

Abstract
Reversible photoswitching of individual molecules has been demonstrated for a number of mutants of the green fluorescent protein (GFP). To date, however, a limited number of switching events with slow response to light have been achieved at the single-molecule level. Here, we report reversible photoswitching characteristics observed in individual molecules of Dronpa, a mutant of a GFP-like fluorescent protein that was cloned from a coral Pectiniidae. Ensemble spectroscopy shows that intense irradiation at 488 nm changes Dronpa to a dim protonated form, but even weak irradiation at 405 nm restores it to the bright deprotonated form. Although Dronpa exists in an acid–base equilibrium, only the photoinduced protonated form shows the switching behavior. At the single-molecule level, 488- and 405-nm lights can be used to drive the molecule back and forth between the bright and dim states. Such reversible photoswitching could be repeated >100 times. The response speed to irradiation depends almost linearly on the irradiation power, with the response time being in the order of milliseconds. The perfect reversibility of the Dronpa photoswitching allows us to propose a detailed model, which quantitatively describes interconversion among the various states. The fast response of Dronpa to light holds great promise for following fast diffusion or transport of signaling molecules in live cells.
Keywords: photochromism, protonation/deprotonation, fluorescence microscopy
Photoinduced alteration of chemical and physical properties of photochromic molecules is of great interest because of its potential applications for optoelectronic devices, such as optical memory and optical switches (1). Photoinduced switching of fluorescent properties is one of the most attractive concepts for the realization of a nondestructive read-out system (2–4). Apart from this application, the photoswitching behavior of green fluorescent proteins (GFPs) or GFP-like proteins is being recognized as new methodology of optical marking (5, 6). Intracellular dynamics of selected molecules can be followed by activating the fluorescent proteins to their fluorescent state (7–11). Realization of photoswitching at the single-molecule level will open up exciting opportunities in the field of optoelectronics and biological imaging, where it could provide molecular-scale devices as well as detection of fast dynamics of individual proteins in living cells. Reversible photoswitching at the single-molecule level, however, has not yet been well characterized (12–17). Dickson et al. (12) reported reversible photoswitching of a mutant of GFP. Although they demonstrated a few photoswitching events at the single-molecule level, minutes of illumination was required to achieve the switching. Irie and coworkers (13, 14) also reported reversible photoswitching of diarylethene derivatives, which occurred relatively slowly with a response time of seconds. Although the switching can be repeated >104 times at the ensemble level (1), the number of switching events obtained at the single-molecule level is limited. More recently, reversible photoswitching of carbocyanine dyes at the single-molecule level was reported by Heilemann et al. (15) and Bates et al. (16). In proper conditions, >100 switching events were reported (15), although the response times are still slow (15, 16). Therefore, photochromic substances that show durable and prompt switching even at the single-molecule level have been the topic of intense research.
Recently, Ando et al. (18) developed a reversibly photoswitchable fluorescent protein named Dronpa. Although the protein has its absorption maximum at 503 nm and emits bright fluorescence peaking at 518 nm, it can be converted on intense irradiation at 488 nm into a dim state that has an absorption spectrum peaking at 390 nm, and the dim state is switched back to the original emissive state with minimal irradiation at 405 nm. By using this protein, fast protein dynamics, such as nucleocytoplasmic shuttling of signaling proteins, was examined at multiple time points in individual cells. In this work, we demonstrate the reversible photoswitching behavior of Dronpa at the single-molecule level. Individual Dronpa molecules were photoswitched reversibly >100 times. The switching occurred in a millisecond time scale. Based on the results of both ensemble and single-molecule spectroscopic experiments, we propose a scheme explaining the reversible photoswitching behavior of Dronpa.
Experimental Procedures
Sample Preparation. Two milliliters of 3 × 10-6 M Dronpa solution in a phosphate buffer (10 mM KH2PO4/10 mM K2HPO4/138 mM NaCl/2.7 mM KCl, pH 7.4) or a phosphate-citrated buffer (10 mM sodium phosphate/5 mM citrate, pH 5.0) were used for ensemble measurements. Samples for single-molecule measurements were prepared by spin-coating Dronpa (1 × 10-10 M) in PBS (pH 7.4) containing 1% (wt) poly(vinyl alcohol) (PVA) on a cover glass at 3,000 rpm. The sample preparation included careful cleaning of the glassware used for sample preparation as well as a subsequent cleaning of the cover glasses by sonication in acetone, sodium hydroxide (10%), and MilliQ water (Millipore).
