

Abstract
Antibody‐drug conjugates (ADCs), chemotherapeutic agents conjugated to an antibody to enhance their targeted delivery to tumors, represent a significant advancement in cancer therapy. ADCs combine the precise targeting capabilities of antibodies and the potent cell‐killing effects of chemotherapy, allowing for enhanced cytotoxicity to tumors while minimizing damage to healthy tissues. Here, we provide an overview of the current clinical landscape of ADCs, highlighting 11 U.S. Food and Drug Administration (FDA)‐approved products and discussing over 500 active clinical trials investigating newer ADCs. We also discuss some key challenges associated with the clinical translation of ADCs and highlight emerging strategies to overcome these hurdles. Our discussions will provide useful guidelines for the future development of safer and more effective ADCs for a broader range of indications.
Keywords: ADC, antibody, antibody–drug conjugate, cancer, cancer treatment, chemotherapy, clinic, clinical translation, clinical trial, drug delivery, FDA
Translational Impact Statement.
This review aims to provide an overview of the current clinical landscape of antibody‐drug conjugates (ADCs), an emerging modality for targeted cancer therapy. We discuss Food and Drug Administration‐approved ADC products and highlight the diversity of new investigative ADCs in active clinical trials based on their indication, antibody type, target antigen, and payload while also outlining the challenges in ADC development. Together, this review provides an understanding of the current state of ADCs in the clinic while fostering research initiatives to improve ADC development.
1. INTRODUCTION
Cancer has long been a global challenge, recognized as the second leading cause of death worldwide, accounting for one in six deaths. 1 , 2 Traditional treatment methods—surgery, radiotherapy, and chemotherapy—have been the cornerstone of cancer management for decades. However, their effectiveness is hampered by several factors such as the stage of cancer at diagnosis (limiting surgery's viability), the damage to healthy cells, organs, and tissues (a consequence of radiotherapy and chemotherapy), and the development of drug resistance (a challenge for chemotherapy). 1 , 2 Moreover, these treatments tend to focus on the cancer's location or histological features rather than on specific molecular changes. 3 Recent advances in molecular and tumor biology have shifted cancer treatment from these broad approaches to more personalized and precise therapies. 3 Inspired by Paul Ehrlich's “magic bullet” concept, new cancer treatment options aim to minimize toxicity by targeting specific molecular markers of cancer. Targeted therapies, which include monoclonal antibodies (mAbs) and small‐molecule inhibitors, have transformed the management of various cancers, such as those affecting the breast, colon, lungs, and digestive tract, enhancing the efficacy of traditional chemotherapy. 4 , 5 , 6
The foundation for antibody‐based therapies was laid in the 1960s with the identification of tumor antigen expression and the development of antibodies in the late 19th century. mAbs have proven effective in both diagnosing and treating hematological malignancies and solid tumors. 7 , 8 They work by targeting tumor‐associated antigens, either inhibiting cell growth and angiogenesis or stimulating a long‐lasting immune response against the tumor. 9 , 10 This led to the creation of Antibody‐Drug Conjugates (ADCs), which merge the targeted approach of mAbs with the cell‐killing power of chemotherapy, sparing healthy tissue and thus representing a significant advancement in cancer therapy. 11 , 12 , 13 Over the past few decades, clinical studies of ADCs have been increasingly active. To date, the US Food and Drug Administration (FDA) has approved 11 ADCs, with two additional approvals by other regulatory agencies. Numerous ADCs are under clinical investigation, promising to expand the range of treatable cancers. Ongoing trials are also exploring the most effective treatment combinations using approved ADCs. In this review, we provide an overview of the clinical landscape of ADCs. We discuss the design considerations and mechanism of actions of ADCs, highlight approved products, and review >500 active clinical trials involving both approved and new investigative ADCs. We also discuss the challenges for clinical translation of ADCs and provide a prospect for the future development of more effective and safer ADCs.
2. KEY COMPONENTS AND MECHANISM OF ACTIONS OF ADCs
2.1. Key components
An ADC is composed of an antibody conjugated to the cytotoxic payload by a chemically stable linker. While this sounds simple, the complexity of the ideal properties of each of these components has impacted the progress of ADC research. 14 Here, we discuss key considerations related to the design of each ADC component.
2.1.1. Antibody
mAbs, which have specificity to a particular antigenic epitope, are more commonly used to formulate ADCs. 15 , 16 The antibody can be considered the driver that facilitates the specific delivery of the payload to tumor cells. The generally recommended properties of the antibody component include: (i) high selectivity for cancer antigens over healthy cells, and (ii) high target binding affinity. 15 Other desirable properties include strong retention after binding, low immunogenicity, and minimal cross‐reactivity. Earlier generations of ADCs were formulated using murine mAbs, which were problematic due to immunogenicity that reduced efficacy. However, newer generations of ADCs employ humanized antibodies, which have a lower risk of immune activation. 14 , 15
A key aspect in designing the antibody component is the selection of antigenic targets. Ideally, the target should be exclusively expressed on tumor cells. 15 , 17 , 18 However, a more realistic goal is to identify a target that (i) has high expression on tumor cells and low expression on healthy cells, with a minimum target antigen threshold of >10,000 copies/cell, 18 , 19 (ii) is displayed on the surface of tumor cells with minimal shedding to enable efficient antibody binding, and (iii) has the ability to be internalized to aid the transport of ADC into the cell. 14 , 15 , 17 , 20 There are over 50 known antigens used in ADCs, and common antigens in approved ADC products include HER2, Trop2, B‐cell maturation antigen (BCMA), Nectin4, CD19, CD22, CD30, CD33, and CD79b. 15 More recently, research focus has also shifted to the identification of antigens beyond the tumor cells. Antigens expressed in the tumor microenvironment, such as on the stroma, vasculature, extracellular matrix, and tumor matrix, have the potential to broaden the target antigen scope of ADCs. Additionally, antigens expressed in these areas are less susceptible to mutations and could prevent the development of drug resistance. 17
The size of the antibody in an ADC is also important. 17 Immunoglobulin G (IgG) antibodies (IgG1, IgG2, IgG3, IgG4) are commonly used in ADCs. 15 IgG1 is the most commonly employed subtype due to its abundance in the serum and strong effector functions, while IgG3 is rarely used due to its short half‐life in the blood. 18 While IgG antibodies are the most common in the serum, their large size often limits penetration through the blood capillaries and tumor tissue. To overcome this, newer ADCs are formed with miniaturized antibodies by removing the fragment crystallizable (Fc) segment. This has made ADCs more applicable to solid tumors but also comes with the problem of reduced half‐life. 17
In the design of ADCs, a careful balance between the antibody's binding affinity and internalization is important. Often, higher binding affinity results in rapid internalization of the antibody. However, in the case for solid tumors, the rapid internalization of ADCs mostly occurs at the tumor periphery only. 17 , 19 This effect is because of the binding site barrier, which causes the trapping of ADCs near the blood vessels in solid tumors hindering their penetration to distant tumor cells. 17 , 21
Aside from enabling delivery of the payload, the antibody also plays some cytotoxic functions such as antibody‐dependent cytotoxicity (ADCC), antibody‐dependent cellular phagocytosis (ADCP), and complement‐dependent cytotoxicity (CDC), 14 which will be further discussed in a later section.
2.1.2. Linker
The linker is an important component that influences the stability, payload release, pharmacokinetics (PK), toxicity, and overall therapeutic efficacy of ADCs. 14 , 22 , 23 Most recent advances in ADCs are due to improvements in drug‐linker technologies. 22 An ideal ADC linker should be stable enough in circulation to prevent premature drug release while also being sufficiently sensitive to the release stimuli at the target site. 19 , 24 , 25 , 26
ADC Linkers are broadly classified as either cleavable or noncleavable. Cleavable linkers can be chemical cleavage linkers (such as hydrazone or disulfide bond based) or enzyme cleavage linkers (such as glucuronide or peptide bond based). 17 , 19 , 24 Upon internalization of ADCs into target cells, such linkers are degraded through several mechanisms such as proton lysis, thiol reduction, proteolysis, or carbohydrate hydrolysis, resulting in the release of the cytotoxic payload. 24 This cleavage occurs in the endosomal‐lysosomal compartment of the tumor cells. Because the stimuli responsible for the cleavage of these linkers are not exclusively restricted to tumor cells, some of these linkers are also susceptible to chemicals and enzymes in the blood or tumor microenvironment, increasing the risk of systemic toxicity. 19
Conversely, noncleavable linkers enable to release the payload by enzymatic degradation of the antibody in the endosome/lysosome. 27 The linker remains conjugated to the payload along with some amino acid residues, which restricts the diffusion of the payload across the cells, thus reducing systemic toxicity. 18 , 26 These linkers are less susceptible to the physiological environment, resulting in increased plasma stability 16 , 17 and specific drug release. 27 However, the persistence of the linker and amino acid residue could affect the function of the payload; hence, only small molecules that tolerate chemical modifications are suitable for these linkers. 17 , 18
Other considerations for the ADC linker include its length and hydrophobicity. Accumulating evidence indicates that shorter linkers improve the stability of ADCs as the payloads benefit from the steric shield provided by the antibody. 23 Simultaneously, hydrophilic linkers increase the solubility and improve the PK of ADCs and are more beneficial for ADCs with hydrophobic payloads. 19 , 22 , 25
2.1.3. Payload
ADCs are formulated with highly potent payloads that possess picomolar or nanomolar IC50 to ensure cytotoxic efficacy. 15 Earlier generations of ADCs utilized conventional chemotherapeutic drugs; however, due to the limited amount (1%–2%) of the payload reaching the target site, the efficacy of these moderately potent agents was suboptimal. 14 , 17 , 19 The new generation of approved ADCs deploys more potent payloads that inhibit microtubules necessary for cell division or inflict damage on cell DNA. The potency of these agents exceeds that of traditional chemotherapy by more than 100‐ to 1000‐fold. 18 , 21 , 28 Examples of such microtubule‐targeting agents include Dolastatin10‐based auristatin analogs and maytansinoids, and commonly used DNA‐damaging payloads include Calicheamin analogs (inducing DNA double‐strand breaks), Duocarmycin analogs (promoting DNA alkylation), and topoisomerase 1 inhibitors (causing DNA intercalation). 18 Having an intracellular target is an important requirement for these payloads, as they are designed to be released within the tumor cells. 28 A thorough review of ADC payloads can be found in recent reviews published elsewhere. 21 , 28 , 29
Beyond their high potency, ADC payloads should also exhibit several other key properties including stability in systemic circulation, resistance to degradation within endosomes/lysosomes, minimal immunogenicity, a relatively low molecular weight, and chemical groups amendable to conjugation with the linker. Additionally, an appropriate hydrophobicity of the payload is needed to balance solubility for successful conjugation to the ADC with good cellular permeability while preventing rapid clearance. 15 , 17 , 19 , 28
2.2. Mechanisms of action
Figure 1 provides an overview of the mechanism of actions of ADCs. After intravenous administration, ADCs are distributed throughout the body and accumulate in the tumors. The circulation of ADCs is facilitated by the long half‐life of the antibody component, while their accumulation within tumor is driven by the binding of the fragment antigen‐binding (Fab) segment of the antibody to the antigenic target. 30 The large size of mAbs limits the diffusion of ADCs through tumor vasculature, resulting in only a small fraction (0.0003%–0.08% per gram of tumor 31 ) of the administered dose eventually accumulating at the target site, underscoring the need for a highly potent payload. 32 The binding of the antibody to its target triggers the internalization of the ADC, which can occur via clathrin‐mediated endocytosis (CME), caveolar‐mediated endocytosis, or pinocytosis. 15 , 27 Subsequently, the ADC is packed into an early endosome, which matures and fuses with a lysosome where the payload is released upon endosomal/lysosomal degradation of the ADC. The type of linker determines the payload release mechanism postinternalization. 32 Noncleavable linkers require ADC localization in the lysosome for proteolytic degradation, whereas the payload release from cleavable linkers is triggered by intracellular stimuli (such as pH sensitivity, protease sensitivity, or glutathione sensitivity), bypassing the need for lysosomal trafficking. 27 , 33
FIGURE 1.
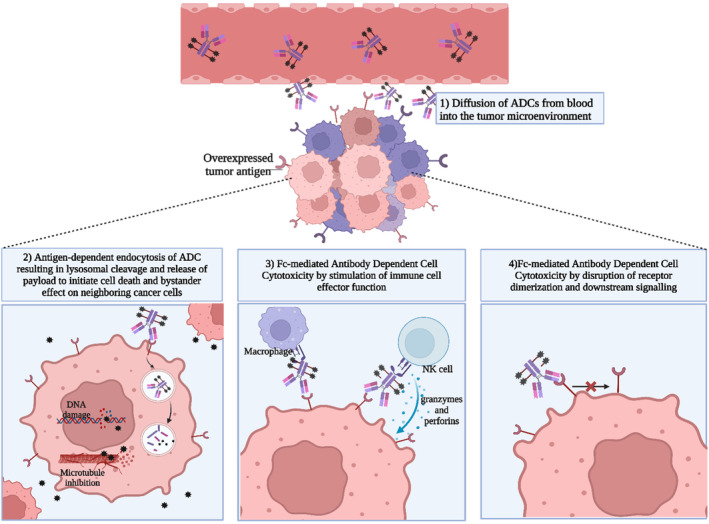
Schematic representation of the mechanisms of action of antibody‐drug conjugates (ADCs). Created with BioRender. Fc, fragment crystallizable.
Once the payload is released into the cytoplasm, it disrupts cellular functions through microtubule inhibition or DNA damage, leading to cancer cell death. This is the primary mechanism of action of ADCs. In addition to enhanced payload delivery to targeted cells, some ADCs can display bystander‐killing effect where the released payload permeates through the cell membrane, diffusing to and killing neighboring tumor cells. Moreover, CDC, ADCC, and ADCP are additional mechanisms to enhance ADC's effect. These mechanisms are medicated by the interaction of the Fc region of the antibody with the neonatal Fc receptors present on effector immune cells. 34 In ADCC, the Fc region's interaction with the FcƔ receptor activates immune cells like natural killer (NK) cells, which release cytotoxic molecules (e.g., perforins and granzymes). ADCP involves macrophages engulfing cancer cells following a similar interaction. 9 However, these Fc‐mediated actions can also reduce ADC efficacy by causing ADCs to be sequestered by immune cells, leading to off‐target toxicity. 35 However, these effector functions are mainly associated with IgG1, as IgG2 and IgG4 are less effective in medicating Fc‐dependent activities. 18 , 34 Furthermore, another mechanism of action of ADC involves its inhibition of downstream signaling pathways. This is particularly observed with antigenic targets upstream of the oncogenic pathway, where antibody binding prevents the dimerization of the receptors. 32 , 34
3. FDA‐APPROVED ADC PRODUCTS
Currently, there are 11 FDA‐approved ADC products and two more approved by other regulatory agencies (NMPA and PMDA). Another two ADCs (Moxetumomab pasudotox and Belantamab mafodotin‐blmf) were previously approved by the FDA but were withdrawn due to limited clinical use or failure to meet primary efficacy endpoints. All the approved products are administered intravenously and are intended to treat a specific type of cancer. Key information for each approved product is summarized in Table 1.
TABLE 1.
