

Abstract
Acute myeloid leukemia (AML) is an aggressive hematological malignancy with a heterogeneous molecular landscape. In the pediatric context, the NUP98 gene is a frequent target of chromosomal rearrangements that are linked to poor prognosis and unfavorable treatment outcomes in different AML subtypes. The translocations fuse NUP98 to a diverse array of partner genes, resulting in fusion proteins with novel functions. NUP98 fusion oncoproteins induce aberrant biomolecular condensation, abnormal gene expression programs, and re‐wired protein interactions which ultimately cause alterations in the cell cycle and changes in cellular structures, all of which contribute to leukemia development. The extent of these effects is steered by the functional domains of the fusion partners and the influence of concomitant somatic mutations. In this review, we discuss the complex characteristics of NUP98 fusion proteins and potential novel therapeutic approaches for NUP98 fusion‐driven AML.
INTRODUCTION
Leukemia is the most common childhood cancer, with acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML) accounting for 80% and 15%–20% of all cases, respectively. Overall, pediatric AML has a long‐term survival rate of 60%–80%; however, for cytogenetic high‐risk subgroups survival rates drop to 30% and less. 1 , 2 , 3 , 4 , 5 AML is a genetically heterogeneous disorder, that is characterized by uncontrolled clonal growth of immature hematopoietic cells with impaired differentiation. Genetic alterations can lead to the formation of chimeric proteins that play a critical role in the pathophysiology of leukemia. The genes involved in these rearrangements can encode transcription factors, epigenetic writers that possess histone posttranslational modification (PTM) activity, or epigenetic readers, which may recruit effector proteins to aberrant genomic sites. Histone PTMs are important determinants of gene regulation, and many oncogenic fusion proteins in AML can write, erase, or read activating or repressive histone marks, thus utilizing this mechanism to shape their own distinct transcriptional programs and oncogenic changes. Some chimeric fusion proteins can also interact with and recruit other regulatory proteins to alter the expression of hematopoiesis‐related genes. 6 , 7
Recurrent chromosomal rearrangements involving the Nucleoporin 98 (NUP98) gene are observed in 5%−10% of pediatric AML cases and in approximately 2%–4% of adult AML cases, categorizing it as a high‐risk subtype in both childhood and adult leukemias. 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 Children and young adults with this rearrangement show a complete remission rate of 50% after one course of induction therapy, with overall poor survival rates of 25%–35%, and they face a substantially high risk of disease relapse, which ranges from 64% to 68%. 11 , 18 NUP98::NSD1 and NUP98::KDM5A represent the most prevalent fusion events. 18 , 19 Specifically, NUP98::NSD1 is detected in 8% in children and young adults with AML cases, marking a substantial occurrence within this group. Although the NUP98::KDMA5 fusion is less frequent, occurring in only 1.4% of pediatric AML cases, its relevance increases in specific subtypes, such as acute megakaryoblastic leukemia (AMKL), where it is present in 10% of patients. 11 , 18 , 19 NUP98 fusions are predominantly linked to myeloid malignancies such as AML, chronic myeloid leukemia in blast crisis and mixed phenotype acute leukemia. 18 , 20 , 21 , 22 , 23 , 24 , 25 Although rare in B‐cell malignancies, about 12% of patients with NUP98 fusions are associated with T‐cell acute lymphoblastic leukemia (T‐ALL). 22 Since the presence of NUP98 fusions in leukemia patients is associated with high induction failure and a low survival rate, understanding the molecular landscape of this leukemia subtype is critical for improving therapeutic options for this group of AMLs.
Herein we review the physiological and pathological molecular mechanisms of wild‐type and fusion forms of the NUP98 protein. Furthermore, we discuss the genomic landscape of NUP98 fusion‐driven leukemia and highlight prospective treatment options for this AML subtype.
ROLES OF WILD‐TYPE NUP98 IN NORMAL CELL PHYSIOLOGY
The NUP98 protein is a component of the nuclear pore complex (NPC), which is composed of over 30 different proteins. NPCs are transport channels that mediate the bidirectional transport of molecules (ions, polypeptides, mRNA, and proteins) between the nucleus and the cytoplasm by diffusion and active transport. NUP98 and NUP98‐NUP96 are two mRNA splice variants that are encoded by the NUP98 gene. The NUP98‐NUP96 polypeptide undergoes cleavage, leading to the generation of a 90 kDa N‐terminal peptide and a 96 kDa C‐terminal peptide. Likewise, the precursor NUP98 polypeptide undergoes autoproteolytic cleavage, resulting in the generation of a 90 kDa N‐terminal peptide and an 8 kDa C‐terminal peptide (Figure 1). 21 , 26 , 27
Figure 1.
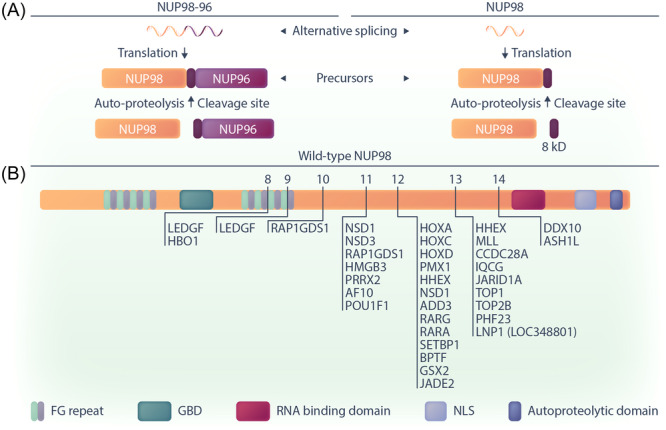
Schematic representation of NUP98 expression and structure in cells. (A) The expression involves alternative splicing and autoproteolytic cleavage, leading to the production of mature NUP98 and NUP96 proteins, as well as an 8 kD fragment. (B) The structure of the wild‐type NUP98 protein and position of NUP98 fusion breakpoints in leukemia. Arrows indicate exon numbers. GBD, Gle2‐binding domain; NLS, nuclear localization signal.
The presence of tandem phenylalanine‐glycine (FG) repeat domains is a hallmark of many NPC proteins. 28 These intrinsically disordered regions (IDRs) form a mesh‐like structure and act as a selective barrier in the central channel of the NPC, preventing free exchange of molecules larger than 5 nm. 29 In addition, by interacting with soluble nuclear transport receptors (NTRs) that bind FG repeats and cargo, these IDRs facilitate selective transport of molecules through the NPC. The N‐terminus of the NUP98 protein contains two FG/GLFG (Gly‐Leu‐Phe‐Gly) repeat regions with 38 repeats in total that are bisected by a Gle2‐binding sequence (GLEBS) domain. 30 The FG repeats of NUP98 are essential for the maintenance of the NPC's entropic barrier and nucleocytoplasmic trafficking, primarily by interacting with NTRs such as CRM1 (chromosomal maintenance 1; also called Exportin 1 or Xpo1). 31 , 32 , 33 , 34 , 35
While the NUP98 protein primarily resides in and interacts with components of the NPC including scaffold NUPs, fluorescence recovery after photobleaching (FRAP) experiments have shown that it is highly mobile and can also be detected in the nucleoplasm. 36 The mobility of NUP98 mainly depends on its association with RNA polymerase II (RNAP II). 13 , 14 , 15 The nucleoplasmic fraction of NUP98 is situated in self‐aggregated intranuclear clusters known as “GLFG” bodies and participates in gene expression or cell cycle regulation. 37 , 38 , 39 , 40 , 41 The formation of GLFG bodies is linked to the involvement of FG repeats in the assembly of phase‐separated biomolecular condensates (explained further below), in which NUP98 proteins define their own protein interactome and engage in the regulation of gene expression regulation. 28 , 42 , 43 , 44 The involvement of NUP98 in transcriptional regulation occurs through its co‐localization with RNAP II, where NUP98 acts as a co‐transcriptional activator or repressor. This dual functionality arises from the association of the NUP98 FG repeats with various chromatin regulatory proteins. 23 , 30 , 45 , 46 , 47 , 48 The GLEBS domain of NUP98 is involved in RNA binding and transport through the nuclear envelope as well as cell cycle regulation and mitotic spindle formation mediated by the RNA export factor RAE1 (Gle2). 41 , 49 The C‐terminus of wild‐type NUP98 contains an RNA binding domain and a unique autoproteolytic cleavage site that is required for the production of the mature protein and is crucial for proper localization of the NUP98 protein in the NPC. 21 , 49 Using a C‐terminally truncated NUP98 variant that cannot bind to the NPC and is present in the nucleoplasm, Franks et al. showed that the Wdr82–Set1A/COMPASS (complex of proteins associated with Set1; WSC) is a binding partner for the NUP98 protein. 48 WSC is a protein complex with roles in epigenetic regulation that promotes H3K4 trimethylation of specific genes. 50 WSC is thought to be recruited by NUP98 to developmentally regulated genes to deposit H3K4me3 marks and induce gene expression. 48 , 51 In summary, the intricate balance maintained by NUP98 in cellular processes emphasizes its importance, and any shifts in this balance can have profound effects on normal cell physiology.
NUP98 FUSIONS ENCOMPASS MULTIPLE FUSION PARTNERS IN HEMATOLOGICAL MALIGNANCIES
The NUP98 gene can be involved in genomic rearrangements with various partner loci. 18 These fusions result from the junction of the 5′‐end of NUP98 on chromosome 11p15 to the 3′‐end of the fusion gene partners. 18 NUP98::HOXA9 was the first described NUP98 fusion in hematological malignancies. 52 , 53 NUP98::NSD1 is the most common NUP98 fusion protein, found in approximately 8% of pediatric AML patients. 54 Along with NUP98::HOXA9 and NUP98::KDM5A (JARID1A) it is the most intensively studied NUP98 fusion protein. NUP98‐rearranged (NUP98‐r) AML is associated with poor prognosis, treatment failure, and high relapse rates. 8 , 9 , 14 , 18 The NUP98 translocations can occur at any age but are more commonly observed in children, adolescents and young adults with a higher incidence in males with about 60% of cases. 8 , 18 Morphologically, NUP98‐r AMLs are linked to the M2/M4 FAB subtypes. 18 , 55 However, some of the NUP98 fusions have a distinct phenotype and propensity to manifest in specific disease subsets. For example, AMLs harboring the NUP98::KDM5A fusion typically fall into the M6/7 category with no expression of the pluripotent markers CD34 and CD123. 18 This particular fusion is linked to approximately 20% of childhood and adult cases with acute erythroid leukemia (AEL), 56 and is also present in 10% of infant AMKL. 20 , 57 The NUP98::NSD1 fusion is more common in childhood AML, accounting for 16% of normal karyotype pediatric leukemia 14 and is predominantly found in M4/M5 AML subtypes. 18 , 55 When NUP98 fusions occur alongside the FLT3‐ITD mutation, which happens in very high frequency in NUP98::NSD1, aberrant blasts often show signs of monocytic maturation, as evidenced by the expression of CD11b, CD36, and CD64. 18 Certain NUP98 fusions, such as those with JADE2, RARA, or RARG fusion partners, are associated with an acute promyelocytic leukemia phenotype (M3). 55 , 58 , 59 Other NUP98 fusions, while lacking a consistent immunophenotype, predominantly express markers characteristic of early progenitors. 18 Notably, quantification of mRNA transcripts of the NUP98 fusions can provide a targeted method for detecting molecular residual disease (MRD). 60 , 61
So far, more than 30 distinct fusion partners of NUP98 have been reported (Figure 2 and Table 1). Considering the sub‐telomeric location of the NUP98 breakpoint, traditional karyotyping may miss some of the NUP98 fusions such as NUP98::NSD1, leading to their misclassification as AMLs with a cytogenetically normal karyotype. 14 , 21 Due to the rapid improvement of sequencing technologies more NUP98 fusions have recently been revealed in monocytic, megakaryoblastic, and erythroid AML (French‐American‐British FAB M4, M5, M6, M7). 55
Figure 2.