Ensemble Spectroscopy. Steady-state spectroscopy was performed on a PerkinElmer Lambda 40 spectrophotometer and a Fluorolog 1500 fluorimeter (Spex Industries, Metuchen, NJ). A Dronpa solution was irradiated with the 488-nm light provided by a continuous wave Ar-Kr ion laser (Stabilete 2018-RM, Spectra-Physics) at a power of 10–80 mW/cm2. The photoswitched sample was irradiated again with the 405-nm light provided by frequency doubling the output of a mode-locked picosecond Ti:Sapphire laser (Tsunami; 82-mHz repetition rate; full width at half maximum = 2 ps; Spectra-Physics) at a power of 0.25–2 mW/cm2. During the irradiation experiment, absorption spectra were recorded. Time-resolved fluorescence measurements were performed by using the time-correlated single photon counting method (19) (excitation at 390 and 488 nm). The temperature of the samples was controlled by using a thermoelectric temperature controller (LFI-3751, Wavelength Electronics, Bozeman, MT).
Single-Molecule Spectroscopy. Samples were mounted on a inverted microscope (IX-70, Olympus) equipped with a scanning stage (Physik Instrumente, Karlsruhe/Palmbach, Germany). Circularly polarized 488-nm light from a continuous wave Ar-Kr ion laser was introduced into the microscope and focused by an oil immersion objective lens (Olympus, numerical aperture 1.4, ×60). For two-color excitation measurements, the 488- and 405-nm light from a frequency doubled mode-locked picosecond Ti:Sapphire laser was introduced coaxially into the microscope. Excitation power at the sample was set to 80–600 nW for one-color excitation and 220 nW (488 nm) and 37 nW (405 nm) for two-color excitation, respectively. Mechanical shutters (response time: 16–20 ms) were inserted in the optical path. Fluorescence was collected by the same objective, passed through a dichroic mirror (495DCLP, Chroma Technology, Rockingham, VT), filtered through a notch filter (488 nm, Kaiser Optical Systems, Ann Arbor, MI) and long-pass filters (450LP, 505LP, Chroma Technology), and focused through a 100-μm pinhole, on an avalanche photodiode (SPCM 15, EG & G, Salem, MA). Fluorescence from single Dronpa molecules was registered by a time-correlated single photon counting PC card (SPC 630, Becker & Hickl, Berlin) using FIFO (first-in, first-out) mode (20). All of the single-molecule data were measured at room temperature. Fast on–off blinking was analyzed with an autocorrelation function computed from the data set with a homemade program. Details of the autocorrelation analysis are given in Supporting Text, which is published as supporting information on the PNAS web site.
Results and Discussion
Steady-State Ensemble Spectroscopy. The absorption spectrum of Dronpa in PBS (pH 7.4) displays a major peak at 503 nm and a minor peak at 388 nm (Fig. 1a). Based on the pH dependence of the absorption spectrum, the 503- and 388-nm bands can be attributed to the deprotonated and protonated forms of the chromophore, respectively (18) (see Supporting Text). The fluorescence spectrum shows a major peak at 518 nm independent of the excitation wavelength (Fig. 1a, green and red). When excited at 390 nm, an additional emission band peaking at 450 nm was observed (Fig. 1a, red). This band is assigned to the fluorescence that originated from the excited state of the protonated form. The fluorescence quantum yield (ϕfl) on excitation at 390 nm (ϕfl = 0.02) is much smaller than ϕfl excited at 488 nm (ϕfl = 0.85), suggesting that nonradiative deactivation is dominant for the protonated form. The fluorescence excitation spectrum of Dronpa detected at 540 nm is similar to the absorption spectrum, except for the absence of the 388-nm band. The difference between the absorption and excitation spectra was more prominent at acidic pH (phosphate–citrate buffer, pH 5.0; see Fig. 6, which is published as supporting information on the PNAS web site). Although the presence of the 518-nm band in the fluorescence spectrum excited at 390 nm would indicate a connection between the protonated and deprotonated forms, probably through excited-state proton transfer (ESPT), the absence of the 388-nm peak from the excitation spectrum suggests that the efficiency of ESPT is extremely low (<0.01). These data suggest that the contribution of the direct excitation of the deprotonated form at 390 nm cannot be ignored even if the absorption of the deprotonated form at the wavelength is much smaller than that of the protonated form.
Fig. 1.

