

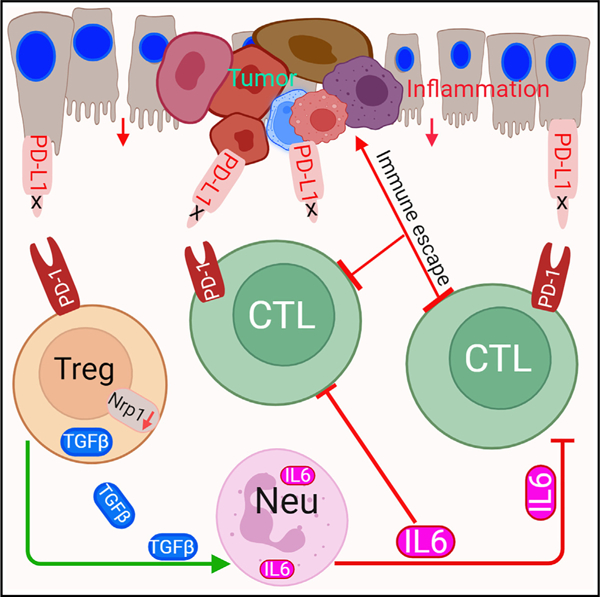
Graphical abstract
SUMMARY
T cells function not only as an essential component of host cancer immunosurveillance but also as a regulator of colonic inflammation, a process that promotes colorectal cancer. Programmed death-ligand 1 (PD-L1) is a T cell-negative regulator, but its role in regulation of T cell functions in the context of colorectal cancer is unknown. We report that global deletion of Cd274 results in increased colonic inflammation, PD-1+ T cells, and inflammation-driven colorectal tumorigenesis in mice. Single-cell RNA sequencing (scRNA-seq) analysis revealed that PD-L1 suppresses subpopulations of programmed cell death protein 1 (PD-1)+Nrp1lo regulatory T (Treg) cells and interleukin (IL) 6+ neutrophils in colorectal tumor. Treg cells produce transforming growth factor (TGF) β to recruit IL6+ neutrophils. Neutrophils produce IL6 to inhibit activation of tumor-specific cytotoxic T lymphocytes (CTLs) and primary CTLs. Accordingly, IL6 blockade immunotherapy increases CTL activation and suppresses colon tumor growth in vivo. Our findings determine that PD-L1 restrains PD-1+Nrp1loTGFβ+ Treg cells to suppress IL6+ neutrophil tumor recruitment to sustain CTL activation to control inflammation-driven colorectal tumorigenesis.
In brief
Poschel et al. observed that loss of host PD-L1 function leads to increased inflammation-driven colorectal tumorigenesis in mice. Mechanistically, loss of PD-L1 expands the PD-1+Nrp1loTGFβ+ Treg to recruit IL6+ neutrophils to inhibit CTL activation. PD-L1 therefore functions as a suppressor of inflammation-driven colorectal tumor via restraining PD-1+Nrp1loTGFβ+ Treg cells.
INTRODUCTION
The programmed death-ligand 1 (PD-L1) binds to the programmed cell death protein 1 (PD-1) to inhibit T and myeloid cell activation.1–5 In human cancer patients and tumor-bearing mice, PD-L1 expression is elevated and engages PD-1 expressed on T cells to activate intrinsic SH2 domain-containing tyrosine phosphatase 2 (SHP2) to dephosphorylate T cell receptor (TCR) and co-stimulatory receptor CD28 to inhibit T cell activation.6,7 PD-L1 also binds to PD-1 expressed on myeloid cells to reprogram myeloid cell metabolism, regulate myeloid cell differentiation, and repress type I interferon (IFN-I) expression to impair CTL tumor recruitment.2–4 The PD-(L)1 pathway therefore acts as an immune checkpoint that negatively regulates T cell activation and recruitment in the tumor microenvironment to promote tumor immune escape. PD-(L)1 immune checkpoint inhibitor (ICI) immunotherapy has been developed to re-activate the dysfunctional T cells and has shown durable efficacy in human cancer immunotherapy.8 However, human colorectal cancer, except for the small subset of microsatellite instability high (MSI-H) colorectal cancer,9 does not respond to PD-(L)1 ICI immunotherapy.8 The mechanism underlying colorectal cancer non-response to PD-(L)1 ICI immunotherapy is currently unknown.
It has been shown that tumor cell PD-L1 (tPD-L1) alone is sufficient to suppress T cell activation to promote tumor growth in subcutaneous colon tumor-bearing mice.10 It has also been shown that host immune cell PD-L1, including PD-L1 of dendritic cells (DCs) and macrophages, plays the dominant role in suppressing T cell activation to promote tumor immune evasion in subcutaneous tumor-bearing mice.11–15 These findings thus indicate that the response of T cells to PD-L1 may depend on the host tumor microenvironment.16–19 Furthermore, these findings in preclinical subcutaneous tumor mouse models do not correlate with human cancer patient response to ICI immunotherapy. Meta-analysis of 17 phase III randomized clinical trials of 11,166 patients indicates that tPD-L1 level is the strongest predictor of a better overall survival of squamous cell carcinoma to ICI immunotherapy,20 but the combined tPD-L1 and immune cell PD-L1 level is the strongest predictor of better response to ICI immunotherapy in patients with adenocarcinoma.20 Meta-analysis of 17 clinical trials of 1,746 patients with metastatic breast cancer revealed that tPD-L1-positive patient had a longer overall survival after ICI immunotherapy.21 These findings thus indicate that the response of T cells to PD-L1 may depend on the tumor type and microenvironment.16–19
A unique feature of colorectal cancer is the anatomical location of the tumor in the colon epithelium, which is in a continual state of low-grade inflammation,22 a process that promotes tumor development. Furthermore, colonic inflammation and colitis are immune-related adverse effects of the PD-(L)1 ICI immunotherapy in human cancer patients.23–25 Administering Fc-fused PD-L1 protein abrogates colonic inflammation and colitis in experimental colitis mouse models26 and inflammation-inducing agents cause significantly more severe colonic inflammation and colitis in PD-L1 knockout (KO) mice than in wild-type (WT) mice.27 PD-L1 therefore also functions as an essential suppressor of colonic inflammation and colitis.
We report here that PD-L1 functions as a tumor suppressor under inflammatory conditions in the cellular context of colorectal cancer. We determine that PD-L1 restrains the PD-1+Nrp1loTGFβ+Treg/IL6+ neutrophils cellular pathway to sustain T cell activation and host cancer immunosurveillance to suppress inflammation-driven colorectal tumorigenesis.
RESULTS
Global PD-L1 deficiency promotes inflammation-driven colorectal tumorigenesis
Analysis of single-cell RNA sequencing (scRNA-seq) datasets of non-neoplastic human tissues revealed that PD-L1 is expressed in both epithelial cells and immune cells with the highest expression level in Langerhans cells, syncytiotrophoblasts, granulocytes, and extravillous trophoblasts (Figure 1A). To determine the specific cell types that express PD-L1 in human colon cancer, three representative human colon tumor tissues were stained with PD-L1-specific antibody by immunohistochemical analysis. PD-L1 is expressed in both tumor cells and tumor-infiltrating leukocytes (Figure S1A). We then extracted scRNA-seq dataset of human colon cancer patients28 and analyzed PD-L1 expression profile in human colon tumor in the single-cell level. Low level of PD-L1 is detected in subsets of immune cells, including B, T, natural killer (NK), innate lymphoid cells (ILCs), and stromal cells. Tumor cells are the most heterogeneous cell population that express PD-L1. Mast cells account for a small fraction of tumor-infiltrating immune cells but express high level of PD-L1. Myeloid cells constitute a large population of tumor-infiltrating immune cells and express the highest level of PD-L1 (Figure 1B). Among the myeloid cell subpopulations, monocytes, macrophages and mregDC express high level of PD-L1 (Figure 1C).
Figure 1. Loss of global PD-L1 promotes inflammation-driven colorectal tumorigenesis.
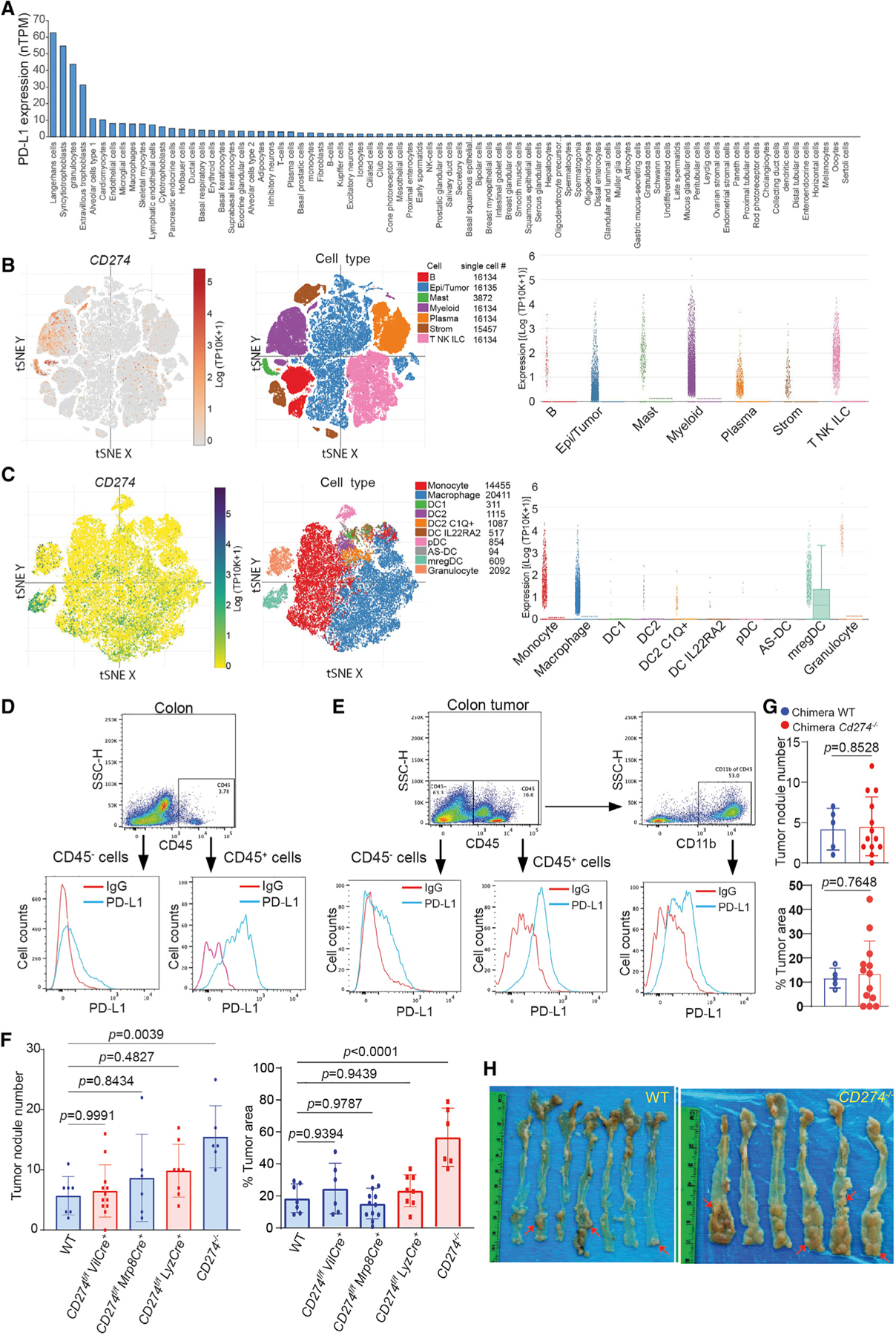
(A) The human CD274 mRNA level in the indicated human cell types in the single-cell level. The CD274 transcript datasets were downloaded from the Human Protein Atlas and analyzed.
(B and C) PD-L1 expression level in major cell types (B) and myeloid cell subpopulations (C) of human colon tumor tissues in the single-cell level. The human colon cancer patient scRNA-seq datasets (GEO: GSE178341) were analyzed for the indicated cell types. tSNE: t-distributed Stochastic Neighbor Embedding’ mregDC: mature dendritic cells enriched in immunoregulatory molecules (mregDCs).
(D) PD-L1 expression in colon epithelial cells (CD45−) and colon resident immune cells (CD45+) of tumor-free mice. Representative data of one of three mice.
(E) PD-L1 expression in tumor cells (CD45−), tumor-infiltrating total immune cells (CD45+), and myeloid cells (CD11b+) of colon tumor-bearing mice. Representative data of one of three tumor-bearing mice.
(F) Colorectal tumor nodule number and tumor size in WT mice and mice with Cd274 deletion in the indicated cell types. Column, mean; bar, standard deviation (SD). p value was determined by Student’s t test. Each dot represents data from one mouse.
(G) Colorectal tumor nodule number and tumor size in PD-L1 KO chimera mice and WT chimera control mice. Column, mean; bar, SD. p value was determined by Student’s t test. Each dot represents data from one mouse.
(H) Colorectal tumorigenesis in WT mice and mice with Cd274 global deletion. The red arrows point to tumor nodules.
