

Abstract
Aminoacyl-tRNA synthetases (aaRSs) are modular enzymes globally conserved in the three kingdoms of life. All catalyze the same two-step reaction, i.e., the attachment of a proteinogenic amino acid on their cognate tRNAs, thereby mediating the correct expression of the genetic code. In addition, some aaRSs acquired other functions beyond this key role in translation. Genomics and X-ray crystallography have revealed great structural diversity in aaRSs (e.g., in oligomery and modularity, in ranking into two distinct groups each subdivided in 3 subgroups, by additional domains appended on the catalytic modules). AaRSs show huge structural plasticity related to function and limited idiosyncrasies that are kingdom or even species specific (e.g., the presence in many Bacteria of non discriminating aaRSs compensating for the absence of one or two specific aaRSs, notably AsnRS and/or GlnRS). Diversity, as well, occurs in the mechanisms of aaRS gene regulation that are not conserved in evolution, notably between distant groups such as Gram-positive and Gram-negative Bacteria. The review focuses on bacterial aaRSs (and their paralogs) and covers their structure, function, regulation, and evolution. Structure/function relationships are emphasized, notably the enzymology of tRNA aminoacylation and the editing mechanisms for correction of activation and charging errors. The huge amount of genomic and structural data that accumulated in last two decades is reviewed, showing how the field moved from essentially reductionist biology towards more global and integrated approaches. Likewise, the alternative functions of aaRSs and those of aaRS paralogs (e.g., during cell wall biogenesis and other metabolic processes in or outside protein synthesis) are reviewed. Since aaRS phylogenies present promiscuous bacterial, archaeal, and eukaryal features, similarities and differences in the properties of aaRSs from the three kingdoms of life are pinpointed throughout the review and distinctive characteristics of bacterium-like synthetases from organelles are outlined.
INTRODUCTION
Key Role of Aminoacyl-tRNA Synthetases in Biology and Focus of the Review
Aminoacyl-tRNA synthetases are ancient proteins present in all living organisms whose origin intermingles with that of life. They are responsible for correct expression of the genetic code at the translational level, where they catalyze the attachment of amino acids on tRNA. For biological necessity, this seminal function has to be specific (although errors often occur) and is governed by rules mainly conserved in evolution that act as an operational second genetic code (1, 2). Because of their pivotal role, synthetases were the subject of intensive research covered in various reviews (3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14). Extant synthetases were shaped by evolution and show structural diversity in the tree of life, but amazingly they also show new functions beyond their key role in translation (15, 16, 17).
This essay focuses on bacterial synthetases and their paralogs, emphasizes how structures account for functional properties, outlines the consequences of their evolutionary history, and highlights enzymologic aspects, including the editing function that ensures correction of aminoacylation errors. The regulation of synthetase expression in bacteria is also covered, and approaches to inhibit their enzymatic activities and to engineer their structure are outlined. Since the phylogeny of synthetases presents complex patterns with promiscuous bacterial, archaeal, and eukaryal features (3, 14, 18), similarities and differences in properties of synthetases from the three kingdoms of life will be pinpointed all along the review and distinctive characteristics of bacterium-like synthetases from organelles will be outlined. Finally, perspectives toward future discoveries and deeper understanding are proposed.
Abbreviations and Visualization of Three-Dimensional Structures
Main abbreviations
aaRS, aminoacyl-tRNA synthetase, with the three-letter code for amino acids, e.g., AlaRS for alanyl-tRNA synthetase; ND-aaRSs, Non-Discriminating aaRSs, namely ND-AspRS or ND-GluRS that mischarge tRNAAsn or tRNAGln in organisms lacking AsnRS or GlnRS—a contrario, D-AspRS or D-GluRS for the Discriminating enzymes, i.e., that are specific for their cognate transfer RNAs and mt-aaRS for mitochondrial-aaRS (if not otherwise indicated, aaRSs are discriminating); tRNA, transfer RNA (e.g., tRNAAla for a molecule specific for alanine and alanyl-tRNAAla when this tRNA is charged with alanine) and ASL for Anticodon Stem Loop; mRNAaaRS, messenger RNA of aaRS. Most cited aaRS domains are abbreviated as follows: ABD, Acceptor-stem-Binding Domain; ACB, AntiCodon Binding; CP, Connective Peptide (in CP1 and CP2 versions); EMAP, Endothelial Monocyte Activating Polypeptide; OB-fold, Oligonucleotides/oligosaccharides Binding fold and SC-fold for Stem-Contact fold. Trbp stands for TrNA-binding protein. The nomenclature of tRNA is as given in the tRNA database (19), with, e.g., A76 for amino acid accepting 3′-terminal adenosine and N34N35N36 for anticodon triplet; abbreviations of modified nucleosides cited in the text are as follows: m2A, 2-methyl-adenosine; t6A, N6-threonyl-carbamoyl-adenosine; k2C, lysidine (2-lysyl-cytidine); Q, queuosine (a 7-deazaguanosine derivative with a dihydroxy-cyclopentene ring bound at its C7 carbon); s2U, 2-thiouridine; and mnm5s2U, 5-methyl-aminomethyl-2-thiouridine.
Visualization of structures
To facilitate understanding of aaRS architectures, readers can visualize and manipulate their three-dimensional (3D) structures on the websites of the RCSB PDB (Protein Data Bank of the Research Collaboratory for Structural Bioinformatics consortium) (http://www.pdb.org) (20) or the user-friendly Proteopedia free encyclopedia (http://www.proteopedia.org) (21), using the four-digit accession codes given in the text (in italics and bold); as an example, visualize the structure of Escherichia coli MetRS (3h99). Readers may also use the tools of PyMol (Delano Scientific, http://www.pymol.org).
Historical Background
The first aaRS was discovered in 1958 as an amino acid activation enzyme (22), and it was rapidly suggested that the aaRS enzymes are partners of protein synthesis, where they have a dual role—first to activate amino acids and second to transfer the activated amino acids on tRNA (23, 24):
For a long time, aaRSs were considered as a family of 20 functionally homogeneous proteins, although they were characterized by a huge diversity in sequence, subunit size, and oligomeric structure. The surprise was great when it was realized in 1990, after analysis of the accessible aaRS sequences and early crystallographic work, that these enzymes fall into two groups of 10 members each that would aminoacylate tRNAs by different mechanisms (25, 26). This dichotomy is a result of evolution; it is not completely understood and is still under debate (27, 28, 29, 30, 31, 32, 33), although recent data favor the view of a common origin of both aaRS classes as suggested by the synthesis of functional class I and class II aaRS-mimics (in fact amino acid activating enzymes) that could have been coded by opposite strands of the same gene (34).
Understanding how aaRSs recognize tRNA was and remains an important issue approached by all available theoretical and experimental tools, often applied to bacterial systems. Early data pinpointed the importance of the tRNA-accepting end and of the anticodon for recognition by aaRSs (reviewed in reference 5) and contributed to the progress of genetic methods for the in vivo search of tRNA identity determinants (35, 36). On the other hand, it was realized that specificity of aaRSs for both amino acid activation and tRNA aminoacylation is rather low in vitro (reviewed in references 37 and 38). For biological necessity, it was then conjectured that editing mechanisms should exist to correct activation and aminoacylation errors. Today, these precursory seminal suggestions find support from structural biology and genomics as reported below.
Overview of the aaRS World
General features of aaRSs and peculiarities in Bacteria
The world of aaRSs shows a large diversity with enzymes of modular structure ranking in two evolutionarily distinct classes (Figure 1) and featuring significant variations in sequence, length, and oligomery (Table 1). In addition to the two types of class-specific catalytic domains and the ACB domains needed to ensure the seminal tRNA aminoacylation function, aaRSs contain additional modules of diverse structure (N- or C-terminally located or inserted into the protein core formed by the catalytic and ACB domains) that participate in varied functions (e.g., editing in the three kingdoms of life and kingdom-specific ancillary activities). Structural and functional complexity of aaRSs increases in Eukarya with the systematic addition of new domains and motifs (see below). Distinctive features in bacterial aaRSs are diverse, such as the large insertion in the catalytic domain of AspRSs (18, 39) and many discrete elements revealed by computational methods in other aaRSs (40). Furthermore, bacterial aaRSs differ from other aaRSs by physico-chemical characteristics, such as isoelectric points (pI) in the mild acidic range (pI ~5 to 6, as compiled for E. coli and Thermus thermophilus aaRSs), in contrast to human cytosolic and mitochondrial aaRSs, where pI are shifted toward alkaline values (pI ~5.5 to 8.5) (41).
Figure 1.
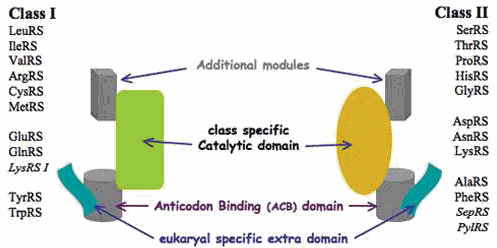
Modular architecture of aminoacyl-tRNA synthetases and their partition in two classes with subclasses. Modularity in aaRS structure was first revealed after analysis of the E. coli AlaRS sequence (50) and was confirmed by crystallography of many other aaRSs (51). SepRS (O-phosphoseryl-tRNA synthetase) and PylRS (pyrrolysyl-tRNA synthetase) are noncanonical aaRSs found in Archaea, except a few bacterial PylRSs (see “The ambiguous status of archaeal aaRSs—a short synopsis,” below, for details).
Table 1.
Overall structural characteristics of aminoacyl-tRNA synthetases, with emphasis on E. coli members
| Class I aaRSs | Class II aaRSs | ||||
|---|---|---|---|---|---|
| Subclasses | Oligomeric structure | No. aa/subunit (E. coli aaRSs) | Subclasses | Oligomeric structure | No. aa/subunit (E. coli aaRSs) |
| Class Ia | Class IIa | ||||
| LeuRS | α or αβa | 860 | SerRS | α2 | 430 |
| IleRSl | α | 937b | ThrRS | α2 | 642 |
| ValRS | α | 951 | ProRS | α2 | 572 or 590 |
| ArgRS | α | 577 | HisRS | α2 | 424 |
| CysRS | α | 461 | GlyRS | α2β2c | 303 & 689 |
| MetRS | α2 or α | 642 | |||
| Class Ib | Class IIb | ||||
| GluRS | αd | 471 | AspRS | α2 | 590 |
| GlnRS | α | 551 | AsnRS | α2 | 467 |
| LysRS I e | α | –f | LysRS | α2 | 505 |
| Class Ic | Class IIc | ||||
| TyrRS | α2 | 424 | AlaRS | α4 | 875 |
| TrpRS | α2 | 334 | PheRSg | α2β2h | 327 & 795 |
| PylRSi | α2 | –j | |||
| SepRSh | α4 | –k | |||
Can be αβ heterodimeric in a few Bacteria, such as Aquifex aeolicus from the phylum Aquificae (see “Structures of aminoacyl-tRNA synthetases,” below).
Revised amino acid sequence by means of mass spectrometry peptide mapping (42).
Tetrameric α2β2 GlyRSs are exclusively found in Bacteria; however, a few Bacteria contain dimeric α2 GlyRSs (of archaeal/eukaryal-type). A dimeric α2-type GlyRS in Eukarya, was as first found in baker’s yeast (43).
A catalytically active homodimeric form of Mycobacterium tuberculosis GluRS has been characterized that is in equilibrium with the monomer (44).
Present in a few Bacteria, but mainly in Archaea.
523 amino acids in archaeal Pyrococcus horikoshii (39).
Small catalytic α-subunit is the class II characteristic domain in PheRSs.
Monomers in mitochondria.
Noncanonical archaeal aaRSs (PylRS and SepRS); note the presence of PylRS in some Bacteria, such as in Desulfitobacteria hafniense (46) from the phylum Firmicutes.
454 amino acids in the subunit of the Methanococcus mazei PylRS (47).
Underlined aaRSs: protein sequence available.
For additional references on aaRS sequences, see reference 10.
The first known aaRS sequence was that of dimeric TrpRS from Bacillus stearothermophilus (or Geobacillus stearothermophilus), the smallest aaRS, with a subunit of 327 amino acids (52). The first amino acid sequence deduced from DNA sequencing was that of tetrameric AlaRS from E. coli, with a subunit of 875 amino acids (53). Note that genome sequencing revealed strain-dependent differences in E. coli proteins, notably in ProRS, with 572 amino acids in an enterohemorrhagic strain (54) and 590 amino acids in an uropathogenic strain (55). Altogether, the E. coli aaRSs reveal a complex evolutionary history with unexpected connections with other protein families and intricate horizontal gene transfer events (39). Surprisingly, several aaRSs are even encoded in viral genomes (56, 57).
Partition of aaRSs in two families (Figure 1) is based on different structures of their active sites. It was revealed by sequence peculiarities in E. coli ProRS (25, 26) and unprecedented crystallographic features in E. coli apo-SerRS (25) (not in PDB; see [1ses] for a structure with a seryl-adenylate analog) and the yeast AspRS:tRNAAsp complex (1asy) (58) (also commented upon in references 18, 59, 60, and 61). While in class I aaRSs the ATP-binding domain is a Rossmann fold similar to the dinucleotide-binding fold discovered in glyceraldehyde-3-phosphate dehydrogenases (62), it is a seven-stranded antiparallel β-sheet in class II.
In class I aaRSs, the signature motifs HIGH and KMSKS (in fact, HiGh and kmSKs, with amino acids in lowercase letters being less conserved) are responsible for the interaction with the universal substrate ATP. These signatures are always located close to the α-phosphate of ATP and assist catalysis. In class II aaRSs, residues of the so-called motifs 2 (fRxe) and 3 (gxgxgfd/eR) play this role. A third signature motif, called motif 1 (gΦxxΦxxP, with Φ for hydrophobic residues), is responsible for dimerization. Class I aaRSs are mostly monomers, and class II aaRSs are mostly dimers (Table 1). It is noteworthy that class I TrpRS (63) and TyrRS (64) are dimers and recognize tRNA as do class II aaRSs, thereby making a link with dimeric class II enzymes. From the side of function, aaRS partition explains the regiospecific tRNA acylation on either the 2′-OH group (class I and class II PheRS) or the 3′-OH group (class II, except PheRS) of the ribose at terminal A76 (65, 66, 67) and the differential ATP- and tRNA-binding modes (see “Aminoacylation of tRNA,” below).
The high degeneracy of the three motifs in class II aaRSs has to be noticed. With only one proline and two arginine residues strictly conserved, these signatures could not be discovered on a sole sequence inspection but required knowledge of 3D structures in which the single-signature amino acids are similarly located in specific architectural frameworks. This means that conservation of 3D structure is not necessarily accompanied by strict conservation of sequence, since 15% sequence identity can be sufficient to account for conservation of the core structure of aaRSs (68). This has implications for exploring the evolution and phylogeny of aaRSs that should be structure based (40, 68). In this view, the discovery by advanced bioinformatic tools of hidden protein motifs that connect structurally and functionally unrelated aaRSs should be stressed. Thus, Rossmannoid motifs (i.e., structural motifs derived from the classical dinucleotide-binding fold or Rossmann fold that are present in a variety of proteins) could be identified in the ACB domain of class II aaRSs, notably in ProRS and GlyRS (69).
For a long time it was believed that each amino acid has its own aaRS and, consequently, that all organisms should contain 20 aaRSs. However, it was observed early on that some Bacteria require a misacylating GluRS and an amidotransferase for formation of glutaminyl-tRNAGln (70, 71). This pathway remained mysterious for a long time, although it was readily suggested that it might serve to compensate for a defective GlnRS (71). Today we know that GlnRS, and also AsnRS, are missing in some Bacteria and in most Archaea, where their absence is compensated for by the presence of ND-GluRS or ND-AspRS. These nondiscriminating aaRSs are actors in an indirect pathway of aminoacyl-tRNA synthesis via transamidation of mischarged tRNAGln or tRNAAsp (7, 72). Notice, by the way, that some ND-aaRSs have lost the capability to efficiently charge their cognate tRNA and specialized to be only misacylating enzymes. This is the case of GluRS-2 from Helicobacter pylori that specialized to misacylate tRNAGln with glutamate (73). (See “Alternative functions of bacterial aminoacyl-tRNA synthetases,” below, for details on the biology of ND-aaRSs.)
It was also found that two distinct genes coding for distinct aaRSs of the same specificity could coexist in a same bacterium. Such duplicated aaRSs are known for various specificities (e.g., Arg, Asp, Cys, Glu, Ile, Lys, Ser, Thr, Trp, and Tyr). Thus, two forms of ArgRS occur in Oenococcus oeni (74), two of AspRS occur in T. thermophilus (75), two of CysRS occur in Mycobacterium smegmatis (76), two of GluRS occur in H. pylori and Acidithiobacillus ferrooxidans (77, 78, 79), two of IleRS occur in Staphylococcus aureus (80), two of LysRS occur in E. coli (81), two of SerRS occur in Streptomyces taxa (82), two of ThrRS and TyrRS occur in Bacillus subtilis (83), and two of TrpRS occur in various bacterial genera, including the Salmonella taxa (84). Duplications also occur in Archaea and Eukarya. Different reasons can account for this fact. In the case of the two E. coli LysRS forms, their differential regulation could provide a metabolic strategy to adapt the microorganism to different physiological conditions (85). For duplicated AspRS, regulation of the D and ND forms keeps protein synthesis efficient when the direct pathway of aspartic acid biosynthesis is repressed. On the other hand, duplicated aaRS genes were found to code for paralogous proteins that have acquired new functionalities. Thus, the second CysRS in M. smegmatis was identified as the product of the mshC gene encoding an ATP-dependent Cys:GlncN-Ins ligase (76). Likewise, a putative LysRS gene in Salmonella codes for the PoxA protein (86) that lysylates a protein mimic of tRNA (see “Paralogs of bacterial aminoacyl-tRNA synthetases,” below, for comments).
As a last point, notice that a single aaRS can aminoacylate isoaccepting tRNA species. The degeneracy of the genetic code often accounts for this property, a prominent example being the SerRSs that serylate the five tRNASer isoacceptors needed to read the six serine codons (87).
Peculiarities of eukaryal aaRSs and differences with the bacterial orthologs—a short synopsis
Eukaryal cytosolic aaRSs have an enhanced structural complexity with the presence of additional domains and motifs in comparison with the bacterial orthologs (18, 88). These extrastructures exhibit a variety of functions, some indirectly related to tRNA aminoacylation but most without apparent connection, and they were progressively incorporated during evolution into virtually all aaRSs following a scenario correlating well with the progressive evolution and complexity of eukaryal organisms (15, 88). Thus, N-terminal appended domains are found in both class I (e.g., CysRS, GlnRS, and MetRS) and class II (e.g., GlyRS, AspRS, AsnRS, and LysRS) aaRSs. Likewise, C-terminal extensions are present in several class I aaRSs (e.g., CysRS, MetRS, and TyrRS) and in class II SerRS. As an example, the 70-residue-long N-terminal extension in Saccharomyces cerevisiae AspRS (see “Structure of aminoacyl-tRNA synthetases,” below) contains a lysine-rich helical domain that recognizes the anticodon stem-helix of tRNAAsp and thereby enhances significantly its binding (89). Such N-terminal appendices encompassing helices with RNA-binding motifs are present in other eukaryal AspRSs (89) and in mammalian LysRSs (90). Four other extradomains (EMAP II and the associated ELR tripeptide, leucine zipper, glutathione S-transferase, and WHEP domains) are shared by several aaRSs in higher Eukarya (GluRS, GlyRS, HisRS, MetRS, ProRS, and TrpRS), and, among them, the WHEP domain is of special interest. This WHEP domain (so named because it was discovered in WRS [TrpRS], HRS [HisRS], and EPRS [GluProRS, a fused aaRS build by GluRS and ProRS]), besides having regulatory functions in TrpRS, serves also as the linker that joins GluRS to ProRS in EPRS (15). The origin of EPRS in Eukarya is ancient and dates back to the most primitive Metazoa (91). There are also motifs specific to only one eukaryal aaRS and not found in other proteins. To date, eight such motifs have been characterized in AspRS, CysRS, GluRS, IleRS, LeuRS, PheRS, SerRS, and ThrRS (15). As noticed by the authors who discovered them in databases, once added to a given aaRS in the course of evolution, they were irreversibly retained until the appearance of humans. Mutations in these domains that are not concerned with the tRNA aminoacylation function can cause severe pathologies in humans (92, 93, 94). Furthermore, eukaryal aaRSs often exist under various isoforms that can be produced in vivo either through alternative mRNA splicing or by natural proteolytic fragmentation, as occurs for human TrpRS (95, 96) and TyrRS (97), as well as by posttranslational modifications (98). This structural diversity reflects functional diversity. Thus, human mini-TrpRS has antiproliferative and antiangiogenic activity (95). The mini-TyrRS is a cytokine that activates angiogenic signal transduction pathways (97), and the EPRS duplex regulates inflammatory gene expression via phosphorylation of two serine residues in the WHEP domain connecting GluRS and ProRS (91). Another fascinating property of the eukaryal aaRSs, found in higher Eukarya, is their assemblage in a large supramolecular architecture, named the MARS complex. This complex comprises nine aaRSs (ArgRS, AspRS, GlnRS, GluRS, IleRS, LeuRS, LysRS, MetRS, and ProRS) and three auxiliary proteins (p18, p38, and p43) (99, 100). Although its structure and functions are not fully elucidated, the newest evidence indicates a dynamic organization, a role in cellular channeling, and participation in regulatory pathways (99, 101, 102).
Most aaRSs from eukaryal organelles show bacterium-like features (for a short survey, see “Bacterium-like aminoacyl-tRNA synthetases,” below). However, some of them deviate from this scheme and present unprecedented idiosyncratic properties. This is the case of mt-PheRSs that are small monomers, in contrast to the large (αβ)2 heterotetramers in Bacteria, Archaea, and the cytosol of Eukarya (103, 104). It is noteworthy that the human mt-PheRS is one of the smallest aaRSs with a chimerical architecture (3cmq) combining the catalytic domain of the α-subunit with the ACB domain of the β-subunit of canonical large PheRSs (103) (see also “Structure of aminoacyl-tRNA synthetases,” below).
