

Abstract
Primary vitreoretinal lymphoma (PVRL) is a rare malignant lymphoma subtype with an unfavorable prognosis due to frequent central nervous system (CNS) progression. Thus, identifying factors associated with CNS progression is essential for improving the prognosis of PVRL patients. Accordingly, we conducted a comprehensive genetic analysis using archived vitreous humor samples of 36 PVRL patients diagnosed and treated at our institution and retrospectively examined the relationship between genetic alterations and CNS progression. Whole-exome sequencing (N=2) and amplicon sequencing using a custom panel of 107 lymphomagenesis-related genes (N=34) were performed to assess mutations and copy number alterations. The median number of pathogenic genetic alterations per case was 12 (range, 0-22). Pathogenic genetic alterations of CDKN2A, MYD88, CDKN2B, PRDM1, PIM1, ETV6, CD79B, and IGLL5, as well as aberrant somatic hypermutations, were frequently detected. The frequency of ETV6 loss and PRDM1 alteration (mutation and loss) was 23% and 49%, respectively. Multivariate analysis revealed ETV6 loss (hazard ratio [HR]=3.26, 95% confidence interval [CI]: 1.08–9.85) and PRDM1 alteration (HR=2.52, 95% CI: 1.03–6.16) as candidate risk factors associated with CNS progression of PVRL. Moreover, these two genetic factors defined slow-, intermediate-, and rapid-progression groups (0, 1, and 2 factors, respectively), and the median period to CNS progression differed significantly among them (52 vs. 33 vs. 20 months, respectively). Our findings suggest that genetic factors predict the CNS progression of PVRL effectively, and the genetics-based CNS progression model might lead to stratification of treatment.
Introduction
Primary vitreoretinal lymphoma (PVRL) is a malignant lymphoma subtype with lesions limited to the vitreous humor, retina, and optic nerve.1 The pathological classification of PVRL is typically diffuse large B-cell lymphoma (DLBCL)2 with MYD88 L265P and/or CD79B mutations.3,4
Intravitreal chemotherapy, such as methotrexate (MTX), and local radiotherapy have been reported to achieve intraocular complete response and improve visual symptoms.5-7 Additionally, systemic chemotherapy is often administered following local treatment in an effort to prevent subsequent central nervous system (CNS) progression. We previously reported a single-arm prospective study on newly diagnosed PVRL patients who received an intravitreal MTX injection followed by systemic high-dose MTX (HD-MTX). All patients achieved intraocular complete response, and the adverse events were generally tolerable.8 However, the high rate of CNS progression indicated that these prophylactic strategies did not improve PVRL prognosis.9,10 Therefore, the identification of factors associated with CNS progression is essential to improve the prognosis of PVRL patients.
In recent years, comprehensive genetic analyses using next-generation sequencing have revealed numerous genetic alterations in systemic DLBCL and provided solid evidence to newly classify DLBCL based on genetic alterations;11-13 PVRL is classified into MCD/cluster 5 subtype with MYD88 and CD79B mutations. Furthermore, genetic subtype-guided immunochemotherapy was reported to show better efficacy than conventional chemotherapy in DLBCL.14,15 Thus, this genetic approach is expected to be applied in clinical settings, such as exploring new target therapies and prognostic stratification.
We recently performed a retrospective analysis of PVRL patients diagnosed and treated at our hospital to identify the clinical factors associated with CNS progression, revealing bilateral disease and the detection of B-cell clonality confirmed via flow cytometry at diagnosis as risk factors.16 Previously, we conducted direct sequencing and allele-specific polymerase chain reaction (PCR) to check the mutation of CD79B Y196 and MYD88 L265P on the archived vitreous humor samples from 17 patients with PVRL and argued that CD79B Y196 potentially has a prognostic potential for patients with PVRL.3 In the present study, we performed a comprehensive and massive genetic analysis of archived vitreous humor samples from 36 PVRL patients to identify genetic alterations strongly associated with CNS progression.