Information on ADC products approved by the FDA or other regulatory agencies.
| Name | Payload (payload class), payload action | Linker type | Target antigen | Monoclonal antibody isotype | Approved indication | Approval year | |
|---|---|---|---|---|---|---|---|
| Hematological tumors | Gemtuzumab ozogamicin/Mylotarg® (Pfizer/Wyeth) | Ozogamicin (Calicheamicin), DNA cleavage | Acid cleavable | CD33 | IgG4 | Relapsed acute myelogenous leukemia | FDA (2000, 2017) |
| Brentuximab vedotin/ADCETRIS® (Seattle Genetics) | MMAF (Auristatin), Microtubule inhibitor | Cleavable | CD30 | IgG1 | Relapsed and/or refractory systemic anaplastic large cell lymphoma (2011); Relapsed and/or refractory primary cutaneous anaplastic large cell lymphoma or CD30+ mycosis fungoides (2017); Classical Hodgkin lymphoma, systemic anaplastic large cell lymphoma, or CD30+ peripheral T‐cell lymphoma (2018) | FDA (2011, 2017, 2018); EMA (2012) | |
| Inotuzumab ozogamicin/Besponsa® (Pfizer/Wyeth) | Ozogamicin (Calicheamicin), DNA cleavage | Acid cleavable | CD22 | IgG4 | Relapsed or refractory B‐cell precursor acute lymphoblastic leukemia | FDA (2017) | |
| Moxetumomab pasudotox/Lumoxiti® (Astrazeneca) | Pseudomonas exotoxin (Pseudotox), Inhibitor of protein synthesis | Recombinant covalently fused (linkerless) | CD22 | IgG4 | Relapsed or refractory hairy cell leukemia | FDA (2018)‐ withdrawn | |
| Polatuzumab vedotin‐piiq/Polivy® (Genentech, Roche) | MMAE (Auristatin), Microtubule inhibitor | Enzyme cleavable | CD79b | IgG1 | Relapsed or refractory diffuse large B‐cell lymphoma | FDA (2019) | |
| Belantamab mafodotin‐blmf/Blenrep® (GlaxoSmithKline (GSK)) | MMAF (Auristatin), Microtubule inhibitor | Noncleavable | BCMA | IgG1 | Relapsed or refractory multiple myeloma | FDA (2020)‐withdrawn | |
| Loncastuximab tesirine‐lpyl/Zynlonta® (ADC Therapeutics) | SG3199 (PBD Dimer), DNA cleavage | Enzyme cleavable | CD19 | IgG1 | Relapsed or refractory diffuse large B‐cell lymphoma | FDA (2021) | |
| Solid tumors | Ado‐trastuzumab emtansine/Kadcyla® (Genentech, Roche) | DM1 (Maytansinoid), Microtubule inhibitor | Noncleavable | HER2 | IgG1 | HER2+ metastatic breast cancer previously treated with trastuzumab and a taxane (2013); HER2+ early breast cancer after neoadjuvant taxane & trastuzumab‐based treatment (2019) | FDA (2013, 2019) |
| Fam‐trastuzumab deruxtecan‐nxki/Enhertu® (AstraZeneca/Daiichi Sankyo) | DXd (Camptothecin), TOP1 inhibitor | Enzyme cleavable | HER2 | IgG1 | Unresectable or metastatic HER2+ breast cancer after 2 or more anti‐HER2 regimens (2019) d; locally advanced or metastatic HER2+ gastric or gastroesophageal junction adenocarcinoma after a trastuzumab‐based regimen (2021) | FDA (2019, 2021) | |
| Enfortumab vedotin‐ejfv/Padcev® (Astellas/Seattle Genetics) | MMAE (Auristatin), Microtubule inhibitor | Enzyme cleavable | Nectin4 | IgG1 | Locally advanced or metastatic urothelial cancer after a PD‐1 or PD‐L1 inhibitor and a Pt‐containing chemotherapy (2019) or are ineligible for cisplatin‐containing chemotherapy and previously received 1 or more lines of therapy (2021) | FDA (2019, 2021,2023) | |
| Sacituzumab govitecan‐hziy/Trodelvy® (Immunomedics) | SN‐38 | Acid cleavable | TROP2 | IgG1 | Locally advanced or metastatic triple‐negative breast cancer; locally advanced or metastatic urothelial cancer | FDA (2020, 2021) | |
| Tisotumab vedotin‐tftv/Tivdak® (Seagen Inc) | MMAE (Auristatin), Microtubule inhibitor | Enzyme cleavable | Tissue Factor (TF) | IgG1 | Recurrent or metastatic cervical cancer | FDA (2021) | |
| Mirvetuximab soravtansine/ELAHERE™ (ImmunoGen) | DM4 (Maytansinoid), Microtubule inhibitor | Cleavable | FRα | Undisclosed | Ovarian cancer, fallopian tube cancer, primary peritoneal cancer | FDA (2022) | |
| Cetuximab sarotalocan/Akalux® (Rakuten Medical) | IRDye700DX, photoimmunotherapy | None | EGFR | IgG1 | Unresectable locally advanced or recurrent HNSCC | PMDA (2020) | |
| Disitamab vedotin/Aidixi® (RemeGen) | MMAE (Auristatin), Microtubule inhibitor | Enzyme cleavable | HER2 | IgG1 | Locally advanced or metastatic gastric cancer | NMPA (2021) |
Abbreviations: ADC, antibody‐drug conjugates; FDA, Food and Drug Administration; IgG, Immunoglobulin G; MMAE, monomethyl auristatin E; MMAF, monomethyl auristatin F.
3.1. Approved ADCs for hematological cancers
To date, seven ADCs have been approved by the FDA for treating hematological cancers, with two of them having been withdrawn due to limited clinical efficacy or use. Mylotarg® (Gemtuzumab Ozogamicin, GO) is the first ADC approved for the treatment of acute myelogenous leukemia (AML), characterized by poor bone hematopoiesis. 36 GO was initially approved in 2000 for AML but was then voluntarily withdrawn from the market because of failing to demonstrate clinical benefits and excessive fatal toxicities. Before the approval of GO, the standard of care for AML was a 7 + 3 regimen, involving a 7‐day treatment with cytarabine followed by a 3‐day treatment with daunorubicin. 36 , 37 GO was reapproved in 2017 with new data showing safety and efficacy after dose adjustment for CD33+ AML in patients of 2 years and older. 38 , 39 It consists of a humanized anti‐CD33 IgG4 mAb linked to DNA‐damaging calicheamicin via a covalent linker. The linker is acid‐cleavable and enables the release of the payload in the endosome/lysosome of myeloblasts. The treatment is not considered intensive and hence is suitable for elderly patients and patients with comorbidities. 37 In the Phase 3 trial of 280 patients in France (ALFA‐0701 Trial), GO demonstrated superior event‐free survival for patients with newly diagnosed AML, 39 and a meta‐analysis of 3325 adult patients also showed an overall improvement in survival. 38 However, Mylotarg® comes with a blackbox warning for hepatotoxicity, and other warnings include the risk of severe hemorrhage and infusion‐related reactions.
ADCETRIS® (Brentuximab vedotin) is the standard of care for treating patients with refractory or relapsed classical Hodgkin Lymphoma (cHL). It was most recently approved by the FDA in 2018 and consists of a human chimeric anti‐CD30 IgG1 antibody covalently linked with monomethyl auristatin E (MMAE) via a valine‐citrulline cleavable linker. 40 Upon binding to and internalization by CD30+ cancer cells, the linker undergoes cleavage by endosomal/lysosomal proteases to release MMAE, which induces cell death via apoptosis. 41 Brentuximab vedotin has also been reported to be active in diseases with low CD30 expression, due to its bystander effect where free MMAE diffuses to and kills adjacent cancer cells. 41 , 42 Before its approval, the frontline treatment of cHL involved a combination of chemotherapy agents: doxorubicin, bleomycin, vinblastine, and dacarbazine, but this treatment regimen is associated with relapse in up to 40% of patients. The Phase 3 ECHELON‐1 trial revealed that replacing bleomycin with brentuximab vedotin in this treatment regimen led to superior progression‐free survival in patients with stage III and IV cHL, 43 and long‐term follow‐up showed that this benefit is sustained. 41 , 44 However, ADCETRIS® comes with a blackbox warning for progressive multifocal leukoencephalopathy. 45
Besponsa® (Inotuzumab ozogamicin) is the only ADC approved for the treatment of relapsed or refractory B‐cell precursor acute lymphoblastic leukemia (R/R ALL). It is a humanized anti‐CD22 IgG4 antibody covalently conjugated to calicheamicin via a butanoic acid liable linker. 46 , 47 In this case, the cytotoxicity is mediated only by the payload upon release, and its effectiveness is thus dependent on effective internalization and sensitivity to calicheamicin. 46 Inotuzumab ozogamicin has been reported to improve clinical outcomes compared with salvage chemotherapy. 48 The INO‐VATE Phase 3 trial revealed that Inotuzumab ozogamicin led to a higher response rate than standard‐of‐care chemotherapy. 46 , 49 The overall response rate of Inotuzumab ozogamicin is 60%–80% in patients with R/R ALL. 47
Polivy® (Polatuzumab vedotin‐piiq) received accelerated FDA approval in 2019 for the treatment of R/R diffuse large B‐cell lymphoma (DLBCL) in combination with bendamustine and rituximab (BR). 50 The Phase 2 trials of Polatuzumab vedotin‐piiq demonstrated a higher complete response (CR) rate and reduced the risk of death in patients with transplantation‐ineligible R/R DLBCL by 58% in patients treated with a combination of Polatuzumab vedotin with BR compared with BR alone. 51 Polatuzumab vedotin consists of a humanized anti‐CD79b IgG1 mAb linked to MMAE via a protease cleavable linker. This formulation uses an engineered cysteine (THIOMABs) to achieve the efficient and homogenous conjugation of antibody with MMAE. 52
Zynlonta® (Loncastuximab tesirine, SG3199), consisting of a humanized anti‐CD19 IgG1 antibody conjugated to a pyrrolobenzodiazepine (PDB) dimer cytotoxin, is another approved ADC for the treatment of DLBCL. Upon endosomal/lysosomal cleavage, SG3199 forms inter‐strand crosslinks within the cell's DNA leading to cell death. SG3199 also exhibits a bystander‐killing effect. 53 , 54 In July 2023, further clinical trials on Zynlonta® were terminated due to FDA's hold on this ADC, stemming from concerns over excessive fatal toxicities.
LUMOXITI® (moxetumomab pasudotox‐tdfk) was initially approved in 2018 for the treatment of adult patients with R/R hairy cell leukemia. This ADC comprises the Fv fragment of a CD22‐targeting antibody conjugated to an immunotoxin. Once released, the immunotoxin induces apoptosis through the catalysis of ADP‐ribosylation of the diphthamide residue in elongation factor‐2. LUMOXITI's approval was based on a Phase 3 study that showed up to 90% of circulating CD19+ B cells were depleted by Day 8 of treatment. 55 Although this study reported a generally acceptable tolerability profile, LUMOXITI was withdrawn from the market in 2023 due to inadequate clinical use.
Blenrep® (Belantamab mafodotin‐blmf) was approved in 2020 for the treatment of R/R multiple myeloma in adult patients. 56 Belantamab mafodotin‐blmf was a first‐in‐class ADC with an anti‐BCMA antibody and the first ADC with the microtubule inhibitor, monomethyl auristatin F (MMAF) payload to receive approval. Its approval was based on the DREAMM‐2 global trial, which demonstrated an overall response rate of 31%. However, 77% of patients receiving the treatment of Belantamab mafodotin‐blmf experienced ocular toxicity, leading to a black‐box label by the FDA on this product. 57 In 2020, GSK announced the withdrawal of Blenrep® from the US market as it failed to meet the primary endpoint in the DREAMM‐3 confirmatory clinical trial. 58
3.2. Approved ADCs for solid tumors
To date, six ADCs have been approved by the FDA and two more by other regulatory agencies for treating solid tumors. Kadcyla® (Ado‐trastuzumab emtansine) is the first ADC approved by the FDA for the treatment of HER2‐positive metastatic breast cancer. 59 Ado‐trastuzumab emtansine (T‐DM1) consists of a humanized anti‐HER2 IgG1 antibody known as trastuzumab, which was introduced in 1998 for the treatment of HER2+ breast cancers. Up to 25% of breast cancer patients exhibit HER2 overexpression, which is associated with poor prognosis. 60 , 61 However, a significant portion of patients under trastuzumab treatment did not respond or experienced relapse. Ado‐trastuzumab emtansine is a combination of trastuzumab and the microtubule‐inhibiting maytansinoid, linked via a nonreducible thioether linker. Endosomal/lysosomal degradation of the antibody leads to the release of maytansinoid causing apoptosis. There is also additional antibody‐mediated cytotoxicity due to the downregulation of HER2, inhibition of HER2 dimerization, activation of immune response, and ADCC. 61 , 62 Various clinical studies have shown improvement in overall survival and quality of life in patients treated with Ado‐trastuzumab emtansine. 59 , 63 Despite the improvement in outcome noted with T‐DM1, there are concerns about the development of drug resistance observed in initial responders. 62 Kadcyla® also comes with FDA‐boxed warnings for hepatotoxicity, cardiotoxicity, pulmonary toxicity, and embryo‐fetal toxicity. 64
Padcev® (Enfortumab vedotin‐ejfv) is a first‐in‐class ADC for the treatment of metastatic urothelial carcinoma (UC), an aggressive cancer with a poor prognosis. Enfortumab vedotin‐ejfv (EV) consists of a fully humanized anti‐Nectin‐4 IgG1 antibody linked to MMAE. 65 Enfortumab vedotin‐ejfv received accelerated FDA approval in 2019 based on the Phase 1 and 2 trials indicating that EV had a high response rate, disease control rate, and improved overall survival in UC patients. The Phase 2 (EV‐201) trial, in which 90% of enrolled patients had metastatic visceral disease, showed that EV led to an overall response rate of 44% and complete remission rate of 12%. A Phase 3 (EV‐301) trial with 608 patients demonstrated the superior efficacy of EV compared with single‐agent chemotherapy. 66 , 67 This facilitated the approval of EV in 2021 by the FDA for the treatment of UC in two adult populations, including patients who had previously received a PD‐L1 inhibitor and platinum‐based chemotherapy and patients who are ineligible for cisplatin‐based chemotherapy. 67 Padcev® comes with an FDA‐boxed warning for serious skin reactions.
Enhertu® (Fam‐trastuzumab deruxtecan‐nxki, T‐DXd) is the first ADC approved for HER2‐low breast cancer, accounting for about 40%–50% of HER2‐negative breast cancers. T‐DXd is composed of a humanized anti‐HER2 IgG1 linked to a topoisomerase‐1 inhibiting exatecan derivative (DXd) via a stable tetrapeptide linker. Like trastuzumab emtansine, the payload is released within the cancer cell. However, with a drug‐to‐antibody ratio (DAR) of 8, T‐DXd led to an efficient delivery of DXd even to tumors with low HER2‐expression. 68 , 69 T‐DXd was granted accelerated approval in 2019 for the treatment of patients with unresectable or metastatic breast cancer. This expansion of indication to cover all HER2‐expressing tumors allows for flexibility in the use of the medication. Its initial approval was based on a Phase 2 trial indicating an overall response rate of 60.3%. 68 Following a Phase 3 trial indicating meaningful improvement in progression‐free survival and overall survival, T‐DXd received regular approval from the FDA in 2022. 69 , 70 Based on the DESTINY‐Gastric01, TXd was also approved in 2021 for the treatment of locally advanced or metastatic HER2‐positive gastric or gastroesophageal junction adenocarcinoma in patients who had received a prior trastuzumab‐based regimen. Again in 2022, following results from the DESTINY‐Lung02 trial, T‐DXd received FDA approval for the treatment of unresectable or metastatic nonsmall cell lung cancer in adult patients with HER2‐overexpression or HER2 mutations. 71 Enhertu® comes with an FDA‐boxed warning related to the risk of interstitial lung disease, pneumonitis, and embryo‐fetal toxicity. 64
Trodelvy® (Sacituzumab govitecan) is the only approved ADC targeting TROP2 as its antigen. It received its first approval in 2020 for treating metastatic triple‐negative breast cancer in adult patients who had undergone at least two prior therapies for metastatic disease. 72 In 2021, it also gained approval for treating metastatic urothelial cancer. Sacituzumab govitecan comprises a humanized anti‐TROP2 IgG1 antibody linked by a hydrolyzable hydrazone linker to SN‐38, a topoisomerase‐1 inhibitor and the active metabolite of irinotecan. Besides its DNA‐damaging effect within the internalized cell, SN‐38 demonstrates a bystander effect due to its high membrane permeability. 53 , 72 In 2023, Sacituzumab govitecan received extended FDA approval for treating patients with hormone‐positive and HER‐2/NEU‐negative metastatic breast cancer with a boxed warning for neutropenia and diarrhea. 53 , 73
Tivdak® (Tisotumab vedotin‐tftv [TV]), is a first‐in‐class tissue factor (TF)‐directed ADC approved in 2021 for treating recurrent or metastatic cervical cancer in adult patients. About 10%–20% of patients with early‐stage disease and 70% patients with locally advanced disease experience relapse within 2 years of diagnosis. Only a small fraction of these patients are responsive to curative treatment, necessitating the need for more targeted treatment alternatives. 74 TV, a humanized IgG1 antibody conjugated to MMAE via a protease‐cleavable linker, 74 demonstrated clinically meaningful and durable antitumor activity in a Phase 2 clinical study, with target lesions reduced in 79% of treated patients. 75 , 76 With more than 50% of patients in the innovaTV 201 and innova TV 204 trials developing ocular related adverse effects, Tivdak® comes with an FDA blackbox warning for ocular toxicity. 64
ELAHERE™ (mirvetuximab soravtansine‐gynx) is another first‐in‐class ADC approved for treating adult patients with a folate receptor‐α (FRα)‐positive, platinum‐resistant epithelial ovarian, fallopian tube, or primary peritoneal cancer. 77 , 78 Approved for patients not responding to platinum‐based chemotherapy who have undergone other types of chemotherapy, it features a chimeric anti‐FRα IgG1 antibody conjugated to a maytansine derivative (DM4) via a cleavable disulfide linker. 78 Upon endosomal/lysosomal cleavage, DM4 causes cell cycle arrest and apoptosis. DM4 also exhibits a bystander‐killing effect. 53 ELAHERE™ also comes with an FDA‐boxed warning for ocular toxicity.