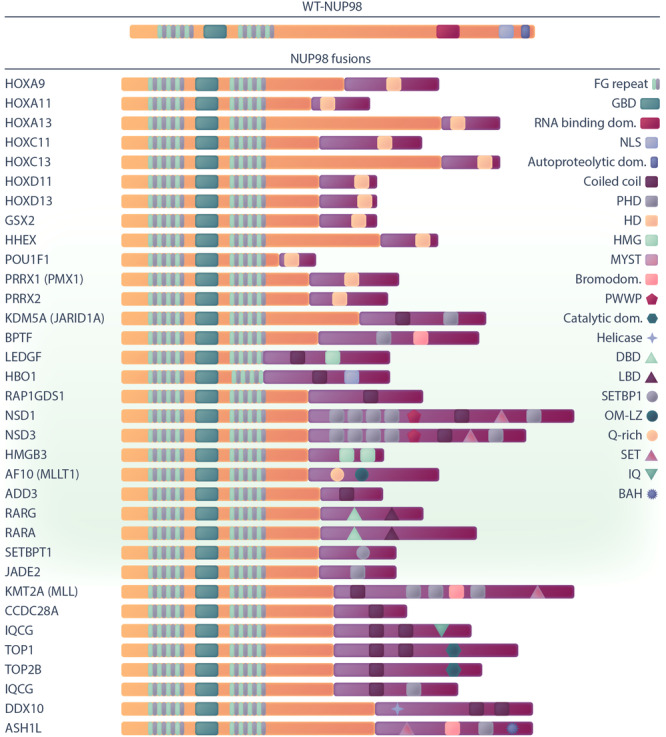
Graphical representation of wild type NUP98 and its fusion partner structures, which involve functional domains for each fusion partner. BAH, bromo‐adjacent homology; DBD, DNA binding domain; GBD, Gle2‐binding domain; HMG, high mobility group; LBD, ligand‐binding domain; NLS, nuclear localization signal; OM‐LZ, octapeptide motif‐leucine‐zipper; PWWP, Pro‐Trp‐Trp‐Pro; Q‐rich, glutamine‐rich; RNB, RNA‐binding domain. Functional domains for VRK1, LOC348801 (LNP1), FN1, and ANKRD28 have not been identified so far.
Table 1.
NUP98 fusions in leukemia.
| Fusion partner | Chromosome | Domain | Disease | Outcome | References |
|---|---|---|---|---|---|
| HOXA9 | 7p15 | HD | AML, CML, MDS, CMML |
Median OS: 13.5 months Median RFS: 6 months N fusion: 11 N total = 493 |
[53, 62, 63, 64, 65, 66] |
| HOXA11 | 7p15 | HD | CML, JMML | [67, 68] | |
| HOXA13 | 7p15 | HD | AML, CML, MDS | [62, 67, 69] | |
| HOXC11 | 12q13 | HD | AML | [70] | |
| HOXC13 | 12q13 | HD | AML | [70, 71] | |
| HOXD11 | 2q31 | HD | AML | [72] | |
| HOXD13 | 2q31 | HD | AML, CML | [62, 73, 74] | |
| GSX2 (Gsh2) | 4q12 | HD | AML | [75] | |
| PRRX1 (PMX1) | 1q23 | HD | AML, CML, MDS | [23, 76, 77] | |
| PRRX2 | 9q34 | HD | AML | [78, 79] | |
| POU1F1 | 3p11 | HD | AML | [80] | |
| JADE2 | 5q31 | PHD | APL‐AML, MDS‐MPN, JMML | [81, 82] | |
| PHF23 | 17p13 | Coiled domain‐PHD | AML | [83] | |
| HHEX | 10q23 | HD | AML | [84] | |
| NSD1 | 5q35 | SET‐PHD‐PWWP‐Coiled domain | MDS/MPN, AML, MPAL |
CR: 38% OS: 36% EFS: 17% RR: 64% N fusion: 108 N total = 2235 |
[14, 18, 85, 86, 87, 88] |
| NSD3 | 8p11 | SET‐PHD‐PWWP‐Coiled domain | MDS, AML | [24, 89] | |
| MLL (KMT2A) | 11q23 | SET‐PHD‐Ciled domain‐SET binding domain‐Bromodoamin | AML‐MDS | [90] | |
| ASH1L | SET, Bromo, PHD, BHD | AML, MDS | [91] | ||
| SETBP1 | 18q12 | SET binding domain | T‐ALL | [92] | |
| 17q23 | PHD‐Bromodomain‐CC | AMKL‐ T‐ALL | [93, 94] | ||
| KDM5A (JARID1A) | 12p13 | PHD‐Jumonji (Demethylase) | AMKL |
CR: 81% OS: 30% EFS: 25% RR: 68% N fusion = 32 N total = 2235 |
[18, 95] |
| MLLT10 (AF10) | 10p12 | OM‐LZ, Q‐rich | MDS | [75] | |
| ANKRD28* | 3p25 | Not identified | AML‐MDS | [96] | |
| DDX10 | 11q22 | Helicase domains‐CC | MDS, AML, CML | [97, 98] | |
| HMGB3 | Xq28 | HMG box‐CC | AML | [99] | |
| KAT7 (HBO1) | 17q21 | MYST‐CC | CMML | [62, 100] | |
| PSIP1 (LEDGF) | 9p22 | HMG box‐CC | MDS, AML, CML | [21, 101, 102, 103, 104] | |
| RAP1GDS1 | 4q21 | CC | T‐ALL, AML | [105, 106] | |
| RARA | 17q21 | DBD‐LBD | APL | [59] | |
| RARG | 12q13 | DBD‐LBD | AML | [58] | |
| TOP1 | 20q11 | Topoisomerase‐Coiled domain | AML, MDS | [107, 108] | |
| TOP2B | 3p24 | Topoisomerase‐ Coiled domain | AML | [109] | |
| VRK1 | 14q32 | Not identified | T‐ALL | [110] | |
| CCDC28A (C6orf80) | 6q24 | Coiled domain | T‐ALL, AML | [111] | |
| IQCG | 3q29 | Coiled domain, IQ | MPAL‐ T‐ALL | [25, 112] | |
| LOC348801 (LNP1) | 3q12 | Not identified | AML | [113] | |
| ADD3 | 10q25 | Coiled domain | T‐ALL, AML | [114] | |
| FN1 | 2q31 | Not identified | AML | [115] |
Abbreviations: AML, acute myeloid leukaemia; CR, complete remission; MDS, myelodysplastic syndrome; N, number; OS, overall survival; RFS, relapse‐free survival; RR, relapse risk.
The fusion of NUP98 with other proteins often leads to a gain of function and the resulting fusion oncoproteins inherits parts of the properties and functionalities of both NUP98 and its partner. Although functional domains have not been identified in all fusion partners, many of them encode homeodomain DNA‐binding motifs (HD), including clustered (HOXA9/11/13, HOXC11/13, HOXD11/13) and nonclustered (HHX, GSX2, PRRX1, PRRX2, POU1F) HOX genes. Additionally, there are various non‐HOX fusion protein partners for NUP98, which possess DNA‐ or chromatin‐binding domains or domains with other functions (detailed in Table 1). Notably, certain fusion partners such as NSD1, NSD3, and KMT2A (MLL) harbor multiple functional domains. These encompass both the chromatin‐binding plant homeodomain (PHD), enabling H3K4me3 binding, and the catalytic lysine methyltransferase SET (Su(var)3‐9, Enhancer‐of‐zeste, and Trithorax) domain, mediating histone methylation. Additionally, NUP98 fusions frequently co‐occur with a set of other recurrent somatic mutations, the most prevalent being FLT3‐ITD and WT1. For example, 74% of patients with NUP98::NSD1 harbor the FLT3‐ITD mutation, and 42% possess the WT1 mutation. 18 These additional mutations can confer proliferative advantages to the leukemic cells and contribute to leukemogenesis (Table 2).
Table 2.
Common co‐occurrent mutations with NUP98 fusions in AML.
| Co‐occurrent mutations | Fusion partner | References |
|---|---|---|
| FLT3‐ITD | NSD1, KDM5A, LNP1, HOXC11, HOXA9, HOXA13, HOXD13, NSD3 | [14, 18, 116, 117, 118] |
| WT1 | NSD1, KDM5A, TOP1, HOXC11, HOXA9, HOXD11, NSD3, HOXA13, HOXA11 | [14, 18, 116, 118] |
| NRAS | NSD1, HOXA9, HOXA11, DDX10 | [14, 56, 118] |
| KRAS | HOXA9, HOXD11 | [11, 118] |
| RB1 | KDM5A | [62, 119] |
| NOTCH1 | RAP1GDS1 | [117] |
| MYC | NSD1 | [62, 120, 121] |
| KIT | HOXC11, HOXA9, NSD3, NSD1 | [118] |
| RUNX1 | NSD1, RARG | [122, 123] |
| ASXL1 | NSD1 | [124] |
| MYB | RAP1GDS1 | [117] |
NUP98 FUSION PROTEINS FORM ABERRANT BIOMOLECULAR CONDENSATES
Despite intensive research, the molecular mechanisms underlying NUP98 fusion‐induced oncogenesis have remained incompletely understood. Recent results indicate that NUP98 fusion proteins show a different subcellular distribution than wild‐type NUP98 and form aberrant biomolecular condensates that affect chromatin architecture and gene expression, ultimately leading to AML development. 125 , 126 Various studies have demonstrated that NUP98 fusion proteins localize to nuclear puncta and promote the formation of aberrant biomolecular condensates. 125 , 127 , 128 , 129 This altered cellular distribution has been attributed to the lack of a C‐terminally located NPC‐targeting signal in the fusion protein (Figure 3). 30 Fluorescence recovery after photobleaching (FRAP) experiments performed with NUP98::HOXA9 and NUP98::NSD1 demonstrated the liquid‐like nature of the phase‐separated droplets formed by these two fusion proteins. 128 , 129
Figure 3.
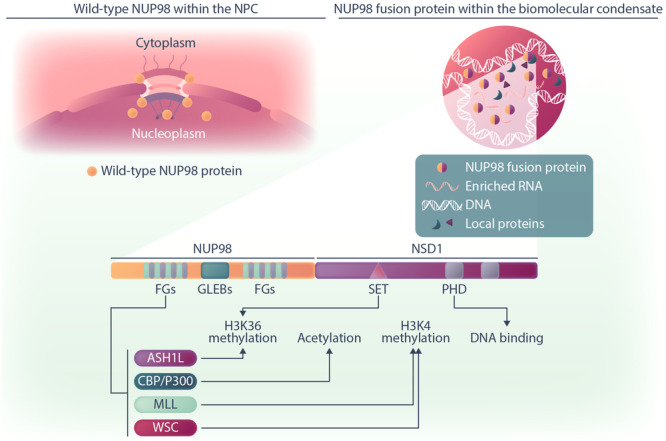
Schematic illustration of the NUP98 protein in healthy cells and interactions of NUP98 fusion proteins in AML cells. The mature wild‐type NUP98 protein is predominantly located in both the nucleus and cytoplasmic regions of the NPC, as well as in the nucleoplasm. The NUP98 fusion proteins are predominantly observed within nuclear punctate structures, known as biomolecular condensates, distributed throughout the nucleoplasm. Within these condensates, NUP98 fusions form their distinct protein interactome, where the N‐terminus FG repeats engage with various regulatory proteins. Specifically, the FG repeats interact with MLL, WSC, CBP/P300, and ASH1L protein complexes, all of which play roles in epigenetic regulation. Simultaneously, functional domains such as PHD and SET in fusion partners like NSD1 collaborate with this regulatory function or engage with chromatin. Figure Made in BioRender.com.
Given the important roles of NUP98 fusion proteins in the context of biomolecular condensates, the investigation of the fusion protein interactome has provided exciting new insights into the molecular mechanisms of leukemogenesis driven by NUP98 fusion proteins. Affinity purification coupled to mass spectrometry (AP‐MS) experiments revealed a minimal overlap between the interactors of NUP98 fusions and those of the wild‐type NUP98 protein. 125 While NUP98 was interacting with components of the NPC and proteins involved in nucleocytoplasmic transport, NUP98 fusion proteins were predominately interacting with RNA‐binding proteins and RNA helicases. In line with this, the core interactome of distinct NUP98 fusion proteins featured factors with roles in RNA splicing, ribosome biogenesis and transcriptional control. In addition, the core interactome of NUP98 fusion proteins was highly enriched for biomolecular condensation‐related proteins, including FUS, HNRNPA1 and GAR1. 125 Terlecki‐Zaniewicz et al. showed that expression of NUP98 fusion proteins significantly changed the composition of biomolecular condensates. Proteins that were recruited into phase‐separated structures in a NUP98 fusion‐dependent fashion were enriched for complexes involved in transcriptional activation and chromatin organization. 125
Notably, the nature of NUP98 fusion protein‐containing biomolecular condensates and their core interactome are dependent on the FG repeats in the N‐terminal part of NUP98. Deletion of FG repeats in NUP98::HOXA9 abrogated puncta formation and the ability of leukemic transformation of mouse hematopoietic stem and progenitor cells (HSPC) in vitro and in vivo, emphasizing the essentiality of the FG repeat regions for oncogenesis. 127 , 128 Remarkably, replacing the NUP98 N‐terminus by an artificial stretch of 39 FG repeats fused to the C‐terminus of KDM5A (artFG‐KDM5A) preserved its localization to nuclear puncta and maintained its oncogenic function by inducing leukemia‐associated gene expression programs similar to the original NUP98::KDM5A fusion protein. 125
Within the setting of biomolecular condensates induced by NUP98 fusions, it has been shown that mutation of the FG motifs in the NUP98 N‐terminus led to the loss of the interaction between NUP98::NSD1 and the SMARCA5 protein, a core component of the Nucleosome Remodeling Factor (NURF) complex. 129 Furthermore, the interaction of SMARCA5 with NUP98::NSD1 was shown to be crucial for NUP98::NSD1‐driven leukemic transformation. These data highlight the importance of IDR‐mediated phase separation for the maintenance of leukemogenic transcriptional programs by NUP98 fusion proteins.