Methods
Patients
We enrolled 36 PVRL patients diagnosed from April 2012 to March 2022 who were treated at our hospital and had archived vitreous humor samples. Some of them have been included in previous studies,3,8,16 and eight of 36 patients herein were identical to those in the previous study.3 PVRL was defined as VRL localized to the eyes and was diagnosed as previously described.8,9,16 Details of PVRL patients in this study are shown in Figure 1. The methods of FCM analysis, PCR analysis of IGH rearrangement, and cytokine measurement were previously described3,8 and shown in the Online Supplementary Appendix. Treatment for the patients were also described in the Online Supplementary Appendix. This study was performed in accordance with the Declaration of Helsinki and approved by the Ethics Committee of Tokyo Medical and Dental University (approval number: M2017-341). All patients provided written informed consent.
DNA extraction and next-generation sequencing
Genomic DNA was extracted from vitreous humor or from formalin-fixed paraffin-embedded (FFPE) brain tissue biopsies of PVRL patients using an EZ1 Virus Mini Kit v2.0 (QIAGEN, Hilden, Germany) or a QIAamp DNA FFPE Advanced Kit (QIAGEN), respectively. EZ1 Virus Mini Kit v2.0 usually extracts cell-free DNA; however, Zong et al. reported17 that genomic DNA with enough quality and quantity was extracted from cells. Library preparation for amplicon-based targeted sequencing was performed as previously described18 using a custom gene panel of 107 genes frequently mutated in lymphoma, particularly in PVRL (Online Supplementary Table S1). Briefly, the library was prepared using AmpliSeq Library Plus for Illumina (Illumina, San Diego, CA, USA). Further, synthesized libraries were sequenced in Miseq (Illumina) paired-end runs. The details of library synthesis method are described in the Online Supplementary Appendix.
Gene alteration analysis
We used a mutational analysis pipeline based on previously reported method.18 The data handling step and used tools were described in the Online Supplementary Appendix. Variants considered pathogenic were identified according to previous reports (Online Supplementary Table S1). We also identified and counted aberrant somatic hypermutation (aSHM) to specify hypermutated cases, although their pathogenicity could not be determined. Copy number alterations (CNA) were calculated based on a previously described method.19 Detection methods of CNA were described in the Online Supplementary Appendix in detail.
Figure 1.

Classification of primary vitreoretinal lymphoma patients in this study. *Vitreous humor opacity and/or retinal or sub-retinal proliferative lesions. PVRL: primary vitreoretinal lymphoma; FCM: flow cytometry; PCR: polymerase chain reaction; IL: interleukin.
Statistical analysis
Fisher exact test was used for categorical variable analysis and Mann-Whitney U test for continuous variable analysis. The cumulative incidence of CNS progression was calculated in the presence of death as a competing event, and the difference was tested using Gray test, although all PVRL patients who died had CNS progression before death. Factors used for the multivariate analysis were selected using the stepwise Akaike information criterion (AIC) method from the factors that revealed significant differences in the univariate analysis. Multivariate analysis was performed using Fine and Gray proportional hazard modeling. AIC was used as the selection criteria. All statistical analyses were performed using R 4.2.0 software (The R Foundation for Statistical Computing), and statistical significance was defined as P<0.05 based on a two-sided test.
Results
Patient characteristics
We evaluated 36 patients diagnosed with PVRL. The median follow-up period was 29 months (range, 2-119 months). Patient characteristics are summarized in Table 1. Ocular involvement was unilateral in 16 and bilateral in 20 patients. The median time from the onset of initial visual symptoms to diagnosis was 8 months (range, 1-29 months). Cytopathology, flow cytometry analysis, and IGH rearrangement were positive for PVRL in 42%, 74%, and 80% of patients, respectively. All 36 patients received intravitreal MTX injections following PVRL diagnosis, and 20 of 36 patients were treated with systemic HD-MTX thereafter. During the observation period, 19 patients developed CNS progression. Among the patients suspected with PVRL, one patient had CNS progression and ocular relapse, and one patient had ocular relapse.
Landscape of pathogenic genetic alterations in primary vitreoretinal lymphoma
Whole-exome sequencing (N=2) and amplicon sequencing using a custom panel containing 107 lymphomagenesis-related genes (N=34) were performed on vitreous humor samples to assess mutations and CNA. Average coverage depths of whole-exome sequencing were 105.8 and 143.5, and the mean coverage depth of amplicon sequencing was 621.8 (range, 74.8-1,098.0). One sample had low-quality DNA and could not be evaluated for CNA. The detected pathogenic gene mutations and CNA are presented in Online Supplementary Tables S2 and S3, respectively.
