

Abstract
Aldosterone exerts profound effects on renal and cardiovascular physiology. In the kidney, aldosterone acts to preserve electrolyte and acid-base balance in response to changes in dietary sodium (Na+) or potassium (K+) intake. These physiological actions, principally through activation of mineralocorticoid receptors (MRs), have important effects particularly in patients with renal and cardiovascular disease as demonstrated by multiple clinical trials. Multiple factors, be they genetic, humoral, dietary, or otherwise, can play a role in influencing the rate of aldosterone synthesis and secretion from the adrenal cortex. Normally, aldosterone secretion and action respond to dietary Na+ intake. In the kidney, the distal nephron and collecting duct are the main targets of aldosterone and MR action, which stimulates Na+ absorption in part via the epithelial Na+ channel (ENaC), the principal channel responsible for the fine-tuning of Na+ balance. Our understanding of the regulatory factors that allow aldosterone, via multiple signaling pathways, to function properly clearly implicates this hormone as central to many pathophysiological effects that become dysfunctional in disease states. Numerous pathologies that affect blood pressure (BP), electrolyte balance and overall cardiovascular health are due to abnormal secretion of aldosterone, mutations in MR, ENaC, or effectors and modulators of their action. Study of the mechanisms of these pathologies has allowed researchers and clinicians to create novel dietary and pharmacological targets to improve human health. This review covers the regulation of aldosterone synthesis & secretion, receptors, effector molecules, and signaling pathways that modulate its action in the kidney. We also consider the role of aldosterone in disease and the benefit of mineralocorticoid antagonists.
Graphical Abstract
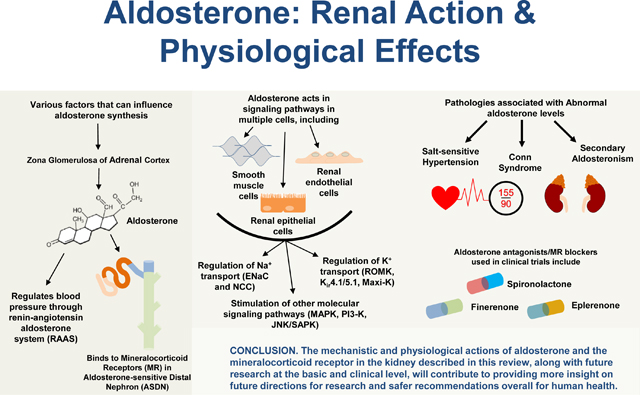
Introduction
Discovery and perspective
In 1953, Simpson et al. reported the crystallization of a substance synthesized by the adrenal cortex that was, and still remains today, the most potent sodium (Na+)-conserving corticosteroid known (745). Initially designated as electrocortin, this hormone was subsequently renamed aldosterone when its structure was determined (Figure 1) (165, 745, 746). In 1961, Jean Crabbe reported that one hour after of exposure to aldosterone, short-circuit current (SCC)1 increased in the toad urinary bladder, the amphibian analogue of the renal collecting duct (CD). The SCC provided an index of Na+ reabsorption indicating that aldosterone acted directly to increase Na+ absorption in the target tissue, rather than by secondary hormonal action (146). These studies were consistent with work by Edelman and colleagues who reported a significant lag time before the effect of aldosterone could be detected (187).
Figure 1: Mineralocorticoid and Glucocorticoid synthesis pathways.
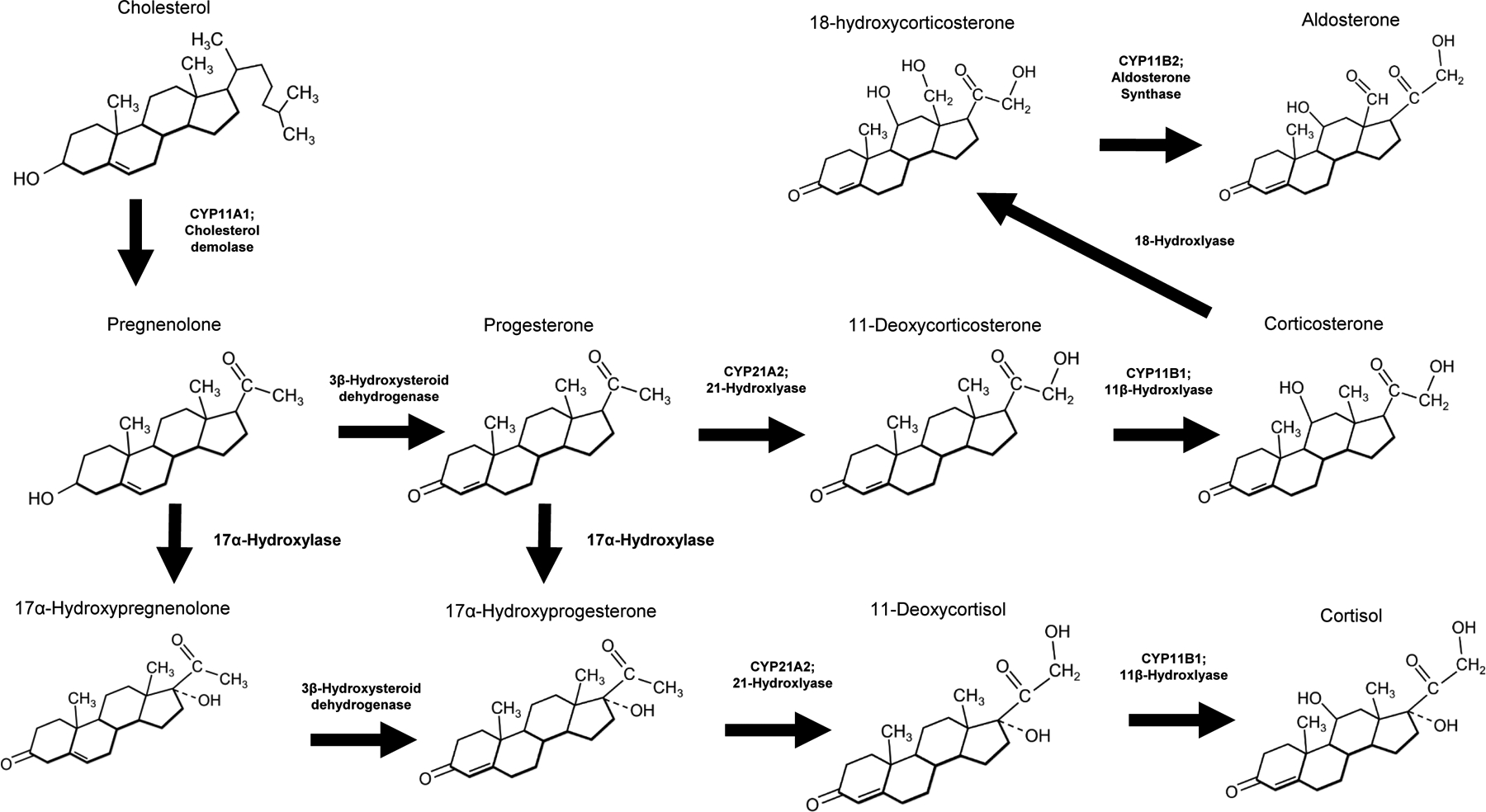
Enzymes involved in pathway reactions are in bold
Approximately 62,000 PubMed articles have been published that indexed “aldosterone” or “mineralocorticoid” since its discovery. In the past decade, the number of articles pertaining to aldosterone has exceeded 1,000 per year. Consequently, our understanding of this hormone, its mechanism of action, and physiological and pathophysiological effects continue to evolve. This review focuses primarily on major recent developments in our understanding of aldosterone’s function. We also review critical experiments that have been poorly appreciated but are essential for the proper understanding of the action of this hormone. Given the size of this topic, frequent reference to other comprehensive reviews will be cited.
This review focuses on the action in the kidney with a primary discussion of the action of aldosterone to stimulate ion transport in the renal tubules. Aldosterone also exerts important effects on the vasculature, central nervous system (CNS), immune system, and the circadian clock. Aldosterone action on the systemic vasculature and the CNS has been the subject of intense investigation, and recent reviews document the interdependence of renal function on these systems (48, 83, 183–185, 220, 261, 267, 467, 530, 558, 566, 572, 676, 685, 742, 854, 872, 884, 938, 962).
Classic and evolving understanding of aldosterone effects
Within the kidney, aldosterone acts primarily to promote net Na+ retention and net acid excretion. Globally, it acts to preserve potassium (K+) homeostasis. Although the classical effects of aldosterone are mediated through the mineralocorticoid receptor (discussed in the section “Aldosterone Nuclear Hormone Receptors”), aldosterone also acts by non-genomic mechanisms that are more rapid than the classic genomic-mediated mechanisms (discussed in “Non-Genomic Mechanisms of Action of Aldosterone”). In addition, aldosterone has other important actions besides its effect on electrolyte transport (discussed in the section “Role of Aldosterone in Salt-Sensitive Hypertension”), and recent electrolyte balance studies have prompted a review of previous studies (discussed in the section “Aldosterone and K+ Homeostasis” and “Clinical Trials Involving MR Blockade & Challenges”) (657).
Aldosterone Synthesis, Secretion, and Metabolism Aldosterone synthesis
The adrenal cortex accounts for the vast majority of circulating plasma aldosterone. Some evidence suggests that local tissue aldosterone production occurs and may contribute to pathophysiology, but this issue remains debated (899). The adrenal cortex has three anatomically and functionally distinct regions: zona glomerulosa (ZG), zona fasciculata, and zona reticularis. Aldosterone synthesis occurs almost exclusively in the most superficial ZG layer as illustrated in Figure 2. Aldosterone biosynthesis requires the mitochondrial and microsomal electron transport systems (Figure 1), and two reactions normally regulate its synthesis (499, 759). The first is the conversion of cholesterol to pregnenolone by the cholesterol side chain-cleaving enzyme, cytochrome P-450scc (encoded by the gene CYP11A1). This enzyme resides at the matrix side of the inner mitochondrial membrane (759). The second step catalyzes the sequential conversion of deoxycorticosterone to corticosterone and thence to aldosterone, (88, 150, 348, 411, 662) via aldosterone synthase (AS, encoded by the gene CYP11B2, also known as cytochrome P-450aldo or P-450c11AS), which is selectively expressed in the zona glomerulosa (330, 526, 734). Hormones and factors that affect aldosterone production principally regulate one of these two enzymatic conversions (83, 653). However, disruption of the circadian clock proteins Cry1 & Cry2 in mice suggest that 3-β-hydroxysteroid dehydrogenase (Mus-Hsd3b6 or Hum-HSD3B1) may become rate limiting under some conditions (171). The d-aldosterone stereoisomer is the active isoform synthesized and metabolized in humans. (504, 833) Cortisol and corticosterone, the primary glucocorticoid in humans and rodents, respectively, are largely bound to plasma proteins (~96% bound, ~4% free) with the greatest fraction bound to corticosteroid binding globulin (also known as transcortin) In contrast, aldosterone has a greater fraction that is not bound (60% bound, 40% free). Corticosteroid binding globulin and albumin are the principal carrier proteins for aldosterone, 22% and 38%, respectively. Mutations leading to deficiency of corticosteroid binding globulin have been linked to human diseases and impaired aldosterone responsiveness, an area that deserves further investigation. Normal values are listed in Table 1.
Figure 2: The adrenal gland.
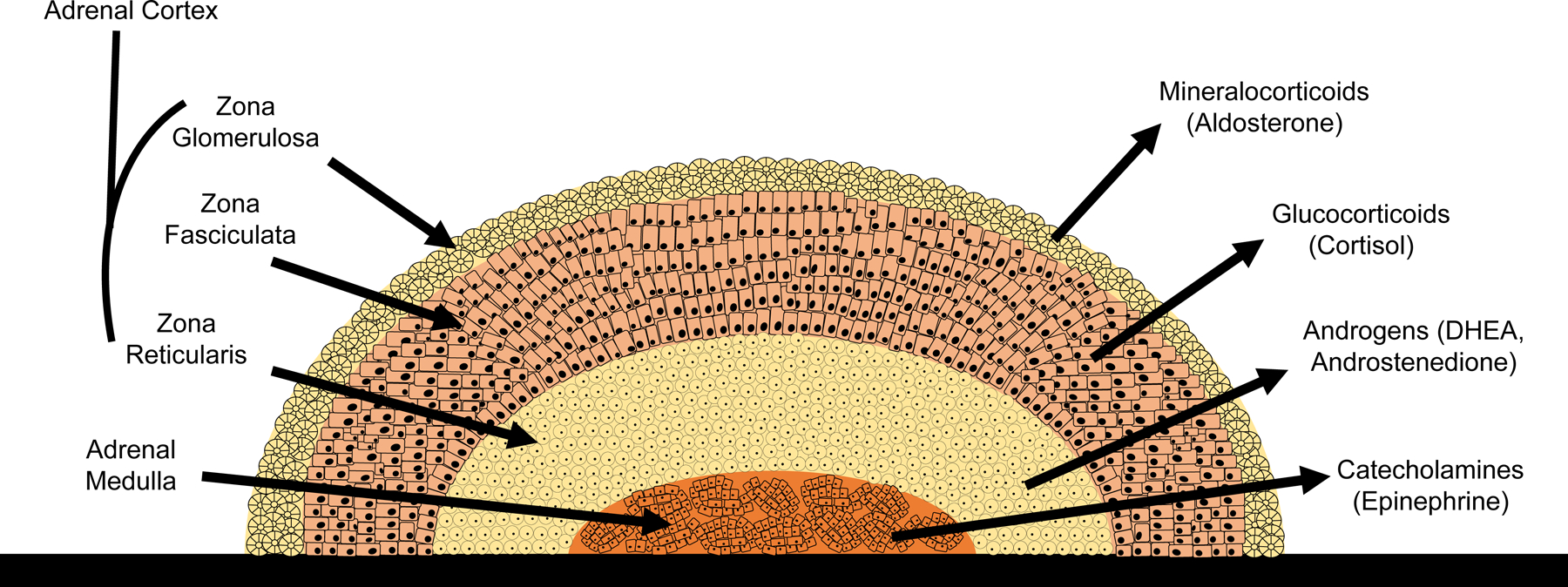
The adrenal cortex is made up of three layers: the outermost layer is known as the zona glomerulosa and is the main area for mineralocorticoid production. The middle layer of the cortex is called the zona fasciculata, and mainly synthesizes glucocorticoids. The inner layer of the cortex is known as the zona reticularis, and this section produces androgens. The core of the adrenal gland is known as the adrenal medulla, which is the site of catecholamine production.
Table 1 -.
Normal Circulating Levels of Hormones and Associated Pathologies
| Hormone | Ranges | References | ||
|---|---|---|---|---|
| Aldosterone |
|
(198, 213, 305) | ||
| Renin |
|
(416, 592) | ||
| Cortisol |
|
(66, 415) | ||
| Aldosterone Level | Renin Level | Cortisol Level | Pathology | References |
| High | Low | Normal | Conn’s syndrome/primary hyperaldosteronism | (131, 214) |
| High | High | Normal | Secondary hyperaldosteronism | (131, 214) |
| Low | High | Low | Addison’s disease/primary adrenal insufficiency | (131, 214) |
Hormones and factors that physiologically control aldosterone secretion and concentration
The role of aldosterone in Na+, K+, and acid-base homeostasis is best appreciated by understanding the factors that stimulate aldosterone secretion. When ZG cells are studied in vitro, many hormones stimulate aldosterone secretion. The sensitivity, specificity, magnitude, and duration of the stimulation, and the conditions under which such stimulation occurs has been extensively reviewed (361, 764, 785, 881, 899).
The major physiological regulators of aldosterone secretion in vivo are angiotensin II (Ang II), extracellular K+ concentration, and, to a lesser extent, natriuretic hormones (759). Aside from the major stimuli, the contribution of others to physiologically modulate aldosterone secretion in vivo is less clearly defined. The presence of specific receptors in ZG cells for adrenocorticotropic hormone (ACTH), vasopressin, serotonin, dopamine, atrial natriuretic peptide, somatostatin, and other compounds would suggest the potential for these substances to modulate aldosterone under specific conditions (653). Importantly, aldosterone synthesis requires multiple steps with the potential for different rate limiting reactions under different conditions (Figure 1),
Early studies recognized the importance of a volume sensitive mechanism, or mechanisms, K+ intake or extracellular [K+], and ACTH to stimulate aldosterone secretion (160, 247). The kidney exhibited a critical role for aldosterone secretion as well in response to hypovolemia (159, 248). Such studies, (7) and others reviewed extensively elsewhere (83, 653), provided evidence for the critical role of Ang II in physiological regulation of aldosterone secretion by the renin-angiotensin-aldosterone system.
Multiple mechanisms control adrenal aldosterone synthesis that are not necessarily additive. Early studies identified that part of this regulation involved changes in Ang II receptor binding sites, that was itself regulated by Ang II (331). Notably, studies in rats show that short-term Na+ restriction resulted in an increase in Ang II adrenal receptors. However, studies in primates suggest that this may not occur in all species (635). With the current recognition of at least two types of Ang II receptors (Ang II type 1 [AT1] and type 2 [AT2]) it would be instructive to determine whether sub-types of Ang II receptors are regulated differently in rodents and in primates.
The requirement of extracellular calcium (Ca2+) for sustained aldosterone secretion is a consistent observation (5, 52, 435, 653–655) and the contribution of extracellular [K+] has been systematically studied (169). Notably, continuous sampling studies in awake unstressed humans show that aldosterone, like many hormones, is secreted in bursts, occurring predominantly in the latter part of sleep and early morning. This secretion correlates with cortisol secretion but the correlation with renin was less consistent (409). Unfortunately, the correlation coefficients were not reported, so the degree of variation in aldosterone values explained by cortisol (and by inference ACTH) and renin remains to be determined. The role of ACTH in aldosterone secretion is discussed below (83, 264, 759, 899).
Angiotensin II-induced aldosterone secretion
Ang II is the penultimate product of the classical Renin-Angiotensin-Aldosterone System (RAAS), which is depicted in Figure 3. Renal baroreceptors and the macula densa of the juxtaglomerular apparatus, sense two distinct signals, arteriolar pressure and luminal sodium chloride (NaCl) concentration (or osmolality), respectively. These signals lead to the secretion of renin, an aspartic protease that cleaves angiotensinogen to the decapeptide angiotensin I, which is further processed by angiotensin converting enzyme (ACE) to the octapeptide Ang II. Ang II acts as a primary regulator of adrenal aldosterone secretion by binding to Ang II type1 (AT1) receptors located in the cells of the adrenal ZG (309, 315, 404, 518, 857). Ang II-induced secretion occurs by AT1 G-coupled receptor activation that causes not only membrane depolarization, but also a cascade of other cellular events. These include activation of non-selective cation channels, phosphoinositide-specific phospholipase (PLC), and inositol 1,4,5-trisphosphate (IP(3))-Ca2+/calmodulin-mediated signaling (505, 759). The net overall result is an increased activity of the rate-limiting enzymes for aldosterone synthesis.
Figure 3: The renin-angiotensin aldosterone system (RAAS).
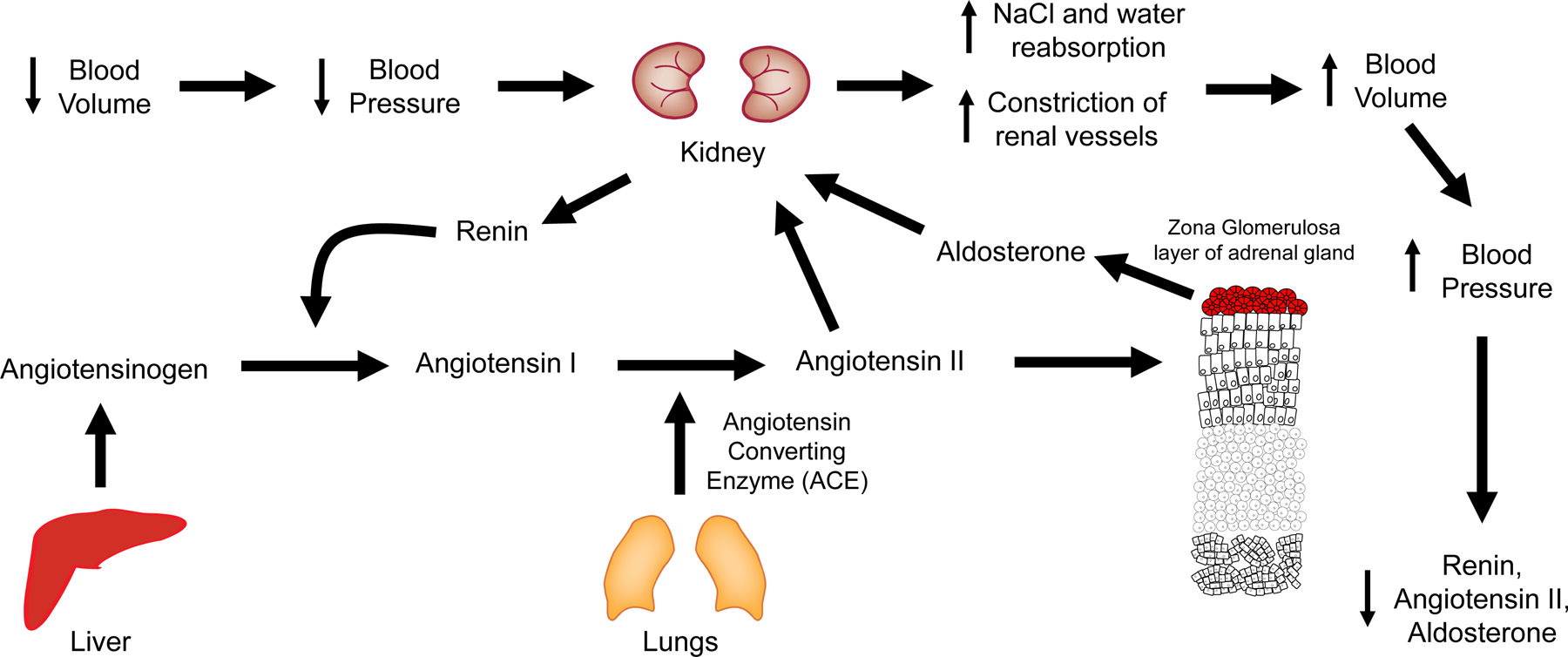
In response to a decrease in blood volume, BP decreases, which elicits a response from the kidney to increase BP. This renal response is mediated by the production and secretion of renin by the juxtaglomerular cells in response to decreased arterial pressure sensed by renal baroreceptors and decreased luminal NaCl concentration sensed by the macula densa. The enzyme renin cleaves angiotensinogen, also known as renin substrate to produce angiotensin I, which is further processed to angiotensin II. Angiotensin II increases blood volume by two mechanisms: directly constricting systemic and renal arteries and arterioles, and by stimulating the production of aldosterone from the adrenal gland. The subsequent increase in vascular resistance and the NaCl and water reabsorption restores blood volume and BP toward normal and reduces RAAS activity.
Ang II results in plasma membrane depolarization, similar to physiological increases in plasma [K+] (see below), which can lead to activation of voltage-dependent Ca2+ channels independent of cyclic adenosine monophosphate (cyclic AMP; cAMP) formation (499, 653). However, Ang II binding to AT1 receptor produces additional cellular responses that are independent of changes in the plasma membrane voltage that coordinate the stimulation of aldosterone production and secretion from the adrenal zona glomerulosa cells. In addition, activation of T-type Ca2+ channels by Ang ll at physiological concentrations appears to require the presence of intracellular GTP signaling (501, 529). Whether membrane depolarization per se, however, contributes significantly to Ca2+ channel activation has been questioned by studies that found only small effects of Ang II to depolarize membrane voltage (Vm) in bovine glomerulosa cells (124).
K+-induced aldosterone secretion
The ZG cells of the adrenal cortex express five classes of K+ channels and at their resting membrane potential or voltage (Vmr) are nearly exclusively conductive to K+. Although not all these K+ channels may contribute to the resting membrane potential in rat ZG cells (498, 500, 652, 841), at least two families of K+ channels are known to contribute to K+ conductance: 1) inwardly rectifying K+ (Kir) channels and 2) background K+ current channels (473). Mutations in some, notably Kir3.4 (KCNJ5) and Task3 (KCNK9), lead to membrane depolarization and increased aldosterone secretion (132, 621, 720). Quinn et al. studied aldosterone secretion by patch clamp analysis on rat and bovine ZG cells and demonstrated the presence of voltage-sensitive Ca2+ channels with a small activation threshold that are activated by changes in extracellular [K+] ([K+]e) (651).
Although chronic increases or decreases in K+ intake are associated with corresponding changes in plasma aldosterone, the variation of plasma K+ concentration ([K+]p) throughout the day is normally ~10% (< 0.4 mEq/L)(729) whereas aldosterone exhibits a robust circadian pattern of secretion with typically a 2–4 fold variation throughout the day. These considerations suggest that Ang II, circadian rhythm, or other factors, perhaps modulated by [K+]e, are more important in controlling the daily variation in aldosterone concentration (785). Indeed, evidence indicates that ZG cells exhibit slow periodic voltage spikes and coordinated bursts of intracellular Ca2+ oscillations that can be modulated by angiotensin II and [K+]e (360). Whether the frequency of these voltage spikes and Ca2+ oscillations predict the rate of ZG cell aldosterone secretion, as inferred from indirect evidence (53) remains a promising area of investigation.
The role of [K+]e as a mechanism responsible for physiological regulation of aldosterone synthesis requires that the changes in [K+]e occur within the range normally observed in vivo. Spät (758) emphasized the distinction between physiological and supra-physiological [K+] changes because separate mechanisms likely mediate each. Abrupt increases in [K+]e will cause plasma Vm to depolarize like the effect of Ang II on Vm via inhibition of TWIK-related acid-sensitive K+ channels (153). Thus, [K+]e -induced and Ang II-induced mechanisms of aldosterone secretion have elements in common, but they exhibit significant differences. In contrast to the multiple actions of Ang II to orchestrate aldosterone secretion, the effect of [K+]e is primarily mediated by Ca2+ influx.
The role of the cellular membrane potential or voltage to influence aldosterone secretion, however, is supported by studies that have dissected the genetic causes of primary hyperaldosteronism, with important contributions made by Lifton and coworkers. Choi et al. provided compelling evidence that somatic mutations in the potassium channel KCNJ5 were responsible for aldosterone-producing adrenal adenomas in eight of 22 cases that they analyzed. The mutations were predicted to result in amino acid changes in (G151R) or near (L168R, T158A) the selectivity filter of KCNJ5. When expressed in 293T cells, the mutations resulted in a loss of channel selectivity, increased sodium conductance, and cell membrane depolarization. Such characteristics would be predicted to increase calcium entry and aldosterone production. (132) Other mutations have been reported for the CACNA1D calcium channel and CLCN2 chloride channel that are also consistent with a voltage or calcium mediated stimulation of aldosterone secretion and implicated in hyperaldosteronism. (718, 719, 721, 727) Thus, several somatic or germline mutations in ion channels appear to be responsible for a significant number of cases of hyperaldosteronism, although other mechanisms of hyperaldosteronism exist. (257, 479, 554, 611) Several excellent reviews provide additional analysis. (232, 720, 727)
The previous considerations are relevant to the work of Lotshaw, who has examined the role of membrane depolarization, and the channels involve in K+-stimulated aldosterone secretion. Physiological changes in [K+]e appear to activate predominantly T-type channels, but supra-physiological concentrations of K+ trigger Ca2+ influx through L-type Ca2+ channels (499). Since T-type channels activate between −80 and −70 mV (841), and an increase in [K+]e by 1 mM induces a small depolarization, it is likely that the effect of changes in [K+]e are not solely due to changes in Vm. A positive feed-back mechanism of Ca2+ signaling appears to amplify the initial signal by activating CaMKII, which reduces the activation threshold of T-type channels (54). In addition, small changes in [K+]e produce changes in cytoplasmic [Ca2+] (646). The sensitivity of ZG cells to changes in [K+]e may also involve cellular swelling to amplify the mechanism of Ca2+ signaling and sensitize T-type channels to depolarization. Thus, a role for Ca2+ signaling, and an increase in cell volume may be important cytosolic events (510). Further study of the genetic and cellular mechanisms that underlie aldosterone secretion under physiological and pathophysiological conditions are needed and will likely be highly rewarding.
Adrenocorticotropic hormone (ACTH)
ACTH stimulates aldosterone production, but this effect appears to be subject to the experimental conditions. For example, in collagenase-dispersed adrenal glomerulosa cells, both Ang II and ACTH evoked marked increases in steroid production (175). Ang II concentrations as low as 30 pM, similar to normal rat blood Ang II concentrations, significantly increased aldosterone and corticosterone production, with 1000 pM Ang II increasing aldosterone production by 6–7 times that of basal values. However, dispersed capsular cells were also highly sensitive to ACTH, with a threshold of ~3 pM and a maximum aldosterone response of 20-fold greater than basal values.
Oelkers et al. examined the effect of ACTH on the RAAS in humans. Using normal male subjects, 10 IU of ACTH was infused for 34 hours. ACTH infusions led to a significant increase in plasma renin activity and Ang II concentration with a maximum response after 24 hours. This effect was not seen with sham or hydrocortisone infusions and was not mediated by a rise in plasma renin substrate (588). These results led to the suggestion that ACTH may be a physiological regulator of renin secretion, and that ACTH may stimulate aldosterone secretion on the second day of treatment through renin and Ang II. In a follow-up study, the same dose of ACTH infusion led to an increase in systolic BP on the second day of infusion in eight normal men (589). Treating the subjects with propranolol or indomethacin led the researchers to the conclusion that the effect of ACTH was not mediated by renal beta-adrenergic receptors, and that ACTH may be physiologically involved in the regulation of renin secretion based on the infusion rate (589).
In contrast, in vivo adrenal transplantation experiments in sheep showed greater Ang II specificity to selectively increase aldosterone. Ang II produced large increases in aldosterone secretion without a consistent effect on cortisol or corticosterone (75). ACTH infusion resulted in large increases of cortisol and corticosterone secretion with little increase in aldosterone secretion rate until secretion of cortisol and corticosterone exceeded 1,000 and 45 μg per hour.
Moreover, the effect of ACTH is time-dependent. Acute ACTH administration stimulates aldosterone production, but assessment of its contribution to the physiological regulation of aldosterone secretion in vivo depends on the experimental design of the studies used to assess this contribution. If ACTH is administered by continuous infusion its action is transient and tachyphylaxis develops (83, 653). On the other hand, pulsatile administration, which more closely simulates endogenous ACTH release does not result in suppression of aldosterone secretion, at least over 48 hours (726). Regarding the chronic infusion experiments, some studies (6, 229), but not all (570) support the suppression of the renin-angiotensin axis as an explanation. However, this suppression is also observed in patients with hyperaldosteronism (71), and when renin activity is stimulated by a low Na+ diet and diuretic treatment (587).
More recent studies in mice provide insight for the tachyphylaxis (182) which appears to be, at least in part, due to a transcriptional mechanism in the adrenal gland (540) that results in a shift in the biosynthetic pathway from aldosterone towards glucocorticoids with expansion of the zona fasciculata. However, these studies employed chronic infusion, and thus the fundamental role of ACTH remains open to interpretation depending on the experimental design. It is tempting to speculate that the difference between continuous and pulsatile stimulation of aldosterone by ACTH reflect the role of mechanisms related to periodic oscillating genes such as those described for the voltage spikes and changes cytosolic Ca2+ oscillation described above.
Natriuretic peptides
The natriuretic peptides, atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP) and C-type natriuretic peptide (CNP), represent a class of hormones that have all been shown to inhibit aldosterone secretion in humans and laboratory animals (138, 380, 505, 699, 709, 959). Receptors for ANP are present on adrenal ZG cells, and physiological rates of ANP infusion result in significantly reduced aldosterone secretion without an effect on plasma renin activity. Unfortunately, the role of ANP to inhibit aldosterone secretion, although well documented experimentally, has not received sufficient attention to resolve whether it represents a bona fide in vivo regulator of aldosterone (786). It is possible that some of the heterogeneity of hyperaldosteronism may be related to differences in ANP action (699). Resolution of this issue will require systematic studies in normal human subjects and patients with hyperaldosteronism on different levels of well-defined Na+ and K+ intake and accurate plasma renin, aldosterone and natriuretic peptide levels performed under uniform conditions noting time of day, sex, age, and with rates of aldosterone production before and after saline suppression. With sufficiently large populations of normal individuals and patients with hyperaldosteronism according to specific etiology, the predictive value of aldosterone and renin can be compared to that observed with aldosterone, renin, and ANP. If ANP is an important determinant of aldosterone production, it should improve the accuracy of distinguishing normal and pathological states.
Circadian control of aldosterone secretion and plasma aldosterone concentration
Plasma aldosterone concentration is not constant but exhibits a characteristic circadian variation with the greatest values occurring during the latter part of sleeping and shortly after awaking in humans (409, 410). Although upright posture and activation of renin secretion are frequently cited as the predominant regulators of daily fluctuations in plasma aldosterone, when plasma aldosterone concentration is measured frequently (every 10 or 30 minutes) over 24 hours in the same individual, rapid increases in plasma aldosterone concentration occur in secretory bursts (266, 409, 410). Moreover, simultaneous measurements of renin, aldosterone and cortisol revealed that the predominant correlation was between aldosterone and cortisol. Both hormones exhibited a consistent circadian rhythm (409, 410). These observations suggest that disruption of the molecular components of the circadian clock would be expected to affect aldosterone secretion.
Indeed, work involving circadian clock gene knockout mouse models indicates that aldosterone production is directly controlled by the molecular clock. For example, Doi et al. discovered a salt-sensitive hypertension phenotype in mice lacking Cry1 and Cry2 accompanied by dramatically high plasma aldosterone values (171). This effect was directly linked to significant overexpression of Hsd3b6 in the ZG of the adrenal gland. Expression of the human homolog has been linked to hypertension. Interestingly, the opposite phenotype was observed in Period 1 (Per1) heterozygous mice (666). Richards et al. showed that mice with reduced Per1 expression exhibited reduced plasma aldosterone levels compared to WT mice and this effect was associated with reduced expression of Hsd3b6 in the adrenal gland. Clock knockout mice exhibit reduced blood pressure (BP) and altered circadian rhythmicity in plasma aldosterone levels (576).The first kidney specific knockout of a clock gene, Bmal1, was generated by Firsov and colleagues (820). Deletion of Bmal1 from renin-producing cells also resulted in decreased Bmal1 expression in the juxtaglomerular apparatus, the thick ascending limb (TAL) and CD. These Bmal1 knockout mice had lower BP and reduced aldosterone levels than controls, providing additional evidence that the clock controls aldosterone production. Wang et al. evaluated the cardiovascular phenotype of mice lacking three circadian transcription factors, DBP, HLF, and TEF, and these mice also exhibited reduced plasma aldosterone levels relative to WT mice (869). This effect was associated with reduced BP and cardiac hypertrophy. Taken together, these studies support a role for the molecular clock components in the regulation of aldosterone levels with implications for renal electrolyte handling and BP control.
Influence of sex on aldosterone and MR
Sex is another factor that should be considered when discussing aldosterone. Plasma aldosterone levels in premenopausal women are lower than in men of similar age (547), and premenopausal women have lower levels than age-matched men (96, 179). Plasma aldosterone also varies throughout the menstrual cycle. Women with high levels of circulating 17β-estradiol (E2) (greater than 300 pmol/liter) were found to have significantly higher levels of plasma aldosterone than women with low E2 levels (less than 100 pmol/liter)(547). The sex difference in plasma aldosterone concentration is lost when postmenopausal women are compared with age-matched men (723). E2 treatment was shown to reduce aldosterone secretion after ovariectomy in rats, an experimental technique which replicates the effects of menopause (508, 673). However when translated to humans, the effect of E2 treatment was not as profound. Postmenopausal women that were given estrogen replacement therapy (ERT) had slightly lower levels of aldosterone compared with postmenopausal women without ERT, but the difference was not statistically significant (723). Caroccia et al. found that in the adrenal cortex, estrogen receptor β (ERβ) is the predominant subtype, followed by G protein-coupled estrogen receptor 1 (GPER-1) (112). In vitro experiments involving the HAC15 adrenocortical cell line found that E2 inhibits aldosterone production through ERβ, and stimulates aldosterone production through GPER-1 activation (112). Another study examined the sex-specific expression of mineralocorticoid target genes encoding αENaC (Scnn1a), serum and glucocorticoid kinase 1 (Sgk1), and Gilz in the kidneys of adult mice, along with 11ß-hydroxysteroid dehydrogenase type 2 (11ß-HSD2), an enzyme that regulates and metabolizes non-mineralocorticoids. The relative mRNA expression of MR and αENaC was not different between sexes in these mice at three months of age. However, in females, gene expression of Gilz was less and Sgk1 and 11ß-HSD2 were higher than in male mice (181). There is also evidence that sex may influence the action of aldosterone in extra-renal tissues (545).
In clinical studies, sex-specific responses exist to the mineralocorticoid antagonist spironolactone in an African American population with hypertension. Spironolactone reduced BP more effectively in hypertensive women than men (140). Thus, the sex of the patient may also determine the type of antihypertensive treatment. In a cross-sectional study including 17,856 patients in France, researchers found that women were more frequently treated with aldosterone receptor blockers than men, along with other antihypertensive treatments (164). Animal and clinical studies emphasize the effect of sex hormones on aldosterone production and MR function, but more work is needed to determine the contribution and significance of these interactions to pathophysiological states.
Aldosterone metabolism
Although plasma aldosterone is primarily determined by the rate of secretion by the ZG, other factors including posture, K+ intake, and hepatic aldosterone metabolism can affect plasma values (117). In addition, aldosterone metabolism occurs in target tissues and may significantly modify the action of aldosterone (190, 344). In early studies, Tait et al. used a multi-compartment model to estimate the plasma half-life of aldosterone as approximately 35 minutes (796). They concluded that aldosterone distributes into a much greater volume and has a higher rate of metabolism than that of cortisol or corticosterone. Further work noted effects of ACTH on aldosterone metabolism (647). Evidence that large changes in dietary Na+ intake failed to consistently affect aldosterone metabolic clearance rate suggested that if aldosterone metabolic clearance rate is an important factor, it was not related to the major feedback pathway (dietary Na+) that it regulates (648). Although the reported metabolic clearance rate of aldosterone varies substantially (89, 796) two studies observed no change in metabolic clearance rate between hypertensive individuals and controls (972) (89), but one study reported significantly higher aldosterone secretion rates in low-renin versus normal-renin hypertensive patients (89). Whether differences in aldosterone metabolism in renal target cells, ether in vivo or in vitro, contributes to its physiological or pathophysiological effects remains an issue to be considered.
Aldosterone Nuclear Hormone Receptors
The superfamily of nuclear hormone receptors
The canonical receptor for aldosterone is the mineralocorticoid receptor (MR). MR is a nuclear receptor and specifically a member of the NR3C subfamily of steroid hormone receptors that also includes the glucocorticoid receptor (GR), the androgen receptor, and the progesterone receptor. Aldosterone binds to receptors, primarily MR, which act largely through genomic mechanisms. Additionally, aldosterone binds to receptors that act, as currently understood, through non-genomic mechanisms. Some non-genomic mechanisms of aldosterone action are mediated by MR, but also by less extensively studied receptors and pathways. This section discusses the properties of MR and related proteins that mediate the genomic actions of aldosterone. Non-genomic signaling via MR and other aldosterone receptors are discussed in the section “Non-Genomic Mechanisms of Action of Aldosterone.”
Molecular cloning of the glucocorticoid and mineralocorticoid receptors
In 1985, Hollenberg and coworkers reported the primary structure and functional expression of the human GR (350). These investigators based their molecular analysis on highly purified protein preparations for the rat and the human GR. Two deduced primary amino acid sequences were discovered by analysis of cDNA derived from human lymphocytes and fibroblasts. The sequences, designated αhGR and ßhGR, differed by 50 and 15 amino acids at C termini of the primary common sequence, respectively. Arriza and coworkers subsequently identified MR using a strategy based on homology with GR (28). Tissue analysis of mRNA by Northern blot showed similar mRNAs that were most abundant in hippocampus followed by kidney, whole brain, and less abundant signals in heart, pituitary, muscle, and spleen. In vitro studies of the expressed protein demonstrated that the affinity of MR for aldosterone and for cortisol were very similar, despite the fact that cortisol is present in the plasma at concentrations ~three orders of magnitude greater than that of aldosterone (28). These studies substantially changed the conceptualization of the action of aldosterone mediated by MR (28) as described under Aldosterone Action & Signaling Pathways.
Genomic action of aldosterone & mineralocorticoids
The very high-affinity binding of aldosterone is a property unique to MR (28, 374), but deoxycorticosterone, cortisol, and dexamethasone exhibit equally high affinities (Table 2), which prompted significant revision of the mechanism for corticosteroid specificity. However, aldosterone’s action can be mediated by other steroid hormone receptors, especially GR. GR and MR are expressed together in many cell types in the kidney (2). On one hand, the physiological circulating concentration of aldosterone is in the very low nanomolar range, well below the apparent GR affinity for aldosterone. However, the circulating concentration of aldosterone in patients with primary hyperaldosteronism remains chronically elevated, opening the possibility that persistently high concentrations may exert pathological effects by virtue of persistent MR or GR activation. In vitro experiments revealed that aldosterone activation of GR is readily detectable at concentrations as low as 10 nmol (782). The presence of GR in kidney cells shown to produce abundant levels of 11β-HSD2 suggested a role for GR independent of cortisol. Moreover, conditional overexpression of GR in the murine renal CD resulted in increased expression of well-established aldosterone target genes, such as glucocorticoid-induced leucine zipper protein 1 (gilz1) and the α subunit of ENaC (αENaC; Na+ channel epithelial 1 alpha subunit; encoded by scnn1a), and altered Na+ handling (573). The present section will focus on the general characteristics of the broader family of steroid hormone receptors with an emphasis on the features that differentiate MR from its cousins.
Table 2 –
Comparison of affinities of mineralocorticoid receptor (MR), glucocorticoid receptor (GR), and estrogen receptor ß (ERß) for cognate ligands and other corticosteroids
| Receptor | Ligand | Affinity |
|---|---|---|
| Mineralocorticoid Receptor (MR) | Aldosterone | 0.5–3 nM |
| Deoxycorticosterone (DOC) | Similar to aldosterone in vitro; 2% of aldosterone in vivo | |
| Cortisol | ~0.5 nM | |
| Dexamethasone | ~0.7 nM | |
| Progesterone | High, but lower than corticosteroids | |
| Estradiol | Very poor | |
| Glucocorticoid Receptor (GR) | Aldosterone | ~ 14 nM |
| Cortisol | ~ 12 nM | |
| Dexamethasone | ~ 4nM | |
| Estrogen Receptor ß (Erß) | Estradiol | 1nM for ERß1; 8nM for ERß2 |
The contribution of aldosterone via MR to mineralocorticoid activity may also require revision given the discrepancy in the phenotype of the aldosterone synthase (AS) versus the MR KO mouse. Although MR null mice have normal prenatal development, during the first week after birth they lose weight, develop hyponatremia, hyperkalemia, and significant elevation of plasma renin, Ang II, and aldosterone concentrations, with greater than eight-fold increase in renal fractional Na+ excretion (FeNa). These mice die within two weeks after birth from dehydration and renal Na+ and water loss consistent with pseudohypoaldosteronism as originally reported (61).
AS null mice are born in expected Mendelian ratios from heterozygous parents and also failed to thrive. However, mortality between 7 and 28 days of birth is substantially less (30%) and hyponatremia and salt wasting are absent on a normal diet but hyperkalemia is present (464, 511, 818, 930). Plasma [Cl−] was significantly reduced in AS null mice, but plasma [Na+] was not statistically different from wild type. Stimulation of glucocorticoid production acting through MR may explain some of the differences in phenotype as originally proposed (464). However, unless 11ßHSD activity is suppressed in the AS null mice, this enzyme should prevent glucocorticoid occupancy of MR. Further studies with 11ßHSD null mice may be informative (see section “Specificity of Aldosterone as a Mineralocorticoid”). In addition, GR and MR are expressed together in many cell types in the kidney (2), and heterodimerization of MR and GR occurs and would be predicted to exhibit properties distinct from either homodimer. Ligand-dependent conformational changes are likely important as has been discussed for GR (412).
In the absence of mineralocorticoids, MR resides in the cytoplasm in a large chaperone complex containing heat shock protein Hsp90, Hsp70 and other proteins (241, 252). The Hsp90 complex stabilizes MR in the cytoplasm in a conformation that is competent for ligand binding but sequestered away from its site of genomic action in the nucleus. The role of the Hsp90 complex in MR stability became apparent in an experiment designed to inactivate Hsp90 (205). The antibiotic 17-allylamino-17-demethoxygeldanamycin binds to Hsp90, rendering MR susceptible to rapid ubiquitin-dependent turnover. In response to aldosterone activation of MR, a second immunophilin FBK52 replaces FBK51 facilitating MR rapid translocation to the nucleus via dynein/dynactin dependent movement along the microtubule network (238, 239). Essentially the same process occurs with GR, and this has been directly demonstrated by dexamethasone stimulation of GR in podocytes (302).
Genomic action of MR most often involves the formation of a MR:MR homodimer. Sophisticated resonance energy transfer experiments indicate that MR dimerization occurred after translocation to the nucleus, rather than as a cytoplasmic event (297). Although MR clearly forms a homodimer, studies have long suggested the possibility of a MR:GR heterodimer (706). This concept received strong support from fluorescence energy transfer measurements suggesting physical interaction between MR and GR forming a heterodimeric MR:GR receptor (577). Additional evidence in the form of transcription activation reporter gene assays supported a role for GR as either a subunit of a MR:GR heterodimer, or perhaps a coactivator for MR:MR (828). Based on the DNA binding properties of MR:MR and GR:GR (see below), it seems reasonable to expect that MR:GR binds the same response elements. However, it also seems likely that each receptor complex has unique transcription activation/repression properties.
For gene activation to occur, a dimeric steroid hormone receptor binds to a sequence generically referred to as hormone response element (HRE) located within a target gene resulting in the recruitment of cofactors that drive assembly of the transcription preinitiation complex. However, steroid hormone receptors are not strictly transcriptional activators but can act as transcriptional repressors, as well. Previous work established that the transcriptional activities of a number of aldosterone target genes such as sgk1 and glucocorticoid-induced leucine zipper 1 (gilz1) were stimulated by either MR or GR (568, 739) (See section “Aldosterone-Induced Transcripts & Aldosterone-Induced Proteins.”). Surveys were conducted of gene expression following short term exposure of kidney cell lines to aldosterone with the goal of identifying early aldosterone response genes under direct steroid hormone receptor control. Both serial analysis of gene expression (670) and gene expression microarray studies (306) in aldosterone-stimulated kidney cell lines yielded extensive lists of possible target genes in kidney cell lines. Notably, the most highly responsive early target gene in murine inner medullary CD (mIMCD-3) cells turned out to be the circadian clock gene per1 (304, 308). Subsequently, it became apparent that per1 plays a direct role in regulation of ENaC and BP (783). Aldosterone induction of the endothelin-1 (ET-1) gene (edn1) via both MR- and GR-dependent mechanisms was an interesting finding in view of the role of ET-1 in renal Na+ regulation (782, 784, 912). Expression profiles were altered by expression of mutant forms of MR in renal cells (212). Fakitsas et al. (203) applied the microarray approach to investigate mRNA levels in the microdissected distal nephron from mice injected with 10 μg/kg aldosterone. Nine mRNAs were upregulated by at least two-fold while expression of 13 others decreased. A particularly interesting observation was increased expression of the ubiquitin-specific protease Usp2-45. The enzyme functions to stabilize ENaC in the nephron. The mitogen activated protein kinase (MAPK) signaling system scaffold gene cnksr3 was also found to be an aldosterone target gene in the cortical collecting duct (CCD) (971). The implications of these early response genes on the action(s) of aldosterone are discussed in the section “Aldosterone Action in Tight Epithelia & Target Genes.”
State of the art technologies, such as ChIP-Seq, allow for comprehensive identification of genes directly regulated by MR in specific mineralocorticoid-responsive tissues. Ueda et al. examined this issue in the murine distal convoluted tubule (mDCT) cell line overexpressing a FLAG epitope tagged human MR (832). More than 1000 MR binding sites were detected, but it is likely that a much smaller number actually contribute to gene regulation. Nevertheless, these studies confirmed previous findings which identified sgk1, per1, edn1, and connective tissue growth factor (ctgf) as early response genes to the action of aldosterone (306) and added 22 genes to the list of candidate targets of MR in this cell line. Recently, Le Billan et al. performed a similar study employing Chip-Seq to identify MR binding sites in the genome of human renal tubular cells stably transfected with a recombinant (GFP)-MR vector (462). Again, the number of MR binding sites approached 1000. Four genes already established as aldosterone targets contained the top-six highest scoring MR binding sites: zinc-finger and BTB domain containing 16 (zbtb16), FK506 binding protein 5 (fkbp5), scnn1a, and per1. Eight other genes not previously known to be subject to aldosterone regulation displayed strong MR binding and at least some evidence of altered mRNA levels in response to aldosterone. Developing an atlas of aldosterone-regulated genes for the kidney by tubule segment is a technically achievable goal with the potential to provide novel insights into the regulation of ion balance. Thus far, there has not been a comprehensive assessment of genes uniquely responsive to MR as opposed to those also subject to activation by the other steroid hormone receptors in the kidney.
Modular structure and function
The steroid hormone receptors share common modular domain architectures (for review see (231, 265, 339, 446). Family members have an N-terminal domain (NTD), a central DNA binding domain (DBD), and a carboxyl terminal ligand binding domain (LBD). A flexible “hinge” links the DBD and LBD facilitating the major ligand-dependent conformational changes that free the nuclear receptors from the Hsp90 complex, promote dimerization, and make the receptors available for association with cofactors (for review see (927, 929).Although the literature contains reports of excellent preparations of purified MR (141), the structure of the intact protein has not been reported. The inherent flexibility of the protein along with solubility issues apparently complicate crystallization.
MR transcription and translation
The human MR gene (NR3C2) is located at 4q31.23 and extends across approximately 400 kilobase pairs (UCSC Genome Browser). Transcription can initiate at any of three promoters (612). As a result, three different exons can serve as the templates for the 5’ end of the primary MR transcript. None of these exons contains coding sequence. The regulatory role of such alternative transcriptional initiation mechanisms requires further study. The intact primary transcript includes nine exons that are spliced to generate mRNA approximately 5750 base pairs in length that encodes the canonical 984 amino acid, ~107 kD MR protein (28). Like other nuclear receptors, MR is clearly a participant in the large multi-protein complexes that direct gene transcription. Phage display technology using intact MR as bait for binding partners captured no less than 30 peptides representing potential binding partners (928). Follow-up reporter gene assays revealed that many of these peptides exerted either positive or negative influence on transactivation activity of MR. Beyond the canonical receptor, several normal MR variants have been reported. “MRA” and “MRB” proteins result from differential selection of start codons at met-1 or met-15 during translation initiation (612). Both isoforms directed transcriptional activation, but curiously when MRA and MRB were expressed individually, both displayed reduced efficiency as a result of an unknown mechanism. In humans and rats, an in-frame variant occurs as a result of inclusion of 12 extra bases added at the exon 3–4 splice junction (79) and makes up a minority of total MR (669, 895). The sequence of the intronic alternative 5’ splice site that gives rise to MR+4 displays strong evolutionary conservation suggesting that there may be some unknown functional importance for this variant (669). The transcription activation properties of this “MR+4” variant appeared to be essentially the same as MR (38). Evidence also exists for two meaningful MR variants generated by alternative splicing. The “hMRΔ56” variant arises from exon skipping of both exons 5 and 6 resulting in a 75 kDa MR protein lacking the entire LBD (963). The hMRΔ56 variant bound DNA and activated transcription in a hormone-independent manner. This variant was readily detectable in kidney RNA, but its influence on transcription has not been investigated in depth. Such findings raise important questions that deserve further study. In addition, a MR transcript lacking exon 5 was reported in the RNA database (139, 416), and one might speculate that “hMRΔ5” could exhibit properties comparable to hMRΔ56. There are of course many mutations and polymorphisms and some of them result in clinically significant pathologies (255, 256, 471, 538, 579, 582).
N-Terminal Domain (NTD)
The NTDs of steroid hormone receptors are primary regions of interaction with coregulatory proteins (for review see (927, 929)). Like other steroid hormone receptors, a fragment of MR consisting of only the NTD-DBD was sufficient for constitutive transactivation function in reporter gene assays (216). There is little sequence conservation among NTDs within the steroid hormone receptor family, so each receptor functions by protein-protein interactions between its NTD and a unique set of regulatory proteins. MR contains the largest and most complex NTD of all the steroid hormone receptors. In humans, the MR-NTD is defined as the first 602 amino acids and is subdivided into three functional domains. Activation function 1a (AF1a, amino acids 1–169) and AF1b (amino acids 451–602) flank the middle domain. A high resolution structure of MR-NTD has not yet been reported, probably because primary sequence analysis suggests that much of the MR-NTD exists in an intrinsically disordered state (459). As a result, MR apparently exists as an ensemble of differing conformations rather than in a fixed ordered structure. Fischer et al. (216) provided a singular contribution to the field by expressing and characterizing polypeptides modeled on all three MR-NTD sub-domains. Only the AF1b polypeptide showed significant structural stability in aqueous solution.
Comparison of primary MR sequence from differing species indicates localized segments of conservation within the NTD. These sites are thought to interact with cofactors (459). Yang (927) provided a listing of transcription factors known to bind to MR. An established example of a transcriptional activator known to specifically interact with the MR-NTD include the elongation factor 11–19 lysine rich leukemia “ELL” that binds specifically to AF1b (613). A direct tie of MR to the transcriptional preinitiation complex may occur via protein-protein contact between an LxxIL motif within AF1b and the general transcription factor TATA binding protein (TBP) (216). The steroid hormone coactivator 1 (SRC1) apparently binds to sites located in both AF1 in the NTD and activation function 2 (AF2) in the LBD. Murai-Takeda et al. (559) applied multiple independent approaches to demonstrate a specific interaction between MR AF-1 and NF-YC, a subunit of transcription factor NF-Y. Luciferase reporter gene assays suggested that NF-YC acts as a transcriptional corepressor for MR. One might envision the NF-YC corepressor binding to a NTD conformation in the ensemble and stabilizing a form of MR that is inappropriate for coactivator SRC1. In view of the size and extent of sequence conservation, it is very likely that additional MR-associated co-regulatory proteins will be identified. Together the evidence suggests that the MR-NTD assumes varying conformations that provide surfaces available to accommodate the protein-protein interactions with many cofactors.
DNA Binding Domain (DBD)
The DBDs of steroid hormone receptors account for sequence specific binding to HREs in target genes and contribute to the interface for receptor dimerization. The MR-DBD is 66 amino acids long and has 94% sequence identity with the GR-DBD. Not surprisingly, both receptors recognize an identical palindromic HRE consensus sequence (AGAACAN3TGTTCT). Direct aldosterone-dependent binding of both MR and GR to HREs located in a growing number of target genes has been observed in mammalian kidney cells by chromatin immunoprecipitation (782, 971). High resolution structures of the GR-DBD (539) and MR-DBD (363) bound to the consensus HRE have been determined by x-ray crystallography. Both DBDs consists of two zinc finger motifs. Only one of the zinc fingers actually dictates sequence-specific binding to DNA while the other serves as a structural feature. Each subunit of a nuclear receptor dimer contacts one half-site of a HRE through the zinc-finger. The chemical contacts between GR and MR with DNA have been described at the molecular level. Small structural differences exist between the two receptor DBDs that may contribute to selectivity in target gene activation between GR and MR (363).
Typically, one half-site is closely related to the consensus sequence, but significant variation from the consensus is often observed within the other half-site (541). Indeed, few authentic HREs possess half-sites where both are identical to the consensus sequence. As a result, receptors display considerable differences in receptor affinity for individual HREs, and this in turn affects the probability and stability of receptor binding to any given HRE. A wide variety of HRE sequences are indeed recognized by MR (462). Of note, at least some negative gene regulation occurs in instances involving the binding of a monomeric steroid hormone receptor to a single HRE half site. Sequence variations within an HRE can be an effector of the transcriptional response by imparting subtle conformational changes through the DBD (258). Therefore, simply detecting receptor binding to a HRE located within a gene should not be viewed as sufficient to predict the nature of the aldosterone response.
Ligand Binding Domain (LBD)
Due to the importance of receptor specificity for hormonal action, the LBDs of steroid hormone receptors have been the subjects of intense investigation (for reviews see (230, 231, 238, 374). The MR-LBD is 251 amino acids in length and shares approximately 55% primary sequence homology with the GR-LBD. The specific determinants of aldosterone binding to the MR-LBD have been defined at the level of bonds between various ligands and individual amino acids in the MR-LBD by a combination of biochemical, genetic and structural studies. In addition to high affinity recognition of cognate ligands, the LBDs house the transcriptional AF2 domain, contain the site for interaction with the HSP90 complex, and contribute much of the dimerization interface. Three groups independently solved high resolution structures of mutant forms of the MR-LBD with a variety of different ligands bound. Most of the structures were solved by including either the C808S or the S810L substitution to provide solubility and stability that facilitated crystallization. To date, structures of the LBD have been solved with aldosterone (77), corticosterone (478), deoxycorticosterone (77, 202), progesterone (77, 202), cortisone (77), spironolactone (77, 329, 373) and a non-steroid derivative of Benzoxazin-3 (329). Although the LBD structures have only 11 α-helices, the overall fold is very similar to the 12-helix bundle characteristic of other steroid hormone receptor LBDs. Typically, the ligand binding was seen deep within the fold that completely occludes the steroid. Together these papers helped to define the structural determinants that dictate ligand specificity and the actions of MR agonists and antagonists. The importance of the ligand-dependence response was illustrated by structure-function studies of the MR-LBD containing the pseudohyperaldosterone mutation S810L (77, 202). The amino acid substitution altered the transcriptional response such that MR with progesterone bound assumed an activated rather than repressed state. This appears to account for the severity of hypertension in affected individuals during pregnancy. The hypertension in men and non-pregnant women with this mutation has been attributed to MR activation by cortisone (See section “Specificity of Aldosterone as a Mineralocorticoid”). In addition, the structures have opened a window on rational drug design. Fagart et al. (201) reported molecular docking experiments that led to the identification of BR-4628 as a potent non-steroid inhibitor of MR.
In the activated ligand bound state, MR-LBD, the carboxyl-terminal helix-12 is compressed against the bundle sealing a hydrophobic pocket. In the Li structure (478), the pocket was filled with a polypeptide modeled on the NR boxes (LxxLL motif) of the SRC1 coactivator. Together with transcription activation experiments using mutant forms of SRC1, the structural study firmly established the pocket as the AF2 domain. The MR antagonist finerenone affects nuclear translocation and blocks MR protein-protein interaction with SRC1 (21). Hultman et al. (367) used a two-hybrid system in HEK293 cells to show polypeptides modeling NR boxes in SRC1, peroxisome proliferator-activated receptor γ coactivator 1 (PGC-1) and activating signal cointegrator 2 (ASC2) all displayed strong association with the MR-LBD. Similarly, a yeast two-hybrid screen detected another LxxLL protein, tesmin, bound to the MR LBD (675). Additional experiments demonstrated that tesmin coactivation was specific to mineralocorticoid stimulation of MR. This appears to be the first example of a ligand-discriminant coactivation of MR. Importantly, binding of these proteins was inhibited by the MR antagonist eplerenone.
The flexible “hinge” linking the MR-DBD and MR-LBD facilitates a global hormone-dependent conformation shift (for review see (927). This conformational change involves bringing the MR-NTD and MR-LBD into proximity resulting in direct intra-molecular protein-protein interactions (629, 674). The nature of the interaction is dependent on the specific ligand bound.
Posttranslational Modifications of MR
The stability, nuclear translocation and transactivation functions of steroid hormone receptors including MR are known to be regulated via posttranslational modification (for review see (211, 852)). Unfortunately, aside from phosphorylation, limited information exists on specific MR modifications. Examination of the primary sequence revealed consensus modification sites for phosphorylation, ubiquitination, sumoylation and acetylation (613). Although apparent phosphorylation of the LBD has been reported (347), the primary sites for physiologically significant modification of MR cluster within the NTD. Phosphorylation of MR has been observed to affect transcription both positively and negatively. For example, in human colon carcinoma cells co-transfected with MR and cyclin dependent kinase 5, MR became phosphorylated on ser128, ser250 and thr159 in the NTD (418). In this instance, phosphorylation suppressed aldosterone-dependent transcription. In contrast, several reports demonstrated that the Cyclic AMP-dependent protein kinase (Protein Kinase A; PKA), protein kinase D1 (PKD1) and extracellular signal-related kinase 1/2 (ERK1/2) all phosphorylated the MR NTD leading to increased transcription (294, 444, 480, 522, 536).
More recently, Shibata et al. reported that phosphorylation of ser843 in the ligand binding domain (LBD) reduces aldosterone binding and MR functional transcriptional activity (738). Using in vitro reporter assays to assess transcriptional activity, the substitution of glutamate for serine at S843 to mimic phosphorylation was interpreted to render MR incompetence for transcription. MR-S843E was localized to the cytoplasm in intercalated cells. Adaptation to a low salt diet reduced the phosphorylated MR abundance, as did the with no lysine kinase 4 (WNK4, discussed also in sections “PI3-Akt-mTOR2-OSR1”, “Aldosterone Regulation of the NaCl Cotransporter”, and “NCC-WNK1-WNK4 pathway, Cl−ic and Aldosterone Signaling”) and Ang II, the latter acting through a signaling mechanism that dephosphorylated MR-S843. The effect of Ang II was blocked by the Ang II blocker losartan.
In ICs, reduced phosphorylation of MR was associated with increased pendrin and H+-ATPase activity at the apical membrane. Both WNK4 and protein phosphatase 1 (PP1) appeared to be involved in the regulation of MR phosphorylation at S843 localized to ICs (738) and WNK 4 knockout mice exhibit significantly increased MR S843-P (115). Evidence suggests that c-Src regulates the interaction of WNK 4 and sgk1 by increasing PP1 binding WNK4 and modulating WNK4 activity (483). In contrast to the effects of a low Na+ diet, angiotensin II, and WNK4 to reduce MR S843-P values, a high K+ diet for 7 days resulted in an increase in MR S843-P. IC MR was predominantly cytoplasmic on a high K+ diet, but predominantly nuclear in the in NaCl cotransporter (NCC) knockout. In contrast, the localization of MR in CD PCs was nuclear during high K+ diet or in NCC knockout mice. The authors interpret these data to conclude that MR is inhibited in ICs during a high K+ diet but is in an active state under conditions of maximal Na+ conservation. Subsequently Jimenez-Canino et al. reported that phosphorylation of serine 843 resulted in significant reduction in ligand dependent nuclear translocation and inability to mediate ligand dependent gene trans-activation, with impaired interaction with the steroid receptor co-activator-1 (SRC-1) (398). Although MR-S843-P could dimerize it showed reduced nuclear translocation, which suggested impaired coupling between ligand binding and receptor activation.
Other post-translational modifications of MR have been reported. MR is modified by acetylation that has been reported to modify transcriptional activity in some but not all cases (398, 465, 516). Polyubiquitination of MR marks the receptor for turnover by the proteasome (933). Recently, Faresse et al. linked ERK1/2 phosphorylation to ubiquitination of MR. MR was found to be in a stable mono-ubiquitinated state in mCCDc11 cells (206). This mono-ubiquitin moiety was lost upon phosphorylation resulting in MR polyubiquitination and rapid proteasome dependent degradation. MR appears to be subject to this form of regulation through five putative sumoylation sites located within the NTD. Coimmunoprecipitation assays, GST pulldowns and yeast two-hybrid experiments consistently detected the SUMO-1 conjugating enzyme Ubc9 and other sumoylation pathway proteins in association with MR (799, 815, 816, 934). The reported change in function was enhanced transcription from a promoter construct containing several tandem MRE sequences in the absence of sumoylation (815). On the other hand, Ubc9 also appeared to act as a positive coactivator protein for MR in the presence or absence of receptor sumoylation (934). Like the results from phosphorylation studies, the observations should not necessarily be viewed as incompatible. Sumoylation of the MR interacting protein 11ßHSD-2 can also modify MR activity by modifying MR nuclear translocation (discussed below in the section “Specificity of Aldosterone as a Mineralocorticoid”) (398). Clearly, there is much that remains unknown about the action of posttranslational modifications on regulation MR function.
Renal distribution of MR
The distribution of MR in the kidney has been extensively examined using different techniques. The results are generally consistent and indicate that aldosterone’s principal action is in the distal nephron and CD. In the rabbit, rat, and Xenopus kidney, MR localization has been examined by Doucet and Katz (173) and Farman et al. using 3H-aldosterone (209, 210, 263). Initial studies demonstrated that binding affinity in the rabbit kidney was similar to that previously described for the high affinity or Type I receptor. Competition experiments with other corticosteroids were similar to that subsequently described by Arriza (28) at high (30 nM) but not low concentrations of labeled aldosterone. In microdissected rabbit proximal tubule (PT), TAL, DCT, CCD and medullary collecting duct (MCD) segments, aldosterone binding was greatest in the DCT and CCD. Specific localization was high and similar in the DCT and CCD and lesser degrees of binding in more proximal segments. The specific aldosterone binding in the DCT and CCD was approximately four times the binding of the TAL and >20 times the binding in the PT (209). Moreover, the specific labeling studies showed no concentration dependence in the PT, but similar concentration dependence in the cortical and medullary TAL as the high affinity sites present in the DCT, connecting tubule (CNT; DCT-granular), and CCD. The MCD also exhibited high capacity aldosterone binding equivalent to the DCT, CNT and CCD. These investigators characterized the temperature dependence and segment– specific aldosterone metabolites in the rabbit kidney, and high capacity specific binding in rat kidney segments were similar to that of the rabbit. Studies in isolated glomeruli revealed only a small amount of specific binding (less that one tenth of DCT/CCD segments) but only at high physiological concentrations of labeled aldosterone. These studies illustrate one of the potential reasons for discrepancies between studies with different techniques, namely the level of expression of MR in different segments.
Despite the low level of MR expression in isolated glomeruli increasing evidence supports the functional importance of MR in glomerular podocytes. Twenty patients with diabetic nephropathy receiving recommended renoprotective treatment experienced a 32% reduction in proteinuria with 25 mg daily spironolactone versus placebo in a double-masked randomized crossover trial for two months that was well tolerated. (712) Two recent reviews concluded that MR antagonism significantly reduced proteinuria and improved blood pressure control with a favorable risk profile in selected patients with renal disease and proteinuria, but more long-term studies are needed. (48, 151)
Additionally, the recent Fidelio-DKD trial showed a benefit of MR antagonism in patients with Type 2 diabetes mellitus. (44) Such findings support a role for MR in the pathogenesis of proteinuria, renal fibrosis, and progression of CKD. This topic is discussed in the section on clinical trials, but for a more thorough discussion the reader is directed to several recent reviews. (4, 48–51, 742)
Immunohistochemical studies using anti-MR antibodies reported immunoreactivity in the DCT, CNT and CCDs. Most studies reported no staining in the glomerulus or proximal tubules, in agreement with previous auto-radiographic studies. In most studies, immunoreactivity was present in the TAL and more distal segments. Some observed staining in medullary papillary CDs and in interstitial cells of the papilla and the epithelium lining the papilla. Differences between immunoreactivity pattern in the cytoplasm and nuclei were variable (86, 207, 443, 495, 671, 686, 700). Recent studies have proposed a role for MR in the action in the proximal tubule that was not detected in earlier studies. The level of expression of MR in various nephron segments may, in part, explain these discrepancies, as judged by the lower level of specific aldosterone binding in proximal tubules than in more distal segments noted earlier (690). More recently studies by Ackermann (2) examined the distribution of MR and GR in the rodent kidney. Under control conditions MR and GR was expressed in the TAL, DCT, CNT and CD cell nuclei. GR was also observed in nuclei and subapical compartments of proximal tubular cells. A high NaCl diet reduced GR expression in 11ßHSD2 positive cells, defined as the ASDN, without changes in nuclear MR signal. Adrenalectomy markedly reduced MR and GR nuclear signal in all epithelia, whereas aldosterone administration resulted in nuclear re-localization. In the TAL, interesting, aldosterone only regulated subcellular localization of MR in adrenalectomized animals, when glucocorticoids were nominally absent, which suggests that physiological variations of aldosterone in vivo are not sufficient to produce aldosterone-specific MR mediated transcriptional effects in this segment. During low-dose administration of corticosterone, nuclear localization of MR, but not GR was observed in ASDN cells expressing high levels of 11ßHSD2. However, cells with little 11ßHSD2 expression (proximal tubule, TAL, early DCT, and intercalated cells) had GR redistributed to the nuclei. With high dose corticosterone administration, nuclear GR localization occurred in cells with high expression of 11ßHSD2 in the ASDN. MR was also identified in intercalated cells, consistent with an effect of aldosterone on proton secretion. These studies support the concept of ligand-induced nuclear translocation of MR and GR as part of their regulation in a cell-type specific manner modulated by 11ßHSD2. The authors conclude from these data, however, that in the kidney variations in plasma aldosterone levels primarily affect the subcellular localization of GR, not MR, and that 11ß-HSD2 primarily protects GR from circulating corticosteroids. Whether previous work suggesting regulation of GR by MR is relevant to these studies requires further investigation. (343, 546) As noted below, further study of the role of 11ß-HSD2 and its regulation will likely provide important new information.
Specificity of aldosterone as a mineralocorticoid
Early studies of aldosterone binding to nuclear extracts identified both high affinity and low affinity receptors that bound aldosterone in concentrations similar to the concentrations in plasma for aldosterone and cortisol or corticosterone. Logically, the high affinity MR receptor was presumed to bind aldosterone and the low affinity should be the cognate receptor for GR. Although some previous evidence questioned this thesis, (442) this concept changed with the cloning and functional expression of MR. Interestingly, the affinity of corticosteroid binding to MR was not correlated with aldosterone selective binding to MR (28). Expression of MR in CVl and COS-1 monkey kidney cell lines yielded high levels peptide expression, with cortisol and aldosterone exhibiting nearly identical binding affinities (Figure 4). The proposal to explain this apparent paradox was local cellular metabolism of non-mineralocorticoids, such as cortisol and corticosterone, by the enzyme 11ß-HSD, subsequently denoted 11ß-HSD1. (189, 234) Further study identified a distinct isoform, 11ß-HSD2, that exhibited optimal activity in the presence of nicotinamide adenine dinucleotide (NAD) as the high-affinity cortisol- (and corticosteroid-) metabolizing enzyme, which was cloned from sheep and human kidney. (3, 15, 90, 687) This enzyme co-localizes to cells expressing MR in tissues that exhibit mineralocorticoid-specific effects on ion transport. Stewart and co-workers noted that glycyrrhizinic acid, a component of natural licorice, was a potent inhibitor of 11ß-HSD2 and ingestion of this compound could result in a phenotype that was characteristic of mineralocorticoid excess (775). A recent clinical-pathological conference illustrates the potentially severe phenotype of 11ß-HSD2 inhibition. (186) These observations support the concept that during inhibition of the enzyme 11ß-HSD2, cortisol and corticosterone acted as endogenous mineralocorticoids. These findings also provided an explanation for the genetic defect in 11ß-HSD2 in children with the syndrome of apparent mineralocorticoid excess originally reported by New et al. (569), and further characterized by Ulick et al. (834) Subsequent study showed that individuals with this syndrome exhibited loss of enzymatic function and suffered from a severe phenotype with systemic hypertension, hypokalemia, and metabolic alkalosis. Cloning of this enzyme allowed study of this concept in animals that had a disruption of 11ß-HSD2 with a recapitulation of the human phenotype (314, 351, 439). Further work has established that mutations that result in less severe loss of enzymatic function may present in adults. (460, 776, 892) Biller et al. examined the effect of intravenous carbenoxolone (6 mg/h) to inhibit renal 11ß-HSD2 activity in the rat on electrolyte transport by free-flow micropuncture of distal tubule segments. Interestingly, carbenoxolone reduced fractional sodium excretion, but sodium reabsorption in the accessible distal tubule was similar in control and carbenoxolone treated animals. However, distal tubular potassium secretion was increased by carbenoxolone. (72) Bailey et al. studied the effect of carbenoxolone on unidirectional 22Na+ absorptive flux in adrenalectomized rats infused with high-dose corticosterone and observed a significant increase in sodium absorptive flux, providing direct evidence supporting the role of 11ß-HSD2 to inhibit the action of corticosterone. Subsequent work has further validated the importance of 11ß-HSD2 in the activation of ENaC and NCC. (40, 41, 371) The distribution of this enzyme largely coincides with the site of aldosterone-responsive ion transport (Figure 5). Nevertheless, as noted by Bhargava & Pearce (69), such a mechanism would not provide differential gene regulation by MR relative to GR. As previously noted (See section “Modular structure and function), work from Zennaro et al. also suggests that in certain tissues, notably the heart, the specificity of steroid action deserves further study (963). Steroid-specific gene transcription involves multiple mechanisms, including differences in MR or GR interactions with other transcription factors, co-activators or inhibitors, and cis-acting or DNA elements, all in a context-specific manner (for further review see (42, 230, 534, 878)). In addition, Jimenez-Canino et al. have provided evidence that 11β-HSD2 sumoylation at lysine 266 modulates cortisol-mediated MR nuclear translocation. (400). Thus, whereas the role of 11ß-HSD2 as a major element in steroid specificity is well established, the discrimination of MR to evoke a mineralocorticoid-specific response deserves further study and will likely be rewarding. For further consideration of the role of 11β-HSD2, see Chapman, Holmes, and Seckl (120).