The recent interest in aaRSs from unicellular Protozoa (e.g., amoeboids, flagellates, apicoplasts, and kinetoplastids) that are often pathogens for humans deserves a comment. One reason lies in the presence of distinctive structural features in these aaRSs, absent in the orthologs from other eukaryal genera, particularly in human aaRSs, making these proteins potential new targets in the search for new antipathogen drugs. Three examples illustrate the point. First, a specific insertion was found in the ACB domain of the AspRS from apicomplexan pathogens that are the vectors of malaria (105). Another insertion is present between the catalytic and ACB domains in the AspRS from the parasite Entamoeba histolytica. Second, analysis of its 3D structure (3i7f) (106) suggests that the insertion enhances tRNAAsp binding and thereby plays a role similar to the RNA-binding motif from the N-terminal extensions in the AspRSs from lower Eukarya that are absent in Entamoeba. The third example comes from the knowledge of the HisRS structures from Trypanosoma brucei (3hri) and Trypanosoma cruzi (3hrk), the vectors of leishmaniases. In this aaRS, the binding pocket for the adenine moiety of ATP differs substantially both from the binding site in bacterial structures and from the homologous pocket in human HisRS (107).
The ambiguous status of archaeal aaRSs—a short synopsis
In contrast to all Eukarya and most Bacteria that are equipped with a full set of the 20 canonical aaRSs, Archaea are lacking one or several aaRSs (i.e., AsnRS, CysRS, GlnRS and/or LysRS) whose absence is compensated for by the presence of noncanonical aaRSs, namely ND-aaRSs, SepRS (O-phosphoseryl-tRNA synthetase) and/or class I LysRS-1 (108). In addition, some methanogenic Archaea encode a SepRS and/or a PylRS (pyrrolysyl-tRNA synthetase), two atypical and strictly archaeal aaRSs. PylRS charges the unusual 22nd proteinogenic amino acid pyrrolysine on cognate tRNAPyl (109), and, in those Archaea lacking CysRS, SepRS charges O-phosphoserine to tRNACys (108). PylRSs and SepRSs belong to class II aaRSs, where they are ranked into class IIc because of structural similarities with PheRSs (Table 1). The dimeric α2 PylRSs (110) show close relationship with the β-subunit of PheRSs, as seen by crystallography (e.g., 2zcd [46, 47]) and the tetrameric α4 SepRSs (111) resemble the catalytic α-subunit of PheRSs, as seen in structures of apo- (2odr) (48) and tRNA-bound 2du3 (112) versions of these proteins. The fact that the monomer of α4 SepRS shares strong resemblance to the catalytic domain of α2β2 PheRS supports the idea of a common origin of these two class II aaRSs (113). The structural similarity of the SepRS and PheRS catalytic domains accounts for the idiosyncratic aminoacylation of tRNASep and tRNAPhe at the 2′-OH position instead the 3′-OH position normally recognized by class II aaRSs including PylRS (114).
GlnRS is absent from all Archaea, and AsnRS is absent from most Archaea, where they are substituted by ND-GluRS and ND-AspRS that mischarge noncognate tRNAGln and tRNAAsn. Then, synthesis of asparaginyl-tRNAAsn and glutaminyl-tRNAGln occurs by an indirect route similar to that elucidated for Bacteria (see “Alternative functions of aminoacyl-tRNA synthetases,” below). Cysteinyl-tRNACys, as well, is produced indirectly when CysRS is lacking in a pathway utilizing a Sep-tRNA:Cys-tRNA synthase that converts phosphoseryl-tRNACys to cysteinyl-tRNACys (48). As to noncanonical class Ib LysRS-1, also found in a few Bacteria (Table 1 and “Structure of aminoacyl-tRNA synthetases,” below), it is the lysine-specific aaRS in most Archaea, and, in a few cases, it is present together with a canonical class II LysRS.
For reasons not completely understood, archaeal aaRSs of a given amino acid specificity can be of strictly archaeal type (e.g., GlyRSs, MetRSs, and most ThrRSs) and/or can encompass structural features of bacterial and/or eukaryal aaRSs (18). The first known example of an archaeal aaRS with bacterial features was AspRS from a Pyrococcus strain (115). At opposite, archaeal aaRSs can lack domains present in bacterial enzymes, as found in the crenarchaeal ThrRSs from Sulfolobus solfataricus (116) and Aeropyrum pernix (3a32) (117) that are deprived of a cis-editing domain. In the majority of other archaeal ThrRSs, as in Pyrococcus abyssi, the bacterial editing domain (also found in eukaryal ThrRSs) is replaced by a D-aminoacyl-tRNA deacylase-like domain (2hl1) (118) (see “Error correction” under “Aminoacylation of tRNA,” below, for details). Remarkably, the crenarchaeal ThrRSs are of the strictly archaeal-type, as well as the freestanding editing domain found in this group of Archaea (115). On the other hand, the editing domain of archaeal PheRSs resembles that of eukaryal PheRSs (119). Altogether, these intricate properties confer an ambiguous status to archaeal aaRSs, reflecting their ancient origin and their subsequent intricate evolutionary history.
Conserved aaRS properties and the challenge to understand their global biology
The preceding short survey on the aaRS world has highlighted their universal presence in all living organisms where they have the pivotal role in protein synthesis in mediating the correct expression of the genetic code. For each proteinogenic amino acid, evolution shaped a specific aaRS that specifically charges that amino acid on a cognate tRNA molecule. Amazingly, this universal function is accompanied by great structural diversity, as illustrated in Figure 1 and Table 1. Most intriguing is the partition of the aaRSs into two classes of 10 members each with class-specific catalytic domains. Likewise, the architectural modularity of aaRSs with a huge variety of modules appended to or inserted within their catalytic domains and a trend of larger structures in Eukarya is puzzling. This survey has also sketched other fascinating facts, notably the ubiquitous presence of aaRS paralogs in living organisms and the participation of aaRSs in various biological processes unrelated to protein synthesis. Furthermore, it was shown that aaRSs are of interest for human medicine (e.g., as targets to inhibit pathogens or when mutated as causative agents of various diseases). A general belief argues that all of these features reflect the evolutionary history of aaRSs and, more generally, that of the genetic code and even of life. Altogether, this calls for a global interdisciplinary approach where information gathered on aaRSs from the three kingdoms of life and even from viruses must be compared. In this context, the bacterial aaRSs are of interest because of the ancient origin of Bacteria and the relative ease of experimental studies. Thus, the search for primordial features accounting for aaRS function is facilitated in Bacteria. Likewise, the relative ease of access to pure bacterial aaRSs and the availability of sophisticated tools for their study turned out to be essential to gather a huge and diversified corpus of structural and functional characteristics, some idiosyncratic to bacterial aaRSs, others shared by archaeal and eukaryal aaRSs. How do we rationalize data that are seemingly unrelated? How does genomics meet structure-function problems and evolution in aaRS research? How should we assemble the pieces of the puzzle? It is the aim of this essay to bring answers to these questions with focus on bacterial aaRSs.
STRUCTURE OF AMINOACYL-tRNA SYNTHETASES
Toward a Structural Understanding of Bacterial aaRSs
Crystallogenesis of aaRSs and aaRS:tRNA complexes
In 1971, a monomeric active fragment of E. coli MetRS obtained by limited trypsin digestion of the native enzyme was the first crystallized aaRS (120). It took 30 years to get satisfying structures of a free monomeric MetRS, in apo-form or with bound methionyl-adenylate analogs, either from E. coli (121, 122, 123) (1p7p) or T. thermophilus (124) (1a8h). The first crystals of a complex between aaRS and tRNA (the yeast aspartate complex) were grown in 1980 (125, 126) when it was realized that tRNA interaction with aaRS and aminoacylation capacity were maintained above 1.0 M ammonium sulfate (125, 126, 127, 128). This finding, restricted to ammonium sulfate, was a surprise because salts at high concentration were known to inhibit tRNA aminoacylation and to disrupt protein/nucleic acid interactions, particularly in the aaRS field (129, 130). At present, ammonium sulfate is widely used as a major crystallant of aaRS:tRNA complexes (51). As to Bacteria, the first crystallization successes concerned the E. coli GlnRS:tRNAGln complex (131) followed by the T. thermophilus SerRS:tRNASer (132) and PheRS:tRNAPhe (133) complexes (both with native tRNAs from T. thermophilus). Note that many crystals were grown with aaRSs (sometimes truncated but still active) from thermophilic Bacteria or Archaea that crystallize more readily than the orthologs from mesophiles. Today, representatives of all aaRS families, either free or with ligands, have been crystallized, leading to ~630 crystal structures (including aaRS fragments and paralogs) deposited in the RCSB Protein Data Bank.
A panel of bacterial crystal structures
Although bacterial aaRSs represent the majority of solved structures (48% versus 20%, 31%, and <1% for Archaea, Eukarya [cytosol], and mitochondria), their distribution in Bacteria is uneven (Table 2). Structures from E. coli and T. thermophilus aaRSs are the most represented with ~100 and ~70 structures, respectively, corresponding to 12 amino acid specificities for E. coli and 15 for T. thermophilus. In the case of E. coli aaRSs, ~60 structures correspond to apo-proteins or proteins in interaction with small ligands and 33 to aaRS:tRNA complexes (including 18 GlnRS:tRNAGln complexes) in different functional states. Figure 2 is a gallery of bacterial aaRS structures representative of the 20 amino acid identities, displaying when available structures of aaRS:tRNA complexes. It is noteworthy that the structure of Aquifex aeolicus AlaRS (1yfr) corresponds to a truncated molecule restricted to the catalytic and tRNA-recognition domains (with 453 instead of 866 amino acids for the complete protein) (134). This truncated molecule alanylates tRNAAla and is monomeric and homologous to a fragment from E. coli AlaRS so far not crystallized. A full-size tetrameric α4-structure of an AlaRS is still missing, but a model based on crystal structures of the two halves of archaeal Archaeoglobus fulgidus AlaRS, namely AlaRSΔC (2ztg, comprising catalytic, tRNA recognition, and editing domains) and AlaRSC (2zvf, dimerization domain) allowed to propose a dimeric butterfly-like organization (135).
Table 2.
Crystallographic structures of free or ligand-bound native bacterial aminoacyl-tRNA synthetases, with emphasis on E. coli membersa
| aaRSs | E. coli structurea | Other bacterialc structures | ||
|---|---|---|---|---|
| apo-aaRSs or in complex with small ligandsb | aaRS:tRNAb | apo-aaRSs or in complex with small ligands | aaRS:tRNA | |
| Class I aminoacyl-tRNA synthetases | ||||
| Class Ia | ||||
| LeuRS | – | 3zit, 4aq7, 4ari, 4as1, 4cqn, 3zgz, 4arc | Mmo, Tth | Tth |
| IleRS | – | – | Tth | Sau |
| ValRS | – | – | Tth | Tth |
| ArgRS | 4oby | – | Tth, Kpn, Cje | – |
| CysRS | 1li5, 1li7 | 1u0b | – | – |
| MetRS | 1qqt, 3h97, 3h9c, 1f4l, 1p7p, 1pfu, 1pg2, 1pg0, 1pfy, 1pfw, 1pfv, 3h99, 3h9b | – | Aae, Bab, Bme, Msm, Sta, Tth | Aae |
| Class Ib | ||||
| GluRS | – | – | Bbu, Bth, Mtu, Sel, Tma, Tth | Pae, Tma, Tth |
| GlnRS | 1nyl, 2rd2, 2re8 | 1gsg, 1gts, 1qrs, 1qru, 1qrt, 1qtq, 1euq, 1euy, 1exd, 1o0b, 1o0c, 1zjw, 1gtr, 4jyz, 4jxx, 4jyz | Dra, Pae | – |
| Class Ic | ||||
| TyrRS | 1x8x, 1wq4, 1vbm, 1vbn, 1wq3, 2yxn, 4oud | – | Bst, Mtu, Sau, Tth | Tth |
| TrpRS | – | – | Bst, Bsu, Cje, Dra, Mpn, Tma, Tth, Vch, Ype | – |
| Class II aminoacyl-tRNA synthetases | ||||
| Class IIa | ||||
| SerRS | not in PDB | – | Aae, Tth | Tth |
| ThrRS | 1evk, 1tje, 1tke, 1tkg, 1tky, 1evl, 1fyf, 4hwo, 4hwp, 4hwr, 4hws, 4p3o, 4p3p | 1qf6, 1kogd | Sau | – |
| ProRS | – | – | Efa, Rpa, Tth | Tth |
| HisRS | 1kmn, 1kmm, 2el9, 1htt | – | Aba, Bth, Nostoc, Sau,Tth | Tth |
| GlyRS | – | – | Cje, Tma, Tth | – |
| Class IIb | ||||
| AspRS | 1eqr | 1c0a, 1il2 | Msm, Mtu, Tth | Pae, Tth |
| AsnRS | – | Tth (not in PDB) | – | |
| LysRS | 1lyl, 1bbu, 1bbw, 1e1o, 1e1t, 1e22, 1e24 | – | Bst, Bth | – |
| Class IIc | ||||
| AlaRS | —e | – | Aae | – |
| PheRS | 3pco | – | Pae, Sha, Tth | Tth |
E. coli aaRSs (~200 structures) are designated by their PDB accession code (identifiers of apo-forms without bound ligands are in italics).
Small ligands bound to aaRSs (or aaRS:tRNA complexes) can be inhibitors.
Bacteria abbreviated as follows: Aba, Acinetobacter baumannii; Aae, Aquifex aeolicus; Bab, Brucella abortus; Bme, Brucella melitensis; Bst, Bacillus (Geobacillus) stearothermophilus; Bsu, Bacillus subtilis; Bth, Burkholderia thailandensis; Bbu, Borrelia burgdorferi; Cje, Campylobacter jejuni; Cbu, Coxiella burnetii; Dra, Deinococcus radiodurans; Efa, Enterococcus faecalis; Kpn, Klebsiella pneumoniae; Mmo, Mycoplasma mobile; Msm, Mycobacterium smegmatis; Mtu, Mycobacterium tuberculosis; Nostoc, Nostoc sp. PPC 7120; Pae, Pseudomonas aeruginosa; Rpa, Rhodopseudomonas palustris; Sau, Staphylococcus aureus; Sha, Staphylococcus haemolyticus; Sel, Synechococcus elongatus; Tma, Thermotoga maritima; Tth, Thermus thermophilus; Vch, Vibrio cholerae; Ype, Yersinia pestis. For more details and PDB identifiers see reference (51). Note the presence in Borrelia burgdorferi and other Spirochetes, and in a few α-Proteobacteria, of an atypical class I LysRS (136), which structure has been solved for the archaeal P. horikoshii enzyme (1irx) (45). For the remaining ~200 known bacterial structures, the Table gives only their phylogenetic origin.
ThrRS with bound tRNA-like domain of mRNAThrRS.
Figure 2.
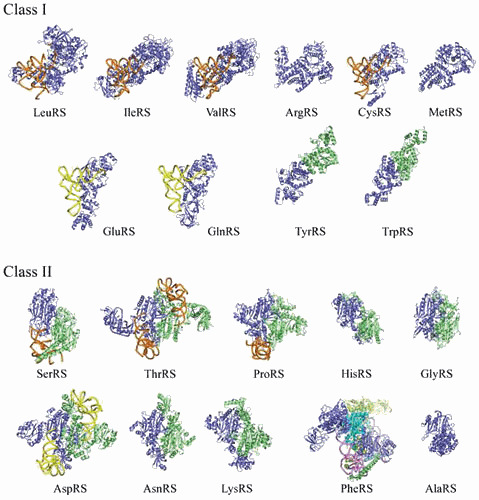
A gallery of canonical bacterial aminoacyl-tRNA synthetase structures. AaRSs in each class are displayed with their catalytic domain in the same orientation. Subclass distribution is indicated by tRNA colors, with orange, yellow, and pink backbones for subclasses a, b, and c, respectively (see Table 2 for subclass distribution). For dimeric aaRSs of classes Ic, IIa, and IIb, the second monomer is green. For tetrameric class IIc PheRS, one αβ-heterodimer is shown in blue and the other is shown in green; for AlaRS, the structure corresponds to a monomeric active fragment (see text). Identification of the displayed structures: class I T. thermophilus LeuRS with tRNALeu in posttransfer editing conformation (2bte) (137), S. aureus IleRS with tRNAIle (1qu2) (138), T. thermophilus ValRS with tRNAVal (1gax) (139), T. thermophilus ArgRS (1iq0) (140); E. coli CysRS with tRNACys (1u0b) (141), E. coli MetRS (1qqt) (122), T. thermophilus GluRS with tRNAGlu (2dxi) (142), E. coli GlnRS with tRNAGln (1o0b) (143), E. coli TyrRS (1x8x) (144), and T. thermophilus TrpRS (2el7) (unpublished from RIKEN Structural Genomics Initiative); class II E. coli SerRS with tRNASer (not in PDB) (25), E. coli ThrRS with tRNAThr (1qf6) (145), T. thermophilus ProRS with tRNAPro (1h4s) (146), E. coli HisRS (1kmn) (147), T. thermophilus GlyRS (1ati) (148), E. coli AspRS with tRNAAsp (1c0a) (149), T. thermophilus AsnRS (not in PDB) (150), E. coli LysRS (1e1o) (151), T. thermophilus PheRS with tRNAPhe (2iy5) (152), and A. aeolicus AlaRS (1yfr) (134).
Besides the large panel of aaRS structures from E. coli (closely related to Salmonella, for which no crystallographic structure is yet known, except a putative LysRS structure [3g1z] from Salmonella enterica serovar Typhimurium), 3D structures are also available for 10 other Proteobacteria (Acinetobacter baumannii, Brucella abortus, Brucella melitensis, Campylobacter jejuni, Coxiella burnetii, Klebsiella pneumoniae, Pseudomonas aeroginosa, Rhodopseudomonas palustris, Vibrio cholerae, Yersinia pestis). Additional structures come from 14 other Bacteria, notably from four species closely rooted with Archaea (A. aeolicus, Deinococcus radiodurans, Thermotoga maritima, T. thermophilus), five Bacilli from the Firmicutes phylum (B. stearothermophilus, Bacillus subtilis, Enterococcus faecalis, S. aureus, Staphylococcus haemolyticus), two Gram-positive Actinobacteria (M. mobile, Mycobacterium tuberculosis), two Cyanobacteria (Synechococcus elongatus, Nostoc), and one spirochete (Borrelia burgdorferi) (Table 2).
Many structures correspond to snapshots of conformational states occurring during the tRNA aminoacylation step, including the editing step, and thus inform about aaRS functioning. This is the case of E. coli LeuRS, MetRS, GlnRS, TyrRS, ThrRS, HisRS, AspRS, and LysRS and B. stearothermophilus TrpRS (see “Aminoacylation of tRNA,” below, for mechanistic details). For other aaRSs, the motivation was pharmacological with the aim of drug/antibiotics development. This concerns E. coli (153, 154, 155) and T. thermophilus (156) LeuRS, S. aureus (138) and T. thermophilus (157) IleRS, M. smegmatis MetRS (158), E. coli ThrRS (159), and S. aureus TyrRS (160) in complex with inhibitors acting as potential antipathogens (see “Inhibition and engineering of bacterial aminoacyl-tRNA synthetases,” below, for details). Note the increasing number of aaRS structures in complex with inhibitors and the structures solved recently by structural genomics consortia (e.g., by the Seattle Center for Structural Genomics of Infectious Disease).
The structure of the ND-AspRS from T. thermophilus (1n9w) that aspartylates both tRNAAsp and tRNAAsn, is of interest (161). This atypical AspRS is an archaeal-type with strong similarity to Pyrococcus kodakaraensis AspRS (1b8a) (162) and misses the bacteria-specific insertion found in the catalytic core of T. thermophilus (1l0w, 1g51) and E. coli (1eqr) AspRSs (163, 164, 165). Interestingly, this AspRS has strong structural resemblance with yeast AspRS (166), although it is missing the eukaryal N-terminal extension (Figure 3). Generalizing this observation, most bacterial aaRSs show overall conserved structures in evolution, but differ from their archaeal and eukaryal orthologs by kingdom-specific appendices and/or inserted or deleted domains (18).
Figure 3.

Variability of aminoacyl-tRNA synthetase structures during evolution: the case of AspRS. The figure shows the organization of dimeric AspRS in the three kingdoms of life with bacterial-type (E. coli) (1eqr) (149) AspRS in the middle surrounded by archaeal-type (P. horikoshii) (1b8a) (162) and eukaryal-type (S. cerevisiae) (1eov) (166) AspRS at left and right, respectively. AspRSs are displayed with the anticodon-binding domain in blue, the hinge region in yellow, and the catalytic domain in orange. The second monomer is in light green, and the kingdom-specific domains or extensions are in pink. The exact fold of the N-terminal extension of yeast AspRS is unknown, and the present model is based on structure predictions (89).
The resolution of structures is comprised between 1.4 and 3.3 Å with a tendency of the highest resolutions for aaRSs interacting with ligands, e.g., 1.4 Å for a mutant MetRS from E. coli complexed with methionine (3h99) (167). All structures systematically contain regions not well resolved and characterized by high crystallographic B-factors (these factors, also known as temperature factors, are correlated with mobility/disorder of atoms in crystals). In early reports, this fact was completely overlooked. At present, the structural plasticity of aaRSs is partly understood and reflects important functional features. Indeed, disordered domains not seen in the apo-proteins, or in complexes with small ligands, mostly consist of loops that make contacts with ligands. This intrinsic plasticity of aaRSs, often reflected by induced-fit/allosteric conformational changes, is required for their biological functioning either during the tRNA aminoacylation process or the interplay with other cellular partners (see “Aminoacylation of tRNA,” below, for details on aaRS plasticity).
Subclass Characteristics and Other Remarkable Anatomies of Bacterial aaRSs
All extant aaRSs have a class-defining catalytic domain and an ACB domain of variable architecture, both being decorated by idiosyncratic additional domains (Figure 1), often defining the subclasses. ACB domains are mainly conserved in evolution and correspond either to α-helical bundles, sometimes called DALR motifs in the seven class Ia aaRSs (because of conservation of D, A, L, and R amino acids; note that in the ArgRS nomenclature the domain is called Add2); α-helical folds (α-ACB in GluRSs, TyrRSs, TrpRSs, and LysRSs); β-sheeted folds (β-ACB in GlnRSs); α/β-mixed architecture (α/β-ACB in ThrRSs, HisRSs, and ProRSs); OB-folds (OB-ACB in AspRSs, AsnRSs, and LysRSs); or ferredoxin folds (FDX-ACB in PheRS β-subunits) (39). Additional details on the structure of the 20 aaRS families can be found in reference 18; for functional aspects see the next sections and reference 18.
Class Ia ArgRS, CysRS, IleRS, LeuRS, MetRS, and ValRS
These are monomeric proteins, with the exception of MetRSs that can be dimeric, as is native E. coli MetRS. This enzyme, however, remains fully active as a monomer after proteolytic C-terminal truncation (168). Class Ia aaRSs contain three subclass characteristic domains, namely, a flexibly linked CP1 domain of variable length (~150 to 200 amino acids) inserted into the Rossmann fold, except in CysRS and ArgRS (called Ins-2 in ArgRS), where it is half this size, a smaller CP2 also inserted in the Rossmann fold, and, in the C-terminal location, an α-helical bundle domain for recognition of tRNA anticodon. ACB domains are connected to the Rossmann domain by an SC-fold of β-α-α-β-α topology that contains the class I-specific KMSKS motif (1a8h) (124). Four class Ia members (IleRS, LeuRS, CysRS, and MetRS) contain one or two Zn-binding motifs.
