

Abstract
The aggregation of amyloid-beta peptide and tau protein dysregulation are implicated to play key roles in Alzheimer’s disease pathogenesis and are considered the main pathological hallmarks of this devastating disease. Physiologically, these two proteins are produced and expressed within the normal human body. However, under pathological conditions, abnormal expression, post-translational modifications, conformational changes, and truncation can make these proteins prone to aggregation, triggering specific disease-related cascades. Recent studies have indicated associations between aberrant behavior of amyloid-beta and tau proteins and various neurological diseases, such as Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis, as well as retinal neurodegenerative diseases like Glaucoma and age-related macular degeneration. Additionally, these proteins have been linked to cardiovascular disease, cancer, traumatic brain injury, and diabetes, which are all leading causes of morbidity and mortality. In this comprehensive review, we provide an overview of the connections between amyloid-beta and tau proteins and a spectrum of disorders.
Keywords: amyloid-beta, cancer, cardiovascular diseases, diabetes, neurodegeneration, Tau, traumatic brain injury
Introduction
Extracellular amyloid plaques or senile plaques composed of the amyloid-beta (Aβ) and intracellular neurofibrillary tangles (NFT) comprising phosphorylated tau protein are known as the main pathological hallmarks of Alzheimer’s disease (AD) since the early 1900s when Alois Alzheimer first published his historical treatise that formally introduced the disease (Zilka and Novak, 2006; d‘Errico and Meyer-Luehmann, 2020). While senile plaques and NFT are well-established pathological hallmarks of AD, the presence of one or both of these has also been reported in other diseases. In this review, we provide an overview of the physiological role of amyloid-beta and tau proteins, mechanisms underlying their accumulation, and pathogenesis in diseases including cardiovascular diseases (CVD), cerebral amyloid angiopathy (CAA) and stroke, cancer, diabetes, retinal diseases, Parkinson’s disease (PD), traumatic brain injury (TBI) In addition, we briefly discuss the role of Aβ and tau protein in other conditions such as Autism, multiple sclerosis (MS), motor neuron disease, Huntington’s disease (HD), Creutzfeldt-Jakob disease (CJD) and Wilson’s disease (WD) where dysregulation of these two proteins has been reported (Figure 1).
Figure 1.
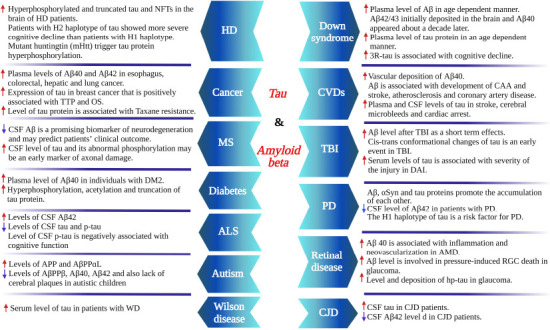
Various diseases associated with tau protein and amyloid beta and some main related points.
Created with BioRender.com. Aβ: Amyloid-beta; ALS: amyotrophic lateral sclerosis; AMD: age-related macular degeneration; APP: amyloid precursor protein; CAA: cerebral amyloid angiopathy; CJD: Creutzfeldt-Jakob disease; CSF: cerebrospinal fluid; CVD: cardiovascular disease; DAI: diffuse axonal injury; HD: Huntington’s disease; MS: multiple sclerosis; NTF: neurofibrillary tangles; OS: overall survival; PD: Parkinson’s disease; p-tau: phosphorylated tau; TBI: traumatic brain injury; TM2: type 2 diabetes; TTP: time to progression; WD: Wilson disease.
Search Strategy
We conducted a comprehensive literature search using various search engines, such as PubMed, Scopus, and Web of Science. The search was performed without any year restrictions, and we utilized keywords such as “amyloid-beta”, “amyloid plaques”, “senile plaques”, “tau”, and “neurofibrillary tangles”. Additionally, we manually screened the references of selected studies to identify potentially relevant articles for inclusion in this narrative review. Only English-language documents were considered.
Amyloid-β Formation and Aggregation
Aβ refers to peptides with 36–43 amino acids that derive from amyloid-β precursor protein (AβPP, APP), APP is a single-pass transmembrane glycoprotein expressed in many tissues, especially in the brain in both neuronal and non-neuronal cells (Marsden et al., 2011; Chen et al., 2017). APP is located on chromosome 21q21.3 and belongs to a larger gene family in humans which has two other members including the APP-like protein-1 (APLP1) and the APP-like protein-2 (APLP2). APP and APLP2 are expressed in several tissues, while APLP1 expression is limited to neural tissue (Pandey et al., 2016; Chen et al., 2017). These members have similar structures and are processed in the same manner; however, the Aβ sequence, which is involved in senile plaques is specific to APP (Chitranshi et al., 2021). Differential mRNA splicing of exons 7, and 8 results in the expression of three isoforms including the 695 amino acid isoform, which is the main isoform in the brain, and 751 and 770 amino acid isoforms that are mainly expressed in peripheral cells and platelets (Figure 2). APP is first cleaved by ɑ- or β-secretase, which starts two different pathways, named non-amyloidogenic and amyloidogenic pathways, respectively (Kojro and Fahrenholz, 2005; Sun et al., 2015). In a non-amyloidogenic pathway, cleavage by ɑ-secretase results in the amino-terminal fragment named secreted APP α (sAPP α) and the 83 amino acid long carboxyterminal fragments (CTF83 or C83), then CTF83 subjected to γ-secretase cleavage that produced P3 (3 kDa) and amino-terminal APP intracellular domain; however, in the amyloidogenic pathway, β-secretase particularly beta-secretase 1 (BACE1) cleavage liberates the amino-terminal fragment named secreted APPβ (sAPPβ) and the 99 amino acid long carboxyterminal fragment (CTF99 or C99) that produced Aβ (4 kDa) and APP intracellular domain following cleavage by γ-secretase (Figure 3; Kojro and Fahrenholz, 2005; Zhang et al., 2011). Generated Aβ has 39–43 amino acids but Aβ with 40 amino acids is relatively more abundant (Murphy and LeVine, 2010), while Aβ 42 is the predominant protein component in senile plaques probably due to faster aggregation of Aβ-42 compared to Aβ-40, and might be more toxic (Meisl et al., 2014; Wang et al., 2021). Several factors, including aging, inflammation, renal dysfunction, ischemia, genetic polymorphisms, and drugs, increase tissue deposition of Aβ by augmenting APP production or by decreasing Aβ clearance and degradation (Mawuenyega et al., 2010; Sadigh-Eteghad et al., 2015; Abyadeh et al., 2023a).
Figure 2.
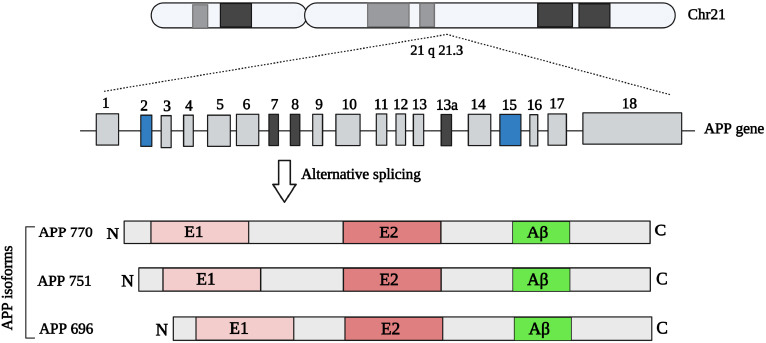
APP gene and different isoforms resulted from alternative splicing.
Created with BioRender.com. Aβ: Amyloid-beta; APP: Aβ precursor protein.
Figure 3.
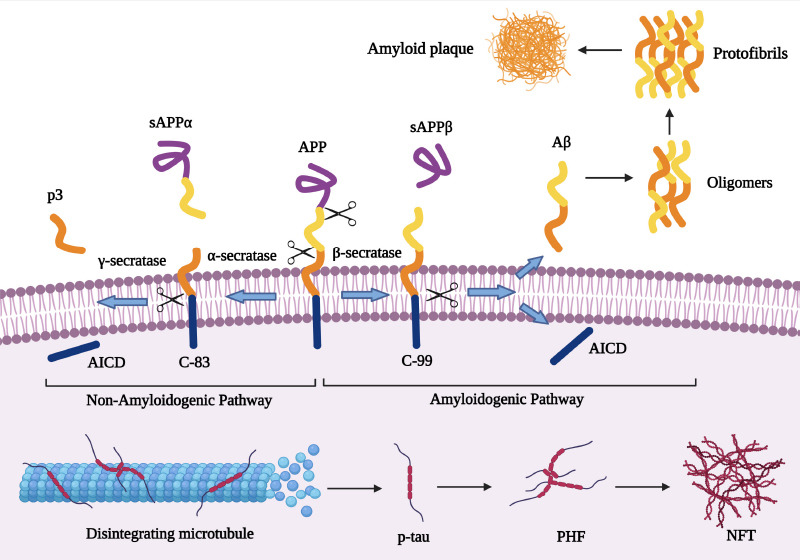
Two main pathological hallmarks of AD include the amyloid plaque and the NFT formation process including APP proteolysis in the non-amyloidogenic and amyloidogenic pathway, and also tau protein aggregation and NFT formation.
Created with BioRender.com. Aβ: Amyloid-beta; AICD: intracellular domain; APP: amyloid precursor protein; NFT: neurofibrillary tangles; PHF: paired helical filaments; p-tau: phosphorylated tau; sAPP: soluble APP beta protein.
Senile plaque formation is a four-step process including (1) primary nucleation, where Aβ monomers interact with each other molecules (lipids, alpha-synuclein) and form small soluble aggregates also called oligomers that are highly toxic and suggested to play a main role in cell and tissue toxicity; (2) elongation, in this step Aβ monomers add to existing soluble aggregates and increase aggregate length; (3) secondary nucleation, existing aggregates trigger the formation of new small soluble aggregates; (4) fragmentation, in which formed fibrils break down into several fibrils (Santos et al., 2016; Chen et al., 2017). Senile plaques have been observed in several diseases as mentioned before, particularly in the brain of AD patients, however, these plaques have also been observed in some cognitively normal older individuals (Murray and Dickson, 2014; Mormino and Papp, 2018).
Tau Protein
Tau is a microtubule (MT) associated protein (MAP) that is involved in the assembly and stabilization of MTs and is also a key player in the DNA and RNA protection (Violet et al., 2014). Tau is encoded by the microtubule-associated protein tau (MAPT) gene with a size of about 50 kb located on chromosome 17q21 and contains 16 exons, Alternative splicing of exons 2, 3, and 10 leads to the formation of six different isoforms of 352–441 amino acids tau proteins, which can be divided into two groups namely 3E and 4R based on whether they have three or four carboxy-terminal microtubule-binding repeat domains (Figure 4; Kolarova et al., 2012; Huda and Pan, 2018; Barbier et al., 2019). All of these six isoforms can be found in the brain and are mostly expressed by neurons and to some extent by astrocytes and oligodendrocytes (Mietelska-Porowska et al., 2014; Maté de Gérando et al., 2021). Tau interacts with the C-terminus of tubulin and increases their assembly into the MTs, which are involved in the formation and stability of the neuronal cytoskeleton, axonal transport, neurite outgrowth, and cell division (Rodríguez-Martín et al., 2013; Barbier et al., 2019). Physiologically, tau is a soluble and unfolded protein; however, in pathological conditions, it becomes insoluble and aggregates into paired helical filaments and NFTs (Figure 3). MT binding ability and DNA protection feature of tau protein is affected by its gene mutation, conformational changes, and post-translational modifications (PTMs), particularly phosphorylation (Violet et al., 2014). Tau protein undergoes several PTMs including phosphorylation, acetylation, truncation, nitration, glycation, glycosylation, and ubiquitination; however, as the most abundant PTM, phosphorylation of tau protein has been more studied and suggested as the key PTM in the pathological aggregation of tau (Mietelska-Porowska et al., 2014; Abyadeh et al., 2020; Zhao and Zlokovic, 2021). Tau protein has about 85 serine (S), threonine (T), and tyrosine (Y) sites of potential phosphorylation and is mainly phosphorylated by glycogen synthase kinase (GSK)-3β, cyclin-dependent kinase 5, mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK), and c-Jun N-terminal kinase in both physiological and pathological conditions and also dephosphorylated by the protein phosphatase 2A. Tau phosphorylation at specific sites is required for its normal function, however abnormal phosphorylation or hyperphosphorylation (hp-tau) triggers its conversion to a form that plays a pathological role (Mietelska-Porowska et al., 2014; Hobday and Parmar, 2021; Samimi et al., 2021; Abyadeh et al., 2022a). Abnormal phosphorylation and toxic tau formation are believed to be affected by several proteins such as Aβ, Fyn kinase, peptidylprolyl cis/trans isomerase, NIMA-interacting 1 (Pin1), heat shock cognate 70, heat shock protein 90, immunophilins FKBP51 and FKBP52, α-synuclein (α-Syn) or actin interacting protein PACSIN1 (Prots et al., 2013; Mietelska-Porowska et al., 2014).
Figure 4.
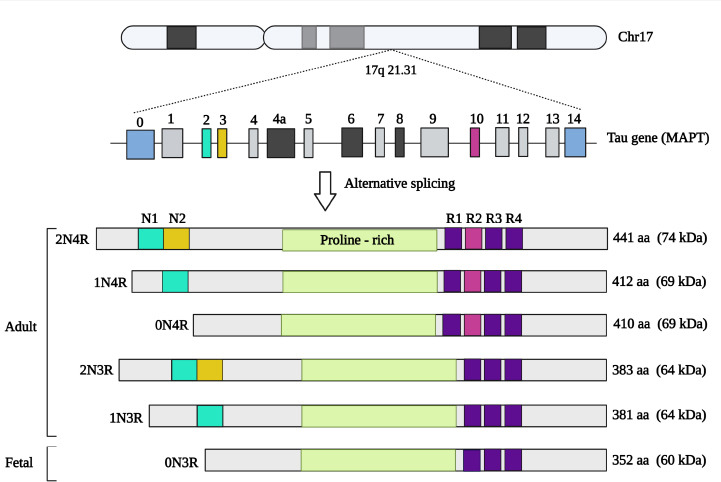
Tau gene (MAPT) and different isoforms resulted from alternative splicing.
Created with BioRender.com. MAPT: Microtubule-associated protein tau.
The pathological role of tau protein has been investigated in several disorders including brain neurodegenerative diseases, retinal diseases, PD, cancer, and TBI (Anderson et al., 2008; Baquero et al., 2011; Barbier et al., 2019). However, it has been studied more extensively in a group of neurodegenerative diseases termed tauopathies. These disorders are histopathologically characterized by tau protein aggregates in neurons or glial cells, or both (Samimi et al., 2021). Tauopathies are divided into two groups: primary and secondary tauopathies. In primary tauopathies, tau is the main contributing factor of the neurodegenerative process such as Pick’s disease, progressive supranuclear palsy, and chronic traumatic encephalopathy (CTE), however, in secondary tauopathies, tau aggregation is not the primary cause of neurodegeneration (Josephs, 2017; Chung et al., 2021). Interestingly toxic forms of tau protein have been suggested to spread from cell to cell in a prion-like fashion, and injection of tau aggregates obtained from AD brain into the mouse brain was shown to induce endogenous tau aggregation, which indicated that toxic tau can acquire the ability to self-propagate, like the prion proteins that are responsible for CJD pathology (Walker, 2018). However, prion-like propagation of tau protein was shown to require isoform pairing between the infecting prion and the recipient cells and even tau aggregates from AD and CTE patients that have two isoforms (3R and 4R) did not significantly infect either 3R- or 4R-expressing cells (Woerman et al., 2016; Wood, 2018).
Cardiovascular Diseases
CVDs are the leading cause of death globally, which takes over 17.9 million lives each year and is estimated to reach 22.2 million death annually by 2030 (Ruan et al., 2018). CVDs and AD share several risk factors (particularly aging) and pathological mechanisms. There is strong evidence indicating a causal association between CVDs and dementias; and individuals with CVDs are at higher risk of developing AD (Attems and Jellinger, 2014; Santos et al., 2017; Tini et al., 2020). A growing body of experimental and clinical evidence suggests that Aβ, the pathological hallmark of AD, constitutes a risk factor for CVDs (Tublin et al., 2019; Stakos et al., 2020). Here, we summarize current knowledge on mechanisms underpinning Aβ pathogenesis in CVDs that are currently centralized around the pathogenesis of Aβ in vascular components.
CAA is a specific cerebrovascular disease in which Aβ plays a key pathological role, CAA is a cerebrovascular disorder that is caused by deposition of Aβ peptide (mainly Aβ1–40) within the capillaries, arterioles, leptomeninges and small to medium-sized cerebral blood vessels. It is believed to result from a defective drainage of neuronal Aβ from these vessels and faulty Aβ clearance but not overproduction of Aβ (Biffi and Greenberg, 2011; Goulay et al., 2020; Stakos et al., 2020). The source of Aβ in CAA is mainly neuronal cells as observed in transgenic mice models and also blood plasma and the muscular layer of vessel walls (Herzig et al., 2006; Auriel and Greenberg, 2012; Goulay et al., 2020). Aβ40 is the predominant type of Aβ in vascular deposition. Although vascular deposition of Aβ1–42 is limited and it is the main Aβ species in parenchymal lesions of AD, its presence facilitates Aβ1–40 vascular deposition (McGowan et al., 2005; Schaich et al., 2019). Aβ deposition also impairs perivascular space drainage leading to perivascular space enlargement in the cortical grey matter and the underlying white matter, which can be seen in brain images and is a potential biomarker of neurovascular disease (Ramirez et al., 2016; Charidimou et al., 2017; Goulay et al., 2020). CAA results in hemorrhagic and ischemic lesions, blood-brain barrier breakdown, neurological deficits, cognitive impairment, stroke, dementia, and death (DeSimone et al., 2017; Goulay et al., 2020). While CAA increases the risk of stroke, in turn, stroke-induced hypoxia increases the expression of APP in vascular smooth muscle cells and thereby increases CAA development (Rensink et al., 2003; Goulay et al., 2020). Moreover, hypoxia condition stabilizes hypoxia-inducible factor-1ɑ, which bind to hypoxia-responsive element on the BACE1 gene promoter and leads to increased Aβ processing in both endothelial cells and macrophages and result in increased levels of Aβ particularly Aβ1–40 in patients with acute ischemic stroke (Schaich et al., 2019). Hypertension as a common risk factor between AD and stroke was shown to increase Aβ-induced neurovascular dysfunction, β-secretase activity, and amyloidogenic processing of APP (Faraco et al., 2016).
The presence of Aβ also has been reported in atherosclerotic plaque; atherosclerosis is a multifactorial disease and several risk factors contribute to atherosclerotic lesion formation (Mundi et al., 2018; Markin et al., 2020). Inflammation plays a central role in the initiation and progression of atherosclerosis and currently, this disease is also known as an inflammatory disease (Spagnoli et al., 2007; Raggi et al., 2018). Aβ presence has been reported in human atherosclerotic plaques in the vicinity of activated macrophages and platelets (Tibolla et al., 2010; Lathe et al., 2014). In this regard, an in vitro investigation using human and murine cells indicated that platelet phagocytosis by perivascular macrophages leads to the processing of platelet-derived APP towards Aβ production, as mRNA of β-secretase has been found in macrophages. Subsequently, Aβ evokes macrophage activation as indicated by up-regulation of inducible nitric oxide synthase (a marker of macrophage activation), while phagocytosis of platelets from APP knockout mice did not stimulate macrophage activation (De Meyer et al., 2002; Jans et al., 2006; Spitzer et al., 2020). Moreover, Aβ is shown to be involved in macrophage inflammatory responses including increased reactive oxygen species (ROS) production and tumor necrosis factor-α expression via CD36-dependent signaling cascade. CD36 is a cell surface receptor on macrophages, platelets, and microvascular endothelium that promotes inflammation and thereby atherogenesis (Canton et al., 2013). Further studies showed that Aβ1-40 is the major form of Aβ within the human aortic atherosclerotic lesions and plasma levels of Aβ1–40 are associated with atherosclerosis progression (Jans et al., 2006). Moreover, APP overexpression in transgenic mice causes endothelial dysfunction through increasing oxidative stress and reducing the availability of nitric oxide; however, activation of peroxisome proliferator-activated receptor-delta can prevent APP-mediated endothelial dysfunction and modulate the level of nitric oxide (Kokjohn et al., 2011). The association of Aβ1–40 with vascular aging has been reported by both in vitro and in vivo studies (Bonda et al., 2011; Laina et al., 2018). Aβ1–40 mediated vascular aging is decreased by sirtuin 1, also known as NAD-dependent deacetylase, through increasing the expression of ADAM10 (ADAM metallopeptidase domain 10), which is an important α-secretase, and promoting the non-amyloidogenic pathway (Laina et al., 2018). Results of in vivo studies also showed impaired endothelial function, vascular development, angiogenesis, telomerase activity, and increased cellular senescence following exposure to Aβ (Donnini et al., 2010; Wang et al., 2015; Laina et al., 2018). However, increased circulating level of Aβ1–40 has been also observed in healthy elderly subjects, which might be due to decreased degradation of the peptide (Silverberg et al., 2010; Li et al., 2016). Current data are inconsistent with respect to the roles of Aβ in angiogenesis. While, in vitro studies indicate both anti-and pro-angiogenic features of Aβ in a dose-dependent manner, where higher concentration impairs angiogenesis and low concentration promotes angiogenesis through increasing cell proliferation, migration, and tube formation; in vivo studies showed increased cerebral vascularization in human AD brains and also APP transgenic animals (Biron et al., 2011; Cameron et al., 2012; Ristori et al., 2020).
There is also evidence indicating that Aβ interacts with endothelial cells of blood vessels and promotes the generation of superoxide radicals leading to impaired endothelial structure and function and a disrupted cerebrovascular autoregulation (Thomas et al., 1996; Niwa et al., 2002). In addition, a vasoactive role has been suggested for Aβ, which reduces acetylcholine-induced relaxation, enhances contraction of blood vessels, and reduces cerebral blood flow, and pretreatment with superoxide dismutase significantly resolved Aβ-related effects (Thomas et al., 1996; Iadecola et al., 1999). Interestingly, NADPH oxidase 2 (Nox2) inactivation also resulted in reduced APP-mediated vascular dysfunction without changing brain Aβ load and amyloid plaques, which indicated the key role of Nox2-derived ROS in APP pathogenesis (Park et al., 2008). Plasma concentration of Aβ1–40 is reported as an independent marker of aortic stiffness and also is associated with the severity of coronary artery calcium deposition score and coronary artery disease (Stamatelopoulos et al., 2018a).
In acute coronary syndrome, Aβ metabolism is increased and associated with clinical presentation, moreover, Aβ peptides possibly derived from platelet also accumulate in the myocardium with ischemic heart failure (Kitazume et al., 2012; Stamatelopoulos et al., 2018b; Inyushin et al., 2020). Interestingly, Aβ1–40 but not Aβ1–42 was reported to be increased and associated with coronary artery disease, and diabetes mellitus (DM) type 2 (Roeben et al., 2016). Moreover, increased Aβ metabolism, inflammation, and cognitive dysfunction were observed in mice models of myocardial infarction induced by ligation of the left anterior descending artery, which indicated a causal association of infarction with AD (Hong et al., 2013). This hypothesis also has been confirmed in another study that showed increased ROS, Aβ deposition, tau protein phosphorylation, and activated microglia in the brain of myocardial infarction mouse models (Zhang and Luo, 2020). Intriguingly, a retrospective cross-sectional study found the intramyocardial aggregates of Aβ1–40 and Aβ1–42 in AD patients, in addition, these patients showed diastolic dysfunction, suggesting AD as a systemic disease that may lead to failure of several organs (Troncone et al., 2016).
The expression of tau protein has been reported in the heart, and mouse modeling studies have shown that loss of this protein impaired cardiac function leading to elevated blood pressure, cardiac hypertrophy, and decreased left atrial contractility that was exacerbated with aging (Gu et al., 1996; Betrie et al., 2017). However, like Aβ, dysfunctional tau protein is also reported in vascular disease, especially cerebrovascular disease, and in this regards several tau protein modifications have been reported including tau hyperphosphorylation, de-phosphorylation, and truncation. The association of plasma and cerebrospinal fluid (CSF) levels of tau protein has been reported to be associated with the risk of developing, severity, and outcome of stroke and the presence of cerebral microbleeds, which increase the risk of stroke and dementia (De Vos et al., 2017; Romero et al., 2020). Furthermore, its serum level was also shown to be elevated in patients with cardiac arrest, possibly released into the serum due to brain hypoxia, and was negatively associated with neurological outcomes after 6 months (Mörtberg et al., 2011; Randall et al., 2013). Overexpression of tau protein-induced blood vessel abnormalities in the mouse cortex such as abnormal and spiraling morphologies, increased density, and reduced size of blood vessels; these changes were associated with cortical atrophy and overexpression of angiogenesis-related genes including Serpine1, Vegfa, and Plau in CD31-positive endothelial cells (Bennett et al., 2018). Collectively, these results indicated that tauopathy adversely affects brain endothelial cells and the integrity of the brain’s microvasculature, resulting in hypoperfusion of the cerebral cortex (Thomas et al., 2015; Bennett et al., 2018). In turn, hypoperfusion may increase p-tau through the down-regulation of neuroglobin, a scavenger of ROS, increased free radicals, and the induction of neuroinflammatory cascade, ultimately leading to blood-brain barrier compromised permeability and neuronal cell death (Raz et al., 2019).
