would first become evident in this hallmark region. We hypothesized that the metabolites exhibiting any correlations would be those related to synaptic and neuronal health, i.e., Glx and NAA, respectively. Glx is comprised of the combined Glu and Gln signals, and is therefore regarded as a marker of overall glutamatergic neurotransmission (Ramadan et al., 2013). Reduced Glx has been linked to memory decline (Nikolova et al., 2017), perhaps caused by deficits in glutamatergic signaling related to synapse loss (Zeydan and Kantarci, 2021; Motegi et al., 2021) or reductions in the number of glutamatergic transporters (Wang and Reddy, 2017), which have been shown to co-localize with phosphorylated tau in patients with AD dementia (Sasaki et al., 2009). NAA is a well-known marker of neuronal health, whose levels decline in most neurological disorders, likely due to compromised mitochondrial function (Moffett et al., 2007). We therefore hypothesized that lower Glx and lower NAA would correlate with higher CSF p-tau181 (T) and with smaller hippocampal volume (N).
These hypotheses were further motivated by a recent study of the PCC, a “hotspot” for Aß deposition (Ali et al., 2022), hypometabolism (Mosconi et al., 2008), hypoperfusion (Wierenga et al., 2014), and atrophy (Choo et al., 2010). In a cohort of cognitively unimpaired elderly, with an identical proportion of APOE4 carriers as in our study, lower Glu/Cr and lower NAA/Cr were found to correlate with higher 18F-flortaucipir PET signal (Kara et al., 2022). Therefore, as in the hippocampus, we hypothesized that lower Glx and lower NAA would correlate with higher CSF p-tau181 in the PCC as well, but with the added expectation that the correlations would be weaker than those in the earlier-affected hippocampus. We also expected correlations between these metabolites and parietal cortical thickness.
Given the nature of our cohort (mean age ~70 years with normal cognition) it was reasonable not to expect that, on average, T/N pathology would be found in regions affected in the last Braak stages (V and VI) (Braak and Braak, 1991). We therefore designated the precuneus and precentral gyrus as negative control regions, where correlations between NAA, Glx, and T/N were not expected. The precuneus and the precentral gyrus were chosen on the basis of having volumes and morphology suitable for the relatively large (compared to structural MRI) MRS voxels, and for being situated in locations which generally yield high-quality MRS signal.
All correlations were expected to be moderated by APOE genotype, in line with previous 1H MRS results (Voevodskaya et al., 2016; Gomar et al., 2014), and with the known associations of APOE4 with Aß aggregation (Yamazaki et al., 2019; Kanekiyo et al., 2014), increased tau burden (T) (Shi et al., 2017; Young et al., 2023) and accelerated hippocampal atrophy (N) (Abushakra et al., 2020; Gorbach et al., 2020). We therefore hypothesized stronger associations with T and N in APOE4 carriers than in non-carriers.
4.2. Technical approach
The hippocampus is one of the most challenging structures to interrogate with MRS, as it is situated deep in the temporal lobe, close to the skull base and nasal sinuses (Bartsch, 2012). This local environment introduces spatial field inhomogeneity and susceptibility effects, leading to loss of spectral resolution and low SNR (Dixon et al., 2002). The whole-brain shimming used by the MRSI acquisition, therefore, is not well-suited to ameliorate these effects, because it optimizes B0 homogeneity over a much larger volume. Indeed, we had found the MRSI hippocampal data obtained by spectral averaging (as done for the precuneus and precentral gyrus) to be of poor quality and therefore chose not to analyze hippocampal voxels from the whole-brain MRSI acquisition. Better B0 homogeneity, which yields narrower linewidths and higher SNR, can instead be achieved by local shimming with single-voxel MRS (Allaili et al., 2015). The widely used point-resolved spectroscopy (PRESS) sequence, however, incurs chemical shift displacement errors (CSDEs) of 30 % or more (Öz et al., 2021), and these localization inaccuracies propagate downstream to metabolite quantification errors (Öz et al., 2021; Watanabe and Takaya, 2018). Recent MRS consensus statements (Öz et al., 2021; Wilson et al., 2019), therefore, have recommended sLASER localization for its high-bandwidth adiabatic pulses, which minimize CSDEs, as well as for other advantages.
For all of the above reasons, we recently optimized a sLASER sequence for use in the hippocampus (Gajdosik et al., 2021). We also aimed to improve the accuracy of metabolite quantification by minimizing the macromolecular baseline that is present at short TEs. We found that a TE of 120 ms yielded both a flat macromolecular baseline and adequate SNR for the quantification of all metabolites, including the J-coupled mI and Glx species (Gajdosik et al., 2021). The current study uses the described approach without any modifications.
Located in the inferior parietal lobe, the PCC is, in contrast to the hippocampus, much less vulnerable to magnetic susceptibility effects. Single-voxel MRS, however, can be affected by variations in the voxel’s location, size, and tissue composition. For example, many studies use a large single voxel which bilaterally covers the posterior cingulate gyri and inferior precunei, and therefore refer to their region-of-interest as “PCC/precuneus” (Kantarci et al., 2011; Voevodskaya et al., 2016; Nedelska et al., 2017; Spotorno et al., 2022; Voevodskaya et al., 2019; Suri et al., 2017). The amount of WM partial volume in single voxels can also be variable and cannot be accounted for in post-processing. Given such limitations and the important fact that the PCC and precuneus are classified under separate PET-based Braak stages (IV and V, respectively), we applied a MRSI approach, which allows for a segmentation-based definition of cortical regions. This results in minimal WM partial volume and provides more accurate estimates of metabolite levels in these individual structures.
Two important approaches common to the post-processing of both the MRS and MRSI data, were absolute quantification and correction for CSF partial volume. The former avoids the use of metabolic ratios, which can introduce ambiguity in interpreting the results (Wilson et al., 2019). For example, the assumption that levels of Cr (the frequent denominator) are stable may be incorrect, as its levels may be altered in neurological disorders (Swanberg et al., 2019; Bartnik-Olson et al., 2021), including AD (Tumati et al., 2013). The latter ensures that inter-subject differences in voxel CSF content (due to age- or pathology-related atrophy) are taken into consideration.