At least one pathogenic genetic alteration was detected in 31 of 36 samples, and the median number of pathogenic genetic alterations per case was 12 (range, 0-22) (Figure 2A).
The landscape of pathogenic genetic alteration is shown in Figure 2B. The top three altered genes were CDKN2A (25/36, 69%), MYD88 (23/36, 64%), and CDKN2B (21/36, 58%). Of the 25 cases with altered CDKN2A, 23 showed copy number loss, two of which also showed mutation, and mutation only was observed in two cases. All CDKN2B alterations were copy number loss, and all MYD88 alterations were mutations in p.Leu265. The other frequently altered genes were PRDM1 (47%), PIM1 (44%), ETV6 (42%), CD79B (42%), and IGLL5 (42%). In some cases, mutation and copy number loss were observed in the same genes. The cases with gene mutations showing >50% variant allele frequencies in the presence of copy number loss were identified. These alterations occurred in different alleles, indicating deletion of the normal allele.
Aberrant somatic hypermutation
Activation-induced deaminase mediates SHM and class-switch recombination by converting cytosine residues into uracil residues. aSHM arises from errors during SHM and occurs in genes other than IGV such as PIM1 and IGLL5. aSHM is frequently detected in DLBCL, a subtype that accounts for most PVRL cases. However, the association between aSHM and DLBCL initiation has yet to be verified.20,21 We picked up eight genes (PIM1, OSBPL10, MPEG1, IGLL5, BTG1, BTG2, ETV6, and IRF4), which were reportedly related to aSHM and examined the impact of aSHM in our study. Figure 3A shows the number of mutations per gene per case. One or more mutations in PIM1, OSBPL10, MPEG1, IGLL5, BTG1, BTG2, ETV6, and IRF4 were found in 24, 14, 12, 20, 10, 12, 12, and two of the 36 PVRL cases, respectively. Figure 3B shows the total number of mutations detected in these eight genes per case. The median number of mutations per case was ten (range, 0-35). There was no clear correlation between the number of pathogenic genetic alterations and the number of mutations detected in these eight genes related to aSHM per case.
Table 1.
Patient characteristics at primary vitreoretinal lymphoma diagnosis (N=36, unless otherwise indicated).
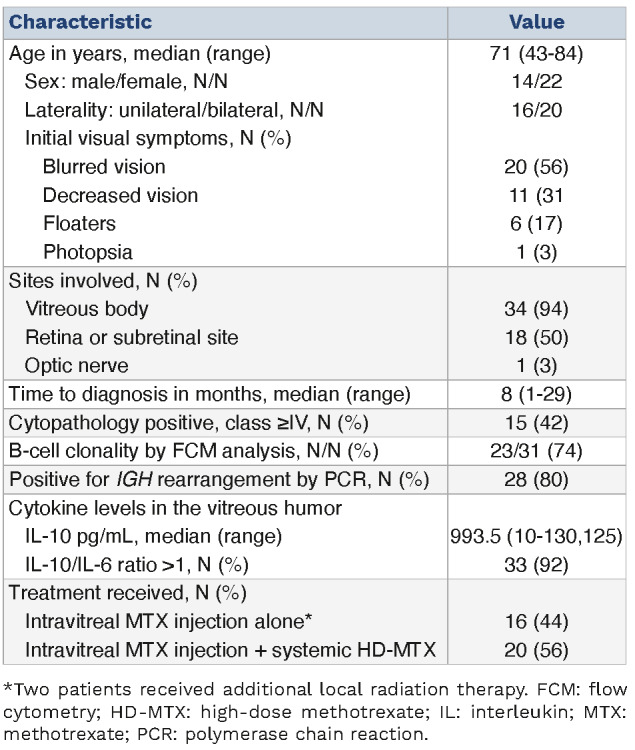
Figure 2.