4. ADCs IN ACTIVE CLINICAL TRIALS
Since the approval of the first ADC in 2000, continued efforts have focused on designing new ADCs with improved efficacy and reduced toxicity. These efforts are evident by the number of ADCs currently in active trials, which represent only a small portion of all ADC research. We conducted a search on clinicaltrials.gov to identify active clinical trials for both approved and newer investigative ADCs. For trials related to approved ADCs, we conducted the search by inputting the drug name in the “Other terms” category for each approved ADC, while checking off “interventional studies,” in active status (“not yet recruiting,” “recruiting,” “enrolling by invitation,” and “active, not recruiting”). For trials related to new ADCs, we conducted the search by using the keywords “antibody drug conjugate OR antibody‐drug conjugate OR ADC OR ADCs OR antibody drug conjugates OR antibody‐drug conjugates” in the “Other terms” category on clinicaltrials.gov, and also checked off “interventional studies,” in active status (“not yet recruiting,” “recruiting,” “enrolling by invitation,” and “active, not recruiting”). All the collected trials were then manually screened to only include trials focusing on IgG‐based ADCs bearing pan‐cytotoxic payloads. Our search identified a total of 551 active clinical trials as of October 2023. Here, we discuss these active trials and highlight new trends emerging from the investigative ADCs in active trials compared with approved products. Tables 2 and 3 present representative active trails for approved ADCs and new investigative ADCs.
TABLE 2.
Representative active clinical trials for approved ADCs.
| ADC name | mAB | Antigen | Payload | Payload class | Approved indication | No. of active trials | Examples of active trials | ||
|---|---|---|---|---|---|---|---|---|---|
| Indication | ID | ||||||||
| Hematological tumors | Brentuximab vedotin | IgG1 | CD30 | MMAF | Auristatin | Relapsed and/or refractory (R/R) systemic anaplastic large cell lymphoma (2011); R/R primary cutaneous anaplastic large cell lymphoma or CD30+ mycosis fungoides (2017); Classical Hodgkin lymphoma, systemic anaplastic large cell lymphoma, or CD30+ peripheral T‐cell lymphoma (2018) | 79 | Hodgkin lymphoma | NCT02166463 |
| T‐cell lymphoma | NCT05442554 | ||||||||
| Mycosis fungoides, Sezary Syndrome, Lymphomatoid Papulosis | NCT03587844 | ||||||||
| Gemtuzumab ozogamicin | IgG4 | CD33 | Ozogamicin | Calicheamicin | Relapsed acute myelogenous leukemia (2017) | 30 | R/R CD33+ acute myeloid leukemia | NCT04070768 | |
| Acute myelogenous leukemia and myelodysplastic syndrome | NCT02221310 | ||||||||
| Acute myeloid leukemia | NCT03568994 | ||||||||
| Belantamab mafodotin‐blmf | IgG1 | BCMA | MMAF | Auristatin | R/R multiple myeloma after at least 4 prior therapies including an anti‐CD38 mAb, a proteasome inhibitor, and an immunomodulatory agent (2020) | 14 | Recurrent plasma cell myeloma, refractory plasma cell myeloma | NCT05847569 | |
| Multiple myeloma | NCT05064358 | ||||||||
| Transplant ineligible newly diagnosed multiple myeloma | NCT05573802 | ||||||||
| Loncastuximab tesirine‐lpyl | IgG1 | CD19 | SG3199 | PBD dimer | R/R large B‐cell lymphoma after 2 or more lines of systemic therapy, including diffuse large B‐cell lymphoma (DLBCL) not otherwise specified, DLBCL arising from low‐grade lymphoma, and high‐grade B‐cell lymphoma (2021) | 10 | Waldenstrom macroglobulinemia | NCT05190705 | |
| R/R B‐cell non‐Hodgkin lymphoma | NCT04970901 | ||||||||
| B‐cell lymphoid malignancies | NCT05270057 | ||||||||
| Moxetumomab pasudotox | IgG4 | CD22 | Pseudomonas exotoxin | Pseudotox | Adults with R/R hairy cell leukemia (2018) | 1 | Hairy cell leukemia | NCT03805932 | |
| Polatuzumab vedotin‐piiq | IgG1 | CD79b | MMAE | Auristatin | R/R DLBCL (2019) | 10 | Richter's transformation | NCT04679012 | |
| R/R mantle cell lymphoma | NCT04659044 | ||||||||
| High‐risk diffuse large b‐cell lymphoma | NCT04323956 | ||||||||
| Inotuzumab ozogamicin | IgG4 | CD22 | Ozogamicin | Calicheamicin | R/R CD22‐positive B‐cell precursor acute lymphoblastic leukemia (2017) | 26 | Acute lymphocytic leukemia | NCT05456698 | |
| B‐cell acute lymphoblastic leukemia | NCT05016947 | ||||||||
| Precursor cell lymphoblastic leukemia | NCT03460522 | ||||||||
| Solid tumors | Ado‐trastuzumab emtansine | IgG1 | HER2 | DM1 | Maytansinoid | HER2+ metastatic breast cancer previously treated with trastuzumab and a taxane (2013); HER2+ early breast cancer after neoadjuvant taxane & trastuzumab‐based treatment (2019) | 75 | Brain metastases | NCT05323955 |
| HER2‐positive breast cancer, ER‐negative breast cancer, PR‐negative breast cancer, Node‐negative breast cancer | NCT04675827 | ||||||||
| HER2‐positive Salivary Gland Carcinomas | NCT04620187 | ||||||||
| Disitamab vedotin | IgG1 | MMAE | Auristatin | Patients with locally advanced or metastatic gastric cancer (including gastroesophageal junction adenocarcinoma) who have received at least 2 types of systemic chemotherapy (2021) | 23 | Her2 overexpressing high‐risk nonmuscle invasive bladder urothelial carcinoma | NCT05495724 | ||
| Colorectal Neoplasms | NCT05493683 | ||||||||
| Nonsmall cell lung cancer, ERBB2 mutation‐related tumors | NCT05847764 | ||||||||
| Fam‐trastuzumab deruxtecan‐nxki | IgG1 | DXd | Camptothecin | Unresectable or metastatic HER2+ breast cancer after 2 or more anti‐HER2 regimens (2019) d; locally advanced or metastatic HER2+ gastric or gastroesophageal junction adenocarcinoma after a trastuzumab‐based regimen (2021) | 19 | Advanced solid tumor | NCT05097599 | ||
| HER2+ breast cancer with brain metastasis | NCT05376878 | ||||||||
| Locally advanced breast cancer, metastatic breast cancer | NCT05744375 | ||||||||
| Tisotumab vedotin‐tftv | IgG1 | Tissue factor | MMAE | Auristatin | Recurrent or metastatic cervical cancer with disease progression on or after chemotherapy (2021) | 3 | Colorectal neoplasms, carcinoma, nonsmall‐cell lung, exocrine pancreatic cancer, carcinoma squamous cell of head and neck | NCT03485209 | |
| Cervical cancer | NCT03786081 | ||||||||
| Cervical cancer | NCT04697628 | ||||||||
| Sacituzumab govitecan‐hziy | IgG1 | TROP2 | SN‐38 | Camptothecin | Locally advanced or metastatic TNBC after at least two prior therapies (2020); locally advanced or metastatic urothelial cancer after a Pt‐containing chemotherapy and a PD‐1 or PD‐L1 inhibitor (2021) | 35 | Nonsmall cell lung cancer | NCT05089734 | |
| Metastatic solid tumor | NCT04319198 | ||||||||
| Cervical cancer | NCT05838521 | ||||||||
| Mirvetuximab soravtansine | Undisclosed | FRα | DM4 | Maytansinoid | Ovarian cancer (2022) | 13 | Endometrial cancer | NCT03835819 | |
| Epithelial ovarian cancer, peritoneal cancer, fallopian tube cancer | NCT05622890 | ||||||||
| Platinum‐resistant ovarian cancers | NCT05483933 | ||||||||
| Enfortumab vedotin‐ejfv | IgG1 | Nectin‐4 | MMAE | Auristatin | Locally advanced or metastatic urothelial cancer after a PD‐1 or PD‐L1 inhibitor and a Pt‐containing chemotherapy (2019) or are ineligible for cisplatin‐containing chemotherapy and previously received 1 or more lines of therapy (2021) | 15 | Metastatic urothelial carcinoma | NCT04963153 | |
| Muscle invasive bladder cancer | NCT04960709 | ||||||||
| Nonmuscle invasive bladder cancer | NCT05014139 | ||||||||
Abbreviations: ADCs, antibody‐drug conjugates; IgG, Immunoglobulin G; mAB, Monoclonal antibody; MMAE, monomethyl auristatin E; MMAE, monomethyl auristatin E; MMAF, monomethyl auristatin F; TNBC, triple‐negative breast cancer.
TABLE 3.
Representative active clinical trials for new investigative ADCs.
| ADC name | mAB | Antigen | Payload | Payload class | Linker type | No. of trials | Examples of trials | ||
|---|---|---|---|---|---|---|---|---|---|
| Indication | ID | ||||||||
| Hematological tumor | IMGN632 | IgG1 | CD123 | IGN | Monoimine | Cleavable peptide | 2 | Leukemia | NCT03386513 |
| ADCT‐301 | CD25 | SG3199 | Tesirine | Cleavable | 2 | Relapsed hodgkin lymphoma; refractory hodgkin lymphoma | NCT04052997 | ||
| SAR3419 | CD19 | DM4 | Maytansinoid | Cleavable | 1 | Acute lymphocytic leukemia | NCT01440179 | ||
| BN301 | CD74 | Maytansoid | Maytansinoid | Noncleavable | 1 | Lymphoma | NCT05611853 | ||
| STRO‐001 | CD74 | Maytansinoid | Maytansinoid | Noncleavable | 1 | Lymphoma | NCT03424603 | ||
| INA03 | IgG4 | CD71 | MMAE | auristatin | Undisclosed | 1 | Leukemia | NCT03957915 | |
| ADCT‐602 | IgG2 | CD22 | SG3249 | PBD | Cleavable | 1 | Leukemia | NCT03698552 | |
| STI‐6129 | Unspecified | CD38 | Duostatin | Duostatin | Noncleavable (C‐lock chemical linker) | 4 | Multiple myeloma | NCT05308225 | |
| F0002‐ADC | CD30 | DM1 | Maytansinoid | Noncleavable (Stable SMCC linker) | 1 | Refractory or recurrent CD30+ hematologic malignancies | NCT03894150 | ||
| CC‐99712 | BCMA | Undisclosed | Undisclosed | Noncleavable | 1 | Multiple myeloma | NCT04036461 | ||
| JBH492 | CCR7 | DM4 | Maytansinoid | Cleavable | 1 | Non‐Hodgkins lymphoma; chronic lymphocytic leukemia | NCT04240704 | ||
| IKS03 | CD19 | Femtogenix's sequence‐selective DNA‐interactive payload molecule | Pyrrolobenzodiazepine | Cleavable | 1 | Lymphoma | NCT05365659 | ||
| CS5001 | ROR1 | Undisclosed | PBD | Cleavable | 1 | Advanced lymphoma | NCT05279300 | ||
| MRG001 | CD20 | MMAE | Auristatin | Cleavable (Valine‐citrulline) | 1 | Relapsed or refractory B‐cell non‐Hodgkin lymphoma | NCT05155839 | ||
| Solid tumors | BYON3521 | IgG1 | c‐MET | Duocarmycin | Duocarmycin | Cleavable | 1 | Solid tumor | NCT05323045 |
| STRO‐002 | Folate receptor alpha (FolRα) | Dibenzocyclooctyne | 3‐aminophenyl‐hemiasterlin | Cleavable | 2 | Ovarian cancer|; fallopian tube cancer; primary peritoneal carcinoma | NCT05200364 | ||
| U3‐1402 | HER3 | Deruxtecan | TOP1i | Cleavable | 5 | Breast cancer | NCT04610528 | ||
| XMT‐1536 | NaPi2b | AF‐HPA | Auristatin | Cleavable | 3 | High grade serous ovarian cancer; fallopian tube cancer; primary peritoneal cancer | NCT05329545 | ||
| ARX788 | HER2 | AS269 | Amberstatin/Auristatin | Noncleavable | 5 | Breast neoplasms | NCT04983121 | ||
| MORAb‐202 | FolRα | Eribulin | Halichondrin | Cleavable | 3 | Solid tumor | NCT04300556 | ||
| SYD1875 | 5T4 | Duocarmycin | Duocarmycin | Cleavable | 1 | Solid tumor | NCT04202705 | ||
| DS‐8201a | HER2 | Deruxtecan | TOP1i | Cleavable | 4 | Breast tumors | NCT04132960 | ||
| CAB‐AXL‐ADC | AXL | MMAE | Auristatin | Cleavable | 2 | Nonsmall‐cell lung cancer (NSCLC) | NCT04681131 | ||
| DS‐7300A | B7‐H3 | Deruxtecan | TOP1i | Cleavable | 1 | Extensive‐stage small‐cell lung cancer | NCT05280470 | ||
| IMGC936 | ADAM9 | DM21 | Maytansinoid | Cleavable | 1 | Advanced solid tumor | NCT04622774 | ||
| OBT076 | Ly75/CD205 | DM4 | Maytansinoid | Cleavable | 1 | Solid tumor | NCT04064359 | ||
| CAB‐ROR2‐ADC | ROR2 | MMAE | Auristatin | Cleavable | 1 | NSCLC; triple negative breast cancer; melanoma; head and neck cancer | NCT03504488 | ||
| NBE‐002 | ROR1 | PNU | TOP1i | Noncleavable | 1 | Advanced solid tumor; triple negative breast cancer | NCT04441099 | ||
| DS‐1062a | TROP2 | Exatecan | TOP1i | Cleavable | 1 | Metastatic lung cancer | NCT04940325 | ||
| GQ1001 | HER2 | DM1 | Maytansinoid | Undisclosed | 2 | HER2‐positive breast cancer; HER2‐positive gastric cancer; advanced solid tumor | NCT04450732 | ||
| SYD985 | HER2 | Duocarmycin | Duocarmycin | Cleavable | 5 | Endometrial cancer | NCT04205630 | ||
| HS‐20093 | B7‐H3 | TOP1i | TOP1i | Cleavable | 2 | Advanced solid tumor | NCT05276609 | ||
| OBI‐999 | anti‐globo H | MMAE | Auristatin | Cleavable | 1 | Locally advanced solid tumor | NCT04084366 | ||
| XMT‐1592 | NaPi2b | AF‐HPA | Auristatin | Undisclosed | 1 | Ovarian cancer; NSCLC | NCT04396340 | ||
| ABBV‐399 | cMET | MMAE | Auristatin | Cleavable | 1 | Advanced solid tumors cancer | NCT02099058 | ||
| ARX517 | PSMA | AS269 | Amberstatin | Noncleavable | 1 | Advanced solid tumor; solid neoplasm | NCT04662580 | ||
| Dato‐DXd | TROP2 | Deruxtecan | TOP1i | Cleavable | 6 | Carcinoma, nonsmall‐cell lung; triple negative breast cancer | NCT05460273 | ||