Figure 4. Steroid-binding properties of the human mineralocorticoid receptor (MR) expressed in cultured cells.
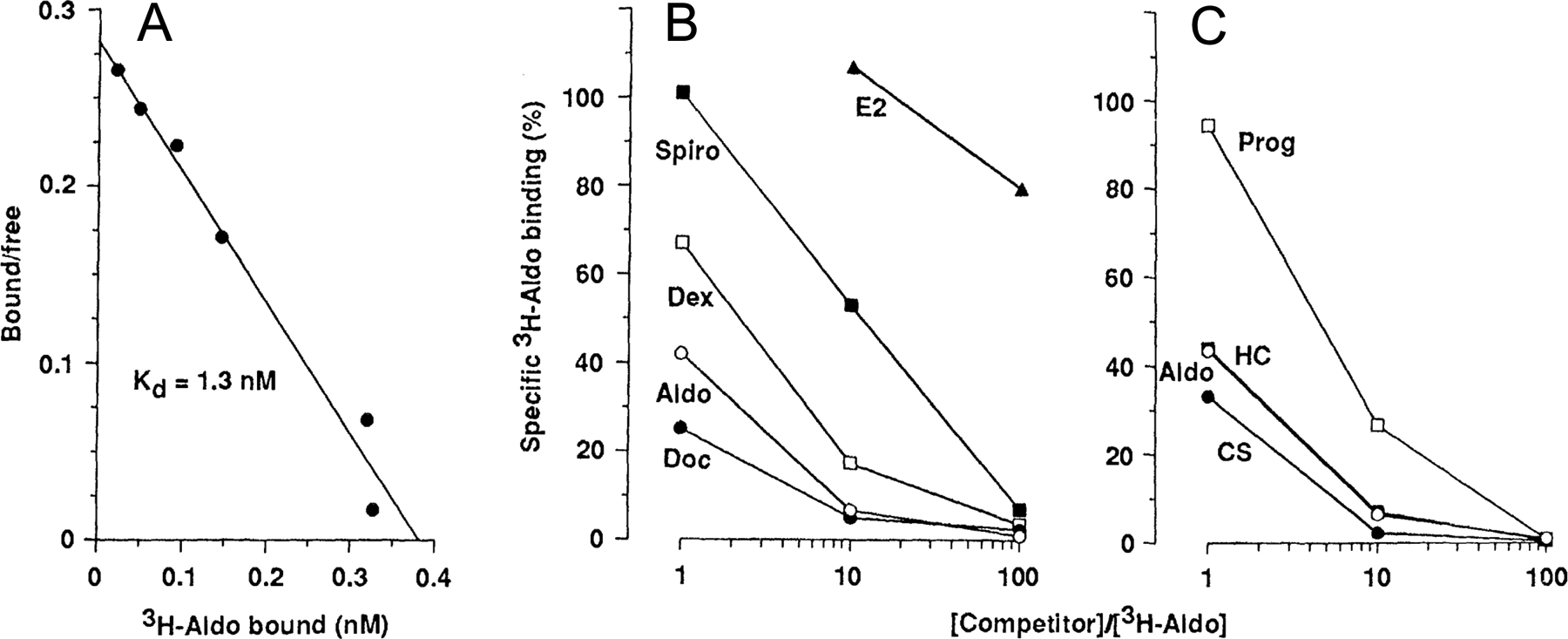
(A) Scatchard analysis of tritiated aldosterone binding in extracts prepared from pRShMR-transfected COS cells (B and C) Competition of unlabeled steroids for binding with 5 nM [3H] aldosterone in transfected COS cell extracts Abbreviations: Aldo, aldosterone, Doc, deoxycorticosterone; Dex, dexamethasone; Spiro, spironolactone; E2, 17β-estradiol; CS, corticosterone; HC, hydrocortisone; and Prog, progesterone. Figure adapted from Arriza et al (28).
Figure 5: Cellular Mechanisms of Na+ and K+ Transport in the Aldosterone-sensitive Distal Nephron (ASDN).
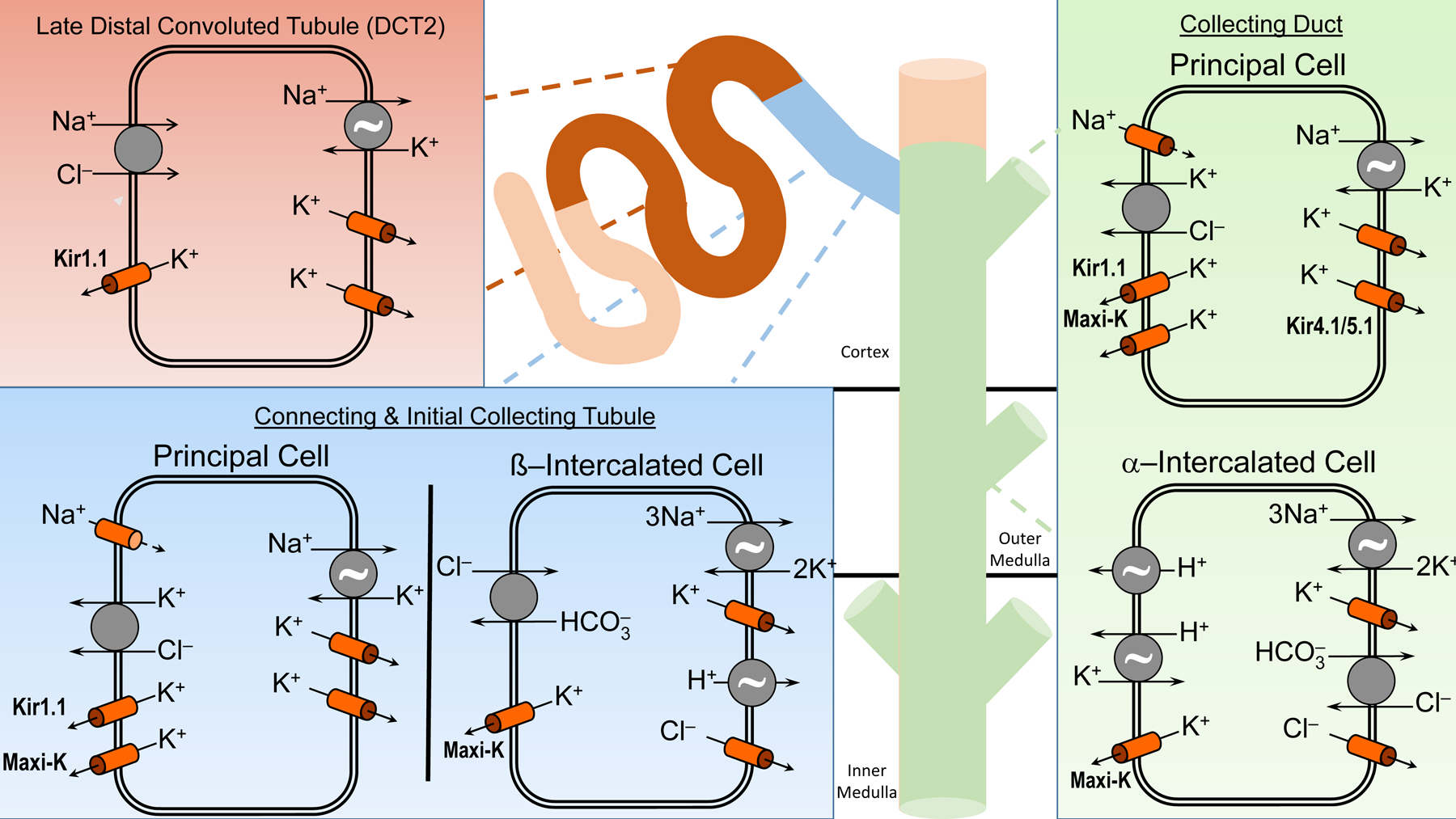
The ASDN, consisting of the late distal convoluted tubule (DCT2), the connecting (CNT) and initial collecting tubule (ICT), and the collecting duct (CD), express the mineralocorticoid receptor (MR) and the high affinity enzyme 11ß-hydroxysteroid dehydrogenase type 2 (11ß-HSD2) which oxidizes cortisol to cortisone and is important in conferring mineralocorticoid specificity to the ASDN. Na+ reabsorption occurs predominantly by the electroneutral NaCl cotransporter in the DCT2, with progressive increasing the proportion of electrogenic Na+ absorption occurring in the CNT, ICT and CD. K+ in the ASDN is secreted by two classes of K+ channels, inwardly-rectifying K+ channels (Kir1.1; also known as the renal outer medullary channel or ROMK) and large conductance Ca2+-activated K+ channels (also known as BK or Maxi-K channels). The apical KCl cotransporter in principal cells is involved in non-conductive K+ secretion. K+ absorption is an active process driven by an apical HKα1 H+K+-ATPase and basolateral K+ channels in intercalated cells and in the principal cells by HKα2 H+K+-ATPase (not shown). The ~ symbol indicates an ATPase.
Aldosterone Action and Signaling Pathways
The signaling pathways that mediate aldosterone’s actions are very diverse. For example, the binding of aldosterone to MR modulates transcription, either directly or indirectly, of genes encoding a number of cellular kinases including sgk1, phosphatidylinositol 3-kinase (PI3-kinase), Protein Kinase B (AKT), PKA, WNK1, the epidermal growth factor receptor (EGFR), and the Janus kinases (Jak kinases). However, aldosterone binding to MR (or other receptors that have not been fully delineated, see below) may also result in direct activation of signaling pathways, by non-genomic mechanisms. Here, we review aldosterone’s action on ion homeostasis and the aldosterone-dependent signaling pathways within the kidney, renal vasculature, and endothelial cells. As noted previously, aldosterone has additional inflammatory effects mediated by MR in multiple tissues, including the heart and the kidney, with evidence of MR expression in the podocyte. Recent clinical trials suggest beneficial effects of MR antagonists in patients with diabetic kidney disease as discuss in “Clinical Trials involving MR Blockade & Challenges”.
Aldosterone action on cellular physiology
Renal Na+ retention was the classical action of aldosterone that led to its original discovery and isolation (745). Nevertheless, it is now apparent that by its action through MR (and other receptors yet to be fully characterized), aldosterone has significant effects besides electrolyte transport, including effects on immune-mediated inflammation, circadian clock function, and other neuro-humoral and paracrine-autocrine signaling systems. Here, we review the classic action of aldosterone on Na+ and electrolyte transport.
Until the early 1990s three hypotheses prevailed regarding the mechanism of predominant action of aldosterone to promote Na+ absorption, without a general consensus which was the predominant mechanism of action, and each was supported by experimental data (519). The Permease Theory proposed that the major site of action of aldosterone was to increase Na+ permeability through the apical membrane. The Metabolic (or Energy) Theory asserted that aldosterone’s primary action was to increase metabolic adenosine triphosphate (ATP) production. The Pump Theory hypothesized that aldosterone increased transepithelial Na+ flux primarily because of an increase in the rate and capacity of the basolateral Na+ pump, or Na,K-ATPase.
However, it is now apparent that aldosterone acts on multiple cellular processes to increase Na+ reabsorption. Early events will lead to adaptive cellular changes which exert their effect on different time scales. Moreover, aldosterone’s action on ion transport may require one or more early acting signals that may not produce a change in transepithelial ion flux. Such considerations may help to reconcile the reported early non-genomic effects with more extensively studied genomic actions of aldosterone. Additionally, since the effect of aldosterone is known to depend on the specific target tissue studied, (780, 905) it is likely that the identification of immediate aldosterone responsive genes will also depend on the cell type examined. Although a significant number of early aldosterone-responsive genes are presently known, whether a core set of conserved genes invariant of cell type is consistently modulated by aldosterone, remains an issue to be addressed.
Time frame for aldosterone’s effect on Na+ transport: model systems
The initial or acute effects of aldosterone on Na+ transport can be broadly classified into three phases that are best characterized by changes in SCC in cultured cell preparations or toad or turtle urinary bladders (355). More prolonged studies in the mammalian kidney reveal a fourth phase with persistent mineralocorticoid stimulation (337, 581). The use of SCC instead of direct measurement of Na+ flux, has the advantage of convenience and resolution of the time of onset of aldosterone’s action, but should be validated by direct Na+ flux measurements for the specific experimental conditions. In the initial phase, no change in SCC is apparent. The earliest changes in SCC or Na+ flux typically occur 30 minutes to four hours after aldosterone treatment. In this second phase, Na+ absorption or SCC typically increase by two to six-fold with a corresponding increase in transepithelial conductance (795). The late phase is not evident for at least three hours. During this time, SCC will typically increase by two to more than four times the rate of the early stimulation with little change in conductance and such changes depend on gene transcription. Mineralocorticoid stimulation for several days decreases chloride (Cl−) conductance in the mammalian CD. Since Cl− conductance represents the majority of total transepithelial CD conductance, changes in Cl− conductance will exert major effects on total conductance (337). Aldosterone’s action to reduce the Cl− conductance, frequently attributed to the paracellular or shunt pathway in part explains the substantial increase in transepithelial voltage (VT) observed with chronic aldosterone stimulation that has been reported, largely in the rabbit CD.
Many of the signaling events exert their principal influence at specific time points either early or late in the signaling cascade. In particular, the late phase aldosterone’s action is blocked by actinomycin D and cycloheximide, supporting the requirement of transcription and de novo protein synthesis for this last phase. For example, in the toad bladder aldosterone (70 nM) induced new polyadenylated RNAs during the first 30–60 min after application, and by 90–240 min newly synthesized 18S and 28S cytoplasmic rRNA were induced. SCC increased modestly (1.7 X control) over 12 hrs and this action on SCC and new RNA synthesis were totally abolished by Actinomycin D. However, inhibition of rRNA but not mRNA synthesis with 3'deoxycytidine did not impair the mineralocorticoid response during the initial three hours (679). Thus, the specific experimental conditions may have a substantial effect on the latency period and the magnitude of the response.
The earliest studies of aldosterone’s effect on Na+ transport were conducted in the urinary bladder of the toad, and subsequently the turtle. These epithelia exhibit substantial morphological and physiological similarities to the CD of the mammalian kidney. Changes in SCC were not evident until 20 minutes to four hours after aldosterone administration. Crabbe’s original observation in the toad bladder, pertaining to the time required before an appreciable increase in Na+ transport occurs, is frequently cited (146). However, two additional observations from these studies are less well appreciated and deserve emphasis. First, the in vitro response to aldosterone differed substantially in bladders from animals maintained in distilled water from the response in toads maintained in saline. In fact, bladders in the latter case did not exhibit an absolute, but only a relative increase in SCC when compared to time controls (Figure 6). Second, the relative increase in Na+ current with respect to aldosterone concentration was much greater than the effect of this concentration of aldosterone in most vertebrates in vivo, and also greater than that necessary for aldosterone to saturate binding of MR in vitro (Figure 4)(28). Thus, time, temperature, cytosolic aldosterone concentration, the target tissue, and the experimental conditions all influence the duration of this delay and the magnitude of the response.
Figure 6: Stimulation by aldosterone of active sodium transport across the toad bladder in vitro.
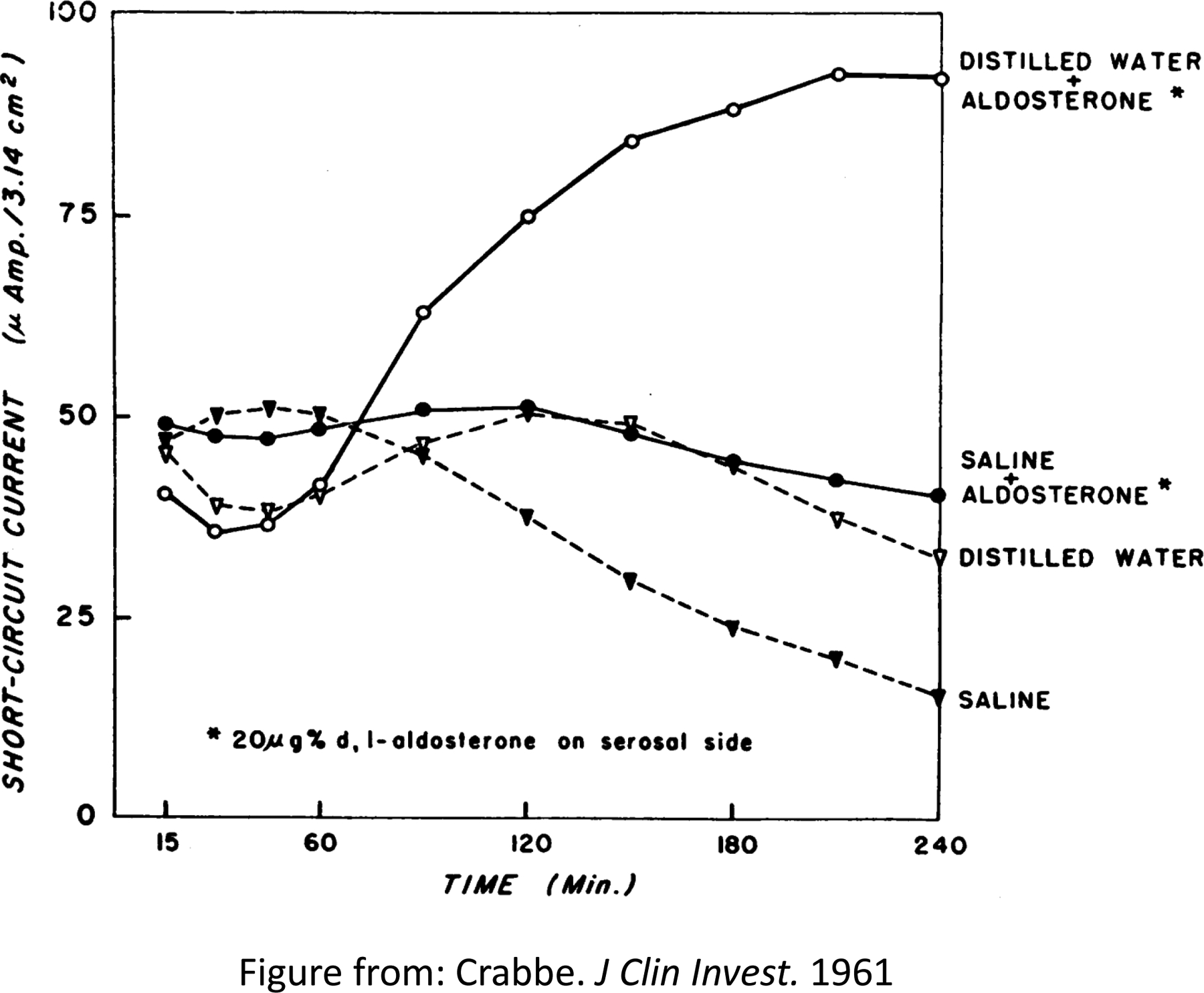
Experiments performed on paired membranes. The serosal surface of one bladder half was exposed to aldosterone and the corresponding half served as control. Eight toads had been maintained in distilled water, eight in saline, prior to these incubations. Figure adapted from Crabbé et al (146).
The question of which distal nephron segments were responsive to aldosterone was studied by Gross, Imai, and Kokko (292) in the rabbit CCD and DCT perfused in vitro after dietary conditioning, or in vivo after chronic MC treatment. They noted that on a normal Na+ diet, VT of the CCD was slightly lumen-positive, whereas in the DCT from rabbits on this same control diet, the VT was substantially lumen-negative. Chronic adaptation of rabbits to a low Na+ /high K+ diet and deoxycorticosterone (DOC), or DOC alone resulted in the development of a substantially lumen-negative VT in the CCD, consistent with the stimulation of electrogenic Na+ absorption. In contrast, the morphologically distinct DCT exhibited a similar lumen negative voltage that was not affected by dietary conditioning or by exogenous DOC.2 Addition of basolateral ouabain or luminal amiloride reduced VT toward zero in the CCD, further suggesting that lumen-negative voltage was due to electrogenic Na+ absorption. These authors suggested that aldosterone influenced electrogenic Na+ transport in the CCD but not the DCT. Studies by four other investigators, O’Neil & Helman (581) and Schwartz and Burg (724), systematically examined the effects of dietary adaptation and the chronic action of mineralocorticoids to stimulate the lumen-negative VT and Na+ absorption in the CCD and observed qualitatively similar findings. Collectively these three studies supported an effect of chronic mineralocorticoid stimulation to increase electrogenic Na+ absorption in the CCD. The effect of dietary adaptation was assumed, but not established, to occur by the action of aldosterone. Subsequently, Gross & Kokko (293) reported that 200 nM aldosterone in the perfusate or 1 μM in the bath resulted in an increase in the lumen-negative approximately 20–30 min after the addition of the hormone that had a latency period that was temperature-dependent. However, in the mammalian CD, similar to the studies of Crabbé in the toad urinary bladder, the in vivo condition of the animal substantially affected the results. When the action of aldosterone was studied in cortical CDs obtained from adrenalectomized rabbits, an effect of aldosterone was observed on Na+ absorption after a delay of ~90–120 min (Figure 7) (905), but without a significant change in VT or in K+ secretion. The above description, however, refers to studies largely done in the rabbit, and extrapolation to similar anatomic segments in other species including the rat and mouse, must be done with caution since there are distinct species differences, particularly the distal cortical nephron and CD
Figure 7: Effect of aldosterone on urine Na excretion.
Results of a representative clearance experiment. Urine sodium excretion (UNaV), urinary Na:K concentration ratio (Una/UK), and glomerular filtration rate (GFR) are graphed over time before and after intravenous aldosterone (arrow). Adapted from Wingo et al (905).
Frindt and Burg (223) also observed that VT in the CCD became more lumen negative when rabbits were conditioned to a high K+ and low Na+ diet. Such conditions would induce secondary hyperaldosteronism, and were consistent with the findings of O’Neil & Helman (581). These investigators noted that chronic mineralocorticoid stimulation decreases paracellular Cl− conductance, and hence low mineralocorticoid states (high Na+ diet or adrenalectomy) would exhibit a large paracellular Cl− conductance. Similar to the Crabbé observations, the in vivo conditions substantially affect the cellular (tissue) response to aldosterone (146). Thus, aldosterone exerts effects on paracellular conductance (20), and the effect of aldosterone in the CCD from adrenalectomized animals to increase Na+ absorption without a change in VT or increase in K+ secretion may reflect the large paracellular conductance of the adrenalectomized state. Recent studies are consistent with this interpretation (702). Such findings may reflect part of the spectrum of the action of aldosterone that is both time-dependent and tissue-specific as illustrated in Figure 8.
Figure 8: Effect of low versus high mineralocorticoid levels on paracellular chloride (Cl−) permeability and conductance.
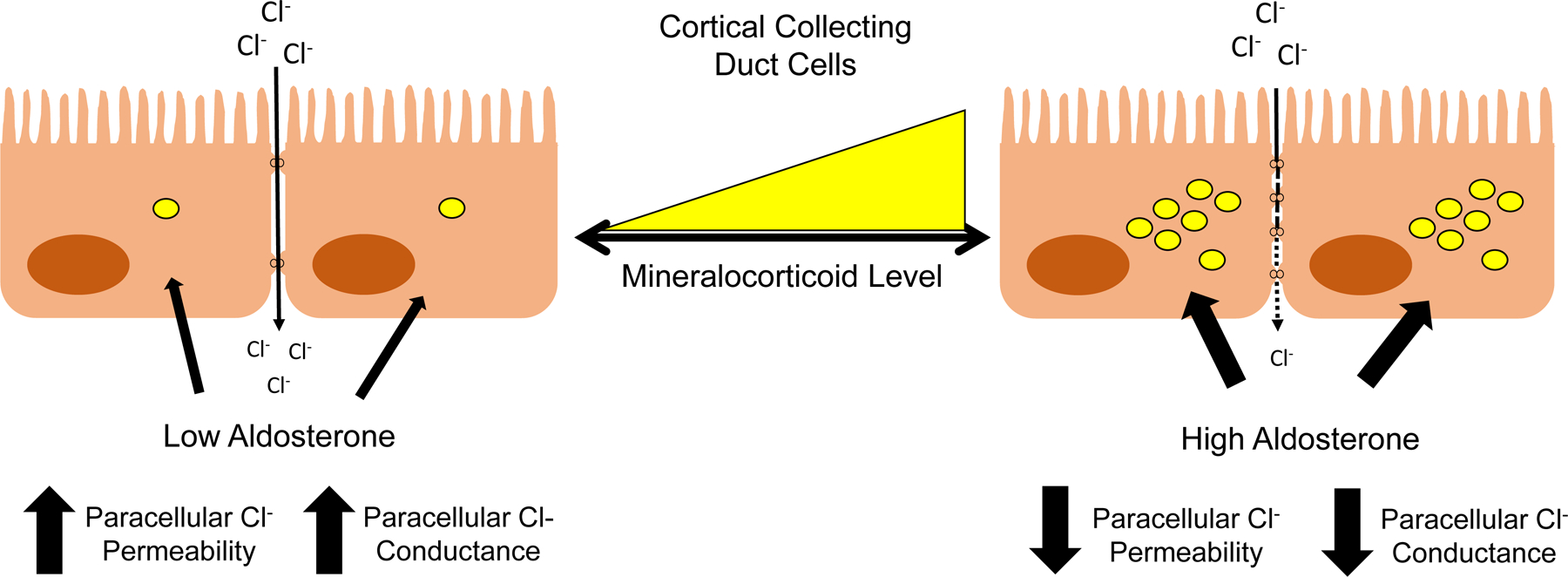
The low tissue conductance in response to high mineralocorticoid stimulation correlates with a high resistance. At low mineralocorticoid levels, tissue conductance increases to modest levels.
It is now apparent that aldosterone has primary effects to increase Na+-K+-ATPase pump number and activity that are independent of effects mediated by increased Na+ transport. Thus, the distinction between a primary permease, energy, or pump hypothesis is likely an oversimplification in view of plasticity of eukaryotic cells to compensate to stress or the environment. A time-dependent increase in pump activity after three hours by aldosterone was not observed in the presence of amiloride to block Na+ entry suggesting that the early action of aldosterone on pump activity depended on apical Na+ influx (625). Similarly, the rate of 35S-methionine incorporation was unchanged at three hours by aldosterone, a time when changes in Na+ transport were observed (146, 250), also suggested early events were not dictated by pump activity. However, after 18 hours aldosterone produced dose-dependent (0.2 and 20 nM aldosterone) increase in α and β subunit signal even in the presence of amiloride, suggesting both time and dose dependent direct effects on pump action. Elegant studies of the role of the receptor occupancy (of 0.8 nM to 100 nM aldosterone) further emphasized this time and dose dependence (250), which appeared to depend on occupancy of the type I (high affinity) receptor (Kd 0.3 nM Nmax 23 fmol/mg protein), suggesting that the Type I receptor was involved in both the early and late responses. Other studies support the role of both Type I (MR) and Type II (GR) receptors to produce the full action of aldosterone, with < 10 nM increasing Na+ absorption at three hours, and increased biosynthesis of Na+-K+-ATPase subunits after six hours, but new pump synthesis was not necessary for the early mineralocorticoid response. (250)
Interestingly, increased Na+ transport corresponded to nuclear occupation of the lower affinity (Type 2, GR) receptors, but distinct aldosterone effects on Na+-K+-ATPase synthesis were more closely related to occupation of type 1 receptors (250). Thus, aldosterone had late (~18 hours) effects directly on Na+-K+-ATPase expression, but this likely reflects direct actions on the pump and secondary actions due to increased Na+ entry and transport. Studies in other systems, such as the Xenopus amphibian epithelial kidney cell line (A6) also support a primary action of aldosterone on the pump and not as consequence of increased apical Na+ entry. Aldosterone produced a four-fold increase in the abundance of steady-state mRNA for NaK β1 and a twofold increase NaK α1 mRNA over a period of six hours. Nuclear run on assays confirmed that this increase was largely due to an increased rate of transcription after 15 minutes (850). Heterologous expression studies suggested that the action of aldosterone may be isoform specific. Xenopus A6 cells stably expressing either rat α1, α2 or α3 isoforms aldosterone produced an increase in pump current for the endogenous pump and for the α1 subunit but not for α2, and the very small currents with α3 isoform made the results inconclusive (626). In summary, the weight of the evidence would suggest that aldosterone exerts direct effects on the Na+ pump that are a relatively late event (18–24hrs.) and not observed at three hours, but that this augmentation in activity can be further increased by a load dependent effect of Na+ transport.
Role of nuclear receptor occupancy to transport action
Gaeggeler et al. further addressed the contribution of GR versus MR to sodium absorption in systematic studies of the of aldosterone-, corticosterone-, and dexamethasone-mediated sodium transport time- and dose-dependence of short circuit current (SCC, Isc) and the role of 11ß-HSD2 in a cultured CCD cell line (mCCDcl1). Equilibrium binding assays revealed that aldosterone and corticosterone both bound a high-affinity, low-capacity site, but dexamethasone bound a single site. By modelling the data, they concluded that MR occupancy principally controls sodium transport during circadian (physiological) changes in aldosterone concentration. However, both MR and GR occupancy affected sodium transport during salt restriction or acute stress. (237) The contribution of both MR and GR to sodium transport is also supported in studies by Bailey et al. in a mouse model of Cushing syndrome. (39) The relevance of GR and the activity of 11ß-HSD2 to sodium retention and salt-sensitive hypertension is also supported by the work of Craigie et al. (147)
More recently, Canonica et al. (108) have examined the contribution of MR and GR by cell-specific inhibition of the expression of these proteins in the kidney using an inducible Pax8-Cre in adult mice. Disruption of MR resulted in a failure to thrive phenotype and a severe pseudohypoaldosteronism type 1 (PHA-1) phenotype that became lethal when mice are fed a Na+-deficient diet. Expression of the NaCl cotransporter (NCC) (gene SLC12a3), its phosphorylated form (NCC-P), and αENaC expression were reduced with MR disruption, but GR expression increased. When fed a high Na diet these mice correct plasma electrolytes and body weight without correction of plasma aldosterone.
Interesting, disruption of GR (gene Nr3c1) in adult mice with Pax8-Cre did not impair growth, but reduced NCC activity with normal plasma aldosterone concentrations and produced only mild phenotypic changes (107). Although these studies suggest a predominant role for MR for the transport and phenotypic actions of aldosterone, the contribution of GR particularly as a MR-GR heterodimer and also as a function of 11ßHSD2 activity needs further study.
From permease to discovery of ENaC
Extensive work by many individuals culminated in the identification of the deduced primary structure of the subunits composing the highly selective amiloride-sensitive ENaC and led to a molecular connection to disease phenotypes (See below section). Because of the importance of these findings, this section summarizes experiments leading to this discovery, its molecular characterization, and structure-function studies vital to disease phenotypes.
Prior to the molecular cloning of the three subunits of the highly selective epithelial Na+ channel (known as ENaC or HSC), investigators had recognized three distinct types of Na+ channels in various Na+ transporting epithelia. Palmer (594, 595) classified these channels in three categories based on their biophysical properties (Na+ selectivity, single channel conductance, gating kinetics) and their pharmacological inhibition by amiloride:
Type 1 channels were highly Na+ selective (PNa/PK > 20), exhibited small single channel or unitary conductance (4–5 pS), with time constants for open and closed states (τ) > 1–2 sec, high amiloride sensitivity (Ki < 1μM) (594), and were expressed in the apical membrane of CCD and the distal colon.
Type 2 channels were moderately Na+ selective (PNa/PK ~ 3 – 4), with larger single channel conductances (~9–14 pS), rapid gating kinetics (τ ~10 to 100 ms), high amiloride sensitivity (Ki < 1μM) and were reported in sweat duct cells.
Type 3 channels were not selective for Na+ over K+ (a cation non-selective channel with PNa/PK~1), exhibited the largest single channel conductance (22–28 pS), rapid gating kinetics (τ ~100 ms), high amiloride sensitivity (Ki < 1μM), and were observed in OMCD and IMCD of the kidney, vascular endothelial cells and thyroid (594).
As discussed in the section “Aldosterone Action in Tight Epithelia and Target Genes,” distinction should be made between the highly selective, small conductance, amiloride sensitive, heterotrimeric epithelial Na+ channel denoted as ENaC (or HSC for highly selective channel), and other Na+-permeable cation channels that are also amiloride sensitive. The elegant work of Trac et al. (826) demonstrates that both the HSC (ENaC) and NSC are composed of αENaC subunits but have very different properties. Thus, tissue expression of the αENaC subunit should not be equated with the expression of the holo-channel complex αßγ-ENaC.
Initial characterization in the toad urinary bladder and the mammalian CCD suggested a similar electrophysiological signature of a putative Na+ channel that exhibited a small unitary conductance, was highly Na+ selective, and was amiloride-sensitive. The toad bladder exhibited a highly Na+ selective channel (selectivity of Na+ to K+ of ~ 1000:1) (596, 598) sensitive to intracellular Na+, Ca2+ and pH (597), and the channel density was modulated by aldosterone (605). Aldosterone increased the apical Na+ permeability (PNa) by 140% and basolateral Na+ pump current (INa) by 130% at five hours, and by 680% and 500%, respectively, at 18 hours, which suggests stimulatory effects on both apical Na+ entry and basolateral Na+ exit (607).
Patch clamp studies of the rat CCD apical membrane suggested a similar channel was present that had a small conductance (~5pS) and was highly Na+ selective with a conductance that saturated as a function of external Na+. The channel exhibited cation selectivity of Na+/K+ using inside-out patches of >= 10:1, had long (3–4 sec) open and closed states, and was sensitive to apical amiloride (0.5 μM) (603).
A similar electrophysiological signature was observed from the apical surface of cultured Xenopus A6 cells. The characteristics of the channel included a small unitary conductance of approximately 7–10 pS with a current-voltage relationship that exhibited little non-linearity from ± 80 mV and selectivity for Na+ to K+ of 3–4:1. Amiloride reduced the mean open time of the channel but not the single-channel conductance or IV relationship (317, 318). These investigators subsequently described two types of amiloride-sensitive channels in A6 cells. The first was similar to that reported previously (318), and the second had a single channel conductance of 1–3 pS with a nonlinear IV relationship with Na+/K+ selectivity of > 20:1. The two channels exhibited differences in the mean open and closed times with the smaller channel open only 10% of the time and the larger about 30% of the time (319).
The amphibian A6 cell line also provided insight into the mechanism of aldosterone signaling even though dexamethasone was the most potent, on a molar basis, agonist to stimulate SCC. Patch clamp analysis allowed measurement of the number (N), the open probability (Po), and the activity (NPo) of a small conductance (4pS) highly Na+-selective, amiloride-sensitive ion channel in response to aldosterone that could be compared with aldosterone’s effect on SCC. With removal of aldosterone, NPo and SCC decreased to a minimum by 48 hours with a pronounced reduction in mean open time (To) of ~ 40-fold. Upon re-addition of 1.5 μM aldosterone, NPo was significantly increased by two hours and SCC was near control values by 7.5 hours. During the first hour after aldosterone addition little change in channel activity was observed, but between 60 to 180 min channel activity dramatically increased so that by 180 minutes most recordings detected channel activity. Notably, the effect of re-addition of aldosterone appeared to depend on the duration of aldosterone removal with the most pronounced effect resulting in cell deprived of the hormone for the longest time. The results indicated that aldosterone increases SCC by increasing the Po, and hence the kinetics, of this channel (415). More recent work has supported the notion that increases in SCC are related to aldosterone-dependent increase in apical membrane ENaC expression via induction of sgk1. Frindt et al. performed quantitative analysis of the relative role for aldosterone-dependent increases in SCC via channel activation or kinetics versus the apical ENaC expression (226) and provided evidence for multiple complementary mechanism that are discussed in “Aldosterone Action and Signaling Pathways.”
Benzamil inhibition of Na+ transport in bovine kidney cortex membrane vesicles provided a biochemical approach. Benzamil inhibited Na+ transport with an IC50 of 4 nM, and labeled benzamil bound to a single high affinity species and bromo-benzamil was photo-incorporated into three groups of proteins with a relative molecular weight of 176,000, 77,000 and 47,000 Da (422). Using amiloride to isolate and purify proteins from bovine renal papilla and cultured A6 cells, an estimated molecular weight of 730,000 Da was identified and resolved into five principle polypeptide components of molecular weights 315,000, 149,000, 95,000, 71,000 and 55,000 Da (60). Other studies proposed one protein, GP 70 from toad urinary bladders, as either a component or modulator of ENaC (794). Asher et al. reported Na+ channels in membrane vesicles from cultured toad bladder cells with 50%-70% of labeled uptake inhibited by amiloride with a Ki of less than 0.1 μmol (31).
A molecular approach using Xenopus oocyte expression system examined mRNA from A6 cells. Fractionated mRNA from treated oocytes produced an amiloride-sensitive current under voltage clamp conditions after a period of one to three days whereas water injected oocytes had no detectable current. Size fractionation of the mRNA identified a species with a sedimentation coefficient of 17 S. Although treatment of A6 cells for 24 hours with 300 nM aldosterone resulted in a several-fold increase in Na+ current, no such increase in current was observed from mRNA isolated from A6 cells without treatment (600).
In 1993 two groups reported the identification of components of an amiloride-sensitive highly Na+-selective channel. Rossier and colleagues isolated a cDNA from a rat colonic library that contained a 2094 bp open reading frame encoding a deduced peptide of 698 amino acids (104). Northern blot analysis revealed expression of the corresponding mRNA from distal colon, rat kidney medulla and cortex, and stomach, in order of abundance. Injection of complementary RNA into oocytes resulted in an amiloride-sensitive Na+ current that was inhibited by amiloride with a Ki of 0.1 μM (104). Lingueglia et al. reported the identification of an amiloride-sensitive epithelial Na+ channel using expression cloning also from rat colon (487). Subsequently, both groups reported that ENaC was a heteromeric protein composed of three distinct but homologous subunits (106, 485), α, ß, and γ, expressed at the apical membrane in the aldosterone-sensitive nephron and collecting duct (106), and regulated differently by aldosterone (104, 486). A fourth subunit, δ, was described subsequently (862). The α subunit in humans is expressed as one of two transcripts (531, 855) and further work by Thomas et al. showed the presence of at least four transcripts, three of which do not change the proteins sequence. However, one transcript (ENaC 2) encoded an additional 59 amino acids to the αENaC 1 protein (810). Snyder et al. assessed the topology of the α subunit using in vitro transcription, translation, and translocation into microsomal membranes and concluded that the N- and C-termini were intracellular with two transmembrane domains and a large extracellular loop that was n-glycosylated. Based on the homology of the ß and γ to α, they suggested that these and related subunits likely exhibited the same topology (748). Canessa et al. examined the topology of the three subunits and proposed a minimal model as a heterotrimer (105). As noted in the upcoming section “ENaC Subunit Structure”, Stockand and co-workers subsequently provided evidence for a 1:1:1 stoichiometry of the ENaC subunits in two cell lines (769). More recently Baconguis and coworkers confirmed the heterotrimeric structure of ENaC using cryo-electron microscopy (580). Nevertheless, conclusions based on functional and biochemical studies have suggested four or more subunits. (215)
The cellular distribution of ENaC subunits at the level of mRNA and protein by in situ hybridization and by immunocytochemistry (ICC) in the rat kidney, colon, salivary, and sweat glands demonstrated their expression in aldosterone-responsive tissue (180). The DCT, CNT, CCD, and OMCD, demonstrated specific expression of α, ß, and γ ENaC. Most cells of CCD and OMCD were labeled at the apical membrane by IHC. In addition, the superficial rat colonic epithelial cells and in the secretory ducts of salivary and sweat glands were positive. Since all these tissues expressed an amiloride sensitive Na+ flux, the data further suggested that channels composed of these subunits as responsible for amiloride-sensitive Na+ absorption. The electrogenic Na+ absorption in the IMCD may reflect α, ß, and γ ENaC (856) or another mechanism (180). The species differences noted previously are also relevant for the distribution of α, ß, and γ ENaC subunits in the mouse kidney as clearly described by Loffing et al (493). In addition, dietary Na+ content affects the cellular distribution of ENaC, with low Na+ diets increasing the extent and apical staining for all three subunits (491, 492).
However, tissue specific regulation may also contribute to the diversity of aldosterone’s response in various target epithelia. Studies in the lung demonstrated that the steroid regulation of ENaC subunits differs between tissues. Whether this represents an alternatively spliced isoform, or other tissue specific regulatory differences from that in the kidney deserves additional consideration. Tchepichev et al. found that a pattern of expression in the lung epithelial cells which suggested that glucocorticoids, and not aldosterone were important in subunit expression in this tissue (802).
Moreover, Farman et al. observed expression of α and γ ENaC did not appear to be coordinated with ß ENaC (208). Abundant expression of α and γ was detected in populations of nasal, tracheal and lung alveolar cells by in situ hybridization, whereas equal levels of all three subunits were observed in bronchiolar epithelial cells and nasal gland ducts. Such observations indicate that caution should be exercised regarding the presence of a single HSC/ENaC subunit. This may have direct relevance to some of the more unusual observations on the induction and expression of “ENaC” in different aldosterone responsive epithelial cells (see section “Aldosterone Regulation of ENaC”) (208).
Aldosterone action in tight epithelia and target genes.
Studies for aldosterone-induced transcripts (AITs) and aldosterone-induced proteins (AIPs) in mammalian primary culture of immunodissected rabbit CNT and CCD cells have identified a number of genes that respond acutely after aldosterone administration. Bindels et al. examined the action of 100 nM aldosterone on transcellular Na+ flux, assessed as benzamil-sensitive SCC and correlated it with changes in protein expression of AIPs (73). An increase in current was observed after two hours with a plateau after six hours. Analysis of the expressed proteins that were increased by 15 hours by 2-dimensional PAGE led to the identification of three aldosterone-induced proteins of relative molecular weights of 100, 70–77, and 46–50 kDa in the membrane and microsomal fractions, with one heterogeneous protein observed in the soluble fraction of relative mass 77 Kda. In this initial phase, aldosterone significantly decreased transepithelial resistance.
The time of onset of aldosterone’s action on ENaC subunit mRNA and protein expression in primary cultures of rabbit CNT and CCD cells was consistent with previous reports by SCC or net transport. After a delay of ~two hours SCC started to increase, and by three hours aldosterone increased benzamil-sensitive SCC ~180%, and by 16 hours was ~2.5-fold greater than control (166). Semi-quantitative mRNA for each subunit did not increase at three hours but was increased two-fold by six hours. The level of subunit protein expression by immunoprecipitation of 35S labeled cultures revealed an increase in α and ß subunits at six and 16 hours but no increase in γ subunit at either time points (166). The regulation of Na+ flux raised the question of its feedback and was studied by stimulation of apical Na+ entry using electrophysiological means. This suppressed aldosterone-stimulated benzamil-sensitive SCC and the effect on SCC were reflected in ßENaC subunit expression, suggesting a negative feedback inhibition that coincided with decreased synthesis or increased degradation of ßENaC (167).
The identification of AITs was a logical line of investigation since MR, a ligand-gated transcription factor, mediates aldosterone’s genomic action. Thus, it might be expected that the key signals that induce aldosterone’s action would occur by MR induced gene transcription with a several fold change in the rate of transcription of these key genes. Spindler and co-workers took two approaches to identify aldosterone early response genes in a clonal amphibian kidney cell line, A6 cells (761). The first was a candidate gene approach, including trimeric G protein, energy metabolism genes, calmodulin, and Na+-K+-ATPase α and ß subunits. A modest induction in the Na+ pump subunits occurred without appreciable early or late changes in the other candidate genes. The second approach produced interesting results. Initial studies identified several potential candidates by differential display polymerase chain reaction (PCR). Of these candidates, five were studied in greater detail including one for an amino acid transporter, but most notable among these was the identification of the Xenopus homolog for the A splice variant of K-Ras2 (761). Given aldosterone’s action to increase Na+ absorption in sites of the distal nephron and collecting duct, and the strategy for the identification of ENaC subunits, it would be natural to assume that one or more of these subunits would be AIPs. Indeed, this was subsequently confirmed, but as noted in the previous section patch clamp studies provided strong support that aldosterone increased SCC by increasing the channel kinetics (415).
In 1993, the same year as the identification of the molecular composition of ENaC α, β, and γ subunits, the laboratory of Firestone cloned a novel serine/threonine protein kinase. The kinase was novel because it was transcriptionally induced by treatment with either serum or glucocorticoids (875). The mRNA for sgk was stimulated by serum and glucocorticoids within 30 min independent of de novo protein synthesis. The serum-stimulated mRNA induction was brief and followed by a rapid decrease in mRNA. The sgk isoform sgk1 was found to be expressed in several tissues including the thymus, ovary, lung, and kidney.
The electrophysiological studies suggested that the early action of aldosterone to increase SCC and Na+ reabsorption was due to stimulation of the activity or quantity, or both, of existing channels. Sgk was identified subsequently as an AIT by several groups that represented a major breakthrough in our understanding of the action of aldosterone (18, 123, 568, 739). Collectively, these studies provide persuasive evidence that aldosterone acting through MR rapidly increases sgk1 expression, at least in the kidney which in turn increases amiloride-sensitive current. As noted below, however, the mechanism for the increase in current does not appear to be due to channel phosphorylation. Further evidence did not support a change in channel open probability, but that the increased current was due to a larger number of active channels at the plasma membrane (18, 123, 568, 739).
Using a subtraction cDNA library generated after a one-hour exposure to 100 nM dexamethasone3 or vehicle from A6 amphibian cells, (123) Chen et al. identified sgk as an aldosterone-induced modulator of ENaC activity from several lines of evidence. The first was that dexamethasone rapidly increased the mRNA and protein for sgk by 30 min in A6 cells. Second, the mammalian orthologue (sgk1) was expressed abundantly in rat kidney, particularly in the inner medulla, and was rapidly induced by aldosterone by four hours in distal tubule and collecting duct cells as detected by in situ hybridization. In addition, co-expression of rat sgk1 with ENaC α, β, and γ subunits in a Xenopus oocytes expression system markedly increased amiloride-sensitive Na+ current. However, ROMK current (Kir1.1) was not stimulated by sgk1 (123).
A combined strategy of subtractive hybridization and differential display in primary cultures of rabbit CCD cells identified a ~350 bp cDNA fragment transcript was increased within 30 min of exposure to aldosterone. This led to the identification of rabbit sgk1 that was stimulated equally by 1μM RU28362, a glucocorticoid agonist, and 10 nM aldosterone that was most consistent with high affinity, MR mediated stimulation of sgk. When sgk1 was co-expressed in Xenopus oocytes for 36 hours with α, β, and γ ENaC subunits, amiloride-sensitive Na+ current was increased (568). Co-expression studies in Xenopus oocytes supported the notion that the increase in current was not due to direct phosphorylation of the channel. Nor was there evidence for the role of tyrosine residues necessary for endocytosis. Patch-clamp analysis showed no effect of sgk to increase Po or alter the kinetics of ENaC, although sgk expression resulted in a three-fold increase in the abundance of ENaC in the plasma membrane, suggesting that sgk mediates its effect on channel current by increasing the number of channels at the plasma membrane (18, 568). Other work confirmed that aldosterone (30 nM) produced a rapid ~five-fold increase in the steady-state sgk mRNA in rat kidney and colon, and the induction was blocked by 30 μM RU 26752, a specific MR antagonist (739). In contrast, aldosterone did not induce sgk perceptibly in the lung, whereas dexamethasone caused a small increase in sgk. The increase in mRNA occurred in both kidney cortex and medulla, and expression was greatest in the renal papilla. Co-expression of sgk and ENaC in Xenopus oocytes increased the amiloride-sensitive current fourfold after 48–96 hours, and this current was unaffected by the mutation (Y618A) that affected Nedd4-2 interaction (739). The mechanism for the increase in channel activity and the role of Nedd4-2 is discussed subsequently. Additional evidence links the action of aldosterone to stimulate αENaC via sgk1: Zhang et al. showed that aldosterone-induced Sgk1 relieves the transcriptional repression mediated by Dot1a-Af9 of αENaC as discussed in the section “Aldosterone-Induced Transcripts (AITs) & Aldosterone-Induced Proteins (AIPs) (967).”
Aldosterone acts in large part by MR, so it would be expected to increase steady-state mRNA by stimulating transcriptional mechanisms rather than by altering mRNA stability. Although each of the ENaC subunits may be rate limiting for assembly of the functional channel under different conditions or in different tissues, αENaC is generally rate limiting in the kidney (769). Aldosterone acted directly on the αENaC subunit to increase its rate of transcription (544). Aldosterone stimulated αENaC expression via either GR or MR in heterologous expression studies using a cell line that did not endogenously express either MR or GR, but there was a greater affinity of aldosterone to stimulate through MR than through GR. This stimulation appears to be due to enhanced transcription because aldosterone did not affect αENaC mRNA stability. This response was mediated by an imperfect HRE in the 5’-flanking region of the gene and this HRE was required to produce aldosterone responsiveness to the αENaC gene promoter (544). As noted subsequently, activation of MR by aldosterone appears to be modulated by small GTPase proteins whose action is further regulated by Guanosine nucleotide Dissociation Inhibitors (GDIs). Such observations are discussed in section “Principal Cell-Intercalated Cell MR interactions, MR phosphorylation & localization, and MR-Rac1 interactions.
It is instructive to consider the differences between the genes for the α subunit and other ENaC subunits (810). The human gene encoding the γ subunit of ENaC (813) has a similar genomic organization to the rat γ subunit (811). Interestingly, attempts to identify functional GRE motifs were unsuccessful although a minimal promoter was identified (811). Recent genomic analysis of MR binding did not find MR binding to the regulatory elements of the γ ENaC gene. The analysis of the functional channels from expression studies is further complicated by the presence of at least four potential interacting subunits that may pair with a yet to be fully defined stoichiometry (aside from the well-defined highly Na+-selective α:ß:γ-ENaC). In addition, one of these subunits, δENaC may not be functional in all species, and in particular the mouse, which adds to the complexity of analyzing the molecular characterization and functional properties of Na+ permeable channels in humans. The evidence that the δ subunit is likely a pseudogene in rodents has limited investigation of its function. Detailed reviews provide further consideration of this issue (326, 394, 395, 921).
The complexity of potential interacting partners, at least for αENaC, is further emphasized by the recent identification that certain non-selective cation channels are composed, in part, by αENaC subunits with the acid sensing ion channel 1a (ASIC 1a). (826) This channel has similar electrophysiological characteristics as the NSC described in the OMCD (919). Hence, inference of HSC/ENaC activity should not be based solely on the identification of alpha ENaC expression in tissue.
Aldosterone-induced transcripts (AITs) & aldosterone-induced proteins (AIPs)
Since the early work of Edelman and coworkers demonstrated that aldosterone increased Na+ absorption (as SCC),required new protein synthesis, and was blocked by actinomycin D (187) and cycloheximide, (453) studies focused on the identification of aldosterone induced protein (AIPs). With the advent and development of molecular biology, it became apparent that aldosterone had additional effects not only on RNA transcripts (AITs) that were not translated into proteins (non-coding RNAs) but also chromatin structure, as discussed in “Aldosterone Action on Chromatin Structure and non-coding RNAs.” In this section we discuss some of the well identified AIPs that are encoded by early response genes and their role in ion transport.
The identification of sgk1 as a reliable marker of aldosterone-mediated activation of MR prompted investigators to examine what other transcripts were rapidly induced by mineralocorticoid activation. Several groups have used either candidate gene or unbiased approaches, such as serial analysis of gene expression (SAGE) or microarrays with the discovery of novel transcripts (33, 306, 414, 567, 670, 851, 971). These have been complemented by analysis of aldosterone responsive genes in other tissues (458). In 2003, Gumz et al. examined what genes were acutely regulated in a mouse renal collecting duct cell line derived from the inner medulla. Using sgk1 as a positive control they determine that this cell line expressed αENaC, 11ß-HSD2, and MR. A limited collection of transcripts was either stimulated or repressed by aldosterone, including 14-3-3 isoforms and PI3K subunits, which subsequent studies corroborate. Four transcripts, Per1 (PER1, ET-1(EDN1), sgk1 (SGK1), and CTGF (CCN2), were examined by multiple assays and found to exhibit rapid stimulation in response to aldosterone. Of these four transcripts, three have been directly linked to ion transport or BP control whereas CTGF has implications for aldosterone’s role in inflammation.
Further studies indicate a role for Per1 in the regulation of renal ENaC (308). Treatment with aldosterone induced rhythmic expression of Bmal1 and Per2 in cultured cardiomyocytes (801), and aldosterone treatment induced expression of per1 mRNA, an effect blocked by spironolactone, and similar to its effect in the kidney (306, 308). The effect of aldosterone via the circadian clock appears in part related to an interaction of MR and Per1 with the αENaC promoter, an effect that is increased in renal collecting duct cells in response to aldosterone (667). Tissue-specific knockouts of clock genes and MR, along with an investigation of the interplay between these signaling pathways should shed significant light on how aldosterone and the molecular circadian clock interact to regulate physiological functions.
Aldosterone’s action on the edn1 gene to stimulate ET-1 expression may be relevant to aldosterone’s long-term effects. ET-1 acts by multiple signaling pathways through two G protein-coupled receptors which are encoded by separate genes (136, 620, 797, 874, 923, 924). Endothelin type A receptors (ETA) predominantly act through a Ca2+ mediated mechanism to induce vasoconstriction in vascular smooth muscle and stimulate aldosterone release from the adrenal zona glomerulosa. Endothelin type B receptors (ETB) are principally coupled to nitric oxide production by endothelial cells and cells of the renal collecting duct. This causes vasorelaxation of arteries and arterioles and decreases Na+ absorption within the collecting duct (431, 433). ETB predominate in the renal medulla, the site of the highest production of ET-1 in the body. The predominant endothelin isoform expressed in the kidney is ET-1 (431). For an in-depth discussion of the endothelin isoforms, see Davenport et al (157). The highest level of expression of ET-1 is in the renal medulla (431, 433). Renal medullary ET-1 is well known to modify renal Na+ excretion (91, 100, 375–379, 401, 402, 431, 433, 507, 565, 636, 642–645, 701, 715, 716, 760, 791, 843). Disruption of edn1 specifically in the collecting duct aquaporin-2 expressing cells results in salt-sensitive hypertension (8).
The identification of edn1 as an early aldosterone response gene is particularly interesting because ET-1 inhibits Na+ reabsorption in the CD (306). The mechanism for aldosterone regulation of the edn1 was systematically examined by Stow et al (782). Aldosterone acutely stimulated edn1 mRNA and ET-1 peptide and heterogeneous nuclear RNA synthesis confirmed that the mRNA stimulation occurred at the level of transcription. This action of aldosterone to induce edn1 mRNA occurred in cell lines from the cortex, outer medulla, and inner medulla of the collecting duct. These findings led to the proposal that ET-1 serves as a direct inhibitor of aldosterone action in the CD (306, 782). This hypothesis was directly examined by Lynch et al. who examined the effect of a high salt diet and mineralocorticoid stimulation in complete metabolic and electrolyte balance studies in normal mice and in mice with selective disruption of ET-1 expression in the CD (CD ET-1 KO) (506). In both groups, aldosterone stimulated Na+ absorption, however CD ET-1 KO exhibited continued Na+ retention throughout the study, whereas controls exhibited the typical response of aldosterone escape (Figure 9). Importantly, this persistent net positive Na+ balance occurred despite the greater BP in the CD ET-1 KO mice compared to WT mice. Thus, the loss of ET-1 expression in the CD produced Na+ retention in the presence of greater systemic BP indicating inappropriate renal Na+ regulation. As illustrated in Figure 10, such data support an important role of aldosterone/mineralocorticoid-induced ET-1 expression as an essential signal for normal regulation of Na+ balance.
Figure 9: The role for the renal aldosterone endothelin feedback system (RAEFS) on mineralocorticoid-stimulated Na+ retention.
Normal mice are shown in black lines and squares. Mice not expressing ET-1 in the collecting duct (CD ET-1 KO) are shown in dashed lines and open circles. Normal mice exhibit transient Na+ retention before escaping from progressive positive Na+ balance. CD ET-1 KO fail to undergo aldosterone escape, with persistent positive daily Na+ balance. Daily Na+ balance over days 1–19. Solid lines are controls, and dotted lines are CD ET-1 KO. Control, n = 9; CD ET-1 KO, n = 6. Data is shown as means + SE. In B, *P < 0.05 within control, day 1 vs. days 2, 3, 6, and 7, Tukey's test; †P < 0.05 within control, day 11 vs. days 9–12 and 14–19, Tukey's test; ‡P < 0.05, significant effect of genotype, repeated-measures ANOVA. Adapted from Lynch et al (506).
Figure 10: Diagram of a theoretical renal aldosterone-endothelin feedback system (RAEFS) in the collecting duct.
In response to a low Na+ intake, high aldosterone levels activate mineralocorticoid receptors (MR), but the production of endothelin-1 (ET-1) is attenuated by reduced distal luminal Na+ delivery and flow. Consequently, ET-1 mediated inhibition of ENaC in the collecting duct (CD) is reduced and the CD is poised to enhance Na+ absorption. When aldosterone is inappropriately high for the dietary Na+ content, high luminal Na and flow serve to enhance aldosterone mediated ET-1 activity which reduces Na absorption. Evidence for a sex-dependent natriuretic effect of ET-1 via endothelin A (ETA) receptors has been shown previously by Nakano et al (565). Dashed line indicates attenuation of pathway. UNaV, urinary sodium excretion; ETB, endothelin B receptors; NOS1, nitric oxide synthase 1; NO, nitric oxide.
In addition to sgk1, Per1, and edn1 as AITs and AIPs, four additional genes – gilz, the histone H3 lysine-79 methyltransferase disruptor of telomeric silencing splice variant “a” (dot1a), promyelocytic leukemia zinc finger protein (plzfp), and connector enhancer of kinase suppressor of Ras 3 (cnksr3 [CNKSR3 or CNK3]) are among the many genes that in various systems are stimulated or repressed by aldosterone and modulate Na+ transport. Several of the encoded proteins stimulate Na+ absorption by a similar theme. They do not stimulate Na+ absorption directly. They instead reduce the action of an inhibitory mechanism. In other words, they increase Na+ absorption by disinhibition. As noted above and described in section “Serum- and Glucocorticoid-induced Kinase,” Sgk1 acts similarly to inhibit the retrieval of ENaC from the plasma membrane by the action of Nedd4-2.
Aldosterone stimulated gilz expression rapidly in mpkCCDc14 cells (757). Gilz appears to be involved in cell signaling and may induce signaling through the ERK pathway. Expression of gilz in mpkCCDc14 cells reduces phospho-ERK expression with increased Na+ absorption (757). In addition, gilz has been reported to modulate renal Na+ and K+ excretion and NCC activity in the distal nephron (661). The work of Pearce and colleagues provides further insight into the mechanism of action of aldosterone and supports the role of gilz to stimulate Na absorption by aldosterone through disinhibition. Moreover, their evidence suggests that ENaC exists in part as an aldosterone-dependent dynamic regulatory complex. Soundararajan et al. provided evidence that the Raf-MAPK/ERK kinase and the ubiquitin ligase, Nedd4-2, are tonic inhibitory components of ENaC regulation that serve to decrease the expression of, cell surface ENaC. Their evidence indicates that gilz and the PI3K-dependent kinase sgk1, which are both stimulated by aldosterone, inhibit Raf-1- and Nedd4-2-dependent activities in a cooperative manner, supporting disinhibition as a mechanism for aldosterone stimulation of Na absorption.(753) They further suggest that cnksr3 (cnk3) participates in this dynamic assembly in response to aldosterone. (752, 755) Further work in this area should be rewarding.
A similar disinhibition appears to explain the action of dot1a. A novel aldosterone-signaling network involves the interaction of a histone methyltransferase and its interacting partner to affect αENaC expression and may have more general action (966). The histone H3 lysine-79 methyltransferase, mDot1a is a histone modification enzyme, which acts through hypermethylating histone H3 K79 associated with the αENaC promoter, as a negative regulator of αENaC transcription. Aldosterone downregulated mdot1a mRNA values in mIMCD-3 cells associated with histone H3 K79 hypomethylation and trans-activation of αENaC. mDot1a overexpression resulted in the converse with hypermethylation of histone H3 K79 at the promoter of αENaC and suppression of its mRNA (965). Dot1a interacts with the putative transcription factor (ALL1-fused gene from chromosome 9) (AF9) both in vitro and in vivo, and increased expression of AF9 produces hypermethylation of histone H3 Lys-79 of most of the endogenous αENaC promoter and reduction in mRNA expression. Knockdown of AF9 causes the opposite effect. Chromatin immunoprecipitation assays reveal that overexpressed endogenous AF9 and Dot1a are each associated with the αENaC promoter. Aldosterone inhibits both AF9 mRNA and protein expression and AF9 overexpression also inhibited other genes that were inducible by aldosterone (966). Interestingly and like Nedd 4–2 inhibition, sgk1 phosphorylates AF9, reducing its interaction with Dot1a, which leads to hypermethylation of the ENaC promoter (967). Dot1a appears to have significant interactions with other proteins, notably AF17 which competes for interaction with AF9 and facilitates Dot1a nuclear export (663, 664, 914, 968).
Plzfp was identified as an AIP from a CCD cell line stably expressing MR and acts to inhibit Na+ transport and reduces mRNA for β and γ ENaC subunits. Further study of its in vivo action on aldosterone-mediated Na+ transport remains to be determined (567). Finally, Ang II acts directly to stimulate ENaC activity in the collecting duct, so that activation of the RAAS has coordinate actions that may act synergistically on renal Na+ reabsorption (624).
Aldosterone cellular effects on ion transport
As noted above, a consistent effect of aldosterone is to stimulate Na+ reabsorption by relieving tonic inhibitory mechanisms that suppress Na+ reabsorption(67). In addition, aldosterone exerts effects via alteration of chromatin structure and the de novo expression of non-coding RNAs.
Aldosterone and control of ion transport by inhibitory mechanisms
In addition to ET-1, several systems act to reduce Na+ absorption in the CD by signals that are only partially elucidated and some of these are stimulated by aldosterone. For example, nitric oxide-, EGF- and purinergic- mechanisms all inhibit Na+ absorption in the CD, and the study of their contribution to aldosterone-stimulated Na+ absorption will likely be informative. Whether these systems have common stimulatory signals, and thus are functionally part of the same regulatory pathway or have distinct and independent activating mechanisms should be systematically investigated. For example, ET-1 signaling acts in part through nitric oxide to inhibit Na+ absorption, and the same stimulus, increased luminal flow, increases both ET-1 and nitric oxide production. Additional studies are needed to determine the relative role of ET-1 signaling via nitric oxide-dependent and independent mechanisms of feedback control (100, 375, 376, 643). The quantitative contribution of ET-1-dependent and nitric oxide dependent signaling to feedback control of aldosterone’s action may allow assessment of whether their effects can be distinguished and under what circumstances, given their similar actions in the setting of DOC-salt hypertension (643). Likewise, the stimulation of EGF by aldosterone and the effect of EGF to inhibit Na+ absorption suggest that other mechanisms exist to ensure a robust attenuation of aldosterone’s chronic effect and thereby limit Na+ absorption.
Aldosterone action on chromatin structure and non-coding RNAs
Aldosterone exerts early actions via the homodimer MR (or MR/GR heterodimer) mediated effects by binding to cis-acting regulatory elements within genes or intergenic sites, and transcription and translation of additional products acting in trans. Not all of these effects enhance Na+ reabsorption. For example, aldosterone increases the transcription of the endothelin-1 gene (edn1) and protein (ET-1) which acts to inhibit Na+ reabsorption and thus restraints the action of aldosterone. Another level of regulation was uncovered by Welch et al. who studied changes in the chromatin structure of the edn1 gene. This action may help to explain the later actions of aldosterone. The edn1 gene has a CpG island that encompasses the transcription start site and four sites in the 5' regulatory region that have been previously linked to transcriptional regulation (883). Using a quantitative PCR-based DNaseI hypersensitivity assay they studied edn1 structure in murine hepatocyte (AML12), renal cortical collecting duct (mpkCCDC14), outer medullary collecting duct1 (OMCD1), and inner medullary collecting duct-3 (IMCD-3) cell lines. The CpG island was uniformly accessible, suggesting a state of the gene which was competent for transcription. In all cell lines one NFAT element remained at low chromatin accessibility under all conditions tested. The second Ca2+ responsive NFAT element located at −1563bp upstream became markedly more accessible in IMCD-3 cells exposed to aldosterone.
Of the two established aldosterone hormone response elements, the HRE at −671bp relative to the transcription start site remained highly accessible, the other HRE (-551bp) became more accessible in response to aldosterone treatment in two target epithelial cell lines (IMCD-3 and OMCD1 cells). Thus, it appears that aldosterone activation of MR produces a MR-hormone complex binding at HRE at −671bp that opens chromatin structure in other regulatory elements of the edn1 gene. These studies provide not only evidence of additional regulatory mechanisms, but also have implications for the understanding of other ligand-gated nuclear transcription factors. Finally, recent work has emphasized that aldosterone may affect non-coding RNAs that serve to integrate hormonal responses in specific tissues (98, 461). Investigations into this area may provide more understanding of the global mechanism of action of aldosterone and is worthy of further study.
Aldosterone action via miRNAs
There is also evidence that aldosterone signaling is modulated by the action of a microRNA (miRNA)-mediated mechanism (98). Evidence supports a role for aldosterone regulation of a miRNA that affects ankyrin G expression and ENaC surface expression (97, 99). Further studies in this area will likely be rewarding. For example, microarray profiling of a mouse CCD cell line identified upregulated and down-regulated miRs by a single 24-h aldosterone stimulation that were similarly regulated in CCD cells isolated from kidneys of mice placed on a low sodium diet. The downregulated miRs, miR-335–3p, miR-290–5p and miR-1983, targeted ankyrin-3, a membrane regulatory protein. Aldosterone stimulated CCD cells exhibited increased ankrin-3 expression and when miR-335–3p, miR-290–5p and miR-1983 were inhibited, which mimicked the effect of aldosterone. Furthermore, ankrin-3 accelerated the delivery of ENaC to the apical surface of mCCD cells to increase membrane surface ENaC abundance and sodium reabsorption. (98)
Aldosterone treatment of mCCD cells also resulted in upregulation of a miR cluster that was predicted to target the 30UTR of intersectin-2, and this regulation was validated in vitro and in vivo following aldosterone stimulation. An increase in ENaC-mediated sodium transport occurred when intersectin-2 protein expression was reduced by RNAi. (98)
Microarray studies of Jacobs et al. examined the role of miRNA action on edn1 mRNA expression in renal distal collecting duct cells to provide a comprehensive assessment of miRNAs present in a mIMCD-3 cells and the effect of aldosterone (385). Argonaute immunoprecipitation experiments investigated the binding of the RNA-induced silencing complex (RISC) to edn1 mRNA, with 34 miRNAs identified as being expressed at very high abundance, and many others were present at lower levels. Quantitative PCR analysis confirmed the microarray experiments of selected miRNAs. These studies and in silico examination of the edn1 3' UTR provided a panel of candidate miRNAs that could affect edn1 expression. Co-immunoprecipitation of edn1 mRNA with an Argonaute protein antibody indicated involvement of RISC, and this interaction was inhibited by anti-miR-709 oligonucleotides, all suggesting that edn1 mRNA is a target of RISC. This area remains an active subject of research and as summarized in a recent review aldosterone regulates multiple miRNAs that serve to modulate effector proteins of the RAAS (99).
Aldosterone and Purinergic Signaling
Purinergic regulation of Na+ balance has not been specifically linked to aldosterone signaling although evidence support the importance of both systems in Na+ excretion and their connection (92, 148, 468, 551, 552, 777, 824, 896). However, it is quite likely that the magnitude of purinergic inhibition is also modulated by mineralocorticoid activity. Indeed, there is increasing evidence for crosstalk between aldosterone signaling and purinergic regulation of ENaC. (668, 777, 838) For example, H+,K+-ATPase activity in the collecting duct, which is modulated by mineralocorticoids (281) has been implicated in the regulation of purinergic signaling of ENaC (550). Further evidence supporting this point is discussed in the section “Aldosterone and K+ Homeostasis”. An in-depth discussion is beyond the present scope and the interested reader is referred to recent comprehensive reviews (148, 469, 839).
There is also evidence that aldosterone signaling may be modulated by the action of a microRNA (miRNA)-mediated mechanism. Evidence supports a role for aldosterone regulation of a miRNA that affects ankyrin G expression and ENaC surface expression (97–99). Further studies in this area will likely be rewarding. For example, microarray studies of Jacobs et al. examined the role of miRNA action on edn1 mRNA expression in renal distal collecting duct cells to provide a comprehensive assessment of miRNAs present in a mIMCD-3 cells and the effect of aldosterone (385). Argonaute immunoprecipitation experiments investigated the binding of the RISC to edn1 mRNA, with 34 miRNAs identified as being expressed at very high abundance, and many others were present at lower levels. Quantitative PCR analysis confirmed the microarray experiments of selected miRNAs. These studies and in silico examination of the edn1 3’ UTR provided a panel of candidate miRNAs that could affect edn1 expression. Co-immunoprecipitation of edn1 mRNA with an Argonaute protein antibody indicated involvement of RISC, and this interaction was inhibited by anti-miR-709 oligonucleotides, all suggesting that edn1 mRNA is a target of RISC. This area remains an active subject of research and as summarized in a recent review aldosterone regulates multiple miRNAs that serve to modulate effector proteins of the RAAS (99).
Mechanism of action on Na+ transport
ENaC subunit structure
The critical role of the kidney in BP regulation has been well demonstrated by the work of Arthur Guyton and colleagues (310, 311). The discovery of the molecular structure of the subunits of αβγENaC, that are expressed in the aldosterone-sensitive distal nephron (ASDN) and CD4 also provided information on the genes that encode these proteins. Genetic analysis demonstrated complete linkage of genes encoding specific ENaC subunits with severe hypertensive or hypotensive phenotypes. This in turn provided a mechanism to connect the physiological and pathophysiological function of aldosterone with the molecular events that underlie its action.
The role of ENaC (and NCC) is central to the understanding of the action of aldosterone in the ASDN & CD. Only a brief discussion of the structural-functional relationships of this protein is provided. For more in-depth analysis the reader is referred to several further reviews (85, 325, 420, 835). Early reports suggested a stoichiometry of two α subunits with one ß and one γ. More recently Staruschenko et al. combined fluorescence intensity ratio analysis and total internal reflection fluorescence microscopy to examine the stoichiometry of the ENaC subunits in Chinese Hamster Ovary (CHO) and COS-7 cells (769). The former provided an estimate of relative subunit stoichiometry, whereas the latter allows isolation of plasma membrane fluorescence signals. The conclusions of these studies were that ENaC expressed at the plasma membrane had equal numbers of each type of α, ß, and γ, subunits in fixed stoichiometry under steady state conditions. These studies are consistent with the crystal structure of the closely related ASIC of the chicken ASIC1 deletion mutant that was determined at a resolution of 1.9 Å (391). This crystal structure shows three subunits forming a homotrimer with two transmembrane helices in each subunit. Recently Baconguis and co-workers confirmed trimeric structure of ENaC using cryo-electron microscopy (580). These studies show that ENaC assembles as an equal number of α:ß:γ: subunits in a 1:1:1 stoichiometry arranged in a counter-clockwise configuration.
Although α, ß, and γ ENaC subunits exhibit much in common, notable differences in the primary structure of each subunit exists. For example, both α and γ subunits have tracts of embedded amino acids that are inhibitory (420). These inhibitory tracts are cleaved by proteases as discussed later. (See section “Post-translational Processing of ENaC: Proteolytic Cleavage and Paracrine Signaling Mechanisms”). Both the ß and γ subunits, but not αENaC, have discrete phosphatidylinositol binding motifs beneath the inner surface of the plasma membrane that modulate PI(3,4,5)P2-dependent ENaC activity (641), and aldosterone stimulates ENaC activity via actions of PI(3,4,5)P2 on γENaC (338, 807, 808, 846, 918). Sequences in the N- and C-terminus of αENaC contain motifs important for channel function, with a proline-rich sequence in the C-terminus that binds to an SH3 domain of α-spectrin and mediates the localization of ENaC to the apical membrane. (681) Structural motifs in the N-terminus of ENaC α subunit have been implicated in gating of the channel and polymorphisms in the C-terminus have been linked to a hypertensive phenotype (299, 300). Thus, phosphatidylinositols act to increase ENaC activity by increases in Po. Other differences between subunits include palmitoylation of specific cytoplasmic cysteines on the ß and γ subunits, but not alpha, that affect channel Po (346, 640, 956).
Additional studies, reviewed in (918), provide evidence for the regulation of ENaC by cytokines and chemokines, and other studies report that ENaC activity is differentially affected by changes in intracellular [Ca2+] in discrete apical and basolateral cytoplasmic domains that are dependent on mitochondrial Ca2+ sequestration (808). There is also evidence in Xenopus A6 cells that a basolateral P2X4 channel stimulates ENaC activity (807), and phosphatidylinositol 3,4,5-trisphosphate mediation of aldosterone stimulation of ENaC via its interaction with γENaC (338). The complexity of the regulation of this family of interacting subunits is further underscored by the presence of alternative splice variants of human ENaC subunits (139, 326, 810).