4.3. Hippocampal metabolites and their relationships with tau pathology and atrophy
The whole-group analysis of all subjects revealed no relationships between CSF p-tau181 (T) and metabolite levels from the left hippocampus. The stratified-group analysis demonstrated a correlation between higher levels of CSF p-tau181 and higher levels of hippocampal Glx, in APOE4 carriers only, but this finding did not survive adjustment for years of education (Fig. 5, Supplementary Table 5). We had hypothesized an association between CSF p-tau181 and hippocampal Glx, given that tau neurofibrillary tangle pathology is described in the hippocampus during the earliest stages of AD (i.e., Braak II) (Braak and Braak, 1991), but we expected to find an indirect, rather than direct, relationship (Section 4.1). Elevated Glx may reflect excitotoxic injury through calcium (Ca2+) dyshomeostasis and increased cell death (Camacho and Massieu, 2006), or disrupted neural connectivity and functioning due to the role of Glu in regulating myelination (Turan et al., 2021). Specifically, Lee et al. (Lee et al., 2004) demonstrated an upregulation of neuronal group II metabotropic glutamate receptors in the hippocampal postmortem brain tissue of patients with AD compared to age-matched controls, which has been shown to increase Aß deposition (Stephenson and Clemens, 1998) and tau phosphorylation (Bruno et al., 2001) downstream. Therefore, upregulation of hippocampal glutamatergic neurotransmission may accompany, or contribute to, tau accumulation in those with genetic risk for AD. Interestingly, our finding fell just below the threshold for statistical significance (r = 0.75, p = 0.054) after adjusting for years of education.
The left hippocampus was preferentially interrogated over the right hippocampus based on studies examining the functional asymmetry of left and right hippocampi. Left hippocampal volume, often derived from structural MRI, has been cited as a stronger predictor of AD progression (Ezzati et al., 2016; Uysal and Ozturk, 2020), with faster rates of atrophy (Thompson et al., 2003), greater levels of atrophy (Feng et al., 2021), and stronger correlations with cognitive decline (Feng et al., 2021). As expected, and in line with results from previous hippocampal MRS studies of AD (Dixon et al., 2002; Schuff et al., 1997), we found an association between smaller left hippocampal volume (N) and lower concentrations of left hippocampal NAA (Supplementary Table 6). Again, this relationship appeared to be driven by APOE4 carriers, as non-carriers exhibited no correlations between any metabolite and volume (Fig. 6, Supplementary Table 7). Given that we accounted for variable amount of voxel CSF (Section 4.2), the observed correlation suggests compromised neuronal health in the remaining hippocampal neurons.
A more specific interpretation of this association can also be formulated. NAA, a marker of neuronal health and mitochondrial function (Moffett et al., 2007), is involved in various cellular processes, including osmoregulation, energy metabolism, and myelin lipid synthesis, although its exact role is unknown (Moffett et al., 2007). Reduced NAA, observed in most neurodegenerative diseases, can be interpreted as reduced neuronal density (Moffett et al., 2007; Baslow, 2003), loss of neuronal cytoarchitecture (Baslow, 2003), or as damage to the neuronal mitochondria (Moffett et al., 2007; Li et al., 2013; Vagnozzi et al., 2010). In the context of AD, a greater load of neurotoxic Aß or tau pathology, particularly in vulnerable APOE4 carriers (Kantarci et al., 2012), may impair synaptic transmission (Mucke and Selkoe, 2012) and contribute to eventual synaptic and neuronal loss. Energy metabolism is also impaired in AD, and mitochondrial dysfunction is well-documented (Wang et al., 2020). All these processes would give rise to lower levels of NAA and are eventually associated with irreversible neuronal loss reflected in smaller hippocampal volumes.
Adjusting for age, sex, years of education, Fazekas score, and months from CSF collection to MRI rendered the association between left hippocampal NAA and left hippocampal volume no longer significant, only within the stratified-group analysis. This finding may be due to the lower power of the stratified-group analysis (smaller sample sizes), or can be explained by two other reasons: (i) the full set of covariates likely included extraneous factors that can randomly influence the partial p values associated with other factors within the model; or (ii) LASSO may have missed one or more meaningful cofactors that, when adjusted for, explained enough variance to nullify the significant association.
4.4. Parietal cortex metabolites and their relationships with tau pathology and atrophy
In contrast to previous studies that found associations between lower NAA/Cr (Kara et al., 2022) or higher mI/Cr (Spotorno et al., 2022) and greater levels of tau deposition in the PCC of cognitively unimpaired elderly, we report no associations between any metabolite within the PCC and CSF p-tau181 (T) (Supplementary Table 4). We propose three possible explanations: (i) tau pathology is absent from the PCC; (ii) tau pathology is present, but at sub-threshold levels; or (iii) tau pathology is present but independent of regional metabolic changes. Similarly, we report no associations between CSF p-tau181 and any metabolite from the precuneus or precentral gyrus, the negative control regions that were selected for their involvement in even later Braak stages (Braak and Braak, 1991) (Supplementary Table 4).
We had hypothesized the same types of correlations in both the hippocampus and the PCC, but we expected stronger correlations when Glx and NAA were measured from the hippocampus than from the PCC. Instead, we saw no associations with any metabolite in the PCC, failing to replicate the results from Kara et al. (Kara et al., 2022). Accordingly, mixed model regression yielded negative findings (Supplementary Table 10). However, the significant findings that we did observe in the current study supported our more general hypothesis, i.e., the hippocampus may be a more sensitive MRS biomarker region than the PCC. Specifically, the findings are consistent with the topographic and temporal ordering of pathological events modelled by both the Braak staging scheme and the A/T/N biomarker framework (Jack et al., 2010): T and N markers were associated with Glx and NAA only in the hippocampus, and exclusively in APOE4 carriers. Absence of correlations with T and N in other regions, particularly in the PCC, is characteristic of an early phenotype along the AD continuum. Therefore, for augmenting A/T/N in the early stages of AD pathological change, the hippocampus may be a better MRS target than the PCC.