Analysis of colorectal tissues of tumor-free mice determined that PD-L1 protein is located on the surfaces of colorectal epithelial cells and colorectal resident immune cells (Figure 1D). To determine the relative functions of cell type-specific PD-L1 in colorectal cancer, we made use of the azoxymethane (AOM) and dextran sodium sulfate (DSS)-induced inflammation-driven colorectal cancer mouse model.29 In the mouse colorectal tumor tissue, PD-L1 protein is present on the surfaces of both tumor cells and tumor-infiltrating immune cells (Figure 1E). Further analysis revealed that myeloid cells account for a large fraction of the tumor-infiltrating immune cells and express PD-L1 (Figure 1E). Taken together, these findings determine that this inflammation-driven colorectal tumor mouse model resembles human colorectal cancer in the PD-L1 expression profile.
Myeloid cell PD-L1 has been shown to promote tumor immune evasion in transplanted subcutaneous tumor mouse models.12,28,30–32 Subcutaneous tumor models are excellent models but may not resemble the colon tumor microenvironment since it is distant from the colon. To determine myeloid PD-L1 function in colorectal tumorigenesis under pathophysiological conditions, we created mice with PD-L1 deletion only in myeloid cells by crossing Cd274 floxed mouse33 to Lyz-Cre and Mrp8-Cre mice, respectively. Lyz-Cre generated mice with PD-L1 deletion only in myeloid cells and Mrp8-Cre mice generate mice with PD-L1 deletion only in neutrophils. Colorectal tumors were then induced in these mice using AOM-DSS procedure. Analysis of the colorectal tissues determined that knocking out PD-L1 in myeloid cells or neutrophils has no significant effect on colorectal tumorigenesis (Figure 1F). We then sought to knock out PD-L1 in all immune cells using adoptive transfer of bone marrow (BM) cells from the PD-L1 KO mice13 to lethally irradiated WT recipient mice to create PD-L1 KO chimera mice.34 Similar to what was observed in myeloid and neutrophil PD-L1 KO mice, deletion of PD-L1 in immune cells has no significant effect on colorectal tumorigenesis (Figure 1G). It has also been reported that tumor cell PD-L1 alone is sufficient to promote tumor immune evasion in a subcutaneous transplanted colon tumor mouse model.10 We then created mice with PD-L1 deletion only in colorectal epithelial cells. PD-L1 deletion in colorectal epithelial cells also has no significant effect on colorectal tumorigenesis (Figure 1F). It has also been reported that both tumor cells and host PD-L1 are required for suppression of host anti-tumor immune response,13 and our above findings suggest that colon epithelial cell/tumor cell PD-L1 and host immune cell PD-L1 may compensate each other to promote tumor immune evasion. To test this hypothesis, we induced colorectal tumor in mice with global PD-L1 deletion. Strikingly, instead of the expected decrease in tumorigenesis, global deletion of PD-L1 significantly increased colorectal tumor nodule number and tumor size as compared to WT mice in this sporadic and inflammation-driven colorectal tumorigenesis mouse model (Figures 1F and 1H).
PD-L1 controls colonic inflammation and restrains PD-1+ T cell expansion in colorectal tumor-bearing mice
PD-L1 KO mice exhibit severe colonic inflammation and inflammation-associated colitis when treated with the inflammation-inducing agent DSS.27 PD-L1 protein inhibits colonic inflammation and colitis in mice.26 A major adverse effect of PD-(L)1 ICI immunotherapy in human cancer patients is colonic inflammation.23 We observed that PD-L1 KO mice exhibit significant greater body weight loss (Figure 2A) as compared to WT mice when treated with AOM and DSS. Histological analysis of colorectal tissues revealed that PD-L1 KO mice exhibit a greater degree of colonic inflammation as measured by inflammation score (Figure 2B).34 At day 21, colorectal adenomas form in WT mice, whereas adenomas with high-grade dysplasia (HGD) and adenocarcinoma in situ (AIS) were observed in PD-L1 KO mice (Figure 2B). At day 79, adenomas with HGD and AIS were observed in WT mice (Figure 2B). These observations indicate that host PD-L1 controls colonic inflammation to suppress inflammation-driven colorectal tumorigenesis in mice.
Figure 2. PD-L1 suppresses colonic inflammation during colon tumorigenesis.
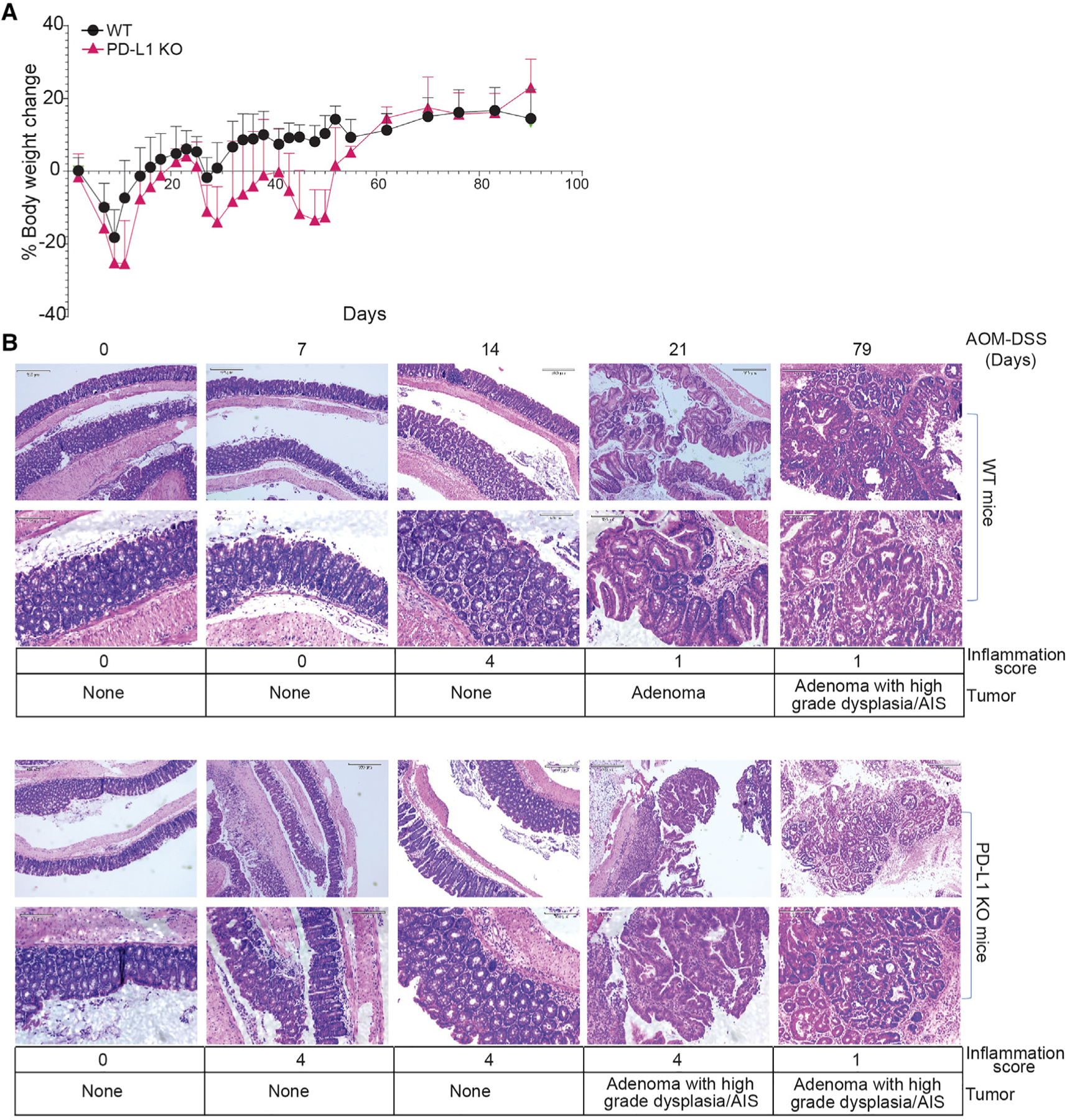
(A) Mouse body weight change kinetics after AOM-DSS treatment.
(B) Colon tissues at the indicated time points were stained by H&E and analyzed for inflammation and tumor development. Bottom panels show magnified images (scale bar: 320 μM) of the top panels (scale bar: 130 μM) in both WT and PD-L1 KO panels. Inflammation scores are defined as grade 0, normal colonic mucosa; grade 1, loss of one-third of the crypts; grade 2, loss of two-thirds of the crypts; grade 3, lamina propria is covered with a single layer of epithelium, mild inflammatory cell infiltrate present; grade 4, erosions and marked inflammatory cell infiltration present. AIS, adenocarcinoma in situ.
We then analyzed PD-1 level in T cells in tumor-bearing mice. Flow cytometry analysis indicates that tumor-bearing WT and PD-L1 KO mice have significantly more PD-1+ cells in both CD4+ and CD8+ T cell populations in the spleens (Figures S1B and S1C). This observation indicates that loss of PD-L1 leads to a significant expansion of PD-1+ T cells in colorectal tumor-bearing mice.
A high-resolution cellular landscape of PD-L1-deficient mouse colorectal tumor
The above findings determine that loss of PD-L1 function promotes inflammation-driven colorectal tumorigenesis despite its well-known function in activating functionally exhausted T cells. To elucidate the mechanisms underlying PD-L1 function as a suppressor in inflammation-driven colorectal tumor, we dissected tumor tissues from WT and PD-L1 KO mice and analyzed the tumor by scRNA-seq (Figure 3A). A total of 24,000 single cells passed quality control and were annotated using canonical lineage markers. This high-level annotation was further confirmed using reference gene signatures.35 Uniform manifold approximation and projection (UMAP) visualization identified 28 unique cell clusters/population in the colorectal tumor tissues (Figure 3B) with unique gene signatures (Figure S2A). PD-1 transcripts are present in two cell clusters (Figure S2B). Consistent with the higher PD-1 protein level in T cells of PD-L1 KO mice than in WT mice (Figure S1), PD-1 transcript level is also higher in these two clusters in PD-L1 KO mice than in WT mice (Figure S2B). Only one exon in the Cd274 gene was deleted in the PD-L1 KO mice13 and PD-L1 transcripts are detected in 18 cell clusters. PD-L2 transcripts are detected in the same 18 cell clusters as PD-L1 (Figure S2B). Tumors from the WT and PD-L1 KO mice exhibit similar cellular clusters (Figure 3B). Nearest-neighbor clustering identified 12 cell populations (Figure 3C) and each cell population has unique gene signatures (Figures 3D and S3A). Among these 12 cell populations, B cell abundance decreased dramatically whereas neutrophils and T cells increased. Further analysis of the immune cell population validated the decreased B cells and increased neutrophils and T cell populations in colorectal tumors from PD-L1 KO mice (Figure 3D). PD-1 transcript is primarily detected in NK/T cells, and PD-L1 and PD-L2 are expressed in all major immune cell subpopulations (Figure S3B). PD-1 level is higher in NK/T cells in PD-L1 KO tumor-bearing mice than in WT tumor-bearing mice (Figure S3B). Our findings thus determine that decreased B cells, and increased neutrophils and T cells, are hallmarks of inflammation-driven colon cancer in mice.
Figure 3. Tumor cell and immune cell landscapes of WT and PD-L1 KO mouse colon tumor.
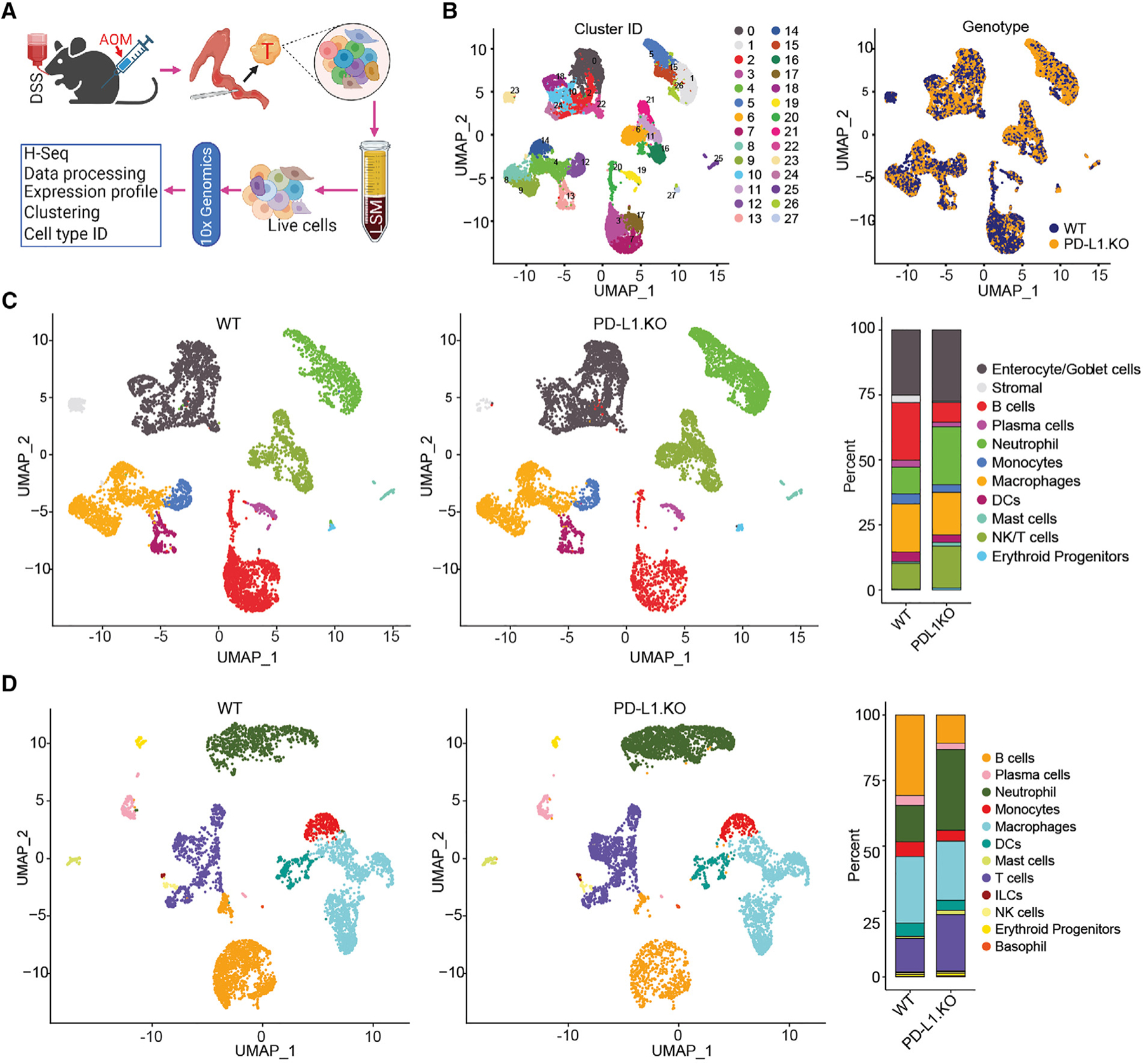
(A) scRNA-seq experimental scheme.