MetRS: Bacterial MetRSs have a C-terminal extension that can occur as an autonomous paralog in some organisms, notably the Trbp111 protein (comprising 111 amino acids) in A. aeolicus, the bacterial CsaA proteins (acronym derived from the name of the B. subtilis csaA gene), and mammalian EMAP II cytokines (see “Paralogs of bacterial aminoacyl-tRNA synthetases,” below, for details). In E. coli MetRS, the C-terminal extension is a Trbp111-like domain, while in Gram-negative Borrelia burgdorferi it is closely related to the EMAP II domain (18). Remarkably, MetRSs can be classified in four groups on the basis of their Zn-binding features (the presence of one or two identical Zn-binding knuckles either containing or void of zinc) (122). These knuckles, located far away from the catalytic site, are inserted into the short CP1 domain connecting the two halves of the Rossmann fold. Likely, they have an intrinsic fold similar to that of the isolated E. coli MetRS-derived knuckle solved by NMR (169). Interestingly, monomeric MetRS from M. smegmatis revealed a new conformation of the KMSKS domain triggered by the binding of adenosine (2x1l, 2x1m) (158).
IleRS, LeuRS, and ValRS: These aaRSs are large proteins of elongated shape (Figure 2) that are related to evolution. They have a large and flexible CP1 globular insertion that protrudes from the catalytic domain and a smaller CP2 insertion that binds Zn2+ ions in certain IleRSs. The CP1 domain participates in editing activity (this is not the case in MetRSs, where the CP1 insertion is of smaller size and without editing function). A similar domain named ABD is present in class Ib enzymes, where it serves for recognition of the tRNA acceptor end.
The CP1 insertion reveals a conserved core and conserved residues at the editing site, suggesting a common editing mechanism in class Ia enzymes (see “Error correction,” below, for details). The location of CP1 in the sequence of the catalytic domain, subsequent to the first Zn-binding insert, is exclusive to bacterial and mitochondrial LeuRSs. Another peculiarity of bacterial LeuRSs is an additional insertion of ~50 amino acids in the catalytic core prior to the conserved KMSKS signature. This insertion was first identified in T. thermophilus LeuRS (1h3n) (170). This well-ordered and flexibly linked extradomain protrudes out of the protein core and participates in recognition of the tRNALeu acceptor stem (137, 170). Activity of deletion mutants and variants with chimerical swaps within the LeuRS-specific domain of E. coli confirms the critical role of this insertion in aminoacylation and excludes a direct participation in editing (171). Finally, bacterial LeuRSs have an exclusive C-terminal RNA-binding extension (~60 amino acids), only visible in the LeuRS:tRNALeu complex, where it is close to the tertiary G19•C56 pair that links together the D- and T-loops of tRNA (2byt) (137). A flexible peptide tether (of length more important than sequence) controls accessibility of this extension and facilitates its rigid-body movements (172). Note the peculiar αβ-heterodimeric LeuRS from the deep-rooted bacterium A. aeolicus whose structure is split in two parts in the catalytic domain, with part of the catalytic domain and the CP1 module constituting the large α-subunit and the remaining part of the catalytic domain with the KMSKS signature and the tRNA-binding domain constituting the small β-subunit (173). Note also the peculiar structure of Mycoplasma LeuRSs that have highly degenerate CP1 modules and in the case of M. mobile LeuRS is lacking the CP1 domain (3ziu) (155).
IleRS and ValRS structures mainly stem from studies of the T. thermophilus enzymes, either in apo-versions or in complexes with small ligands and/or tRNA. Structures of S. aureus IleRS:tRNAIle complexes are also available (18, 51). These structures were essential for deciphering the mechanism of tRNA isoleucylation and valylation, as well as the editing mechanism of mischarged tRNAIle or tRNAVal (174), notably for understanding the role of conformational changes (see “Aminoacylation of tRNA,” below, for details on catalysis and editing). Note that the ACB domain of IleRSs contains additional Zn-binding and α/β modules and that of ValRSs includes a α/β module.
CysRS and ArgRS: These two aaRSs deserve special attention because of unusual structural and functional properties (18). CysRSs exhibit a small size resulting either from the absence of an insertion in the catalytic domain or from the addition of small insertions within the Rossmann fold as is apparent in the E. coli crystal structure (1li5) (175). This enzyme does not need editing to reject serine and alanine (176), in agreement with the small size (75 amino acids) of its CP1 insertion (175).
ArgRSs are quite large proteins and show a remarkable structural modularity mainly conserved in evolution with several domains appended around their catalytic core (140, 177). Add1 and Add2 (Additional domains) are the two nucleic acid-binding modules attached at the N- and C-terminal sides of the catalytic domain. Ins-1 and Ins-2 (Insertion domains) are inserted modules in the first and second half of the Rossmann fold (RF1 and RF2). Add1 has the topology of a motif from the ribosome-recycling factor (RRF). Add2 corresponds to the class Ia characteristic ACB domain and is located in ArgRSs, as are the CP1 domains in the class Ia editing aaRSs. This structural organization is explicitly seen in E. coli ArgRS (4oby) (178). The crystal structure of T. thermophilus ArgRS reveals an additional insertion within RF2 (Ins-3) that is bacterial specific (1iq0) (140). Finally, and of great functional significance, ArgRSs belong to the few aaRSs that recognize the D-stem and loop of tRNA (via idiosyncratic Add1) and require cognate tRNA for amino acid activation.
Class Ib GlnRS, GluRS, and LysRS-1
These aaRSs need their cognate tRNA for amino acid activation in the first step of the aminoacylation reaction (in fact for the pyrophosphate exchange reaction). This functional feature is shared with ArgRS, that in addition shows resemblance with structural modules from GluRS and GlnRS. This confers an ambiguous status to ArgRSs that are sometimes ranked in class Ib aaRSs (see e.g., reference 3).
GlnRS and GluRS: These two aaRSs are evolutionarily linked and form the GlxRS superfamily of intricate evolutionary history with gene duplications and lateral gene transfers between Archaea, Eukarya, and Bacteria (18). GlxRS enzymes contain five structural domains, namely the class I-specific Rossmann fold (domain 1), the ABD insertion (domain 2), the SC-fold (domain 3), and two distal domains (4 and 5) interacting with the tRNA anticodon arm, as was explicitly visualized in the crystal structures of the E. coli GlnRS:tRNAGln complex (1gsg) (179) and that of native T. thermophilus GluRS (1gln) (180). While the N-terminal halves (domains 1 to 3) in bacterial GlnRSs and GluRSs present a high degree of similarity, the C-terminal halves (domains 4 and 5) are fundamentally different, being two α-helix bundles in GluRSs and antiparallel β-sheets forming a β-barrel in GlnRSs (1gtr). As in class Ia aaRSs, the catalytic domain of GlxRS enzymes is split in two parts, namely, the Rossmann fold (domain 1) and the ABD insertion designed to recognize the acceptor end of tRNA (domain 2). In E. coli GluRS, this ABD insertion contains a Zn2+ ion sequestered by a Cys-x-Cys-xn-Cys-x-His (n = 6 to 25) signature typical of the SWIM domain (a Zn-chelating domain designated after SWI2/SNF2 and MuDR proteins that are predicted to interact with DNA and/or proteins [181]), which finds its homolog in E. coli GlnRS where a leucine residue stabilizes the tRNAGln acceptor end (182). On the other hand, T. thermophilus GluRS does not contain zinc (180), and several other bacterial GluRSs do not contain in their ABD insertion the residues needed to bind zinc (182). Note that ABD insertions in class Ib aaRSs have structural features in common with the CP1 domains present in class Ia aaRSs. Note also a structural similarity of E. coli GlnRS with MetRS (183).
Extant GlxRSs likely originate from an ancestral ND-GluRS (184). In modern ND-GluRSs, discrete amino acid substitutions in domain 4 likely abolish the ability to discriminate between glutamine (34CUG36 and 34UUG36) and glutamate (34CUC36 and 34UUC36) anticodons, respectively. This assumption is supported by data on T. thermophilus GluRS (2dxi) that become nondiscriminating after replacement of Arg358, the residue recognizing C36 in tRNAGlu, by smaller glutamine (185). As could be anticipated, the overall architecture of the complex with tRNA is conserved in the cocrystal structure of ND-GluRS from T. maritima with noncognate tRNAGln and a stable analog of glutamyl-adenylate (3akz) (186). It is noteworthy that comparison with the binary complex of T. thermophilus D-GluRS with cognate tRNAGlu reveals a quite different structure in the outer corner of the L-shaped tRNAGlu and tRNAGln, especially in their D-loops (Figure 4). This feature is important for the recognition of tRNAGln by GatCAB in the glutamine transamidosome (186) (see “Alternative functions of bacterial aminoacyl-tRNA synthetases,” below, for details).
Figure 4.
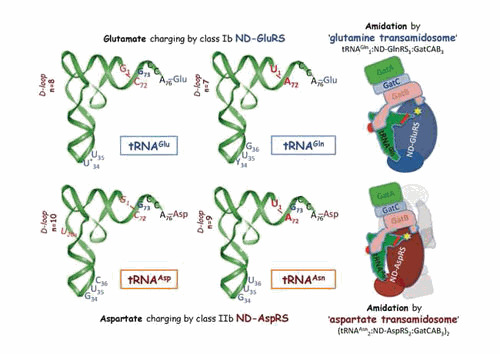
Dual tRNA aminoacylation by ND-GluRS and ND-AspRS and tRNA-dependent amino acid amidation, the two steps of the indirect pathway of glutaminyl-tRNAGln and asparaginyl-tRNAAsn formation. (Left) The two tRNA couples (tRNAGlu / tRNAGln and tRNAAsp / tRNAAsn) aminoacylated by ND-GluRS and ND-AspRS, respectively, and (Right) schematized representations of the T. maritima glutamine (3al0) (186) and T. thermophilus aspartate (3kfu) (187) transamidosomes. The main structural and functional features important for tRNA aminoacylation by ND-aaRSs and for tRNA-dependent conversion of the glutamyl and aspartyl residues into glutamine and asparagine are shown, as well as the amidation site (yellow star) in the transamidosomes. The major identity determinants for aminoacylation of the four tRNAs by their cognate aaRSs are shown in blue (6, 188). In Bacteria, position 34 is a modified U in tRNAGlu and a pyrimidine (Y) in tRNAGln. Notice the quite similar identity sets in the tRNAGlu / tRNAGln and tRNAAsp / tRNAAsn couples, in agreement with their dual aminoacylation by the ND-aaRSs. In bold red: U1–A72, the major identity determinants for amidation in tRNAGln and tRNAAsn; in red italics: notably the antideterminant G1–C72 pair that prevents glutamate and aspartate amidation in charged tRNAGlu and tRNAAsp (186, 189). The longer length of the D-loop in tRNAGlu and tRNAAsp (as compared to tRNAGln and tRNAAsn, a feature conserved in Bacteria [19]) is a further antideterminant that prevents amidation. Transamidosomes show an overall conserved organization based on the association of ND-aaRS, tRNA, and heterotrimeric GatCAB. Notice the Yqey domain of GatB that contacts the D-loop of tRNA and thereby plays a key role in transamidation. Notice further the different sizes of the two transamidosomes. While the glutamine transamidosome is formed by five entities (as seen in the figure), the much larger aspartate transamidosome is formed by 14 macromolecular entities (for clarity, only half of the structure is shown, with the second subunit of AspRS and its tRNA ligand shown in light grey). This architectural variation is due to structural differences in ND-GluRSs (class Ib monomers) and ND-AspRSs (class IIb dimers) and the correlated mechanistic differences in the aminoacylation and transamidation steps occurring within the two types of transamidosome (see Fig. 6 in “Aminoacylation of tRNA” and “Indirect pathways of specific tRNA aminoacylation for ribosome-mediated translation,” below, for details).
LysRS-1: Bacterial class Ib LysRS (i.e., LysRS-1) has a special status. This aaRS is of archaeal origin and is present in only a few Bacteria, notably in α-Proteobacteria and in spirochetes such as B. burgdorferi. Like the other class Ib members, it requires cognate tRNALys for adenylate formation (190). The 3D structures of bacterial class Ib LysRSs resemble that of LysRS-1 from archaeal P. horikoshii (1irx) (45). Ranking of LysRS-1 proteins in class Ib is justified because of their striking architectural similarity to GluRSs. Remarkably, comparison of class I and class II LysRSs suggests similar strategies for substrate recognition within their unrelated catalytic site topologies (45).
Class Ic TyrRS and TrpRS
TyrRSs and TrpRSs are small dimeric proteins of elongated shape and atypical properties that share strong structural resemblance despite low sequence identity (191). Bacterial TrpRSs and TyrRSs, however, differ from their eukaryal and archaeal orthologs in that they are missing extensions whose functions are unrelated with aminoacylation but play a role, e.g., in signal transduction pathways, notably for stimulating or inhibiting angiogenesis, as is the case, respectively, for fragments of human TyrRS and TrpRS (192) (see “An overview of the aaRS world,” below). Both TyrRSs and TrpRSs bind the tRNA-accepting stem from the major groove side in a way reminiscent of class II aaRSs and with the tRNA molecule spanning across the dimer, as was first proposed for B. stearothermophilus TyrRS on the basis of protein engineering data (193) and later confirmed by crystallography for both enzymes. The structural biology of bacterial TyrRSs and TrpRSs is well documented and covers 12 organisms (see Table 2). About 20 crystal structures were solved for each (http://www.pdb.org), but only one structure of a complex with tRNA, that of TyrRS from T. thermophilus (1h3e) (64), is available. Note the absence of amino acid editing by the two aaRSs, despite the presence of a CP1-like insertion in the catalytic domain (note that typical CP1 domains are inserted in the catalytic domain of those class Ia aaRSs with editing activity, namely, IleRSs, LeuRSs, and ValRSs). This insertion is of small size (~50 amino acids) in comparison with that from class I editing aaRSs and, instead of editing, is involved in dimer interface interactions and binding of the tRNA-accepting ends (64, 194, 195).
TyrRS: These aaRSs present an overall conserved organization in the three kingdoms of life, with a N-terminal catalytic domain and a C-terminal region of variable architecture that contains the ACB domain (18, 194). In Bacteria, this C-terminal region has a bipartite architecture with a α-helical domain (α-ACB) followed by an S4-like domain (as present in the superfamily of RNA-binding proteins homologous to the ribosomal protein S4) specific to Bacteria and mitochondria. The crystal structure of B. stearothermophilus TyrRS shows well-ordered catalytic and α-ACB domains, but the C-terminal domain is not seen in the electron density map (3ts1) (196). This domain is mobile and as determined by NMR has a well-defined S4-fold (1jh3). It consists of a five-stranded β-sheet packed against two α-helices on one side and one α-helix on the other side (197). Mobility concerns also the C-terminal EMAP II-like domain in eukaryal TyrRSs. In crystallized TyrRSs, the C terminus is often resected or when present not seen in the crystal structures. This structural organization and the functional implications are fully confirmed by the crystal structure of the T. thermophilus TyrRS:tRNATyr complex (64). Thus, the anticodon of tRNATyr contacts the α-ACB domain of one subunit while its accepting arm interacts with the catalytic domain of the other subunit. The mobile C-terminal S4 domain is required for contacting and anchoring the Bacteria-specific long variable arm of the tRNA on the protein and thus is visible in the complex. In Eukarya and Archaea, tRNATyr has a small variable region, and TyrRS has a different type of ACB domain that is homologous to that of TrpRS, implying protein-RNA contacts in the TyrRS:tRNATyr complexes from eukaryal and archaeal organisms different from those occurring in Bacteria (194).
TrpRS: Bacterial TrpRSs have the smallest subunit in the aaRS world (e.g., 328 amino acids for B. stearothermophilus TrpRS) and represent good approximations of a minimalist aaRS (see “Mimicry of catalytic domains,” below, for details on the engineering of an artificial aaRS named the TrpRS urzyme [a neologism, with the prefix ur- emphasizing the possible ancestral nature of the enzyme] [198]). They are excellent model systems for exploring conformational changes during the functional cycle of aaRSs (199). A large set of crystal structures is known for B. stearothermophilus TrpRS (191, 195, 199, 200) showing the bipartite organization of the protein with the catalytic domain followed by a small C-terminal ACB domain (called SD, for Small Domain, in the TrpRS literature) as well as different functional states of the enzyme. It is noteworthy that, when ATP and tryptophan bind to TrpRS, relative rigid-body movements of the ACB/SD domain occur (1d2r, 1maw) (195, 199). Note that the complex of TrpRS with an ATP analog (2ov4) reveals an untwisting of the ACB/SD domain relative to the Rossmann-fold domain during aminoacylation, in contrast to TrpRS under ATP/PPi exchange conditions in which no conformational change takes place (200). The topology of the last C-terminal ~60 amino acids (forming a long discontinuous α-helix running from one extremity of a monomer to the dimer axis) is idiosyncratic to TrpRS (195). Remarkably and unforeseen, the structure of TrpRS from the thermophilic bacterium T. maritima (2g36) differs from other TrpRSs by the presence of an iron-sulfur cluster [4Fe–4S] coordinated in its C-terminal ACB domain by a 4-cysteine motif (201). The discovery of such a cluster in the aaRS world is unprecedented and its biological role remains elusive. However, since the tRNA-modifying enzyme MiaB from T. maritima is involved in thiolation and methylation of A37 in tRNA anticodon loops (including that of tRNATrp) and contains, as well, an iron-sulfur cluster sequestered in a 4-cysteine motif (202), it is conceivable that the cluster in the thermophilic TrpRS participates in the specific recognition of the anticodon loop from tRNATrp (201).
Class IIa SerRS, ThrRS, ProRS, HisRS, and GlyRS
Class IIa aaRSs constitute a rather heterogeneous family of homodimeric α2-proteins (except GlyRS that is either of α2- or of α2β2-type) showing important subunit size variations (see Table 1). These aaRSs charge small and polar amino acids (glycine, serine, threonine, proline, and histidine) on tRNA. Overall, the core of the monomers comprises two main modules that are the catalytic domain with the canonical class II signature sequences and a C-terminal ACB domain of mixed α/β-architecture. This ACB domain is the distinctive feature of class IIa aaRSs, except in SerRS that lacks this domain (either lost or not appended during evolution). Despite this absence, ranking of SerRS in class IIa is justified since it is structurally and phylogenetically related to ThrRS and ProRS (14). Idiosyncratic insertions and/or appended domains distinguish the different enzymes of this subclass, which otherwise show large functional diversity (e.g., only ThrRS and ProRS are editing enzymes encompassing distinct editing domains within class IIa aaRSs). Remarkably, GlyRS and ProRS have bipartite and evolutionarily distinct distributions in Bacteria, that is, of bacterial- or archaeal/eukaryal-type for GlyRSs and of bacterial- or eukaryal-type for ProRSs.
SerRS, ThrRS, and ProRS: These three aaRSs are structurally and phylogenetically related (14), despite unique structural and functional features characterizing each of them. Interestingly, the structural relatedness concerns also the three amino acids, with serine and threonine capable of forming an internal H-bonded five-membered ring structure that mimics the ring structure of proline (14).
The crystal structure of E. coli SerRS reveals an unprecedented N-terminal domain forming an antiparallel α-helical coiled-coil conserved among SerRSs that is stretching 60 Å out into the solvent and is stabilized by interhelical hydrophobic interactions (25). The antiparallel coiled-coil domain is well seen on the structure of the SerRS:tRNASer complex from T. thermophilus (1set) (203). It is also visible on Figure 2 in the structure of the E. coli complex (not in PDB) but becomes more obvious under a different orientation. Note that tRNASer spans over the two subunits of SerRS and that the extended coiled-coil domain of one subunit makes contacts with the long variable arm and the T-loop of tRNASer and thereby directs the acceptor stem into the active site of the other subunit (203). Interestingly, the tRNA anticodon is not recognized by SerRS; this is also the case for AlaRS (class IIc) and LeuRS (class Ia). SerRSs lack an editing domain, but, as shown with the yeast enzyme, possess a hydrolytic activity toward noncognate aminoacyl-adenylates (204).
The ThrRS enzymes, and particularly E. coli ThrRS, are interesting for other reasons. They are editing aaRSs and, in the case of E. coli and in Bacteria closely related to E. coli, were shown to recognize both tRNA and a tRNA-like domain encrypted in the operator region of its own mRNA. This recognition is governed by the identity rules (as described below), with threonine identity elements in tRNAThr and mRNAThrRS recognized in an equivalent manner by the ThrRS (145, 205, 206) (see “Regulation strategies,” below, for other details).
ThrRSs belong to the TGS superfamily (after ThrRS, GTPase, and SpoT guanosine polyphosphate hydrolase) because of the presence of the small TGS subdomain (~50 amino acids) in their N-terminal part (39). TGS features are common to many enzymes, including AlaRSs. ThrRSs have a conserved overall architecture with a N-terminal editing domain located ~39 Å away from the threonylation site and the protein core formed by a central catalytic domain and a C-terminal ACB domain similar to that of the GlyRS, HisRS, and ProRS families. The N-terminal domain, linked to the core by a long α-helix, is divided into two subdomains, named N1 and N2. This editing domain has strong sequence and folding analogy with the editing domain of AlaRSs (145, 207, 208). Its small N1 subdomain has the topology of proteins from the ubiquitin family, while the N2 subdomain that hydrolyzes mischarged seryl-tRNAThr has a new fold consisting of a α-helix surrounded by antiparallel β-sheets. Several crystal structures of the isolated editing domain (residues 1 to 224) show how serine is recognized and threonine rejected (209). As seen in the structure of the ThrRS:tRNAThr complex (1qf6), subdomain N2 contacts the acceptor arm of tRNA on its minor groove side, so that the tRNA is clamped between the catalytic and N2 domains (145). Structures with small ligands from S. aureus ThrRS (1nyq) confirm the structural scheme found for the E. coli protein and reveal conformational changes important for activity (210).
As for ProRSs, phylogeny and crystallography indicate that they belong to two evolutionary groups of bacterial- and eukaryal/archaeal-types and show structures that fold in at least five distinct architectures (211). Thus, E. coli ProRS (no crystal structure available), E. faecalis (2j3l), and R. palustris (2i4l) ProRSs (211) are bacterium-like and T. thermophilus (1hc7) ProRS is eukarya-/archaea-like (212). The main differences between the two groups are the presence in the eukarya/archaea-like enzymes of a C-terminal Zn-binding module appended to the class IIa-specific ACB domain and in bacterium-like ProRSs of a large INS insertion domain (~180 amino acids) in the catalytic domain. Although ProRSs are editing enzymes, the editing INS domain can be missing, as in R. palustris ProRS, but, in that case, its absence is compensated for by freestanding homologs of the INS domain acting in trans (see “Error correction” under “Aminoacylation of tRNA” below).