| BB‐1705 | EGFR | Eribulin | Halichondrin | Cleavable | 1 | Solid tumor | NCT05217693 | ||
| MRG002 | HER2 | MMAE | Auristatin | Cleavable | 9 | Breast cancer with liver metastases | NCT05263869 | ||
| ADC‐1013 | CD40 | Undisclosed | Undisclosed | Undisclosed | 1 | Metastatic pancreatic ductal adenocarcinoma | NCT04888312 | ||
| ADCT‐301 | CD25 | SG3199 | Tesirine | Cleavable | 1 | Advanced solid tumors; head and neck cancer squamous cell carcinoma; NSCLC; gastrointestinal cancers; bladder cancer; renal cell carcinoma; melanoma; triple‐negative breast cancer; ovarian cancer; fallopian tube cancer | NCT03621982 | ||
| MRG003 | EGFR | MMAE | Auristatin | Cleavable (Valine‐citrulline) | 7 | Advanced or metastatic gastric cancer; advanced or metastatic gastroesophageal junction carcinoma | NCT05188209 | ||
| BA3021 | Ror2 | MMAE | Auristatin | Cleavable | 1 | Head and neck cancer | NCT05271604 | ||
| SGN‐LIV1A | LIV‐1 | MMAE | Auristatin | Protease‐Cleavable | 1 | HER2 positive breast neoplasms; hormone receptor‐positive breast neoplasms; triple negative breast neoplasms | NCT01969643 | ||
| MYTX‐011 | c‐MET | MMAE | Auristatin | Cleavable | 1 | NSCLC | NCT05652868 | ||
| PYX‐201 | Fibronectin extra‐domain B (ED‐B) | Aur0101 | Auristatin | Cleavable | 1 | Solid tumor; advanced solid tumor | NCT05720117 | ||
| HuMax‐AXL‐ADC (Enapotamab vedotin) | AXL | MMAE | Auristatin | Protease Cleavable | 1 | Ovarian cancer; cervical cancer; endometrial cancer; NSCLC; thyroid cancer; melanoma; sarcoma; solid tumors | NCT02988817 | ||
| SHR‐A1811 | EGFR | SHR9265 | TOP1i | Cleavable | 5 | Breast cancer | NCT05824325 | ||
| SGN‐CD228A | CD228 | MMAE | Auristatin | Cleavable (Novel glucuronide linker) | 1 | Cutaneous melanoma; pleural mesothelioma; HER2 negative breast neoplasms; NSCLC; colorectal cancer; pancreatic ductal adenocarcinoma | NCT04042480 | ||
| ABT‐414 | EGFR | MMAF | Auristatin | Noncleavable | 1 | Glioblastoma; gliosarcoma | NCT02573324 | ||
| ADCT‐901 | KAAG1 | SG3199 | Tesirine | Cleavable | 1 | Advanced solid tumors | NCT04972981 | ||
| BAY 94–9343 | MF‐T | DM4 | Maytansinoid | Cleavable | 1 | NSCLC | NCT03455556 | ||
| SOT102 | Claudin‐18.2 | PNU‐159682 | TOPi | Noncleavable | 1 | Gastric cancer; pancreatic cancer; gastro‐esophageal junction cancer | NCT05525286 | ||
| BMS‐986148 | Mesothelin | Tubulysin | Tubulysin | Cleavable (Valine‐citrulline) | 1 | Advanced cancer | NCT02341625 | ||
| RC48‐ADC | IgG4 | HER2 | MMAE | Auristatin | Cleavable | 21 | Upper urinary tract urothelial carcinoma | NCT05912816 | |
| REGN5093‐M114 | MET | M114 | Maytansinoid | Protease Cleavable | 1 | Advanced NSCLC | NCT04982224 | ||
| ASN004 | 5T4 | Dolaflexin | Auristatin | Noncleavable (Dolaflexin‐drug linker) | 1 | Breast cancers; |NSCLC; colorectal cancer; ovarian cancer | NCT04410224 | ||
| BGB‐A317 | PD‐1 | Undisclosed | Undisclosed | Undisclosed | 1 | HER2‐positive or mutated advanced colorectal cancer | NCT05350917 | ||
| TR1801‐ADC | IgG2 | c‐MET | SG3249 | PBD | Cleavable | 1 | Unspecified adult solid tumor, protocol‐specific | NCT03859752 | |
| SHR‐A1403 | c‐MET | Undisclosed | Undisclosed | Noncleavable | 1 | Advanced solid tumor | NCT03856541 | ||
| FDA022 | Unspecified | HER2 | Undisclosed | Undisclosed | Undisclosed | 1 | Advanced solid tumors | NCT05564858 | |
| IMGN151 | FolRα | DM21 | Maytansinoid | Cleavable, peptide | 1 | Endometrial cancer; ovarian cancer; primary peritoneal carcinoma; fallopian tube cancer | NCT05527184 | ||
| RC108 | c‐MET | MMAE | Auristatin | Undisclosed | 1 | Digestive cancer | NCT05628857 | ||
| TQB2102 | HER2 | TOP1i | TOP1i | Cleavable, enzyme | 1 | Advanced cancer | NCT05735496 | ||
| JSKN003 | HER2 | TOP1i | TOP1i | Cleavable (Dibenzocyclooctyne tetrapeptide linker) | 1 | Advanced solid tumors; metastatic solid tumors | NCT05494918 | ||
| M9140 | CEACAM5 | DM4 | Maytansinoid | Cleavable | 1 | Colorectal cancer | NCT05464030 | ||
| RC118‐ADC | Claudin‐18.2 | MMAE | Auristatin | Cleavable | 1 | Advanced solid tumor | NCT05205850 | ||
| SKB264 | TROP2 | Belotecan | Camptothecin | Cleavable (Sulfonyl pyrimidine‐CL2A‐carbonate) | 3 | Ovarian epithelial cancer; gastric adenocarcinoma; breast cancer; urothelial carcinoma; NSCLC; small‐cell lung cancer | NCT04152499 | ||
| RC88 | MSLN | MMAE | Auristatin | Cleavable | 1 | Solid tumor | NCT04175847 | ||
| AZD8205 | B7‐H4 | TOP1i | TOP1i | Cleavable | 1 | Breast cancer; cholangiocarcinoma; ovarian cancer; endometrial cancer | NCT05123482 | ||
| PF‐06647020 | PTK7 | Aur0101 | Auristatin | Cleavable | 1 | Cancer; NSCLC | NCT04189614 | ||
| W0101 | IGF‐R1 | Auristatin | Auristatin | Noncleavable | 1 | Advanced/metastatic solid tumors | NCT03316638 | ||
| FDA018‐ADC | TROP2 | Undisclosed | Undisclosed | undisclosed | 1 | Advanced/metastatic solid tumors | NCT05174637 | ||
| TORL‐2‐307‐ADC | Claudin‐18.2 | MMAE | Auristatin | Undisclosed | 1 | Advanced solid tumor; gastric cancer; Pancreas cancer; Gastroesophageal Junction Adenocarcinoma | NCT05156866 | ||
| STI‐3258 | TROP2 | Undisclosed | Undisclosed | Undisclosed | 1 | Solid tumor | NCT05060276 | ||
| M1231 | EGFR, MUC1 | Hemiasterlin | Hemiasterlin | Cleavable | 1 | Metastatic solid tumors; esophageal cancer; NSCLC | NCT04695847 | ||
| YL201 | TAA | Undisclosed | Undisclosed | Cleavable (TMALIN) | 1 | Advanced solid tumor | NCT05434234 | ||
| TORL‐1‐23 | Claudin 6 | MMAE | Auristatin | Cleavable | 1 | Advanced solid tumor; ovarian cancer; endometrial cancer | NCT05103683 | ||
| CPO102 | Claudin‐18.2 | MMAE | Auristatin | Noncleavable | 1 | Pancreatic cancer; gastric cancer | NCT05043987 | ||
| A166 | HER2 | Undisclosed | Auristatin | Cleavable | 1 | Breast cancer | NCT05311397 | ||
| A166 | HER2 | Undisclosed | Auristatin | Cleavable | 2 | Breast cancer; gastrointestinal cancer; salivary gland cancer; |lung cancer; colo‐rectal cancer; head and neck cancer; bladder cancer; cervical cancer; liver cancer; bile duct cancer; prostate cancer; ovarian carcinoma | NCT03602079 | ||
| FOR46 | CD46 | MMAE | Auristatin | Undisclosed | 2 | Prostate cancer metastatic | NCT03575819 | ||
| IMMU‐132 | TROP2 | SN‐38 | Camptothecin | Undisclosed | 1 | Prostate cancer | NCT03725761 | ||
| SKB315 | Claudin‐18.2 | TOP1i | TOP1i | Uncleavable | 1 | Advanced solid tumors | NCT05367635 | ||
| ADCT‐601 | AXL | SG3199 | Tesirine | Cleavable | 1 | Advanced solid tumors | NCT05389462 | ||
| HER3‐DXd | HER3 | DXd | TOP1i | Cleavable | 3 | Metastatic Breast Cancer | NCT02980341 | ||
| XB002 | TF | MMAE | Auristatin | Cleavable | 1 | NSCLC; urothelial cancer; ovarian cancer; cervical cancer; pancreatic cancer; prostate cancer; breast cancer | NCT04925284 | ||
| BIO‐106 | TROP2 | Undisclosed | Undisclosed | Undisclosed | 1 | Advanced solid tumor| | NCT05320588 | ||
| MRG004A | TF | MMAE | Auristatin | Cleavable | 1 | Advanced or metastatic solid tumors | NCT04843709 | ||
| CX‐2029 | CD71 | MMAE | Auristatin | Cleavable | 1 | Solid tumor, head and neck cancer; NSCLC; diffuse large B‐cell lymphoma; esophageal cancer | NCT03543813 | ||
| SKB264 | TROP2 | Belotecan | Camptothecin | Cleavable (Sulfonyl pyrimidine‐CL2A‐carbonate) | 3 | NSCLC | NCT05351788 | ||
| IBI354 | HER2 | Undisclosed | Camptothecin | Undisclosed | 1 | Locally advanced unresectable or metastatic solid tumors | NCT05636215 | ||
| TQB2103 | Claudin‐18.2 | DDDXD | TOP1i | Cleavable | 1 | ADVANCED malignant neoplasm | NCT05867563 | ||
| FZ‐AD004 | TROP2 | TOP1i | TOP1i | Undisclosed | 1 | Advanced and metastatic solid tumor | NCT05914545 | ||
| DS‐3939a | TA‐MUC1 | DxD | TOP1i | Cleavable | 1 | Advanced/ metastatic solid tumor | NCT05875168 | ||
| AMT‐151 | FolRα | Undisclosed | Undisclosed | Undisclosed | 1 | Advanced solid tumor; ovarian cancer; endometrial cancer; lung adenocarcinoma; triple negative breast cancer; pancreatic ductal adenocarcinoma | NCT05498597 | ||
| BAT8008 | TROP2 | Exatecan | TOP1i | Cleavable | 1 | Advanced solid tumors | NCT05620017 | ||
| B003 | HER2 | DM1 | Maytansinoid | Noncleavable (Nonreduceable thioether linkage) | 1 | HER2‐positive breast cancer | NCT03953833 | ||
| STI‐6129 | CD38 | Duostatin | Duostatin | Noncleavable (C‐lock chemical linker) | 1 | Advanced solid tumor | NCT05584709 | ||
| BAT8009 | B7‐H3 | Exatecan | TOP1i | Cleavable | 1 | Locally advanced/metastatic solid tumors | NCT05405621 | ||
| OMTX705 | MTX5 | TAM558 | Tubulysin | Cleavable | 1 | Advanced solid tumor | NCT05547321 | ||
| KM501 | HER2 | Undisclosed | Undisclosed | Undisclosed | 1 | Advanced solid tumors | NCT05804864 | ||
| EBC‐129 | CEACAM5 | MMAE | Auristatin | Undisclosed | 1 | Advanced solid tumors | NCT05701527 | ||
| BAT8007 | Nectin‐4 | TOP1i | TOP1i | Cleavable | 1 | Advanced solid tumors | NCT05879627 | ||
| AZD5335 | FolRα | TOP1i | TOP1i | Undisclosed | 1 | Ovarian cancer; lung adenocarcinoma | NCT05797168 | ||
| AZD9592 | EGFR, c‐MET | TOP1i | TOP1i | Cleavable | 1 | Advanced solid tumors; carcinoma nonsmall cell lung; head and neck neoplasms | NCT05647122 | ||
| MHB088C | B7‐H3 | TOP1i | TOP1i | Cleavable | 1 | Advanced or metastatic solid tumors | NCT05652855 | ||
| PRO1184 | FolRα | Exatecan | TOP1i | Cleavable | 1 | Ovarian cancer; primary peritoneal carcinoma; fallopian tube cancer; endometrial cancer; NSCLC; mesothelioma; breast cancer | NCT05579366 | ||
| IKS014 | HER2 | MMAE | Auristatin | Cleavable (Beta‐glucuronide) | 1 | Breast cancer; gastric cancer; gastroesophageal‐junction cancer | NCT05872295 | ||
| HS‐20089 | B7‐H4 | TOP1i | Undisclosed | Undisclosed | 1 | Advanced solid tumor | NCT05263479 | ||
| 9 MW2821 | Nectin‐4 | MMAE | Auristatin | Undisclosed | 2 | Advanced malignant solid tumors | NCT05216965 | ||
| Other diseases | STI‐6129 | CD38 | Duostatin | Duostatin | Noncleavable (C‐lock chemical linker) | 2 | Light chain (al) amyloidosis | NCT04316442 | |
| Light chain (AL) amyloidosis | NCT05692908 | ||||||||
Abbreviations: ADCs, antibody‐drug conjugates; AF‐HPA, auristatin F‐hydroxypropylamide; DxD, dexrutecan; IgG, Immunoglobulin G; IGN, indolinobenzodiazepine; mAB, Monoclonal antibody; MMAE, monomethyl auristatin E; MMAE, monomethyl auristatin E; MMAF, monomethyl auristatin F; TOP1i, topoisomerase 1 inhibitor.
4.1. Scope of disease indications
A large portion of ADC‐focused active trials are geared toward solid tumors, with breast cancer being the most investigated indication, featuring 140 trials. Figure 2 provides an overview of the scope of indications addressed in ADC‐focused active clinical trials. The majority (64%) of these trials aim to expand the clinical application of currently approved products. The ADCs in these trials have the same components as the marketed product but vary in the scope of indications under investigation. Approximately 36% of trials focus on new ADCs that have not yet been approved (Figure 2). When comparing the two groups of interest—trials for new ADCs versus trials related to approved ADCs—a significant shift toward solid tumor applications is noted in the trials for new ADCs, with solid tumor applications representing about 90% of the trials. A detailed breakdown of disease indications for the identified active ADC‐related trials is shown in Figure 2. Another notable observation is the shift in antigen targets, with the second group (trials for new ADCs) showcasing a broader diversity in antigen targets than the first group (Figure 3). There is also a broader diversity in the antibody and drug payload components used in the trials for new ADCs, which all contribute to the wider range of disease indications covered by this group. This diversification is likely driven by the growing understanding of target expression patterns in cancers.
FIGURE 2.
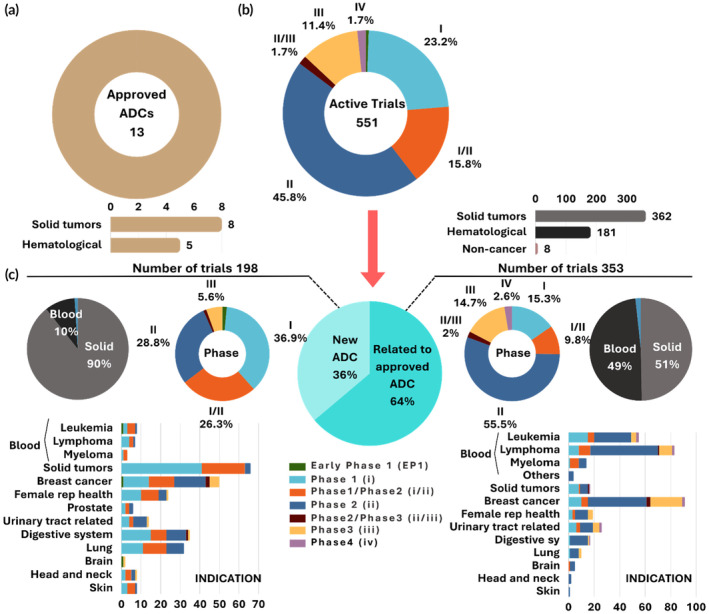
Overview of antibody‐drug conjugates (ADCs) in active clinical trials. (a) Approved ADCs (11 approved by the FDA and 2 approved by other regulatory agencies) in the market showing their scope of disease indications. (b) Phase and disease scope of ADCs in active clinical trials. (c) In‐depth analysis of ADC trials showing the ratio of trials based on new ADC products (left) to trials based on approved ADCs (right) analyzed on phase and indications.