The fusion partners of NUP98 also affect condensate formation and composition. Mutation of the DNA‐binding homeodomain of HOXA9 in the context of the NUP98::HOXA9 fusion oncoprotein resulted in fewer but larger condensates that showed less overlap with DNA. This indicates that not only homotypic interactions between the intrinsically disordered regions (IDRs) of the NUP98 N‐terminus are important for proper formation of oncogenic condensates, but also heterotypic interactions between the HOXA9 DNA‐binding domain and chromatin. 127 , 128 However, the PHD chromatin‐binding domains of NUP98::NSD1 might play a less important role, as their deletion did not affect puncta formation or global gene expression patterns. 129
The formation of aberrant condensates has functional consequences for chromatin organization. Ahn and colleagues investigated the effect of NUP98‐IDR‐mediated phase separation on NUP98::HOXA9 chromatin occupancy and global chromatin architecture. 127 Chromatin immunoprecipitation followed by high‐throughput sequencing (ChIP‐seq) showed that mutations in the NUP98 IDR impaired chromatin binding of NUP98::HOXA9. While the non‐mutated NUP98::HOXA9 fusion protein bound to genes associated with developmental processes and leukemia, inducing HOX genes, PBX3 and MEIS1 in the context of regions decorated with the activating H3K27ac histone mark, this pattern was lost when the NUP98 N‐terminal IDR was mutated. Replacing the NUP98 N‐terminus with the phase‐separation‐prone IDR of the FUS protein induced chromatin binding patterns similar to those of NUP98::HOXA9. Furthermore, analysis of changes in the three‐dimensional chromatin structure revealed 232 specific chromatin loops that were dependent on the intact FG repeat regions of the NUP98::HOXA9 fusion protein. Interestingly, the respective loop anchors overlapped with NUP98::HOXA9 binding sites but not with CTCF binding sites. These observations support a phase separation‐driven mechanism of NUP98‐fusion chromatin binding and chromatin looping that is independent of CTCF. 127 Altogether, these studies suggest that the conserved N‐terminus of NUP98 fusion oncoproteins plays a pivotal role in the initiation and maintenance of leukemogenesis via the formation of phase separation‐mediated biomolecular condensates. However, as several studies used ectopic expression of fusion genes at potentially nonphysiological levels and/or nonhematopoietic cell models, further validation of this concept is warranted.
Several studies have shown that interactions between NUP98 fusion proteins and other chromatin binders result in the recruitment of NUP98 fusion proteins to defined genomic regions such as the HOXA locus. Although not explored in the context of biomolecular condensates, these interactions are highly likely to occur within condensed cellular structures. For instance, the FG repeats of the NUP98::HOXA9 fusion protein interact with chromatin pre‐bound CRM1, resulting in selective recruitment of the fusion protein to the HOXA cluster region and subsequent activation of gene expression. This induction of HOX gene expression has been attributed to the ability of the NUP98::HOXA9‐CRM1 complex to change chromatin structure. 130 Moreover, the FG repeats of multiple NUP98 fusion proteins can interact with the KMT2A protein complex, and this interaction is essential for the oncogenic activity of the fusion proteins (Figure 3). 30 , 46 , 131 The KMT2A protein complex recruits NUP98 fusion proteins to its target loci and induces trimethylation of H3K4 through the C‐terminal SET domain of KMT2A, thereby converting promoters into an active state. 30 These findings are consistent with previous work showing the association of NUP98‐FG repeats with the CBP/p300 protein complex on active genes. 47 In line with distinct intracellular distribution, wild‐type NUP98 cannot interact with KMT2A. Given that both KMT2A and NUP98 fusion oncoproteins induce similar gene signatures in AML and that the KMT2A protein plays a role in recruiting NUP98 fusion proteins to particular gene loci, more investigation is required to determine whether and to what extent the identity of NUP98 fusion protein target genes is defined by the KMT2A protein complex or NUP98 fusions. In summary, NUP98 fusion proteins establish a complex network of protein interactions, likely within biomolecular condensates, which contributes to the specific transcriptional output that drives leukemic transformation.
SHAPING TRANSCRIPTIONAL PROGRAMS BY NUP98 FUSION PROTEINS
Phase separation can regulate transcription by concentrating IDR‐containing transcription factors and coactivators and inducing proximity between super‐enhancers and promoters. 132 , 133 These and similar observations contributed to the emergence of models that link phase separation to transcriptional factories. This implies that transcription occurs in non‐random locations throughout the nucleus and might be controlled by the local concentration of the transcriptional machinery in subnuclear compartments. 133 , 134 Numerous studies have consistently associated NUP98‐r AML with the overexpression of a specific subset of pivotal genes with important functions in leukemia. This set includes HOXA/B, MEIS1, MEF2C, PBX3, CDK6, FLT3 and IGF2BP2, all of which play critical roles in the regulation of proliferation and differentiation processes in hematopoietic cells. 18 , 54 , 116 , 135 , 136 , 137 There is also a notable overlap in the transcriptional program of NUP98‐r AMLs with those found in NPM1‐mut, KMT2A‐r, and UBTF‐TD AMLs. This overlap contributes to the co‐clustering of NUP98‐r AMLs with these specific AML subtypes, suggesting shared pathways and disease mechanisms. 116 , 136 , 138 , 139 , 140 In line with the model of condensation‐dependent transcriptional regulation, mutation of the N‐terminal IDR in NUP98 fusion oncoproteins has significant effects on global gene expression. While the expression of NUP98::HOXA9 caused differential expression of almost 900 genes, including HOXA genes, PBX1 and MEIS1, expression of the IDR‐mutated NUP98::HOXA9 only induced the deregulation of 61 genes. 127
Moreover, based on their global gene expression, AMLs with NUP98 fusions cluster in two groups, comprising either AMKLs or myelo‐monocytic AML types. 140 As described before, interaction with proteins such as CRM1 and KMT2A direct NUP98 fusion proteins to their target gene loci, including the HOXA gene cluster, where their association with chromatin is enhanced through the functional domains in the C‐terminus of the fusion protein. 30 , 141 Many NUP98 fusion partners possess DNA or chromatin binding domains such as HD (homeodomain), PHD (plant homeodomain), or PWWP (Pro‐Trp‐Trp‐Pro). For example, NUP98 fusions such as NUP98::NSD1, NUP98::KDM5A and NUP98::PHF23 have a PHD domain at the C‐terminus which can recognize and bind to H3K4me2/3, a mark of open active chromatin. 21 , 142 In this context, deletion or mutation of the PHD domain inhibits H3K4me binding activity and reduces the expression of critical transcriptional factor genes such as HOX genes, GATA3, MEIS1, EYA1, and PBX1. This, in turn, leads to the inhibition of leukemogenesis in an AML mouse model. 142 The H3K4me3 binding ability of PHD domains may serve as a seed for recruiting other proteins with roles in epigenetic regulation to induce spreading of the H3K4me3 mark on chromatin. Simultaneously, regulatory domains within the fusion protein partners can also actively participate in gene activation. For example, the catalytic SET domain of the NSD1 protein in the NUP98::NSD1 fusion is involved in the mono‐ and dimethylation of H3K36 in intergenic regions, a feature of actively transcribed genes such as the HOXA gene cluster. 143 , 144 Similarly, the DNA binding activity of the HD fusion partners of NUP98 such as HOXA9 has been highlighted as a crucial factor influencing aberrant gene expression. 145 , 146
The transcriptional programs downstream of NUP98 fusion proteins are induced and/or re‐enforced by transcription factors with important functions in hematopoiesis and leukemia. Recent studies in this context underscore the crucial role of MEIS1 in NUP98‐r AML. MEIS1 is an early target gene of NUP98 fusion expression. 116 , 136 Together with HOXA9 and PBX3, MEIS1 forms a trimeric protein complex that is highly expressed in immature hematopoietic stem cells. Overexpression of MEIS1/PBX3 or MEIS1/HOXA9 is sufficient for leukemic transformation of mouse hematopoietic stem cells and is linked to the development of various AMLs with poor prognosis. 139 , 147 , 148 , 149 , 150 An important function of MEIS1 is to regulate expression of FLT3, another gene that is highly expressed in NUP98‐r AMLs. 137 , 151 Signaling via the FLT3 receptor is a crucial molecular pathway that actively supports survival and stimulates cell proliferation of hematopoietic stem and progenitor cells. Previous findings indicate that AML patients with FLT3‐ITD had significantly different outcomes based on co‐occurring mutations. 152 Individuals with FLT3‐ITD and favorable risk mutations in NPM1, CEBPA, t(8;21), or inv(16) had a superior 5‐year Event‐Free Survival (EFS) of 64% compared to those of 22% for patients harboring poor‐risk mutations such as WT1, UBTF, or NUP98::NSD1. 152 The prevalence of the FLT3‐ITD mutation in NUP98‐r AML is associated with poor prognosis and induction failure. 54 , 137 This observation hints at a potential collaboration and interplay between MEIS1 and FLT3 in the context of NUP98 fusion‐driven leukemia.
In summary, differences in the transcriptional programs in cells harboring NUP98 fusions versus cells expressing wild‐type NUP98 can be attributed to aberrant distribution of the fusion oncoprotein within the nucleus and the presence of a chromatin binding and/or ‐regulatory moiety such as PHD, HD, and SET domains in the C‐terminus of the fusion partners. These domains mediate a strong and stable association of the NUP98 fusion protein with chromatin, which is absent in the wild‐type NUP98 protein. Strong association with chromatin increases the recruitment of other regulatory protein partners of NUP98 fusions, leading to activation or repression of target genes. 23 , 48 , 146 , 153 However, the mechanism through which different NUP98 fusions, involving partners possessing chromatin binding domains like NSD1 or lacking such domains like DDX10, execute a similar transcriptional program has remained unclear. 97 , 144
THERAPEUTIC OPPORTUNITIES FOR NUP98‐r AML
Despite significant progress in understanding the molecular mechanism of NUP98 fusions and their concomitant genetic alterations in oncogenesis, finding effective therapeutic strategies for this AML subtype is still a significant challenge. Genomic heterogeneity and somatically acquired mutations are major factors that may impact treatment outcome. The presence of NUP98 fusions is associated with an adverse clinical outcome. Within a pediatric AML cohort, it has been observed that 72% of AML patients with NUP98 fusions were refractory to induction therapy. 8 Notably, the outcomes for NUP98‐r AML patients can vary depending on the type of fusion partner involved. A study of 2235 children and young adults showed that while the overall CR rate for NUP98‐r AML patients was 50% after initial induction therapy, patients with NUP98::NSD1 fusions had a substantially lower CR rate of 38%. This was in contrast to an 80% CR rate for patients with NUP98::KDM5A fusions. 18 After achieving CR in the first or second course of induction therapy, these AML patients often undergo HSCT to reduce the risk of relapse. However, various studies have reported that more than 25% (25%–70%) of NUP98‐r AML patients experience relapse even after HSCT. 8 , 95 , 154 , 155 Notably, it has been observed that patients with NUP98::HOXA9 fusions have lower relapse rates when receiving HSCT after their first CR (25%) compared to those undergoing transplantation after their second CR (45%). 154 Considering the toxicity of chemotherapy and the high rate of relapse in this AML subgroup, novel targeted approaches may provide interesting options to improve patient outcomes. Insights into the composition of NUP98 fusion protein complexes, the interaction with secondary mutations such as FLT3‐ITD and the fusion oncoprotein‐induced transcriptional networks have identified multiple candidates for a targeted intervention.