4.5. Limitations
As outlined in Section 3.1, of the 34 subjects recruited and scanned, 27 provided CSF p-tau181 data within ±5 years of their MRI exam. This criterion was chosen to conduct analyses with sufficient power, as only 17 subjects would have provided a sample within ±2 years. We demonstrated that the delay between CSF collection and MRI exam was not a significant covariate for CSF p-tau181 in the following ways: First, LASSO analysis revealed years of education to be the only predictor of CSF p-tau181, and findings failed to achieve statistical significance after adjustment for this variable (Section 3.2, Supplementary Section 1 [Part C], Supplementary Tables 4 and 5). Second, a more conservative statistical analysis, which adjusted correlations for a consistent set of covariates including key moderators such as age, sex, and delay, revealed the same result (Section 3.2, Supplementary Tables 4 and 5). Thus, while we observed that APOE4 carriers received a lumbar puncture that was closer in time to their MRI exam compared to non-carriers (Student’s t-test, p = 0.01; Table 1), our statistics were not impacted, providing insufficient evidence to support substantial CSF p-tau181 changes over a period of five years.
Given that absolute levels of CSF p-tau181 reflect global, rather than regional, tau burden, it is difficult to assess whether tau has reached any of the four studied regions; a limitation which may be addressed with PET-based measures of tau deposition. One large tau-PET study of 576 cognitively unimpaired individuals over age 50 (average 71 ± 11 years old) observed abnormal medial temporal lobe (MTL) and/or extra-MTL signal in 334 (58 %) subjects (Lowe et al., 2018). This finding suggests the likely presence of tau pathology in our comparable cohort of cognitively unimpaired elderly (who are also over age 50; average 69 ± 9 years old). Moreover, we note that CSF p-tau181 and tau-PET, while both markers of T, are sensitive to different species of tau, at different times along the AD continuum. The hyperphosphorylation of soluble tau isoforms, detected by CSF phosphorylated tau markers, such as p-tau181 (Alonso et al., 2001), have been shown to self-assemble into insoluble paired helical and straight filaments (Alonso et al., 2001; Hirata-Fukae et al., 2009), the major constituents of intraneuronal neurofibrillary tangles, which are detected by tau-PET (Alonso et al., 2001). This sequence of events is, in turn, consistent with discordant observations of normal tau-PET but abnormal CSF phosphorylated tau in cognitively unimpaired individuals (Therriault et al., 2022; Mattsson-Carlgren et al., 2020). Therefore, despite its inherent lack of regional specificity, CSF p-tau181 may be better suited than tau-PET for measuring early changes in tau metabolism, particularly within our cohort of cognitively unimpaired elderly.
The lack of an A marker (e.g., CSF Aß42 or amyloid-PET) precludes characterizing subjects according to either the International Working Group’s (IWG) or the National Institute on Aging–Alzheimer’s Association’s (NIA-AA) classification of preclinical AD (Jack et al., 2016). We can posit, however, that since carriership of APOE4 is the strongest genetic risk factor for AD (Yamazaki et al., 2019), and is consistently associated with increased Aß burden in MCI and AD dementia (Baek et al., 2020), categorizing subjects into APOE4 carrier and non-carrier subgroups provides a useful examination of early metabolic changes that are likely related to AD, rather than to normal aging.
While we enrolled fewer subjects in comparison to the five other MRS studies of cognitively unimpaired individuals (Kantarci et al., 2011; Voevodskaya et al., 2016; Nedelska et al., 2017; Kara et al., 2022; Spotorno et al., 2022) (≥40 subjects), we examined, for the first time, AD-related regions other than the PCC. This was enabled by our two-pronged, advanced methodological approach. First, we used a consensus-recommended MRS sequence, which we recently optimized for hippocampal use with a reproducibility study. This enabled increased precision of metabolite quantification within the hippocampus, a structure critical to the development and progression of AD, but understudied with MRS due to technical challenges. Second, a whole-brain MRSI acquisition enabled the study of multiple parietal cortex regions, including some which served as negative controls.
4.6. Conclusions
The present study tested hypotheses related to Braak stage-dependent relationships between MRS and T/N markers in cognitively unimpaired elderly. Correlations were found only in the left hippocampus (Braak II), which concurs with the spatially- and temporally-resolved Braak staging model of pathological severity in specific cortical regions along the course of AD-pathological change. The specific correlations were for the synaptic marker Glx with T, and for the neuronal marker NAA with N. Because both correlations were found exclusively within APOE4 carriers (i.e., in those with high risk for Aß deposition), we speculate that they may be related to AD-pathological change, rather than to normal aging. We therefore conclude that by providing cortical metabolic biomarkers, MRS has the potential to augment A/T/N staging, and that the hippocampus may be a more sensitive MRS target than the PCC. The next step is to corroborate these findings and conclusions using regional A and T markers from PET imaging.
Supplementary Material
Acknowledgements
We thank all subjects for taking part in this study.
Funding
This research was supported by a pilot grant to Dr. Kirov from the NYU Alzheimer’s Disease Research Center, under award number P30AG066512 from the National Institute on Aging (NIA) of the National Institutes of Health (NIH). Support for the CSF studies was provided by the NIA under award numbers R01AG013616, R01AG012101, R01AG022374 to Dr. de Leon; and R21AG049348, R21AG055002, R01AG056031, R01AG056531 to Dr. Osorio. Additional support was provided by the National Heart, Lung, and Blood Institute (NHLBI) under award number R01HL118624 to Dr. Osorio and by the Steven A. and Alexandra M. Cohen Foundation to Dr. de Leon.
The following funding sources are also acknowledged: NIA awards R01AG080672 to Dr. Kirov, P30AG066512 and P01AG060882 to Dr. Wisniewski, as well as NHLBI award HL111724 to Dr. Glodzik.
The funding sources were not involved in the collection, analysis, and interpretation of data; in the writing of the report; and in the decision to submit the article for publication. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
List of abbreviations
- AD
Alzheimer’s disease
- APOE
Apolipoprotein E
- Cho
Choline
- Cr
Creatine
- CRLB
Cramér-Rao lower bound
- FLAIR
Fluid-Attenuated Inversion Recovery
- FOV
Field-of-view
- Glx
Glutamate-plus-glutamine
- GM
Gray matter
- 1H MRS
Proton magnetic resonance spectroscopy
- 1H MRSI
Proton magnetic resonance spectroscopic imaging
- MCI
Mild Cognitive Impairment
- mI
myo-inositol
- MPRAGE
Magnetization Prepared Rapid Gradient Echo
- NAA
N-acetyl-aspartate
- PET
Positron emission tomography
- sLASER
Semi-Adiabatic Localization by Adiabatic Selective Refocusing
- SNR
Signal-to-noise ratio
- TE
Echo time
- TI
Inversion time
- TR
Repetition time
- VOI
Volume-of-interest
- WM
White matter
Footnotes
Informed consent
All subjects provided written informed consent prior to their participation.