Pathogenic genetic alterations in primary vitreoretinal lymphoma. (A) Number of pathogenic genetic alterations per primary vitreoretinal lymphoma (PVRL) case. (B) Landscape of pathogenic genetic alterations in PVRL cases. Each column represents a case, and each row represents a recurrently altered gene. The bar graph on the right represents the frequency of pathogenic genetic alteration in each gene.
Genetic risk factors associated with central nervous system progression
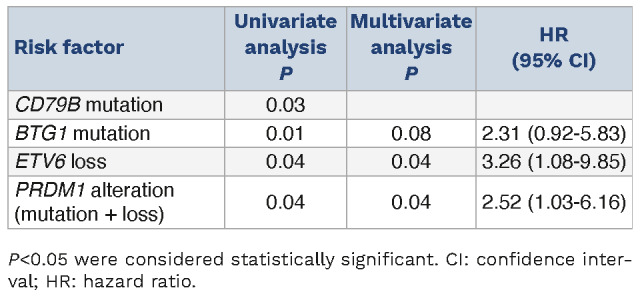
Figure 4A shows the cumulative incidence of CNS progression, and the 5-year cumulative incidence of CNS progression was 78.3%. We investigated possible genetic alterations associated with CNS progression. The univariate analysis identified CD79B mutation, BTG1 mutation, ETV6 loss, and PRDM1 alteration (mutation and copy number loss) as candidate risk factors (Table 2). Factors used for the multivariate analysis were selected using the stepwise AIC method from these four factors, and CD79B mutation was excluded. ETV6 loss and PRDM1 alteration remained significant in the multivariate analysis (Table 2). The number of pathogenic genetic alterations and the number of mutations detected in eight genes related to aSHM were not associated with CNS progression. We also investigated the association between ETV6 loss/PRDM1 alteration and clinical findings of PVRL, but there was no significant correlation (Online Supplementary Tables S4 and S5, respectively).
Figure 3.

Aberrant somatic hypermutations of primary vitreoretinal lymphoma. (A) Number of mutations per gene per case. Each dot represents a case. (B) Total number of mutations detected in these 8 genes per case. The order of cases in the column is the same as in Figure 2A.
Figure 4.

Genetic model of central nervous system progression in primary vitreoretinal lymphoma. (A) Cumulative incidence of central nervous system (CNS) progression in primary vitreoretinal lymphoma (PVRL). (B) Genetic model using ETV6 loss and PRDM1 alteration to define slow-, intermediate-, and rapid-progression groups (0, 1, and 2 factors, respectively). Cumulative incidence of CNS progression in the 3 groups is shown.
Genetic model of central nervous system progression in primary vitreoretinal lymphoma
ETV6 loss and PRDM1 alteration were identified as risk factors for CNS progression in PVRL. We created a genetics-based CNS progression model using these two factors to define the slow-, intermediate-, and rapid-progression groups (0, 1, and 2 factors, respectively) (Figure 4B). The median period to CNS progression differed significantly among the three groups (52 vs. 33 vs. 20 months, respectively).
Genetic comparison between primary vitreous humor and brain samples
CNS progression occurred in 19 of 36 PVRL patients, four of whom underwent brain biopsy, and the tissue was processed using FFPE. The genetic-based group of the four patients was two in the intermediate-progression group and one in the slow- and rapid-progression groups. The period of CNS progression was 39 months (slow-progression group), 11 and 32 months (intermediate-progression group), and 20 months (rapid-progression group). We performed amplicon sequencing and analysis to compare pathogenic genetic alterations in the brain tissue samples with those in the vitreous humor samples taken at diagnosis. All four patients had at least one concordant alteration and had additional alterations that were found in the brain tissue samples but not in the vitreous humor samples (Figure 5). Details of detected pathogenic gene mutations and CNA in the brain tissue and vitreous humor samples are presented in Online Supplementary Table S6.
Discussion
We conducted a comprehensive genetic analysis of 36 PVRL patients using vitreous humor samples taken at diagnosis and determined the genetic alterations related to CNS progression in PVRL.