(B) UMAP plot of all cells isolated from colon tumor, colored by identified cell clusters (right). The cell cluster overlap of tumors by tumor-bearing mouse genotypes are shown at the right panel.
(C) UMAP plot (left) and barplot (right) of identities of major cell subpopulations of total colon tumor cells as shown in (B).
(D) UMAP plot (left) and barplot (right) of identities of the subpopulation of CD45+ cells in the colon tumor.
PD-L1 suppresses Nrp1lo regulatory T cell accumulation and TGFβ production
Given the function of PD-L1 as a potent inhibitor of T cells in the tumor microenvironment1,36,37 and our above observation that PD-L1 KO tumor-bearing mice have increased PD-1+ T cell accumulation, we then analyzed T cell subpopulations in the single-cell level. Clustering analysis demarcated populations of CD4+, CD8+, γδ T, and regulatory T (Treg) cells. Surprisingly, the levels of CD4+ and CD8+ T cells are not significantly increased in the PD-L1 KO colorectal tumor (Figures 4A and 4B). Treg and γδ T cells are the major populations of T cells, and Treg cell level increased dramatically in the PD-L1 KO colorectal tumor (Figures 4A and S1D). Among the Treg cells, the major change is increase of Nrp1lo subset in PD-L1 KO mice as compared to WT mice (Figure 4B). These Nrp1lo Treg cells co-express PD-1 (Figures 4C and 4D). Flow cytometry analysis of the AOM-DSS-induced mouse colorectal tumors in PD-L1 KO mice determined that only 0%–11.2% of Treg cells are Nrp1+. Among the Nrp1lo/− Treg cells, 93.2%–96.7% are PD-1+ (Figures 4E–4G). Analysis of human colon tumor scRNA-seq datasets revealed that 0.29% of tumor-infiltrating Treg cells are Nrp1+ and 11.48% of Treg cells express PD-1 (Figures 4H–4J). Flow cytometry analysis shows that 3.8%–33.9% tumor-infiltrating Treg cells are Nrp1+ and 18.9%–87.4% of tumor-infiltrating NRP1lo Treg cells are PD-1+ in human colon cancer patients (Figures 4K and 4L). A consequence of PD-(L)1 blockade is increased Treg cell accumulation.38–40 Consistent with this phenomenon, the level of PD-1+ Treg cells is significantly higher in colorectal tumor-bearing PD-L1 KO mice than in WT mice (Figure S1D).
Figure 4. PD-L1 suppresses PD-1-expressing Nrp1lo Treg cell accumulation in colon tumor.
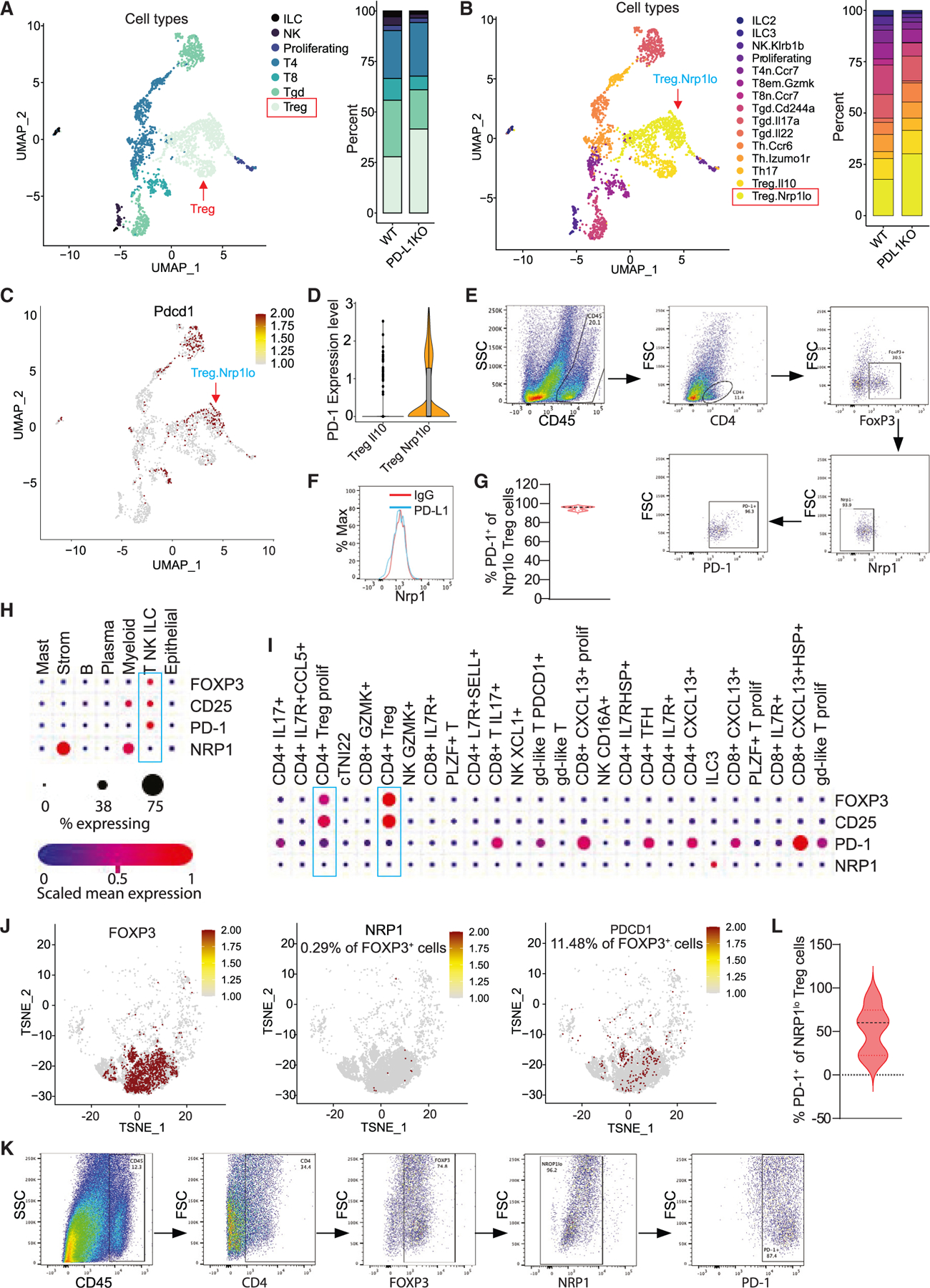
(A) UMAP projection (left) and barplot (right) of T, NK, and ILC in colon tumor at day 106. Treg, T regulatory cells; Tgd, γδ T cells; T8, CD8+ T cells; T4, CD4+ T cells; proliferating, proliferating cells; NK, NK cells; and ILC, innate lymphoid cells.
(B) UMAP projection (left) and barplot (right) of subpopulations of T, NK, and ILCs.
(C) UMAP projection showing PD-1 expression level in the indicated cell subpopulations as shown in (B).
(D) Expression of PD-1 in Treg subpopulations.
(E–G) Flow cytometry analysis of AOM-DSS-induced mouse colon tumors. Shown are representative gating strategies of one of five mice (E), representative Nrp1 expression level in Treg cells of one of five mice (F), and quantification of PD-1+ cells in Nrp1lo Treg cells (G) (n = 5).
(H and I) Human colon cancer patient scRNA-seq datasets (GEO: GSE178341) were analyzed for expression of the indicated genes in major cell types (H) and T cell subpopulations (I). The correlations are shown in dot plots.
(J) TSNE projection showing NRP1 and PDCD1 expression in POXP3+ cells in human colon tumor as shown in (H) and (I).
(K and L) Flow cytometry analysis of colon tumor tissues from human colon cancer patients. Shown are representative gating strategy of one of four mice (K) and quantification of PD-1+ cells in NRP1lo Treg cells (L) (n = 4).
PD-L1 deficiency increases neutrophil accumulation and IL6 production
Neutrophils are a highly heterogeneous population in human cancer patients and tumor-bearing mice.41,42 Our above finding revealed that loss of PD-L1 leads to expansion of tumor-associated neutrophils in colorectal tumor. To determine whether PD-L1 regulates neutrophil subpopulation differentiation, neutrophil subsets were further identified. Five unique neutrophil clusters were identified (Figures 5A and 5B). However, PD-L1 deficiency did not significantly change neutrophil subset clusters (Figures 5A and 5B), indicating that PD-L1 deficiency leads to expansion of the total neutrophils in colorectal tumor but not unique subsets of neutrophils. Analysis of spleen cells of the AOM-DSS-induced colorectal tumor validated the significant increase of neutrophils in mice with global Cd274 deletion (Figure 5C) and in the chimera mice (Figure 5D). However, mice with Cd274 deletion only in colorectal epithelial cells or myeloid cells have similar levels of neutrophils to the WT mice (Figure 5C).
Figure 5. Neutrophils express high levels of IL6 in mouse colorectal tumor.
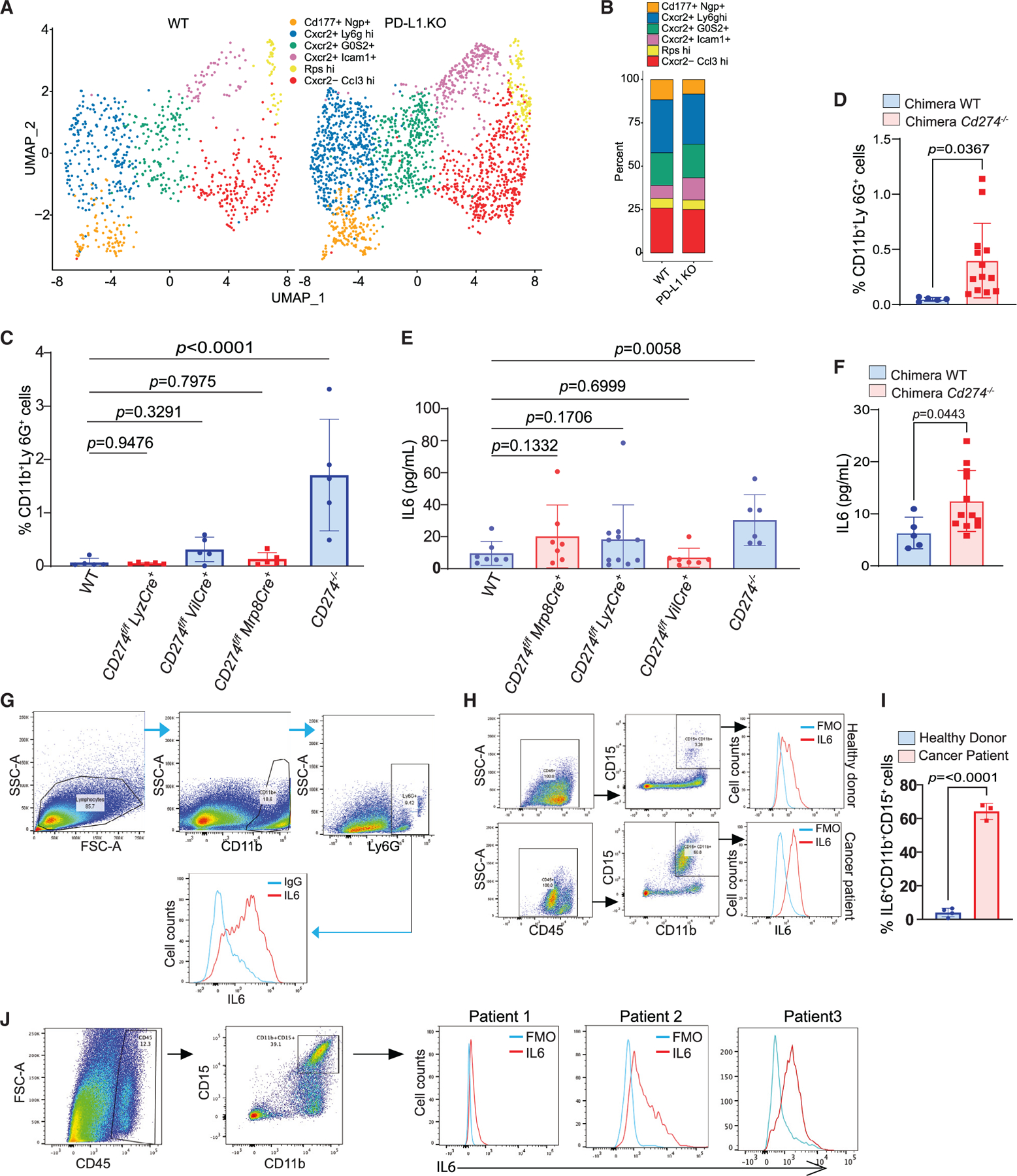
(A and B) UMAP projection (A) and barplot (B) of neutrophil subpopulations in colon tumor of WT and PD-L1 KO mice.
