

Abstract
The attachment protein (glycoprotein) of respiratory syncytial virus (RSV) has long been associated with disease potentiation and respiratory symptoms. The glycoprotein has a conserved cysteine-rich region (GCRR) whose function is unknown and which is not necessary for efficient viral replication. In this report, we show that the GCRR is a powerful inhibitor of the innate immune response against RSV, and that early secretion of glycoprotein is critical to modulate inflammation after RSV infection. Importantly, the GCRR is also a potent inhibitor of cytokine production mediated by several TLR agonists, indicating that this peptide sequence displays broad antiinflammatory properties. These findings have important implications for RSV pathogenesis and describe an inhibitor of TLR-mediated inflammatory responses that could have clinical applications.
Keywords: glycoprotein, innate immunity, Toll-like receptor 4
Early detection of pathogens during infection relies on pattern-recognition receptors that allow the immune system an immediate response (1). The first of these receptors to be described in humans was Toll-like receptor 4 (TLR4) (1). TLR4 activates innate inflammation by promoting nuclear translocation of the NF-κB transcription factor through a conserved signal transduction pathway. NF-κB induces production of inflammatory cytokines, chemokines, vasoactive agents, adhesion molecules, proteases, and antiproteases involved in host defense (1). Activation of TLR4 can be elicited by endotoxin (LPS), and its effects are associated with a variety of illnesses, ranging from Gram-negative sepsis to asthma. Respiratory syncytial virus (RSV) also can activate TLR4 through interaction with the viral fusion (F) glycoprotein (2, 3).
RSV is the leading viral respiratory cause of hospitalization in infants and young children in the United States and in the world (4). More than 50% of infants experience an RSV infection during their first year of life, and >90% have become infected by the end of their second season of RSV exposure. Most primary infections are symptomatic, 30–71% as bronchiolitis and/or pneumonia, but only a small percentage of infected infants require hospitalization (4). The mechanisms that determine whether healthy infants develop mild or severe lung disease after primary infection are not well understood.
An important mechanism known to determine severity of disease during RSV infection is the immune response, of which innate immunity is an important component (2, 3). Although host immunity clearly is important for restricting and resolving RSV infection, it also is thought to contribute to RSV disease. The RSV F protein interacts with the CD14+/TLR4 complex in monocytes and stimulates production of proinflammatory cytokines, such as IL-6, IL-1β, and IL-8 (2), by promoting nuclear translocation of NF-κB (5). These proinflammatory cytokines play an important role in neutrophil and macrophage chemotaxis and activation during RSV disease. Furthermore, the cellular inflammatory response during severe lung disease in RSV-infected infants is composed overwhelmingly of neutrophils and macrophages (6), and loss-of-function mutations or polymorphisms in TLR4 affect severity of disease in mice and are associated with severe disease in humans (3, 7). The interaction of RSV with CD14+/TLR4 also promotes increased pulmonary infiltration with natural killer (NK) cells and is important for viral clearance after infection (3).
In addition to stimulating innate immunity, F elicits neutralizing antibody against RSV (4), has cytotoxic T lymphocyte (CTL) epitopes in mice and humans (8, 9) and has been associated with increased production of T helper 1 (Th1) cytokines (10). Conversely, the other neutralization antigen, RSV attachment protein (glycoprotein) (2), is not a strong agonist of the CD14+/TLR4 complex, does not stimulate CTL activity (8, 9), and primes for a Th2 response upon RSV infection (10, 11). No role is currently recognized for the glycoprotein in innate immunity. However, addition of glycoprotein to F protein during immunization in mice decreases production of IFN-γ by up to 70-fold upon RSV challenge compared to immunization with F alone (10). Furthermore, infection of BALB/c mice with an RSV mutant lacking the glycoprotein and small hydrophobic (SH) genes increases NK and neutrophil trafficking to the lungs compared to control mice infected with a strain of RSV that has glycoprotein and SH (12). These findings suggest that RSV glycoprotein may modulate innate inflammation.