TARGETING NUP98 FUSION PARTNER MOIETIES
Several studies have shown that the fusion protein partners of NUP98 play important roles in leukemogenesis. 21 , 142 , 144 To this end, researchers have attempted to block the activity of the functional domains in the NUP98 fusion partner sequences. One of the challenges is that most transcriptional regulators which serve a critical role in driving leukemia are difficult to drug due to a lack of deep protein pockets or large protein–protein interaction (PPI) interfaces. 156 However, covalent inhibition of the NSD1 histone methyltransferase has been shown to exert an antileukemic effect against NUP98::NSD1 leukemia cells. 144 The NSD1 inhibitor BT5 blocked the histone methyltransferase activity of the NSD1 SET domain and suppressed H3K36 dimethylation in HOXA genes, subsequently impairing the expression of target genes, cell proliferation, and colony‐forming activity. 144 Another strategy is to target the PHD domain, which is located in the C‐terminus of different NUP98 fusion oncoproteins. Blocking the PHD domain of NUP98::PHF23 and NUP98::KDM5A fusion proteins by the FDA‐approved drug disulfiram has been shown to disrupt the H3K4me3 binding potential of fusion proteins, impair expression of HOXA/B, MEIS1 genes and increase cell death in AML mouse cells (Figure 4). 83 It has been previously shown that disulfiram also impairs leukemia progression in KMT2A‐r AML by inhibiting the CXXC domain (Cysteine, X, X, Cysteine), a crucial DNA‐binding motif found in KMT2A fusion proteins. 157 Consequently, Disulfiram might be useful for the treatment of a wider group of AML subtypes driven by oncoproteins that possess chromatin or DNA binding domains.
Figure 4.
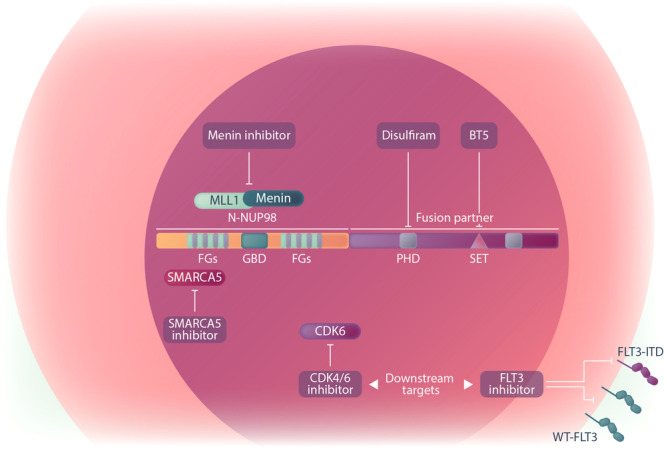
Graphical depiction of treatment opportunities for NUP98‐r AML.
TARGETING NUP98 FUSION PROTEIN INTERACTIONS
Another strategy for the treatment of NUP98‐r AML is the targeting of NUP98 fusion protein interactions. In NUP98‐r, KMT2A‐r, and NPM1‐mutant leukemia, all of which induce and depend on the MEIS1/HOXA transcriptional program, small molecule inhibitors that disrupt the Menin‐KMT2A interaction have demonstrated potent antileukemic effects. 116 , 136 , 158 Generally, the KMT2A cofactor Menin plays a crucial role in recruiting KMT2A to the promoter regions of key target genes, such as MEIS1. Similarly, NUP98 fusion proteins also interact with the KMT2A protein complex at its target genes, including MEIS1 and HOXA9. In NPM1‐mutant AML, the Menin‐KMT2A interaction is also vital in shaping the leukemic gene signature. 136 , 158 , 159 , 160 Additionally, UBTF‐TD AML demonstrates transcriptional profiles similar to those observed in NUP98‐r, KMT2A‐r, and NPM1‐mutant AMLs, marked by a significant overlap of overexpressed genes. 138 , 140 , 161 , 162 Thus, these AML (KMT2A‐r, NUP98‐r, UBTF‐TD, and NPM1‐mutant) subtypes share a similar transcriptional program with Menin playing the role of a conductor. Sensitivity of KMT2A‐r and NPM1‐mut AMLs to Menin inhibition has been documented previously. 158 , 159 , 160 , 163 , 164 Notably, this sensitivity is abolished by mutations in the MEN1 gene, further confirming the critical interaction between Menin and KMT2A. 165 In a study, we recently demonstrated that AMLs harboring UBTF‐TD are also sensitive to Menin inhibition, and that this manifests in similar gene expression changes. 162 Heikamp et al. showed that NUP98‐r AML cells are Menin‐dependent and disrupting the Menin‐KMT2A interaction with the Menin inhibitor VTP50469 induced anti‐proliferative effects and survival benefits in mouse AML models driven by NUP98::NSD1 and NUP98::KDM5A. 136 By using in vivo and ex vivo models, we also showed that Menin inhibition via revumenib is effective in halting leukemic progression of NUP98::NSD1 (FLT3‐ITD+) and NUP98::TOP1 (WT1+) AMLs, regardless of concomitant mutations. Furthermore, combined Menin and FLT3 inhibition exerted a synergistic effect in suppressing NUP98::NSD1 expressing AML cells. 116 However, the effectiveness of Menin inhibition in NUP98‐r AMLs has only been studied in a limited number of NUP98 fusions. Further investigation is necessary to fully assess the potential therapeutic impact of Menin inhibitors across a broad spectrum of NUP98 fusion variants and AML subtypes. The promising preclinical results with Menin inhibitors have led to the initiation of first‐in‐human clinical trials for revumenib, ziftomenib, and JNJ‐75276617 in adults with relapsed/refractory (R/R) leukemia featuring KMT2A‐r and NPM−1 mutations. Subsequently, NUP98‐r AMLs have also been included in some of these clinical trials (Table 3). The early result of phase I/II study investigating oral combination of revumenib with decitabine/cedazuridine (ASTX727) and venetoclax has demonstrated high efficacy in AML patients with KMT2A‐r, NPM1 mutations, and NUP98‐r. 166 A phase I trial is currently underway to assess the safety and tolerability of the Menin inhibitor JNJ‐75276617 (developed by Janssen Pharmaceuticals) as both monotherapy and in combination with chemotherapy (NCT05521087) for AML patients with KMT2A‐r, NUP98‐r, and NPM1 mutations. Ziftomenib (KO‐539) is also under evaluation as monotherapy or in combination with chemotherapy for assessing its efficacy in KMT2A‐r and NPM1‐mutant AML. Notably, however, Menin inhibitors in clinical use may give rise to toxicities, as exemplified by the differentiation syndrome observed in trials all Menin inhibitors when used as monotherapy. 167
Table 3.
Menin inhibitors in clinical trials.
| Compound | Trial | Combination | Eligibility | Participant | Status | Phase |
|---|---|---|---|---|---|---|
| Revumenib (Syndax Pharmaceuticals) | NCT04065399 (Augment‐101) | Monotherapy | R/R KMT2A‐r AML/ALL/MPAL, NPM1c, NUP98‐r AML | Child, Adult, Older Adult | Recruiting | I–II |
| NCT05326516 (AUGMENT‐102) | Combination with FLA chemotherapy | R/R KMT2A‐r, NPM1c, NUP98‐r AML/ALL/MPAL | Child, Adults | Not yet recruiting | I | |
| NCT06226571 | Chemotherapy, HiDAC | KMT2A‐r, NPM1c, NUP98‐r AML | Adult, Older Adult | Not yet recruiting | I | |
| NCT06222580 | Gilteritinib | R/R FLT3 mutated AML with KMT2A‐r, NUP98‐r and NPM1c alterations | Adult, Older Adult | Not yet recruiting | I | |
| NCT05360160 | Decitabine/cedazuridine (ASTX727) and venetoclax | R/R MPAL/AML with KMT2Ar, or NUP98r, or NPM1c | Child, Adult, Older Adult | Recruiting | I–II | |
| NCT06229912 | Monotherapy | Leukemia Associated With Upregulation of HOX genes, including NUP98‐r AML | Child, Adult, Older Adult | Not yet recruiting | II | |
| NCT06177067 | Revumenib, azacitidine, and venetoclax | R/R KMT2A‐r, NPM1c, NUP98‐r, PICALM::MLLT10, DEK::NUP214, UBTF‐TD, KAT6A::CREBBP, SET::NUP214 AML | Child, Adult | Not yet recruiting | I | |
| Ziftomenib (Kura Oncology) | NCT06376162 | FLA chemotherapy | R/R KMT2A‐r/NUP98‐r/NPMc Acute leukemia | Child 0–21 years | Not yet recruiting | I |
| JNJ‐75 276 617 (Janssen) | NCT05521087 | AML: JNJ‐75 276 617/FLA ALL: JNJ‐75 276 617/DEX/VCR/PEG | R/R KMT2A‐r, NPM1c, NUP98 or NUP214 alterations Acute leukemia | Child, Adult | Not yet recruiting | I |
| NCT04811560 | Monotherapy | R/R KMT2A‐r, NPM1c, NUP98 or NUP214 alterations Acute leukemia | Adult, Older Adult | Recruiting | I |
Abbreviations: chemotherapy, Cytarabine and Daunorubicin; DEX, Dexamethasone; FLA, Fludarabine, Cytarabine; HiDAC, high‐dose cytarabine; MPAL, mixed‐phenotype acute leukemia; PEG, PEG‐Asparaginase; VCR, Vincristine.
In this context, AML subtypes that display a transcriptional similarity and co‐cluster with KMT2A‐r, NPM1‐mutant, and NUP98‐r AMLs, including those harboring UBTF‐TD and DEK::NUP214 may also show increased sensitivity towards Menin inhibition. 140 This hypothesis is further supported by our recent finding that UBTF‐TD AML is highly sensitive to Menin inhibition. 162
While the exact mechanisms by which NUP98 fusions regulate protein interactions in the nucleoplasm or biomolecular condensates are not fully understood, disrupting the formation of nuclear foci or the functional interactions of associated proteins presents another potential avenue for targeting leukemia cells. In NUP98::NSD1 AML cells, targeting the transcriptional co‐regulator SMARCA5, which has been reported as a critical partner of NUP98 fusions in biomolecular condensates, has already been addressed. 129 As describe previously, CRM1 is involved in recruiting NUP98 fusions to its target sites and providing access chromatin for HOX gene regulation. CRM1 inhibitors such as leptomycin B and selinexor, which has obtained FDA approval for the treatment of adult patients with multiple myeloma, could potentially be effective in NUP98‐r AML. 62 , 129 , 168
TARGETING NUP98 FUSION PROTEIN DOWNSTREAM FACTORS AND CO‐OPERATIVE MUTATIONS
Another potential treatment strategies for NUP98‐r is targeting factors that are induced by NUP98 fusion proteins, as the majority of NUP98 fusions exploits similar downstream targets to fuel leukemia.
Among NUP98 target genes, loss of CDK6 has a significant impact on NUP98 fusion‐driven leukemogenesis, a feature shared with other AML subtypes such as KMT2A‐rearranged or RUNX1::RUNX1T1‐positive AMLs. 135 , 169 , 170 The CDK4/6 inhibitor palbociclib, which is approved for the treatment of breast cancer, demonstrated potent anti‐proliferative activity and differentiation induction in murine AML cells driven by NUP98::KDM5A, NUP98::DDX10, and NUP98::NSD1, as well as in human NUP98‐r leukemic cells. 135 Furthermore, CDK6 is reported as a critical factor for the survival of AML cells with FLT3‐ITD mutations, where FLT3‐ITD signaling is the primary cause of CDK6 overexpression through a pathway involving the SRC‐family kinase HCK. 171 As FLT3‐ITD is a common recurrent mutation in NUP98‐r AML, targeting CDK6 may be a promising therapy option for this AML subtype.