Photoswitching of Dronpa at the ensemble level. (a) Steady-state spectra of Dronpa at pH 7.4; absorption spectrum (blue line), fluorescence spectrum excited at 488 nm (green line) and 390 nm (red line), and excitation spectrum detected at 540 nm (black line). a.u., arbitrary units. (b and c) Fluorescence decays of Dronpa (pH 7.4) excited at 488 nm and detected at 520 nm (b) and excited at 390 nm and detected at 440 nm (c). (d) Time evolution of the absorption spectrum of Dronpa (pH 7.4) on irradiation with a 488-nm laser (P = 10 mW/cm2). (e and f) Time evolution of the peak absorbance of the deprotonated (e) and protonated (f) form on irradiation with a 488-nm laser. The solid lines show the fitting with a first-order kinetic model. (g) Time evolution of the absorption spectrum of the photoswitched Dronpa (488-nm irradiation) on irradiation with a 405-nm laser (P = 1 mW/cm2). (h and i) Time evolution of the peak absorbance of the deprotonated (h) and protonated (i) form on irradiation with a 405-nm laser. The solid lines show the fitting with a first-order kinetic model.
Time-Resolved Ensemble Spectroscopy. Excitation of Dronpa into the absorption band of the deprotonated form (488 nm) led to a single-exponential decay of the detected fluorescence with a time constant of 3.6 ns, independent of the detection wavelength from 515 to 560 nm (Fig. 1b). This result indicates the presence of only one emitting species for the deprotonated form of the protein. Excitation of Dronpa into the absorption band of the protonated form (390 nm) led to a fast decay of the fluorescence when monitored at 440 nm (Fig. 1c). The decay showed a multiexponential behavior with an average decay time of 14 ps. The dominant contribution is a 10-ps component, which can be attributed to the fast nonradiative deactivation to the ground state of the protonated form (21).
Photoswitching at Ensemble Level. The photoswitching behavior of Dronpa was outlined in a previous work (18). Here, the kinetics of the switching is discussed in more detail. Irradiation of the Dronpa solution at 488 nm resulted in a decrease in the absorption band of the deprotonated form and a concomitant increase in the absorption band of the protonated form (Fig. 1d). The absorption maximum of the two species remained constant during the irradiation. An isosbestic point at 430 nm was observed, indicating a simple photoswitching from the deprotonated to protonated forms on irradiation without occurrence of photobleaching. Time evolutions of the peak absorbencies of the deprotonated and protonated forms are depicted in Fig. 1 e and f, respectively. They could be fitted well with a first-order kinetic model that gave similar rate constants of 6.7 × 10-4 and 6.9 × 10-4 s-1 for the disappearance of the deprotonated form and the appearance of the protonated form, respectively.∥ Then, the photoswitched protonated sample was irradiated with a 405-nm laser. The absorption band of the protonated form diminished, and concomitantly the absorption band of the deprotonated form increased (Fig. 1g). Again, an isosbestic point at 430 nm was observed. The time evolutions were fitted with a first-order kinetic model, giving similar rate constants of 9.6 × 10-3 and 9.0 × 10-3 s-1 for the appearance of the deprotonated form (Fig. 1h) and the disappearance of the protonated form (Fig. 1i), respectively. Similar rate constants determined from the deprotonated and protonated forms for both the forward (the deprotonated to the protonated form) and the backward (the protonated to the deprotonated form) reactions clearly demonstrate the direct connection between the two forms through photoswitching. After a single round of photoswitching, the absorption of the deprotonated form was recovered completely, indicating the perfect reversibility of the Dronpa photochromism. This result is in contrast to other photochromic mutants of GFP in which only a small fraction (10–40%) of the original intensity could be restored. The rate constants for the photoswitching increased linearly with the irradiation power (data not shown), suggesting that both the protonation and deprotonation reactions occur through a one-photon excitation process.