Several recent reviews provide detailed information about the structure-function relationships of ENaC, its interaction with intracellular regulatory proteins, and its regulation by extracellular and intracellular factors (420, 619). Much of the analysis of structure-function relationships have focused on α, ß, and γ ENaC subunits (with the assumption that the assembled αβγ-ENaC holo-channel is the only, or the predominant, conductive pathway for Na+ absorption in the ASDN). However, Waldmann et al. reported the cloning and functional expression of a novel amiloride-sensitive Na+ channel subunit termed δ from a human kidney cDNA library that was abundantly expressed in brain, pancreas, testis, lung, colon, and ovary. When functionally expressed alone in Xenopus oocytes it produced a small amiloride sensitive current with distinct properties from expression of the α subunit alone. The channel was more conductive to Na+ than Li+ (in contrast to αβγ-ENaC), and co-expression with ß and γ subunits resulted in a 50-fold increase in current. In contrast to αβγ-ENaC channels, proteases do not activate δβγ-ENaC channels, nor did these channels exhibited Na self-inhibition. Moreover, single channel conductance, ion selectivity and amiloride sensitivity also distinguish δβγ-ENaC channels from αβγ-ENaC channels (325, 326, 395, 862, 920, 922). Thus, the potential for significant heterogeneity of the response to amiloride, or its analogues, and the functional significance of these distinct heteromeric channels requires further study.
Syndromes of ENaC dysregulation: Liddle’s Syndrome
In 1963, Liddle and coworkers reported a familial renal disorder resulting in severe hypertension at an early age (second decade of life) that was associated with the features of hyperaldosteronism but with negligible aldosterone secretion. Liddle proposed that the syndrome was due to the synthesis of an unknown mineralocorticoid. The pathophysiology of this condition, however, awaited the identification of the primary structure of the three subunits to the highly Na-selective epithelial Na+ channel (HSC or αβγ-ENaC), and studies of genetic linkage analysis of Liddle’s original and other kindreds to mutations in genes encoding specific ENaC subunits. These findings and studies of the properties of the normal and mutated proteins in vivo and in vitro, collectively implicated mutations of the PY motif in the ß or γ subunit as explanations for this hereditary cause of hypertension (Liddle’s Syndrome) (180, 322, 323, 413, 710, 711, 800).
Seminal studies by Shimkets et al. reported complete linkage of the gene encoding the ß subunit of ENaC to individuals with early-onset severe hypertension in Liddle’s original pedigree (741). Further analysis of four additional families with characteristic features of Liddle’s syndrome showed either premature termination or frame shift mutations in the C terminus of the β subunit, providing convincing evidence of a direct linkage between the β subunit and this form of hypokalemic, early-onset severe hypertension. The finding of another pedigree of Liddle’s Syndrome with a truncation mutation of the last 76 amino acids of the C terminus in the γ subunit, and expression studies of human β and γ mutations in Xenopus oocytes suggested a critical PY motif present in β and γ subunits as a plausible explanation for the increased channel activity. A series of studies provided further evidence that altered cell surface expression in mutations that produced the characteristics of Liddle’s Syndrome were responsible for this phenotype. Schild et al. noted that expression of the Liddle ENaC mutation in Xenopus resulted in an increase in channel activity, (710) and subsequently identified a PY motif in ENaC that was mutated or deleted in patients with Liddle’s Syndrome (711). These mutations reduced the ability to downregulate the channel (413, 711). Thus, shortly after the identification of the core molecular components of ENaC, a direct connection between mutations in ENaC subunits and functional disease states established a crucial role for this channel subunit in rare disease states of hypertension. The plausible explanation was an unrestrained over activity of ENaC lead to inappropriate Na reabsorption, simulating the renal phenotype of hyperaldosteronism. Such a connection was further supported by loss of function of ENaC and states of hypotension as described below.
The elucidation of Liddle’s Syndrome also demonstrated the importance of the ubiquitin E3 ligase Neural precursor cell expressed developmentally down-regulated4-2 (Nedd4-2), and related ligases, in the regulation of ENaC cell surface expression by controlling channel retrieval from the plasma membrane. Studies by Staub et al. (773) showed that the tryptophan motifs (WW) in Nedd4-2 bound the PY motifs in the cytoplasmic tail of β and γ ENaC subunits. Studies by Dinudom et al. (168) of the control of ENaC in the salivary duct, provided a further understanding of the kinetics of ENaC ubiquitination. These investigators showed that increases in intracellular Na+, as with increase ENaC activity, results in ligase binding and ubiquitination of ENaC with down-regulation of ENaC activity, thus forming an intracellular feedback loop. As noted in the section “Aldosterone Action and Signaling Pathways,” additional levels of regulation of Nedd4-2 – ENaC interaction exist mediated by Sgk1 in response to aldosterone stimulated increases in Sgk1 and Na+ absorption (See (80) for a detailed consideration of the role of Nedd4-2 and related ligases in the regulation of ion channels and membrane transporters.). Further consideration of Nedd4-2 role in salt-dependent hypertension and chronic kidney disease (CKD) is discussed in the section “Role of Aldosterone in Salt-sensitive Hypertension (342, 677).” Also, as noted previously (Genomic action of aldosterone and mineralocorticoids), aldosterone not only modulates the rate of retrieval of channels from the plasma membrane by Sgk1-Nedd4-2 mechanisms, but also the rate of insertion into the plasma membrane by the action of Usp2-45, a ubiquitin-specific protease (203).
Quantification of the number of epitope-tagged ENaC subunits expressed at the cell surface of Xenopus oocytes compared with the whole cell amiloride-sensitive current for WT and channels expressing the Liddle mutation showed that channels composed of α, β, and γ ENaC subunits were expressed at the plasma membrane with greatest efficiency. Moreover, the ENaC open probability for the ensemble of channels expressed at the cell surface was about one-tenth that reported in previous single-channel recordings. The Liddle’s Syndrome mutant (ß R564stop) increased channel activity by increasing channel cell surface expression and by altering channel open probability (215).
Syndromes of ENaC dysregulation: Pseudohypoaldosteronism (Type I)
In 1956, Cheek and Perry described the case of an infant that exhibited a renal salt wasting phenotype refractory to mineralocorticoid therapy but corrected by large quantities of oral NaCl. Since then, other reports have documented cases in which plasma aldosterone was markedly elevated in the presence of renal salt wasting, hypotension, and other evidence of intravascular volume depletion. In 1996, Chang et al. reported in five kindreds that had loss of function mutations in either the α or ß ENaC subunits resulting in either frameshift, premature termination, or missense mutations as responsible for pseudohypoaldosteronism type 1 (119). Hummler et al. and Grunder et al. provided additional evidence that mutations in ENaC subunits could result in pseudohypoaldosteronism by analysis of structure-function relationships of these subunits (298, 368). Subsequently, Bonny et al. reported two cases from the same family of autosomal recessive pseudohypoaldosteronism type 1 that had an R492stop mutation of the α subunit that resulted in markedly reduced ENaC current as measured in a heterologous expression in Xenopus laevis oocytes (84).
Additional evidence supports the assertion that a hypomorphic phenotype similar to PHA type1 can be observed in mice that do not express various ENaC subunits in selected nephron segments. Genetically modified mice harboring loss of function mutations have confirmed a severe PHA type I phenotype in some studies. However, selective knockout of subunit expression in different nephron segments have revealed a range of phenotypes from severe to mild defects in Na+ conservation. The reasons for these differences are not apparent. For example, Rubera et al. (684), found that cell-specific knockout of αENaC in just the CD without deleting its expression in the late DCT and CNT (using a Hoxb7 promoter to express Cre recombinase) resulted in a mild phenotype with little difference in the response of lox/lox/Cre- and lox/lox/Cre+ mice to a low Na+, high K+ (2.6%), or water deprivation. In contrast, using a Pax8-inducible model of αENaC KO, Perrier et al. observed a severe Na-wasting phenotype (622). Canonica et al. and Terker et al. noted a severe phenotype with an adult nephron-specific deletion of MR (108, 805). Whether the completeness of the CD KO of αENaC, the time of the gene deletion (in development versus as adult), compensatory mechanism, or other factors account for these apparent discrepancies will require further study. Studies with another conditional KO model are likely to be instructive.
Aldosterone regulation of ENaC
The regulation of ENaC occurs at multiple levels including, transcription and post-translational of ENaC subunits, assembly of subunits, processing to a functional channel, insertion into and retrieval from the plasma membrane, and post-translational modifications that affect channel gating and activity. In addition, the holo-enzyme complex is also the target of various proteases that alter its activity and kinetics (364, 419, 421, 556, 614, 733, 754).
Several groups examined the contribution of aldosterone to the regulation of the three subunits that comprised the functional channel, either by direct administration of aldosterone in vivo, or by indirect stimulation of aldosterone via activation of the RAAS by a low Na+ diet. This latter maneuver would increase plasma aldosterone, by stimulating plasma renin and Ang II levels, and likely affected other hormones. These studies were made prior to the discovery of the notable effect of aldosterone on sgk1, GILZ, Per1, or ET-1, which is described subsequently under the section “Aldosterone Signaling in Renal Epithelial Cells.”
Renard et al. examined the expression of α ß and γ ENaC mRNA and protein in lung, colon and kidney and the effect of dexamethasone and a low Na+ diet by Northern blot and immunohistochemistry (IHC) (665). They found low levels of immunohistochemical expression in the lung alveoli but staining of most cells of the trachea and bronchioles. In the colon, dexamethasone and a low Na+ diet stimulated the level of ß and γ mRNA, whereas α was observed to be constitutively expressed. In the lung, adrenalectomy markedly reduced immunohistochemical staining for all three subunits, but dexamethasone or a low Na+ diet did not increase staining intensity. Intriguingly, in the kidney they observed no significant effect of either dexamethasone or a low Na+ diet on α, β, or γ subunit mRNA expression and speculated that if regulated in kidney it would require different regulatory mechanisms than in lung or colon (665). Asher et al. (33) examined the increase in ENaC subunits in response to aldosterone and found that in the kidney cortex both aldosterone and dexamethasone produced a 2.5–2.7-fold increase in α with no change in ß or γ subunits. In contrast, both aldosterone and dexamethasone produced a marked (>7–10 fold) induction of ß and γ in the distal colon with no change in the level of α. Interestingly, dexamethasone appeared equally effective as aldosterone to regulate these subunits. The aldosterone induced changes generally were after 3 hours to 24 hours on a low Na+ diet. Similar disparate effect of aldosterone in kidney and colon on other proteins have been observed by Jaisser et al. (386) who reported an induction of (HKa2) subunit of H+K+-ATPase in colon but no increase in the renal medulla. In addition CHIF, an epithelial protein that induces K+ channel activity, was observed to be induced in the colon but not the kidney by mineralocorticoids (861). The reasons for the marked tissue-specific induction by mineralocorticoids and glucocorticoids of specific subunits remains an open question (33).
Using isoform specific antibodies, Masilamani and coworkers examined role of aldosterone stimulation for 10 days to increase each of the subunits of ENaC in the rat kidney. Increasing plasma aldosterone by dietary NaCl restriction, and secondary activation of the RAAS, resulted in a ~four-fold increase in plasma aldosterone concentration (0.75±0.23 nM to 3.5±1.3 nM), and an increase in the steady-state expression of αENaC by 285±28% relative to control values. However, there was no change in the relative abundance of either the ß or γ ENaC, but there was a qualitative change in the molecular weight of γENaC due to proteolytic processing. In separate studies, plasma aldosterone infusion at a rate of 200 μg/day resulted in a ~ 50-fold increase in plasma aldosterone concentration (from 1.8 ± 0.5 nM to 80.4 ± 3.3 nM) after 10 days and αENaC protein abundance was increased approximately fivefold (472 ± 84%) compared with control, but neither the steady-state expression of ß nor γ ENaC was affected. However, a qualitative change in γENaC due to proteolytic processing occurred, similar to that seen with dietary Na+ restriction, with a decrease in the 85 kDa band and an increase in a 70 kDa band (521). Subsequent studies examined the time course of the acute and chronic effects of aldosterone as well as characterization of the mechanisms involved in both phases, using a combination of electrophysiology of rat cortical collecting ducts in conjunction with renal cortical immunoblots and surface labeling techniques (226, 228, 606, 615).
Frindt et al. examined the effect of secondary hyperaldosteronism induced by 15 hours of Na+ deprivation in rats that did not reduce glomerular filtration rate (GFR) and produced a ~2.5-fold increase in plasma aldosterone. They observed post translational processing of the γ subunit with no quantitative change in the abundance of any ENaC subunit. Specifically, Na+ deprivation was associated with an increase in proteolytic processing and expression of a smaller version of the γ subunit (225).
Whereas chronic studies resulted in an increase in ENaC current to approximately 300–800 pA/cell, (225, 226) acute administration of aldosterone in rats resulted in a significant, but clearly smaller, amiloride-sensitive whole cell current of approximately 100 pA/cell (approximately 20–30% of chronic values). (226) This value of 100 pA/cell was, however, significantly greater than control values in the salt replete state. Commensurate with the increase in amiloride sensitive current there were qualitative changes in the expression of ENaC subunits in the collecting duct. Although there was not a significant increase in full-length αENaC by three hours, the cleaved forms of α ENaC and γ ENaC judged by immunoblot increased, and the surface expression of β and γ ENaC as measured by surface protein biotinylation also increased. (226) These studies demonstrated that aldosterone increases proteolytic processing of both the α and γ ENaC subunits. Among the family of channel-activating serine proteases (CAPs) (303, 836, 858), convincing experimental evidence supports the role of furin to activate ENaC (364). These studies are consistent with an acute effect of aldosterone to increase plasma membrane surface expression mediated in part by sgk1 as discussed in “Aldosterone Signaling in Renal Epithelial Cells.”
In the kidney, aldosterone appears to preferentially stimulate increases in the α subunit. However, this physiological regulation of Na+ channel activity by aldosterone does not appear to be shared by all tissues. Stokes and Sigmund examined the effect of a low Na+ diet and exogenous mineralocorticoid or glucocorticoid to stimulate steady-state mRNA for α, ß, and γ ENaC subunits (778). Interestingly, a low Na+ (0.13 %) diet for 2–3 weeks did not alter the qualitative distribution of mRNA by Northern blot analysis, nor did it affect quantitatively mRNA for ß or γ in the cortex or outer medulla. However, αENaC mRNA was selectively increased (with no change in ß or γ) in the inner medulla by a low Na+ diet. In contrast, this same maneuver elicited an increase in ß and γ subunits without an effect on αENaC in the descending colon (778). The regulation by a low Na+ diet, which would chronically increase aldosterone, angiotensin II, and possibly other relevant hormones, was contrasted with the effect of acute corticosteroid action. The acute (six hour) effect of mineralocorticoid (aldosterone 1.5 mg/kg, plus a glucocorticoid antagonist RU-38486, 1.8 mg/kg) or the glucocorticoid agonist was designed to selectively stimulate either MR or GR, respectively. The response in the kidney was strikingly different from that observed in the lung or colon: αENaC was only increased by glucocorticoid in the lung, but in the colon both mineralocorticoid and glucocorticoid stimulation increased ß and γ ENaC without affecting αENaC. In the kidney, however, mineralocorticoid or glucocorticoid stimulated increased αENaC, but not ß or γ ENaC, in the cortex, outer medulla, and inner medulla. These observations suggest very distinct temporal and tissue-specific responses (778).
Other studies have examined and contrasted the effect of glucocorticoid on amiloride-sensitive SCC and α ENaC expression and the molecular events associated with this induction in pulmonary, renal and colonic cell lines. Dexamethasone increased amiloride-sensitive SCC ~two-fold at 24 hours in H441 lung cells and increased αENaC expression by five-fold. This induction of αENaC was blocked by RU38486 and inhibited by actinomycin D but not by cycloheximide. Two promoters were identified with the 5’ (P1) promotor exhibiting greater rates of transcription. A directional-independent fragment (~289–143INV) upstream of both promoters conferred glucocorticoid-responsiveness to both promoters. Separate constructs from the α ENaC 5’-flanking region identified a region ~ 150 to 500 bps upstream of the transcriptional start site that contained two imperfect hormone response elements (HREs). However, only the downstream HRE produced glucocorticoid-dependent transcription. Dexamethasone increased αENaC expression in a collecting duct cell line in a dose-dependent manner starting at 10 nM. By testing glucocorticoid-mediated induction of αENaC gene transcription in a renal cell line derived from the CCD they found similar αENaC transcription in pulmonary and renal epithelial cells. Expression of GR in a colonic cell line restored glucocorticoid–stimulated αENaC transcription These studies support a role for glucocorticoids to stimulate αENaC in lung and kidney via activation of HRE located ~142–487 bp upstream from the transcriptional start site (708).
Aldosterone Regulation of the NaCl Cotransporter
In 1993, Gamba and coworkers reported the identification of a thiazide-sensitive electroneutral Na+-Cl− cotransporter from the urinary bladder of the winter flounder (246), initially denoted as TSC, and more recently as NCC, (gene slc12a3). Subsequently other members of this family of solute-linked cotransport family were identified, including the mammalian homologue of the flounder (118, 524, 525) and the structurally related apical and basolateral bumetanide-sensitive Na+-K+-2Cl cotransporters (245). The SLC12A family of transporters share (with few exceptions) much in common, including primary sequence homology, transmembrane structure, and several transport properties (all described are electroneutral). NCC (SLC12A3) is recognized as an important transporter in the kidney and mutations of both alleles produce a significant phenotype, Gitelman’s Syndrome, resulting in relative hypotension and hypokalemia. Mutations in WNK kinases within the regulatory cascade responsible for modulating the activity of this transporter give rise to a phenotype of salt-sensitive and thiazide-sensitive hypertension with hyperkalemia. For further discussion, the reader is directed to recent comprehensive reviews (29, 243, 244, 357, 358, 425). In addition to stimulating the expression of ENaC, studies by Knepper and coworkers also determined that chronic aldosterone stimulation induces the expression of NCC, which is induced in the DCT, CNT, initial collecting tubule (ICT), and CCD (708). This transporter is discussed under the section “Aldosterone Signaling in Renal Epithelial Cells,” and comprehensively in recent reviews (29, 357, 358, 425).
The role of NCC as a mechanism for aldosterone-mediated NaCl absorption was noted by Chen et al. (125) after noting the effects of diuretic treatment and dietary Na on 3H-metolazone binding (126). Velazquez et al. (844) administered aldosterone, dexamethasone, or both for 3–5 days by mini-pump to adrenalectomized rats that stimulated thiazide-sensitive NaCl transport in distal tubules perfused in vivo by more than fivefold. The investigators quantified NCC cotransporter abundance in the renal cortex by [3H]-metolazone binding. Both aldosterone and dexamethasone increased specific binding sites in kidney cortex more than threefold, and they concluded that stimulation of GR and MR can increase NCC activity and abundance.
Kim and coworkers (417) used an affinity-purified antibody to NCC to examine its expression in response to chronic mineralocorticoid stimulation. Immunoblots detected a protein of approximately 165 kDa by electrophoretic mobility from membrane fractions of the renal cortex, but not the renal medulla. IHC detected labeling restricted to the DCT. When rats were fed a low NaCl diet for 10 days expression by immunoblotting increased ~ two-fold with more intense IHC staining. When aldosterone was administered by osmotic minipumps for 10 days NCC expression was increased nearly four-fold (3.8-fold). Administration of fludrocortisone to rats for one week resulted in an approximately 6.5-fold increase in NCC expression. These studies demonstrated that chronic increases in either exogenous or endogenous aldosterone or treatment with the mineralocorticoid fludrocortisone produced a pronounced stimulation of NCC.
Recent work by Ellison and coworkers suggest that the action of aldosterone may not be direct but secondary to changes in [K+]p that stimulates NCC phosphorylation and activation. Terker et al. examined the selective knockout of MR expression in vivo in the adult mouse kidney using an inducible Cre lox system and found a significant reduction in total and phosphorylated(T53) NCC of ~60% NCC expression (805), which would be consistent with the action of mineralocorticoid stimulation of NCC. However, a chronic reduction in dietary K+ intake with a reduction in [K+]p resulted in a significant increase in total and phosphorylated(T53) NCC to control values prior to the selective knockout of MR. The authors interpret these results to suggest that the predominant effect on NCC and phospho-NCC is mediated by changes in plasma [K+] but note that the data do not exclude a direct effect of aldosterone on NCC.
Canonica, et al. (108) selectively disrupted MR in the kidney except the glomerulus and observed a marked phenotype typical of PHA I on a normal Na+ diet that was accentuated by dietary Na+ restriction and rescued by a high Na+, low K+ diet. αENaC, total and pT53-phospho-NCC were reduced significantly during a normal Na+ diet and Na+-restricted diet. After 2 weeks of a high Na+, low K+ diet, both αENaC and pT53-phospho-NCC remained significantly reduced but Total NCC was not significantly different from controls. The authors interpreted their observations to suggest a predominant effect of the attendant hyperkalemia to reduce NCC expression under normal salt and Na+ restricted conditions. Czogalla et al. (154) concluded that MR is critical for ENaC and Na+K+-ATPase regulation in the connecting segment (CS) but was dispensable for NCC and Na+-K+-ATPase regulation in the DCT. Thus, secondary and cell-specific events are probably complex and only partially understood (see Aldosterone Escape) (417).
Studies performed in Xenopus oocytes or transfected HEK293 cells have also probed the mechanism for NCC regulation by aldosterone. (30, 683) One study proposed a sgk1 WNK4 NCC pathway and another suggested that Nedd4-2 regulates NCC by a novel sgk1-kinase-dependent and Nedd4-2 ubiquitylation-dependent pathway (30, 683). The action of aldosterone to increase NCC is thus inferred in these model systems to have similarities and differences with the mechanism by which aldosterone increase ENaC channel cell surface expression.
The acute action of aldosterone to increase in NCC activity was examined by Hoover and colleagues using a cultured cell line from the DCT (mDCT15), which express the mineralocorticoid receptor (426). NCC activity was significantly increased when first measured at 12 hours by 30% and by 56% at 36 hours. The aldosterone stimulated thiazide sensitive Na+ uptake was discernible at 10 nM and maximal at 100 nM after 24 hours. This action was mediated by MR as antagonized fully by spironolactone, whereas the glucocorticoid receptor antagonist RU-486 did not block the stimulation. The increase in NCC activity was also blocked by the sgk inhibitor GSK-650394 (100 nM). Interestingly, aldosterone did not increase NCC abundance or distribution during the first 24 hours, which suggests that the increase in Na+ uptake reflected a change in NCC activity. Commensurate with this, aldosterone increased phosphorylated threonine 53-NCC (NCC-T53-P) expression at 24 hours in mDCT15 cells and in adrenalectomized rats treated with aldosterone infusion 50 μg (200g body wt)−1(24 h)−1 by minipump. At 24 hours aldosterone increased both SPAK and oxidative stress-responsive kinase 1 (OSR1) expression. SPAK-specific siRNA blocked the effect of aldosterone to increase thiazide sensitive Na+ uptake. Most of these data have been interpreted via the effect of aldosterone via the classic paradigm of aldosterone acting through its cognate nuclear receptor, MR. Recently, however, Cheng et al. provided evidence of an acute action of aldosterone to increase NCC phosphorylation and implicated effects mediated by plasma membrane receptors and MR (127). Clearly, further work is needed and is considered in the section “GPR30 as an Alternate Aldosterone Receptor (127).”
An additional level of complexity that should be considered is the potential for MR or GR to alter the amount of total NCC or modulate its degree of phosphorylation and activity. In this regard, Ivy et al. have provided evidence that the acute administration of corticosterone to adrenal-intact or adrenalectomized male mice increased NCC phosphorylation two-fold without altering total NCC abundance. Corticosterone also increased renal expression of the core clock genes Per1, Per2, cryptochrome 1, and aryl hydrocarbon receptor nuclear translocator-like protein 1 (Bmal1). Chronic blockade of GR with RU486 did not affect total NCC but prevented corticosterone-induced NCC phosphorylation and the activation of clock genes. Spironolactone blockade of MR reduced the total pool of NCC but did not affect stimulation by corticosterone. These studies suggest that both MR and GR are regulators of NCC but act by distinct receptor pathways. (384)
In summary, distinct mechanisms for the action of aldosterone to stimulate NCC have been proposed that may act over different time frames and are not mutually exclusive. Future study should focus on in vivo model systems in the mammalian kidney that can allow quantitative dissection of contributions of each of these proposed mechanisms and the time course over which they operate.
Aldosterone action on other Na+-dependent mechanisms
In addition to ENaC and NCC the ASDN expresses other Na+-dependent transporters. Notable of these is the Na+-driven Cl−/bicarbonate (HCO3−) exchanger (NDCBE), which is expressed in multiple tissues, including neurons, lung, gastrointestinal tract, muscle, and kidney and is sensitive to thiazide diuretics (610). This transporter has been proposed as the mechanism of ENaC-insensitive, thiazide-sensitive Na+ absorption in the CCD and to operate in parallel with the apical anion exchanger pendrin (474). This would result in NaCl reabsorption that is inhibited by thiazide diuretics. Since pendrin has been proposed to contribute to mineralocorticoid-sensitive hypertension and to be increased by mineralocorticoid stimulation, a commensurate increased NDCBE expression or activity would be anticipated (847). Other studies have provided evidence for bi-directional Cl− flux in the CCD during conditions of secondary hyperaldosteronism (a KCl-rich, Na+-deficient diet for 4 to 13 days) (970). The secretory Cl− flux was ouabain-sensitive, and the absorptive Cl− flux was sensitive to the H+K+-ATPase inhibitor Sch28080. The authors proposed that Cl− secretion was due to KCl secretion by an apical KCl transporter or parallel Cl− and K+ channels, and the ouabain-insensitive, Sch28080-sensitive absorptive Cl− flux occurred by parallel action of an apical Cl−/HCO3− exchanger, presumably pendrin, and an apical H+K+-ATPase (970). Whether the effect of mineralocorticoid stimulation on either of these mechanisms (474, 970) is due to a direct or indirect mechanism warrants further investigation.
Aldosterone action on H+ transport
Aldosterone stimulates H+-ATPase and H+-K+-ATPase in the collecting duct
One of the most consistent effects of aldosterone, aside from enhanced renal Na+ absorption, is increased renal net acid excretion, classically defined as the sum of ammonium excretion plus titratable acidity minus the excretion of bicarbonate. In contrast to the most frequent form of metabolic alkalosis, which is responsive to correction by the administration of neutral Cl− salts, such as NaCl or KCl, mineralocorticoid-induced metabolic alkalosis is not corrected, and frequently exacerbated by a diet with ample NaCl content.
The pathways that are responsible for this acid-base disorder are complex and include effects of aldosterone on amino acid, nitrogen, and ammonia metabolism that are considered elsewhere (366, 423, 880). Indirect effects of changes in acid-base balance on K+ metabolism are considered below (see section “Aldosterone on K+ Homeostasis”); and the direct effects of aldosterone on acid-base transporters, that are considered here.
Aldosterone stimulates two classes of proton pump, H+-ATPases and H+-K+-ATPases (281, 780). Whether this is a direct effect of MR to increase gene transcription on cis-acting regulatory elements is less certain as many studies have assessed the specificity of this action using the mineralocorticoid antagonist spironolactone. Recent studies raise questions whether spironolactone may have effects independent of its action to inhibit MR (277). However, acidosis stimulates RAAS and results in increased expression for several subunits of the H+-ATPase as well as both α subunits for the H+K+-ATPase isoforms (129).
Acute and in vitro studies
Aldosterone stimulates net luminal proton secretion in high resistance epithelia such as the toad and turtle urinary bladder and the aldosterone sensitive distal nephron, which includes the late DCT, the connecting segment, the initial CCT, and the CD. Due to this mechanism, aldosterone-mediated stimulation of renal net acid excretion can lead to a metabolic alkalosis that is observed in hyperaldosteronism.
Studies in the mammalian collecting duct and the analogous structure of amphibians, the toad and turtle urinary bladder, reported that net luminal acid secretion increases approximately 60 minutes after aldosterone stimulation. In the turtle bladder this occurs before a discernable increase in Na+ absorption, and does not require mucosal (luminal) Na+ or HCO3− in the mucosal or serosal solutions (12). These studies provided evidence supporting an electrogenic H+-ATPase as a mechanism responsible for luminal acidification. Further studies identified two types of carbonic anhydrase rich cells in the turtle bladder, similar to two types of intercalated cells found in the renal CCD (509).
The sensitivity of this H+-ATPase to various inhibitors has also been used to characterize the properties of this mechanism of acidification. Steinmetz and colleagues (774) concluded that the turtle bladder H+-ATPase exhibits properties that were “virtually identical to those of the H+-ATPases of plasma membranes” of yeast and fungus that are sensitive to dicyclohexylcarbodiimide (DCCD) and vanadate. They distinguished it from the plasma membrane proton pump characterized by Sachs et al. (688) because the latter was non-electrogenic. In these and other studies, however, the concentration required for the direct inhibition of the enzyme, in contrast to the concentration applied in vitro to the tissue, could not be ascertained. Moreover, the nonspecific effects of these inhibitors was acknowledged, but not fully addressable by the preparation studied and methods employed. The vanadate sensitivity of acid secretion and observation of K+ absorption in the urinary bladder suggested that in addition to an H+-ATPase, a component of this acidification was due to an H+-K+-ATPase (372, 940). In addition to an electrogenic H+-ATPase the urinary bladder possesses a vanadate sensitive proton pump (94, 95, 372, 388, 389, 434, 939–941), but the effect of aldosterone on this pump has not been well characterized.
The OMCD of the mammalian kidney is a high-capacity site of urinary acidification (494). Studies in adrenalectomized rabbits and chronic mineralocorticoid stimulation with DOC, as DOC acetate (DOCA), revealed that this segment was highly responsive to long-term changes in mineralocorticoid activity. Adrenalectomy abolished luminal proton secretion and DOC treatment increased it by ~80%. Acute in vitro addition of aldosterone (50 and 1000 nM) revealed dose- and time-dependent effects of aldosterone treatment. Treatment of isolated perfused OMCDi with 50 nM aldosterone added to the bath or peritubular solution resulted in a (nonsignificant) trend toward stimulation of JHCO3 (net HCO3− absorptive flux, equivalent to proton secretion, JH) at 180 minutes post treatment. However, with continued treatment of these same OMCDi for 260 minutes, JHCO3 approximately tripled. With the application of 1000 nM aldosterone to the bath solution, the effect of aldosterone was apparent after 180 minutes of treatment (779, 780). Interestingly, this stimulation of luminal proton secretion was not accompanied by a change in VT, similar to the observations in the CCD (779–781). These studies were prior to the appreciation of an H+-K+-ATPase-mediated mechanism in the CD. Thus, it is unclear whether this lack of change in VT reflected an electroneutral mechanism of acidification, or a large shunt conductance that did not allow a detection of small changes in VT.
Studies in Madin-Darby Canine Kidney (MDCK) cells provided further evidence that aldosterone stimulated proton secretion by an H+-K+ pump (584–586). In these studies, SCC was stimulated by 100 nM aldosterone for 24 hours prior to study. When K+ replaced Na+ in the apical solution, the pH gradient increased, and reducing apical [K+] to 0.5 mM or addition of omeprazole (0.1mM) to the apical solution resulted in ~75% inhibition. Application of apical barium completely abolished SCC. The authors concluded that MDCK cells can express an apical H-K pump that was stimulated by aldosterone and inhibited by omeprazole. The measured current was ascribed to K+ recycling through a K+ channel that operated in conjunction with the pump.
Extracellular K+-dependent proton extrusion after an acute acid load as a measure of H+-K+-ATPase activity of rat CCD intercalated cells (ICs) and ~two weeks of NaCl deprivation increased H+-K+-ATPase activity, consistent with a chronic effect of secondary hyperaldosteronism (743). H+-K+-ATPase activity was increased two-fold by a low-NaCl diet. Sch-28080 (10 μM) fully inhibited H+-K+-ATPase activity, but with adaptation to a low-NaCl diet H+-K+-ATPase activity was partially blocked by Sch-28080 or ouabain (1 mM), suggesting activation of an additional mechanism that required both inhibitors to abolish K+-dependent pHi recovery. Chronic treatment with exogenous aldosterone to increase plasma aldosterone did not support a role for aldosterone to stimulate H+-K+-ATPase activity but longer mineralocorticoid stimulation did stimulate H+-K+-ATPase activity suggesting that time of analysis is an important factor (281).
Chronic action of aldosterone on acid-base transporters
The long-term effects of aldosterone on renal Na+ transport result in fluid retention due to increased NaCl reabsorption, and the development of a metabolic alkalosis due to NaHCO3 retention. Part of the effect of aldosterone is mediated by increased expression of NCC (see section “Aldosterone Regulation of the NaCl Cotransporter”). However, part of the NaCl absorption mediated by aldosterone occurs through coordinated action of ENaC and the anion exchanger pendrin. This transport protein (SLC26a4) was identified by positional cloning as the cause of Pendred syndrome, a recessively inherited disorder characterized by congenital deafness and thyroid goiter. Initially found in the thyroid, this protein is expressed abundantly at the apical membrane of the type B and non-A non-B ICs of the CCD, (847). It appears critical for the correction of Cl− depletion during metabolic alkalosis by serving to exchange luminal Cl− for cytosolic HCO3− (240). Pendrin protein expression and its mRNA are increased with mineralocorticoid stimulation, with a two-fold increase in non-A non-B ICs and six-fold increase in type B ICs. Knockout of the pendrin gene reduces the weight gain and the increase in systemic BP in response to DOC pivalate treatment (847). Thus, the expression of this transporter is under long-term control of aldosterone; whether this represents a primary or secondary effect due to increased ENaC expression deserves further investigation.
In addition, mineralocorticoid stimulation increased both renally expressed H+-K+-ATPases (HKα1/gastric and HKα2 /non-gastric) in a K+-dependent manner. Greenlee et al. examined the dependence of the action of chronic mineralocorticoid stimulation on the presence of either HKα1 or HKα2 –containing H+-K+-ATPases (281). They observed that DOC treatment stimulates the activity and expression of HKα1- and HKα2-containing H+-K+-ATPases, but in a time-dependent manner with HKα1 expression being significantly increased at six days post DOC administration and HKα2 expression significantly increased at eight days post DOC treatment. In addition, they observed that the lack of expression of both H+-K+-ATPase isoforms resulted in significantly less Na+ retention and weight gain and abrogated the action of DOC to produce metabolic alkalosis. The observations suggest that these proton pumps are chronically stimulated by MC and appear to participate in the renal actions on Na+ retention and acid-base disturbances. The action on proton secretion is discussed in the section “Aldosterone Action on H+ Transport: Acute and in vitro Studies.”
The metabolic alkalosis that ensues from chronic mineralocorticoid stimulation, such as DOC-salt or hyperaldosteronism is typically modest in the presence of ample NaCl in the diet. This likely reflects the ability to correct the metabolic alkalosis by activation of pendrin-dependent chloride bicarbonate exchanger in the collecting duct. Notably, however, the magnitude of this metabolic alkalosis is markedly amplified when K+ intake is restricted (366). Moreover, the degree of metabolic alkalosis induction by K+ deprivation is obliterated by co-administration of amiloride (365). Collectively, such observations are consistent with an interaction of renal HKα1 H+-K+-ATPase and ENaC, and recent evidence further supports such an interaction (550). Since K+ restriction has been shown to stimulate H+K+-ATPase α2, it is plausible that this proton pump may be also responsible for a significant component of the metabolic alkalosis developing with mineralocorticoid excess, as supported by studies with knockout mice (281).
Ammonia (NH4+ + NH3) metabolism
An important element in acid-base balance is ammonium excretion. Indeed, ammonium excretion is the most responsive component of net acid excretion in response to acute acid loading experiments (880). Chronic mineralocorticoid stimulation increases the rate of net acid excretion principally by increasing ammonium excretion (366). A discussion of the effect of aldosterone on ammonia metabolism is beyond the scope of this review and the reader is referred to several excellent recent reviews and articles (320, 327, 880).
Aldosterone and renal organic anion transporters
Organic anion transporters (OATs) are members of the SLC22 transporter subfamily, consisting of a group of over 10 transmembrane proteins (574). OAT1 (encoded by Slc22a6) was originally identified in the kidney by Lopez-Nieto et al. as NKT, or novel kidney transporter (497). OAT3 (encoded by Slc22a8) was originally known as Roct (reduced in osteosclerosis [oc] transporter) due to its reduced expression in the kidneys of oc mutant mice compared to wild type mice (87). While OAT1 and OAT3 are both highly expressed in the basolateral membrane of the proximal tubule, OAT3 has been found to be expressed in the distal portion of the nephron as well (436, 490). Nevertheless, both OAT1 and OAT3 have been noted as important transporters for the elimination of drugs and other toxins through the kidney (260).
Information regarding OATs in relation to aldosterone secretion needs further study. OAT3 has been demonstrated by Vallon et al. to play a role in maintaining BP, as Oat3−/− mice display a 10–15% reduction in BP compared to their wild type controls (837). Despite the lowered BP, Oat3−/− mice have elevated plasma renin and aldosterone under basal conditions compared to wild type mice. Giving Oat3−/− mice a low NaCl diet results in blunted levels of aldosterone, suggesting a dysregulation of circulating aldosterone linked to a loss of Oat3. The reduced plasma aldosterone observed in these mice under low NaCl intake was associated with a greater loss in body weight and increased fluid intake (837). In a study by Eraly et al., Oat1 knockout does not appear to have the same BP-lowering effects as Oat3, and aldosterone levels were not measured in the study, warranting future analysis (197).
Aldosterone and K+ Homeostasis
The role of aldosterone in K+ homeostasis is complex and involves its action on renal and extra-renal mechanisms. Proper appreciation of its action requires an understanding of both. David Young and coworkers systematically studied the role of aldosterone on K+ homeostasis in a series of elegant studies primarily in dogs over three decades (527, 608, 944–953). These studies remain a foundation for much of our understanding of how aldosterone affects K+ homeostasis. Young succinctly summarized much that we currently know: “Aldosterone is part of a complex system that regulates plasma K+ concentration by affecting the renal excretion of the ion as well as its distribution within the body (945).”
These studies investigated the steady state inter-relationships between the plasma [K+] and aldosterone, Na+ and K+ intake and excretion, systemic arterial blood pressure, and other indices of cardiovascular function. Pan and Young carefully documented the constellation of cardiovascular and renal changes produced by aldosterone administration, which included a transient increase in renal Na+ retention, an increase in mean circulatory filling pressure, an increase in systemic arterial BP, and a reduction in plasma K+ concentration ([K+]p) (608). Of note, they did not observe a significant increase in urinary K+ excretion (relative to intake). Nevertheless, aldosterone did increase the renal clearance of K+ because it significantly reduced [K+]p. Despite the importance to these studies, however, they do not have the time resolution to address mechanisms. Recent studies have provided insight into these mechanisms and are discussed below (see section “Aldosterone, Flow-induced K+ Secretion, and K+ Recycling).
In comparison to the consistent effect of acute aldosterone administration to reduce Na+ excretion after 30 to 90 min in adrenalectomized female dogs, the magnitude of the change in K+ excretion is modest, (47, 249) and is typically less that the normal circadian variation in K+ excretion. (657) Nevertheless, the effect of chronic exogenous aldosterone administration to reproducibly reduce steady-state plasma K+ concentration along with the notion that K+ secretion was due to the absorption of luminal Na+ in exchange for cytosolic K+ supported the concept that aldosterone was an essential signal for renal K+ excretion. Thus, the observation that the CCD of adrenalectomized rabbits responded appropriately for the prior dietary K+ intake of the animal was considered novel when reported (907).
Together with insulin and ß-adrenergic catecholamines, aldosterone serves as an important regulator of extra-renal K homeostasis and plasma [K+], which is discussed subsequently. Adrenal synthesis and secretion of aldosterone and its plasma concentration increases as dietary K+ intake and plasma [K+] increases (945). Consequently, increasing dietary K+ intake results in little change in plasma K+ concentration in a normal individual (307, 656). This remarkable stability of plasma K+ concentration, essential for normal nerve and muscle function, reflects aldosterone’s role as one important component of the defense against hyperkalemia. Mineralocorticoid stimulation increases the capacity for renal K+ excretion and decreases plasma K+, which serves to reduce K+ excretion. This represents the classic feedback hypothesis for aldosterone’s role in renal K+ regulation whereby increased K+ intake leads to an increase in plasma [K+] and stimulates adrenal aldosterone secretion. Aldosterone then acts to increase cellular K+ uptake, which in the kidney facilitates renal K+ excretion, and a reduction in plasma K+. In contrast, hyperkalemia is a complication of hypoaldosteronism (464).
There is, however, substantial evidence that the concept of feedback control of K+ excretion is only a partial explanation of the regulation of K+ excretion. Notably, studies in humans (102, 649) and in laboratory animals (280, 533, 543, 555, 590, 827, 942, 943) show that ingestion of a K+ load can result in robust renal K excretion without measurable changes in plasma [K+] or plasma aldosterone. These studies support the idea of a rapid reflex arc such as a gut-brain-renal axis that responses quickly to an oral K+ load but remains incompletely defined (307, 357, 533, 658, 943).
Renal nephron regulation of K+ excretion
The renal regulation of K+ excretion is largely confined to the ASDN. Until the classic studies by Berliner and colleagues, renal K+ excretion was viewed to be result of selective reabsorption of K+ that was freely filtered at the glomerulus. Although some early reports suggested that K+ excretion could exceed the filtered K+ load (GFR × [K+]p), it was not until the systematic studies of Berliner and coworkers that it was accepted, based on their deductions from clearance experiments, that the majority of urinary K+ was due to active K+ secretion by the distal nephron (62). Micropuncture studies of rat superficial nephrons by Giebisch and coworkers directly confirmed the conclusions of Berliner et al. and showed that regardless of dietary K+ intake, the majority (80–90%) of K+ filtered at the glomerulus was reabsorbed by the early DCT (513). Malnic, Klose, and Giebisch showed that fractional delivery of K+ increased along the length of the distal convolution in rats fed a normal K+ diet, providing direct evidence for net K+ secretion in surface distal nephrons and the rate of net K+ secretion further increased on a high K+ diet. On a low K+ diet, net K+ secretion was negligible in the cortex and significant net K+ reabsorption occurred between the last accessible surface nephrons and the final urine.
Whether changes in absorptive K+ flux also contributed to the changes in net K+ secretion in the superficial cortical ASDN was first examined by Fowler et al. (221) These investigators studied unidirectional K+ absorption by 42K tracer flux under conditions of K+ loading and K+ depletion and concluded that changes in reabsorptive K+ efflux participate in the regulation of tubular net K+ movement. They observed that 42K efflux from the lumen was stimulated by a low K+ diet and was reduced but still present under conditions of K loading. The implications of these findings are discussed below (See section “Aldosterone, Flow-induced K+ Secretion, and K+ Recycling”).
Wright and coworkers systematically examined the mechanisms of K+ transport in the distal nephron by in vivo microperfusion and characterized net and unidirectional absorptive and secretory K+ fluxes, the role of luminal flow rate, luminal [Na+] and [Cl-] concentration, transepithelial voltage, and the effect of luminal barium, a non-specific K+ channel inhibitor. These investigators established the importance of luminal flow rate as a critical determinant of distal nephron K+ secretion (276), whereas an increase in distal Na+ concentration (from 23 to 95 mM), Na+ delivery, or Na+ absorption did not affect K+ secretion if flow rate was unchanged. When the perfused distal K+ concentration exceeded ~ 30mM, however, net K+ secretion was reversed to net K+ absorption (276). Good, Velazquez, and Wright estimated a luminal half-maximal [Na+] of ~10 mM for K+ secretion by examining the effect of reducing the perfusion fluid [Na+] to 10 or 3 mM. The reduction in net K+ secretion by drastically reducing luminal [Na+] could not be explained solely by changes in electrical driving forces (275). Ellison, Velazquez, and Wright identified the early distal convolution, reflecting the DCT, as the major site of action of thiazide diuretics and noted the importance of luminal Na+ composition on the measured rates of absorptive Na+ flux (195). Velazquez, Ellison, and Wright recognized the heterogeneity of K+ transport mechanisms in the distal surface nephrons with greater rates of K+ secretion occurring in the late distal surface nephrons (CNT and ICT) (845).
Whether an increase in luminal flow or luminal Na+ delivery is primarily responsible for the increased rate of K+ secretion in the ASDN has been addressed indirectly by clearance experiments under normal dietary Na+ intake. (370) These studies also support the primacy of luminal flow over luminal Na+ delivery as the major mechanism which stimulates distal nephron K+ secretion. Hunter et al. studied acute inhibition of NCC by thiazide diuretics and observed no significant acute increase in K+ excretion, consistent with the observations of Good and Wright. (276) Interestingly, Hunter et al. further examined animals after conditioning to a Na+-restricted diet. Under these conditions, hydrochlorothiazide acutely stimulated K+ excretion, but the natriuretic effect of hydrochlorothiazide was not enhanced by coadministration of benzamil. Whether stimulation of angiotensin II or additional signaling mechanisms participated in this acute kaliuretic response deserves further investigation, particularly since acute oral K+ loading, which results in dephosphorylation of NCC and inhibits NCC, results in an acute kaliuresis (750). Grimm et al. observed a similar lack of an effect of acute hydrochlorothiazide on K+ excretion in a constitutively active SPAK mouse with increased NCC phosphorylation and proposed a trans-tubule coupling mechanism. (283) Whether this relates to flow induced K+ secretion (FIKS) or other paracrine systems deserves further investigation.
Wright and co-workers further measured unidirectional rates of K+ secretory and absorptive fluxes in the surface distal nephrons (196) and observed that absorptive K+ flux was ~25–35% of secretory fluxes in K+ replete rats, consistent with the work of Fowler et al. (221). Okusa et al. provided direct evidence for the mechanism of at least one component of this absorptive flux in the superficial distal nephron. They observed that the surface distal nephron of rats fed a low K+ diet exhibited active net K+ reabsorption that was inhibited by the HKα1 H,K-ATPase inhibitor Sch28080 (591). The implications of these studies are discussed subsequently (see section “Aldosterone, Flow-induced K+ Secretion, and K+ Recycling”).
Cellular mechanisms of K+ transport
Aldosterone not only exert effects on the transport mechanisms of the apical membrane but also of the basolateral membrane. These are most well appreciated after chronic mineralocorticoid stimulation which results in the proliferation of the basolateral membrane and an increase in Na±K+-ATPase (767, 859), but electrophysiological studies also demonstrate a role of aldosterone on the conductive properties of the basolateral membrane. As noted by Weinstein, K+ transport in the ASDN has secretory and absorptive components with net absorption predominating in the renal medulla (882). Figure 5 illustrates the predominant mechanisms for active K+ secretion and absorption in the ASDN.
K+ secretion
K+ secretion is an active process that occurs in the principal cells of the DCT2, CNT, ICT, and CCD of the ASDN (Figure 5). Active K+ secretion is driven by a basolateral Na+K+-ATPase and an apical Na+ channel (ENaC) which provides the driving force for conductive K+ secretion (227, 602, 604, 870). Non-conductive K+ secretion requires active K+ uptake by the basolateral Na+K+-ATPase and an apical KCl cotransporter or possibly parallel apical K+ and Cl− channels, a point that deserves further study (196, 845, 902, 903). During K+-replete conditions the rate of net K+ secretion increases from the DCT to the CNT and ICT (845). In the CNT, ICT, and CCD, K+ is secreted by two classes of K+ channels: an inward rectifier K+ channel, Kir1.1 (KCNJ1) (also referred to as the renal outer medullary K+ channel, ROMK), which is expressed as three splice variants, and large conductance Ca2+-activated K+ channels [BK channels (KCNMA1) and SK3 channels (KCNN3 or KCa2.3)] (64, 289, 638, 694) and a KCl cotransporter (196, 903). The inward rectifier K+ channel Kir1.1 is expressed on the apical membrane of principal cells and is viewed as the predominant K+ channel that mediates basal rates of conductive K+ secretion (334). Evidence supports regulation of this channel by tyrosine kinases (482), by clathrin-regulated endocytosis (885–888, 936, 937), and by mTORC signaling (751). Evidence also supports aldosterone-dependent and independent effects on renal K+ transport as discussed below (227, 602, 751, 930).
K+ absorption
K+ absorption is an active process that occurs in part in the intercalated cells of the CNT, ICT, CCD, OMCD, and IMCD of the ASDN in which active K+ absorption is driven by luminal HKα1 (ATP4a) H+K+-ATPase and a basolateral K+ channel (9, 103, 305, 744, 906). In addition, the HKα2 (ATP12a) H+K+-ATPase is expressed in intercalated cells and principal cells of the mouse CNT and CCD (11, 848). Of note, K+ restriction and the resulting hypokalemia increases mRNA for HKα2 in the outer medulla, whereas this same maneuver increases HKα1 mRNA in the cortex, which suggests differential control (10). In addition, functional evidence indicates that HKα1 H+K+-ATPase is important for a component of luminal acidification in the outer and inner medulla. In the OMCD from K+-replete rabbits, removal of luminal K+, 10 μM Sch28080, or a structurally distinct H+K+-ATPase inhibitor A80915a significantly inhibited net proton secretion (25, 26, 829). In the terminal IMCD of acid-loaded rats both luminal K+ removal or 10 μM Sch28080 substantially inhibited net acidification (864). Wall and DuBose further demonstrated that K+ restriction increased total and Sch28080 sensitive proton secretion in the terminal IMCD (863). These data are consistent with an increase in HKα1 and HKα2 H+K+-ATPase activity.
The regulation of HKα2 H+K+-ATPase activity is under tonic inhibitory control by tissue kallikrein (192). Mice with disruption of the tissue kallikrein gene (mouse, klk1, human KLK1) (TK null mice) exhibit a defect in early adaptation to a K+ load with the development of hyperkalemia. This rapid excretion of a K+ load is in part aldosterone-independent (162, 660). TK null mice exhibit enhanced net K+ absorption in the CCD, more rapid pHi recovery from an acid load and increased expression of mRNA for HKα2 indicating that tissue kallikrein inhibits HKα2 H+K+-ATPase activity.
Studies in TRPV4 null mice also indicate that changes in absorptive K+ flux contribute to overall renal K+ homeostasis. TRPV4 null mice fed a low K+ diet did not develop hypokalemia in contrast to WT mice fed this same low K+ diet (822). Moreover, the rate of renal adaptation to a low K+ diet was faster in TRPV4 null mice. This more rapid adaptation in TRPV4 null mice was associated with a significantly increased H+K+-ATPase activity, which suggests that TRPV4 also acts to inhibit H+K+-ATPase activity (822).
Aldosterone, flow-induced K+ secretion, and K+ recycling
Recent studies have clarified the role of aldosterone in K+ handling and help to explain the complex role that aldosterone plays in the regulation of K+ balance. These studies include: the mechanism of flow-induced K+ secretion (FIKS) by BK channels, its role in purinergic signaling, and the regulation of ENaC; the action aldosterone on basolateral transport and conductances of the ASDN; the importance of inward rectifier channels Kir4.1/5.1 as critical determinants of the basolateral membrane resting membrane voltage of the ASDN; the role of NCC, WNK4, and WNK1 in the regulation of basolateral membrane voltage via basolateral Cl− channels and intracellular [Cl−]; and the contribution of aldosterone and high K+ diets to renal K+ recycling.
As noted previously, K+ absorptive flux contributes to tubular net K+ transport (221). Subsequently, Good and Wright (276), Ellison, Velazquez, and Wright (196), Okusa and coworkers (591), and Jamison and coworkers provided further evidence for the regulation of absorptive as well as secretory K+ fluxes in the ASDN. Jamison and coworkers systematically documented that a portion of distal cortical K+ secretion was reabsorbed beyond the late surface distal nephrons, a phenomenon known as K+ recycling (57, 390).
Paradoxically, the degree of K+ recycling increases during conditions of K+ loading. (27, 57) Jamison and colleagues showed that the fractional delivery of K+ to the descending limb of Henle increased when rats were fed a high K+ diet versus a normal K+ diet. Importantly, the fractional delivery of K+ to the thin descending limb of Henle decreased after the administration of amiloride, which indicates that the source for the increased delivery to the thin descending limb of Henle was K+ secretion in the distal nephron. Thus, a portion of distally secreted K+ was recycled from the CD to the loop of Henle (390). Conversely, K+ recycling was reduced on a low K+ diet (170).
The studies of Jamison and coworkers are consistent with observations of Fowler et al who measured bidirectional K+ transport (221). In addition, Field, Stanton, and Giebisch demonstrated that acute aldosterone administration stimulates K+ secretion in the cortical CNT and ICT without increasing urinary K+ excretion (UKV) (213) and studies by Higashihara & Kokko showed that aldosterone increased K+ recycling (345). Collectively, these studies are most consistent with the action of mineralocorticoids to stimulate increases cortical net K+ secretion, but also enhances the capacity of K+ reabsorptive pumps (281) whose activity is normally greatest in the cortex (174).
The role of BK channels in flow-induced K+ secretion, purinergic signaling and ENaC
BK channels are comprised of pore-forming α subunits and regulatory ß subunits and are activated by intracellular Ca2+. The α subunit is expressed in intercalated cells with the β4 accessory subunit and in principal cells with the β1 accessory subunit (284, 289, 638, 639). BK channels are viewed as the predominant channel that mediates flow-dependent K+ secretion (692, 703, 911), and some but not all studies support regulation of this channel by aldosterone (199, 889, 890). Flow sensed by cilia triggers Ca2+ signaling via TRPV4 channels (514) that is important for adaptation to increased dietary K+ intake. The importance of TRPV4 regulation of K+ secretion by BK channels is illustrated in TRPV4 channel null mice that exhibit impaired K+ excretion and hyperkalemia when fed a high K+ diet (514).
BK channel-mediated K+ secretion is coupled with ATP secretion and purinergic signaling. Mice with deletion of the β4 regulatory subunit of the BK channel have reduced urinary ATP response to increases in Na intake, defective purinergic signaling, and impaired regulation of Na excretion (92, 285, 286, 352). Holtzclaw et al. linked flow-sensitive K+ and ATP secretion to a connexin- and BK-sensitive pathway (352), and Mironova et al. demonstrated dysregulation of ENaC activity in HKα1 null but not WT mice (550). ENaC activity was not suppressed by a high Na+ diet and urinary ATP was reduced in HKα1 null mice compared to WT mice. Moreover, HKα1 null mice fed a high-Na+ diet had greater Na+ retention than WT mice and had an impaired dipsogenic response. Such findings suggest a role of H+K+-ATPases as part of a signaling mechanism linking flow induced K+ secretion and BK channel activity with purinergic signaling. Defects in both α and β subunits of the BK channel have been linked to hypertension. Although published studies suggest that the hypertension is linked to a vascular defect, the above and other studies support a renal defect in Na+ excretion and further study is warranted (113, 286, 288, 354, 705).
Analysis and summary
In summary, increased dietary K+ intake stimulates active net K+ secretion in the DCT2, CNT, ICT and CCD by coordinate action to stimulate unidirectional K+ secretory flux and to inhibit unidirectional K+ absorptive flux. Conversely, decreased dietary K+ intake reduces net K+ secretion in the renal cortex and results in net K+ reabsorption in the medullary CD, by coordinate action on unidirectional K+ fluxes (484, 513, 904). However, the work of Jamison and colleagues shows that the quantity of distally secreted K+ that is reabsorbed during conditions of K+ loading exceeds that which is reabsorbed during times of K+ depletion (390). Direct measurements of absorptive K+ flux in the ASDN show that this flux represents a significant fraction of total flux and participates in the regulation of tubular net K+ transport (221). Measurements of the components of this absorptive flux are likely to be informative.
Collectively, these studies suggest that changes in absorptive K+ flux as well as secretory flux contribute to net K+ secretion and these changes are demonstrable in the cortex, not just the medulla. Whether FIKS represents strictly an additional mechanism for K+ secretion or may also participate as part of a system linked to purinergic signaling of ENaC, as supported by the cited studies, deserves further study. Diets rich in K+ are known to reduce BP (55, 155, 193, 533, 689, 804, 817) and diets low in K+ intake exacerbate salt sensitive hypertension (440, 441, 557) that is in part due to renal dysregulation of Na+ handling. The importance of FIKS and K+ recycling may be related to K+-dependent regulation of purinergic signaling. The finding that TRPV4 null mice are better protected from the development of hypokalemia than normal mice on a K+-deplete diet by activation of H+-K+-ATPase activity suggests that the role of these K+ absorptive pumps may be equally important as part of a local signaling mechanism independent of their action on net K+ excretion (822). Further work in this area is likely to be rewarding.
Aldosterone effect on basolateral plasma membrane transport properties of the ASDN
Aldosterone has prominent effects on the apical membrane to increase Na+ conductance via increasing the number and activity of the ENaC (226, 599), but it also has equally pronounced effects on the basolateral membrane (250, 428, 765, 859). The basolateral changes initially presumed to be due to increased transcellular Na+ absorption with the attendant need for increased basolateral Na+-K+ pump activity, but the work of Rossier and colleagues provided convincing evidence that aldosterone directly increase the synthesis of new Na+-K+-ATPase pump subunits. Aldosterone also has selective effects on the morphology and the electrophysiological characteristics of the basolateral membrane of the principal cells of the late distal nephron and CCD. Stanton et al. noted a selective effect of adrenalectomy to decrease, and chronic aldosterone infusion to increase, the basolateral membrane length of the principal cells of the ICT (765). In subsequent studies these investigators extended these observations and demonstrated independent effects of aldosterone and a K+ rich diet on the morphology of the basolateral membrane of the principal cells (766).
Along with the selective effect of aldosterone on basolateral membrane length, aldosterone results in significant changes in the electrophysiological characteristics of the basolateral membrane with a reduction in basolateral membrane resistance (429), which is independent of a K+ rich diet (560). With mineralocorticoid stimulation there is an increase in the relative K+ conductance of the basolateral membrane (428) and ion selective electrode intracellular K+ activity measurements in principal cells of the rabbit CCD show that K+ activity is below electrochemical equilibrium across the basolateral membrane with an increased K+ conductance, which would promote K+ secretion (693).
Role of inwardly rectifying K+ channels 4.1 and 5.1 in determining basolateral membrane conductance
Studies have established an important role for the inward rectifier channel Kir4.1 as the major K+ conductance of the basolateral membrane of the principal cells of the DCT (868, 915), CNT, and CCD (788). Kir4.1 can form functional K+ selective channels as a homotetramer Kir4.1, but this subunit also forms functional K+ selective channels as a heterodimer with Kir5.1, albeit with different properties. Lachheb reported that Kir4.1/5.1 is the major K+ conductance of the mouse CCD principal cells (452).
Kir4.1/5.1 is under hormonal control by insulin and insulin-like growth factor-1 (IGF-1) (958) and is directly or indirectly regulated by Nedd4-2 (964). Kir4.1/5.1 has been implicated in the regulation of renal K+ excretion (787, 916) and modulating ENaC activity (789). Whether aldosterone regulates this K+ channel in the ASDNneeds further study (see below). However, in the retina, another aldosterone target epithelia, Kir4.1 is responsive to aldosterone via MR, which is consistent with the electrophysiological changes observed in the ASDN (969).
The action of aldosterone likely acts in coordination with direct effects of basolateral [K+] as part of a regulatory mechanism for K+ secretion in the ASDN. Sorensen et al. provided convincing evidence that [K+] modulates the activity of the type 2 mTOR complex (mTORC2) via basolateral K+ channel activity to increase SGK1 and indirectly ENaC activity, which suggests that functions at the basolateral membrane of principal cells may act to integrate multiple signals to modulate K+ secretion (751).
Additional work supporting this thesis comes from the studies of Tomilin et al. who found that dietary K+ and Cl− can independently regulate Kir4.1/5.1 and ClC-K2/b in the principal and intercalated cells of the CD, respectively (823). Placing C57BL/6J mice on a high K+, high Cl− diet (6% K+, 5% Cl−) for 1 week resulted in a significant increase in basolateral K+-selective current, single channel Kir4.1/5.1 activity, and a hyperpolarization of basolateral membrane potential in principal cells compared with values in mice on a regular diet (0.9% K+, 0.5% Cl−). However, basolateral Cl− selective current and single channel ClC-K2/b activity decreased in intercalated cells under this high K+, high Cl− diet. When dietary K+ was replaced with Na (High NaCl diet) intercalated cell ClC-K2/b activity was also reduced. Mice fed a high K+, normal Cl− diet (6% K+, 0.5% Cl−) also had an increased basolateral K+-selective current, single channel Kir4.1/5.1 activity, and hyperpolarization of basolateral membrane potential in principal cells without a significant effect on ClC-K2/b activity in intercalated cells. In addition, mice treated with DOCA for three days increased Kir4.1/5.1 activity while ClC-K2/b activity was unaffected, providing more evidence for aldosterone signaling via basolateral K+ conductance in the principal cells of the CD (823).
Collectively, these studies support a role for aldosterone to regulate basolateral properties and conductances in cell expressing Kir4.1/5.1 K+ channels such as principal cells of the ASDN and other MR responsive tissues. These studies are also consistent with the increased clearance of K+ observed with experimental and clinical hyperaldosteronism (608). The precise signaling mechanisms deserve further study.
NCC-WNK1-WNK4 pathway, Cl−ic, and aldosterone signaling
The identification of mutations linked to PHA type II (900) led to the discovery of the NCC WNK signaling pathway (901, 925), subsequent identification of intermediate regulatory mechanisms (853), the connection with Ang II signaling (691) and the potential role of compensatory systems (287, 313) (see section “Aldosterone Regulation of the NaCl Cotransporter”). The regulation of NCC (194, 244) and the role of WNK signaling and intracellular Cl− (312, 627) have been extensively studied and reviewed recently (357, 592) The potential for changes in intracellular Cl− to modulate WNK signaling, and NCC activity, has been explored under conditions of chronic hypokalemia in NCC/SLC12a3 null mice (806) and in KCNJ10 null mice (149) which are null for the mouse homologue of EAST/SeSAME syndrome in humans (81, 717). Children with this disorder exhibit a constellation of symptoms that include seizures, neurosensory deafness, ataxia, mental retardation, and hypokalemia. Conversely, Grimm et al. studied the effect of constituent activation of SPAK which results in hyperkalemia (283). Collectively these studies suggest that alteration in basolateral membrane voltage dictated by K+ and Cl− channels are important for the regulation of solute transport in the ASDN. In view of previous cited studies demonstrating electrophysiological effects of aldosterone on Cl− and K+ conductances (428), further studies are needed to define the interaction of NCC-WNK signaling with mineralocorticoids. It is likely that such studies will provide considerable insight if conducted with attention to Na+ and K+ balance, BP, and apical and basolateral membrane driving forces for K+ transport.
The cooperative and complementary roles of K+ and Cl− channels is illustrated by the study by Pinelli et al (628) of the ClC-K2 Cl− channel expressed in the type B intercalated cells of the ASDN. This ~10-pS channel exhibits a steep voltage dependence so that channel activity increases with membrane depolarization. Intracellular and extracellular pH differentially regulate the voltage dependence curve with alkaline intracellular pH reducing the voltage dependence. Intracellular and extracellular pH and Ca2+ increase channel activity but have less effect on channel open probability. Membrane voltage alters the number of active channels and their open probability, but [Cl−] has little influence. From their analysis and modeling, they propose a model of how the relative Cl conductance (transference ratio) dictates the mode of transport (Cl−/HCO3− exchange versus NaCl absorption) in the type B intercalated cell. Since the other conductance is to K+, voltage changes dictated by ionic K+ conductances in response to aldosterone will be translated into changes in the mode of transport of the type B intercalated cells. Although restricted to the type B intercalated cell the insight from these studies may well be relevant to the interrelationship of Cl− flux and intracellular [Cl−] to Vbl.
Acute action of mineralocorticoids on renal K+ transport: In vivo studies
Numerous investigators have studied the acute effect of aldosterone administration on renal K+ transport under different experimental conditions both in vivo and in vitro and are discussed separately.
Horisberger & Diezi provided evidence that aldosterone not only promotes an anti-natriuesis but also a kaliuresis when administered acutely in adrenalectomized male Wistar rats (359). These investigators examine the effect of the acute administration of aldosterone or vehicle to rats 24 hours after adrenalectomy at a dose of 1 μg per kilogram given intravenously followed by a constant infusion of 1 μg per kilo gram per hour of aldosterone. Urine was collected every 30 minutes, aldosterone was administered after the first 30-minute period, and collections were made every 30 minutes over the next four hours. Seven groups of animals were studied and the effect of increasing delivery of Na+ to the distal nephron was examined using solutions with increasing concentration of NaCl administered intravenously. The initial anti-natriuretic effect of aldosterone was observed in all groups between 30–60 minutes with the maximal effect observed at 120–180 minutes. A kaliuretic effect in the aldosterone treated group, indicated by a significant increase in urinary K+ excretion (UKV) compared with similar animals administered only vehicle, started between 30–60 minutes in the two groups infused with the largest rates of NaCl. However, no significant increase in UKV occurred in the group receiving the smallest rate of NaCl infusion despite an effect of aldosterone to reduce Na+ excretion. This latter finding and the collective observations of Horisberger and Diezi were noted in a subsequent study by Field et al. (213) that is discussed below.
Giebisch and coworkers (213) examined the acute effects of aldosterone, the synthetic glucocorticoid dexamethasone, and hyperkalemia on distal tubule K+ secretion and urinary K+ excretion in adrenalectomized male Sprague Dawley rats. Adrenalectomy was performed 8 to 10 days before the transport studies and corticosteroid replacement was administered by osmotic mini pumps. The aldosterone replacement dose was previously found to produce physiological plasma aldosterone concentrations in awake and unstressed rats. The dose of dexamethasone was previously found to result in normal weight gain and normalized glomerular filtration rate (GFR) and produced normal fasting plasma glucose and insulin concentrations in adrenalectomized rats. Rats were randomly divided into five groups: the first three examined the acute effects of aldosterone and dexamethasone during normokalemic; two groups examined the effect of aldosterone in the presence of hyperkalemia. The first three groups were infused with Ringer solution (Na+ 145 mM, K +5 mM) whereas the latter two groups received, a high K+ solution (Na+ 60 mM, K+ 90 mM). Animals in the first three groups received either vehicle, aldosterone (2 μg per kilogram body weight bolus and then continuous infusion per hour), or dexamethasone (2 μg per kilogram body weight bolus and then continuous infusion per hour). The transport studies were conducted 120–240 minutes after acute hormone or vehicle infusion. BP and plasma electrolyte composition were carefully monitored and the high K+ solution resulted in significant hyperkalemia ([K+] 6.6–7.3 mM).
Aldosterone reduced urinary flow rate significantly in the normokalemic rats and by a non-significant amount in the hyperkalemic animals. In both the normokalemic and the hyperkalemic groups aldosterone reduced the absolute and fractional excretion of Na+ significantly, but they observed no significant effect acute aldosterone infusion on absolute or fractional K+ excretion both during normokalemic and hyperkalemia treatment groups (Figure 11)(213). The authors attributed the lack of effect of aldosterone on UKV to differences in urinary flow.
Figure 11.
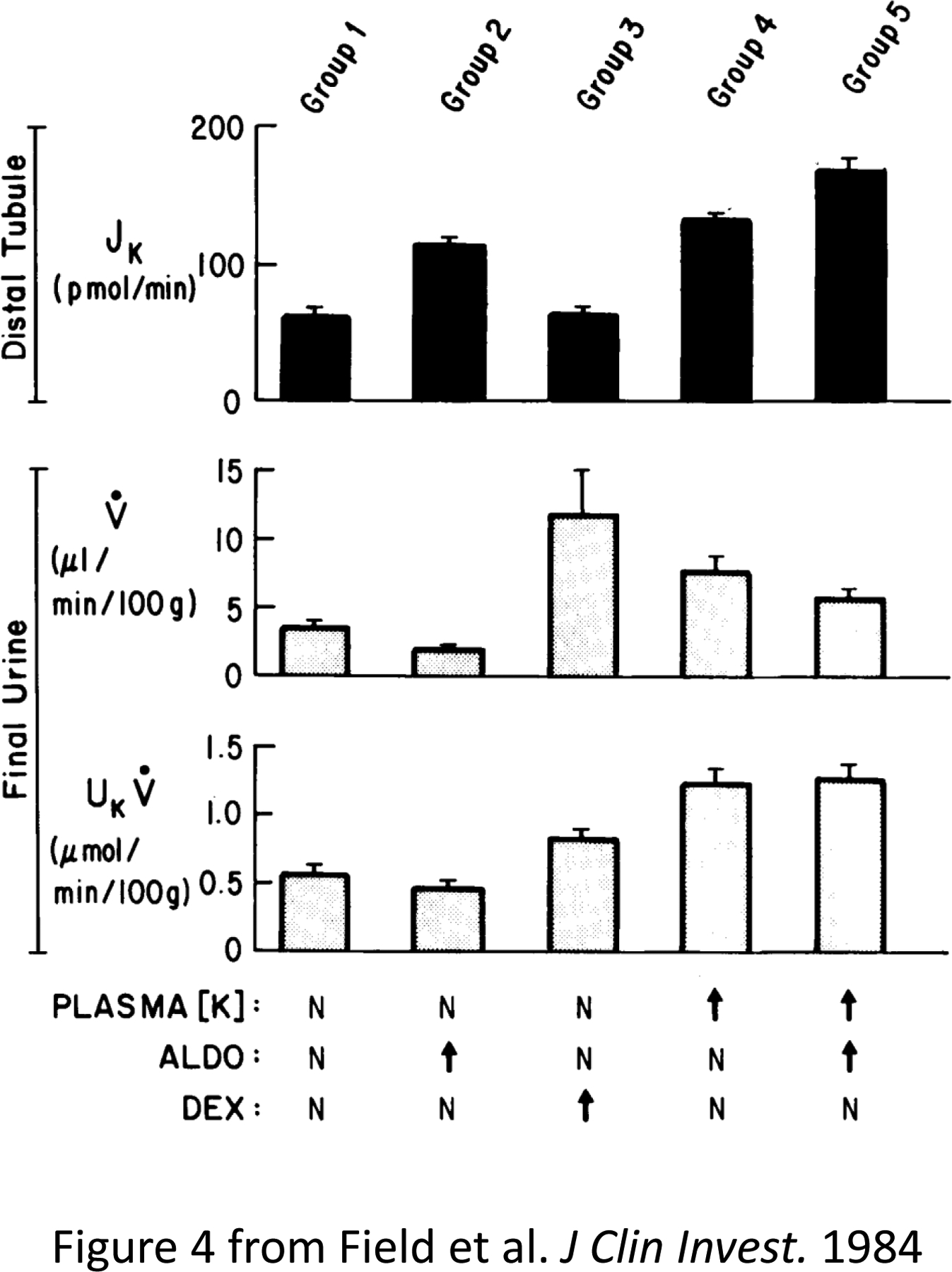
Summary of principal results of simultaneous microperfusion experiments (distal tubular potassium secretion, JK, top) and clearance experiments (urinary flow rate, V, and potassium excretion, UKV, middle and lower, respectively) in each of the five experimental groups studied. The conditions prevailing in each group are shown at the bottom of the figure: N represents normal (for plasma K+), or basal (for hormone levels). ALDO, aldosterone; DEX, dexamethasone. Adapted from Field et al (213).
However, acute aldosterone infusion increased the rate of K+ secretion by in vivo microperfusion of distal tubules nearly 100% in the normokalemic group and approximately a 50% in the hyperkalemic group. The discrepancy between the effect of aldosterone on distal nephron K+ secretion and urinary K+ excretion suggests that aldosterone also increased K+ recycling and the rates of K+ absorptive flux beyond the distal nephron. Alternatively, it is possible that mid-cortical and juxtamedullary nephrons behaved differently to superficial nephrons.
Stanton et al. came to similar conclusions regarding the effect of aldosterone on K+ excretion when they investigated the effect of K+ intake and aldosterone on K+ adaptation in the distal nephron K+ secretion by in vivo microperfusion and clearance studies when KCl was administered by infusion to intact and adrenalectomized rats. These studies are discussed under the section “K+ Adaptation” (766).
Subsequently, Palmer and Frindt studied the action of aldosterone on Na+ and K+ conductances and concluded that aldosterone activated Na+ channels but did not increase the predominant low conductance K+ channel(SK channel) (602, 604). Short term (six hours) feeding with a high K+ diet, which also increased plasma aldosterone, increased the activity of SK channels. However, this effect was not reproduced by administration of aldosterone. They proposed that for increased K+ secretion some unknown second factor other than aldosterone was necessary to increase the number of apical K+ channels and augment K+ secretion. The acute effect of aldosterone was recently re-examined by Frindt and Palmer. These investigators observed that the single dose of aldosterone (20 μg/kg body wt) reduced Na+ excretion over the subsequent three hours. However, they observed no increase in K+ excretion (226).
Analysis
Differences in FeNa and urinary flow likely explain the ostensibly disparate conclusions of these studies. Aldosterone increased K+ excretion when FeNa was large (> 1%), but physiologically it would be expected to exert its most prominent effect during states of low Na+ excretion as Giebisch and coworkers noted.
The role of aldosterone to modulate FIKS mediated by changes in luminal flow rate deserves further investigation and whether this mechanism is related to changes in the activity of other locally acting systems such as the kallikrein kinin, purinergic, or endothelin system or via flow dependent mechanisms may help to resolve some of the controversy regarding the kaliuretic activity of aldosterone. Nevertheless, the acute administration of aldosterone in vivo in non-volume expanded or salt loaded states while increasing distal nephron K+ secretion does not consistently promote an increase in renal K+ excretion in physiological doses or conditions.