Mutation and copy number analyses revealed that pathogenic genetic alterations of CDKN2A, MYD88, CDKN2B, PRDM1, PIM1, ETV6, CD79B, and IGLL5 were common, as well as aSHM in PIM1, OSBPL10, MPEG1, IGLL5, BTG1, and BTG2. Due to the rarity of PVRL, comprehensive genetic analysis is challenging, and this is compounded by the low quantity and quality of DNA extracted from vitreous humor samples.
Table 2.
Genetic risk factors for central nervous system progression.
However, a few groups have recently published exciting reports in this context using small amounts of DNA or cell-free DNA.22-25 Because the results of our genetic analysis were highly consistent with the results of these comprehensive studies, we considered them suitable for the analysis of genetic alterations predictive of CNS progression in PVRL. Notably, there were 36 participants in our study, which is more than previous genetic analyses of PVRL.
We identified ETV6 loss and PRDM1 alteration (mutation and copy number loss) as candidate genetic alterations predicting CNS progression in PVRL. Our study is the first comprehensive genetic analysis to imply the association of genetic risk factors with CNS involvement. Thus, our findings stand out in terms of novelty.
ETV6 is a transcriptional repressor that plays a crucial role in hematopoiesis and is related to various types of hematological malignancies, including DLBCL.11,26-28 ETV6 loss, mutation, and fusion have been reported in primary central nervous system lymphoma (PCNSL).29-31 This is consistent with our finding that ETV6 loss is a factor related to CNS progression in PVRL, although the precise mechanism remains unclear. Notably, the level of ETV6 protein expression is negatively correlated with BIRC5 (survivin) expression and is associated with the antitumor effect of YM155, a BIRC5-specific inhibitor.32 YM155 has shown clinical efficacy as a single agent or in combination with rituximab or bendamustine to treat relapsed/refractory DLBCL.33,34 Future studies on the effectiveness of YM155 treatment for PVRL and its association with ETV6 loss are anticipated.
PRDM1 is also a transcriptional repressor and a key molecule involved in plasma cell differentiation.35 PRDM1 mutation and loss are frequently detected in activated B-cell-like (ABC) DLBCL,11 and conditional knockout of PRDM1 in B cells results in constitutive NF-κB activation and the development of lymphoproliferative disorders resembling ABC-DLBCL in vivo.36 Genetic alteration of PRDM1 frequently occurs in PCNSL.29 Although the precise mechanism of CNS progression remains undefined, considering that PRDM1 alterations are infrequent in systemic extranodal DLBCL,37,38 there may be a CNS-specific genetic pathogenesis. Bruton tyrosine kinase (BTK) inhibitors interfere with B-cell receptor and NF- κB signaling by inhibiting BTK, and ibrutinib (a BTK inhibitor) has shown encouraging clinical activity against lymphomas involving the CNS and intraocular sites.39 Thus, it would be interesting to investigate whether the presence or absence of genetic alterations in PRDM1 affects BTK inhibitor efficacy. Moreover, Pascual et al. reported that constitutive NF-κB activation and impaired differentiation resulting from Blimp1 (a PRDM1 homolog) inactivation downregulated p53 signaling and triggered immune escape in ABC-DLBCL and that simultaneous PD-1 blockade improved the efficacy of anti-CD20 immunotherapy in an ABC-DLBCL-like mouse model.40 In parallel, since the efficacy of PD-1 blockade for CNS lymphoma has been reported,41,42 immune checkpoint modulation for PVRL patients with PRDM1 alteration may be an intriguing therapeutic approach.
Figure 5.

Genetic alterations at initial onset and after central nervous system progression. Number of pathogenic genetic alterations in vitreous humor samples taken at disease onset and brain tissue biopsy samples taken after central nervous system (CNS) progression per case. Concordant alterations (found in both vitreous humor and brain tissue) and discordant alterations (found in either vitreous humor or brain tissue) are indicated.