Similar photoswitching was observed at pH 5.0 (see Fig. 7, which is published as supporting information on the PNAS web site). As discussed previously (18), the photoswitched and acid-induced protonated forms are not interconvertible. The population ratio between the protonated and deprotonated forms, which is determined by the solution pH, could not be modified toward the deprotonated form even with prolonged irradiation at 405 nm. Although the photoswitching between the protonated and deprotonated forms has been suggested for GFP and its mutants (12, 22), the photoswitched and acid-induced protonated forms have not been distinguished (12). Our data clearly demonstrated a different nature of the two protonated forms. The excellent photoswitching properties of Dronpa might be related to the nature of the photoswitched protonated form. The spectroscopic properties of the photoswitched protonated form must be, however, very similar to those of the original one. The spectral shape of both the absorption and the fluorescence of the photoswitched protonated form are identical to the spectra of the protonated form that originally existed (see Fig. 8, which is published as supporting information on the PNAS web site). Moreover, the 388-nm peak is also absent from the excitation spectrum of the photoswitched protonated form, suggesting that the efficiency of direct ESPT from the photoswitched protonated form to the deprotonated form is extremely low (<0.01).** Although fluorescence decay of the photoswitched protonated form could not be measured because of the very efficient photoswitching to the deprotonated form, a nonradiative deactivation to the corresponding ground state would be very fast, taking into account the very low fluorescence signal from the photoswitched protonated form. The extremely low efficiency of the direct ESPT demonstrates that this is not the mechanism involved in the observed reversible photoswitching as the quantum yield of the switching 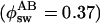 is much higher than that of ESPT (18). ESPT from the photoswitched protonated form to a nonfluorescent intermediate that converts into the deprotonated form in the ground state might account for the photoinduced deprotonation reaction.
is much higher than that of ESPT (18). ESPT from the photoswitched protonated form to a nonfluorescent intermediate that converts into the deprotonated form in the ground state might account for the photoinduced deprotonation reaction.
Recovery of the deprotonated form from the photoswitched protonated form was observed also in the dark. Although almost full recovery of the deprotonated form was observed, the process took place in the order of days at room temperature (22) (data not shown).
Single-Molecule Experiments at 488-nm Irradiation. As expected from the extinction coefficient (95,000 M-1·cm-1 at 503 nm) and the fluorescence quantum yield (ϕfl = 0.85), individual Dronpa molecules immobilized in a PVA film were observed as bright spots upon excitation at 488 nm (see below). A typical single-molecule fluorescence trajectory (excitation power = 220 nW) is depicted in Fig. 2a. Within 140 s, several intense bursts and long off-times (order of tens of seconds) were observed. Two bursts in Fig. 2a are shown in an expanded time scale in Fig. 2 b and c. The burst in Fig. 2b shows fast on–off blinking (≈1 ms). In addition to the fast blinking, a medium-long off-time was observed for the burst in Fig. 2c (order of tens of milliseconds). The medium-long off-state was observed for 35% of 250 bursts analyzed. The rest (65%) contained only the fast on–off blinking. The on-off behavior of 50 molecules was statistically analyzed. The on-times, which include fast blinking attributed to the triplet state, were histogrammed and could be fitted with a single exponential decay with a time constant of 38 ms (Fig. 2d). The medium-long off-state and long off-state were characterized with time constants of 65 ms and 11.0 s, respectively (Fig. 2 e and f). The short off-time during the fast on–off blinking was analyzed using the autocorrelation method. A histogram of the off-times was fitted with a Gaussian function, giving an average value of 1.2 ms (Fig. 2g). The on-time showed a linear dependence on excitation power as shown in Table 1. A high excitation power (600 nW) resulted in a shorter on-time (8.6 ms), whereas a lower power (80 nW) resulted in a longer on-time (85 ms). The survival time of the molecule until it photoswitches into the photoswitched protonated form, which corresponds to the on-time, can be calculated on the basis of the rate k0,1 for excitation from the electronic ground-state S0 to the first excited-state S1 and the quantum yield of the photoswitching from the deprotonated to the photoswitched protonated form 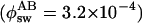 (ref. 18; see also Supporting Text). The survival time is estimated to be 39, 14, and 5.0 ms for 80-, 220-, and 600-nW excitation, respectively. These values agree well with those determined experimentally (85, 38, and 8.6 ms).
(ref. 18; see also Supporting Text). The survival time is estimated to be 39, 14, and 5.0 ms for 80-, 220-, and 600-nW excitation, respectively. These values agree well with those determined experimentally (85, 38, and 8.6 ms).
Fig. 2.

Single-molecule fluorescence measurements of Dronpa on 488-nm excitation. (a) Fluorescence intensity trajectory from a single Dronpa molecule in PVA (pH 7.4) on 488-nm excitation at a power of 220 nW. (b and c) Zoom of the bursts in the trajectory. Blue and red lines show the on-time and the medium-long off-time, respectively. (d–g) Frequency histogram of on-time (d), medium-long off-time (e), long off-time (f), and short off-time (g). Solid lines in d–f show a single-exponential fitting. Solid line in g shows a G fit.