Importantly, the observation that acute aldosterone administration during normo- or hyperkalemic conditions did not increase renal K+ excretion led Giebisch and coworkers (213) to conclude that renal excretion of an acute K+ load does not require an acute increase in plasma aldosterone in excess of basal plasma aldosterone concentration (Figure 11) (213). In contrast, they noted that K+ infusion to intact rats that increased K+ excretion was associate with an increase in plasma aldosterone (768). However, acute aldosterone administration resulted in significantly lower plasma [K+] than the group receiving vehicle, despite identical K+ loads in the two groups. As the authors note, this suggests that acute aldosterone infusion significantly enhanced extrarenal K+ uptake which would increase the animal’s tolerance of the acute K+ load. Such observations are consistent with the studies of Young and coworkers and are discussed in the section “Extra-renal Action of Aldosterone on K+ Homeostasis” and studies by Alexander and Levinsky (16).
In vitro studies
Only two studies have examined the in vitro effect of aldosterone on net ion transport in the mammalian nephron or CD (780, 905). Both examined CD segments from adrenalectomized rabbits, an in vivo condition that was assumed to provide a greater sensitivity to aldosterone. One microperfusion study examined the effect of aldosterone on net total CO2 and VT in the outer medullary collecting duct (OMCD) of adrenalectomized rabbits and was discussed previously in the section “Aldosterone Action on H+ Transport”. The other examined the effect of aldosterone on Na+ and K+ net flux, and VT in the isolated perfused CCD from adrenalectomized rabbits. These studies were discussed previously with respect to Na+ transport (“Mechanism of action on Na+ transport”). Initial clearance experiments established the minimum time that was required for consistent changes in net Na+ and K+ excretion in vivo, and these clearance experiments were used to determine the appropriate time for the perfused tubule studies. Aldosterone consistently reduced renal Na+ excretion by 120 minutes, but without an increase in the rate of K+ excretion by the kidney. In the perfused CCD, aldosterone acutely stimulated Na+ absorption but did not increase K+ secretion.
Chronic action of mineralocorticoids: renal action of aldosterone on K+ transport
In chronic balance studies reproducible effects of long-term mineralocorticoid stimulation on plasma K+ occurred. This effect is well known and has been systematically examined by Young and coworkers, using both metabolic balances and measurement of transcellular K+ distribution with 42K. These investigators demonstrated that although aldosterone did not increase the absolute rate of K excretion aldosterone decreased serum K+ and altered the steady-state distribution of K+ between the intracellular and extracellular compartments (608, 948). From these studies they concluded that aldosterone results in a redistribution of K+ with a greater amount in the intracellular compartment.
Comparison of K+ intake to excretion in metabolic balance experiments revealed no enhanced renal K+ excretion attributable to mineralocorticoid stimulation (i.e. excretion of K+ greater than intake resulting in negative K+ balance) in either dogs (365, 366), pigs (282), rabbits (161), or mice (281, 506). In recent studies mineralocorticoid stimulation resulted in significant positive K+ balance despite significant hypokalemia (506). This is consistent with the observation that mineralocorticoids not only stimulate K+ secretion in the CD but also stimulate the activity of K+ absorbing pumps (281).
The lack of a kaliuretic effect of aldosterone in chronic studies of mineralocorticoid stimulation may in part be explained by the observation that aldosterone increases the activity of H, K-ATPases in the CD (281). Mineralocorticoids increased mRNA for the H+-K+-ATPase α subunit and H+-K+-ATPase activity in a K+-dependent manner, and mice lacking these H-K pumps exhibited negative K+ balance. These studies support a role for H+-K+-ATPase as a reabsorptive mechanism to maintain K+ balance with chronic mineralocorticoid treatment (281). More recently, Lynch et al. examined the effect of chronic DOC on total body Na and K+ balance (506). Whereas the effect of DOC to decrease [K+]p was observed, net K+ balance became positive after DOC administration in normal mice.
K+ Adaptation
It has been known since the early part of the past century (17, 725, 809) that animals could become tolerant to an oral load of K+ that would otherwise be lethal by prolonged feeding of a high K+ diet. Thatcher & Radike reported that “true and specific tolerance to the potassium ion” by increasing the amount of K+ salts given orally (809). In addition, they observed that a significant degree of this tolerance persists for at least seven days. Treatment with adrenal extract or desoxycorticosterone increased K+ tolerance, but both were substantially less effective than the tolerance developed by K+ adaptation to a high K+ load. They suggested that the “primary potassium-tolerance mechanism is based upon a functional change in the kidney” but recognized an important role for the adrenal gland. Such studies have prompted significant investigation into the renal and extra-renal mechanisms of K+ adaptation. K+ adaptation involves adaptive changes in multiple organs including the kidney (332, 353, 659, 766), the adrenal gland (528), skeletal muscle (74), colon (218), vasculature (116, 533, 593), and likely other organs (942). Thus, K+ adaptation involves mechanisms besides aldosterone, but unquestionably includes changes in aldosterone metabolism and action (945).
Giebisch and coworkers (766) examined the effect of K+ intake and aldosterone on distal nephron K+ secretion by in vivo microperfusion when KCl was administered by infusion to intact and adrenalectomized rats. These studies examined the contribution of dietary K+ and aldosterone on K+ adaptation, judged by both the rate of distal nephron K+ secretion and the ultrastructural changes in the degree of increase in the basolateral membrane observed by electron microscopy (766).
A high K+ diet for 10 days induced significant K+ adaptation, defined either functionally (JK) or morphologically in adrenalectomized rats, given fixed dose dexamethasone replacement or low or high dose aldosterone replacement. An increase in aldosterone unaccompanied by a high K+ intake did not enhance the ability of the kidney to excrete an acute K+ load (766). Moreover, on a high K+ diet, an increase in plasma aldosterone was not associated with significant change in urinary K+ excretion (766). These authors concluded that both K+ intake and aldosterone were necessary for full K+ adaptation (766, 767). Moreover, increased plasma aldosterone concentration was not associated with increased K+ excretion. The time required for aldosterone’s adaptive effect are consistent with those of Alexander and Levinsky which are discussed under “Extra-renal action of aldosterone on K+ homeostasis,” subsequently (16).
Extra-renal action of aldosterone on K+ homeostasis
The administration of high-physiological rates of aldosterone administration sufficient to produce hypertension in dogs results in a reduction in steady-state plasma K+ concentration that involves both renal mechanisms, with increased K+ clearance, and extra-renal mechanisms with increased cellular uptake of K+ in tissues other than the kidney (945).
In pathological conditions such as hyperaldosteronism Chobanian et al. reported substantial reductions in total body K+. However, the isolated administration of a mineralocorticoid (9-α-fluorohydrocortisone, DOC), which increased exchangeable 22Na+ and extracellular fluid volume, did not recapitulate the reduction in the total body K+, suggesting that other factors besides isolated mineralocorticoid stimulation contributed to the severe reduction in total body K+ (131).
Whether aldosterone’s physiological renal action results in K+ depletion, is addressed in balance studies and most of these studies do not support a primary effect of aldosterone to promote negative K+ balance. Several balance studies of mineralocorticoid treatment show that initial K+ balance was largely unchanged (158, 281, 282, 365, 366, 678), or became slightly positive despite the development of hypokalemia (161, 506). The latter suggests an increase in tissue K+ content as discussed below (222, 948). It is also unclear whether, in the presence of mineralocorticoid stimulation, the kidney can respond appropriately to a reduction in dietary K+ intake to prevent K+ depletion independent of a smaller steady-state plasma K+ concentration. Does abnormal mineralocorticoid stimulation impair the appropriate adaptation of the kidney and the individual to progressive dietary K+ restriction? This question is likely relevant to the studies of Chobanian et al. that documented substantial K+ depletion in patients with hyperaldosteronism (131) because even small defects in appropriate K+ homeostasis could, with time, result in substantial cumulative K+ deficiency. Given the known potential for K+ depletion to cause or exacerbate hypertension, such studies may provide insight into the development systemic hypertension. For example, if substantial heterogeneity among individuals exists in renal K+ conservation ability, it would be reasonable to determine whether such a defect reflects a propensity for systemic hypertension or CKD. This could have practical importance in all hypertensive patients, and particularly those with hyperaldosteronism.
Studies by Alexander and Levinsky (16) provided important evidence that in addition to its action in the kidney, aldosterone exerted important extra-renal effects on the disposition of a K+ load. The investigators studied normal and nephrectomized rats that were conditioned to a normal or high K+ diet and examined the response of these groups to a fixed amount of KCl administered intraperitoneally. They observed that animals conditioned to the high K+ diet maintained a lower plasma K+ concentration compared to animals conditioned to a regular diet. Moreover, chronic but not acute adrenalectomy abolished this adaptation. When plasma aldosterone was stimulated endogenously by a low Na+ diet, a similar protection to a K+ load was observed and this protection was also observed in adrenalectomized rats after treatment for several days with deoxycorticosterone. The loss of protection that occurred with adrenalectomy did not occur if the operation was performed immediately prior to the administration of a K+ load. Equally important, a single large dose of aldosterone administered acutely just prior to a K+ load failed to elicit the same protective effect they observed with either chronic K+ adaptation to a high K+ diet or several days of deoxycorticosterone administration. Measurement of gastrointestinal K+ content did not reveal evidence that the protection was due to excretion of K+ into the gut. The authors concluded that the protection against a K+ load was due to enhance cellular uptake that required the chronic action of aldosterone (16).
Studies have challenged these findings based on the development of an acute “paradoxical K depletion” in animals fed a high K+ diet after withdrawal of the diet by measurement of the greater K+ excretion (763). Unfortunately, no total measure of K+ content was made, and the studies were not extended to establish whether new steady-state of renal and fecal excretion occurred that was commensurate with intake. Since healthy animals ingesting a very high K+ intake will maintain large rates of K+ excretion to remain in balance, abrupt removal of K+ intake will require time before renal excretion is reduced. Thus, the concept of paradoxical K+ depletion ignores the past large rates of K+ intake to which the kidney must adapt, and that adaptation will not be instantaneous.
Young and Jackson examined the effect of 4–7 days of aldosterone on plasma K+ concentration, exchangeable K (Ke), assessed as 42K, and urinary Na+ and K+ excretion, in dogs ingesting 27 mEq/day of Na+ and K+ (948). They observed that infusion of a high rate (250 μg/day) of aldosterone resulted in a significant one-day reduction in Na excretion, but K+ excretion did not change. By the third and fourth days of aldosterone administration, electrolyte excretion returned to the normal range. Thus, the large rate of aldosterone infusion, which was approximately 3 times the maximum physiological range for the dog resulted in transient Na+ retention during the first day but without a significant increase in K+ excretion. (160) After 24 hours this high rate of aldosterone infusion resulted in a significant decrease in plasma K+ concentration (~11%) and a further ~20% decrease by day 6 (80±1% of control). Ke decreased slightly (~8%), but the discrepancy between the balance measurements that reported no significant negative balance and the effect on Ke showing a small, albeit significant, decrease in Ke was not discussed. Nevertheless, when all values of plasma K+ concentration were plotted against Ke for all high and low dose infusions of aldosterone the relationship between plasma K+ concentration and Ke was shifted so that plasma K+ was less as a function of Ke. The authors concluded that the data “are consistent with the hypothesis that aldosterone alters the distribution of K+ between the intra- and extracellular spaces, a greater portion of total K+ being intracellular at higher levels of aldosterone (948).”
Summary and Conclusions
In summary, under steady-state conditions, states of high aldosterone/mineralocorticoid activity result in a redistribution of K+ with a greater amount intracellular versus extracellular due to mineralocorticoid action to increase the number and abundance of Na pump at the basolateral membrane in the ASDN and in non-polarized MR responsive cells. As such, this increases the extracellular clearance of K+ due to renal and extra-renal mechanisms. Under non-steady-state conditions, this same action of aldosterone to increase Na pump activity allows for more efficient defense against hyperkalemia and renal excretion of K+ when the organism is challenged with a high K+ load.
Aldosterone escape (and breakthrough)
Initial observations and definition
In 1958, George Thorn and his colleagues reported their studies on the administration of supra physiologic dosages of aldosterone to humans given intramuscularly in sesame oil (34). They summarized that “following an initial period of weight gain and Na+ retention, weight gain ceased and Na+ excretion returned approximately to control levels, despite continued administration of the hormone.” The subjects did not develop edema, consistent with the lack of edema typically observed in primary hyperaldosteronism. They concluded that “the ‘paradoxical’ Na+ excretion seen in cases of primary aldosteronism occurs as a result of this same ‘escape’ phenomenon.”
The term “aldosterone escape”, coined by these authors, has been the subject of significant study. However, distinction should be noted between that described by Thorn and his colleagues and a more recently described phenomena, arising from clinical practice in the treatment of hypertension. This phenomenon is the lack of sustained suppression of the RAAS system in patients treated with either an angiotensin-converting enzyme (ACE) inhibitor or angiotensin receptor blocker (ARB) (137). In such cases, plasma aldosterone levels fail to be continuously suppressed and return to baseline (non-suppressed) values after a variable period, and thus exhibit a “breakthrough” from the pharmacological suppression. This second phenomenon, best described as “aldosterone breakthrough” is generally characterized by an unfavorable prognosis, with increased risk of cardiovascular disease. Unfortunately, aldosterone breakthrough has been referred to as “aldosterone escape”, a usage that should be avoided (Table 3).
Table 3:
Aldosterone Escape vs. Aldosterone Breakthrough
| Aldosterone Escape | Aldosterone Breakthrough | |
|---|---|---|
| Definition | The phenomenon of an increase in urinary Na+ excretion (UNaV) following the initial increase in Na+ reabsorption and retention caused by aldosterone. UNaV increases back to baseline levels that occurs despite continued aldosterone action. | The lack of continuing suppression of the renin-angiotensin-aldosterone system (RAAS) despite treatment with RAAS blockers. Plasma aldosterone returns to previously unsuppressed and elevated baseline values, exhibiting a “breakthrough” from the suppression. |
| Mechanisms behind phenomena |
|
|
| Disease state/treatment that induces phenomena | Primary hyperaldosteronism | Treatment with an angiotensin-converting enzyme (ACE) inhibitor or angiotensin receptor blocker (ARB) in patients with hypertension, chronic kidney disease, or chronic heart failure. |
Older studies on aldosterone escape
Since the initial observations of August, Nelson, and Thorn, numerous studies have examined various aspects of this escape phenomenon, including the contribution of systemic BP, renal hemodynamics, specific nephron segments, and signaling mechanisms that are responsible for inhibiting the action of aldosterone or compensating for its action. In particular, the chronic effects of aldosterone on Na+ excretion require appreciate of its effects on systemic BP and renal hemodynamics as emphasized by the studies of Hall et al (316). These investigators showed that if an increase in renal artery pressure was prevented by servo-controlled constriction of the aortic, progressive Na+ retention with the development of ascites occurred.
Early work on the nephron segments responsible for aldosterone escape (hereafter denoted “escape”) suggested contribution of the proximal tubule and the loop of Henle (432, 913). Although prostaglandins and the kallikrein-kinin system were implicated by some pharmacological studies, systematic examination by Ballerman et al. implicated the distal nephron and collecting duct and suggested a role for ANP in the development of escape (46). These investigators observed that mineralocorticoid stimulation resulted in ANP synthesis and release, suggesting a role for ANP to override the action of mineralocorticoids. Yokota et al. provide further support for ANP as a component of this phenomena (935). An antagonist to the guanylate cyclase-coupled natriuretic peptide receptors significantly attenuated, but did not abolish chronic escape phase, and proposed that cardiac ANP initiated escape. Studies by Lee et al. further supported a role of natriuretic peptides in escape, and demonstrated that the rate of renal excretion of ANP increased after two days of mineralocorticoid stimulation with DOCA (466). In rats administered DOCA and a high NaCl diet, they detected an increase in mRNA expression not only in cardiac ventricles, but also the renal cortex as well. They proposed that in addition to the heart, the kidney may possess a paracrine mechanism that contributes to escape. Although endogenous mineralocorticoid stimulation of ANP appeared unlikely, since a low NaCl diet with stimulated plasma renin (aldosterone was not reported) did not increase urinary ANP excretion, the contribution of dietary NaCl intake on DOCA stimulation of ANP was not established since DOCA treatment on a low NaCl intake was not examined. The role of dietary NaCl as a contributing factor necessary for escape is plausible since ANP fails to induce a natriuresis under certain conditions of volume expansion such as pregnancy (520).
Recent studies of the mechanism of aldosterone escape
The sustained action of aldosterone to promote Na+ absorption is known to be attenuated by mechanisms that are only partially understood. The mechanism for aldosterone escape could reflect suppression of Na+ absorption in more proximal segments of the kidney or inhibition of the action of aldosterone in the ASDN & CD. Changes in proximal or distal tubule transport (871), secretion and renal action of natriuretic peptides (37), prostaglandins (397), nitric oxide (830), and purinergic signaling (551, 777) have all been invoked as an explanation for the renal “escape” from the action of aldosterone, and it is likely that many of these signaling systems participate in this escape mechanism. However, most studies are qualitative and quantification of the exact contribution to overall aldosterone escape is frequently not performed.
A proteomics approach to quantify the abundance of specific Na+ transporters at four days after MC stimulation showed that of the transporters tested, the Na+/H+ exchanger 3 (NHE3), NaPi-2, NKCC2, NCC, ENaC subunits (α, ß, γ), or Na+-K+-ATPase, only NCC (encoded by slc12a3) (871) showed a marked reduction in the level of total protein abundance. Other studies have shown evidence for a role of purinergic signaling in the escape by assessing the contribution of P2Y2 receptors as assess in WT and P2Y2 null mice (777). Whereas these studies clearly suggest that purinergic signaling via P2Y2 receptors participates in escape, the magnitude of the contribution of this system is not known. Since release of ATP into the lumen is necessary for purinergic signaling, plausible candidate transporters have been examined. One of these, connexin 30 appears to be important in the ASDN & CD, and, the differential reduction in Na+ excretion due to mineralocorticoid stimulation in the Cx30 null mice versus WT mice (551) suggests that purinergic signaling participates in the aldosterone escape.
Other studies have examined the role of prostaglandins and specifically the microsomal enzyme prostaglandin E synthase-1 (mPGES-1) and suggest a role for this system as a component of MC escape (397). In mice, a 14-day aldosterone infusion (0.35 mg × kg−1 × day−1) via an osmotic minipump on a normal salt intake produced a transient Na+ accumulation on the first two days in WT mice but after 14 days Na+ and water balance were normal. However, the same treatment in mice null for mPGES-1 resulted in increased Na+ and water balance, body weight. During escape, WT mice had a significant increase in urinary PGE(2) excretion increased mPGES-1 in proximal tubules, and reduction in the expression of most major transporters of the proximal tubule (NHE3), TAL (NKCC2), or DCT (NCC) renal Na+ and water transporters. The reduction in NHE3, NKCC2, and NCC expression and the increase in urinary PGE2 was substantially reduced in the mPGES-1 null mice, which had significant increases in Na+ and water intake during aldosterone treatment. These studies strongly suggest that mPGES-1 production of PGE-2 or other prostanoids opposed the Na+-retaining action of aldosterone. The action of aldosterone could reflect renal actions of aldosterone or extra-renal actions, or both.
Since aldosterone is known to stimulate ET-1 expression in the kidney (782) and a high Na+ diet increases renal ET-1 production (430, 431), studies examined the action of MC-stimulated renal ET-1 production to inhibit renal Na+ absorption as a mechanism to explain aldosterone escape. This would be consistent with the effect of ET-1 to attenuate the development of salt-sensitive hypertension (8). Lynch et al. hypothesized that a local renal aldosterone-ET-1 feedback system (RAEFS) exerted considerable influence on the final effect of aldosterone, by attenuating aldosterone’s action, particularly when aldosterone’s action was inappropriate for the level of Na+ intake. They tested whether renal ET-1 expression was necessary for the escape phenomena (506), and observed transient Na+ retention on day 2–3 and the typical escape observed in WT mice to DOC. However, this escape was absent in mice with cell-specific disruption of edn1 in principal and inner medullary collecting duct cells. The lack of compensation by other systems suggests that ET-1 is an essential component of the escape phenomena, and that other systems could not compensate for its loss.
Thus, multiple changes can compensate for the enhanced Na+ absorption induced by long-term mineralocorticoid stimulation such as modulation of transport in more proximal segments, purinergic signaling, and prostaglandins each of which may be sufficient to compensate for defects in common or parallel elements. However, the signals necessary for this down-regulation along the nephron appear to require, or are depend upon, ET-1 expression which is a non-redundant and necessary component for aldosterone escape. The components necessary for operation of this local renal feedback system will require further study.
Aldosterone Signaling in Renal Epithelial Cells
Multiple lines of evidence suggested that aldosterone activation of Na+ absorption was more complex than merely due to a de novo increase in the number of channels. Patch-clamp analysis noted previously suggested that aldosterone’s action was to change the kinetics of the channel via changes in mean open time (Po) (415). In addition, the studies of Spindler et al. did not support a direct and immediate action of aldosterone to increase expression of subunits of ENaC (761). Thus, evidence suggested that aldosterone increased Na+ absorption via signaling due to intermediate molecules and processes, but the nature of these events remained elusive. The nature of cell signaling is likely to be tissue- or cell type-specific, so conclusions across different cell types or systems should be considered provisional and accepted with caution.
Post-translational processing of ENaC: Role of methylation and Ras signaling
Two interrelated pathways appear to be important elements in the early aldosterone signaling mediated by methylation: methylation of Ras and ßENaC. (Palmitoylation, which is another post-translational modification is discuss in the section on “ENaC subunit structure”.) In cell culture systems protein methylation appears to be critical in the early response of aldosterone and the activation of ENaC and the ß subunit of ENaC is one of the targets of this methylation. The amount of Na+ absorption in frog Xenopus kidney A6 cells was substantially increased by agents that methylated membrane proteins. Further studies implicated guanine nucleotide-dependent carboxymethylation as a pathway for aldosterone modulation of apical Na+ permeability (696–698). Aldosterone induced an increase in GTP-stimulated methylation of a 90 kDa polypeptide at the apical membrane in A6 cells (698). Mastroberardino et al. tested whether the rapid induction of K-Ras2A was responsible for the increase in Na+ absorption using a Xenopus oocyte expression system. Overexpressing K-ras2 resulted in stimulation of Na+ absorption, in addition to its effect to induce oocyte maturation (523). Progesterone was used as a control for oocyte maturation and when the effect of K-Ras2A was compared to that of progesterone, the relative effect suggested that K-Ras2A increased the mean endogenous ENaC activity.
The previous identification of K-ras2 as an aldosterone-regulated gene was further examined at the level of protein expression in A6 cells and also in vivo (762). Aldosterone treatment of female frogs (Xenopus laevis) for 150 minutes resulted in a kidney-specific induction of K-ras2 mRNA by 250%. In A6 epithelia, aldosterone treatment for 150 minutes increased p21ras protein synthesis ~six-fold. However, aldosterone had other effects including a modest induction of Xl-ras and fra-2 mRNA, but suppression of mRNA for GR, and time dependent changes in c-fos, c-jun, and c-myc. These findings suggest that aldosterone’s effects are not limited to a single signaling mechanism but are diverse and include mechanisms besides K-Ras2 signaling. However, K-ras2 is a membrane associated small monomeric G protein, and members of this family (K-RasA, K-RasB, H-Ras, and N-Ras) stimulate multiple activators of distal signaling pathways, with the principal immediate effectors being Raf, RalGDS, and PI3-kinase which initiate, respectively, the MAPK 1/2, Ral/Rac/Rho and PI3-kinase cascades (770). K-Ras proteins possess a consensus sequence CAAX (or CAAL) at their carboxy terminus that, with a second signal are sufficient to promote targeting of ras proteins to the plasma membrane (321, 340). The CAAX sequence is a target for protein methylation. Additional studies also suggested a role for a 20 kDa protein small G-proteins in the action of aldosterone. Specifically, antisense inhibition of K-ras markedly reduced Na+ channel activity in A6 cells (13), suggesting that the response was dependent on this protein.
However, and not surprisingly, the mouse CCD cell line and the amphibian A6 cells do exhibit differences in response to aldosterone (188). In the CCD cell line, aldosterone increased Na+ absorption after 60 minutes along with methylase activity but without changes in the expression any ENaC subunit. Generalized inhibition of methylation reactions completely inhibited aldosterone’s effect on SCC but more specific inhibitors of small G-protein methylation did not inhibit the early increase in SCC by aldosterone. Overexpressing the ras-specific methyl transferase increased aldosterone-stimulated SCC in A6 cell whereas similar overexpression in CCD cells affected neither the basal nor aldosterone-stimulate SCC. These studies suggest that aldosterone increases protein methylation, an activity that is necessary to increase SCC/Na+ transport, but without a requirement for ras methylation. Similar to A6 cells, in CCD cells aldosterone increased the methylation of proteins and this action was necessary for the early increase in SCC. In addition, one of the methylated proteins is the ß subunit of ENaC. However, in contrast to A6 cells, in CCD cells k-ras was not detected as an AIP and blocking the action of k-ras failed to inhibit the early response to aldosterone. Thus, in CCD cells the aldosterone-induced increase in SCC/Na+ transport is not mediated via methylation of ras (which is not induced by aldosterone) and overexpression of the enzyme that methylates ras did not directly increase SCC/Na+ transport. Such differences may reflect the level or regulation of k-ras expression, the requirement of additional signaling mechanisms, or other time-dependent differences in amphibian A6 versus mammalian CCD cells. Thus, assuming the specificity of methylation inhibition, in mammalian cells, the early activation of ENaC requires methylation, likely of the ENaC ß-subunit, as has demonstrated in A6 cells (188). Further study of the plasma membrane-integral or associated proteins involved in this action is likely to be profitable.
Post-translational processing of ENaC: Proteolytic cleavage and paracrine signaling mechanisms
Substantial work supports the role for regulation of αβγ-ENaC activity by proteolytic cleavage of ENaC subunits by the serine endoproteases (or endopeptidase) furin, kallikrein, and prostasin. Several recent reviews provide an in-depth analysis of this area (420, 455, 619). Alterations in the activity of endopeptidase by aldosterone may be dependent on dietary Na+ or K+ intake. This statement is not without precedent since there is evidence that tissue kallikrein (and the Kinin-kallikrein system) is regulated by Na+ and K+ intake (192), and other systems may be subject to this regulation. This would provide an additional control mechanism for the fine-tune adjustment of transport in the ASDN by more proximal tubule segments. Other systems that may also exert a paracrine control on aldosterone-mediated transport in the ASDN include changes in bioactive substrates such as ammonia/ammonium, nitric oxide, substrates that are metabolized, and ET-1 (456, 474, 722, 821). Whether aldosterone directly or indirectly regulates the specific cellular mechanisms that result in the maturation of functional channels deserves further study.
In the degenerin/ENaC protein family, αβγ-ENaC is the only constitutively open ion channel, and its regulation must be tightly controlled to avoid excessive Na+ absorption. Indeed, it is well known that ENaC processing, cleavage, maturation, and trafficking to the plasma membrane involves multiple steps, and each have the potential for regulating the mature, fully functional channel, and thus Na+ absorption. Furin and prostasin are channel-activating proteases, and the discovery that αβγ-ENaC activity was regulated by protease activity provided significant new insight into epithelial Na+ transport (836). Both the α and γ subunits are cleaved twice, which releases inhibitory tracts and transitions channels to a state of greater open probability (Po). Whereas furin cleaves αENaC twice, it cleaves γENaC only once. A second protease cleaves γENaC to transition ENaC to a very high Po state. Aldosterone stimulates the cleavage of γENaC by two proteases, (364, 682, 793, 814, 894, 954, 957). Processing occurs in the cytosol, and at the plasma membrane. (792, 793, 831, 957).
In this regard, the findings of Wichmann et al. (894) illustrate the importance and complexity of channel maturation. These investigators examined and contrasted the processing of αβγ-ENaC and δβγ-ENaC in Xenopus laevis as a model expression system. They showed that the δ-subunit enhances the amount of current generated by ENaC due to an increased open probability. The fully functional αβγ-ENaC required proteolytic processing of the α- and γ-subunits, but the δ-subunit did not undergo proteolytic maturation by endogenous furin. The currents generated by δβγ-ENaC were not activated by extracellular chymotrypsin, and equally important expression of the δ-subunit prevented cell surface cleavage of γ-ENaC. They concluded that subunit composition represents an additional level of ENaC regulation. However, the experimental systems need to be noted. Whereas human δβγENaC do not exhibit Na self-inhibition, Xenopus channels have robust inhibition. Further detailed comparison of δβγ-ENaC versus αβγ-ENaC in terms of pharmacological and enzymatic inhibition, channel gating and kinetics, signaling mechanisms, and interacting proteins will likely be highly informative.
Post-translational processing of ENaC: Role of phosphorylation
Evidence for physiologically significant in vivo regulation of ENaC by direct ENaC phosphorylation remains unsettled. Studies in a MDCK cell line that stably expressed ENaC subunits examined whether aldosterone modified ENaC activity at 0, 3, or 16 hours by direct channel phosphorylation (740). Aldosterone increased amiloride-sensitive SCC by ~two-fold by three hours. These studies suggested a mechanism of action of aldosterone working through at least one serine/threonine kinase to modulate the activity of ENaC by direct phosphorylation of the β and γ subunits. Some evidence suggested direct ENaC subunit phosphorylation in Xenopus oocyte expression assays as the Liddle-mutation of the ß-subunit reduced Protein Kinase C (PKC) inhibition of the human ENaC, the Liddle mutation reported in the γ subunit were still sensitive to inhibition by PKC (672), but these results are not entirely consistent with previous reports (740). In both cases phorbol 12-myristate 13-acetate was used to assess PKC action.
Studies have shown that both ERK and casein kinase II can phosphorylate ENaC subunits and these actions appear to be physiologically relevant (63, 732, 931). ERK has been shown to phosphorylate the carboxyl end of ß and γ ENaC at ßThr-613 and γThr623, respectively (731). Inhibition of ERK1/2 phosphorylation using the ERK kinase inhibitor PD-98059 reduced EGF and ATP-dependent inhibition of amiloride-sensitive SCC (204) During a state of hypotonic stress, a p38-dependent induction of MAPK phosphatase 1 (MKP-1) induces β and γ ENaC expression through dephosphorylation of ERK (575). The casein kinase II phosphorylation site in ß ENaC was found more recently by Abd El-Aziz et al. to modulate channel activity. Proper casein kinase II signaling facilitates ankyrin-3 binding to increase ENaC activity by increasing its expression in the plasma membrane (1).
With the discovery of Sgk1 as a rapidly induced kinase that served as a critical signal intermediary in the action of aldosterone, it became apparent that this kinase was responsible for much of the early signaling. Other genes were also identified to be rapidly induced or repressed by aldosterone (see section “Aldosterone-induced Transcripts (AITs) and Aldosterone-Induced Proteins (AIPs)).” Nevertheless, the central importance of Sgk1 is emphasized by its interaction with mTOR, ubiquitin ligases, and the WNK signaling pathway.
Post-translational processing of ENaC: Role of acetylation
Acetylation is another post-translational modification that has been shown to affect MR and Na+ transport. Lysine deacetylase (KDAC) inhibition has been shown to have an antihypertensive effect through studies with spontaneously hypertensive rats. (730)To determine the molecular mechanism behind this antihypertensive response, MR activity was studied in Sprague Dawley rats that underwent uninephrectomy and were given DOCA salt treatment with or without valproate, a KDAC inhibitor. Lee et al found that KDAC inhibition attenuated the transcriptional activity of MR through acetylation along with preventing the development of DOCA-induced hypertension. In the spontaneously hypertensive rat, KDAC inhibition of lysine deacetylase (KDAC) leads to increased acetylation of MR in the kidney cortex. The increased MR acetylation did not affect MR protein expression and was independent of histone acetylation (730). In murine CCD cells, the KDAC inhibitor Trichostatin A suppressed aldosterone-mediated control of Na+ transport by ENaC and SGK1 activity and abundance (516). It is known that Hsp90 helps to influence MR subcellular dynamics (198). Jiménez-Canino et al. went further to analyze the mechanism behind this regulation of subcellular dynamics and transactivation of MR, modifying Hsp90 acetylation levels through HDAC6 expression or activity (399). They found that in transfected COS-7 cells, Hsp90 acetylation shifted the subcellular localization of naïve MR towards the nucleus mediated by HDAC6, and this acetylation occurred at residue K294 in humans (K295 in mouse ortholog) (399). Because ubiquitination also occurs on Lys residues, one additional consequence of acetylation of lysine residues would be to prevent channel ubiquitination and associated channel endocytosis.
Phosphatidylinositol 3-Kinases
PI3-kinases are a family of proteins that phosphorylate the 3’-hydroxyl group of the inositol ring of phosphatidylinositol. The first member of the family, pp60c-src, was immunoprecipitated from polyoma transformed cells (893). Since that initial observation, over a dozen PI3-kinases have been identified. The PI3-kinase family consists of 14 enzymes that Gharbi et al. divide into four groups based on their structural features that catalyze the formation of 3-polyphosphoinositides (259, 840). Class 1A (the most widely expressed and well-studied PI3-kinase (424) are heterodimers of an ~110 kDa enzymatic subunit (α, ß, or δ) and an 85 kDa regulatory subunit (α,ß, or γ) (840). PI3-kinases can be classified on the basis of inhibitor specificities, also (424).
PI3-kinase appears to play a critical role in the action of aldosterone on Na+ absorption. Blazer-Yost et al. examined the in vitro action of aldosterone in A6 cells and reported a requirement for functional PI3-kinase for aldosterone-regulated Na+ absorption (76). Aldosterone at 1μM increased SCC at 60 minutes and monotonically thereafter for the duration of the experiment (300 minutes). A relatively specific inhibitor of PI3-kinase (LY-29004, 50μM), decreased basal and aldosterone-stimulated SCC, as did the PI3-kinase inhibitor wortmannin (100nM). Wortmannin, in contrast to LY-29004, did not inhibit basal SCC. Although accepted as a relatively specific PI3-kinase inhibitor, further study indicated that LY-29004 may have other targets (259). With that caveat, these studies would suggest that the action of aldosterone requires a functional PI3-kinase to stimulate Na+ absorption.
PI3-kinase may also participate in the activation of SGK1. Several groups independently demonstrated that SGK1 is a substrate of PI3-kinase. In particular, SGK1 activation was reported to be completely abolished by pre-treatment with a PI3-kinase inhibitor (427, 609). Furthermore, mutation of the putative PI3-kinase phosphorylation sites on SGK1, Thr256 and Ser422, inhibited SGK1 activation. Thus, by modulating SGK1 activity, PI3-kinase has been implicated as a component of the signaling mechanism for aldosterone’s regulation of Na+ reabsorption. Staruschenko et al. showed that K-Ras could activate ENaC in cells stably expressing α, ß and γ ENaC that was GTP-dependent and required PI3K for this activation (770). Additional evidence indicates that Ras couples phosphoinositide 3-OH kinase to ENaC (770), that phosphatidylinositides are critical to this acute regulation of ENaC (640, 641, 772), and that PI3K acts to acutely regulate ENaC (771). The role of Rac1 as a modulator of ENaC activity was further explored by Staruschenko and colleagues, using the Rac1 inhibitor NSC23766 which markedly decreased ENaC activity in freshly isolated CDs and explored the interaction and regulation of ENaC by Wiskott-Aldrich syndrome protein which participates in the control of the cytoskeletal. These studies, which identified new regulators of ENaC, further link the regulation of ENaC with the cytoskeleton (408).
PI3-Akt-mTOR2-OSR1
Significant interactions exist between the PI3-kinase, Akt, and mTOR (mammalian/mechanistic target of rapamycin) signaling pathways that appears to be aldosterone-sensitive. ENaC regulation involves a multi-protein complex that acts through PI3-kinase, mTOR, and Raf-MEK-ERK-dependent pathway, the action of the latter serves to reduce channel activity by Nedd4-2 mediated endocytosis (752). The convergence of these ostensibly disparate signals in the action of ENaC regulation is further emphasized by Pavlov et al. who studied the mechanism of ENaC regulation in response to insulin. Insulin acutely increased ENaC activity in split open tubules and increased transepithelial flux in MDCK cells that was sensitive to both the PI3-kinase inhibitor LY294002 and the mTOR inhibitor, PP242. Since aldosterone increases amiloride-sensitive flux by a mechanism that is sensitive to PI3-kinase inhibitors, common elements appear to participate in both signaling systems, and further studies and likely to be productive (616). In view of evidence that ENaC and in NCC may physically interact (426, 553), studies by various groups linking NCC and the WNK signaling pathway and OSR1 suggest an intricate and interconnected cellular regulatory system.
Serum- and glucocorticoid-induced kinase
Three mammalian isoforms have been identified, Sgk1, Sgk2, and Sgk3. With the discovery that aldosterone rapidly stimulated the induction of Sgk1 (123, 568), a serine/threonine kinase, and evidence that Sgk1 increased ENaC abundance at the plasma membrane of oocytes (18, 739), this rapidly induced kinase became a highly plausible mediator of some or perhaps most of aldosterone’s action, at least with reference to its action on Na+ transport. Subsequent work identified additional early response genes for the action of aldosterone (68, 123, 306, 308, 617, 756, 757, 782). Pearce and colleagues identified SGK1 and subsequently GILZ as proteins involved in electrolyte transport (123, 756, 757). Moreover, these and further work emphasized the importance of the regulation of cellular trafficking of ENaC to and from the plasma membrane as one mechanism to increase aldosterone-stimulated Na+-current. Nedd4-2, an E3 ubiquitin ligase involved in the regulation of ENaC recycling at the apical membrane, is an example of a protein regulated indirectly by aldosterone via activation of Sgk1 (32, 82, 163, 217, 341, 406, 438, 454, 512, 618, 749).
Within renal tubules, aldosterone induced a significant expression of Sgk1 at both the mRNA and protein levels (123). A major target of SGK1 phosphorylation within renal tissues is Nedd4-2 (66). Because Nedd4-2 targets ENaC for degradation (405), it functions to limit Na+ reabsorption. SGK1 interacts with Nedd4-2 through a PY motif and phosphorylates it on Ser 444 and, to a lesser extent, Ser 338 (163). These post-translational modifications interfere with the interaction between Nedd4-2 and ENaC. This in turn leads to less degradation of ENaC stabilizing it on the apical membrane and consequently increasing Na+ currents (163). These early works defined an initial regulatory pathway for the reabsorption of Na+ and provided a mechanism for aldosterone-dependent ENaC retention at the apical membrane (Figure 12) (163). The aldosterone/SGK1/Nedd4-2 pathway has been reviewed extensively elsewhere (755, 849) and therefore this signaling pathway will not be addressed in detail here.
Figure 12: Molecular pathways through which aldosterone can stimulate ENaC.
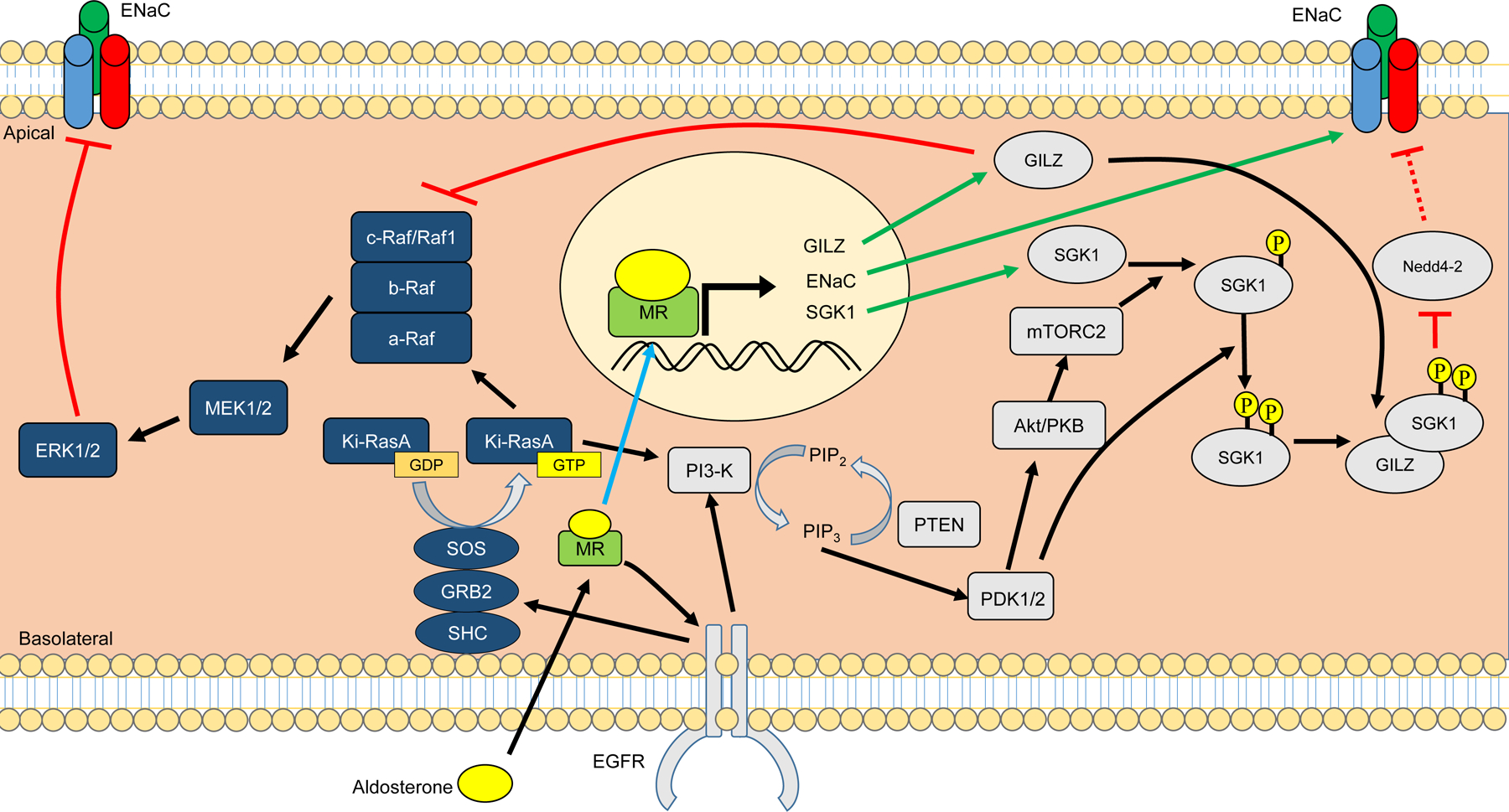
Mineralocorticoid receptor (MR) activation by aldosterone leads to stimulation of the epidermal growth factor receptor (EGFR). Stimulation of EGFR lead to multiple cascading pathways, including the mitogen activated protein kinase (MAPK) and the phosphatidylinositol 3-kinase (PI3-K) pathways. The MAPK pathway leads to an inhibition of ENaC by the ERK1/2, reducing Na+ reabsorption and promoting urinary Na+ excretion. The PI3-K pathway leads to the stimulation of ENaC through the synergistic inhibition of the ubiquitin ligase Nedd4-2 by MR transcription products SGK1 and GILZ. GILZ can also inhibit the ability of the MAPK pathway to inhibit ENaC by inhibiting Raf1 (c-Raf). Black arrows indicate direction of pathways. Blue arrows indicate aldosterone-bound MR relocating to the nucleus. Green arrows indicate MR transcription products. ENaC, epithelial sodium channel; GILZ, glucocorticoid-induced leucine zipper; SGK1, serum- and glucocorticoid-induced kinase 1; PKB, protein kinase B (also known as Akt), PTEN, Phosphatidylinositol-3,4,5-trisphosphate 3-phosphatase; mTORC2, mammalian target of rapamycin complex 2, PDK1/2, phosphoinositide-dependent kinases 1 and 2; ERK1/2, extracellular signal-regulated kinases 1 and 2; MEK, MAPK/ERK kinase or mitogen-activated protein kinase kinase (MAPKK).
Sgk1 exerts other actions, including an increase in CFTR current in Xenopus oocytes (860), and the expression and function of SGK1 (477) is inhibited in IMCD-3 cells by a purinergic receptor- (P2R) mediated mechanism, and the aldosterone induced increase of SGK1 kinase activity was also suppressed by P2R agonists (477). Since P2R stimulation consistently inhibits Na+ absorption, this suggests a potential contribution of Sgk1 to purinergic signaling. Studies in mpkCCDc14 cells extend and validate the mechanism of action described in oocytes and studied the mechanism for aldosterone to increase ENaC activity and current by Nedd 4–2 inhibition and the role of 14-3-3 protein isoforms in this process (66). Tryptophan-rich sequences (WW motifs) in Nedd4-2 interact with a proline-tyrosine PY motif in the C-terminal tail of ENaC and this interaction is necessary for Nedd4-2 inhibition of ENaC. As in oocytes, SGK1 phosphorylates and inhibits the ubiquitin ligase Nedd4-2 and decreases cell surface expression of ENaC. This inhibition occurs in part by a phosphorylation dependent interaction of Nedd4-2 with 14-3-3 regulatory protein isoforms. Nedd4-2-mutants S444A and P446A were not able to interact with 14-3-3 in a SGK1-dependent manner suggesting that Nedd4-2 phosphorylation by SGK1 at Ser444 facilitates interaction Nedd4-2 with a 14-3-3 family member. A likely candidate for this interaction is 14-3-3 γ that has been shown to be rapidly induced by aldosterone (306). Multiple sites appear to be involved in this interaction including a proline downstream of serine 444 (66).
The relative contribution of channel number versus the effect of aldosterone-dependent activation of existing apical membrane channels has been examined by several careful studies. Physiological studies provided evidence that aldosterone modified the activation or kinetics of existing apical membrane channels (415, 601). With the identification of Sgk1 as an aldosterone-inducted protein and its mechanism to increase ENaC trafficking and retention by Sgk1 inhibition of ENaC ubiquitylation (163, 224, 749) performed careful quantitative analysis of apical membrane ENaC subunit expression in response to aldosterone. These investigators concluded that some but not all the action of aldosterone on Na+ current were due to membrane trafficking with a two to five-fold augmentation of surface channel subunit expression, but that Na+ current could vary by as much as 50-fold from Na+ replete–aldosterone-suppressed states to Na+-depleted and high aldosterone states(224). Further work should seek to quantify the contribution of membrane trafficking and channel activity to Na+ absorption, and the mechanisms and time scale over which they operate.
Protein Kinase A/cyclic AMP-dependent Protein Kinase
PKA is activated by increasing levels of cAMP within the cell. When activated, it phosphorylates several diverse cellular targets including enzymes, ion channels, transcription factors, and other regulatory proteins. Initial reports indicating a possible role for PKA in mediating Na+ reabsorption arose when it was demonstrated that elevated cAMP levels increased SGK1 protein expression and activation in both ovarian granulosa cells (272) and COS-7 cells (623). However, the functional ability of these specific signaling events to alter Na+ currents was not confirmed until Mustafa and colleagues showed that the increased SGK1 expression and function results in increased ENaC protein levels and Na+ current (842). Furthermore, these events were sensitive to both PKA and PI3-kinase inhibitors. This indicates that in cells with minimal endogenous α-ENaC expression, SMG-C6 cells, SGK1 is stimulated by DbcAMP via both PI3-K and PKA pathways and that a number of diverse signaling pathways, including the PKA pathway, converge at the level of SGK1. Presently, there is no data indicating that aldosterone, per se, increases PKA activity. However, ADH is fully capable of driving an increase in intracellular cAMP levels, and in turn increasing PKA activity. Thus, in a state of anti-diuresis, the effects of ADH could presumably work in conjunction with aldosterone to retain salt and water via a mechanism that comprises ADH-mediated increases in cAMP levels, subsequent activation of PKA, and ensuing increases in Na+ reabsorption.
Principal Cell-Intercalated Cell MR interactions, MR phosphorylation & localization, and MR-Rac1 interactions
Much work has focused on the action of MR-mediated effects on Na+ transport in principal cells (PCs) of the ASDN (See section “Post-translational Processing: Role of Phosphorylation”). However, MR is also expressed in ICs and recent evidence would suggest that the signaling mechanism is cell type specific, and specific site phosphorylation of MR, MR localization, and MR regulation by small GTPases are important in mineralocorticoid-dependent signal transduction.
MR interacts with the small GTPase protein Rac1 (737), and previous evidence suggested that disruption of the rho GTP dissociation inhibitor α gene (Arhgdia null) was associated with a significant renal phenotype characterized by proteinuria, rapid progression of renal disease, sterility in males, and reduced lifespan (819). Subsequent in vitro work implicated a connection between MR and Rac1, namely, a constituently active mutant Rac1 increased MR transcriptional activity and nuclear localization. Arhgdia null mice had increased Rac1 and MR signaling, and renal pathology was attenuated by either the MR antagonist eplerenone or the Rac-specific inhibitor NSC23766. However, plasma aldosterone concentrations and systemic BP were similar to wild type controls. Collectively, such data implicate Rac1 as a component in proteinuric renal disease responsive to MR antagonism.
Angiotensin II, mTOR, and circadian clock signaling
Aldosterone is also known to increase the expression of numerous genes besides sgk1 including per1, edn1, gilz, scnn1α, nherf2, and ctgf (306, 757). In addition, aldosterone shares common signaling pathways with Ang II including the PI3K/PKC pathway (83) and with other growth factor signaling pathways (83, 109, 270, 515). The interaction of these two important pressors also share common elements with core circadian clock proteins and with mTOR.
mTOR is part of two regulatory multimeric protein complexes: mTORC1 and mTORC2, which appear to govern distinct but overlapping functions of cell physiology. mTORC1 is believed to regulate and sense energy levels, AMP/ATP ratio, amino acid content and regulates autophagy cell cycle progression and growth, whereas evidence suggests that mTORC2 regulates cytoskeletal organization and cell survival. Both are involved in cell metabolism (For review see (14, 457, 707)). mTOR is a large protein kinase initially identified by genetic screens of yeast that exhibited toxic effects to rapamycin. The mammalian homolog (mTOR) is an atypical serine/threonine protein kinase of the PI3-kinase superfamily. It assembles with other proteins as mTORC1 and mTORC2, which each contain the catalytic kinase mTOR subunit. Rapamycin acts to affect a gain of function complex with a small intracellular protein FK506-binding protein (FKBP12) that inhibits mTOR as part of mTORC1 but not mTORC2. mTORC1 senses multiple signals and is regulated by hamartin-tuberin (tuberous sclerosis 1 and tuberous sclerosis 2 complex or TSC 1/2). The hamartin-tuberin heterodimer complex functions as a GTPase activating protein (GAP) for the Ras homolog enriched in brain (Rheb) GTPases. The GTP-bound form of Rheb interacts with mTORC1 to increase its activity. TSC 1/2 inhibits Rheb and acts as a negative regulator of mTORC1. mTORC2 is essential for SGK1 activity (302, 303, 458). mTORC2 phosphorylates akt within its hydrophobic sequence, and similarly phosphorylates and activates SGK1. The proteins Raptor and Rictor confer the pharmacological properties of mTORC1 and mTORC2, respectively. Phosphorylation of SGK1 occurs by mTORC2 that contains rictor whereas mTORC1 does not phosphorylate SGK1. Inhibition of mTORC2 inhibited both the phosphorylation of SGK1 and reduced amiloride sensitive Na+ transport, which indicates this complex affects signaling of transport pathways (502, 503, 752).
Shibata and colleagues identified unc-51-like kinase 1 (ULK1) as a kinase that phosphorylated MR to inhibit its function in ICs (736). Ang II signals through the AT1 receptor and mTOR to phosphorylate ULK1. mTOR has been identified as a kinase that phosphorylates and inhibits ULK1 at S757 (407). The ATP-competitive mTOR inhibitor AZD8055 inhibits the phosphorylation of ULK1 at S757 but this reaction is not inhibited by rapamycin. Ang II inhibits ULK1 via mTOR, and ULK1 phosphorylation of MR at S843 inhibits MR action by reducing ligand-dependent nuclear localization and transcription. Thus, the action of Ang II, acting through mTOR and ULK1 resulted in a decrease in MR phosphorylation at S843 and resulted in an increase in the expression of pendrin. This Ang II effect could be blocked by AZD8055, which prevented the increase in pendrin expression. However, the abundance of ENaC in PCs or NCC in the DCT was not inhibited by ACZ8055.
These studies provide a potential connection between epithelial transport, metabolism, and autophagy. Previous studies have shown that ULK1 is a target protein that is phosphorylated by mTOR and plays a role in autophagy. In addition, ULK1 inhibits cell proliferation by its action to phosphorylate Raptor. ULK1 also phosphorylates hexokinase which results in glucose metabolism by the hexose monophosphate shunt pathway. Additional evidence indicates that mTORC1 phosphorylates SGK1 and mTORC2 modulates the activity of ENaC and apical K+ channels in PCs. Sengupta et al provided evidence that mTOR activates the OSR1, a protein kinase which directly phosphorylates NCC (728).
How Ang II stimulates mTOR needs further investigation. Some work suggests a role for EGF receptor in the Ang II-mediated regulation of mTOR intestinal epithelial cells (130). Additional evidence suggests Ang II mediates its actions in part by WNK4 which acts to inhibit ULK1 expression levels. Specifically, Ang II was shown to increase both the level of expression and kinase activity of WNK kinases (114). Phosphorylation of MR on serine 843 modulates the activity of WNK kinases. Such observations have led to the speculation that WNK kinases act as an intermediate signal of Ang II to mTOR.
mTORC2 phosphorylates and regulates the activity of SGK1, and additional studies indicate that mTORC2 affects ENaC activity in principal cells (262, 279). mTOR also phosphorylates and activates OSR1 (728). Angiotensin II increases mTOR activity possibly by EGF receptor activation and phosphorylation of S6, WNK1, and other kinases (191, 867), and decreased WNK1 activity increases ULK1 expression and phosphorylation of phospho-(Ser555) ULK1 and activation of AMP-activated protein kinase (242). Several reports indicate that Ang II can increase expression and activity of WNK kinases and the degree of phosphorylation of MR on serine 843 (human MR, MR S843-P). Shibata et al have speculated that one or more WNK kinases represent part of the signal transduction of Ang II and mTOR signaling (735, 798, 961).
Further evidence supports a connection between mTOR signaling and the circadian clock in other cell types, which suggests that similar interactions may be observed in the kidney (110, 488, 542, 695, 917). Specifically, the core clock protein PER2 inhibits mTORC1 activity by recruitment of Tsc1 to mTORC1 complex (917), and the core clock protein BMAL1 is reported to regulate translation by S6K1-mediated phosphorylation, thus serving a distinct function from its transcriptional activity (488, 542). This latter observation is particularly intriguing given the evidence that key components of the translational machinery, eukaryotic translation initiation factor 4E and eukaryotic translation initiation factor 4E-binding protein (4EBP), exhibit diurnal oscillation in the mouse hippocampus (695). Whereas such observations have not been demonstrated in the kidney, the common and fundamental nature of protein translation suggests that study of connections in other tissues, including the kidney may be profitable.
Epidermal Growth Factor
Additional evidence supports a role for aldosterone to act through the EGF signaling pathway (295). Growth factors such as EGF produce marked cellular responses via activation of mitogenic pathways. Aldosterone treatment also increased the tyrosine phosphorylation of the EGFR itself and pretreatment with an EGFR receptor tyrosine kinase inhibitor abolished this effect. Using MDCK cells, Silbernagl and colleagues found that nanomolar concentrations of aldosterone induced a rapid increase in ERK1/2 phosphorylation similar to that observed with EGF (253). Aldosterone treatment of these same cells also increased intracellular Ca2+ levels and Na+-H+ exchange activity. Moreover, subsequent studies showed that aldosterone has the capacity to increase both EGFR mRNA and protein levels (444). The sum effect of aldosterone coupling to the EGFR signaling pathway in renal cells appears to be that this rapid and non-genomic mechanism is a means to provide negative feedback to aldosterone signaling in the form of decreased Na+ transport.
Aldosterone Signaling in Renal Endothelial and Vascular Smooth Muscle Cells
In addition to being able to activate signaling pathways within renal epithelial cells, aldosterone modulates many of these same pathways within non-renal cells. Vascular smooth muscle express MR and in the renal vasculature, as well as non-renal vasculature aldosterone exerts important effects that affect vascular compliance. Here, the pathways relevant to the renal parenchyma, namely vascular smooth muscle and endothelia cells and their potential significance in cardiovascular physiology are discussed. For a more in-depth review of the action of aldosterone on the systemic vasculature, the reader is referred to several excellent reviews (58, 59, 93, 122, 156, 183, 396, 558, 650).
Protein Kinase B (AKT)
The importance of AKT in mediating the effects of aldosterone deserves note. As indicated above, AKT plays a key role in several cellular processes with the most notable of these being cell survival and this is especially true in vascular smooth muscle cells (VSMCs). In 2009, Sowers and colleagues reported that the activating phosphorylation of AKT at Ser 473 was impaired in the aortas of rats that overexpress renin, but this critical phosphorylation was restored with MR antagonism (877). In addition, restoration of AKT function was coincident with reduced NADPH oxidase activity, decreased lipid peroxidation, and reduced levels of Ang II. These results suggested that MR antagonism protects the vasculature from aldosterone-induced vascular apoptosis and injury via the rescue of AKT activation. Furthermore, this benefit was independent of marked BP reductions.
AKT action also appears to be important in the heart as well. In particular, the treatment of neonatal rat cardiomyocytes with aldosterone for either a short (10 minutes) or long (24 hours) time period activated AKT (563). The short-term activation of AKT by aldosterone was sensitive to PI3-K and Na+/H+ exchanger inhibitors, but not the MR antagonist eplerenone. Interestingly, the long-term activation of AKT by aldosterone was sensitive to eplerenone, indicating that aldosterone activates AKT signaling via a biphasic mechanism. When taken together, these studies indicate that during the early phase of AKT activation, aldosterone has favorable effects on cardiomyocyte function via a mechanism that is MR-independent. In contrast, the long-term activation of AKT by aldosterone produces pathological effects via a mechanism that is MR-dependent.
Cyclic AMP and adenylate cyclase
The cyclic AMP response element-binding protein (CREB) is an important transcriptional regulatory protein that binds to cyclic AMP responsive elements (CRE) in numerous genes. The soluble adenylate cyclase isoform has the unique property of being stimulated by bicarbonate, and by Ca2+ (714). Evidence indicates that inhibition of soluble adenylate cyclase impairs the rate of transcription in genes regulated in response to either mineralocorticoids or glucocorticoids. Sgk1 provides an example of a gene that is regulated activity by soluble adenylate cyclase-dependent signaling. Expression of sgk1 is substantially stimulated by cyclic AMP and mouse DCT cells (18, 271). Additional studies have provided evidence that soluble adenylate cyclase is involved in Rho/Rac signaling. Rac1 modulates cytoskeletal function and increases MR nuclear accumulation and MR dependent promoter activity (737)
Much less is known about the effect of mineralocorticoid stimulation on adenylate cyclase activity although evidence suggests that MR inhibits CREB signaling by calcineurin activation (296). In contrast, arginine vasopressin (AVP) has been shown to stimulate the expression of 11ß-HSD2. Since 11ß-HSD2 is important in conferring mineralocorticoid specificity these data would suggest that vasopressin signaling may be important in the expression of this enzyme central to mineralocorticoid specificity. Additionally, PKA appears to modulate MR function (522). These investigators provided evidence that PKA modulated the transcriptional activity of MR using transient transfection assays. The analog 8-bromo-cyclic AMP stimulated at a hormone response element that was strictly MR dependent. Aldosterone and cyclic AMP analogues were synergistic in stimulating transcriptional activity (522). These authors concluded that PKA’s effect is indirect possibly by relieving the effect of an MR repressor.
The endothelial Na+ channel, MR, and arterial stiffness
Aldosterone acts directly on endothelial cells and VSMCs via an MR-mediated mechanism (111, 472, 496). Both cell types are heterogenous, but for simplicity will be discussed collectively. Endothelial cells also express an amiloride-sensitive, highly selective Na+ channel and MR activation contributes to arterial stiffness and thus an increase in systemic BP (447, 448, 583), Although the endothelial cell Na+ channel (EnENaC) has many properties in common with the classically characterized Na+ channel of epithelial cells there is evidence that the two should be regarded as distinct (152). Both channels are responsive to MC stimulation with increased activity however, suggesting a coordinated action of aldosterone on renal Na+ absorption and vascular reactivity to preserve BP or increase BP under pathological states (872). Sex differences in vascular responsiveness are also recognized (79). Recent reviews have provided an in-depth analysis of MR signaling in endothelial and VSM cells (93, 156, 177, 178, 183, 392, 532, 558, 872).
Non-genomic Mechanisms of Action of Aldosterone
Historically, aldosterone’s primary action was viewed to modulate the rate of gene transcription via MR, and most data suggest that MR expression is critical for the action of aldosterone.
Although rapid actions may be necessary molecular events for aldosterone’s action (a point that still deserves further study), the global loss of MR expression largely recapitulates the state of aldosterone deficiency (61). Moreover, transport studies of the early action of aldosterone show that this hormone does not produce consistent effects to increase Na+ absorption until ~ 60 to 90 minutes (780, 905).
Nevertheless, non-genomic effects of aldosterone have been reported to occur as early as two minutes following aldosterone exposure in VSMCs (133, 876). Rapid non-genomic effects include changes in Na+ transport, intracellular pH, Ca2+ levels, and reactive oxygen species (470, 537, 680). These effects occur too quickly to be explained by de novo protein synthesis, and they are not inhibited by blocking transcription and translation using actinomycin D or cycloheximide, respectively.
Some studies suggest that the non-genomic effects of aldosterone could be via MR (233). Aldosterone binding to MR can affect non-genomic signaling by activation of either the EGFR or the insulin-like growth factor-1 receptor (IGF-1R). An alternate route for non-genomic effects of aldosterone passes through a G-protein coupled receptor, GPR30, which was previously thought to bind solely estradiol (233, 291, 891). The non-genomic effects of aldosterone have been studied in both renal and non-renal tissue. For additional discussion of the extra-renal non-genomic effects, see (172, 891).
Early studies suggesting a rapid aldosterone-activation of G-coupled receptors via MR were conducted in MDCK cells. Gekle and coworkers noted that nanomolar concentrations of aldosterone induced a rapid increase in ERK1/2 activation and phosphorylation (253). This was accompanied by increases in intracellular Ca2+ that did not appear to be due to an increase in inositol-1,4,5-triphosphate (IP3). This increase was attributed to activation of the EGFR. Aldosterone resulted in increased phosphorylation of EGFR at tyrosine residues as detected by phospho-specific antibodies. In addition, increased intracellular Ca2+ resulted in cell alkalization. The increased cell alkalization occurred with minutes of application of aldosterone and was inhibited by ethyl isopropyl amiloride (EIPA), consistent with increased Na+/H+ exchange activity. The expression of EGF-R was induced in CHO cells by heterologous expression of human MR (444). Evidence suggests a role for aldosterone-induced signaling through the tyrosine kinase cSrc, and direct MR binding to the gene encoding EGF-R by chromatin immunoprecipitation studies. EGF-R and MR have been reported to co-localize at the plasma membrane using florescence resonance energy transfer (FRET) (296). The inference was that this pathway of aldosterone signaling occurred via MR frequently relied on abrogation of the effect by MR antagonists. Whereas this is reasonable given our current knowledge, studies using independent methods to inhibit MR action such as RNAi would be informative.
Role of MR in Non-genomic signaling - transactivation of G-coupled receptors
Aldosterone is known to play both a physiological role and a pathophysiological role in the kidney and VSMC biology. Within the kidney it has been implicated as one of the mechanisms that accelerate the progression to chronic kidney disease (CKD). Unfortunately, few studies have examined this important issue in the kidney, but extensive studies have been done in the heart and vasculature.
Within the VSMC, the EGF signal transduction pathway appears to play a key role in mediating the actions of aldosterone. Specifically, treatment with low dose aldosterone (10−12 M) and low dose Ang II (10−10 M) significantly enhanced DNA synthesis, whereas each ligand by itself was without effect at these same concentrations (549). Not surprisingly, these effects were inhibited by the AT1 receptor blocker, olmesartan, and the mineralocorticoid receptor antagonist, spironolactone. Interestingly, this synergistic effect was blocked with the EGF receptor tyrosine kinase inhibitor, AG1478. Blockade of the EGFR also decreased the early activation of ERK1/2, suggesting that this tyrosine kinase signaling pathway might be rate limiting in terms of mediating the mitogenic effects of aldosterone in VSMC. Another important aldosterone-mediated phenomenon within VSMC that relies on EGFR-dependent signaling is the regulation of 12- and 15-lipoxygenase. These proteins are dioxygenase enzymes that are expressed within VSMC and they incorporate oxygen into unsaturated fatty acids such as arachidonic and linoleic acid. They have also been implicated in pathogenic VSMC signaling. Studies by Stern and colleagues found that aldosterone significantly increased the mRNA and protein levels of 12- and 15-lipoxygenase, and these effects were blocked with spironolactone and eplerenone (481). Moreover, this deleterious effect was blocked with AG 1478. Thus, these studies indicate that aldosterone contributes to lipid oxidation within VSMC and this action is EGFR-dependent.
Transactivation of EGFR
EGFR is a member of the epidermal growth factor family (ErBb) and the receptor tyrosine kinase superfamily (RTK) (reviewed in (301)). EGFR is widely expressed throughout the mammalian kidney, including the glomerular mesangial cells, proximal tubule, cortical, outer, and inner medullary CD, and medullary interstitial cells (960). The protein itself is an integral membrane protein that consists of a globular, cysteine-rich, extracellular domain followed by a single transmembrane domain, and ends in a cytoplasmic domain. The cytoplasmic domain contains a protein tyrosine kinase core that is flanked by regulatory regions (713). EGF or other ligands will induce either homodimerization of EGFR or heterodimerization with another member of the ErBb family such as erbB3. Heterodimerization of the EGFR is thought to allow for a more nuanced cell signal cascade. The dimerization event causes autophosphorylation on the cytoplasmic side of the receptor where one or more tyrosines are phosphorylated. The phosphorylation of these tyrosine residues provides docking sites for cell signaling proteins, such as GRB2.
The ability of the EGFR signaling pathway to mediate the downstream actions of aldosterone is not limited to VSMC. Of interest, aldosterone stimulated glomerular mesangial cell proliferation through an EGFR-dependent mechanism (362). In this work, the authors found that aldosterone increases the levels of reactive oxygen species within glomerular mesangial cells and this leads to EGFR trans-activation. This trans-activation also was dependent upon the PI3-K/Akt signaling pathway. More recently, these authors demonstrated that activation of peroxisome proliferator-activated receptor-γ (PPARγ) inhibits the aldosterone-dependent transactivation of the EGFR in mesangial cells suggesting that PPARγ might be a novel therapeutic target against various glomerular diseases (955). Collectively, these reports underscore the intimate nature by which these signaling pathways are linked.
Interestingly, in the kidney, the EGF ligand, which is an integral membrane protein, is predominantly expressed on the apical membrane, whereas the receptor is present on the basolateral membrane (960). The EGF ligand extracellular domain binds to the EGF receptor and is cleaved by a cell-surface metalloprotease (301). This results in the EGF ligand in the lumen where it is present in the urine at concentrations up to 50 nM (960). This physical separation of the ligand from the receptor is thought to be another layer of regulation of the EGF-R. This review, however, focuses on the events that occur with transactivation of the EGFR with MR. In this model, no EGF ligand is involved in the activation of EGFR. Aldosterone binding to MR releases MR from the inhibitory heat shock proteins. Some of the complexed aldosterone-MR will not translocate to the nucleus and will briefly co-localize with EGFR at the cell membrane (296). This co-localization correlates the activation of the EGFR, which is thought to occur through the tyrosine kinase, c-Src (101, 294). However, the co-localization event only takes place in the short-term, only within 60 minutes of aldosterone exposure. Under conditions of prolonged aldosterone exposure, greater than or equal to 24 hours, the colocalization disappears and almost all of the MR translocates into the nucleus.
Transactivation of IGF-1R
Fewer studies have examined the effect of aldosterone to transactivate other G-coupled receptors. In studies by Holzman using the amphibian cell line A6 cells, application of aldosterone for 10 minutes (1.5μM) resulted in phosphorylation of the IGF receptor as detected by antibodies to the tyrosine 1131 phosphorylated IGF receptor (355). Aldosterone increased short-circuit current, an index of Na+ absorption, in A6 cells and this effect was blocked by inhibitors of PI3-K. In addition, a 10 minute exposure to aldosterone resulted in phosphorylation of Akt comparable to that observed with a 100 μmol IGF-1. These short-term effects were not affected by the presence of actinomycin D although SCC measured after 48 hours was reduced. Notably, the effects could be blocked by either spironolactone (10 μM) or RU-486, (10 μM). The authors propose that aldosterone acting through a MR/GR heterodimer induced a rapid phosphorylation of IGF-1R, which acting through insulin receptor substrate-1 (IRS-1) activated PI3-K then PD K1/2. The latter subsequently was responsible for activation of both Sgk and Akt, leading to the activation of ENaC (355).
GPR30 as an alternate aldosterone receptor
Besides rapid effects of aldosterone that are sensitive to MR antagonists, some effects occur by mechanisms that are insensitive to spironolactone or eplerenone, and presumably mediated by molecules other than MR. Le Moellic et al. found that biotinylated aldosterone could trigger non-genomic events without entering the cell (463). The authors propose a possible alternate receptor for MR might exist at the plasma membrane. Other studies also indicated that MR may not be involved in all non-genomic effects of aldosterone. For example, in aldosterone-treated VSMCs nitric oxide synthase activity is increased via PI3-K dependent pathway that is not inhibited by MR antagonists (56, 489, 561). Gros et al. found the elusive membrane-associated receptor, called GPR30, was responsible for at least some of the non-genomic effects of aldosterone (291). GPR30 is a seven transmembrane domain, G protein-coupled receptor. It was first discovered as a plasma membrane receptor for 17-β-estradiol. At present, the effect of aldosterone on GPR30 has been mainly studied in VSMCs. However, GPR30 is expressed in the DCT, connecting tubule, and inner cortical CD (349). In the presence of aldosterone, GPR30 stimulates the PI3-K dependent signaling pathway. This signaling pathway causes increased phosphorylation of ERK1/2 and of myosin light chain (291). The phosphorylation of the latter may be the cause for contraction seen in rat aortic VSMCs (290). Interestingly, it appears that GPR30 also exerts a genomic effect on MR. As expression of GPR30 increases, the expression of MR decreases. However, currently it is premature to conclude that GPR30 is primarily responsible for any non-genomic effects by aldosterone. In his commentary on the article by Gros et al, Funder notes that the ability for corticosterone to act as an antagonist on GPR30, as it does for MR, has not been evaluated (233). A competition assay between corticosterone and aldosterone would be informative because the circulating concentration of corticosterone is conservatively, two orders of magnitude greater than that of aldosterone. Cheng et al. provided further evidence supporting the role of GPR30 in the kidney as part of an aldosterone signaling pathway that acutely increased the phosphorylation of NCC by studies done in vivo and ex vivo within minutes. The effects were independent of transcription and translation but did not occur in the presence of high [K+]. Signaling appeared to involve MAPK/ERK, PI3/AKT, and cAMP/PKA pathways, and the effect was attenuated in GPR30 null mice (127). The phosphoproteomic, network analysis, and ancillary information from this work will likely be useful for complementary studies.
IGF and EGF signaling pathways
One arm of the signaling cascade that is caused by the transactivation of the EGFR is the MAPK cascade. This signaling cascade is found ubiquitously in mammalian cells and has been extensively studied (449). There are three different MAPK pathways that each consist of three “tiers,” with each tier phosphorylating the next. The three pathways are the classic MAPK (also known as ERK) pathway, the c-Jun N-Terminal Kinase/Stress Activated Protein Kinase (JNK/SAPK) pathway, and the p38 MAPK pathway. The latter two pathways are induced by stress. The most proximal portion of the cascade is the MAPK kinase kinases (MAPKKKs or MAP3Ks) a member of this family is the protein Raf. The kinases in this tier are activated by EGFR. The middle tier is the MAPK kinases or MAPK/Extracellular signal-related Kinases (MEKs). The MEKs are regulated by serine/threonine phosphorylation by the MAP3Ks. The bottom tier is the MAPKs. These proteins act to promote transcription of genes via activation of transcription factors (356, 449).
Aldosterone activates the classical ERK pathway, which is a MAPK pathway (Figure 12). Aldosterone-activated MR binds EGFR to activate the Ki-RasA protein (35). K-RasA is a GTPase that is located at the plasma membrane. The active, GTP-bound form of Ki-RasA will recruit and activate via binding c-Raf1, a MAP3K protein (35). When A6 renal cells are exposed to aldosterone, both the protein abundance and activation of Ki-RasA are increased (825). Thus, aldosterone stimulates c-Raf1 through both genomic and non-genomic mechanisms. In both rat and human mesangial cells, aldosterone causes increases in c-Raf1 phosphorylation, but not protein, levels (362, 803). Detectable amounts of phosphorylation occur in as little as 30 minutes after aldosterone exposure and peak within six hours. This effect dramatically decreases to almost pre-treatment levels at 24 hours, which correlates with the loss of MR co-localization with EGFR (296, 803). The triggering of the cascade quickly activates the MAPKKs followed by the MAPKs. The activation of both ERK1/2 (a MAPK) and MEK1/2 (a MAPKK) follows the same time course as c-Raf1 (803). The activation of ERK1/2 by aldosterone (via EGFR) has been shown in A6 renal cells, MDCK cells, human and rat mesangial cells, the medullary TAL, and renal cortical CD cells (362, 535, 562, 578, 803, 825). To date, only one in vivo study has been performed (747). The effects of low dose (150 μg/kg BW) and high dose (500 μg/kg BW) aldosterone introduced via intraperitoneal injection in rats were evaluated. After 30 minutes both doses showed increased levels of phosphorylated ERK1/2 in the glomerulus, peritubular capillary, TAL of Henle’s loop, outer medullary collecting duct, proximal straight tubule, inner medullar collecting duct, inner medullary vasa recta, and thin limb of Henle’s loop. Alternatively, GPR30 may cause this transactivation in some of these regions or that there is direct aldosterone binding/interaction with EGFR. This response was dose-dependent with the animals receiving the high dose exhibited higher levels of ERK1/2 phosphorylation than the low dose animals.