(C) Quantification of neutrophils in spleens of WT and the indicated tissue-specific and global PD-L1 KO mice by flow cytometry. Column, mean; bar, SD. p value was determined by Student’s t test. Each dot represents data from one mouse.
(D) Quantification of neutrophils in spleens of WT and PD-L1 KO chimera mice. Column, mean; bar, SD. p value was determined by Student’s t test. Each dot represents data from one mouse.
(E) IL6 protein level in serum of WT and the indicated tissue-specific and global PD-L1 KO mice. Column, mean; bar, SD. p value was determined by Student’s t test. Each dot represents data from one mouse.
(F) IL6 protein level in serum of WT and PD-L1 KO chimera mice. Column, mean; bar, SD. p value was determined by Student’s t test. Each dot represents data from one mouse.
(G) IL6 protein level in neutrophils of the orthotopic CT26 colon tumor as analyzed by flow cytometry. Shown are representative data of one of five mice.
(H) IL6 protein level in neutrophils in the peripheral blood of healthy donors (n = 3) and colorectal cancer patients (n = 3). Shown is a representative gating strategy of IL6 MFI of one donor of the three donors.
(I) Quantification of neutrophil levels in the peripheral blood of healthy donors and colorectal cancer patients as shown in (H). Column, mean; bar, SD. p value was determined by Student’s t test. Each dot represents data from one donor.
(J) IL6 protein level in human colon tumor. Shown are representative gating strategies (left) of one of three patients and IL6 MFI (right) in the tumor-infiltrating neutrophils.
IL6 protein level is significantly elevated in the tumor-bearing PD-L1 KO mice and chimera mice as compared to the WT mice (Figures 5E and 5F). The level of inflammatory cytokine IL17 is not significantly different between WT and PD-L1 KO mice (Figure S4). The levels of interferon gamma (IFNγ) and tumor necrosis factor alpha (TNFα), two cytokines produced by activated T cells, are also not significantly different between WT and PD-L1 KO mice (Figure S4). Only the levels of chemokines, including Cxcl1 and T cell chemoattractant Cxcl9 and Cxcl10, are significantly different between WT and PD-L1 KO tumor-bearing mice (Figure S4). Consistent with the neutrophil profiles, tumor-bearing mice with Cd274 deletion only in colorectal epithelial cells or myeloid cells have no significant change in IL6 protein level in the peripheral blood (Figure 5E). These findings establish a correlation between neutrophil accumulation level and IL6 protein level in the WT and PD-L1-deficient tumor-bearing mice, suggesting that neutrophils are producers of IL6 in the colorectal tumor-bearing mice. To test this hypothesis, we injected colon tumor cells to mouse cecal wall to establish orthotopic colon tumor. Analysis of the colon tumor identified a distinct population of neutrophils, and the majority of the neutrophils express IL6 (Figure 5G). Analysis of human peripheral blood samples revealed that CD11b+CD15+ neutrophils express IL6 (Figure 5H) and neutrophils are much more abundant in colorectal cancer patient blood in healthy donor blood (Figure 5I). Analysis of human colon tumor tissues revealed that tumor-infiltrating neutrophils express high levels of IL6 (Figure 5J). We therefore conclude that PD-L1 deficiency results in expansion of neutrophils in colorectal tumor, and tumor-infiltrating neutrophils are major producers of IL6 protein.
Treg cells recruit IL6+ neutrophils via secreting TGFβ
Emerging experimental data indicate that TGFβ works in concert with PD-L1 to maintain T cells in a dysfunctional state.43 TGFβ is a cytokine that not only regulates Treg differentiation but is also expressed in Treg cells.44,45 scRNA-seq analysis of the immune cell subsets revealed that TGFβ transcript is expressed in various cell types in WT and PD-L1 KO mouse colon tumors (Figure S5A). Tgfb1 is expressed in PD-1+ Nrp1lo Treg cells in the AOM-DSS-induced colorectal tumor (Figures 6A and 6B). Flow cytometry analysis determined that 90.4%–94.1% of PD-1+Nrp1lo Treg cells are TGFβ+ in mouse colorectal tumor (Figure 6C).
Figure 6. Treg cells produce TGFb to recruit neutrophils that secret IL6 to inhibit T cell activation in mouse colorectal tumor.
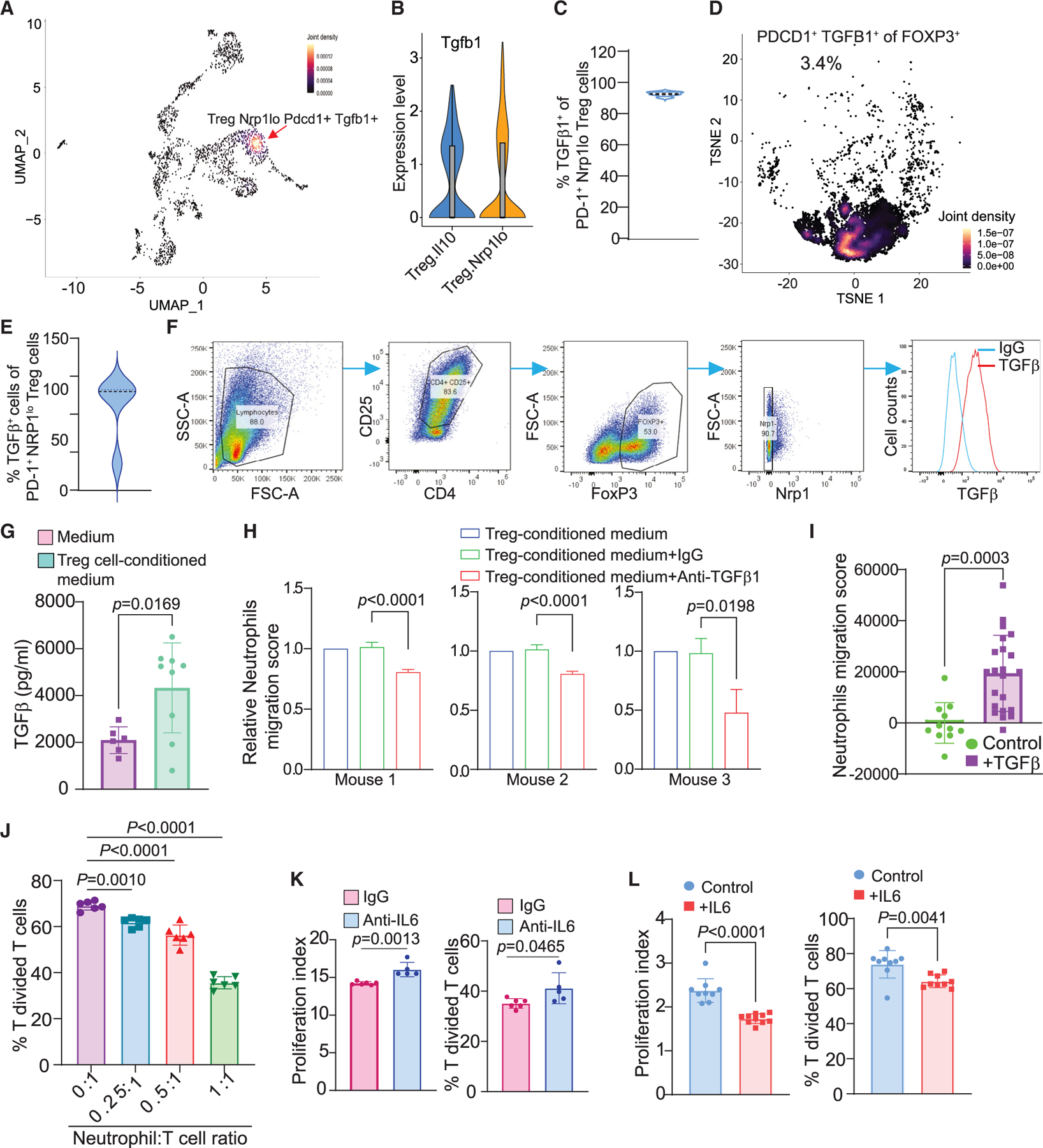
(A) Density plot of UMAP projection showing co-expression of Tgfb1 and PD-1 in Nrp1lo Treg cells in AOM-DSS-induced colon tumor.
(B) Expression of Tgfb1 in Treg cell subpopulations.
(C) Quantification of TGFβ1+ cells in PD-1+Nrp1lo Treg cells in AOM-DSS-induced mouse colon tumor as shown in Figure 4E.
(D) Density plot of PD-1+TGFβ+ cells in FOXP3+ Treg cells in human colon tumor as shown in Figure 4J.
(E) Quantification of TGFβ+ cells in PD-1+NRP1lo Treg cells in human colon tumor tissues (n = 4). The tumor tissues were analyzed by flow cytometry as shown in Figure 4K.
(F) Naive T cells were isolated from mouse spleens and induced to differentiate into Treg cells in vitro and analyzed for TGFβ expression by flow cytometry.
(G) Treg cells secrete TGFβ in vitro. Column, mean; bar, SD. p value was determined by Student’s t test. Each dot represents one replicate of measurement.
(H) Neutrophils were isolated from three tumor-bearing mice and cultured in Transwells with in vitro differentiated Treg cells in the presence of TGFβ1 neutralization mAb. Neutrophil migration was quantified. Column, mean; bar, SD. p value was determined by Student’s t test. Shown are results from three individual mouse-derived neutrophils.
(I) TGFβ protein induces migration of neutrophils in vitro. Column, mean; bar, SD. p value was determined by Student’s t test. Each dot represents one replicate of measurement.
(J) Neutrophils inhibit T cell activation in vitro. Neutrophils and T cells were co-cultured at the indicated ratio in anti-CD3/CD28-coated plates for 3 days. Column, mean; bar, SD. p value was determined by Student’s t test. Each dot represents one replicate of measurement.
(K and L) Neutrophils inhibit T cell activation through IL6. Neutrophils and T cells were co-cultured in a 1:1 ratio in the presence of immunoglobulin G (IgG) and IL6 neutralization mAb (K) and recombinant IL6 protein (L). Column, mean; bar, SD. p value was determined by Student’s t test. Each dot represents one replicate of measurement.
Analysis of human colon cancer patient scRNA-seq datasets28 validated that TGFB1 is expressed in various immune cell populations, including Treg cells (Figures S5B–S5E). Further analysis revealed that TGFB1 is highly expressed in tumor-infiltrating Treg cells in human colon tumor (Figures S5C and S5E). TGFB1 is expressed in the NRP1lo proliferating Treg cells in human colon tumor (Figures S5F and S5G). Correlation analysis indicates that TGFB1 is co-expressed in PD-1+NRP1lo Treg cells (Figures S5F and S5G) and approximately 3.4% PD-1+NRP1lo Treg cells express TGFB1 in human colon tumor (Figure 6D). Flow cytometry analysis shows that 26.7%–100% of tumor-infiltrating PD-1+NRP1lo Treg cells express TGFβ in human colon tumor (Figure 6E).
We then sought to test the hypothesis that Treg cells use TGFβ to recruit neutrophils. Naive T cells were purified from mouse spleens and cultured in vitro to induce differentiation into Treg cells. The in vitro-differentiated Treg cells are mostly Nrp1lo and express high levels of TGFβ (Figure 6F). The cultured Treg cells secrete TGFβ (Figure 6G). Neutrophils were then purified from tumor-bearing mice (Figure S5H) and cultured in Transwells in Treg cell-conditioned medium with or without TGFβ neutralization monoclonal antibody (mAb). Blocking TGFβ significantly decreased neutrophil migration (Figure 6H). Similarly, TGFβ protein significantly increased neutrophil migration (Figure 6I). Tumor-associated neutrophils are known to have potent suppressive activity against T cell activation.17,46–48 To determine whether neutrophils suppress T cell activation through IL6, we first co-cultured neutrophils with the antigen-specific 2/20 CTL line and analyzed T cell proliferation. Neutrophils inhibited T cell proliferation in a cell dose-dependent manner (Figure 6J). To determine whether neutrophils suppress T cell proliferation through IL6, IL6 neutralization mAb was then added to the co-culture. IL6 blockade increased T cell activation in the neutrophil and T cell co-culture (Figure 6K). To further determine IL6 function in T cell activation, we cultured purified splenic T cells in anti-CD3 and anti-CD28-coated plates in the presence of IL6. Analysis of T cell proliferation indicates that IL6 inhibited T cell activation (Figure 6L). Taken together, our findings determine that Treg cells produce TGFβ to recruit neutrophils and neutrophils secrete IL6 to inhibit T cell activation.