Moreover, dephosphorylation and differential re-phosphorylation of tau protein in the canine brain has been observed after CA-induced ischemia and subsequent reperfusion. Immediate dephosphorylation of tau protein was observed after CA-induced cerebral ischemia, which was almost restored 24 hours after reperfusion, apart from phosphorylation of Ser262/356, which is involved in microtubule binding ability of tau protein (Mailliot et al., 2000). Furthermore, accumulation of tau protein and NFT-like formations have been observed in the brain of rodent stroke model that was associated with aberrant activation of Cdk5 and phosphorylation of GSK3, indicating the key role of Cdk5 and GSK3 in ischemia-induced phosphorylation and subsequent aggregation of tau protein (Morioka et al., 2006; Wen et al., 2007).
These studies collectively provide valuable insights into the complex relationship of Aβ and tau protein with cardiovascular and cerebrovascular diseases. They highlight the need for further research to understand the precise mechanisms underlying Aβ and tau protein dysregulation and its implications for cardiovascular and cerebrovascular health. Investigating the effects of Aβ and tau protein accumulation, modifications and their potential as diagnostic markers or therapeutic targets could help develop strategies for early detection and intervention in these diseases and also AD.
Cancer
Cancer is the second leading cause of death globally, which has caused around 10 million loss of lives in 2020 (Sung et al., 2021). APP, APLP2, and gamma synuclein are reported to be overexpressed in gastrointestinal, breast, prostate, and lung cancers (Hansel et al., 2003; Wu et al., 2007; Takagi et al., 2013; Pandey et al., 2015; Pandey et al., 2016; Ito et al., 2019).
Increased expression of APP has been observed in mice and human breast cancer cell lines, particularly in those with higher metastatic potential, moreover, a level of APP was shown to be associated with tumor development. Accordingly, APP knockdown cancer cell lines showed reduced cell growth, migration, and invasion ability through modulating insulin-like growth factor 1/AKT signaling pathway and subsequently AKT/FOXO signaling, and also increased p27kip1 and caspase-3-mediated apoptosis and sensitivity to chemotherapeutic agents (Lim et al., 2014). However, there are also reports indicating intact levels of AKT and p-AKT upon silencing APP in bladder cancer cells, and a significant decrease in levels of RAS, RAF, and phosphorylated-mitogen-activated protein kinase kinase. Moreover, silencing APP resulted in cell cycle arrest in the G2/M phase and inhibited cancer cells proliferation, migration, and invasion (Zhang et al., 2018).
Furthermore, the presence of APP in human prostate cancer cell lines was shown to be triggered with copper, as levels of this metal ion were increased in several cancer tissues. APP mitigates copper-induced growth inhibition possibly through a mechanism mediated by its copper-binding domain located in the E1 extracellular domain and phosphorylation of tyrosine residues within the cytosolic domain (Gupte and Mumper, 2009; Gough et al., 2014). In this regard, APP was shown to play a key role in liver cancer resistance to 5-fluorouracil (5-FU), an important anti-cancer drug; the level of APP in liver cancer cells showed an increase following treatment with 5-FU, and cell lines overexpressing APP were resistant to 5-FU. They showed decreased apoptosis possibly through increasing the expression of two apoptosis suppressor genes such as Bcl-2 and Bcl-xl and down-regulation of mitochondrial apoptotic pathways genes such as BAX and BID, while APP knock-down cells were more sensitive to 5-FU with a higher rate of apoptosis compared to the control cells (Wu et al., 2020a; Sethy and Kundu, 2021).
In prostate cancer cells, increased level of APP was associated with increased proliferation and migration possibly through increasing the expression of metalloproteinase (MMP) genes such as ADAM10 and ADAM17, and epithelial-mesenchymal transition (EMT)-related genes, including VIM, and SNAI2. Interestingly ADAM10 and ADAM17 are reported to act as ɑ-secretases for APP, and increased expression of these metalloproteinases has been found in several cancers, such as breast cancer; Increased expression of ADAM10 and sAPPα has been reported to be associated with worst outcome in non-luminal breast cancer. Moreover, similar functional effects were observed upon the down-regulation of APP or ADAM10. Interestingly, knockdown of ADAM10 resulted in reduced cell migration, which was reserved by adding sAPPα but not APP, suggesting the key role of ADAM10 in APP-mediated toxicity (Tsang et al., 2018; Wozniak and Ludwig, 2018). In addition, high expression of ADAM17 was shown to be associated with a shorter survival rate for breast cancer patients, and blocking this enzyme resulted in reduced proliferation of breast cancer cells (McGowan et al., 2008; Tsang et al., 2018). EMT is a necessary step for tumor metastasis; up-regulation of some other mesenchymal markers including MMP-9, MMP-2, MMP-3, N-cadherin and vimentin, and down-regulation of epidermal-associated markers such as N-cadherin and cytokeratin were also observed in breast cancer cell lines upon treatment with Aβ. In addition, Aβ affected the phosphorylation level of MAPK signaling pathway components including, mitogen-activated protein kinase kinase kinase 11 (MLK3), mitogen-activated protein kinase kinase 4 (MEK4), and mitogen-activated protein kinase 10, interestingly MEK inhibitor significantly reduced the phosphorylation level of MAPK signaling pathway components and expression of EMT genes, suggesting that Aβ activates MAPK signaling pathway. The downstream transcription factor of this pathway may trigger the expression of EMT genes, leading to enhanced migration and invasion of human breast cancer cells (Shi et al., 2014; Zhao et al., 2019; Wu et al., 2020b). In nasopharyngeal carcinoma cells, also inhibition of APP expression caused down-regulation of the MAPK signaling and subsequently decreased expression of EMT genes and resulted in decreased cell viability, migration, and invasion (Xu et al., 2019).
In pancreatic cancer cells, blocking β-secretase activity results in reduced growth and viability, however, did not affect the non-transformed pancreatic cell line (Peters et al., 2012; Pandey et al., 2016). Accumulation of both extracellular and intracellular Aβ in human glioma cells has been reported (Zayas-Santiago et al., 2020). Moreover, plasma levels of Aβ40 and Aβ42 were found to be significantly higher in several cancer types including esophagus cancer, colorectal cancer, hepatic cancer, and lung cancer compared to normal controls, although were slightly lower than AD samples (Jin et al., 2017). Accordingly, in AD patients with a cancer history, no differences were observed compared to AD patients without any history of cancer, but a significantly lower level of paired helical filament was observed in patients with a cancer history compared to control subjects (Yarchoan et al., 2017). Unlike the reported negative association of APP in cancer, increased levels of Aβ42 in tumor cells lead to telomere DNA damage, telomere uncapping, chromosome fusion, and telomere shortening, downregulation of telomerase reverse transcriptase and subsequently cell senescence and apoptosis (Qin et al., 2019). Furthermore, Aβ oligomers showed anti-proliferative effects on different cancer cells including human acute promyelocytic leukemia, human lung cancer, and human breast cancer (Pavliukeviciene et al., 2019).
A growing number of epidemiological studies have indicated both positive and negative associations of tau protein level with the risk of development and progression of several cancers, for example, high tau levels were reported in patients with breast cancer, particularly in estrogen receptor-positive and low-grade cancers and to some extent in estrogen receptor-negative and high-grade tumors, showed to be positively associated with the longer median time to tumor progression and overall survival (Pusztai et al., 2009; Baquero et al., 2011). However, in contrast to breast cancer, the level of tau protein was shown to be negatively associated with overall survival in epithelial ovarian cancer and prostate cancer patients (Smoter et al., 2013; Sekino et al., 2020). Moreover, tau protein expression also affects the response to microtubule-targeting chemotherapeutic agents such as taxanes, a group of drugs that inhibit microtubule depolymerization through binding to the β-subunit of the tubulin heterodimer, leading to impaired microtubule dynamic and thereby inhibit the process of cell division; high levels of tau protein was reported to negatively affect drug response in patients with different types of cancer including ovarian, gastric, prostate, breast, and non-small-cell lung cancer (Mimori et al., 2006; Smoter et al., 2013; Maloney et al., 2020; Papin and Paganetti, 2020). In vitro studies indicated that taxanes have the same tubulin-binding site as tau protein, therefore tau protein interferes with the binding of taxanes to tubulin and showed that the presence of tau protein decrease paclitaxel, a member of taxanes agents, binding and paclitaxel-induced MT polymerization (Rouzier et al., 2005; Gargini et al., 2019; Maloney et al., 2020).
Tau and APP have two conformations including trans and cis-conformations, where trans-conformation is functional and “healthy”, and facilitate their normal functions, while cis-conformation is pathogenic, formed in stress conditions after phosphorylation, is dysfunctional and prone to aggregation (Kondo et al., 2015; Wang et al., 2020). Conformational conversion between cis and trans is mediated by the Pin1 enzyme, which is down-regulated in AD (Wang et al., 2020). Pin1-knockout mice represent AD features such as hp-tau, Aβ accumulation, and neurodegeneration, surprisingly these mice models of AD were resistant to breast cancer induced by oncogene Ras or Neu over-expression (Wulf et al., 2004; Lanni et al., 2021). These results indicated the importance of tau and Aβ up-stream regulators in the development of cancer, as Pin1 was shown to be increased in several types of cancer and promote oncogenesis (Yu et al., 2020), therefore it may be at the crossroad between cancer and neurodegeneration and observed changes in tau and Aβ levels in cancer stem from Pin 1 alteration.
Diabetes Mellitus
In 2019, DM was reported to affect 463 million people worldwide and is estimated to hit 700 million by 2045. DM is also among the top 10 leading causes of death, and a major cause of blindness, kidney failure, heart attacks, stroke, and lower limb amputation (Saeedi et al., 2019; Sinclair, 2021). Diabetes is a disorder characterized by insufficient insulin and resistance to its metabolic effects, with hyperglycemia leading to chronic damage in multiple organ systems (Kottaisamy et al., 2021; Yao et al., 2021). Patients with diabetes show cognitive decline and also brain changes similar to those observed in AD brains and an increased prevalence of AD (nearly 65%) has been reported in diabetic patients, particularly in those with diabetes mellitus type 2 (DM2) (Arvanitakis et al., 2004; Stanciu et al., 2020). AD and DM share many risk factors and pathological changes such as the apolipoprotein E4, higher cholesterol, oxidative stress, mitochondrial dysfunction, inflammation, and resistance to insulin that comprises the core mechanism of DM2, which gave rise to a new term for AD named, type 3 diabetes (Stanciu et al., 2020; Diniz Pereira et al., 2021; Abyadeh et al., 2022). Increased plasma levels of Aβ40 (28%) and Aβ42 (37%) also have been observed in individuals with DM2 (Peng et al., 2020). Insulin dysregulation may affect both the production and degradation of Aβ and lead to increased extracellular levels of Aβ through increasing the activity of β-secretase and decreasing the release of insulin-degrading enzyme (IDE; one of the major Aβ degrading enzyme) into the extracellular space via inhibition of the PI3K-Akt pathway, activation of which promotes non-amyloidogenic processing of APP, furthermore insulin competitively inhibits Aβ degradation via IDE (Gasparini et al., 2002; Shieh et al., 2020). Conversely, expression of IDE is shown to be reduced in both AD and DM2 mice models and the size of Aβ plaques was inversely correlated with IDE activity, therefore reduced levels of IDE in diabetes were suggested as a potential trigger of Aβ accumulation and cognitive decline in both AD and DM2 (Li et al., 2018; Delikkaya et al., 2019). Aβ oligomers were shown to make a substantial loss of neuronal surface insulin receptors (IRs) in hippocampal neuronal culture, which may contribute to insulin resistance condition and in turn insulin resistance increases Aβ production and deposition in cerebral blood vessels leading to increased AD pathology (Zhao et al., 2008). Moreover, BACE1 was shown to degrade IRs in the liver in a glucose concentration-dependent manner and its plasma level and activity increase in diabetic conditions leading to a reduced amount of IRs; however, its effect on neuronal IRs is not still clear (Meakin et al., 2018; Bao et al., 2021). In addition, global deletion of BACE1 was shown to be associated with reduced risk of diet-induced obesity and diabetes in mice, while neuronal BACE1 knock-in resulted in Aβ accumulation, neuroinflammation, and more interestingly systemic diabetes in mice, which indicated the increased central BACE1 activity in AD patients as the potential mechanism underpinning the higher prevalence of metabolic disorders in AD patients (Meakin et al., 2012; Plucińska et al., 2014; Plucińska et al., 2016). While studies showed the association of nutrient-induced insulin resistance with elevated Aβ deposition in the brain of both humans and diabetic AD model mice, genetically induced insulin resistance mice (deficient for either IRS-2 or neuronal insulin-like growth factor 1 receptor) showed a protective effect against brain Aβ deposition, suggesting that the increased Aβ pathology might be due to high-fat diet-induced metabolic stress or inflammation but not direct effects of insulin signaling dysfunction (Wakabayashi et al., 2019). Interestingly, early intranasal insulin administration was suggested as a therapeutic option to improve memory and cognitive performance in people with mild cognitive impairment or early AD. However, a recent report by the same group did not confirm their previous findings (Craft et al., 2012; Suzanne, 2012; Craft et al., 2020). A suggested mechanism for the beneficial effects of insulin on AD symptoms is through decreasing Aβ generation. Insulin was shown to decrease the endocytosis rate of AβPP and increase AβPP O-GlcNAcylation via Akt insulin signaling leading to decreased sAβPPβ and increased sAβPPɑ production, collectively increasing non-amyloidogenic processing of the APP (Kwon et al., 2019).
There is a negative association between the level of tau protein phosphorylation and the insulin signaling pathway, which is interesting given the reports that patients with DM2 have increased CSF levels of p-tau (Moran et al., 2015; Lu et al., 2018). Moreover, the elevated level of tau hyperphosphorylation and its inability for binding to MTs have been observed in the brains of both DM1 and DM2 mice models (Chatterjee and Mudher, 2018).
Insulin regulates the phosphorylation of tau protein through the PI3K/AKT signaling pathway that leads to GSK-3β phosphorylation and inactivation, therefore an impaired insulin signaling pathway results in GSK-3β-mediated tau hyperphosphorylation. In addition, tau protein phosphorylation is also negatively regulated through modification by O-GlcNAcylation and impaired insulin-PI3K-AKT signaling may also lead to abnormal tau hyper-phosphorylation through down-regulation of O-GlcNAcylation (Liu et al., 2011; Hobday and Parmar, 2021). In turn, tau hyperphosphorylation and aggregation could further contribute to insulin signaling impairment through interacting with phosphatase and tensin homolog deleted on chromosome 10 (PTEN) which is a negative regulator of insulin/phosphoinositide 3-kinase signaling. In combination these changes can lead to cognitive dysfunction (Hobday and Parmar, 2021), which can be limited by the inhibition of GSK-3β (King et al., 2013). Moreover, tau acetylation and truncation resulted in disrupted tau-microtubule interactions and hastened aggregation of pathological tau in AD patients. In this respect, increased acetylation and truncation of tau protein have been also observed in diabetic mice models in hyperglycemic conditions, and this may lead to its dysfunction and aggregation (Kim et al., 2009; Chatterjee and Mudher, 2018). Another mechanism that affects tau protein hyperphosphorylation in AD is mTOR/S6K1 signaling, and its abnormal up-regulation is shown to be correlated with tau hyperphosphorylation and NFT formation in AD brains (Tang et al., 2013). mTOR/S6K1 signaling is also involved in glucose metabolism and required for memory formation (Sipula et al., 2006; Krebs et al., 2007; Lana et al., 2017). Interestingly up-regulation of mTOR/S6K1 signaling has been observed in the brain of diabetic mouse models and subsequent inhibition of mTOR signaling via rapamycin reduced the level of hp-tau and decreased DM-induced cognitive decline (Wang et al., 2014). Further analyses by the same group showed that caveolin-1, a transmembrane scaffolding protein that negatively regulates mTOR signaling, is down-regulated in chronic hyperglycemic conditions resulting in overactivity of mTOR/S6K signaling and subsequently tau hyperphosphorylation in neurons of diabetic rats (Wu et al., 2017a).
Interestingly, insulin accumulation as oligomers was observed in the brain of AD patients with hp-tau aggregations. In addition, insulin accumulation was independent of whether the patient had DM or not, indicating that peripheral and brain insulin levels are independently regulated. Further in vitro analyses showed that hp-tau-induced intraneuronal accumulation of insulin may lead to decreased IR levels (Craft et al., 2017). The identified shared risk factors and pathological changes, such as apolipoprotein E4, higher cholesterol, oxidative stress, mitochondrial dysfunction, inflammation, and insulin resistance, provide insights into the intricate interplay between DM and AD. Additionally, the observed increase in plasma levels of Aβ40, Aβ42, and p-tau in individuals with DM suggests a potential role of these proteins in the development and progression of both conditions.
Retinal Disorders
The retina is an extension of the central nervous system with both derived from the neural tube. They are partially protected from the vasculature via blood-retinal and blood-brain barriers respectively. Moreover, upon aging both the retina and the central nervous system show extracellular deposits associated with degenerative pathology such as the drusen and senile plaques respectively (Ratnayaka et al., 2015). The retina is affected by Aβ accumulation in various neurodegenerative disorders. Visual impairment and retinal Aβ deposits have been reported in patients with early AD even before any significant neurodegeneration (Koronyo-Hamaoui et al., 2011; Criscuolo et al., 2018).
Aβ is produced in the retinal ganglion cells (RGC) which along with retinal pigment epithelium (RPE) monolayer and other retinal neurons has been suggested to be the main sources of Aβ generation and secretion (Ohno-Matsui, 2011). Retinal Aβ levels have been found to be increased with aging, for example, cultured RPE cells from geriatric mice showed a higher level of Aβ and β-secretase activity and a lower level of neprilysin (which clears Aβ), compared to the younger controls (Wang et al., 2012; Ratnayaka et al., 2015). In addition, increased accumulation of Aβ in the RPE-Bruch’s membrane interface and retinal/choroidal blood vessels and decreased blood flow rates was observed in C57BL/6 mice with aging (Berisha et al., 2007; Hoh Kam et al., 2010). The above-mentioned changes seem to be more significant in age-related macular degeneration (AMD) and glaucoma.
AMD is the leading cause of severe, irreversible vision loss in people over age 60 with a global prevalence of 170 million, which is estimated to hit 288 million by the year 2040 (Kaarniranta et al., 2011; Pennington and DeAngelis, 2016). Glaucoma is among the top three causes of blindness, affecting about 76 million patients, and estimated to reach 111 million by 2040 (Allison et al., 2020). A pathological role of Aβ have been reported in AMD and glaucoma which share many pathological events with AD, including oxidative stress and neuroinflammation. Furthermore, increased levels of Aβ has been observed in the retina of AD patients (Ning et al., 2008; Kaarniranta et al., 2011; Song et al., 2017; Jonas et al., 2018). AMD, glaucoma, and AD are all age-related diseases and some epidemiological studies have reported that patients with AMD or glaucoma may be at higher risk of developing AD (Lee et al., 2019; Wang and Mao, 2021).
Drusen are one of the pathological hallmarks of AMD, comprising a complex of extracellular deposits of debris located between the basal lamina of the RPE and the inner collagenous layer of Bruch’s membrane (Spaide and Curcio, 2010). Disruption of retinal RPE following drusen formation leads to degeneration of photoreceptor cells that results in central vision loss in AMD patients. Studies have reported Aβ to be one of the major constituents of drusen and also to be present in the RPE of AMD patients, suggesting it may be a key player in AMD progression (Prasad et al., 2017; Wang and Mao, 2021). Recent reports suggest that the most abundant Aβ species within the retina is Aβ40 (Wang and Mao, 2021). Late-stage AMD is associated with progressive RPE degeneration and can lead to either wet/exudative AMD with choroidal neovascularization or dry/non-exudative AMD with larger areas of RPE atrophy also known as geographic atrophy (Arya et al., 2018). Subretinal injection of Aβ42 was shown to be associated with RPE senescence, retinal degeneration, and AMD-like ocular pathology in mice (Liu et al., 2015a). Moreover, treatment of human RPE cells with Aβ40 showed RPE atrophy and basal deposit formation along with increased production of vascular endothelial growth factor, monocyte chemoattractant protein-1, and interleukin 8 (IL-8) by RPE that are key players in the growth of the abnormal blood vessels (i.e., choroidal neovascularization). There was also a significant decrease in pigment epithelium-derived factor, which is a potent inhibitor of neovascularization, thereby promoting choroidal neovascularization formation in AMD (Stellmach et al., 2001; Yoshida et al., 2005; Wu et al., 2017b; Tian et al., 2021).
Aβ-induced mitochondrial ROS were shown to be involved in Aβ-induced secretion of angiogenesis factor by RPE cells (Wu et al., 2017b). Proteomic analysis of RPE-choroid complex tissue samples from Aβ treated mice showed that Aβ impaired mitochondrial function and increased ROS production through up-regulating PU.1 (a transcription factor) which in turn activated NADPH oxidases, particularly NOX4-p22phox (Sun et al., 2020). Impaired mitochondrial function following exposure to Aβ was also observed in photoreceptor cells, with ribosomal machinery and cytoskeletal organization found to be altered upon exposure to Aβ in a time and concentration-dependent manner (Deng et al., 2019, 2023).
RGC apoptosis is a key step causing irreversible vision loss in glaucoma. While elevated intraocular pressure is recognized as the main trigger for RGC death the underlying mechanism is multifactorial and far from clear (Guo et al., 2005). In a rat model of glaucoma, chronic ocular hypertension increased caspase-3 and caspase-8 activation which led to abnormal APP processing and increased Aβ level, which subsequently played a key role in pressure-induced RGC death., Treatment with Aβ antibodies significantly reduced RGC apoptosis (Guo et al., 2007). Moreover, β-secretase inhibitors showed neuroprotective effects against glutamate-induced RGC death in vitro and also on retinal damage induced by optic nerve crush in vivo (Yamamoto et al., 2004). Aβ also disrupts microvilli, the tight junctions, and adhesion of the RPE cells (Bruban et al., 2009).
The effect of Aβ40 and Aβ42 on retinal inflammation have been reported in several studies, highlighting the importance of inflammation in all age-related diseases. RPE, neuroretina, and vitreous analyses of animals (mouse and rat) that received intravitreal injections of Aβ showed overexpression of inflammation cytokines including IL-6, tumor necrosis factor-α, IL-1β, IL-18, caspase-1, NLRP3, and XAF1, microglia activation and ultimately RGC loss, possibly through binding to microglial scavenger receptor CD36, TLR4, and NF-κB signaling pathways (Liu et al., 2013; Chen et al., 2016; Lei et al., 2017; Simons et al., 2021).
Shared pathological events between AD and neurodegenerative diseases of the eye such as AMD and glaucoma, also warrant investigating the involvement of tau proteins, however, there is limited research on the role of tau deposition in AMD and glaucoma.
Tau protein is expressed in RGCs, however, a higher level of this protein can be found in the axons of developing RGCs. Tau protein also plays a key role in proper axon development and the survival of RGCs (Ho et al., 2012). Abnormal tau deposition and the presence of p-tau were observed in the retina of patients with uncontrolled primary and secondary open-angle glaucoma compared to healthy controls, while normal tau was found in the retina of healthy controls but not glaucoma patients (Gupta et al., 2008; Chan et al., 2021). An elevated level of p-tau also has been observed in the lateral geniculate nucleus of the monkey model of glaucoma following increased intraocular pressure (Yan et al., 2017). Interestingly, an in vivo study on a rat model of glaucoma showed both hyper and hypo-phosphorylation of tau protein and also mislocalization of tau protein in the somatodendritic compartment of RGCs, but not in axons. Subsequent down-regulation of tau by short interfering RNA resulted in an increased survival rate of RGCs, confirming the toxicity of tau protein (Chiasseu et al., 2016). Further results suggested the potential role of tauopathy in impaired autophagy and death of RGCs after optic nerve crush which can be ameliorated by silencing the tau gene via short interfering RNA (Oku et al., 2019). In rat RGCs, aggregation of tau protein impaired the anterograde axonal transport and transportation of mitochondria by kinesin-like motors toward the cell periphery leading to deficient energy production and accumulation of ROS (Stamer et al., 2002; Ho et al., 2012). Tau protein also interacts with the C-terminus of the P150 subunit of the dynactin complex. Moreover, the tau protein enhanced the binding of the dynactin complex to microtubules and co-localized with dynactin. Therefore the abnormal distribution of tau in RGCs can result in the mislocalization of dynactin in axons, which can lead to neurodegeneration (Magnani et al., 2007; Ho et al., 2012).
Collectively abnormal accumulation of both Aβ and tau protein appear to contribute to retinal disease pathogenesis, with aggregation of these factors inducing several down-stream events, including activation of retinal astrocytes and microglia and secretion of inflammatory cytokines such as IL-1β, IL-6, and tumor necrosis factor-α, leading to inflammation (Ashok et al., 2020; Baudouin et al., 2020; Tan et al., 2020; Abyadeh et al., 2023b).