HisRS: A series of crystallographic structures at 2.4- to 2.8-Å resolution of bacterial HisRSs either free or with small ligands bound, namely from E. coli (e.g., 1kmn [147, 213] and a structure with a histidyl-adenylate analog [2el9, unpublished from RIKEN Structural Genomics Initiative]), T. thermophilus (1h4v) (146, 214) and S. aureus (1qe0) (215), give a clear picture of the HisRS architecture and its conformational plasticity that is essential for activity. The homodimeric E. coli enzyme has a globular shape, with monomers (424 amino acids) consisting of three domains: first, a N-terminal catalytic core displaying the three class II signatures within a six-stranded antiparallel β-sheet sitting on two α-helices and superposes well with the homologous domain of other class II aaRSs; second, a α-helical insertion (~60 amino acids) interrupting the catalytic domain between motif 2 and motif 3; and third, a C-terminal α/β-domain (100 amino acids) resembling half of a β-barrel and oriented so as to contact the anticodon stem and part of the anticodon loop of tRNAHis. Comparison of the different structures reveals slight conformational changes in subdomains that correlate with different functional states, notably, a significant mobility of the insertion domain, as suggested by poor electron density in the T. thermophilus HisRS structure (1adj, 1ady) (214). Mobility of the insertion domain likely favors contacts with the acceptor stem of tRNAHis and is associated with adenylate formation (216).
GlyRS: These are enzymes of two mutually exclusive α2- and α2β2-types without sequence similarity and a phylogeny that does not correspond to the taxonomic classification of organisms. While Archaea and Eukarya use dimeric α2 GlyRSs, Bacteria use both α2 and α2β2 GlyRSs (14, 68). This reflects an intricate evolutionary history. Crystal structures of bacterial α2β2 GlyRSs, as present in E. coli, are not yet available; however, several structures of archaeal/eukaryal α2-type GlyRSs are known (Table 2). Their architecture is illustrated by the GlyRS from T. thermophilus (1ati) (148). It contains the three class II motifs, but motif 1 does not contain the proline believed to be a class II invariant. Each monomer consists of an active site resembling that of AspRS and SerRS, a C-terminal ACB domain of 100 residues and a third domain unusually inserted between motif 1 and 2, almost certainly interacting with the acceptor arm of tRNAGly. This insertion domain is a rubredoxin-like zinc ribbon (217). The C-terminal domain has a novel five-stranded parallel/antiparallel β-sheet structure with three surrounding helices. The residues responsible for substrate recognition, particularly in the glycine-binding pocket, were readily identified because of the conserved nature of the class II active sites (148).
Class IIb AspRS, AsnRS, and LysRS
These homodimeric aaRSs are closely related in structure and ligand recognition. In Bacteria and Archaea, their monomers have a bipartite structure, with the ACB module joined to the catalytic core by short hinges. N-terminal extensions with different biological functions (e.g., enhancing tRNA binding, important for aaRS regulation) are appended on the ACB modules and characterize the eukaryal enzymes (89). The ACB domains are made of OB-folds (40), a common motif with β-barrel architecture found in many proteins (218). These OB-folds recognize tRNA anticodon loops, notably the related anticodon identity determinants (aspartate GUC, asparagine GUU, and lysine UUU triplets) and thus play a key role in tRNA discrimination for specific aminoacylation. Discrimination relies on the positioning of the so-called L45 loop in the OB-folds of AspRS, AsnRS, and LysRS in the complexes with their cognate tRNA and on peculiar sequence conservation within the L45 loops of these aaRSs (219). Distinction between D- and ND-AspRSs relies also on structural differences in their OB-folds (161, 220, 221). Furthermore, discrete amino acid changes in the aspartate-, asparagine-, or lysine-binding pockets prevent amino acid misrecognition by the class IIb aaRSs (222).
AspRS: The distinctive feature of bacterial AspRSs is the large extradomain (~120 amino acids) inserted in the catalytic domain between class II signature motifs 2 and 3. This domain, discovered in T. thermophilus AspRS (1l0w) (163, 164), has the architecture of a five-stranded antiparallel β-sheet flanked by three α-helices and resembles the so-called ferredoxin fold (163). This distinctive feature of bacterial AspRSs is well seen in the crystal structure of the E. coli enzyme displayed in pink in Figure 3. It also has strong homology with a domain found in archaeal GatB proteins and thus was designated the GAD domain (after GatB/AaRS/Domain) (39). This homology reflects an evolutionary link between bacterial AspRSs and GatB transamidation enzymes and suggests a role of the GAD domain in the indirect pathway of tRNAAsn charging (see also “Alternative functions of bacterial aminoacyl-tRNA synthetases,” below, for details). As in the case of tRNAGlu and tRNAGln charging by ND-GluRS (see above), the structure of tRNA plays a critical role in the mechanism of tRNA aspartylation by ND-AspRS (Figure 4).
The E. coli AspRS:tRNAAsp:aspartyl-adenylate complex (1c0a) shows different binding modes of the two branches of the L-shaped tRNAAsp with AspRS (149). While the anticodon branch, including the three anticodon bases (but not the bacterial Q-modification at position 34 of tRNAAsp), binds the β-barrel of the N-terminal ACB domain exclusively through direct interactions, the water-solvated acceptor arm establishes both direct and water-mediated hydrogen bonds with AspRS. For the recognition of aspartyl-adenylate, Bacteria-specific Gln231 (replaced in Eukarya and Archaea by serine), together with class II-conserved arginine in motif 2, plays the key role in stabilizing the transition state of the aspartylation reaction. Note the closed conformation of the so-called flipping loop within the catalytic domain that anchors aspartic acid or aspartyl-adenylate in its binding pocket and helps correct positioning of the tRNA terminal A76 to facilitate the transfer of aspartic acid to its ribose 3′-OH group. In contrast, when tRNA is absent, this flipping loop has an open conformation (149, 165).
AsnRS: The crystallography of AsnRSs is poorly documented, with only one bacterial structure reported—that of T. thermophilus apo-AsnRS (not in PDB) (150). This structure is remarkably similar to that of eukaryal and archaeal AspRSs and to class II LysRSs but is lacking the large insertion within the catalytic domain characterizing bacterial AspRSs. Its catalytic site is similar to that of AspRSs, but with a notable difference for discrimination of the related aspartic acid and asparagine substrates. The three structures of free and adenylate-bound archaeal P. horikoshii AsnRS at high resolution (1.45 to 1.98 Å) (1x54, 1x55, 1x56) shed light on a peculiar water-assisted asparagine recognition, in contrast to the situation in AspRSs, in which aspartic acid recognition is achieved exclusively through extensive interactions with amino acid residues (223).
LysRS: Crystal structures of E. coli LysRS (the constitutive LysS form), in complex with small ligands (1e22, 1lyl) (151, 224), show as anticipated, similarities with AspRS and AsnRS structures. The LysU form (the product of the lysU gene expressed under extreme physiological conditions, such as heat shock) differs slightly (88% sequence identity) from the major LysS form that is synthesized under normal growth conditions (85). It is interesting to note that the LysU enzyme synthesizes Ap4A and other adenyl-dinucleotide compounds known as alarmones and is involved in the heat shock response (225, 226). Two structures of T. thermophilus LysRS in complex with either homologous unmodified tRNALys transcript or modified E. coli tRNALys are also available (not in PDB) (227). These bacterial LysRS structures show three well-resolved metal ions (Mn2+ mimicking the biological Mg2+ ions) coordinating ATP and lysine with conserved residues of the catalytic site and thereby stabilizing a pentavalent transition state (151). Two structures of B. stearothermophilus LysRS in complex with lysyl-adenylate analogs (3e9h, 3e9i) highlight the functional role of a conserved glutamate residue (Glu411 in B. stearothermophilus) for the nucleophilic attack of PPi in the ATP/PPi exchange reaction (228). As to the LysRS:tRNALys complex, its structure shows an ordered tRNA anticodon branch in contact with the ACB-OB-fold of the protein in a way reminiscent of what is seen in AspRS, with best electron density for the anticodon identity determinants interacting with the aaRS (227). This observation supports a common class IIb interaction mode of tRNA, where interaction of anticodon with the aaRS is the first trigger for productive tRNA aminoacylation (166). Finally it is worth mentioning the structure of PoxA from S. enterica (229) (3g1z), a paralog of LysRS, and to remember the existence in a few Bacteria of a class Ib LysRS that shares similarities with GluRS (see above).
Class IIc AlaRS and PheRS
AlaRS and PheRS are the largest aaRSs and are made of four subunits arranged in intricate topologies. Tetrameric α4-AlaRS and especially α2β2-PheRS have complex multidomain arrangements. Both demonstrate editing activity. Their ranking in class IIc, however, is based not on robust evidence (AlaRS was first ranked in class IIa) but rather on idiosyncrasies that differentiate them from the other class II members. The oligomeric organization of AlaRS and PheRS is conserved during evolution, except in mitochondria, where PheRS is monomeric (see “Bacterium-like aminoacyl-tRNA synthetases,” below). Note that the true topology of cytosolic PheRSs is dimeric of (αβ)2-type with two heterodimers. Interestingly, among aaRSs, AlaRSs have the highest degree of sequence conservation and have limited similarity with other aaRSs (18).
AlaRS: The AlaRS monomers (~800 amino acids) are organized into four functional domains that are, starting from the N terminus, the class-defining catalytic domain, a tRNA recognition domain (that is split into two structural modules), an editing domain, and a C-terminal oligomerization domain. Although a complete crystallographic structure of a native AlaRS is still missing, data on AlaRS fragments give a rather comprehensive view on structure/function relationships. Thus, the catalytic fragments of A. aeolicus AlaRS (453 amino acids) (1yfr) show class II characteristics and a bipartite organization of the tRNA recognition domain made of a helical module with a hairpin motif critical for acceptor-stem recognition and a C-terminal module of mixed α/β-fold. Docking of tRNAAla suggests critical contacts with three AlaRS domains (230). For archaeal A. fulgidus AlaRS, two halves of the enzyme were solved (135), namely, the large active truncation comprising the catalytic/tRNA-recognition domain and the editing domain (AlaRSΔC) and the smaller C-terminal truncation restricted to the dimerization domain (AlaRSC). These structures inform about the insertion of the editing domain in the enzyme core via tight hydrophobic interactions to the catalytic/tRNA-recognition domains, on the side opposite from that in ThrRS. Thus, tripartite AlaRSΔC (2ztg) forms a groove containing the amino acids required for recognition of the alanine G3•U70 identity pair in tRNA. Therefore, this groove appears to be an alternative tRNA-binding site that specifically recognizes the G3•U70 pair in the acceptor stem of tRNAAla. This implies that both tRNA recognition and editing are triggered by the same determinant. The dimerization domain (2zvf) consists of helical and globular modules. The helical module mediates dimerization by forming a helix-loop-helix zipper, while the globular module with its positively charged face suitable for tRNA binding is important for the aminoacylation and editing activities.
PheRS: Extensive sequence analysis and solution studies on various PheRS proteins, together with detailed crystallographic investigations on the PheRS from T. thermophilus (152, 231, 232, 233, 234) and more recently from E. coli (235), provide a good understanding of these enzymes that have both aminoacylation and editing activity. The heterotetrameric (αβ)2 PheRSs have an unprecedented architecture shaped as a leatherback turtle with large flippers (Figure 5) formed by the N-terminal parts of the β-subunits as first revealed by the crystal structure of the T. thermophilus enzyme (1pys) (234). This structure, and refined versions with ligands (231, 233, 235), including tRNA (152, 232) (2iy5), highlights 11 domains (3 in the catalytic α-subunit, namely, A0 to A2—with N-terminal A0 disordered in the T. thermophilus structure—and 8 domains in the β-subunit, namely, B1 to B8). The small catalytic α-subunit (327 to 350 amino acids) comprises the active site (A1 and A2) and domain A0 with a compact triple-helix structure that contains a GxxG RNA-binding sequence for a specific contact with tRNAPhe (232, 235). The large β-subunit (775 to 797 amino acids) contains a heterodimeric domain (B1 and B5) similar to the DNA-binding module of CAP (Catabolite gene Activator Protein); a domain inserted in B1 (B2), similar to the EMAP II and OB-folds found, respectively, in MetRS and class IIb aaRSs, where they contact the tRNA anticodon region; an editing domain of intricate architecture (B3 and B4) distant from the aminoacylation domain; a cryptic “catalytic-like” domain without class II signatures (B6 and B7); and a recognition domain of tRNAPhe anticodon (B8). This domain is missing in eukaryal PheRSs so that recognition of tRNAPhe differs in Bacteria and Eukarya in agreement with differences in the catalytic efficiency of tRNAPhe charging. It is noteworthy, and most intriguing, that B8 shares structural similarity with the U1A spliceosomal protein and B3/B4 shares structural similarity with the biotin synthetase/repressor protein (BirA) from E. coli, with a DNA-binding α/β-motif (B3), and a SH3-like (for SRC Homology) motif found in proteins of signaling pathways (B4) (236). Remarkably, comparison of the T. thermophilus PheRS structure with the E. coli PheRS structure uncovers significant rearrangements of the structural domains involved in tRNAPhe binding/translocation (234). The high-resolution PheRS structure from pathogenic S. haemolyticus (2rhq) (237) and P. aeruginosa (4p71) (238) overall agrees with the architecture of the T. thermophilus and E. coli orthologs but reveals idiosyncratic pockets for drug discovery.
Figure 5.

Shape convergence in biomacromolecular structures and morphology of living organisms, or when macromolecular structures meet zoology. The two panels represent the mimicry of the morphology of a leatherback turtle (Dermochelys coriacea) (left) with the crystal structure of a representative PheRS (e.g., from T. thermophilus [2iy5]) (right). Notice the bilateral symmetry in the shape of both turtle and pseudodimeric (αβ)2 PheRS (with catalytic short α-subunit in green CPK amino acid models displayed on a yellow background and a large β-subunit in cyan). The large front flippers of the turtle show astonishing mimicry with the B1–B5 domains from the β-subunit of the PheRS. Other shape convergences can be found when comparing the structures of dimeric class IIb aaRSs or the dimerization domain of AlaRSC with the symmetric morphology of butterflies (try to find these mimicries, and others, after clicking on the PDB accession codes of aaRS structures given in the text). Whether such shape convergences have biological meaning remains an open question.
General Conclusions on the Similarity and Diversity in aaRS Structures
AaRSs are modular enzymes globally conserved in the three kingdoms of life. All of them catalyze the same two-step reaction, namely, the attachment of a proteinogenic amino acid on their cognate tRNAs. Based on the tertiary structure of their catalytic domains, they are ranked in two distinct groups of 10 enzymes each. On the basis of other structural features, each of the two groups is subdivided into three subgroups. Structural diversity comes from the nature, the number, and the size of various additional domains appended on the catalytic cores—in particular, different types of anticodon-binding domains and, in the case of certain amino acid specificities, of editing domains where noncognate amino acids mischarged on tRNA are hydrolyzed. The oligomery of α-, α2-, α4-, or a2β2-type contributes further to the structural diversity of aaRSs. Genomic and X-ray crystallography methods were the main tools that led to these general conclusions but allowed also the discovery a plasticity of the aaRS architectures related to different functional states, as well as the characterization of faint structural idiosyncrasies within a given group of aaRSs specific for the same amino acid that could be correlated with kingdom and even species functional differences, such as for cognate tRNA recognition or for new functions. In this regard, the recent identification of novel protein domains in cyanobacterial aaRSs deserves attention (239). In addition to these general trends, one should notice the following: (i) the absence of GlnRS and AsnRS in many bacterial and most archaeal organisms, compensated for by the presence of nondiscriminating GluRS and AspRS that aminoacylate, respectively, noncognate tRNAGln and tRNAAsn in addition to their cognate tRNAGlu and tRNAAsp; (ii) the presence in some Bacteria of a class I LysRS (LysRS-1) instead of the otherwise conserved class II LysRS, thus breaking the unicity of the aaRS ranking rule; and (iii) the presence of atypical aaRSs in Archaea (ND-aaRSs, LysRS-1, and the two strictly archaeal PylRS and SepRS).
AMINOACYLATION OF tRNA
aaRS-Class-Specific Features for Substrate Binding
Difference in size and in the architectural organization of the active sites accounts for different binding modes of substrates in the two aaRS classes. Thus, in class I aaRSs, the active-site subdomain is smaller than in class II enzymes as a result of differences in the number of amino acids forming the two class-specific catalytic domains (~150 residues in the class I Rossmann fold and ~250 residues in class II antiparallel β-sheet domain). Moreover, topological differences in the protein fold and in the overall shape of the two types of active sites favor discrimination of amino acids with similar chemical groups, but with a different size (i.e., glutamate and aspartate or arginine and lysine on class I and class II enzymes, respectively). These differences lead to two binding modes for ATP (Figure 6). In class I aaRSs, ATP adopts an extended conformation reminiscent of that found in other proteins containing a Rossmann fold, while in class II enzymes it exhibits a bent conformation with the γ-phosphate folded back over the adenine base. In both cases, Mg2+ ions and strongly conserved amino acids are involved in ATP binding as well as in stabilization of the reaction transition state (i.e., residues from the flexible KMSKS signature loop in class I and the two invariant arginines in motif 2 and 3 in class II aaRSs [240]).
Figure 6.
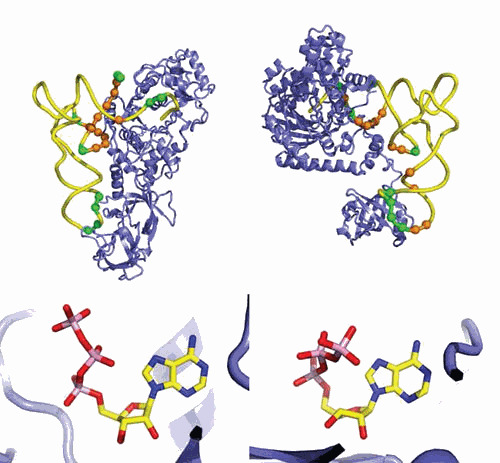
Different substrate recognition modes by class I and class II aminoacyl-tRNA synthetases. The differences are illustrated by the structures of E. coli glutamine (left) and aspartate (right) complexes ([1gsg] [179, 244] and [1c0a] [149], respectively): tRNA recognition (top); ATP recognition (bottom). For clarity, only one subunit of the AspRS:tRNAAsp complex is displayed. The tRNAs are shown as yellow ribophosphate backbones with contact residues represented as colored spheres (contacts with CCA are not shown): identity determinants are in green (a few do not contact the aaRS; see text) and other contact residues in orange. The class II adenylate conformation is from the archaeal AspRS from P. kodakaraensis (1b8a) (162). In the E. coli GlnRS:tRNAGln complex, 13 tRNAGln nucleotides (nt) are both determinants and make contact with GlnRS (nt 1, 2, 3, 10, 34, 35, 36, 37, 38, 70, 71, 72, 73) and 10 other nucleotides make additional contact with GlnRS (nt 4, 5, 6, 7, 8, 11, 12, 13, 14, 15). In the E. coli AspRS:tRNAAsp complex, 8 tRNAAsp nucleotides are both determinants and contact residues (nt 2, 10, 34, 35, 36, 38, 71, 73) and 11 other residues make additional contacts with AspRS (nt 11, 12, 25, 28, 32, 33, 67, 68, 69, 70, 72).
In the two classes, Mg2+ ions play a dual role in amino acid activation by stabilizing the conformation of ATP and participating in adenylate formation (18). They assist catalysis and facilitate adoption of the productive conformation of the PPi moiety in ATP. However, and in contrast with the class-dependent conserved ATP-binding modes, those of Mg2+ ions are diverse and depend more on the aminoacylation system than on the aaRS class (241). For instance, class I E. coli GlnRS has one Mg2+-binding site controlling interaction with the β- and γ-phosphates of ATP (242). In B. stearothermophilus TrpRS, a Mg2+ ion controls tryptophan activation by subtle allosteric effects (243). Several class II enzymes have three Mg2+ sites, notably in E. coli AsnRS (151) and P. kodakaraensis AspRS (162), where the catalytic Mg2+ is located between the α- and β-phosphates and the two others are seated on each side of the γ-phosphate bond. In class II E. coli HisRS, only two Mg2+ ions are observed, with the catalytic ion replaced by an arginine residue, absent in other class II aaRSs (147). This arginine residue, Arg259, is common to all HisRSs and, as a Mg2+ ion, has a catalytic role in maintaining an interaction with the α-phosphate of ATP (1kmn) (147) or histidyl-adenylate (1htt) (213) (see also “Mechanistic of tRNA aminoacylation” for details).
Furthermore, the crystal structures of the glutamine and aspartate aaRS:tRNA complexes have revealed two evolutionary conserved aaRS class-dependent tRNA recognition schemes. In bacterial systems, this is best seen when comparing the structures of E. coli class I GlnRS (179, 244) and class II AspRS (149, 245) in complex with their cognate tRNAs (Figure 6). Thus, class I and class II aaRSs recognize opposite sides of tRNA as a consequence of the different entry mode of their helical acceptor stems in the catalytic sites. Class I aaRSs bind this stem on the minor groove side and class II enzymes on the major groove side. This different tRNA positioning implies conformational adaptation of the acceptor end of tRNA. In class II AspRS, the backbone of the terminal –N73CCA76 sequence keeps a helical conformation, whereas in class I GlnRS it makes a sharp hairpin turn toward the inside of the L, with a disruption of the first U1–A72 pair. In both cases, binding of tRNA maintains the terminal ribose in a position that facilitates attack of the α-phosphorus of the adenylate intermediate. As a final result, the class-dependent entry of the tRNA into the active site accounts for the correct positioning of the 2′-OH (class I) or 3′-OH (class II) of the terminal ribose for receiving the amino acid.
Fidelity of Amino Acid Recognition and Functional Implications
A first prerequisite of faithful tRNA aminoacylation is fidelity of the amino acid activation step. For that, the extant aaRS family evolved to preferentially recognize the L-enantiomers of amino acids and to become selective for the 20 canonical proteinogenic members from the present genetic code. However, the too great relatedness of amino acid structure and shape should probably be fatal for recognition fidelity by the aaRSs with the consequence that these enzymes would make errors, as anticipated in the fifties by Pauling (246) and since then amply documented. To overcome this intrinsic weakness and exclude nondesired amino acids from protein synthesis, evolution developed strategies, both to avoid incorrect amino acid (and tRNA) recognitions and to clear errors due to amino acid misactivation and correlated tRNA mischarging (see, e.g., references 247 and 248 and below for details on tRNA recognition and editing).