IL6 blockade immunotherapy suppresses colon tumor growth in vivo
To determine whether the above in vitro findings can be translated to in vivo tumor growth regulation in vivo, we injected CT26 tumor cells to the cecal wall of syngeneic mice to establish orthotopic colon tumor. CT26 is a microsatellite stable (MSS) subtype of colon tumor cell line.49 The AOM-DSS-induced colorectal tumor has no mutations of the DNA mismatch repair genes and is also a DNA mismatch repair-proficient MSS colorectal tumor subtype.50 To block neutrophil function, the CT26 tumor-bearing mice were treated with Ly6G neutralization mAb. Analysis of tumor tissues from the tumor-bearing mice shows that Ly6G blockade therapy effectively depleted neutrophils (Figures 7A and 7B). However, repeated attempts at neutrophil neutralization did not significantly change CD8+ T cell tumor infiltration level (Figure 7C) and tumor growth (Figure 7D). We then tested IL6 blockade. Treatment of the CT26 tumor-bearing mice with IL6 neutralization mAb significantly increased tumor-infiltrating CD8+ T cells (Figure 7E) and significantly suppressed tumor growth (Figure 7F). We then tested IL6 blockade in PD-L1 KO colon tumor and observed that IL6 neutralization also inhibited PD-L1 Ko colon tumor growth in vivo (Figures 7G and 7H). Next, we treated the AOM-DSS-induced colon tumor-bearing mice with IL6 neutralization mAb. IL6 blockade also increased CD8+ T cell activation and colorectal tumorigenesis (Figures 7I and 7J). We therefore conclude that IL6 inhibits T cell activation to promote colorectal tumor immune evasion.
Figure 7. IL6 blockade immunotherapy activates T cells and suppresses colon tumor growth in vivo.
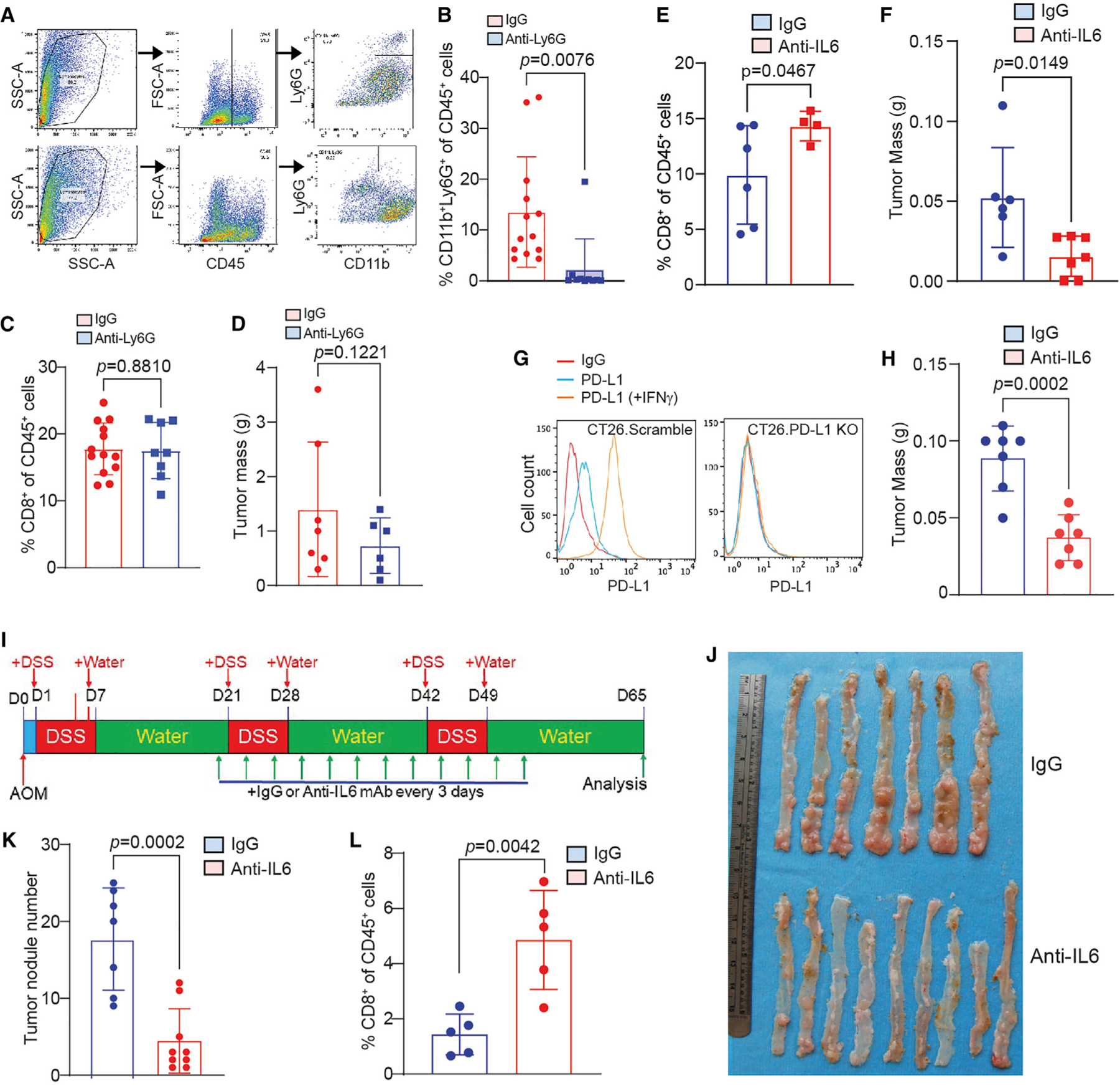
(A–D) CT26 orthotopic tumor-bearing mice were treated with Ly6G neutralization mAb. Shown are flow cytometry analysis representative gating strategy of the tumor of one of 13 mice (A), quantification of tumor-infiltrating neutrophils (B), quantification of tumor-infiltrating CD8+ T cells (C), and tumor weight (D). Column, mean; bar, SD. p value was determined by Student’s t test. Each dot represents data from one mouse.
(E and F) CT26 orthotopic tumor-bearing mice were treated with IL6 neutralization mAb. Shown are quantification of tumor-infiltrating CD8+ T cells by flow cytometry (E), and tumor weight (F). Column, mean; bar, SD. p value was determined by Student’s t test. Each dot represents data from one mouse.
(G) Generation of PD-L1 KO colon tumor CT26 cells.
(H) PD-L1 KO CT26 orthotopic tumor-bearing mice were treated with IL6 neutralization mAb and measured for tumor weight at the endpoint. Column, mean; bar, SD. p value was determined by Student’s t test. Each dot represents data from one mouse.
(I–L) The AOM-DSS-induced colon tumor mice were treated with IL6 neutralization mAb. Shown are treatment scheme (I), tumor-bearing colons (J), colon tumor nodule number (K), and tumor-infiltrating CD8+ T cell level (L). Column, mean; bar, SD. p value was determined by Student’s t test. Each dot represents data from one mouse.
DISCUSSION
Colorectal cancer was the first human neoplasia found to be under immunosurveillance more than a decade ago.51 The type, density, and location of tumor-infiltrating immune cells correlate with tumor initiation, progression, and recurrence.52 Human colorectal tumors are therefore a type of highly immunogenic cancer.51 However, human colorectal cancer is the third most commonly diagnosed human cancer, suggesting that the host immunosurveillance may be impaired in the colon epithelium and colorectal tumor microenvironment, resulting in tumor immune escape during colorectal tumorigenesis despite high tumor immunogenicity. A consequence of PD-(L)1 blockade immunotherapy is increased Treg cell accumulation, which promotes tumor progression.38–40 In this study, we observed that loss of PD-L1 function results in expansion of PD-1+ Treg cells. Treg cells are major suppressors of colonic inflammation and colitis.53,54 However, we observed that, despite increased Treg cells in the inflamed colon and colorectal tumor, PD-L1 KO mice exhibited increased colonic inflammation. It appears that Nrp1 is required for Treg immune suppressive function and stability. Nrp1hi Treg exhibits higher suppressive activity than Nrp1lo Treg cells.55–57 In this study, we observed that loss of host PD-L1 increased PD-1+Nrp1loTGFβ+ Treg cells. The Nrp1lo Treg cells might have no significant activity in suppression of colonic inflammation and colitis55–57 but suppresses tumor-reactive CTLs.
We determined that PD-1+Nrp1loTreg cells secrete TGFβ to recruit IL6+ neutrophils to the colorectal tumor. Neutrophils have both pro- and anti-tumor activity.41,42,58 Consistent with this phenomenon, neutrophil blockade with the Ly6G-specific mAb has no effect on colonic inflammation and colitis.59 It is therefore possible that neutrophil blockade may block both pro- and anti-tumor neutrophils. IL6 is a key suppressor of the anti-tumor immune response.17,48,60,61 We observed that IL6 blockade significantly suppressed colon tumor growth. We further determined that IL6 directly inhibits T cell activation. It is therefore possible that Ly6G− neutrophils may contribute to IL6 production. It is also possible that other cells, such as tumor cells, may also produce IL6, which may compensate neutrophil-produced IL6 function in IL6 blockade immunotherapy. Consistent with this notion, the levels of IFNγ and TNFα proteins, two effectors of activated T cells, are not increased in PD-L1 KO tumor-bearing mice as compared to WT mice. In this regard, loss of PD-L1 in colorectal cancer results in expansion of PD-1+Nrp1loTGFβ+ Treg cells that recruit IL6+ neutrophils, and neutrophils produce IL6 to inhibit activation of CD8+ T cells even though PD-L1 is absent. Our findings therefore indicate that the expanded PD-1+Nrp1lo Treg/IL6+ neutrophil cellular axis apparently overpowers the PD-L1 loss of function-activated CD8+ T cells to permit colorectal tumor immune escape, resulting in PD-L1 function as a suppressor of inflammation-driven colorectal tumor.
Approximately 85%–90% of human colorectal cancer is the MSS type, and only about 10%–15% of human colorectal cancer is the MSI-H subtype.62 Strikingly, membrane-bound PD-L1 protein is only present in MSI-H subset of human colorectal cancer.63 Considering the unique anatomic location of human colon, which is under a constant low-grade inflammation,22,64 the loss of PD-L1 protein in MSS human colorectal cancer and the higher MSS colorectal cancer frequency as compared to MSI-H subtype in humans suggest that PD-L1 might function as a suppressor in human MSS colorectal cancer, which remains a hypothesis to be tested.
Limitations of the study
This study focused on the inflammation-driven colorectal tumor in mice. IL6 is known to suppress T cell activation to promote tumor immune evasion.17,61 It is therefore likely that IL6 inhibition of T cell activation and tumor growth is a general phenomenon for both inflammation-driven and sporadic tumors. However, whether the PD-1+Nrp1loTGFβ+ Treg cells function to suppress T cells and whether PD-L1 acts as a suppressor of this subset of Treg cells in non-inflammation-driven colorectal tumor in mice remain to be determined. In human colon cancer patients, although the majority of tumor-infiltrating Treg cells are NRP1lo, the level of PD-1+ cells in the NRP1lo Treg cells ranges from 18.9% to 87.4%, and the level of TGFβ+ cells in the PD-1+NRP1lo Treg cells ranges from 26.7% to 100%. Considering that the unique anatomic location of colon that is under a constant low-grade inflammation,22,64 it is possible that the PD-1+NRP1loTGFβ+ Treg cells may contribute to colorectal tumor development in humans, especially in the MSS subtype of human colorectal cancer, which remains to be determined.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Kebin Liu: Kliu@augusta.edu.
Materials availability
Materials generated in this study is available upon request to the lead contact.
Data and code availability
scRNA-seq data generated in this studies are deposited in the National Institutes of Health Gene Expression Omnibus (GEO) database (GEO: GSE246038, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE150743) and are publicly available.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
BALB/c and C57BL/6 were purchased from Jackson Laboratory (Bar Harbor, ME) and Charles River Laboratories. Cd274fx/fx mice were created as described previously.33 LyzCre (B6.129P2-Lyz2tm1(cre)Ifo/J) and the Mrp8Cre (B6.Cg-Tg(S100A8-cre,-EGFP)1Ilw/J) were obtained from the Jackson Laboratory (Bar Harbor, ME). Cd274−/− mice were provided by Genentech13 (South San Francisco, CA). Use of animal studies were approved by Augusta University (Protocol #2008–0162) and Charlie Norwood VA Medical Center Institutional Animal Care and Use Committees (Protocol #1314554–16).
Human colorectal tumor specimens
Human colorectal tumor specimens and peripheral blood specimens were obtained from Charlie Norwood VA Medical Center and the Cooperative Human Tissue Network (CHTN) Sothern Division (Duke University School of Medicine). Use of human tumor tissues and peripheral blood was approved by Augusta University and Charlie Norwood VA Medical Center Institutional Review Boards.
Cell lines
CT26 cell line was obtained from American Type Culture Collection (ATCC) (Manassas, VA). CT26.Scramble and CT26.PD-L1 KO cell lines were generated as previously described.4 AH1 antigen-specific T cells (2/20 CTLs) were generated and maintained as previously described.65 The 2/20 CTLs (1.3×105 cells/ml) were cultured with AH1 peptide (SPSYVYHQF, 1 μg/mL), recombinant IL-2 protein (10 Units/ml), and lethally irradiated BALB/c mouse spleen cells (1/36 spleen/ml) weekly in 24-well palte. Cell lines were tested bi-monthly for mycoplasma and were mycoplasma-free at time of experiments.