CRediT authorship contribution statement
Anna M. Chen: Writing – review & editing, Writing – original draft, Visualization, Formal analysis, Data curation. Martin Gajdošík: Visualization, Methodology, Investigation, Formal analysis, Data curation. Wajiha Ahmed: Resources, Investigation. Sinyeob Ahn: Software. James S. Babb: Writing – review & editing, Formal analysis. Esther M. Blessing: Resources. Allal Boutajangout: Writing – review & editing, Resources, Investigation. Mony J. de Leon: Supervision, Resources. Ludovic Debure: Resources, Investigation. Naomi Gaggi: Investigation. Mia Gajdošík: Project administration. Ajax George: Supervision, Resources. Mobeena Ghuman: Writing – review & editing, Resources, Investigation. Lidia Glodzik: Writing – review & editing, Supervision, Resources. Patrick Harvey: Project administration. Christoph Juchem: Supervision. Karyn Marsh: Resources, Investigation. Rosemary Peralta: Project administration. Henry Rusinek: Writing – review & editing, Supervision. Sulaiman Sheriff: Software, Resources. Alok Vedvyas: Resources. Thomas Wisniewski: Supervision, Resources. Helena Zheng: Data curation. Ricardo Osorio: Writing – review & editing, Supervision, Conceptualization. Ivan I. Kirov: Writing – review & editing, Supervision, Methodology, Investigation, Funding acquisition, Conceptualization.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.neuroimage.2024.120742.
Data availability
Data will be made available on request.
References
- Abushakra S, et al. , 2020. APOE ε4/ε4 homozygotes with early Alzheimer’s disease show accelerated hippocampal atrophy and cortical thinning that correlates with cognitive decline. Alzheimer’s Dementia: Trans. Res. Clin. Interventions 6, e12117. 10.1002/trc2.12117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams JN, Maass A, Harrison TM, Baker SL, Jagust WJ, 2019. Cortical tau deposition follows patterns of entorhinal functional connectivity in aging. Elife 8. 10.7554/eLife.49132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aisen PS, et al. , 2017. On the path to 2025: understanding the Alzheimer’s disease continuum. Alzheimers. Res. Ther. 9, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali DG, et al. , 2022. Amyloid-PET levels in the precuneus and posterior cingulate cortices are associated with executive function scores in preclinical alzheimer’s disease prior to overt global amyloid positivity. J. Alzheimers. Dis. 88, 1127–1135. 10.3233/JAD-220294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allaili N, et al. , 2015. Single-voxel (1)H spectroscopy in the human hippocampus at 3 T using the LASER sequence: characterization of neurochemical profile and reproducibility. NMR Biomed. 28, 1209–1217. 10.1002/nbm.3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso A.d.C., Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K, 2001. Hyperphosphorylation induces self-assembly of τ into tangles of paired helical filaments/straight filaments. Proc. National Acad. Sci. 98, 6923–6928. 10.1073/pnas.121119298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andronesi OC, et al. , 2010. Spectroscopic imaging with improved gradient modulated constant adiabaticity pulses on high-field clinical scanners. J. Magn. Reson. 203, 283–293. 10.1016/j.jmr.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antuono PG, Jones JL, Wang Y, Li SJ, 2001. Decreased glutamate + glutamine in Alzheimer’s disease detected in vivo with (1)H-MRS at 0.5 T. Neurology. 56, 737–742. 10.1212/wnl.56.6.737. [DOI] [PubMed] [Google Scholar]
- Ashburner J, Friston KJ, 2005. Unified segmentation. Neuroimage 26, 839–851. 10.1016/j.neuroimage.2005.02.018. [DOI] [PubMed] [Google Scholar]
- Association A.s., 2022. 2022 Alzheimer’s Disease Facts and Figures. Alzheimers. Dement. 18, 700–789. 10.1002/alz.12638. [DOI] [PubMed] [Google Scholar]
- Baek MS, et al. , 2020. Effect of APOE epsilon4 genotype on amyloid-beta and tau accumulation in Alzheimer’s disease. Alzheimers. Res. Ther. 12, 140. 10.1186/s13195-020-00710-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker PB, et al. , 1993. Quantitation of proton NMR spectra of the human brain using tissue water as an internal concentration reference. NMR Biomed. 6, 89–94. [DOI] [PubMed] [Google Scholar]
- Bartnik-Olson BL, et al. , 2021. The clinical utility of proton magnetic resonance spectroscopy in traumatic brain injury: recommendations from the ENIGMA MRS working group. Brain ImAging Behav. 15, 504–525. 10.1007/s11682-020-00330-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartsch T, 2012. The Clinical Neurobiology of the Hippocampus: An integrative View. Oxford University Press. [Google Scholar]
- Baslow MH, 2003. N-acetylaspartate in the vertebrate brain: metabolism and function. Neurochem. Res. 28, 941–953. 10.1023/a:1023250721185. [DOI] [PubMed] [Google Scholar]
- Besson FL, et al. , 2015. Cognitive and brain profiles associated with current neuroimaging biomarkers of preclinical Alzheimer’s disease. J. Neurosci. 35, 10402–10411. 10.1523/JNEUROSCI.0150-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biel D, et al. , 2021. Tau-PET and in vivo Braak-staging as prognostic markers of future cognitive decline in cognitively normal to demented individuals. Alzheimers. Res. Ther. 13, 137. 10.1186/s13195-021-00880-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkenbihl C, Salimi Y, Frohlich H, 2022. Japanese Alzheimer’s Disease Neuroimaging, I. & Alzheimer’s Disease Neuroimaging, I. Unraveling the heterogeneity in Alzheimer’s disease progression across multiple cohorts and the implications for data-driven disease modeling. Alzheimers. Dement. 18, 251–261. 10.1002/alz.12387. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E, 1991. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Bruno V, et al. , 2001. Metabotropic glutamate receptor subtypes as targets for neuroprotective drugs. J. Cereb. Blood Flow Metab. 21, 1013–1033. 10.1097/00004647-200109000-00001. [DOI] [PubMed] [Google Scholar]