The other two pathways are thought to be activated in response to stress but are less well-studied in kidney cells. However, they are stimulated by aldosterone in other cell types (Figure 13). In podocytes and VSMCs, aldosterone can stimulate the p38MAPK pathway (101, 121). The JNK/SAPK pathway was activated in CHO cells by transfected human-MR (294). The JNK/SAPK pathway can also be activated via RhoA-GTPase activation in rat adrenomedullary cells (278). It is most likely that aldosterone-induced RhoA activation occurs via EGFR transactivation (393). However, it should be noted that in this model MR is overexpressed in the CHO cells. Therefore, it is possible that the JNK/SAPK pathway activation could be due to the excess of MR present in this study. Studies in a cell line that natively expresses MR should be informative.
Figure 13: Other pathways stimulated by aldosterone and mineralocorticoid receptor (MR) activation.
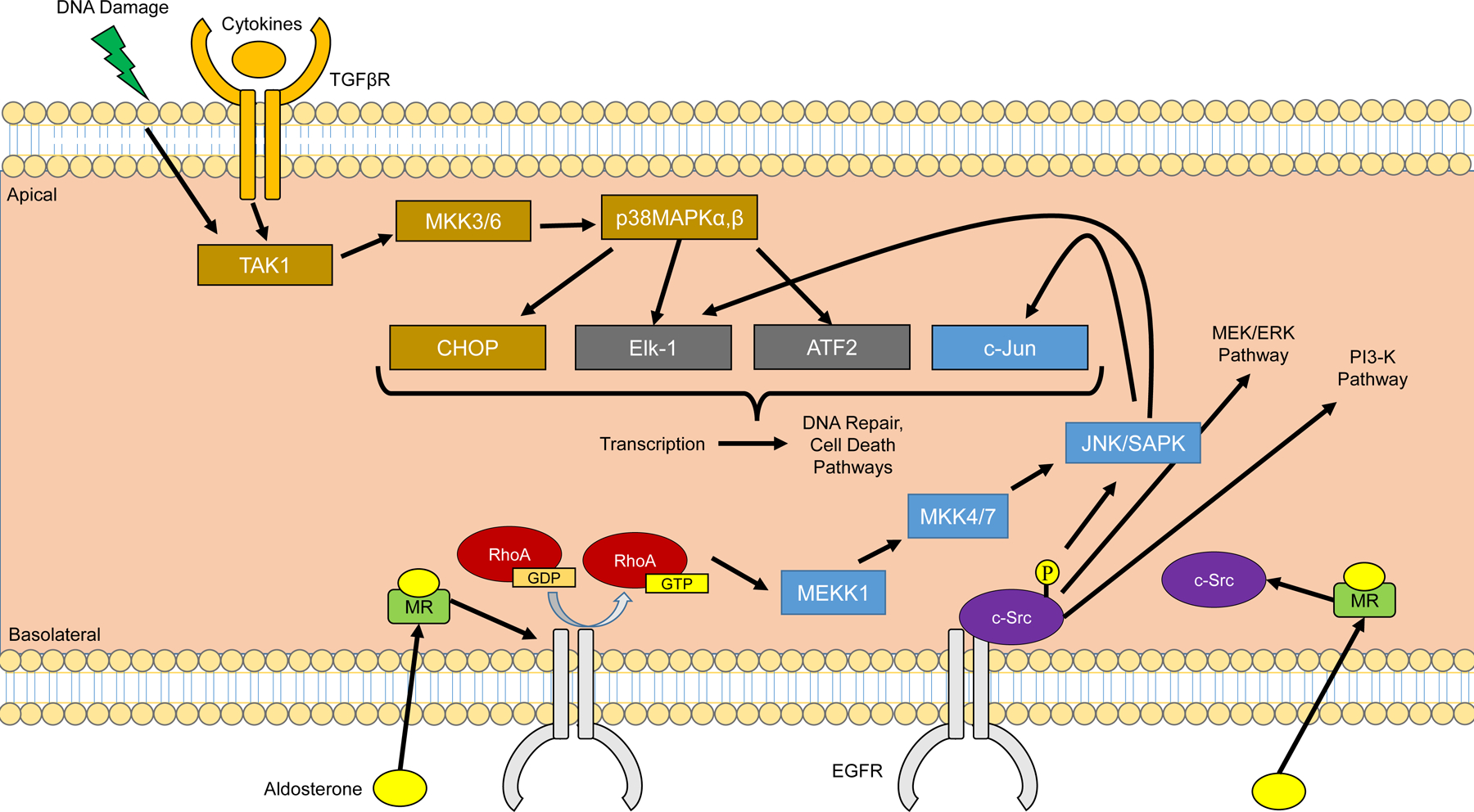
MR activation by aldosterone leads to stimulation of the epidermal growth factor receptor (EGFR), either on its own or through activation of c-Src. Stimulation of EGFR leads to multiple cascading pathways, including the MEK/ERK and PI3K pathways (Figure 12), along with the JNK/SAPK pathways. MR-induced activation of c-Src can also lead to the activation of p38MAPK, a kinase pathway commonly known to be induced by cell stress or inflammatory cytokines. MAPK, mitogen activated protein kinase; ERK, extracellular signal-regulated kinase; MEK, MAPK/ERK kinase or mitogen-activated protein kinase kinase (MAPKK, also known as MKK); MEKK, map kinase kinase kinase; PI3-K, phosphatidylinositol 3-kinase. JNK/SAPK, c-Jun NH2-terminal kinases/stress-activated protein kinase; TGFβR, Transforming growth factor ß receptor.
Other pathways influence the ERK cascade. For instance, PKD1 is required to stabilize the ERK1/2 activation in renal cortical collecting duct cells (535). In mesangial cells, but not podocytes, Ki-RasA can activate the PI3-K pathway. PI3-K, through PKD1 activates AKT) (121, 825). One of the effects of Akt is to activate c-Raf1, which feeds into the ERK signaling cascade (825). Reactive oxygen species generation is required for activation of the MAPK cascade (362, 955). If an antioxidant or NADPH oxidase inhibitor is added, then activation of the pathway is abrogated.
Aldosterone is reported to bind the C2-domain of PKC α in rat CCD cells (19). At low aldosterone concentrations and in very short time periods (less than five minutes), PKC-α inhibitors, but not MR antagonists, block non-genomic events (517). Interestingly, PKC-α phosphorylates MR on serine and threonine residues (463). However, PKC-α does not phosphorylate the glucocorticoid receptor. The PKC pathway interacts with other pathways, as well. Inhibition of both general PKCs and PKC-delta blocked PKD1 activation in CCD cells. In summary, aldosterone is an important signaling molecule that mediates its physiological and pathophysiological actions via a number of different yet linked signaling pathways. Future studies that will determine the points at where these signaling pathways are rate-limiting will prove useful as they may have potential therapeutic implications.
Intracellular Ca2+ as a mediator of aldosterone action
In the proximal S3 segments of the kidney, aldosterone is reported to cause a dose-dependent increase in intracellular Ca2+ concentrations within one minute (470). The same holds true in both MDCK and CCD cells (254, 328). Pre-incubation with aldosterone for six minutes caused an increase of intracellular Ca2+ for an hour. Interestingly, spironolactone did not diminish the effect of aldosterone when pre-incubated for two or fifteen minutes, indicating this effect is unlikely to be mediated by MR. There is some evidence in M-1 CCD cells that the increase in Ca2+ concentration occurs in a PKC dependent mechanism, but the receptor mediating this effect is unclear (328).
Aldosterone modulation of transporter activity
Aldosterone causes an increase in pHi through non-genomic mechanisms (470, 517, 908, 909). The increase in pHi in CCD cells is unaffected by spironolactone, but is reduced by 5’-(N-ethyl-N-isopropyl) amiloride (EIPA) and a PKC-α inhibitor (517). This increase appears due to insertion of ion pumps and exchangers at the plasma membrane. For example, the amount of H-ATPase protein in isolated collecting ducts at the apical membrane increases by 2–3 fold within 15 minutes of aldosterone exposure (909). Changes in activity of Na+/H+ exchangers may also contribute to this effect, as is the case with Na+/H+ exchanger 3 (NHE3) in the medullary TAL. Aldosterone causes a 30% decrease in NHE3 activity, which causes inhibition of bicarbonate absorption, in an ERK1/2 dependent manner (273, 274, 873). However, aldosterone does not universally cause a decrease in NHE3 activity in all regions of the kidney. For example, in renal proximal tubules aldosterone has been reported to cause an increase in NHE3 activity and membrane expression (176, 517).
Many of the pathways cause changes in expression, activity, and membrane presence of ENaC. Aldosterone treatment of CCD cells causes redistribution of ENaC subunits in the cytoplasm in a PDK1 dependent manner (537). Aldosterone may stimulate the activity and gating of ENaC, as well (338, 770, 772). In CHO cells transfected with α, β, and γ ENaC subunits, exposure to aldosterone causes an increase of ENaC activity by 3-fold (772). This occurs through a Ki-Ras to PI3-K dependent pathway and is largely independent of the MAPK pathway (770). Furthermore, it appears that PIP3 is required for this activation of ENaC in CCD and MDCK cells (338). It is unknown at this time if the pathway is dependent upon MR or GPR30. Undoubtedly, MR is involved in the pathway as PDK1 is required for the stable nuclear translocation of the receptor. The likely answer lies in the relative concentration of GPR30 and MR in the specific cell types and needs further investigation.
Cellular trafficking and ion transport in renal and non-renal tissue
Aldosterone mediates Na+ absorption via ENaC in other transporting epithelia that may have relevance to the action of aldosterone in the kidney. Na+ transport in distal airway and alveolar epithelia generates a net driving force to move fluid from the lumen of the lung into the interstitial space. This Na+ transport is mediated via ENaC and is tightly regulated throughout life in order to prevent the lungs from filling with fluid (860). The lung has a high degree of sympathetic innervation that is fully capable of generating cAMP. Studies using the human airway epithelial cell line, H441, found that cAMP induced a rapid and sustained increase in Na+ transport (812). This transport was abolished when the cells were pre-treated with inhibitors of either PI3-K or PKA. The early effect of cAMP on Na+ transport was Brefeldin A-sensitive and mediated via PKA. Thus, these results suggest that the early effect of cAMP is to increase trafficking of Na+ channels to the apical surface of the lung whereas the sustained effect is to mediate the de novo synthesis of α-ENaC. However, this effect appears to be independent of aldosterone.
Other non-classical aldosterone signaling mechanisms
There continues to be intriguing evidence that aldosterone may act by a non-genomic mechanism in the proximal tubule. Older studies by Hierholzer and colleagues suggested that aldosterone stimulated fluid transport in the proximal tubule (344). More recent studies suggested that aldosterone increases in NHE3 expression and function at the apical membrane of the proximal tubule under pathological conditions (cirrhosis) and in primary human cultured proximal tubule cells (176, 445). These aldosterone effects on Na+/H+ exchange have been reported to occur within approximately 15 minutes and are blocked by RU-486 suggesting that they are receptor mediated possibly by the glucocorticoid receptor. However, Salyer and colleagues (690) provide evidence that aldosterone acts in the proximal tubule through the mineralocorticoid receptor. Using a well-respected antibody to MR (Gomez Sanchez) these investigators provided and IHC evidence in human and rat and mouse kidneys that MR was present in the cytoplasm of proximal tubule cells. Rats that were adrenalectomized exhibited decreased expression for MR and Na+-K+-ATPase α-1 subunit and NHE1 in proximal tubule. The authors suggested that aldosterone modulates Na+ transport in proximal tubules via classic MR-mediated mechanism and SGK1 phosphorylation (690).
Role of Aldosterone in Salt-sensitive Hypertension
Aldosterone normally promotes a physiological homeostatic response to volume depletion via activation of RAAS. It contributes to salt dependent hypertension only to the extent that its activity is abnormally increased for the level of Na+ intake. This may reflect an inappropriate degree of aldosterone secretion by cells of the zona glomerulosa of the adrenal cortex for the degree of Na+ intake and renal circulatory filling pressure, or it may reflect an inappropriate and heighten renal response for a normal level of aldosterone relative to Na+ intake. Among the multiple mechanisms responsible for this sensing of appropriate aldosterone secretion and action, the enzyme 11ßHSD2 appears to be particularly important. (147)
As emphasized previously, aldosterone acts in multiple tissues, including the brain and vasculature, that contribute to fluid, volume, and BP homeostasis. In the brain a small group of neurons express the enzyme 11ßHSD2 which confers mineralocorticoid specificity to MR, including cells of the nucleus of the solitary tract (NTS), the subcommissural organ, and the ventromedial nucleus of the hypothalamus. (251) Furthermore, Evans et al. showed that conditional deletion of 11ßHSD2 in the brain stimulates salt appetite and increases systemic blood pressure in mice. They concluded that reduced 11ßHSD2 activity in the brain does not intrinsically cause hypertension, but it promotes enhanced salt appetite and results in a transition from salt resistance to salt sensitivity. (200) These findings are consistent with the studies of Elise and Celso Gomez-Sanchez who observed that infusion of glycyrrhizic acid or carbenoxolone at a sub-pressor systemic dose into the lateral ventricle of the brain produced hypertension and that the development of hypertension with oral administration of either glycyrrhizic acid or carbenoxolone was blocked by intraventricular infusion of the MR antagonist RU28318 at rates that did not affect blood pressure when administered subcutaneously.(268) Further work supported the role of a central benzamil sensitive mechanism. Whether benzamil affects a sodium channel in the brain similar to αβγENaC or has off-target action as it has in the kidney requires further study. (36, 269, 548, 866)
Enhanced mineralocorticoid effect: autonomous aldosterone production, hyperaldosteronism
The initial description of hyperaldosteronism by Conn (142–144) emphasized the potential for autonomous secretion of corticosteroids from the adrenal cortex and in particular aldosterone to produce profound hypertension as well as derangement of electrolytes with hypokalemia and metabolic alkalosis. However, the studies of Chobanian et al. suggest that a significant component of hypertension in early studies may reflect the compounding effect of dietary K+ depletion, per se, which exacerbated an underlying component of hyperaldosteronism. In these studies, Chobanian et al. measured exchangeable K+ as 42K, and observed a marked total body K+ deficiency in patients suffering from hyperaldosteronism. Nevertheless, when they examined the effect of aldosterone excess on total body K+ stores in normal individuals, they observed only a questionable decrease in total body K+ stores, not consistent with that observed in the patients they reported or with other cases of hyperaldosteronism. These observations suggest that the patients with hyperaldosteronism may have secreted adrenal corticosteroids other than aldosterone which provoked the K+ depletion. Equally likely, this select group exhibited exacerbation of their hyperaldosteronism by poor dietary K+ intake, and consequently K+ depletion (131). As noted previously, chronic metabolic balance studies examining the effect of mineralocorticoid excess (either DOC or aldosterone) consistently demonstrate the development of hypokalemia in the absence of discernible evidence of negative K+ balance.
The incidence of hyperaldosteronism, initially proposed to be a major explanation for hypertension by Cohn, was subsequently found to be relatively infrequent cause of hypertension. However, with the advent of more accurate, reliable, and available assays of aldosterone activity, and the more frequent use of screening tests such as the aldosterone to renin ratio, there was a renewed interest in aldosterone’s contribution to hypertension in the general population. Most recent estimates indicate that a substantial portion of hypertensive patients, estimated to be between 5–13%, exhibit hyperaldosteronism. Moreover, the frequency of patients with hyperaldosteronism increases with the severity of the hypertension (879). Clearly, excessive aldosterone production relative to dietary Na+ intake can yield severe hypertension, with all the consequences of stroke, progressive heart and kidney failure that attend thereto.
Enhanced mineralocorticoid effect: defective inhibition of aldosterone action
In clinics focused on the treatment of patients with difficult to manage or refractory hypertension, defined as systemic hypertension in the presence of three antihypertensive medicines in a compliant patient, a significant number of patients will exhibit the features of hyperaldosteronism, but without biochemical evidence of excess aldosterone production. In such cases, treatment with an aldosterone antagonist frequently results in gratifying and dramatic reduction in systemic BP.
Thus, such patienťs exhibit a state of apparent mineralocorticoid excess. The original report of the patients with apparent mineralocorticoid excess lead to the discovery that local corticosteroid metabolism by 11β-HSD was important to prevent inappropriate activation of MR by cortisol. However, deficiency of 11β-HSD is infrequently found to be the explanation for the refractory hypertension. This raises the question as to the cause of the hypertension in patients responsive to spironolactone or eplerenone but with apparently normal plasma aldosterone or rates of adrenal aldosterone secretion. Experimental evidence indicates that aldosterone’s action in IMCD cells, a major target site for the action of aldosterone, activates genes whose products would act to restrain the action of aldosterone, and in particular ET-1 which acts, as noted below, to inhibit the action of aldosterone to promote Na+ retention. Hence defects in the action of this (or additional) inhibitory pathway(s) would be predicted to exhibit a high degree of sensitivity to aldosterone. Further study of the downstream signaling pathways that act to restrain inappropriate aldosterone-mediated Na+ absorption or vasoconstriction in epithelial and vascular tissue, respectively, may be rewarding.
Clinical Trials involving MR blockade & Challenges
There has been a resurgence of interested in aldosterone’s mechanism of action due to clinical trials that have demonstrated as much as a 30–40% reduction in all-cause mortality in selected high risk populations, notably patients with significant heart disease, by the addition of modest doses of aldosterone antagonists or mineralocorticoid receptor blockers (MRBs), starting with the Randomized Aldactone Evaluation Study (RALES). As noted by Jassier and colleagues (387), there are both experimental and clinical evidence that mineralocorticoid blockade reduces mortality in patients with severe cardiac systolic dysfunction, in which most studies have been conducted. There is also evidence that mineralocorticoid blockade may reduce proteinuria and the progression of CKD (65, 134, 135). As discussed below, recent studies further support the benefit of MR blockade as a strategy to slow the progression of CKD. Although there are more than 600 randomized clinical trials that pertain to the clinical aldosterone action, two have had large effects on the practice of clinical medicine, have been validated by additional studies, and have implications for the future (630, 632–634).
The RALES studied the effect of low-dose spironolactone on all-cause mortality in 1663 patients with moderately severe heart failure (634). The patients involved in this study were recruited from 195 centers in 15 countries and were given either spironolactone or placebo treatment. Patients receiving spironolactone treatment showed a 35% reduction in the frequency of hospitalization for worsening heart failure and a 30% in the risk of death compared to the placebo group (634). The Eplerenone Post-AMI Heart Failure Efficacy and Survival Study (EPHESUS) examined whether a more selective mineralocorticoid antagonist, eplerenone, reduced all-cause mortality in patients suffering an acute myocardial infarction with attendant left ventricular dysfunction in approximately 6500 individuals. The investigators reported significant reduction in all-cause mortality and death from cardiovascular disease in patients treated with up to 50 mg per day eplerenone (633).
Both trials were double-blind placebo-controlled studies that effectively demonstrated the beneficial effect of modulating mineralocorticoid activity in patients with significant systolic heart failure, but patients with severe renal disease were excluded. The striking benefit in survival by addition of an MRB is particularly notable since most of these individuals (~85%) were on other medicines to inhibit the RAAS system. Careful adjustment of medicines in these studies resulted in minimal hyperkalemia during the study. With the publication of this study, however, there was a rapid increase in the rate of prescriptions for spironolactone, and a significant increase in the rates of hospital visits for hyperkalemia (403, 910). Other studies suggest that the beneficial effect of mineralocorticoid blockade may extend to other patient populations including those with preserved systolic function, diabetes mellitus or CKD and also occurs with newer nonsteroidal mineralocorticoid blockers (214, 632). The consequence of increased reliance on mineralocorticoid receptor blockers (MRBs) raises the question whether the benefit of mineralocorticoid blockade on the overall risk of cardiovascular disease and mortality persist with the development of mild to moderate hyperkalemia, and if so at what point does the risk of hyperkalemia outweigh the benefit of MRB therapy. Subsequent studies have confirmed the robust risk benefit of MRB therapy, at least in selected populations (632), so it will be important to see whether the benefit of MRB therapy can be extended to patients with CKD or more generally to individuals with minimal cardiac, renal, or other organ pathology. Thus, further studies are needed to determine whether the beneficial effect of MRB treatment outweighs the intendent hyperkalemia that often results as a consequence of this therapy.
The Prevention And Treatment of Hypertension With Algorithm based Therapy (PATHWAY)-2 study of resistant hypertension found that spironolactone was more effective at reducing BP than the α1-adrenoceptor blocker doxazosin, the β1-adrenoceptor blocker bisoprolol, and placebo treatment (897). This was the first randomized controlled trial to compare different BP-lowering treatments in patients with resistant hypertension. This study was also the first to compare an MRB with sympathetic nervous system blockers. A subsequent study assessed the mechanisms that were behind the effectiveness of spironolactone over other treatments in 314 patients with resistant hypertension. Spironolactone reduced thoracic fluid index greater than any other treatment and showed a reduction in clinic systolic and diastolic BP similar to amiloride (898). From these results, the authors suggested that blockade of the MR by spironolactone ameliorates the symptoms of salt retention and resistant hypertension.
A number of novel, non-steroidal MRBs have been used in clinical trials with the intent to reduce the risk of hyperkalemia associated with steroidal MRBs, specifically apararenone, esaxerenone and finerenone (437). Apararenone (also known as MT-3995) is a long-acting non-steroidal MRB that has selective antagonist activity on MRs (564). Although clinical studies using this MRB are scarce, the pharmacokinetic (PK) and pharmacodynamic (PD) profiles of apararenone in both single and multiple doses were tested in 223 healthy adults across a series of three, phase I randomized, double-blinded, placebo-controlled studies. The first study investigated the effects of food, sex, age and race on the PK of a single dose of apararenone while also examining the safety, tolerability and PK of both single and multiple doses in Black and Caucasian men and women. The second study involved Japanese men taking a single oral administration of apararenone at different doses while determining safety and PK. The third study involved healthy Caucasian male volunteers and determined the PD effects of single oral doses after administering fludrocortisone. This study also used eplerenone as a randomized, open-label, active control, and examined the safety and tolerability of apararenone. Overall, the decrease in urinary Na+/K+ that is seen after fludrocortisone loading is suppressed with apararenone. In addition, the authors found that food, sex, age, and race did not have a noticeable effect on PK. The studies supported the mechanism of action of aparenenone as an MRB, providing a solid basis for more clinical studies involving the drug.
Esaxerenone (also known as CS-3150) has a binding affinity for MR that is greater than 1000-fold higher than GR, androgen and progesterone receptors (865). There are a number of research studies in animal models of hypertension that have shown antihypertensive effects of esaxerenone treatment (22, 24, 70, 476). The beneficial effects of esaxerenone treatment in preclinical studies can also be seen with improved renal function (23, 24, 70, 476). In clinical studies, esaxerenone has been used as an intervention for the treatment of different cardiovascular diseases. In two different studies, patients with essential hypertension were given differing doses of esaxerenone, eplerenone or placebo for 12 weeks. Both studies saw decreases in sitting and ambulatory BP compared to baseline, with dose-dependent decreases apparent after esaxerenone treatment(381, 382). These reductions in BP were observed while the incidence of adverse events were similar across all groups. Patients with type 2 diabetes and renal dysfunction were also treated with esaxerenone, and showed reductions in urinary albumin/creatinine ratio in addition to reduced BP (382, 383). The men and women with type 2 diabetes in these two studies were Japanese, so more work is needed in different patient populations with diabetes to reinforce these results. Nevertheless, esaxerenone appears to have beneficial effects as a non-steroidal MRB.
Finerenone, a nonsteroidal MR antagonist (also known as BAY 94–8862), has a high binding affinity for MR and its selectivity for MR is over 1000-fold higher than other steroid receptors (437, 790). This MRB has been used in multiple studies over the years in an effort to find an MRB that is safe to administer without the side effects of hyperkalemia. A study by Pitt et al. examined the safety and tolerability of finerenone in men and women with heart failure and reduced left ventricular ejection fraction and a mild or moderate case of CKD. Administering 5 or 10mg of finerenone led to a reduction in brain natriuretic peptide and urinary albumin/creatinine ratio while showing a lower incidence of hyperkalemia and impaired renal function (631). In older male and female patients with diabetic nephropathy, finerenone again resulted in a dose-dependent reduction in urinary albumin/creatinine ratio with no significant difference in adverse events compared to placebo treatment. This decrease in urinary albumin/creatinine ratio also occurred without any significant change in systolic or diastolic BP (45). Japanese patients with chronic heart failure, diabetes and/or CKD showed a decrease in N-terminal pro-B-type natriuretic peptide while displaying similar levels of safety compared to eplerenone (704). A network meta-analysis examining the efficacy and safety of different MRBs used in treating patients with heart failure with an ejection fraction no more than 45% found that using finerenone had the highest probability of acting as the best alternative among MRBs (932). BLOCK-CKD, a phase 2, randomized, double-blind, placebo-controlled, multicenter study, has been recently announced to assess the non-steroidal MRB KBP-5074 for treating treatment-resistant and uncontrolled hypertension in patients with moderate-to-severe CKD (43).
As noted previously and discussed below, finerenone significantly reduced proteinuria and the progression of CKD in a randomized double-blind multi-center study of 5734 patients with CKD and type 2 diabetes mellitus that were all treated with RAAS blockers. At month 4 the finerenone treated group had a 31% greater reduction in urinary albumin-to-creatinine ratio than the placebo group. However, the reduction in blood pressure attributable to finerenone was modest, amounting to mean systolic blood pressure reduction of 3.0 mmHg at month 1 and 2.1 mmHg at month 12. Whether this reduction in proteinuria despite only modest effects on systolic blood pressure reflects an action of MR blockage to alter cellular physiology of the ASDN, the podocyte, or other MR sensitive targets awaits further investigation.
Overall, such results are encouraging and show great promise, particularly in patients with high-risk of cardiovascular disease and CKD, but more work is needed in other populations to determine the safety and efficacy of these newer therapeutic options.
The role of aldosterone antagonists in the treatment of CKD
CKD is a major health care problem in the United States with current estimates of approximately 15% of the population or 37 million people afflicted with this condition. Unfortunately, the majority of individuals (~ 90%) are unaware that they have CKD and only 50% of patients with severe kidney disease are aware of their condition. The two most frequent causes of CKD are systemic arterial hypertension (high BP) and diabetes mellitus. (https://www.niddk.nih.gov/health-information/health-statistics/kidney-disease; and https://www.cdc.gov/kidneydisease/publications-resources/2019-national-facts.html). Until recently the mainstay for the treatment of CKD has been the treatment of hypertension and the use of RAAS blockade, starting with the studies of Lewis et al. (475) with ACE inhibitors and subsequently the RENAAL study using angiotensin receptor blockers (ARB). These studies demonstrated the superiority of RAAS blockade to slow the progression of kidney disease and reduce proteinuria (as an index of renal damage) over antihypertensive therapy that did not inhibit the renin-angiotensin-aldosterone system. Pharmacological inhibition of RAAS therefore became the standard of care and the benefit of the additional pharmacological intervention required comparison to the standard of care.
MRBs have also been used as a treatment option in patients with CKD. A meta-analysis and review of randomized trials performed by Ng et al. examined a total of 1581 patients across 29 trials. MRBs were able to lower systolic and diastolic BP overall, but there was insufficient data at the time to perform meta-analyses of other cardiovascular effects. The systematic review of the studies consistently found an increase in serum K+ (571). A similar finding was also seen in a study examining the safety and efficacy of spironolactone on patient-centered cardiovascular and renal endpoints. Using 693 patients in stage 3–4 of CKD that took spironolactone compared with 1386 who did not use spironolactone, those that took the MRB had a lower incidence rate of ESRD and a higher incidence rate for hyperkalemia (926). It is clear hyperkalemia is a major risk factor in CKD (369), so the use of MRBs to treat cardiovascular and cardiorenal diseases should assess this risk also for clinical trials moving forward.
Recently, several double-blind placebo-controlled trials have demonstrated benefit of selective endothelin blockers (335), SGLT2 inhibitors (128) and the nonsteroidal aldosterone antagonist finerenone (43, 44). In the study of finerenone, 5734 patients with type II diabetes mellitus were randomized to receive finerenone or placebo. All patients were treated with RAAS blockade that was maximized prior to randomization. All patients had proteinuria and the primary composite endpoint, assessed by the time to event analysis, was kidney failure a sustained decrease of at least 40% and estimated GFR, or death from renal causes. Secondary composite outcomes included death from cardiovascular causes, nonfatal myocardial infarction, nonfatal stroke or hospitalizations for heart failure. The median follow-up was 2.6 years and 504 of 2833 patients (17.8%) in the finerenone group had a primary outcome event. In comparison, 600 out of 2841 patients (21.1%) of placebo had a primary outcome event. Both primary and secondary outcomes were significantly less in the finerenone-treated group compared with placebo. The overall incidence of hyperkalemia-related discontinuation of the trial regiment was greater in the finerenone group (2.3% versus 0.9%, respectively), and patients treated with finerenone had a greater average serum K+ than did patients treated with placebo, with a maximal difference of 0.23 mEq/l at month 4, which persisted throughout the study. Although all-cause mortality was not significantly reduced in the finerenone group, all primary and secondary outcomes, except for nonfatal stroke, were in favor of finerenone treatment. The authors concluded that in patients with type II diabetes treatment with finerenone resulted in lower risk of CKD progression and cardiovascular events than placebo. This well conducted study provides clear evidence that the pharmacological intervention was beneficial in this patient population despite a greater incidence of discontinuation of trial regiment from hyperkalemia. Collectively, the study provides convincing evidence for the benefit of MR blockade in this population. Comparison of MR blockade versus other pharmacological treatment for slowing CKD will be informative.
Summary and Future Directions
Despite more than 50 years of study, our knowledge of the precise mechanisms of action of aldosterone within the kidney, as well as in other target tissues still demands substantial further investigation if we are to fully appreciate the role that this most important and potent adrenal corticosteroid serves in both health and disease. Whether a core set of immediate aldosterone-responsive genes exist across multiple cell type that can be considered a unique “mineralocorticoid responsive signature” will require further study. However, it is becoming progressively clear that not all of aldosterone’s actions are mediated by a single receptor, classically viewed as the MR. Variants for the human mineralocorticoid receptor are known to exist, but the precise role or roles of these separate isoforms warrant further investigation. Moreover, aldosterone regulation in distinct tissues and cell types may be further modified by cellular metabolism, substrate availability, and circadian clock function. The interaction of MR isoforms or heterodimers of MR:GR with various plasma membrane receptors to elicit distinct signaling cascades also deserves further investigation, as is the importance of establishing whether aldosterone binds directly to plasma membrane receptors that are distinct from the nuclear receptor superfamily. The role of aldosterone and MR to regulate gene expression through long-range interactions involving non-coding mRNAs, may prove to be a very fruitful area of investigation. More research in this area can help integrate what is ostensibly a bewildering array of mechanisms and signaling pathways, including cell metabolism and circadian clock regulation, into a more cohesive and understandable model.
The future for clinical research dealing with mineralocorticoid antagonist also will likely continue to provide great insight. With the development of potent inhibitors of the RAAS system we have come to understand the benefits of RAAS blockade to attenuate the development of hypertension, and the progression of cardiovascular and renal disease. Critical issues to consider are whether the benefits of mineralocorticoid antagonists will result in reduction in all-cause mortality or slowing the progression of renal disease, as recent studies have shown in select populations, despite the presence of significant hyperkalemia, and what level of hyperkalemia in patients with high risk for progression to CKD and ESRD can be safely tolerated. Such information would provide the clinician with the assurance that MRB therapy is beneficial and justified as a therapeutic option. In this regard, the role of novel aldosterone antagonists that do not directly block MR mediated transcription should be compared to classic MRBs, preferably in large clinical studies that can assess the effects of each agent on all-cause mortality, as well as on composite and specific outcomes. Higher order structural information about MR, particularly the holoreceptor complex and its interaction with DNA, co-activators or co-repressors, and other transcription factors will allow for more rational drug design. The future of research into this most potent corticosteroid will need to take note of these facts if we are to provide safer and healthier recommendations for the general population.
Acknowledgements
We thank Dr. Rex Jamison for his critical review of sections of the manuscript. The senior author expresses his appreciation to his colleagues who have contributed to this work and to I. Jeanette Matheny (née Lynch) for her outstanding service over the past more than twenty years.
Footnotes
Alphabetical list of abbreviations: 11β-HSD1, 11ß-hydroxysteroid dehydrogenase type 1; 11β-HSD2, 11ß-hydroxysteroid dehydrogenase type 2; ACTH, Adrenocorticotropic hormone; AIT, aldosterone-induced transcript; AKT, protein kinase B; Ang II, angiotensin II; ANP/ANF, atrial natriuretic peptide/factor; AS, aldosterone synthase; ASDN, aldosterone sensitive distal nephron; ASIC, acid sensing ion channel; AT1/2, Ang II type1/2 receptors; ARNTL, aryl hydrocarbon receptor nuclear translocator-like protein 1 (Bmal1); AVP, arginine vasopressin/antidiuretic hormone; Ca2+, Calcium; cAMP, cyclic adenosine monophosphate, cyclic AMP; CHO, Chinese hamster ovary; CKD, chronic kidney disease; CD, collecting duct; CCD, cortical CD; CNT, connecting tubule (or connecting segment); CRY1, Cryptochrome Circadian Regulator 1; DBD, DNA binding domain; Dot1a, disruptor of telomeric silencing splice variant “a”; DCT, distal convoluted tubule, DOC, 11-deoxycorticosterone; E2, 17β-estradiol; ENaC or HSC, epithelial Na+ channel or highly Na+ selective channel); ET-1, endothelin-1; ERK1/2, extracellular signal-related kinase 1/2; FENa, fractional excretion of Na+; FIKS, flow-induced (or dependent) K+ secretion; GDI, Guanosine nucleotide dissociation inhibitor; GFR, glomerular filtration rate; GPER-1, G protein-coupled estrogen receptor 1; GR, glucocorticoid receptor/(gene: NR3C1) HKα1, gastric H+K+-ATPase alpha subunit encoded by ATP4a; HKα2, gastric H+K+-ATPase alpha subunit encoded by ATP12a; HRE, hormone response element; ICT, initial collecting tubule; IGF, insulin-like growth factor-1; IGF-1R, IGF-1 receptor; IHC, immunohistochemistry; IMCD, inner medullary CD; JK, net potassium flux; K+, potassium; [K+]e, exchangeable K+, Kir channel, inwardly rectifying K+ channel; LBD, ligand binding domain; MAPK, mitogen activated protein kinase; MDCK, Madin-Darby Canine Kidney; MR, mineralocorticoid receptor (encoded by NR3C2); mTOR, mammalian/mechanistic target of rapamycin; mTORC2, type 2 mTOR complex; Na+, sodium; NAD, nicotinamide adenine dinucleotide; NHE, Na+/H+ exchanger; NTD, N-terminal domain; NCC, NaCl cotransporter encoded by SLC12A3; NPo, channel activity, (channel number)XPo; NSC, non-selective cation channel; OMCD, outer medullary CD; OSR1, oxidative stress-responsive kinase 1; Per1, Period homolog 1, Per2, Period homolog 2; PHA type II, pseudohypoaldosteronism type II; PI3-kinase, phosphatidylinositol 3-kinase; PKC, protein kinase C; PKD1, protein kinase D1; PLC, phosphoinositide-specific phospholipase; PNa, Na+ permeability; Po, open channel probability; RAAS, renin-angiotensin-aldosterone system; RISC, RNA-induced silencing complex; SCC, short-circuit current; SGK1, serum and glucocorticoid kinase isoform 1; TAL, thick ascending limb; ULK1, unc-51-like kinase 1; UKV, urinary potassium excretion; Vbl, basolateral membrane voltage; Vmr, resting membrane voltage; VT, transepithelial voltage; VSMC, vascular smooth muscle cell; WNK kinase, with no lysine kinase; ZG, zona glomerulosa of adrenal cortex.
Differences between in vivo and in vitro techniques have yielded different absolute values of VT in the DCT.
Na+ transport in A6 cells is glucocorticoid responsive and the effects of aldosterone and dexamethasone on Na+ transport appear indistinguishable.
The CD is embryologically and anatomically distinct from the cells that comprise the nephron. The CD originates from the ureteric bud whereas the cells of the nephron are derived from the metanephric blastema. However, cells of the DCT2, CNT, ICT and CD express MR and respond to aldosterone. We have chosen to extend the definition of the ASDN to include the CD for conciseness, while noting this distinction, which in some cases may be important.
References
- 1.Abd El-Aziz TM, Soares AG, Mironova E, Boiko N, Kaur A, Archer CR, Stockand JD, and Berman JM. Mechanisms and consequences of casein kinase II and ankyrin-3 regulation of the epithelial Na(+) channel. Sci Rep 11: 14600, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ackermann D, Gresko N, Carrel M, Loffing-Cueni D, Habermehl D, Gomez-Sanchez C, Rossier BC, and Loffing J. In vivo nuclear translocation of mineralocorticoid and glucocorticoid receptors in rat kidney: differential effect of corticosteroids along the distal tubule. Am J Physiol Renal Physiol 299: F1473–1485, 2010. [DOI] [PubMed] [Google Scholar]
- 3.Agarwal AK, Mune T, Monder C, and White PC. NAD(+)-dependent isoform of 11 beta-hydroxysteroid dehydrogenase. Cloning and characterization of cDNA from sheep kidney. J Biol Chem 269: 25959–25962, 1994. [PubMed] [Google Scholar]
- 4.Agrawal S, He JC, and Tharaux PL. Nuclear receptors in podocyte biology and glomerular disease. Nat Rev Nephrol 17: 185–204, 2021. [DOI] [PubMed] [Google Scholar]
- 5.Aguilera G, and Catt KJ. Participation of voltage-dependent calcium channels in the regulation of adrenal glomerulosa function by angiotensin II and potassium. Endocrinology 118: 112–118, 1986. [DOI] [PubMed] [Google Scholar]
- 6.Aguilera G, Fujita K, and Catt KJ. Mechanisms of inhibition of aldosterone secretion by adrenocorticotropin. Endocrinology 108: 522–528, 1981. [DOI] [PubMed] [Google Scholar]
- 7.Aguilera G, Hauger RL, and Catt KJ. Control of aldosterone secretion during sodium restriction: adrenal receptor regulation and increased adrenal sensitivity to angiotensin II. Proc Natl Acad Sci U S A 75: 975–979, 1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahn D, Ge Y, Stricklett PK, Gill P, Taylor D, Hughes AK, Yanagisawa M, Miller L, Nelson RD, and Kohan DE. Collecting duct-specific knockout of endothelin-1 causes hypertension and sodium retention. J Clin Invest 114: 504–511, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahn KY, and Kone BC. Expression and cellular localization of mRNA encoding the “gastric” isoform of H(+)-K(+)-ATPase alpha-subunit in rat kidney. Am J Physiol 268: F99–109, 1995. [DOI] [PubMed] [Google Scholar]
- 10.Ahn KY, Park KY, Kim KK, and Kone BC. Chronic hypokalemia enhances expression of the H(+)-K(+)-ATPase alpha 2-subunit gene in renal medulla. Am J Physiol 271: F314–321, 1996. [DOI] [PubMed] [Google Scholar]
- 11.Ahn KY, Turner PB, Madsen KM, and Kone BC. Effects of chronic hypokalemia on renal expression of the “gastric” H(+)-K(+)-ATPase alpha-subunit gene. Am J Physiol 270: F557–566, 1996. [DOI] [PubMed] [Google Scholar]
- 12.Al-Awqati Q, Norby LH, Mueller A, and Steinmetz PR. Characteristics of stimulation of H+ transport by aldosterone in turtle urinary bladder. J Clin Invest 58: 351–358, 1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Al-Baldawi NF, Stockand JD, Al-Khalili OK, Yue G, and Eaton DC. Aldosterone induces ras methylation in A6 epithelia. Am J Physiol Cell Physiol 279: C429–439, 2000. [DOI] [PubMed] [Google Scholar]
- 14.Albert V, and Hall MN. mTOR signaling in cellular and organismal energetics. Curr Opin Cell Biol 33: 55–66, 2015. [DOI] [PubMed] [Google Scholar]
- 15.Albiston AL, Obeyesekere VR, Smith RE, and Krozowski ZS. Cloning and tissue distribution of the human 11 beta-hydroxysteroid dehydrogenase type 2 enzyme. Mol Cell Endocrinol 105: R11–17, 1994. [DOI] [PubMed] [Google Scholar]
- 16.Alexander EA, and Levinsky NG. An extrarenal mechanism of potassium adaptation. J Clin Invest 47: 740–748, 1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Allers WD, Nilson HW, and Kendall EC. Studies on adrenalectomized dogs: the toxic action of potassium. In: Proceedings of Staff Meetings of the Mayo Clinic1936, p. 283–288. [Google Scholar]
- 18.Alvarez dlR, Zhang P, Naray-Fejes-Toth A, Fejes-Toth G, and Canessa CM. The serum and glucocorticoid kinase sgk increases the abundance of epithelial sodium channels in the plasma membrane of Xenopus oocytes. J Biol Chem 274: 37834–37839, 1999. [DOI] [PubMed] [Google Scholar]
- 19.Alzamora R, Brown LR, and Harvey BJ. Direct binding and activation of protein kinase C isoforms by aldosterone and 17beta-estradiol. Mol Endocrinol 21: 2637–2650, 2007. [DOI] [PubMed] [Google Scholar]
- 20.Amasheh S, Milatz S, Krug SM, Bergs M, Amasheh M, Schulzke JD, and Fromm M. Na+ absorption defends from paracellular back-leakage by claudin-8 upregulation. Biochem Biophys Res Commun 378: 45–50, 2009. [DOI] [PubMed] [Google Scholar]
- 21.Amazit L, Le BF, Kolkhof P, Lamribet K, Viengchareun S, Fay MR, Khan JA, Hillisch A, Lombes M, Rafestin-Oblin ME, and Fagart J. Finerenone Impedes Aldosterone-dependent Nuclear Import of the Mineralocorticoid Receptor and Prevents Genomic Recruitment of Steroid Receptor Coactivator-1. J Biol Chem 290: 21876–21889, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arai K, Homma T, Morikawa Y, Ubukata N, Tsuruoka H, Aoki K, Ishikawa H, Mizuno M, and Sada T. Pharmacological profile of CS-3150, a novel, highly potent and selective non-steroidal mineralocorticoid receptor antagonist. Eur J Pharmacol 761: 226–234, 2015. [DOI] [PubMed] [Google Scholar]
- 23.Arai K, Morikawa Y, Ubukata N, Tsuruoka H, and Homma T. CS-3150, a Novel Nonsteroidal Mineralocorticoid Receptor Antagonist, Shows Preventive and Therapeutic Effects On Renal Injury in Deoxycorticosterone Acetate/Salt-Induced Hypertensive Rats. J Pharmacol Exp Ther 358: 548–557, 2016. [DOI] [PubMed] [Google Scholar]
- 24.Arai K, Tsuruoka H, and Homma T. CS-3150, a novel non-steroidal mineralocorticoid receptor antagonist, prevents hypertension and cardiorenal injury in Dahl salt-sensitive hypertensive rats. Eur J Pharmacol 769: 266–273, 2015. [DOI] [PubMed] [Google Scholar]
- 25.Armitage FE, and Wingo CS. Luminal acidification in K-replete OMCDi: contributions of H-K-ATPase and bafilomycin-A1-sensitive H-ATPase. Am J Physiol 267: F450–458, 1994. [DOI] [PubMed] [Google Scholar]
- 26.Armitage FE, and Wingo CS. Luminal acidification in K-replete OMCDi: inhibition of bicarbonate absorption by K removal and luminal Ba. Am J Physiol 269: F116–124, 1995. [DOI] [PubMed] [Google Scholar]
- 27.Arrascue JF, Dobyan DC, and Jamison RL. Potassium recycling in the renal medulla: effects of acute potassium chloride administration to rats fed a potassium-free diet. Kidney Int 20: 348–352, 1981. [DOI] [PubMed] [Google Scholar]
- 28.Arriza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, Housman DE, and Evans RM. Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science 237: 268–275, 1987. [DOI] [PubMed] [Google Scholar]
- 29.Arroyo JP, Kahle KT, and Gamba G. The SLC12 family of electroneutral cation-coupled chloride cotransporters. Mol Aspects Med 34: 288–298, 2013. [DOI] [PubMed] [Google Scholar]
- 30.Arroyo JP, Lagnaz D, Ronzaud C, Vazquez N, Ko BS, Moddes L, Ruffieux-Daidie D, Hausel P, Koesters R, Yang B, Stokes JB, Hoover RS, Gamba G, and Staub O. Nedd4-2 modulates renal Na+-Cl- cotransporter via the aldosterone-SGK1-Nedd4-2 pathway. J Am Soc Nephrol 22: 1707–1719, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Asher C, Moran A, Rossier BC, and Garty H. Sodium channels in membrane vesicles from cultured toad bladder cells. Am J Physiol 254: C512–C518, 1988. [DOI] [PubMed] [Google Scholar]
- 32.Asher C, Sinha I, and Garty H. Characterization of the interactions between Nedd4-2, ENaC, and sgk-1 using surface plasmon resonance. Biochim Biophys Acta 1612: 59–64, 2003. [DOI] [PubMed] [Google Scholar]
- 33.Asher C, Wald H, Rossier BC, and Garty H. Aldosterone-induced increase in the abundance of Na+ channel subunits. Am J Physiol 271: C605–C611, 1996. [DOI] [PubMed] [Google Scholar]
- 34.August JT, Nelson DH, and Thorn GW. Response of normal subjects to large amounts of aldosterone. J Clin Invest 37: 1549–1555, 1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Avruch J, Khokhlatchev A, Kyriakis JM, Luo Z, Tzivion G, Vavvas D, and Zhang XF. Ras activation of the Raf kinase: tyrosine kinase recruitment of the MAP kinase cascade. Recent Prog Horm Res 56: 127–155, 2001. [DOI] [PubMed] [Google Scholar]
- 36.Ayasse N, Berg P, Andersen JF, Svendsen SL, Sorensen MV, Fedosova NU, Lynch IJ, Wingo CS, and Leipziger J. Benzamil-mediated urine alkalization is caused by the inhibition of H(+)-K(+)-ATPases. Am J Physiol Renal Physiol 320: F596–F607, 2021. [DOI] [PubMed] [Google Scholar]
- 37.Bae EH, Kim IJ, Ma SK, and Kim SW. Altered regulation of renal sodium transporters and natriuretic peptide system in DOCA-salt hypertensive rats. Regul Pept 157: 76–83, 2009. [DOI] [PubMed] [Google Scholar]
- 38.Bahr V, Bumke-Vogt C, Gotze J, Pfeiffer AF, and Diederich S. Function of human mineralocorticoid receptor splice variant. Eur J Endocrinol 151: 295, 2004. [DOI] [PubMed] [Google Scholar]
- 39.Bailey MA, Mullins JJ, and Kenyon CJ. Mineralocorticoid and glucocorticoid receptors stimulate epithelial sodium channel activity in a mouse model of Cushing syndrome. Hypertension 54: 890–896, 2009. [DOI] [PubMed] [Google Scholar]
- 40.Bailey MA, Paterson JM, Hadoke PW, Wrobel N, Bellamy CO, Brownstein DG, Seckl JR, and Mullins JJ. A switch in the mechanism of hypertension in the syndrome of apparent mineralocorticoid excess. J Am Soc Nephrol 19: 47–58, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bailey MA, Unwin RJ, and Shirley DG. In vivo inhibition of renal 11beta-hydroxysteroid dehydrogenase in the rat stimulates collecting duct sodium reabsorption. Clin Sci (Lond) 101: 195–198, 2001. [PubMed] [Google Scholar]
- 42.Bain DL, De Angelis RW, Connaghan KD, Yang Q, Degala GD, and Lambert JR. Dissecting Steroid Receptor Function by Analytical Ultracentrifugation. Methods Enzymol 562: 363–389, 2015. [DOI] [PubMed] [Google Scholar]
- 43.Bakris G, Yang YF, and Pitt B. Mineralocorticoid Receptor Antagonists for Hypertension Management in Advanced Chronic Kidney Disease: BLOCK-CKD Trial. Hypertension 76: 144–149, 2020. [DOI] [PubMed] [Google Scholar]
- 44.Bakris GL, Agarwal R, Anker SD, Pitt B, Ruilope LM, Rossing P, Kolkhof P, Nowack C, Schloemer P, Joseph A, and Filippatos G. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N Engl J Med 383: 2219–2229, 2020. [DOI] [PubMed] [Google Scholar]
- 45.Bakris GL, Agarwal R, Chan JC, Cooper ME, Gansevoort RT, Haller H, Remuzzi G, Rossing P, Schmieder RE, Nowack C, Kolkhof P, Joseph A, Pieper A, Kimmeskamp-Kirschbaum N, and Ruilope LM. Effect of Finerenone on Albuminuria in Patients With Diabetic Nephropathy: A Randomized Clinical Trial. JAMA 314: 884–894, 2015. [DOI] [PubMed] [Google Scholar]
- 46.Ballermann BJ, Bloch KD, Seidman JG, and Brenner BM. Atrial natriuretic peptide transcription, secretion, and glomerular receptor activity during mineralocorticoid escape in the rat. J Clin Invest 78: 840–843, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barger AC, Berlin RD, and Tulenko JF. Infusion of Aldosterone, 9-a-Fluorohydrocortisone and Antidiuretic Hormone into the Renal Artery of Normal and Adrenalectomized, Unanesthetized Dogs. Endocrinology 62: 804–815, 1958. [DOI] [PubMed] [Google Scholar]
- 48.Barrera-Chimal J, Girerd S, and Jaisser F. Mineralocorticoid receptor antagonists and kidney diseases: pathophysiological basis. Kidney Int 96: 302–319, 2019. [DOI] [PubMed] [Google Scholar]
- 49.Barrera-Chimal J, and Jaisser F. Vascular and inflammatory mineralocorticoid receptors in kidney disease. Acta Physiol (Oxf) e13390, 2019. [DOI] [PubMed] [Google Scholar]
- 50.Barrera-Chimal J, and Jaisser F. Vascular mineralocorticoid receptor activation and disease. Exp Eye Res 188: 107796, 2019. [DOI] [PubMed] [Google Scholar]
- 51.Barrera-Chimal J, Lima-Posada I, Bakris GL, and Jaisser F. Mineralocorticoid receptor antagonists in diabetic kidney disease - mechanistic and therapeutic effects. Nat Rev Nephrol 18: 56–70, 2022. [DOI] [PubMed] [Google Scholar]
- 52.Barrett PQ, Bollag WB, Isales CM, McCarthy RT, and Rasmussen H. Role of calcium in angiotensin II-mediated aldosterone secretion. Endocr Rev 10: 496–518, 1989. [DOI] [PubMed] [Google Scholar]
- 53.Barrett PQ, Guagliardo NA, and Bayliss DA. Ion Channel Function and Electrical Excitability in the Zona Glomerulosa: A Network Perspective on Aldosterone Regulation. Annu Rev Physiol 83: 451–475, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barrett PQ, Lu HK, Colbran R, Czernik A, and Pancrazio JJ. Stimulation of unitary T-type Ca(2+) channel currents by calmodulin-dependent protein kinase II. Am J Physiol Cell Physiol 279: C1694–C1703, 2000. [DOI] [PubMed] [Google Scholar]
- 55.Barri YM, and Wingo CS. The effects of potassium depletion and supplementation on blood pressure: a clinical review. Am J Med Sci 314: 37–40, 1997. [DOI] [PubMed] [Google Scholar]
- 56.Batenburg WW, Jansen PM, van den Bogaerdt AJ, and Ah JD. Angiotensin II-aldosterone interaction in human coronary microarteries involves GPR30, EGFR, and endothelial NO synthase. Cardiovasc Res 94: 136–143, 2012. [DOI] [PubMed] [Google Scholar]
- 57.Battilana CA, Dobyan DC, Lacy FB, Bhattacharya J, Johnston PA, and Jamison RL. Effect of chronic potassium loading on potassium secretion by the pars recta or descending limb of the juxtamedullary nephron in the rat. J Clin Invest 62: 1093–1103, 1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bender SB, DeMarco VG, Padilla J, Jenkins NT, Habibi J, Garro M, Pulakat L, Aroor AR, Jaffe IZ, and Sowers JR. Mineralocorticoid receptor antagonism treats obesity-associated cardiac diastolic dysfunction. Hypertension 65: 1082–1088, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bender SB, McGraw AP, Jaffe IZ, and Sowers JR. Mineralocorticoid receptor-mediated vascular insulin resistance: an early contributor to diabetes-related vascular disease? Diabetes 62: 313–319, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Benos DJ, Saccomani G, and Sariban-Sohraby S. The epithelial sodium channel. Subunit number and location of the amiloride binding site. J Biol Chem 262: 10613–10618, 1987. [PubMed] [Google Scholar]
- 61.Berger S, Bleich M, Schmid W, Cole TJ, Peters J, Watanabe H, Kriz W, Warth R, Greger R, and Schutz G. Mineralocorticoid receptor knockout mice: pathophysiology of Na+ metabolism. Proc Natl Acad Sci U S A 95: 9424–9429, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Berliner RW. Renal mechanisms of potassium excretion. In: Harvey Lecture, Series 55. New York: Academic Press, 1961, p. 141–171. [Google Scholar]
- 63.Berman JM, Mironova E, and Stockand JD. Physiological regulation of the epithelial Na(+) channel by casein kinase II. Am J Physiol Renal Physiol 314: F367–f372, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Berrout J, Mamenko M, Zaika OL, Chen L, Zhang W, Pochynyuk O, and O'Neil RG. Emerging role of the calcium-activated, small conductance, SK3 K+ channel in distal tubule function: regulation by TRPV4. PLoS One 9: e95149, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bertocchio JP, Warnock DG, and Jaisser F. Mineralocorticoid receptor activation and blockade: an emerging paradigm in chronic kidney disease. Kidney Int 79: 1051–1060, 2011. [DOI] [PubMed] [Google Scholar]
- 66.Bhalla V, Daidie D, Li H, Pao AC, LaGrange LP, Wang J, Vandewalle A, Stockand JD, Staub O, and Pearce D. Serum- and glucocorticoid-regulated kinase 1 regulates ubiquitin ligase neural precursor cell-expressed, developmentally down-regulated protein 4–2 by inducing interaction with 14-3-3. Mol Endocrinol 19: 3073–3084, 2005. [DOI] [PubMed] [Google Scholar]
- 67.Bhalla V, Soundararajan R, Pao AC, Li H, and Pearce D. Disinhibitory pathways for control of sodium transport: regulation of ENaC by SGK1 and GILZ. Am J Physiol Renal Physiol 291: F714–F721, 2006. [DOI] [PubMed] [Google Scholar]
- 68.Bhargava A, Fullerton MJ, Myles K, Purdy TM, Funder JW, Pearce D, and Cole TJ. The serum- and glucocorticoid-induced kinase is a physiological mediator of aldosterone action. Endocrinology 142: 1587–1594, 2001. [DOI] [PubMed] [Google Scholar]
- 69.Bhargava A, and Pearce D. Mechanisms of mineralocorticoid action: determinants of receptor specificity and actions of regulated gene products. Trends Endocrinol Metab 15: 147–153, 2004. [DOI] [PubMed] [Google Scholar]
- 70.Bhuiyan AS, Rafiq K, Kobara H, Masaki T, Nakano D, and Nishiyama A. Effect of a novel nonsteroidal selective mineralocorticoid receptor antagonist, esaxerenone (CS-3150), on blood pressure and renal injury in high salt-treated type 2 diabetic mice. Hypertens Res 42: 892–902, 2019. [DOI] [PubMed] [Google Scholar]
- 71.Biglieri EG, Shambelan M, and Slaton PE, Jr. Effect of adrenocorticotropin on desoxycorticosterone, corticosterone and aldosterone excretion. J Clin Endocrinol Metab 29: 1090–1101, 1969. [DOI] [PubMed] [Google Scholar]
- 72.Biller KJ, Unwin RJ, and Shirley DG. Distal tubular electrolyte transport during inhibition of renal 11beta-hydroxysteroid dehydrogenase. Am J Physiol Renal Physiol 280: F172–F179, 2001. [DOI] [PubMed] [Google Scholar]
- 73.Bindels RJ, Engbersen AM, Hartog A, and Blazer-Yost BL. Aldosterone-induced proteins in primary cultures of rabbit renal cortical collecting system. Biochim Biophys Acta 1284: 63–68, 1996. [DOI] [PubMed] [Google Scholar]
- 74.Blachley JD, Crider BP, and Johnson JH. Extrarenal potassium adaptation: role of skeletal muscle. Am J Physiol 251: F313–318, 1986. [DOI] [PubMed] [Google Scholar]
- 75.Blair-West JR, Coghlan JP, Denton DA, Goding JR, Munro JA, Peterson RE, and Wintour M. Humoral stimulation of adrenal cortical secretion. J Clin Invest 41: 1606–1627, 1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Blazer-Yost BL, Butterworth M, Hartman AD, Parker GE, Faletti CJ, Els WJ, and Rhodes SJ. Characterization and imaging of A6 epithelial cell clones expressing fluorescently labeled ENaC subunits. Am J Physiol Cell Physiol 281: C624–C632, 2001. [DOI] [PubMed] [Google Scholar]
- 77.Bledsoe RK, Madauss KP, Holt JA, Apolito CJ, Lambert MH, Pearce KH, Stanley TB, Stewart EL, Trump RP, Willson TM, and Williams SP. A ligand-mediated hydrogen bond network required for the activation of the mineralocorticoid receptor. J Biol Chem 280: 31283–31293, 2005. [DOI] [PubMed] [Google Scholar]
- 78.Blocka K Cortisol Level Test: Purpose, Procedure, and Risks. Healthline https://www.healthline.com/health/cortisol-urine#results. [January 4, 2022].
- 79.Bloem LJ, Guo C, and Pratt JH. Identification of a splice variant of the rat and human mineralocorticoid receptor genes. J Steroid Biochem Mol Biol 55: 159–162, 1995. [DOI] [PubMed] [Google Scholar]
- 80.Boase NA, and Kumar S. NEDD4: The founding member of a family of ubiquitin-protein ligases. Gene 557: 113–122, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bockenhauer D, Feather S, Stanescu HC, Bandulik S, Zdebik AA, Reichold M, Tobin J, Lieberer E, Sterner C, Landoure G, Arora R, Sirimanna T, Thompson D, Cross JH, vanť Hoff W, Al Masri O, Tullus K, Yeung S, Anikster Y, Klootwijk E, Hubank M, Dillon MJ, Heitzmann D, Arcos-Burgos M, Knepper MA, Dobbie A, Gahl WA, Warth R, Sheridan E, and Kleta R. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med 360: 1960–1970, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Boehmer C, Wilhelm V, Palmada M, Wallisch S, Henke G, Brinkmeier H, Cohen P, Pieske B, and Lang F. Serum and glucocorticoid inducible kinases in the regulation of the cardiac sodium channel SCN5A. Cardiovasc Res 57: 1079–1084, 2003. [DOI] [PubMed] [Google Scholar]
- 83.Bollag WB. Regulation of aldosterone synthesis and secretion. Compr Physiol 4: 1017–1055, 2014. [DOI] [PubMed] [Google Scholar]
- 84.Bonny O, Chraibi A, Loffing J, Jaeger NF, Grunder S, Horisberger JD, and Rossier BC. Functional expression of a pseudohypoaldosteronism type I mutated epithelial Na+ channel lacking the pore-forming region of its alpha subunit. J Clin Invest 104: 967–974, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Boscardin E, Alijevic O, Hummler E, Frateschi S, and Kellenberger S. The function and regulation of acid-sensing ion channels (ASICs) and the epithelial Na(+) channel (ENaC): IUPHAR Review 19. Br J Pharmacol 173: 2671–2701, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bostanjoglo M, Reeves WB, Reilly RF, Velázquez H, Robertson N, Litwack G, Morsing P, Dørup J, Bachmann S, and Ellison DH. 11Beta-hydroxysteroid dehydrogenase, mineralocorticoid receptor, and thiazide-sensitive Na-Cl cotransporter expression by distal tubules. J Am Soc Nephrol 9: 1347–1358, 1998. [DOI] [PubMed] [Google Scholar]
- 87.Brady KP, Dushkin H, Förnzler D, Koike T, Magner F, Her H, Gullans S, Segre GV, Green RM, and Beier DR. A novel putative transporter maps to the osteosclerosis (oc) mutation and is not expressed in the oc mutant mouse. Genomics 56: 254–261, 1999. [DOI] [PubMed] [Google Scholar]
- 88.Brixius-Anderko S, and Scott EE. Structural and functional insights into aldosterone synthase interaction with its redox partner protein adrenodoxin. J Biol Chem 296: 100794, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brown RD. Aldosterone metabolic clearance is normal in low-renin essential hypertension. J Clin Endocrinol Metab 42: 661–666, 1976. [DOI] [PubMed] [Google Scholar]
- 90.Brown RW, Chapman KE, Edwards CR, and Seckl JR. Human placental 11 beta-hydroxysteroid dehydrogenase: evidence for and partial purification of a distinct NAD-dependent isoform. Endocrinology 132: 2614–2621, 1993. [DOI] [PubMed] [Google Scholar]
- 91.Bugaj V, Pochynyuk O, Mironova E, Vandewalle A, Medina JL, and Stockand JD. Regulation of the epithelial Na+ channel by endothelin-1 in rat collecting duct. Am J Physiol Renal Physiol 295: F1063–1070, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bugaj V, Sansom SC, Wen D, Hatcher LI, Stockand JD, and Mironova E. Flow-sensitive K+-coupled ATP secretion modulates activity of the epithelial Na+ channel in the distal nephron. J Biol Chem 287: 38552–38558, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Buonafine M, Bonnard B, and Jaisser F. Mineralocorticoid Receptor and Cardiovascular Disease. Am J Hypertens 31: 1165–1174, 2018. [DOI] [PubMed] [Google Scholar]
- 94.Burnay M, Crambert G, Kharoubi-Hess S, Geering K, and Horisberger JD. Bufo marinus bladder H-K-ATPase carries out electroneutral ion transport. Am J Physiol Renal Physiol 281: F869–F874, 2001. [DOI] [PubMed] [Google Scholar]
- 95.Burnay M, Crambert G, Kharoubi-Hess S, Geering K, and Horisberger JD. Electrogenicity of Na,K- and H,K-ATPase activity and presence of a positively charged amino acid in the fifth transmembrane segment. J Biol Chem 278: 19237–19244, 2003. [DOI] [PubMed] [Google Scholar]
- 96.Burt VL, Whelton P, Roccella EJ, Brown C, Cutler JA, Higgins M, Horan MJ, and Labarthe D. Prevalence of hypertension in the US adult population. Results from the Third National Health and Nutrition Examination Survey, 1988–1991. Hypertension 25: 305–313, 1995. [DOI] [PubMed] [Google Scholar]
- 97.Butterworth MB. MicroRNAs and the regulation of aldosterone signaling in the kidney. Am J Physiol Cell Physiol 308: C521–C527, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Butterworth MB. Role of microRNAs in aldosterone signaling. Curr Opin Nephrol Hypertens 27: 390–394, 2018. [DOI] [PubMed] [Google Scholar]
- 99.Butterworth MB, and Alvarez de la Rosa D. Regulation of Aldosterone Signaling by MicroRNAs. Vitam Horm 109: 69–103, 2019. [DOI] [PubMed] [Google Scholar]
- 100.Cai Z, Xin J, Pollock DM, and Pollock JS. Shear stress-mediated NO production in inner medullary collecting duct cells. Am J Physiol Renal Physiol 279: F270–F274, 2000. [DOI] [PubMed] [Google Scholar]
- 101.Callera GE, Touyz RM, Tostes RC, Yogi A, He Y, Malkinson S, and Schiffrin EL. Aldosterone activates vascular p38MAP kinase and NADPH oxidase via c-Src. Hypertension 45: 773–779, 2005. [DOI] [PubMed] [Google Scholar]
- 102.Calò L, Borsatti A, Favaro S, and Rabinowitz L. Kaliuresis in normal subjects following oral potassium citrate intake without increased plasma potassium concentration. Nephron 69: 253–258, 1995. [DOI] [PubMed] [Google Scholar]
- 103.Campbell-Thompson ML, Verlander JW, Curran KA, Campbell WG, Cain BD, Wingo CS, and McGuigan JE. In situ hybridization of H-K-ATPase beta-subunit mRNA in rat and rabbit kidney. Am J Physiol 269: F345–354, 1995. [DOI] [PubMed] [Google Scholar]
- 104.Canessa CM, Horisberger JD, and Rossier BC. Epithelial sodium channel related to proteins involved in neurodegeneration. Nature 361: 467–470, 1993. [DOI] [PubMed] [Google Scholar]
- 105.Canessa CM, Merillat AM, and Rossier BC. Membrane topology of the epithelial sodium channel in intact cells. Am J Physiol 267: C1682–C1690, 1994. [DOI] [PubMed] [Google Scholar]
- 106.Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD, and Rossier BC. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature 367: 463–467, 1994. [DOI] [PubMed] [Google Scholar]
- 107.Canonica J, Frateschi S, Boscardin E, Ebering A, Sergi C, Jäger Y, Peyrollaz T, Mérillat AM, Maillard M, Klusonova P, Odermatt A, Koesters R, Debonneville A, Staub O, Verouti SN, and Hummler E. Lack of Renal Tubular Glucocorticoid Receptor Decreases the Thiazide-Sensitive Na(+)/Cl(-) Cotransporter NCC and Transiently Affects Sodium Handling. Front Physiol 10: 989, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Canonica J, Sergi C, Maillard M, Klusonova P, Odermatt A, Koesters R, Loffing-Cueni D, Loffing J, Rossier B, Frateschi S, and Hummler E. Adult nephron-specific MR-deficient mice develop a severe renal PHA-1 phenotype. Pflugers Arch 468: 895–908, 2016. [DOI] [PubMed] [Google Scholar]
- 109.Cantley LC. The phosphoinositide 3-kinase pathway. Science 296: 1655–1657, 2002. [DOI] [PubMed] [Google Scholar]
- 110.Cao R mTOR Signaling, Translational Control, and the Circadian Clock. Front Genet 9: 367, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Caprio M, Newfell BG, la Sala A, Baur W, Fabbri A, Rosano G, Mendelsohn ME, and Jaffe IZ. Functional mineralocorticoid receptors in human vascular endothelial cells regulate intercellular adhesion molecule-1 expression and promote leukocyte adhesion. Circ Res 102: 1359–1367, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Caroccia B, Seccia TM, Campos AG, Gioco F, Kuppusamy M, Ceolotto G, Guerzoni E, Simonato F, Mareso S, Lenzini L, Fassina A, and Rossi GP. GPER-1 and estrogen receptor-β ligands modulate aldosterone synthesis. Endocrinology 155: 4296–4304, 2014. [DOI] [PubMed] [Google Scholar]
- 113.Carrisoza-Gaytan R, Ray EC, Flores D, Marciszyn AL, Wu P, Liu L, Subramanya AR, Wang W, Sheng S, Nkashama LJ, Chen J, Jackson EK, Mutchler SM, Heja S, Kohan DE, Satlin LM, and Kleyman TR. Intercalated cell BKα subunit is required for flow-induced K+ secretion. JCI Insight 5: 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Castaneda-Bueno M, Arroyo JP, Zhang J, Puthumana J, Yarborough O III, Shibata S, Rojas-Vega L, Gamba G, Rinehart J, and Lifton RP. Phosphorylation by PKC and PKA regulate the kinase activity and downstream signaling of WNK4. Proc Natl Acad Sci U S A 114: E879–E886, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Castaneda-Bueno M, Cervantes-Perez LG, Vazquez N, Uribe N, Kantesaria S, Morla L, Bobadilla NA, Doucet A, Alessi DR, and Gamba G. Activation of the renal Na+:Cl- cotransporter by angiotensin II is a WNK4-dependent process. Proc Natl Acad Sci U S A 109: 7929–7934, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Castro H, and Raij L. Potassium in hypertension and cardiovascular disease. Semin Nephrol 33: 277–289, 2013. [DOI] [PubMed] [Google Scholar]
- 117.Chan JC. Control of aldosterone secretion. Nephron 23: 79–83, 1979. [DOI] [PubMed] [Google Scholar]
- 118.Chang H, Tashiro K, Hirai M, Ikeda K, Kurokawa K, and Fujita T. Identification of a cDNA encoding a thiazide-sensitive sodium-chloride cotransporter from the human and its mRNA expression in various tissues. Biochem Biophys Res Commun 223: 324–328, 1996. [DOI] [PubMed] [Google Scholar]
- 119.Chang SS, Grunder S, Hanukoglu A, Rösler A, Mathew PM, Hanukoglu I, Schild L, Lu Y, Shimkets RA, Nelson-Williams C, Rossier BC, and Lifton RP. Mutations in subunits of the epithelial sodium channel cause salt wasting with hyperkalaemic acidosis, pseudohypoaldosteronism type 1. Nat Genet 12: 248–253, 1996. [DOI] [PubMed] [Google Scholar]
- 120.Chapman K, Holmes M, and Seckl J. 11β-hydroxysteroid dehydrogenases: intracellular gate-keepers of tissue glucocorticoid action. Physiol Rev 93: 1139–1206, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chen C, Liang W, Jia J, van GH, Singhal PC, and Ding G. Aldosterone induces apoptosis in rat podocytes: role of PI3-K/Akt and p38MAPK signaling pathways. Nephron Exp Nephrol 113: e26–e34, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chen CW, Jaffe IZ, and Karumanchi SA. Pre-eclampsia and cardiovascular disease. Cardiovasc Res 101: 579–586, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chen SY, Bhargava A, Mastroberardino L, Meijer OC, Wang J, Buse P, Firestone GL, Verrey F, and Pearce D. Epithelial sodium channel regulated by aldosterone-induced protein sgk. Proc Natl Acad Sci U S A 96: 2514–2519, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chen XL, Bayliss DA, Fern RJ, and Barrett PQ. A role for T-type Ca2+ channels in the synergistic control of aldosterone production by ANG II and K+. Am J Physiol 276: F674–F683, 1999. [DOI] [PubMed] [Google Scholar]
- 125.Chen Z, Vaughn DA, Blakely P, and Fanestil DD. Adrenocortical steroids increase renal thiazide diuretic receptor density and response. J Am Soc Nephrol 5: 1361–1368, 1994. [DOI] [PubMed] [Google Scholar]
- 126.Chen ZF, Vaughn DA, Beaumont K, and Fanestil DD. Effects of diuretic treatment and of dietary sodium on renal binding of 3H-metolazone. J Am Soc Nephrol 1: 91–98, 1990. [DOI] [PubMed] [Google Scholar]
- 127.Cheng L, Poulsen SB, Wu Q, Esteva-Font C, Olesen ETB, Peng L, Olde B, Leeb-Lundberg LMF, Pisitkun T, Rieg T, Dimke H, and Fenton RA. Rapid Aldosterone-Mediated Signaling in the DCT Increases Activity of the Thiazide-Sensitive NaCl Cotransporter. J Am Soc Nephrol 30: 1454–1470, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cherney DZI, Dekkers CCJ, Barbour SJ, Cattran D, Abdul Gafor AH, Greasley PJ, Laverman GD, Lim SK, Di Tanna GL, Reich HN, Vervloet MG, Wong MG, Gansevoort RT, and Heerspink HJL. Effects of the SGLT2 inhibitor dapagliflozin on proteinuria in non-diabetic patients with chronic kidney disease (DIAMOND): a randomised, double-blind, crossover trial. Lancet Diabetes Endocrinol 8: 582–593, 2020. [DOI] [PubMed] [Google Scholar]
- 129.Cheval L, Morla L, Elalouf JM, and Doucet A. Kidney collecting duct acid-base “regulon”. Physiol Genomics 27: 271–281, 2006. [DOI] [PubMed] [Google Scholar]
- 130.Chiu T, Santiskulvong C, and Rozengurt E. EGF receptor transactivation mediates ANG II-stimulated mitogenesis in intestinal epithelial cells through the PI3-kinase/Akt/mTOR/p70S6K1 signaling pathway. Am J Physiol Gastrointest Liver Physiol 288: G182–G194, 2005. [DOI] [PubMed] [Google Scholar]
- 131.Chobanian AV, Burrows BA, and Hollander W. Body fluid and electrolyte composition in arterial hypertension. II. Studies in mineralocorticoid hypertension. J Clin Invest 40: 416–422, 1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Choi M, Scholl UI, Yue P, Bjorklund P, Zhao B, Nelson-Williams C, Ji W, Cho Y, Patel A, Men CJ, Lolis E, Wisgerhof MV, Geller DS, Mane S, Hellman P, Westin G, Akerstrom G, Wang W, Carling T, and Lifton RP. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science 331: 768–772, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Christ M, Meyer C, Sippel K, and Wehling M. Rapid aldosterone signaling in vascular smooth muscle cells: involvement of phospholipase C, diacylglycerol and protein kinase C alpha. Biochem Biophys Res Commun 213: 123–129, 1995. [DOI] [PubMed] [Google Scholar]
- 134.Chrysostomou A, and Becker G. Spironolactone in addition to ACE inhibition to reduce proteinuria in patients with chronic renal disease. N Engl J Med 345: 925–926, 2001. [DOI] [PubMed] [Google Scholar]
- 135.Chrysostomou A, Pedagogos E, MacGregor L, and Becker GJ. Double-blind, placebo-controlled study on the effect of the aldosterone receptor antagonist spironolactone in patients who have persistent proteinuria and are on long-term angiotensin-converting enzyme inhibitor therapy, with or without an angiotensin II receptor blocker. Clin J Am Soc Nephrol 1: 256–262, 2006. [DOI] [PubMed] [Google Scholar]
- 136.Chu TS, Tsuganezawa H, Peng Y, Cano A, Yanagisawa M, and Alpern RJ. Role of tyrosine kinase pathways in ETB receptor activation of NHE3. Am J Physiol 271: C763–C771, 1996. [DOI] [PubMed] [Google Scholar]
- 137.Cicoira M, Zanolla L, Rossi A, Golia G, Franceschini L, Cabrini G, Bonizzato A, Graziani M, Anker SD, Coats AJ, and Zardini P. Failure of aldosterone suppression despite angiotensin-converting enzyme (ACE) inhibitor administration in chronic heart failure is associated with ACE DD genotype. J Am Coll Cardiol 37: 1808–1812, 2001. [DOI] [PubMed] [Google Scholar]
- 138.Clark BA, Brown RS, and Epstein FH. Effect of atrial natriuretic peptide on potassium-stimulated aldosterone secretion: potential relevance to hypoaldosteronism in man. J Clin Endocrinol Metab 75: 399–403, 1992. [DOI] [PubMed] [Google Scholar]
- 139.Clawson H, Raney B, Kuhn R, Karolchik D, and Heitner S. UCSC Genome Browser Univ. Calif. at Santa Cruz https://genome.ucsc.edu/. [Dec. 2013 initial release; Dec. 2017 patch release 12.