METHOD DETAILS
The AOM-DSS spontaneous colorectal tumor model
Mice were injected with azoxymethane (AOM) 10 mg/kg body weight, intraperitoneally, once. The day after AOM injection, mice were given dextran sodium sulfate (DSS), 2%, in the drinking water for 1 week. The DSS was then replaced with drinking water for 2 weeks. The DSS was repeated twice for a total of 3 cycles. Mice were maintained on normal drinking water until being sacrificed. Mice were weighed every 3–4 days and checked daily for survival. Colon tissues were harvested and cleaned thoroughly with PBS. The colons were cut open longitudinally. Both male and female mice were used for all genotypes. All mice were genotyped prior to the experiment.
Tumor cell transplant
Tumor cells were cultured in RPMI160 with 10% FBS for 24 h, and then harvested with trypsin and washed three times with PBS to remove residual media. Tumor cells were resuspended in PBS and injected subcutaneously into the right flank or orthotopically into the cecal wall of mice.
In vivo blockade
Tumor cells were implanted orthotopically to the cecum using an insulin syringe (5×104 cells/mouse). Treatment began on days 3–10. Mice received 200 μg of the blockade mAbs every 3 days via ip injection.
scRNA-sequencing
Colons were collected from the tumor-bearing mice. Tumor nodules were dissected from the colon and digested with collagenase/hyaluronidase/DNaseI solution to RPMI1640 medium at 37°C for 30 min with continuous agitation by stir bar and needle aspiration. Live cells were subsequently isolated by lymphocyte separation medium gradient centrifugation. Single cell isolation and library generation were performed by 10xGenomic protocol. Single-cell RNA sequencing reads were mapped to the mouse reference genome and processed into gene expression matrices with CellRanger (l0x Genomics; version 6.1.2). Subsequent analysis was conducted with Seurat (v4.3) in R. Cells were subsetted to those with <15% mitochondrial reads and 200–6000 RNA features. Doublets were identified and discarded using DoubletFinder. Four scRNA-seq data sets were merged and integrated using Seurat Canonical Correlation Analysis (CCA) algorithm. Cells then underwent nearest-neighbor clustering; clusters were annotated by CelliD and manual review. For immune cell subtypes-specific analysis, cells of each subtype were subsetted and re-integrated using CCA or Harmony (for T, NK and ILC cells). Seurat and scCustomize were used to generate various plots for visualization.
Tumor and colon digestion
Tumors were manually dissected and digested using a collagenase/hyaluronidase/DNase I solution in scintillation vials with magnetic stir rods to break up the tissue. Tissues are digested for 25 min at 37°C or at room temperature for 1 h.
Human non-neoplastic tissue and colon tumor tissue scRNA-seq dataset analysis
The non-neoplastic human tissue scRNA-Seq datasets were retrieved from the Human Protein Atlas database (HPA)(https://v19.proteinatlas.org) that contains 81 cell types from 31 datasets from HPA and Genotype-Tissue Expression (GTEx) (https://gtexportal.org) project. Human colon cancer datasets were retrieved from the GEO database (Accession # GSE178341)28 and the Broad Institute single-cell portal (https://singlecell.broadinstitute.org/single_cell). T, NK, and ILC cells were extracted, and the original cell type annotation and tSNE embedding were used to generate plots using scCustomize or Seurat.
Flow cytometry
Cells resuspended in PBS or FACS buffer. Antibodies were added to appropriate concentration and stained at 4°C for 30–60 min, then washed with PBS+0.5% BSA. Cells were fixed in in 2% paraformaldehyde. For Zombie UV staining: Cells resuspended in PBS or FACS buffer. Zombie UV (1:1000) added, incubate for 10 min at room temperature in the dark. Antibodies then added at appropriate concentrations and stain as above. Intracellular staining was carried out using BD Cytofix/Cytoperm kit. Cells were resuspended in FACS buffer with Golgiplug for 2 h. Stained for surface markers per above protocol. Washed 2x with FACS buffer. Resuspended in permeabilization solution, then washed 2x in perm/wash buffer. Resuspended in perm/wash buffer. Antibodies for intracellular proteins of interest added, incubate for 20 min at room temperature in the dark. Washed in perm/wash buffer, resuspended in 2% paraformaldehyde.
Analysis of colonic inflammation
Colon tissues were prepared as Swiss rolls and fixed in 10% formalin overnight. The fixed tissues were processed into paraffin blocks and cut into 10-μmicron sections. For the inflammation score, each grade represents the following: Grade 0: normal colonic mucosa; Grade 1: loss of 1/3 of the crypts; Grade 2: loss of 2/3 of the crypts; Grade 3: lamina propria is covered with a single layer of epithelium, mild inflammatory cell infiltrate present; Grade 4: erosions and marked inflammatory cell infiltration present. The slides were evaluated by a board-certified pathologist (A.B.).
ELISA
A 96-well assay plate was coated overnight with capture antibody. The plate was then washed 3 times with 0.05% Tween 20 in PBS and blocked for 1–3 h with 1x assay diluent. Standards and samples diluted as appropriate in assay diluent were then added to the wells, in duplicate/triplicate. The plate was then incubated 1–3 h, and washed 3 times with wash buffer. Capture antibody was then added for 1 h, and the plate was again washed times. Avidin-HRP was added for 30 min, and then washed 3 times. 1:1 TMB substrate was then added to each well, and the plate was left to incubate in the dark for 10–15 min (based on manufacturer protocol). The reaction was stopped with 1M H2SO4 and plate was read for absorbance in a plate reader.
Serum cytokine analysis
Blood samples were collected either via cardia bleed or via a submandibular vein bleed into serum gel tubes. Tubes were allowed to clot for at least 30 min, then centrifuged to collect the serum. Simultaneous quantification of cytokines in murine serum was performed using LEGENDplex Mouse Inflammation Panel according to manufacturer’s instructions. In brief, serum samples were diluted 2-fold with assay buffer and standards were mixed with matrix solution (Biolegend) to account for additional components in the serum samples. Standards and samples were plated with capture beads and incubated for 2 h at room temperature on plate shaker (800 rpm). After washing the plate with wash buffer, detection antibodies were added to each well. The plate was incubated on shaker for 1h at room temperature. Finally, without washing, SA-PE was added and incubated for 30 min. Samples were acquired on Novocyte Quanteon flow cytometer (Agilent Technologies). Standard curves and protein concentration were calculated using R package DrLumi installed on R 3.5.2. The limit of detection was calculated as an average of background samples plus 3xSD. Assay and data calculations were performed at Immune Monitoring Shared Resource laboratory.
T cell proliferation assay
CD3+ T cells were purified from BALB/c mouse spleen cells with the MojoSort mouse CD3 T cell isolation kit (Biolegend, San Diego, CA) according to the manufacturer’s instructions. Isolated cells are incubated in a 37°C water bath in prewarmed PBS+0.15–0.30μM CFSE (Life Technologies, Carlsbad, CA, USA)for 15 min, vortexing every 5 min. The cells are then washed and incubated in RPMI 1640 medium with 10% FBS at a volume of 5x the volume used for labelling for 30 min, vortexing every 10 min. The cells are again washed 2 times with medium and plated. CFSE intensity is analyzed using a flow cytometer. For T cell proliferation assay, a 96-well culture plate was coated with anti-mouse CD3 (8 μg/mL), anti-mouse CD28 MAbs (10 μg/mL), and re. The purified T cells were labeled with and then seeded in the plate at a density of 1.5 × 105 cells/well in 150 μL medium for 3 days. Cells were analyzed by flow cytometry. Proliferation index is calculated using the proliferation tool in FlowJo program and expressed as the total number of divisions divided by the number of cells that went into division.
Treg differentiation
24-well plates were coated with anti-CD3 mAb (8 μg/mL) overnight in 4°C. Single cell suspension was prepared from mouse spleen and CD4+ cells were isolated using Biolegend CD4 isolation kit. Treg were differentiated from the purified CD4+ cells using ImmunoCult Mouse Treg Differentiation Supplement (Stemcell Technologies) according to the manusfacturer’s instructions. Briefly, cells were suspended in Treg differentiation media, then added to the anti-CD3 mAb-coated plate. The cells were incubated for 6 days, then analyzed via flow cytometry. The culture supernatant was collected, centrifuged at 13,000 RPM for 5 min and used as Treg-conditioned medium.
Neutrophil isolation and migration assay
Neutrophils were isolated using Biolegend Neutrophil isolation kit, per manufacturer protocols. Neutropihls were isolated from the spleen of AT3 tumor bearing mice. The CytoSelect 96-Well Cell Migration Assay Kit (Cell Biolabs) was used for migration assay. The migration plate was brought to room temperature. 250 μL of cells (2×106/well) were added to each well insert. 400μL of serum-free media and TGFβ1, or Treg cell-conditioned medium was added to the lower chamber. The cells were incubated for 18–24 h. On day 2, 225 μL of the lower well was transferred to a white walled 96 well plate for fluorescence after following manufacturer instructions for adherent cells. The CyQuant dye was diluted 1:75 with 4x lysis buffer. 75 μL of the dye/lysis buffer mixture was added to each well of the 96 well plate. Flourescence was read on 48/520nm channel.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were analyzed using GraphPad Prism 10.2.2. p value was calculated by student t test with p < 0.05 as being significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| BV711-CD3 (OKT3) | Biolegend | 317327, RRID AB_11219592 |
| CD3e (Clone 145–2C11) | Biolegend | 100302; RRID AB_312667 |
| PE-CD4 (RM4–4) | Biolegend | 116005, RRID AB_313690 |
| AF700 CD4 (RPA-T4) | Biolegend | 300526, RRID AB_493743 |
| CD4 (4SM95) | ThermoFisher Scientific | 14–9766-82, RRID AB_2573008 |
| AF700-CD8 (53–6.7) | Biolegend | 100729, RRID AB_493702 |
| PE-CD8 | Biolegend | 100707, RRID AB_312746 |
| FITC-CD11b (M1/70) | Biolegend | 101206, RRID AB_312789 |
| BV711-CD11b (M1/70) | Biolegend | 101241, RRID AB_11218791 |
| CD25 (EPR22588–18) | Abcam | Ab227834 |
| PerCP-CD25 (M-A251) | Biolegend | 356131, RRID AB_2563591 |
| PerCP-CD25 (PC61) | Biolegend | 102027, RRID AB_893290 |
| Mouse anti CD28 | Biolegend | 102102, RRID AB_312867 |
| FITC-CD45 (30-F11) | Biolegend | 103107, RRID AB_312972 |
| AF647-FOXP3 (206D) | Biolegend | 320113, RRIDAB_439753 |
| AF647-FOXP3 (MF-14) | Biolegend | 126407, RRID AB_1089116 |
| PE-FOXP3 | Biolegend | 126403, RRID AB_1089118 |
| Hamster IgG | Bioxcell | BE0091, RRID AB_1107773 |
| Mouse IgG (MOPC-21) | Bioxcell | BP0083, RRID AB_1107784 |
| Rat IgG (2A3) | Bioxcell | BP0089, RRID AB_1107769 |
| IL-6 (MP5–20F3) | Bioxcell | BE0046, RRID AB _1107709 |
| FITC-IL6 (MQ2–13A5) | Biolegend | 501103, RRID AB_315151 |
| PE-IL6 (MP5–20F3) | Biolegend | 504504 RRID AB_315338 |
| PerCP-Ly6C (HK1.4) | Biolegend | 128028, RRID AB_10897805 |
| AF700-Ly6C (HK1.4) | Biolegend | 128024, RRID AB_10643270 |
| PerCP-Ly6G (1A8) | Biolegend | 127654, RRID AB_2616999 |
| Ly6G (1A8) | BioXCell | BP0075, RRID AB_1107721 |
| APC-Ly6G (1A8) | Biolegend | 127613, RRID AB_1877163 |
| APC-Nrp1 (3E12) | Biolegend | 145206, RRID AB_2562032 |
| PE-Nrp1 (3E12) | Biolegend | 145203, RRID AB_2561927 |
| PE/Cy7-Nrp1 (3e12) | Biolegend | 145211, RRID AB_2562359 |
| PE-PD-1 (29F.1A12) | Biolegend | 135205, RRID AB_1877232 |
| FITC-PD-1 (29F.1A12) | Biolegend | 135213, RRID AB_10689633 |
| PE/Cy7-PD-L1 (10F.9G2) | Biolegend | 124314, AB_10643573 |
| PD-L1 (28 8) | Abcam | 205921, N/A |
| TGF-β1 (19D8) | Biolegend | 521708, AB_2810654 |
| FITC-TGFβ1 (TW7–16B4) | Biolegend | 141413, RRID AB_2721327 |