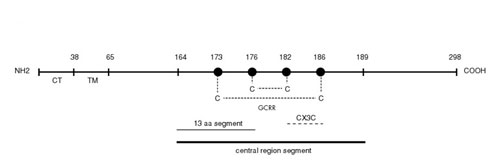
The RSV glycoprotein is produced as a transmembrane form with an N-terminal cytoplasmatic tail and an N-terminally proximal hydrophobic signal anchor, and as an N-terminally truncated soluble form that is rapidly secreted (4, 13). Although the secreted form accounts for no more than 20% of the total glycoprotein synthesized in cell culture through the course of infection, secreted glycoprotein represents ≈80% of the protein released into the medium early in infection, during the first 24 h (13). The secreted form was hypothesized to serve as a decoy to saturate the anti-RSV antibody response (4), but its timing also suggests that it might be targeted to modulate a very early event, like TLR4 mediated innate immunity. The ectodomain of the glycoprotein consists of two mucin-like domains, with divergent amino acid sequences between isolates, separated by a short, circumscribed central region that is highly conserved between RSV antigenic subgroups A and B (13). This conserved region includes a 13-aa segment (amino acid 164–176) that is identical in all wild-type isolates of RSV and overlaps four cysteine residues (positions 173, 176, 182, and 186) held by disulfide bonds between 173–186 and 176–182 (14) (see Fig. 6, which is published as supporting information on the PNAS web site). The conservation of the 13-aa segment and the cysteine-rich region (GCRR) among RSV isolates originally had been interpreted as indicating a role in receptor binding, but recent data have shown that they are not required for efficient infection in vitro and in mice (15, 16). As a consequence, the reason for early secretion of RSV glycoprotein and the role of its conserved GCRR remain unexplained. We now show that RSV glycoprotein, through its GCRR, antagonizes the proinflammatory effect of RSV F regulating the innate immune response. Furthermore, the glycoprotein has a similar effect on the unrelated TLR4 agonist LPS, indicating that the GCRR has broad antiinflammatory properties.
Methods
Virus Infection and Sampling. Four- to 6-week-old female C57BL/10 mice (The Jackson Laboratories) were used for these experiments. Intranasal infection was performed with 106 plaque-forming units (pfu) of live RSV, ΔG, and mG. RSV titers in the lungs of mice were determined as described (17). A severity scoring system was used to characterize the degree of pulmonary infiltration. Briefly, for innate inflammation, the lung parenchyma was scored as: 0, absence of inflammation; 1, <20% of field with focal polymorphonuclear (PMN) and macrophage inflammation; 2, 20% or more of the field with focal PMN and macrophage inflammation; and 3, diffuse PMN and macrophage inflammation. Scores were assigned by blinded examiners (five to six mice per group).
Monocyte Stimulation. Human peripheral blood mononuclear cells were isolated from leukopaks by using Histopaque (Sigma). Monocytes were isolated by using the Monocyte Isolation kit II (positive selection for CD3, CD7, CD16, CD19, CD56, CD123, and glycophorin A, Miltenyi Biotec) with Macs LS separation columns. Remaining cells were >90% monocytes by anti-CD14 staining and forward- and side-light scatter analysis using FACScan (Becton Dickinson).
Purified monocytes were stimulated with LPS (1 μg) or purified proteins F or glycoprotein (3 μg; kindly provided by V. Randolph, Wyeth Lederle, NY). All rRSV variants were grown in Hep-2 cells and Vero cells as described (15, 16), and used at 105 pfu [multiplicity of infection (MOI) = 1] for stimulations. Lysate of Hep-2 cells and Vero cells were used as control stimuli. UV-inactivation was performed as described (5), and lack of virus replication was confirmed in cell culture. For additional experiments, three RSV (GCys182Arg, GCys186Arg, and control), selected by using glycoprotein-specific monoclonal antibodies as described, were used (14). Peptides used for stimulations included the GCRR 164HFEVFNFVPCSICSNNPTCWAICKRI189, a cysteine to serine control peptide 164HFEVFNFVPSSISSNNPTSWAISKRI189 (GSRR), and a 321YFARGPGIHIRKRKN307 reverse-oriented HIV V3 loop. An additional peptide encoding the conserved 13-aa region upstream of the GCRR (164–176; ref. 4) was also used in add-back experiments. All peptides were synthesized by 9-fluorenylmethoxycarbonyl solid-phase chemistry (SynPep, Dublin, CA). Selective formation of disulfide bonds in GCRR was accomplished by protection of two selective cysteine residues (176 and 182) with acid-labile groups, and two with non-acid-labile groups (173 and 186). Sequential removal of the acid-labile protection groups followed by oxidation and disulfide bond formation, and subsequent deprotection of non-acid-labile groups followed by the same process led to selective formation of the native 1–4/2–3 bridges. Peptides were tested by analytical HPLC, mass spectrometry and LC/MS (SynPep). The GCRR and control peptides included biotin-SGSG at the N termini. The anti-CX3CR1 antibody was kindly provided by P. Murphy (National Institute of Allergy and Infectious Diseases, Bethesda) and used as described (18). For supplemental experiments, we used peptidoglycan (PGN; Fluka, Sigma) at 10 μg and CpGDNA (GTCGTT; HyCult Biotechnology) at 1 mM. Cytokines were measured in supernatant fluids 18 h after stimulation by immunoassay following manufacturer's instructions (Biosource Europe).
Flow Cytometry. Alveolar macrophages were obtained by BAL followed by magentic bead depletion (Militenyi Biotec). Macrophages were incubated with brefeldin A for 6 h, fixed with Cytofix/Cytoperm (Becton Dickinson), and stained with phycoerythrin-conjugated anti-IL-6 antibody (Becton Dickinson). Data were analyzed by using side and forward scatter plots and FACScan (Becton Dickinson).