Parkinson’s Disease
PD is the second most common brain neurodegenerative disease after AD. In 2016 about 6.1 million individuals had PD globally (Dorsey et al., 2018). It is pathologically characterized by progressive loss of dopaminergic neurons in the substantia nigra pars compacta and the formation of Lewy bodies (LBs). LBs contain the protein α-Syn (Surmeier, 2018; Pan et al., 2021); however, it constitutes only about 9% of the LB (Mccormack et al., 2016). α-Syn is a neuronal protein that is considered a major contributor to several neurodegenerative disorders known as synucleinopathies, including PD, dementia with LBs, and multiple system atrophy (Gonçalves and Outeiro, 2017). Recently a study reported using CSF α-Syn seeded assay as a novel biomarker for PD together with an olfactory test, producing a sensitivity of 98.6% (Siderowf et al., 2023).
There are multiple lines of evidence linking α-Syn and tau to Parkinson’s disease. MAPT gene (tau protein) and SNCA (alpha-synuclein) have been identified as risk genes for PD in several genome-wide association studies (Wray and Lewis, 2010; Davis et al., 2016). Mutations in tau can result in FTD and Parkinsonism in a mouse model (Dawson et al., 2007). Consistent with the idea that the two proteins interact, mutations in SNCA A53T in humans exhibited exacerbated tau pathology (Markopoulou et al., 2008).
Interestingly α-Syn deposition is also observed in AD patients and increased deposition of insoluble of α-Syn has been linked to cognitive dysfunction in a transgenic AD mouse (Clinton et al., 2010; Vasili et al., 2019). Coincident with this, elevated aggregation of Aβ and tau have been reported in PD patients, although the association of AD pathological hallmarks with PD is not consistently reported (Jendroska et al., 1996; Siderowf et al., 2010; Bäckström et al., 2015; Liu et al., 2015b; Winer et al., 2018; Melzer et al., 2019). Experimental studies have linked α-Syn mainly to tau hyperphosphorylation, however, some studies have indicated the role of Aβ in inducing aggregation of α-Syn and tau protein in synucleinopathies (Twohig and Nielsen, 2019; Bassil et al., 2020).
Aβ plaques have been found in PD patients along with the typical LB deposition and a direct association has been reported between these two pathological proteins (Lashley et al., 2008). Analysis of post-mortem brain from PD patients with dementia identified three pathologic types of PD, including (1) Predominant synucleinopathy; (2) Predominant synucleinopathy with Aβ plaques and minimal or no cortical tauopathy; (3) Synucleinopathy and Aβ plaques and at least moderate neocortical tau aggregations. This study also indicated Aβ deposition and synucleinopathy but not tauopathy as the main contributors to PD development and Aβ accumulation was shown to be associated with poor survival rate in PD patients with dementia (Kotzbauer et al., 2012). Early experimental studies indicated that α-Syn and Aβ have distinct and convergent pathogenic effects on brain function in a transgenic mouse model of AD with LBs. Further in vitro studies showed that Aβ, particularly Aβ42 even at low concentration, may promote the intraneuronal accumulation of α-Syn, but α-Syn did not affect overall Aβ level or plaque formation (Hamilton, 2000; Masliah et al., 2001; Köppen et al., 2020). These results were confirmed in post-mortem analysis of PD brain, which showed the higher presence of LBs and increased level of insoluble α-Syn in brains with Aβ deposits compared to those without Aβ deposits, and suggested the Aβ enhanced development of α-Syn lesions in PD patients (Pletnikova et al., 2005). In addition, further studies indicated that Aβ, α-Syn, and tau proteins may interact synergistically to promote their mutual accumulation (Clinton et al., 2010). In this regard, in vivo, results showed that Aβ plaques promote α-Syn seeding and spread throughout the brain, and subsequently, α-Syn enhances tau aggregation (Bassil et al., 2020).
In summary, studies have reported a significant association between Aβ, tau, and α-Syn proteins that synergistically contribute to PD and AD pathogenesis. Interestingly, a meta-analysis in 2017 shows reduced CSF levels of Aβ42 in PD with cognitive impairment (Hu et al., 2017b). This is in agreement with more recent studies and suggested to occur because the Aβ42 may be sequestered away from CSF due to increased intracellular Aβ42 accumulation (Lewczuk et al., 2021; Nabizadeh et al., 2023).
Genetic studies have indicated the association of several gene polymorphisms with the risk of PD, including SNCA (synuclein alpha, encoding α-Syn), GBA (glucosylceramidase beta, encoding GBA protein), LRRK2 (leucine-rich repeat kinase 2, encoding LRRK2 protein), and MAPT (Bras and Singleton, 2009; Pan et al., 2021). MAPT that encodes tau protein has two haplotypes namely H1 and H2 and genome-wide association studies have repeatedly shown the association of H1 with increased risk of PD and AD, although results from different populations are not consistent. H1 is debated as a risk factor for PD and this inconsistency is possibly due to the effects of genetic background and environmental factors (epigenetic) (Sánchez-Juan et al., 2019; Pan et al., 2021). Meta-analyses of total tau (t-tau) and p-tau in CSF show increased levels in PD dementia cohorts (Liu et al., 2015b; Hu et al., 2017a).
Cell transplantation has emerged as a potential treatment for PD, interestingly p-tau and NFTs have been observed in transplanted cells 18 months to 21 years after transplantation (Cisbani et al., 2017; Ornelas et al., 2020). The presence of toxic tau in healthy transplanted cells is possibly through prion-like propagation (Clavaguera et al., 2017; Mudher et al., 2017). α-Syn accumulates and forms fibrillary assemblies in PD that play a key role in the pathogenesis of PD. Of interest is that tau is colocalized with α-Syn in LBs (Hu et al., 2010; Pan et al., 2021), and in vivo studies showed impairment and subsequent cognitive decline in a mouse model of familial PD (A53T mutant α-synuclein) shown to be mediated by tau protein (Teravskis et al., 2018; Singh et al., 2019).
Experimental studies have shown that tau monomers interact with the c-terminal region of α-Syn and promote its aggregation. In addition, α-Syn monomers and aggregates also increase tau aggregation (Dasari et al., 2019). Further studies showed that extracellular α-Syn increases the phosphorylation of GSK-3β at Tyr216 and decreases its phosphorylation at Ser9. This enhances its activity and in turn, increases tau phosphorylation at Ser396 leading to microtubular destabilization (Giasson et al., 2003; Gąssowska et al., 2014; Credle et al., 2015). Conversely, GSK-3β dysregulation contributes to Parkinson’s-like pathophysiology with associated region-specific phosphorylation and accumulation of tau and alpha-synuclein (Credle et al., 2015). Another study indicated that aberrant expression of human P301L mutant tau increased phosphorylation and aggregation of α-Syn via GSK-3β in rTg4510 mice (Takaichi et al., 2020). Overall, these studies highlight the intricate relationship between Aβ, tau, and α-Syn proteins in PD and AD. Understanding these interactions is essential to unravel the mechanisms underlying these neurodegenerative diseases and develop effective therapeutic strategies.
Traumatic Brain Injury
TBI, the “silent epidemic” is an increasingly recognized global health problem that is estimated to affect about 69 million people worldwide each year (Dewan et al., 2018). TBI is divided into two types: focal TBI, which affects only a specific area such as epidural or subdural hematoma and parenchymal contusions, and diffuse TBI which affects more widespread areas with traumatic axonal injury and diffuse cerebral edema (Andriessen et al., 2010). TBI is not isolated to a single event and has long-term consequences. Multiple concussions or even a single moderate to severe TBI increases the risk of developing several neurodegenerative diseases including CTE, AD, and PD at an early age (Katsumoto et al., 2019). Currently, no effective treatments are available for TBI or TBI-related dementia. Although the underlying mechanisms of the association of TBI with neurodegenerative disease remained unclear, the presence of hp-tau, tau aggregates, and Aβ have been reported in the brain months after TBI (Roberts et al., 1994; Smith et al., 2003; Katsumoto et al., 2019).
Increased levels of Aβ and its aggregation have been reported as a consequence of TBI and suggested as a major contributor to neurodegeneration and cognitive decline (Johnson et al., 2010; Acosta et al., 2017). In addition, increased level of BACE1 and γ-secretase following TBI has been reported in animal studies and suggested as two therapeutic targets for the treatment of TBI (Blasko et al., 2004; Loane et al., 2009). It has been reported that about 30% of individuals after a severe TBI with a survival time of between four hours and 2.5 years shows Aβ deposition and that presence of Aβ accumulation following TBI increased with age (Roberts et al., 1994). These findings also have been observed in another study that showed the presence of NFTs and Aβ plaques in the brain of single moderate to severe TBI cases after 1–47 years (Johnson et al., 2012). However, in contrast to these results, the presence of Aβ plaques in short-term post-TBI cases but not long-term survivors (up to 3 years) was reported, suggesting that increased level of neprilysin after TBI may be the reason for the reduced level of Aβ plaques (Chen et al., 2009). Decreased level of Aβ plaque was also observed in aged plaque-forming APP transgenic mice 16 weeks after TBI, which indicated that plaque pathology may be reversible (Nakagawa et al., 2000). Although most of the animal studies have indicated that Aβ level was initially increased after TBI as a short-term effect but was resolved over time, and is not a long-term effect (Tsitsopoulos and Marklund, 2013; Bird et al., 2016), there are some reports indicating increased Aβ accumulation as a long-term sequela after TBI. For example, the presence of Aβ plaques in injured axons without increased gene expression of APP was observed in a non-transgenic rat model of TBI even after 1 year (Iwata et al., 2002), and also increased levels of Aβ, BACE, presenilin-1 and caspase-3 in swollen axons of a non-transgenic pig model of TBI after 6 months (Chen et al., 2004). The discrepancy in these results may be attributed due to the studies being carried out in different TBI models.
The association of TBI with tauopathies has been suggested in several studies. A single incidence of severe brain trauma was shown to induce widespread hyperphosphorylated tau pathology in individuals surviving more than a year after injury (Zanier et al., 2018). Similar results were also observed in mice with a single severe TBI-induced tau pathology that reflected late post-TBI tau pathology in humans. Further analyses showed that induced tau toxicity can spread from the site of injury to other brain regions and also injection of toxic tau from TBI mice to wild-type mice can induce tau pathology, memory deficits, and synaptic alterations, which indicate prion-like behavior of tau protein (Woerman et al., 2016; Zanier et al., 2018). A proteome comparison between the frontal cortex of diffuse and focal TBI cases revealed a significantly higher level of proteins involved in neurodegeneration such as tau protein in diffuse TBI cases compared to focal TBI cases, however, no differences were found in the level of Aβ40 and Aβ42 between two types of TBI (Hamdeh et al., 2018). The results of this study indicated that even a single TBI can induce long-term progressive tau pathology and subsequent neurodegeneration, especially in the presence of diffused axonal injury. Further, repeated mild TBI also has been shown to be associated with NFT formation, neurodegeneration, and cognitive decline. In this regard, studies on American football players, boxers, and wrestlers that are repeatedly exposed to mild TBI, showed an increased level of hp-tau, NFTs, and cognitive impairment and developed CTE (Omalu et al., 2011; Katsumoto et al., 2019). Moreover, serum levels of tau protein were shown to be positively associated with the severity of the injury in a rat model of diffuse axonal injury, suggesting it as a potential diagnostic biomarker to assess the severity of diffuse axonal injury in the early phase (Tomita et al., 2020).
There are two conformational forms of phosphorylated tau at Thr231 reported, including trans p-tau which is a physiological conformation, and cis p-tau as the toxic form which is more physiological, but is converted to trans p-tau by Pin1 (Figure 5; Nakamura et al., 2013; Kondo et al., 2015). In TBI reduced activity of Pin1 results in an increased level of cis p-tau which appears prior to tau oligomerization and aggregation, resulting in impaired axonal microtubule networks and mitochondrial transport. This toxic form can spread to other neurons and induce apoptosis leading to neurodegeneration and cognitive impairment, but this could be relatively ameliorated upon using a cis p-tau antibody (Kondo et al., 2015; Albayram et al., 2017). Thus, the results of these studies indicated that cis p-tau contributes to both short-term and long-term consequences of TBI that can be effectively neutralized by cis p-tau antibody treatment.
Figure 5.
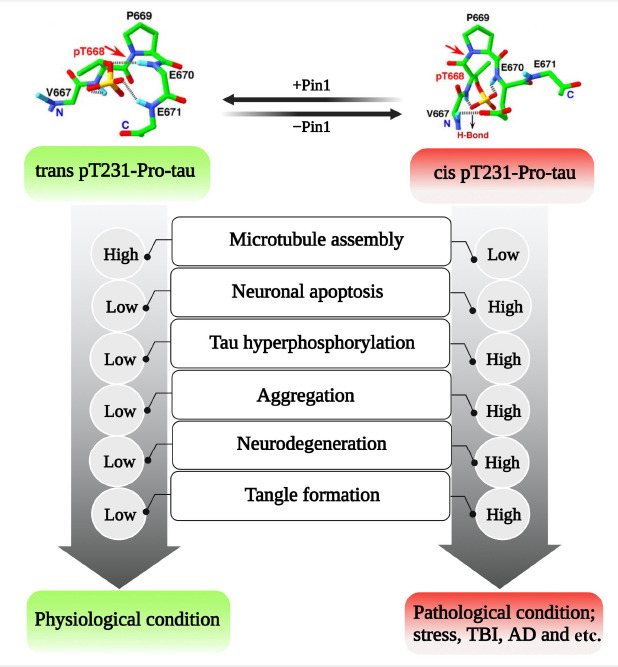
Conformational changes of tau protein between trans and cis-conformations are mediated by Pin1; trans p-tau is considered as the physiological form of tau protein but cis p-tau is the toxic form that is increased in pathological condition due to impaired Pin 1 activity.
Created with BioRender.com. AD: Alzheimer’s disease; Pin1: peptidyl-prolyl cis-trans isomerase NIMA-interacting 1; TBI: traumatic brain injury.
These findings support the notion that TBI can initiate neurodegenerative processes resembling those seen in AD and CTE. The accumulation of Aβ and tau proteins, along with their pathological consequences, suggests a complex interplay between TBI, protein aggregation, and neurodegeneration. Further research is needed to unravel the intricate mechanisms underlying these processes, which can pave the way for the development of targeted interventions and improved management strategies for individuals with TBI, reducing the risk of long-term cognitive decline and neurodegenerative diseases.
Down Syndrome
Down syndrome (DS) is the leading genetic cause of intellectual disability with an incidence rate of about 14 in 10,000 live births in the United States (Dierssen, 2012). The disorder is caused by trisomy of chromosome 21 and is therefore known as trisomy 21. Individuals with DS have common phenotypes such as short stature, muscle hypotonia, mental retardation, small head, short neck, protruding tongue, and flat faces; moreover, they are more prone to develop certain health conditions, such as AD, CVDs, and diabetes (Van Goor et al., 1997; Sobey et al., 2015; Antonarakis et al., 2020).
Individuals with DS also showed higher blood levels of Aβ and tau protein and its phosphorylation forms compared to healthy individuals at young ages. Plasma level of Aβ showed a progressive age-dependent manner that almost all individuals with DS show sufficient neuropathology for a diagnosis of AD by the age of 40 (Head and Lott, 2004; Lee et al., 2017). As described above, the APP gene is located on chromosome 21 and thus triplicated in DS, resulting in an increased level of APP. In addition, increased activity of β-secretase and decreased activity of α-secretase especially after 40 years of age have been reported in individuals with DS. It is reported that Aβ42/43 is initially deposited in the brain with Aβ40 appearing about a decade later (Iwatsubo et al., 1995; Mori et al., 2002; Head et al., 2018). Interestingly intracellular Aβ accumulation has been reported in DS that appeared prior to extracellular Aβ aggregation and NFTs and was shown to be accumulated in endosomes causing endocytic pathway abnormalities. These could be prevented in a mouse model of DS by partial inhabitation of BACE1(Cataldo et al., 2000; Jiang et al., 2016). However, it is important to note that BACE1 inhibition reduced the level of APP-β c terminal fragment but did not alter Aβ40 and Aβ42 peptide levels. Therefore, endocytic abnormalities may be due to a higher level of APP-βCTF but not Aβ levels (Jiang et al., 2016). Although the exact mechanism of intracellular Aβ aggregation is not still clear, it has been suggested that AβPP overexpression in DS may disrupt mitochondrial function, which in turn triggers intracellular Aβ aggregation that can further contribute to mitochondrial dysfunction (Busciglio et al., 2002; Abyadeh et al., 2021). Both intracellular and extracellular Aβ may contribute to NFTs formation and trigger caspase activation and accumulation of caspase cleavage products resulting in apoptotic pathways activation and neuronal loss in the DS brain. These observations were found in the brain of DS individuals aged over 40 years (Head et al., 2002; Head et al., 2018). In addition, age-dependent deposition of phosphorylated Aβ at serine residue 8 has been observed in extracellular plaques and within the vasculature of the brain of DS (Kumar et al., 2020).
The presence of an insoluble and phosphorylated form of tau protein and also NFTs have been reported in the brain of individuals with DS, even in the presence of low Aβ burden (Hanger et al., 1991; Zammit et al., 2021). Similar to Aβ, the plasma level of tau protein is higher in DS patients and showed a progressive age-dependent manner (Kasai et al., 2017; Lee et al., 2017). Although like AD, deposition of tau protein is observed in the DS brain, it may occur in different brain regions of adult DS compared to AD (Lemoine et al., 2020).
The balance between 3R-tau and 4R-tau levels is critical for proper neuronal function and increased expression of either 3R-tau or 4R-tau was shown to be correlated with several tauopathies such as Pick’s disease (3R-tau), corticobasal degeneration and progressive supranuclear palsy (4R-tau) (Barron et al., 2020). Expression of 3R-tau is increased in the DS brain and shown to be related to increased expression of dual-specificity tyrosine-phosphorylated and regulated kinase 1A (Dyrk1A), located on the chr21 and triplicated in DS. In this regard, inhibition of Dyrk1A expression resulted in decreased expression of 3R-tau and improved impaired general behaviors in mice models of DS (Shi et al., 2008; Yin et al., 2017). Further studies have indicated that PI3K/Akt/mTOR axis is hyper-activated in DS, resulting in decreased autophagy, IRS1, and GSK3β activity. However, tau was found to be hyperphosphorylated and associated with elevated expression of Dyrk1A, also enhanced expression of regulator of calcineurin 1 (RCAN1), which is linked to tau hyper-phosphorylation (Perluigi et al., 2014; Di Domenico et al., 2018). Collectively these results suggested the key role mTOR pathway and RCAN1 in the hyper-phosphorylation of tau protein in DS (Figure 6). Consistent with the hypothesis of prion-like behavior of tau protein, injection of plasma-derived neuron-derived small extracellular vesicles of DS-AD cases into the dorsal hippocampus of wild-type mice resulted in an increased level of hp-tau even in the corpus callosum and the mediolateral axis (Ledreux et al., 2021). Therefore, these results indicated prion-like behavior of tau protein that can spread within the brain. These findings collectively shed light on the complex neuropathological mechanisms underlying DS and its association with AD. Understanding these mechanisms may help in the development of targeted therapeutic approaches for DS-related cognitive decline and neurodegeneration.
Figure 6.
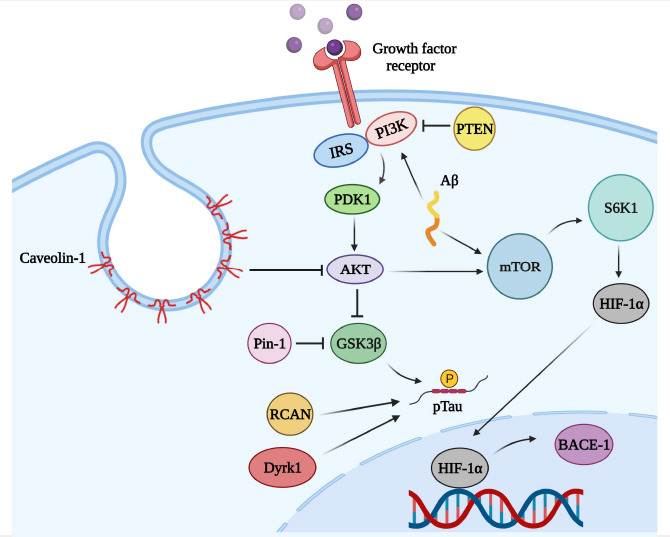
Interplay between mTOR, Aβ, and Tau.
Created with BioRender.com. Aβ: Amyloid beta; AKT: protein kinase B (Akt); BACE-1: beta-secretase 1; BCAN: brevican; Dyrk1: dual-specificity tyrosine-phosphorylation-regulated kinase 1; GSK3β: glycogen synthase kinase 3 beta; HIF-1α: hypoxia-inducible factor 1 alpha; IRS: insulin receptors substrate; mTOR: mechanistic target of rapamycin; PDK1: phosphoinositide-dependent kinase-1; PI3K: phosphoinositide 3-kinase; Pin-1: peptidyl-prolyl cis-trans isomerase NIMA-interacting 1; p-tau: phosphorylated Tau protein; PTEN: phosphatase and tensin homolog deleted on chromosome 10; S6K1: ribosomal protein S6 kinase 1.
Motor Neuron Disease
Motor neuron disease including amyotrophic lateral sclerosis (ALS), the eponymous Lou Gehrig’s disease, is a fatal progressive neurodegenerative disorder characterized by the degeneration of both the upper motor neurons of the motor cortex, and the lower motor neurons of the brainstem and spinal cord (Shadfar et al., 2022). The most distinguishing symptoms of ALS are muscle atrophy and weakness, and progressive paralysis (Masrori and Van Damme, 2020). The pathogenic mechanisms leading to neurodegeneration in ALS are not fully defined. Hence, at present, there is no effective cure for the disease, and patients have an average lifespan of three years following diagnosis, with death occurring due to respiratory failure (Masrori and Van Damme, 2020). The only currently approved Food and Drug Administration (FDA) therapeutics for ALS are relyvrio (AMX0035), riluzole, and edaravone, although these drugs only extend the life span of patients by two to three months. Furthermore, they do not halt the neurodegenerative process in ALS (Mora, 2017). Therefore, there is an urgent need to develop novel therapeutics for the treatment of ALS, which target the underlying disease mechanisms.
A growing body of evidence indicates the role of Aβ and tau in ALS pathology. Increased levels of APP have been observed in ALS patients’ spinal cord anterior horn neurons even with moderate motor neuronal loss (Calingasan et al., 2005). Similarly, compared to controls, increased levels of Aβ in the skin of ALS patients were demonstrated using enzyme-linked immunosorbent assays (Tamaoka et al., 2000).
The Caspase family is known to mediate alternative proteolysis of APP (Salvesen and Dixit, 1999). Among the members of the Caspase family, Caspase-3 predominantly participates in APP cleavage (François et al., 1999). Caspase-3 can directly cleave APP through apoptosis, leading to increased formation of Aβ (François et al., 1999; Salvesen and Dixit, 1999). Accumulating evidence suggests that caspase-3 plays a significant role in initiating neurodegenerative processes in transgenic mouse models of ALS (Salvesen and Dixit, 1999; Pasinelli et al., 2000), which may contribute to the elevated levels of Aβ formation.
Higher Aβ42 immunoreactivity was observed in motor neurons in the anterior horn in the postmortem lumbar spinal cord of ALS patients (Calingasan et al., 2005). There was colocalization of the Aβ42 with the oxidative damage markers including heme oxygenase-1, 8-hydroxydeoxyguanosine, and nitrotyrosine in these tissues (Calingasan et al., 2005). Remarkably, enhanced cleaved caspase-3 immunoreactivity was observed within the neurons with intracellular Aβ42 accumulation (Calingasan et al., 2005).
Approximately 4% of fALS and approximately 1% of sALS cases worldwide are caused by mutations in TARDBP, the majority of which are missense and autosomal dominant mutations (Sreedharan et al., 2008; Renton et al., 2014). Importantly, however, almost all cases of ALS (97%) and FTD cases (50%) are characterized by the presence of TDP-43 pathology (Chou et al., 2018). TDP-43 pathology is one of the primary features of ALS and FTLD-TDP, and is characterized by loss of TDP-43 function in the nucleus and enhanced deposition into cytoplasmic inclusion bodies in the brain and spinal cord neurons (Arai et al., 2006; Konopka et al., 2020). Redistribution of TDP-43 from the nucleus to the cytoplasm is recognized as a key characteristic of ALS patient motor neurons (Chou et al., 2018). Significantly higher levels (200%) of TDP-43 were observed in cortical autopsies of late-stage AD patients (Herman et al., 2011). Additionally, TDP-43 inclusions have been found in up to 57% of AD cases (Josephs et al., 2014; James et al., 2016). Elevated TDP-43 pathology was detected in the rat motor cortex, following lentiviral expression of Aβ1–42 (Herman et al., 2011). In addition, there was a correlation between Aβ1–42 expression and increased phosphorylation of TDP-43 and its accumulation in the cytosol (Herman et al., 2011). Compared to wild-type mice, TDP-43 modifications were detected in 3xTransgenic AD (3×Tg-AD); however, these modifications were reduced in parkin-injected hippocampi, despite the presence of Tau pathology. This indicated that Aβ triggers TDP-43 pathology, even in the absence of Tau (Herman et al., 2011).
