

Abstract
Killer lymphocytes release perforin and granzymes from cytotoxic granules into the immunological synapse to destroy target cells as a critical mechanism in the defense against viruses and cancer. Perforin, a Ca2+-dependent pore-forming protein that multimerizes in membranes, delivers granzymes into the target cell cytosol. The original model for perforin (acting by forming a cell membrane channel through which granzymes pass) does not fit the experimental data. Recently, an alternative model has been proposed that involves active target cell collaboration with perforin to deliver granzymes and direct the target cell to an apoptotic, rather than necrotic, death.
Introduction
Cytotoxic T lymphocytes and natural killer cells are important effector cells in the immune response to viruses, intracellular bacteria and tumors [1,2]. These cells dump the contents of their cytotoxic granules into the immunological synapse formed with a specifically recognized target cell to trigger its apoptosis [3,4]. Although bathed in the same cell death-inducing mix on the other side of the synapse, the killer cells escape unharmed and can then, like the serial killers they are, seek and destroy another target [5,6]. Activated CD8 T cells and some TH1 and Treg CD4 T cells [7,8] can synthesize cytotoxic granules and acquire the capacity to kill, but because of the inherent potential danger of unleashing apoptosis the activation of cytolytic function is tightly controlled. Cytotoxic granules contain perforin and a group of serine proteases called granzymes in a proteoglycan matrix [1,2]. The most abundant granzymes are granzyme A and granzyme B [2,9,10]. The granzymes are redundant, each of which is capable of proteolytically activating independent cell death pathways, although the pathways activated by granzymes other than granzyme A and granzyme B (the ‘orphan’ granzymes) are just beginning to be described [1,2,11–16]. Individual killer cells only express a subset of cytolytic molecules, and the expression of each of these molecules appears to be regulated differently [17]. Perforin expression is controlled by an extended 150 kilobase domain that includes a locus control region that regulates the developmental and activation-specific expression of perforin in T cells and natural killer cells [18•]. Perforin, a Ca2+-dependent pore-forming protein that has homology to complement components, is the only molecule that can deliver granzymes into the target cell. Therefore, mice deficient in perforin are profoundly immunodeficient and have enhanced susceptibility to viral infection and cancer, whereas mice deficient in either granzyme A or B are generally able to handle most infections although they display subtle impairment in defending against some viruses [19–30]. Humans that have familial hemophagocytic lymphohistiocytosis (FHL) caused by biallelic perforin mutations are also severely immunocompromised [31–33].
Although almost twenty years have passed since perforin was first cloned [34–36], how perforin works remains a puzzle. The original simple model for perforin — that it acts by forming a cell membrane pore through which granzymes pass — has been questioned. This review will discuss recent studies that bring us closer to understanding the molecular basis for how this crucial immune defense molecule functions, but the story is still a work in progress.
Most studies of the mechanism for perforin delivery of granzymes have been performed using purified native perforin added to cells at the same time as granzyme B, using apoptosis induction as the readout for granzyme delivery to the cytosol (where it must be delivered to induce cell death). A few caveats need to be kept in mind when interpreting these loading experiments. First, the dose-response curve for perforin is very steep: if the amount added is too low (sub-threshold) it does not deliver granzymes; if it is too high (lytic) it triggers necrosis independently of the granzymes, and adding granzymes does not lead to apoptosis because apoptosis is a slower process than necrosis that requires the active participation of a functioning cell [37•]. The ‘just-right’ or sublytic concentration causes about 10% necrosis on its own and delivers granzymes for apoptosis induction. The sublytic dose varies between cells. In some cases, the subthreshold and lytic concentration can differ by only a few-fold. Because perforin is not very stable and its activity is altered by freeze-thawing, it is important to titrate the perforin dose and verify that it is sublytic for each cell type and experiment. It is also important to bear in mind that what happens during experiments loading granzymes and perforin into cells might not accurately recapitulate what happens when granzymes and perforin are released into the tight space of the immunological synapse, in which only a small portion of the target cell membrane is exposed. A back-of-the-envelope estimate of the perforin concentration in the immunological synapse (calculated based on the yield of perforin from natural killer [NK] cells, the fact that only about a sixth to a third of granules are exocytosed during a single attack [6,38], and volume estimates of the synapse) suggests that the perforin concentration at the synapse may be 2–3 orders of magnitude higher than the sublytic concentration used in loading experiments. Therefore, it is important that any results obtained using purified perforin are verified during cytotoxic T lymphocyte (CTL) or NK cell lysis.
Perforin structure and function
Perforin is a tricky molecule to purify that is difficult to maintain in solution in an active form, and a recombinant form has only recently been reported [13]. Even so, expression of recombinant active perforin has not been reproduced by other laboratories. This has stymied research. The identification of perforin mutations as a cause of FHL has led to the sequencing of many perforin alleles and to the identification of nonsense, frameshift and missense mutations that disrupt perforin activity. The importance of some of these has been validated by expressing the mutants in rat basophilic leukemia cells together with a granzyme and then testing for cytolytic function, as originally described by Henkart and co-workers [20,39,40]. A comprehensive recent review discusses this growing literature [41].
Perforin multimerizes in a Ca2+-dependent manner in the plasma membrane of cells to form 5–20 nm pores [42–44]. It is still not known whether a fixed number of perforin molecules form a well-defined pore of a fixed size or whether pores of varying sizes might be formed if more perforin is present or longer times are allowed for multimerization. (Some bacterial pore-forming proteins form well-defined pores of a fixed size, whereas others form variable-sized pores.) The precursor of human perforin is a 555 amino acid protein that is synthesized with a 21 amino acid leader sequence (Figure 1). The perforin precursor contains two glycosylation sites. En route to or in the granule, a glycosylated carboxy-terminal peptide is removed from human (but possibly not mouse) perforin at an indeterminate site by an undefined cysteine protease to produce the mature active protein [45]. The A91V mutation in certain FHL patients inhibits this processing, causing reduced cytotoxicity [46]. The carboxy terminus of the mature protein (amino acids 395–478) contains a C2 domain, implicated in Ca2+-dependent phospholipid binding in a variety of proteins including protein kinase C, phospholipase Cδ (PLCδ) and synaptotagmins (the rapid Ca2+-dependent oligomerization of which is required for vesicle exocytosis). Structures for the C2 domains of these molecules in the presence or absence of Ca2+ show a β-sandwich formed by eight β-strands, with a Ca2+-binding domain at one end of the sandwich [47–52]. Generally, three Ca2+ ions bind in proximity to each C2 domain. Upon Ca2+ binding, the β-sandwich might open up to enable binding to a phospholipid head group and membrane docking by way of the Ca2+-binding loops [53]. Alternatively, Ca2+ binding could change the surface charge of the molecule to facilitate electrostatic interactions [54]. The Griffiths laboratory modeled the perforin C2 domain on PLCδ and identified putative Ca2+-binding aspartic acid residues at residues 409, 415, 435, 463, 465 and 471 [45]. Moreover, they expressed a perforin C2–glutathione S-transferase fusion protein in Escherichia coli and showed that it binds in a Ca2+-dependent manner to liposomes. FHL-associated mutations affect this domain; the G428E mutant is impaired in Ca2+-dependent membrane-binding and cytotoxicity [40]. Therefore, the perforin C2 domain is probably responsible for Ca2+-dependent membrane binding — a first step in pore formation.