NF-κB Nuclear Translocation Immunoassay. Human monocytes were stimulated with purified RSV F and/or glycoprotein (1 μg each) or indicated viruses for 60 min. After stimulation, nuclear extracts were obtained by using a hypotonic lysis buffer (10 mM Hepes, pH 7.9/1.5 mM MgCl2/10 mM KCl/0.5 mM DTT/0.1% Triton X-100 and protease inhibitors) and an extraction buffer (20 mM Hepes, pH 7.9/1.5 mM MgCl2/0.42 M NaCl/0.5 mM DTT/0.2 mM EDTA/1.0% Igepal CA-630/25% vol/vol glycerol, and protease inhibitors). NF-κB subunits p50 and p65 were detected by a modified immunoassay using a double-stranded byotinylated oligonucleotide containing the consensus sequence for NF-κB binding (5′-GGGACTTTCC-3′) (Chemicon).
IκBα Western Blot. Purified human monocytes were incubated with the corresponding recombinant RSV for 60 min at 37°C, collected and lysed in isotonic buffer (10 mM Hepes-KOH, pH 7.2/142.5 mM KCl/5 mM MgCl2/1 mM EGTA/0.3% Nonidet P-40). Proteins were separated by SDS/PAGE, transferred onto poly(vinylidene difluoride) membranes (Millipore, Bedford, MA), and blocked with 5% milk in PBS-T (1X PBS, 0.1% Tween-20). IκBα was detected with a rabbit anti-IκBα (Santa Cruz Biotechnology), followed by a horseradish peroxidase-conjugated anti-rabbit IgG (Amersham Pharmacia) and developed with SuperSignal Pico Chemiluminescent Substrate (Pierce).
Statistical Analysis. Data were analyzed with statistical software (statview). Comparisons were made by using the Mann–Whitney U test where appropriate. All experimentation was approved by The Johns Hopkins Medical Institutions.
Results
The Central Region of RSV Glycoprotein Inhibits Production of Inflammatory Cytokines in Vitro. To determine whether the RSV glycoprotein can inhibit F-mediated monocyte production of proinflammatory cytokines, we incubated purified human monocytes with purified protein F, purified glycoprotein from subgroup A, or a combination of F plus glycoprotein A and examined supernatant fluids for production of IL-6 (Fig. 1a). Incubation of monocytes with F elicited high levels of IL-6, whereas levels were low after incubation with glycoprotein. Monocytes incubated with both proteins decreased IL-6 production by ≈1.5 log compared to monocytes incubated only with F (P < 0.01). In contrast, addition of equivalent amounts of BSA or Hep-2 cell lysate to F-treated monocytes had no effect on IL-6 production. An inhibitory effect similar to that observed for glycoprotein on F-induced IL-6 production also was detected when IL-1β or IL-10 were measured in supernatant fluids (data not shown), and when the glycoprotein from RSV antigenic subgroup B was used instead of the glycoprotein from the RSV A2 strain of subgroup A (Fig. 1a).
Fig. 1.

Production of IL-6 by monocytes after stimulation with purified proteins (a) or live RSV (b). (a) Purified human monocytes were stimulated with RSV F protein (filled bar), RSV glycoprotein from antigenic subgroup A (GA protein, open bar) or antigenic subgroup B (GB protein, gray bar), RSV F and GA proteins (horizontal striped bar), RSV F and GB proteins (vertical striped bar), RSV F protein and GCRR peptide (3 μg, dotted bar and 15 μg, crossed bar), RSV F protein and BSA (dark gray bar), and RSV F protein and Hep-2 cell lysate (black stripped bar). (b) Dose–response curves to RSV (filled squares) or the ΔG mutant (open squares). IL-6 and IL-1β were measured by immunoassay in supernatant fluids 18 h after incubation for in a and b. Results are mean ± SEM and are representative of three independent experiments.
Inhibition of inflammatory cytokine production elicited by glycoproteins A and B despite the overall divergence in amino acid sequence between the two proteins (4, 13) suggested that the domain exerting the modulatory effect could localize to the conserved central segment of both proteins (Fig. 6). To determine whether the modulatory effect on cytokine production was elicited by this conserved central region, we incubated purified human monocytes with F protein in combination with increasing concentrations of a synthetic peptide representing amino acids 164–189 of the RSV glycoprotein A, which includes the conserved 13-aa segment and the GCRR (Fig. 1a). Interestingly, increasing concentrations of this peptide led to a dose-dependent inhibition of F-mediated IL-6 production. In contrast, the inhibitory effect was not observed when another glycoprotein A peptide (residues 273–288) within the glycoprotein mucin-like domain or an unrelated V3 loop peptide from HIV type 1 (residues 307–321) were added with F as controls (data not shown).
The Glycoprotein Inhibits Monocyte Production of Inflammatory Cytokines upon Exposure to RSV. To determine whether the RSV glycoprotein also inhibited cytokine production during RSV infection, purified monocytes were incubated with increasing concentrations of wild-type RSV or with a live recombinant (r) RSV that does not express the glycoprotein (ΔG) (16), and supernatant fluids were assayed for inflammatory cytokines 18 h later (Fig. 1b). Exposure of monocytes to live ΔG increased production of IL-6 and IL-1β in a dose-dependent manner compared to exposure to an identical RSV with normal intact glycoprotein (Fig. 1b). Differences between viruses were highest at an MOI of 1, which was therefore used for subsequent experiments.