The basis of selectivity for the L-enantiomers is intriguing and was only investigated in a few instances. For E. coli AspRS and TyrRS, molecular dynamics simulations and energy calculations support this preference (249). A similar approach shows the unfavorable electrostatic surrounding in the active site of E. coli HisRS for D-amino acid binding (250).
Amino acid recognition by aaRSs was studied in depth for E. coli AspRS by structure-based computer simulations and site-directed mutations (222, 251). They revealed the existence of a network of electrostatic interactions and a charge distribution in the active site accounting for optimal binding of charged aspartate. Binding of aspartate is stronger than for related compounds, particularly for neutral asparagine. Importantly, the intricate interplay between the amino acids constituting the aspartate-binding pocket protects AspRS against most binding errors and renders its engineering, e.g., in view of specificity switches, difficult (251).
For class Ia ArgRS and class Ib aaRSs, binding of amino acids in the catalytic site requires the presence of tRNA (reviewed in reference 252). This unique property is explained by crystallography. As seen in the structure of the T. thermophilus GluRS:tRNAGlu:Glu ternary complex (2cv0), tRNA and aaRS collaborate to form a specific binding pocket for L-glutamate. This cooperation occurs by subtle conformational changes allowing specific amino acid binding and activation. In contrast, in the GluRS:Glu binary complex (2cuz), when tRNAGlu is absent, this conformational change does not occur. As a consequence, the amino acid binding site is defective, thus accounting for binding of incorrect amino acids and lack of glutamate activation (142). Further evidence comes from a mutational study with E. coli GluRS. Thus, when tRNAGlu is present, a Cys100Tyr variant (with an altered Zn-binding motif in the SWIM-fold involved in recognition of the acceptor-end of tRNA, see above) has a lower affinity for L-glutamate than the wild-type enzyme, while in the absence of tRNAGlu, glutamate binds with the same affinity to both variant and to wild-type GluRS (253). Such effects likely apply as well for amino acid activation by GlnRSs (254, 255). It is noteworthy that site-directed mutagenesis on the wild-type E. coli GlnRS explicitly demonstrated that amino acid selectivity relies on the collective remodeling of the active site and not on direct amino acid contacts with the enzyme, since switching the activation specificity from glutamine to glutamate necessitates 22 amino acid substitutions and one deletion in the Rossmann fold of the wild-type GlnRS (256). It is anticipated that tRNA-dependent activation of arginine by ArgRSs (257, 258) and lysine by LysRS-1 (45) follows similar schemes. Note that aaRSs requiring cognate tRNA for activation can tightly bind their amino acid substrates in the absence of tRNA, thereby leading to inactive conformers as seen, e.g., in E. coli ArgRS (4oby) (178) and B. burgdorferi GluRS (4gri) (unpublished from the Seattle Structure Genomics Center for Infectious Disease). In these enzymes, the functional catalytic site is shaped by subtle conformational changes triggered by cognate tRNA. In summary, RNA is the specificity effector for amino acid recognition and activation.
Fidelity of tRNA Recognition and the Identity Problem
General concepts and definitions
Specific recognition of tRNA by aaRSs depends on thermodynamic and kinetic parameters, with a first discrimination by KD for binding (259) and additional discrimination by Vmax brought by the aminoacylation reaction (260). Identity rules account for these effects and are presently phenomenologically understood (6, 188, 261, 262, 263), although many mechanistic aspects remain elusive (264). These rules are referred to as the second genetic code (1) and rely in each aaRS–tRNA system on a limited number of tRNA determinants that contact the aaRS or act by indirect effects, but also on less known antideterminants that prevent false tRNA interactions with noncognate aaRSs. Determinants and antideterminants are defined as nucleosides (more precisely, as chemical groups on these nucleosides) and form the so-called recognition/identity sets. They are located mainly at the two distal ends of the tRNA molecule and, in most cases, contact identity amino acids on aaRSs. Identity sets can be completed by permissive elements. Such elements, only characterized in a few tRNAs, depend on the nucleotidic context (265). In a more structural perspective, crystallographic data show a limited number of contact points between aaRS and tRNA with some of them involving identity elements (see, e.g., reference 18 for specific references). Note that tRNA backbone interactions, or the related concept of indirect readout of tRNA (recognition by aaRSs of sequence-dependent conformations of tRNA via the sugar-phosphate backbone or nonspecific portions of the bases) (266), can have a functional role as first explicitly seen in the recognition of the acceptor stem of tRNAAsp by E. coli AspRS (149, 267) and documented for a few other systems (266). Table 3 gives an outlook of the structural features in tRNA that are important for recognition and specificity toward aaRSs.
Table 3.
Structural features within tRNA involved in interactions with aminoacyl-tRNA synthetases and/or important for the specificity of tRNA aminoacylation
| Structural features | Effecta on | Comments and references | ||
|---|---|---|---|---|
| Interaction | Specificity | |||
| Shape of tRNA recognized by an aaRS | ||||
| Canonical L-shape | +++ | ± | *Under special conditions, all tRNAs can be recognized by certain aaRSs, but tRNA mischarging efficiency is low (37) | |
| Atypical shapes tRNAs with large variable region, tRNA-like domains, many mitochondrial tRNAs, etc | +++ | ++ | *For tRNAs with large variable region (e.g., tRNALeu) or atypical tertiary interaction networks (e.g., tRNACys) (268) *For tRNA mimics in mRNA, e.g., in E. coli mRNAThr (206)*Aminoacylation of tRNA mimics can be efficient, e.g., in viral tRNA-like structures (264) *For atypical mitochondrial tRNAs, e.g., reference (269) | |
| Determinants in tRNA for specific tRNA aminoacylation directly read by an aaRS | ||||
| Bases Restricted number (2–11) in identity sets; mainly in single-stranded regions; sometimes in WC pairs | +++ | +/+++++ | *System-specific contacts, e.g., in E. coli aspartate and glutamine system (149, 244) and identity sets (6, 188) *Atomic determinants in E. coli alanine system (270) | |
| Sugar (from identity bases) | + | + | *Few examples of direct contacts of ribose O2′ with aaRSs, e.g., in the E. coli aspartate system (149)*Only a few examples of ribose as identity determinant (271, 272) | |
| Modified residues | + | ++ | *Few documented system-specific examples (273) | |
| Determinants in tRNA for specific tRNA aminoacylation indirectly read by an aaRS | ||||
| Sequence-dependent Individual or collective | +/++ | +++ | *Recognition of idiosyncratic conformations in cognate tRNAs (266)*Water mediated recognition of individual identity determinants, e.g., in E. coli aspartate system (149) | |
| Antideterminants in tRNA for noncognate tRNA rejections by an aaRS | ||||
| Individual nucleosides WC or modified nucleosides; possibly idiosyncratic tRNA domains | − | ++++ | *Not systematically searched; only a few cases supported by experimental validation (188, 274, 275) | |
| Other nucleotidic constituents for stability of a given aaRS:tRNA complex and for tuning its specificity | ||||
| Bases | +/+++ | ±/++ | *System-specific contacts contributing to overall binding affinity, e.g., in the E. coli glutamine system (149, 244) | |
| Ribophosphate backbones | +/+++ | ±/++ | *System-specific contacts (as above), e.g., reference 266 | |
Effects: +++++, strongest;++++, very important; +++, important; ++, medium; +, weak; ±, low. WC, Watson-Crick; n.d., not determined. See the text for more detail. Note that the structural features in tRNA are mirrored by proteic elements in the aaRSs (less well defined and characterized than their counterparts on tRNA), notably amino acids that contact identity nucleotides (e.g., in the E. coli AspRS Arg76, Glu93, and Gln46 make hydrogen-bond interactions with identity determinants Q34 and U35 from the anticodon of cognate tRNAAsp [149]). Two of these amino acids (Gln46 and Glu93) are conserved through evolution in AspRSs (276) and therefore can be considered as aspartate identity amino acids.
Because anticodon bases specify correct reading of the genetic message on mRNAs and form the relationship between the amino acids forming proteins and the trinucleotides of the genetic code, they were soon considered to be prime candidates for specifying tRNA recognition by aaRSs. This assumption turned out to be correct in many systems but does not apply to the alanine, leucine, and serine identities (6, 188, 261). Based on other considerations, participation of tRNA residues near the amino acid CCA-acceptor end was assumed to be crucial. A wealth of experimental data and thoughts on the origin of tRNA aminoacylation confirmed the importance of the anticodon and accepting branch in tRNA identity, and at present it is accepted that tRNA identity rules are universal, with nevertheless idiosyncratic distinctions in the three kingdoms of life and even species-dependent subtleties (262, 264, 277). In a few cases, experimental evidence shows an initial role of the accepting branch when contacting the catalytic site of aaRSs (e.g., in class Ib, see above) or of the tRNA anticodon domain as for AspRS (166), as well as of other structural features (e.g. the relationship between D-loop, variable region, and T-loop [278]). The importance of the tRNA shape, as well, should not be overlooked because it provides the structural framework that dictates the correct positioning of the identity elements for optimized interaction with the aaRSs (see “Role of tRNA and identity determinants in tRNA aminoacylation,” below).
Three main strategies were used to find identity determinants. In its simplest version, the first strategy consists in searching consensus sequences of all tRNAs charged by a same aaRS. In a more elaborate version, the search is computer-assisted as first applied for E. coli tRNAs (279). This strategy was recently ameliorated thanks to novel computational tools (280, 281, 282). The second strategy, widely used, is the in vitro transcription of artificial tRNA genes that can readily produce any type of tRNA transcript for aminoacylation assays (283). This method, however, does not evaluate the role of posttranscriptional modifications in identity. Atomic mutagenesis (removal or replacement of chemical groups in tRNA) would be the method of choice to discover the chemical signals in tRNA nucleosides important in identity (284). But, because of methodological difficulties, the approach was restricted to those systems where aaRSs can aminoacylate small RNA substrates (285). The third strategy, also widely used, especially for E. coli tRNAs, allows the in vivo study of mutated suppressor tRNAs with a reporter system based, for instance, on the reading by engineered suppressors of an amber mutation at position 10 of a dihydrofolate reductase gene (286, 287).
The strength of an identity determinant is given by the functional effect produced by its mutation (kcat/Km of aminoacylation for in vitro methods or suppression strength for in vivo methods). The strongest determinants are located mainly at both extremities of the L-shaped tertiary structure of tRNA and are essentially conserved in evolution. Completion of an identity set is verified by transplantation of the putative identity set into the background of a tRNA with another identity. This conceptual framework allowed characterization of most of the strongest determinants and also a gross understanding of the role of tRNA architecture but not to unravel subtleties underlying expression of identities (6, 188, 261, 264).
Determinants and antideterminants
The distribution of identity determinants in E. coli tRNAs is listed in Table 4. It is reminiscent of that observed for S. cerevisiae and a few other eukaryal and archaeal organisms (6, 188, 262, 288). The most striking difference between bacterial systems and other systems concerns tyrosine identity given by G1–C72 in Bacteria (and organelles) and C1–G72 in Eukarya and Archaea (194). This differential expression of tyrosine identity in evolution correlates with kingdom-specific features in tRNATyr, namely, large variable regions in Bacteria and small ones in Eukarya and Archaea (19, 194). The crystal structure of both T. thermophilus and Methanococcus jannaschii TyrRS:tRNATyr complexes explains the functional idiosyncrasies that are mainly due to different acceptor stem recognitions and involvement of the S4-like domain of bacterial TyrRSs in recognition of the large variable region of bacterial tRNATyr (64, 289). Note that the human mitochondrial TyrRS, closely related with bacterial TyrRSs, disobeys the universal identity rules since the G1–C72 pair is not needed (290). In contrast, however, E. coli tRNAs make ample use of residues in the tRNA acceptor helix. For instance, in the aspartate system, the two first base pairs in this helix are determinants (291, 292). Whether idiosyncratic identity determinants within the acceptor stem of mitochondrial tRNAs contribute to aminoacylation specificity, e.g., in the human tyrosine system, remains to be tested.
Table 4.
Distribution in tRNA of identity determinants for aminoacylation by bacterial aminoacyl-tRNA synthetases and aminoacylation capacity of tRNA minihelicesa
| tRNA domains | Aminoacyl-tRNA synthetases | |
|---|---|---|
| Class I | Class II | |
| Amino acid accepting branch | ||
| nt-1 (1 identity) | – | H |
| Discriminator base 73 (18 identities) | C, E, I, L, M, Q, R, V, W, Y | A, D, F, G, H, K, N, P, S |
| Acceptor stemb (12 identities) | C, E, I, M, Q, V, W | A, D, G, H, T, S |
| Core regionc (13 identities) | C, E, L, M, Q, R, W | A, D, F, G, P, S |
| Anticodon stem-loop branch | ||
| Anticodon stemd (2 identities) | Q | K |
| Anticodon loope (3 identities) | E, M, Q | – |
| Anticodon tripletf (17 identities) | C, E, I, M, Q, R, V, W, Y | D, F, G, H, K, N, P, T |
| tRNA structure (collective participation)g | C, E, I | F, K |
| Amino acid accepting minihelices | ||
| Identity of accepting RNA | I, Lh, M, Q, V | A, D, G, H, P, S |
AaRSs are abbreviated by the one-letter code; underscored letters mean participation in identity of modified nucleotides (273).
Base pair (bp) 1–72, 2–71, 3–70, 4–69.
Concerns mainly conserved or semiconserved residues important for tRNA architecture, such as the atypical Levitt G15•G48 pair for cysteine identity (293).
bp 29–41, 30–40, 31–39.
Residues 37 and 38.
Triplet 34, 35, 36 (numbering of tRNA residues is according to reference 19).
As a result of decreased catalytic aminoacylation efficiency of unmodified tRNA transcripts in comparison with the efficiency of fully modified native tRNAs (data only available for six E. coli systems, with AlaRS not sensitive to the presence of tRNA modifications [273]). Most data on identities were retrieved from references 6 and 188; see also newer literature for the importance of the tRNA core region in cysteine identity (294), of N73 and other elements in leucine and proline identities (295, 296, 297), and of anticodon stem elements for tRNA recognition by E. coli LysRS and GlnRS (298). For data on minihelices, see references 270, 285, 299, and 300.
Only A. aeolicus LeuRS.
Antideterminants preventing false recognition of tRNA are not well known, and so far only a few have been discovered. The hypermodified lysidine residue (k2C or 2-lysyl-cytidine) at the first anticodon position of minor E. coli tRNA2Ile is responsible for the rejection of this tRNA by MetRS and was the first bacterial antideterminant discovered (274). Likewise, A36 in E. coli tRNAArg is an antideterminant against TrpRS (301), and the G2•U71 pair in B. burgdorferi tRNALys (cognate for class Ib LysRS) prevents misrecognition by class IIb LysRS (190).
Specificity determinants and antideterminants exist at the protein level but were not systematically searched and identified by mutagenesis analyses. Several examples are worth mentioning. Thus, Arg83 in AsnRS is involved in the recognition of U36 at the third identity position of tRNAAsn anticodon (223). Likewise, identity amino acids have been described in bacterial TyrRSs. They consist of conserved or semiconserved residues recognizing anticodon (Asp259, Asp423) and acceptor arm (Glu154, Arg198) identity bases (194, 290). On the other hand, amino acids acting as antideterminants were found in E. coli MetRS, B. stearothermophilus TyrRS, and H. pylori GluRS. Thus, Asp449 and Asp456 from the C-terminal ACB-domain of MetRS are negative signals that reject noncognate tRNA anticodons (302). Likewise, Glu152 of TyrRS rejects noncognate tRNAs by electrostatic and steric repulsion (303), and Arg350 in the ACB region of GluRS rejects tRNAGln (77).
Role of modified nucleosides
Five modified nucleosides were explicitly characterized as identity determinants in E. coli tRNAs, namely, k2C34 (in tRNA2Ile), s2U34 (in tRNA1Gln and tRNAGlu), mnm5s2U34 (in tRNALys), Q34 (in tRNATyr), and t6A37 (in tRNAIle), all located in anticodon loops (273) (for the meaning of modification symbols, see the Introduction, above). The role of k2C34 in minor E. coli tRNA2Ile is remarkable since this residue is both an antideterminant against MetRS (see above) and a determinant for IleRS (274). However, the determinant role could be indirect, since the major tRNA1Ile has a G at position 34 that is structurally different from k2C, which could mean that E. coli IleRS does not make a direct functional contact at position 34. On the other hand, the drastic reduction of the kcat-dependent isoleucylation capacity of E. coli tRNA1Ile after replacement of t6A37 by A37 shows that t6A37 is an isoleucine identity determinant and probably is involved in a direct interaction with E. coli IleRS (304). The case of the E. coli glutamate system is interesting since isolated hypomodified tRNAGlu species (modivariants) have variable aminoacylation capacities (305). The strongest impairment concerns the modivariants lacking mnm5s2U34 and m2A37. The fact that another modivariant solely containing s2U34 is efficiently aminoacylated conclusively indicates that the mnm5 group (5-methyl-aminomethyl group) is not important for aminoacylation, which in turn indicates that the s2 group acts as a determinant for tRNAGlu aminoacylation (305).
Minimalist tRNAs as means for understanding aaRS functions and evolution
The aminoacylation capacity of minimalist tRNAs restricted to an accepting branch, or part of it, is so far documented in 11 systems (Table 4). This property allowed discovery of novel identity elements in tRNA, yielded information on the mechanism of identity expression, and provided insights into how evolution established the identity rules (2, 270, 285). Note that the ability to prepare charged or mischarged minihelices turned out to be useful for studying editing (299, 306, 307). Below is a short survey of significant outcomes from these studies.
The first data came from the E. coli alanine system and indicated that native L-shaped tRNA is not a prerequisite for the aaRS aminoacylation function (308, 309). By atomic mutagenesis it was conclusively shown that the unpaired exocyclic 2-amino group of G3 positioned in the minor groove of the wobble G3•U70 alanine identity pair is required for aminoacylation by AlaRS (309). Moreover, it was shown that the A73 discriminator base of minihelixAla is a determinant of the transfer step of aminoacylation and that substitution of the exocyclic amino group of A73 with a keto-oxygen results in negative discrimination (310).
Likewise, RNA hairpin helices based on the acceptor stem of tRNAMet and tRNAHis were shown to be specifically aminoacylated by MetRS and HisRS (311). Other studies with a tRNATyr-derived microhelix gave the clue of the tyrosylation species specificity that is solely determined by the N1–N72 base pair (312). Furthermore, and most interesting, it was shown that a single atomic group in an RNA helix based on E. coli tRNAPro (the 6-keto group of C72) is needed for positive discrimination by cognate ProRS and is an antideterminant for negative discrimination by noncognate AlaRS (313). As for serine identity, serylation rates for minihelixSer variants revealed that E. coli SerRS recognizes five base pairs (1–72 to 5–68) of the tRNASer acceptor stem with major recognition elements clustered between base pair positions 2–71 and 4–69 (314). Serylation efficiency of these minihelices revealed the role of functional groups from the major groove, in agreement with the structure of the T. thermophilus SerRS:tRNASer complex (132). This allows SerRS to recognize the set of tRNASer isoacceptors that present sequence variations within the five last base pairs in the accepting arms. Other studies on the charging ability of tRNAAsp- and tRNAVal-derived minihelices shed new light on tRNA recognition by ValRS and AspRS (315, 316). Furthermore, in the E. coli histidine system, atomic mutagenesis probed the exact role of the additional G-1–C73 base pair in tRNAHis (a characteristic of all tRNAHis species) in histidylation by E. coli HisRS (317). Results indicated that G-1 serves to position the 5′-monophosphate, which is critical for aminoacylation, and additionally that C73 and G-1 contain exocyclic atomic groups located in the major groove of the accepting RNA helix that contribute to HisRS recognition.
As a last point, note that tRNA fragments restricted to an anticodon stem loop (ASL) can interact with aaRSs. Thus, an ASLVal containing the anticodon 34CAC36 valine identity determinants stimulates somewhat valylation of a free-standing tRNAVal-derived minihelix (315). Likewise, an ASL molecule derived from E. coli tRNAGlu containing the s2U34 identity determinant (see above) binds to GluRS and inhibits aminoacylation of native tRNAGlu (318). Most interesting in this context is the glutamylation of bacterial tRNAAsp on the Q-base at position 34 of its anticodon by a mimic of the catalytic domain of bacterial GluRS (comments in [319] and [320], and see “Mimicry of Catalytic Domains”, below for details and implications).
In the perspective of evolution, it is important to know that the functionality of minimalist tRNAs (mini- or microhelices, ASLs) depends on the presence of identity determinants, since their absence impairs or completely abolishes their aminoacylation or inhibition capacities. This strongly suggests that primordial aminoacylation systems consisted of pairs of minimalist aaRS and tRNA (2, 198, 321, 322), whose specificity was governed by recognition rules excluding evolutionarily more recent signals from ACB domains in aaRSs and ASLs in tRNAs. Also related to evolution is the question of the coding properties of tRNA identity bases in the acceptor and ASL domains. An answer comes from a recent physico-chemical-based study showing that the anticodon encodes the hydrophobicity of each amino acid side chain and the acceptor stem codes preferentially for the surface area or size of each side chain (323). These orthogonal properties suggest that genetic coding of protein 3D structures evolved in distinct stages, based initially on the size of the amino acid and later on its compatibility with globular folding in water (323).
Mechanism of tRNA Aminoacylation
Catalytic mechanisms of tRNA aminoacylation have been discussed in depth in several reviews and, altogether, an overall common mechanism emerged (6, 7, 240, 324, 325). In this section, emphasis is given to landmark features of the reactions catalyzed by emblematic representatives of the two classes of bacterial aaRSs (remenber that the partition of aaRSs in two classes is based on different structures of their catalytic sites). Other aspects, either atypical or idiosyncratic (e.g., editing and activity of aaRS fragments) are covered in “Error correction” and “Truncated aaRSs,” below.
Amino acid activation
After entry of amino acids and ATP to their respective binding pockets, with ATP in the conformation characteristic of each aaRS class, the α-carboxylate group of the amino acid substrates attacks the α-phosphate of ATP, leading to intermediates stabilized by the aaRS class-conserved residues and Mg2+-assisted hydrolysis of the phosphate bond followed by PPi release. Binding of the adenylate on the protein (that can be enhanced by tRNA binding) facilitates the amino acid transfer on tRNA and prevents the activated amino acid from reacting with other nucleophiles present in the solvent (e.g., water) or on the surface of the aaRS (e.g., the side chain of lysine residues). In all systems so far investigated, conformational changes occur in the aaRSs upon amino acid binding, e.g., in E. coli LysRS (1e1o) (326), bacterial-type ProRSs (2j3m) (221), and B. stearothermophilus TrpRS (1m83, 1man) (327).