FLT3‐ITD itself is a clinically highly relevant mutation that frequently co‐occurs with NUP98 rearrangements. Currently, different FLT3 inhibitors are used for the treatment of patients with FLT3 mutations, including midostaurin, sorafenib, quizartinib, crenolanib, and gilteritinib. Based on type and generation, FLT3 inhibitors have different specificities and target active or inactive states of the FLT3 protein in cells with FLT3‐ITD or FLT3 kinase domain point mutations (FLT3‐TKD). A type 1 inhibitor such as gilteritinib inhibits both the TKD and the ITD mutations, whereas a type 2 inhibitor such as quizartinib solely targets the ITD mutation, but not the TKD. 172 , 173 A study using mouse bone marrow progenitor cells co‐expressing NUP98::NSD1 and FLT3‐ITD reported that these cells are more sensitive to midostaurin compared to cells expressing either aberration alone, highlighting the defining role of the FLT3 signaling pathway in NUP98::NSD1‐driven AML. 174 However, during monotherapy with FLT3 inhibitors loss of the FLT3‐ITD mutation has been reported in relapsed patients, indicating the selective outgrowth of minor clones without FLT3 mutations under selective pressure. 175 Furthermore, after treatment with FLT3 inhibitors, de novo resistance mutations may arise in the FLT3 molecule. 175 , 176 , 177 This process may result in the development of resistance followed by relapse. Thus, the best strategy may be to use inhibitors with a more narrow resistance profile (e.g., gilteritinib) or applying combination therapy with chemotherapeutic agents, HSCT, MEK inhibitors, hypo‐methylating agents (HMA), or CDK6 inhibitors to maximize the survival benefit for AML patients. 171 , 178 , 179 , 180 , 181 , 182 , 183 , 184
Combination therapies that include BCL2 inhibitors, such as venetoclax, paired with cytotoxic or hypomethylating agents, present a promising treatment strategy for NUP98‐r AML, despite the limited clinical data available for this subgroup. Venetoclax has shown significant efficacy in AMLs with KMT2A‐r and NPM1 mutation. 185 , 186 , 187 This effectiveness is partly attributed to the link between BCL‐2 inhibitor efficacy and the overexpression of HOXA/B genes. 188 Although applied in a limited number of cases, it has been observed that patients with NUP98::NSD1 (and FLT3‐ITD+) who respond poorly to chemotherapy may benefit from the inclusion of venetoclax and FLT3 inhibitors in their treatment regimens. 189 , 190 , 191 The transcriptional similarities between KMT2A‐r and NPM1‐mutant AMLs and NUP98‐r AMLs suggest that venetoclax combinations might also be effective in treating AMLs with NUP98 fusions, particularly those with high BCL2 expression such as NUP98::NSD1, which are typically resistant to conventional chemotherapy. Currently, the efficacy of combining the Menin inhibitor revumenib, venetoclax, and a hypomethylating agent is being investigated in children with relapsed/refractory AML in a phase I/II, investigator‐initiated trial (NCT05360160). 166 Furthermore, It has been demonstrated that the combination of the BCL2‐inhibitor navitoclax and the SRC/ABL‐inhibitor dasatinib has synergistic effects on patients and engineered cell models with NUP98::NSD1 and FLT3‐ITD. 183 The authors of this study speculate that enhanced expression of LCK, FGR, and BCL2A1 in NUP98::NSD1+, FLT3‐ITD+ patients may be the cause of this synergistic effect. In addition, there is a report of a patient with the NUP98::NSD1 fusion, along with concomitant IDH1 and GATA2 mutations, who did not achieve CR despite two courses of chemotherapy, but the combination of venetoclax and decitabine led to complete remission. 191 Taken together, recent advancements in targeted therapy offer encouraging strategies to address the complexities associated with treatment of NUP98‐r AML.
Currently, various preclinical studies are exploring the unique properties of NUP98‐r AMLs, providing valuable insights into potential therapeutic strategies. These investigations suggest that FLT3, Menin, and BCL2 inhibitors, either alone or in combination with chemotherapy or hypomethylating agents, may offer therapeutic benefits for NUP98‐r AML patients. Encouraged by promising preclinical findings, these drugs and their combinations are now being evaluated in clinical trials that involve AML patients with NUP98 rearrangements (Table 3).
SUMMARY AND CONCLUSIONS
NUP98 translocations are common genetic alterations in pediatric AML that are associated with dismal prognosis. NUP98 fusions often co‐occur with a set of additional somatic mutations that confer a proliferative advantage to AML cells and add more complexity to the disease, demanding more effective therapeutics. Though novel insights into the molecular mechanism of NUP98 fusion protein‐driven leukemia have led to various new therapeutic opportunities, many questions remain. As most targeted AML agents have been studied and approved in adult AML, there is a critical need to establish their efficacy and pharmacokinetics specifically for pediatric patients. Thus, it is of vital importance to understand the molecular mechanisms underlying oncogenic transformation by NUP98 fusion oncoproteins which are prevalent in pediatric leukemia. Detailed analysis of individual samples with different NUP98 fusions and comparison of their transcriptional landscape together with mechanistic investigations will open new and more efficient therapeutic avenues towards efficient treatments for patients suffering from these highly aggressive AMLs.
AUTHOR CONTRIBUTIONS
Milad Rasouli and Selina Troester contributed equally as first authors. Olaf Heidenreich, Florian Grebien, Bianca F. Goemans, and C. Michel Zwaan contributed equally as last authors.
CONFLICT OF INTEREST STATEMENT
Olaf Heidenreich received research funding from Syndax and Roche. The other authors declare no conflicts of interest.
FUNDING
This work was supported by KiKa program grant 329 to Olaf Heidenreich. Work in the Grebien laboratory was supported by the Austrian Science Fund (FWF, grants no. TAI‐490 and P35628). Selina Troester is the recipient of a DOC fellowship of the Austrian Academy of Sciences at the University of Veterinary Medicine.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Huang BJ, Smith JL, Farrar JE, et al. Integrated stem cell signature and cytomolecular risk determination in pediatric acute myeloid leukemia. Nat Commun. 2022;13(1):5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hoffman AE, Schoonmade LJ, Kaspers GJ. Pediatric relapsed acute myeloid leukemia: a systematic review. Expert Rev Anticancer Ther. 2021;21(1):45‐52. [DOI] [PubMed] [Google Scholar]
- 3. Klein K, de Haas V, Kaspers GJL. Clinical challenges in de novo pediatric acute myeloid leukemia. Expert Rev Anticancer Ther. 2018;18(3):277‐293. [DOI] [PubMed] [Google Scholar]
- 4. Zwaan CM, Kolb EA, Reinhardt D, et al. Collaborative efforts driving progress in pediatric acute myeloid leukemia. J Clin Oncol. 2015;33(27):2949‐2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schulpen M, Goemans BF, Kaspers GJL, Raaijmakers MHGP, Zwaan CM, Karim‐Kos HE. Increased survival disparities among children and adolescents & young adults with acute myeloid leukemia: a Dutch population‐based study. Int J Cancer. 2022;150(7):1101‐1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. van Dijk AD, de Bont ESJM, Kornblau SM. Targeted therapy in acute myeloid leukemia: current status and new insights from a proteomic perspective. Expert Rev Proteomics. 2020;17(1):1‐10. [DOI] [PubMed] [Google Scholar]
- 7. Zhang J, Gao X, Yu L. Roles of histone deacetylases in acute myeloid leukemia with fusion proteins. Front Oncol. 2021;11:3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Struski S, Lagarde S, Bories P, et al. NUP98 is rearranged in 3.8% of pediatric AML forming a clinical and molecular homogenous group with a poor prognosis. Leukemia. 2017;31(3):565‐572. [DOI] [PubMed] [Google Scholar]
- 9. Niktoreh N, Walter C, Zimmermann M, et al. Mutated WT1, FLT3‐ITD, and NUP98‐NSD1 fusion in various combinations define a poor prognostic group in pediatric acute myeloid leukemia. J Oncol. 2019;2019:1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Akiki S, Dyer SA, Grimwade D, et al. NUP98‐NSD1 fusion in association with FLT3‐ITD mutation identifies a prognostically relevant subgroup of pediatric acute myeloid leukemia patients suitable for monitoring by real time quantitative PCR. Genes Chromosom Cancer. 2013;52(11):1053‐1064. [DOI] [PubMed] [Google Scholar]
- 11. Bisio V, Zampini M, Tregnago C, et al. NUP98‐fusion transcripts characterize different biological entities within acute myeloid leukemia: a report from the AIEOP‐AML group. Leukemia. 2017;31(4):974‐977. [DOI] [PubMed] [Google Scholar]
- 12. Umeda M, Ma J, Westover T, et al. A new genomic framework to categorize pediatric acute myeloid leukemia. Nat Genet. 2024;56:281‐293. 10.1038/s41588-023-01640-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhu H, Zhao Y, Cheng W, et al. Clinical characteristics, genetic profile and prognosis of adult acute myeloid leukemia with NUP98 rearrangement. Blood. 2023;142:2867. [Google Scholar]
- 14. Hollink IHIM, van den Heuvel‐Eibrink MM, Arentsen‐Peters STCJM, et al. NUP98/NSD1 characterizes a novel poor prognostic group in acute myeloid leukemia with a distinct HOX gene expression pattern. Blood. 2011;118(13):3645‐3656. [DOI] [PubMed] [Google Scholar]
- 15. Chou WC, Chen C‐Y, Hou HA, et al. Acute myeloid leukemia bearing t (7; 11)(p15; p15) is a distinct cytogenetic entity with poor outcome and a distinct mutation profile: comparative analysis of 493 adult patients. Leukemia. 2009;23(7):1303‐1310. [DOI] [PubMed] [Google Scholar]
- 16. Xie W, Raess PW, Dunlap J, et al. Adult acute myeloid leukemia patients with NUP98 rearrangement have frequent cryptic translocations and unfavorable outcome. Leuk Lymphoma. 2022;63(8):1907‐1916. [DOI] [PubMed] [Google Scholar]
- 17. Tian J, Zhu Y, Li J, et al. The landscape of NUP98 rearrangements clinical characteristics and treatment response from 1491 acute leukemia patients. Blood Cancer J. 2024;14(1):81. 10.1038/s41408-024-01066-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bertrums EJM, Smith JL, Harmon L, et al. Comprehensive molecular and clinical characterization of NUP98 fusions in pediatric acute myeloid leukemia. Haematologica. 2023;108(8):2044‐2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. De Rooij JDE, Masetti R, van den Heuvel‐Eibrink MM, et al. Recurrent abnormalities can be used for risk group stratification in pediatric AMKL: a retrospective intergroup study. Blood. 2016;127(26):3424‐3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. de Rooij JDE, Hollink IHIM, Arentsen‐Peters STCJM, et al. NUP98/JARID1A is a novel recurrent abnormality in pediatric acute megakaryoblastic leukemia with a distinct HOX gene expression pattern. Leukemia. 2013;27(12):2280‐2288. [DOI] [PubMed] [Google Scholar]
- 21. Gough SM, Slape CI, Aplan PD. NUP98 gene fusions and hematopoietic malignancies: common themes and new biologic insights. Blood. 2011;118(24):6247‐6257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Romana SP, Radford‐Weiss I, Ben Abdelali R, et al. NUP98 rearrangements in hematopoietic malignancies: a study of the Groupe Francophone de Cytogénétique Hématologique. Leukemia. 2006;20(4):696‐706. [DOI] [PubMed] [Google Scholar]
- 23. Bai X‐T, Gu B‐W, Yin T, et al. Trans‐repressive effect of NUP98‐PMX1 on PMX1‐regulated c‐FOS gene through recruitment of histone deacetylase 1 by FG repeats. Cancer Res. 2006;66(9):4584‐4590. [DOI] [PubMed] [Google Scholar]
- 24. Taketani T, Taki T, Nakamura H, Taniwaki M, Masuda J, Hayashi Y. NUP98–NSD3 fusion gene in radiation‐associated myelodysplastic syndrome with t (8; 11)(p11; p15) and expression pattern of NSD family genes. Cancer Genet Cytogenet. 2009;190(2):108‐112. [DOI] [PubMed] [Google Scholar]
- 25. Pan Q, Zhu YJ, Gu BW, et al. A new fusion gene NUP98‐IQCG identified in an acute T‐lymphoid/myeloid leukemia with at (3; 11)(q29q13; p15) del (3)(q29) translocation. Oncogene. 2008;27(24):3414‐3423. [DOI] [PubMed] [Google Scholar]