To determine whether the secreted form of the glycoprotein was responsible for the modulatory effect observed during live RSV infection, we incubated purified monocytes with a rRSV in which the glycoprotein gene was modified to express only the membrane-anchored (mG) form (ref. 16; Fig. 2a). Incubation of monocytes with mG enhanced IL-6 production to similar levels compared to ΔG, demonstrating that the secreted form of glycoprotein is required to modulate production of inflammatory cytokines during live RSV infection. Importantly, the IL-6 response was restored to levels similar to those elicited by RSV when soluble glycoprotein was added back to the culture media of mG-infected cells (Fig. 2a).
Fig. 2.

Production of IL-6 after incubation with viruses. (a) Purified monocytes were incubated with RSV (closed bar), Vero cell lysate (gray bar), ΔG (open bar), mG (horizontal striped bar), mG with soluble G (black stripped bar), GΔ172–187 (gray horizontal stripped bar), or ΔG + G164–176 peptide (10 μg) (dotted bar). All viruses were inoculated at 105 pfu (MOI = 1). (b) Purified monocytes were incubated with UV-inactivated RSV (closed bar) or ΔG (open bar). (c) Purified monocytes were incubated with RSV lacking one of four cysteines in the GCRR and therefore the corresponding disulfide bridge (GCys182Arg and GCys186Arg), in addition to the correspondent control virus (14), to examine cytokine production. IL-6 was measured by immunoassay in supernatant fluids 18 h after infection. Results are mean ± SEM and are representative of two to three independent experiments.
Subsequently, to determine whether live RSV infection is required for the observed modulatory effects, we incubated human monocytes with UV-inactivated RSV and ΔG and measured cytokine production (Fig. 2b). Again, ΔG led to enhanced IL-6 production compared to RSV, demonstrating that live infection is not necessary for glycoprotein modulation of the monocyte inflammatory response.
The Conserved GCRR Is Critical for the Inhibitory Effect. The 13-aa segment between positions 164 and 176 (G164–176) is a conserved segment in the ectodomain of the glycoprotein, and it overlaps a GCRR located between positions 173–186, whose cysteine residues are invariant in both RSV subgroups A and B (4). To further elucidate the inhibitory role of the conserved central segment of the glycoprotein, purified human monocytes were incubated with a rRSV that lacked the GCRR (GΔ172–187) (Fig. 2a). Incubation with GΔ172–187 led to higher levels of IL-6 than incubation with RSV, demonstrating that the GCRR is critical to modulate innate inflammatory responses in this model. Similar results were obtained with UV-inactivated viruses (data not shown). We then further examined the importance of the cysteine residues on the modulatory effect exerted by glycoprotein by incubating human monocytes with two RSV mutants containing substitutions at cysteine residues 182 (GCys182Arg) or 186 (GCys186Arg) (Fig. 2c). Each of these viruses, GCys182Arg and GCys186Arg, present substitutions disrupting one of the two disulfide bridges in the GCRR (Fig. 6). Interestingly, both GCys182Arg and GCys186Arg elicited higher IL-6 levels that their RSV control (14) at every MOI tested, confirming that the presence of the cysteine residues is essential for the modulatory effect displayed by the GCRR.
To investigate whether the fractalkine motif in the GCRR (Fig. 6) played a role in the modulatory effect on cytokine production, human monocytes were incubated with RSV and a polyclonal anti-CX3CR1 antibody known to block binding of the fractalkine motif in the glycoprotein to the CX3CR1 chemokine receptor (18). Production of IL-6 was identical in presence or absence of anti-CX3CR1 antibody (data not shown). Finally, to explore the role of the conserved 13-aa segment upstream of the GCRR in modulation of inflammatory cytokines, we incubated monocytes with ΔG in presence of a glycoprotein peptide encompassing amino acids 164–176 (G164–176) (Fig. 2a). G164–176 failed to modulate IL-6 production by ΔG, indicating that the 13-aa peptide upstream of the GCRR provided in trans cannot complement the inhibitory effect of the glycoprotein during live infection. Similar results were observed when using G164–176 with mG (data not shown).
The Glycoprotein Decreases Nuclear Translocation of the NF-κB Transcription Factor. To determine whether the inhibitory effect of the glycoprotein on inflammatory cytokine production is associated with decreased nuclear translocation of NF-κB, we incubated human monocytes with F protein, glycoprotein, F plus glycoprotein, and with either RSV or ΔG and extracted the nuclei to measure translocation of NF-κB (Fig. 3 a and b). NF-κB nuclear translocation was decreased in the presence of the glycoprotein either after purified protein or live virus stimulation. In addition, IκBα blots from extracts of human monocytes after stimulation with RSV, ΔG, or GΔ172–187 showed that degradation of IκBα was greater in cells stimulated with ΔG or GΔ172–187 than in those incubated with RSV (Fig. 3c). Taken together, these results demonstrate that RSV glycoprotein, through its GCRR, inhibits nuclear translocation of NF-κB to modulate the innate inflammatory response during RSV infection.