FIGURE 3.
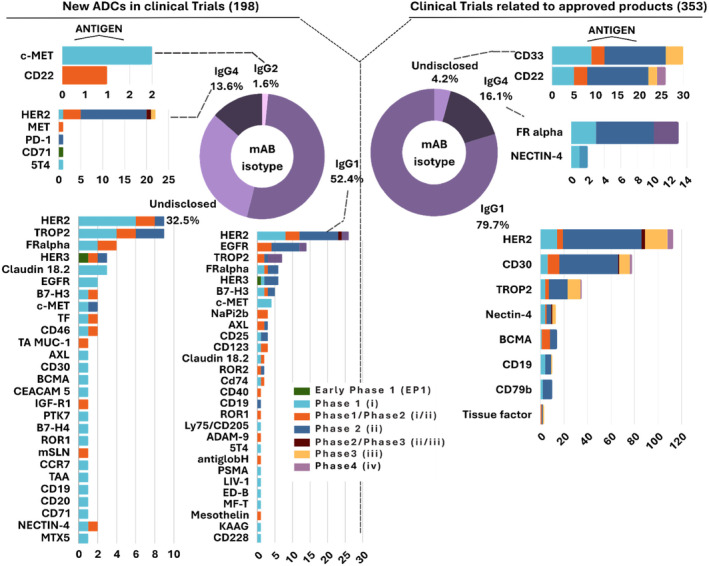
Scope of antibody used in antibody‐drug conjugates (ADCs) under active clinical trials. Comparison between trials for new ADCs (right) and trials for approved ADCs (left) based on mAb isotype. Each isotype is further analyzed (bar chats) to show the range of target‐antigen in different phases of clinical trials.
4.2. Scope of antibody
The antibody is an essential component of ADCs, as it determines target specificity, thereby enhancing the on‐target cytotoxic effect of the payload. 79 Moreover, the antibody component influences the plasma concentration, immunogenicity, and immune functions of the ADC and can also contribute to direct or indirect cytotoxic effects. 17 IgGs are the most used antibodies in ADCs, with the IgG1 subclass being the most prevalent (Figure 3). 79 About 70% (383 trials) of active clinical trials utilize IgG1, mostly humanized, and likely because of its abundance in serum and high binding affinity to IgG‐binding Fc‐gamma receptors compared with other subclasses, resulting in enhanced antibody‐dependent cytotoxicity and phagocytosis. 17 , 79 Following is IgG4, accounting for 15% of active trials. A detailed breakdown of antibody subtypes used in active ADC trials is shown in Figure 3. Additionally, there are three trials involving ADCs made of the IgG2 subclass.
4.3. Target antigens
Antigen selection is crucial for the effectiveness and safety of ADCs, as ADCs carry highly potent cytotoxic payloads that require precise delivery to minimize off‐target toxicity. Key considerations in antigen selections include (i) the exclusive or predominant expression of the target on tumor cells for selectivity, 17 , 79 (ii) the target antigens' surface expression on tumor cells without their secretion, which could lead to nonspecific drug release, 17 , 80 and (iii) the target's ability to trigger cellular internalization of ADCs, crucial for payload delivery. 17 , 81
Currently, ADCs approved by the FDA and other regulatory agencies target 11 distinct antigens for hematological malignancies and solid tumors Of the active clinical trials, about 80% (447 trials) focus on these established antigens used in approved products; a detailed breakdown of this is given in Table 2. The leading antigen targets in trials are HER2 (32%), CD30 (14%), and TROP2 (9%). Yet, more novel antigen targets were found in trials for new ADCs, with novel targets accounting for about 20% (104 trials) of total active trials. A detailed breakdown of these novel targets used in active ADC trials is shown in Figure 4. The pursuit of novel targets is a major driver for the extension of ADCs to solid tumors, as the use of these novel targets could potentially reduce ADC's toxicity to normal tissues. While most of these antigens are tumor‐associated rather than tumor‐specific, there is also an extension from typical tumor cell antigens to antigens found in the tumor microenvironment and neovasculature.
FIGURE 4.
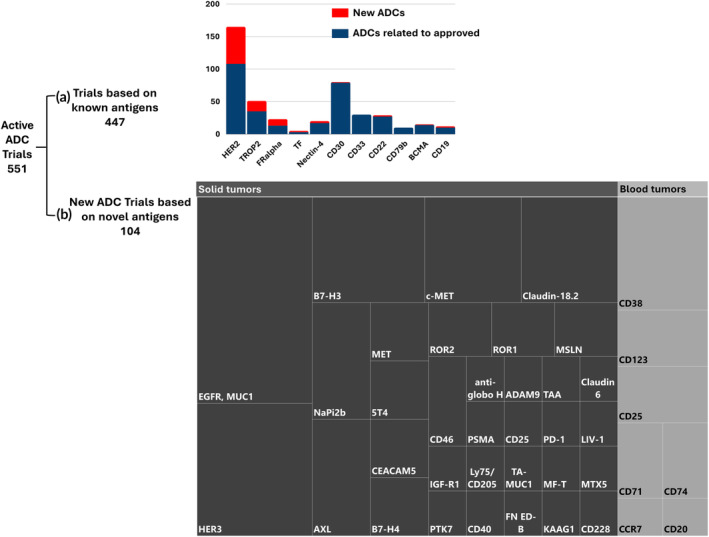
The scope of antigen targets in active antibody‐drug conjugate (ADC)‐focused clinical trials. (a) Bar chart showing the number of trials for new investigative and approved ADCs focusing on known antigens. (b) Tree chart showing the scope of novel antigen targets in trials for new ADC for solid tumors or hematological tumors.
4.3.1. Novel targets in active clinical trials—Hematological cancer antigens
Hematological cancers are considered more accessible than solid tumors. This explains why most antigens used in ADCs for treating hematological cancers often pertain to both neoplastic and non‐neoplastic cells, given ADC's direct access to diseased cells. Additionally, the absence of these targets on hematopoietic stem cells and nonhematopoietic tissues allows for the continuous replenishment of blood cells and reduces cytotoxicity, respectively. Classic antigens used in approved ADC formulations for hematological cancers include CD19, CD22, CD30, CD33, CD79b, and BCMA, with detailed reviews available elsewhere. 53 , 82 A significant portion of ADC‐focused active clinical trials (32%) targeting these antigens aims to expand the indications of approved ADCs or enhance their efficacy through combinations with other chemotherapeutics (e.g., doxorubicin, cyclophosphamide, gemcitabine) or immunotherapy (e.g., pembrolizumab, nivolumab, rituximab). Currently, there are 18 new ADC trials (3% of total active trials) focusing on hematological tumors, with 14 targeting novel antigens. Details about the scope of these antigens in clinical trials can be found in Table 3. These antigens include CD74, CD20, CCR7, and CD25 for lymphomas; CD71, CD123, CD25, and CD38 for leukemias; and CD38 for myeloma and light chain amyloidosis. These antigens are broadly expressed in immune cells (e.g., B cells, T cells, NK cells, dendritic cells, monocytes, macrophages), erythroid lineage cells, and other tissues as well.
4.3.2. Novel targets in active clinical trials—Solid tumor antigens
Unlike hematological cancer antigens, solid tumor antigens are not lineage‐specific and are mostly tumor‐associated. This means these antigens are mainly overexpressed in tumor cells but may also be expressed at lower levels in healthy cells, raising concerns about off‐target toxicity and reduced intratumoral drug delivery. 82 Therefore, identifying targets with limited expression in healthy tissues is crucial to improve the therapeutic effectiveness of ADCs. Classical antigens targeted by FDA‐approved ADCs for solid tumor include HER2, TROP2, TF, nectin‐4, and FRα. Currently, 32 novel solid tumor antigens are being investigated in clinical trials for new investigative ADCs, such as B7 family proteins, EGFR, HER3, mesenchymal–epithelial transition factor (c‐MET), AXL, Claudin‐18.2, and NaPi2b accounting for 15% of active trials. Further details about these new antigens and related trials are shown in Figure 4 and Table 4.
TABLE 4.
Representative active ADC trials involving novel antigens (more than four trials) for solid tumors.
| Antigen | ADC name | Condition | Payload | Phase | ID |
|---|---|---|---|---|---|
| B7‐H3 | DS‐7300A | Extensive‐stage small‐cell lung cancer | Deruxtecan | 2 | NCT05280470 |
| MGC018 | Cancer: solid tumors | Duocarmycin | 1/2 (in combination with retifanlimab) | NCT03729596 | |
| 1 (in combination with Lorigerlimab) | NCT05293496 | ||||
| HS‐20093 | Advanced solid tumor | TOP1i | 1 | NCT05276609 | |
| MHB088C | Advanced or metastatic solid tumors | TOP1i | 1/2 | NCT05652855 | |
| BAT8009 | Locally advanced/metastatic solid tumors | TOP1i | 1 | NCT05405621 | |
| HS‐20093 | Osteosarcoma/sarcoma | TOP1i | 2 | NCT05830123 | |
| EGFR | M1231 | Metastatic solid tumors; esophageal cancer; nonsmall cell lung cancer (NSCLC) | Hemiasterlin | 1 | NCT04695847 |
| BB‐1705 | Solid tumor | Eribulin | 1/2 | NCT05217693 | |
| MRG003 | Advanced or metastatic gastric cancer; advanced or metastatic gastroesophageal junction carcinoma | MMAE | 2 | NCT05188209 | |
| Recurrent or metastatic nasopharyngeal carcinoma | 2 | NCT05126719 | |||
| Recurrent or metastatic squamous cell carcinoma of head and neck | 2 | NCT04868162 | |||
| Advanced or metastatic biliary tract cancer | 2 | NCT04838964 | |||
| Carcinoma, nonsmall‐cell lung | 2 | NCT04838548 | |||
| Advanced solid tumors | 1/2 | NCT05688605 | |||
| Squamous cell carcinoma of the head and neck | 3 | NCT05751512 | |||
| AZD9592 | advanced solid tumors; carcinoma nonsmall cell lung; head and neck neoplasms | TOP1i | 1 | NCT05647122 | |
| SHR‐A1811 | Breast cancer | SHR9265 | 1/2 | NCT05824325 | |
| Triple‐negative breast cancer (TNBC) | 2 | NCT05749588 | |||
| HER2 low breast carcinoma | 2 | NCT05911958 | |||
| Breast neoplasm; breast cancer; hormone receptor positive tumor|HER2‐negative breast cancer; advanced breast cancer | 2 | NCT05594095 | |||
| Breast neoplasm; breast cancer; breast tumors; TNBC; HER2‐positive breast cancer; HER2‐negative breast cancer; hormone receptor positive tumor; hormone receptor negative tumor; early‐stage breast cancer; locally advanced breast cancer | 1/2 | NCT05582499 | |||
| ABT‐414 | Glioblastoma; gliosarcoma | MMAF | 3 | NCT02573324 | |
| HER3 | U3‐1402 | Breast cancer | Deruxtecan | Early Phase 1 | NCT04610528 |
| Metastatic breast cancer | 2 | NCT04965766 | |||
| Metastatic breast cancer; locally advanced breast cancer | 2 | NCT04699630 | |||
| Metastatic colorectal cancer | 2 | NCT04479436 | |||
| NSCLC | 1 | NCT03260491 | |||
| HER3‐DXd | Metastatic breast cancer | TOP1i | 1 | NCT02980341 | |
| Metastatic breast cancer; advanced nonsmall cell squamous lung cancer; solid tumor | 2 | NCT05865990 | |||
| Brain metastases | Early Phase 1 | NCT05620914 | |||
| c‐MET | BYON3521 | Solid tumor | duocarmycin | 1 | NCT05323045 |
| RC108 | Solid tumor | Undisclosed Microtubule | 1 | NCT04617314 | |
| Digestive cancer | Inhibitor | 2 | NCT05628857 | ||
| TR1801‐ADC | Unspecified adult solid tumor, protocol‐specific | SG3249 | 1 | NCT03859752 | |
| BYON3521 | Solid Tumor | duocarmycin | 1 | NCT05323045 | |
| MYTX‐011 | NSCLC; NSCLC Stage IV|NSCLC Stage IIIB; NSCLC; advanced nonsmall cell squamous lung cancer; advanced NSCLC; advanced nonsmall cell nonsquamous lung cancer | MMAE | 1 | NCT05652868 | |
| SHR‐A1403 | Advanced solid tumor | Undisclosed Microtubule Inhibitor | 1 | NCT03856541 | |
| AXL | CAB‐AXL‐ADC | Nonsmall‐cell lung cancer | MMAE | 2 | NCT04681131 |
| Solid tumor; NSCLC; melanoma; sarcoma; sarcoma, ewing; osteosarcoma; leiomyosarcoma; synovial sarcoma; liposarcoma; soft tissue sarcoma; bone sarcoma; refractory sarcoma | 1/2 | NCT03425279 | |||
| ADCT‐601 | Advanced solid tumors | SG3199 | 1 | NCT05389462 | |
| HuMax‐AXL‐ADC (Enapotamab vedotin) | Ovarian cancer; cervical cancer; endometrial cancer; NSCLC|thyroid cancer; melanoma; sarcoma; solid tumors | MMAE | 1/2 | NCT02988817 | |
| Claudin 18.2 | TORL‐2‐307‐ADC | Advanced solid tumor; gastric cancer; pancreas cancer; gastroesophageal junction adenocarcinoma | MMAE | 1 | NCT05156866 |
| RC118‐ADC | Advanced solid tumor | MMAE | 1/2 | NCT05205850 | |
| SKB315 | Advanced solid tumors | TOP1i | 1 | NCT05367635 | |
| TQB210 | Advanced malignant neoplasm | DDDXD | 1 | NCT05867563 | |
| SOT102 | Gastric cancer; pancreatic cancer; gastro‐esophageal junction cancer | PNU‐159682 | 1/2 | NCT05525286 | |
| NaPi2b | XMT‐1536 | High grade serous ovarian cancer; fallopian tube cancer; primary peritoneal cancer | AF‐HPA | 3 | NCT05329545 |
| Platinum‐sensitive ovarian cancer (UPGRADE‐A) | 1/2 | NCT04907968 | |||
| Platinum‐resistant ovarian cancer; NSCLC metastatic | 1/2 | NCT03319628 | |||
| XMT‐1592 | Ovarian cancer; NSCLC | Undisclosed | 1/2 | NCT04396340 |
Abbreviations: ADC, antibody‐drug conjugates; IgG, Immunoglobulin G; MMAE, monomethyl auristatin E; MMAF, monomethyl auristatin F.
4.4. Diversity of payload and linker in active clinical trials
Due to their specificity for tumor cells or tissues, ADCs can minimize the off‐target effects associated with the parent chemotherapeutic drugs. 12 , 83 The currently approved ADCs utilize DNA‐damaging and microtubule‐inhibiting payloads, such as auristatins, maytansinoid, camptothecin, and calicheamicin, effective at sub‐nanomolar concentrations. 82 These payloads are not suitable for systemic administration alone due to their high cytotoxicity. ADCs present a valuable tool for repurposing small‐molecule drugs previously limited by off‐target toxicity. 12
The ongoing expansion of indications for ADCs in active clinical trials also reflects the inclusion of new payloads into investigative ADCs. Of the 200 active clinical trials for new ADCs, about a quarter involve new payloads. Figure 5 provides a breakdown of the payloads in current ADC trials, with microtubule inhibitors constituting 51% of these payloads. Auristatins, which inhibit tubulin polymerization, dominate with 81 trials, possibly attributed to their favorable biochemical properties. 82 Topoisomerase inhibitors, which cause DNA damage through DNA intercalation, represent the second major payload class in ADC trials, totaling 44 trials. Some examples of the new payloads being explored in investigative ADCs include the DNA alkylating clas—duocarmycin (10 trials), PBD dimers and pyridinobenzodiazepines (7 trials), and monoamine indolinobenzodiazepines (7 trials). These molecules are known as highly potent antitumor agents. 84 Detailed chemical properties of these payloads can be found in recent reviews published elsewhere. 28
FIGURE 5.