Modifications in tau metabolism have been also reported in ALS (Strong et al., 2020), the most conspicuous tau alteration is its pathological phosphorylation at Thr175 (pThr175tau) (Strong et al., 2020). pThr175tau has also been identified in CTE with ALS and in both in vivo and in vitro experimental paradigms, emphasizing the key role of tau phosphorylation in the pathobiology of ALS (Strong et al., 2020). Experimental rodent models suggest the presence of phosphorylated tau and alterations in the metabolism of TDP-43 and tau act synergistically to deteriorate the pathology of either (Chornenkyy et al., 2019; Strong et al., 2020).
Furthermore, in extracts of both the brain and ventral spinal cord of sporadic ALS patients, neurotoxic tau fragment (tau45–230) has been identified (Lang et al., 2014; Vintilescu et al., 2016). Pathological forms of phosphorylated tau, together with tau immunoreactive inclusions have been detected in ALS patients (Yang et al., 2003; Yang et al., 2005; Yang and Strong, 2012). Prominent tau deposition (pThr175tau) was observed in motor neurons following TDP-43 pathology (Yang and Strong, 2012).
Alterations in tau metabolism were also detected in cortical and spinal motor neurons in sporadic ALS patients in antemortem or postmortem obtained tissues (McKee et al., 2009; McKee et al., 2016; Mez et al., 2017). pThr231tau, pThr175tau, and oligomeric tau (T22) were predominantly observed in the samples of sporadic ALS patients (Moszczynski et al., 2018). A recent study showed the increased levels of phosphorylated tau (p-tau) in CSF of ALS patients with or without cognitive impairment (Gong et al., 2022). Although the CSF p-tau level and p-tau:t-tau ratio were lower in patients with ALS than in ALS patients with cognition impairment, CSF p-tau could be used as a reliable index of cognition impairment in patients with ALS (Gong et al., 2022).
Amyloid-β and Tau Protein Signatures in Other Diseases
Autism
Autism spectrum disorders (ASD), are a diverse group of conditions characterized by challenges with social skills, communication, and repetitive behaviors. The prevalence of autism has increased in recent decades and according to the Centers for Disease Control and Prevention 2016 data, about 1 in 54 children in the USA are diagnosed with autism. Males have a higher risk of autism and related disorders than females (Palmer et al., 2017). The exact mechanism underlying autism is not clear, however, environmental and genetic factors are supposed to play a causative role (Palmer et al., 2017; Emberti Gialloreti et al., 2019). Children with autism commonly showed brain overgrowth, which is coincidental with the onset of symptoms. Initial studies in patients with severe autism observed an increased level of APP and AβPPɑ but decreased levels of AβPPβ, Aβ40, Aβ42 and also lack of cerebral plaques, therefore indicating the aberrant hyperactivity of non-amyloidogenic pathway (Bauman, 1994; Sokol et al., 2006; Ray et al., 2011). Data regarding the serum level of tau protein and its phosphorylation are inconsistent and both increased and decreased levels of this protein have been reported in both autistic humans and mice (Kadak et al., 2015; Barón-Mendoza et al., 2018; Ayaydın et al., 2020; Grigg et al., 2020). However recently it has been shown that genetically decreasing tau protein level prevents behavioral signs of autism in mice models possibly through the increasing levels of phosphatase and tensin homolog deleted on chromosome 10 (PTEN). This then suppresses the PI3K/Akt/mTOR signaling pathway that is overactivated in autism and is described as a shared altered mechanism between autism and AD (Tai et al., 2020; Mencer et al., 2021). The understanding of autism’s underlying mechanisms is still evolving, and further research is necessary to uncover the complex interactions between genetic and environmental factors. Investigating the roles of amyloid metabolism, tau protein, and shared pathways with AD may provide further insights. The heterogeneity of ASD requires a personalized approach to diagnosis, treatment, and support, taking into account the individual’s unique needs and characteristics.
Multiple sclerosis
MS is an autoimmune inflammatory disease of the central nervous system, that is the leading cause of acquired neurological disability in young adults worldwide and currently affects about 300,000 to 400,000 individuals in the US and over 2.5 million people worldwide (Van Schependom et al., 2019; Wallin et al., 2019). MS is pathologically characterized by the formation of demyelinating lesions in the brain and spinal cord, which induce inflammation and subsequently neurodegeneration (Filippi et al., 2018; Mirzaei et al., 2022). Myelin dysfunction also has been shown to drive the Aβ deposition in AD mouse models (Depp et al., 2023). Brain atrophy is extensively observed in MS patients and is recognized as an imaging marker of neurodegeneration in MS patients; however, early neurodegeneration may appear in the absence of brain atrophy (Van Schependom et al., 2019). The presence of amyloid plaques has been reported in MS patients; moreover, CSF levels of soluble Aβ1–42 were shown to be decreased and associated with cognitive decline in MS patients. It was suggested as a potential biomarker of neurodegeneration that may predict clinical outcomes (Mai et al., 2011; Mori et al., 2011; Augutis et al., 2013; Pietroboni et al., 2017). In contrast to Aβ, the level of tau protein and its abnormal phosphorylation was found to be increased in MS patients in the neurodegenerative phase of tissue injury, which was associated with neuronal and axonal loss and suggested as an early marker of axonal damage in MS patients (Brettschneider et al., 2005; Terzi et al., 2007; Anderson et al., 2008). The presence of both amyloid plaques and abnormal tau protein levels in MS highlights the complex nature of the disease and suggests overlapping mechanisms with other neurodegenerative disorders such as AD. Further research is needed to fully understand the role of these biomarkers in the progression of MS and their potential implications for diagnosis, monitoring, and treatment of the disease.
Huntington’s disease
HD is a rare genetic neurodegenerative disorder with a prevalence of 2.71 in 100,000 live birth globally (Pringsheim et al., 2012; Bates et al., 2015). HD patients commonly experience uncontrolled body movement, cognitive decline, and psychiatric disturbances (Li and Li, 2004; Żukiewicz-Sobczak et al., 2014). Patients with HD showed amyloid-like inclusions in their brain and heart that may cause neurodegeneration and cardiomyopathy (Melkani et al., 2013). Currently, available data about the level of Aβ in HD patients are inconsistent, while some researchers indicated a higher level of Aβ in HD patients, others failed to find significant differences with healthy controls (Jellinger, 1998; McGowan et al., 2000; Mollenhauer et al., 2006; Gratuze et al., 2016). However, high levels of hyperphosphorylated and truncated tau and NFTs have been observed in the brain of HD patients (Masnata et al., 2020). Moreover, tau haplotype was shown to affect the cognitive function of HD patients, whereas patients with the H2 haplotype showed more severe cognitive decline than patients with H1 haplotype (Vuono et al., 2015; Gratuze et al., 2016). In addition, mutant huntingtin, the HD protein, triggers tau protein hyperphosphorylation and alters its subcellular distribution (Blum et al., 2015). The level of caspase-2 is increased in HD patients and its inhibition improved cognitive and motor deficits in mice model of HD, interestingly a recent study suggested that caspase-2-mediated tau cleavage results in a soluble truncated tau that adversely affects synaptic function and cognitive performance in HD patients (Liu et al., 2019). Understanding the mechanisms underlying their interactions and contributions to disease progression is crucial for developing targeted therapies for HD.
Creutzfeldt-Jakob disease
CJD is a rare and fatal neurodegenerative disorder and the most common human prion disease with a prevalence of one individual per million per year worldwide (Iwasaki, 2017; Sitammagari and Masood, 2022). Prion diseases, previously known as transmissible spongiform encephalopathy are a group of neurodegenerative diseases that affect almost all mammals. In prion diseases, the pathological form of prion protein is able to transmit the disease (Gough and Maddison, 2010). A significantly increased CSF tau protein has been reported in CJD patients (58-fold) compared to healthy controls, while CSF Aβ42 level was decreased in CJD patients (Kapaki et al., 2001). Moreover, abundant Aβ and marginal prion deposits (an inverse association) also have been reported in some CJD patients; in addition, CSF tau level showed a negative association with cognitive performance and a positive correlation with disease severity in CJD patients, indicating the potential use of CSF tau as a biomarker of neuronal damage (Debatin et al., 2008; Cohen et al., 2016). These findings indicate that while CJD shares some pathological features with other neurodegenerative diseases, such as the presence of tau pathology, it also exhibits unique characteristics, including decreased levels of Aβ42. The abnormal accumulation of prion proteins and the associated neurodegeneration are the key hallmarks of CJD. Moreover, further research is needed to better understand the mechanisms of CJD and to explore the potential of CSF tau and other biomarkers as diagnostic and prognostic tools.
Wilson’s disease
WD is a rare genetic disorder with a prevalence of 1 in 30,000 (based on limited available data) (Sandahl et al., 2020). WD is characterized by abnormal accumulation of copper (Cu) in the liver, brain, and other vital organs and with several symptoms ranging from fatigue to difficulty with speech, controlling movements, or muscle stiffness. Disordered Cu metabolism is also associated with other neurodegenerative diseases such as AD and PD (Bandmann et al., 2015). Studies reported that Cu binds to Aβ and decreases its clearance from blood-brain barrier, therefore leading to Aβ deposition; moreover, Cu also increases Aβ production and inflammation (Syme et al., 2004; Singh et al., 2013). Cu also can bind to tau protein and may induce its aggregation, moreover, serum levels of tau protein also have been found to be increased in patients with WD, and are believed to be a promising biomarker of axonal impairment and possible neuronal damage in patients with WD (Soragni et al., 2008; Lekomtseva et al., 2019). These findings highlight the complex interactions between copper metabolism and the accumulation of Aβ and tau proteins in WD. Further research is needed to better understand the underlying mechanisms linking copper dysregulation to Aβ and tau pathology in WD, as well as its implications for disease progression and potential therapeutic interventions that may involve targeting copper homeostasis.
Conclusion
Aβ and tau protein are well-established pathogenic factors in several neurodegenerative diseases, particularly in AD; however, a growing body of evidence has indicated the presence of these factors in other diseases and provided molecular mechanisms underlying their aggregation and pathogenesis. However, research about the role of these pathological factors in other diseases is limited. The reasons for the alteration of protein expression and modification in these diseases are multifactorial and may vary depending on the specific condition. One possible explanation is that Aβ and tau proteins are involved in various cellular processes beyond their roles in neurodegeneration. They participate in signaling pathways, synaptic function, and neuronal plasticity, and their dysregulation can contribute to pathological processes in different diseases. For example, in cardiovascular diseases, Aβ and tau may be involved in vascular dysfunction and contribute to cerebrovascular pathology. In diabetes, these proteins may contribute to neuronal damage and cognitive impairment associated with the disease. The presence of Aβ and tau protein alterations in these diseases may indicate the potential involvement of neurodegenerative mechanisms, but their presence alone does not appear to be specific. However, these proteins may play a critical role in cellular dysfunction and contribute to the progression of pathology specific to each condition.
Most studies are focused on altered levels of Aβ and tau protein and also phosphorylation of tau protein in disease; however, other PTMs such as acetylation and truncation also play a critical role. Moreover, conformational changes including trans and cis conformation may induce different levels of toxicity, which is not well investigated in these diseases. Regarding their potential as diagnostic and therapeutic biomarkers, further research is needed. While Aβ and tau have been extensively studied in the context of AD, their use as biomarkers in other diseases is still being explored. It is important to consider the specificity and sensitivity of these biomarkers in different conditions, as well as their correlation with disease progression and response to treatment. Utilizing Aβ and tau as diagnostic and therapeutic markers in non-neurodegenerative diseases may require tailored approaches and comprehensive evaluation to establish their clinical utility. Along with Aβ and tau protein, upstream regulators of these factors such as mTOR signaling and Pin1 have been suggested as other potential therapeutic targets to slow or prevent disease progression and ameliorate disease symptoms. Overall, the presence of Aβ and tau protein alterations in diseases beyond AD suggests shared mechanisms and potential contributions to pathogenesis. Further research is necessary to unravel the specific roles of these proteins in different conditions, assess their diagnostic value, and explore their potential as therapeutic targets.
Additional file: Open peer review report 1 (79KB, pdf) .
Footnotes
Conflicts of interest: The authors declare no conflicts of interest. No conflicts of interest exist between ProGene Technologies Pty Ltd. and the publication of this paper.
Data availability statement: Not applicable.
Open peer reviewer: Mikhail Inyushin, Universidad Central Del Caribe, USA.
P-Reviewer: Inyushin M; C-Editors: Zhao M, Liu WJ, Qiu Y; T-Editor: Jia Y
References
- 1.Abyadeh M, Meyfour A, Gupta V, Zabet Moghaddam M, Fitzhenry MJ, Shahbazian S, Hosseini Salekdeh G, Mirzaei M. Recent advances of functional proteomics in gastrointestinal cancers-a path towards the identification of candidate diagnostic prognostic and therapeutic molecular biomarkers. Int J Mol Sci. (2020);21:8532. doi: 10.3390/ijms21228532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abyadeh M, Gupta V, Chitranshi N, Gupta V, Wu Y, Saks D, Wander Wall R, Fitzhenry MJ, Basavarajappa D, You Y. Mitochondrial dysfunction in Alzheimer's disease-a proteomics perspective. Expert Rev Proteomics. (2021);18:295–304. doi: 10.1080/14789450.2021.1918550. [DOI] [PubMed] [Google Scholar]
- 3.Abyadeh M, Tofigh N, Hosseinian S, Hasan M, Amirkhani A, Fitzhenry MJ, Gupta V, Chitranshi N, Salekdeh GH, Haynes PA. Key genes and biochemical networks in various brain regions affected in Alzheimer's disease. Cells. (2022);11:987. doi: 10.3390/cells11060987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abyadeh M, Yadav V, Kaya A. Common molecular signatures between coronavirus infection and Alzheimer's disease reveal targets for drug development. bioRxiv. (2023a) doi: 10.3233/JAD-230684. doi:10.1101/2023.06.14.544970. [DOI] [PubMed] [Google Scholar]
- 5.Abyadeh M, Gupta V, Paulo JA, Sheriff S, Shadfar S, Fitzhenry M, Amirkhani A, Gupta V, Salekdeh GH, Haynes PA, Graham SL, Mirzaei M. Apolipoprotein εin brain and retinal neurodegenerative diseases. Aging Dis. (2023b);14:1311–1330. doi: 10.14336/AD.2023.0312-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Acosta SA, Tajiri N, Sanberg PR, Kaneko Y, Borlongan CV. Increased amyloid precursor protein and tau expression manifests as key secondary cell death in chronic traumatic brain injury. J Cell Physiol. (2017);232:665–677. doi: 10.1002/jcp.25629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Albayram O, Kondo A, Mannix R, Smith C, Tsai CY, Li C, Herbert MK, Qiu J, Monuteaux M, Driver J. Cis P-tau is induced in clinical and preclinical brain injury and contributes to post-injury sequelae. Nat Commun. (2017);8:1000. doi: 10.1038/s41467-017-01068-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allison K, Patel D, Alabi O. Epidemiology of glaucoma:the past present and predictions for the future. Cureus. (2020);12:e11686. doi: 10.7759/cureus.11686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anderson J, Hampton D, Patani R, Pryce G, Crowther R, Reynolds R, Franklin R, Giovannoni G, Compston D, Baker D. Abnormally phosphorylated tau is associated with neuronal and axonal loss in experimental autoimmune encephalomyelitis and multiple sclerosis. Brain. (2008);131:1736–1748. doi: 10.1093/brain/awn119. [DOI] [PubMed] [Google Scholar]
- 10.Andriessen TM, Jacobs B, Vos PE. Clinical characteristics and pathophysiological mechanisms of focal and diffuse traumatic brain injury. J Cell Mol Med. (2010);14:2381–2392. doi: 10.1111/j.1582-4934.2010.01164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Antonarakis SE, Skotko BG, Rafii MS, Strydom A, Pape SE, Bianchi DW, Sherman SL, Reeves RH. Down syndrome. Nat Rev Dis Primers. (2020);6:9. doi: 10.1038/s41572-019-0143-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. (2006);351:602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- 13.Arvanitakis Z, Wilson RS, Bienias JL, Evans DA, Bennett DA. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol. (2004);61:661–666. doi: 10.1001/archneur.61.5.661. [DOI] [PubMed] [Google Scholar]
- 14.Arya M, Sabrosa AS, Duker JS, Waheed NK. Choriocapillaris changes in dry age-related macular degeneration and geographic atrophy:a review. Eye Vis. (2018);5:22. doi: 10.1186/s40662-018-0118-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ashok A, Singh N, Chaudhary S, Bellamkonda V, Kritikos AE, Wise AS, Rana N, McDonald D, Ayyagari R. Retinal degeneration and Alzheimer's disease:an evolving link. Int J Mol Sci. (2020);21:7290. doi: 10.3390/ijms21197290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Attems J, Jellinger KA. The overlap between vascular disease and Alzheimer's disease-lessons from pathology. BMC Med. (2014);12:206. doi: 10.1186/s12916-014-0206-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Augutis K, Axelsson M, Portelius E, Brinkmalm G, Andreasson U, Gustavsson MK, Malmeström C, Lycke J, Blennow K, Zetterberg H. Cerebrospinal fluid biomarkers of β-amyloid metabolism in multiple sclerosis. Mult Scler. (2013);19:543–552. doi: 10.1177/1352458512460603. [DOI] [PubMed] [Google Scholar]
- 18.Auriel E, Greenberg SM. The pathophysiology and clinical presentation of cerebral amyloid angiopathy. Curr Atheroscler Rep. (2012);14:343–350. doi: 10.1007/s11883-012-0254-z. [DOI] [PubMed] [Google Scholar]
- 19.Ayaydın H, Kirmit A, Çelik H, Akaltun İ, Koyuncu İ, Ulgar ŞB. High serum levels of serum 100 beta protein neuron-specific enolase. Tau active caspase-3 M30 and M65 in children with autism spectrum disorders. Clin Psychopharmacol Neurosci. (2020);18:270–278. doi: 10.9758/cpn.2020.18.2.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bäckström DC, Domellöf ME, Linder J, Olsson B, Öhrfelt A, Trupp M, Zetterberg H, Blennow K, Forsgren L. Cerebrospinal fluid patterns and the risk of future dementia in early incident. Parkinson disease. JAMA Neurol. (2015);72:1175–1182. doi: 10.1001/jamaneurol.2015.1449. [DOI] [PubMed] [Google Scholar]
- 21.Bandmann O, Weiss KH, Kaler SG. Wilson's disease and other neurological copper disorders. Lancet Neurol. (2015);14:103–113. doi: 10.1016/S1474-4422(14)70190-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bao H, Liu Y, Zhang M, Chen Z, Zhang W, Ge Y, Kang D, Gao F, Shen Y. Increased β-site APP cleaving enzyme 1-mediated insulin receptor cleavage in type 2 diabetes mellitus with cognitive impairment. Alzheimers Dement. (2021);17:1097–1108. doi: 10.1002/alz.12276. [DOI] [PubMed] [Google Scholar]
- 23.Baquero MT, Lostritto K, Gustavson MD, Bassi KA, Appia F, Camp RL, Molinaro AM, Harris LN, Rimm DL. Evaluation of prognostic and predictive value of microtubule associated protein tau in two independent cohorts. Breast Cancer Res. (2011);13:R85. doi: 10.1186/bcr2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barbier P, Zejneli O, Martinho M, Lasorsa A, Belle V, Smet-Nocca C, Tsvetkov PO, Devred F, Landrieu I. Role of tau as a microtubule-associated protein:structural and functional aspects. Front Aging Neurosci. (2019);11:204. doi: 10.3389/fnagi.2019.00204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barón-Mendoza I, García O, Calvo-Ochoa E, Rebollar-García JO, Garzón-Cortés D, Haro R, González-Arenas A. Alterations in neuronal cytoskeletal and astrocytic proteins content in the brain of the autistic-like mouse strain C58/J. Neurosci Lett. (2018);682:32–38. doi: 10.1016/j.neulet.2018.06.004. [DOI] [PubMed] [Google Scholar]
- 26.Barron MR, Gartlon J, Dawson LA, Atkinson PJ, Pardon MC. Increasing Tau 4R Tau levels exacerbates hippocampal Tau hyperphosphorylation in the hTau model of tauopathy but also Tau dephosphorylation following acute systemic inflammation. Front Immunol. (2020);11:293. doi: 10.3389/fimmu.2020.00293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bassil F, Brown HJ, Pattabhiraman S, Iwasyk JE, Maghames CM, Meymand ES, Cox TO, Riddle DM, Zhang B, Trojanowski JQ. Amyloid-beta (Aβ) plaques promote seeding and spreading of alpha-synuclein and tau in a mouse model of Lewy body disorders with Aβpathology. Neuron. (2020);105:260–275. doi: 10.1016/j.neuron.2019.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, Nance M, Ross CA, Scahill RI, Wetzel R. Huntington disease. Nat Rev Dis Primers. (2015);1:15005. doi: 10.1038/nrdp.2015.5. [DOI] [PubMed] [Google Scholar]
- 29.Baudouin C, Kolko M, Melik-Parsadaniantz S, Messmer EM. Inflammation in Glaucoma:from the back to the front of the eye and beyond. Prog Retin Eye Res. (2020);83:100916. doi: 10.1016/j.preteyeres.2020.100916. [DOI] [PubMed] [Google Scholar]
- 30.Bauman M. Neuroanatomic observations of the brain in autism. In: Bauman ML, Kemper TL, editors. In:The neurobiology of autism. Baltimore: Johns Hopkins University Press; (1994). pp. 119–145. [Google Scholar]
- 31.Bennett RE, Robbins AB, Hu M, Cao X, Betensky RA, Clark T, Das S, Hyman BT. Tau induces blood vessel abnormalities and angiogenesis-related gene expression in P301L transgenic mice and human Alzheimer's disease. Proc Natl Acad Sci U S A. (2018);115:E1289–1298. doi: 10.1073/pnas.1710329115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berisha F, Feke GT, Trempe CL, McMeel JW, Schepens CL. Retinal abnormalities in early Alzheimer's disease. Invest Ophthalmol Vis Sci. (2007);48:2285–2289. doi: 10.1167/iovs.06-1029. [DOI] [PubMed] [Google Scholar]
- 33.Betrie AH, Ayton S, Bush AI, Angus JA, Lei P, Wright CE. Evidence of a cardiovascular function for microtubule-associated protein tau. J Alzheimers Dis. (2017);56:849–860. doi: 10.3233/JAD-161093. [DOI] [PubMed] [Google Scholar]
- 34.Biffi A, Greenberg SM. Cerebral amyloid angiopathy:a systematic review. J Clin Neurol. (2011);7:1–9. doi: 10.3988/jcn.2011.7.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bird SM, Sohrabi HR, Sutton TA, Weinborn M, Rainey-Smith SR, Brown B, Patterson L, Taddei K, Gupta V, Carruthers M. Cerebral amyloid-βaccumulation and deposition following traumatic brain injury—a narrative review and meta-analysis of animal studies. Neurosci Biobehav Rev. (2016);64:215–228. doi: 10.1016/j.neubiorev.2016.01.004. [DOI] [PubMed] [Google Scholar]