Fig. 3.

Nuclear translocation of transcription factor NF-κB in human monocytes. (a and b) Monocytes were stimulated for 60 min with F and G protein individually and in combination (a) or RSV or ΔG (MOI = 1) (b), and NF-κB subunits p50 and p65 were detected in purified nuclei by a modified immunoassay. (c) Monocytes were stimulated for 60 min with RSV, ΔG, and GΔ172–187 (MOI = 1), and cytoplasmatic IκBα was detected by Western blot.
RSV Glycoprotein Decreases Inflammation During RSV Infection in Vivo. To determine whether the modulatory effect of RSV glycoprotein affected the innate immune response in vivo, we first obtained alveolar macrophages from naïve mice and incubated them with purified F, glycoprotein and recombinant viruses (Fig. 4 a and b). Cytokine production in murine alveolar macrophages mimicked the response previously observed in human monocytes (Fig. 1), with the glycoprotein modulating the innate responses elicited by both purified F and RSV. We subsequently infected mice intranasally with RSV or ΔG and measured intracellular production of IL-6 in pulmonary macrophages after infection. ΔG increased production of intracellular IL-6 in pulmonary macrophages compared to RSV (79% vs. 21% of purified macrophages in Fig. 4d) 24 h after infection.
Fig. 4.

The role of RSV glycoprotein during infection in vivo. Purified murine alveolar macrophages were stimulated with RSV F protein (closed bar), RSV glycoprotein from antigenic subgroup A (GA protein, open bar) or RSV F and glycoproteinA (horizontal stripped bar) (a); or RSV (dark gray bar), ΔG (vertical stripped bar), or GΔ172–187 (black horizontal stripped bar) (b). (c) Virus titration in lungs of mice 4 days after infection. Viruses were inoculated intranasally at 106 pfu (n = 5 per group). (d) Intracellular expression of IL-6 by alveolar macrophages from mice infected with RSV, ΔG, or placebo analyzed by flow cytometry 24 h after infection. Viruses were inoculated intranasally at 106 pfu. (e) Pulmonary histopathology score assessing PMN and macrophage infiltration after infection with RSV (filled squares), ΔG (open squares), or mG (gray squares). (f) Pulmonary histopathology in mice 24 h after infection with the indicated virus (PAS, ×10).
To determine whether this increase in production of inflammatory cytokines after ΔG infection was associated with increased replication of ΔG in the lungs of mice compared to RSV, we measured lung titers of both viruses in infected animals (Fig. 4c). Consistent with previous findings (15, 16), RSV replicated to higher titers in lungs of mice than ΔG (P = 0.001). Thus, the proinflammatory effect of ΔG is not associated with heightened replication in the respiratory tract, but rather occurred despite a dramatic decrease in replication. Conversely, replication of mG was similar to RSV (P > 0.05) (Fig. 4c) despite eliciting different inflammatory responses (see below, Fig. 4 e and f).
To further examine the effect of the glycoprotein in vivo on the innate immune response, we stained lung sections of mice infected with RSV, ΔG, and mG with periodic acid Schiff (PAS) to compare the degree of pulmonary granulocyte and mononuclear cell infiltration on days 1, 4, and 7 after infection (Fig. 4 e and f). Considering that replication levels may affect histopathology (19), we selected mG in addition to ΔG because, unlike the highly restricted replication of ΔG, replication of mG is not restricted in lungs of mice (16). Furthermore, the majority of glycoprotein early after infection is secreted and therefore absent in mG (13, 16). As shown in Fig. 4 e and f, early after infection, granulocyte and macrophage alveolar infiltration was increased in mice infected with mG and ΔG compared to mice infected with RSV. Mice infected with ΔG had focal areas of increased alveolar inflammation, whereas the neutrophil and macrophage infiltration in mG recipients was more diffuse. The inflammatory response elicited by the three viruses leveled 7 days after infection, when adaptive responses are an important component of the immune response.