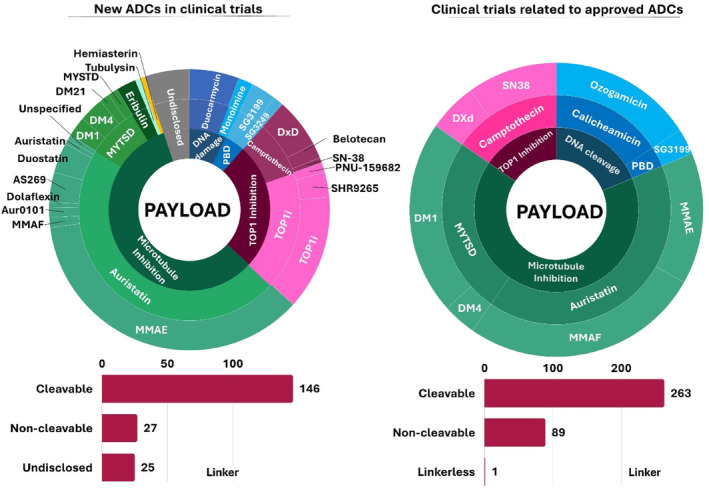
Payload and linker diversity in antibody‐drug conjugate (ADC)‐focused active clinical trials. Pie‐charts showing the drug action (tier 1), class (tier 2), and payload (tier 3) in trials for new ADCs (left) and trials for approved ADCs (right).
The linker in an ADC significantly impacts the ADC's safety and efficacy. In ADC design, an ideal linker should be stable enough to prevent premature drug release while being sensitive to enable site‐specific drug release. 84 ADCs are largely designed with either cleavable or noncleavable linkers. The cleavable linkers are responsive to pH changes, glutathione/disulfide isomerase, or proteases present in the TME, while noncleavable linkers rely on the endosomal/lysosomal degradation of the ADCs for drug release. 53 , 84 Reflecting the preference in approved ADC products, cleavable linkers are favored in new investigative ADCs in current clinical trials (Figure 5). Examples of such cleavable linkers include glutathione‐sensitive disulfide linkers, protease‐sensitive linkers (phenylalanine‐lysine, valine‐citrulline), and acid‐sensitive hydrazone linkers.
5. CHALLENGES AND OUTLOOK FOR CLINICAL TRANSLATION OF ADCs
The principle behind the efficacy of ADCs is straightforward. However, the development of an effective ADC is challenging. To date, improvements such as the use of humanized antibodies, highly potent payloads, and the development of highly stable linkers have driven the development of more effective ADCs with many promising candidates in clinical trials. 20
A pressing challenge in the development of ADCs is optimizing drug loading. This includes determining the optimal DAR and achieving a homogenous drug conjugation. The DAR is an important property that influences the PK, stability, and efficacy of ADCs. 21 Different studies have shown the need to link a certain number of drug units to each mAb to optimize its effectiveness. However, a higher DAR does not necessarily imply better efficacy. While ADCs conjugated with a high number of payloads demonstrate increased in vitro potency, several in vivo studies have revealed a negative correlation between high DAR and toxicity and aggregation. 85 , 86 An ADC with a DAR of 4 showed equivalent in vivo antitumor activity to that with a DAR of 8, and a further reduction to a DAR of 2 improved in vivo activity. 21 , 87 , 88 An average DAR of four is often recommended, as higher DAR values increase plasma clearance and antibody aggregation, reducing the therapeutic index of ADCs. 87 , 89 However, contrary to this general recommendation, a good number of new ADCs have a DAR higher than 4 and are showing promising results in clinical trials. For example, XMT‐1536, a dolaflexin‐based ADC targeting SLC34A2/NaPi2b in solid tumors, has a DAR of up to 15. Although this contradicts the typical DAR recommendations, the payload is a prodrug of auristatin F that exhibits notable bystander cytotoxicity, increasing its antitumor efficacy. Once metabolized intratumorally, auristatin F becomes impermeable to cell membranes, further reducing the systemic drug exposure and improving the overall tolerability. 90 Thus, this prodrug approach holds potential in improving ADC efficacy, as its overall PK profile is comparable to those of other clinically investigated ADCs with lower DARs.
Early approved ADCs are based on random/heterogeneous conjugation of the payload to the antibody. This approach has been shown to negatively impact the therapeutic index of ADCs. 89 , 91 Heterogenicity could translate to each ADC containing an amount of material above the nominal DAR 86 and/or ADCs with both unconjugated and overloaded antibodies. 89 Hence, the current research focus is on achieving homogenous ADCs that have the same site of drug attachment between individual mAbs. This goal is technologically challenging and depends on the method of conjugation of the linker to the mAb. The most common method of conjugation is via the lysine side‐chain amines or cysteine sulfhydryl groups. 87 , 89 This results in a mixture of species (>100 species) with different DARs, linked at different sites, and each species displaying distinct in vivo PK and efficacy patterns. 87 , 91 Purification can be used to eliminate species with different DARs, but this still leaves a heterogeneous mixture with payloads attached at different sites, resulting in batch‐to‐batch variations in ADC production. 91
In addition, the site of payload attachment and the coupling technique also influence the PK and stability of ADCs. 85 , 89 , 92 Coupling of payloads can induce both physical and chemical instability. For example, in cysteine conjugation, there is a reduction of the interchain disulfide bond on the mAb's free cysteine residue. These cysteines play an important role in maintaining the structure of the antibody, and such disturbance increases the risk of instability. 93 To overcome these challenges, an emerging approach is to use a site‐specific attachment strategy. This approach involves conjugating payloads to sites that individually minimize the density and solvent accessibility to the hydrophobic payloads. 92 , 94 It has been shown that while ADCs with a DAR of 8, only achieved minor tumor inhibition in vivo, using a site‐specific ADC, still with a DAR of 8, resulted in superior tumor inhibition, showing that location matters. 85 This necessitates investigating mAbs to find specific sites that display plasma exposure equivalent to the unconjugated antibody, thereby improving the therapeutic efficacy of ADCs. Alternatively, another emerging strategy is to adopt other methods of conjugation. 91 , 95 A few of the new ADCs in clinical trials adopt novel conjugation methods. SKB264, a TROP‐2 targeting ADC, utilizes a novel coupling strategy that permits the conjugation of seven to eight payloads on the reduced interchain disulfide bonds via a covalent sulfonyl pyrimidine‐CL2A‐carbonate linker. This strategy improves the stability of the ADC and increases its plasma half‐life to up to 57 h in mice. When compared with TRODELVY (an approved TROP‐2 targeting ADC), SKB264 at the same dose demonstrates improved antitumor efficacy and reduced adverse effects. 96 STI‐6129, another new ADC with six ongoing clinical trials, utilizes this disulfide re‐bridging approach to achieve site‐specific conjugation of five duostatin molecules to an anti‐CD38 mAb. Using this strategy, STI‐6129 shows an internalization rate comparable to that of the unconjugated antibody. 97 Other strategies to improve conjugation include the introduction of an additional cysteine group at strategic points on the mAbs to preserve the innate cysteine. ADCT‐602 and IMGN632, which contain a cysteine‐engineered anti‐CD22 mAB 98 and anti‐CD123 mAb, 99 , 100 respectively, are notable examples for this strategy. This modification retains antigen binding and specificity and yields homogeneous conjugates. 89 , 101 Choosing the right site can thus improve drug loading and reduce clearance. 92 Other studies have suggested that lysine conjugation could be more beneficial than site‐specific cysteine conjugation, 102 highlighting the need for a case‐by‐case optimization of conjugation methods in ADC development. There are a few notable ADCs with other site‐specific conjugation strategies in clinical trials. ARX‐788, an amberstatin‐bearing anti‐HER2 ADC with a DAR of 1.4, utilizes a noncleavable linker based on a non‐natural amino acid technology. This results in a homogeneous ADC with high serum stability, outperforming T‐DM1 in preclinical studies. 103 Likewise, ADCT‐601 utilizes an N‐glycosylation site to achieve site‐specific conjugation of SG3199 to an anti‐AXL mAb via a cleavable linker. 104 , 105
Another notable challenge affecting the development and clinical application of ADCs is the development of resistance. The advantage of having multiple mechanisms of action also sets ADCs up for resistance, as it can occur at any of these steps. 106 Resistance to ADCs can be antigen‐related, payload‐related, or tumor‐cell‐related.
Antigen‐related resistance could be due to the reduced expression of antigen or truncated forms of antigen ectodomain, leading to reduced binding of antibodies to the cell surface. 106 , 107 , 108 It has been shown that months of treatment with anti‐HER 2 trastuzumab‐maytansinoid ADC (TM‐ADC) resulted in 16‐fold resistance to TM‐ADC and cross‐resistance to other trastuzumab ADCs, partly due to decreased HER2 expression. 109 A similar trend in the downregulation of antigen (CD30) has also been reported for resistance to Brentuximab Vedotin. 110 Tumor heterogenicity 111 , 112 and genomic alterations 113 of antigens can also lead to varied expression of antigens in different parts of the tumor, contributing to treatment failures. Such resistance is typically overcome by switching to other ADCs or standard‐of‐care chemotherapeutics. 109 , 114
Payload‐related resistance is mostly due to the overexpression of drug efflux pumps on the tumor cells. Efflux pumps, such as MDR1, also known as permeability glycoprotein1, are responsible for the development of resistance to many small‐molecule drugs and likewise ADCs. 106 , 109 , 110 The overexpression of ABC transporters has been reported to be responsible for resistance to T‐Dxd, T‐DM1, gemtuzumab ozogamicin, and an anti‐CD33‐calicheamicin ADC. 107 , 115 Strategies to overcome such resistance include the diversification of payloads, which account for the number of new payloads under ADC trials (Figure 4). 108 Replacing the tubulin inhibitor DM1 with a topoisomerase inhibitor was reported to effectively overcome T‐DM1 resistance, 115 and switching from auristatin‐based ADCs to anthracycline‐based ADCs also showed a similar effect. 116 Other strategies include optimizing DAR and conjugation techniques and developing more hydrophilic ADCs, as MDR1 has a higher preference for hydrophobic compounds. 117 , 118
The genetic instability of cancer cells enables them to continually develop mechanisms to evade treatment. Tumor‐cell‐related resistance mechanisms involve changes in trafficking pathways, lysosomal dysfunction, and alterations in apoptotic signaling pathways. Effective internalization of ADCs is central to its mechanism of action (Figure 1) and predominantly occurs via CME. 117 The use of alternative routes like caveolar‐mediated endocytosis could result in the accumulation of ADCs in caveolin‐1 (CAV1)‐coated vesicles, reduced lysosomal colocalization, and overall reduced efficacy. 117 , 119 It was reported that while HER2 expression in some T‐DM1‐resistant cell lines remained normal, intracellular traffic, lysosomal pH, and proteolytic activity were abnormal. Increased lysosomal pH and deranged protease activity result in the accumulation of intact ADC within the cell. 120 This is particularly noted in ADCs with linkers that require complete proteolysis of the antibody to release payload. Hence, switching to using linkers that only require one proteolytic event could help overcome this mechanism of resistance. 117 Other strategies involve the use of ADCs with alternative cleavage mechanisms, 121 , 122 the use of nanoparticles and other drugs to stabilize lysosomal pH, 123 and more recently, the use of dual‐drug ADCs and bispecific ADCs. Leveraging insights from these resistance mechanisms, some new ADCs in clinical trials feature new designs to improve their efficacy. Notably, SYD985 and DS‐8201a, ADCs based on trastuzumab, show promise in overcoming T‐DM1 drug resistance in HER2‐positive breast cancers. These second‐generation ADCs employ more potent cytotoxic payloads and have shifted from covalent to cleavable linkers. SYD985, currently in 5 active trials, utilizes a duocarmycin‐derived payload, whose cytotoxicity is not cell cycle dependent, conjugated using a valine‐citrulline linker. Unlike T‐DM1, which requires the complete degradation of the antibody for payload release, SYD985's payload is released through cathepsin‐mediated cleavage of its linker. 124 , 125 As another example, DS‐8201a contains a topoisomerase‐inhibiting DXd payload conjugated via a stable tert‐butoxycarbonyl‐glycyl‐glycyl‐phenylalanyl‐glycine linker to trastuzumab. DS‐8201a can circumvent T‐DM1 resistance as DXd is resistant to the effect of p‐glycoproteins, and the stable linker allows for the conjugation of up to eight drug molecules, enhancing its cytotoxicity. 126 Furthermore, both ADCs exhibit significant bystander cytotoxicity due to the high membrane permeability of the payloads. 126
Bispecific biparatopic ADCs are an emerging strategy to overcome tumor‐cell‐related ADC resistance. These ADCs are designed to have antibodies with different nanobodies that can simultaneously recognize either different targets or the same targets but different and nonoverlapping epitopes. This helps to improve internalization, lysosomal trafficking, and subsequent degradation of the ADC. A notable example is M1231, a first‐in‐class bispecific ADC targeting MUC‐1 and EGFR, demonstrating superior efficiency in cell internalization and lysosomal trafficking compared with monospecific mAbs. 127 , 128 By recognizing different epitopes, this strategy is useful in overcoming resistance due to antigen downregulation. 129 Furthermore, combining the specificity of two antibodies can also improve ADC potency by blocking targets central to disease progression and/or initiating ADCC or CDC. 130 It was reported that engaging the overexpressed MET in lung cancers with a biparatopic ADC, METxMET‐M114, provided more benefits than merely blocking the MET function and had the potential to overcome acquired resistance to MET‐selective tyrosine kinase inhibitors. 131 Numerous other studies have demonstrated the potential of bispecific ADCs, suggesting that this could indeed be a future direction for ADC development. 130 , 132 , 133 , 134 , 135 However, it is worth noting that the functioning of biparatopic antibody requires a minimal number of antigen expressions, thus limiting its function below that threshold. 129
Another emerging strategy to address the problem of ADC resistance incorporates the concept of polypharmacy, but without the complexity of multiple drug regimens. This involves the use of dual‐drug ADCs. These ADCs commonly consist of payloads with different physiochemical properties or mechanisms of action attached to the mAb via the same linker. 136 By coupling MMAE and MMAF, Levengood et al. 137 reported a 3:5 cure rate, compared with a 1:5 cure rate achieved with only MMAF. Similarly, another study designed an MMAE and MMAF dual ADC using a click chemistry‐based linker, which allowed for effective control of DAR and more effective tumor killing. 138 Similarly, it was demonstrated that a dual ADC bearing MMAE and a PBD dimer could achieve tumor killing via different mechanisms. 139 ADCs designed in this manner have been reported to have increased efficacy compared with the single‐drug formulations and represent a future direction in ADC development. 136
6. CONCLUSION
The surge in the clinical adoption of ADCs since their initial approval is attributed to their superior therapeutic effectiveness over traditional cytotoxic therapies. ADCs have emerged as the primary treatment option for certain blood and solid tumors that are unresponsive to conventional chemotherapy, underscoring their potential in targeting cancers with identifiable markers. Currently, more than 500 clinical trials are exploring numerous new ADCs, suggesting an anticipated increase in ADC approvals across a broader range of indications in the forthcoming years. Insights gained from the clinical use of existing ADCs have spurred the creation of next‐generation ADCs, which promise enhanced efficacy and fewer adverse effects. Innovations including the discovery of novel targets, the refinement of conjugation techniques, optimization of the DAR and the diversification of cytotoxic agents are poised to improve the PK and safety profiles of ADCs significantly. Despite facing challenges such as drug resistance and tumor heterogeneity, ongoing advancements in ADC technology offer optimism for overcoming these obstacles.
AUTHOR CONTRIBUTIONS
Edidiong Udofa: Data curation; validation; writing–original draft; writing–review and editing. Disha Sankholkar: Data curation; formal analysis; methodology; writing–original draft; writing–review and editing. Samir Mitragotri: Conceptualization; supervision; writing–review and editing. Zongmin Zhao: Conceptualization; supervision; writing–review and editing.
CONFLICT OF INTEREST STATEMENT
SM is a shareholder of Aarvik Therapeutics.
PEER REVIEW
The peer review history for this article is available at https://www.webofscience.com/api/gateway/wos/peer-review/10.1002/btm2.10677.
ACKNOWLEDGMENTS
ZZ acknowledges support from the College of Pharmacy at University of Illinois Chicago. SM acknowledges support from the John A. Paulson School of Engineering & Applied Sciences and Wyss Institute at Harvard University. Schematic representations were generated with the use of Biorender (©BioRender – biorender.com).