- Camacho A, Massieu L, 2006. Role of glutamate transporters in the clearance and release of glutamate during ischemia and its relation to neuronal death. Arch. Med. Res. 37, 11–18. 10.1016/j.arcmed.2005.05.014. [DOI] [PubMed] [Google Scholar]
- Chen Y, Fu AKY, Ip NY, 2019. Synaptic dysfunction in Alzheimer’s disease: mechanisms and therapeutic strategies. Pharmacol. Ther. 195, 186–198. 10.1016/j.pharmthera.2018.11.006. [DOI] [PubMed] [Google Scholar]
- Chen AM, et al. , 2023. Replicability of proton MR spectroscopic imaging findings in mild traumatic brain injury: implications for clinical applications. Neuroimage Clin. 37, 103325 10.1016/j.nicl.2023.103325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo IH, et al. , 2010. Posterior cingulate cortex atrophy and regional cingulum disruption in mild cognitive impairment and Alzheimer’s disease. Neurobiol. Aging 31, 772–779. 10.1016/j.neurobiolaging.2008.06.015. [DOI] [PubMed] [Google Scholar]
- Conway ME, 2020. Alzheimer’s disease: targeting the glutamatergic system. Biogerontology. 21, 257–274. 10.1007/s10522-020-09860-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damulina A, et al. , 2019. White Matter Hyperintensities in Alzheimer’s Disease: a Lesion Probability Mapping Study. J. Alzheimers. Dis. 68, 789–796. 10.3233/JAD-180982. [DOI] [PubMed] [Google Scholar]
- de Leon MJ, et al. , 2018. The nonlinear relationship between cerebrospinal fluid Aβ42 and tau in preclinical Alzheimer’s disease. PLoS. One 13, e0191240. 10.1371/journal.pone.0191240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, Karran E, 2016. The Cellular Phase of Alzheimer’s Disease. Cell 164, 603–615. 10.1016/j.cell.2015.12.056. [DOI] [PubMed] [Google Scholar]
- den Heijer T, et al. , 2006. MR spectroscopy of brain white matter in the prediction of dementia. Neurology. 66, 540–544. 10.1212/01.wnl.0000198256.54809.0e. [DOI] [PubMed] [Google Scholar]
- Devi G, Scheltens P, 2018. Heterogeneity of Alzheimer’s disease: consequence for drug trials? Alzheimers. Res. Ther. 10, 122. 10.1186/s13195-018-0455-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding XQ, et al. , 2015. Reproducibility and reliability of short-TE whole-brain MR spectroscopic imaging of human brain at 3T. Magn. Reson. Med. 73, 921–928. 10.1002/mrm.25208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon RM, Bradley KM, Budge MM, Styles P, Smith AD, 2002. Longitudinal quantitative proton magnetic resonance spectroscopy of the hippocampus in Alzheimer’s disease. Brain 125, 2332–2341. [DOI] [PubMed] [Google Scholar]
- Dubois B, et al. , 2021. Clinical diagnosis of Alzheimer’s disease: recommendations of the International Working Group. Lancet Neurol. 20, 484–496. 10.1016/S1474-4422(21)00066-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebel A, Maudsley AA, Schuff N, 2007. Correction of local B0 shifts in 3D EPSI of the human brain at 4 T. Magn. Reson. ImAging 25, 377–380. 10.1016/j.mri.2006.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezzati A, et al. , 2016. Differential association of left and right hippocampal volumes with verbal episodic and spatial memory in older adults. Neuropsychologia 93, 380–385. 10.1016/j.neuropsychologia.2016.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayed N, Modrego PJ, Rojas-Salinas G, Aguilar K, 2011. Brain glutamate levels are decreased in Alzheimer’s disease:a magnetic resonance spectroscopy study. Am. J. Alzheimer’s Disease Other Dementias® 26, 450–456. 10.1177/1533317511421780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazekas F, Chawluk JB, Alavi A, Hurtig HI, Zimmerman RA, 1987. MR signal abnormalities at 1.5 T in Alzheimer’s dementia and normal aging. AJR Am. J. Roentgenol. 149, 351–356. 10.2214/ajr.149.2.351. [DOI] [PubMed] [Google Scholar]
- Feng F, et al. , 2021. Altered volume and structural connectivity of the hippocampus in Alzheimer’s disease and amnestic mild cognitive impairment. Front. Aging Neurosci. 13, 705030 10.3389/fnagi.2021.705030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl B, 2012. FreeSurfer. Neuroimage 62, 774–781. 10.1016/j.neuroimage.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foy CML, et al. , 2011. Hippocampal proton MR spectroscopy in early Alzheimer’s disease and mild cognitive impairment. Brain Topogr. 24, 316–322. 10.1007/s10548-011-0170-5. [DOI] [PubMed] [Google Scholar]
- Gajdosik M, et al. , 2021a. Hippocampal single-voxel MR spectroscopy with a long echo time at 3 T using semi-LASER sequence. NMR Biomed. 34, e4538. 10.1002/nbm.4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajdosik M, Landheer K, Swanberg KM, Juchem C, 2021b. INSPECTOR: free software for magnetic resonance spectroscopy data inspection, processing, simulation and analysis. Sci. Rep. 11, 2094. 10.1038/s41598-021-81193-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomar JJ, et al. , 2014. APOE genotype modulates proton magnetic resonance spectroscopy metabolites in the aging brain. Biol. Psychiatry 75, 686–692. 10.1016/j.biopsych.2013.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbach T, et al. , 2020. Longitudinal association between hippocampus atrophy and episodic-memory decline in non-demented APOE ε4 carriers. Alzheimers. Dement. (Amst) 12, e12110. 10.1002/dad2.12110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryawala MZ, Sheriff S, Maudsley AA, 2016. Regional distributions of brain glutamate and glutamine in normal subjects. NMR Biomed. 29, 1108–1116. 10.1002/nbm.3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryawala MZ, Sheriff S, Stoyanova R, Maudsley AA, 2018. Spectral decomposition for resolving partial volume effects in MRSI. Magn. Reson. Med. 79, 2886–2895. 10.1002/mrm.26991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff-Radford J, Kantarci K, 2016. Magnetic Resonance Spectroscopy of Degenerative Brain Diseases. Springer International Publishing, pp. 55–70. Gülin Öz. [Google Scholar]
- Hirata-Fukae C, et al. , 2009. Levels of soluble and insoluble tau reflect overall status of tau phosphorylation in vivo. Neurosci. Lett. 450, 51–55. 10.1016/j.neulet.2008.11.023. [DOI] [PubMed] [Google Scholar]