- 140.Clemmer JS, Faulkner JL, Mullen AJ, Butler KR, and Hester RL. Sex-specific responses to mineralocorticoid receptor antagonism in hypertensive African American males and females. Biol Sex Differ 10: 24, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Clyne CD, Chang CY, Safi R, Fuller PJ, McDonnell DP, and Young MJ. Purification and characterization of recombinant human mineralocorticoid receptor. Mol Cell Endocrinol 302: 81–85, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Conn JW. Presidential address. I. Painting background. II. Primary aldosteronism, a new clinical syndrome. J Lab Clin Med 45: 3–17, 1955. [PubMed] [Google Scholar]
- 143.Conn JW. Primary aldosteronism. J Lab Clin Med 45: 661–664, 1955. [PubMed] [Google Scholar]
- 144.Conn JW, and Louis LH. Primary aldosteronism: a new clinical entity. Trans Assoc Am Physicians 68: 215–231; discussion, 231–213, 1955. [PubMed] [Google Scholar]
- 145.Cooper J What is an Aldosterone Test? WebMD. https://www.webmd.com/a-to-z-guides/what-is-an-aldosterone-test. [January 4, 2022].
- 146.Crabbe J Stimulation of active sodium transport by the isolated toad bladder with aldosterone in vitro. J Clin Invest 40: 2103–2110, 1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Craigie E, Evans LC, Mullins JJ, and Bailey MA. Failure to downregulate the epithelial sodium channel causes salt sensitivity in Hsd11b2 heterozygote mice. Hypertension 60: 684–690, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Craigie E, Menzies RI, Larsen CK, Jacquillet G, Carrel M, Wildman SS, Loffing J, Leipziger J, Shirley DG, Bailey MA, and Unwin RJ. The renal and blood pressure response to low sodium diet in P2X4 receptor knockout mice. Physiol Rep 6: e13899, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cuevas CA, Su XT, Wang MX, Terker AS, Lin DH, McCormick JA, Yang CL, Ellison DH, and Wang WH. Potassium Sensing by Renal Distal Tubules Requires Kir4.1. J Am Soc Nephrol 28: 1814–1825, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Curnow KM, Tusie-Luna MT, Pascoe L, Natarajan R, Gu JL, Nadler JL, and White PC. The product of the CYP11B2 gene is required for aldosterone biosynthesis in the human adrenal cortex. Mol Endocrinol 5: 1513–1522, 1991. [DOI] [PubMed] [Google Scholar]
- 151.Currie G, Taylor AH, Fujita T, Ohtsu H, Lindhardt M, Rossing P, Boesby L, Edwards NC, Ferro CJ, Townend JN, van den Meiracker AH, Saklayen MG, Oveisi S, Jardine AG, Delles C, Preiss DJ, and Mark PB. Effect of mineralocorticoid receptor antagonists on proteinuria and progression of chronic kidney disease: a systematic review and meta-analysis. BMC Nephrol 17: 127, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Czikora I, Alli AA, Sridhar S, Matthay MA, Pillich H, Hudel M, Berisha B, Gorshkov B, Romero MJ, Gonzales J, Wu G, Huo Y, Su Y, Verin AD, Fulton D, Chakraborty T, Eaton DC, and Lucas R. Epithelial Sodium Channel-α Mediates the Protective Effect of the TNF-Derived TIP Peptide in Pneumolysin-Induced Endothelial Barrier Dysfunction. Front Immunol 8: 842, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Czirják G, Petheo GL, Spät A, and Enyedi P. Inhibition of TASK-1 potassium channel by phospholipase C. Am J Physiol Cell Physiol 281: C700–708, 2001. [DOI] [PubMed] [Google Scholar]
- 154.Czogalla J, Vohra T, Penton D, Kirschmann M, Craigie E, and Loffing J. The mineralocorticoid receptor (MR) regulates ENaC but not NCC in mice with random MR deletion. Pflugers Arch 468: 849–858, 2016. [DOI] [PubMed] [Google Scholar]
- 155.Dahl LK, Leitl G, and Heine M. Influence of dietary potassium and sodium/potassium molar ratios on the development of salt hypertension. J Exp Med 136: 318–330, 1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Davel AP, Jaffe IZ, Tostes RC, Jaisser F, and Belin de Chantemèle EJ. New roles of aldosterone and mineralocorticoid receptors in cardiovascular disease: translational and sex-specific effects. Am J Physiol Heart Circ Physiol 315: H989–H999, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Davenport AP, Hyndman KA, Dhaun N, Southan C, Kohan DE, Pollock JS, Pollock DM, Webb DJ, and Maguire JJ. Endothelin. Pharmacol Rev 68: 357–418, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Davis JO. Some aspects of the physiology of aldosterone. J Natl Med Assoc 49: 42–50, 1957. [PMC free article] [PubMed] [Google Scholar]
- 159.Davis JO, Carpenter CC, Ayers CR, Holman JE, and Bahn RC. Evidence for secretion of an aldosterone-stimulating hormone by the kidney. J Clin Invest 40: 684–696, 1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Davis JO, Urquhart J, and Higgins JT Jr. The effects of alterations of plasma sodium and potassium concentration on aldosterone secretion. J Clin Invest 42: 597–608, 1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Dawborn JK, and Ross EJ. The effect of prolonged administration of aldosterone on sodium and potassium turnover in the rabbit. Clin Sci 32: 559–570, 1967. [PubMed] [Google Scholar]
- 162.de Mello-Aires M, Giebisch G, and Malnic G. Kinetics of potassium transport across single distal tubules of rat kidney. J Physiol 232: 47–70, 1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Debonneville C, Flores SY, Kamynina E, Plant PJ, Tauxe C, Thomas MA, Münster C, Chraïbi A, Pratt JH, Horisberger JD, Pearce D, Loffing J, and Staub O. Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na(+) channel cell surface expression. EMBO J 20: 7052–7059, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Deborde T, Amar L, Bobrie G, Postel-Vinay N, Battaglia C, Tache A, Chedid A, Dhib MM, Chatellier G, Plouin PF, Burgun A, Azizi M, and Jannot AS. Sex differences in antihypertensive treatment in France among 17 856 patients in a tertiary hypertension unit. J Hypertens 36: 939–946, 2018. [DOI] [PubMed] [Google Scholar]
- 165.Deming QB, and Luetscher JA Jr. Bioassay of desoxycorticosterone-like material in urine. Proc Soc Exp Biol Med 73: 171–175, 1950. [Google Scholar]
- 166.Dijkink L, Hartog A, Deen PM, van Os CH, and Bindels RJ. Time-dependent regulation by aldosterone of the amiloride-sensitive Na+ channel in rabbit kidney. Pflugers Arch 438: 354–360, 1999. [DOI] [PubMed] [Google Scholar]
- 167.Dijkink L, Hartog A, Van Os CH, and Bindels RJ. Modulation of aldosterone-induced stimulation of ENaC synthesis by changing the rate of apical Na+ entry. Am J Physiol Renal Physiol 281: F687–F692, 2001. [DOI] [PubMed] [Google Scholar]
- 168.Dinudom A, Harvey KF, Komwatana P, Young JA, Kumar S, and Cook DI. Nedd4 mediates control of an epithelial Na+ channel in salivary duct cells by cytosolic Na+. Proc Natl Acad Sci U S A 95: 7169–7173, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Dluhy RG, Axelrod L, Underwood RH, and Williams GH. Studies of the control of plasma aldosterone concentration in normal man. II. Effect of dietary potassium and acute potassium infusion. J Clin Invest 51: 1950–1957, 1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Dobyan DC, Lacy FB, and Jamison RL. Suppression of potassium-recycling in the renal medulla by short-term potassium deprivation. Kidney Int 16: 704–709, 1979. [DOI] [PubMed] [Google Scholar]
- 171.Doi M, Takahashi Y, Komatsu R, Yamazaki F, Yamada H, Haraguchi S, Emoto N, Okuno Y, Tsujimoto G, Kanematsu A, Ogawa O, Todo T, Tsutsui K, van der Horst GT, and Okamura H. Salt-sensitive hypertension in circadian clock-deficient Cry-null mice involves dysregulated adrenal Hsd3b6. Nat Med 16: 67–74, 2010. [DOI] [PubMed] [Google Scholar]
- 172.Dooley R, Harvey BJ, and Thomas W. Non-genomic actions of aldosterone: from receptors and signals to membrane targets. Mol Cell Endocrinol 350: 223–234, 2012. [DOI] [PubMed] [Google Scholar]
- 173.Doucet A, and Katz AI. Mineralocorticoid receptors along the nephron: [3H]aldosterone binding in rabbit tubules. Am J Physiol 241: F605–F611, 1981. [DOI] [PubMed] [Google Scholar]
- 174.Doucet A, and Marsy S. Characterization of K-ATPase activity in distal nephron: stimulation by potassium depletion. Am J Physiol 253: F418–423, 1987. [DOI] [PubMed] [Google Scholar]
- 175.Douglas J, Aguilera G, Kondo T, and Catt K. Angiotensin II receptors and aldosterone production in rat adrenal glomerulosa cells. Endocrinology 102: 685–696, 1978. [DOI] [PubMed] [Google Scholar]
- 176.Drumm K, Kress TR, Gassner B, Krug AW, and Gekle M. Aldosterone stimulates activity and surface expression of NHE3 in human primary proximal tubule epithelial cells (RPTEC). Cell Physiol Biochem 17: 21–28, 2006. [DOI] [PubMed] [Google Scholar]
- 177.Drummond HA. The (F)low down on the endothelial epithelial sodium channel: epithelial sodium channel as a brake on flow-mediated vasodilation. Hypertension 53: 903–904, 2009. [DOI] [PubMed] [Google Scholar]
- 178.Drummond HA, Price MP, Welsh MJ, and Abboud FM. A molecular component of the arterial baroreceptor mechanotransducer. Neuron 21: 1435–1441, 1998. [DOI] [PubMed] [Google Scholar]
- 179.Dubey RK, Oparil S, Imthurn B, and Jackson EK. Sex hormones and hypertension. Cardiovasc Res 53: 688–708, 2002. [DOI] [PubMed] [Google Scholar]
- 180.Duc C, Farman N, Canessa CM, Bonvalet JP, and Rossier BC. Cell-specific expression of epithelial sodium channel alpha, beta, and gamma subunits in aldosterone-responsive epithelia from the rat: localization by in situ hybridization and immunocytochemistry. J Cell Biol 127: 1907–1921, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Dumeige L, Storey C, Decourtye L, Nehlich M, Lhadj C, Viengchareun S, Kappeler L, Lombès M, and Martinerie L. Sex-Specificity of Mineralocorticoid Target Gene Expression during Renal Development, and Long-Term Consequences. Int J Mol Sci 18: 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Dunbar DR, Khaled H, Evans LC, Al-Dujaili EA, Mullins LJ, Mullins JJ, Kenyon CJ, and Bailey MA. Transcriptional and physiological responses to chronic ACTH treatment by the mouse kidney. Physiol Genomics 40: 158–166, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.DuPont JJ, Hill MA, Bender SB, Jaisser F, and Jaffe IZ. Aldosterone and vascular mineralocorticoid receptors: regulators of ion channels beyond the kidney. Hypertension 63: 632–637, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.DuPont JJ, and Jaffe IZ. 30 YEARS OF THE MINERALOCORTICOID RECEPTOR: The role of the mineralocorticoid receptor in the vasculature. J Endocrinol 234: T67–T82, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.DuPont JJ, Kenney RM, Patel AR, and Jaffe IZ. Sex differences in mechanisms of arterial stiffness. Br J Pharmacol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Edelman ER, Butala NM, Avery LL, Lundquist AL, and Dighe AS. Case 30–2020: A 54-Year-Old Man with Sudden Cardiac Arrest. N Engl J Med 383: 1263–1275, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Edelman IS, Bogoroch R, and Porter GA. On the Mechanism of Action of Aldosterone on Sodium Transport: The Role of Protein Synthesis. Proc Natl Acad Sci U S A 50: 1169–1177, 1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Edinger RS, Yospin J, Perry C, Kleyman TR, and Johnson JP. Regulation of epithelial Na+ channels (ENaC) by methylation: a novel methyltransferase stimulates ENaC activity. J Biol Chem 281: 9110–9117, 2006. [DOI] [PubMed] [Google Scholar]
- 189.Edwards CRW, Burt D, McIntyre MA, de Kloet ER, Stewart PM, Brett L, Sutanto WS, and Monder C. Localisation of 11b-hydroxysteroid dehydrogenase-tissue specific protector of the mineralocorticoid receptor. Lancet October 29: 986–989, 1988. [DOI] [PubMed] [Google Scholar]
- 190.Egfjord M, Dahl HB, Kayser C, and Blaehr H. Aldosterone metabolism in cultures of rat renal cortical and medullary cells. Scand J Clin Lab Invest 59: 107–113, 1999. [DOI] [PubMed] [Google Scholar]
- 191.Eguchi S, Iwasaki H, Ueno H, Frank GD, Motley ED, Eguchi K, Marumo F, Hirata Y, and Inagami T. Intracellular signaling of angiotensin II-induced p70 S6 kinase phosphorylation at Ser(411) in vascular smooth muscle cells. Possible requirement of epidermal growth factor receptor, Ras, extracellular signal-regulated kinase, and Akt. J Biol Chem 274: 36843–36851, 1999. [DOI] [PubMed] [Google Scholar]
- 192.El Moghrabi S, Houillier P, Picard N, Sohet F, Wootla B, Bloch-Faure M, Leviel F, Cheval L, Frische S, Meneton P, Eladari D, and Chambrey R. Tissue kallikrein permits early renal adaptation to potassium load. Proc Natl Acad Sci U S A 107: 13526–13531, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Ellison DH, and Terker AS. Why Your Mother Was Right: How Potassium Intake Reduces Blood Pressure. Trans Am Clin Climatol Assoc 126: 46–55, 2015. [PMC free article] [PubMed] [Google Scholar]
- 194.Ellison DH, Terker AS, and Gamba G. Potassium and Its Discontents: New Insight, New Treatments. J Am Soc Nephrol 27: 981–989, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Ellison DH, Velázquez H, and Wright FS. Thiazide-sensitive sodium chloride cotransport in early distal tubule. Am J Physiol 253: F546–554, 1987. [DOI] [PubMed] [Google Scholar]
- 196.Ellison DH, Velázquez H, and Wright FS. Unidirectional potassium fluxes in renal distal tubule: effects of chloride and barium. Am J Physiol 250: F885–894, 1986. [DOI] [PubMed] [Google Scholar]
- 197.Eraly SA, Vallon V, Vaughn DA, Gangoiti JA, Richter K, Nagle M, Monte JC, Rieg T, Truong DM, Long JM, Barshop BA, Kaler G, and Nigam SK. Decreased renal organic anion secretion and plasma accumulation of endogenous organic anions in OAT1 knock-out mice. J Biol Chem 281: 5072–5083, 2006. [DOI] [PubMed] [Google Scholar]
- 198.Erlejman AG, Lagadari M, Toneatto J, Piwien-Pilipuk G, and Galigniana MD. Regulatory role of the 90-kDa-heat-shock protein (Hsp90) and associated factors on gene expression. Biochim Biophys Acta 1839: 71–87, 2014. [DOI] [PubMed] [Google Scholar]
- 199.Estilo G, Liu W, Pastor-Soler N, Mitchell P, Carattino MD, Kleyman TR, and Satlin LM. Effect of aldosterone on BK channel expression in mammalian cortical collecting duct. Am J Physiol Renal Physiol 295: F780–788, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Evans LC, Ivy JR, Wyrwoll C, McNairn JA, Menzies RI, Christensen TH, Al-Dujaili EA, Kenyon CJ, Mullins JJ, Seckl JR, Holmes MC, and Bailey MA. Conditional Deletion of Hsd11b2 in the Brain Causes Salt Appetite and Hypertension. Circulation 133: 1360–1370, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Fagart J, Hillisch A, Huyet J, Barfacker L, Fay M, Pleiss U, Pook E, Schafer S, Rafestin-Oblin ME, and Kolkhof P. A new mode of mineralocorticoid receptor antagonism by a potent and selective nonsteroidal molecule. J Biol Chem 285: 29932–29940, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Fagart J, Huyet J, Pinon GM, Rochel M, Mayer C, and Rafestin-Oblin ME. Crystal structure of a mutant mineralocorticoid receptor responsible for hypertension. Nat Struct Mol Biol 12: 554–555, 2005. [DOI] [PubMed] [Google Scholar]
- 203.Fakitsas P, Adam G, Daidie D, van Bemmelen MX, Fouladkou F, Patrignani A, Wagner U, Warth R, Camargo SM, Staub O, and Verrey F. Early aldosterone-induced gene product regulates the epithelial sodium channel by deubiquitylation. J Am Soc Nephrol 18: 1084–1092, 2007. [DOI] [PubMed] [Google Scholar]
- 204.Falin R, Veizis IE, and Cotton CU. A role for ERK1/2 in EGF- and ATP-dependent regulation of amiloride-sensitive sodium absorption. Am J Physiol Cell Physiol 288: C1003–1011, 2005. [DOI] [PubMed] [Google Scholar]
- 205.Faresse N, Ruffieux-Daidie D, Salamin M, Gomez-Sanchez CE, and Staub O. Mineralocorticoid receptor degradation is promoted by Hsp90 inhibition and the ubiquitin-protein ligase CHIP. Am J Physiol Renal Physiol 299: F1462–F1472, 2010. [DOI] [PubMed] [Google Scholar]
- 206.Faresse N, Vitagliano JJ, and Staub O. Differential ubiquitylation of the mineralocorticoid receptor is regulated by phosphorylation. FASEB J 26: 4373–4382, 2012. [DOI] [PubMed] [Google Scholar]
- 207.Farman N, Oblin ME, Lombes M, Delahaye F, Westphal HM, Bonvalet JP, and Gasc JM. Immunolocalization of gluco- and mineralocorticoid receptors in rabbit kidney. Am J Physiol 260: C226–C233, 1991. [DOI] [PubMed] [Google Scholar]
- 208.Farman N, Talbot CR, Boucher R, Fay M, Canessa C, Rossier B, and Bonvalet JP. Noncoordinated expression of alpha-, beta-, and gamma-subunit mRNAs of epithelial Na+ channel along rat respiratory tract. Am J Physiol 272: C131–C141, 1997. [DOI] [PubMed] [Google Scholar]
- 209.Farman N, Vandewalle A, and Bonvalet JP. Aldosterone binding in isolated tubules I. Biochemical determination in proximal and distal parts of the rabbit nephron. Am J Physiol 242: F63–F68, 1982. [DOI] [PubMed] [Google Scholar]
- 210.Farman N, Vandewalle A, and Bonvalet JP. Autoradiographic study of aldosterone and dexamethasone binding in isolated glomeruli of rabbit kidney. Am J Physiol 243: F235–F242, 1982. [DOI] [PubMed] [Google Scholar]
- 211.Faus H, and Haendler B. Post-translational modifications of steroid receptors. Biomed Pharmacother 60: 520–528, 2006. [DOI] [PubMed] [Google Scholar]
- 212.Fernandes-Rosa FL, Hubert EL, Fagart J, Tchitchek N, Gomes D, Jouanno E, Benecke A, Rafestin-Oblin ME, Jeunemaitre X, Antonini SR, and Zennaro MC. Mineralocorticoid receptor mutations differentially affect individual gene expression profiles in pseudohypoaldosteronism type 1. J Clin Endocrinol Metab 96: E519–E527, 2011. [DOI] [PubMed] [Google Scholar]
- 213.Field MJ, Stanton BA, and Giebisch GH. Differential acute effects of aldosterone, dexamethasone, and hyperkalemia on distal tubular potassium secretion in the rat kidney. J Clin Invest 74: 1792–1802, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Filippatos G, Anker SD, Bohm M, Gheorghiade M, Kober L, Krum H, Maggioni AP, Ponikowski P, Voors AA, Zannad F, Kim SY, Nowack C, Palombo G, Kolkhof P, Kimmeskamp-Kirschbaum N, Pieper A, and Pitt B. A randomized controlled study of finerenone vs. eplerenone in patients with worsening chronic heart failure and diabetes mellitus and/or chronic kidney disease. Eur Heart J 37: 2105–2114, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Firsov D, Schild L, Gautschi I, Merillat AM, Schneeberger E, and Rossier BC. Cell surface expression of the epithelial Na channel and a mutant causing Liddle syndrome: a quantitative approach. Proc Natl Acad Sci U S A 93: 15370–15375, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Fischer K, Kelly SM, Watt K, Price NC, and McEwan IJ. Conformation of the mineralocorticoid receptor N-terminal domain: evidence for induced and stable structure. Mol Endocrinol 24: 1935–1948, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Flores SY, Debonneville C, and Staub O. The role of Nedd4/Nedd4-like dependant ubiquitylation in epithelial transport processes. Pflugers Arch 446: 334–338, 2003. [DOI] [PubMed] [Google Scholar]
- 218.Foster ES, Jones WJ, Hayslett JP, and Binder HJ. Role of aldosterone and dietary potassium in potassium adaptation in the distal colon of the rat. Gastroenterology 88: 41–46, 1985. [DOI] [PubMed] [Google Scholar]
- 219.Foundation PA. Step 1: Aldosterone Renin Ratio (ARR). Primary Aldosteronism Foundation; https://www.primaryaldosteronism.org/step-1-aldosterone-renin-ratio-arr/. [January 4, 2022]. [Google Scholar]
- 220.Fourkiotis VG, Hanslik G, Hanusch F, Lepenies J, and Quinkler M. Aldosterone and the kidney. Horm Metab Res 44: 194–201, 2012. [DOI] [PubMed] [Google Scholar]
- 221.Fowler N, Giebisch G, and Whittembury G. Distal tubular tracer microinjection study of renal tubular potassium transport. Am J Physiol 229: 1227–1233, 1975. [DOI] [PubMed] [Google Scholar]
- 222.French IW, and Manery JF. The effect of aldosterone on electrolytes in muscle, kidney cortex, and serum. Can J Biochem 42: 1459–1476, 1964. [DOI] [PubMed] [Google Scholar]
- 223.Frindt G, and Burg MB. Effect of vasopressin on sodium transport in renal cortical collecting tubules. Kidney Int 1: 224–231, 1972. [DOI] [PubMed] [Google Scholar]
- 224.Frindt G, Ergonul Z, and Palmer LG. Surface expression of epithelial Na channel protein in rat kidney. J Gen Physiol 131: 617–627, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Frindt G, Masilamani S, Knepper MA, and Palmer LG. Activation of epithelial Na channels during short-term Na deprivation. Am J Physiol Renal Physiol 280: F112–F118, 2001. [DOI] [PubMed] [Google Scholar]
- 226.Frindt G, and Palmer LG. Acute effects of aldosterone on the epithelial Na channel in rat kidney. Am J Physiol Renal Physiol 308: F572–578, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Frindt G, and Palmer LG. K+ secretion in the rat kidney: Na+ channel-dependent and -independent mechanisms. Am J Physiol Renal Physiol 297: F389–396, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Frindt G, and Palmer LG. Regulation of epithelial Na+ channels by adrenal steroids: mineralocorticoid and glucocorticoid effects. Am J Physiol Renal Physiol 302: F20–26, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Fuchs-Hammoser R, Schweiger M, and Oelkers W. The effect of chronic low-dose infusion of ACTH (1–24) on renin, renin-substrate, aldosterone and other corticosteroids in sodium replete and deplete man. Acta Endocrinol (Copenh) 95: 198–206, 1980. [DOI] [PubMed] [Google Scholar]
- 230.Fuller PJ. Novel interactions of the mineralocorticoid receptor. Mol Cell Endocrinol 408: 33–37, 2015. [DOI] [PubMed] [Google Scholar]
- 231.Fuller PJ, Yao Y, Yang J, and Young MJ. Mechanisms of ligand specificity of the mineralocorticoid receptor. J Endocrinol 213: 15–24, 2012. [DOI] [PubMed] [Google Scholar]
- 232.Funder JW. The genetic basis of primary aldosteronism. Curr Hypertens Rep 14: 120–124, 2012. [DOI] [PubMed] [Google Scholar]
- 233.Funder JW. GPR30, mineralocorticoid receptors, and the rapid vascular effects of aldosterone. Hypertension 57: 370–372, 2011. [DOI] [PubMed] [Google Scholar]
- 234.Funder JW, Pearce PT, Smith R, and Smith AI. Mineralocorticoid action: target tissue specificity is enzyme, not receptor, mediated. Science 242: 583–585, 1988. [DOI] [PubMed] [Google Scholar]
- 235.Funding HR. Explanation of Aldosterone Blood Test Results. HealthResearchFunding.org. https://healthresearchfunding.org/explanation-of-aldosterone-blood-test-results/. [January 4, 2022]. [Google Scholar]
- 236.Funding HR. Renin Blood Test Results Interpreted. HealthResearchFunding.org. https://healthresearchfunding.org/renin-blood-test-results-interpreted/. [January 4, 2022]. [Google Scholar]
- 237.Gaeggeler HP, Gonzalez-Rodriguez E, Jaeger NF, Loffing-Cueni D, Norregaard R, Loffing J, Horisberger JD, and Rossier BC. Mineralocorticoid versus glucocorticoid receptor occupancy mediating aldosterone-stimulated sodium transport in a novel renal cell line. J Am Soc Nephrol 16: 878–891, 2005. [DOI] [PubMed] [Google Scholar]
- 238.Galigniana MD, Erlejman AG, Monte M, Gomez-Sanchez C, and Piwien-Pilipuk G. The hsp90-FKBP52 complex links the mineralocorticoid receptor to motor proteins and persists bound to the receptor in early nuclear events. Mol Cell Biol 30: 1285–1298, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Galigniana NM, Ballmer LT, Toneatto J, Erlejman AG, Lagadari M, and Galigniana MD. Regulation of the glucocorticoid response to stress-related disorders by the Hsp90-binding immunophilin FKBP51. J Neurochem 122: 4–18, 2012. [DOI] [PubMed] [Google Scholar]
- 240.Galla JH, Bonduris DN, and Luke RG. Effects of chloride and extracellular fluid volume on bicarbonate reabsorption along the nephron in metabolic alkalosis in the rat. Reassessment of the classical hypothesis of the pathogenesis of metabolic alkalosis. J Clin Invest 80: 41–50, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Gallo LI, Ghini AA, Piwien PG, and Galigniana MD. Differential recruitment of tetratricorpeptide repeat domain immunophilins to the mineralocorticoid receptor influences both heat-shock protein 90-dependent retrotransport and hormone-dependent transcriptional activity. Biochemistry 46: 14044–14057, 2007. [DOI] [PubMed] [Google Scholar]
- 242.Gallolu Kankanamalage S, Lee AY, Wichaidit C, Lorente-Rodriguez A, Shah AM, Stippec S, Whitehurst AW, and Cobb MH. Multistep regulation of autophagy by WNK1. Proc Natl Acad Sci U S A 113: 14342–14347, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Gamba G Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol Rev 85: 423–493, 2005. [DOI] [PubMed] [Google Scholar]
- 244.Gamba G The thiazide-sensitive Na+-Cl- cotransporter: molecular biology, functional properties, and regulation by WNKs. Am J Physiol Renal Physiol 297: F838–848, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Gamba G, Miyanoshita A, Lombardi M, Lytton J, Lee WS, Hediger MA, and Hebert SC. Molecular cloning, primary structure, and characterization of two members of the mammalian electroneutral sodium-(potassium)-chloride cotransporter family expressed in kidney. J Biol Chem 269: 17713–17722, 1994. [PubMed] [Google Scholar]
- 246.Gamba G, Saltzberg SN, Lombardi M, Miyanoshita A, Lytton J, Hediger MA, Brenner BM, and Hebert SC. Primary structure and functional expression of a cDNA encoding the thiazide-sensitive, electroneutral sodium-chloride cotransporter. Proc Natl Acad Sci U S A 90: 2749–2753, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Gann DS, Cruz JF, Casper AG, and Bartter FC. Mechanism by which potassium increases aldosterone secretion in the dog. Am J Physiol 202: 991–996, 1962. [DOI] [PubMed] [Google Scholar]
- 248.Ganong WF, and Mulrow PJ. Evidence of secretion of an aldosterone-stimulating substance by the kidney. Nature 190: 1115–1116, 1961. [DOI] [PubMed] [Google Scholar]
- 249.Ganong WF, and Mulrow PJ. Rate of Change in Sodium and Potassium Excretion After Injection of Aldosterone Into the Aorta and Renal Artery of the Dog. Am J Physiol 195: 337–342, 1958. [DOI] [PubMed] [Google Scholar]
- 250.Geering K, Claire M, Gaeggeler HP, and Rossier BC. Receptor occupancy vs. induction of Na+-K+-ATPase and Na+ transport by aldosterone. Am J Physiol 248: C102–C108, 1985. [DOI] [PubMed] [Google Scholar]
- 251.Geerling JC, and Loewy AD. Aldosterone in the brain. Am J Physiol Renal Physiol 297: F559–576, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Gekle M, Bretschneider M, Meinel S, Ruhs S, and Grossmann C. Rapid mineralocorticoid receptor trafficking. Steroids 81: 103–108, 2014. [DOI] [PubMed] [Google Scholar]
- 253.Gekle M, Freudinger R, Mildenberger S, and Silbernagl S. Aldosterone interaction with epidermal growth factor receptor signaling in MDCK cells. Am J Physiol Renal Physiol 282: F669–F679, 2002. [DOI] [PubMed] [Google Scholar]
- 254.Gekle M, Golenhofen N, Oberleithner H, and Silbernagl S. Rapid activation of Na+/H+ exchange by aldosterone in renal epithelial cells requires Ca2+ and stimulation of a plasma membrane proton conductance. Proc Natl Acad Sci U S A 93: 10500–10504, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Geller DS, Farhi A, Pinkerton N, Fradley M, Moritz M, Spitzer A, Meinke G, Tsai FT, Sigler PB, and Lifton RP. Activating mineralocorticoid receptor mutation in hypertension exacerbated by pregnancy. Science 289: 119–123, 2000. [DOI] [PubMed] [Google Scholar]
- 256.Geller DS, Rodriguez-Soriano J, Vallo BA, Schifter S, Bayer M, Chang SS, and Lifton RP. Mutations in the mineralocorticoid receptor gene cause autosomal dominant pseudohypoaldosteronism type I. Nat Genet 19: 279–281, 1998. [DOI] [PubMed] [Google Scholar]
- 257.Geller DS, Zhang J, Wisgerhof MV, Shackleton C, Kashgarian M, and Lifton RP. A novel form of human mendelian hypertension featuring nonglucocorticoid-remediable aldosteronism. J Clin Endocrinol Metab 93: 3117–3123, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Geserick C, Meyer HA, and Haendler B. The role of DNA response elements as allosteric modulators of steroid receptor function. Mol Cell Endocrinol 236: 1–7, 2005. [DOI] [PubMed] [Google Scholar]
- 259.Gharbi SI, Zvelebil MJ, Shuttleworth SJ, Hancox T, Saghir N, Timms JF, and Waterfield MD. Exploring the specificity of the PI3K family inhibitor LY294002. Biochem J 404: 15–21, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, Dahlin A, Evers R, Fischer V, Hillgren KM, Hoffmaster KA, Ishikawa T, Keppler D, Kim RB, Lee CA, Niemi M, Polli JW, Sugiyama Y, Swaan PW, Ware JA, Wright SH, Yee SW, Zamek-Gliszczynski MJ, and Zhang L. Membrane transporters in drug development. Nat Rev Drug Discov 9: 215–236, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Gilbert KC, and Brown NJ. Aldosterone and inflammation. Curr Opin Endocrinol Diabetes Obes 17: 199–204, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Gleason CE, Frindt G, Cheng CJ, Ng M, Kidwai A, Rashmi P, Lang F, Baum M, Palmer LG, and Pearce D. mTORC2 regulates renal tubule sodium uptake by promoting ENaC activity. J Clin Invest 125: 117–128, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Gnionsahe A, Claire M, Koechlin N, Bonvalet JP, and Farman N. Aldosterone binding sites along nephron of Xenopus and rabbit. Am J Physiol 257: R87–R95, 1989. [DOI] [PubMed] [Google Scholar]
- 264.Gomez-Sanchez CE, Kuppusamy M, and Gomez-Sanchez EP. Of Mice and Man and the Regulation of Aldosterone Secretion. Hypertension 70: 240–242, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Gomez-Sanchez CE, and Oki K. Minireview: potassium channels and aldosterone dysregulation: is primary aldosteronism a potassium channelopathy? Endocrinology 155: 47–55, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Gomez-Sanchez CE, Rossi GP, Fallo F, and Mannelli M. Progress in primary aldosteronism: present challenges and perspectives. Horm Metab Res 42: 374–381, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Gomez-Sanchez E, and Gomez-Sanchez CE. The multifaceted mineralocorticoid receptor. Compr Physiol 4: 965–994, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Gomez-Sanchez EP, and Gomez-Sanchez CE. Central hypertensinogenic effects of glycyrrhizic acid and carbenoxolone. Am J Physiol 263: E1125–1130, 1992. [DOI] [PubMed] [Google Scholar]
- 269.Gomez-Sanchez EP, and Gomez-Sanchez CE. Effect of central amiloride infusion on mineralocorticoid hypertension. Am J Physiol 267: E754–758, 1994. [DOI] [PubMed] [Google Scholar]
- 270.Goncalves MD, Hopkins BD, and Cantley LC. Phosphatidylinositol 3-Kinase, Growth Disorders, and Cancer. N Engl J Med 379: 2052–2062, 2018. [DOI] [PubMed] [Google Scholar]
- 271.Gonzalez-Nunez D, Morales-Ruiz M, Leivas A, Hebert SC, and Poch E. In vitro characterization of aldosterone and cAMP effects in mouse distal convoluted tubule cells. Am J Physiol Renal Physiol 286: F936–F944, 2004. [DOI] [PubMed] [Google Scholar]
- 272.Gonzalez-Robayna IJ, Falender AE, Ochsner S, Firestone GL, and Richards JS. Follicle-Stimulating hormone (FSH) stimulates phosphorylation and activation of protein kinase B (PKB/Akt) and serum and glucocorticoid-lnduced kinase (Sgk): evidence for A kinase-independent signaling by FSH in granulosa cells. Mol Endocrinol 14: 1283–1300, 2000. [DOI] [PubMed] [Google Scholar]
- 273.Good DW, George T, and Watts BA III. Aldosterone inhibits HCO absorption via a nongenomic pathway in medullary thick ascending limb. Am J Physiol Renal Physiol 283: F699–F706, 2002. [DOI] [PubMed] [Google Scholar]
- 274.Good DW, George T, and Watts BA III. Nongenomic regulation by aldosterone of the epithelial NHE3 Na(+)/H(+) exchanger. Am J Physiol Cell Physiol 290: C757–C763, 2006. [DOI] [PubMed] [Google Scholar]
- 275.Good DW, Velázquez H, and Wright FS. Luminal influences on potassium secretion: low sodium concentration. Am J Physiol 246: F609–619, 1984. [DOI] [PubMed] [Google Scholar]
- 276.Good DW, and Wright FS. Luminal influences on potassium secretion: sodium concentration and fluid flow rate. Am J Physiol 236: F192–205, 1979. [DOI] [PubMed] [Google Scholar]
- 277.Good ME, Chiu YH, Poon IKH, Medina CB, Butcher JT, Mendu SK, DeLalio LJ, Lohman AW, Leitinger N, Barrett E, Lorenz UM, Desai BN, Jaffe IZ, Bayliss DA, Isakson BE, and Ravichandran KS. Pannexin 1 Channels as an Unexpected New Target of the Anti-Hypertensive Drug Spironolactone. Circ Res 122: 606–615, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Goto J, Otsuka F, Yamashita M, Suzuki J, Otani H, Takahashi H, Miyoshi T, Mimura Y, Ogura T, and Makino H. Enhancement of aldosterone-induced catecholamine production by bone morphogenetic protein-4 through activating Rho and SAPK/JNK pathway in adrenomedullar cells. Am J Physiol Endocrinol Metab 296: E904–E916, 2009. [DOI] [PubMed] [Google Scholar]
- 279.Grahammer F, Nesterov V, Ahmed A, Steinhardt F, Sandner L, Arnold F, Cordts T, Negrea S, Bertog M, Ruegg MA, Hall MN, Walz G, Korbmacher C, Artunc F, and Huber TB. mTORC2 critically regulates renal potassium handling. J Clin Invest 126: 1773–1782, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Greenlee M, Wingo CS, McDonough AA, Youn JH, and Kone BC. Narrative review: evolving concepts in potassium homeostasis and hypokalemia. Ann Intern Med 150: 619–625, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Greenlee MM, Lynch IJ, Gumz ML, Cain BD, and Wingo CS. Mineralocorticoids stimulate the activity and expression of renal H+,K+-ATPases. J Am Soc Nephrol 22: 49–58, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Grekin RJ, Terris JM, and Bohr DF. Electrolyte and hormonal effects of deoxycorticosterone acetate in young pigs. Hypertension 2: 326–332, 1980. [DOI] [PubMed] [Google Scholar]
- 283.Grimm PR, Coleman R, Delpire E, and Welling PA. Constitutively Active SPAK Causes Hyperkalemia by Activating NCC and Remodeling Distal Tubules. J Am Soc Nephrol 28: 2597–2606, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Grimm PR, Foutz RM, Brenner R, and Sansom SC. Identification and localization of BK-beta subunits in the distal nephron of the mouse kidney. Am J Physiol Renal Physiol 293: F350–359, 2007. [DOI] [PubMed] [Google Scholar]
- 285.Grimm PR, Irsik DL, Liu L, Holtzclaw JD, and Sansom SC. Role of BKbeta1 in Na+ reabsorption by cortical collecting ducts of Na+-deprived mice. Am J Physiol Renal Physiol 297: F420–428, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Grimm PR, Irsik DL, Settles DC, Holtzclaw JD, and Sansom SC. Hypertension of Kcnmb1−/− is linked to deficient K secretion and aldosteronism. Proc Natl Acad Sci U S A 106: 11800–11805, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Grimm PR, Lazo-Fernandez Y, Delpire E, Wall SM, Dorsey SG, Weinman EJ, Coleman R, Wade JB, and Welling PA. Integrated compensatory network is activated in the absence of NCC phosphorylation. J Clin Invest 125: 2136–2150, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Grimm PR, and Sansom SC. BK channels and a new form of hypertension. Kidney Int 78: 956–962, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Grimm PR, and Sansom SC. BK channels in the kidney. Curr Opin Nephrol Hypertens 16: 430–436, 2007. [DOI] [PubMed] [Google Scholar]
- 290.Gros R, Ding Q, Davis M, Shaikh R, Liu B, Chorazyczewski J, Pickering JG, and Feldman RD. Delineating the receptor mechanisms underlying the rapid vascular contractile effects of aldosterone and estradiol. Can J Physiol Pharmacol 89: 655–663, 2011. [DOI] [PubMed] [Google Scholar]
- 291.Gros R, Ding Q, Sklar LA, Prossnitz EE, Arterburn JB, Chorazyczewski J, and Feldman RD. GPR30 expression is required for the mineralocorticoid receptor-independent rapid vascular effects of aldosterone. Hypertension 57: 442–451, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Gross JB, Imai M, and Kokko JP. A functional comparison of the cortical collecting tubule and the distal convoluted tubule. J Clin Invest 55: 1284–1294, 1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Gross JB, and Kokko JP. Effects of aldosterone and potassium-sparing diuretics on electrical potential differences across the distal nephron. J Clin Invest 59: 82–89, 1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Grossmann C, Benesic A, Krug AW, Freudinger R, Mildenberger S, Gassner B, and Gekle M. Human mineralocorticoid receptor expression renders cells responsive for nongenotropic aldosterone actions. Mol Endocrinol 19: 1697–1710, 2005. [DOI] [PubMed] [Google Scholar]
- 295.Grossmann C, Freudinger R, Mildenberger S, Krug AW, and Gekle M. Evidence for epidermal growth factor receptor as negative-feedback control in aldosterone-induced Na+ reabsorption. Am J Physiol Renal Physiol 286: F1226–F1231, 2004. [DOI] [PubMed] [Google Scholar]
- 296.Grossmann C, Husse B, Mildenberger S, Schreier B, Schuman K, and Gekle M. Colocalization of mineralocorticoid and EGF receptor at the plasma membrane. Biochim Biophys Acta 1803: 584–590, 2010. [DOI] [PubMed] [Google Scholar]
- 297.Grossmann C, Ruhs S, Langenbruch L, Mildenberger S, Strätz N, Schumann K, and Gekle M. Nuclear shuttling precedes dimerization in mineralocorticoid receptor signaling. Chem Biol 19: 742–751, 2012. [DOI] [PubMed] [Google Scholar]
- 298.Gründer S, Firsov D, Chang SS, Jaeger NF, Gautschi I, Schild L, Lifton RP, and Rossier BC. A mutation causing pseudohypoaldosteronism type 1 identifies a conserved glycine that is involved in the gating of the epithelial sodium channel. EMBO J 16: 899–907, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Gründer S, Jaeger NF, Gautschi I, Schild L, and Rossier BC. Identification of a highly conserved sequence at the N-terminus of the epithelial Na+ channel alpha subunit involved in gating. Pflugers Arch 438: 709–715, 1999. [DOI] [PubMed] [Google Scholar]
- 300.Gründer S, Zagato L, Yagil C, Yagil Y, Sassard J, and Rossier BC. Polymorphisms in the carboxy-terminus of the epithelial sodium channel in rat models for hypertension. J Hypertens 15: 173–179, 1997. [DOI] [PubMed] [Google Scholar]
- 301.Gschwind A, Fischer OM, and Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer 4: 361–370, 2004. [DOI] [PubMed] [Google Scholar]
- 302.Guess A, Agrawal S, Wei CC, Ransom RF, Benndorf R, and Smoyer WE. Dose- and time-dependent glucocorticoid receptor signaling in podocytes. Am J Physiol Renal Physiol 299: F845–F853, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Guipponi M, Vuagniaux G, Wattenhofer M, Shibuya K, Vazquez M, Dougherty L, Scamuffa N, Guida E, Okui M, Rossier C, Hancock M, Buchet K, Reymond A, Hummler E, Marzella PL, Kudoh J, Shimizu N, Scott HS, Antonarakis SE, and Rossier BC. The transmembrane serine protease (TMPRSS3) mutated in deafness DFNB8/10 activates the epithelial sodium channel (ENaC) in vitro. Hum Mol Genet 11: 2829–2836, 2002. [DOI] [PubMed] [Google Scholar]
- 304.Gumz ML, Cheng KY, Lynch IJ, Stow LR, Greenlee MM, Cain BD, and Wingo CS. Regulation of αENaC expression by the circadian clock protein Period 1 in mpkCCD(c14) cells. Biochim Biophys Acta 1799: 622–629, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Gumz ML, Lynch IJ, Greenlee MM, Cain BD, and Wingo CS. The renal H+-K+-ATPases: physiology, regulation, and structure. Am J Physiol Renal Physiol 298: F12–21, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Gumz ML, Popp MP, Wingo CS, and Cain BD. Early transcriptional effects of aldosterone in a mouse inner medullary collecting duct cell line. Am J Physiol Renal Physiol 285: F664–F673, 2003. [DOI] [PubMed] [Google Scholar]
- 307.Gumz ML, Rabinowitz L, and Wingo CS. An Integrated View of Potassium Homeostasis. N Engl J Med 373: 60–72, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Gumz ML, Stow LR, Lynch IJ, Greenlee MM, Rudin A, Cain BD, Weaver DR, and Wingo CS. The circadian clock protein Period 1 regulates expression of the renal epithelial sodium channel in mice. J Clin Invest 119: 2423–2434, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Gupta P, Franco-Saenz R, and Mulrow PJ. Locally generated angiotensin II in the adrenal gland regulates basal, corticotropin-, and potassium-stimulated aldosterone secretion. Hypertension 25: 443–448, 1995. [DOI] [PubMed] [Google Scholar]
- 310.Guyton AC, Coleman TG, Cowley AV Jr., Scheel KW, Manning RD Jr., and Norman RA Jr. Arterial pressure regulation. Overriding dominance of the kidneys in long-term regulation and in hypertension. Am J Med 52: 584–594, 1972. [DOI] [PubMed] [Google Scholar]
- 311.Guyton AC, Coleman TG, Young DB, Lohmeier TE, and DeClue JW. Salt balance and long-term blood pressure control. Annu Rev Med 31: 15–27, 1980. [DOI] [PubMed] [Google Scholar]
- 312.Hadchouel J, Ellison DH, and Gamba G. Regulation of Renal Electrolyte Transport by WNK and SPAK-OSR1 Kinases. Annu Rev Physiol 78: 367–389, 2016. [DOI] [PubMed] [Google Scholar]
- 313.Hadchouel J, Soukaseum C, Büsst C, Zhou XO, Baudrie V, Zürrer T, Cambillau M, Elghozi JL, Lifton RP, Loffing J, and Jeunemaitre X. Decreased ENaC expression compensates the increased NCC activity following inactivation of the kidney-specific isoform of WNK1 and prevents hypertension. Proc Natl Acad Sci U S A 107: 18109–18114, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Hadoke PW, Christy C, Kotelevtsev YV, Williams BC, Kenyon CJ, Seckl JR, Mullins JJ, and Walker BR. Endothelial cell dysfunction in mice after transgenic knockout of type 2, but not type 1, 11beta-hydroxysteroid dehydrogenase. Circulation 104: 2832–2837, 2001. [DOI] [PubMed] [Google Scholar]
- 315.Hajnóczky G, Csordás G, Bagó A, Chiu AT, and Spät A. Angiotensin II exerts its effect on aldosterone production and potassium permeability through receptor subtype AT1 in rat adrenal glomerulosa cells. Biochem Pharmacol 43: 1009–1012, 1992. [DOI] [PubMed] [Google Scholar]
- 316.Hall JE, Granger JP, Smith MJ Jr., and Premen AJ. Role of renal hemodynamics and arterial pressure in aldosterone “escape”. Hypertension 6: I183–192, 1984. [DOI] [PubMed] [Google Scholar]
- 317.Hamilton KL, and Eaton DC. Regulation of single sodium channels in renal tissue: a role in sodium homeostasis. Fed Proc 45: 2713–2717, 1986. [PubMed] [Google Scholar]
- 318.Hamilton KL, and Eaton DC. Single-channel recordings from amiloride-sensitive epithelial sodium channel. Am J Physiol 249: C200–C207, 1985. [DOI] [PubMed] [Google Scholar]
- 319.Hamilton KL, and Eaton DC. Single-channel recordings from two types of amiloride-sensitive epithelial Na+ channels. Membr Biochem 6: 149–171, 1986. [DOI] [PubMed] [Google Scholar]
- 320.Hamm LL, Hering-Smith KS, and Nakhoul NL. Acid-base and potassium homeostasis. Semin Nephrol 33: 257–264, 2013. [DOI] [PubMed] [Google Scholar]
- 321.Hancock JF, Cadwallader K, Paterson H, and Marshall CJ. A CAAX or a CAAL motif and a second signal are sufficient for plasma membrane targeting of ras proteins. EMBO J 10: 4033–4039, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Hansson JH, Nelson-Williams C, Suzuki H, Schild L, Shimkets R, Lu Y, Canessa C, Iwasaki T, Rossier B, and Lifton RP. Hypertension caused by a truncated epithelial sodium channel gamma subunit: genetic heterogeneity of Liddle syndrome. Nat Genet 11: 76–82, 1995. [DOI] [PubMed] [Google Scholar]
- 323.Hansson JH, Schild L, Lu Y, Wilson TA, Gautschi I, Shimkets R, Nelson-Williams C, Rossier BC, and Lifton RP. A de novo missense mutation of the beta subunit of the epithelial sodium channel causes hypertension and Liddle syndrome, identifying a proline-rich segment critical for regulation of channel activity. Proc Natl Acad Sci U S A 92: 11495–11499, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Hanstein B, Liu H, Yancisin MC, and Brown M. Functional analysis of a novel estrogen receptor-beta isoform. Mol Endocrinol 13: 129–137, 1999. [DOI] [PubMed] [Google Scholar]
- 325.Hanukoglu I ASIC and ENaC type sodium channels: conformational states and the structures of the ion selectivity filters. FEBS J 284: 525–545, 2017. [DOI] [PubMed] [Google Scholar]
- 326.Hanukoglu I, and Hanukoglu A. Epithelial sodium channel (ENaC) family: Phylogeny, structure-function, tissue distribution, and associated inherited diseases. Gene 579: 95–132, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Harris AN, Grimm PR, Lee HW, Delpire E, Fang L, Verlander JW, Welling PA, and Weiner ID. Mechanism of Hyperkalemia-Induced Metabolic Acidosis. J Am Soc Nephrol 29: 1411–1425, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Harvey BJ, and Higgins M. Nongenomic effects of aldosterone on Ca2+ in M-1 cortical collecting duct cells. Kidney Int 57: 1395–1403, 2000. [DOI] [PubMed] [Google Scholar]
- 329.Hasui T, Matsunaga N, Ora T, Ohyabu N, Nishigaki N, Imura Y, Igata Y, Matsui H, Motoyaji T, Tanaka T, Habuka N, Sogabe S, Ono M, Siedem CS, Tang TP, Gauthier C, De Meese LA, Boyd SA, and Fukumoto S. Identification of benzoxazin-3-one derivatives as novel, potent, and selective nonsteroidal mineralocorticoid receptor antagonists. J Med Chem 54: 8616–8631, 2011. [DOI] [PubMed] [Google Scholar]
- 330.Hattangady NG, Olala LO, Bollag WB, and Rainey WE. Acute and chronic regulation of aldosterone production. Mol Cell Endocrinol 350: 151–162, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Hauger RL, Aguilera G, and Catt KJ. Angiotensin II regulates its receptor sites in the adrenal glomerulosa zone. Nature 271: 176–178, 1978. [DOI] [PubMed] [Google Scholar]
- 332.Hayslett JP, and Binder HJ. Mechanism of potassium adaptation. Am J Physiol 243: F103–112, 1982. [DOI] [PubMed] [Google Scholar]
- 333.Healthmatters. Aldosterone. Healthmatters. https://healthmatters.io/understand-blood-test-results/aldosterone. [January 4, 2022]. [Google Scholar]
- 334.Hebert SC, Desir G, Giebisch G, and Wang W. Molecular diversity and regulation of renal potassium channels. Physiol Rev 85: 319–371, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Heerspink HJL, Parving HH, Andress DL, Bakris G, Correa-Rotter R, Hou FF, Kitzman DW, Kohan D, Makino H, McMurray JJV, Melnick JZ, Miller MG, Pergola PE, Perkovic V, Tobe S, Yi T, Wigderson M, and de Zeeuw D. Atrasentan and renal events in patients with type 2 diabetes and chronic kidney disease (SONAR): a double-blind, randomised, placebo-controlled trial. Lancet 393: 1937–1947, 2019. [DOI] [PubMed] [Google Scholar]
- 336.Hellal-Levy C, Couette B, Fagart J, Souque A, Gomez-Sanchez C, and Rafestin-Oblin M. Specific hydroxylations determine selective corticosteroid recognition by human glucocorticoid and mineralocorticoid receptors. FEBS Lett 464: 9–13, 1999. [DOI] [PubMed] [Google Scholar]
- 337.Helman SI, and O'Neil RG. Model of active transepithelial Na and K transport of renal collecting tubules. Am J Physiol 233: F559–571, 1977. [DOI] [PubMed] [Google Scholar]
- 338.Helms MN, Liu L, Liang YY, Al-Khalili O, Vandewalle A, Saxena S, Eaton DC, and Ma HP. Phosphatidylinositol 3,4,5-trisphosphate mediates aldosterone stimulation of epithelial sodium channel (ENaC) and interacts with gamma-ENaC. J Biol Chem 280: 40885–40891, 2005. [DOI] [PubMed] [Google Scholar]
- 339.Helsen C, and Claessens F. Looking at nuclear receptors from a new angle. Mol Cell Endocrinol 382: 97–106, 2014. [DOI] [PubMed] [Google Scholar]
- 340.Henis YI, Hancock JF, and Prior IA. Ras acylation, compartmentalization and signaling nanoclusters (Review). Mol Membr Biol 26: 80–92, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Henry PC, Kanelis V, O'Brien CM, Kim B, Gautschi I, Forman-Kay J, Schild L, and Rotin D. Affinity and specificity of interactions between Nedd4 isoforms and ENaC. J Biol Chem.: 2003. [DOI] [PubMed] [Google Scholar]
- 342.Henshall TL, Manning JA, Alfassy OS, Goel P, Boase NA, Kawabe H, and Kumar S. Deletion of Nedd4-2 results in progressive kidney disease in mice. Cell Death Differ 24: 2150–2160, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Herman JP, and Spencer R. Regulation of hippocampal glucocorticoid receptor gene transcription and protein expression in vivo. J Neurosci 18: 7462–7473, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Hierholzer K, Schoneshofer M, Siebe H, Tsiakiras D, and Weskamp P. Corticosteroid metabolism in isolated rat kidney in vitro. I. Formation of lipid soluble metabolites from corticosterone (B) in renal tissue from male rats. Pflugers Arch 400: 363–371, 1984. [DOI] [PubMed] [Google Scholar]
- 345.Higashihara E, and Kokko JP. Effects of aldosterone on potassium recycling in the kidney of adrenalectomized rats. Am J Physiol 248: F219–227, 1985. [DOI] [PubMed] [Google Scholar]
- 346.Hilgemann DW, Feng S, and Nasuhoglu C. The complex and intriguing lives of PIP2 with ion channels and transporters. Sci STKE 2001: re19, 2001. [DOI] [PubMed] [Google Scholar]
- 347.Hirschberg D, Jagerbrink T, Samskog J, Gustafsson M, Stahlberg M, Alvelius G, Husman B, Carlquist M, Jornvall H, and Bergman T. Detection of phosphorylated peptides in proteomic analyses using microfluidic compact disk technology. Anal Chem 76: 5864–5871, 2004. [DOI] [PubMed] [Google Scholar]
- 348.Hobler A, Kagawa N, Hutter MC, Hartmann MF, Wudy SA, Hannemann F, and Bernhardt R. Human aldosterone synthase: recombinant expression in E. coli and purification enables a detailed biochemical analysis of the protein on the molecular level. J Steroid Biochem Mol Biol 132: 57–65, 2012. [DOI] [PubMed] [Google Scholar]
- 349.Hofmeister MV, Damkier HH, Christensen BM, Olde B, Fredrik Leeb-Lundberg LM, Fenton RA, Praetorius HA, and Praetorius J. 17beta-Estradiol induces nongenomic effects in renal intercalated cells through G protein-coupled estrogen receptor 1. Am J Physiol Renal Physiol 302: F358–F368, 2012. [DOI] [PubMed] [Google Scholar]
- 350.Hollenberg SM, Weinberger C, Ong ES, Cerelli G, Oro A, Lebo R, Thompson EB, Rosenfeld MG, and Evans RM. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature 318: 635–641, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Holmes MC, Kotelevtsev Y, Mullins JJ, and Seckl JR. Phenotypic analysis of mice bearing targeted deletions of 11beta-hydroxysteroid dehydrogenases 1 and 2 genes. Mol Cell Endocrinol 171: 15–20, 2001. [DOI] [PubMed] [Google Scholar]
- 352.Holtzclaw JD, Cornelius RJ, Hatcher LI, and Sansom SC. Coupled ATP and potassium efflux from intercalated cells. Am J Physiol Renal Physiol 300: F1319–1326, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Holtzclaw JD, Grimm PR, and Sansom SC. Intercalated cell BK-alpha/beta4 channels modulate sodium and potassium handling during potassium adaptation. J Am Soc Nephrol 21: 634–645, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Holtzclaw JD, Grimm PR, and Sansom SC. Role of BK channels in hypertension and potassium secretion. Curr Opin Nephrol Hypertens 20: 512–517, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Holzman JL, Liu L, Duke BJ, Kemendy AE, and Eaton DC. Transactivation of the IGF-1R by aldosterone. Am J Physiol Renal Physiol 292: F1219–F1228, 2007. [DOI] [PubMed] [Google Scholar]
- 356.Hommes DW, Peppelenbosch MP, and van Deventer SJ. Mitogen activated protein (MAP) kinase signal transduction pathways and novel anti-inflammatory targets. Gut 52: 144–151, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Hoorn EJ, Gritter M, Cuevas CA, and Fenton RA. Regulation of the Renal NaCl Cotransporter and Its Role in Potassium Homeostasis. Physiol Rev 100: 321–356, 2020. [DOI] [PubMed] [Google Scholar]
- 358.Hoorn EJ, Nelson JH, McCormick JA, and Ellison DH. The WNK kinase network regulating sodium, potassium, and blood pressure. J Am Soc Nephrol 22: 605–614, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Horisberger JD, and Diezi J. Effects of mineralocorticoids on Na+ and K+ excretion in the adrenalectomized rat. Am J Physiol 245: F89–99, 1983. [DOI] [PubMed] [Google Scholar]
- 360.Hu C, Rusin CG, Tan Z, Guagliardo NA, and Barrett PQ. Zona glomerulosa cells of the mouse adrenal cortex are intrinsic electrical oscillators. J Clin Invest 122: 2046–2053, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Huan Y, Deloach S, Keith SW, Goodfriend TL, and Falkner B. Aldosterone and aldosterone: renin ratio associations with insulin resistance and blood pressure in African Americans. J Am Soc Hypertens 6: 56–65, 2012. [DOI] [PubMed] [Google Scholar]
- 362.Huang S, Zhang A, Ding G, and Chen R. Aldosterone-induced mesangial cell proliferation is mediated by EGF receptor transactivation. Am J Physiol Renal Physiol 296: F1323–F1333, 2009. [DOI] [PubMed] [Google Scholar]
- 363.Hudson WH, Youn C, and Ortlund EA. Crystal structure of the mineralocorticoid receptor DNA binding domain in complex with DNA. PLoS One 9: e107000, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Hughey RP, Bruns JB, Kinlough CL, Harkleroad KL, Tong Q, Carattino MD, Johnson JP, Stockand JD, and Kleyman TR. Epithelial sodium channels are activated by furin-dependent proteolysis. J Biol Chem 279: 18111–18114, 2004. [DOI] [PubMed] [Google Scholar]
- 365.Hulter HN, Licht JH, and Sebastian A. K+ deprivation potentiates the renal acid excretory effect of mineralocorticoid: obliteration by amiloride. Am J Physiol 236: F48–57, 1979. [DOI] [PubMed] [Google Scholar]
- 366.Hulter HN, Sigala JF, and Sebastian A. K+ deprivation potentiates the renal alkalosis-producing effect of mineralocorticoid. Am J Physiol 235: F298–309, 1978. [DOI] [PubMed] [Google Scholar]
- 367.Hultman ML, Krasnoperova NV, Li S, Du S, Xia C, Dietz JD, Lala DS, Welsch DJ, and Hu X. The ligand-dependent interaction of mineralocorticoid receptor with coactivator and corepressor peptides suggests multiple activation mechanisms. Mol Endocrinol 19: 1460–1473, 2005. [DOI] [PubMed] [Google Scholar]
- 368.Hummler E, Barker P, Talbot C, Wang Q, Verdumo C, Grubb B, Gatzy J, Burnier M, Horisberger JD, Beermann F, Boucher R, and Rossier BC. A mouse model for the renal salt-wasting syndrome pseudohypoaldosteronism. Proc Natl Acad Sci U S A 94: 11710–11715, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Hunter RW, and Bailey MA. Hyperkalemia: pathophysiology, risk factors and consequences. Nephrol Dial Transplant 34: iii2–iii11, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Hunter RW, Craigie E, Homer NZ, Mullins JJ, and Bailey MA. Acute inhibition of NCC does not activate distal electrogenic Na+ reabsorption or kaliuresis. Am J Physiol Renal Physiol 306: F457–467, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Hunter RW, Ivy JR, Flatman PW, Kenyon CJ, Craigie E, Mullins LJ, Bailey MA, and Mullins JJ. Hypertrophy in the Distal Convoluted Tubule of an 11beta-Hydroxysteroid Dehydrogenase Type 2 Knockout Model. J Am Soc Nephrol 26: 1537–1548, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Husted RF, and Steinmetz PR. Mechanisms of K+ transport in isolated turtle urinary bladder. Induction of active K+ secretion in a K+-absorbing epithelium. J Clin Invest 70: 832–834, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Huyet J, Pinon GM, Fay MR, Fagart J, and Rafestin-Oblin ME. Structural basis of spirolactone recognition by the mineralocorticoid receptor. Mol Pharmacol 72: 563–571, 2007. [DOI] [PubMed] [Google Scholar]
- 374.Huyet J, Pinon GM, Fay MR, Rafestin-Oblin ME, and Fagart J. Structural determinants of ligand binding to the mineralocorticoid receptor. Mol Cell Endocrinol 350: 187–195, 2012. [DOI] [PubMed] [Google Scholar]
- 375.Hyndman KA, Boesen EI, Elmarakby AA, Brands MW, Huang P, Kohan DE, Pollock DM, and Pollock JS. Renal collecting duct NOS1 maintains fluid-electrolyte homeostasis and blood pressure. Hypertension 62: 91–98, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Hyndman KA, Bugaj V, Mironova E, Stockand JD, and Pollock JS. NOS1-dependent negative feedback regulation of the epithelial sodium channel in the collecting duct. Am J Physiol Renal Physiol 308: F244–F251, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Hyndman KA, Ho DH, Sega MF, and Pollock JS. Histone deacetylase 1 reduces NO production in endothelial cells via lysine deacetylation of NO synthase 3. Am J Physiol Heart Circ Physiol 307: H803–H809, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Hyndman KA, and Pollock JS. Nitric oxide and the A and B of endothelin of sodium homeostasis. Curr Opin Nephrol Hypertens 22: 26–31, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Hyndman KA, Xue J, MacDonell A, Speed JS, Jin C, and Pollock JS. Distinct regulation of inner medullary collecting duct nitric oxide production from mice and rats. Clin Exp Pharm Physiol 40: 233–239, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Igaki T, Itoh H, Suga SI, Hama N, Ogawa Y, Komatsu Y, Yamashita J, Doi K, Chun TH, and Nakao K. Effects of intravenously administered C-type natriuretic peptide in humans: comparison with atrial natriuretic peptide. Hypertens Res 21: 7–13, 1998. [DOI] [PubMed] [Google Scholar]
- 381.Ito S, Itoh H, Rakugi H, Okuda Y, and Yamakawa S. Efficacy and safety of esaxerenone (CS-3150) for the treatment of essential hypertension: a phase 2 randomized, placebo-controlled, double-blind study. J Hum Hypertens 33: 542–551, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Ito S, Itoh H, Rakugi H, Okuda Y, Yoshimura M, and Yamakawa S. Double-Blind Randomized Phase 3 Study Comparing Esaxerenone (CS-3150) and Eplerenone in Patients With Essential Hypertension (ESAX-HTN Study). Hypertension 75: 51–58, 2020. [DOI] [PubMed] [Google Scholar]
- 383.Ito S, Shikata K, Nangaku M, Okuda Y, and Sawanobori T. Efficacy and Safety of Esaxerenone (CS-3150) for the Treatment of Type 2 Diabetes with Microalbuminuria: A Randomized, Double-Blind, Placebo-Controlled, Phase II Trial. Clin J Am Soc Nephrol 14: 1161–1172, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Ivy JR, Jones NK, Costello HM, Mansley MK, Peltz TS, Flatman PW, and Bailey MA. Glucocorticoid receptor activation stimulates the sodium-chloride cotransporter and influences the diurnal rhythm of its phosphorylation. Am J Physiol Renal Physiol 317: F1536–F1548, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Jacobs ME, Jeffers LA, Welch AK, Wingo CS, and Cain BD. MicroRNA regulation of endothelin-1 mRNA in renal collecting duct cells. Life Sci 118: 195–199, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Jaisser F, Escoubet B, Coutry N, Eugene E, Bonvalet JP, and Farman N. Differential regulation of putative K(+)-ATPase by low-K+ diet and corticosteroids in rat distal colon and kidney. Am J Physiol 270: C679–C687, 1996. [DOI] [PubMed] [Google Scholar]
- 387.Jaisser F, and Farman N. Emerging Roles of the Mineralocorticoid Receptor in Pathology: Toward New Paradigms in Clinical Pharmacology. Pharmacol Rev 68: 49–75, 2016. [DOI] [PubMed] [Google Scholar]
- 388.Jaisser F, Horisberger JD, Geering K, and Rossier BC. Mechanisms of urinary K+ and H+ excretion: primary structure and functional expression of a novel H,K-ATPase. J Cell Biol 123: 1421–1429, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Jaisser F, Horisberger JD, and Rossier BC. Primary sequence and functional expression of a novel beta subunit of the P-ATPase gene family. Pflugers Arch 425: 446–452, 1993. [DOI] [PubMed] [Google Scholar]
- 390.Jamison RL. Potassium recycling. Kidney Int 31: 695–703, 1987. [DOI] [PubMed] [Google Scholar]
- 391.Jasti J, Furukawa H, Gonzales EB, and Gouaux E. Structure of acid-sensing ion channel 1 at 1.9 A resolution and low pH. Nature 449: 316–323, 2007. [DOI] [PubMed] [Google Scholar]
- 392.Jernigan NL, Speed J, LaMarca B, Granger JP, and Drummond HA. Angiotensin II regulation of renal vascular ENaC proteins. Am J Hypertens 22: 593–597, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Ji H, Meng Y, Zhang X, Luo W, Wu P, Xiao B, Zhang Z, and Li X. Aldosterone induction of hepatic stellate cell contraction through activation of RhoA/ROCK-2 signaling pathway. Regul Pept 169: 13–20, 2011. [DOI] [PubMed] [Google Scholar]
- 394.Ji HL, and Benos DJ. Degenerin sites mediate proton activation of deltabetagamma-epithelial sodium channel. J Biol Chem 279: 26939–26947, 2004. [DOI] [PubMed] [Google Scholar]
- 395.Ji HL, Zhao RZ, Chen ZX, Shetty S, Idell S, and Matalon S. δ ENaC: a novel divergent amiloride-inhibitable sodium channel. Am J Physiol Lung Cell Mol Physiol 303: L1013–1026, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Jia G, Habibi J, Aroor AR, Hill MA, Yang Y, Whaley-Connell A, Jaisser F, and Sowers JR. Epithelial Sodium Channel in Aldosterone-Induced Endothelium Stiffness and Aortic Dysfunction. Hypertension 72: 731–738, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Jia Z, Aoyagi T, Kohan DE, and Yang T. mPGES-1 deletion impairs aldosterone escape and enhances sodium appetite. Am J Physiol Renal Physiol 299: F155–F166, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Jiménez-Canino R, Fernandes MX, and Alvarez de la Rosa D. Phosphorylation of Mineralocorticoid Receptor Ligand Binding Domain Impairs Receptor Activation and Has a Dominant Negative Effect over Non-phosphorylated Receptors. J Biol Chem 291: 19068–19078, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Jimenez-Canino R, Lorenzo-Diaz F, Jaisser F, Farman N, Giraldez T, and Alvarez de la Rosa D. Histone Deacetylase 6-Controlled Hsp90 Acetylation Significantly Alters Mineralocorticoid Receptor Subcellular Dynamics But Not its Transcriptional Activity. Endocrinology 157: 2515–2532, 2016. [DOI] [PubMed] [Google Scholar]
- 400.Jiménez-Canino R, Lorenzo-Díaz F, Odermatt A, Bailey MA, Livingstone DEW, Jaisser F, Farman N, and Alvarez de la Rosa D. 11β-HSD2 SUMOylation Modulates Cortisol-Induced Mineralocorticoid Receptor Nuclear Translocation Independently of Effects on Transactivation. Endocrinology 158: 4047–4063, 2017. [DOI] [PubMed] [Google Scholar]
- 401.Jin C, Speed JS, Hyndman KA, O'Connor PM, and Pollock DM. Sex differences in ET-1 receptor expression and Ca2+ signaling in the IMCD. Am J Physiol Renal Physiol 305: F1099–1104, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Johnston JG, Speed JS, Jin C, and Pollock DM. Loss of endothelin B receptor function impairs sodium excretion in a time- and sex-dependent manner. Am J Physiol Renal Physiol 311: F991–f998, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Juurlink DN, Mamdani MM, Lee DS, Kopp A, Austin PC, Laupacis A, and Redelmeier DA. Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N Engl J Med 351: 543–551, 2004. [DOI] [PubMed] [Google Scholar]
- 404.Kakiki M, Morohashi K, Nomura M, Omura T, and Horie T. Expression of aldosterone synthase cytochrome P450 (P450aldo) mRNA in rat adrenal glomerulosa cells by angiotensin II type 1 receptor. Endocr Res 23: 277–295, 1997. [DOI] [PubMed] [Google Scholar]
- 405.Kamynina E, Debonneville C, Bens M, Vandewalle A, and Staub O. A novel mouse Nedd4 protein suppresses the activity of the epithelial Na+ channel. FASEB J 15: 204–214, 2001. [DOI] [PubMed] [Google Scholar]
- 406.Kamynina E, and Staub O. Concerted action of ENaC, Nedd4-2, and Sgk1 in transepithelial Na(+) transport. Am J Physiol Renal Physiol 283: F377–F387, 2002. [DOI] [PubMed] [Google Scholar]
- 407.Kang SA, Pacold ME, Cervantes CL, Lim D, Lou HJ, Ottina K, Gray NS, Turk BE, Yaffe MB, and Sabatini DM. mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science 341: 1236566, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Karpushev AV, Levchenko V, Ilatovskaya DV, Pavlov TS, and Staruschenko A. Novel role of Rac1/WAVE signaling mechanism in regulation of the epithelial Na+ channel. Hypertension 57: 996–1002, 2011. [DOI] [PubMed] [Google Scholar]
- 409.Katz FH, Romfh P, and Smith JA. Diurnal variation of plasma aldosterone, cortisol and renin activity in supine man. J Clin Endocrinol Metab 40: 125–134, 1975. [DOI] [PubMed] [Google Scholar]
- 410.Katz FH, Romfh P, and Smith JA. Episodic secretion of aldosterone in supine man; relationship to cortisol. J Clin Endocrinol Metab 35: 178–181, 1972. [DOI] [PubMed] [Google Scholar]
- 411.Kawamoto T, Mitsuuchi Y, Toda K, Yokoyama Y, Miyahara K, Miura S, Ohnishi T, Ichikawa Y, Nakao K, Imura H, Ulick S, and Shizuta Y. Role of steroid 11b-hydroxylase and steroid 18-hydroxylase in the biosynthesis of glucocorticoids and mineralocorticoids in humans. Proc Natl Acad Sci U S A 89: 1458–1462, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Keenan CR, Lew MJ, and Stewart AG. Biased signalling from the glucocorticoid receptor: Renewed opportunity for tailoring glucocorticoid activity. Biochem Pharmacol 112: 6–12, 2016. [DOI] [PubMed] [Google Scholar]
- 413.Kellenberger S, Gautschi I, Rossier BC, and Schild L. Mutations causing Liddle syndrome reduce sodium-dependent downregulation of the epithelial sodium channel in the Xenopus oocyte expression system. J Clin Invest 101: 2741–2750, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Kellner M, Peiter A, Hafner M, Feuring M, Christ M, Wehling M, Falkenstein E, and Losel R. Early aldosterone up-regulated genes: new pathways for renal disease? Kidney Int 64: 1199–1207, 2003. [DOI] [PubMed] [Google Scholar]
- 415.Kemendy AE, Kleyman TR, and Eaton DC. Aldosterone alters the open probability of amiloride-blockable sodium channels in A6 epithelia. Am J Physiol 263: C825–837, 1992. [DOI] [PubMed] [Google Scholar]