Udofa E, Sankholkar D, Mitragotri S, Zhao Z. Antibody drug conjugates in the clinic. Bioeng Transl Med. 2024;9(6):e10677. doi: 10.1002/btm2.10677
Edidiong Udofa and Disha Sankholkar contributed equally to this study.
Contributor Information
Samir Mitragotri, Email: mitragotri@seas.harvard.edu.
Zongmin Zhao, Email: zhaozm@uic.edu.
DATA AVAILABILITY STATEMENT
All data are available in the main article.
REFERENCES
- 1. Padma VV. An overview of targeted cancer therapy. Biomedicine (Taipei). 2015;5(4):19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Debela DT, Muzazu SG, Heraro KD, et al. New approaches and procedures for cancer treatment: current perspectives. SAGE Open Med. 2021;9:20503121211034366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zugazagoitia J, Guedes C, Ponce S, Ferrer I, Molina‐Pinelo S, Paz‐Ares L. Current challenges in cancer treatment. Clin Ther. 2016;38(7):1551‐1566. [DOI] [PubMed] [Google Scholar]
- 4. Hou J, He Z, Liu T, et al. Evolution of molecular targeted cancer therapy: mechanisms of drug resistance and novel opportunities identified by CRISPR‐Cas9 screening. Front Oncol. 2022;12:755053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Keefe DMK, Bateman EH. Potential successes and challenges of targeted cancer therapies. J Natl Cancer Inst Monogr. 2019;2019(53):lgz008. [DOI] [PubMed] [Google Scholar]
- 6. Gerber DE. Targeted therapies: a new generation of cancer treatments. Am Fam Physician. 2008;77(3):311‐319. [PubMed] [Google Scholar]
- 7. Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer. 2012;12(4):278‐287. [DOI] [PubMed] [Google Scholar]
- 8. Parakh S, King D, Gan HK, Scott AM. Current development of monoclonal antibodies in cancer therapy. In: Theobald M, ed. Current Immunotherapeutic Strategies in Cancer. Springer International Publishing; 2020:1‐70. [DOI] [PubMed] [Google Scholar]
- 9. Zahavi D, Weiner L. Monoclonal antibodies in cancer therapy. Antibodies. 2020;9(3):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shuel SL. Targeted cancer therapies: clinical pearls for primary care. Can Fam Physician. 2022;68(7):515‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Strebhardt K, Ullrich A. Paul Ehrlich's magic bullet concept: 100 years of progress. Nat Rev Cancer. 2008;8(6):473‐480. [DOI] [PubMed] [Google Scholar]
- 12. Dumontet C, Reichert JM, Senter PD, Lambert JM, Beck A. Antibody–drug conjugates come of age in oncology. Nat Rev Drug Discov. 2023;22(8):641‐661. [DOI] [PubMed] [Google Scholar]
- 13. Fuentes‐Antrás J, Genta S, Vijenthira A, Siu LL. Antibody–drug conjugates: in search of partners of choice. Trends Cancer. 2023;9(4):339‐354. [DOI] [PubMed] [Google Scholar]
- 14. Diamantis N, Banerji U. Antibody‐drug conjugates—an emerging class of cancer treatment. Br J Cancer. 2016;114(4):362‐367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Khongorzul P, Ling CJ, Khan FU, Ihsan AU, Zhang J. Antibody–drug conjugates: a comprehensive review. Mol Cancer Res. 2020;18(1):3‐19. [DOI] [PubMed] [Google Scholar]
- 16. Pettinato MC. Introduction to antibody‐drug conjugates. Antibodies (Basel). 2021;10(4):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fu Z, Li S, Han S, Shi C, Zhang Y. Antibody drug conjugate: the “biological missile” for targeted cancer therapy. Signal Transduct Target Ther. 2022;7(1):93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Perez HL, Cardarelli PM, Deshpande S, et al. Antibody–drug conjugates: current status and future directions. Drug Discov Today. 2014;19(7):869‐881. [DOI] [PubMed] [Google Scholar]
- 19. Maecker H, Jonnalagadda V, Bhakta S, Jammalamadaka V, Junutula JR. Exploration of the antibody‐drug conjugate clinical landscape. MAbs. 2023;15(1):2229101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chau CH, Steeg PS, Figg WD. Antibody–drug conjugates for cancer. Lancet. 2019;394(10200):793‐804. [DOI] [PubMed] [Google Scholar]
- 21. Baah S, Laws M, Rahman KM. Antibody‐drug conjugates‐a tutorial review. Molecules. 2021;26(10):2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lyon RP, Bovee TD, Doronina SO, et al. Reducing hydrophobicity of homogeneous antibody‐drug conjugates improves pharmacokinetics and therapeutic index. Nat Biotechnol. 2015;33(7):733‐735. [DOI] [PubMed] [Google Scholar]
- 23. Su D, Zhang D. Linker design impacts antibody‐drug conjugate pharmacokinetics and efficacy via modulating the stability and payload release efficiency. Front Pharmacol. 2021;12:687926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mehrling T, Soltis D. Challenges in Optimising the successful construction of antibody drug conjugates in cancer therapy. Antibodies. 2018;7(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Su Z, Xiao D, Xie F, et al. Antibody‐drug conjugates: recent advances in linker chemistry. Acta Pharm Sin B. 2021;11(12):3889‐3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Matikonda SS, McLaughlin R, Shrestha P, Lipshultz C, Schnermann MJ. Structure–activity relationships of antibody‐drug conjugates: a systematic review of chemistry on the Trastuzumab scaffold. Bioconjug Chem. 2022;33(7):1241‐1253. [DOI] [PubMed] [Google Scholar]
- 27. Kalim M, Chen J, Wang S, et al. Intracellular trafficking of new anticancer therapeutics: antibody–drug conjugates. Drug des Devel Ther. 2017;11:2265‐2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang Z, Li H, Gou L, Li W, Wang Y. Antibody–drug conjugates: recent advances in payloads. Acta Pharm Sin B. 2023;13(10):4025‐4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Anderl J, Faulstich H, Hechler T, Kulke M. Antibody–drug conjugate payloads. In: Ducry L, ed. Antibody‐Drug Conjugates. Humana Press; 2013:51‐70. [DOI] [PubMed] [Google Scholar]
- 30. Birrer MJ, Moore KN, Betella I, Bates RC. Antibody‐drug conjugate‐based therapeutics: state of the science. J Natl Cancer Inst. 2019;111(6):538‐549. [DOI] [PubMed] [Google Scholar]
- 31. Carter PJ, Senter PD. Antibody‐drug conjugates for cancer therapy. Cancer J. 2008;14(3):154‐169. [DOI] [PubMed] [Google Scholar]
- 32. Passaro A, Jänne PA, Peters S. Antibody‐drug conjugates in lung cancer: recent advances and implementing strategies. J Clin Oncol. 2023;41(21):3747‐3761. [DOI] [PubMed] [Google Scholar]
- 33. Lambert JM, Berkenblit A. Antibody–drug conjugates for cancer treatment. Annu Rev Med. 2018;69(1):191‐207. [DOI] [PubMed] [Google Scholar]
- 34. Samantasinghar A, Sunildutt NP, Ahmed F, et al. A comprehensive review of key factors affecting the efficacy of antibody drug conjugate. Biomed Pharmacother. 2023;161:114408. [DOI] [PubMed] [Google Scholar]
- 35. Hoffmann RM, Coumbe BGT, Josephs DH, et al. Antibody structure and engineering considerations for the design and function of antibody drug conjugates (ADCs). Onco Targets Ther. 2018;7(3):e1395127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Swaminathan M, Cortes JE. Update on the role of gemtuzumab‐ozogamicin in the treatment of acute myeloid leukemia. Ther Adv Hematol. 2023;14:20406207231154708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Selby C, Yacko LR, Glode AE. Gemtuzumab ozogamicin: back again. J Adv Pract Oncol. 2019;10(1):68‐82. [PMC free article] [PubMed] [Google Scholar]
- 38. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta‐analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986‐996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lambert J, Pautas C, Terré C, et al. Gemtuzumab ozogamicin for de novo acute myeloid leukemia: final efficacy and safety updates from the open‐label, phase III ALFA‐0701 trial. Haematologica. 2019;104(1):113‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Van Der Weyden C, Dickinson M, Whisstock J, Prince HM. Brentuximab vedotin in T‐cell lymphoma. Expert Rev Hematol. 2019;12(1):5‐19. [DOI] [PubMed] [Google Scholar]
- 41. Scott LJ. Brentuximab vedotin: a review in CD30‐positive Hodgkin lymphoma. Drugs. 2017;77(4):435‐445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ansell SM. Brentuximab vedotin. Blood. 2014;124(22):3197‐3200. [DOI] [PubMed] [Google Scholar]
- 43. Straus DJ, Długosz‐Danecka M, Alekseev S, et al. Brentuximab vedotin with chemotherapy for stage III/IV classical Hodgkin lymphoma: 3‐year update of the ECHELON‐1 study. Blood. 2020;135(10):735‐742. [DOI] [PubMed] [Google Scholar]
- 44. Ansell SM, Radford J, Connors JM, et al. Overall survival with Brentuximab Vedotin in stage III or IV Hodgkin's lymphoma. N Engl J Med. 2022;387(4):310‐320. [DOI] [PubMed] [Google Scholar]
- 45. Carson KR, Newsome SD, Kim EJ, et al. Progressive multifocal leukoencephalopathy associated with brentuximab vedotin therapy: a report of 5 cases from the southern network on adverse reactions (SONAR) project. Cancer. 2014;120(16):2464‐2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Aujla A, Aujla R, Liu D. Inotuzumab ozogamicin in clinical development for acute lymphoblastic leukemia and non‐Hodgkin lymphoma. Biomark Res. 2019;7:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Savoy JM, Welch MA, Nasnas PE, Kantarjian H, Jabbour E. Inotuzumab ozogamicin for the treatment of acute lymphoblastic leukemia. Ther Adv Hematol. 2018;9(12):347‐356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Uy N, Nadeau M, Stahl M, Zeidan AM. Inotuzumab ozogamicin in the treatment of relapsed/refractory acute B cell lymphoblastic leukemia. J Blood Med. 2018;9:67‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. 2016;375(8):740‐753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Deeks ED. Polatuzumab vedotin: first global approval. Drugs. 2019;79(13):1467‐1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sehn LH, Herrera AF, Flowers CR, et al. Polatuzumab vedotin in relapsed or refractory diffuse large B‐cell lymphoma. J Clin Oncol. 2020;38(2):155‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dornan D, Bennett F, Chen Y, et al. Therapeutic potential of an anti‐CD79b antibody–drug conjugate, anti‐CD79b‐vc‐MMAE, for the treatment of non‐Hodgkin lymphoma. Blood. 2009;114(13):2721‐2729. [DOI] [PubMed] [Google Scholar]
- 53. Gogia P, Ashraf H, Bhasin S, Xu Y. Antibody‐drug conjugates: a review of approved drugs and their clinical level of evidence. Cancers (Basel). 2023;15(15):3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xu B. Loncastuximab tesirine: an effective therapy for relapsed or refractory diffuse large B‐cell lymphoma. Eur J Clin Pharmacol. 2022;78(5):707‐719. [DOI] [PubMed] [Google Scholar]
- 55. Dhillon S. Moxetumomab Pasudotox: first global approval. Drugs. 2018;78(16):1763‐1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ketchum EB, Clarke A, Clemmons AB. Belantamab mafodotin‐BLMF: a novel antibody‐drug conjugate for treatment of patients with relapsed/refractory multiple myeloma. J Adv Pract Oncol. 2022;13(1):77‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Baines AC, Ershler R, Kanapuru B, et al. FDA approval summary: belantamab Mafodotin for patients with relapsed or refractory multiple myeloma. Clin Cancer Res. 2022;28(21):4629‐4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. GlaxoSmithKline . GSK provides an update on Blenrep (belantamab mafodotin‐blmf) US marketing authorisation; 2022. Accessed January 2. 2024.