The GCRR Inhibits Endotoxin-Mediated Cytokine Production. The ability of the GCRR to antagonize the production of proinflammatory cytokines in RSV-infected or F protein-exposed monocytes suggested that this protein region could inhibit inflammatory responses elicited by other CD14+/TLR4 agonists. Therefore, we examined whether addition of the GCRR peptide inhibited production of cytokines in monocytes stimulated by LPS (Fig. 5). For this purpose, we incubated purified human monocytes with LPS and increasing concentrations of the GA central region peptide (amino acids 164–189) and measured cytokine production. As expected, incubation of monocytes with LPS alone elicited high levels of IL-6 and IL-1β in supernatant fluid. Remarkably, addition of the GCRR peptide to the LPS-treated monocytes caused a dose-dependent inhibition of cytokine production. Addition of an unrelated HIV V3 loop control peptide or a modified central region peptide in which the four cysteine residues had been replaced by serines (GSRR), and therefore lacked disulfide bridges, had no effect (Fig. 5). Similar results were observed when incubating LPS with a scrambled peptide containing the same amino acid composition as GCRR, but in random order (VFNHFECSIFVPCSNRICWANPTICK, data not shown). Addition of the GCRR peptide to monocytes incubated with LPS also affected production of IL-10 (data not shown). These findings demonstrate that the GCRR not only modulates RSV-mediated inflammation, but also antagonizes LPS-mediated production of inflammatory cytokines.
Fig. 5.

Production of IL-6 (a) and IL-1β (b) by monocytes after stimulation with endotoxin. Cells were incubated with LPS (1 μg; filled bar), LPS and GCRR peptide (3 μg, dotted bar, and 15 μg, hatched bar), and LPS and control HIV V3 loop peptide (3 μg, horizontal stripped bar, and 15 μg, vertical stripped bar) or LPS and GSRR peptide lacking the four cysteines and disulfide bridges (15 μg, open bar). Supernatants were collected 18 h after addition of stimulants. IL-6 and IL-1β were measured by immunoassay. Results are given as mean ± SEM and are representative of three independent experiments.
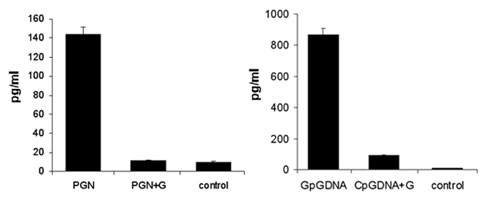
Finally, we examined whether the glycoprotein affected the inflammatory response elicited by other molecules involved in innate inflammation. Therefore, we incubated human monocytes with the TLR2 agonist PGN and the TLR9 agonist CpGDNA and examined whether coincubation with the glycoprotein affected cytokine production (Fig. 7, which is published as supporting information on the PNAS web site). As previously observed with LPS, cytokine production was inhibited by the glycoprotein, showing that this molecule is able to counteract a variety of proinflammatory stimuli.
Discussion
This study identified a role for the conserved GCRR of RSV, a region that is highly conserved in wild-type isolates (4, 13, 14). As shown here, the RSV glycoprotein modulates monocyte/macrophage cytokine production by inhibiting nuclear translocation of NF-κB, and decreases inflammation in the lungs early after RSV infection. In addition, our data show that the GCRR modulates inflammatory responses elicited by other TLR agonists, indicating that this sequence displays broad antiinflammatory properties.
Previous studies proposed a variety of immunological effects associated with the RSV glycoprotein. Preferential priming with glycoprotein, due to disruption of protein F during formalin inactivation of RSV, had been hypothesized to be the basis for an enhanced form of RSV disease that occurred in recipients of a formalin-inactivated RSV vaccine after subsequent natural exposure to RSV in the community (20). However, reports from others and our laboratory demonstrated that this protein is not necessary for disease enhancement by showing similar severity of illness after RSV challenge in mice immunized with inactivated RSV vaccines with or without glycoprotein (17, 21). In addition, the glycoprotein has been reported to induce leukocyte chemotaxis in vitro (18), and has been associated with an adaptive immune response that contributes to wheezing and asthma (11). Our results show that RSV glycoprotein inhibits production of inflammatory cytokines early after infection, thereby modulating the innate inflammatory response to the virus. These findings may have important implications on adaptive immunity and deserve further study, as the glycoprotein may affect cytotoxic T lymphocyte responses and other mechanisms of viral clearance (3), along with its effect on the Th bias of the immune response (11).
Several proteins and drugs modulate the innate inflammatory response. Glucocorticoids affect NF-κB dependent gene induction, presumably by interfering with direct contacts between p65 and the transcriptional machinery (22). Some additional mechanisms that may silence NF-κB dependent genes in different cell lines are associated with the RBP-Jκ corepressor complex, Foxj1, the single Ig IL-1R-related molecule, and the p38 and ERK inhibitors (23, 24). The wide specificity of GCRR modulation suggests that its effects are exerted directly or indirectly through pathways common to a variety of proinflammatory agents. Interestingly, the conserved GCRR has a structural homology with the fourth subdomain of TNFR1, suggesting that its modulatory effect may be associated with binding and inactivation of TNF or an unknown TNF homologue (25). TNF also mediates endotoxin-induced shock, and TNFR1-deficient mice are resistant to lethal dosages of endotoxin (26, 27). Furthermore, shedding of the TNFR1 modulates innate immune activation (28). Indeed, early secretion of G may bind TNF-α and contribute to delay RSV clearance, as early production of TNF-α is protective against RSV infection in mice (29).