| MACH 2 Rabbit HRP-Polymer | Biocare Medical | RHRP520G |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Azoxymethane | Sigma-Aldrich | A5486 |
| β-mercaptoethanol | Sigma-Aldrich | 63689 |
| 3,3′-Diaminobenzidine (DAB) | Sigma-Aldrich | D5905 |
| DAPI | Sigma-Aldrich | D9542 |
| Dextran sodium sulfate (SDS) | MP Biomedicals | 160110 |
| DMEM medium | Corning | 10–013-CV |
| EDTA pH8.0 | Invitrogen | 15575–038 |
| Eosin | Sigma-Aldrich | 45260 |
| Fetal Bovine Serum (FBS) | Hyclone | SH30396.03 |
| H2SO4 | Sigma-Aldrich | 335741 |
| Haematoxylin | Sigma-Aldrich | H3136 |
| HEPES pH 7.3 | ThermoFisher Scientific | J16924AE |
| Paraformaldehyde | Sigma-Aldrich | 158127 |
| RPMI 1640 medium | Corning | 10–040-CV |
| Slide mounting media (diamond) | ThermoFisher Scientific | P36961 |
| Sodium pyruvate | LONZA | 13–115E |
| Sodium bicarbonate | Sigma-Aldrich | S5761 |
| Tween 20 | Signa-Aldrich | P1379 |
| Universal-Agarose, peqGOLD | Peqlab | 351020 |
| Xylenes | Sigma-Aldrich | 214736 |
| Zombie UV | Biolegend | 423107 |
| Collagenase | Sigma-Aldrich | C0130 |
| DNase I | Sigma-Aldrich | D4263–5VL |
| Fc blocking reagent | Biolegend | 101319 |
| Hyaluronidase | Sigma-Aldrich | H3506 |
| Mouse IL-6 | StemCell | 78052 |
| TGF-β1 | Biolegend | 763104 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Antigen retrieval solution pH 6.0 | Vector Laboratories | H-3300–250 |
| Antigen retrieval solution pH 9.0 | Vector Laboratories | H3301–250 |
| BD Inracellular staining kit | BD Biosciences | 555028 |
| CD4 cell isolation kit MojoSort | Biolegend | 480033 |
| ImmunoCult Mouse Treg Differentiation Supplement | Stemcell Technologies | 10957 |
| Chromium Single Cell 3′ Reagent Kit v3 | 10x Genomics | N/A |
| The CytoSelect 96-Well Cell Migration Assay Kit | Cell Biolabs | CBA-104 |
| QuantiLuc 4 Reagent | Invivogen | QLCA-45–03 |
| MojoSort mouse neutrophil isolation kit | Biolegend | 480057 |
| Penicillin-Streptomycin solution | Hyclone | SV30010 |
| ZymoPURE II Plasmid Maxiprep Kit | Zymo Research | D4203 |
| Quick-DNA miniprep Plus Kit | Zymo Research | D4069 |
|
| ||
| Deposited data | ||
|
| ||
| Single-cell RNA sequencing | GEO/NCBI/NIH | Accession # GSE246038 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: BALB/c | Jackson Laboratory and Charles River Laboratories | N/A |
| Mouse: C57/BLJ6 | Jackson Laboratory and Charles River Laboratories | N/A |
| Mouse: Cd274f/f | Schwartz et al., 201726 | N/A |
| Mouse: LyzMCre | Jackson Laboratory | N/A |
| Mouse: Mrp8Cre | Jackson Laboratory | N/A |
| Mouse: Cd274−/− | Genentech | Oh et al. 202021 |
| Mouse: VilinCre | Jackson Laboratory | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
|
Cd274 sgRNA 5′ TCCAAAGGACTTGTACGTGG 3′ |
Genscript | N/A |
| Scramble sgRNA 5′ CTCGTATCTTTTCCCACGGC 3′ |
Genscript | N/A |
|
LyzMCre-Mutant: 5′CCC AGA AAT GCC AGA TTA CG 3′ |
IDT | N/A |
|
LyzMCre-Common: 5′ CTTGGGCTGCCAGAATTTCTC 3′ |
IDT | N/A |
|
LyzMCre-WT: 5′ TTACAGTCGGCCAGGCTGAC 3′ |
IDT | N/A |
|
Vil1-Cre-Mutant: 5′ AGGCAAATTTTGGTGTACGG 3′ |
IDT | N/A |
|
Vil1-Cre-Common: 5′ GCCTTCTCCTCTAGGCTCGT 3′ |
IDT | N/A |
|
Vil1-Cre-WT: 5′ TATAGGGCAGAGCTGGAGGA 3′ |
IDT | N/A |
|
Mrp8-Cre-F: 5′GCGGTCTGGCAGTAAAAACTATC 3′ |
IDT | N/A |
|
Mrp8-Cre-R: 5′ GTGAAACAGCATTGCTGTCACTT 3′ |
IDT | N/A |
|
Cd274f/f-F: 5′ AGAGGAGATACTCAGTGTTTGCC 3′ |
IDT | N/A |
|
Cd274f/f-R: 5′ TTCAAACTCAGCCAAGGACC 3′ |
IDT | N/A |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| CT26 | ATCC | CRL-2638 |
| 2/20 CTL | Ryan et al., 200135 | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| BD Diva 8.01 | BD Biosciences | N/A |
| CelliD | Cortal et al.35 | https://github.com/RausellLab/CelliD |
| CellQuestPro | BD Biosciences | N/A |
| FlowJo v10.6.0 | BD Biosciences | N/A |
| GraphPad Prism 10.2.2 | GraphPad Prism | N/A |
| Harmony (V0.1.1) | The Comprehensive R Archive Network (CRAN) | https://github.com/immunogenomics/harmony |
| LASX software | Leica Microsystems | N/A |
| Monocle3 (V1.2.9 | The Comprehensive R Archive Network (CRAN) | https://cole-trapnell-lab.github.io/monocle3/ |
| R (V4.2.3) | The R Foundation | https://www.r-project.org/ |
| scCustomize | Marsh SE | https://samuel-marsh.github.io/scCustomize/ |
| Seurat (V4.3.0.1) | Hao et al. | https://satijalab.org/seurat/ |
| VISION | – | https://github.com/YosefLab/VISION |
Highlights.
Global PD-L1 knockout promotes inflammation-driven colorectal tumorigenesis
Loss of PD-L1 function expands PD-1+Nrp1lo Treg cells in inflammation-driven colorectal tumor
PD-1+Nrp1lo Treg cells secrete TGFβ to recruit IL6+ neutrophils to inhibit T cell activation
IL6 blockade therapy increases CD8+ T cell tumor infiltration and suppresses colon tumor
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health, United States grants R01CA278852 (to K.L.), R01CA264983 (to H.S.), F30CA236436 (to J.D.K.), and F31CA257212 (to A.D.M.), and VA Merit Review Award I01CX001364 (to K.L.). We thank Dr. Rafal Pacholczyk at the Immune Monitoring Core Lab in Georgia Cancer Center for flow-bead array analysis. We thank Dr. Sam Chang-Sheng at the Integrated Genomic and High-Performance Computing core facility at Georgia Cancer Center for technical assistance for performing the 10x Genomics single-cell sequencing analysis. Penny Roon and Donna Kumiski at the Medical College of Georgia Electron Microscopy and Histology Core provided technical assistance in tissue preparation and histology work. Megan Griffin and Elizabeth Howington at the Cooperative Human Tissue Network (Duke University School of Medicine) provided human colon tumor specimens. The graphic abstract was prepared using BioRender.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.114819.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al. (2002). Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat. Med 8, 793–800. 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 2.Christofides A, Katopodi XL, Cao C, Karagkouni D, Aliazis K, Yenyuwadee S, Aksoylar HI, Pal R, Mahmoud MAA, Strauss L, et al. (2023). SHP-2 and PD-1-SHP-2 signaling regulate myeloid cell differentiation and antitumor responses. Nat. Immunol 24, 55–68. 10.1038/s41590-022-01385-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Strauss L, Mahmoud MAA, Weaver JD, Tijaro-Ovalle NM, Christofides A, Wang Q, Pal R, Yuan M, Asara J, Patsoukis N, and Boussiotis VA (2020). Targeted deletion of PD-1 in myeloid cells induces anti-tumor immunity. Sci. Immunol 5, eaay1863. 10.1126/sciimmunol.aay1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klement JD, Redd PS, Lu C, Merting AD, Poschel DB, Yang D, Savage NM, Zhou G, Munn DH, Fallon PG, and Liu K (2023). Tumor PD-L1 engages myeloid PD-1 to suppress type I interferon to impair cytotoxic T lymphocyte recruitment. Cancer Cell 41, 620–636.e9. 10.1016/j.ccell.2023.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, Gupta R, Tsai JM, Sinha R, Corey D, et al. (2017). PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 545, 495–499. 10.1038/nature22396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, Sasmal DK, Huang J, Kim JM, Mellman I, and Vale RD (2017). T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 355, 1428–1433. 10.1126/science.aaf1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, Konieczny BT, Daugherty CZ, Koenig L, Yu K, et al. (2017). Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science 355, 1423–1427. 10.1126/science.aaf0683science.aaf0683[pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brahmer JR, Tykodi SS, Chow LQM, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. (2012). Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med 366, 2455–2465. 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cercek A, Lumish M, Sinopoli J, Weiss J, Shia J, Lamendola-Essel M, El Dika IH, Segal N, Shcherba M, Sugarman R, et al. (2022). PD-1 Blockade in Mismatch Repair-Deficient, Locally Advanced Rectal Cancer. N. Engl. J. Med 386, 2363–2376. 10.1056/NEJMoa2201445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Juneja VR, McGuire KA, Manguso RT, LaFleur MW, Collins N, Haining WN, Freeman GJ, and Sharpe AH (2017). PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J. Exp. Med 214, 895–904. 10.1084/jem.20160801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oh SA, Wu DC, Cheung J, Navarro A, Xiong H, Cubas R, Totpal K, Chiu H, Wu Y, Comps-Agrar L, et al. (2020). PD-L1 expression by dendritic cells is a key regulator of T-cell immunity in cancer. Nat. Can. (Ott.) 1, 681–691. 10.1038/s43018-020-0075-x. [DOI] [PubMed] [Google Scholar]
- 12.Peng Q, Qiu X, Zhang Z, Zhang S, Zhang Y, Liang Y, Guo J, Peng H, Chen M, Fu YX, and Tang H (2020). PD-L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade. Nat. Commun 11, 4835. 10.1038/s41467-020-18570-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lau J, Cheung J, Navarro A, Lianoglou S, Haley B, Totpal K, Sanders L, Koeppen H, Caplazi P, McBride J, et al. (2017). Tumour and host cell PD-L1 is required to mediate suppression of anti-tumour immunity in mice. Nat. Commun 8, 14572. 10.1038/ncomms14572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin H, Wei S, Hurt EM, Green MD, Zhao L, Vatan L, Szeliga W, Herbst R, Harms PW, Fecher LA, et al. (2018). Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J. Clin. Invest 128, 805–815. 10.1172/JCI96113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang H, Liang Y, Anders RA, Taube JM, Qiu X, Mulgaonkar A, Liu X, Harrington SM, Guo J, Xin Y, et al. (2018). PD-L1 on host cells is essential for PD-L1 blockade-mediated tumor regression. J. Clin. Invest 128, 580–588. 10.1172/JCI96061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dammeijer F, van Gulijk M, Mulder EE, Lukkes M, Klaase L, van den Bosch T, van Nimwegen M, Lau SP, Latupeirissa K, Schetters S, et al. (2020). The PD-1/PD-L1-Checkpoint Restrains T cell Immunity in Tumor-Draining Lymph Nodes. Cancer Cell 38, 685–700.e8. 10.1016/j.ccell.2020.09.001. [DOI] [PubMed] [Google Scholar]
- 17.Mace TA, Shakya R, Pitarresi JR, Swanson B, McQuinn CW, Loftus S, Nordquist E, Cruz-Monserrate Z, Yu L, Young G, et al. (2018). IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut 67, 320–332. 10.1136/gutjnl-2016-311585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perry CJ, Muñoz-Rojas AR, Meeth KM, Kellman LN, Amezquita RA, Thakral D, Du VY, Wang JX, Damsky W, Kuhlmann AL, et al. (2018). Myeloid-targeted immunotherapies act in synergy to induce inflammation and antitumor immunity. J. Exp. Med 215, 877–893. 10.1084/jem.20171435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zanker DJ, Owen KL, Baschuk N, Spurling AJ, and Parker BS (2021). Loss of type I IFN responsiveness impairs natural killer cell anti-tumor activity in breast cancer. Cancer Immunol. Immunother 70, 2125–2138. 10.1007/s00262-021-02857-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoon HH, Jin Z, Kour O, Kankeu Fonkoua LA, Shitara K, Gibson MK, Prokop LJ, Moehler M, Kang YK, Shi Q, and Ajani JA (2022). Association of PD-L1 Expression and Other Variables With Benefit From Immune Checkpoint Inhibition in Advanced Gastroesophageal Cancer: Systematic Review and Meta-analysis of 17 Phase 3 Randomized Clinical Trials. JAMA Oncol 8, 1456–1465. 10.1001/jamaoncol.2022.3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zou Y, Zou X, Zheng S, Tang H, Zhang L, Liu P, and Xie X (2020). Efficacy and predictive factors of immune checkpoint inhibitors in metastatic breast cancer: a systematic review and meta-analysis. Ther. Adv. Med. Oncol 12, 1758835920940928. 10.1177/1758835920940928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arthur JC, Perez-Chanona E, Mühlbauer M, Tomkovich S, Uronis JM, Fan TJ, Campbell BJ, Abujamel T, Dogan B, Rogers AB, et al. (2012). Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 338, 120–123. 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. (2015). Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med 373, 23–34. 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang DY, Mooradian MJ, Kim D, Shah NJ, Fenton SE, Conry RM, Mehta R, Silk AW, Zhou A, Compton ML, et al. (2019). Clinical characterization of colitis arising from anti-PD-1 based therapy. OncoImmunology 8, e1524695. 10.1080/2162402X.2018.1524695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goodman RS, Lawless A, Woodford R, Fa’ak F, Tipirneni A, Patrinely JR, Yeoh HL, Rapisuwon S, Haydon A, Osman I, et al. (2023). Extended Follow-Up of Chronic Immune-Related Adverse Events Following Adjuvant Anti-PD-1 Therapy for High-Risk Resected Melanoma. JAMA Netw. Open 6, e2327145. 10.1001/jamanetworkopen.2023.27145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song MY, Hong CP, Park SJ, Kim JH, Yang BG, Park Y, Kim SW, Kim KS, Lee JY, Lee SW, et al. (2015). Protective effects of Fc-fused PD-L1 on two different animal models of colitis. Gut 64, 260–271. 10.1136/gutjnl-2014-307311. [DOI] [PubMed] [Google Scholar]