- 36.Biron KE, Dickstein DL, Gopaul R, Jefferies WA. Amyloid triggers extensive cerebral angiogenesis causing blood brain barrier permeability and hypervascularity in Alzheimer's disease. PLoS One. (2011);6:e23789. doi: 10.1371/journal.pone.0023789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blasko I, Beer R, Bigl M, Apelt J, Franz G, Rudzki D, Ransmayr G, Kampfl A, Schliebs R. Experimental traumatic brain injury in rats stimulates the expression production and activity of Alzheimer's disease β-secretase (BACE-1) J Neural Transm. (2004);111:523–536. doi: 10.1007/s00702-003-0095-6. [DOI] [PubMed] [Google Scholar]
- 38.Blum D, Herrera F, Francelle L, Mendes T, Basquin M, Obriot H, Demeyer D, Sergeant N, Gerhardt E, Brouillet E. Mutant huntingtin alters Tau phosphorylation and subcellular distribution. Hum Mol Genet. (2015);24:76–85. doi: 10.1093/hmg/ddu421. [DOI] [PubMed] [Google Scholar]
- 39.Bonda DJ, Lee HG, Camins A, Pallàs M, Casadesus G, Smith MA, Zhu X. The sirtuin pathway in ageing and Alzheimer disease:mechanistic and therapeutic considerations. Lancet Neurol. (2011);10:275–279. doi: 10.1016/S1474-4422(11)70013-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bras JM, Singleton A. Genetic susceptibility in Parkinson's disease. Biochim Biophys Acta. (2009);1792:597–603. doi: 10.1016/j.bbadis.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brettschneider J, Maier M, Arda S, Claus A, Süssmuth S, Kassubek J, Tumani H. Tau protein level in cerebrospinal fluid is increased in patients with early multiple sclerosis. Mult Scler. (2005);11:261–265. doi: 10.1191/1352458505ms1159oa. [DOI] [PubMed] [Google Scholar]
- 42.Bruban J, Glotin AL, Dinet V, Chalour N, Sennlaub F, Jonet L, An N, Faussat AM, Mascarelli F. Amyloid-β(1-42) alters structure and function of retinal pigmented epithelial cells. Aging Cell. (2009);8:162–177. doi: 10.1111/j.1474-9726.2009.00456.x. [DOI] [PubMed] [Google Scholar]
- 43.Busciglio J, Pelsman A, Wong C, Pigino G, Yuan M, Mori H, Yankner BA. Altered metabolism of the amyloid βprecursor protein is associated with mitochondrial dysfunction in Down's syndrome. Neuron. (2002);33:677–688. doi: 10.1016/s0896-6273(02)00604-9. [DOI] [PubMed] [Google Scholar]
- 44.Calingasan NY, Chen J, Kiaei M, Beal MF. β-amyloid 42 accumulation in the lumbar spinal cord motor neurons of amyotrophic lateral sclerosis patients. Neurobiol Dis. (2005);19:340–347. doi: 10.1016/j.nbd.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 45.Cameron DJ, Galvin C, Alkam T, Sidhu H, Ellison J, Luna S, Ethell DW. Alzheimer's-related peptide amyloid-βplays a conserved role in angiogenesis. PLoS One. (2012);7:e39598. doi: 10.1371/journal.pone.0039598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Canton J, Neculai D, Grinstein S. Scavenger receptors in homeostasis and immunity. Nat Rev Immunol. (2013);13:621–634. doi: 10.1038/nri3515. [DOI] [PubMed] [Google Scholar]
- 47.Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA. Endocytic pathway abnormalities precede amyloid βdeposition in sporadic Alzheimer's disease and Down syndrome:differential effects of APOE genotype and presenilin mutations. Am J Pathol. (2000);157:277–286. doi: 10.1016/s0002-9440(10)64538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chan JW, Chan NC, Sadun AA. Glaucoma as neurodegeneration in the brain. Eye Brain. (2021);13:21–28. doi: 10.2147/EB.S293765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Charidimou A, Boulouis G, Pasi M, Auriel E, van Etten ES, Haley K, Ayres A, Schwab KM, Martinez-Ramirez S, Goldstein JN. MRI-visible perivascular spaces in cerebral amyloid angiopathy and hypertensive arteriopathy. Neurology. (2017);88:1157–1164. doi: 10.1212/WNL.0000000000003746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chatterjee S, Mudher A. Alzheimer's disease and type 2 diabetes:a critical assessment of the shared pathological traits. Front Neurosci. (2018);12:383. doi: 10.3389/fnins.2018.00383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen GF, Xu TH, Yan Y, Zhou YR, Jiang Y, Melcher K, Xu HE. Amyloid beta:structure biology and structure-based therapeutic development. Acta Pharmacol Sin. (2017);38:1205–1235. doi: 10.1038/aps.2017.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen L, Bai Y, Zhao M, Jiang Y. TLR4 inhibitor attenuates amyloid-β-induced angiogenic and inflammatory factors in ARPE-19 cells:implications for age-related macular degeneration. Mol Med Rep. (2016);13:3249–3256. doi: 10.3892/mmr.2016.4890. [DOI] [PubMed] [Google Scholar]
- 53.Chen XH, Siman R, Iwata A, Meaney DF, Trojanowski JQ, Smith DH. Long-term accumulation of amyloid-β β-secretase presenilin-1 and caspase-3 in damaged axons following brain trauma. Am J Pathol. (2004);165:357–371. doi: 10.1016/s0002-9440(10)63303-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen XH, Johnson VE, Uryu K, Trojanowski JQ, Smith DH. A lack of amyloid βplaques despite persistent accumulation of amyloid βin axons of long-term survivors of traumatic brain injury. Brain Pathol. (2009);19:214–223. doi: 10.1111/j.1750-3639.2008.00176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chiasseu M, Vargas JLC, Destroismaisons L, Velde CV, Leclerc N, Di Polo A. Tau accumulation altered phosphorylation and missorting promote neurodegeneration in glaucoma. J Neurosci. (2016);36:5785–5798. doi: 10.1523/JNEUROSCI.3986-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chitranshi N, Kumar A, Sheriff S, Gupta V, Godinez A, Saks D, Sarkar S, Shen T, Mirzaei M, Basavarajappa D. Identification of novel cathepsin B inhibitors with implications in Alzheimer's disease:Computational refining and biochemical evaluation. Cells. (2021);10:1946. doi: 10.3390/cells10081946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chornenkyy Y, Fardo DW, Nelson PT. Tau and TDP-43 proteinopathies:kindred pathologic cascades and genetic pleiotropy. Lab Invest. (2019);99:993–1007. doi: 10.1038/s41374-019-0196-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chou CC, Zhang Y, Umoh ME, Vaughan SW, Lorenzini I, Liu F, Sayegh M, Donlin-Asp PG, Chen YH, Duong DM, Seyfried NT, Powers MA, Kukar T, Hales CM, Gearing M, Cairns NJ, Boylan KB, Dickson DW, Rademakers R, Zhang YJ, et al. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat Neurosci. (2018);21:228–239. doi: 10.1038/s41593-017-0047-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chung DC, Roemer S, Petrucelli L, Dickson DW. Cellular and pathological heterogeneity of primary tauopathies. Mol Neurodegener. (2021);16:57. doi: 10.1186/s13024-021-00476-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cisbani G, Maxan A, Kordower JH, Planel E, Freeman TB, Cicchetti F. Presence of tau pathology within foetal neural allografts in patients with Huntington's and Parkinson's disease. Brain. (2017);140:2982–2992. doi: 10.1093/brain/awx255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Clavaguera F, Tolnay M, Goedert M. The prion-like behavior of assembled tau in transgenic mice. Cold Spring Harb Perspect Med. (2017);7:a024372. doi: 10.1101/cshperspect.a024372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Clinton LK, Blurton-Jones M, Myczek K, Trojanowski JQ, LaFerla FM. Synergistic interactions between Aβ tau and α-synuclein:acceleration of neuropathology and cognitive decline. J Neurosci. (2010);30:7281–7289. doi: 10.1523/JNEUROSCI.0490-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cohen O, Chapman J, Korczyn A, Warman-Alaluf N, Nitsan Z, Appel S, Kahana E, Rosenmann H. CSF tau correlates with CJD disease severity and cognitive decline. Acta Neurol Scand. (2016);133:119–123. doi: 10.1111/ane.12441. [DOI] [PubMed] [Google Scholar]
- 64.Craft S, Baker LD, Montine TJ, Minoshima S, Watson GS, Claxton A, Arbuckle M, Callaghan M, Tsai E, Plymate SR. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment:a pilot clinical trial. Arch Neurol. (2012);69:29–38. doi: 10.1001/archneurol.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Craft S, Claxton A, Baker LD, Hanson AJ, Cholerton B, Trittschuh EH, Dahl D, Caulder E, Neth B, Montine TJ. Effects of regular and long-acting insulin on cognition and Alzheimer's disease biomarkers:a pilot clinical trial. J Alzheimers Dis. (2017);57:1325–1334. doi: 10.3233/JAD-161256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Craft S, Raman R, Chow TW, Rafii MS, Sun C-K, Rissman RA, Donohue MC, Brewer JB, Jenkins C, Harless K. Safety efficacy and feasibility of intranasal insulin for the treatment of mild cognitive impairment and Alzheimer disease dementia:a randomized clinical trial. JAMA Neurol. (2020);77:1099–1109. doi: 10.1001/jamaneurol.2020.1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Credle JJ, George JL, Wills J, Duka V, Shah K, Lee YC, Rodriguez O, Simkins T, Winter M, Moechars D. GSK-3βdysregulation contributes to parkinson's-like pathophysiology with associated region-specific phosphorylation and accumulation of tau and α-synuclein. Cell Death Differ. (2015);22:838–851. doi: 10.1038/cdd.2014.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Criscuolo C, Cerri E, Fabiani C, Capsoni S, Cattaneo A, Domenici L. The retina as a window to early dysfunctions of Alzheimer's disease following studies with a 5xFAD mouse model. Neurobiol Aging. (2018);67:181–188. doi: 10.1016/j.neurobiolaging.2018.03.017. [DOI] [PubMed] [Google Scholar]
- 69.d'Errico P, Meyer-Luehmann M. Mechanisms of pathogenic tau and Aβprotein spreading in Alzheimer's disease. Front Aging Neurosci. (2020);12:265. doi: 10.3389/fnagi.2020.00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dasari AK, Kayed R, Wi S, Lim KH. Tau interacts with the C-terminal region of α-synuclein promoting formation of toxic aggregates with distinct molecular conformations. Biochemistry. (2019);58:2814–2821. doi: 10.1021/acs.biochem.9b00215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Davis AA, Andruska KM, Benitez BA, Racette BA, Perlmutter JS, Cruchaga C. Variants in GBA, SNCA and MAPT influence Parkinson disease risk age at onset and progression. Neurobiol Aging. (2016);37:209. doi: 10.1016/j.neurobiolaging.2015.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dawson HN, Cantillana V, Chen L, Vitek MP. The tau N279K exon 10 splicing mutation recapitulates frontotemporal dementia and parkinsonism linked to chromosome 17 tauopathy in a mouse model. J Neurosci. (2007);27:9155–9168. doi: 10.1523/JNEUROSCI.5492-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.De Meyer GR, De Cleen DM, Cooper S, Knaapen MW, Jans DM, Martinet W, Herman AG, Bult H, Kockx MM. Platelet phagocytosis and processing of β-amyloid precursor protein as a mechanism of macrophage activation in atherosclerosis. Circ Res. (2002);90:1197–1204. doi: 10.1161/01.res.0000020017.84398.61. [DOI] [PubMed] [Google Scholar]
- 74.De Vos A, Bjerke M, Brouns R, De Roeck N, Jacobs D, Van den Abbeele L, Guldolf K, Zetterberg H, Blennow K, Engelborghs S. Neurogranin and tau in cerebrospinal fluid and plasma of patients with acute ischemic stroke. BMC Neurol. (2017);17:170. doi: 10.1186/s12883-017-0945-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Debatin L, Streffer J, Geissen M, Matschke J, Aguzzi A, Glatzel M. Association between deposition of beta-amyloid and pathological prion protein in sporadic Creutzfeldt-Jakob disease. Neurodegener Dis. (2008);5:347–354. doi: 10.1159/000121389. [DOI] [PubMed] [Google Scholar]
- 76.Delikkaya B, Moriel N, Tong M, Gallucci G, Suzanne M. Altered expression of insulin-degrading enzyme and regulator of calcineurin in the rat intracerebral streptozotocin model and human apolipoprotein E-ε4–associated Alzheimer's disease. Alzheimers Dement. (2019);11:392–404. doi: 10.1016/j.dadm.2019.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Deng L, Pushpitha K, Joseph C, Gupta V, Rajput R, Chitranshi N, Dheer Y, Amirkhani A, Kamath K, Pascovici D. Amyloid βinduces early changes in the ribosomal machinery cytoskeletal organization and oxidative phosphorylation in retinal photoreceptor cells. Front Mol Neurosci. (2019);12:24. doi: 10.3389/fnmol.2019.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Deng L, Gupta V, Abyadeh M, Chitranshi N, Pushpitha K, Wu Y, Gupta V, You Y, Paulo JA, Graham SL. Oxidative stress induced dysfunction of protein synthesis in 661W mice photoreceptor cells. Proteomes. (2023);11:12. doi: 10.3390/proteomes11020012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Depp C, Sun T, Sasmita AO, Spieth L, Berghoff SA, Nazarenko T, Overhoff K, Steixner-Kumar AA, Subramanian S, Arinrad S. Myelin dysfunction drives amyloid-βdeposition in models of Alzheimer's disease. Nature. (2023);618:349–357. doi: 10.1038/s41586-023-06120-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.DeSimone CV, Graff-Radford J, El-Harasis MA, Rabinstein AA, Asirvatham SJ, Holmes DR. Cerebral amyloid angiopathy:diagnosis clinical implications and management strategies in atrial fibrillation. J Am Coll Cardiol. (2017);70:1173–1182. doi: 10.1016/j.jacc.2017.07.724. [DOI] [PubMed] [Google Scholar]
- 81.Dewan MC, Rattani A, Gupta S, Baticulon RE, Hung YC, Punchak M, Agrawal A, Adeleye AO, Shrime MG, Rubiano AM. Estimating the global incidence of traumatic brain injury. J Neurosurg. (2018);130:1080–1097. doi: 10.3171/2017.10.JNS17352. [DOI] [PubMed] [Google Scholar]
- 82.Di Domenico F, Tramutola A, Foppoli C, Head E, Perluigi M, Butterfield DA. mTOR in Down syndrome:role in Aßand tau neuropathology and transition to Alzheimer disease-like dementia. Free Radic Biol Med. (2018);114:94–101. doi: 10.1016/j.freeradbiomed.2017.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dierssen M. Down syndrome:the brain in trisomic mode. Nat Rev Neurosci. (2012);13:844–858. doi: 10.1038/nrn3314. [DOI] [PubMed] [Google Scholar]
- 84.Diniz Pereira J, Gomes Fraga V, Morais Santos AL, Carvalho MdG, Caramelli P, Braga Gomes K. Alzheimer's disease and type 2 diabetes mellitus:a systematic review of proteomic studies. J Neurochem. (2021);156:753–776. doi: 10.1111/jnc.15166. [DOI] [PubMed] [Google Scholar]
- 85.Donnini S, Solito R, Cetti E, Corti F, Giachetti A, Carra S, Beltrame M, Cotelli F, Ziche M. Aßpeptides accelerate the senescence of endothelial cells in vitro and in vivo impairing angiogenesis. FASEB J. (2010);24:2385–2395. doi: 10.1096/fj.09-146456. [DOI] [PubMed] [Google Scholar]
- 86.Dorsey ER, Elbaz A, Nichols E, Abd-Allah F, Abdelalim A, Adsuar JC, Ansha MG, Brayne C, Choi JYJ, Collado-Mateo D. Global regional and national burden of Parkinson's disease 1990–2016:a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. (2018);17:939–953. doi: 10.1016/S1474-4422(18)30295-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Emberti Gialloreti L, Mazzone L, Benvenuto A, Fasano A, Garcia Alcon A, Kraneveld A, Moavero R, Raz R, Riccio MP, Siracusano M. Risk and protective environmental factors associated with autism spectrum disorder:evidence-based principles and recommendations. J Clin Med. (2019);8:217. doi: 10.3390/jcm8020217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Faraco G, Park L, Zhou P, Luo W, Paul SM, Anrather J, Iadecola C. Hypertension enhances A β-induced neurovascular dysfunction promotes β-secretase activity and leads to amyloidogenic processing of APP. J Cereb Blood Flow Metab. (2016);36:241–252. doi: 10.1038/jcbfm.2015.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Filippi M, Bar-Or A, Piehl F, Preziosa P, Solari A, Vukusic S, Rocca MA. Multiple sclerosis. Nat Rev Dis Primers. (2018);4:43. doi: 10.1038/s41572-018-0041-4. [DOI] [PubMed] [Google Scholar]
- 90.François GG, Daigen X, George SR, John PV, Yanxia Z, JingQi H, Andréa L, David S, Michael R, Mark SS. Involvement of caspases in proteolytic cleavage of Alzheimer's amyloid-βprecursor protein and amyloidogenic Aβpeptide formation. Cell. (1999);97:395–406. doi: 10.1016/s0092-8674(00)80748-5. [DOI] [PubMed] [Google Scholar]
- 91.Gargini R, Segura-Collar B, Sánchez-Gómez P. Novel functions of the neurodegenerative-related gene tau in cancer. Front Aging Neurosci. (2019);11:231. doi: 10.3389/fnagi.2019.00231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gasparini L, Netzer WJ, Greengard P, Xu H. Does insulin dysfunction play a role in Alzheimer's disease? Trends Pharmacol Sci. (2002);23:288–293. doi: 10.1016/s0165-6147(02)02037-0. [DOI] [PubMed] [Google Scholar]
- 93.Gąssowska M, Czapski GA, Pająk B, Cieślik M, Lenkiewicz AM, Adamczyk A. Extracellular α-synuclein leads to microtubule destabilization via GSK-3β-dependent Tau phosphorylation in PC12 cells. PLoS One. (2014);9:e94259. doi: 10.1371/journal.pone.0094259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, Trojanowski JQ, Lee VMY. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science. (2003);300:636–640. doi: 10.1126/science.1082324. [DOI] [PubMed] [Google Scholar]
- 95.Gonçalves SA, Outeiro TF. Traffic jams and the complex role of α-Synuclein aggregation in Parkinson disease. Small GTPases. (2017);8:78–84. doi: 10.1080/21541248.2016.1199191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gong Z, Gao L, Lu Y, Wang Z. CSF p-tau as a potential cognition impairment biomarker in ALS. Front Neurol. (2022);13:991143. doi: 10.3389/fneur.2022.991143. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 97.Gough KC, Maddison BC. Prion transmission:prion excretion and occurrence in the environment. Prion. (2010);4:275–282. doi: 10.4161/pri.4.4.13678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gough M, Blanthorn-Hazell S, Delury C, Parkin E. The E1 copper binding domain of full-length amyloid precursor protein mitigates copper-induced growth inhibition in brain metastatic prostate cancer DU145 cells. Biochem Biophys Res Commun. (2014);453:741–747. doi: 10.1016/j.bbrc.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Goulay R, Romo LM, Hol EM, Dijkhuizen RM. From stroke to dementia:a comprehensive review exposing tight interactions between stroke and Amyloid-βformation. Transl Stroke Res. (2020);11:601–614. doi: 10.1007/s12975-019-00755-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gratuze M, Cisbani G, Cicchetti F, Planel E. Is Huntington's disease a tauopathy? Brain. (2016);139:1014–1025. doi: 10.1093/brain/aww021. [DOI] [PubMed] [Google Scholar]
- 101.Grigg I, Ivashko-Pachima Y, Hait TA, Korenková V, Touloumi O, Lagoudaki R, Van Dijck A, Marusic Z, Anicic M, Vukovic J. Tauopathy in the young autistic brain:novel biomarker and therapeutic target. Transl Psychiatry. (2020);10:228. doi: 10.1038/s41398-020-00904-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gu Y, Oyama F, Ihara Y. τis widely expressed in rat tissues. J Neurochem. (1996);67:1235–1244. doi: 10.1046/j.1471-4159.1996.67031235.x. [DOI] [PubMed] [Google Scholar]
- 103.Guo L, Moss SE, Alexander RA, Ali RR, Fitzke FW, Cordeiro MF. Retinal ganglion cell apoptosis in glaucoma is related to intraocular pressure and IOP-induced effects on extracellular matrix. Invest Ophthalmol Vis Sci. (2005);46:175–182. doi: 10.1167/iovs.04-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Guo L, Salt TE, Luong V, Wood N, Cheung W, Maass A, Ferrari G, Russo-Marie F, Sillito AM, Cheetham ME. Targeting amyloid-βin glaucoma treatment. Proc Natl Acad Sci U S A. (2007);104:13444–13449. doi: 10.1073/pnas.0703707104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gupta N, Fong J, Ang LC, Yücel YH. Retinal tau pathology in human glaucomas. Can J Ophthalmol. (2008);43:53–60. doi: 10.3129/i07-185. [DOI] [PubMed] [Google Scholar]
- 106.Gupte A, Mumper RJ. Elevated copper and oxidative stress in cancer cells as a target for cancer treatment. Cancer Treat Rev. (2009);35:32–46. doi: 10.1016/j.ctrv.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 107.Hamdeh SA, Shevchenko G, Mi J, Musunuri S, Bergquist J, Marklund N. Proteomic differences between focal and diffuse traumatic brain injury in human brain tissue. Sci Rep. (2018);8:6807. doi: 10.1038/s41598-018-25060-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hamilton RL. Lewy bodies in Alzheimer's disease:a neuropathological review of 145 cases using α-synuclein immunohistochemistry. Brain Pathol. (2000);10:378–384. doi: 10.1111/j.1750-3639.2000.tb00269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hanger D, Brion JP, Gallo J, Cairns N, Luthert P, Anderton B. Tau in Alzheimer's disease and Down's syndrome is insoluble and abnormally phosphorylated. Biochem J. (1991);275:99–104. doi: 10.1042/bj2750099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hansel DE, Rahman A, Wehner S, Herzog V, Yeo CJ, Maitra A. Increased expression and processing of the Alzheimer amyloid precursor protein in pancreatic cancer may influence cellular proliferation. Cancer Res. (2003);63:7032–7037. [PubMed] [Google Scholar]
- 111.Head E, Lott I, Cribbs DH, Cotman CW, Rohn TT. β-Amyloid deposition and neurofibrillary tangle association with caspase activation in Down syndrome. Neurosci Lett. (2002);330:99–103. doi: 10.1016/s0304-3940(02)00705-x. [DOI] [PubMed] [Google Scholar]
- 112.Head E, Lott IT. Down syndrome and beta-amyloid deposition. Curr Opin Neurol. (2004);17:95–100. doi: 10.1097/00019052-200404000-00003. [DOI] [PubMed] [Google Scholar]
- 113.Head E, Helman AM, Powell D, Schmitt FA. Down syndrome beta-amyloid and neuroimaging. Free Radic Biol Med. (2018);114:102–109. doi: 10.1016/j.freeradbiomed.2017.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Herman AM, Khandelwal PJ, Stanczyk BB, Rebeck GW, Moussa CEH. β-amyloid triggers ALS-associated TDP-43 pathology in AD models. Brain Res. (2011);1386:191–199. doi: 10.1016/j.brainres.2011.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Herzig MC, Van Nostrand WE, Jucker M. Mechanism of cerebral β-amyloid angiopathy:murine and cellular models. Brain Pathol. (2006);16:40–54. doi: 10.1111/j.1750-3639.2006.tb00560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ho WL, Leung Y, Tsang AWT, So KF, Chiu K, Chang RCC. Tauopathy in the retina and optic nerve:does it shadow pathological changes in the brain? Mol Vis. (2012);18:2700–2710. [PMC free article] [PubMed] [Google Scholar]
- 117.Hobday AL, Parmar MS. The link between diabetes mellitus and Tau hyperphosphorylation:implications for risk of Alzheimer's disease. Cureus. (2021);13:e18362. doi: 10.7759/cureus.18362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hoh Kam J, Lenassi E, Jeffery G. Viewing ageing eyes:diverse sites of amyloid Beta accumulation in the ageing mouse retina and the up-regulation of macrophages. PLoS One. (2010);5:e13127. doi: 10.1371/journal.pone.0013127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hong X, Bu L, Wang Y, Xu J, Wu J, Huang Y, Liu J, Suo H, Yang L, Shi Y. Increases in the risk of cognitive impairment and alterations of cerebral β-amyloid metabolism in mouse model of heart failure. PLoS One. (2013);8:e63829. doi: 10.1371/journal.pone.0063829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hu WT, Chen-Plotkin A, Arnold SE, Grossman M, Clark CM, Shaw LM, McCluskey L, Elman L, Karlawish J, Hurtig HI. Biomarker discovery for Alzheimer's disease frontotemporal lobar degeneration and Parkinson's disease. Acta Neuropathol. (2010);120:385–399. doi: 10.1007/s00401-010-0723-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hu X, Yang Y, Gong D. Changes of cerebrospinal fluid Aβ42 t-tau and p-tau in Parkinson's disease patients with cognitive impairment relative to those with normal cognition:a meta-analysis. Neurol Sci. (2017a);38:1953–1961. doi: 10.1007/s10072-017-3088-1. [DOI] [PubMed] [Google Scholar]