Sequencing studies have shown that the glycoprotein is the most variable protein between the RSV subgroups, with only 53% identity between the proteins of subgroup A and B prototypical strains (4, 14). The inhibitory effect described here was characteristic of both RSV subgroups A and B and was elicited by the conserved GCRR, in which the cysteine residues forming two disulfide bonds between positions 173–186 and 176–182 (4, 13) played a critical role. Even though amino acid sequences of the A and B GCCRs are not identical, this inhibitory effect may likely be associated with its conformation rather than with conservation of the exact sequence between serogroups. The conserved central region of the G protein also contains a 13-aa segment that is immediately upstream of the GCRR and is conserved among human isolates. This segment did not appear to be involved in modulating innate immunity, at least not on its own, and its significance to RSV replication and biology remains to be identified.
An important observation in this manuscript is that the modulatory effect of the glycoprotein on inflammation is also observed when using inactivated RSV. Therefore, the modulatory effect of glycoprotein on NF-κB nuclear translocation is probably exerted by secreted glycoprotein already present in supernatant fluids containing the RSV inoculum (15, 16). Supporting this notion, reconstitution of cultures of human monocytes inoculated with mG with purified soluble glycoprotein led to responses identical to those observed with wild-type RSV. Interestingly, because infection in vivo is a sequential process involving multiple rounds of replication and spreading from infected to uninfected cells, the first cycle of RSV replication upon infection is probably not affected early on by the modulatory effect, but provides secreted glycoprotein to areas where the virus is expanding and affects the immune response directed against it.
An important finding of these studies is that the GCRR can also modulate LPS-mediated cytokine production. Genetic polymorphisms in TLR4 but not in CD14 appear to affect severity of disease both during Gram-negative sepsis and RSV infection (7, 30). The inhibitory effect of the GCRR on LPS-mediated inflammation may have implications for the treatment of severe diseases. LPS plays a critical role in many illnesses, among others septic shock due to Gram-negative bacteria and development of childhood asthma (30, 31). In addition, inflammation elicited by other proinflammatory agents through other TLR receptors is also affected by this protein region.
In summary, we have described a role for the RSV GCRR, a conserved domain present in all wild type isolates, offered a significance to the early secretion of glycoprotein after RSV infection, and revealed an increased complexity of the regulation of the host immune response during RSV infection.
Supplementary Material
Acknowledgments
We thank Maria del Carmen Puggioli for excellent technical assistance. This work was supported in part by a National Institute of Environmental Health Sciences contract mechanism with Johns Hopkins University and Fundacion INFANT and National Institutes of Health Grant AI-054952 (to F.P.P.).
Author contributions: F.P.P., P.M.I., and S.R.K. designed research; F.P.P., P.M.I., S.J.H., M.P.S., G.A.M., M.F.D., F.R.L., and B.T. performed research; B.T., R.M.H., J.A.M., R.A.K., and P.L.C. contributed new reagents/analytic tools; F.P.P., P.M.I., and S.R.K. analyzed data; and F.P.P. and P.M.I. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: TLR4, Toll-like receptor 4; RSV, respiratory syncytial virus; NK, natural killer; Thn, T helper n; pfu, plaque-forming unit; PMN, polymorphonuclear; MOI, multiplicity of infection.
Note Added in Proof. While this paper was being reviewed, Arnold et al. (32) reported a modulator effect for secreted glycoprotein during RSV infection of human epithelial cells in vitro.
References
- 1.Medzhitov, R. (2001) Nat. Rev. Immunol. 1, 135–145. [DOI] [PubMed] [Google Scholar]
- 2.Kurt-Jones, E. A., Popova, L., Kwinn, L., Haynes, L. M., Jones, L. P., Tripp, R. A., Walsh, E. E., Freeman, M. W., Golenbock, D. T., Anderson, L. J., et al. (2000) Nat. Immunol. 1, 398–401. [DOI] [PubMed] [Google Scholar]
- 3.Haynes, L. M., Moore, D. D., Kurt-Jones, E. A., Finberg, R. W., Anderson, L. J. & Tripp, R. A. (2001) J. Virol. 75, 10730–10737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Collins, P. L., Chanock, R. M. & Murphy, B. R. (2001) in Fields Virology, eds. Knipe, D. M. & Howley, P. M. (Lippincott Williams and Wilkins, Philadelphia), pp. 1443–1486.