Kinetic investigations and isotope-exchange methods combined with crystal structure analysis suggest an overall similar in-line mechanism by nucleophilic attack for four class I aaRSs (IleRS, MetRS, TrpRS, and TyrRS) (240). Refined views emerged when function was interpreted in the light of crystallographic and molecular dynamics results. In the case of E. coli class II HisRS, energy variation during the mutual approach of histidine and ATP to form adenylate shows that the surrounding nanospace of the protein confines the reactants in geometry suitable for the in-line nucleophilic attack. Electrostatic potential data indicate a role of Mg2+ and Arg259 in the active site facilitating the process by reducing the negative charge distributed over the oxygen atoms of the ATP α-phosphate in a way mimicking that of the two Mg2+ cations from the catalytic site of other class II aaRSs (250). For B. stearothermophilus TrpRS, a conformational transition state accompanies tryptophan activation (327). Moreover, linking new TrpRS crystal structures (with bound AMP, PPi, and/or tryptophan) and molecular simulations techniques (i) gave support to the existence of a transiently covalently linked adenosine for stabilizing closed TrpRS conformations, (ii) showed high affinity of the KMSKS loop for the β-phosphate of ATP, (iii) revealed a conformational free-energy barrier early to the induced-fit phase of the catalytic process (328), and (iv) showed the importance of Mg2+ in the catalytic mechanism (329). For E. coli MetRS, recent structural and functional studies (high-resolution structures at 1.6-Å resolution combined with an advanced mutagenesis analysis) elucidated the mechanism of amino acid selectivity by this aaRS. Interestingly, this selectivity switches from an induced fit in the native protein to a lock-and-key mechanism in a MetRS with altered amino acid specificity (167).
Amino acid attachment on tRNA
Once tRNA is bound to its cognate aaRS and A76 is properly located, the second step of the aminoacylation reaction can proceed. The 2′- or 3′-hydroxyl of the terminal tRNA ribose donates its proton to the phosphate of the adenylate, thereby forming a second transition state. Then, one of the two free-oxygen atoms of the α-phosphate attracts the proton from the attacking OH of the terminal ribose, forming a cyclic intermediate that converts into the ester linkage between the amino acid and the tRNA ribose. This general mechanism was proposed for E. coli GlnRS and S. cerevisiae AspRS, the two archetypes of class I and class II enzymes (18, 149, 242). As usual for catalysts, the main function of the aaRS is to correctly orient the substrates and to stabilize the intermediate transition states. Juxtaposition of the reactive groups of the substrates triggers the reaction, and the structure of the intermediates promotes by itself its forward progression. The site of amino acid charging on the tRNA seems to be the only difference between the two classes (65, 66, 67, 330). This difference is explained in part by the mode of tRNA entry into the catalytic site that dictates proper positioning of the acceptor OH. The significance of the conservation of this 2′ or 3′ specificity through evolution remains obscure, since a rapid isomerization of the amino acid occurs between the 2′- and 3′-hydroxyls after release from the enzyme (331). For acylations on 2′-OH, an isomerization is required to produce the 3′ species used for protein synthesis. This step could be assisted in vivo by elongation factor EF-Tu, since this protein was shown to stabilize the orthoester isomerization intermediate (332).
Asymmetric functioning of oligomeric aaRSs
Half-of-sites reactivity and asymmetric functioning were proposed for dimeric class Ic aaRSs. A large number of mechanistic studies on B. stearothermophilus TyrRS demonstrated that this protein acts as an asymmetric dimer (only one site in action) in charging tRNA (333). Likewise, structural studies on B. stearothermophilus TrpRS under different conformational states (i.e., after ligand binding) led to similar conclusions and showed open complexes of the enzyme interacting with tryptophan and ATP that are asymmetric in the manner observed in apo-TrpRS (199).
Functional and structural asymmetry is also documented for class II aaRSs, notably dimeric bacterial AspRS, HisRS, LysRS, and ThrRS, and tetrameric GlyRS. For E. coli AspRS, the tRNA-dependent structural asymmetry of the dimer is enhanced when heterologous yeast tRNAAsp is bound (291) and correlates with the functional asymmetry found in the yeast AspRS, revealed by the existence of two different affinity constants and Km-values for ATP (334). For HisRS and ThrRS from E. coli, amino acid activation (as well as misactivation of serine by ThrRS) occurs at different rates in the two active sites when tRNA is absent (335, 336), but half-of-sites reactivity is not observed in HisRS (334). To clarify the basis of this asymmetry, fluorescence resonance energy transfer experiments using differential labeling of the two HisRS monomers were undertaken. They allowed measurement of similar adenylate formation rates, but they were asymmetric with respect to the two active sites of the dimer. Furthermore, these experiments revealed rigid-body rotation of the HisRS α-helical insertion domain, suggesting that conformational changes are rate limiting for product formation. This “alternating site” model for HisRS catalysis may be common to other class II aaRSs (216). Likewise, asymmetry for nucleotide binding was predicted in the LysU isoform of E. coli LysRS on the basis of molecular dynamics simulations (338). Enzymology of GlyRSs dates back to the 1960s and suggested soon an asymmetric functioning of these heterotetrameric aaRSs. However, the exact nature of the functional asymmetry (by half-of-sites or flip-flop mechanisms) remains elusive (339).
Role of tRNA and identity determinants in tRNA aminoacylation
Optimal aminoacylation efficiency depends both on tRNA shape and full sets of identity determinants in tRNA, the relative contribution of each being system dependent. Thus, the specificity of serylation depends principally on recognition of the shape of tRNASer. This shape is peculiar because of the presence of a large variable hairpin region protruding from the canonical L-shape that is contacted by SerRS. Large variable regions occur in all tRNASer species (except the mitochondrial ones), as well as in tRNALeu and bacterial tRNATyr species (19), conferring a peculiar tripod-like shape to these tRNAs that can be considered as an identity determinant in serine, leucine, and tyrosine systems. Likewise, most tRNACys species present a structural idiosyncrasy because of the noncanonical nature of their N15•N48 tertiary pair, and, interestingly, recognition of tRNACys by E. coli CysRS was shown to be shape dependent (141). This conclusion finds robust support in the crystal structure of the SerRS:tRNASer complex from T. thermophilus that explicitly shows that recognition of tRNA principally relies on backbone contacts with SerRS and secondarily on sequence-specific interactions (203).
In the other systems where tRNAs have a small variable region, efficient aminoacylation predominantly relies on full identity sets (6, 188). As an example, in the E. coli histidine system, anticodon and core regions in tRNAHis play critical roles in the initial binding/discrimination between cognate and noncognate tRNAs, whereas acceptor stem residues, particularly at identity position 73, influence the reaction after tRNA binding (340). Rapid kinetics using tRNAHis and HisRS mutants more precisely defined the functional role of the identity elements. Thus, mutations at identity positions in histidine anticodon preferentially affect the thermodynamics of initial complex formation; in contrast, mutations in the acceptor stem of tRNAHis or the conserved signature motif 2 in HisRS impose a specific kinetic block on aminoacyl transfer and decrease tRNA-mediated kinetic control of amino acid activation (334). As a second example, in the E. coli tryptophan system, stopped-flow fluorimetric investigations indicate that the identity determinant A73 of tRNATrp contributes to stabilization of the transition state during tryptophanyl-tRNATrp synthesis (341). In general, however, the functional relationship between identity determinants is poorly understood. For instance, the structural basis underlying the cooperative, additive, or anticooperative relationships of identity determinants in the aspartate system remains to be explicitly elucidated (342). Thus, pairs of determinants located far apart in the 3D structure of tRNAAsp behave cooperatively, while those clustered in the GUC anticodon act additively or anticooperatively. It is noteworthy that the strong anticooperative effect of the triple anticodon mutant CAU (with a global effect that is less severe than the sum of individual effects) can be explained by the loss of all tRNA contacts with the ACB domain of AspRS, making this mutant a mimic of an aspartate minihelix marked by the sole G73 determinant (316). A structural interpretation of additive or cooperative effects is less straightforward and implies transfer of chemical information from the anticodon region to the catalytic site. Only few such studies have been undertaken (see, e.g., another example in the glutamine system [343]). It can be anticipated that such long-range effects, such as those occurring between distant regions in the aaRS structure, are widespread (see below).
A last point of practical importance concerns incomplete charging (e.g., tRNA aminoacylation plateaus less than 100% that can be as low as ~1%), frequently observed with noncognate tRNAs (occurs also with cognate and mutant tRNAs, as well as in reactions catalyzed by mutant aaRSs). Incomplete tRNA charging is aaRS dependent, and this phenomenon remains puzzling for many researchers. In fact, it reflects the equilibrium between forward acylation and reverse deacylation and is linked to the fragility of the ester bond the amino acids make with the terminal ribose of tRNA (344). Thus, incomplete charging does not necessarily mean the presence of inactive tRNA molecules but mainly reflects the functioning of charging reactions with low acylation and high aminoacyl-tRNA deacylation rates.
Exit of charged tRNA from aaRSs
Early steady-state kinetic investigations that determined the rate-determining step for tRNA aminoacylation gave a phenomenological view on the exit of charged tRNA from aaRSs. No common picture emerged, and dissociation of charged tRNA from the enzyme is not always rate limiting as could be expected (345). The problem is still under debate, and it is not excluded that rate-limiting steps are modulated by experimental conditions. Nevertheless, more recent data on GluRS and GlnRS resulting from global approaches combining crystallographic, physicochemical, and functional analyses rejuvenated the question and brought structural understanding. Thus, for E. coli GlnRS, exit of charged tRNA was actually found to be rate limiting according to pre-steady-state kinetics that revealed a rapid burst of product formation followed by a slower linear increase of glutaminyl-tRNAGln formation. In addition, the data conclusively demonstrated the existence of long-distance pathways of communication through the GlnRS:tRNAGln complex (i.e., by allosteric phenomena; see below). In support of this assumption, mutation of U35 in the tRNAGln anticodon loop decreases the aminoacylation rate and weakens glutamine-binding affinity, indicating that the active-site configuration depends on enzyme-tRNA contacts ~40 Å apart (343). For T. thermophilus GluRS, a computational study examined factors affecting release of charged tRNAGlu (protonation states of amino acids and substrates present in the active site and the presence and absence of AMP and EF-Tu) that gave a more intricate picture of the exit mechanism of charged tRNA. Thus, distinct nonequilibrium posttransfer states were identified, and the undocking of AMP or charged tRNA was shown to proceed along thermodynamically competitive pathways. Release of the tRNA acceptor stem appeared to be further accelerated by the deprotonation of the α-ammonium group on the charging amino acid. It is noteworthy that the addition of EF-Tu to the aaRS:tRNA complex stimulated the dissociation of the tRNA core and the tRNA acceptor stem (346).
Structural and functional plasticity in aaRS:ligand systems—motions and allosteric phenomena
Disorders in early crystallographic structures of aaRSs (196) and mechanistic studies on the amino acid activation step (reviewed e.g., in references 4, 12, and 13) indicated enzyme flexibility and induced-fit mechanisms during catalysis. On the other hand, one can conjecture that aaRSs are allosteric enzymes since they are contacted outside their catalytic site by identity determinants of tRNA during the aminoacylation process (342, 347). This fact implies conformational rearrangements that would occur upon tRNA binding, as documented for classical allosteric enzymes when interacting with their allosteric effectors (see, e.g., reference 348 for a review). In the early phenomenological picture of the functioning of aaRS:tRNA systems, it was suggested that tRNA binding takes place through discrete kinetic-dependent steps involving scattered recognition sites, with best mutual adaptation of the two interactants in the cognate complexes (5). From the standpoint of allostery (and induced-fit phenomena), tRNA-triggered conformational changes in active aaRSs, as well, were soon suggested (5). However, despite intensive work, the allosteric effector role of aaRS ligands during the aminoacylation process remained elusive for years and it is only in the last decade that advanced physico-chemical, mutagenesis, and computational approaches brought new insight into the fine mechanistic understanding of these enzymes.
Structural plasticity in aaRSs and in their macromolecular tRNA ligands, and long-range domain-domain communication in their structures (263, 264, 349), are functional necessities to ensure the specificity of tRNA aminoacylation and other aaRS functions. Indeed, functional aaRS complexes must process the chemical information brought by the interaction of the small (amino acid, ATP, aminoacyl-adenylate, PPi, and Mg2+) and macromolecular (tRNA) substrates. Contacts with substrates necessarily occur in the catalytic site and also in remote domains, mainly in ACB domains located ~50 to 70 Å apart from the catalytic site and in editing domains (see “Error correction,” below). These contacts are accompanied by induced fit/allosteric phenomena. On the other hand, the fact that aaRSs are multidomain proteins implies the existence of communication between domains and of coupled domain motions. In other words, functional aaRS:tRNA complexes can be considered as “signal transduction” systems in which specific conformational changes occur, which can be subtle or dramatic (264, 350, 351). A few examples taken from bacterial systems illustrate the point.
Subtle conformational changes in aaRSs essential for tRNA aminoacylation are well documented in the aspartate system. Thus, the mutual adaptability of E. coli AspRS with its substrates is accompanied by flexibility in the N-terminal ACB domain and great mobility of the so-called flipping loop (that controls the proper positioning of aspartate) not seen in the crystal structure when tRNA is absent (165). Interestingly, all regions of AspRS that contact tRNAAsp show local flexibility in the apo-enzyme, which suggests that analyzing the flexibility of apo-aaRSs may be informative about tRNA binding (165). Other examples, based on functional and molecular dynamics methods, come from E. coli and M. smegmatis MetRSs, E. coli ThrRS, and B. stearothermophilus TrpRS. For the MetRSs, flexibility of the amino acid-binding pocket is required to accommodate methionine (121) and adenosine (158). Moreover, domain-domain communication and motion (352) as well as rigid-body rotation of the catalytic and ACB domains occur upon tRNA binding (347) in the E coli MetRS. For E. coli ThrRS, a study of the active-site dynamics during the aminoacyl transfer step, when threonyl-adenylate and tRNAThr are bound, explains the catalytic mechanism in which a histidine residue plays a key role (353). For B. stearothermophilus TrpRS, domain motion closes and twists the ACB and catalytic domains of the aaRS, with the ACB domain moving as a rigid-body with both catalytic HIGH and KMSKS class I signatures (with HIGH replaced by TIGN in B. stearothermophilus TrpRS) to deliver ATP to the tryptophan carboxyl group in the active site within the Rossmann fold (328) (see “Overview of the aaRS world,” above, for the meaning of the features defining class I aaRSs). In this process, ATP acts as an allosteric effector for TrpRS (199). Selection of the amino acid requires also domain motion in the catalytic domain triggered by a remote structural motif (354). Other allosteric effects are triggered by Mg2+ when the metal ion assists catalysis of tryptophan activation (329).
The crucial question, still unsolved, is how chemical signals in aaRS and tRNA (i.e., from identity elements) trigger specific aminoacylation. Phenomenological evidence clearly demonstrates that mutations in anticodon identity elements differentially affect catalytic efficiency of tRNA aminoacylation (342). On the other hand, crystallographic data indicate conformational changes in aaRS:tRNA complexes associated with different functional states of the aaRSs or generated by mutations in the aaRSs. Attempts to understand how signals are transduced from the aaRS anticodon domain to the catalytic site were approached by molecular dynamics simulations on E. coli MetRS (352, 355, 356). Four communication pathways between the active site and the ACB domain ~50 Å away were identified in the MetRS body (355). When comparing the intramolecular mobility of native MetRS with that of a variant deficient in aminoacylation after mutating Trp461 (a conserved residue in contact with the anticodon of cognate tRNAMet), significant differences were found. While native MetRS shows mobility in all motifs important for catalysis and correlated motions between distant residues (e.g., residues from the active site or the Zn-binding motif and residues from the ACB domain), mobility and correlated motions decrease significantly but not uniformly in the aminoacylation-deficient variant (352). Other simulations on the bacterial GluRS:tRNAGlu complex from T. thermophilus found intricate dynamical communication networks between identity elements of tRNAGlu and residues within the GluRS catalytic sites, with differences between pretransfer and posttransfer networks (357). The general conclusion is that residues within both GluRS and tRNAGlu are essential for information transduction. Unexpectedly, the allosteric networks in GluRS display considerable similarities with those in an archaeal LeuRS:tRNALeu complex (357) despite the remarkably different interactions GluRS (1n78) and LeuRS (2v0c) make with their cognate tRNAs (358, 359). In the case of the E. coli GlnRS:tRNAGln complex, pre-steady-state kinetics on GlnRS variants (with contact points with tRNA mutated) were used to discover allosteric signaling pathways in the aaRS body that would regulate glutamine binding and glutaminyl-tRNA formation. Interestingly, data suggest long-range signal propagation from the tRNA anticodon to the catalytic site and reveal protein contacts that weaken glutamine-binding affinity across distances up to 40 Å (360).
Coupled motions also occur between catalytic and editing domains of bacterial aaRSs. For instance, normal mode analyses in T. thermophilus LeuRS suggest that sparsely distributed amino acid clusters are critical for long-range mechanochemical motions in this enzyme (361). In bacterial-like E. coli ProRS, coupled dynamics occur between the INS region in the editing domain and protein segments containing the catalytically important proline-binding loop. Interestingly, it was suggested that multiple pathways are possible between the editing and catalytic domains and that the amino acids engaged in the motions are evolutionarily conserved and/or have co-evolved (362).
On the other hand, in the context of the multidomain architecture of aaRSs, it is worth mentioning the extreme case of structural mobility occurring in the GlnRS from D. radiodurans (2hz7). Indeed, this large aaRS (852 amino acids) contains a well-structured C-terminal extension of 215 residues (a paralog of Yqey proteins, the freestanding proteins of elusive function consisting of two different α-helical bundles) appending the ACB domain not seen at all in the 2.3-Å crystal structure of this GlnRS because of mobility in the crystal lattice (363, 364). The function of the Yqey appendix likely is to enhance the stability of the complex with tRNA by contacts in cis (363, 364). It is noteworthy that Yqey constitutes also the C-terminal domain of GatB, a subunit of the trimeric aminoacyl-tRNA amidotransferases, where it contacts the D-loop of misacylated tRNA. Because of the structural similarity with GatB (see “Alternative functions of bacterial aminoacyl-tRNA synthetases,” below), it can be suggested that Yqey proteins participate in amidation pathways.
In summary, the allosteric phenomena in the aaRS field that for a long time were based on speculation have received robust experimental support in the past decade. The existence of molecular communication pathways in aaRSs and aaRS:tRNA complexes is presently demonstrated for ∼15 aaRS specificities (reviewed in reference 351). Today the large body of results demonstrating the plasticity of aaRS structures and the structural mobility of their constitutive domains, together with thermodynamics informing about cooperativity and anticooperativity in aaRS reactions, provide the experimental background for further efforts to understand the physical basis of signal communication, in particular, between identity determinants in tRNA or aaRS and residues from the aaRS catalytic sites. This needs accurate analyses of both the local and extended networks of amino acids participating in allostery, as investigated e.g., for TrpRSs (365, 366). However, despite the recent breakthroughs, many open questions remain. For instance, can the conclusions claiming alternative communication networks, as seen e.g., in E. coli ProRS (362), be generalized to the ensemble of aaRSs or are system-specific idiosyncrasies possible? Likewise, the idea of evolutionary conserved amino acids in allosteric communication pathways needs clarification. Also better correlations between thermal fluctuations, as reflected by the crystallographic B-factors, and functional dynamics are needed (367). For that, more high resolution structures of aaRSs under different functional states are awaited, as well as more enzymologic investigations combined with enhanced computational methods for monitoring the mechanochemistry of these enzymes.
Error Correction
Amino acid misactivation and tRNA mischarging
Fidelity of translation results from the accuracy of three processes involving tRNA, namely, (i) tRNA aminoacylation (7), (ii) selection of aminoacylated tRNAs by initiation (368) and elongation (369) factors, and (iii) decoding of the genetic message on mRNA by tRNA anticodons (370). It has been estimated that the summed error frequency of these processes does not exceed 1/3,000 in vivo (371). Faithful tRNA aminoacylation depends primarily on the successful discrimination between cognate and noncognate amino acids and tRNA substrates. Inaccuracy in amino acid selection (10−4 to 10−5) is more frequent than tRNA selection (10−6) because of the larger surface area of the tRNA molecules and the resulting greater structural diversity of the contact regions. Half of the bacterial aaRSs (class Ia LeuRS, IleRS, ValRS, and MetRS and class II SerRS, ThrRS, ProRS, LysRS, AlaRS, and PheRS) misactivate noncognate amino acids that are similar in shape to their cognate substrates (Table 5), and many aaRSs catalyze tRNA mischarging (as a result of noncognate tRNA recognition followed by the attachment of the cognate amino acid) (37). Also worth emphasizing (and often overlooked) is the nonabsolute discrimination between L- and D-amino acids by aaRSs as first observed ~40 years ago in the case of E. coli and B. subtilis TyrRSs (372). Today, significant D-aminoacylation of tRNA has been characterized for E. coli AspRS, HisRS, LysRS, TrpRS, and TyrRS and was suggested for a few others (373, 374, 375, 376).
Table 5.
Error correction by bacterial editing aminoacyl-tRNA synthetasesa
| aaRS | Amino acids misactivatedb | Editing domainc | Pretransfer editing | Posttransfer editing | trans editing |
|---|---|---|---|---|---|
| Class I | |||||
| IleRS | V, T, C | CP1 | + d | + | No |
| ValRS | I, T, C, S, A | CP1 | + d | + | No |
| LeuRS | I, M, V, D, N | CP1 | + | + | No |
| MetRS | Only nonstandarde | Aminoacylation site | + d | ? | No |
| Class II | |||||
| SerRS | T, C | No separate editing domain | + | – | No |
| ThrRS | S, V | N2 subdomain or paralog | – f | + | + |
| ProRS | A, C | INS insertion or paralog | + | + | + |
| LysRS | M, L, C, A, T | Aminoacylation site | + | – | No |
| AlaRS | G, S, C | C-terminal domain or paralog | + d, g | + | + |
| PheRS | Y, I, L, M | B3/B4 subdomain | + | + | No |
Only standard amino acids are given; for amino acids in italics, the relative rate of activation is extremely low (38, 377).
Only mischarging with misactivated amino acids is given – note that most of the editing aaRSs also can misrecognize tRNA and catalyze mischarging with their cognate amino acid (mostly under particular conditions, either in vitro and in vivo, e.g., reference 37).
On the basis of increased rates of the ATP–PPi exchange reaction.