The genetics-based CNS progression model that we proposed in this study used two genetic alterations, namely, ETV6 loss and PRDM1 alterations, to successfully define three statistically significant groups for CNS progression in PVRL patients. To break through this intractable lymphoma, therapeutic strategies should be adapted using conventional HD-MTX-based chemotherapy regimens in potential combination with novel agents, such as BTK inhibitors, to the CNS progression risk of each patient. Our genetics-based CNS progression model might help this stratified treatment. Since a comparison between the genetic alterations in PVRL at disease onset and after CNS progression had never been reported, we performed a longitudinal comparison of pathogenic genetic alterations identified using amplicon sequencing of FFPE brain tissue samples from four PVRL patients with CNS progression and their vitreous humor samples at diagnosis. All four patients with PVRL had at least one concordant alteration. Balikov et al. conducted target sequencing of matched brain and vitreous samples in two PCNSL patients with VRL and showed shared genetic alterations, suggesting the same origin.43 Similarly, our results described that brain lesions were of the same origin as the vitreous lesions at diagnosis. Moreover, all four patients had additional pathogenic genetic alterations that were absent at disease onset. Thus, future analysis with a larger number of PVRL patients may facilitate the identification of additional genetic alterations associated with CNS lesion development.
This study has some limitations. First, as a single-institute, retrospective analysis, selection bias cannot be ignored. Second, although we considered the number of lymphomagenesis-related genes (N=107) examined in the amplicon sequences sufficient to cover most genetic alterations in the context of PVRL, it is possible that other (untested) genetic alterations are involved in CNS progression. Third, although our study enrolled 36 PVRL patients, which is the largest in number to date for a comprehensive genetic analysis of this rare disease, the sample size was small. During the analysis of rare diseases, the small sample size might reduce the power of detection.44 Therefore, we did not use multiplicity correction methods for the results of regression tests because they would have further reduced the detection power. Studies with large sample sizes using multiple comparison correction methods in multivariate analysis will enable a detailed investigation of the biological characteristics of PVRL. Finally, we could not validate our results in another cohort. We seek to test the validity of this genetics-based CNS progression model in a prospective and large cohort through international collaborations in the future.
To summarize, our comprehensive genetic analysis identified ETV6 loss and PRDM1 alterations as candidate genetic risk factors related to CNS progression in PVRL. Subsequently, we created a new model for CNS progression using these two genetic risk factors. A prospective and large study is necessary to validate this model. With proven validity, interventions with new drugs targeting these genetic alterations in possible combination with other available therapeutic options based on this model may improve the outcome of PVRL.
Supplementary Material
Acknowledgments
The authors thank Enago (https://www.enago.com/) for English language editing.
Funding Statement
Funding: This work was supported by JSPS KAKENHI (grant numbers JP22K20785, JP23K15296, and JP22K09763).
Data sharing statement
Requests for original data should be addressed to the corresponding author.
References
- 1.Soussain C, Malaise D, Cassoux N. Primary vitreoretinal lymphoma: a diagnostic and management challenge. Blood. 2021;138(17):1519-1534. [DOI] [PubMed] [Google Scholar]
- 2.Chan CC, Rubenstein JL, Coupland SE, et al. Primary vitreoretinal lymphoma: a report from an International Primary Central Nervous System Lymphoma Collaborative Group symposium. Oncologist. 2011;16(11):1589-1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yonese I, Takase H, Yoshimori M, et al. CD79B mutations in primary vitreoretinal lymphoma: Diagnostic and prognostic potential. Eur J Haematol. 2019;102(2):191-196. [DOI] [PubMed] [Google Scholar]
- 4.Raja H, Salomão DR, Viswanatha DS, Pulido JS. Prevalence of MYD88 L265P mutation in histologically proven, diffuse large B-cell vitreoretinal lymphoma. Retina. 2016;36(3):624-628. [DOI] [PubMed] [Google Scholar]