- 26. Fontoura BMA, Blobel G, Matunis MJ. A conserved biogenesis pathway for nucleoporins: proteolytic processing of a 186‐kilodalton precursor generates Nup98 and the novel nucleoporin, Nup96. J Cell Biol. 1999;144(6):1097‐1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Franks TM, Hetzer MW. The role of Nup98 in transcription regulation in healthy and diseased cells. Trends Cell Biol. 2013;23(3):112‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shinkai Y, Kuramochi M, Miyafusa T. New Family Members of FG Repeat Proteins and Their Unexplored Roles During Phase Separation. Front Cell Dev Biol. 2021;9:1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Onischenko E, Tang JH, Andersen KR, et al. Natively unfolded FG repeats stabilize the structure of the nuclear pore complex. Cell. 2017;171(4):904‐917.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xu H, Valerio DG, Eisold ME, et al. NUP98 fusion proteins interact with the NSL and MLL1 complexes to drive leukemogenesis. Cancer Cell. 2016;30(6):863‐878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Terry LJ, Wente SR. Flexible gates: dynamic topologies and functions for FG nucleoporins in nucleocytoplasmic transport. Eukaryotic Cell. 2009;8(12):1814‐1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ghavami A, Van Der Giessen E, Onck PR. Energetics of transport through the nuclear pore complex. PLoS One. 2016;11(2):e0148876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Frey S, Görlich D. A saturated FG‐repeat hydrogel can reproduce the permeability properties of nuclear pore complexes. Cell. 2007;130(3):512‐523. [DOI] [PubMed] [Google Scholar]
- 34. Schmidt HB, Görlich D. Nup98 FG domains from diverse species spontaneously phase‐separate into particles with nuclear pore‐like permselectivity. eLife. 2015;4:e04251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ng SC, Görlich D. A simple thermodynamic description of phase separation of Nup98 FG domains. Nat Commun. 2022;13(1):6172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Griffis ER, Altan N, Lippincott‐Schwartz J, Powers MA. Nup98 is a mobile nucleoporin with transcription‐dependent dynamics. Mol Biol Cell. 2002;13(4):1282‐1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Capelson M, Liang Y, Schulte R, Mair W, Wagner U, Hetzer MW. Chromatin‐bound nuclear pore components regulate gene expression in higher eukaryotes. Cell. 2010;140(3):372‐383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Laurell E, Beck K, Krupina K, et al. Phosphorylation of Nup98 by multiple kinases is crucial for NPC disassembly during mitotic entry. Cell. 2011;144(4):539‐550. [DOI] [PubMed] [Google Scholar]
- 39. Kalverda B, Pickersgill H, Shloma VV, Fornerod M. Nucleoporins directly stimulate expression of developmental and cell‐cycle genes inside the nucleoplasm. Cell. 2010;140(3):360‐371. [DOI] [PubMed] [Google Scholar]
- 40. Light WH, Freaney J, Sood V, et al. A conserved role for human Nup98 in altering chromatin structure and promoting epigenetic transcriptional memory. PLoS Biol. 2013;11(3):e1001524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jeganathan KB, Malureanu L, Van Deursen JM. The Rae1–Nup98 complex prevents aneuploidy by inhibiting securin degradation. Nature. 2005;438(7070):1036‐1039. [DOI] [PubMed] [Google Scholar]
- 42. Boija A, Klein IA, Sabari BR, et al. Transcription factors activate genes through the phase‐separation capacity of their activation domains. Cell. 2018;175(7):1842‐1855.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schmidt HB, Barreau A, Rohatgi R. Phase separation‐deficient TDP43 remains functional in splicing. Nat Commun. 2019;10(1):4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. King JT, Shakya A. Phase separation of DNA: from past to present. Biophys J. 2021;120(7):1139‐1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hou C, Corces VG. Nups take leave of the nuclear envelope to regulate transcription. Cell. 2010;140(3):306‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shima Y, Yumoto M, Katsumoto T, Kitabayashi I. MLL is essential for NUP98‐HOXA9‐induced leukemia. Leukemia. 2017;31(10):2200‐2210. [DOI] [PubMed] [Google Scholar]
- 47. Kasper LH, Brindle PK, Schnabel CA, Pritchard CEJ, Cleary ML, Van Deursen JMA. CREB binding protein interacts with nucleoporin‐specific FG repeats that activate transcription and mediate NUP98‐HOXA9 oncogenicity. Mol Cell Biol. 1999;19(1):764‐776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Franks TM, McCloskey A, Shokirev MN, Benner C, Rathore A, Hetzer MW. Nup98 recruits the Wdr82–Set1A/COMPASS complex to promoters to regulate H3K4 trimethylation in hematopoietic progenitor cells. Genes Dev. 2017;31(22):2222‐2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xu S, Powers MA. Nuclear Pore Proteins and Cancer. Elsevier; 2009:620‐630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wu M, Wang PF, Lee JS, et al. Molecular regulation of H3K4 trimethylation by Wdr82, a component of human Set1/COMPASS. Mol Cell Biol. 2008;28(24):7337‐7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sump B, Brickner JH. Nup98 regulation of histone methylation promotes normal gene expression and may drive leukemogenesis. Genes Dev. 2017;31(22):2201‐2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nakamura T, Largaespada DA, Lee MP, et al. Fusion of the nucleoporin gene NUP98 to HOXA9 by the chromosome translocation t (7; 11)(p15; p15) in human myeloid leukaemia. Nat Genet. 1996;12(2):154‐158. [DOI] [PubMed] [Google Scholar]
- 53. Borrow J, Shearman AM, Stanton VP, et al. The t (7; 11)(p15; p15) translocation in acute myeloid leukaemia fuses the genes for nucleoporin NUP96 and class I homeoprotein HOXA9. Nat Genet. 1996;12(2):159‐167. [DOI] [PubMed] [Google Scholar]
- 54. Ostronoff F, Othus M, Gerbing RB, et al. NUP98/NSD1 and FLT3/ITD coexpression is more prevalent in younger AML patients and leads to induction failure: a COG and SWOG report. Blood. 2014;124(15):2400‐2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mohanty S. NUP98 Rearrangements in AML: molecular mechanisms and clinical implications. Onco. 2023;3(3):147‐164. [Google Scholar]
- 56. Iacobucci I, Wen J, Meggendorfer M, et al. Genomic subtyping and therapeutic targeting of acute erythroleukemia. Nat Genet. 2019;51(4):694‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Calvo C, Fenneteau O, Leverger G, Petit A, Baruchel A, Méchinaud F. Infant acute myeloid leukemia: a unique clinical and biological entity. Cancers. 2021;13(4):777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Such E, Cervera J, Valencia A, et al. A novel NUP98/RARG gene fusion in acute myeloid leukemia resembling acute promyelocytic leukemia. Blood. 2011;117(1):242‐245. [DOI] [PubMed] [Google Scholar]
- 59. Zhu H‐H, Yang M‐C, Wang F, et al. Identification of a novel NUP98‐RARA fusion transcript as the 14th variant of acute promyelocytic leukemia. Am J Hematol. 2020;95(7):E184‐E186. [DOI] [PubMed] [Google Scholar]
- 60. Gallego Hernanz MP, Torregrosa Diaz JM, Sorel N, et al. Long‐term molecular remission in a patient with acute myeloid leukemia harboring a new NUP98‐LEDGF rearrangement. Cancer Med. 2019;8(4):1765‐1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dillon R, Potter N, Freeman S, Russell N. How we use molecular minimal residual disease (MRD) testing in acute myeloid leukaemia (AML). Br J Haematol. 2021;193(2):231‐244. [DOI] [PubMed] [Google Scholar]
- 62. Michmerhuizen NL, Klco JM, Mullighan CG. Mechanistic insights and potential therapeutic approaches for NUP98‐rearranged hematologic malignancies. Blood. 2020;136(20):2275‐2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Slape C, Lin YW, Hartung H, Zhang Z, Wolff L, Aplan PD. NUP98‐HOX translocations lead to myelodysplastic syndrome in mice and men. JNCI Monographs. 2008;2008(39):64‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sloma I, Beer P, Desterke C, et al. NUP98‐HOXA9‐initiates molecular and biologic features of disease progression in a humanized model of CML. Blood. 2019;134:4139. [Google Scholar]
- 65. Burillo‐Sanz S, Morales‐Camacho RM, Caballero‐Velázquez T, et al. NUP 98–HOXA 9 bearing therapy‐related myeloid neoplasm involves myeloid‐committed cell and induces HOXA 5, EVI 1, FLT 3, and MEIS 1 expression. Int J Lab Hematol. 2016;38(1):64‐71. [DOI] [PubMed] [Google Scholar]
- 66. Wei S, Wang S, Qiu S, et al. Clinical and laboratory studies of 17 patients with acute myeloid leukemia harboring t (7; 11)(p15; p15) translocation. Leuk Res. 2013;37(9):1010‐1015. [DOI] [PubMed] [Google Scholar]
- 67. Fujino T, Suzuki A, Ito Y, et al. Single‐translocation and double‐chimeric transcripts: detection of NUP98‐HOXA9 in myeloid leukemias with HOXA11 or HOXA13 breaks of the chromosomal translocation t (7; 11)(p15; p15). Blood. 2002;99(4):1428‐1433. [DOI] [PubMed] [Google Scholar]
- 68. Mizoguchi Y, Fujita N, Taki T, Hayashi Y, Hamamoto K. Juvenile myelomonocytic leukemia with t (7; 11)(p15; p15) and NUP98‐HOXA11 fusion. Am J Hematol. 2009;84(5):295‐297. [DOI] [PubMed] [Google Scholar]
- 69. Taketani T, Taki T, Ono R, Kobayashi Y, Ida K, Hayashi Y. The chromosome translocation t (7; 11)(p15; p15) in acute myeloid leukemia results in fusion of the NUP98 gene with a HOXA cluster gene, HOXA13, but not HOXA9. Genes Chromosom Cancer. 2002;34(4):437‐443. [DOI] [PubMed] [Google Scholar]
- 70. Taketani T, Taki T, Shibuya N, Kikuchi A, Hanada R, Hayashi Y. Novel NUP98‐HOXC11 fusion gene resulted from a chromosomal break within exon 1 of HOXC11 in acute myeloid leukemia with t (11; 12)(p15; q13). Cancer Res. 2002;62(16):4571‐4574. [PubMed] [Google Scholar]
- 71. Panagopoulos I, Isaksson M, Billström R, Strömbeck B, Mitelman F, Johansson B. Fusion of the NUP98 gene and the homeobox gene HOXC13 in acute myeloid leukemia with t (11; 12)(p15; q13). Genes Chromosom Cancer. 2003;36(1):107‐112. [DOI] [PubMed] [Google Scholar]
- 72. Taketani T, Taki T, Shibuya N, et al. The HOXD11 gene is fused to the NUP98 gene in acute myeloid leukemia with t (2; 11)(q31; p15). Cancer Res. 2002;62(1):33‐37. [PubMed] [Google Scholar]
- 73. Raza‐Egilmez SZ, Jani‐Sait SN, Grossi M, Higgins MJ, Shows TB, Aplan PD. NUP98‐HOXD13 gene fusion in therapy‐related acute myelogenous leukemia. Cancer Res. 1998;58(19):4269‐4273. [PubMed] [Google Scholar]
- 74. Kundu S, Park ES, Chung YJ, et al. Thymic precursor cells generate acute myeloid leukemia in NUP98‐PHF23/NUP98‐HOXD13 double transgenic mice. Sci Rep. 2019;9(1):17213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Soler G, Kaltenbach S, Dobbelstein S, et al. Identification of GSX2 and AF10 as NUP98 partner genes in myeloid malignancies. Blood Cancer J. 2013;3(7):e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Nakamura T, Yamazaki Y, Hatano Y, Miura I. NUP98 is fused to PMX1 homeobox gene in human acute myelogenous leukemia with chromosome translocation t (1; 11)(q23; p15). Blood. 1999;94(2):741‐747. [PubMed] [Google Scholar]
- 77. Zhang L, Alsabeh R, Mecucci C, et al. Rare t (1; 11)(q23; p15) in therapy‐related myelodysplastic syndrome evolving into acute myelomonocytic leukemia: a case report and review of the literature. Cancer Genet Cytogenet. 2007;178(1):42‐48. [DOI] [PubMed] [Google Scholar]
- 78. Chonabayashi K, Yoshida Y, Kitawaki T, et al. Acute myeloid leukemia with a cryptic NUP98/PRRX2 rearrangement developing after low‐dose methotrexate therapy for rheumatoid arthritis. Ann Hematol. 2019;98:2841‐2843. [DOI] [PubMed] [Google Scholar]
- 79. Gervais C, Mauvieux L, Perrusson N, et al. A new translocation t (9; 11)(q34; p15) fuses NUP98 to a novel homeobox partner gene, PRRX2, in a therapy‐related acute myeloid leukemia. Leukemia. 2005;19(1):145‐148. [DOI] [PubMed] [Google Scholar]