Figure 1.
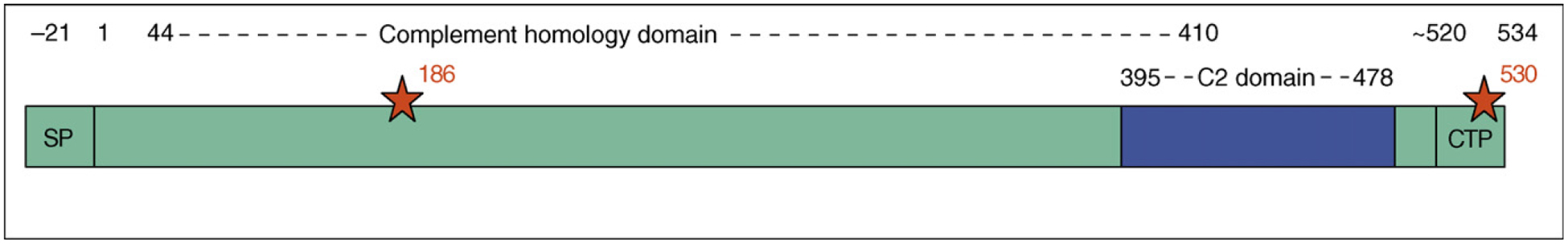
Perforin sequence. Mature human perforin is produced by removal of the signal peptide (SP) and a poorly defined carboxy-terminal peptide (CTP). The C2 domain is thought to be crucial for Ca2+-dependent membrane binding and the complement homology domain is needed for membrane insertion. Glycosylation sites are indicated by red stars. Numbering corresponds to that of the mature human protein.
Perforin does not contain a stretch of neutral amino acids capable of forming a transmembrane domain. Residues 44–410 share some homology (~20%) with the terminal C7–9 subunits of complement. The homologous C9 component of complement forms amphipathic helices that are believed to self-associate to form hydrophobic outer domains capable of membrane insertion [55]. Therefore, the complement homology domain of perforin is probably responsible for perforin membrane insertion and multimerization. Although it has been suggested that the amino-terminal 22 or 34 amino acid perforin peptide might have pore-forming activity [56,57], this idea is not widely accepted.
Relatively few of nearly 30 missense mutations identified in FHL patients that have been analyzed appear to render perforin non-functional without reducing its expression in cells [41,58•]. This makes it difficult to determine whether impaired cytotoxicity is caused by lower perforin expression or its reduced function after exocytosis. Thus, continued efforts to improve methods to express recombinant perforin molecules that are active in vitro will be crucial for dissecting its domain structure and function and to understand the biological consequences of mutated perforin in FHL patients.
Protecting the killer cell from its own perforin
The biosynthesis and storage of perforin in killer cells is carefully designed to protect killer cells from the potentially lethal effect of perforin. Upon synthesis in the endoplasmic reticulum, perforin probably binds to its inhibitor calreticulin [59–62]. It is then transported, presumably bound to calreticulin, via the trans-Golgi to cytotoxic granules — modified secretory lysosomes [63]. The cytotoxic granules are acidic (pH 5.1–5.4) and contain, in addition to perforin and granzymes, calreticulin and the proteoglycan serglycin (named for its many Ser–Gly repeats), as well as enzymes and membrane-associated molecules typically found in lysosomes (such as cathepsins, CD63, CD107a and CD107b). Perforin and granzymes bind to serglycin in the granule [64,65]. Perforin is inactive at the acidic pH of the cytolytic granules, but perforin protein stability in the granules requires the acidic environment [66]. Perforin levels in cells treated with concanamycin, an inhibitor of the vacuolar H+-ATPase, are so diminished that concanamycin-treated CTLs are not cytolytic. Perforin also needs to be activated by a cysteine protease to remove a carboxy-terminal glycosylated peptide [45]. Proteolytic cleavage probably occurs in the granule because it requires an acidic environment. Therefore, during its biogenesis and storage, many safeguards protect the killer cell from perforin. The precursor protein is probably bound to an inhibitor before it gets to the cytotoxic granule and is inactive until it is processed in the cytotoxic granule; once in the granule, perforin is inactive at its acidic pH in the absence of free Ca2+ (bound by the granule calreticulin) and is not free to multimerize because it is complexed with serglycin.
When cytotoxicity is triggered, perforin is released into the synapse. At neutral pH, perforin is released from serglycin [64,67] and is free to do its job. Although the pH of the immunological synapse has never been measured, it is likely that perforin dissociates from serglycin in the synapse. But if perforin is free to act on the target cell membrane, how is the killer cell membrane protected from perforin? One attractive hypothesis, proposed by Henkart and co-workers, is that the granule membrane protein cathepsin B, transferred to the killer cell plasma membrane when the cytotoxic granule membrane fuses to the plasma membrane, inactivates by proteolysis any perforin redirected toward the killer cell [68]. However, killer cells genetically deficient in cathepsin B survive unscathed when they kill targets [69]. A possible explanation for these seemingly contradictory results would be that other membrane-bound granule cathepsins besides cathepsin B (or perhaps other CTL surface proteases or perforin inhibitors) could also proteolytically inactivate perforin redirected at the killer cell.
How does perforin deliver granzymes?
How perforin delivers granzymes into the cytosol of target cells has been the subject of intense recent scrutiny and debate and is still unresolved. Based on its homology to complement and the pores seen in perforin-exposed cells by electron microscopy, perforin was originally hypothesized to multimerize in the plasma membrane to form pores through which granzymes passed (Figure 2). However, perforin pores might be too small to allow passage of globular molecules as big as granzymes. In fact, small dyes that ought to be able to pass through perforin-sized pores do not seem to get into the cytosol of perforin-treated cells [65,70].
Figure 2.