- Hoch SE, Kirov II, Tal A, 2017. When are metabolic ratios superior to absolute quantification? A statistical analysis. NMR Biomed. 30. 10.1002/nbm.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Morris JC, Goate AM, 2011. Alzheimer’s disease: the challenge of the second century. Sci. Transl. Med. 3, 77sr71. 10.1126/scitranslmed.3002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglewicz B, Hoaglin DC, 1993. Volume 16: How to Detect and Handle Outliers. Quality Press. [Google Scholar]
- Jack CR Jr., et al. , 1999. Prediction of AD with MRI-based hippocampal volume in mild cognitive impairment. Neurology. 52, 1397–1403. 10.1212/wnl.52.7.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., et al. , 2010. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 9, 119–128. 10.1016/s1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., et al. , 2016. A/T/N: an unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology. 87, 539–547. 10.1212/WNL.0000000000002923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., et al. , 2018. NIA-AA Research Framework: toward a biological definition of Alzheimer’s disease. Alzheimers. Dement. 14, 535–562. 10.1016/j.jalz.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekiyo T, Xu H, Bu G, 2014. ApoE and Aβ in Alzheimer’s disease: accidental encounters or partners? Neuron 81, 740–754. 10.1016/j.neuron.2014.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarci K, et al. , 2011. Magnetic resonance spectroscopy, β-amyloid load, and cognition in a population-based sample of cognitively normal older adults. Neurology. 77, 951–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarci K, et al. , 2012. APOE modifies the association between Abeta load and cognition in cognitively normal older adults. Neurology. 78, 232–240. 10.1212/WNL.0b013e31824365ab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kara F, et al. , 2022. 1H MR spectroscopy biomarkers of neuronal and synaptic function are associated with tau deposition in cognitively unimpaired older adults. Neurobiol. Aging 112, 16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreis R, 2004. Issues of spectral quality in clinical 1H-magnetic resonance spectroscopy and a gallery of artifacts. NMR Biomed. 17, 361–381. [DOI] [PubMed] [Google Scholar]
- Kynast J, et al. , 2018. White matter hyperintensities associated with small vessel disease impair social cognition beside attention and memory. J. Cereb. Blood Flow Metab. 38, 996–1009. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landheer K, Swanberg KM, Juchem C, 2021. Magnetic resonance Spectrum simulator (MARSS), a novel software package for fast and computationally efficient basis set simulation. NMR Biomed. 34, e4129. 10.1002/nbm.4129. [DOI] [PubMed] [Google Scholar]
- Lee HG, et al. , 2004. Aberrant expression of metabotropic glutamate receptor 2 in the vulnerable neurons of Alzheimer’s disease. Acta Neuropathol. 107, 365–371. 10.1007/s00401-004-0820-8. [DOI] [PubMed] [Google Scholar]
- Leitão MJ, et al. , 2019. Clinical validation of the Lumipulse G cerebrospinal fluid assays for routine diagnosis of Alzheimer’s disease. Alzheimers. Res. Ther. 11, 91. 10.1186/s13195-019-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li BS, Wang H, Gonen O, 2003. Metabolite ratios to assumed stable creatine level may confound the quantification of proton brain MR spectroscopy. Magn. Reson. ImAging 21, 923–928. 10.1016/s0730-725x(03)00181-4. [DOI] [PubMed] [Google Scholar]
- Li S, et al. , 2013. Decreased NAA in gray matter is correlated with decreased availability of acetate in white matter in postmortem multiple sclerosis cortex. Neurochem. Res. 38, 2385–2396. 10.1007/s11064-013-1151-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A, et al. , 2021. Minimum Reporting Standards for in vivo Magnetic Resonance Spectroscopy (MRSinMRS): experts’ consensus recommendations. NMR Biomed. 34, e4484. 10.1002/nbm.4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe VJ, et al. , 2018. Elevated medial temporal lobe and pervasive brain tau-PET signal in normal participants. Alzheimers. Dement. (Amst) 10, 210–216. 10.1016/j.dadm.2018.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, et al. , 2005. Routine clinical brain MRI sequences for use at 3.0 Tesla. J. Magn. Reson. Imaging 22, 13–22. 10.1002/jmri.20356. [DOI] [PubMed] [Google Scholar]
- Mattsson-Carlgren N, et al. , 2020. Aβ deposition is associated with increases in soluble and phosphorylated tau that precede a positive Tau PET in Alzheimer’s disease. Sci. Adv. 6, eaaz2387. 10.1126/sciadv.aaz2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maudsley AA, et al. , 2006. Comprehensive processing, display and analysis for in vivo MR spectroscopic imaging. NMR Biomed. 19, 492–503. 10.1002/nbm.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maudsley AA, et al. , 2009. Mapping of brain metabolite distributions by volumetric proton MR spectroscopic imaging (MRSI). Magn. Reson. Med. 61, 548–559. 10.1002/mrm.21875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maudsley AA, Domenig C, Sheriff S, 2010. Reproducibility of serial whole-brain MR spectroscopic imaging. NMR Biomed. 23, 251–256. 10.1002/nbm.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDougall GJ, 1990. A review of screening instruments for assessing cognition and mental status in older adults. Nurse Pract. 15, 18–28. [PMC free article] [PubMed] [Google Scholar]