Table 1. Excitation power dependence of the on–off times at 488-nm irradiation.
| Excitation power, nW | On-time, ms | Long off-time, s | Medium-long off-time, ms | Short off-time, ms |
|---|---|---|---|---|
| 80 | 85 | 6.9 | 57 | 1.7 |
| 220 | 38 | 11.0 | 65 | 1.2 |
| 600 | 8.6 | 10.0 | 59 | 1.0 |
By contrast, the three off-times were independent of the excitation power (Table 1). The fastest on–off blinking can be attributed to transitions into triplet states with a triplet lifetime of 1.2 ms. Similar values for the triplet lifetime have been reported for GFP-like protein embedded in a PVA film (23). The medium-long off-time (65 ms) can be ascribed to transitions between the deprotonated form and an unknown dark state. The long off-time (11.0 s) could be related to a spontaneous switching from the photoswitched protonated to the deprotonated form (see Supporting Text).
Single-Molecule Experiments at Simultaneous 488- and 405-nm Irradiation. Fig. 3 displays two sets of single-molecule fluorescence images of Dronpa excited at 488 nm. The molecules changed to the dim state (photoswitched protonated form) during continuous scanning (images 1–4). Then, the same area was scanned once with a 405-nm laser line (between images 4 and 5). Next, the recovery of the original fluorescence was observed with the 488-nm laser scanning (image 5). This procedure was repeated four times (405-nm light was applied between image 8/9, 12/13, and 16/17). These results indicate the photoinduced switching of Dronpa at a single-molecule level (12).
Fig. 3.

Time series of single-molecule fluorescence images of Dronpa in PVA (pH 7.4). A and B denote two different molecules. The images were obtained with a 488-nm excitation at a power of 220 nW. The integration time was 1 ms/pixel with the pixel size of 78 × 78 nm. The same area was scanned with a 405-nm laser at a power of 220 nW (1 ms/pixel with the pixel size of 78 × 78 nm) before scanning images 5, 9, 13, and 17 with a 488-nm laser.
Reversible photoswitching of Dronpa at a single-molecule level was investigated further with simultaneous dual-color excitation measurements in which individual molecules were continuously illuminated with a 488-nm laser line (excitation power = 220 nW), while a 405-nm laser line (excitation power = 37 nW) was applied every other second. A typical fluorescence trajectory is depicted in Fig. 4a. Intense bursts appeared several times within a second while the 405-nm light was on (Fig. 4b). Because the nonradiative deactivation is dominant for the photoswitched protonated form, excitation of Dronpa into the absorption band of the protonated form (405 nm) does not give a detectable fluorescence signal. Hence, the observed signals correspond to the molecule in the deprotonated state. The fluorescence trajectory with the dual-color excitation demonstrated that the molecule was driven into the photoswitched protonated form when the 405-nm light was absent. Perfect recovery of the fluorescence signal on irradiation with the 405-nm light proves the reversible photoswitching of the molecule. The fluorescence appeared and disappeared within the response time of the shutter (16–20 ms) for the on and off switching of the 405-nm light (Fig. 4c). Based on the ensemble data, the photoswitching speed in the order of milliseconds to tens of milliseconds was expected under the illumination conditions. The observed photoswitching speed as fast as tens of milliseconds thus demonstrates that Dronpa can hold its excellent photoswitching property even at the single-molecule level.
Fig. 4.

Photoswitching of Dronpa at the single-molecule level. (a) Fluorescence intensity trajectory from a single Dronpa molecule in PVA (pH 7.4) on two-color excitation at a power of 220 and 37 nW at 488 and 405 nm, respectively. Lower shows the on–off state of the 488-nm (cyan line) and 405-nm (blue line) light. (b and c) Zoom of the bursts in the trajectory. (d–f) Frequency histogram of on-time (d), medium-long off-time (e), and short off-time (f). Solid lines in d and f show a single-exponential fitting. Solid line in f shows a G fit. (g) Fluorescence intensity trajectory from a single Dronpa molecule in PVA (pH 7.4) on two-color excitation at a power of 320 and 37 nW at 488 and 405 nm, respectively. Lower shows the on–off state of the 488-nm (cyan line) and 405-nm (blue line) light.
As observed for single-color excitation at 488 nm, a zoom into the trace of the dual-color excitation experiment shows similar on–off behavior (Fig. 4 b and c), resulting in an on-time of 52 ms and two off-times of 65 and 1.3 ms (Fig. 4 d–f). The medium-long off-time (65 ms) was detected in both single- and dual-color excitation experiments. The short off-time (1.3 ms) was similar to the value (1.2 ms) obtained with excitation at 488 nm (Fig. 2g).