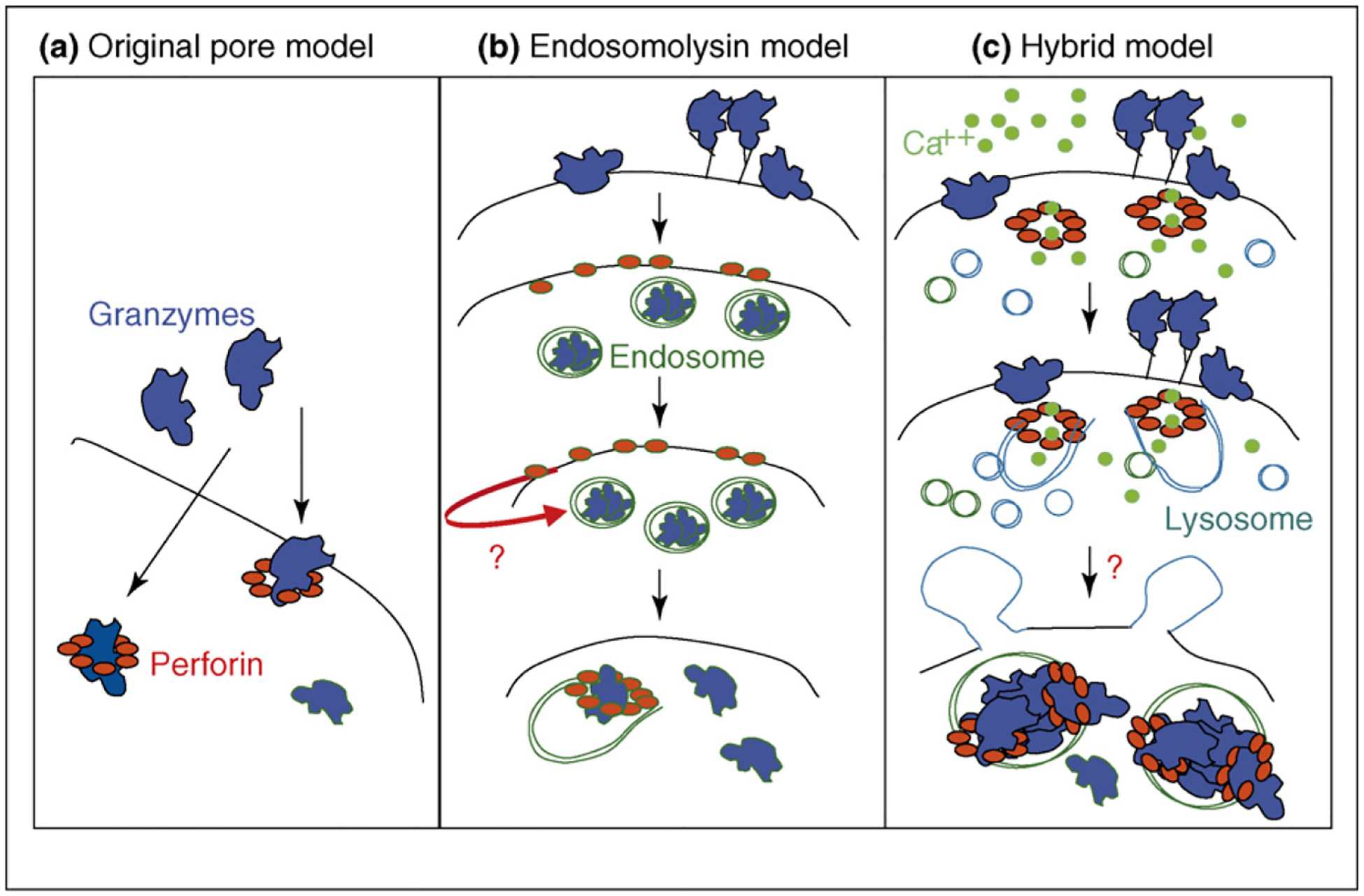
Three models for how perforin delivers granzymes. (a) The original model for perforin delivery of granzymes was via multimerization in the cell membrane to form pores large enough for granzymes to pass through. (b) This model was revised by Froelich [71] to propose that granzymes are endocytosed independently of perforin and that perforin then acts as an endosomolysin. (c) We propose a hybrid model in which perforin forms small pores in the cell membrane that trigger a Ca2+ influx, which in turn activates a membrane repair response in which internal vesicles donate their membranes to patch the holes. The next step involves rapid co-endocytosis of granzymes and perforin into giant endosomes, followed by perforin-mediated release of granzymes to the cytosol. We do not know what triggers the rapid endocytosis or whether perforin pores in endosomal membranes destabilize the endosome (causing it to burst) or whether perforin forms endosomal membrane pores large enough to allow granzymes to escape. We think it is unlikely that granzymes enter the cell through plasma membrane pores, but that remains possible. In the figure the plasma membrane is black, endosome membranes are green and lysosomal membranes are blue.
The original plasma membrane pore model [43,44] was questioned when it was found that granzyme B can be endocytosed on its own without perforin [71–74] and that apoptosis can be triggered when perforin is added to washed cells that have endocytosed granzyme B in the absence of perforin [71,74]. Based on these results, Froelich and co-workers [65,71] proposed that perforin does not act at the plasma membrane, but rather at the endosomal membrane, to release granzymes from endosomes, presumably by forming pores in the endosomal membrane (Figure 2). This idea was supported by the finding that bacterial and viral pore-forming proteins, such as streptolysin O and listeriolysin, could substitute for perforin and effectively deliver granzymes to activate apoptosis [71,75]. However, the topology of how perforin acting outside the cell membrane could trigger release of granzymes within cytosolic membrane-bound endosomes was difficult to understand.
Our group looked carefully at the data that formed the basis for Froelich’s revised model of perforin acting as an endosomolysin and questioned the interpretation of these experiments [76]. First we found that, because the granzymes are highly basic, they stick to the cell membrane by charge and are not washed off with the medium used for the granzyme endocytosis experiment [76]. The Bleackley and Bird groups also identified cell surface receptors, including the cation-independent mannose-6-phosphate receptor (CI-MPR) and heparin receptors, respectively, on the cell surface that enhance granzyme B binding and killing [73,77,78]. However, receptor-mediated binding does not appear to be required for granzyme uptake [79,80]. Second, when granzyme B-preincubated cells are washed using medium that contains a high concentration of charged molecules to inhibit ionic interactions or are treated with trypsin to remove all cell surface granzyme B, subsequent addition of perforin does not trigger apoptosis, suggesting that perforin and granzymes need to be co-endocytosed to trigger granzyme delivery for apoptosis [76]. Third, the uptake of granzymes into cells in the absence of perforin, probably by macropinocytosis, is much slower and less efficient than with perforin [37•]. When cells are incubated with sublytic perforin, granzymes are endocytosed much more rapidly and efficiently (Figure 3).