- 59. Patel KC, Hageman K, Cooper MR. Ado‐trastuzumab emtansine for the treatment of human epidermal growth factor receptor 2–positive metastatic breast cancer. Am J Health‐Syst Pharm. 2014;71(7):537‐548. [DOI] [PubMed] [Google Scholar]
- 60. Corrigan PA, Cicci TA, Auten JJ, Lowe DK. Ado‐trastuzumab emtansine:a HER2‐positive targeted antibody‐drug conjugate. Ann Pharmacother. 2014;48(11):1484‐1493. [DOI] [PubMed] [Google Scholar]
- 61. Lewis Phillips GD, Li G, Dugger DL, et al. Targeting HER2‐positive breast cancer with trastuzumab‐DM1, an antibody–cytotoxic drug conjugate. Cancer Res. 2008;68(22):9280‐9290. [DOI] [PubMed] [Google Scholar]
- 62. Hunter FW, Barker HR, Lipert B, et al. Mechanisms of resistance to trastuzumab emtansine (T‐DM1) in HER2‐positive breast cancer. Br J Cancer. 2020;122(5):603‐612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Verma S, Miles D, Gianni L, et al. Trastuzumab emtansine for HER2‐positive advanced breast cancer. N Engl J Med. 2012;367(19):1783‐1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gouda MA, Subbiah V. Strategies for mitigating antibody‐drug conjugate related adverse events for precision therapy. The Cancer Journal. 2022;28(6):496‐507. [DOI] [PubMed] [Google Scholar]
- 65. Mantia CM, Sonpavde G. Enfortumab vedotin‐ejfv for the treatment of advanced urothelial carcinoma. Expert Rev Anticancer Ther. 2022;22(5):449‐455. [DOI] [PubMed] [Google Scholar]
- 66. Chang E, Weinstock C, Zhang L, et al. FDA approval summary: enfortumab vedotin for locally advanced or metastatic urothelial carcinoma. Clin Cancer Res. 2021;27(4):922‐927. [DOI] [PubMed] [Google Scholar]
- 67. Halford Z, Anderson MK, Clark MD. Enfortumab vedotin‐ejfv: a first‐in‐class anti–nectin‐4 antibody‐drug conjugate for the management of urothelial carcinoma. Annals of Pharmacotherapy. 2021;55(6):772‐782. [DOI] [PubMed] [Google Scholar]
- 68. Narayan P, Osgood CL, Singh H, et al. FDA approval summary: fam‐trastuzumab deruxtecan‐Nxki for the treatment of unresectable or metastatic HER2‐positive breast cancer. Clin Cancer Res. 2021;27(16):4478‐4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Nguyen X, Hooper M, Borlagdan JP, Palumbo A. A review of fam‐trastuzumab deruxtecan‐nxki in HER2‐positive breast cancer. Ann Pharmacother. 2021;55(11):1410‐1418. [DOI] [PubMed] [Google Scholar]
- 70. Narayan P, Dilawari A, Osgood C, et al. US Food and Drug Administration approval summary: fam‐Trastuzumab Deruxtecan‐nxki for human epidermal growth factor receptor 2‐low Unresectable or metastatic breast cancer. J Clin Oncol. 2023;41(11):2108‐2116. [DOI] [PubMed] [Google Scholar]
- 71. Li BT, Smit EF, Goto Y, et al. Trastuzumab deruxtecan in HER2‐mutant non‐small‐cell lung cancer. N Engl J Med. 2022;386(3):241‐251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Syed YY. Sacituzumab govitecan: first approval. Drugs. 2020;80(10):1019‐1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rugo HS, Bardia A, Marmé F, et al. Overall survival with sacituzumab govitecan in hormone receptor‐positive and human epidermal growth factor receptor 2‐negative metastatic breast cancer (TROPiCS‐02): a randomised, open‐label, multicentre, phase 3 trial. Lancet. 2023;402(10411):1423‐1433. [DOI] [PubMed] [Google Scholar]
- 74. Heitz N, Greer SC, Halford Z. A review of tisotumab Vedotin‐tftv in recurrent or metastatic cervical cancer. Ann Pharmacother. 2023;57(5):585‐596. [DOI] [PubMed] [Google Scholar]
- 75. Coleman RL, Lorusso D, Gennigens C, et al. Efficacy and safety of tisotumab vedotin in previously treated recurrent or metastatic cervical cancer (innovaTV 204/GOG‐3023/ENGOT‐cx6): a multicentre, open‐label, single‐arm, phase 2 study. Lancet Oncol. 2021;22(5):609‐619. [DOI] [PubMed] [Google Scholar]
- 76. Aschenbrenner DS. New drug treats cervical cancer. Am J Nurs. 2022;122(1):21. [DOI] [PubMed] [Google Scholar]
- 77. Dilawari A, Shah M, Ison G, et al. FDA approval summary: mirvetuximab soravtansine‐Gynx for FRα‐positive, platinum‐resistant ovarian cancer. Clin Cancer Res. 2023;29(19):3835‐3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Moore KN, Angelergues A, Konecny GE, et al. Mirvetuximab soravtansine in FRα‐positive, platinum‐resistant ovarian cancer. N Engl J Med. 2023;389(23):2162‐2174. [DOI] [PubMed] [Google Scholar]
- 79. Ponziani S, Di Vittorio G, Pitari G, et al. Antibody‐drug conjugates: the new frontier of chemotherapy. Int J Mol Sci. 2020;21(15):5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Damelin M, Zhong W, Myers J, Sapra P. Evolving strategies for target selection for antibody‐drug conjugates. Pharm Res. 2015;32(11):3494‐3507. [DOI] [PubMed] [Google Scholar]
- 81. Singh S, Serwer L, DuPage A, et al. Nonclinical efficacy and safety of CX‐2029, an anti‐CD71 Probody‐drug conjugate. Mol Cancer Ther. 2022;21(8):1326‐1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Criscitiello C, Morganti S, Curigliano G. Antibody–drug conjugates in solid tumors: a look into novel targets. J Hematol Oncol. 2021;14(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Drago JZ, Modi S, Chandarlapaty S. Unlocking the potential of antibody–drug conjugates for cancer therapy. Nat Rev Clin Oncol. 2021;18(6):327‐344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Conilh L, Sadilkova L, Viricel W, Dumontet C. Payload diversification: a key step in the development of antibody–drug conjugates. J Hematol Oncol. 2023;16(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Strop P, Delaria K, Foletti D, et al. Site‐specific conjugation improves therapeutic index of antibody drug conjugates with high drug loading. Nat Biotechnol. 2015;33(7):694‐696. [DOI] [PubMed] [Google Scholar]
- 86. Sun X, Ponte JF, Yoder NC, et al. Effects of drug–antibody ratio on pharmacokinetics, biodistribution, efficacy, and tolerability of antibody–Maytansinoid conjugates. Bioconjug Chem. 2017;28(5):1371‐1381. [DOI] [PubMed] [Google Scholar]
- 87. Hamblett KJ, Senter PD, Chace DF, et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res. 2004;10(20):7063‐7070. [DOI] [PubMed] [Google Scholar]
- 88. Duerr C, Friess W. Antibody‐drug conjugates‐stability and formulation. Eur J Pharm Biopharm. 2019;139:168‐176. [DOI] [PubMed] [Google Scholar]
- 89. Sochaj AM, Świderska KW, Otlewski J. Current methods for the synthesis of homogeneous antibody‐drug conjugates. Biotechnol Adv. 2015;33(6 Pt 1):775‐784. [DOI] [PubMed] [Google Scholar]
- 90. Bodyak ND, Mosher R, Yurkovetskiy AV, et al. The dolaflexin‐based antibody–drug conjugate XMT‐1536 targets the solid tumor lineage antigen SLC34A2/NaPi2b. Mol Cancer Ther. 2021;20(5):896‐905. [DOI] [PubMed] [Google Scholar]
- 91. Junutula JR, Raab H, Clark S, et al. Site‐specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol. 2008;26(8):925‐932. [DOI] [PubMed] [Google Scholar]
- 92. Strop P, Liu S‐H, Dorywalska M, et al. Location matters: site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem Biol. 2013;20(2):161‐167. [DOI] [PubMed] [Google Scholar]
- 93. Adem YT, Schwarz KA, Duenas E, Patapoff TW, Galush WJ, Esue O. Auristatin antibody drug conjugate physical instability and the role of drug payload. Bioconjug Chem. 2014;25(4):656‐664. [DOI] [PubMed] [Google Scholar]
- 94. Shen B‐Q, Xu K, Liu L, et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody‐drug conjugates. Nat Biotechnol. 2012;30(2):184‐189. [DOI] [PubMed] [Google Scholar]
- 95. Junutula JR, Flagella KM, Graham RA, et al. Engineered thio‐trastuzumab‐DM1 conjugate with an improved therapeutic index to target human epidermal growth factor receptor 2–positive breast cancer. Clin Cancer Res. 2010;16(19):4769‐4778. [DOI] [PubMed] [Google Scholar]
- 96. Cheng Y, Yuan X, Tian Q, et al. Preclinical profiles of SKB264, a novel anti‐TROP2 antibody conjugated to topoisomerase inhibitor, demonstrated promising antitumor efficacy compared to IMMU‐132. Front Oncol. 2022;12:951589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Li L, Hau A, Tong W, et al. Abstract LB‐227: preclinical development and characterization of STI‐6129, an anti‐CD38 antibody‐drug conjugate, as a new therapeutic agent for multiple myeloma. Cancer Res. 2020;80(16_Supplement):LB227. [Google Scholar]
- 98. Ceci C, Lacal PM, Graziani G. Antibody‐drug conjugates: resurgent anticancer agents with multi‐targeted therapeutic potential. Pharmacol Ther. 2022;236:108106. [DOI] [PubMed] [Google Scholar]
- 99. Adams S, Wilhelm A, Harvey L, et al. IMGN632: a CD123‐targeting antibody‐drug conjugate (ADC) with a novel DNA‐alkylating payload, is highly active and prolongs survival in acute myeloid leukemia (AML) xenograft models. Blood. 2016;128(22):2832. [Google Scholar]
- 100. Walsh SJ, Bargh JD, Dannheim FM, et al. Site‐selective modification strategies in antibody–drug conjugates. Chem Soc Rev. 2021;50(2):1305‐1353. [DOI] [PubMed] [Google Scholar]
- 101. Beck A, Goetsch L, Dumontet C, Corvaïa N. Strategies and challenges for the next generation of antibody–drug conjugates. Nat Rev Drug Discov. 2017;16(5):315‐337. [DOI] [PubMed] [Google Scholar]
- 102. Yoder NC, Bai C, Tavares D, et al. A case study comparing heterogeneous lysine‐ and site‐specific cysteine‐conjugated maytansinoid antibody‐drug conjugates (ADCs) illustrates the benefits of lysine conjugation. Mol Pharm. 2019;16(9):3926‐3937. [DOI] [PubMed] [Google Scholar]
- 103. Skidmore L, Sakamuri S, Knudsen NA, et al. ARX788, a site‐specific anti‐HER2 antibody‐drug conjugate, demonstrates potent and selective activity in HER2‐low and T‐DM1‐resistant breast and gastric cancers. Mol Cancer Ther. 2020;19(9):1833‐1843. [DOI] [PubMed] [Google Scholar]
- 104. Wijdeven MA, van Geel R, Hoogenboom JH, et al. Enzymatic glycan remodeling‐metal free click (GlycoConnect™) provides homogenous antibody‐drug conjugates with improved stability and therapeutic index without sequence engineering. MAbs. 2022;14(1):2078466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Zammarchi F, Corbett S, Adams L, et al. hLL2‐Cys‐PBD, a new site‐specifically conjugated, Pyrrolobenzodiazepine (PBD) dimer‐based antibody drug conjugate (ADC) targeting CD22‐expressing B‐cell malignancies. Blood. 2016;128(22):4176. [Google Scholar]
- 106. Khoury R, Saleh K, Khalife N, et al. Mechanisms of resistance to antibody‐drug conjugates. Int J Mol Sci. 2023;24(11):9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Loganzo F, Sung M, Gerber H‐P. Mechanisms of resistance to antibody–drug conjugates. Mol Cancer Ther. 2016;15(12):2825‐2834. [DOI] [PubMed] [Google Scholar]
- 108. Chang HL, Schwettmann B, McArthur HL, Chan IS. Antibody‐drug conjugates in breast cancer: overcoming resistance and boosting immune response. J Clin Invest. 2023;133(18):e172156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Loganzo F, Tan X, Sung M, et al. Tumor cells chronically treated with a trastuzumab‐maytansinoid antibody‐drug conjugate develop varied resistance mechanisms but respond to alternate treatments. Mol Cancer Ther. 2015;14(4):952‐963. [DOI] [PubMed] [Google Scholar]
- 110. Chen R, Hou J, Newman E, et al. CD30 downregulation, MMAE resistance, and MDR1 upregulation are all associated with resistance to Brentuximab Vedotin. Mol Cancer Ther. 2015;14(6):1376‐1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Filho OM, Viale G, Stein S, et al. Impact of HER2 heterogeneity on treatment response of early‐stage HER2‐positive breast cancer: phase II Neoadjuvant clinical trial of T‐DM1 combined with Pertuzumab. Cancer Discov. 2021;11(10):2474‐2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Gebhart G, Lamberts LE, Wimana Z, et al. Molecular imaging as a tool to investigate heterogeneity of advanced HER2‐positive breast cancer and to predict patient outcome under trastuzumab emtansine (T‐DM1): the ZEPHIR trial. Ann Oncol. 2016;27(4):619‐624. [DOI] [PubMed] [Google Scholar]
- 113. Coates JT, Sun S, Leshchiner I, et al. Parallel genomic alterations of antigen and payload targets mediate polyclonal acquired clinical resistance to sacituzumab govitecan in triple‐negative breast cancer. Cancer Discov. 2021;11(10):2436‐2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Ogitani Y, Aida T, Hagihara K, et al. DS‐8201a, a novel HER2‐targeting ADC with a novel DNA topoisomerase I inhibitor, demonstrates a promising antitumor efficacy with differentiation from T‐DM1. Clin Cancer Res. 2016;22(20):5097‐5108. [DOI] [PubMed] [Google Scholar]
- 115. Takegawa N, Nonagase Y, Yonesaka K, et al. DS‐8201a, a new HER2‐targeting antibody‐drug conjugate incorporating a novel DNA topoisomerase I inhibitor, overcomes HER2‐positive gastric cancer T‐DM1 resistance. Int J Cancer. 2017;141(8):1682‐1689. [DOI] [PubMed] [Google Scholar]
- 116. Yu SF, Zheng B, Go M, et al. A novel anti‐CD22 Anthracycline‐based antibody‐drug conjugate (ADC) that overcomes resistance to auristatin‐based ADCs. Clin Cancer Res. 2015;21(14):3298‐3306. [DOI] [PubMed] [Google Scholar]
- 117. García‐Alonso S, Ocaña A, Pandiella A. Resistance to antibody–drug conjugates. Cancer Res. 2018;78(9):2159‐2165. [DOI] [PubMed] [Google Scholar]
- 118. Kovtun YV, Audette CA, Mayo MF, et al. Antibody‐maytansinoid conjugates designed to bypass multidrug resistance. Cancer Res. 2010;70(6):2528‐2537. [DOI] [PubMed] [Google Scholar]
- 119. Sung M, Tan X, Lu B, et al. Caveolae‐mediated endocytosis as a novel mechanism of resistance to trastuzumab emtansine (T‐DM1). Mol Cancer Ther. 2018;17(1):243‐253. [DOI] [PubMed] [Google Scholar]
- 120. Ríos‐Luci C, García‐Alonso S, Díaz‐Rodríguez E, et al. Resistance to the antibody–drug conjugate T‐DM1 is based in a reduction in lysosomal proteolytic activity. Cancer Res. 2017;77(17):4639‐4651. [DOI] [PubMed] [Google Scholar]
- 121. Tumey LN. An overview of the current ADC discovery landscape. In: Tumey LN, ed. Antibody‐Drug Conjugates: Methods and Protocols. Springer; 2020:1‐22. [DOI] [PubMed] [Google Scholar]
- 122. Wang H, Wang W, Xu Y, et al. Aberrant intracellular metabolism of T‐DM1 confers T‐DM1 resistance in human epidermal growth factor receptor 2‐positive gastric cancer cells. Cancer Sci. 2017;108(7):1458‐1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Trudeau KM, Colby AH, Zeng J, et al. Lysosome acidification by photoactivated nanoparticles restores autophagy under lipotoxicity. J Cell Biol. 2016;214(1):25‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Singh D, Dheer D, Samykutty A, Shankar R. Antibody drug conjugates in gastrointestinal cancer: from lab to clinical development. J Control Release. 2021;340:1‐34. [DOI] [PubMed] [Google Scholar]
- 125. Trail PA, Dubowchik GM, Lowinger TB. Antibody drug conjugates for treatment of breast cancer: novel targets and diverse approaches in ADC design. Pharmacol Ther. 2018;181:126‐142. [DOI] [PubMed] [Google Scholar]
- 126. Xu Z, Guo D, Jiang Z, et al. Novel HER2‐targeting antibody‐drug conjugates of Trastuzumab beyond T‐DM1 in breast cancer: trastuzumab deruxtecan(DS‐8201a) and (Vic‐)trastuzumab duocarmazine (SYD985). Eur J Med Chem. 2019;183:111682. [DOI] [PubMed] [Google Scholar]
- 127. Knuehl C, Toleikis L, Dotterweich J, et al. Abstract 5284: M1231 is a bispecific anti‐MUC1xEGFR antibody‐drug conjugate designed to treat solid tumors with MUC1 and EGFR co‐expression. Cancer Res. 2022;82(12_Supplement):5284. [Google Scholar]
- 128. Zutshi A, Neuteboom B, Kumar S, et al. Abstract 5423: translational PK/PD/efficacy modeling and efficacious human dose prediction for a first‐in‐class MUC1‐EGFR (M1231) bispecific antibody drug conjugate. Cancer Res. 2022;82(12_Supplement):5423. [Google Scholar]
- 129. Li JY, Perry SR, Muniz‐Medina V, et al. A Biparatopic HER2‐targeting antibody‐drug conjugate induces tumor regression in primary models refractory to or ineligible for HER2‐targeted therapy. Cancer Cell. 2016;29(1):117‐129. [DOI] [PubMed] [Google Scholar]
- 130. Weisser NE, Sanches M, Escobar‐Cabrera E, et al. An anti‐HER2 biparatopic antibody that induces unique HER2 clustering and complement‐dependent cytotoxicity. Nat Commun. 2023;14(1):1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. DaSilva JO, Yang K, Surriga O, et al. A Biparatopic antibody‐drug conjugate to treat MET‐expressing cancers, including those that are unresponsive to MET pathway blockade. Mol Cancer Ther. 2021;20(10):1966‐1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Kast F, Schwill M, Stüber JC, et al. Engineering an anti‐HER2 biparatopic antibody with a multimodal mechanism of action. Nat Commun. 2021;12(1):3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Spangler JB, Neil JR, Abramovitch S, et al. Combination antibody treatment down‐regulates epidermal growth factor receptor by inhibiting endosomal recycling. Proc Natl Acad Sci U S A. 2010;107(30):13252‐13257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Pegram MD, Hamilton EP, Tan AR, et al. First‐in‐human, phase 1 dose‐escalation study of biparatopic anti‐HER2 antibody‐drug conjugate MEDI4276 in patients with HER2‐positive advanced breast or gastric cancer. Mol Cancer Ther. 2021;20(8):1442‐1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Fan J, Zhuang X, Yang X, et al. A multivalent biparatopic EGFR‐targeting nanobody drug conjugate displays potent anticancer activity in solid tumor models. Signal Transduct Target Ther. 2021;6(1):320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Nervig CS, Owen SC. Advances in the development of dual‐drug antibody drug conjugates. ADC Rev J Antibody Drug Conjug. 2023. doi: 10.14229/jadc.2023.01.05.001 [DOI] [Google Scholar]
- 137. Levengood MR, Zhang X, Hunter JH, et al. Orthogonal cysteine protection enables homogeneous multi‐drug antibody‐drug conjugates. Angew Chem Int Ed Engl. 2017;56(3):733‐737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Yamazaki CM, Yamaguchi A, Anami Y, et al. Antibody‐drug conjugates with dual payloads for combating breast tumor heterogeneity and drug resistance. Nat Commun. 2021;12(1):3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Kumar A, Kinneer K, Masterson L, et al. Synthesis of a heterotrifunctional linker for the site‐specific preparation of antibody‐drug conjugates with two distinct warheads. Bioorg Med Chem Lett. 2018;28(23):3617‐3621. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are available in the main article.