Homocysteine, norleucine, ethionine, selenomethionine (activated selenomethionine is transferred to tRNAMet and not edited, a useful property for crystallography since this allows production of selenomethionine-labeled proteins used for phasing by the Multiwavelength Anomalous Dispersion, or MAD, method [see references 378 and 379]).
A Zn2+ ion prevents misactivation of valine (isosteric with threonine) but does not prevent serine activation (see reference 380).
No definitive and direct structural proof.
Loose specificity was certainly advantageous in primitive aaRSs when chemical evolution established life on Earth because it generated diversity in an emerging protein world. It still remains a biological necessity in modern ND-aaRSs that catalyze tRNA mischarging in organisms lacking GlnRS or AsnRS (72). Unspecific tRNA aminoacylation became toxic when the genetic code and the translation machinery were fixed because high levels of mischarged tRNA would lead to coding ambiguity with codons translated by statistical distributions of amino acids rather than by the specific amino acid. Thus, aaRSs became more accurate and some, stepwise, acquired proofreading/editing capability to correct activation and aminoacylation errors. The implication in modern life is that defective corrections would produce cellular disorders. This conjecture was verified with E. coli cells harboring editing-defective aaRSs that show perturbed viability (381, 382) and, in the case of mammalian cells, editing-defective aaRSs (AlaRS, GlyRS, ValRS) that produce diverse cellular disorders, including human neurodegenerative pathologies (383, 384, 385).
A recurrent question concerns the functional specificity and distinction of editing-defective aaRSs, since some of them catalyze amino acid misactivation (e.g., in plant ArgRSs [386]) and/or tRNA mischarging (e.g., in ArgRS, GluRS, GlnRS of different origins [37]), while others achieve high amino acid specificity (e.g., in E. coli CysRS [387]). The point is of interest for CysRS and ArgRS that both belong to the same structure-based group of aaRSs (class Ia) as the editing IleRS, ValRS, LeuRS, and MetRS. It concerns also class Ib and class IIb aaRSs that recognize structurally related amino acids (note that ND-GluRSs and ND-GlnRSs are mischarging enzymes). For these aaRSs, three mechanisms were shaped by evolution to achieve the specificity level compatible with fidelity of protein synthesis: (i) kinetic driven specificity with high catalytic discrimination between the correct and the incorrect reactions (this concerns essentially tRNA mischarging [37] but also misactivation followed by tRNA charging of the false amino acid, as observed with plant ArgRSs [386]), (ii) tRNA-induced remodeling of the catalytic site for enhanced amino acid binding (see below), and (iii) tuning a proper balance of tRNA and aaRS concentrations (388). The direct implication is cellular toxicity in the case of perturbed homeostasis due to elevated aaRS concentrations or, in other words, high levels of mischarged tRNAs that produce errors in protein synthesis (often observed when overproducing aaRS genes).
Editing by deacylases
Since certain aaRSs can mischarge their cognate tRNA with D-amino acids (see above), it was anticipated that cells should be equipped with enzymes capable to clear the D-aminoacyl-tRNAs. D-Tyrosyl-tRNATyr deacylase is the enzyme capable of fulfilling this task (372). It has wide amino acid specificity, and the deletion of its gene is toxic for E. coli as shown by an accumulation of intracellular D-tyrosyl-tRNA (373, 375). Distribution of this dimeric tRNA-dependent deacylase is universal in Bacteria and Eukarya (376). (B. subtilis is an exception and lacks a gene encoding a functional D-aminoacyl-tRNA deacylase; see “Regulation in aminoacyl-tRNA synthetase gene expression in E. coli,” below, for details). The crystal structure of the E. coli D-tyrosyl-tRNATyr deacylase (1jke) reveals a β-barrel architecture, assembled together with its two subunits, that is closed on one side by a β-sheet lid (373). The ortholog from Haemophilus influenzae (1j7g) shows the same structural organization (389). The active site, at the interface of the two subunits, accounts for the broad editing specificity that likely occurs by a conserved mechanism in D-tyrosyl-tRNATyr deacylases (390). It is noteworthy that a D-aminoacyl-tRNA-like domain of a structure similar to the E. coli deacylase was added in the course of evolution to most archaeal ThrRSs for posttransfer editing of mischarged seryl-tRNAThr (118, 373, 391, 392). This domain differs from the freestanding editing domain in Crenarchaea, such as S. solfataricus, that hydrolyzes seryl-tRNAThr (116).
Editing by aaRSs
General concepts: To ensure the viability of organisms, cellular aaRS expression is generally regulated by growth rates to keep levels of tRNA misrecognition and hence mischarging low (393) (see “Regulation of aminoacyl-tRNA synthetase gene expression in E. coli,” below for details). Correction mechanisms that clear mistakes before mischarged L-amino acids would be misincorporated into proteins therefore enhance accuracy of translation. First indications on corrections mediated by IleRS, ValRS, or PheRS came from observations on aaRS-dependent deacylation of correctly or incorrectly charged tRNAs (344, 394, 395, 396) and led to the proposal of “kinetic proofreading” (174) and the generalization of “chemical proofreading” in the prevention of tRNA mischarging (397). Altogether, 10 editing aaRSs act in Bacteria (Table 5).
A realistic “double-sieve” model rationalized the phenomenology of editing (398). This is a “steric-exclusion” mechanism that received strong structural support with the discovery of a separate editing site in many aaRSs distinct from the catalytic site ensuring tRNA aminoacylation. Thus, a “coarse sieve” corresponds to the catalytic/synthetic site and would reject the amino acids that are larger than the cognate one but would bind smaller and isosteric amino acids. A second “fine sieve” corresponds to a distinct editing site that would hydrolyze the noncognate amino acids that were misactivated (pretransfer editing) or mischarged on tRNA (posttransfer editing). This scheme was explicitly visualized by crystal structures of class I T. thermophilus IleRS (1ile) in complex with L-isoleucine or L-valine (399) and of S. aureus IleRS in complex with tRNAIle and mupirocin (an analog of isoleucyl-adenylate; see “Inhibition and engineering of bacterial aminoacyl-tRNA synthetases,” below, for details) (1qu2) (138) in conjunction with functional data on E. coli IleRS (400, 401). As a result, the misactivated adenylate or the mischarged acceptor strand of tRNA bound to editing IleRS via its anticodon stem-loop have to shuttle between the catalytic/synthetic and editing sites ~30 Å apart. This scheme is also valid for ValRS and LeuRS. The situation is more intricate for MetRS and class II editing aaRSs in which the “double-sieve” model does not apply stricto sensu because amino acids larger than or dissimilar to the correct ones can be misactivated, transferred to tRNA, and cleaved. Although posttransfer editing domains distinct from the synthetic domains have been characterized in most class II editing aaRSs, editing by these enzymes proceeds by different mechanisms and can even be catalyzed in trans by freestanding editing proteins. This was first described for YbaK, a homolog to the editing domain of H. influenzae ProRS (299) followed by AlaX homologous to that of archaeal AlaRSs (402) and led to the proposal of a “triple-sieve” mechanism with the third sieve being the separate protein (403). The present understanding of the diverse aaRS-mediated editing types (Table 5) is based on extensive biochemical, genetic, and structural data that are covered in several reviews (248, 325, 404, 405, 406). The prominent and newest facts are summarized below.
Pretransfer editing: This editing mode is difficult to demonstrate because of the connected mechanistic features not easy to separate by experiment, such as the inherent lability of aminoacyl-adenylates, tRNA dependence, and occurrence of this editing possibility in posttransfer editing background (e.g., with GlnRS [407], IleRS [401, 408, 409, 410], LeuRS [297, 411], and ValRS [410, 412]). For E. coli LeuRS with posttransfer editing capability, activity of variants with either point mutations in CP1 or entire CP1 resected suggests a pretransfer translocation step that moves misactivated adenylates from the activation site to that for editing (413) and supports the conclusion that a latent pretransfer editing mechanism is activated upon deletion of CP1 (the large connective peptide inserted in the catalytic domain of LeuRS) (414). In A. aeolicus LeuRS pretransfer editing is favored (411), while in an editing-defective enzyme mutated in CP1 a tRNA-independent editing activity likely occurs in the synthetic site (415). This mechanism is exclusively used in Mycoplasma pathogens whose LeuRSs are missing or contain a degenerate CP1 domain (416). For E. coli ValRS, explicit evidence of pretransfer editing is lacking, although mutational analysis revealed a direct relationship between the ability of a tRNA to be valylated and its ability to stimulate editing activity (412). In support to pretransfer editing, molecular dynamics simulations indicate that noncognate threonyl-AMP and threonyl-A76 substrates bind more strongly than cognate valyl-AMP and valyl-A76 in both pre- and posttransfer editing sites of T. thermophilus ValRS and that mutations in the CP1 domain decrease the binding ability of the pretransfer threonyl-AMP substrate (417). For E. coli IleRS, novel kinetic approaches show a tRNA-dependent hydrolysis of valyl-adenylate that is largely insensitive to mutations in the CP1 editing domain and a pretransfer editing in IleRS likely residing in its catalytic/synthetic site, suggesting a kinetically controlled partitioning of the noncognate aminoacyl-adenylate between the editing and synthetic sites (418). This result indicates that both pre- and posttransfer editing are important in IleRS, in contrast to ValRS and LeuRS, where editing occurs nearly exclusively by posttransfer hydrolysis in the editing domain (418, 419). Finally, computational methods applied to A. aeolicus MetRS, strongly suggest existence of substrate-assisted pretransfer editing of homocysteine and homoserine in which the carboxylate from an aspartate residue (Asp259) in the active site acts as a base in the hydrolytic mechanism (420). This mechanism may occur in IleRS, LeuRS, and ValRS since similarly located aspartate or glutamate residues are found in the synthetic site of these aaRSs (420).
Pretransfer editing in CP1-containing aaRSs (IleRS, LeuRS, MetRS, ValRS) probably is ancient and would have been present in primitive aaRSs before addition of the CP1 domain. In modern aaRSs, however, it is not the major route and, as seen in A. aeolicus and E. coli LeuRS, represents only 5%, the remaining 95% being CP1-dependent posttransfer editing (415, 419). Importantly, this conclusion concerns also norvaline, a nonstandard amino acid naturally found in Bacteria that is efficiently misactivated by A. aeolicus LeuRS and edited in the posttransfer pathway (415).
The case of E. coli GlnRS, a nonediting class I aaRS, deserves a special comment since this enzyme hydrolyzes glutaminyl-adenylate by a tRNA-dependent mechanism. This hydrolysis is analogous to pretransfer editing of noncognate aminoacyl-adenylates by editing aaRSs such as IleRS. Because GlnRS does not possess a spatially separate editing domain, this absence shows that a pretransfer editing-like reaction can occur within a class I catalytic site (407).
Pretransfer editing is also found in class II aaRSs. Thus E. coli ProRS misactivates alanine and hydrolyzes noncognate alanyl-adenylate before alanine transfer to tRNAPro (421), likely within the prolylation active site (422). LysRS as well uses pretransfer editing with methionine, leucine, cysteine, alanine, threonine, and a few nonstandard amino acids (377). The situation in ThrRS is peculiar since valine, isosteric with threonine, is expected to be misactivated but is not. This is explained by the presence of a Zn2+ ion in the active site that participates in threonine recognition and prevents binding of the methyl group of valine (380). Thus, the Zn2+ ion acts as the “first sieve” against valine. ThrRS, however, activates serine, but there is no evidence of pretransfer editing of seryl-adenylate (423).
The editing abilities of MetRS and class II LysRS are unique for different reasons. MetRS misactivates only nonstandard amino acids, preferentially homocysteine, the cellular precursor of methionine, but also unnatural norleucine, selenomethionine, and even telluromethionine that can be incorporated in proteins under nonphysiological conditions (38, 424). Editing of homocysteinyl-adenylate involves a cyclized homocysteine thiolactone intermediate and occurs within the MetRS synthetic site that appears partitioned for aminoacylation and editing activities. The case of LysRS is a priori intriguing since, besides efficiently misactivating ornithine and a series of standard amino acids (although less efficiently), it mischarges tRNALys with these noncognate amino acids (377). Ornithine is readily edited via a cyclization pathway, so that the charging extent of tRNALys is less than 1%. Mischarging with standard amino acids can reach plateau levels of 90% (e.g., for arginyl-tRNALys) and is quasi-abolished when aminoacylations are conducted in the presence of lysine (377) and thus is tolerated for cellular life.
Finally, the pretransfer editing status of PheRS remains puzzling since it has to select between phenylalanine and larger tyrosine by a mechanism differing from the “double-sieve” model. Explicit evidence for tyrosine discrimination came from fast kinetic data obtained with yeast PheRS (425) and was extended to E. coli PheRS (426). The observation of PheRS-dependent tyrosyl-adenylate pyrophosphorolysis (425) suggests a kinetically driven process that could occur within the catalytic site, an interpretation consistent with the plasticity of the catalytic site seen in crystal structures of T. thermophilus PheRS in complex with substrates larger than phenylalanine (2akw, 2aly) (233). As for LeuRS (see below), this editing activity likely is a remnant of the early evolution of PheRS that acquired later posttransfer editing capacity.
Posttransfer editing: Occurrence of posttransfer editing is documented for seven aaRSs (class I IleRS, LeuRS, ValRS and class II AlaRS, PheRS, ProRS, ThrRS) and relies on specialized editing sites different from the synthetic aminoacylation sites. This necessitates motion of tRNA on the aaRSs. In class I enzymes, the flexible 3′-end of the misacylated tRNA is translocated from the aminoacylation site to the hydrolytic site on their appended CP1 domains, implying that tRNA interactions are partially distinct in the two catalytic steps, as illustrated by a tRNA-dependent translocation of ∼25 Å of misactivated valine on E. coli IleRS (409). This translocation of the tRNA amino acid accepting end is accompanied by a clear segregation of nucleotide determinants for the editing and aminoacylation functions of tRNA. In class II aaRSs, the situation is more intricate since different types of editing domains, and hence different editing mechanisms, have been identified and that editing can be achieved in trans by helper proteins.
In closely related IleRS and LeuRS, posttransfer editing likely occurs by a similar mechanism. This conclusion is supported by the presence in their homologous CP1 editing domains of a conserved threonine-rich peptide and of a second conserved region separated by ~100 amino acids that make up each part of the editing site in both E. coli enzymes (427). However, some idiosyncratic positions in LeuRS and/or IleRS account for specific substrate recognition (427, 428). Crystallography and computational analysis suggest that during the editing pathway the CP1 domain rotates via its flexible β-strand linker relative to the main aaRS body with the end of the N-terminal β-strand acting as a hinge. From the viewpoint of physiology, the vital importance of editing is illustrated by the toxicity of many amino acids in E. coli when LeuRS editing is inactivated (429). From the viewpoint of structure, two complexes of T. thermophilus LeuRS with analogs of pre- or posttransfer editing substrate show binding of the pretransfer editing analog in both the catalytic/synthetic and editing active sites (1obh), while the binding of the mischarged tRNA posttransfer editing analog occurs solely in the editing site (1obc) (430). Moreover, LeuRS structure of an isolated CP1 editing domain of A. aeolicus (3pz6) (431) and mutagenesis of the C-terminal domain of M. tuberculosis LeuRS indicate the importance of these two domains in regulating quality control of leucyl-tRNALeu synthesis (432). Most interesting, crystal structures of E. coli LeuRS in the aminoacylation (4arc) and editing conformation (4aq7) show correlated rotations of four flexibly linked LeuRS domains and the unexpected role of the editing domain that stabilizes tRNA during aminoacylation (433). Besides the conserved CP1 editing domain, the related IleRS, LeuRS, and ValRS contain, in addition, a bipartite CP2 domain that is spatially close to the acceptor stem of tRNA (137, 138, 139) and thus could play a role in editing. Studies with engineered LeuRS molecules bring support to this expectation and show that CP2 indeed is needed for posttransfer editing and leucine activation (434). In addition, CP2 helps correct orientation of the mischarged tRNAs into the CP1 editing site since deletion mutants missing CP2 are editing defective (434).
Note the unusual editing properties of deep-rooted A. aeolicus LeuRS that is capable of editing the complete set of mischarged tRNAs that can be generated by LeuRS, IleRS, and ValRS (307). Interestingly, this conclusion comes from studies on the editing of charged or mischarged RNA minihelices with triple leucine, isoleucine, and valine identity, suggesting that the editing domain of A. aeolicus LeuRS preserved properties of an ancestral editing domain (307).
Posttransfer editing used by class II aaRSs follows different routes and appears fundamentally different from the editing of class I enzymes. In E. coli ProRS, it is the 180-amino-acid-long INS (INSertion) domain located between class II signature motif 2 and 3 that catalyzes deacylation of mischarged alanyl- or cysteinyl-tRNAPro (435), with the single Cys443 residue within motif 3 (dispensable for ProRS aminoacylation activity) critical for the hydrolytic editing of alanyl-tRNAPro (421). It is noteworthy that the INS domain can act as an independent protein (299) and finds paralogs in nature (403). Posttransfer editing by ThrRS and AlaRS deserves particular attention because of intriguing similarities, but also peculiarities, that have their origin in the different evolutionary histories of the two aaRSs. Thus, both bacterial enzymes have structurally related editing sites on which the overall catalytic domain is fused C-terminally in ThrRS and N-terminally in AlaRS (401). More precisely, in E. coli ThrRS, posttransfer editing is associated with a N-terminal domain and is independent from aminoacylation since a resected variant missing this domain can misacylate tRNAThr with serine, but is unable to hydrolyze seryl-tRNAThr. The editing site is located in a cleft of subdomain N2 and is triggered by conserved amino acids (Asp180, His73, His77) in the bacterial, yeast, and human ThrRSs (423). This editing domain found in Bacteria and Eukarya is absent in Archaea, in contrast to the similar AlaRS editing domain present in the three branches of the tree of life (207).
The case of AlaRS is paradoxical since glycine and serine, sterically smaller and larger than alanine, are both misactivated. Editing of glycyl-tRNAAla is in line with the idea that the aaRS sieves out amino acids that are smaller than alanine, but the editing of seryl-tRNAAla does not fit this scheme. The crystal structures of an active fragment of A. aeolicus AlaRS in complex, separately, with Mg2+/ATP, alanine, glycine, or serine shed light on the paradox: while alanine and glycine are bound in similar orientations in a pocket where they are stabilized by salt bridges and H-bonding with class II conserved Asn194, serine in contrast forces pocket expansion and has its binding reinforced by that of Mg2+/ATP in the active site and by coordination with Asn194, thereby breaking the sieve for serine exclusion (134). On the other hand, serine exclusion could involve participation of the AlaXp protein (a trans-editing protein homologous to the editing domain of AlaRS) widely represented in Bacteria (402). Importantly, the small C-terminal C-Ala domain universally tethered to the editing domain promotes cooperative binding of both AlaRS aminoacylation and editing domains to tRNAAla (436).
Biological necessity of posttransfer editing of tyrosyl-tRNAPhe is well illustrated by tyrosine incorporation into proteins at phenylalanine positions when an editing-defective PheRS is overproduced in vivo (437). Its mechanism in Bacteria is partly deciphered based on structural and functional results (233, 438, 439, 440). Thus, the posttransfer editing site is localized within the large β-subunit of PheRS at the B3/B4 interface (B3 and B4 are two of the eight subdomains constituting the β-subunit) (439) ~35 Å apart from the catalytic site on the small α-subunit. Importantly, binding of tyrosine in the editing site is seen by crystallography (2amc) (233). Moreover, the activity of deletion mutants indicates that the adjacent B2 subdomain on the β-subunit, similar to an EMAP-fold with tRNA-binding capacity in other aaRSs, acts as a secondary tRNA-binding site that could contribute to editing by promoting the translocation of mischarged tRNA to the editing site (440). Note that the EMAP-fold, also present in MetRSs and a few eukaryal aaRSs, is a distinct version of the OB-fold. Its acronym derived from “Endothelial Monocyte Activating Polypeptide” reminds that the fold was discovered in a cytokine bearing no structural resemblance with other cytokines but with aaRSs, thus accounting for the cytokine activity of fragments of eukaryal aaRSs or of proteins associated with aaRSs such as p43 or Arc1p (10). Finally, a new crystal structure of T. thermophilus PheRS complexed with puromycin (4tva) and molecular mechanics calculations revealed the architecture of the enzyme liganded with a mimic of the tyrosyl-A76 moiety of misacylated tRNAPhe and allowed to propose a universal hydrolytic editing mechanism (441).
Relative contributions of pre- and posttransfer editing: The relative contribution of both editing modes in bacterial aaRSs has been evaluated for class I LeuRS, IleRS, and ValRS and class II ThrRS. It is noteworthy that, in T. thermophilus LeuRS, the editing CP1 domain binds the pre- and posttransfer substrates in largely overlapping sites, suggesting a similar mechanism of hydrolysis for both editing substrates (430). As a result, the contributions of the two editing modes of E. coli LeuRS are similar, but, for reasons not yet understood, the posttransfer editing mode is disfavored (29%) by the A. aeolicus LeuRS (411). Furthermore, in either E. coli or A. aeolicus LeuRS, preediting is low and does not exceed 10% of the total editing activity (411). In contrast, tRNA-dependent pretransfer editing reaches 35% in E. coli LeuRS and up to 65% in A. aeolicus LeuRS and appears to depend on the interaction of the tRNA acceptor end with the CP1 editing domain (411). For E. coli IleRS and ValRS, kinetic effects and the terminal A76 of tRNA control partitioning of pre- and posttransfer editing (418, 442). For E. coli ThrRS, it is the rate of aminoacyl transfer that modulates the relative contribution of pre- and posttransfer editing, and pretransfer hydrolysis of seryl-tRNAThr contributes to editing only when the rate of transfer is low (332).
Motion and plasticity of editing domains: Dramatic structural changes occur in editing systems and imply movement of ~30 Å of the accepting end of tRNA during translocation of mischarged tRNAs from the aminoacylation site to the hydrolytic editing sites. This translocation is transient and its molecular understanding remains elusive, although it is likely accompanied by conformational changes in the whole body of both RNA and protein. In E. coli IleRS it utilizes two amino acids from the hinge region between the core of the enzyme and the separate editing domain (401). In E. coli LeuRS it is a peptide within the editing domain (443) and a tRNA-dependent communication during pretransfer editing that adjust the conformational rearrangements at the disordered interface between the synthetic and editing domains (444). As for ThrRS, the bipartite architecture of its subunits (with the C-terminal core comprising the catalytic domain connected by a linker helix of 18 residues to the N-terminal domain with the N2 editing site) makes the whole protein flexible as revealed by different crystal structures of S. aureus and E. coli ThrRSs. In particular, variations of the relative orientation of the two ThrRS parts could be seen as well as four mobile regions participating in tRNA binding (210). This overall flexibility of ThrRS most likely facilitates the needed shuttling of the mischarged CCA end of tRNAThr from the catalytic site to the N2 editing site.