Figure 3.
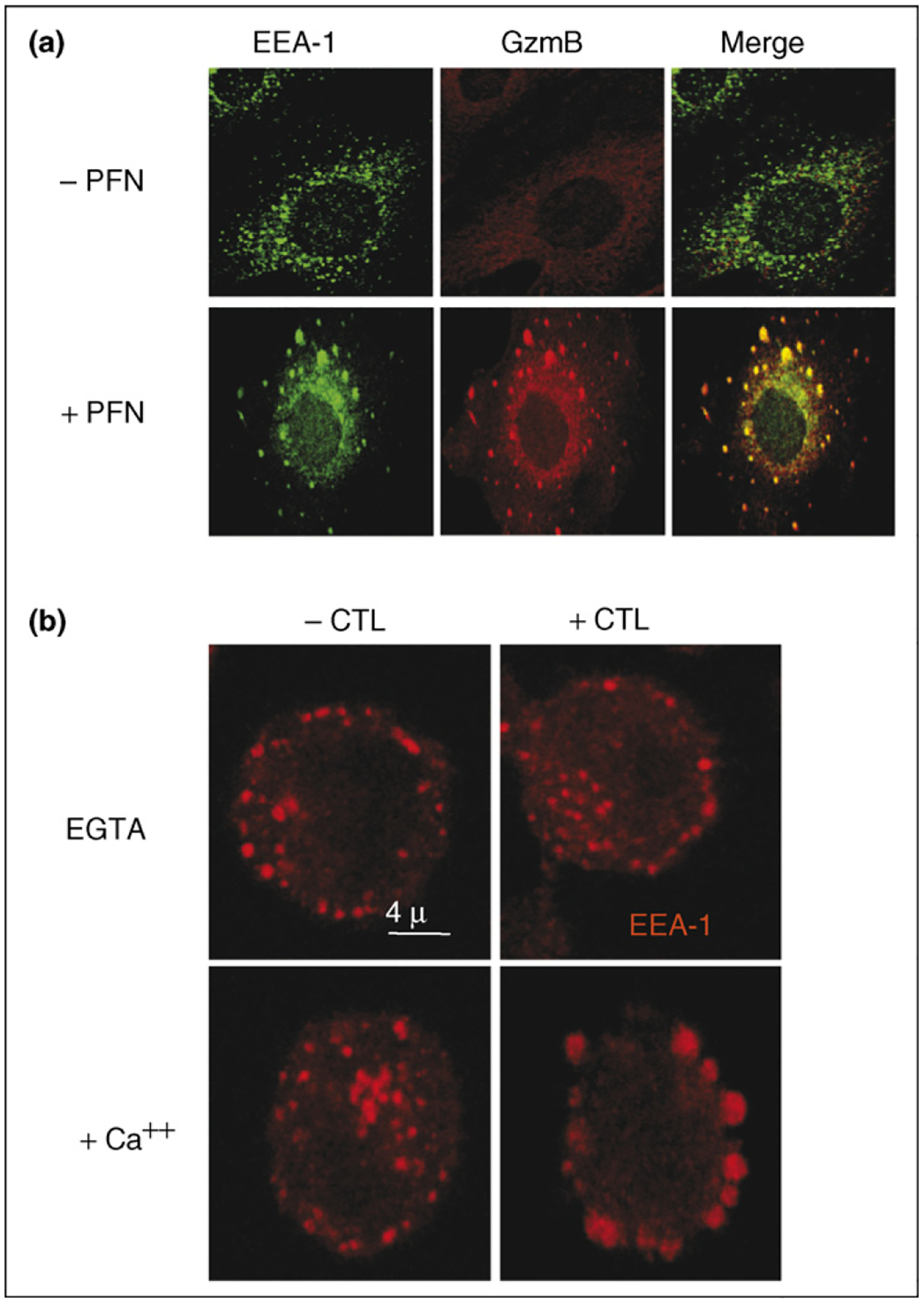
Perforin triggers the rapid endocytosis of granzyme B into giant endosomes. (a) Target cells loaded with granzyme B (GzmB) and perforin (PFN) are rapidly taken up into gigantic early endosome antigen 1 (EEA-1)-staining endosomes. In the absence of perforin, some granyzme B is taken up into endosomes, but uptake is much less efficient. Images are obtained 2 min after incubating cells with granzyme B and perforin. (b) Large EEA-1-staining endosomes are also seen in target cells attacked by lymphokine-activated killer cells. Cells were preincubated in medium containing ethylene glycol tetraacetic acid (EGTA), and then granule exocytosis was triggered by adding Ca2+. Images reproduced with permission from [37•].
During sublytic perforin loading experiments and cytotoxic T-cell attack, perforin does form cell membrane pores [37•] (Figure 2). Ca2+ from the extracellular fluid rapidly and transiently enters the cell. Moreover, small molecule dyes get in too, but they are difficult to see in the target cell. This was missed in earlier experiments because not much dye enters and the dye that does enter does not diffuse throughout the cytoplasm but is sequestered in juxtamembrane vesicles. These observations led us to show that the target cell actively participates in perforin-induced granzyme delivery in an unexpected way. Because levels of cytosolic Ca2+ are normally low whereas the extracellular milieu is rich in Ca2+ the cell senses a rise in cytosolic Ca2+ above ~100 μM as evidence of a damaged membrane and immediately triggers a stereotypic damaged-membrane response, sometimes called the ‘cellular wound-healing response’, because it is also activated by mechanical trauma to the plasma membrane [81]. Intracellular vesicles, including endosomes and lysosomes, are mobilized within seconds to donate their membranes to reseal the damaged plasma membrane [81–85]. The areas of fused membrane can be seen as giant blebs that form rapidly on the surface of cells treated with sublytic perforin. A hallmark of cellular wound-healing is finding lysosomal membrane proteins, such as CD107a (also known as Lamp-1), on the cell membrane. When sublytic perforin (as well as CTL attack) triggers a rapid damaged membrane repair response, plasma membrane integrity is restored, allowing co-delivered granzymes to induce the slow process of apoptosis. When the perforin dose is lytic, the repair response is unable to cope with the membrane damage, the Ca2+ flux persists and the cell dies rapidly by necrosis. When the repair response is inhibited, cells treated with granzyme B and sublytic perforin are more likely to die by necrosis than by apoptosis. Interfering with the repair response in target cells during CTL attack also shifts the balance of target cell death from apoptosis towards necrosis. The rapid membrane repair response explains why small molecule dyes that enter from the extracellular space are hard to see in target cells — the pores are open only for seconds and the dye that does get in is contained in membrane-bound compartments that isolate and patch the damaged cell membrane. Therefore, the target cell membrane repair response seals off perforin pores and allows the cell to undergo the slow, but controlled, death of apoptosis. Because apoptotic cells are rapidly recognized by scavenger receptors on macrophages and are engulfed, but necrotic cells trigger inflammation, directing the dying cell towards apoptosis is thought to be an essential feature of cell-mediated death to limit bystander cell damage.