- 27.Scandiuzzi L, Ghosh K, Hofmeyer KA, Abadi YM, Lázár-Molnár E, Lin EY, Liu Q, Jeon H, Almo SC, Chen L, et al. (2014). Tissue-expressed B7-H1 critically controls intestinal inflammation. Cell Rep 6, 625–632. 10.1016/j.celrep.2014.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pelka K, Hofree M, Chen JH, Sarkizova S, Pirl JD, Jorgji V, Bejnood A, Dionne D, Ge WH, Xu KH, et al. (2021). Spatially organized multicellular immune hubs in human colorectal cancer. Cell 184, 4734–4752.e20. 10.1016/j.cell.2021.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neufert C, Becker C, and Neurath MF (2007). An inducible mouse model of colon carcinogenesis for the analysis of sporadic and inflammation-driven tumor progression. Nat. Protoc 2, 1998–2004. 10.1038/nprot.2007.279. [DOI] [PubMed] [Google Scholar]
- 30.Gopalakrishnan V, Helmink BA, Spencer CN, Reuben A, and Wargo JA (2018). The Influence of the Gut Microbiome on Cancer, Immunity, and Cancer Immunotherapy. Cancer Cell 33, 570–580. 10.1016/j.ccell.2018.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y, Liang X, Dong W, Fang Y, Lv J, Zhang T, Fiskesund R, Xie J, Liu J, Yin X, et al. (2018). Tumor-Repopulating Cells Induce PD-1 Expression in CD8(+) T Cells by Transferring Kynurenine and AhR Activation. Cancer Cell 33, 480–494.e7. 10.1016/j.ccell.2018.02.005. [DOI] [PubMed] [Google Scholar]
- 32.Petty AJ, Dai R, Lapalombella R, Baiocchi RA, Benson DM, Li Z, Huang X, and Yang Y (2021). Hedgehog-induced PD-L1 on tumor-associated macrophages is critical for suppression of tumor-infiltrating CD8+ T cell function. JCI Insight 6, e146707. 10.1172/jci.insight.146707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schwartz C, Khan AR, Floudas A, Saunders SP, Hams E, Rodewald HR, McKenzie ANJ, and Fallon PG (2017). ILC2s regulate adaptive Th2 cell functions via PD-L1 checkpoint control. J. Exp. Med 214, 2507–2521. 10.1084/jem.20170051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Redd PS, Lu C, Klement JD, Ibrahim ML, Zhou G, Kumai T, Celis E, and Liu K (2018). H3K4me3 mediates the NF-kappaB p50 homodimer binding to the pdcd1 promoter to activate PD-1 transcription in T cells. OncoImmunology 7, e1483302. 10.1080/2162402X.2018.1483302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cortal A, Martignetti L, Six E, and Rausell A (2021). Gene signature extraction and cell identity recognition at the single-cell level with Cell-ID. Nat. Biotechnol 39, 1095–1102. 10.1038/s41587-021-00896-6. [DOI] [PubMed] [Google Scholar]
- 36.Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, Krzysiek R, Knutson KL, Daniel B, Zimmermann MC, et al. (2003). Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat. Med 9, 562–567. 10.1038/nm863. [DOI] [PubMed] [Google Scholar]
- 37.Gato-Canas M, Zuazo M, Arasanz H, Ibanez-Vea M, Lorenzo L, Fernandez-Hinojal G, Vera R, Smerdou C, Martisova E, Arozarena I, et al. (2017). PDL1 Signals through Conserved Sequence Motifs to Overcome Interferon-Mediated Cytotoxicity. Cell Rep 20, 1818–1829. 10.1016/j.celrep.2017.07.075. [DOI] [PubMed] [Google Scholar]
- 38.Wakiyama H, Kato T, Furusawa A, Okada R, Inagaki F, Furumoto H, Fukushima H, Okuyama S, Choyke PL, and Kobayashi H (2022). Treg-Dominant Tumor Microenvironment Is Responsible for Hyperprogressive Disease after PD-1 Blockade Therapy. Cancer Immunol. Res 10, 1386–1397. 10.1158/2326-6066.CIR-22-0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kamada T, Togashi Y, Tay C, Ha D, Sasaki A, Nakamura Y, Sato E, Fukuoka S, Tada Y, Tanaka A, et al. (2019). PD-1(+) regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc. Natl. Acad. Sci. USA 116, 9999–10008. 10.1073/pnas.1822001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kumagai S, Togashi Y, Kamada T, Sugiyama E, Nishinakamura H, Takeuchi Y, Vitaly K, Itahashi K, Maeda Y, Matsui S, et al. (2020). The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat. Immunol 21, 1346–1358. 10.1038/s41590-020-0769-3. [DOI] [PubMed] [Google Scholar]
- 41.Veglia F, Hashimoto A, Dweep H, Sanseviero E, De Leo A, Tcyganov E, Kossenkov A, Mulligan C, Nam B, Masters G, et al. (2021). Analysis of classical neutrophils and polymorphonuclear myeloid-derived suppressor cells in cancer patients and tumor-bearing mice. J. Exp. Med 218, e20201803. 10.1084/jem.20201803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Vlerken-Ysla L, Tyurina YY, Kagan VE, and Gabrilovich DI (2023). Functional states of myeloid cells in cancer. Cancer Cell 41, 490–504. 10.1016/j.ccell.2023.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Castiglioni A, Yang Y, Williams K, Gogineni A, Lane RS, Wang AW, Shyer JA, Zhang Z, Mittman S, Gutierrez A, et al. (2023). Combined PD-L1/TGFbeta blockade allows expansion and differentiation of stem cell-like CD8 T cells in immune excluded tumors. Nat. Commun 14, 4703. 10.1038/s41467-023-40398-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li MO, Wan YY, and Flavell RA (2007). T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity 26, 579–591. 10.1016/j.immuni.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 45.Turner JA, Stephen-Victor E, Wang S, Rivas MN, Abdel-Gadir A, Harb H, Cui Y, Fanny M, Charbonnier LM, Hung Fong JJ, et al. (2020). Regulatory T Cell-Derived TGF-beta1 Controls Multiple Checkpoints Governing Allergy and Autoimmunity. Immunity 53, 1331–1332. 10.1016/j.immuni.2020.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Singel KL, Emmons TR, Khan ANH, Mayor PC, Shen S, Wong JT, Morrell K, Eng KH, Mark J, Bankert RB, et al. (2019). Mature neutrophils suppress T cell immunity in ovarian cancer microenvironment. JCI Insight 4, e122311. 10.1172/jci.insight.122311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hailemichael Y, Johnson DH, Abdel-Wahab N, Foo WC, Bentebibel SE, Daher M, Haymaker C, Wani K, Saberian C, Ogata D, et al. (2022). Interleukin-6 blockade abrogates immunotherapy toxicity and promotes tumor immunity. Cancer Cell 40, 509–523.e6. 10.1016/j.ccell.2022.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huseni MA, Wang L, Klementowicz JE, Yuen K, Breart B, Orr C, Liu LF, Li Y, Gupta V, Li C, et al. (2023). CD8(+) T cell-intrinsic IL-6 signaling promotes resistance to anti-PD-L1 immunotherapy. Cell Rep. Med 4, 100878. 10.1016/j.xcrm.2022.100878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Castle JC, Loewer M, Boegel S, de Graaf J, Bender C, Tadmor AD, Boisguerin V, Bukur T, Sorn P, Paret C, et al. (2014). Immunomic, genomic and transcriptomic characterization of CT26 colorectal carcinoma. BMC Genom 15, 190. 10.1186/1471-2164-15-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pan Q, Lou X, Zhang J, Zhu Y, Li F, Shan Q, Chen X, Xie Y, Su S, Wei H, et al. (2017). Genomic variants in mouse model induced by azoxymethane and dextran sodium sulfate improperly mimic human colorectal cancer. Sci. Rep 7, 25. 10.1038/s41598-017-00057-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kroemer G, Galluzzi L, Zitvogel L, and Fridman WH (2015). Colorectal cancer: the first neoplasia found to be under immunosurveillance and the last one to respond to immunotherapy? OncoImmunology 4, e1058597, [pii]. 10.1080/2162402X.2015.10585971058597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, Tosolini M, Camus M, Berger A, Wind P, et al. (2006). Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313, 1960–1964. [DOI] [PubMed] [Google Scholar]
- 53.Sujino T, Kanai T, Ono Y, Mikami Y, Hayashi A, Doi T, Matsuoka K, Hisamatsu T, Takaishi H, Ogata H, et al. (2011). Regulatory T cells suppress development of colitis, blocking differentiation of T-helper 17 into alternative T-helper 1 cells. Gastroenterology 141, 1014–1023. 10.1053/j.gastro.2011.05.052. [DOI] [PubMed] [Google Scholar]
- 54.Hadis U, Wahl B, Schulz O, Hardtke-Wolenski M, Schippers A, Wagner N, Müller W, Sparwasser T, Förster R, and Pabst O (2011). Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity 34, 237–246. 10.1016/j.immuni.2011.01.016. [DOI] [PubMed] [Google Scholar]
- 55.Chen W, Huang W, Xue Y, Chen Y, Qian W, Ma J, August A, Wang J, Zheng SG, and Lin J (2022). Neuropilin-1 Identifies a New Subpopulation of TGF-beta-Induced Foxp3(+) Regulatory T Cells With Potent Suppressive Function and Enhanced Stability During Inflammation. Front. Immunol 13, 900139. 10.3389/fimmu.2022.900139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Campos-Mora M, Contreras-Kallens P, Gálvez-Jirón F, Rojas M, Rojas C, Refisch A, Cerda O, and Pino-Lagos K (2019). CD4+Foxp3+T Regulatory Cells Promote Transplantation Tolerance by Modulating Effector CD4+ T Cells in a Neuropilin-1-Dependent Manner. Front. Immunol 10, 882. 10.3389/fimmu.2019.00882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chuckran CA, Liu C, Bruno TC, Workman CJ, and Vignali DA (2020). Neuropilin-1: a checkpoint target with unique implications for cancer immunology and immunotherapy. J. Immunother. Cancer 8, e000967. 10.1136/jitc-2020-000967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim R, Hashimoto A, Markosyan N, Tyurin VA, Tyurina YY, Kar G, Fu S, Sehgal M, Garcia-Gerique L, Kossenkov A, et al. (2022). Ferroptosis of tumour neutrophils causes immune suppression in cancer. Nature 612, 338–346. 10.1038/s41586-022-05443-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shin AE, Tesfagiorgis Y, Larsen F, Derouet M, Zeng PYF, Good HJ, Zhang L, Rubinstein MR, Han YW, Kerfoot SM, et al. (2023). F4/80(+)Ly6C(high) Macrophages Lead to Cell Plasticity and Cancer Initiation in Colitis. Gastroenterology 164, 593–609.e13. 10.1053/j.gastro.2023.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bent EH, Millán-Barea LR, Zhuang I, Goulet DR, Fröse J, and Hemann MT (2021). Microenvironmental IL-6 inhibits anti-cancer immune responses generated by cytotoxic chemotherapy. Nat. Commun 12, 6218. 10.1038/s41467-021-26407-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ware MB, Phillips M, McQuinn C, Zaidi MY, Knochelmann HM, Greene E, Robinson B, Herting CJ, Mace TA, Chen Z, et al. (2023). Dual IL-6 and CTLA-4 blockade regresses pancreatic tumors in a T cell- and CXCR3-dependent manner. JCI Insight 8, e155006. 10.1172/jci.insight.155006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boland CR, and Goel A (2010). Microsatellite instability in colorectal cancer. Gastroenterology 138, 2073–2087.e3. 10.1053/j.gastro.2009.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. (2015). PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med 372, 2509–2520. 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tjalsma H, Boleij A, Marchesi JR, and Dutilh BE (2012). A bacterial driver-passenger model for colorectal cancer: beyond the usual suspects. Nat. Rev. Microbiol 10, 575–582. 10.1038/nrmicro2819. [DOI] [PubMed] [Google Scholar]
- 65.Ryan MH, Bristol JA, McDuffie E, and Abrams SI (2001). Regression of extensive pulmonary metastases in mice by adoptive transfer of antigen-specific CD8(+) CTL reactive against tumor cells expressing a naturally occurring rejection epitope. J. Immunol 167, 4286–4292. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
scRNA-seq data generated in this studies are deposited in the National Institutes of Health Gene Expression Omnibus (GEO) database (GEO: GSE246038, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE150743) and are publicly available.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.