- McRae-McKee K, et al. , 2019. Combining hippocampal volume metrics to better understand Alzheimer’s disease progression in at-risk individuals. Sci. Rep. 9, 7499. 10.1038/s41598-019-42632-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffett JR, Ross B, Arun P, Madhavarao CN, Namboodiri AM, 2007. N-Acetylaspartate in the CNS: from neurodiagnostics to neurobiology. Prog. Neurobiol. 81, 89–131. 10.1016/j.pneurobio.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L, Pupi A, De Leon MJ, 2008. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1147, 180–195. 10.1196/annals.1427.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motegi T, et al. , 2021. Glutamine + glutamate level predicts the magnitude of microstructural organization in the gray matter in the healthy elderly. Int. Psychogeriatr. 33, 21–29. 10.1017/S1041610219001418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, Selkoe DJ, 2012. Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold. Spring. Harb. Perspect. Med. 2, a006338 10.1101/cshperspect.a006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Near J, et al. , 2021. Preprocessing, analysis and quantification in single-voxel magnetic resonance spectroscopy: experts’ consensus recommendations. NMR Biomed. 34, e4257. 10.1002/nbm.4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedelska Z, et al. , 2017. 1H-MRS metabolites and rate of β-amyloid accumulation on serial PET in clinically normal adults. Neurology. 89, 1391–1399. 10.1212/wnl.0000000000004421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neeb H, Zilles K, Shah NJ, 2006. Fully-automated detection of cerebral water content changes: study of age- and gender-related H2O patterns with quantitative MRI. Neuroimage 29, 910–922. 10.1016/j.neuroimage.2005.08.062. [DOI] [PubMed] [Google Scholar]
- Nerland S, et al. , 2022. A comparison of intracranial volume estimation methods and their cross-sectional and longitudinal associations with age. Hum. Brain Mapp. 43, 4620–4639. 10.1002/hbm.25978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolova S, Stark SM, Stark CEL, 2017. 3T hippocampal glutamate-glutamine complex reflects verbal memory decline in aging. Neurobiol. Aging 54, 103–111. 10.1016/j.neurobiolaging.2017.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossenkoppele R, et al. , 2022. Amyloid and tau PET-positive cognitively unimpaired individuals are at high risk for future cognitive decline. Nat. Med. 28, 2381–2387. 10.1038/s41591-022-02049-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Öz G, et al. , 2021. Advanced single voxel 1H magnetic resonance spectroscopy techniques in humans: experts’ consensus recommendations. NMR Biomed. 34, e4236. 10.1002/nbm.4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennanen C, et al. , 2004. Hippocampus and entorhinal cortex in mild cognitive impairment and early AD. Neurobiol. Aging 25, 303–310. 10.1016/S0197-4580(03)00084-8. [DOI] [PubMed] [Google Scholar]
- Petrone PM, et al. , 2019. Prediction of amyloid pathology in cognitively unimpaired individuals using voxel-wise analysis of longitudinal structural brain MRI. Alzheimers. Res. Ther. 11, 72. 10.1186/s13195-019-0526-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porsteinsson AP, Isaacson RS, Knox S, Sabbagh MN, Rubino I, 2021. Diagnosis of early alzheimer’s disease: clinical practice in 2021. J. Prev. Alzheimers. Dis. 8, 371–386. 10.14283/jpad.2021.23. [DOI] [PubMed] [Google Scholar]
- Price JL, et al. , 2009. Neuropathology of nondemented aging: presumptive evidence for preclinical Alzheimer disease. Neurobiol. Aging 30, 1026–1036. 10.1016/j.neurobiolaging.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramadan S, Lin A, Stanwell P, 2013. Glutamate and glutamine: a review of in vivo MRS in the human brain. NMR Biomed. 26, 1630–1646. 10.1002/nbm.3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupsingh R, Borrie M, Smith M, Wells JL, Bartha R, 2011. Reduced hippocampal glutamate in Alzheimer disease. Neurobiol. Aging 32, 802–810. 10.1016/j.neurobiolaging.2009.05.002. [DOI] [PubMed] [Google Scholar]
- Sabati M, et al. , 2015. Multivendor implementation and comparison of volumetric whole-brain echo-planar MR spectroscopic imaging. Magn. Reson. Med. 74, 1209–1220. 10.1002/mrm.25510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki K, et al. , 2009. Excitatory amino acid transporter 2 associates with phosphorylated tau and is localized in neurofibrillary tangles of tauopathic brains. FEBS Lett. 583, 2194–2200. 10.1016/j.febslet.2009.06.015. [DOI] [PubMed] [Google Scholar]
- Schuff N, et al. , 1997. Changes of hippocampal N-acetyl aspartate and volume in Alzheimer’s disease. A proton MR spectroscopic imaging and MRI study. Neurology. 49, 1513–1521. 10.1212/wnl.49.6.1513. [DOI] [PubMed] [Google Scholar]
- Seiger R, Ganger S, Kranz GS, Hahn A, Lanzenberger R, 2018. Cortical thickness estimations of freesurfer and the CAT12 Toolbox in patients with Alzheimer’s disease and healthy controls. J. Neuroimaging 28, 515–523. 10.1111/jon.12521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, et al. , 2017. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527. 10.1038/nature24016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva MVF, et al. , 2019. Alzheimer’s disease: risk factors and potentially protective measures. J. Biomed. Sci. 26, 33. 10.1186/s12929-019-0524-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soher BJ, Young K, Govindaraju V, Maudsley AA, 1998. Automated spectral analysis III: application to in vivo proton MR spectroscopy and spectroscopic imaging. Magn. Reson. Med. 40, 822–831. [DOI] [PubMed] [Google Scholar]
- Spotorno N, et al. , 2022. Astrocytic function is associated with both amyloid-β and tau pathology in non-demented APOE ϵ4 carriers. Brain Commun. 4, fcac135. 10.1093/braincomms/fcac135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg M, et al. , 2008. Point and 5-year period prevalence of neuropsychiatric symptoms in dementia: the Cache County Study. Int. J. Geriatric Psychiatry: A J. Psychiatry Late Life Allied Sci. 23, 170–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson DT, Clemens JA, 1998. Metabotropic glutamate receptor activation in vivo induces intraneuronal amyloid immunoreactivity in guinea pig hippocampus. Neurochem. Int. 33, 83–93. 10.1016/s0197-0186(05)80012-9. [DOI] [PubMed] [Google Scholar]