Perforin triggers the rapid uptake of granzymes into enormous vesicles that stain with endosomal markers [37•] (Figure 3). Similar gigantic endosomes are also seen in cells targeted by CTLs [37•]. The mechanisms for triggering endocytosis and formation of giant endosomes are unknown. The cellular membrane repair response activates promiscuous heterotypic and homotypic membrane-fusion events that might contribute to either endocytosis or formation of giant endosomes [81]. However, the granzyme-containing vesicles do not stain for Lamp-1, which is on many of the membrane patches [37•]. Therefore, the giant endosomes do not form from internalized blebs. Moreover, triggering wound-healing with a Ca2+ ionophore does not activate granzyme uptake [37•]. This suggests that the granzyme vesicles are not formed as part of cellular membrane repair.
At this point it is still uncertain whether granzymes are internalized through the same plasma membrane pores as Ca2+ and small dyes. Therefore, the original membrane-pore hypothesis for perforin delivery of granzymes still remains a viable model. However, in our view, this is unlikely because if entry were through plasma membrane pores, granzymes would be expected to be found diffusely in the cytoplasm rather than in endosomes. Moreover, other positively charged molecules, irrespective of size, stuck to the cell membrane are co-internalized with granzymes into giant endosomes and are then co-released into the cytosol (JL, unpublished). This includes mega-dalton lysine-coupled dextrans, which would not be able to squeeze through the plasma membrane pores of the size that have been seen on electron micrographs.
We hypothesize that perforin is coendocytosed with granzymes and that perforin perturbs the endosomal membrane to release endosomal contents [37•,76]. In fact, granzymes are released from the giant endosomes about 10–15 min after loading (JL, unpublished). Therefore, we would expect to see perforin costaining with granzymes in giant endosomes. Unfortunately, no one has been able to see perforin in target cells to date. The mechanism by which perforin perturbs the endosome is also a matter of conjecture: does it form small pores that disrupt the acidification of the endosome and somehow destabilize it and cause it to burst or does perforin form large pores (possibly bigger than the ones previously seen in the plasma membrane) that allow granzymes to exit into the cytosol to unleash their cell death programs? In the next few years, we hope that these questions will be answered and a clear model will emerge.
Acknowledgement
This work was supported by National Institutes of Health grant AI063430 (JL). MEP was supported by NIH grants AI040127 and AI044432 to Anjana Rao. JL wishes to thank collaborators, Lianfa Shi, Dennis Keefe, Jerome Thiery, Ramiro Massol, Alan Rigby and Tom Kirchhausen, for their experimental work and many discussions on perforin.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Lieberman J: Cell death and immunity: The ABCs of granule-mediated cytotoxicity: new weapons in the arsenal. Nat Rev Immunol 2003, 3:361–370. [DOI] [PubMed] [Google Scholar]
- 2.Russell JH, Ley TJ: Lymphocyte-mediated cytotoxicity. Annu Rev Immunol 2002, 20:323–370. [DOI] [PubMed] [Google Scholar]
- 3.Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, Dustin ML: The immunological synapse: a molecular machine controlling T cell activation. Science 1999, 285:221–227. [DOI] [PubMed] [Google Scholar]
- 4.Stinchcombe JC, Bossi G, Booth S, Griffiths GM: The immunological synapse of CTL contains a secretory domain and membrane bridges. Immunity 2001, 15:751–761. [DOI] [PubMed] [Google Scholar]
- 5.Catalfamo M, Henkart PA: Perforin and the granule exocytosis cytotoxicity pathway. Curr Opin Immunol 2003, 15:522–527. [DOI] [PubMed] [Google Scholar]
- 6.Isaaz S, Baetz K, Olsen K, Podack E, Griffiths GM: Serial killing by cytotoxic T lymphocytes: T cell receptor triggers degranulation, re-filling of the lytic granules and secretion of lytic proteins via a non-granule pathway. Eur J Immunol 1995, 25:1071–1079. [DOI] [PubMed] [Google Scholar]
- 7.Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ: Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity 2004, 21:589–601. [DOI] [PubMed] [Google Scholar]
- 8.Qin HY, Mukherjee R, Lee-Chan E, Ewen C, Bleackley RC, Singh B: A novel mechanism of regulatory T cell-mediated down-regulation of autoimmunity. Int Immunol 2006, 18:1001–1015. [DOI] [PubMed] [Google Scholar]
- 9.Lieberman J, Fan Z: Nuclear war: the granzyme A-bomb. Curr Opin Immunol 2003, 15:553–559. [DOI] [PubMed] [Google Scholar]
- 10.Lord SJ, Rajotte RV, Korbutt GS, Bleackley RC: Granzyme B: a natural born killer. Immunol Rev 2003, 193:31–38. [DOI] [PubMed] [Google Scholar]
- 11.Grossman WJ, Revell PA, Lu ZH, Johnson H, Bredemeyer AJ, Ley TJ: The orphan granzymes of humans and mice. Curr Opin Immunol 2003, 15:544–552. [DOI] [PubMed] [Google Scholar]
- 12.Johnson H, Scorrano L, Korsmeyer SJ, Ley TJ: Cell death induced by granzyme C. Blood 2003, 101:3093–3101. [DOI] [PubMed] [Google Scholar]
- 13.Kelly JM, Waterhouse NJ, Cretney E, Browne KA, Ellis S, Trapani JA, Smyth MJ: Granzyme M mediates a novel form of perforin-dependent cell death. J Biol Chem 2004, 279:22236–22242. [DOI] [PubMed] [Google Scholar]
- 14.Lu H, Hou Q, Zhao T, Zhang H, Zhang Q, Wu L, Fan Z: Granzyme M directly cleaves inhibitor of caspase-activated DNase (CAD) to unleash CAD leading to DNA fragmentation. J Immunol 2006, 177:1171–1178. [DOI] [PubMed] [Google Scholar]
- 15.Zhao T, Zhang H, Guo Y, Fan Z: Granzyme K directly processes Bid to release cytochrome c and endonuclease G leading to mitochondria-dependent cell death. J Biol Chem 2007, in press. [DOI] [PubMed] [Google Scholar]
- 16.Zhao T, Zhang H, Guo Y, Zhang Q, Hua G, Lu H, Hou Q, Liu H, Fan Z: Granzyme K cleaves the nucleosome assembly protein SET to induce single-stranded DNA nicks of target cells. Cell Death Differ 2007, 14:489–499. [DOI] [PubMed] [Google Scholar]
- 17.Kelso A, Costelloe EO, Johnson BJ, Groves P, Buttigieg K, Fitzpatrick DR: The genes for perforin, granzymes A–C and IFN-γ are differentially expressed in single CD8+ T cells during primary activation. Int Immunol 2002, 14:605–613. [DOI] [PubMed] [Google Scholar]
- 18.•.Pipkin ME, Ljutic B, Cruz-Guilloty F, Nouzova M, Rao A, Zuniga-Pflucker JC, Lichtenheld MG: Chromosome transfer activates and delineates a locus control region for perforin. Immunity 2007, 26:29–41. [DOI] [PubMed] [Google Scholar]; This study defines the locus control region that governs perforin expression in T cells and NK cells.