- 122.Hu X, Yang Y, Gong D. Changes of cerebrospinal fluid Aβ42 t-tau and p-tau in Parkinson's disease patients with cognitive impairment relative to those with normal cognition:a meta-analysis. Neurol Sci. (2017b);38:1953–1961. doi: 10.1007/s10072-017-3088-1. [DOI] [PubMed] [Google Scholar]
- 123.Huda MN, Pan CH. Tau in Tauopathies that leads to cognitive disorders and in cancer. In:Cognitive disorders:IntechOpen. (2018) doi:10.5772/INTECHOPEN.74025. [Google Scholar]
- 124.Iadecola C, Zhang F, Niwa K, Eckman C, Turner SK, Fischer E, Younkin S, Borchelt DR, Hsiao KK, Carlson GA. SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nat Neurosci. (1999);2:157–161. doi: 10.1038/5715. [DOI] [PubMed] [Google Scholar]
- 125.Inyushin M, Zayas-Santiago A, Rojas L, Kucheryavykh L. On the role of platelet-generated amyloid beta peptides in certain amyloidosis health complications. Front Immunol. (2020);11:571083. doi: 10.3389/fimmu.2020.571083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ito S, Miki Y, Saito R, Inoue C, Okada Y, Sasano H. Amyloid precursor protein and its phosphorylated form in non-small cell lung carcinoma. Pathol Res Pract. (2019);215:152463. doi: 10.1016/j.prp.2019.152463. [DOI] [PubMed] [Google Scholar]
- 127.Iwasaki Y. Creutzfeldt-Jakob disease. Neuropathology. (2017);37:174–188. doi: 10.1111/neup.12355. [DOI] [PubMed] [Google Scholar]
- 128.Iwata A, Chen XH, McIntosh TK, Browne KD, Smith DH. Long-term accumulation of amyloid-βin axons following brain trauma without persistent upregulation of amyloid precursor protein genes. J Neuropathol Exp Neurol. (2002);61:1056–1068. doi: 10.1093/jnen/61.12.1056. [DOI] [PubMed] [Google Scholar]
- 129.Iwatsubo T, Mann DM, Odaka A, Suzuki N, Ihara Y. Amyloid βprotein (Aβ) deposition:Aβ42 (43) precedes Aβ40 in Down syndrome. Ann Neurol. (1995);37:294–299. doi: 10.1002/ana.410370305. [DOI] [PubMed] [Google Scholar]
- 130.James BD, Wilson RS, Boyle PA, Trojanowski JQ, Bennett DA, Schneider JA. TDP-43 stage mixed pathologies and clinical Alzheimer's-type dementia. Brain. (2016);139:2983–2993. doi: 10.1093/brain/aww224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Jans D, Martinet W, Van De Parre T, Herman A, Bult H, Kockx M, De Meyer G. Processing of amyloid precursor protein as a biochemical link between atherosclerosis and Alzheimer's disease. Cardiovasc Hematol Disord Drug Targets. (2006);6:21–34. doi: 10.2174/187152906776092695. [DOI] [PubMed] [Google Scholar]
- 132.Jellinger K. Alzheimer-type lesions in Huntington's disease. J Neural Transm. (1998);105:787–799. doi: 10.1007/s007020050095. [DOI] [PubMed] [Google Scholar]
- 133.Jendroska K, Lees AJ, Poewe W, Daniel SE. Amyloid β-peptide and the dementia of Parkinson's diease. Mov Disord. (1996);11:647–653. doi: 10.1002/mds.870110609. [DOI] [PubMed] [Google Scholar]
- 134.Jiang Y, Rigoglioso A, Peterhoff CM, Pawlik M, Sato Y, Bleiwas C, Stavrides P, Smiley JF, Ginsberg SD, Mathews PM. Partial BACE1 reduction in a Down syndrome mouse model blocks Alzheimer-related endosomal anomalies and cholinergic neurodegeneration:role of APP-CTF. Neurobiol Aging. (2016);39:90–98. doi: 10.1016/j.neurobiolaging.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jin WS, Bu XL, Liu YH, Shen LL, Zhuang ZQ, Jiao SS, Zhu C, Wang QH, Zhou HD, Zhang T. Plasma amyloid-beta levels in patients with different types of cancer. Neurotox Res. (2017);31:283–288. doi: 10.1007/s12640-016-9682-9. [DOI] [PubMed] [Google Scholar]
- 136.Johnson VE, Stewart W, Smith DH. Traumatic brain injury and amyloid-βpathology:a link to Alzheimer's disease? Nat Rev Neurosci. (2010);11:361–370. doi: 10.1038/nrn2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Johnson VE, Stewart W, Smith DH. Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol. (2012);22:142–149. doi: 10.1111/j.1750-3639.2011.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jonas JB, Wei WB, Zhu LP, Xu L, Wang YX. Cognitive function and ophthalmological diseases:the Beijing Eye Study. Sci Rep. (2018);8:4816. doi: 10.1038/s41598-018-23314-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Josephs KA, Whitwell JL, Weigand SD, Murray ME, Tosakulwong N, Liesinger AM, Petrucelli L, Senjem ML, Knopman DS, Boeve BF. TDP-43 is a key player in the clinical features associated with Alzheimer's disease. Acta Neuropathol. (2014);127:811–824. doi: 10.1007/s00401-014-1269-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Josephs KA. Current understanding of neurodegenerative diseases associated with the protein tau. Mayo Clin Proc. (2017);92:1291–1303. doi: 10.1016/j.mayocp.2017.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kaarniranta K, Salminen A, Haapasalo A, Soininen H, Hiltunen M. Age-related macular degeneration (AMD):Alzheimer's disease in the eye? J Alzheimers Dis. (2011);24:615–631. doi: 10.3233/JAD-2011-101908. [DOI] [PubMed] [Google Scholar]
- 142.Kadak MT, Cetin I, Tarakçıoğlu MC, Özer ÖF, Kaçar S, Çimen B. Low serum level α-synuclein and tau protein in autism spectrum disorder compared to controls. Neuropediatrics. (2015);46:410–415. doi: 10.1055/s-0035-1565273. [DOI] [PubMed] [Google Scholar]
- 143.Kapaki E, Kilidireas K, Paraskevas G, Michalopoulou M, Patsouris E. Highly increased CSF tau protein and decreased β-amyloid (1–42) in sporadic CJD:a discrimination from Alzheimer's disease? J Neurol Neurosurg Psychiatry. (2001);71:401–403. doi: 10.1136/jnnp.71.3.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kasai T, Tatebe H, Kondo M, Ishii R, Ohmichi T, Yeung WTE, Morimoto M, Chiyonobu T, Terada N, Allsop D. Increased levels of plasma total tau in adult Down syndrome. PLoS One. (2017);12:e0188802. doi: 10.1371/journal.pone.0188802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Katsumoto A, Takeuchi H, Tanaka F. Tau pathology in chronic traumatic encephalopathy and Alzheimer's disease:similarities and differences. Front Neurol. (2019);10:980. doi: 10.3389/fneur.2019.00980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kim B, Backus C, Oh S, Hayes JM, Feldman EL. Increased tau phosphorylation and cleavage in mouse models of type 1 and type 2 diabetes. Endocrinology. (2009);150:5294–5301. doi: 10.1210/en.2009-0695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.King MR, Anderson NJ, Guernsey LS, Jolivalt CG. Glycogen synthase kinase-3 inhibition prevents learning deficits in diabetic mice. J Neurosci Res. (2013);91:506–514. doi: 10.1002/jnr.23192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kitazume S, Yoshihisa A, Yamaki T, Oikawa M, Tachida Y, Ogawa K, Imamaki R, Hagiwara Y, Kinoshita N, Takeishi Y. Soluble amyloid precursor protein 770 is released from inflamed endothelial cells and activated platelets:a novel biomarker for acute coronary syndrome. J Biol Chem. (2012);287:40817–40825. doi: 10.1074/jbc.M112.398578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kojro E, Fahrenholz F. The non-amyloidogenic pathway:structure and function of α-secretases. Subcell Biochem. (2005);38:105–127. doi: 10.1007/0-387-23226-5_5. [DOI] [PubMed] [Google Scholar]
- 150.Kokjohn TA, Van Vickle GD, Maarouf CL, Kalback WM, Hunter JM, Daugs ID, Luehrs DC, Lopez J, Brune D, Sue LI, Beach TG, Castaño EM, Roher AE. Chemical characterization of pro-inflammatory amyloid-beta peptides in human atherosclerotic lesions and platelets. Biochim Biophys Acta. (2011);1812:1508–1514. doi: 10.1016/j.bbadis.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kolarova M, García-Sierra F, Bartos A, Ricny J, Ripova D. Structure and pathology of tau protein in Alzheimer disease. Int J Alzheimers Dis. (2012);2012:731526. doi: 10.1155/2012/731526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kondo A, Shahpasand K, Mannix R, Qiu J, Moncaster J, Chen CH, Yao Y, Lin YM, Driver JA, Sun Y. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature. (2015);523:431–436. doi: 10.1038/nature14658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Konopka A, Whelan DR, Jamali MS, Perri E, Shahheydari H, Toth RP, Parakh S, Robinson T, Cheong A, Mehta P. Impaired NHEJ repair in amyotrophic lateral sclerosis is associated with TDP-43 mutations. Mol Neurodegener. (2020);15:51. doi: 10.1186/s13024-020-00386-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Köppen J, Schulze A, Machner L, Wermann M, Eichentopf R, Guthardt M, Hähnel A, Klehm J, Kriegeskorte MC, Hartlage-Rübsamen M. Amyloid-beta peptides trigger aggregation of alpha-synuclein in vitro. Molecules. (2020);25:580. doi: 10.3390/molecules25030580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Koronyo-Hamaoui M, Koronyo Y, Ljubimov AV, Miller CA, Ko MK, Black KL, Schwartz M, Farkas DL. Identification of amyloid plaques in retinas from Alzheimer's patients and noninvasive in vivo optical imaging of retinal plaques in a mouse model. Neuroimage. (2011);54:S204–S217. doi: 10.1016/j.neuroimage.2010.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kottaisamy CPD, Raj DS, Prasanth Kumar V, Sankaran U. Experimental animal models for diabetes and its related complications—a review. Lab Anim Res. (2021);37:23. doi: 10.1186/s42826-021-00101-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kotzbauer PT, Cairns NJ, Campbell MC, Willis AW, Racette BA, Tabbal SD, Perlmutter JS. Pathologic accumulation of α-synuclein and Aβin Parkinson disease patients with dementia. Arch Neurol. (2012);69:1326–1331. doi: 10.1001/archneurol.2012.1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Krebs M, Brunmair B, Brehm A, Artwohl M, Szendroedi J, Nowotny P, Roth E, Fürnsinn C, Promintzer M, Anderwald C. The Mammalian target of rapamycin pathway regulates nutrient-sensitive glucose uptake in man. Diabetes. (2007);56:1600–1607. doi: 10.2337/db06-1016. [DOI] [PubMed] [Google Scholar]
- 159.Kumar S, Lemere CA, Walter J. Phosphorylated Aβpeptides in human Down syndrome brain and different Alzheimer's-like mouse models. Acta Neuropathol Commun. (2020);8:118. doi: 10.1186/s40478-020-00959-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kwon OH, Cho YY, Kim TW, Chung S. O-GlcNAcylation of amyloid-βprotein precursor by insulin signaling reduces amyloid-βproduction. J Alzheimers Dis. (2019);69:1195–1211. doi: 10.3233/JAD-190060. [DOI] [PubMed] [Google Scholar]
- 161.Laina A, Stellos K, Stamatelopoulos K. Vascular ageing:Underlying mechanisms and clinical implications. Exp Gerontol. (2018);109:16–30. doi: 10.1016/j.exger.2017.06.007. [DOI] [PubMed] [Google Scholar]
- 162.Lana D, Di Russo J, Mello T, Wenk G, Giovannini M. Rapamycin inhibits mTOR/p70S6K activation in CA3 region of the hippocampus of the rat and impairs long term memory. Neurobiol Learn Mem. (2017);137:15–26. doi: 10.1016/j.nlm.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lang AE, Methner DR, Ferreira A. Neuronal degeneration synaptic defects and behavioral abnormalities in tau45-230 transgenic mice. Neuroscience. (2014);275:322–339. doi: 10.1016/j.neuroscience.2014.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lanni C, Masi M, Racchi M, Govoni S. Cancer and Alzheimer's disease inverse relationship:an age-associated diverging derailment of shared pathways. Mol Psychiatry. (2021);26:280–295. doi: 10.1038/s41380-020-0760-2. [DOI] [PubMed] [Google Scholar]
- 165.Lashley T, Holton JL, Gray E, Kirkham K, O'Sullivan SS, Hilbig A, Wood NW, Lees AJ, Revesz T. Cortical α-synuclein load is associated with amyloid-βplaque burden in a subset of Parkinson's disease patients. Acta Neuropathol. (2008);115:417–425. doi: 10.1007/s00401-007-0336-0. [DOI] [PubMed] [Google Scholar]
- 166.Lathe R, Sapronova A, Kotelevtsev Y. Atherosclerosis and Alzheimer-diseases with a common cause?Inflammation oxysterols vasculature. BMC Geriatr. (2014);14:36. doi: 10.1186/1471-2318-14-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ledreux A, Thomas S, Hamlett ED, Trautman C, Gilmore A, Hager ER, Paredes DA, Margittai M, Fortea J, Granholm AC. Small neuron-derived extracellular vesicles from individuals with Down syndrome propagate tau pathology in the wildtype mouse brain. J Clin Med. (2021);10:3931. doi: 10.3390/jcm10173931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Lee CS, Larson EB, Gibbons LE, Lee AY, McCurry SM, Bowen JD, McCormick WC, Crane PK. Associations between recent and established ophthalmic conditions and risk of Alzheimer's disease. Alzheimers Dement. (2019);15:34–41. doi: 10.1016/j.jalz.2018.06.2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lee NC, Yang SY, Chieh JJ, Huang PT, Chang LM, Chiu YN, Huang AC, Chien YH, Hwu WL, Chiu MJ. Blood beta-amyloid and tau in Down syndrome:a comparison with Alzheimer's disease. Front Aging Neurosci. (2017);8:316. doi: 10.3389/fnagi.2016.00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lei C, Lin R, Wang J, Tao L, Fu X, Qiu Y, Lei B. Amelioration of amyloid β-induced retinal inflammatory responses by a LXR agonist TO901317 is associated with inhibition of the NF-κB signaling and NLRP3 inflammasome. Neuroscience. (2017);360:48–60. doi: 10.1016/j.neuroscience.2017.07.053. [DOI] [PubMed] [Google Scholar]
- 171.Lekomtseva Y, Voloshyn-Gaponov I, Tatayna G. Targeting higher levels of tau protein in Ukrainian patients with Wilson's disease. Neurol Ther. (2019);8:59–68. doi: 10.1007/s40120-019-0134-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lemoine L, Ledreux A, Mufson EJ, Perez SE, Simic G, Doran E, Lott I, Carroll S, Bharani K, Thomas S. Regional binding of tau and amyloid PET tracers in Down syndrome autopsy brain tissue. Mol Neurodegener. (2020);15:68. doi: 10.1186/s13024-020-00414-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lewczuk P, Wiltfang J, Kornhuber J, Verhasselt A. Distributions of Aβ42 and Aβ42/40 in the cerebrospinal fluid in view of the probability theory. Diagnostics. (2021);11:2372. doi: 10.3390/diagnostics11122372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Li H, Zhu H, Wallack M, Mwamburi M, Abdul-Hay SO, Leissring MA, Qiu WQ. Age and its association with low insulin and high amyloid-βpeptides in blood. J Alzheimers Dis. (2016);49:129–137. doi: 10.3233/JAD-150428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Li H, Wu J, Zhu L, Sha L, Yang S, Wei J, Ji L, Tang X, Mao K, Cao L. Insulin degrading enzyme contributes to the pathology in a mixed model of Type 2 diabetes and Alzheimer's disease:possible mechanisms of IDE in T2D and AD. Biosci Rep. (2018);38:BSR20170862. doi: 10.1042/BSR20170862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Li SH, Li XJ. Huntingtin–protein interactions and the pathogenesis of Huntington's disease. Trends Genet. (2004);20:146–154. doi: 10.1016/j.tig.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 177.Lim S, Yoo BK, Kim HS, Gilmore HL, Lee Y, Lee HP, Kim SJ, Letterio J, Lee HG. Amyloid-βprecursor protein promotes cell proliferation and motility of advanced breast cancer. BMC Cancer. (2014);14:928. doi: 10.1186/1471-2407-14-928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Liu C, Cao L, Yang S, Xu L, Liu P, Wang F, Xu D. Subretinal injection of amyloid-βpeptide accelerates RPE cell senescence and retinal degeneration. Int J Mol Med. (2015a);35:169–176. doi: 10.3892/ijmm.2014.1993. [DOI] [PubMed] [Google Scholar]
- 179.Liu C, Cholerton B, Shi M, Ginghina C, Cain KC, Auinger P, Zhang J, Investigators PSGD. CSF tau and tau/Aβ42 predict cognitive decline in Parkinson's disease. Parkinsonism Relat Disord. (2015b);21:271–276. doi: 10.1016/j.parkreldis.2014.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Liu P, Smith BR, Huang ES, Mahesh A, Vonsattel JPG, Petersen AJ, Gomez-Pastor R, Ashe KH. A soluble truncated tau species related to cognitive dysfunction and caspase-2 is elevated in the brain of Huntington's disease patients. Acta Neuropathol Commun. (2019);7:111. doi: 10.1186/s40478-019-0764-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Liu RT, Gao J, Cao S, Sandhu N, Cui JZ, Chou CL, Fang E, Matsubara JA. Inflammatory mediators induced by amyloid-beta in the retina and RPE in vivo:implications for inflammasome activation in age-related macular degeneration. Invest Ophthalmol Vis Sci. (2013);54:2225–2237. doi: 10.1167/iovs.12-10849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Liu Y, Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Deficient brain insulin signalling pathway in Alzheimer's disease and diabetes. J Pathol. (2011);225:54–62. doi: 10.1002/path.2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Loane DJ, Pocivavsek A, Moussa CE, Thompson R, Matsuoka Y, Faden AI, Rebeck GW, Burns MP. Amyloid precursor protein secretases as therapeutic targets for traumatic brain injury. Nat Med. (2009);15:377–379. doi: 10.1038/nm.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Lu Y, Jiang X, Liu S, Li M. Changes in cerebrospinal fluid tau and β-amyloid levels in diabetic and prediabetic patients:a meta-analysis. Front Aging Neurosci. (2018);10:271. doi: 10.3389/fnagi.2018.00271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Magnani E, Fan J, Gasparini L, Golding M, Williams M, Schiavo G, Goedert M, Amos LA, Spillantini MG. Interaction of tau protein with the dynactin complex. EMBO J. (2007);26:4546–4554. doi: 10.1038/sj.emboj.7601878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Mai W, Hu X, Lu Z, Peng F, Wang Y. Cerebrospinal fluid levels of soluble amyloid precursor protein and β-amyloid 42 in patients with multiple sclerosis neuromyelitis optica and clinically isolated syndrome. J Int Med Res. (2011);39:2402–2413. doi: 10.1177/147323001103900641. [DOI] [PubMed] [Google Scholar]
- 187.Mailliot C, Podevin-Dimster V, Rosenthal RE, Sergeant N, Delacourte A, Fiskum G, Buée L. Rapid tau protein dephosphorylation and differential rephosphorylation during cardiac arrest-induced cerebral ischemia and reperfusion. J Cereb Blood Flow Metab. (2000);20:543–549. doi: 10.1097/00004647-200003000-00013. [DOI] [PubMed] [Google Scholar]
- 188.Maloney SM, Hoover CA, Morejon-Lasso LV, Prosperi JR. Mechanisms of taxane resistance. Cancers. (2020);12:3323. doi: 10.3390/cancers12113323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Markin AM, Sobenin IA, Grechko AV, Zhang D, Orekhov AN. Cellular mechanisms of human atherogenesis:focus on chronification of inflammation and mitochondrial mutations. Front Pharmacol. (2020);11:642. doi: 10.3389/fphar.2020.00642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Markopoulou K, Dickson D, McComb R, Wszolek Z, Katechalidou L, Avery L, Stansbury M, Chase B. Clinical neuropathological and genotypic variability in SNCA A53T familial Parkinson's disease:Variability in familial Parkinson's disease. Acta Neuropathol. (2008);116:25–35. doi: 10.1007/s00401-008-0372-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Marsden IT, Minamide LS, Bamburg JR. Amyloid-β-induced amyloid-βsecretion:a possible feed-forward mechanism in Alzheimer's disease. J Alzheimers Dis. (2011);24:681–691. doi: 10.3233/JAD-2011-101899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Masliah E, Rockenstein E, Veinbergs I, Sagara Y, Mallory M, Hashimoto M, Mucke L. β-Amyloid peptides enhance α-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer's disease and Parkinson's disease. Proc Natl Acad Sci U S A. (2001);98:12245–12250. doi: 10.1073/pnas.211412398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Masnata M, Salem S, de Rus Jacquet A, Anwer M, Cicchetti F. Targeting Tau to treat clinical features of Huntington's disease. Front Neurol. (2020);11:580732. doi: 10.3389/fneur.2020.580732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Masrori P, Van Damme P. Amyotrophic lateral sclerosis:a clinical review. Eur J Neurol. (2020);27:1918–1929. doi: 10.1111/ene.14393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Matéde Gérando A, d'Orange M, Augustin E, Joséphine C, Aurégan G, Gaudin-Guérif M, Guillermier M, Hérard AS, Stimmer L, Petit F. Neuronal tau species transfer to astrocytes and induce their loss according to tau aggregation state. Brain. (2021);144:1167–1182. doi: 10.1093/brain/awab011. [DOI] [PubMed] [Google Scholar]
- 196.Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS β-amyloid in Alzheimer's disease. Science. (2010);330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Mccormack A, Chegeni N, Chegini F, Colella A, Power J, Keating D, Chataway T. Purification of α-synuclein containing inclusions from human post mortem brain tissue. J Neurosci Methods. (2016);266:141–150. doi: 10.1016/j.jneumeth.2016.03.016. [DOI] [PubMed] [Google Scholar]
- 198.McGowan D, van Roon-Mom W, Holloway H, Bates G, Mangiarini L, Cooper G, Faull R, Snell R. Amyloid-like inclusions in Huntington's disease. Neuroscience. (2000);100:677–680. doi: 10.1016/s0306-4522(00)00391-2. [DOI] [PubMed] [Google Scholar]
- 199.McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, Skipper L, Murphy MP, Beard J, Das P. Aβ42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. (2005);47:191–199. doi: 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.McGowan P, McKiernan E, Bolster F, Ryan B, Hill A, McDermott E, Evoy D, O'Higgins N, Crown J, Duffy M. ADAM-17 predicts adverse outcome in patients with breast cancer. Ann Oncol. (2008);19:1075–1081. doi: 10.1093/annonc/mdm609. [DOI] [PubMed] [Google Scholar]
- 201.McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, Santini VE, Lee HS, Kubilus CA, Stern RA. Chronic traumatic encephalopathy in athletes:progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. (2009);68:709–735. doi: 10.1097/NEN.0b013e3181a9d503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.McKee AC, Alosco ML, Huber BR. Repetitive head impacts and chronic traumatic encephalopathy. Neurosurg Clin N Am. (2016);27:529–535. doi: 10.1016/j.nec.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Meakin PJ, Harper AJ, Hamilton DL, Gallagher J, McNeilly AD, Burgess LA, Vaanholt LM, Bannon KA, Latcham J, Hussain I. Reduction in BACE1 decreases body weight protects against diet-induced obesity and enhances insulin sensitivity in mice. Biochem J. (2012);441:285–296. doi: 10.1042/BJ20110512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Meakin PJ, Mezzapesa A, Benabou E, Haas ME, Bonardo B, Grino M, Brunel J-M, Desbois-Mouthon C, Biddinger SB, Govers R. The beta secretase BACE1 regulates the expression of insulin receptor in the liver. Nat Commun. (2018);9:1306. doi: 10.1038/s41467-018-03755-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Meisl G, Yang X, Hellstrand E, Frohm B, Kirkegaard JB, Cohen SI, Dobson CM, Linse S, Knowles TP. Differences in nucleation behavior underlie the contrasting aggregation kinetics of the Aβ40 and Aβ42 peptides. Proc Natl Acad Sci U S A. (2014);111:9384–9389. doi: 10.1073/pnas.1401564111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Melkani GC, Trujillo AS, Ramos R, Bodmer R, Bernstein SI, Ocorr K. Huntington's disease induced cardiac amyloidosis is reversed by modulating protein folding and oxidative stress pathways in the Drosophila heart. PLoS Genet. (2013);9:e1004024. doi: 10.1371/journal.pgen.1004024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Melzer TR, Stark MR, Keenan RJ, Myall DJ, MacAskill MR, Pitcher TL, Livingston L, Grenfell S, Horne KL, Young BN. Beta amyloid deposition is not associated with cognitive impairment in Parkinson's disease. Front Neurol. (2019);10:391. doi: 10.3389/fneur.2019.00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Mencer S, Kartawy M, Lendenfeld F, Soluh H, Tripathi MK, Khaliulin I, Amal H. Proteomics of autism and Alzheimer's mouse models reveal common alterations in mTOR signaling pathway. Transl Psychiatry. (2021);11:480. doi: 10.1038/s41398-021-01578-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Mez J, Daneshvar DH, Kiernan PT, Abdolmohammadi B, Alvarez VE, Huber BR, Alosco ML, Solomon TM, Nowinski CJ, McHale L. Clinicopathological evaluation of chronic traumatic encephalopathy in players of American football. JAMA. (2017);318:360–370. doi: 10.1001/jama.2017.8334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Mietelska-Porowska A, Wasik U, Goras M, Filipek A, Niewiadomska G. Tau protein modifications and interactions:their role in function and dysfunction. Int J Mol Sci. (2014);15:4671–4713. doi: 10.3390/ijms15034671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Mimori K, Sadanaga N, Yoshikawa Y, Ishikawa K, Hashimoto M, Tanaka F, Sasaki A, Inoue H, Sugimachi K, Mori M. Reduced tau expression in gastric cancer can identify candidates for successful Paclitaxel treatment. Br J Cancer. (2006);94:1894–1897. doi: 10.1038/sj.bjc.6603182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Mirzaei M, Abyadeh M, Turner AJ, Wall RV, Chick JM, Paulo JA, Gupta VK, Basavarajappa D, Chitranshi N, Mirshahvaladi SSO. Fingolimod effects on the brain are mediated through biochemical modulation of bioenergetics autophagy and neuroinflammatory networks. Proteomics. (2022);22:2100247. doi: 10.1002/pmic.202100247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Mollenhauer B, Bibl M, Esselmann H, Steinacker P, Trenkwalder C, Brechlin P, Wiltfang J, Otto M. Selective reduction of amyloid β42 discriminates Alzheimer's disease from Huntington's disease:indication for distinct pathological events in amyloid βpeptide aggregation. J Neurol Neurosurg Psychiatry. (2006);77:1201–1203. doi: 10.1136/jnnp.2005.084673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Mora JS. Edaravone for treatment of early-stage ALS. Lancet Neurol. (2017);16:772. doi: 10.1016/S1474-4422(17)30289-2. [DOI] [PubMed] [Google Scholar]