- 5.Fishburne BC, Wilson DJ, Rosenbaum JT, Neuwelt EA. Intravitreal methotrexate as an adjunctive treatment of intraocular lymphoma. Arch Ophthalmol. 1997;115(9):1152-1156. [DOI] [PubMed] [Google Scholar]
- 6.Habot-Wilner Z, Frenkel S, Pe’er J. Efficacy and safety of intravitreal methotrexate for vitreo-retinal lymphoma - 20 years of experience. Br J Haematol. 2021;194(1):92-100. [DOI] [PubMed] [Google Scholar]
- 7.Ferreri AJ, Blay JY, Reni M, et al. Relevance of intraocular involvement in the management of primary central nervous system lymphomas. Ann Oncol. 2002;13(4):531-538. [DOI] [PubMed] [Google Scholar]
- 8.Akiyama H, Takase H, Kubo F, et al. High-dose methotrexate following intravitreal methotrexate administration in preventing central nervous system involvement of primary intraocular lymphoma. Cancer Sci. 2016;107(10):1458-1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takase H, Arai A, Iwasaki Y, et al. Challenges in the diagnosis and management of vitreoretinal lymphoma - clinical and basic approaches. Prog Retin Eye Res. 2022;90:101053. [DOI] [PubMed] [Google Scholar]
- 10.Castellino A, Pulido JS, Johnston PB, et al. Role of systemic high-dose methotrexate and combined approaches in the management of vitreoretinal lymphoma: a single center experience 1990-2018. Am J Hematol. 2019;94(3):291-298. [DOI] [PubMed] [Google Scholar]
- 11.Schmitz R, Wright GW, Huang DW, et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. N Engl J Med. 2018;378(15):1396-1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chapuy B, Stewart C, Dunford AJ, et al. Author correction: Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med. 2018;24(8):1290-1291. [DOI] [PubMed] [Google Scholar]
- 13.Wright GW, Huang DW, Phelan JD, et al. A probabilistic classification tool for genetic subtypes of diffuse large B cell lymphoma with therapeutic implications. Cancer Cell. 2020;37(4):551-568.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilson WH, Wright GW, Huang DW, et al. Effect of ibrutinib with R-CHOP chemotherapy in genetic subtypes of DLBCL. Cancer Cell. 2021;39(12):1643-1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang MC, Tian S, Fu D, et al. Genetic subtype-guided immunochemotherapy in diffuse large B cell lymphoma: the randomized GUIDANCE-01 trial. Cancer Cell. 2023;41(10):1705-1716. [DOI] [PubMed] [Google Scholar]
- 16.Motomura Y, Yoshifuji K, Tachibana T, et al. Clinical factors for central nervous system progression and survival in primary vitreoretinal lymphoma. Br J Haematol. 2024;204(4):1279-1287. [DOI] [PubMed] [Google Scholar]
- 17.Zong Y, Kamoi K, Kurozumi-Karube H, Zhang J, Yang M, Ohno-Matsui K. Safety of intraocular anti-VEGF antibody treatment under in vitro HTLV-1 infection. Front Immunol. 2022;13:1089286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sadato D, Hirama C, Kaiho-Soma A, et al. Archival bone marrow smears are useful in targeted next-generation sequencing for diagnosing myeloid neoplasms. PLoS One. 2021;16(7):e0255257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagao T, Yoshifuji K, Sadato D, et al. Establishment and characterization of a new activated B-cell-like DLBCL cell line, TMD12. Exp Hematol. 2022;116:37-49. [DOI] [PubMed] [Google Scholar]
- 20.Pasqualucci L, Neumeister P, Goossens T, et al. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature. 2001;412(6844):341-346. [DOI] [PubMed] [Google Scholar]
- 21.Pasqualucci L, Bhagat G, Jankovic M, et al. AID is required for germinal center-derived lymphomagenesis. Nat Genet. 2008;40(1):108-112. [DOI] [PubMed] [Google Scholar]
- 22.Wang X, Su W, Gao Y, et al. A pilot study of the use of dynamic cfDNA from aqueous humor and vitreous fluid for the diagnosis and treatment monitoring of vitreoretinal lymphomas. Haematologica. 2022;107(9):2154-2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bonzheim I, Sander P, Salmerón-Villalobos J, et al. The molecular hallmarks of primary and secondary vitreoretinal lymphoma. Blood Adv. 2022;6(5):1598-1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee J, Kim B, Lee H, et al. Whole exome sequencing identifies mutational signatures of vitreoretinal lymphoma. Haematologica. 2020;105(9):e458-460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cani AK, Hovelson DH, Demirci H, Johnson MW, Tomlins SA, Rao RC. Next generation sequencing of vitreoretinal lymphomas from small-volume intraocular liquid biopsies: new routes to targeted therapies. Oncotarget. 