- 19.Nakajima H, Park HL, Henkart PA: Synergistic roles of granzymes A and B in mediating target cell death by rat basophilic leukemia mast cell tumors also expressing cytolysin/perforin. J Exp Med 1995, 181:1037–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shiver JW, Su L, Henkart PA: Cytotoxicity with target DNA breakdown by rat basophilic leukemia cells expressing both cytolysin and granzyme A. Cell 1992, 71:315–322. [DOI] [PubMed] [Google Scholar]
- 21.Kagi D, Ledermann B, Burki K, Seiler P, Odermatt B, Olsen KJ, Podack ER, Zinkernagel RM, Hengartner H: Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature 1994, 369:31–37. [DOI] [PubMed] [Google Scholar]
- 22.Lowin B, Hahne M, Mattmann C, Tschopp J: Cytolytic T-cell cytotoxicity is mediated through perforin and Fas lytic pathways. Nature 1994, 370:650–652. [DOI] [PubMed] [Google Scholar]
- 23.Heusel JW, Wesselschmidt RL, Shresta S, Russell JH, Ley TJ: Cytotoxic lymphocytes require granzyme B for the rapid induction of DNA fragmentation and apoptosis in allogeneic target cells. Cell 1994, 76:977–987. [DOI] [PubMed] [Google Scholar]
- 24.Ebnet K, Hausmann M, Lehmann-Grube F, Mullbacher A, Kopf M, Lamers M, Simon MM: Granzyme A-deficient mice retain potent cell-mediated cytotoxicity. EMBO J 1995, 14:4230–4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mullbacher A, Ebnet K, Blanden RV, Hla RT, Stehle T, Museteanu C, Simon MM: Granzyme A is critical for recovery of mice from infection with the natural cytopathic viral pathogen, ectromelia. Proc Natl Acad Sci USA 1996, 93:5783–5787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kagi D, Seiler P, Pavlovic J, Ledermann B, Burki K, Zinkernagel RM, Hengartner H: The roles of perforin- and Fas-dependent cytotoxicity in protection against cytopathic and noncytopathic viruses. Eur J Immunol 1995, 25:3256–3262. [DOI] [PubMed] [Google Scholar]
- 27.van den Broek ME, Kagi D, Ossendorp F, Toes R, Vamvakas S, Lutz WK, Melief CJ, Zinkernagel RM, Hengartner H: Decreased tumor surveillance in perforin-deficient mice. J Exp Med 1996, 184:1781–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Topham DJ, Cardin RC, Christensen JP, Brooks JW, Belz GT, Doherty PC: Perforin and Fas in murine gammaherpesvirus-specific CD8+ T cell control and morbidity. J Gen Virol 2001, 82:1971–1981. [DOI] [PubMed] [Google Scholar]
- 29.Rossi CP, McAllister A, Tanguy M, Kagi D, Brahic M: Theiler’s virus infection of perforin-deficient mice. J Virol 1998, 72:4515–4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smyth MJ, Street SE, Trapani JA: Cutting edge: granzymes A and B are not essential for perforin-mediated tumor rejection. J Immunol 2003, 171:515–518. [DOI] [PubMed] [Google Scholar]
- 31.Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA, Henter JI, Bennett M, Fischer A, de Saint Basile G et al. : Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science 1999, 286:1957–1959. [DOI] [PubMed] [Google Scholar]
- 32.Goransdotter Ericson K, Fadeel B, Nilsson-Ardnor S, Soderhall C, Samuelsson A, Janka G, Schneider M, Gurgey A, Yalman N, Revesz T et al. : Spectrum of perforin gene mutations in familial hemophagocytic lymphohistiocytosis. Am J Hum Genet 2001, 68:590–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Katano H, Ali MA, Patera AC, Catalfamo M, Jaffe ES, Kimura H, Dale JK, Straus SE, Cohen JI: Chronic active Epstein-Barr virus infection associated with mutations in perforin that impair its maturation. Blood 2004, 103:1244–1252. [DOI] [PubMed] [Google Scholar]
- 34.Lichtenheld MG, Olsen KJ, Lu P, Lowrey DM, Hameed A, Hengartner H, Podack ER: Structure and function of human perforin. Nature 1988, 335:448–451. [DOI] [PubMed] [Google Scholar]
- 35.Shinkai Y, Takio K, Okumura K: Homology of perforin to the ninth component of complement (C9). Nature 1988, 334:525–527. [DOI] [PubMed] [Google Scholar]
- 36.Shinkai Y, Yoshida MC, Maeda K, Kobata T, Maruyama K, Yodoi J, Yagita H, Okumura K: Molecular cloning and chromosomal assignment of a human perforin (PFP) gene. Immunogenetics 1989, 30:452–457. [DOI] [PubMed] [Google Scholar]
- 37.•.Keefe D, Shi L, Feske S, Massol R, Navarro F, Kirchhausen T, Lieberman J: Perforin triggers a plasma membrane-repair response that facilitates CTL induction of apoptosis. Immunity 2005, 23:249–262. [DOI] [PubMed] [Google Scholar]; This study identified an active role for the target cell membrane repair response in allowing perforin to deliver granzymes to activate apoptosis rather than necrosis.