- 215.Moran C, Beare R, Phan TG, Bruce DG, Callisaya ML, Srikanth V. Type 2 diabetes mellitus and biomarkers of neurodegeneration. Neurology. (2015);85:1123–1130. doi: 10.1212/WNL.0000000000001982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Mori C, Spooner ET, Wisniewski KE, Wisniewski TM, Yamaguchi H, Saido TC, Tolan DR, Selkoe DJ, Lemere CA. Intraneuronal Aβ42 accumulation in Down syndrome brain. Amyloid. (2002);9:88–102. [PubMed] [Google Scholar]
- 217.Mori F, Rossi S, Sancesario G, Codecà C, Mataluni G, Monteleone F, Buttari F, Kusayanagi H, Castelli M, Motta C. Cognitive and cortical plasticity deficits correlate with altered amyloid-βCSF levels in multiple sclerosis. Neuropsychopharmacology. (2011);36:559–568. doi: 10.1038/npp.2010.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Morioka M, Kawano T, Yano S, Kai Y, Tsuiki H, Yoshinaga Y, Matsumoto J, Maeda T, Hamada JI, Yamamoto H. Hyperphosphorylation at serine 199/202 of tau factor in the gerbil hippocampus after transient forebrain ischemia. Biochem Biophys Res Commun. (2006);347:273–278. doi: 10.1016/j.bbrc.2006.06.096. [DOI] [PubMed] [Google Scholar]
- 219.Mormino EC, Papp KV. Amyloid accumulation and cognitive decline in clinically normal older individuals:implications for aging and early Alzheimer's disease. J Alzheimers Dis. (2018);64:S633–S646. doi: 10.3233/JAD-179928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Mörtberg E, Zetterberg H, Nordmark J, Blennow K, Catry C, Decraemer H, Vanmechelen E, Rubertsson S. Plasma tau protein in comatose patients after cardiac arrest treated with therapeutic hypothermia. Acta Anaesthesiol Scand. (2011);55:1132–1138. doi: 10.1111/j.1399-6576.2011.02505.x. [DOI] [PubMed] [Google Scholar]
- 221.Moszczynski AJ, Strong W, Xu K, McKee A, Brown A, Strong MJ. Pathologic Thr175 tau phosphorylation in CTE and CTE with ALS. Neurology. (2018);90:e380–387. doi: 10.1212/WNL.0000000000004899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Mudher A, Colin M, Dujardin S, Medina M, Dewachter I, Naini SMA, Mandelkow EM, Mandelkow E, Buée L, Goedert M. What is the evidence that tau pathology spreads through prion-like propagation? Acta Neuropathol Commun. (2017);5:99. doi: 10.1186/s40478-017-0488-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Mundi S, Massaro M, Scoditti E, Carluccio MA, Van Hinsbergh VW, Iruela-Arispe ML, De Caterina R. Endothelial permeability, LDL deposition and cardiovascular risk factors—a review. Cardiovasc Res. (2018);114:35–52. doi: 10.1093/cvr/cvx226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Murphy M, LeVine H. Alzheimer's disease and the amyloid-beta peptide. J Alzheimers Dis. (2010);19:311–323. doi: 10.3233/JAD-2010-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Murray ME, Dickson DW. Is pathological aging a successful resistance against amyloid-beta or preclinical Alzheimer's disease? Alzheimers Res Ther. (2014);6:24. doi: 10.1186/alzrt254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Nabizadeh F, Sodeifian F, Kargar A. Cerebrospinal fluid alpha-synuclein amyloid beta total tau and phosphorylated tau in tremor-dominant Parkinson's disease. Acta Neurol Belg. (2023);123:1429–1437. doi: 10.1007/s13760-023-02251-9. [DOI] [PubMed] [Google Scholar]
- 227.Nakagawa Y, Reed L, Nakamura M, McIntosh TK, Smith DH, Saatman KE, Raghupathi R, Clemens J, Saido TC, Lee VMY. Brain trauma in aged transgenic mice induces regression of established Aβdeposits. Exp Neurol. (2000);163:244–252. doi: 10.1006/exnr.2000.7375. [DOI] [PubMed] [Google Scholar]
- 228.Nakamura K, Zhou XZ, Lu KP. Distinct functions of cis and trans phosphorylated tau in Alzheimer's disease and their therapeutic implications. Curr Mol Med. (2013);13:1098–1109. doi: 10.2174/1566524011313070001. [DOI] [PubMed] [Google Scholar]
- 229.Ning A, Cui J, To E, Ashe KH, Matsubara J. Amyloid-βdeposits lead to retinal degeneration in a mouse model of Alzheimer disease. Invest Ophthalmol Vis Sci. (2008);49:5136–5143. doi: 10.1167/iovs.08-1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Niwa K, Kazama K, Younkin L, Younkin SG, Carlson GA, Iadecola C. Cerebrovascular autoregulation is profoundly impaired in mice overexpressing amyloid precursor protein. Am J Physiol Heart Circ Physiol. (2002);283:H315–323. doi: 10.1152/ajpheart.00022.2002. [DOI] [PubMed] [Google Scholar]
- 231.Ohno-Matsui K. Parallel findings in age-related macular degeneration and Alzheimer's disease. Prog Retin Eye Res. (2011);30:217–238. doi: 10.1016/j.preteyeres.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 232.Oku H, Kida T, Horie T, Taki K, Mimura M, Kojima S, Ikeda T. Tau is involved in death of retinal ganglion cells of rats from optic nerve crush. Invest Ophthalmol Vis Sci. (2019);60:2380–2387. doi: 10.1167/iovs.19-26683. [DOI] [PubMed] [Google Scholar]
- 233.Omalu B, Bailes J, Hamilton RL, Kamboh MI, Hammers J, Case M, Fitzsimmons R. Emerging histomorphologic phenotypes of chronic traumatic encephalopathy in American athletes. Neurosurgery. (2011);69:173–183. doi: 10.1227/NEU.0b013e318212bc7b. [DOI] [PubMed] [Google Scholar]
- 234.Ornelas AS, Adler CH, Serrano GE, Curry JR, Shill HA, Kopyov O, Beach TG. Co-existence of tau and α-synuclein pathology in fetal graft tissue at autopsy:a case report. Parkinsonism Relat Disord. (2020);71:36–39. doi: 10.1016/j.parkreldis.2019.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Palmer N, Beam A, Agniel D, Eran A, Manrai A, Spettell C, Steinberg G, Mandl K, Fox K, Nelson SF. Association of sex with recurrence of autism spectrum disorder among siblings. JAMA Pediatr. (2017);171:1107–1112. doi: 10.1001/jamapediatrics.2017.2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Pan L, Meng L, He M, Zhang Z. Tau in the pathophysiology of Parkinson's disease. J Mol Neurosci. (2021);71:2179–2191. doi: 10.1007/s12031-020-01776-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Pandey P, Rachagani S, Das S, Seshacharyulu P, Sheinin Y, Naslavsky N, Pan Z, Smith BL, Peters HL, Radhakrishnan P. Amyloid precursor-like protein 2 (APLP2) affects the actin cytoskeleton and increases pancreatic cancer growth and metastasis. Oncotarget. (2015);6:2064. doi: 10.18632/oncotarget.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Pandey P, Sliker B, Peters HL, Tuli A, Herskovitz J, Smits K, Purohit A, Singh RK, Dong J, Batra SK. Amyloid precursor protein and amyloid precursor-like protein 2 in cancer. Oncotarget. (2016);7:19430. doi: 10.18632/oncotarget.7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Papin S, Paganetti P. Emerging evidences for an implication of the neurodegeneration-associated protein TAU in cancer. Brain Sci. (2020);10:862. doi: 10.3390/brainsci10110862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Park L, Zhou P, Pitstick R, Capone C, Anrather J, Norris EH, Younkin L, Younkin S, Carlson G, McEwen BS. Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc Natl Acad Sci U S A. (2008);105:1347–1352. doi: 10.1073/pnas.0711568105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Pasinelli P, Houseweart MK, Brown RH, Jr, Cleveland DW. Caspase-1 and-3 are sequentially activated in motor neuron death in Cu Zn superoxide dismutase-mediated familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. (2000);97:13901–13906. doi: 10.1073/pnas.240305897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Pavliukeviciene B, Zentelyte A, Jankunec M, Valiuliene G, Talaikis M, Navakauskiene R, Niaura G, Valincius G. Amyloid βoligomers inhibit growth of human cancer cells. PLoS One. (2019);14:e0221563. doi: 10.1371/journal.pone.0221563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Peng X, Xu Z, Mo X, Guo Q, Yin J, Xu M, Peng Z, Sun T, Zhou L, Peng X. Association of plasma β-amyloid 40 and 42 concentration with type 2 diabetes among Chinese adults. Diabetologia. (2020);63:954–963. doi: 10.1007/s00125-020-05102-x. [DOI] [PubMed] [Google Scholar]
- 244.Pennington KL, DeAngelis MM. Epidemiology of age-related macular degeneration (AMD):associations with cardiovascular disease phenotypes and lipid factors. Eye Vis. (2016);3:34. doi: 10.1186/s40662-016-0063-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Perluigi M, Pupo G, Tramutola A, Cini C, Coccia R, Barone E, Head E, Butterfield DA, Di Domenico F. Neuropathological role of PI3K/Akt/mTOR axis in Down syndrome brain. Biochim Biophys Acta. (2014);1842:1144–1153. doi: 10.1016/j.bbadis.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Peters HL, Tuli A, Wang X, Liu C, Pan Z, Ouellette MM, Hollingsworth MA, MacDonald RG, Solheim JC. Relevance of amyloid precursor-like protein 2 C-terminal fragments in pancreatic cancer cells. Int J Oncol. (2012);41:1464–1474. doi: 10.3892/ijo.2012.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Pietroboni AM, Schiano di Cola F, Scarioni M, Fenoglio C, Spanò B, Arighi A, Cioffi SM, Oldoni E, De Riz MA, Basilico P. CSF β-amyloid as a putative biomarker of disease progression in multiple sclerosis. Mult Scler. (2017);23:1085–1091. doi: 10.1177/1352458516674566. [DOI] [PubMed] [Google Scholar]
- 248.Pletnikova O, West N, Lee MK, Rudow GL, Skolasky RL, Dawson TM, Marsh L, Troncoso JC. Aβdeposition is associated with enhanced cortical α-synuclein lesions in Lewy body diseases. Neurobiol Aging. (2005);26:1183–1192. doi: 10.1016/j.neurobiolaging.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 249.Plucińska K, Crouch B, Koss D, Robinson L, Siebrecht M, Riedel G, Platt B. Knock-in of human BACE1 cleaves murine APP and reiterates Alzheimer-like phenotypes. J Neurosci. (2014);34:10710–10728. doi: 10.1523/JNEUROSCI.0433-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Plucińska K, Dekeryte R, Koss D, Shearer K, Mody N, Whitfield PD, Doherty MK, Mingarelli M, Welch A, Riedel G. Neuronal human BACE1 knockin induces systemic diabetes in mice. Diabetologia. (2016);59:1513–1523. doi: 10.1007/s00125-016-3960-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Prasad T, Zhu P, Verma A, Chakrabarty P, Rosario AM, Golde TE, Li Q. Amyloid βpeptides overexpression in retinal pigment epithelial cells via AAV-mediated gene transfer mimics AMD-like pathology in mice. Sci Rep. (2017);7:1–15. doi: 10.1038/s41598-017-03397-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Pringsheim T, Wiltshire K, Day L, Dykeman J, Steeves T, Jette N. The incidence and prevalence of Huntington's disease:a systematic review and meta-analysis. Mov Disord. (2012);27:1083–1091. doi: 10.1002/mds.25075. [DOI] [PubMed] [Google Scholar]
- 253.Prots I, Veber V, Brey S, Campioni S, Buder K, Riek R, Böhm KJ, Winner B. α-Synuclein oligomers impair neuronal microtubule-kinesin interplay. J Biol Chem. (2013);288:21742–21754. doi: 10.1074/jbc.M113.451815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Pusztai L, Jeong JH, Gong Y, Ross JS, Kim C, Paik S, Rouzier R, Andre F, Hortobagyi GN, Wolmark N. Evaluation of microtubule-associated protein-Tau expression as a prognostic and predictive marker in the NSABP-B 28 randomized clinical trial. J Clin Oncol. (2009);27:4287. doi: 10.1200/JCO.2008.21.6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Qin H, Wang J, Ren J, Qu X. Amyloid βand tumorigenesis:amyloid β-induced telomere dysfunction in tumor cells. CCS Chemistry. (2019);1:313–325. [Google Scholar]
- 256.Raggi P, Genest J, Giles JT, Rayner KJ, Dwivedi G, Beanlands RS, Gupta M. Role of inflammation in the pathogenesis of atherosclerosis and therapeutic interventions. Atherosclerosis. (2018);276:98–108. doi: 10.1016/j.atherosclerosis.2018.07.014. [DOI] [PubMed] [Google Scholar]
- 257.Ramirez J, Berezuk C, McNeely AA, Gao F, McLaurin J, Black SE. Imaging the perivascular space as a potential biomarker of neurovascular and neurodegenerative diseases. Cell Mol Neurobiol. (2016);36:289–299. doi: 10.1007/s10571-016-0343-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Randall J, Mörtberg E, Provuncher GK, Fournier DR, Duffy DC, Rubertsson S, Blennow K, Zetterberg H, Wilson DH. Tau proteins in serum predict neurological outcome after hypoxic brain injury from cardiac arrest:results of a pilot study. Resuscitation. (2013);84:351–356. doi: 10.1016/j.resuscitation.2012.07.027. [DOI] [PubMed] [Google Scholar]
- 259.Ratnayaka JA, Serpell LC, Lotery AJ. Dementia of the eye:the role of amyloid beta in retinal degeneration. Eye. (2015);29:1013–1026. doi: 10.1038/eye.2015.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Ray B, Long JM, Sokol DK, Lahiri DK. Increased secreted amyloid precursor protein-α(sAPPα) in severe autism:proposal of a specific anabolic pathway and putative biomarker. PLoS One. (2011);6:e20405. doi: 10.1371/journal.pone.0020405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Raz L, Bhaskar K, Weaver J, Marini S, Zhang Q, Thompson JF, Espinoza C, Iqbal S, Maphis NM, Weston L. Hypoxia promotes tau hyperphosphorylation with associated neuropathology in vascular dysfunction. Neurobiol Dis. (2019);126:124–136. doi: 10.1016/j.nbd.2018.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Rensink AA, de Waal RM, Kremer B, Verbeek MM. Pathogenesis of cerebral amyloid angiopathy. Brain Res Brain Res Rev. (2003);43:207–223. doi: 10.1016/j.brainresrev.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 263.Renton AE, Chio A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. (2014);17:17–23. doi: 10.1038/nn.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Ristori E, Donnini S, Ziche M. New insights into blood-brain barrier maintenance:the homeostatic role of β-amyloid precursor protein in cerebral vasculature. Front Physiol. (2020);11:1056. doi: 10.3389/fphys.2020.01056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Roberts G, Gentleman S, Lynch A, Murray L, Landon M, Graham D. Beta amyloid protein deposition in the brain after severe head injury:implications for the pathogenesis of Alzheimer's disease. J Neurol Neurosurg Psychiatry. (1994);57:419–425. doi: 10.1136/jnnp.57.4.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Rodríguez-Martín T, Cuchillo-Ibáñez I, Noble W, Nyenya F, Anderton BH, Hanger DP. Tau phosphorylation affects its axonal transport and degradation. Neurobiol Aging. (2013);34:2146–2157. doi: 10.1016/j.neurobiolaging.2013.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Roeben B, Maetzler W, Vanmechelen E, Schulte C, Heinzel S, Stellos K, Godau J, Huber H, Brockmann K, Wurster I. Association of plasma Aβ40 peptides but not Aβ42 with coronary artery disease and diabetes mellitus. J Alzheimers Dis. (2016);52:161–169. doi: 10.3233/JAD-150575. [DOI] [PubMed] [Google Scholar]
- 268.Romero JR, Demissie S, Beiser A, Himali JJ, DeCarli C, Levy D, Seshadri S. Relation of plasma β-amyloid clusterin and tau with cerebral microbleeds:Framingham Heart Study. Ann Clin Transl Neurol. (2020);7:1083–1091. doi: 10.1002/acn3.51066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Rouzier R, Rajan R, Wagner P, Hess KR, Gold DL, Stec J, Ayers M, Ross JS, Zhang P, Buchholz TA. Microtubule-associated protein tau:a marker of paclitaxel sensitivity in breast cancer. Proc Natl Acad Sci U S A. (2005);102:8315–8320. doi: 10.1073/pnas.0408974102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Ruan Y, Guo Y, Zheng Y, Huang Z, Sun S, Kowal P, Shi Y, Wu F. Cardiovascular disease (CVD) and associated risk factors among older adults in six low-and middle-income countries:results from SAGE Wave 1. BMC Public Health. (2018);18:778. doi: 10.1186/s12889-018-5653-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Sadigh-Eteghad S, Sabermarouf B, Majdi A, Talebi M, Farhoudi M, Mahmoudi J. Amyloid-beta:a crucial factor in Alzheimer's disease. Med Princ Pract. (2015);24:1–10. doi: 10.1159/000369101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, Colagiuri S, Guariguata L, Motala AA, Ogurtsova K. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045:results from the International Diabetes Federation Diabetes Atlas. Diabetes Res Clin Pract. (2019);157:107843. doi: 10.1016/j.diabres.2019.107843. [DOI] [PubMed] [Google Scholar]
- 273.Salvesen GS, Dixit VM. Caspase activation:the induced-proximity model. Proc Natl Acad Sci U S A. (1999);96:10964–10967. doi: 10.1073/pnas.96.20.10964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Samimi N, Sharma G, Kimura T, Matsubara T, Huo A, Chiba K, Saito Y, Murayama S, Akatsu H, Hashizume Y. Distinct phosphorylation profiles of tau in brains of patients with different tauopathies. Neurobiol Aging. (2021);108:72–79. doi: 10.1016/j.neurobiolaging.2021.08.011. [DOI] [PubMed] [Google Scholar]
- 275.Sánchez-Juan P, Moreno S, de Rojas I, Hernández I, Valero S, Alegret M, Montrreal L, García González P, Lage C, López-García S. The MAPT H1 haplotype is a risk factor for Alzheimer's disease in APOE ε4 non-carriers. Front Aging Neurosci. (2019);11:327. doi: 10.3389/fnagi.2019.00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Sandahl TD, Laursen TL, Munk DE, Vilstrup H, Weiss KH, Ott P. The prevalence of Wilson's disease:an update. Hepatology. (2020);71:722–732. doi: 10.1002/hep.30911. [DOI] [PubMed] [Google Scholar]
- 277.Santos CY, Snyder PJ, Wu WC, Zhang M, Echeverria A, Alber J. Pathophysiologic relationship between Alzheimer's disease cerebrovascular disease and cardiovascular risk:a review and synthesis. Alzheimers Dement. (2017);7:69–87. doi: 10.1016/j.dadm.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Santos R, Bulteau A-L, Gomes CM. Neurodegeneration neurogenesis and oxidative stress 2015. Oxid Med Cell Longev. (2016);2016:7632025. doi: 10.1155/2016/7632025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Schaich CL, Maurer MS, Nadkarni NK. Amyloidosis of the brain and heart:two sides of the same coin? JACC Heart Fail. (2019);7:129–131. doi: 10.1016/j.jchf.2018.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Sekino Y, Han X, Babasaki T, Goto K, Inoue S, Hayashi T, Teishima J, Shiota M, Takeshima Y, Yasui W. Microtubule-associated protein tau (MAPT) promotes bicalutamide resistance and is associated with survival in prostate cancer. Urol Oncol. (2020);795:e791–795. doi: 10.1016/j.urolonc.2020.04.032. [DOI] [PubMed] [Google Scholar]
- 281.Sethy C, Kundu CN. 5-Fluorouracil (5-FU) resistance and the new strategy to enhance the sensitivity against cancer:Implication of DNA repair inhibition. Biomed Pharmacother. (2021);137:111285. doi: 10.1016/j.biopha.2021.111285. [DOI] [PubMed] [Google Scholar]
- 282.Shadfar S, Brocardo M, Atkin JD. The complex mechanisms by which neurons die following DNA damage in neurodegenerative diseases. Int J Mol Sci. (2022);23:2484. doi: 10.3390/ijms23052484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Shi C, Zhu X, Wang J, Long D. Estrogen receptor αpromotes non-amyloidogenic processing of platelet amyloid precursor protein via the MAPK/ERK pathway. J Steroid Biochem Mol Biol. (2014);144:280–285. doi: 10.1016/j.jsbmb.2014.06.010. [DOI] [PubMed] [Google Scholar]
- 284.Shi J, Zhang T, Zhou C, Chohan MO, Gu X, Wegiel J, Zhou J, Hwang YW, Iqbal K, Grundke-Iqbal I, Gong CX, Liu F. Increased dosage of Dyrk1A alters alternative splicing factor (ASF)-regulated alternative splicing of Tau in Down Syndrome. J Biol Chem. (2008);283:28660–28669. doi: 10.1074/jbc.M802645200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Shieh JCC, Huang PT, Lin YF. Alzheimer's disease and diabetes:Insulin signaling as the bridge linking two pathologies. Mol Neurobiol. (2020);57:1966–1977. doi: 10.1007/s12035-019-01858-5. [DOI] [PubMed] [Google Scholar]
- 286.Siderowf A, Xie S, Hurtig H, Weintraub D, Duda J, Chen-Plotkin A, Shaw L, Van Deerlin V, Trojanowski J, Clark C. CSF amyloid β1-42 predicts cognitive decline in Parkinson disease. Neurology. (2010);75:1055–1061. doi: 10.1212/WNL.0b013e3181f39a78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Siderowf A, Concha-Marambio L, Lafontant D-E, Farris CM, Ma Y, Urenia PA, Nguyen H, Alcalay RN, Chahine LM, Foroud T. Assessment of heterogeneity among participants in the Parkinson's Progression Markers Initiative cohort using α-synuclein seed amplification:a cross-sectional study. Lancet Neurol. (2023);22:407–417. doi: 10.1016/S1474-4422(23)00109-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Silverberg GD, Messier AA, Miller MC, Machan JT, Majmudar SS, Stopa EG, Donahue JE, Johanson CE. Amyloid efflux transporter expression at the blood-brain barrier declines in normal aging. J Neuropathol Exp Neurol. (2010);69:1034–1043. doi: 10.1097/NEN.0b013e3181f46e25. [DOI] [PubMed] [Google Scholar]
- 289.Simons E, Smith M, Dengler-Crish C, Crish S. Retinal ganglion cell loss and gliosis in the retinofugal projection following intravitreal exposure to amyloid-beta. Neurobiol Dis. (2021);147:105146. doi: 10.1016/j.nbd.2020.105146. [DOI] [PubMed] [Google Scholar]
- 290.Sinclair AJ. Managing older people with diabetes—we need better evidence with wise interpretation! Age Ageing. (2021);50:1896–1898. doi: 10.1093/ageing/afab165. [DOI] [PubMed] [Google Scholar]
- 291.Singh B, Covelo A, Martell-Martínez H, Nanclares C, Sherman MA, Okematti E, Meints J, Teravskis PJ, Gallardo C, Savonenko AV. Tau is required for progressive synaptic and memory deficits in a transgenic mouse model of α-synucleinopathy. Acta Neuropathol. (2019);138:551–574. doi: 10.1007/s00401-019-02032-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Singh I, Sagare AP, Coma M, Perlmutter D, Gelein R, Bell RD, Deane RJ, Zhong E, Parisi M, Ciszewski J. Low levels of copper disrupt brain amyloid-βhomeostasis by altering its production and clearance. Proc Natl Acad Sci U S A. (2013);110:14771–14776. doi: 10.1073/pnas.1302212110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Sipula IJ, Brown NF, Perdomo G. Rapamycin-mediated inhibition of mammalian target of rapamycin in skeletal muscle cells reduces glucose utilization and increases fatty acid oxidation. Metabolism. (2006);55:1637–1644. doi: 10.1016/j.metabol.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 294.Sitammagari KK, Masood W. In:StatPearls [Internet] Treasure Island (FL): StatPearls Publishing; (2022). Creutzfeldt Jakob Disease. [PubMed] [Google Scholar]