Fig. 4g shows another single-molecule fluorescence trajectory in a dual-color excitation experiment, in which a molecule was illuminated continuously with 488-nm light (excitation power = 320 nW) while a 30-ms pulse of 405-nm laser (excitation power = 37 nW) was applied every second. Multiple switching events within a burst could be avoided under this condition because the average survival time of the deprotonated state was longer than the pulse width of 405-nm laser line (30 ms). Because each peak corresponds to single switching event, the trace of Fig. 4g displays 42 switching events.†† We were able to achieve up to 170 photoswitching events at the single-molecule level.
The fast and reliable reversible photoswitching of Dronpa at a single-molecule level is in contrast with other GFP mutants (12) and diarylethene derivatives (13, 14). No appreciable photochromism was observed for the yellow-emitting variants of Aequorea GFP when measured in bulk (12). The nature of the photoswitched protonated form probably is crucial for the efficient reversible photoswitching. Although the photoswitching property of the diarylethene derivatives at the ensemble level is comparable with that of Dronpa, the number of the photoswitching at the single-molecule level is limited, probably because of relatively low quantum yields of the switching of the compounds. The lower quantum yield of the switching limits the number of the switching before the molecule photobleach. Furthermore, histograms of the on- and off-times obtained for diarylethene derivatives show a Gaussian-like distribution, suggesting that the photoswitching can be hardly treated as a random process. The large structural change of the molecule that is required for the switching might complicate the photoswitching behavior (14). In the case of Dronpa, the protonation/deprotonation is the origin of the switching, which requires minimal structural change of the molecule. Moreover, the chromophore is buried in the protein β-barrel, which reduces the influences of the environments on the switching behavior. These conditions together with the large quantum yield of the switching can account for the fast and efficient photoswitching observed for individual Dronpa molecules. Photoswitching of carbocyanine dyes at the single-molecule level in aqueous solution reported very recently revealed a repeatability similar to Dronpa (15). The reversible photoswitching in aqueous solution is an advantage of this system for in vivo application. However, efficient photoswitching required oxygen removal and the addition of the triplet quencher, which could limit its application.
Scheme of the Photoswitching. The reliable reversible photoswitching of Dronpa at the single-molecule level allows us to propose detailed models for the photoswitching (Fig. 5). Upon excitation of Dronpa molecules into the absorption band of the deprotonated form (B), the excited state decays into the corresponding ground-state with a decay time of 3.6 ns (ϕfl = 0.85) and into the ground-states of the photoswitched protonated form (A2) and unknown dark-state (D) 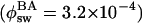 . The D state is metastable and converts into the B state spontaneously with the lifetime of 65 ms (time constant of 15 s-1). Although A2 is the protonated form of the chromophore, this form is not interconvertible with the protonated form that originally exists at a given pH (A1). On excitation of the protonated forms of Dronpa molecules at 405 nm (A1 and A2), they show very efficient nonradiative deactivation. Furthermore, the excited state of A2 can switch into the ground-state of B with the quantum yield of 0.37. Taking into account the fact that direct ESPT from A2 to B is an extremely inefficient process together with the acidic nature of the chromophore in the excited state, the large quantum yield of the switching could be explained by ESPT from A2 to a nonfluorescent intermediate that converts into B in the ground-state. Most of the GFP-like fluorescent proteins have more than one deprotonated state (24, 25), also supporting this model. The very efficient nonradiative deactivation of A2 suggests that the photoswitching takes place within a few picoseconds. To study structural aspects of the observed photoswitching in Dronpa in more detail, NMR, Raman, and femtosecond time-resolved absorption measurements should be carried out. For in vivo applications, it is also important to perform photoswitching measurements at the single-molecule level in a physiological condition to evaluate the influence of the PVA matrix, such as limited oxygen permeability and matrix-protein interaction, on the photoswitching properties.
. The D state is metastable and converts into the B state spontaneously with the lifetime of 65 ms (time constant of 15 s-1). Although A2 is the protonated form of the chromophore, this form is not interconvertible with the protonated form that originally exists at a given pH (A1). On excitation of the protonated forms of Dronpa molecules at 405 nm (A1 and A2), they show very efficient nonradiative deactivation. Furthermore, the excited state of A2 can switch into the ground-state of B with the quantum yield of 0.37. Taking into account the fact that direct ESPT from A2 to B is an extremely inefficient process together with the acidic nature of the chromophore in the excited state, the large quantum yield of the switching could be explained by ESPT from A2 to a nonfluorescent intermediate that converts into B in the ground-state. Most of the GFP-like fluorescent proteins have more than one deprotonated state (24, 25), also supporting this model. The very efficient nonradiative deactivation of A2 suggests that the photoswitching takes place within a few picoseconds. To study structural aspects of the observed photoswitching in Dronpa in more detail, NMR, Raman, and femtosecond time-resolved absorption measurements should be carried out. For in vivo applications, it is also important to perform photoswitching measurements at the single-molecule level in a physiological condition to evaluate the influence of the PVA matrix, such as limited oxygen permeability and matrix-protein interaction, on the photoswitching properties.