- 38.Lyubchenko TA, Wurth GA, Zweifach A: Role of calcium influx in cytotoxic T lymphocyte lytic granule exocytosis during target cell killing. Immunity 2001, 15:847–859. [DOI] [PubMed] [Google Scholar]
- 39.Shiver JW, Henkart PA: A noncytotoxic mast cell tumor line exhibits potent IgE-dependent cytotoxicity after transfection with the cytolysin/perforin gene. Cell 1991, 64:1175–1181. [DOI] [PubMed] [Google Scholar]
- 40.Voskoboinik I, Thia MC, De Bono A, Browne K, Cretney E, Jackson JT, Darcy PK, Jane SM, Smyth MJ, Trapani JA: The functional basis for hemophagocytic lymphohistiocytosis in a patient with co-inherited missense mutations in the perforin (PFN1) gene. J Exp Med 2004, 200:811–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Voskoboinik I, Smyth MJ, Trapani JA: Perforin-mediated target-cell death and immune homeostasis. Nat Rev Immunol 2006, 6:940–952. [DOI] [PubMed] [Google Scholar]
- 42.Sauer H, Pratsch L, Tschopp J, Bhakdi S, Peters R: Functional size of complement and perforin pores compared by confocal laser scanning microscopy and fluorescence microphotolysis. Biochim Biophys Acta 1991, 1063:137–146. [DOI] [PubMed] [Google Scholar]
- 43.Millard PJ, Henkart MP, Reynolds CW, Henkart PA: Purification and properties of cytoplasmic granules from cytotoxic rat LGL tumors. J Immunol 1984, 132:3197–3204. [PubMed] [Google Scholar]
- 44.Tschopp J, Masson D, Stanley KK: Structural/functional similarity between proteins involved in complement- and cytotoxic T-lymphocyte-mediated cytolysis. Nature 1986, 322:831–834. [DOI] [PubMed] [Google Scholar]
- 45.Uellner R, Zvelebil MJ, Hopkins J, Jones J, MacDougall LK, Morgan BP, Podack E, Waterfield MD, Griffiths GM: Perforin is activated by a proteolytic cleavage during biosynthesis which reveals a phospholipid-binding C2 domain. EMBO J 1997, 16:7287–7296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trambas C, Gallo F, Pende D, Marcenaro S, Moretta L, De Fusco C, Santoro A, Notarangelo L, Arico M, Griffiths GM: A single amino acid change, A91V, leads to conformational changes that can impair processing to the active form of perforin. Blood 2005, 106:932–937. [DOI] [PubMed] [Google Scholar]
- 47.Sutton RB, Davletov BA, Berghuis AM, Sudhof TC, Sprang SR: Structure of the first C2 domain of synaptotagmin I: a novel Ca2+/phospholipid-binding fold. Cell 1995, 80:929–938. [DOI] [PubMed] [Google Scholar]
- 48.Grobler JA, Essen LO, Williams RL, Hurley JH: C2 domain conformational changes in phospholipase C-delta 1. Nat Struct Biol 1996, 3:788–795. [DOI] [PubMed] [Google Scholar]
- 49.Nalefski EA, Falke JJ: The C2 domain calcium-binding motif: structural and functional diversity. Protein Sci 1996, 5:2375–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Essen LO, Perisic O, Lynch DE, Katan M, Williams RL: A ternary metal binding site in the C2 domain of phosphoinositide-specific phospholipase C-delta1. Biochemistry 1997, 36:2753–2762. [DOI] [PubMed] [Google Scholar]
- 51.Xu GY, McDonagh T, Yu HA, Nalefski EA, Clark JD, Cumming DA: Solution structure and membrane interactions of the C2 domain of cytosolic phospholipase A2. J Mol Biol 1998, 280:485–500. [DOI] [PubMed] [Google Scholar]
- 52.Zhang X, Rizo J, Sudhof TC: Mechanism of phospholipid binding by the C2A-domain of synaptotagmin I. Biochemistry 1998, 37:12395–12403. [DOI] [PubMed] [Google Scholar]
- 53.Nalefski EA, Falke JJ: Location of the membrane-docking face on the Ca2+-activated C2 domain of cytosolic phospholipase A2. Biochemistry 1998, 37:17642–17650. [DOI] [PubMed] [Google Scholar]
- 54.Rizo J, Sudhof TC: C2-domains, structure and function of a universal Ca2+-binding domain. J Biol Chem 1998, 273:15879–15882. [DOI] [PubMed] [Google Scholar]
- 55.Peitsch MC, Amiguet P, Guy R, Brunner J, Maizel JV Jr, Tschopp J: Localization and molecular modelling of the membrane-inserted domain of the ninth component of human complement and perforin. Mol Immunol 1990, 27:589–602. [DOI] [PubMed] [Google Scholar]
- 56.Ojcius DM, Persechini PM, Zheng LM, Notaroberto PC, Adeodato SC, Young JD: Cytolytic and ion channel-forming properties of the N terminus of lymphocyte perforin. Proc Natl Acad Sci USA 1991, 88:4621–4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu CC, Persechini PM, Young JD: Expression and characterization of functionally active recombinant perforin produced in insect cells. J Immunol 1996, 156:3292–3300. [PubMed] [Google Scholar]
- 58.•.Voskoboinik I, Thia MC, Trapani JA: A functional analysis of the putative polymorphisms A91V and N252S and 22 missense perforin mutations associated with familial hemophagocytic lymphohistiocytosis. Blood 2005, 105:4700–4706. [DOI] [PubMed] [Google Scholar]; This study used rat basophilic leukemia cells expressing mutant forms of perforin as a way of probing the functional significance of perforin mutations identified in FHL patients.