- 80. Lisboa S, Cerveira N, Bizarro S, et al. POU1F1 is a novel fusion partner of NUP98 in acute myeloid leukemia with t (3; 11)(p11; p15). Mol Cancer. 2013;12:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Cheng C‐K, Chan H‐Y, Yung Y‐L, et al. A novel NUP98‐JADE2 fusion in a patient with acute myeloid leukemia resembling acute promyelocytic leukemia. Blood Adv. 2022;6(2):410‐415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Schwartz JR, Ma J, Lamprecht T, et al. The genomic landscape of pediatric myelodysplastic syndromes. Nat Commun. 2017;8(1):1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Gough SM, Lee F, Yang F, et al. NUP98–PHF23 is a chromatin‐modifying oncoprotein that causes a wide array of leukemias sensitive to inhibition of PHD histone reader function. Cancer Discovery. 2014;4(5):564‐577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Sorel N, Raimbault A, Brizard F, et al. Identification and genetic characterization of a NUP98‐HHEX molecular rearrangement in a pediatric acute myeloid leukemia. Leuk Lymphoma. 2021;62(14):3531‐3535. [DOI] [PubMed] [Google Scholar]
- 85. Thol F, Kölking B, Hollink IHIM, et al. Frequency and prognostic impact of NUP98/NSD1 translocations in adult AML and MDS patients. Blood. 2012;120(21):1402. [Google Scholar]
- 86. Behnert A, Lee AG, Young EP, et al. NUP98‐NSD1 driven MDS/MPN in childhood masquerading as JMML. J Pediatr Hematol Oncol. 2021;43(6):e808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Pasillas MP, Shah M, Kamps MP. NSD1 PHD domains bind methylated H3K4 and H3K9 using interactions disrupted by point mutations in human sotos syndrome. Hum Mutat. 2011;32(3):292‐298. [DOI] [PubMed] [Google Scholar]
- 88. Ganapathi SS, Raikar SS, Yatsenko SA, Djokic M, Bukowinski A. Mixed phenotype acute leukemia in a child associated with a NUP98‐NSD1 fusion and NRAS p. Gly61Arg mutation. Cancer Rep. 2021;4(4):e1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Rosati R, La Starza R, Veronese A, et al. NUP98 is fused to the NSD3 gene in acute myeloid leukemia associated with t (8; 11)(p11. 2; p15). Blood. 2002;99(10):3857‐3860. [DOI] [PubMed] [Google Scholar]
- 90. Fisher JN, Thanasopoulou A, Juge S, et al. Transforming activities of the NUP98‐KMT2A fusion gene associated with myelodysplasia and acute myeloid leukemia. Haematologica. 2020;105(7):1857‐1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Tembrink M, Gerding WM, Wieczorek S, et al. Novel NUP98:: ASH1L gene fusion in acute myeloid leukemia detected by optical genome mapping. Cancers. 2023;15(11):2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Panagopoulos I, Kerndrup G, Carlsen N, Strömbeck B, Isaksson M, Johansson B. Fusion of NUP98 and the SET binding protein 1 (SETBP1) gene in a paediatric acute T cell lymphoblastic leukaemia with t (11; 18)(p15; q12). Br J Haematol. 2007;136(2):294‐296. [DOI] [PubMed] [Google Scholar]
- 93. Roussy M, Bilodeau M, Jouan L, et al. NUP98‐BPTF gene fusion identified in primary refractory acute megakaryoblastic leukemia of infancy. Genes Chromosom Cancer. 2018;57(6):311‐319. [DOI] [PubMed] [Google Scholar]
- 94. Kim JC, Zuzarte PC, Murphy T, et al. Cryptic genomic lesions in adverse‐risk acute myeloid leukemia identified by integrated whole genome and transcriptome sequencing. Leukemia. 2020;34(1):306‐311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Noort S, Priscilla Wander W, Todd A. Alonzo A, et al. The clinical and biological characteristics of NUP98‐KDM5A in pediatric acute myeloid leukemia. Haematologica. 2021;106(2):630‐634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Ishikawa M, Yagasaki F, Okamura D, et al. A novel gene, ANKRD28 on 3p25, is fused with NUP98 on 11p15 in a cryptic 3‐way translocation of t (3; 5; 11)(p25; q35; p15) in an adult patient with myelodysplastic syndrome/acute myelogenous leukemia. Int J Hematol. 2007;86:238‐245. [DOI] [PubMed] [Google Scholar]
- 97. Yassin ER, Abdul‐Nabi AM, Takeda A, Yaseen NR. Effects of the NUP98–DDX10 oncogene on primary human CD34+ cells: role of a conserved helicase motif. Leukemia. 2010;24(5):1001‐1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Arai Y, Hosoda F, Kobayashi H, et al. The inv (11)(p15q22) chromosome translocation of de novo and therapy‐related myeloid malignancies results in fusion of the nucleoporin gene, NUP98, with the putative RNA helicase gene, DDX10. Blood. 1997;89(11):3936‐3944. [PubMed] [Google Scholar]
- 99. Petit A, Ragu C, Della‐Valle V, et al. NUP98–HMGB3: a novel oncogenic fusion. Leukemia. 2010;24(3):654‐658. [DOI] [PubMed] [Google Scholar]
- 100. Hayashi Y, Harada Y, Kagiyama Y, et al. NUP98‐HBO1–fusion generates phenotypically and genetically relevant chronic myelomonocytic leukemia pathogenesis. Blood Adv. 2019;3(7):1047‐1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Grand FH, Koduru P, Cross NCP, Allen SL. NUP98‐LEDGF fusion and t (9; 11) in transformed chronic myeloid leukemia. Leuk Res. 2005;29(12):1469‐1472. [DOI] [PubMed] [Google Scholar]
- 102. Hussey DJ, Moore S, Nicola M, Dobrovic A. Fusion of the NUP98 gene with the LEDGF/p52 gene defines a recurrent acute myeloid leukemia translocation. BMC Genet. 2001;2:1‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Yamamoto K, Nakamachi Y, Yakushijin K, et al. Expression of the novel NUP98/PSIP1 fusion transcripts in myelodysplastic syndrome with t (9; 11)(p22; p15). Eur J Haematol. 2012;88(3):244‐248. [DOI] [PubMed] [Google Scholar]
- 104. Ahuja HG, Hong J, Aplan PD, Tcheurekdjian L, Forman SJ, Slovak ML. t (9; 11)(p22; p15) in acute myeloid leukemia results in a fusion between NUP98 and the gene encoding transcriptional coactivators p52 and p75‐lens epithelium‐derived growth factor (LEDGF). Cancer Res. 2000;60(22):6227‐6229. [PubMed] [Google Scholar]
- 105. Mecucci C, La Starza R, Negrini M, et al. t (4; 11)(q21; p15) translocation involving NUP98 and RAP1GDS1 genes: characterization of a new subset of T acute lymphoblastic leukaemia. Br J Haematol. 2000;109(4):788‐793. [DOI] [PubMed] [Google Scholar]
- 106. van Zutven LJCM, Önen E, Velthuizen SCJM, et al. Identification of NUP98 abnormalities in acute leukemia: JARID1A (12p13) as a new partner gene. Genes Chromosom Cancer. 2006;45(5):437‐446. [DOI] [PubMed] [Google Scholar]
- 107. Gurevich RM, Aplan PD, Humphries RK. NUP98‐topoisomerase I acute myeloid leukemia‐associated fusion gene has potent leukemogenic activities independent of an engineered catalytic site mutation. Blood. 2004;104(4):1127‐1136. [DOI] [PubMed] [Google Scholar]
- 108. Yamamoto K, Minami Y, Yakushijin K, et al. Coexpression of NUP98/TOP1 and TOP1/NUP98 in de novo Acute Myeloid Leukemia with t (11; 20)(p15; q12) and t (2; 5)(q33; q31). Cytogenet Genome Res. 2017;150(3‐4):287‐292. [DOI] [PubMed] [Google Scholar]
- 109. Nebral K, Schmidt HH, Haas OA, Strehl S. NUP98 is fused to topoisomerase (DNA) IIβ 180 kDa (TOP2B) in a patient with acute myeloid leukemia with a new t (3; 11)(p24; p15). Clin Cancer Res. 2005;11(18):6489‐6494. [DOI] [PubMed] [Google Scholar]
- 110. Chen B, Jiang L, Zhong M‐L, et al. Identification of fusion genes and characterization of transcriptome features in T‐cell acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 2018;115(2):373‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Petit A, Ragu C, Soler G, et al. Functional analysis of the NUP98‐CCDC28A fusion protein. Haematologica. 2012;97(3):379‐387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Yan L, Ping N, Zhu M, et al. Clinical, immunophenotypic, cytogenetic, and molecular genetic features in 117 adult patients with mixed‐phenotype acute leukemia defined by WHO‐2008 classification. Haematologica. 2012;97(11):1708‐1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Gorello P, Brandimarte L, La Starza R, et al. t (3; 11)(q12; p15)/NUP98‐LOC348801 fusion transcript in acute myeloid leukemia. Haematologica. 2008;93(9):1398‐1401. [DOI] [PubMed] [Google Scholar]
- 114. Lahortiga I, Vizmanos JL, Agirre X, et al. NUP98 is fused to adducin 3 in a patient with T‐cell acute lymphoblastic leukemia and myeloid markers, with a new translocation t (10; 11)(q25; p15). Cancer Res. 2003;63(12):3079‐3083. [PubMed] [Google Scholar]
- 115. Arai Y, Kyo T, Miwa H, et al. Heterogeneous fusion transcripts involving the NUP98 gene and HOXD13 gene activation in a case of acute myeloid leukemia with the t (2; 11)(q31; p15) translocation. Leukemia. 2000;14(9):1621‐1629. [DOI] [PubMed] [Google Scholar]
- 116. Rasouli M, Blair H, Troester S, et al. The MLL–menin interaction is a therapeutic vulnerability in NUP98‐rearranged AML. HemaSphere. 2023;7(8):e935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Crescenzi B, Nofrini V, Barba G, et al. NUP98/11p15 translocations affect CD34+ cells in myeloid and T lymphoid leukemias. Leuk Res. 2015;39(7):769‐772. [DOI] [PubMed] [Google Scholar]
- 118. Taketani T, Taki T, Nakamura T, et al. High frequencies of simultaneous FLT3‐ITD, WT1 and KIT mutations in hematological malignancies with NUP98‐fusion genes. Leukemia. 2010;24(11):1975‐1977. [DOI] [PubMed] [Google Scholar]
- 119. De Rooij JDE, Branstetter C, Ma J, et al. Pediatric non–Down syndrome acute megakaryoblastic leukemia is characterized by distinct genomic subsets with varying outcomes. Nat Genet. 2017;49(3):451‐456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Lavallée VP, Lemieux S, Boucher G, et al. Identification of MYC mutations in acute myeloid leukemias with NUP98–NSD1 translocations. Leukemia. 2016;30(7):1621‐1624. [DOI] [PubMed] [Google Scholar]
- 121. McNeer NA, Philip J, Geiger H, et al. Genetic mechanisms of primary chemotherapy resistance in pediatric acute myeloid leukemia. Leukemia. 2019;33(8):1934‐1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Wei W, Liu Q, Song F, et al. Alkaloid‐based regimen is beneficial for acute myeloid leukemia resembling acute promyelocytic leukemia with NUP98/RARG fusion and RUNX1 mutation: a case report. Medicine. 2020;99(40):e22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Umeda M, Michmerhuizen N, Hiltenbrand R, et al. Genetic and functional characterization of NUP98‐rearranged leukemia. Blood. 2023;142:1562. [Google Scholar]
- 124. Cui J, Xie J, Qin L, Chen S, Zhao Y, Wu D. A unique acute myeloid leukemia patient with cryptic NUP98‐NSD1 gene and ASXL1 mutation. Leuk Lymphoma. 2016;57(1):196‐198. [DOI] [PubMed] [Google Scholar]
- 125. Terlecki‐Zaniewicz S, Eder T, Schmöllerl J, et al. Biomolecular condensation drives leukemia caused by NUP98‐fusion proteins. Nat Struct Mol Biol. 2021;28(2):190‐201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Jevtic Z, Allram M, Grebien F, Schwaller J. Biomolecular condensates in myeloid leukemia: what do they tell us? HemaSphere. 2023;7(7):e923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Ahn JH, Davis ES, Daugird TA, et al. Phase separation drives aberrant chromatin looping and cancer development. Nature. 2021;595(7868):591‐595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Chandra B, Michmerhuizen NL, Shirnekhi HK, et al. Phase separation mediates NUP98 fusion oncoprotein leukemic transformation. Cancer Discovery. 2022;12(4):1152‐1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Jevtic Z, Matafora V, Casagrande F, et al. SMARCA5 interacts with NUP98‐NSD1 oncofusion protein and sustains hematopoietic cells transformation. J Exp Clin Cancer Res. 2022;41(1):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Oka M, Mura S, Yamada K, et al. Chromatin‐prebound Crm1 recruits Nup98‐HoxA9 fusion to induce aberrant expression of Hox cluster genes. eLife. 2016;5:e09540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Pascual‐Garcia P, Jeong J, Capelson M. Nucleoporin Nup98 associates with Trx/MLL and NSL histone‐modifying complexes and regulates Hox gene expression. Cell Rep. 2014;9(2):433‐442. [DOI] [PubMed] [Google Scholar]
- 132. Hnisz D, Shrinivas K, Young RA, Chakraborty AK, Sharp PA. A phase separation model for transcriptional control. Cell. 2017;169(1):13‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Sabari BR, Dall'Agnese A, Boija A, et al. Coactivator condensation at super‐enhancers links phase separation and gene control. Science. 2018;361(6400):eaar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Kim S, Shendure J. Mechanisms of interplay between transcription factors and the 3D genome. Mol Cell. 2019;76(2):306‐319. [DOI] [PubMed] [Google Scholar]
- 135. Schmoellerl J, Barbosa IAM, Eder T, et al. CDK6 is an essential direct target of NUP98 fusion proteins in acute myeloid leukemia. Blood. 2020;136(4):387‐400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Heikamp EB, Henrich JA, Perner F, et al. The Menin‐MLL1 interaction is a molecular dependency in NUP98‐rearranged AML. Blood. 2022;139(6):894‐906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Staffas A, Arabanian LS, Wei SY, et al. Upregulation of Flt3 is a passive event in Hoxa9/Meis1‐induced acute myeloid leukemia in mice. Oncogene. 2017;36(11):1516‐1524. [DOI] [PubMed] [Google Scholar]