Fig. 5.

Schematic diagram of the photoswitching of Dronpa. B, deprotonated form; D, an unknown dark state; A1, the protonated form that originally exists in the sample; A2, the protonated form that is formed by the photoswitching; and I, a nonfluorescent intermediate. The photoswitching pathway from B to A2 is shown by blue arrows, and that from A2 to B is shown by red arrows.
Conclusion
In the present study, we investigated the photoswitching of the GFP-like fluorescent protein Dronpa. Ensemble spectroscopy showed that the protonated and the deprotonated form of the chromophore can be reversibly photoswitched on irradiation with 488- and 405-nm light. Although the deprotonated form is very bright (ϕfl = 0.85; τfl = 3.6 ns), the protonated form is almost nonfluorescent due to the fast nonradiative deactivation (τfl = 14 ps) and the efficiency of ESPT is extremely low. The protonated form that is formed by photoswitching cannot interconvert with the protonated form that is originally existing in the sample at a given pH. Single-molecule two-color excitation measurement showed that the molecule is driven into the protonated form very efficiently when 405-nm light is absent. The fluorescence signal can be recovered perfectly on irradiation with 405-nm light, demonstrating the reversible photoswitching at the single-molecule level. Individual proteins could be photoswitched >100 times. Furthermore, the measured survival time of the molecule in the deprotonated state agrees well with the calculated value on the basis of the quantum yield of the switching and the rate constant for excitation from S0 to S1. Single-molecule spectroscopy also revealed that the molecule has an unknown dark-state with a lifetime of 65 ms.
Supplementary Material
Acknowledgments
This work was supported by the Fonds voor Wetenschappelijk Onderzoek–Vlaanderen, the Flemish Ministry of Education (GOA01/2), the Federal Science Policy of Belgium (IUAP-V-03), the Fonds National de la Recherche Scientifique, and the Fonds Spéciaux de Recherche. This work was partly supported by grants from the Human Frontier Science Program.
Author contributions: S.H., A.M., and J.H. designed research; S.H. performed research; R.A., H.M., and A.M. contributed new reagents/analytic tools; S.H., W.V., and P.D. analyzed data; S.H., A.M., and J.H. wrote the paper; and H.M. provided helpful discussion.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: ESPT, excited-state proton transfer; PVA, poly(vinyl alcohol).
See Commentary on page 9433.
Footnotes
The photoswitched protonated form and the protonated form that is in equilibrium with the deprotonated form are the different species, as discussed later. A detailed analysis of the time evolution of the absorption spectrum that takes into account the acid–base equilibrium is given in Supporting Text.
Here, ESPT is defined as proton transfer between the protonated and the deprotonated form in the excited state. The photoswitching from the protonated and the deprotonated form would, therefore, take place from the excited state of the protonated form to the ground state of the deprotonated form. More likely, the process goes over an intermediate in the excited state and from the intermediate form to the ground state.
Recovery of the fluorescence on 405-nm irradiation is not perfect in Fig. 2g (≈90%). This result is probably due to insufficient irradiation of 405-nm light to recover the deprotonated state or insufficient time that the molecule spent in the deprotonated state to get detectable photon numbers.