- 416.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, and Haussler D. The human genome browser at UCSC. Genome Res 12: 996–1006, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Kim GH, Masilamani S, Turner R, Mitchell C, Wade JB, and Knepper MA. The thiazide-sensitive Na-Cl cotransporter is an aldosterone-induced protein. Proc Natl Acad Sci U S A 95: 14552–14557, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Kino T, Jaffe H, Amin ND, Chakrabarti M, Zheng YL, Chrousos GP, and Pant HC. Cyclin-dependent kinase 5 modulates the transcriptional activity of the mineralocorticoid receptor and regulates expression of brain-derived neurotrophic factor. Mol Endocrinol 24: 941–952, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Kleyman TR, Carattino MD, and Hughey RP. ENaC at the cutting edge: regulation of epithelial sodium channels by proteases. J Biol Chem 284: 20447–20451, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Kleyman TR, Kashlan OB, and Hughey RP. Epithelial Na(+) Channel Regulation by Extracellular and Intracellular Factors. Annu Rev Physiol 80: 263–281, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Kleyman TR, Myerburg MM, and Hughey RP. Regulation of ENaCs by proteases: An increasingly complex story. Kidney Int 70: 1391–1392, 2006. [DOI] [PubMed] [Google Scholar]
- 422.Kleyman TR, Yulo T, Ashbaugh C, Landry D, Cragoe E Jr., Karlin A, and Al-Awqati Q. Photoaffinity labeling of the epithelial sodium channel. J Biol Chem 261: 2839–2843, 1986. [PubMed] [Google Scholar]
- 423.Knepper MA, Packer R, and Good DW. Ammonium transport in the kidney. Physiol Rev 69: 179–249, 1989. [DOI] [PubMed] [Google Scholar]
- 424.Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth B, Balla T, Weiss WA, Williams RL, and Shokat KM. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell 125: 733–747, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Ko B, and Hoover RS. Molecular physiology of the thiazide-sensitive sodium-chloride cotransporter. Curr Opin Nephrol Hypertens 18: 421–427, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Ko B, Mistry AC, Hanson L, Mallick R, Wynne BM, Thai TL, Bailey JL, Klein JD, and Hoover RS. Aldosterone acutely stimulates NCC activity via a SPAK-mediated pathway. Am J Physiol Renal Physiol 305: F645–F652, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Kobayashi T, and Cohen P. Activation of serum- and glucocorticoid-regulated protein kinase by agonists that activate phosphatidylinositide 3-kinase is mediated by 3-phosphoinositide-dependent protein kinase-1 (PDK1) and PDK2. Biochem J 339 (Pt 2): 319–328, 1999. [PMC free article] [PubMed] [Google Scholar]
- 428.Koeppen BM, Biagi BA, and Giebisch GH. Intracellular microelectrode characterization of the rabbit cortical collecting duct. Am J Physiol 244: F35–47, 1983. [DOI] [PubMed] [Google Scholar]
- 429.Koeppen BM, and Giebisch GH. Mineralocorticoid regulation of sodium and potassium transport by the cortical collecting duct. Soc Gen Physiol Ser 39: 89–104, 1985. [PubMed] [Google Scholar]
- 430.Kohan DE. Endothelin, hypertension and chronic kidney disease: new insights. Curr Opin Nephrol Hypertens 19: 134–139, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Kohan DE, Inscho EW, Wesson D, and Pollock DM. Physiology of endothelin and the kidney. Compr Physiol 1: 883–919, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Kohan DE, and Knox FG. Localization of the nephron sites responsible for mineralocorticoid escape in rats. Am J Physiol 239: F149–F153, 1980. [DOI] [PubMed] [Google Scholar]
- 433.Kohan DE, Rossi NF, Inscho EW, and Pollock DM. Regulation of blood pressure and salt homeostasis by endothelin. Physiol Rev 91: 1–77, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Kohn OF, Mitchell PP, and Steinmetz PR. Sch-28080 inhibits bafilomycin-sensitive H+ secretion in turtle bladder independently of luminal [K+]. Am J Physiol 265: F174–F179, 1993. [DOI] [PubMed] [Google Scholar]
- 435.Kojima I, Kojima K, and Rasmussen H. Role of calcium fluxes in the sustained phase of angiotensin II-mediated aldosterone secretion from adrenal glomerulosa cells. J Biol Chem 260: 9177–9184, 1985. [PubMed] [Google Scholar]
- 436.Kojima R, Sekine T, Kawachi M, Cha SH, Suzuki Y, and Endou H. Immunolocalization of multispecific organic anion transporters, OAT1, OAT2, and OAT3, in rat kidney. J Am Soc Nephrol 13: 848–857, 2002. [DOI] [PubMed] [Google Scholar]
- 437.Kolkhof P, and Barfacker L. 30 YEARS OF THE MINERALOCORTICOID RECEPTOR: Mineralocorticoid receptor antagonists: 60 years of research and development. J Endocrinol 234: T125–T140, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Konstas AA, Shearwin-Whyatt LM, Fotia AB, Degger B, Riccardi D, Cook DI, Korbmacher C, and Kumar S. Regulation of the epithelial sodium channel by N4WBP5A, a novel Nedd4/Nedd4-2-interacting protein. J Biol Chem 277: 29406–29416, 2002. [DOI] [PubMed] [Google Scholar]
- 439.Kotelevtsev Y, Brown RW, Fleming S, Kenyon C, Edwards CR, Seckl JR, and Mullins JJ. Hypertension in mice lacking 11beta-hydroxysteroid dehydrogenase type 2. J Clin Invest 103: 683–689, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Krishna GG, Chusid P, and Hoeldtke RD. Mild potassium depletion provokes renal sodium retention. J Lab Clin Med 109: 724–730, 1987. [PubMed] [Google Scholar]
- 441.Krishna GG, Miller E, and Kapoor S. Increased blood pressure during potassium depletion in normotensive men. N Engl J Med 320: 1177–1182, 1989. [DOI] [PubMed] [Google Scholar]
- 442.Krozowski ZS, and Funder JW. Renal mineralocorticoid receptors and hippocampal corticosterone-binding species have identical intrinsic steroid specificity. Proc Natl Acad Sci U S A 80: 6056–6060, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Krozowski ZS, Rundle SE, Wallace C, Castell MJ, Shen JH, Dowling J, Funder JW, and Smith AI. Immunolocalization of renal mineralocorticoid receptors with an antiserum against a peptide deduced from the complementary deoxyribonucleic acid sequence. Endocrinology 125: 192–198, 1989. [DOI] [PubMed] [Google Scholar]
- 444.Krug AW, Grossmann C, Schuster C, Freudinger R, Mildenberger S, Govindan MV, and Gekle M. Aldosterone stimulates epidermal growth factor receptor expression. J Biol Chem 278: 43060–43066, 2003. [DOI] [PubMed] [Google Scholar]
- 445.Krug AW, Papavassiliou F, Hopfer U, Ullrich KJ, and Gekle M. Aldosterone stimulates surface expression of NHE3 in renal proximal brush borders. Pflugers Arch 446: 492–496, 2003. [DOI] [PubMed] [Google Scholar]
- 446.Kumar R, and Thompson EB. Folding of the glucocorticoid receptor N-terminal transactivation function: dynamics and regulation. Mol Cell Endocrinol 348: 450–456, 2012. [DOI] [PubMed] [Google Scholar]
- 447.Kusche-Vihrog K, Callies C, Fels J, and Oberleithner H. The epithelial sodium channel (ENaC): Mediator of the aldosterone response in the vascular endothelium? Steroids 75: 544–549, 2010. [DOI] [PubMed] [Google Scholar]
- 448.Kusche-Vihrog K, Sobczak K, Bangel N, Wilhelmi M, Nechyporuk-Zloy V, Schwab A, Schillers H, and Oberleithner H. Aldosterone and amiloride alter ENaC abundance in vascular endothelium. Pflugers Arch 455: 849–857, 2008. [DOI] [PubMed] [Google Scholar]
- 449.Kyriakis JM, and Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev 92: 689–737, 2012. [DOI] [PubMed] [Google Scholar]
- 450.Laboratories MC. CORT - Overview: Cortisol, Serum. Mayo Foundation for Medical Education and Research. https://www.mayocliniclabs.com/test-catalog/overview/8545#Clinical-and-Interpretive. [January 4, 2022]. [Google Scholar]
- 451.Laboratories MC. PRA - Overview: Renin Activity, Plasma. Mayo Foundation for Medical Education and Research. https://www.mayocliniclabs.com/test-catalog/overview/8060#Clinical-and-Interpretive. [January 4, 2022]. [Google Scholar]
- 452.Lachheb S, Cluzeaud F, Bens M, Genete M, Hibino H, Lourdel S, Kurachi Y, Vandewalle A, Teulon J, and Paulais M. Kir4.1/Kir5.1 channel forms the major K+ channel in the basolateral membrane of mouse renal collecting duct principal cells. Am J Physiol Renal Physiol 294: F1398–1407, 2008. [DOI] [PubMed] [Google Scholar]
- 453.Lahav M, Dietz T, and Edelman IS. The action of aldosterone on sodium transport: Further studies with inhibitors of RNA and protien synthesis. Endocrinology 92: 1685–1699, 1973. [DOI] [PubMed] [Google Scholar]
- 454.Lang F, Henke G, Embark HM, Waldegger S, Palmada M, Bohmer C, and Vallon V. Regulation of channels by the serum and glucocorticoid-inducible kinase - implications for transport, excitability and cell proliferation. Cell Physiol Biochem 13: 41–50, 2003. [DOI] [PubMed] [Google Scholar]
- 455.Lang F, and Pearce D. Regulation of the epithelial Na+ channel by the mTORC2/SGK1 pathway. Nephrol Dial Transplant 31: 200–205, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Lang F, and Shumilina E. Regulation of ion channels by the serum- and glucocorticoid-inducible kinase SGK1. FASEB J 27: 3–12, 2013. [DOI] [PubMed] [Google Scholar]
- 457.Laplante M, and Sabatini DM. mTOR signaling in growth control and disease. Cell 149: 274–293, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Latouche C, Sainte-Marie Y, Steenman M, Castro CP, Naray-Fejes-Toth A, Fejes-Toth G, Farman N, and Jaisser F. Molecular signature of mineralocorticoid receptor signaling in cardiomyocytes: from cultured cells to mouse heart. Endocrinology 151: 4467–4476, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Lavery DN, and McEwan IJ. Structure and function of steroid receptor AF1 transactivation domains: induction of active conformations. Biochem J 391: 449–464, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Lavery GG, Ronconi V, Draper N, Rabbitt EH, Lyons V, Chapman KE, Walker EA, McTernan CL, Giacchetti G, Mantero F, Seckl JR, Edwards CR, Connell JM, Hewison M, and Stewart PM. Late-onset apparent mineralocorticoid excess caused by novel compound heterozygous mutations in the HSD11B2 gene. Hypertension 42: 123–129, 2003. [DOI] [PubMed] [Google Scholar]
- 461.Le Billan F, Amazit L, Bleakley K, Xue QY, Pussard E, Lhadj C, Kolkhof P, Viengchareun S, Fagart J, and Lombès M. Corticosteroid receptors adopt distinct cyclical transcriptional signatures. FASEB J 32: 5626–5639, 2018. [DOI] [PubMed] [Google Scholar]
- 462.Le Billan F, Khan JA, Lamribet K, Viengchareun S, Bouligand J, Fagart J, and Lombès M. Cistrome of the aldosterone-activated mineralocorticoid receptor in human renal cells. FASEB J 29: 3977–3989, 2015. [DOI] [PubMed] [Google Scholar]
- 463.Le MC, Ouvrard-Pascaud A, Capurro C, Cluzeaud F, Fay M, Jaisser F, Farman N, and Blot-Chabaud M. Early nongenomic events in aldosterone action in renal collecting duct cells: PKCalpha activation, mineralocorticoid receptor phosphorylation, and cross-talk with the genomic response. J Am Soc Nephrol 15: 1145–1160, 2004. [PubMed] [Google Scholar]
- 464.Lee G, Makhanova N, Caron K, Lopez ML, Gomez RA, Smithies O, and Kim HS. Homeostatic responses in the adrenal cortex to the absence of aldosterone in mice. Endocrinology 146: 2650–2656, 2005. [DOI] [PubMed] [Google Scholar]
- 465.Lee HA, Lee DY, Cho HM, Kim SY, Iwasaki Y, and Kim IK. Histone deacetylase inhibition attenuates transcriptional activity of mineralocorticoid receptor through its acetylation and prevents development of hypertension. Circ Res 112: 1004–1012, 2013. [DOI] [PubMed] [Google Scholar]
- 466.Lee YJ, Shin SJ, Tan MS, Hsieh TJ, and Tsai JH. Increased renal atrial natriuretic peptide synthesis in rats with deoxycorticosterone acetate-salt treatment. Am J Physiol 271: F779–789, 1996. [DOI] [PubMed] [Google Scholar]
- 467.Lefranc C, Friederich-Persson M, Palacios-Ramirez R, and Nguyen Dinh Cat A. Mitochondrial oxidative stress in obesity: role of the mineralocorticoid receptor. J Endocrinol 238: R143–R159, 2018. [DOI] [PubMed] [Google Scholar]
- 468.Leipziger J Luminal nucleotides are tonic inhibitors of renal tubular transport. Curr Opin Nephrol Hypertens 20: 518–522, 2011. [DOI] [PubMed] [Google Scholar]
- 469.Leipziger J, and Praetorius H. Renal Autocrine and Paracrine Signaling: A Story of Self-protection. Physiol Rev 100: 1229–1289, 2020. [DOI] [PubMed] [Google Scholar]
- 470.Leite-Dellova DC, Malnic G, and Mello-Aires M. Genomic and nongenomic stimulatory effect of aldosterone on H+-ATPase in proximal S3 segments. Am J Physiol Renal Physiol 300: F682–F691, 2011. [DOI] [PubMed] [Google Scholar]
- 471.Lema I, Amazit L, Lamribet K, Fagart J, Blanchard A, Lombes M, Cherradi N, and Viengchareun S. HuR-Dependent Editing of a New Mineralocorticoid Receptor Splice Variant Reveals an Osmoregulatory Loop for Sodium Homeostasis. Sci Rep 7: 4835, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Leopold JA, Dam A, Maron BA, Scribner AW, Liao R, Handy DE, Stanton RC, Pitt B, and Loscalzo J. Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nat Med 13: 189–197, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Lesage F, and Lazdunski M. Molecular and functional properties of two-pore-domain potassium channels. Am J Physiol Renal Physiol 279: F793–F801, 2000. [DOI] [PubMed] [Google Scholar]
- 474.Leviel F, Hübner CA, Houillier P, Morla L, El Moghrabi S, Brideau G, Hassan H, Parker MD, Kurth I, Kougioumtzes A, Sinning A, Pech V, Riemondy KA, Miller RL, Hummler E, Shull GE, Aronson PS, Doucet A, Wall SM, Chambrey R, and Eladari D. The Na+-dependent chloride-bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. J Clin Invest 120: 1627–1635, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Lewis EJ, Hunsicker LG, Bain RP, and Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med 329: 1456–1462, 1993. [DOI] [PubMed] [Google Scholar]
- 476.Li L, Guan Y, Kobori H, Morishita A, Kobara H, Masaki T, Nakano D, and Nishiyama A. Effects of the novel nonsteroidal mineralocorticoid receptor blocker, esaxerenone (CS-3150), on blood pressure and urinary angiotensinogen in low-renin Dahl salt-sensitive hypertensive rats. Hypertens Res 42: 769–778, 2019. [DOI] [PubMed] [Google Scholar]
- 477.Li L, Wingo CS, and Xia SL. Downregulation of SGK1 by nucleotides in renal tubular epithelial cells. Am J Physiol Renal Physiol 293: F1751–F1757, 2007. [DOI] [PubMed] [Google Scholar]
- 478.Li Y, Suino K, Daugherty J, and Xu HE. Structural and biochemical mechanisms for the specificity of hormone binding and coactivator assembly by mineralocorticoid receptor. Mol Cell 19: 367–380, 2005. [DOI] [PubMed] [Google Scholar]
- 479.Lifton RP, Dluhy RG, Powers M, Rich GM, Cook S, Ulick S, and Lalouel JM. A chimaeric 11 beta-hydroxylase/aldosterone synthase gene causes glucocorticoid-remediable aldosteronism and human hypertension. Nature 355: 262–265, 1992. [DOI] [PubMed] [Google Scholar]
- 480.Lim-Tio SS, and Fuller PJ. Intracellular signaling pathways confer specificity of transactivation by mineralocorticoid and glucocorticoid receptors. Endocrinology 139: 1653–1661, 1998. [DOI] [PubMed] [Google Scholar]
- 481.Limor R, Kaplan M, Sharon O, Knoll E, Naidich M, Weisinger G, Keidar S, and Stern N. Aldosterone up-regulates 12- and 15-lipoxygenase expression and LDL oxidation in human vascular smooth muscle cells. J Cell Biochem 108: 1203–1210, 2009. [DOI] [PubMed] [Google Scholar]
- 482.Lin DH, Sterling H, Yang B, Hebert SC, Giebisch G, and Wang WH. Protein tyrosine kinase is expressed and regulates ROMK1 location in the cortical collecting duct. Am J Physiol Renal Physiol 286: F881–892, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Lin DH, Yue P, Rinehart J, Sun P, Wang Z, Lifton R, and Wang WH. Protein phosphatase 1 modulates the inhibitory effect of With-no-Lysine kinase 4 on ROMK channels. Am J Physiol Renal Physiol 303: F110–F119, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484.Linas SL, Peterson LN, Anderson RJ, Aisenbrey GA, Simon FR, and Berl T. Mechanism of renal potassium conservation in the rat. Kidney Int 15: 601–611, 1979. [DOI] [PubMed] [Google Scholar]
- 485.Lingueglia E, Renard S, Voilley N, Waldmann R, Chassande O, Lazdunski M, and Barbry P. Molecular cloning and functional expression of different molecular forms of rat amiloride-binding proteins. Eur J Biochem 216: 679–687, 1993. [DOI] [PubMed] [Google Scholar]
- 486.Lingueglia E, Renard S, Waldmann R, Voilley N, Champigny G, Plass H, Lazdunski M, and Barbry P. Different homologous subunits of the amiloride-sensitive Na+ channel are differently regulated by aldosterone. J Biol Chem 269: 13736–13739, 1994. [PubMed] [Google Scholar]
- 487.Lingueglia E, Voilley N, Waldmann R, Lazdunski M, and Barbry P. Expression cloning of an epithelial amiloride-sensitive Na+ channel. A new channel type with homologies to Caenorhabditis elegans degenerins. FEBS Lett 318: 95–99, 1993. [DOI] [PubMed] [Google Scholar]
- 488.Lipton JO, Yuan ED, Boyle LM, Ebrahimi-Fakhari D, Kwiatkowski E, Nathan A, Guttler T, Davis F, Asara JM, and Sahin M. The Circadian Protein BMAL1 Regulates Translation in Response to S6K1-Mediated Phosphorylation. Cell 161: 1138–1151, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Liu SL, Schmuck S, Chorazcyzewski JZ, Gros R, and Feldman RD. Aldosterone regulates vascular reactivity: short-term effects mediated by phosphatidylinositol 3-kinase-dependent nitric oxide synthase activation. Circulation 108: 2400–2406, 2003. [DOI] [PubMed] [Google Scholar]
- 490.Ljubojevic M, Herak-Kramberger CM, Hagos Y, Bahn A, Endou H, Burckhardt G, and Sabolic I. Rat renal cortical OAT1 and OAT3 exhibit gender differences determined by both androgen stimulation and estrogen inhibition. Am J Physiol Renal Physiol 287: F124–138, 2004. [DOI] [PubMed] [Google Scholar]
- 491.Loffing J, Loffing-Cueni D, Macher A, Hebert SC, Olson B, Knepper MA, Rossier BC, and Kaissling B. Localization of epithelial sodium channel and aquaporin-2 in rabbit kidney cortex. Am J Physiol Renal Physiol 278: F530–539, 2000. [DOI] [PubMed] [Google Scholar]
- 492.Loffing J, Pietri L, Aregger F, Bloch-Faure M, Ziegler U, Meneton P, Rossier BC, and Kaissling B. Differential subcellular localization of ENaC subunits in mouse kidney in response to high- and low-Na diets. Am J Physiol Renal Physiol 279: F252–258, 2000. [DOI] [PubMed] [Google Scholar]
- 493.Loffing J, Summa V, Zecevic M, and Verrey F. Mediators of aldosterone action in the renal tubule. Curr Opin Nephrol Hypertens 10: 667–675, 2001. [DOI] [PubMed] [Google Scholar]
- 494.Lombard WE, Kokko JP, and Jacobson HR. Bicarbonate transport in cortical and outer medullary collecting tubules. Am J Physiol 244: F289–F296, 1983. [DOI] [PubMed] [Google Scholar]
- 495.Lombes M, Farman N, Oblin ME, Baulieu EE, Bonvalet JP, Erlanger BF, and Gasc JM. Immunohistochemical localization of renal mineralocorticoid receptor by using an anti-idiotypic antibody that is an internal image of aldosterone. Proc Natl Acad Sci U S A 87: 1086–1088, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496.Lombès M, Oblin ME, Gasc JM, Baulieu EE, Farman N, and Bonvalet JP. Immunohistochemical and biochemical evidence for a cardiovascular mineralocorticoid receptor. Circ Res 71: 503–510, 1992. [DOI] [PubMed] [Google Scholar]
- 497.Lopez-Nieto CE, You G, Bush KT, Barros EJ, Beier DR, and Nigam SK. Molecular cloning and characterization of NKT, a gene product related to the organic cation transporter family that is almost exclusively expressed in the kidney. J Biol Chem 272: 6471–6478, 1997. [DOI] [PubMed] [Google Scholar]
- 498.Lotshaw DP. Characterization of angiotensin II-regulated K+ conductance in rat adrenal glomerulosa cells. J Membr Biol 156: 261–277, 1997. [DOI] [PubMed] [Google Scholar]
- 499.Lotshaw DP. Role of membrane depolarization and T-type Ca2+ channels in angiotensin II and K+ stimulated aldosterone secretion. Mol Cell Endocrinol 175: 157–171, 2001. [DOI] [PubMed] [Google Scholar]
- 500.Lotshaw DP, and Li F. Angiotensin II activation of Ca(2+)-permeant nonselective cation channels in rat adrenal glomerulosa cells. Am J Physiol 271: C1705–1715, 1996. [DOI] [PubMed] [Google Scholar]
- 501.Lu HK, Fern RJ, Luthin D, Linden J, Liu LP, Cohen CJ, and Barrett PQ. Angiotensin II stimulates T-type Ca2+ channel currents via activation of a G protein, Gi. Am J Physiol 271: C1340–C1349, 1996. [DOI] [PubMed] [Google Scholar]
- 502.Lu M, Wang J, Ives HE, and Pearce D. mSIN1 protein mediates SGK1 protein interaction with mTORC2 protein complex and is required for selective activation of the epithelial sodium channel. J Biol Chem 286: 30647–30654, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503.Lu M, Wang J, Jones KT, Ives HE, Feldman ME, Yao LJ, Shokat KM, Ashrafi K, and Pearce D. mTOR complex-2 activates ENaC by phosphorylating SGK1. J Am Soc Nephrol 21: 811–818, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Luetscher JA, Dowdy A, Lew W, and Callaghan AM. Comparison of effects of d- and l-aldosterone on excretion of sodium and potassium by the adrenalectomized rat. Endocrinology 70: 445–447, 1962. [DOI] [PubMed] [Google Scholar]
- 505.Lumbers ER. Angiotensin and aldosterone. Regul Pept 80: 91–100, 1999. [DOI] [PubMed] [Google Scholar]
- 506.Lynch IJ, Welch AK, Gumz ML, Kohan DE, Cain BD, and Wingo CS. Effect of mineralocorticoid treatment in mice with collecting duct-specific knockout of endothelin-1. Am J Physiol Renal Physiol 309: F1026–1034, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507.Lynch IJ, Welch AK, Kohan DE, Cain BD, and Wingo CS. Endothelin-1 inhibits sodium reabsorption by ET(A) and ET(B) receptors in the mouse cortical collecting duct. Am J Physiol Renal Physiol 305: F568–573, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508.Macova M, Armando I, Zhou J, Baiardi G, Tyurmin D, Larrayoz-Roldan IM, and Saavedra JM. Estrogen reduces aldosterone, upregulates adrenal angiotensin II AT2 receptors and normalizes adrenomedullary Fra-2 in ovariectomized rats. Neuroendocrinology 88: 276–286, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509.Madsen KM, and Tisher CC. Structural-functional relationship along the distal nephron. Am J Physiol 250: F1–15, 1986. [DOI] [PubMed] [Google Scholar]
- 510.Makara JK, Koncz P, Petheö GL, and Spät A. Role of cell volume in K+-induced Ca2+ signaling by rat adrenal glomerulosa cells. Endocrinology 144: 4916–4922, 2003. [DOI] [PubMed] [Google Scholar]
- 511.Makhanova N, Lee G, Takahashi N, Sequeira Lopez ML, Gomez RA, Kim HS, and Smithies O. Kidney function in mice lacking aldosterone. Am J Physiol Renal Physiol 290: F61–69, 2006. [DOI] [PubMed] [Google Scholar]
- 512.Malbert-Colas L, Nicolas G, Galand C, Lecomte MC, and Dhermy D. Identification of new partners of the epithelial sodium channel alpha subunit. C R Biol 326: 615–624, 2003. [DOI] [PubMed] [Google Scholar]
- 513.Malnic G, Klose RM, and Giebisch G. MICROPUNCTURE STUDY OF RENAL POTASSIUM EXCRETION IN THE RAT. Am J Physiol 206: 674–686, 1964. [DOI] [PubMed] [Google Scholar]
- 514.Mamenko MV, Boukelmoune N, Tomilin VN, Zaika OL, Jensen VB, O'Neil RG, and Pochynyuk OM. The renal TRPV4 channel is essential for adaptation to increased dietary potassium. Kidney Int 91: 1398–1409, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515.Manning BD, and Cantley LC. AKT/PKB signaling: navigating downstream. Cell 129: 1261–1274, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516.Mansley MK, Roe AJ, Francis SL, Gill JH, Bailey MA, and Wilson SM. Trichostatin A blocks aldosterone-induced Na(+) transport and control of serum- and glucocorticoid-inducible kinase 1 in cortical collecting duct cells. Br J Pharmacol 176: 4708–4719, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517.Markos F, Healy V, and Harvey BJ. Aldosterone rapidly activates Na+/H+ exchange in M-1 cortical collecting duct cells via a PKC-MAPK pathway. Nephron Physiol 99: 1–9, 2005. [DOI] [PubMed] [Google Scholar]
- 518.Marrero MB, Schieffer B, Paxton WG, Heerdt L, Berk BC, Delafontaine P, and Bernstein KE. Direct stimulation of Jak/STAT pathway by the angiotensin II AT1 receptor. Nature 375: 247–250, 1995. [DOI] [PubMed] [Google Scholar]
- 519.Marver D Regulation of Na+,K(+)-ATPase by aldosterone. Semin Nephrol 12: 56–61, 1992. [PubMed] [Google Scholar]
- 520.Masilamani S, Castro L, and Baylis C. Pregnant rats are refractory to the natriuretic actions of atrial natriuretic peptide. Am J Physiol 36: R1611–R1616, 1994. [DOI] [PubMed] [Google Scholar]
- 521.Masilamani S, Kim GH, Mitchell C, Wade JB, and Knepper MA. Aldosterone-mediated regulation of ENaC alpha, beta, and gamma subunit proteins in rat kidney. J Clin Invest 104: R19–R23, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 522.Massaad C, Houard N, Lombes M, and Barouki R. Modulation of human mineralocorticoid receptor function by protein kinase A. Mol Endocrinol 13: 57–65, 1999. [DOI] [PubMed] [Google Scholar]
- 523.Mastroberardino L, Spindler B, Forster I, Loffing J, Assandri R, May A, and Verrey F. Ras pathway activates epithelial Na+ channel and decreases its surface expression in Xenopus oocytes. Mol Biol Cell 9: 3417–3427, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524.Mastroianni N, Bettinelli A, Bianchetti M, Colussi G, De FM, Sereni F, Ballabio A, and Casari G. Novel molecular variants of the Na-Cl cotransporter gene are responsible for Gitelman syndrome. Am J Hum Genet 59: 1019–1026, 1996. [PMC free article] [PubMed] [Google Scholar]
- 525.Mastroianni N, De FM, Zollo M, Arrigo G, Zuffardi O, Bettinelli A, Ballabio A, and Casari G. Molecular cloning, expression pattern, and chromosomal localization of the human Na-Cl thiazide-sensitive cotransporter (SLC12A3). Genomics 35: 486–493, 1996. [DOI] [PubMed] [Google Scholar]
- 526.Matsukawa N, Nonaka Y, Ying Z, Higaki J, Ogihara T, and Okamoto M. Molecular cloning and expression of cDNAS encoding rat aldosterone synthase: variants of cytochrome P-450(11 beta). Biochem Biophys Res Commun 169: 245–252, 1990. [DOI] [PubMed] [Google Scholar]
- 527.McCabe RD, Bakarich MA, Srivastava K, and Young DB. Potassium inhibits free radical formation. Hypertension 24: 77–82, 1994. [DOI] [PubMed] [Google Scholar]
- 528.McCabe RD, Smith MJ, and Dwyer TM. Aldosterone secretion and the mechanism of potassium adaptation in rats. Steroids 58: 305–313, 1993. [DOI] [PubMed] [Google Scholar]
- 529.McCarthy RT, Isales C, and Rasmussen H. T-type calcium channels in adrenal glomerulosa cells: GTP-dependent modulation by angiotensin II. Proc Natl Acad Sci U S A 90: 3260–3264, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 530.McCurley A, and Jaffe IZ. Mineralocorticoid receptors in vascular function and disease. Mol Cell Endocrinol 350: 256–265, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531.McDonald FJ, Snyder PM, McCray PB Jr., and Welsh MJ. Cloning, expression, and tissue distribution of a human amiloride-sensitive Na+ channel. Am J Physiol 266: L728–734, 1994. [DOI] [PubMed] [Google Scholar]
- 532.McDonald FJ, Yang B, Hrstka RF, Drummond HA, Tarr DE, McCray PB Jr., Stokes JB, Welsh MJ, and Williamson RA. Disruption of the beta subunit of the epithelial Na+ channel in mice: hyperkalemia and neonatal death associated with a pseudohypoaldosteronism phenotype. Proc Natl Acad Sci U S A 96: 1727–1731, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533.McDonough AA, and Youn JH. Potassium Homeostasis: The Knowns, the Unknowns, and the Health Benefits. Physiology (Bethesda) 32: 100–111, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534.McDowell IC, Barrera A, D'Ippolito AM, Vockley CM, Hong LK, Leichter SM, Bartelt LC, Majoros WH, Song L, Safi A, Koçak DD, Gersbach CA, Hartemink AJ, Crawford GE, Engelhardt BE, and Reddy TE. Glucocorticoid receptor recruits to enhancers and drives activation by motif-directed binding. Genome Res 28: 1272–1284, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.McEneaney V, Dooley R, Harvey BJ, and Thomas W. Protein kinase D stabilizes aldosterone-induced ERK1/2 MAP kinase activation in M1 renal cortical collecting duct cells to promote cell proliferation. J Steroid Biochem Mol Biol 118: 18–28, 2010. [DOI] [PubMed] [Google Scholar]
- 536.McEneaney V, Dooley R, Yusef YR, Keating N, Quinn U, Harvey BJ, and Thomas W. Protein kinase D1 modulates aldosterone-induced ENaC activity in a renal cortical collecting duct cell line. Mol Cell Endocrinol 325: 8–17, 2010. [DOI] [PubMed] [Google Scholar]
- 537.McEneaney V, Harvey BJ, and Thomas W. Aldosterone regulates rapid trafficking of epithelial sodium channel subunits in renal cortical collecting duct cells via protein kinase D activation. Mol Endocrinol 22: 881–892, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538.Meijer OC, Buurstede JC, and Schaaf MJM. Corticosteroid Receptors in the Brain: Transcriptional Mechanisms for Specificity and Context-Dependent Effects. Cell Mol Neurobiol 39: 539–549, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539.Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, and Yamamoto KR. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science 324: 407–410, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540.Menzies RI, Zhao X, Mullins LJ, Mullins JJ, Cairns C, Wrobel N, Dunbar DR, Bailey MA, and Kenyon CJ. Transcription controls growth, cell kinetics and cholesterol supply to sustain ACTH responses. Endocr Connect 6: 446–457, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541.Merkulov VM, and Merkulova TI. Structural variants of glucocorticoid receptor binding sites and different versions of positive glucocorticoid responsive elements: Analysis of GR-TRRD database. J Steroid Biochem Mol Biol 115: 1–8, 2009. [DOI] [PubMed] [Google Scholar]
- 542.Michael AK, Asimgil H, and Partch CL. Cytosolic BMAL1 moonlights as a translation factor. Trends Biochem Sci 40: 489–490, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 543.Michell AR, Debnam ES, and Unwin RJ. Regulation of renal function by the gastrointestinal tract: potential role of gut-derived peptides and hormones. Annu Rev Physiol 70: 379–403, 2008. [DOI] [PubMed] [Google Scholar]
- 544.Mick VE, Itani OA, Loftus RW, Husted RF, Schmidt TJ, and Thomas CP. The alpha-subunit of the epithelial sodium channel is an aldosterone-induced transcript in mammalian collecting ducts, and this transcriptional response is mediated via distinct cis-elements in the 5'-flanking region of the gene. Mol Endocrinol 15: 575–588, 2001. [DOI] [PubMed] [Google Scholar]
- 545.Mihailidou AS, and Ashton AW. Cardiac effects of aldosterone: does gender matter? Steroids 91: 32–37, 2014. [DOI] [PubMed] [Google Scholar]
- 546.Miller AH, Spencer RL, Husain A, Rhee R, McEwen BS, and Stein M. Differential expression of type-I adrenal steroid receptors in immune tissues is associated with tissue-specific regulation of type-II receptors by aldosterone. Endocrinology 133: 2133–2140, 1993. [DOI] [PubMed] [Google Scholar]
- 547.Miller JA, Anacta LA, and Cattran DC. Impact of gender on the renal response to angiotensin II. Kidney Int 55: 278–285, 1999. [DOI] [PubMed] [Google Scholar]
- 548.Mills NJ, Sharma K, Haque M, Moore M, and Teruyama R. Aldosterone Mediated Regulation of Epithelial Sodium Channel (ENaC) Subunits in the Rat Hypothalamus. Neuroscience 390: 278–292, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549.Min LJ, Mogi M, Li JM, Iwanami J, Iwai M, and Horiuchi M. Aldosterone and angiotensin II synergistically induce mitogenic response in vascular smooth muscle cells. Circ Res 97: 434–442, 2005. [DOI] [PubMed] [Google Scholar]
- 550.Mironova E, Lynch IJ, Berman JM, Gumz ML, Stockand JD, and Wingo CS. ENaC activity in the cortical collecting duct of HKalpha1 H(+),K(+)-ATPase knockout mice is uncoupled from Na(+) intake. Am J Physiol Renal Physiol 312: F1073–f1080, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 551.Mironova E, Peti-Peterdi J, Bugaj V, and Stockand JD. Diminished paracrine regulation of the epithelial Na+ channel by purinergic signaling in mice lacking connexin 30. J Biol Chem 286: 1054–1060, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552.Mironova E, Suliman F, and Stockand JD. Renal Na(+) excretion consequent to pharmacogenetic activation of Gq-DREADD in principal cells. Am J Physiol Renal Physiol 316: F758–f767, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553.Mistry AC, Wynne BM, Yu L, Tomilin V, Yue Q, Zhou Y, Al-Khalili O, Mallick R, Cai H, Alli AA, Ko B, Mattheyses A, Bao HF, Pochynyuk O, Theilig F, Eaton DC, and Hoover RS. The sodium chloride cotransporter (NCC) and epithelial sodium channel (ENaC) associate. Biochem J 473: 3237–3252, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 554.Miyahara K, Kawamoto T, Mitsuuchi Y, Toda K, Imura H, Gordon RD, and Shizuta Y. The chimeric gene linked to glucocorticoid-suppressible hyperaldosteronism encodes a fused P-450 protein possessing aldosterone synthase activity. Biochem Biophys Res Commun 189: 885–891, 1992. [DOI] [PubMed] [Google Scholar]
- 555.Morita H, Fujiki N, Miyahara T, Lee K, and Tanaka K. Hepatoportal bumetanide-sensitive K(+)-sensor mechanism controls urinary K(+) excretion. Am J Physiol Regul Integr Comp Physiol 278: R1134–1139, 2000. [DOI] [PubMed] [Google Scholar]
- 556.Morla L, Brideau G, Fila M, Crambert G, Cheval L, Houillier P, Ramakrishnan S, Imbert-Teboul M, and Doucet A. Renal proteinase-activated receptor 2, a new actor in the control of blood pressure and plasma potassium level. J Biol Chem 288: 10124–10131, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557.Morris RC Jr., Sebastian A, Forman A, Tanaka M, and Schmidlin O. Normotensive salt sensitivity: effects of race and dietary potassium. Hypertension 33: 18–23, 1999. [DOI] [PubMed] [Google Scholar]
- 558.Moss ME, and Jaffe IZ. Mineralocorticoid Receptors in the Pathophysiology of Vascular Inflammation and Atherosclerosis. Front Endocrinol (Lausanne) 6: 153, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 559.Murai-Takeda A, Shibata H, Kurihara I, Kobayashi S, Yokota K, Suda N, Mitsuishi Y, Jo R, Kitagawa H, Kato S, Saruta T, and Itoh H. NF-YC functions as a corepressor of agonist-bound mineralocorticoid receptor. J Biol Chem 285: 8084–8093, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560.Muto S, Sansom S, and Giebisch G. Effects of a high potassium diet on electrical properties of cortical collecting ducts from adrenalectomized rabbits. J Clin Invest 81: 376–380, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 561.Mutoh A, Isshiki M, and Fujita T. Aldosterone enhances ligand-stimulated nitric oxide production in endothelial cells. Hypertens Res 31: 1811–1820, 2008. [DOI] [PubMed] [Google Scholar]
- 562.Nagai Y, Miyata K, Sun GP, Rahman M, Kimura S, Miyatake A, Kiyomoto H, Kohno M, Abe Y, Yoshizumi M, and Nishiyama A. Aldosterone stimulates collagen gene expression and synthesis via activation of ERK1/2 in rat renal fibroblasts. Hypertension 46: 1039–1045, 2005. [DOI] [PubMed] [Google Scholar]
- 563.Nagoshi T, Date T, Fujisaki M, Yoshino T, Sekiyama H, Ogawa K, Kayama Y, Minai K, Komukai K, Ogawa T, and Yoshimura M. Biphasic action of aldosterone on Akt signaling in cardiomyocytes. Horm Metab Res 44: 931–937, 2012. [DOI] [PubMed] [Google Scholar]
- 564.Nakamura T, and Kawaguchi A. Phase 1 Studies to Define the Safety, Tolerability, and Pharmacokinetic and Pharmacodynamic Profiles of the Nonsteroidal Mineralocorticoid Receptor Antagonist Apararenone in Healthy Volunteers. Clin Pharmacol Drug Dev 10: 353–365, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 565.Nakano D, and Pollock DM. Contribution of endothelin A receptors in endothelin 1-dependent natriuresis in female rats. Hypertension 53: 324–330, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 566.Namsolleck P, and Unger T. Aldosterone synthase inhibitors in cardiovascular and renal diseases. Nephrol Dial Transplant 29 Suppl 1: i62–i68, 2014. [DOI] [PubMed] [Google Scholar]
- 567.Naray-Fejes-Toth A, Boyd C, and Fejes-Toth G. Regulation of epithelial sodium transport by promyelocytic leukemia zinc finger protein. Am J Physiol Renal Physiol 295: F18–F26, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568.Naray-Fejes-Toth A, Canessa C, Cleaveland ES, Aldrich G, and Fejes-Toth G. sgk is an aldosterone-induced kinase in the renal collecting duct. Effects on epithelial na+ channels. J Biol Chem 274: 16973–16978, 1999. [DOI] [PubMed] [Google Scholar]
- 569.New MI, Levine LS, Biglieri EG, Pareira J, and Ulick S. Evidence for an unidentified steroid in a child with apparent mineralocorticoid hypertension. J Clin Endocrinol Metab 44: 924–933, 1977. [DOI] [PubMed] [Google Scholar]
- 570.Newton MA, and Laragh JH. Effect of corticotropin on aldosterone excretion and plasma renin in normal subjects, in essential hypertension and in primary aldosteronism. J Clin Endocrinol Metab 28: 1006–1013, 1968. [DOI] [PubMed] [Google Scholar]
- 571.Ng KP, Arnold J, Sharif A, Gill P, Townend JN, and Ferro CJ. Cardiovascular actions of mineralocorticoid receptor antagonists in patients with chronic kidney disease: A systematic review and meta-analysis of randomized trials. J Renin Angiotensin Aldosterone Syst 16: 599–613, 2015. [DOI] [PubMed] [Google Scholar]
- 572.Nguyen Dinh CA, and Jaisser F. Extrarenal effects of aldosterone. Curr Opin Nephrol Hypertens 21: 147–156, 2012. [DOI] [PubMed] [Google Scholar]
- 573.Nguyen Dinh CA, Ouvrard-Pascaud A, Tronche F, Clemessy M, Gonzalez-Nunez D, Farman N, and Jaisser F. Conditional transgenic mice for studying the role of the glucocorticoid receptor in the renal collecting duct. Endocrinology 150: 2202–2210, 2009. [DOI] [PubMed] [Google Scholar]
- 574.Nigam SK, Bush KT, Martovetsky G, Ahn SY, Liu HC, Richard E, Bhatnagar V, and Wu W. The organic anion transporter (OAT) family: a systems biology perspective. Physiol Rev 95: 83–123, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575.Niisato N, Ohta M, Eaton DC, and Marunaka Y. Hypotonic stress upregulates β- and γ-ENaC expression through suppression of ERK by inducing MKP-1. Am J Physiol Renal Physiol 303: F240–252, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 576.Nikolaeva S, Pradervand S, Centeno G, Zavadova V, Tokonami N, Maillard M, Bonny O, and Firsov D. The circadian clock modulates renal sodium handling. J Am Soc Nephrol 23: 1019–1026, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 577.Nishi M, and Kawata M. Dynamics of glucocorticoid receptor and mineralocorticoid receptor: implications from live cell imaging studies. Neuroendocrinology 85: 186–192, 2007. [DOI] [PubMed] [Google Scholar]
- 578.Nishiyama A, Yao L, Fan Y, Kyaw M, Kataoka N, Hashimoto K, Nagai Y, Nakamura E, Yoshizumi M, Shokoji T, Kimura S, Kiyomoto H, Tsujioka K, Kohno M, Tamaki T, Kajiya F, and Abe Y. Involvement of aldosterone and mineralocorticoid receptors in rat mesangial cell proliferation and deformability. Hypertension 45: 710–716, 2005. [DOI] [PubMed] [Google Scholar]
- 579.Noda S, Aoyama K, Kondo Y, Okamura J, Suzuki A, Yamaguchi N, Yoshida A, and Miyake Y. An infant case of pseudohypoaldosteronism type1A caused by a novel NR3C2 variant. Hum Genome Var 8: 41, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 580.Noreng S, Bharadwaj A, Posert R, Yoshioka C, and Baconguis I. Structure of the human epithelial sodium channel by cryo-electron microscopy. Elife 7: 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 581.O'Neil RG, and Helman SI. Transport characteristics of renal collecting tubules: influences of DOCA and diet. Am J Physiol 233: F544–F558, 1977. [DOI] [PubMed] [Google Scholar]
- 582.Oakley RH, Ramamoorthy S, Foley JF, Busada JT, Lu NZ, and Cidlowski JA. Glucocorticoid receptor isoform-specific regulation of development, circadian rhythm, and inflammation in mice. FASEB J 32: 5258–5271, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 583.Oberleithner H Aldosterone makes human endothelium stiff and vulnerable. Kidney Int 67: 1680–1682, 2005. [DOI] [PubMed] [Google Scholar]
- 584.Oberleithner H, Steigner W, Silbernagl S, Vogel U, Gstraunthaler G, and Pfaller W. Madin-Darby canine kidney cells. III. Aldosterone stimulates an apical H+/K+ pump. Pflugers Arch 416: 540–547, 1990. [DOI] [PubMed] [Google Scholar]
- 585.Oberleithner H, Vogel U, and Kersting U. Madin-Darby canine kidney cells. I. Aldosterone-induced domes and their evaluation as a model system. Pflugers Arch 416: 526–532, 1990. [DOI] [PubMed] [Google Scholar]
- 586.Oberleithner H, Vogel U, Kersting U, and Steigner W. Madin-Darby canine kidney cells. II. Aldosterone stimulates Na+/H+ and Cl-/HCO3- exchange. Pflugers Arch 416: 533–539, 1990. [DOI] [PubMed] [Google Scholar]
- 587.Oelkers W Prolonged ACTH infusion suppresses aldosterone secretion in spite of high renin activity. Acta Endocrinol (Copenh) 108: 91–97, 1985. [DOI] [PubMed] [Google Scholar]
- 588.Oelkers W, Köhler A, Belkien L, Fuchs-Hammoser R, Maiga M, Scherer B, and Weber PC. ACTH stimulates plasma renin and angiotensin II in man. Clin Sci (Lond) 61 Suppl 7: 273s–275s, 1981. [DOI] [PubMed] [Google Scholar]
- 589.Oelkers W, Köhler A, Belkien L, Fuchs-Hammoser R, Maiga M, Scherer B, and Weber PC. Studies on the mechanism by which ACTH stimulates renin activity and angiotensin II formation in man. Acta Endocrinol (Copenh) 100: 573–580, 1982. [DOI] [PubMed] [Google Scholar]
- 590.Oh YT, Kim J, and Youn JH. Role of pituitary in K+ homeostasis: impaired renal responses to altered K+ intake in hypophysectomized rats. Am J Physiol Regul Integr Comp Physiol 304: R1166–1174, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 591.Okusa MD, Unwin RJ, Velázquez H, Giebisch G, and Wright FS. Active potassium absorption by the renal distal tubule. Am J Physiol 262: F488–493, 1992. [DOI] [PubMed] [Google Scholar]
- 592.Ostrosky-Frid M, Castaneda-Bueno M, and Gamba G. Regulation of the renal NaCl cotransporter by the WNK/SPAK pathway: lessons learned from genetically altered animals. Am J Physiol Renal Physiol 316: F146–F158, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 593.Ozolua RI, Omogbai EK, Famodu AB, Ebeigbe AB, and Ajayi OI. Haematological influences of potassium adaptation in normotensive and renally-hypertensive Wistar rats. Br J Biomed Sci 59: 80–84, 2002. [DOI] [PubMed] [Google Scholar]
- 594.Palmer LG. Epithelial Na channels: function and diversity. Annu Rev Physiol 54: 51–66, 1992. [DOI] [PubMed] [Google Scholar]
- 595.Palmer LG. Ion selectivity of epithelial Na channels. J Membr Biol 96: 97–106, 1987. [DOI] [PubMed] [Google Scholar]
- 596.Palmer LG. Ion selectivity of the apical membrane Na channel in the toad urinary bladder. J Membr Biol 67: 91–98, 1982. [DOI] [PubMed] [Google Scholar]
- 597.Palmer LG. Modulation of apical Na permeability of the toad urinary bladder by intracellular Na, Ca, and H. J Membr Biol 83: 57–69, 1985. [DOI] [PubMed] [Google Scholar]
- 598.Palmer LG. Na+ transport and flux ratio through apical Na+ channels in toad bladder. Nature 297: 688–690, 1982. [DOI] [PubMed] [Google Scholar]
- 599.Palmer LG, Antonian L, and Frindt G. Regulation of apical K and Na channels and Na/K pumps in rat cortical collecting tubule by dietary K. J Gen Physiol 104: 693–710, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 600.Palmer LG, Corthesy-Theulaz I, Gaeggeler HP, Kraehenbuhl JP, and Rossier B. Expression of epithelial Na channels in Xenopus oocytes. J Gen Physiol 96: 23–46, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 601.Palmer LG, and Edelman IS. Control of apical sodium permeability in the toad urinary bladder by aldosterone. Ann N Y Acad Sci 372: 1–14, 1981. [DOI] [PubMed] [Google Scholar]
- 602.Palmer LG, and Frindt G. Aldosterone and potassium secretion by the cortical collecting duct. Kidney Int 57: 1324–1328, 2000. [DOI] [PubMed] [Google Scholar]
- 603.Palmer LG, and Frindt G. Amiloride-sensitive Na channels from the apical membrane of the rat cortical collecting tubule. Proc Natl Acad Sci U S A 83: 2767–2770, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 604.Palmer LG, and Frindt G. Regulation of apical K channels in rat cortical collecting tubule during changes in dietary K intake. Am J Physiol 277: F805–812, 1999. [DOI] [PubMed] [Google Scholar]
- 605.Palmer LG, Li JH, Lindemann B, and Edelman IS. Aldosterone control of the density of sodium channels in the toad urinary bladder. J Membr Biol 64: 91–102, 1982. [DOI] [PubMed] [Google Scholar]
- 606.Palmer LG, Patel A, and Frindt G. Regulation and dysregulation of epithelial Na+ channels. Clin Exp Nephrol 16: 35–43, 2012. [DOI] [PubMed] [Google Scholar]
- 607.Palmer LG, and Speez N. Stimulation of apical Na permeability and basolateral Na pump of toad urinary bladder by aldosterone. Am J Physiol 250: F273–F281, 1986. [DOI] [PubMed] [Google Scholar]
- 608.Pan YJ, and Young DB. Experimental aldosterone hypertension in the dog. Hypertension 4: 279–287, 1982. [DOI] [PubMed] [Google Scholar]
- 609.Park J, Leong ML, Buse P, Maiyar AC, Firestone GL, and Hemmings BA. Serum and glucocorticoid-inducible kinase (SGK) is a target of the PI 3-kinase-stimulated signaling pathway. EMBO J 18: 3024–3033, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 610.Parker MD, and Boron WF. The divergence, actions, roles, and relatives of sodium-coupled bicarbonate transporters. Physiol Rev 93: 803–959, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 611.Pascoe L, Curnow KM, Slutsker L, Connell JMC, Speiser PW, New MI, and White PC. Glucocorticoid-suppressible hyperaldosteronism results from hybrid genes created by unequal crossovers between CYP11B1 and CYP11B2. Proc Natl Acad Sci U S A 89: 8327–8331, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 612.Pascual-Le TL, Demange C, and Lombes M. Human mineralocorticoid receptor A and B protein forms produced by alternative translation sites display different transcriptional activities. Eur J Endocrinol 150: 585–590, 2004. [DOI] [PubMed] [Google Scholar]
- 613.Pascual-Le TL, and Lombes M. The mineralocorticoid receptor: a journey exploring its diversity and specificity of action. Mol Endocrinol 19: 2211–2221, 2005. [DOI] [PubMed] [Google Scholar]
- 614.Passero CJ, Mueller GM, Myerburg MM, Carattino MD, Hughey RP, and Kleyman TR. TMPRSS4-dependent activation of the epithelial sodium channel requires cleavage of the gamma-subunit distal to the furin cleavage site. Am J Physiol Renal Physiol 302: F1–F8, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 615.Patel AB, Frindt G, and Palmer LG. Feedback inhibition of ENaC during acute sodium loading in vivo. Am J Physiol Renal Physiol 304: F222–232, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 616.Pavlov TS, Ilatovskaya DV, Levchenko V, Li L, Ecelbarger CM, and Staruschenko A. Regulation of ENaC in mice lacking renal insulin receptors in the collecting duct. FASEB J 27: 2723–2732, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 617.Pearce D The role of SGK1 in hormone-regulated sodium transport. Trends Endocrinol Metab 12: 341–347, 2001. [DOI] [PubMed] [Google Scholar]
- 618.Pearce D SGK1 regulation of epithelial sodium transport. Cell Physiol Biochem 13: 13–20, 2003. [DOI] [PubMed] [Google Scholar]
- 619.Pearce D, Soundararajan R, Trimpert C, Kashlan OB, Deen PM, and Kohan DE. Collecting duct principal cell transport processes and their regulation. Clin J Am Soc Nephrol 10: 135–146, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 620.Peng Y, Moe OW, Chu T, Preisig PA, Yanagisawa M, and Alpern RJ. ETB receptor activation leads to activation and phosphorylation of NHE3. Am J Physiol 276: C938–C945, 1999. [DOI] [PubMed] [Google Scholar]
- 621.Penton D, Bandulik S, Schweda F, Haubs S, Tauber P, Reichold M, Cong LD, El WA, Budde T, Lesage F, Lalli E, Zennaro MC, Warth R, and Barhanin J. Task3 potassium channel gene invalidation causes low renin and salt-sensitive arterial hypertension. Endocrinology 153: 4740–4748, 2012. [DOI] [PubMed] [Google Scholar]
- 622.Perrier R, Boscardin E, Malsure S, Sergi C, Maillard MP, Loffing J, Loffing-Cueni D, Sorensen MV, Koesters R, Rossier BC, Frateschi S, and Hummler E. Severe Salt-Losing Syndrome and Hyperkalemia Induced by Adult Nephron-Specific Knockout of the Epithelial Sodium Channel alpha-Subunit. J Am Soc Nephrol 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 623.Perrotti N, He RA, Phillips SA, Haft CR, and Taylor SI. Activation of serum- and glucocorticoid-induced protein kinase (Sgk) by cyclic AMP and insulin. J Biol Chem 276: 9406–9412, 2001. [DOI] [PubMed] [Google Scholar]
- 624.Peti-Peterdi J, Warnock DG, and Bell PD. Angiotensin II directly stimulates ENaC activity in the cortical collecting duct via AT(1) receptors. J Am Soc Nephrol 13: 1131–1135, 2002. [DOI] [PubMed] [Google Scholar]
- 625.Petty KJ, Kokko JP, and Marver D. Secondary effect of aldosterone on Na-KATPase activity in the rabbit cortical collecting tubule. J Clin Invest 68: 1514–1521, 1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 626.Pfeiffer R, Beron J, and Verrey F. Regulation of Na+ pump function by aldosterone is alpha-subunit isoform specific. J Physiol 516 (Pt 3): 647–655, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 627.Piala AT, Moon TM, Akella R, He H, Cobb MH, and Goldsmith EJ. Chloride sensing by WNK1 involves inhibition of autophosphorylation. Sci Signal 7: ra41, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 628.Pinelli L, Nissant A, Edwards A, Lourdel S, Teulon J, and Paulais M. Dual regulation of the native ClC-K2 chloride channel in the distal nephron by voltage and pH. J Gen Physiol 148: 213–226, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 629.Pippal JB, Yao Y, Rogerson FM, and Fuller PJ. Structural and functional characterization of the interdomain interaction in the mineralocorticoid receptor. Mol Endocrinol 23: 1360–1370, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 630.Pitt B Effect of aldosterone blockade in patients with systolic left ventricular dysfunction: implications of the RALES and EPHESUS studies. Mol Cell Endocrinol 217: 53–58, 2004. [DOI] [PubMed] [Google Scholar]
- 631.Pitt B, Kober L, Ponikowski P, Gheorghiade M, Filippatos G, Krum H, Nowack C, Kolkhof P, Kim SY, and Zannad F. Safety and tolerability of the novel non-steroidal mineralocorticoid receptor antagonist BAY 94–8862 in patients with chronic heart failure and mild or moderate chronic kidney disease: a randomized, double-blind trial. Eur Heart J 34: 2453–2463, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 632.Pitt B, Pfeffer MA, Assmann SF, Boineau R, Anand IS, Claggett B, Clausell N, Desai AS, Diaz R, Fleg JL, Gordeev I, Harty B, Heitner JF, Kenwood CT, Lewis EF, O'Meara E, Probstfield JL, Shaburishvili T, Shah SJ, Solomon SD, Sweitzer NK, Yang S, and McKinlay SM. Spironolactone for heart failure with preserved ejection fraction. N Engl J Med 370: 1383–1392, 2014. [DOI] [PubMed] [Google Scholar]
- 633.Pitt B, Remme W, Zannad F, Neaton J, Martinez F, Roniker B, Bittman R, Hurley S, Kleiman J, and Gatlin M. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 348: 1309–1321, 2003. [DOI] [PubMed] [Google Scholar]
- 634.Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, Palensky J, and Wittes J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 341: 709–717, 1999. [DOI] [PubMed] [Google Scholar]
- 635.Platia MP, Catt KJ, Hodgen GD, and Aguilera G. Regulation of primate angiotensin II receptors during altered sodium intake. Hypertension 8: 1121–1126, 1986. [DOI] [PubMed] [Google Scholar]
- 636.Plato CF, Pollock DM, and Garvin JL. Endothelin inhibits thick ascending limb chloride flux via ET(B) receptor-mediated NO release. Am J Physiol Renal Physiol 279: F326–333, 2000. [DOI] [PubMed] [Google Scholar]
- 637.Plus M Renin Blood Test. National Library of Medicine. https://medlineplus.gov/ency/article/003698.htm. [January 4, 2022]. [Google Scholar]
- 638.Pluznick JL, and Sansom SC. BK channels in the kidney: role in K(+) secretion and localization of molecular components. Am J Physiol Renal Physiol 291: F517–529, 2006. [DOI] [PubMed] [Google Scholar]
- 639.Pluznick JL, Wei P, Grimm PR, and Sansom SC. BK-{beta}1 subunit: immunolocalization in the mammalian connecting tubule and its role in the kaliuretic response to volume expansion. Am J Physiol Renal Physiol 288: F846–854, 2005. [DOI] [PubMed] [Google Scholar]
- 640.Pochynyuk O, Bugaj V, and Stockand JD. Physiologic regulation of the epithelial sodium channel by phosphatidylinositides. Curr Opin Nephrol Hypertens 17: 533–540, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 641.Pochynyuk O, Tong Q, Medina J, Vandewalle A, Staruschenko A, Bugaj V, and Stockand JD. Molecular determinants of PI(4,5)P2 and PI(3,4,5)P3 regulation of the epithelial Na+ channel. J Gen Physiol 130: 399–413, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 642.Pollock DM. Renal endothelin in hypertension. Curr Opin Nephrol Hypertens 9: 157–164, 2000. [DOI] [PubMed] [Google Scholar]
- 643.Pollock DM, Allcock GH, Krishnan A, Dayton BD, and Pollock JS. Upregulation of endothelin B receptors in kidneys of DOCA-salt hypertensive rats. Am J Physiol Renal Physiol 278: F279–F286, 2000. [DOI] [PubMed] [Google Scholar]
- 644.Pollock DM, Keith TL, and Highsmith RF. Endothelin receptors and calcium signaling. FASEB J 9: 1196–1204, 1995. [DOI] [PubMed] [Google Scholar]
- 645.Pollock DM, and Pollock JS. Evidence for endothelin involvement in the response to high salt. Am J Physiol Renal Physiol 281: F144–F150, 2001. [DOI] [PubMed] [Google Scholar]
- 646.Pralong WF, Hunyady L, Várnai P, Wollheim CB, and Spät A. Pyridine nucleotide redox state parallels production of aldosterone in potassium-stimulated adrenal glomerulosa cells. Proc Natl Acad Sci U S A 89: 132–136, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 647.Pratt JH, Dale SL, and Melby JC. The effect of administered ACTH on aldosterone metabolism and secretion. J Clin Endocrinol Metab 42: 355–360, 1976. [DOI] [PubMed] [Google Scholar]
- 648.Pratt JH, Luft FC, Parkinson CA, and Rankin LI. The effect of sodium on aldosterone metabolic clearance. Steroids 37: 1–6, 1981. [DOI] [PubMed] [Google Scholar]
- 649.Preston RA, Afshartous D, Rodco R, Alonso AB, and Garg D. Evidence for a gastrointestinal-renal kaliuretic signaling axis in humans. Kidney Int 88: 1383–1391, 2015. [DOI] [PubMed] [Google Scholar]
- 650.Pruthi D, McCurley A, Aronovitz M, Galayda C, Karumanchi SA, and Jaffe IZ. Aldosterone promotes vascular remodeling by direct effects on smooth muscle cell mineralocorticoid receptors. Arterioscler Thromb Vasc Biol 34: 355–364, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 651.Quinn SJ, Brauneis U, Tillotson DL, Cornwall MC, and Williams GH. Calcium channels and control of cytosolic calcium in rat and bovine zona glomerulosa cells. Am J Physiol 262: C598–C606, 1992. [DOI] [PubMed] [Google Scholar]
- 652.Quinn SJ, Cornwall MC, and Williams GH. Electrophysiological responses to angiotensin II of isolated rat adrenal glomerulosa cells. Endocrinology 120: 1581–1589, 1987. [DOI] [PubMed] [Google Scholar]
- 653.Quinn SJ, and Williams GH. Regulation of aldosterone secretion. Annu Rev Physiol 50: 409–426, 1988. [DOI] [PubMed] [Google Scholar]
- 654.Quinn SJ, Williams GH, and Tillotson DL. Calcium oscillations in single adrenal glomerulosa cells stimulated by angiotensin II. Proc Natl Acad Sci U S A 85: 5754–5758, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 655.Quinn SJ, Williams GH, and Tillotson DL. Calcium response of single adrenal glomerulosa cells to external potassium. Am J Physiol 255: E488–495, 1988. [DOI] [PubMed] [Google Scholar]
- 656.Rabelink TJ, Koomans HA, Hené RJ, and Dorhout Mees EJ. Early and late adjustment to potassium loading in humans. Kidney Int 38: 942–947, 1990. [DOI] [PubMed] [Google Scholar]
- 657.Rabinowitz L. Aldosterone and potassium homeostasis. Kidney Int 49: 1738–1742, 1996. [DOI] [PubMed] [Google Scholar]
- 658.Rabinowitz L, and Aizman RI. The central nervous system in potassium homeostasis. Front Neuroendocrinol 14: 1–26, 1993. [DOI] [PubMed] [Google Scholar]
- 659.Rabinowitz L, and Jackson CA. Rapid potassium adaptation In the rat. Kidney Int 37: 569–569, 1990. [Google Scholar]
- 660.Rabinowitz L, Sarason RL, and Yamauchi H. Effects of KCl infusion on potassium excretion in sheep. Am J Physiol 249: F263–271, 1985. [DOI] [PubMed] [Google Scholar]
- 661.Rashmi P, Colussi G, Ng M, Wu X, Kidwai A, and Pearce D. Glucocorticoid-induced leucine zipper protein regulates sodium and potassium balance in the distal nephron. Kidney Int 91: 1159–1177, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 662.Reddish MJ, and Guengerich FP. Human cytochrome P450 11B2 produces aldosterone by a processive mechanism due to the lactol form of the intermediate 18-hydroxycorticosterone. J Biol Chem 294: 12975–12991, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 663.Reisenauer MR, Anderson M, Huang L, Zhang Z, Zhou Q, Kone BC, Morris AP, Lesage GD, Dryer SE, and Zhang W. AF17 competes with AF9 for binding to Dot1a to up-regulate transcription of epithelial Na+ channel alpha. J Biol Chem 284: 35659–35669, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 664.Reisenauer MR, Wang SW, Xia Y, and Zhang W. Dot1a contains three nuclear localization signals and regulates the epithelial Na+ channel (ENaC) at multiple levels. Am J Physiol Renal Physiol 299: F63–F76, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 665.Renard S, Voilley N, Bassilana F, Lazdunski M, and Barbry P. Localization and regulation by steroids of the alpha, beta and gamma subunits of the amiloride-sensitive Na+ channel in colon, lung and kidney. Pflugers Arch 430: 299–307, 1995. [DOI] [PubMed] [Google Scholar]
- 666.Richards J, All S, Skopis G, Cheng KY, Compton B, Srialluri N, Stow L, Jeffers LA, and Gumz ML. Opposing actions of Per1 and Cry2 in the regulation of Per1 target gene expression in the liver and kidney. Am J Physiol Regul Integr Comp Physiol 305: R735–R747, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 667.Richards J, Jeffers LA, All SC, Cheng KY, and Gumz ML. Role of Per1 and the mineralocorticoid receptor in the coordinate regulation of αENaC in renal cortical collecting duct cells. Front Physiol 4: 253, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 668.Rieg T, Bundey RA, Chen Y, Deschenes G, Junger W, Insel PA, and Vallon V. Mice lacking P2Y2 receptors have salt-resistant hypertension and facilitated renal Na+ and water reabsorption. FASEB J 21: 3717–3726, 2007. [DOI] [PubMed] [Google Scholar]
- 669.Rivers C, Flynn A, Qian X, Matthews L, Lightman S, Ray D, and Norman M. Characterization of conserved tandem donor sites and intronic motifs required for alternative splicing in corticosteroid receptor genes. Endocrinology 150: 4958–4967, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 670.Robert-Nicoud M, Flahaut M, Elalouf JM, Nicod M, Salinas M, Bens M, Doucet A, Wincker P, Artiguenave F, Horisberger JD, Vandewalle A, Rossier BC, and Firsov D. Transcriptome of a mouse kidney cortical collecting duct cell line: effects of aldosterone and vasopressin. Proc Natl Acad Sci U S A 98: 2712–2716, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 671.Robertson NM, Schulman G, Karnik S, Alnemri E, and Litwack G. Demonstration of nuclear translocation of the mineralocorticoid receptor (MR) using an anti-MR antibody and confocal laser scanning microscopy. Mol Endocrinol 7: 1226–1239, 1993. [DOI] [PubMed] [Google Scholar]
- 672.Robins GG, and Sandle GI. Liddle-Mutation of the beta-Subunit, but not the gamma-Subunit, Attenuates Protein Kinase C-Mediated Inhibition of Human Epithelial Sodium Channels (hENaC). J Membr Biol 249: 271–279, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 673.Roesch DM, Tian Y, Zheng W, Shi M, Verbalis JG, and Sandberg K. Estradiol attenuates angiotensin-induced aldosterone secretion in ovariectomized rats. Endocrinology 141: 4629–4636, 2000. [DOI] [PubMed] [Google Scholar]
- 674.Rogerson FM, Brennan FE, and Fuller PJ. Dissecting mineralocorticoid receptor structure and function. J Steroid Biochem Mol Biol 85: 389–396, 2003. [DOI] [PubMed] [Google Scholar]
- 675.Rogerson FM, Yao YZ, Young MJ, and Fuller PJ. Identification and characterization of a ligand-selective mineralocorticoid receptor coactivator. FASEB J 28: 4200–4210, 2014. [DOI] [PubMed] [Google Scholar]
- 676.Ronconi V, Turchi F, Appolloni G, di Tizio V, Boscaro M, and Giacchetti G. Aldosterone, mineralocorticoid receptor and the metabolic syndrome: role of the mineralocorticoid receptor antagonists. Curr Vasc Pharmacol 10: 238–246, 2012. [DOI] [PubMed] [Google Scholar]
- 677.Ronzaud C, Loffing-Cueni D, Hausel P, Debonneville A, Malsure SR, Fowler-Jaeger N, Boase NA, Perrier R, Maillard M, Yang B, Stokes JB, Koesters R, Kumar S, Hummler E, Loffing J, and Staub O. Renal tubular NEDD4–2 deficiency causes NCC-mediated salt-dependent hypertension. J Clin Invest 123: 657–665, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 678.Ross EJ, and Hurst PE. Effect of prolonged administration of aldosterone and corticosterone on plasma and urinary electrolytes in man. Clin Sci 28: 91–98, 1965. [PubMed] [Google Scholar]
- 679.Rossier BC. Role of RNA in the action of aldosterone on Na+ transport. J Membr Biol 40 Spec No: 187–197, 1978. [DOI] [PubMed] [Google Scholar]
- 680.Rossier MF, Pagano S, Python M, Maturana AD, James RW, Mach F, Roux-Lombard P, and Vuilleumier N. Antiapolipoprotein A-1 IgG chronotropic effects require nongenomic action of aldosterone on L-type calcium channels. Endocrinology 153: 1269–1278, 2012. [DOI] [PubMed] [Google Scholar]
- 681.Rotin D, Bar-Sagi D, O'Brodovich H, Merilainen J, Lehto VP, Canessa CM, Rossier BC, and Downey GP. An SH3 binding region in the epithelial Na+ channel (alpha rENaC) mediates its localization at the apical membrane. EMBO J 13: 4440–4450, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 682.Rotin D, and Schild L. ENaC and its regulatory proteins as drug targets for blood pressure control. Curr Drug Targets 9: 709–716, 2008. [DOI] [PubMed] [Google Scholar]
- 683.Rozansky DJ, Cornwall T, Subramanya AR, Rogers S, Yang YF, David LL, Zhu X, Yang CL, and Ellison DH. Aldosterone mediates activation of the thiazide-sensitive Na-Cl cotransporter through an SGK1 and WNK4 signaling pathway. J Clin Invest 119: 2601–2612, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 684.Rubera I, Loffing J, Palmer LG, Frindt G, Fowler-Jaeger N, Sauter D, Carroll T, McMahon A, Hummler E, and Rossier BC. Collecting duct-specific gene inactivation of alphaENaC in the mouse kidney does not impair sodium and potassium balance. J Clin Invest 112: 554–565, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 685.Ruhs S, Nolze A, Hubschmann R, and Grossmann C. 30 YEARS OF THE MINERALOCORTICOID RECEPTOR: Nongenomic effects via the mineralocorticoid receptor. J Endocrinol 234: T107–T124, 2017. [DOI] [PubMed] [Google Scholar]
- 686.Rundle SE, Smith AI, Stockman D, and Funder JW. Immunocytochemical demonstration of mineralocorticoid receptors in rat and human kidney. J Steroid Biochem 33: 1235–1242, 1989. [DOI] [PubMed] [Google Scholar]
- 687.Rusvai E, and Naray-Fejes-Toth A. A new isoform of 11 á-hydroxysteroid dehydrogenase in aldosterone target cells. J Biol Chem 268: 10717–10720, 1993. [PubMed] [Google Scholar]
- 688.Sachs G, Spenney JG, and Lewin M. H+ transport: regulation and mechanism in gastric mucosa and membrane vesicles. Physiol Rev 58: 106–173, 1978. [DOI] [PubMed] [Google Scholar]
- 689.Sacks FM, Svetkey LP, Vollmer WM, Appel LJ, Bray GA, Harsha D, Obarzanek E, Conlin PR, Miller ER 3rd, Simons-Morton DG, Karanja N, and Lin PH. Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. DASH-Sodium Collaborative Research Group. N Engl J Med 344: 3–10, 2001. [DOI] [PubMed] [Google Scholar]
- 690.Salyer SA, Parks J, Barati MT, Lederer ED, Clark BJ, Klein JD, and Khundmiri SJ. Aldosterone regulates Na(+), K(+) ATPase activity in human renal proximal tubule cells through mineralocorticoid receptor. Biochim Biophys Acta 1833: 2143–2152, 2013. [DOI] [PubMed] [Google Scholar]
- 691.San-Cristobal P, Pacheco-Alvarez D, Richardson C, Ring AM, Vazquez N, Rafiqi FH, Chari D, Kahle KT, Leng Q, Bobadilla NA, Hebert SC, Alessi DR, Lifton RP, and Gamba G. Angiotensin II signaling increases activity of the renal Na-Cl cotransporter through a WNK4-SPAK-dependent pathway. Proc Natl Acad Sci U S A 106: 4384–4389, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 692.Sansom SC. Reemergence of the maxi K+ as a K+ secretory channel. Kidney Int 71: 1322–1324; author reply 1324–1325, 2007. [DOI] [PubMed] [Google Scholar]
- 693.Sansom SC, Agulian S, Muto S, Illig V, and Giebisch G. K activity of CCD principal cells from normal and DOCA-treated rabbits. Am J Physiol 256: F136–142, 1989. [DOI] [PubMed] [Google Scholar]
- 694.Sansom SC, and Welling PA. Two channels for one job. Kidney Int 72: 529–530, 2007. [DOI] [PubMed] [Google Scholar]
- 695.Saraf A, Luo J, Morris DR, and Storm DR. Phosphorylation of eukaryotic translation initiation factor 4E and eukaryotic translation initiation factor 4E-binding protein (4EBP) and their upstream signaling components undergo diurnal oscillation in the mouse hippocampus: implications for memory persistence. J Biol Chem 289: 20129–20138, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 696.Sariban-Sohraby S, Burg M, Wiesmann WP, Chiang PK, and Johnson JP. Methylation increases sodium transport into A6 apical membrane vesicles: possible mode of aldosterone action. Science 225: 745–746, 1984. [DOI] [PubMed] [Google Scholar]
- 697.Sariban-Sohraby S, and Fisher RS. Guanine nucleotide-dependent carboxymethylation: a pathway for aldosterone modulation of apical Na+ permeability in epithelia. Kidney Int 48: 965–969, 1995. [DOI] [PubMed] [Google Scholar]
- 698.Sariban-Sohraby S, Fisher RS, and Abramow M. Aldosterone-induced and GTP-stimulated methylation of a 90-kDa polypeptide in the apical membrane of A6 epithelia. J Biol Chem 268: 26613–26617, 1993. [PubMed] [Google Scholar]