- Suri S, et al. , 2017. Effect of age and the APOE gene on metabolite concentrations in the posterior cingulate cortex. Neuroimage 152, 509–516. 10.1016/j.neuroimage.2017.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanberg KM, Landheer K, Pitt D, Juchem C, 2019. Quantifying the metabolic signature of multiple sclerosis by in vivo proton magnetic resonance spectroscopy: current challenges and future outlook in the translation from proton signal to diagnostic biomarker. Front. Neurol. 10, 1173. 10.3389/fneur.2019.01173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweigart B, et al. , 2021. APOE E2/E2 is associated with slower rate of cognitive decline with age. J. Alzheimers Dis. 83, 853–860. 10.3233/jad-201205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therriault J, et al. , 2022. Biomarker modeling of Alzheimer’s disease using PET-based Braak staging. Nat. Aging 2, 526–535. 10.1038/s43587-022-00204-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson PM, et al. , 2003. Dynamics of gray matter loss in Alzheimer’s disease. J. Neurosci. 23, 994–1005. 10.1523/JNEUROSCI.23-03-00994.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumati S, Martens S, Aleman A, 2013. Magnetic resonance spectroscopy in mild cognitive impairment: systematic review and meta-analysis. Neurosci. Biobehav. Rev. 37, 2571–2586. 10.1016/j.neubiorev.2013.08.004. [DOI] [PubMed] [Google Scholar]
- Turan F, et al. , 2021. Effect of modulating glutamate signaling on myelinating oligodendrocytes and their development-A study in the zebrafish model. J. Neurosci. Res. 99, 2774–2792. 10.1002/jnr.24940. [DOI] [PubMed] [Google Scholar]
- Tustison NJ, et al. , 2014. Large-scale evaluation of ANTs and FreeSurfer cortical thickness measurements. Neuroimage 99, 166–179. 10.1016/j.neuroimage.2014.05.044. [DOI] [PubMed] [Google Scholar]
- Uysal G, Ozturk M, 2020. Hippocampal atrophy based Alzheimer’s disease diagnosis via machine learning methods. J. Neurosci. Methods 337, 108669. 10.1016/j.jneumeth.2020.108669. [DOI] [PubMed] [Google Scholar]
- Vagnozzi R, et al. , 2010. Assessment of metabolic brain damage and recovery following mild traumatic brain injury: a multicentre, proton magnetic resonance spectroscopic study in concussed patients. Brain 133, 3232–3242. 10.1093/brain/awq200. [DOI] [PubMed] [Google Scholar]
- Veitch DP, et al. , 2019. Understanding disease progression and improving Alzheimer’s disease clinical trials: recent highlights from the Alzheimer’s Disease Neuroimaging Initiative. Alzheimers. Dement. 15, 106–152. 10.1016/j.jalz.2018.08.005. [DOI] [PubMed] [Google Scholar]
- Vernooij MW, Smits M, 2012. Structural neuroimaging in aging and Alzheimer’s disease. NeuroimAging Clin. N. Am. 22, 33–55. 10.1016/j.nic.2011.11.007 vii-viii. [DOI] [PubMed] [Google Scholar]
- Voevodskaya O, et al. , 2014. The effects of intracranial volume adjustment approaches on multiple regional MRI volumes in healthy aging and Alzheimer’s disease. Front. Aging Neurosci. 6, 264. 10.3389/fnagi.2014.00264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voevodskaya O, et al. , 2016. Myo-inositol changes precede amyloid pathology and relate to APOE genotype in Alzheimer disease. Neurology. 86, 1754–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voevodskaya O, et al. , 2019. Brain myoinositol as a potential marker of amyloid-related pathology: a longitudinal study. Neurology. 92, e395–e405. 10.1212/WNL.0000000000006852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Reddy PH, 2017. Role of glutamate and NMDA receptors in Alzheimer’s disease. J. Alzheimers. Dis. 57, 1041–1048. 10.3233/jad-160763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Zhao F, Ma X, Perry G, Zhu X, 2020. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Mol. Neurodegener. 15, 30. 10.1186/s13024-020-00376-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wansapura JP, Holland SK, Dunn RS, Ball WS Jr., 1999. NMR relaxation times in the human brain at 3.0 tesla. J. Magn. Reson. Imaging 9, 531–538. . [DOI] [PubMed] [Google Scholar]
- Watanabe H, Takaya N, 2018. Quantitation error in (1)H MRS caused by B(1) inhomogeneity and chemical shift displacement. Magn. Reson. Med. Sci. 17, 244–250. 10.2463/mrms.mp.2017-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westman E, Aguilar C, Muehlboeck JS, Simmons A, 2013. Regional magnetic resonance imaging measures for multivariate analysis in Alzheimer’s disease and mild cognitive impairment. Brain Topogr. 26, 9–23. 10.1007/s10548-012-0246-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierenga CE, Hays CC, Zlatar ZZ, 2014. Cerebral blood flow measured by arterial spin labeling MRI as a preclinical marker of Alzheimer’s disease. J. Alzheimers. Dis. 42 (Suppl 4), S411–S419. 10.3233/JAD-141467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson M, et al. , 2019. Methodological consensus on clinical proton MRS of the brain: review and recommendations. Magn. Reson. Med. 82, 527–550. 10.1002/mrm.27742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki Y, Zhao N, Caulfield TR, Liu C-C, Bu G, 2019. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nature Rev. Neurol. 15, 501–518. 10.1038/s41582-019-0228-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z, et al. , 2015. APOE genotype and age modifies the correlation between cognitive status and metabolites from hippocampus by a 2D (1)H-MRS in non-demented elders. PeerJ. 3, e1202. 10.7717/peerj.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young CB, et al. , 2023. APOE effects on regional tau in preclinical Alzheimer’s disease. Mol. Neurodegener. 18, 1. 10.1186/s13024-022-00590-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeydan B, Kantarci K, 2021. Decreased glutamine and glutamate: an early biomarker of neurodegeneration. Int. Psychogeriatr. 33, 1–2. 10.1017/S1041610219001807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeydan B, et al. , 2017. Decreased glutamate levels in patients with amnestic mild cognitive impairment: an sLASER proton mr spectroscopy and pib-pet study. J. Neuroimaging 27, 630–636. 10.1111/jon.12454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, et al. , 2018a. Comparison of reproducibility of single voxel spectroscopy and whole-brain magnetic resonance spectroscopy imaging at 3T. NMR Biomed. 31, e3898. 10.1002/nbm.3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Reinikainen J, Adeleke KA, Pieterse ME, Groothuis-Oudshoorn CGM, 2018b. Time-varying covariates and coefficients in Cox regression models. Ann. Transl. Med. 6, 121. 10.21037/atm.2018.02.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.