References
- 1.Irie, M. (2000) Chem. Rev. 100, 1685-1716. [DOI] [PubMed] [Google Scholar]
- 2.Liang, Y. C., Dvornikov, A. S. & Rentzepis, P. M. (2003) Proc. Natl. Acad. Sci. USA 100, 8109-8112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Giordano, L., Jovin, T. M., Irie, M. & Jares-Erijman, E. A. (2002) J. Am. Chem. Soc. 124, 7481-7489. [DOI] [PubMed] [Google Scholar]
- 4.Fernández-Acebes, A. & Lehn, J.-M. (1999) Chem. Eur. J. 5, 3285-3292. [Google Scholar]
- 5.Lippincott-Schwartz, J., Altan-Bonnet, N. & Patterson, G. H. (2003) Nat. Cell. Biol., Suppl., S7-S14. [PubMed]
- 6.Lippincott-Schwartz, J. & Patterson, G. H. (2003) Science 300, 87-91. [DOI] [PubMed] [Google Scholar]
- 7.Patterson, G. H. & Lippincott-Schwartz, J. (2002) Science 297, 1873-1877. [DOI] [PubMed] [Google Scholar]
- 8.Ando, R., Hama, H., Yamamoto-Hino, M., Mizuno, H. & Miyawaki, A. (2002) Proc. Natl. Acad. Sci. USA 99, 12651-12656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chudakov, D. M., Belousov, V. V., Zaraisky, A. G., Novoselov, V. V., Staroverov, D. B., Zorov, D. B., Lukyanov, S. & Lukyanov, K. A. (2003) Nat. Biotechnol. 21, 191-194. [DOI] [PubMed] [Google Scholar]
- 10.Chudakov, D. M., Verkhusha, V. V., Staroverov, D. B., Souslova, E. A., Lukyanov, S. & Lukyanov, K. A. (2004) Nat. Biotechnol. 22, 1435-1439. [DOI] [PubMed] [Google Scholar]
- 11.Wiedenmann, J., Ivanchenko, S., Oswald, F., Schmitt, F., Röcker, C., Salih, A., Spindler, K.-D. & Nienhaus, G. U. (2004) Proc. Natl. Acad. Sci. USA 101, 15905-15910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dickson, R. M., Cubitt, A. B., Tsien, R. Y. & Moerner, W. E. (1997) Nature 388, 355-358. [DOI] [PubMed] [Google Scholar]
- 13.Irie, M., Fukaminato, T., Sasaki, T., Tamai, N. & Kawai, T. (2002) Nature 420, 759-760. [DOI] [PubMed] [Google Scholar]
- 14.Fukaminato, T., Sasaki, T., Kawai, T., Tamai, N. & Irie, M. (2004) J. Am. Chem. Soc. 126, 14843-14849. [DOI] [PubMed] [Google Scholar]
- 15.Heilemann, M., Margeat, E., Kasper, R., Sauer, M. & Tinnefeld, P. (2005) J. Am. Chem. Soc. 127, 3801-3806. [DOI] [PubMed] [Google Scholar]
- 16.Bates, M., Blosser, T. R. & Zhuang, X. (2005) Phys. Rev. Lett. 94, 108101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jung, G., Wiehler, J., Steipe, B., Bräuchle, C. & Zumbusch, A. (2001) ChemPhysChem. 6, 392-396. [DOI] [PubMed] [Google Scholar]
- 18.Ando, R., Mizuno, H. & Miyawaki, A. (2004) Science 306, 1370-1373. [DOI] [PubMed] [Google Scholar]
- 19.Maus, M., Rousseau, E., Cotlet, M., Hofkens, J., Schweitzer, G., Van der Auweraer, M., De Schryver, F. C. & Kruegen, A. (2001) Rev. Sci. Instrum. 72, 36-40. [Google Scholar]
- 20.Cotlet, M., Hofkens, J., Habuchi, S., Dirix, G., Van Guyse, M., Michiels, J., Vanderleyden, J. & De Schryver, F. C. (2001) Proc. Natl. Acad. Sci. USA 98, 14398-14403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Habuchi, S., Cotlet, M., Hofkens, J., Dirix, G., Michiels, J., Vanderleyden, J., Subramaniam, V. & De Schryver, F. C. (2002) Biophys. J. 83, 3499-3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chattoraj, M., King, B. A., Bublitz, G. U. & Boxer, S. G. (1996) Proc. Natl. Acad. Sci. USA 93, 8362-8367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cotlet, M., Hofkens, J., Köhn, F., Michiels, J., Dirix, G., Van Guyse, M., Vanderleyden, J. & De Schryver, F. C. (2001) Chem. Phys. Lett. 336, 415-423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cotlet, M., Hofkens, J., Maus, M., Gensch, T., Van der Auweraer, M., Michiels, J., Dirix, G., Van Guyse, M., Vanderleyden, J., Visser, A. J. W. G., et al. (2001) J. Phys. Chem. B. 105, 4999-5006. [Google Scholar]
- 25.Creemers, T. M. H., Lock, A. J., Subramaniam, V., Jovin, T. M. & Völker, S. (2000) Proc. Natl. Acad. Sci. USA 97, 2974-2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.