- 138. Georgi J‐A, Stasik S, Eckardt J‐N, et al. UBTF tandem duplications are rare but recurrent alterations in adult AML and associated with younger age, myelodysplasia, and inferior outcome. Blood Cancer J. 2023;13(1):88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Liu J, Qin Y‐Z, Yang S, et al. Meis1 is critical to the maintenance of human acute myeloid leukemia cells independent of MLL rearrangements. Ann Hematol. 2017;96(4):567‐574. [DOI] [PubMed] [Google Scholar]
- 140. Umeda M, Ma J, Huang BJ, et al. Integrated genomic analysis identifies UBTF tandem duplications as a recurrent lesion in pediatric acute myeloid leukemia. Blood Cancer Discovery. 2022;3(3):194‐207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Oka M, Asally M, Yasuda Y, Ogawa Y, Tachibana T, Yoneda Y. The mobile FG nucleoporin Nup98 is a cofactor for Crm1‐dependent protein export. Mol Biol Cell. 2010;21(11):1885‐1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Wang GG, Song J, Wang Z, et al. Haematopoietic malignancies caused by dysregulation of a chromatin‐binding PHD finger. Nature. 2009;459(7248):847‐851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Brennan K, Zheng H, Fahrner JA, et al. NSD1 mutations deregulate transcription and DNA methylation of bivalent developmental genes in Sotos syndrome. Hum Mol Gen. 2022;31:2164‐2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Wang GG, Cai L, Pasillas MP, Kamps MP. NUP98–NSD1 links H3K36 methylation to Hox‐A gene activation and leukaemogenesis. Nat Cell Biol. 2007;9(7):804‐812. [DOI] [PubMed] [Google Scholar]
- 145. Rio‐Machin A, Gómez‐López G, Muñoz J, et al. The molecular pathogenesis of the NUP98‐HOXA9 fusion protein in acute myeloid leukemia. Leukemia. 2017;31(9):2000‐2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Ghannam G, Takeda A, Camarata T, Moore MA, Viale A, Yaseen NR. The oncogene Nup98‐HOXA9 induces gene transcription in myeloid cells. J Biol Chem. 2004;279(2):866‐875. [DOI] [PubMed] [Google Scholar]
- 147. Li Z, Chen P, Su R, et al. PBX3 and MEIS1 cooperate in hematopoietic cells to drive acute myeloid leukemias characterized by a core transcriptome of the MLL‐rearranged disease. Cancer Res. 2016;76(3):619‐629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Heinrichs S, Brenner CJ, Kulkarni RV, Thomas Look A. MEIS1 Is a Required Transcription Factor for AML Cell Proliferation. Blood. 2006;108(11):1944. [Google Scholar]
- 149. Collins C, Wang J, Miao H, et al. C/EBPα is an essential collaborator in Hoxa9/Meis1‐mediated leukemogenesis. Proc Natl Acad Sci USA. 2014;111(27):9899‐9904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Wang GG, Pasillas MP, Kamps MP. Persistent transactivation by meis1 replaces hox function in myeloid leukemogenesis models: evidence for co‐occupancy of meis1‐pbx and hox‐pbx complexes on promoters of leukemia‐associated genes. Mol Cell Biol. 2006;26(10):3902‐3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Palmqvist L, Argiropoulos B, Pineault N, et al. The Flt3 receptor tyrosine kinase collaborates with NUP98‐HOX fusions in acute myeloid leukemia. Blood. 2006;108(3):1030‐1036. [DOI] [PubMed] [Google Scholar]
- 152. Tarlock K, Gerbing RB, Ries RE, et al. Prognostic impact of co‐occurring mutations in FLT3‐ITD pediatric acute myeloid leukemia. Blood Adv. 2024;8:2094‐2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Yang W. Nuclear‐Cytoplasmic Transport, vol. 33. Springer; 2018. [Google Scholar]
- 154. Harada K, Doki N, Aoki J, et al. Outcomes after allogeneic hematopoietic stem cell transplantation in patients with acute myeloid leukemia harboring t (7; 11)(p15; p15). Haematologica. 2018;103(2):e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Shen Y, Zhang T, Zhang L, et al. Allogeneic stem cell transplantation can prolong the survival of patients with NUP98‐rearranged acute myeloid leukemia. Bone Marrow Transplant. 2023;58(10):1149‐1151. [DOI] [PubMed] [Google Scholar]
- 156. Dang CV, Reddy EP, Shokat KM, Soucek L. Drugging the 'undruggable' cancer targets. Nat Rev Cancer. 2017;17(8):502‐508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Cantilena S, Gasparoli L, Pal D, et al. Direct targeted therapy for MLL‐fusion‐driven high‐risk acute leukaemias. Clin Transl Med. 2022;12(6):e933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Fiskus W, Daver N, Boettcher S, et al. Activity of menin inhibitor ziftomenib (KO‐539) as monotherapy or in combinations against AML cells with MLL1 rearrangement or mutant NPM1. Leukemia. 2022;36:2729‐2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Krivtsov AV, Evans K, Gadrey JY, et al. A menin‐MLL inhibitor induces specific chromatin changes and eradicates disease in models of MLL‐rearranged leukemia. Cancer Cell. 2019;36(6):660‐673. e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Klossowski S, Miao H, Kempinska K, et al. Menin inhibitor MI‐3454 induces remission in MLL1‐rearranged and NPM1‐mutated models of leukemia. J Clin Invest. 2020;130(2):981‐997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Duployez N, Vasseur L, Kim R, et al. UBTF tandem duplications define a distinct subtype of adult de novo acute myeloid leukemia. Leukemia. 2023;37(6):1245‐1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Barajas JM, Rasouli M, Umeda M, et al. Acute myeloid leukemias with UBTF tandem duplications are sensitive to Menin inhibitors. Blood J. 2024;143(7):619‐630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Grembecka J, He S, Shi A, et al. Menin‐MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat Chem Biol. 2012;8(3):277‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Issa GC, Aldoss I, DiPersio J, et al. The menin inhibitor revumenib in KMT2A‐rearranged or NPM1‐mutant leukaemia. Nature. 2023;615(7954):920‐924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Perner F, Stein EM, Wenge DV, et al. MEN1 mutations mediate clinical resistance to menin inhibition. Nature. 2023;615(7954):913‐919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Issa GC, Cuglievan B, DiNardo CD, et al. Early results of the phase I/II study investigating the all‐oral combination of the menin inhibitor revumenib (SNDX‐5613) with decitabine/cedazuridine (ASTX727) and venetoclax in acute myeloid leukemia (SAVE). Blood. 2023;142:58. [Google Scholar]
- 167. Fathi A, Wang E, Issa G, et al. P504: updated data for Ziftomenib in patients with NPM1‐mutated relapsed or refractory acute myeloid leukemia. HemaSphere. 2023;7(S3):e19161da. [Google Scholar]
- 168. Podar K, Shah J, Chari A, Richardson PG, Jagannath S. Selinexor for the treatment of multiple myeloma. Expert Opin Pharmacother. 2020;21(4):399‐408. [DOI] [PubMed] [Google Scholar]
- 169. Placke T, Faber K, Nonami A, et al. Requirement for CDK6 in MLL‐rearranged acute myeloid leukemia. Blood. 2014;124(1):13‐23. 10.1182/blood-2014-02-558114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Martinez‐Soria N, McKenzie L, Draper J, et al. The oncogenic transcription factor RUNX1/ETO corrupts cell cycle regulation to drive leukemic transformation. Cancer Cell. 2018;34(4):626‐642. 10.1016/j.ccell.2018.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Lopez S, Voisset E, Tisserand JC, et al. An essential pathway links FLT3‐ITD, HCK and CDK6 in acute myeloid leukemia. Oncotarget. 2016;7(32):51163‐51173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Jones LM, Tarlock K, Cooper T. Targeted therapy in pediatric AML: an evolving landscape. Pediatric Drugs. 2021;23(5):485‐497. [DOI] [PubMed] [Google Scholar]
- 173. Larrosa‐Garcia M, Baer MR. FLT3 inhibitors in acute myeloid leukemia: current status and future directions. Mol Cancer Ther. 2017;16(6):991‐1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Thanasopoulou A, Tzankov A, Schwaller J. Potent co‐operation between the NUP98‐NSD1 fusion and the FLT3‐ITD mutation in acute myeloid leukemia induction. Haematologica. 2014;99(9):1465‐1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Schmalbrock LK, Dolnik A, Cocciardi S, et al. Clonal evolution of acute myeloid leukemia with FLT3‐ITD mutation under treatment with midostaurin. Blood. 2021;137(22):3093‐3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Heidel F, Solem FK, Breitenbuecher F, et al. Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn‐676 in the FLT3 tyrosine kinase domain. Blood. 2006;107(1):293‐300. [DOI] [PubMed] [Google Scholar]
- 177. Man CH, Fung TK, Ho C, et al. Sorafenib treatment of FLT3‐ITD+ acute myeloid leukemia: favorable initial outcome and mechanisms of subsequent nonresponsiveness associated with the emergence of a D835 mutation. Blood. 2012;119(22):5133‐5143. [DOI] [PubMed] [Google Scholar]
- 178. Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377(5):454‐464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Wei AH, Kennedy GA, Morris KL, et al. Results of a phase 2, randomized, double‐blind study of sorafenib versus placebo in combination with intensive chemotherapy in previously untreated patients with FLT3‐ITD acute myeloid leukemia (ALLG AMLM16). Blood. 2020;136:36‐38.32430502 [Google Scholar]
- 180. Pollard JA, Alonzo TA, Brown PA, et al. Sorafenib in combination with standard chemotherapy for children with high allelic ratio FLT3/ITD+ AML improves event‐free survival and reduces relapse risk: a report from the Children's Oncology Group Protocol AAML1031. Blood. 2019;134:292. [Google Scholar]
- 181. Burchert A, Bug G, Fritz LV, et al. Sorafenib maintenance after allogeneic hematopoietic stem cell transplantation for acute myeloid leukemia with FLT3–internal tandem duplication mutation (SORMAIN). J Clin Oncol. 2020;38(26):2993‐3002. [DOI] [PubMed] [Google Scholar]
- 182. Uras IZ, Sexl V, Kollmann K. CDK6 inhibition: a novel approach in AML management. Int J Mol Sci. 2020;21(7):2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Kivioja JL, Thanasopoulou A, Kumar A, et al. Dasatinib and navitoclax act synergistically to target NUP98‐NSD1+/FLT3‐ITD+ acute myeloid leukemia. Leukemia. 2019;33(6):1360‐1372. [DOI] [PubMed] [Google Scholar]
- 184. Friedman R. The molecular mechanisms behind activation of FLT3 in acute myeloid leukemia and resistance to therapy by selective inhibitors. Biochim Biophys Acta. 2022;1877(1):188666. [DOI] [PubMed] [Google Scholar]
- 185. Trabal A, Gibson A, He J, et al. Venetoclax for acute myeloid leukemia in pediatric patients: a Texas Medical Center Experience. Cancers. 2023;15(7):1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Karol SE, Alexander TB, Budhraja A, et al. Venetoclax in combination with cytarabine with or without idarubicin in children with relapsed or refractory acute myeloid leukaemia: a phase 1, dose‐escalation study. Lancet Oncol. 2020;21(4):551‐560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Masetti R, Baccelli F, Leardini D, et al. Venetoclax‐based therapies in pediatric advanced MDS and relapsed/refractory AML: a multicenter retrospective analysis. Blood Adv. 2023;7(16):4366‐4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Kontro M, Kumar A, Majumder MM, et al. HOX gene expression predicts response to BCL‐2 inhibition in acute myeloid leukemia. Leukemia. 2017;31(2):301‐309. [DOI] [PubMed] [Google Scholar]
- 189. Sun H, Yan H, Yan Z, Zhu Y, Yang G, Zhang S. Acute myeloid leukemia patients with NUP98::NSD1 showing initially poor treatment response can benefit from FLT3 inhibitors and venetoclax as well as HSCT. Ann Hematol. 2023;102(2):473‐475. [DOI] [PubMed] [Google Scholar]
- 190. Wen X, Wu Y, Huang P, Zheng H. Combined treatment with venetoclax, dasatinib, and FLT3 inhibitors for NUP98‐NSD1+/FLT3‐ITD+ acute myeloid leukemia: a pediatric case report. Pediatr Blood Cancer. 2023;70(7):e30308. [DOI] [PubMed] [Google Scholar]
- 191. Xu H, Yu H, Xu J, et al. Refractory pediatric acute myeloid leukemia expressing NUP98‐NSD1 fusion gene responsive to chemotherapy combined with venetoclax and decitabine. Pediatr Blood Cancer. 2023;70(3):e30021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.