- 699.Sarzani R, Opocher G, Paci MV, Belloni AS, Mantero F, Dessi-Fulgheri P, and Rappelli A. Natriuretic peptides receptors in human aldosterone-secreting adenomas. J Endocrinol Invest 22: 514–518, 1999. [DOI] [PubMed] [Google Scholar]
- 700.Sasano H, Fukushima K, Sasaki I, Matsuno S, Nagura H, and Krozowski ZS. Immunolocalization of mineralocorticoid receptor in human kidney, pancreas, salivary, mammary and sweat glands: a light and electron microscopic immunohistochemical study. J Endocrinol 132: 305–310, 1992. [DOI] [PubMed] [Google Scholar]
- 701.Sasser JM, Pollock JS, and Pollock DM. Renal endothelin in chronic angiotensin II hypertension. Am J Physiol Regul Integr Comp Physiol 283: R243–248, 2002. [DOI] [PubMed] [Google Scholar]
- 702.Sassi A, Wang Y, Chassot A, Komarynets O, Roth I, Olivier V, Crambert G, Dizin E, Boscardin E, Hummler E, and Feraille E. Interaction between Epithelial Sodium Channel γ-Subunit and Claudin-8 Modulates Paracellular Sodium Permeability in Renal Collecting Duct. J Am Soc Nephrol 31: 1009–1023, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 703.Satlin LM, Sheng S, Woda CB, and Kleyman TR. Epithelial Na(+) channels are regulated by flow. Am J Physiol Renal Physiol 280: F1010–1018, 2001. [DOI] [PubMed] [Google Scholar]
- 704.Sato N, Ajioka M, Yamada T, Kato M, Myoishi M, Yamada T, Kim SY, Nowack C, Kolkhof P, and Shiga T. A Randomized Controlled Study of Finerenone vs. Eplerenone in Japanese Patients With Worsening Chronic Heart Failure and Diabetes and/or Chronic Kidney Disease. Circ J 80: 1113–1122, 2016. [DOI] [PubMed] [Google Scholar]
- 705.Sausbier M, Arntz C, Bucurenciu I, Zhao H, Zhou XB, Sausbier U, Feil S, Kamm S, Essin K, Sailer CA, Abdullah U, Krippeit-Drews P, Feil R, Hofmann F, Knaus HG, Kenyon C, Shipston MJ, Storm JF, Neuhuber W, Korth M, Schubert R, Gollasch M, and Ruth P. Elevated blood pressure linked to primary hyperaldosteronism and impaired vasodilation in BK channel-deficient mice. Circulation 112: 60–68, 2005. [DOI] [PubMed] [Google Scholar]
- 706.Savory JG, Prefontaine GG, Lamprecht C, Liao M, Walther RF, Lefebvre YA, and Hache RJ. Glucocorticoid receptor homodimers and glucocorticoid-mineralocorticoid receptor heterodimers form in the cytoplasm through alternative dimerization interfaces. Mol Cell Biol 21: 781–793, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 707.Saxton RA, and Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell 168: 960–976, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 708.Sayegh R, Auerbach SD, Li X, Loftus RW, Husted RF, Stokes JB, and Thomas CP. Glucocorticoid induction of epithelial sodium channel expression in lung and renal epithelia occurs via trans-activation of a hormone response element in the 5'-flanking region of the human epithelial sodium channel alpha subunit gene. J Biol Chem 274: 12431–12437, 1999. [DOI] [PubMed] [Google Scholar]
- 709.Schiebinger RJ, Kem DC, and Brown RD. Effect of atrial natriuretic peptide on ACTH, dibutyryl cAMP, angiotensin II and potassium-stimulated aldosterone secretion by rat adrenal glomerulosa cells. Life Sci 42: 919–926, 1988. [DOI] [PubMed] [Google Scholar]
- 710.Schild L, Canessa CM, Shimkets RA, Gautschi I, Lifton RP, and Rossier BC. A mutation in the epithelial sodium channel causing Liddle disease increases channel activity in the Xenopus laevis oocyte expression system. Proc Natl Acad Sci U S A 92: 5699–5703, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 711.Schild L, Lu Y, Gautschi I, Schneeberger E, Lifton RP, and Rossier BC. Identification of a PY motif in the epithelial Na channel subunits as a target sequence for mutations causing channel activation found in Liddle syndrome. EMBO J 15: 2381–2387, 1996. [PMC free article] [PubMed] [Google Scholar]
- 712.Schjoedt KJ, Rossing K, Juhl TR, Boomsma F, Tarnow L, Rossing P, and Parving HH. Beneficial impact of spironolactone on nephrotic range albuminuria in diabetic nephropathy. Kidney Int 70: 536–542, 2006. [DOI] [PubMed] [Google Scholar]
- 713.Schlessinger J. Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell 110: 669–672, 2002. [DOI] [PubMed] [Google Scholar]
- 714.Schmitz B, Brand SM, and Brand E. Aldosterone signaling and soluble adenylyl cyclase-a nexus for the kidney and vascular endothelium. Biochim Biophys Acta 1842: 2601–2609, 2014. [DOI] [PubMed] [Google Scholar]
- 715.Schneider MP, Boesen EI, and Pollock DM. Contrasting actions of endothelin ET(A) and ET(B) receptors in cardiovascular disease. Annu Rev Pharmacol Toxicol 47:731–59.: 731–759, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 716.Schneider MP, Ge Y, Pollock DM, Pollock JS, and Kohan DE. Collecting duct-derived endothelin regulates arterial pressure and Na excretion via nitric oxide. Hypertension 51: 1605–1610, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 717.Scholl UI, Choi M, Liu T, Ramaekers VT, Häusler MG, Grimmer J, Tobe SW, Farhi A, Nelson-Williams C, and Lifton RP. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci U S A 106: 5842–5847, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 718.Scholl UI, Goh G, Stolting G, de Oliveira RC, Choi M, Overton JD, Fonseca AL, Korah R, Starker LF, Kunstman JW, Prasad ML, Hartung EA, Mauras N, Benson MR, Brady T, Shapiro JR, Loring E, Nelson-Williams C, Libutti SK, Mane S, Hellman P, Westin G, Akerstrom G, Bjorklund P, Carling T, Fahlke C, Hidalgo P, and Lifton RP. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat Genet 45: 1050–1054, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 719.Scholl UI, Healy JM, Thiel A, Fonseca AL, Brown TC, Kunstman JW, Horne MJ, Dietrich D, Riemer J, Kucukkoylu S, Reimer EN, Reis AC, Goh G, Kristiansen G, Mahajan A, Korah R, Lifton RP, Prasad ML, and Carling T. Novel somatic mutations in primary hyperaldosteronism are related to the clinical, radiological and pathological phenotype. Clin Endocrinol (Oxf) 83: 779–789, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 720.Scholl UI, and Lifton RP. New insights into aldosterone-producing adenomas and hereditary aldosteronism: mutations in the K+ channel KCNJ5. Curr Opin Nephrol Hypertens 22: 141–147, 2013. [DOI] [PubMed] [Google Scholar]
- 721.Scholl UI, Stolting G, Schewe J, Thiel A, Tan H, Nelson-Williams C, Vichot AA, Jin SC, Loring E, Untiet V, Yoo T, Choi J, Xu S, Wu A, Kirchner M, Mertins P, Rump LC, Onder AM, Gamble C, McKenney D, Lash RW, Jones DP, Chune G, Gagliardi P, Choi M, Gordon R, Stowasser M, Fahlke C, and Lifton RP. CLCN2 chloride channel mutations in familial hyperaldosteronism type II. Nat Genet 50: 349–354, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 722.Schulz C, Fork C, Bauer T, Golz S, Geerts A, Schomig E, and Grundemann D. SLC22A13 catalyses unidirectional efflux of aspartate and glutamate at the basolateral membrane of type A intercalated cells in the renal collecting duct. Biochem J 457: 243–251, 2014. [DOI] [PubMed] [Google Scholar]
- 723.Schunkert H, Danser AH, Hense HW, Derkx FH, Kurzinger S, and Riegger GA. Effects of estrogen replacement therapy on the renin-angiotensin system in postmenopausal women. Circulation 95: 39–45, 1997. [DOI] [PubMed] [Google Scholar]
- 724.Schwartz GJ, and Burg MB. Mineralocorticoid effects on cation transport by cortical collecting tubules in vitro. Am J Physiol 235: F576–F585, 1978. [DOI] [PubMed] [Google Scholar]
- 725.Schwartz WB. Potassium and the kidney. N Engl J Med 253: 601–608, 1955. [DOI] [PubMed] [Google Scholar]
- 726.Seely EW, Conlin PR, Brent GA, and Dluhy RG. Adrenocorticotropin stimulation of aldosterone: prolonged continuous versus pulsatile infusion. J Clin Endocrinol Metab 69: 1028–1032, 1989. [DOI] [PubMed] [Google Scholar]
- 727.Seidel E, Schewe J, and Scholl UI. Genetic causes of primary aldosteronism. Exp Mol Med 51: 1–12, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 728.Sengupta S, Lorente-Rodriguez A, Earnest S, Stippec S, Guo X, Trudgian DC, Mirzaei H, and Cobb MH. Regulation of OSR1 and the sodium, potassium, two chloride cotransporter by convergent signals. Proc Natl Acad Sci U S A 110: 18826–18831, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 729.Sennels HP, Jorgensen HL, Goetze JP, and Fahrenkrug J. Rhythmic 24-hour variations of frequently used clinical biochemical parameters in healthy young males--the Bispebjerg study of diurnal variations. Scand J Clin Lab Invest 72: 287–295, 2012. [DOI] [PubMed] [Google Scholar]
- 730.Seok YM, Lee HA, Park KM, Hwangbo MH, and Kim IK. Lysine deacetylase inhibition attenuates hypertension and is accompanied by acetylation of mineralocorticoid receptor instead of histone acetylation in spontaneously hypertensive rats. Naunyn Schmiedebergs Arch Pharmacol 389: 799–808, 2016. [DOI] [PubMed] [Google Scholar]
- 731.Shi H, Asher C, Chigaev A, Yung Y, Reuveny E, Seger R, and Garty H. Interactions of beta and gamma ENaC with Nedd4 can be facilitated by an ERK-mediated phosphorylation. J Biol Chem 277: 13539–13547, 2002. [DOI] [PubMed] [Google Scholar]
- 732.Shi H, Asher C, Yung Y, Kligman L, Reuveny E, Seger R, and Garty H. Casein kinase 2 specifically binds to and phosphorylates the carboxy termini of ENaC subunits. Eur J Biochem 269: 4551–4558, 2002. [DOI] [PubMed] [Google Scholar]
- 733.Shi S, Carattino MD, Hughey RP, and Kleyman TR. ENaC regulation by proteases and shear stress. Curr Mol Pharmacol 6: 28–34, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 734.Shibata H, Ogishima T, Mitani F, Suzuki H, Murakami M, Saruta T, and Ishimura Y. Regulation of aldosterone synthase cytochrome P-450 in rat adrenals by angiotensin II and potassium. Endocrinology 128: 2534–2539, 1991. [DOI] [PubMed] [Google Scholar]
- 735.Shibata S, Arroyo JP, Castaneda-Bueno M, Puthumana J, Zhang J, Uchida S, Stone KL, Lam TT, and Lifton RP. Angiotensin II signaling via protein kinase C phosphorylates Kelch-like 3, preventing WNK4 degradation. Proc Natl Acad Sci U S A 111: 15556–15561, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 736.Shibata S, Ishizawa K, Wang Q, Xu N, Fujita T, Uchida S, and Lifton RP. ULK1 Phosphorylates and Regulates Mineralocorticoid Receptor. Cell Rep 24: 569–576, 2018. [DOI] [PubMed] [Google Scholar]
- 737.Shibata S, Nagase M, Yoshida S, Kawarazaki W, Kurihara H, Tanaka H, Miyoshi J, Takai Y, and Fujita T. Modification of mineralocorticoid receptor function by Rac1 GTPase: implication in proteinuric kidney disease. Nat Med 14: 1370–1376, 2008. [DOI] [PubMed] [Google Scholar]
- 738.Shibata S, Rinehart J, Zhang J, Moeckel G, Castaneda-Bueno M, Stiegler AL, Boggon TJ, Gamba G, and Lifton RP. Mineralocorticoid receptor phosphorylation regulates ligand binding and renal response to volume depletion and hyperkalemia. Cell Metab 18: 660–671, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 739.Shigaev A, Asher C, Latter H, Garty H, and Reuveny E. Regulation of sgk by aldosterone and its effects on the epithelial Na(+) channel. Am J Physiol Renal Physiol 278: F613–F619, 2000. [DOI] [PubMed] [Google Scholar]
- 740.Shimkets RA, Lifton R, and Canessa CM. In vivo phosphorylation of the epithelial sodium channel. Proc Natl Acad Sci U S A 95: 3301–3305, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 741.Shimkets RA, Warnock DG, Bositis CM, Nelson-Williams C, Hansson JH, Schambelan M, Gill JR Jr., Ulick S, Milora RV, Findling JW, Canessa CM, Rossier BC, and Lifton RP. Liddle's syndrome: heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell 79: 407–414, 1994. [DOI] [PubMed] [Google Scholar]
- 742.Shrestha A, Che RC, and Zhang AH. Role of Aldosterone in Renal Fibrosis. Adv Exp Med Biol 1165: 325–346, 2019. [DOI] [PubMed] [Google Scholar]
- 743.Silver RB, Choe H, and Frindt G. Low NaCl diet increases H-K-ATPase in intercalated cells from rat cortical collecting duct. Am J Physiol 44: F94–F102, 1998. [DOI] [PubMed] [Google Scholar]
- 744.Silver RB, and Frindt G. Functional identification of H-K-ATPase in intercalated cells of cortical collecting tubule. Am J Physiol 264: F259–266, 1993. [DOI] [PubMed] [Google Scholar]
- 745.Simpson SA, Tait JF, Wettstein A, Neher R, Von Euw J, and Reichstein T. [Isolation from the adrenals of a new crystalline hormone with especially high effectiveness on mineral metabolism]. Experientia 9: 333–335, 1953. [DOI] [PubMed] [Google Scholar]
- 746.Simpson SA, Tait JF, Wettstein A, Neher R, Von Euw J, Schindler O, and Reichstein T. [Constitution of aldosterone, a new mineralocorticoid]. Experientia 10: 132–133, 1954. [DOI] [PubMed] [Google Scholar]
- 747.Sinphitukkul K, Eiam-Ong S, and Manotham K. Nongenomic effects of aldosterone on renal protein expressions of pEGFR and pERK1/2 in rat kidney. Am J Nephrol 33: 111–120, 2011. [DOI] [PubMed] [Google Scholar]
- 748.Snyder PM, McDonald FJ, Stokes JB, and Welsh MJ. Membrane topology of the amiloride-sensitive epithelial sodium channel. J Biol Chem 269: 24379–24383, 1994. [PubMed] [Google Scholar]
- 749.Snyder PM, Olson DR, and Thomas BC. Serum and glucocorticoid-regulated kinase modulates Nedd4-2-mediated inhibition of the epithelial Na+ channel. J Biol Chem 277: 5–8, 2002. [DOI] [PubMed] [Google Scholar]
- 750.Sorensen MV, Grossmann S, Roesinger M, Gresko N, Todkar AP, Barmettler G, Ziegler U, Odermatt A, Loffing-Cueni D, and Loffing J. Rapid dephosphorylation of the renal sodium chloride cotransporter in response to oral potassium intake in mice. Kidney Int 83: 811–824, 2013. [DOI] [PubMed] [Google Scholar]
- 751.Sorensen MV, Saha B, Jensen IS, Wu P, Ayasse N, Gleason CE, Svendsen SL, Wang WH, and Pearce D. Potassium acts through mTOR to regulate its own secretion. JCI Insight 5: 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 752.Soundararajan R, Lu M, and Pearce D. Organization of the ENaC-regulatory machinery. Crit Rev Biochem Mol Biol 47: 349–359, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 753.Soundararajan R, Melters D, Shih IC, Wang J, and Pearce D. Epithelial sodium channel regulated by differential composition of a signaling complex. Proc Natl Acad Sci U S A 106: 7804–7809, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 754.Soundararajan R, Pearce D, Hughey RP, and Kleyman TR. Role of epithelial sodium channels and their regulators in hypertension. J Biol Chem 285: 30363–30369, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 755.Soundararajan R, Pearce D, and Ziera T. The role of the ENaC-regulatory complex in aldosterone-mediated sodium transport. Mol Cell Endocrinol 350: 242–247, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 756.Soundararajan R, Wang J, Melters D, and Pearce D. Glucocorticoid-induced Leucine zipper 1 stimulates the epithelial sodium channel by regulating serum- and glucocorticoid-induced kinase 1 stability and subcellular localization. J Biol Chem 285: 39905–39913, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 757.Soundararajan R, Zhang TT, Wang J, Vandewalle A, and Pearce D. A novel role for glucocorticoid-induced leucine zipper protein in epithelial sodium channel-mediated sodium transport. J Biol Chem 280: 39970–39981, 2005. [DOI] [PubMed] [Google Scholar]
- 758.Spät A Glomerulosa cell--a unique sensor of extracellular K+ concentration. Mol Cell Endocrinol 217: 23–26, 2004. [DOI] [PubMed] [Google Scholar]
- 759.Spät A, and Hunyady L. Control of aldosterone secretion: a model for convergence in cellular signaling pathways. Physiol Rev 84: 489–539, 2004. [DOI] [PubMed] [Google Scholar]
- 760.Speed JS, Fox BM, Johnston JG, and Pollock DM. Endothelin and renal ion and water transport. Semin Nephrol 35: 137–144, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 761.Spindler B, Mastroberardino L, Custer M, and Verrey F. Characterization of early aldosterone-induced RNAs identified in A6 kidney epithelia. Pflugers Arch 434: 323–331, 1997. [DOI] [PubMed] [Google Scholar]
- 762.Spindler B, and Verrey F. Aldosterone action: induction of p21(ras) and fra-2 and transcription- independent decrease in myc, jun, and fos. Am J Physiol 276: C1154–C1161, 1999. [DOI] [PubMed] [Google Scholar]
- 763.Spital A, and Sterns RH. Paradoxical potassium depletion: a renal mechanism for extrarenal potassium adaptation. Kidney Int 30: 532–537, 1986. [DOI] [PubMed] [Google Scholar]
- 764.Spyroglou A, Sabrautzki S, Rathkolb B, Bozoglu T, Hrabe de AM, Reincke M, Bidlingmaier M, and Beuschlein F. Gender-, strain-, and inheritance-dependent variation in aldosterone secretion in mice. J Endocrinol 215: 375–381, 2012. [DOI] [PubMed] [Google Scholar]
- 765.Stanton B, Janzen A, Klein-Robbenhaar G, DeFronzo R, Giebisch G, and Wade J. Ultrastructure of rat initial collecting tubule. Effect of adrenal corticosteroid treatment. J Clin Invest 75: 1327–1334, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 766.Stanton B, Pan L, Deetjen H, Guckian V, and Giebisch G. Independent effects of aldosterone and potassium on induction of potassium adaptation in rat kidney. J Clin Invest 79: 198–206, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 767.Stanton BA. Regulation of Na+ and K+ transport by mineralocorticoids. Semin Nephrol 7: 82–90, 1987. [PubMed] [Google Scholar]
- 768.Stanton BA, and Giebisch GH. Potassium transport by the renal distal tubule: effects of potassium loading. Am J Physiol 243: F487–493, 1982. [DOI] [PubMed] [Google Scholar]
- 769.Staruschenko A, Adams E, Booth RE, and Stockand JD. Epithelial Na+ channel subunit stoichiometry. Biophys J 88: 3966–3975, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 770.Staruschenko A, Patel P, Tong Q, Medina JL, and Stockand JD. Ras activates the epithelial Na(+) channel through phosphoinositide 3-OH kinase signaling. J Biol Chem 279: 37771–37778, 2004. [DOI] [PubMed] [Google Scholar]
- 771.Staruschenko A, Pochynyuk O, Vandewalle A, Bugaj V, and Stockand JD. Acute Regulation of the Epithelial Na+ Channel by Phosphatidylinositide 3-OH Kinase Signaling in Native Collecting Duct Principal Cells. J Am Soc Nephrol 18: 1652–1661, 2007. [DOI] [PubMed] [Google Scholar]
- 772.Staruschenko A, Pochynyuk OM, Tong Q, and Stockand JD. Ras couples phosphoinositide 3-OH kinase to the epithelial Na+ channel. Biochim Biophys Acta 1669: 108–115, 2005. [DOI] [PubMed] [Google Scholar]
- 773.Staub O, Dho S, Henry P, Correa J, Ishikawa T, McGlade J, and Rotin D. WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle's syndrome. EMBO J 15: 2371–2380, 1996. [PMC free article] [PubMed] [Google Scholar]
- 774.Steinmetz PR, Husted RF, Mueller A, and Beauwens R. Coupling between H+ transport and anaerobic glycolysis in turtle urinary bladder: effect of inhibitors of H+ ATPase. J Membr Biol 59: 27–34, 1981. [DOI] [PubMed] [Google Scholar]
- 775.Steward PM, and Edwards CR. Specificity of the mineralocorticoid receptor: crucial role of 11beta-hydroxysteroid dehydrogenase. Trends Endocrinol Metab 1: 225–230, 1990. [DOI] [PubMed] [Google Scholar]
- 776.Stewart PM, Corrie JE, Shackleton CH, and Edwards CR. Syndrome of apparent mineralocorticoid excess. A defect in the cortisol-cortisone shuttle. J Clin Invest 82: 340–349, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 777.Stockand JD, Mironova E, Bugaj V, Rieg T, Insel PA, Vallon V, Peti-Peterdi J, and Pochynyuk O. Purinergic inhibition of ENaC produces aldosterone escape. J Am Soc Nephrol 21: 1903–1911, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 778.Stokes JB, and Sigmund RD. Regulation of rENaC mRNA by dietary NaCl and steroids: organ, tissue, and steroid heterogeneity. Am J Physiol 274: C1699–C1707, 1998. [DOI] [PubMed] [Google Scholar]
- 779.Stone DK, Seldin DW, Kokko JP, and Jacobson HR. Anion dependence of rabbit medullary collecting duct acidification. J Clin Invest 71: 1505–1508, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 780.Stone DK, Seldin DW, Kokko JP, and Jacobson HR. Mineralocorticoid modulation of rabbit medullary collecting duct acidification. A sodium-independent effect. J Clin Invest 72: 77–83, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 781.Stone DK, Xie XS, and Racker E. An ATP-driven proton pump in clathrin-coated vesicles. J Biol Chem 258: 4059–4062, 1983. [PubMed] [Google Scholar]
- 782.Stow LR, Gumz ML, Lynch IJ, Greenlee MM, Rudin A, Cain BD, and Wingo CS. Aldosterone modulates steroid receptor binding to the endothelin-1 gene (edn1). J Biol Chem 284: 30087–30096, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 783.Stow LR, Richards J, Cheng KY, Lynch IJ, Jeffers LA, Greenlee MM, Cain BD, Wingo CS, and Gumz ML. The circadian protein period 1 contributes to blood pressure control and coordinately regulates renal sodium transport genes. Hypertension 59: 1151–1156, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 784.Stow LR, Voren GE, Gumz ML, Wingo CS, and Cain BD. Dexamethasone stimulates endothelin-1 gene expression in renal collecting duct cells. Steroids 77: 360–366, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 785.Stowasser M, Ahmed AH, Pimenta E, Taylor PJ, and Gordon RD. Factors affecting the aldosterone/renin ratio. Horm Metab Res 44: 170–176, 2012. [DOI] [PubMed] [Google Scholar]
- 786.Stowasser M, and Gordon RD. Primary Aldosteronism: Changing Definitions and New Concepts of Physiology and Pathophysiology Both Inside and Outside the Kidney. Physiol Rev 96: 1327–1384, 2016. [DOI] [PubMed] [Google Scholar]
- 787.Su XT, Ellison DH, and Wang WH. Kir4.1/Kir5.1 in the DCT plays a role in the regulation of renal K(+) excretion. Am J Physiol Renal Physiol 316: F582–f586, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 788.Su XT, and Wang WH. The expression, regulation, and function of Kir4.1 (Kcnj10) in the mammalian kidney. Am J Physiol Renal Physiol 311: F12–15, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 789.Su XT, Zhang C, Wang L, Gu R, Lin DH, and Wang WH. Disruption of KCNJ10 (Kir4.1) stimulates the expression of ENaC in the collecting duct. Am J Physiol Renal Physiol 310: F985–993, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 790.Sueta D, Yamamoto E, and Tsujita K. Mineralocorticoid Receptor Blockers: Novel Selective Nonsteroidal Mineralocorticoid Receptor Antagonists. Curr Hypertens Rep 22: 21, 2020. [DOI] [PubMed] [Google Scholar]
- 791.Sullivan JC, Goodchild TT, Cai Z, Pollock DM, and Pollock JS. Endothelin(A) (ET(A)) and ET(B) receptor-mediated regulation of nitric oxide synthase 1 (NOS1) and NOS3 isoforms in the renal inner medulla. Acta Physiol (Oxf) 191: 329–336, 2007. [DOI] [PubMed] [Google Scholar]
- 792.Svenningsen P, Andersen H, Nielsen LH, and Jensen BL. Urinary serine proteases and activation of ENaC in kidney-implications for physiological renal salt handling and hypertensive disorders with albuminuria. Pflugers Arch 2014. [DOI] [PubMed] [Google Scholar]
- 793.Svenningsen P, Friis UG, Bistrup C, Buhl KB, Jensen BL, and Skott O. Physiological regulation of epithelial sodium channel by proteolysis. Curr Opin Nephrol Hypertens 20: 529–533, 2011. [DOI] [PubMed] [Google Scholar]
- 794.Szerlip HM, Weisberg L, Clayman M, Neilson E, Wade JB, and Cox M. Aldosterone-induced proteins: purification and localization of GP65,70. Am J Physiol 256: C865–C872, 1989. [DOI] [PubMed] [Google Scholar]
- 795.Szerlip HM, Weisberg L, Geering K, Rossier BC, and Cox M. Aldosterone-induced glycoproteins: electrophysiological-biochemical correlation. Biochim Biophys Acta 940: 1–9, 1988. [DOI] [PubMed] [Google Scholar]
- 796.Tait JF, Tait SA, Little B, and Laumas KR. The disappearance of 7-H-3-d-aldosterone in the plasma of normal subjects. J Clin Invest 40: 72–80, 1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 797.Takuwa N, Takuwa Y, Yanagisawa M, Yamashita K, and Masaki T. A novel vasoactive peptide endothelin stimulates mitogenesis through inositol lipid turnover in Swiss 3T3 fibroblasts. J Biol Chem 264: 7856–7861, 1989. [PubMed] [Google Scholar]
- 798.Talati G, Ohta A, Rai T, Sohara E, Naito S, Vandewalle A, Sasaki S, and Uchida S. Effect of angiotensin II on the WNK-OSR1/SPAK-NCC phosphorylation cascade in cultured mpkDCT cells and in vivo mouse kidney. Biochem Biophys Res Commun 393: 844–848, 2010. [DOI] [PubMed] [Google Scholar]
- 799.Tallec LP, Kirsh O, Lecomte MC, Viengchareun S, Zennaro MC, Dejean A, and Lombes M. Protein inhibitor of activated signal transducer and activator of transcription 1 interacts with the N-terminal domain of mineralocorticoid receptor and represses its transcriptional activity: implication of small ubiquitin-related modifier 1 modification. Mol Endocrinol 17: 2529–2542, 2003. [DOI] [PubMed] [Google Scholar]
- 800.Tamura H, Schild L, Enomoto N, Matsui N, Marumo F, and Rossier BC. Liddle disease caused by a missense mutation of beta subunit of the epithelial sodium channel gene. J Clin Invest 97: 1780–1784, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 801.Tanaka K, Ashizawa N, Kawano H, Sato O, Seto S, Nishihara E, Terazono H, Isomoto S, Shinohara K, and Yano K. Aldosterone induces circadian gene expression of clock genes in H9c2 cardiomyoblasts. Heart Vessels 22: 254–260, 2007. [DOI] [PubMed] [Google Scholar]
- 802.Tchepichev S, Ueda J, Canessa C, Rossier BC, and O'Brodovich H. Lung epithelial Na channel subunits are differentially regulated during development and by steroids. Am J Physiol 269: C805–C812, 1995. [DOI] [PubMed] [Google Scholar]
- 803.Terada Y, Kobayashi T, Kuwana H, Tanaka H, Inoshita S, Kuwahara M, and Sasaki S. Aldosterone stimulates proliferation of mesangial cells by activating mitogen-activated protein kinase 1/2, cyclin D1, and cyclin A. J Am Soc Nephrol 16: 2296–2305, 2005. [DOI] [PubMed] [Google Scholar]
- 804.Terker AS, and Ellison DH. Renal mineralocorticoid receptor and electrolyte homeostasis. Am J Physiol Regul Integr Comp Physiol 309: R1068–1070, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 805.Terker AS, Yarbrough B, Ferdaus MZ, Lazelle RA, Erspamer KJ, Meermeier NP, Park HJ, McCormick JA, Yang CL, and Ellison DH. Direct and Indirect Mineralocorticoid Effects Determine Distal Salt Transport. J Am Soc Nephrol 27: 2436–2445, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 806.Terker AS, Zhang C, McCormick JA, Lazelle RA, Meermeier NP, Siler DA, Park HJ, Fu Y, Cohen DM, Weinstein AM, Wang WH, Yang CL, and Ellison DH. Potassium Modulates Electrolyte Balance and Blood Pressure through Effects on Distal Cell Voltage and Chloride. Cell Metab 21: 39–50, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 807.Thai TL, Yu L, Eaton DC, Duke BJ, Al-Khalili O, Lam HY, Ma H, and Bao HF. Basolateral P2X₄ channels stimulate ENaC activity in Xenopus cortical collecting duct A6 cells. Am J Physiol Renal Physiol 307: F806–813, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 808.Thai TL, Yu L, Galarza-Paez L, Wu MM, Lam HY, Bao HF, Duke BJ, Al-Khalili O, Ma HP, Liu B, and Eaton DC. The Polarized Effect of Intracellular Calcium on the Renal Epithelial Sodium Channel Occurs as a Result of Subcellular Calcium Signaling Domains Maintained by Mitochondria. J Biol Chem 290: 28805–28811, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 809.Thatcher JS, and Radike AW. Tolerance to potassium intoxication in the albino rat. Am J Physiol 151: 138–146, 1947. [DOI] [PubMed] [Google Scholar]
- 810.Thomas CP, Auerbach S, Stokes JB, and Volk KA. 5' heterogeneity in epithelial sodium channel alpha-subunit mRNA leads to distinct NH2-terminal variant proteins. Am J Physiol Cell Physiol 274: C1312–C1323, 1998. [DOI] [PubMed] [Google Scholar]
- 811.Thomas CP, Auerbach SD, Zhang C, and Stokes JB. The structure of the rat amiloride-sensitive epithelial sodium channel gamma subunit gene and functional analysis of its promoter. Gene 228: 111–122, 1999. [DOI] [PubMed] [Google Scholar]
- 812.Thomas CP, Campbell JR, Wright PJ, and Husted RF. cAMP-stimulated Na+ transport in H441 distal lung epithelial cells: role of PKA, phosphatidylinositol 3-kinase, and sgk1. Am J Physiol Lung Cell Mol Physiol 287: L843–L851, 2004. [DOI] [PubMed] [Google Scholar]
- 813.Thomas CP, Doggett NA, Fisher R, and Stokes JB. Genomic organization and the 5' flanking region of the gamma subunit of the human amiloride-sensitive epithelial sodium channel. J Biol Chem 271: 26062–26066, 1996. [DOI] [PubMed] [Google Scholar]
- 814.Thomas CP, and Itani OA. New insights into epithelial sodium channel function in the kidney: site of action, regulation by ubiquitin ligases, serum- and glucocorticoid-inducible kinase and proteolysis. Curr Opin Nephrol Hypertens 13: 541–548, 2004. [DOI] [PubMed] [Google Scholar]
- 815.Tirard M, Almeida OF, Hutzler P, Melchior F, and Michaelidis TM. Sumoylation and proteasomal activity determine the transactivation properties of the mineralocorticoid receptor. Mol Cell Endocrinol 268: 20–29, 2007. [DOI] [PubMed] [Google Scholar]
- 816.Tirard M, Jasbinsek J, Almeida OF, and Michaelidis TM. The manifold actions of the protein inhibitor of activated STAT proteins on the transcriptional activity of mineralocorticoid and glucocorticoid receptors in neural cells. J Mol Endocrinol 32: 825–841, 2004. [DOI] [PubMed] [Google Scholar]
- 817.Tobian L, Lange J, Ulm K, Wold L, and Iwai J. Potassium reduces cerebral hemorrhage and death rate in hypertensive rats, even when blood pressure is not lowered. Hypertension 7: I110–114, 1985. [DOI] [PubMed] [Google Scholar]
- 818.Todkar A, Picard N, Loffing-Cueni D, Sorensen MV, Mihailova M, Nesterov V, Makhanova N, Korbmacher C, Wagner CA, and Loffing J. Mechanisms of renal control of potassium homeostasis in complete aldosterone deficiency. J Am Soc Nephrol 26: 425–438, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 819.Togawa A, Miyoshi J, Ishizaki H, Tanaka M, Takakura A, Nishioka H, Yoshida H, Doi T, Mizoguchi A, Matsuura N, Niho Y, Nishimune Y, Nishikawa S, and Takai Y. Progressive impairment of kidneys and reproductive organs in mice lacking Rho GDIalpha. Oncogene 18: 5373–5380, 1999. [DOI] [PubMed] [Google Scholar]
- 820.Tokonami N, Mordasini D, Pradervand S, Centeno G, Jouffe C, Maillard M, Bonny O, Gachon F, Gomez RA, Sequeira-Lopez ML, and Firsov D. Local renal circadian clocks control fluid-electrolyte homeostasis and BP. J Am Soc Nephrol 25: 1430–1439, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 821.Tokonami N, Morla L, Centeno G, Mordasini D, Ramakrishnan SK, Nikolaeva S, Wagner CA, Bonny O, Houillier P, Doucet A, and Firsov D. alpha-Ketoglutarate regulates acid-base balance through an intrarenal paracrine mechanism. J Clin Invest 123: 3166–3171, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 822.Tomilin V, Mamenko M, Zaika O, Wingo CS, and Pochynyuk O. TRPV4 deletion protects against hypokalemia during systemic K(+) deficiency. Am J Physiol Renal Physiol 316: F948–f956, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 823.Tomilin VN, Zaika O, Subramanya AR, and Pochynyuk O. Dietary K(+) and Cl(-) independently regulate basolateral conductance in principal and intercalated cells of the collecting duct. Pflugers Arch 470: 339–353, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 824.Toney GM, Vallon V, and Stockand JD. Intrinsic control of sodium excretion in the distal nephron by inhibitory purinergic regulation of the epithelial Na(+) channel. Curr Opin Nephrol Hypertens 21: 52–60, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 825.Tong Q, Booth RE, Worrell RT, and Stockand JD. Regulation of Na+ transport by aldosterone: signaling convergence and cross talk between the PI3-K and MAPK1/2 cascades. Am J Physiol Renal Physiol 286: F1232–F1238, 2004. [DOI] [PubMed] [Google Scholar]
- 826.Trac PT, Thai TL, Linck V, Zou L, Greenlee M, Yue Q, Al-Khalili O, Alli AA, Eaton AF, and Eaton DC. Alveolar nonselective channels are ASIC1a/α-ENaC channels and contribute to AFC. Am J Physiol Lung Cell Mol Physiol 312: L797–l811, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 827.Tsuchiya Y, Nakashima S, Banno Y, Suzuki Y, and Morita H. Effect of high-NaCl or high-KCl diet on hepatic Na+- and K+-receptor sensitivity and NKCC1 expression in rats. Am J Physiol Regul Integr Comp Physiol 286: R591–596, 2004. [DOI] [PubMed] [Google Scholar]
- 828.Tsugita M, Iwasaki Y, Nishiyama M, Taguchi T, Shinahara M, Taniguchi Y, Kambayashi M, Nishiyama A, Gomez-Sanchez CE, Terada Y, and Hashimoto K. Glucocorticoid receptor plays an indispensable role in mineralocorticoid receptor-dependent transcription in GR-deficient BE(2)C and T84 cells in vitro. Mol Cell Endocrinol 302: 18–25, 2009. [DOI] [PubMed] [Google Scholar]
- 829.Tsuruoka S, and Schwartz GJ. Adaptation of rabbit cortical collecting duct HCO3- transport to metabolic acidosis in vitro. J Clin Invest 97: 1076–1084, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 830.Turban S, Wang XY, and Knepper MA. Regulation of NHE3, NKCC2, and NCC abundance in kidney during aldosterone escape phenomenon: role of NO. Am J Physiol Renal Physiol 285: F843–F851, 2003. [DOI] [PubMed] [Google Scholar]
- 831.Uchimura K, Kakizoe Y, Onoue T, Hayata M, Morinaga J, Yamazoe R, Ueda M, Mizumoto T, Adachi M, Miyoshi T, Shiraishi N, Sakai Y, Tomita K, and Kitamura K. In vivo contribution of serine proteases to the proteolytic activation of gammaENaC in aldosterone-infused rats. Am J Physiol Renal Physiol 303: F939–F943, 2012. [DOI] [PubMed] [Google Scholar]
- 832.Ueda K, Fujiki K, Shirahige K, Gomez-Sanchez CE, Fujita T, Nangaku M, and Nagase M. Genome-wide analysis of murine renal distal convoluted tubular cells for the target genes of mineralocorticoid receptor. Biochem Biophys Res Commun 445: 132–137, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 833.Ulick S Stereospecificity in the metabolism of aldosterone in man. J Biol Chem 236: 680–684, 1961. [PubMed] [Google Scholar]
- 834.Ulick S, Levine LS, Gunczler P, Zanconato G, Ramirez LC, Rauh W, Rosler A, Bradlow HL, and New MI. A syndrome of apparent mineralocorticoid excess associated with defects in the peripheral metabolism of cortisol. J Clin Endocrinol Metab 49: 757–764, 1979. [DOI] [PubMed] [Google Scholar]
- 835.Valinsky WC, Touyz RM, and Shrier A. Aldosterone and Ion Channels. Vitam Horm 109: 105–131, 2019. [DOI] [PubMed] [Google Scholar]
- 836.Vallet V, Chraibi A, Gaeggeler HP, Horisberger JD, and Rossier BC. An epithelial serine protease activates the amiloride-sensitive sodium channel. Nature 389: 607–610, 1997. [DOI] [PubMed] [Google Scholar]
- 837.Vallon V, Eraly SA, Wikoff WR, Rieg T, Kaler G, Truong DM, Ahn SY, Mahapatra NR, Mahata SK, Gangoiti JA, Wu W, Barshop BA, Siuzdak G, and Nigam SK. Organic anion transporter 3 contributes to the regulation of blood pressure. J Am Soc Nephrol 19: 1732–1740, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 838.Vallon V, and Rieg T. Regulation of renal NaCl and water transport by the ATP/UTP/P2Y2 receptor system. Am J Physiol Renal Physiol 301: F463–475, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 839.Vallon V, Unwin R, Inscho EW, Leipziger J, and Kishore BK. Extracellular Nucleotides and P2 Receptors in Renal Function. Physiol Rev 100: 211–269, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 840.Vanhaesebroeck B, and Waterfield MD. Signaling by distinct classes of phosphoinositide 3-kinases. Exp Cell Res 253: 239–254, 1999. [DOI] [PubMed] [Google Scholar]
- 841.Várnai P, Petheö GL, Makara JK, and Spät A. Electrophysiological study on the high K+ sensitivity of rat glomerulosa cells. Pflugers Arch 435: 429–431, 1998. [DOI] [PubMed] [Google Scholar]
- 842.Vasquez MM, Castro R, Seidner SR, Henson BM, Ashton DJ, and Mustafa SB. Induction of serum- and glucocorticoid-induced kinase-1 (SGK1) by cAMP regulates increases in alpha-ENaC. J Cell Physiol 217: 632–642, 2008. [DOI] [PubMed] [Google Scholar]
- 843.Vassileva I, Mountain C, and Pollock DM. Functional role of ETB receptors in the renal medulla. Hypertension 41: 1359–1363, 2003. [DOI] [PubMed] [Google Scholar]
- 844.Velázquez H, Bartiss A, Bernstein P, and Ellison DH. Adrenal steroids stimulate thiazide-sensitive NaCl transport by rat renal distal tubules. Am J Physiol 270: F211–219, 1996. [DOI] [PubMed] [Google Scholar]
- 845.Velázquez H, Ellison DH, and Wright FS. Chloride-dependent potassium secretion in early and late renal distal tubules. Am J Physiol 253: F555–562, 1987. [DOI] [PubMed] [Google Scholar]
- 846.Venema RC, Ju H, Venema VJ, Schieffer B, Harp JB, Ling BN, Eaton DC, and Marrero MB. Angiotensin II-induced association of phospholipase Cgamma1 with the G-protein-coupled AT1 receptor. J Biol Chem 273: 7703–7708, 1998. [DOI] [PubMed] [Google Scholar]
- 847.Verlander JW, Hassell KA, Royaux IE, Glapion DM, Wang ME, Everett LA, Green ED, and Wall SM. Deoxycorticosterone upregulates PDS (Slc26a4) in mouse kidney: role of pendrin in mineralocorticoid-induced hypertension. Hypertension 42: 356–362, 2003. [DOI] [PubMed] [Google Scholar]
- 848.Verlander JW, Moudy RM, Campbell WG, Cain BD, and Wingo CS. Immunohistochemical localization of H-K-ATPase alpha(2c)-subunit in rabbit kidney. Am J Physiol Renal Physiol 281: F357–365, 2001. [DOI] [PubMed] [Google Scholar]
- 849.Verrey F, Fakitsas P, Adam G, and Staub O. Early transcriptional control of ENaC (de)ubiquitylation by aldosterone. Kidney Int 73: 691–696, 2008. [DOI] [PubMed] [Google Scholar]
- 850.Verrey F, Kraehenbuhl JP, and Rossier BC. Aldosterone induces a rapid increase in the rate of Na,K-ATPase gene transcription in cultured kidney cells. Mol Endocrinol 3: 1369–1376, 1989. [DOI] [PubMed] [Google Scholar]
- 851.Verrey F, Schaerer E, Zoerkler P, Paccolat MP, Geering K, Kraehenbuhl JP, and Rossier BC. Regulation by aldosterone of Na+,K+-ATPase mRNAs, protein synthesis, and sodium transport in cultured kidney cells. J Cell Biol 104: 1231–1237, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 852.Viengchareun S, Le MD, Martinerie L, Munier M, Pascual-Le TL, and Lombes M. The mineralocorticoid receptor: insights into its molecular and (patho)physiological biology. Nucl Recept Signal 5: e012, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 853.Vitari AC, Deak M, Morrice NA, and Alessi DR. The WNK1 and WNK4 protein kinases that are mutated in Gordon's hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem J 391: 17–24, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 854.Vitellius G, Trabado S, Bouligand J, Delemer B, and Lombes M. Pathophysiology of Glucocorticoid Signaling. Ann Endocrinol (Paris) 79: 98–106, 2018. [DOI] [PubMed] [Google Scholar]
- 855.Voilley N, Lingueglia E, Champigny G, Mattéi MG, Waldmann R, Lazdunski M, and Barbry P. The lung amiloride-sensitive Na+ channel: biophysical properties, pharmacology, ontogenesis, and molecular cloning. Proc Natl Acad Sci U S A 91: 247–251, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 856.Volk KA, Sigmund RD, Snyder PM, McDonald FJ, Welsh MJ, and Stokes JB. rENaC is the predominant Na+ channel in the apical membrane of the rat renal inner medullary collecting duct. J Clin Invest 96: 2748–2757, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 857.Volpe M, Rubattu S, Gigante B, Ganten D, Porcellini A, Russo R, Romano M, Enea I, Lee MA, and Trimarco B. Regulation of aldosterone biosynthesis by adrenal renin is mediated through AT1 receptors in renin transgenic rats. Circ Res 77: 73–79, 1995. [DOI] [PubMed] [Google Scholar]
- 858.Vuagniaux G, Vallet V, Jaeger NF, Hummler E, and Rossier BC. Synergistic activation of ENaC by three membrane-bound channel-activating serine proteases (mCAP1, mCAP2, and mCAP3) and serum- and glucocorticoid-regulated kinase (Sgk1) in Xenopus Oocytes. J Gen Physiol 120: 191–201, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 859.Wade JB, O'Neil RG, Pryor JL, and Boulpaep EL. Modulation of cell membrane area in renal collecting tubules by corticosteroid hormones. J Cell Biol 81: 439–445, 1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 860.Wagner CA, Ott M, Klingel K, Beck S, Melzig J, Friedrich B, Wild KN, Broer S, Moschen I, Albers A, Waldegger S, Tummler B, Egan ME, Geibel JP, Kandolf R, and Lang F. Effects of the serine/threonine kinase SGK1 on the epithelial Na(+) channel (ENaC) and CFTR: implications for cystic fibrosis. Cell Physiol Biochem 11: 209–218, 2001. [DOI] [PubMed] [Google Scholar]
- 861.Wald H, Goldstein O, Asher C, Yagil Y, and Garty H. Aldosterone induction and epithelial distribution of CHIF. Am J Physiol 271: F322–F329, 1996. [DOI] [PubMed] [Google Scholar]
- 862.Waldmann R, Champigny G, Bassilana F, Voilley N, and Lazdunski M. Molecular cloning and functional expression of a novel amiloride-sensitive Na+ channel. J Biol Chem 270: 27411–27414, 1995. [DOI] [PubMed] [Google Scholar]
- 863.Wall SM, Mehta P, and DuBose TD Jr. Dietary K+ restriction upregulates total and Sch-28080-sensitive bicarbonate absorption in rat tIMCD. Am J Physiol 275: F543–549, 1998. [DOI] [PubMed] [Google Scholar]
- 864.Wall SM, Truong AV, and DuBose TD Jr. H(+)-K(+)-ATPase mediates net acid secretion in rat terminal inner medullary collecting duct. Am J Physiol 271: F1037–1044, 1996. [DOI] [PubMed] [Google Scholar]
- 865.Wan N, Rahman A, and Nishiyama A. Esaxerenone, a novel nonsteroidal mineralocorticoid receptor blocker (MRB) in hypertension and chronic kidney disease. J Hum Hypertens 35: 148–156, 2021. [DOI] [PubMed] [Google Scholar]
- 866.Wang H, Huang BS, and Leenen FH. Brain sodium channels and ouabainlike compounds mediate central aldosterone-induced hypertension. Am J Physiol Heart Circ Physiol 285: H2516–2523, 2003. [DOI] [PubMed] [Google Scholar]
- 867.Wang HX, Yang H, Han QY, Li N, Jiang X, Tian C, Du J, and Li HH. NADPH oxidases mediate a cellular “memory” of angiotensin II stress in hypertensive cardiac hypertrophy. Free Radic Biol Med 65: 897–907, 2013. [DOI] [PubMed] [Google Scholar]
- 868.Wang MX, Cuevas CA, Su XT, Wu P, Gao ZX, Lin DH, McCormick JA, Yang CL, Wang WH, and Ellison DH. Potassium intake modulates the thiazide-sensitive sodium-chloride cotransporter (NCC) activity via the Kir4.1 potassium channel. Kidney Int 93: 893–902, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 869.Wang Q, Maillard M, Schibler U, Burnier M, and Gachon F. Cardiac hypertrophy, low blood pressure, and low aldosterone levels in mice devoid of the three circadian PAR bZip transcription factors DBP, HLF, and TEF. Am J Physiol Regul Integr Comp Physiol 299: R1013–R1019, 2010. [DOI] [PubMed] [Google Scholar]
- 870.Wang WH, and Giebisch G. Regulation of potassium (K) handling in the renal collecting duct. Pflugers Arch 458: 157–168, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 871.Wang XY, Masilamani S, Nielsen J, Kwon TH, Brooks HL, Nielsen S, and Knepper MA. The renal thiazide-sensitive Na-Cl cotransporter as mediator of the aldosterone-escape phenomenon. J Clin Invest 108: 215–222, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 872.Warnock DG, Kusche-Vihrog K, Tarjus A, Sheng S, Oberleithner H, Kleyman TR, and Jaisser F. Blood pressure and amiloride-sensitive sodium channels in vascular and renal cells. Nat Rev Nephrol 10: 146–157, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 873.Watts BA III, George T, and Good DW. Aldosterone inhibits apical NHE3 and. Am J Physiol Renal Physiol 291: F1005–F1013, 2006. [DOI] [PubMed] [Google Scholar]
- 874.Webb DJ, Monge JC, Rabelink TJ, and Yanagisawa M. Endothelin: new discoveries and rapid progress in the clinic. Trends Pharmacol Sci 19: 5–8, 1998. [DOI] [PubMed] [Google Scholar]
- 875.Webster MK, Goya L, Ge Y, Maiyar AC, and Firestone GL. Characterization of sgk, a novel member of the serine/threonine protein kinase gene family which is transcriptionally induced by glucocorticoids and serum. Mol Cell Biol 13: 2031–2040, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 876.Wehling M, Spes CH, Win N, Janson CP, Schmidt BM, Theisen K, and Christ M. Rapid cardiovascular action of aldosterone in man. J Clin Endocrinol Metab 83: 3517–3522, 1998. [DOI] [PubMed] [Google Scholar]
- 877.Wei Y, Whaley-Connell AT, Habibi J, Rehmer J, Rehmer N, Patel K, Hayden M, DeMarco V, Ferrario CM, Ibdah JA, and Sowers JR. Mineralocorticoid receptor antagonism attenuates vascular apoptosis and injury via rescuing protein kinase B activation. Hypertension 53: 158–165, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 878.Weikum ER, Knuesel MT, Ortlund EA, and Yamamoto KR. Glucocorticoid receptor control of transcription: precision and plasticity via allostery. Nat Rev Mol Cell Biol 18: 159–174, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 879.Weiner ID. Endocrine and hypertensive disorders of potassium regulation: primary aldosteronism. Semin Nephrol 33: 265–276, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 880.Weiner ID, and Verlander JW. Ammonia Transporters and Their Role in Acid-Base Balance. Physiol Rev 97: 465–494, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 881.Weiner ID, and Wingo CS. Endocrine Causes of Hypertension - Aldosterone. In: Comprehensive Clinical Nephrology, edited by Freehally J, Johnson RJ, and Floege J. St. Louis: Saunders, 2014, p. 469–476. [Google Scholar]
- 882.Weinstein AM. Potassium excretion during antinatriuresis: perspective from a distal nephron model. Am J Physiol Renal Physiol 302: F658–F673, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 883.Welch AK, Jeanette LI, Gumz ML, Cain BD, and Wingo CS. Aldosterone alters the chromatin structure of the murine endothelin-1 gene. Life Sci 159: 121–126, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 884.Weldon SM, and Brown NF. Inhibitors of Aldosterone Synthase. Vitam Horm 109: 211–239, 2019. [DOI] [PubMed] [Google Scholar]
- 885.Welling PA. Rare mutations in renal sodium and potassium transporter genes exhibit impaired transport function. Curr Opin Nephrol Hypertens 23: 1–8, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 886.Welling PA. Regulation of renal potassium secretion: molecular mechanisms. Semin Nephrol 33: 215–228, 2013. [DOI] [PubMed] [Google Scholar]
- 887.Welling PA. Roles and Regulation of Renal K Channels. Annu Rev Physiol 78: 415–435, 2016. [DOI] [PubMed] [Google Scholar]
- 888.Welling PA, and Ho K. A comprehensive guide to the ROMK potassium channel: form and function in health and disease. Am J Physiol Renal Physiol 297: F849–F863, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 889.Wen D, Cornelius RJ, and Sansom SC. Interacting influence of diuretics and diet on BK channel-regulated K homeostasis. Curr Opin Pharmacol 15: 28–32, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 890.Wen D, Cornelius RJ, Yuan Y, and Sansom SC. Regulation of BK-α expression in the distal nephron by aldosterone and urine pH. Am J Physiol Renal Physiol 305: F463–476, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 891.Wendler A, Albrecht C, and Wehling M. Nongenomic actions of aldosterone and progesterone revisited. Steroids 77: 1002–1006, 2012. [DOI] [PubMed] [Google Scholar]
- 892.White PC. 11beta-hydroxysteroid dehydrogenase and its role in the syndrome of apparent mineralocorticoid excess. Am J Med Sci 322: 308–315, 2001. [DOI] [PubMed] [Google Scholar]
- 893.Whitman M, Kaplan DR, Schaffhausen B, Cantley L, and Roberts TM. Association of phosphatidylinositol kinase activity with polyoma middle-T competent for transformation. Nature 315: 239–242, 1985. [DOI] [PubMed] [Google Scholar]
- 894.Wichmann L, Vowinkel KS, Perniss A, Manzini I, and Althaus M. Incorporation of the delta-subunit into the epithelial sodium channel (ENaC) generates protease-resistant ENaCs in Xenopus laevis. J Biol Chem 293: 6647–6658, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 895.Wickert L, Selbig J, Watzka M, Stoffel-Wagner B, Schramm J, Bidlingmaier F, and Ludwig M. Differential mRNA expression of the two mineralocorticoid receptor splice variants within the human brain: structure analysis of their different DNA binding domains. J Neuroendocrinol 12: 867–873, 2000. [DOI] [PubMed] [Google Scholar]
- 896.Wildman SS, Kang ES, and King BF. ENaC, renal sodium excretion and extracellular ATP. Purinergic Signal 5: 481–489, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 897.Williams B, MacDonald TM, Morant S, Webb DJ, Sever P, McInnes G, Ford I, Cruickshank JK, Caulfield MJ, Salsbury J, Mackenzie I, Padmanabhan S, Brown MJ, and British Hypertension Society's PSG. Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug-resistant hypertension (PATHWAY-2): a randomised, double-blind, crossover trial. Lancet 386: 2059–2068, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 898.Williams B, MacDonald TM, Morant SV, Webb DJ, Sever P, McInnes GT, Ford I, Cruickshank JK, Caulfield MJ, Padmanabhan S, Mackenzie IS, Salsbury J, and Brown MJ. Endocrine and haemodynamic changes in resistant hypertension, and blood pressure responses to spironolactone or amiloride: the PATHWAY-2 mechanisms substudies. Lancet Diabetes Endocrinol 6: 464–475, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 899.Williams GH. Aldosterone biosynthesis, regulation, and classical mechanism of action. Heart Fail Rev 10: 7–13, 2005. [DOI] [PubMed] [Google Scholar]
- 900.Wilson FH, Disse-Nicodeme S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, Gunel M, Milford DV, Lipkin GW, Achard JM, Feely MP, Dussol B, Berland Y, Unwin RJ, Mayan H, Simon DB, Farfel Z, Jeunemaitre X, and Lifton RP. Human hypertension caused by mutations in WNK kinases. Science 293: 1107–1112, 2001. [DOI] [PubMed] [Google Scholar]
- 901.Wilson FH, Kahle KT, Sabath E, Lalioti MD, Rapson AK, Hoover RS, Hebert SC, Gamba G, and Lifton RP. Molecular pathogenesis of inherited hypertension with hyperkalemia: the Na-Cl cotransporter is inhibited by wild-type but not mutant WNK4. Proc Natl Acad Sci U S A 100: 680–684, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 902.Wingo CS. Potassium secretion by the cortical collecting tubule: effect of Cl gradients and ouabain. Am J Physiol 256: F306–313, 1989. [DOI] [PubMed] [Google Scholar]
- 903.Wingo CS. Reversible chloride-dependent potassium flux across the rabbit cortical collecting tubule. Am J Physiol 256: F697–704, 1989. [DOI] [PubMed] [Google Scholar]
- 904.Wingo CS, and Cain BD. The renal H-K-ATPase: physiological significance and role in potassium homeostasis. Annu Rev Physiol 55: 323–347, 1993. [DOI] [PubMed] [Google Scholar]
- 905.Wingo CS, Kokko JP, and Jacobson HR. Effects of in vitro aldosterone on the rabbit cortical collecting tubule. Kidney Int 28: 51–57, 1985. [DOI] [PubMed] [Google Scholar]
- 906.Wingo CS, Madsen KM, Smolka A, and Tisher CC. H-K-ATPase immunoreactivity in cortical and outer medullary collecting duct. Kidney Int 38: 985–990, 1990. [DOI] [PubMed] [Google Scholar]
- 907.Wingo CS, Seldin DW, Kokko JP, and Jacobson HR. Dietary modulation of active potassium secretion in the cortical collecting tubule of adrenalectomized rabbits. J Clin Invest 70: 579–586, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 908.Winter C, Kampik NB, Vedovelli L, Rothenberger F, Paunescu TG, Stehberger PA, Brown D, John H, and Wagner CA. Aldosterone stimulates vacuolar H(+)-ATPase activity in renal acid-secretory intercalated cells mainly via a protein kinase C-dependent pathway. Am J Physiol Cell Physiol 301: C1251–1261, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 909.Winter C, Schulz N, Giebisch G, Geibel JP, and Wagner CA. Nongenomic stimulation of vacuolar H+-ATPases in intercalated renal tubule cells by aldosterone. Proc Natl Acad Sci U S A 101: 2636–2641, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 910.Witham MD, Gillespie ND, and Struthers AD. Hyperkalemia after the publication of RALES. N Engl J Med 351: 2448–2450; author reply 2448–2450, 2004. [PubMed] [Google Scholar]
- 911.Woda CB, Bragin A, Kleyman TR, and Satlin LM. Flow-dependent K+ secretion in the cortical collecting duct is mediated by a maxi-K channel. Am J Physiol Renal Physiol 280: F786–793, 2001. [DOI] [PubMed] [Google Scholar]
- 912.Wong S, Brennan FE, Young MJ, Fuller PJ, and Cole TJ. A direct effect of aldosterone on endothelin-1 gene expression in vivo. Endocrinology 148: 1511–1517, 2007. [DOI] [PubMed] [Google Scholar]
- 913.Wright FS, Knox FG, Howards SS, and Berliner RW. Reduced sodium reabsorption by the proximal tubule of Doca-escaped dogs. Am J Physiol 216: 869–875, 1969. [DOI] [PubMed] [Google Scholar]
- 914.Wu H, Chen L, Zhou Q, and Zhang W. AF17 facilitates Dot1a nuclear export and upregulates ENaC-mediated Na+ transport in renal collecting duct cells. PLoS One 6: e27429, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 915.Wu P, Gao ZX, Su XT, Wang MX, Wang WH, and Lin DH. Kir4.1/Kir5.1 Activity Is Essential for Dietary Sodium Intake-Induced Modulation of Na-Cl Cotransporter. J Am Soc Nephrol 30: 216–227, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 916.Wu P, Gao ZX, Zhang DD, Su XT, Wang WH, and Lin DH. Deletion of Kir5.1 Impairs Renal Ability to Excrete Potassium during Increased Dietary Potassium Intake. J Am Soc Nephrol 30: 1425–1438, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 917.Wu R, Dang F, Li P, Wang P, Xu Q, Liu Z, Li Y, Wu Y, Chen Y, and Liu Y. The Circadian Protein Period2 Suppresses mTORC1 Activity via Recruiting Tsc1 to mTORC1 Complex. Cell Metab 2018. [DOI] [PubMed] [Google Scholar]
- 918.Wynne BM, Zou L, Linck V, Hoover RS, Ma HP, and Eaton DC. Regulation of Lung Epithelial Sodium Channels by Cytokines and Chemokines. Front Immunol 8: 766, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 919.Xia SL, Noh SH, Verlander JW, Gelband CH, and Wingo CS. Apical membrane of native OMCD(i) cells has nonselective cation channels. Am J Physiol Renal Physiol 281: F48–55, 2001. [DOI] [PubMed] [Google Scholar]
- 920.Yamamura H, Ugawa S, Ueda T, Nagao M, Joh T, and Shimada S. Epithelial Na+ channel delta subunit is an acid sensor in the human oesophagus. Eur J Pharmacol 600: 32–36, 2008. [DOI] [PubMed] [Google Scholar]
- 921.Yamamura H, Ugawa S, Ueda T, Nagao M, and Shimada S. Protons activate the delta-subunit of the epithelial Na+ channel in humans. J Biol Chem 279: 12529–12534, 2004. [DOI] [PubMed] [Google Scholar]
- 922.Yamamura H, Ugawa S, Ueda T, and Shimada S. Expression analysis of the epithelial Na+ channel delta subunit in human melanoma G-361 cells. Biochem Biophys Res Commun 366: 489–492, 2008. [DOI] [PubMed] [Google Scholar]
- 923.Yanagisawa M, Inoue A, Takuwa Y, Mitsui Y, Kobayashi M, and Masaki T. The human preproendothelin-1 gene: possible regulation by endothelial phosphoinositide turnover signaling. J Cardiovasc Pharmacol 13 Suppl 5:S13–7; discussion S18.: S13-S17, 1989. [PubMed] [Google Scholar]
- 924.Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, Yazaki Y, Goto K, and Masaki T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 332: 411–415, 1988. [DOI] [PubMed] [Google Scholar]
- 925.Yang CL, Angell J, Mitchell R, and Ellison DH. WNK kinases regulate thiazide-sensitive Na-Cl cotransport. J Clin Invest 111: 1039–1045, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 926.Yang CT, Kor CT, and Hsieh YP. Long-Term Effects of Spironolactone on Kidney Function and Hyperkalemia-Associated Hospitalization in Patients with Chronic Kidney Disease. J Clin Med 7: 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 927.Yang J, and Fuller PJ. Interactions of the mineralocorticoid receptor--within and without. Mol Cell Endocrinol 350: 196–205, 2012. [DOI] [PubMed] [Google Scholar]
- 928.Yang J, Fuller PJ, Morgan J, Shibata H, McDonnell DP, Clyne CD, and Young MJ. Use of phage display to identify novel mineralocorticoid receptor-interacting proteins. Mol Endocrinol 28: 1571–1584, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 929.Yang J, and Young MJ. The mineralocorticoid receptor and its coregulators. J Mol Endocrinol 43: 53–64, 2009. [DOI] [PubMed] [Google Scholar]
- 930.Yang L, Frindt G, Xu Y, Uchida S, and Palmer LG. Aldosterone-dependent and -independent regulation of Na(+) and K(+) excretion and ENaC in mouse kidneys. Am J Physiol Renal Physiol 319: F323–f334, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 931.Yang LM, Rinke R, and Korbmacher C. Stimulation of the epithelial sodium channel (ENaC) by cAMP involves putative ERK phosphorylation sites in the C termini of the channel's beta- and gamma-subunit. J Biol Chem 281: 9859–9868, 2006. [DOI] [PubMed] [Google Scholar]
- 932.Yang P, Shen W, Chen X, Zhu D, Xu X, Wu T, Xu G, and Wu Q. Comparative efficacy and safety of mineralocorticoid receptor antagonists in heart failure: a network meta-analysis of randomized controlled trials. Heart Fail Rev 24: 637–646, 2019. [DOI] [PubMed] [Google Scholar]
- 933.Yokota K, Shibata H, Kobayashi S, Suda N, Murai A, Kurihara I, Saito I, and Saruta T. Proteasome-mediated mineralocorticoid receptor degradation attenuates transcriptional response to aldosterone. Endocr Res 30: 611–616, 2004. [DOI] [PubMed] [Google Scholar]
- 934.Yokota K, Shibata H, Kurihara I, Kobayashi S, Suda N, Murai-Takeda A, Saito I, Kitagawa H, Kato S, Saruta T, and Itoh H. Coactivation of the N-terminal transactivation of mineralocorticoid receptor by Ubc9. J Biol Chem 282: 1998–2010, 2007. [DOI] [PubMed] [Google Scholar]
- 935.Yokota N, Bruneau BG, Kuroski de Bold ML, and de Bold AJ. Atrial natriuretic factor significantly contributes to the mineralocorticoid escape phenomenon. Evidence for a guanylate cyclase-mediated pathway. J Clin Invest 94: 1938–1946, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 936.Yoo D, Fang L, Mason A, Kim BY, and Welling PA. A phosphorylation-dependent export structure in ROMK (Kir 1.1) channel overrides an endoplasmic reticulum localization signal. J Biol Chem 280: 35281–35289, 2005. [DOI] [PubMed] [Google Scholar]
- 937.Yoo D, Flagg TP, Olsen O, Raghuram V, Foskett JK, and Welling PA. Assembly and trafficking of a multiprotein ROMK (Kir 1.1) channel complex by PDZ interactions. J Biol Chem 279: 6863–6873, 2004. [DOI] [PubMed] [Google Scholar]
- 938.Yoshimoto T, and Hirata Y. Aldosterone as a cardiovascular risk hormone. Endocr J 54: 359–370, 2007. [DOI] [PubMed] [Google Scholar]
- 939.Youmans SJ, and Barry CR. Bafilomycin A1 at nanomolar concentrations saturably inhibits a portion of turtle bladder acidification current. J Exp Biol 204: 2911–2919, 2001. [DOI] [PubMed] [Google Scholar]
- 940.Youmans SJ, and Barry CR. Effects of valinomycin on vanadate-sensitive and vanadate-resistant H+ transport in vesicles from turtle bladder epithelium: evidence for a K+/H+ exchanger. Biochem Biophys Res Commun 176: 1285–1290, 1991. [DOI] [PubMed] [Google Scholar]
- 941.Youmans SJ, and Barry CR. Physiological role for vanadate-inhibitable active H+ transport: a new model for distal urinary acidification. Biochem Biophys Res Commun 180: 1505–1512, 1991. [DOI] [PubMed] [Google Scholar]
- 942.Youn JH. Gut sensing of potassium intake and its role in potassium homeostasis. Semin Nephrol 33: 248–256, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 943.Youn JH, and McDonough AA. Recent advances in understanding integrative control of potassium homeostasis. Annu Rev Physiol 71: 381–401, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 944.Young DB. Analysis of long-term potassium regulation. Endocr Rev 6: 24–44, 1985. [DOI] [PubMed] [Google Scholar]
- 945.Young DB. Quantitative analysis of aldosterone's role in potassium regulation. Am J Physiol 255: F811–822, 1988. [DOI] [PubMed] [Google Scholar]
- 946.Young DB. Relationship between plasma potassium concentration and renal potassium excretion. Am J Physiol 242: F599–F603, 1982. [DOI] [PubMed] [Google Scholar]
- 947.Young DB, and Guyton AC. Steady state aldosterone dose-response relationships. Circ Res 40: 138–142, 1977. [DOI] [PubMed] [Google Scholar]
- 948.Young DB, and Jackson TE. Effects of aldosterone on potassium distribution. Am J Physiol 243: R526–530, 1982. [DOI] [PubMed] [Google Scholar]
- 949.Young DB, Jackson TE, Tipayamontri U, and Scott RC. Effects of sodium intake on steady-state potassium excretion. Am J Physiol 246: F772–F778, 1984. [DOI] [PubMed] [Google Scholar]
- 950.Young DB, Lin H, and McCabe RD. Potassium's cardiovascular protective mechanisms. Am J Physiol 268: R825–R837, 1995. [DOI] [PubMed] [Google Scholar]
- 951.Young DB, and Ma G. Vascular protective effects of potassium. Semin Nephrol 19: 477–486, 1999. [PubMed] [Google Scholar]
- 952.Young DB, McCaa RE, Pan YJ, and Guyton AC. The natriuretic and hypotensive effects of potassium. Circ Res 38: 84–89, 1976. [DOI] [PubMed] [Google Scholar]
- 953.Young DB, and Paulsen AW. Interrelated effects of aldosterone and plasma potassium on potassium excretion. Am J Physiol 244: F28–F34, 1983. [DOI] [PubMed] [Google Scholar]
- 954.Yu L, Helms MN, Yue Q, and Eaton DC. Single-channel analysis of functional epithelial sodium channel (ENaC) stability at the apical membrane of A6 distal kidney cells. Am J Physiol Renal Physiol 295: F1519–F1527, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 955.Yuan Y, Zhang A, Huang S, Ding G, and Chen R. A PPARgamma agonist inhibits aldosterone-induced mesangial cell proliferation by blocking ROS-dependent EGFR intracellular signaling. Am J Physiol Renal Physiol 300: F393–F402, 2011. [DOI] [PubMed] [Google Scholar]
- 956.Yue G, Malik B, Yue G, and Eaton DC. Phosphatidylinositol 4,5-bisphosphate (PIP2) stimulates epithelial sodium channel activity in A6 cells. J Biol Chem 277: 11965–11969, 2002. [DOI] [PubMed] [Google Scholar]
- 957.Zachar RM, Skjodt K, Marcussen N, Walter S, Toft A, Nielsen MR, Jensen BL, and Svenningsen P. The Epithelial Sodium Channel gamma-Subunit Is Processed Proteolytically in Human Kidney. J Am Soc Nephrol 26: 95–106, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 958.Zaika O, Palygin O, Tomilin V, Mamenko M, Staruschenko A, and Pochynyuk O. Insulin and IGF-1 activate Kir4.1/5.1 channels in cortical collecting duct principal cells to control basolateral membrane voltage. Am J Physiol Renal Physiol 310: F311–321, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 959.Zeidel ML. Renal actions of atrial natriuretic peptide: regulation of collecting duct sodium and water transport. Annu Rev Physiol 52: 747–759, 1990. [DOI] [PubMed] [Google Scholar]
- 960.Zeng F, Singh AB, and Harris RC. The role of the EGF family of ligands and receptors in renal development, physiology and pathophysiology. Exp Cell Res 315: 602–610, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 961.Zeniya M, Sohara E, Kita S, Iwamoto T, Susa K, Mori T, Oi K, Chiga M, Takahashi D, Yang SS, Lin SH, Rai T, Sasaki S, and Uchida S. Dietary salt intake regulates WNK3-SPAK-NKCC1 phosphorylation cascade in mouse aorta through angiotensin II. Hypertension 62: 872–878, 2013. [DOI] [PubMed] [Google Scholar]
- 962.Zennaro MC, Caprio M, and Feve B. Mineralocorticoid receptors in the metabolic syndrome. Trends Endocrinol Metab 20: 444–451, 2009. [DOI] [PubMed] [Google Scholar]
- 963.Zennaro MC, Souque A, Viengchareun S, Poisson E, and Lombes M. A new human MR splice variant is a ligand-independent transactivator modulating corticosteroid action. Mol Endocrinol 15: 1586–1598, 2001. [DOI] [PubMed] [Google Scholar]
- 964.Zhang DD, Duan XP, Xiao Y, Wu P, Gao ZX, Wang WH, and Lin DH. Deletion of renal Nedd4-2 abolishes the effect of high sodium intake (HS) on Kir4.1, ENaC, and NCC and causes hypokalemia during high HS. Am J Physiol Renal Physiol 320: F883–f896, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 965.Zhang W, Xia X, Jalal DI, Kuncewicz T, Xu W, Lesage GD, and Kone BC. Aldosterone-sensitive repression of ENaCalpha transcription by a histone H3 lysine-79 methyltransferase. Am J Physiol Cell Physiol 290: C936–C946, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 966.Zhang W, Xia X, Reisenauer MR, Hemenway CS, and Kone BC. Dot1a-AF9 complex mediates histone H3 Lys-79 hypermethylation and repression of ENaCalpha in an aldosterone-sensitive manner. J Biol Chem 281: 18059–18068, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 967.Zhang W, Xia X, Reisenauer MR, Rieg T, Lang F, Kuhl D, Vallon V, and Kone BC. Aldosterone-induced Sgk1 relieves Dot1a-Af9-mediated transcriptional repression of epithelial Na+ channel alpha. J Clin Invest 117: 773–783, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 968.Zhang X, Zhou Q, Chen L, Berger S, Wu H, Xiao Z, Pearce D, Zhou X, and Zhang W. Mineralocorticoid receptor antagonizes Dot1a-Af9 complex to increase alphaENaC transcription. Am J Physiol Renal Physiol 305: F1436–F1444, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 969.Zhao M, Valamanesh F, Celerier I, Savoldelli M, Jonet L, Jeanny JC, Jaisser F, Farman N, and Behar-Cohen F. The neuroretina is a novel mineralocorticoid target: aldosterone up-regulates ion and water channels in Müller glial cells. FASEB J 24: 3405–3415, 2010. [DOI] [PubMed] [Google Scholar]
- 970.Zhou X, Xia SL, and Wingo CS. Chloride transport by the rabbit cortical collecting duct: dependence on H,K-ATPase. J Am Soc Nephrol 9: 2194–2202, 1998. [DOI] [PubMed] [Google Scholar]
- 971.Ziera T, Irlbacher H, Fromm A, Latouche C, Krug SM, Fromm M, Jaisser F, and Borden SA. Cnksr3 is a direct mineralocorticoid receptor target gene and plays a key role in the regulation of the epithelial sodium channel. FASEB J 23: 3936–3946, 2009. [DOI] [PubMed] [Google Scholar]
- 972.Zipser RD, LeBoff M, Meidar V, Duke R, and Horton R. Deoxycorticosterone and aldosterone clearance and binding in normal and hypertensive man. J Clin Endocrinol Metab 51: 1085–1088, 1980. [DOI] [PubMed] [Google Scholar]