2017;8(5):7989-7998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ford AM, Palmi C, Bueno C, et al. The TEL-AML1 leukemia fusion gene dysregulates the TGF-beta pathway in early B lineage progenitor cells. J Clin Invest. 2009;119(4):826-836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang J, Ding L, Holmfeldt L, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481(7380):157-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alexander TB, Gu Z, Iacobucci I, et al. The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature. 2018;562(7727):373-379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Radke J, Ishaque N, Koll R, et al. The genomic and transcriptional landscape of primary central nervous system lymphoma. Nat Commun. 2022;13(1):2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bruno A, Boisselier B, Labreche K, et al. Mutational analysis of primary central nervous system lymphoma. Oncotarget. 2014;5(13):5065-5075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chapuy B, Roemer MG, Stewart C, et al. Targetable genetic features of primary testicular and primary central nervous system lymphomas. Blood. 2016;127(7):869-881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marino D, Pizzi M, Kotova I, et al. High ETV6 levels support aggressive B lymphoma cell survival and predict poor outcome in diffuse large B-cell lymphoma patients. Cancers (Basel). 2022;14(2):338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheson BD, Bartlett NL, Vose JM, et al. A phase II study of the survivin suppressant YM155 in patients with refractory diffuse large B-cell lymphoma. Cancer. 2012;118(12):3128-3134. [DOI] [PubMed] [Google Scholar]
- 34.Kaneko N, Mitsuoka K, Amino N, et al. Combination of YM155, a survivin suppressant with bendamustine and rituximab: a new combination therapy to treat relapsed/refractory diffuse large B-cell lymphoma. Clin Cancer Res. 2014;20(7):1814-1822. [DOI] [PubMed] [Google Scholar]
- 35.Shaffer AL, Lin KI, Kuo TC, et al. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity. 2002;17(1):51-62. [DOI] [PubMed] [Google Scholar]
- 36.Mandelbaum J, Bhagat G, Tang H, et al. BLIMP1 is a tumor suppressor gene frequently disrupted in activated B cell-like diffuse large B cell lymphoma. Cancer Cell. 2010;18(6):568-579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li P, Chai J, Chen Z, et al. Genomic mutation profile of primary gastrointestinal diffuse large B-cell lymphoma. Front Oncol. 2021;11:622648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Groen RAL, van Eijk R, Böhringer S, et al. Frequent mutated B2M, EZH2, IRF8, and TNFRSF14 in primary bone diffuse large B-cell lymphoma reflect a GCB phenotype. Blood Adv. 2021;5(19):3760-3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soussain C, Choquet S, Blonski M, et al. Ibrutinib monotherapy for relapse or refractory primary CNS lymphoma and primary vitreoretinal lymphoma: final analysis of the phase II ‘proof-of-concept’ iLOC study by the Lymphoma Study Association (LYSA) and the French Oculo-Cerebral Lymphoma (LOC) network. Eur J Cancer. 2019;117:121-130. [DOI] [PubMed] [Google Scholar]
- 40.Pascual M, Mena-Varas M, Robles EF, et al. PD-1/PD-L1 immune checkpoint and p53 loss facilitate tumor progression in activated B-cell diffuse large B-cell lymphomas. Blood. 2019;133(22):2401-2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nayak L, Iwamoto FM, LaCasce A, et al. PD-1 blockade with nivolumab in relapsed/refractory primary central nervous system and testicular lymphoma. Blood. 2017;129(23):3071-3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ambady P, Szidonya L, Firkins J, et al. Combination immunotherapy as a non-chemotherapy alternative for refractory or recurrent CNS lymphoma. Leuk Lymphoma. 2019;60(2):515-518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Balikov DA, Hu K, Liu CJ, et al. Comparative molecular analysis of primary central nervous system lymphomas and matched vitreoretinal lymphomas by vitreous liquid biopsy. Int J Mol Sci. 2021;22(18):9992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Button KS, Ioannidis JP, Mokrysz C, et al. Power failure: why small sample size undermines the reliability of neuroscience. Nat Rev Neurosci. 2013;14(5):365-376. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Requests for original data should be addressed to the corresponding author.