- 5.Haeberle, H. A., Takizawa, R., Casola, A., Brasier, A. R., Dieterich, H. J., Van Rooijen, N., Gatalica, Z. & Garofalo, R. P., et al. (2002) J. Infect. Dis. 186, 1199–1206. [DOI] [PubMed] [Google Scholar]
- 6.Hull, J., Thomson, A. & Kwiatkowski, D. (2000) Thorax 55, 1023–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tal, G., Mandelberg, A., Dalal, I., Cesar, K., Somekh, E., Tal, A., Oron, A., Itskovich, S, Ballin, A., Houri, S., et al. (2004) J. Infect. Dis. 189, 2057–2063. [DOI] [PubMed] [Google Scholar]
- 8.Chang, J., Srikiatkhachorn, A. & Braciale, T. J. (2001) J. Immunol. 167, 4254–4260. [DOI] [PubMed] [Google Scholar]
- 9.Nicholas, J. A., Rubino, K. L., Levely, M. E, Adams, E. G. & Collins, P. L. (1990) J. Virol. 64, 4232–4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jackson, M. & Scott, R. (1996) J. Med. Virol. 49, 161–169. [DOI] [PubMed] [Google Scholar]
- 11.Hancock, G. E., Scheuer C. A., Sierzega R., Pryharski, K. S., McBride, J. T., Watelet, L. F., Tebbey, P. W. & Smith, J. D. (2001) J. Infect. Dis. 184, 1589–1593. [DOI] [PubMed] [Google Scholar]
- 12.Tripp, R. A., Jones, L. & Anderson, L. J. (2000) J. Virol. 74, 6227–6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson, P. R., Spriggs, M. K., Olmsted, R. A. & Collins, P. L. (1987) Proc. Natl. Acad. Sci. USA 84, 5625–5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rueda, P., García-Barreno, B. & Melero, J. A. (1994) Virology 198, 653–662. [DOI] [PubMed] [Google Scholar]
- 15.Teng, M. N. & Collins, P. L. (2002) J. Virol. 76, 6164–6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Teng, M. N., Whitehead, S. S. & Collins, P. L. (2001) Virology 289, 283–296. [DOI] [PubMed] [Google Scholar]
- 17.Polack, F. P., Teng, M. N., Collins, P. L., Prince, G. A., Exner, M., Regele, H., Lirman, D. D., Rabold, R., Hoffman, S. J., Karp, C. L., et al. (2002) J. Exp. Med. 196, 859–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tripp, R. A., Jones, L. P., Haynes, L. M., Zheng, H., Murphy, P. M. & Anderson, L. J. (2001) Nat. Immunol. 2, 732–738. [DOI] [PubMed] [Google Scholar]
- 19.Johnson, T. R., Parker, R. A., Johnson, J. E. & Graham, B. S. (2003) J. Immunol. 170, 2037–2045. [DOI] [PubMed] [Google Scholar]
- 20.Openshaw, P. J. (1995) Am. J. Resp. Crit. Care. Med. 152, S59–S62. [DOI] [PubMed] [Google Scholar]
- 21.Johnson, T. R., Teng, M. N., Collins, P. L. & Graham, B. S. (2004) J. Virol. 78, 6024–6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Bosscher, K., Schmitz, M. L., Vanden Berghe, W., Plaisance, S., Fiers, W. & Haegeman, G. (1997) Proc. Natl. Acad. Sci. USA 94, 13504–13509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin, L., Spoor, M. S., Gerth, A. J., Brody, S. L. & Peng, S. L. (2004) Science 303, 1017–1020. [DOI] [PubMed] [Google Scholar]
- 24.Wald, D., Qin, J., Zhao, Z., Qian, Y., Naramura, M., Tian, L., Towne, J., Sims, J. E., Stark, G. R. & Li, X. (2003) Nat. Immunol. 4, 920–927. [DOI] [PubMed] [Google Scholar]
- 25.Langedijk, J. P., de Groot, B. L., Berendsen, H. J. & van Oirschot, J. T. (1998) Virology 243, 293–302. [DOI] [PubMed] [Google Scholar]
- 26.Beutler, B., Milsark, I. W. & Cerami, A. (1985) Science 229, 869–871. [DOI] [PubMed] [Google Scholar]
- 27.Pfeffer, K., Matsuyama, Y., Kundig, T. M., Wakeham, A., Kishihara, K., Shahinian, A., Wiegmann, K., Ohashi, P. S., Kronke, M. & Mak, T. W. (1993) Cell 73, 457–467. [DOI] [PubMed] [Google Scholar]
- 28.Xanthoulea, S., Pasparakis, M., Kousteni, S., Brakebusch, C., Wallach, D., Bauer, J., Lassmann, H. & Kollias, G. (2004) J. Exp. Med. 200, 367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rutigliano, J. A. & Graham, B. S. (2004) J. Immunol. 173, 3408–3417. [DOI] [PubMed] [Google Scholar]
- 30.Agnese, D. M., Calvano, J. E., Hahm, S. J., Coyle, S. M., Corbett, S. A., Calvano, S. E. & Lowry, S. F. (2002) J. Infect. Dis. 186, 1522–1525. [DOI] [PubMed] [Google Scholar]
- 31.Braun-Fahrlander, C., Riedler, J., Herz, U., Eder, W., Waser, M., Grize, L., Maisch, S., Carr, D., Gerlach, F., Bufe, A., et al. (2002) N. Engl. J. Med. 347, 869–877. [DOI] [PubMed] [Google Scholar]
- 32.Arnold, R., Konig, B., Werchau, H. & Konig, W. (2004) Virology 330, 384–397. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}