- 295.Smith C, Graham D, Murray L, Nicoll J. Tau immunohistochemistry in acute brain injury. Neuropathol Appl Neurobiol. (2003);29:496–502. doi: 10.1046/j.1365-2990.2003.00488.x. [DOI] [PubMed] [Google Scholar]
- 296.Smoter M, Bodnar L, Grala B, Stec R, Zieniuk K, Kozlowski W, Szczylik C. Tau protein as a potential predictive marker in epithelial ovarian cancer patients treated with paclitaxel/platinum first-line chemotherapy. J Exp Clin Cancer Res. (2013);32:1–8. doi: 10.1186/1756-9966-32-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Sobey CG, Judkins CP, Sundararajan V, Phan TG, Drummond GR, Srikanth VK. Risk of major cardiovascular events in people with Down syndrome. PLoS One. (2015);10:e0137093. doi: 10.1371/journal.pone.0137093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Sokol DK, Chen D, Farlow MR, Dunn DW, Maloney B, Zimmer JA, Lahiri DK. High levels of Alzheimer beta-amyloid precursor protein (APP) in children with severely autistic behavior and aggression. J Child Neurol. (2006);21:444–449. doi: 10.1177/08830738060210062201. [DOI] [PubMed] [Google Scholar]
- 299.Song MK, Bischoff DS, Song AM, Uyemura K, Yamaguchi DT. Metabolic relationship between diabetes and Alzheimer's disease affected by Cyclo (His-Pro) plus zinc treatment. BBA Clin. (2017);7:41–54. doi: 10.1016/j.bbacli.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Soragni A, Zambelli B, Mukrasch MD, Biernat J, Jeganathan S, Griesinger C, Ciurli S, Mandelkow E, Zweckstetter M. Structural characterization of binding of Cu (II) to tau protein. Biochemistry. (2008);47:10841–10851. doi: 10.1021/bi8008856. [DOI] [PubMed] [Google Scholar]
- 301.Spagnoli LG, Bonanno E, Sangiorgi G, Mauriello A. Role of inflammation in atherosclerosis. J Nucl Med. (2007);48:1800–1815. doi: 10.2967/jnumed.107.038661. [DOI] [PubMed] [Google Scholar]
- 302.Spaide RF, Curcio CA. Drusen characterization with multimodal imaging. Retina (Philadelphia, Pa) (2010);30:1441. doi: 10.1097/IAE.0b013e3181ee5ce8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Spitzer P, Walter M, Göth C, Oberstein TJ, Linning P, Knölker HJ, Kornhuber J, Maler JM. Pharmacological inhibition of amyloidogenic APP processing and knock-down of APP in primary human macrophages impairs the secretion of cytokines. Front Immunol. (2020);11:1967. doi: 10.3389/fimmu.2020.01967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. (2008);319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Stakos DA, Stamatelopoulos K, Bampatsias D, Sachse M, Zormpas E, Vlachogiannis NI, Tual-Chalot S, Stellos K. The Alzheimer's disease amyloid-beta hypothesis in cardiovascular aging and disease:JACC focus seminar. J Am Coll Cardiol. (2020);75:952–967. doi: 10.1016/j.jacc.2019.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Stamatelopoulos K, Pol CJ, Ayers C, Georgiopoulos G, Gatsiou A, Brilakis ES, Khera A, Drosatos K, de Lemos JA, Stellos K. Amyloid-beta (1-40) peptide and subclinical cardiovascular disease. J Am Coll Cardiol. (2018a);72:1060–1061. doi: 10.1016/j.jacc.2018.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Stamatelopoulos K, Mueller-Hennessen M, Georgiopoulos G, Sachse M, Boeddinghaus J, Sopova K, Gatsiou A, Amrhein C, Biener M, Vafaie M. Amyloid-β(1-40) and mortality in patients with non–ST-segment elevation acute coronary syndrome:a cohort study. Ann Intern Med. (2018b);168:855–865. doi: 10.7326/M17-1540. [DOI] [PubMed] [Google Scholar]
- 308.Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow E-M. Tau blocks traffic of organelles neurofilaments and APP vesicles in neurons and enhances oxidative stress. J Cell Biol. (2002);156:1051–1063. doi: 10.1083/jcb.200108057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Stanciu GD, Bild V, Ababei DC, Rusu RN, Cobzaru A, Paduraru L, Bulea D. Link between diabetes and Alzheimer's disease due to the shared amyloid aggregation and deposition involving both neurodegenerative changes and neurovascular damages. J Clin Med. (2020);9:1713. doi: 10.3390/jcm9061713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Stellmach V, Crawford SE, Zhou W, Bouck N. Prevention of ischemia-induced retinopathy by the natural ocular antiangiogenic agent pigment epithelium-derived factor. Proc Natl Acad Sci U S A. (2001);98:2593–2597. doi: 10.1073/pnas.031252398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Strong MJ, Donison NS, Volkening K. Alterations in Tau metabolism in ALS and ALS-FTSD. Front Neurol. (2020);11:598907. doi: 10.3389/fneur.2020.598907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Sun J, Chen J, Li T, Huang P, Li J, Shen M, Gao M, Sun Y, Liang J, Li X. ROS production and mitochondrial dysfunction driven by PU 1-regulated NOX4-p22phox activation in Aβ-induced retinal pigment epithelial cell injury. Theranostics. (2020);10:11637. doi: 10.7150/thno.48064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Sun X, Chen WD, Wang YD. β-Amyloid:the key peptide in the pathogenesis of Alzheimer's disease. Front Pharmacol. (2015);6:221. doi: 10.3389/fphar.2015.00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020:GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2021);71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 315.Surmeier DJ. Determinants of dopaminergic neuron loss in Parkinson's disease. FEBS J. (2018);285:3657–3668. doi: 10.1111/febs.14607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Suzanne M. Therapeutic targets of brain insulin resistance in sporadic Alzheimer's disease. Front Biosci (Elite Ed) (2012);4:1582. doi: 10.2741/482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Syme CD, Nadal RC, Rigby SE, Viles JH. Copper binding to the amyloid-β(Aβ) peptide associated with Alzheimer's disease:folding coordination geometry pH dependence stoichiometry and affinity of Aβ-(1–28):insights from a range of complementary spectroscopic techniques. J Biol Chem. (2004);279:18169–18177. doi: 10.1074/jbc.M313572200. [DOI] [PubMed] [Google Scholar]
- 318.Tai C, Chang CW, Yu GQ, Lopez I, Yu X, Wang X, Guo W, Mucke L. Tau reduction prevents key features of autism in mouse models. Neuron. (2020);106:421–437. doi: 10.1016/j.neuron.2020.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Takagi K, Ito S, Miyazaki T, Miki Y, Shibahara Y, Ishida T, Watanabe M, Inoue S, Sasano H, Suzuki T. Amyloid precursor protein in human breast cancer:An androgen-induced gene associated with cell proliferation. Cancer Sci. (2013);104:1532–1538. doi: 10.1111/cas.12239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Takaichi Y, Chambers JK, Inoue H, Ano Y, Takashima A, Nakayama H, Uchida K. Phosphorylation and oligomerization of α-synuclein associated with GSK-3βactivation in the rTg4510 mouse model of tauopathy. Acta Neuropathol Commun. (2020);8:86. doi: 10.1186/s40478-020-00969-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Tamaoka A, Matsuno S, Ono S, Shimizu N, Shoji S. Increased amyloid βprotein in the skin of patients with amyotrophic lateral sclerosis. J Neurol. (2000);247:633–635. doi: 10.1007/s004150070133. [DOI] [PubMed] [Google Scholar]
- 322.Tan W, Zou J, Yoshida S, Jiang B, Zhou Y. The role of inflammation in age-related macular degeneration. Int J Biol Sci. (2020);16:2989. doi: 10.7150/ijbs.49890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Tang Z, Bereczki E, Zhang H, Wang S, Li C, Ji X, Branca RM, Lehtiö J, Guan Z, Filipcik P. Mammalian target of rapamycin (mTor) mediates tau protein dyshomeostasis:implication for Alzheimer disease. J Biol Chem. (2013);288:15556–15570. doi: 10.1074/jbc.M112.435123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Teravskis PJ, Covelo A, Miller EC, Singh B, Martell-Martínez HA, Benneyworth MA, Gallardo C, Oxnard BR, Araque A, Lee MK. A53T mutant alpha-synuclein induces tau-dependent postsynaptic impairment independently of neurodegenerative changes. J Neurosci. (2018);38:9754–9767. doi: 10.1523/JNEUROSCI.0344-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Terzi M, Birinci A, Cetinkaya E, Onar M. Cerebrospinal fluid total tau protein levels in patients with multiple sclerosis. Acta Neurol Scand. (2007);115:325–330. doi: 10.1111/j.1600-0404.2007.00782.x. [DOI] [PubMed] [Google Scholar]
- 326.Thomas T, Thomas G, McLendon C, Sutton T, Mullan M. β-Amyloid-mediated vasoactivity and vascular endothelial damage. Nature. (1996);380:168–171. doi: 10.1038/380168a0. [DOI] [PubMed] [Google Scholar]
- 327.Thomas T, Miners S, Love S. Post-mortem assessment of hypoperfusion of cerebral cortex in Alzheimer's disease and vascular dementia. Brain. (2015);138:1059–1069. doi: 10.1093/brain/awv025. [DOI] [PubMed] [Google Scholar]
- 328.Tian Y, Zhang F, Qiu Y, Wang S, Li F, Zhao J, Pan C, Tao Y, Yu D, Wei W. Reduction of choroidal neovascularization via cleavable VEGF antibodies conjugated to exosomes derived from regulatory T cells. Nat Biomed Eng. (2021);5:968–982. doi: 10.1038/s41551-021-00764-3. [DOI] [PubMed] [Google Scholar]
- 329.Tibolla G, Norata G, Meda C, Arnaboldi L, Uboldi P, Piazza F, Ferrarese C, Corsini A, Maggi A, Vegeto E. Increased atherosclerosis and vascular inflammation in APP transgenic mice with apolipoprotein E deficiency. Atherosclerosis. (2010);210:78–87. doi: 10.1016/j.atherosclerosis.2009.10.040. [DOI] [PubMed] [Google Scholar]
- 330.Tini G, Scagliola R, Monacelli F, La Malfa G, Porto I, Brunelli C, Rosa GM. Alzheimer's disease and cardiovascular disease:a particular association. Cardiol Res Pract. (2020);2020:2617970. doi: 10.1155/2020/2617970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Tomita K, Nakada T-a, Oshima T, Kawaguchi R, Oda S. Serum levels of tau protein increase according to the severity of the injury in DAI rat model. F1000Res. (2020);9:29. doi: 10.12688/f1000research.21132.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Troncone L, Luciani M, Coggins M, Wilker EH, Ho CY, Codispoti KE, Frosch MP, Kayed R, Del Monte F. Aβamyloid pathology affects the hearts of patients with Alzheimer's disease:mind the heart. J Am Coll Cardiol. (2016);68:2395–2407. doi: 10.1016/j.jacc.2016.08.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Tsang JY, Lee MA, Chan TH, Li J, Ni YB, Shao Y, Chan SK, Cheungc SY, Lau KF, Gary M. Proteolytic cleavage of amyloid precursor protein by ADAM10 mediates proliferation and migration in breast cancer. EBioMedicine. (2018);38:89–99. doi: 10.1016/j.ebiom.2018.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Tsitsopoulos PP, Marklund N. Amyloid-βpeptides and tau protein as biomarkers in cerebrospinal and interstitial fluid following traumatic brain injury:a review of experimental and clinical studies. Front Neurol. (2013);4:79. doi: 10.3389/fneur.2013.00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Tublin JM, Adelstein JM, Del Monte F, Combs CK, Wold LE. Getting to the heart of Alzheimer disease. Circ Res. (2019);124:142–149. doi: 10.1161/CIRCRESAHA.118.313563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Twohig D, Nielsen HM. α-synuclein in the pathophysiology of Alzheimer's disease. Mol Neurodegener. (2019);14:23. doi: 10.1186/s13024-019-0320-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Van Goor J, Massa G, Hirasing R. Increased incidence and prevalence of diabetes mellitus in Down's syndrome. Arch Dis Child. (1997);77:183. doi: 10.1136/adc.77.2.183g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Van Schependom J, Guldolf K, D'hooghe MB, Nagels G, D'haeseleer M. Detecting neurodegenerative pathology in multiple sclerosis before irreversible brain tissue loss sets in. Transl Neurodegener. (2019);8:37. doi: 10.1186/s40035-019-0178-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Vasili E, Dominguez-Meijide A, Outeiro TF. Spreading of α-synuclein and tau:a systematic comparison of the mechanisms involved. Front Mol Neurosci. (2019);12:107. doi: 10.3389/fnmol.2019.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Vintilescu CR, Afreen S, Rubino AE, Ferreira A. The neurotoxic TAU45-230 fragment accumulates in upper and lower motor neurons in amyotrophic lateral sclerosis subjects. Mol Med. (2016);22:477–486. doi: 10.2119/molmed.2016.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Violet M, Delattre L, Tardivel M, Sultan A, Chauderlier A, Caillierez R, Talahari S, Nesslany F, Lefebvre B, Bonnefoy E. A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front Cell Neurosci. (2014);8:84. doi: 10.3389/fncel.2014.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Vuono R, Winder-Rhodes S, De Silva R, Cisbani G, Drouin-Ouellet J, Spillantini MG, Cicchetti F, Barker RA. The role of tau in the pathological process and clinical expression of Huntington's disease. Brain. (2015);138:1907–1918. doi: 10.1093/brain/awv107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Wakabayashi T, Yamaguchi K, Matsui K, Sano T, Kubota T, Hashimoto T, Mano A, Yamada K, Matsuo Y, Kubota N. Differential effects of diet-and genetically-induced brain insulin resistance on amyloid pathology in a mouse model of Alzheimer's disease. Mol Neurodegener. (2019);14:15. doi: 10.1186/s13024-019-0315-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Walker LC. Prion-like mechanisms in Alzheimer disease. Handb Clin Neurol. (2018);153:303–319. doi: 10.1016/B978-0-444-63945-5.00016-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Wallin MT, Culpepper WJ, Campbell JD, Nelson LM, Langer-Gould A, Marrie RA, Cutter GR, Kaye WE, Wagner L, Tremlett H. The prevalence of MS in the United States:a population-based estimate using health claims data. Neurology. (2019);92:e1029–1040. doi: 10.1212/WNL.0000000000007035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Wang J, Ohno-Matsui K, Morita I. Elevated amyloid βproduction in senescent retinal pigment epithelium a possible mechanism of subretinal deposition of amyloid βin age-related macular degeneration. Biochem Biophys Res Commun. (2012);423:73–78. doi: 10.1016/j.bbrc.2012.05.085. [DOI] [PubMed] [Google Scholar]
- 347.Wang J, Zhao C, Zhao A, Li M, Ren J, Qu X. New insights in amyloid beta interactions with human telomerase. J Am Chem Soc. (2015);137:1213–1219. doi: 10.1021/ja511030s. [DOI] [PubMed] [Google Scholar]
- 348.Wang L, Zhou Y, Chen D, Lee TH. Peptidyl-prolyl cis/trans isomerase pin1 and alzheimer's disease. Front Cell Dev Biol. (2020);8:355. doi: 10.3389/fcell.2020.00355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Wang L, Mao X. Role of retinal amyloid-βin neurodegenerative diseases:overlapping mechanisms and emerging clinical applications. Int J Mol Sci. (2021);22:2360. doi: 10.3390/ijms22052360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Wang L, Eom K, Kwon T. Different aggregation pathways and structures for Aβ40 and Aβ42 peptides. Biomolecules. (2021);11:198. doi: 10.3390/biom11020198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Wang S, Zhou SL, Min FY, Ma JJ, Shi XJ, Bereczki E, Wu J. mTOR-mediated hyperphosphorylation of tau in the hippocampus is involved in cognitive deficits in streptozotocin-induced diabetic mice. Metab Brain Dis. (2014);29:729–736. doi: 10.1007/s11011-014-9528-1. [DOI] [PubMed] [Google Scholar]
- 352.Wen Y, Yang SH, Liu R, Perez EJ, Brun-Zinkernagel AM, Koulen P, Simpkins JW. Cdk5 is involved in NFT-like tauopathy induced by transient cerebral ischemia in female rats. Biochim Biophys Acta. (2007);1772:473–483. doi: 10.1016/j.bbadis.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 353.Winer JR, Maass A, Pressman P, Stiver J, Schonhaut DR, Baker SL, Kramer J, Rabinovici GD, Jagust WJ. Associations between tau β-amyloid and cognition in Parkinson disease. JAMA Neurol. (2018);75:227–235. doi: 10.1001/jamaneurol.2017.3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Woerman AL, Aoyagi A, Patel S, Kazmi SA, Lobach I, Grinberg LT, McKee AC, Seeley WW, Olson SH, Prusiner SB. Tau prions from Alzheimer's disease and chronic traumatic encephalopathy patients propagate in cultured cells. Proc Natl Acad Sci U S A. (2016);113::E8187–8196. doi: 10.1073/pnas.1616344113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Wood H. Traumatic brain injury induces transmissible tau pathology. Nat Rev Neurol. (2018);14:570–571. doi: 10.1038/s41582-018-0062-3. [DOI] [PubMed] [Google Scholar]
- 356.Wozniak J, Ludwig A. Novel role of APP cleavage by ADAM10 for breast cancer metastasis. EBioMedicine. (2018);38:5–6. doi: 10.1016/j.ebiom.2018.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Wray S, Lewis PA. A tangled web–tau and sporadic Parkinson's disease. Front Psychiatry. (2010);1:150. doi: 10.3389/fpsyt.2010.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Wu J, Zhou SL, Pi LH, Shi XJ, Ma LR, Chen Z, Qu ML, Li X, Nie SD, Liao DF, Pei JJ, Wang S. High glucose induces formation of tau hyperphosphorylation via Cav-1-mTOR pathway:a potential molecular mechanism for diabetes-induced cognitive dysfunction. Oncotarget. (2017a);8:40843–40856. doi: 10.18632/oncotarget.17257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Wu K, Quan Z, Weng Z, Li F, Zhang Y, Yao X, Chen Y, Budman D, Goldberg ID, Shi YE. Expression of neuronal protein synuclein gamma gene as a novel marker for breast cancer prognosis. Breast Cancer Res Treat. (2007);101:259–267. doi: 10.1007/s10549-006-9296-7. [DOI] [PubMed] [Google Scholar]
- 360.Wu L, Tan X, Liang L, Yu H, Wang C, Zhang D, Kijlstra A, Yang P. The role of mitochondria-associated reactive oxygen species in the amyloid βinduced production of angiogenic factors by ARPE-19 cells. Curr Mol Med. (2017b);17:140–148. doi: 10.2174/1566524017666170331162616. [DOI] [PubMed] [Google Scholar]
- 361.Wu X, Chen S, Lu C. Amyloid precursor protein promotes the migration and invasion of breast cancer cells by regulating the MAPK signaling pathway. Int J Mol Med. (2020b);45:162–174. doi: 10.3892/ijmm.2019.4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Wu XL, Chen Y, Kong WC, Zhao ZQ. Amyloid precursor protein regulates 5-fluorouracil resistance in human hepatocellular carcinoma cells by inhibiting the mitochondrial apoptotic pathway. J Zhejiang Univ Sci B. (2020a);21:234–245. doi: 10.1631/jzus.B1900413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Wulf G, Garg P, Liou YC, Iglehart D, Lu KP. Modeling breast cancer in vivo and ex vivo reveals an essential role of Pin1 in tumorigenesis. EMBO J. (2004);23:3397–3407. doi: 10.1038/sj.emboj.7600323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Xu J, Ying Y, Xiong G, Lai L, Wang Q, Yang Y. Amyloid βprecursor protein silencing attenuates epithelial-mesenchymal transition of nasopharyngeal carcinoma cells via inhibition of the MAPK pathway. Mol Med Rep. (2019);20:409–416. doi: 10.3892/mmr.2019.10293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Yamamoto R, Yoneda S, Hara H. Neuroprotective effects of β-secretase inhibitors against rat retinal ganglion cell death. Neurosci Lett. (2004);370:61–64. doi: 10.1016/j.neulet.2004.07.087. [DOI] [PubMed] [Google Scholar]
- 366.Yan Z, Liao H, Chen H, Deng S, Jia Y, Deng C, Lin J, Ge J, Zhuo Y. Elevated intraocular pressure induces amyloid-βdeposition and tauopathy in the lateral geniculate nucleus in a monkey model of glaucoma. Invest Ophthalmol Vis Sci. (2017);58:5434–5443. doi: 10.1167/iovs.17-22312. [DOI] [PubMed] [Google Scholar]
- 367.Yang W, Sopper MM, Leystra-Lantz C, Strong MJ. Microtubule-associated tau protein positive neuronal and glial inclusions in ALS. Neurology. (2003);61:1766–1773. doi: 10.1212/01.wnl.0000099372.75786.f8. [DOI] [PubMed] [Google Scholar]
- 368.Yang W, Ang LC, Strong MJ. Tau protein aggregation in the frontal and entorhinal cortices as a function of aging. Brain Res Dev Brain Res. (2005);156:127–138. doi: 10.1016/j.devbrainres.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 369.Yang W, Strong MJ. Widespread neuronal and glial hyperphosphorylated tau deposition in ALS with cognitive impairment. Amyotroph Lateral Scler. (2012);13:178–193. doi: 10.3109/17482968.2011.622405. [DOI] [PubMed] [Google Scholar]
- 370.Yao M, Teng H, Lv Q, Gao H, Guo T, Lin Y, Gao S, Ma M, Chen L. Anti-hyperglycemic effects of dihydromyricetin in streptozotocin-induced diabetic rats. Food Sci Hum Well. (2021);10:155–162. [Google Scholar]
- 371.Yarchoan M, James BD, Shah RC, Arvanitakis Z, Wilson RS, Schneider J, Bennett DA, Arnold SE. Association of cancer history with Alzheimer's disease dementia and neuropathology. J Alzheimers Dis. (2017);56:699–706. doi: 10.3233/JAD-160977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Yin X, Jin N, Shi J, Zhang Y, Wu Y, Gong CX, Iqbal K, Liu F. Dyrk1A overexpression leads to increase of 3R-tau expression and cognitive deficits in Ts65Dn Down syndrome mice. Sci Rep. (2017);7:619. doi: 10.1038/s41598-017-00682-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Yoshida T, Ohno-Matsui K, Ichinose S, Sato T, Iwata N, Saido TC, Hisatomi T, Mochizuki M, Morita I. The potential role of amyloid βin the pathogenesis of age-related macular degeneration. J Clin Invest. (2005);115:2793–2800. doi: 10.1172/JCI24635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Yu JH, Im CY, Min SH. Function of PIN1 in cancer development and its inhibitors as cancer therapeutics. Front Cell Dev Biol. (2020);8:120. doi: 10.3389/fcell.2020.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Zammit MD, Tudorascu DL, Laymon CM, Hartley SL, Ellison PA, Zaman SH, Ances BM, Johnson SC, Stone CK, Sabbagh MN. Neurofibrillary tau depositions emerge with subthreshold cerebral beta-amyloidosis in down syndrome. Neuroimage Clin. (2021);31:102740. doi: 10.1016/j.nicl.2021.102740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Zanier ER, Bertani I, Sammali E, Pischiutta F, Chiaravalloti MA, Vegliante G, Masone A, Corbelli A, Smith DH, Menon DK. Induction of a transmissible tau pathology by traumatic brain injury. Brain. (2018);141:2685–2699. doi: 10.1093/brain/awy193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Zayas-Santiago A, Díaz-García A, Nuñez-Rodríguez R, Inyushin M. Accumulation of amyloid beta in human glioblastomas. Clin Exp Immunol. (2020);202:325–334. doi: 10.1111/cei.13493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Zhang D, Zhou C, Li Y, Gao L, Pang Z, Yin G, Shi B. Amyloid precursor protein is overexpressed in bladder cancer and contributes to the malignant bladder cancer cell behaviors. Int J Urol. (2018);25:808–816. doi: 10.1111/iju.13726. [DOI] [PubMed] [Google Scholar]
- 379.Zhang W, Luo P. Myocardial infarction predisposes neurodegenerative diseases. J Alzheimers Dis. (2020);74:579–587. doi: 10.3233/JAD-191225. [DOI] [PubMed] [Google Scholar]
- 380.Zhang YW, Thompson R, Zhang H, Xu H. APP processing in Alzheimer's disease. Mol Brain. (2011);4:3. doi: 10.1186/1756-6606-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Zhao WQ, De Felice FG, Fernandez S, Chen H, Lambert MP, Quon MJ, Krafft GA, Klein WL. Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J. (2008);22:246–260. doi: 10.1096/fj.06-7703com. [DOI] [PubMed] [Google Scholar]
- 382.Zhao XW, Zhou JP, Bi YL, Wang JY, Yu R, Deng C, Wang WK, Li XZ, Huang R, Zhang J, Tao DT. The role of MAPK signaling pathway in formation of EMT in oral squamous carcinoma cells induced by TNF-α. Mol Biol Rep. (2019);46:3149–3156. doi: 10.1007/s11033-019-04772-0. [DOI] [PubMed] [Google Scholar]
- 383.Zhao Z, Zlokovic BV. Acetylated tau:a missing link between head injury and dementia. Med. (2021);2:637–639. doi: 10.1016/j.medj.2021.05.005. [DOI] [PubMed] [Google Scholar]
- 384.Zilka N, Novak M. The tangled story of Alois Alzheimer. Bratisl Lek Listy. (2006);107:343. [PubMed] [Google Scholar]
- 385.Żukiewicz-Sobczak W, Król R, Wróblewska P, Piątek J, Gibas-Dorna M. Huntington disease–Principles and practice of nutritional management. Neurol Neurochir Pol. (2014);48:442–448. doi: 10.1016/j.pjnns.2014.10.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.