- 59.Dupuis M, Schaerer E, Krause KH, Tschopp J: The calcium-binding protein calreticulin is a major constituent of lytic granules in cytolytic T lymphocytes. J Exp Med 1993, 177:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fraser SA, Karimi R, Michalak M, Hudig D: Perforin lytic activity is controlled by calreticulin. J Immunol 2000, 164:4150–4155. [DOI] [PubMed] [Google Scholar]
- 61.Fraser SA, Michalak M, Welch WH, Hudig D: Calreticulin, a component of the endoplasmic reticulum and of cytotoxic lymphocyte granules, regulates perforin-mediated lysis in the hemolytic model system. Biochem Cell Biol 1998, 76:881–887. [DOI] [PubMed] [Google Scholar]
- 62.Andrin C, Pinkoski MJ, Burns K, Atkinson EA, Krahenbuhl O, Hudig D, Fraser SA, Winkler U, Tschopp J, Opas M et al. : Interaction between a Ca2+-binding protein calreticulin and perforin, a component of the cytotoxic T-cell granules. Biochemistry 1998, 37:10386–10394. [DOI] [PubMed] [Google Scholar]
- 63.Peters PJ, Borst J, Oorschot V, Fukuda M, Krahenbuhl O, Tschopp J, Slot JW, Geuze HJ: Cytotoxic T lymphocyte granules are secretory lysosomes, containing both perforin and granzymes. J Exp Med 1991, 173:1099–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Masson D, Peters PJ, Geuze HJ, Borst J, Tschopp J: Interaction of chondroitin sulfate with perforin and granzymes of cytolytic T-cells is dependent on pH. Biochemistry 1990, 29:11229–11235. [DOI] [PubMed] [Google Scholar]
- 65.Metkar SS, Wang B, Aguilar-Santelises M, Raja SM, Uhlin-Hansen L, Podack E, Trapani JA, Froelich CJ: Cytotoxic cell granule-mediated apoptosis: perforin delivers granzyme B–serglycin complexes into target cells without plasma membrane pore formation. Immunity 2002, 16:417–428. [DOI] [PubMed] [Google Scholar]
- 66.Kataoka T, Togashi K, Takayama H, Takaku K, Nagai K: Inactivation and proteolytic degradation of perforin within lytic granules upon neutralization of acidic pH. Immunology 1997, 91:493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Persechini PM, Liu CC, Jiang S, Young JD: The lymphocyte pore-forming protein perforin is associated with granules by a pH-dependent mechanism. Immunol Lett 1989, 22:23–27. [DOI] [PubMed] [Google Scholar]
- 68.Balaji KN, Schaschke N, Machleidt W, Catalfamo M, Henkart PA: Surface cathepsin B protects cytotoxic lymphocytes from self-destruction after degranulation. J Exp Med 2002, 196:493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Baran K, Ciccone A, Peters C, Yagita H, Bird PI, Villadangos JA, Trapani JA: Cytotoxic T lymphocytes from cathepsin B-deficient mice survive normally in vitro and in vivo after encountering and killing target cells. J Biol Chem 2006, 281:30485–30491. [DOI] [PubMed] [Google Scholar]
- 70.Kawasaki Y, Saito T, Shirota-Someya Y, Ikegami Y, Komano H, Lee MH, Froelich CJ, Shinohara N, Takayama H: Cell death-associated translocation of plasma membrane components induced by CTL. J Immunol 2000, 164:4641–4648. [DOI] [PubMed] [Google Scholar]
- 71.Froelich CJ, Orth K, Turbov J, Seth P, Gottlieb R, Babior B, Shah GM, Bleackley RC, Dixit VM, Hanna W: New paradigm for lymphocyte granule-mediated cytotoxicity. J Biol Chem 1996, 271:29073–29079. [DOI] [PubMed] [Google Scholar]
- 72.Shi L, Mai S, Israels S, Browne K, Trapani JA, Greenberg AH: Granzyme B (GraB) autonomously crosses the cell membrane and perforin initiates apoptosis and GraB nuclear localization. J Exp Med 1997, 185:855–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Motyka B, Korbutt G, Pinkoski MJ, Heibein JA, Caputo A, Hobman M, Barry M, Shostak I, Sawchuk T, Holmes CF et al. : Mannose 6-phosphate/insulin-like growth factor II receptor is a death receptor for granzyme B during cytotoxic T cell-induced apoptosis. Cell 2000, 103:491–500. [DOI] [PubMed] [Google Scholar]
- 74.Pinkoski MJ, Hobman M, Heibein JA, Tomaselli K, Li F, Seth P, Froelich CJ, Bleackley RC: Entry and trafficking of granzyme B in target cells during granzyme B-perforin-mediated apoptosis. Blood 1998, 92:1044–1054. [PubMed] [Google Scholar]
- 75.Browne KA, Blink E, Sutton VR, Froelich CJ, Jans DA, Trapani JA: Cytosolic delivery of granzyme B by bacterial toxins: evidence that endosomal disruption, in addition to transmembrane pore formation, is an important function of perforin. Mol Cell Biol 1999, 19:8604–8615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shi L, Keefe D, Durand E, Feng H, Zhang D, Lieberman J: Granzyme B binds to target cells mostly by charge and must be added at the same time as perforin to trigger apoptosis. J Immunol 2005, 174:5456–5461. [DOI] [PubMed] [Google Scholar]
- 77.Bird CH, Sun J, Ung K, Karambalis D, Whisstock JC, Trapani JA, Bird PI: Cationic sites on granzyme B contribute to cytotoxicity by promoting its uptake into target cells. Mol Cell Biol 2005, 25:7854–7867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Veugelers K, Motyka B, Goping IS, Shostak I, Sawchuk T, Bleackley RC: Granule-mediated killing by granzyme B and perforin requires a mannose 6-phosphate receptor and is augmented by cell surface heparan sulfate. Mol Biol Cell 2006, 17:623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Trapani JA, Sutton VR, Thia KY, Li YQ, Froelich CJ, Jans DA, Sandrin MS, Browne KA: A clathrin/dynamin- and mannose-6-phosphate receptor-independent pathway for granzyme B-induced cell death. J Cell Biol 2003, 160:223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dressel R, Raja SM, Honing S, Seidler T, Froelich CJ, von Figura K, Gunther E: Granzyme-mediated cytotoxicity does not involve the mannose 6-phosphate receptors on target cells. J Biol Chem 2004, 279:20200–20210. [DOI] [PubMed] [Google Scholar]
- 81.McNeil PL, Steinhardt RA: Plasma membrane disruption: repair, prevention, adaptation. Annu Rev Cell Dev Biol 2003, 19:697–731. [DOI] [PubMed] [Google Scholar]
- 82.Steinhardt RA, Bi G, Alderton JM: Cell membrane resealing by a vesicular mechanism similar to neurotransmitter release. Science 1994, 263:390–393. [DOI] [PubMed] [Google Scholar]
- 83.Bi GQ, Alderton JM, Steinhardt RA: Calcium-regulated exocytosis is required for cell membrane resealing. J Cell Biol 1995, 131:1747–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Miyake K, McNeil PL: Vesicle accumulation and exocytosis at sites of plasma membrane disruption. J Cell Biol 1995, 131:1737–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Reddy A, Caler EV, Andrews NW: Plasma membrane repair is mediated by Ca2+-regulated exocytosis of lysosomes. Cell 2001, 106:157–169. [DOI] [PubMed] [Google Scholar]