Editing polypeptides resected from bacterial aaRSs
The CP1 editing domain (210 to 275 amino acids) from IleRS, LeuRS, and ValRS, and its smaller version in the other class Ia aaRSs, can be resected from the host proteins. Its structural core with a conserved hydrolysis site shows distinct archaeal, bacterial, or eukaryal insertions (2wfe) (445). Active CP1 domains were derived from IleRS, LeuRS, and ValRS. The crystal structure of CP1 from E. coli LeuRS was solved in three versions, either in apo-form (2ajg) or in complexes with methionine (2ajh) and isoleucine (2aji). The structures show a rigid binding pocket formed by conserved amino acids that is best adapted to recognize isoleucine and methionine, but not leucine (446). Likewise, the structure of the fragment from T. thermophilus IleRS was solved in apo-form (1udz) or in complex with valine (1ue0) or adenylate analogs mimicking the substrates in pre- and postediting states (1wnz, 1wk8) (447, 448). Resolution in the 1.6- to 2.0-Å range visualizes the editing site large enough to accommodate valine but not isoleucine. Isolated CP1 domains from E. coli IleRS and B. stearothermophilus ValRS hydrolyze valyl-tRNAIle and threonyl-tRNAVal, respectively, but not correctly charged tRNAs. In contrast, the smaller CP1 insertions of MetRS and CysRS (100 and 50 amino acids, respectively) have no deacylation activity (310).
For E. coli LeuRS, early data suggested an inability of the isolated CP1 domain to catalyze editing (449). However, more recent data obtained with rationally designed E. coli CP1 fragments show that the β-strands, which link the domain to the aminoacylation catalytic core, are required for editing activity (450), thus explaining the apparently contradictory results obtained with domains missing these linkers (449). Activity of the freestanding CP1 domain from A. aeolicus αβ-LeuRS was straightforwardly found using mischarged isoleucyl-tRNALeu and isoleucyl-minihelixLeu. Interestingly, the short β-stranded peptide (20 amino acids) from this freestanding CP1 domain was shown to confer editing capacity after transplantation into the inactive CP1 domain from E. coli LeuRS. Likewise, fusion of the β-subunit (with the tRNA-binding domain) of the αβ-LeuRS to the E. coli editing domain activates its editing function (300). Noticeably, a minihelixLeu with triple leucine, isoleucine, and valine identity, mischarged with isoleucine, valine, or threonine, is edited by the separate CP1 domain from A. aeolicus LeuRS. This contrasts with the editing capacity of the freestanding CP1 domains from IleRS and ValRS that cannot clear mischarged minihelices (307). Therefore, it was proposed that the editing domain from A. aeolicus LeuRS has preserved ambiguous properties from an ancestral editing domain (307).
Editing domains resected from bacterial class II aaRSs (ProRS and AlaRS) have been produced as independent proteins with hydrolytic activity. Thus, an isolated INS domain (a large insertion of ~180 amino acids in the catalytic domain) from E. coli ProRS, when expressed as an independent protein, deacylates mischarged alanyl-microhelixPro (299). In the alanine case, editing of seryl-tRNAAla by resected E. coli AlaRS (438 to 875 fragment) depends on the C-terminal domain (731 to 875) linked to the inactive editing core (438 to 730) (451).
Autonomous bacterial trans-editing proteins
trans-editing proteins homologous to the editing domains inserted into ProRS, AlaRS, and ThrRS have been discovered in many genomes from the three kingdoms of life. When expressed as freestanding proteins, they are able to hydrolyze mischarged alanyl-tRNAPro (299, 402, 403), seryl-tRNAAla (402), or seryl-tRNAThr (115). In Bacteria, autonomous proteins complete the editing action of ProRS and AlaRS and in some organisms compensate for the lack of or poor editing activity of their ProRS or AlaRS that have lost or not acquired the appropriate editing domain. To date, three bacterial trans-editing proteins have been structurally and functionally characterized, namely, YbaK, ProX, and AlaX.
YbaK and ProX proteins have sequence similarity with ProRSs and are widespread in the three kingdoms of life. The crystal structure of H. influenzae YbaK (158 amino acids) solved at 1.8-Å resolution (1dbu, 1dbx) shows a unique globular fold made of a seven-stranded β-sheet surrounded by six short helices (452). A similar fold exists in E. coli ProX (2dxa) (unpublished, from RIKEN Structural Genomics Initiative). YbaK/ProX proteins are aminoacyl-tRNA deacylases with preferential deacylation activity of cysteinyl-tRNAPro (453, 454). They are paralogs of the editing domain inserted into the catalytic core of most ProRSs and catalyze trans-editing in α-Proteobacteria and the higher Eukarya coding for ProRSs missing the internal editing domain (211, 402). A bioinformatics analysis revealed that, in addition to INS and YbaK, there are four other INS-like domains throughout the bacterial kingdom (ProXp-ala, ProXp-x, ProXp-y, and ProXp-z, with the last three of unknown function), as e.g., in Caulobacter crescentus where YbaK and ProXp-ala edit cysteinyl- and alanyl-tRNAPro (455), but without tRNA specificity for YbaK (456). Deletion screens, together with in vitro deacylation assays, showed that the ProXp-x/y/z trans-editing factors recognize multiple tRNAs and prevent mistranslation errors caused by a number of aaRSs, including AlaRS, LysRS, SerRS, and ThrRS (457).
AlaX proteins from Archaea have autonomous editing activity. They are structural homologs of AlaRS editing domains with an architectural organization comprising a N-terminal domain with a glycine-rich motif and a C-terminal domain with two Zn-binding motifs (1wnu, 1v4p, 2e1b) (208, 458, 459). A related AlaXp protein is widely distributed in Bacteria and Eukarya (402). Its serine-editing activity could be demonstrated in vivo by using E. coli cells harboring an editing-defective AlaRS unable to clear seryl-tRNAAla and thus showing serine toxicity that could be rescued by an AlaXp-encoding transgene (460). Likewise, artificial recombinant fragments resected from E. coli AlaRS show hydrolytic editing of seryl-tRNAAla or glycyl-tRNAAla. Furthermore, a fragment of ~110 amino acids from A. aeolicus AlaRS corresponding to the C-Ala domain (the C-terminal domain universally tethered to the editing domain) located N-terminally in AlaRSs and forming a stable crystallizable structure, acts in trans in seryl-tRNAAla editing when tethered to the AlaXp-like editing domain of AlaRSs by a linker of α-helical coiled-coil architecture (3g98) (436). Interestingly, isolated C-Ala binds to the elbow formed by the D- and T-loops of tRNAAla, indicating that the L-shape of tRNA is required for editing and thus explaining why mischarged truncated tRNAAla molecules missing the elbow are not edited (306). Taken together, the accepting branch of tRNA interacts cooperatively with the flexibly tethered editing/C-Ala structure. Binding capacity of C-Ala to tRNA is explained by the resemblance of the distal portion of C-Ala with the DNA binding motif of RecJ DNA repair exonucleases (well conserved in Bacteria). Thus, C-Ala appears to be an ancient single-stranded nucleic acid binding motif that may have played a role in the evolution of AlaRS by coupling aminoacylation to editing (436). On the other hand, a mutational analysis examined the activity of the P. horikoshii AlaX editing domain with seryl-tRNAAla and alanyl-tRNAAla as substrates. Results show that wild-type AlaX preferentially deacylates seryl-tRNAAla but only ~12-fold faster than alanyl-tRNAAla. The impact of mutations was rather limited on seryl-tRNAAla deacylation that was decreased at most ~10-fold and even slightly reversed for two mutants that became more active on alanyl-tRNAAla than on seryl-tRNAAla. This indicates a relatively modest specificity of the AlaRS editing domain and may account for the widespread phylogenetic distribution of AlaX freestanding editing domains, thereby contributing a further mechanism to lower concentrations of misacylated tRNAAla (461). However, it was shown that a single residue in AlaX-S, the smallest of extant AlaX enzymes, is important to determine the deacylation of seryl-tRNAThr and seryl-tRNAAla, suggesting that the AlaX domain was used to maintain translational fidelity in the earlier stages of genetic code evolution when mis-serylation of tRNAs was frequent (462).
Editing determinants
Editing determinants in tRNA have a diverse nature and are less known than aminoacylation determinants. In the alanine system, the tRNA itself is a major editing determinant (306). In the other systems, determinants are discrete nucleosides and they could even be atomic groups from these nucleosides. Thus, in E. coli tRNAIle, major determinants are located in the corner of its L-shape (463), in contrast to aminoacylation where they are found in the anticodon triplet (304), demonstrating a segregation of the signals for editing and aminoacylation. In A. aeolicus tRNALeu, editing determinants are located in the anticodon arm (464), while in the E. coli valine system, atomic mutagenesis suggests that the unprotonated N1 atom in tRNAVal 3′-terminal A76 is a determinant for both editing and aminoacylation (465). However, most of these data remain incomplete and more work is required to define complete sets of editing determinants in the tRNAs concerned. Importantly, for E. coli AlaRS it could be conclusively shown that editing of seryl-tRNAAla is sensitive to the G3•U70 pair that is also the identity determinant for tRNA alanylation (451). This result indicates that two distinct domains in AlaRS can recognize the same base pair in tRNA. A significant breakthrough is the recent characterization of editing determinants in mischarged alanyl- and cysteinyl-tRNAPro that are recognized by the freestanding bacterial trans-editing INS and ProXp-ala proteins: these determinants are the same (G72/A73 and G35/C36) as those recognized by ProRS for aminoacylation (456).
On the protein side, editing signature motifs and determinants have been identified in a few aaRSs. Thus, two conserved HxxxH and CxxxH Zn-binding motifs in the editing domains of E. coli ThrRS and AlaRS dictate amino acid binding and rejection (207, 423). In several E. coli aaRSs, conserved amino acids act as editing determinants, such as (i) Thr252 in the CP1 editing site of LeuRS (466), (ii) Lys279 in the INS domain of ProRS that acts indirectly in alanyl-tRNAPro hydrolysis (467), and (iii) highly conserved Tyr59 in the synthetic site of IleRS that modulates both posttransfer editing and aminoacylation (468).
Additional corrections by elongation factor
Some mischarged tRNAs can escape editing and are released by aaRSs. Consequently they can associate with elongation factor and be conveyed to the ribosome for protein synthesis. Importantly, in view of accuracy, the association with elongation factor is dynamic so that the mischarged species can rebind with aaRSs, allowing resampling by the product-editing pathway. As a result, accuracy of translation mediated by elongation factor was found increased over 10-fold in vitro, supporting the existence of an additional quality control step before translation (469).
Considerations on evolution
Mutations create diversity in macromolecules and are a major driving force in evolution. In such a view, the aaRSs are important actors in producing diversity in proteins because of their rather loose specificity. During chemical evolution toward life, proto-aaRSs likely were in limited number and had minimalist structures restricted to catalytic domains with poor specificity. When the protein synthesis machinery was fixed, the need for diversity declined and the catalytic site of aaRSs became more selective for amino acid recognition. But errorless recognition of amino acids was not possible for amino acids too closely related because of chemical constraints as prophesied by Pauling (246). Therefore, evolution had to invent editing strategies. Although not explicitly proven, this scenario finds support from the phylogenetic analysis of aaRS structures and by the characterization in aaRSs from contemporary organisms of relics of ancestral features. This is the case of redundant editing pathways in LeuRS or ProRS and of different types of posttransfer mechanisms that have evolved independently. This is reflected by the presence in extant aaRSs of different and unrelated internal editing domains and even of freestanding editing domains.
In another perspective, the addition of new amino acids into the early genetic repertoire, such as asparagine, glutamine, and pyrrolysine, was certainly related with mutational tinkering in the aaRS world. A consequence of this tinkering is the structural resemblance of the aaRSs specifying the new amino acids with older aaRSs. This is well seen when comparing the structure of AsnRS or GlnRS with, respectively, AspRS or GluRS. Alternatively, the recent understanding of the mechanisms ensuring specificity and specificity switches in the aaRSs has found unprecedented applications with the design of orthogonal aaRS/tRNA pairs for reprogramming the genetic code and for incorporating unnatural amino acids in proteins (470) (see “Inhibition and engineering of bacterial aminoacyl-tRNA synthetases,” below, for details).
To conclude, it is tempting to propose that the structural complexity of extant aaRSs, together with their rather loose specificity but with error correction potential, are biological necessities that protect these macromolecules against concerted mutational effects that would generate variant aaRSs perturbing the accuracy of protein synthesis and, in the worst case, the universality of the genetic code. Thus, structure and function of aaRSs reflect evolution in action in maintaining an exquisite balance between error and specificity.
Truncated aaRSs
The activity of truncated aaRSs provide strong evidence supporting the modular architecture of these enzymes and informs about the evolutionary history of aaRSs and about their mechanism of action. The first known example of a functional fragment was a truncated E. coli MetRS obtained by mild proteolysis of the native homodimeric (2x676 amino acids) enzyme (168). This truncated enzyme, deprived of its C-terminal domain involved in dimerization, therefore is monomeric (551 amino acids) but fully active. On the other hand, the crystal structure of the isolated C-terminal domain of dimeric MetRS from P. abyssi (1mkh) (471) has an α-helix bundle architecture and resembles that of Trbp111, a dimeric tRNA-binding protein found in many Bacteria and Archaea. This domain displays nonspecific tRNA-binding properties, and functional assays on E. coli MetRS variants show that the presence of the appended C-domain improves binding affinity of tRNAMet. Because the oligomeric state of MetRSs is species dependent, and the monomeric forms (present in the three kingdoms of life) display the shortest sequences, it can be concluded that ancient MetRSs were short monomers.
The other historically important example of active versions of a truncated aaRS came from studies on tetrameric α4-AlaRS and brought the first explicit demonstration of aaRS modularity. Based on structural and functional results, alanine activation, tRNA charging and editing activities were found to depend on the size of the truncature. While the smallest fragment by itself activates alanine (53), aminoacylation, editing, and oligomerization require additional domains (50, 135, 451) (2zvf). Interestingly, these active fragments are monomeric, having lost the oligomerization domain, suggesting, in addition, that oligomerization (as for MetRS) is not essential for enzymatic activity. These observations suggest that the ancestral enzyme was restricted to the active site domain and that extant AlaRSs emerged after simple peptide fusions. This view finds robust support by the alanylation capacity of artificial AlaRS fusions (472) making up the E. coli alanyl-adenylate domain fused to “artificial” peptide sequences (28 amino acids) shown previously to bind to the acceptor arm of tRNAAla (473). Certain fusions were predicted to be functional and indeed could alanylate hairpin microhelices designed after the acceptor stem of tRNAAla (provided they contain the alanine identity G3•U70 base pair), thus demonstrating the chemical logic underlying tRNA aminoacylation (472).
Functional studies on fragments derived from monomeric E. coli GluRS shed light on the evolution of this enzyme and showed its need of tRNAGlu for glutamate activation. Thus, two C-truncated enzymes (GluRS1–313 with domains 1 to 3 and GluRS1–362 with domains 1 to 4) glutamylate specifically tRNAGlu and require tRNAGlu for glutamate activation as does full-length GluRS1–471. The kcat of tRNA glutamylation by the GluRS1–362 variant is ~2,000-fold lower than that determined with the native enzyme but is strongly stimulated by the addition of free domain 5 (GluRS363–471). These functional data indicate that covalent connectivity with domain 5 is not required for activity. Furthermore, since truncated GluRSs form productive complexes with tRNAGlu, these imply that the tRNA acceptor branch interacts first with domains 1 to 3 and that the anticodon branch interacts with domain 5 in a second step. The fact that the decreased catalytic activity of the shorter truncated GluRS1–313 lacking the small domain 4 (49 residues) is not stimulated by its C-complement GluRS314–471 reveals the importance of the covalent connectivity between domain 3 and 4 for the aminoacylation reaction. Altogether, the activity of the truncated versions of GluRS confirms the structural modularity of this aaRS and indicates that proto-GluRS was a minimalist protein restricted to domains 1 to 3 that acquired domains 4 and 5 later in evolution (322).
In summary, studies on truncated aaRSs turned out to be extremely useful in uncoupling the different activities that were encrypted by evolution in extant aaRSs. In this respect, the studies on AlaRS and GluRS were enlightening and informed about the domains essential or dispensable for tRNA aminoacylation. Truncated aaRSs were also essential in many structural biology projects. This was the case of E. coli MetRS and of many other aaRSs that crystallized more readily when rendered structurally more stable after resection of floppy extensions (51) (see “Structure of aminoacyl-tRNA synthetases,” above).
AaRS-Dependent Quality Control of tRNA Aminoacylation
The need of faithful protein synthesis and the apparently contradictory facts showing nonabsolute fidelity of many molecular processes during translation lead to the idea of “quality control” that would account for mistranslation levels compatible with life. This is achieved by two types of mechanisms, namely during aaRS-dependent production of cognate aminoacyl-tRNAs and decoding of the charged tRNAs on the ribosome, so that globally mistranslation levels are minimized and synthesized proteomes functional (see, e.g., references 474, 475, 476, and 477, for reviews).
Two examples on aaRS-dependent production of cognate aminoacyl-tRNAs, namely prolyl-tRNAPro and leucyl-tRNALeu, illustrate the significance of the quality control concept in Bacteria. (i) In a mechanistic perspective, it is known that proofreading of mischarged alanyl- and cysteinyl-tRNAPro is achieved by two types of mechanisms involving the INS domain of ProRS (cis-editing) and the freestanding YbaK protein (trans-editing) (see “Error correction,” above). The poorly understood Ybak-dependent trans-editing pathway of cysteinyl-tRNAPro was elucidated recently, and, remarkably, revealed cyclization of tRNA-bound cysteine prior to hydrolysis and the crucial role of the YbaK backbone atoms in stabilizing the transition state, while the product is stabilized by the 2′-OH of tRNA (478). (ii) In a more physiological perspective, it was shown that the prime biological function of editing by E. coli LeuRS is not, as anticipated, to prevent incorrect isoleucine incorporation into proteins but to prevent misincorporation of the nonstandard amino acid norvaline (479). This quality control mechanism is a key feature for the bacterial adaptive response to oxygen deprivation, and the nonessential role for editing under normal bacterial growth has important implications for the development of resistance to antimicrobial agents targeting the editing site of LeuRS (479).
INHIBITION AND ENGINEERING OF BACTERIAL AMINOACYL-tRNA SYNTHETASES
Adaptation of pathogens to antibiotics calls for new target macromolecules and new strategies to find new antipathogenic agents. AaRSs that acquired subtle kingdom- and sometimes even species-specific idiosyncrasies in their structure, function, or regulation during evolution make them attractive for such strategies. In addition, discovery and design of small molecules that inhibit aaRS function has the advantage of being useful for aaRS crystallography and enzymology (480, 481, 482, 483, 484, 485, 486). On the other hand, engineering of aaRSs is one of the powerful tools to approach their structure/function relationships. Also, the emerging biotechnologies and biology-inspired chemistry that aim to produce proteins having incorporated unnatural amino acids need advanced methods of aaRS engineering. Engineering of orthogonal aaRS/tRNA pairs is the prerequisite to reach this last goal. The present understanding of tRNA identity and knowledge of aaRS structures provides the conceptual background allowing this engineering.
Natural-Product Inhibitors and Derivatives
A number of natural products of bacterial origin inhibit aaRSs, notably pseudomonic acid A (known as mupirocin) and furanomycin (IleRS), granaticin and Agrocin 84 (LeuRS), indolmycin and chuangxinmycin (TrpRS), SB-219383, an atypical dipeptide with the second amino acid bearing a bicyclic scaffold (TyrRS), albomycin derivatives and 4-hydroxyderricin (SerRS), borrelidin (ThrRS), microcin C (AspRS), ascamycin (AlaRS), and ochratoxin A and other natural products (PheRS) (480, 481, 483, 487, 488, 489). These compounds are substrate mimics containing amino acid moieties and compete more or less efficiently for cognate amino acid or aminoacyl-adenylate binding in the catalytic site of the aaRSs. Thus, isoleucyl-adenylate and mupirocin, although of rather hidden mimicry (a methyl terminus of monic acid mimicking the side chain of isoleucine and a pyran ring mimicking adenosine), have a common recognition in the adenylate-binding pocket of IleRS as seen in crystal structures of S. aureus (1qu3, 1ffy) and T. thermophilus (1jzs,1jzq) IleRS. This similar recognition explains why muporicin blocks binding of ATP and isoleucine on IleRS (138, 157). It is noteworthy that a mutagenesis analysis revealed that the selectivity of mupirocin binding in T. thermophilus IleRS over human IleRS comes from differences in only two residues in the aaRS active site (157). Similarly, the active moiety of antibacterial microcin C that inhibits AspRS is an aspartyl-AMP analog with an N-acyl phosphoroamidate linkage generated in vivo by processing its heptapeptide-nucleotidic precursor (490). The panel of natural products inhibiting bacterial aaRSs was recently enlarged to compounds of plant origin. Thus, the bioactive chalcone 4-hydroxyderricin isolated from Angelica keiskei covalently modifies S. aureus SerRS and thereby inhibits serylation of tRNAs (491).
So far, only mupirocin is on the market as a bacteriostatic compound used in dermatology as a topical antibiotic and, more generally, in eradication of methicillin-resistant S. aureus, which is a cause of death in hospitalized patients who have received antibiotic therapy (492, 493). Based on a better understanding of aaRS and inhibitor structures, several of these natural products were rationally modified to improve their inhibition capacities. This is notably the case of dipeptidic SB-219383, a bacterial fermentation product that inhibits TyrRS from staphylococci and streptococci (494) and of AspRS-specific microcin C (495). The crystal structure of S. aureus TyrRS with the dipeptidic inhibitor bound (1jii) (160) shows its interaction in the active site and provides the framework for designing novel antimicrobial agents. Finally, engineering of the mupirocin structure led to molecules with improved potency (496, 497).
On the other hand, resistance to antibiotics is becoming a worldwide public health problem and touches specifically bacterial pathogens that are becoming resistant to aaRS inhibitors (498, 499, 500). For example, sequencing the ileS gene from several strains of S. aureus with various degrees of susceptibility to mupirocin revealed a variety of point mutations clustering all on the surface of the IleRS (501). Interestingly, mutations do not solely lie in the Rossmann fold at positions critical for the antibiotic binding but are also found at the N and C termini of IleRS at positions distal to the tRNA-binding face and, more puzzling, within the CP1-editing domain or even outside the tRNA-binding site (501). Resistance can also occur by antibiotic-mediated aaRS gene induction or amplification. Thus, the second TrpRS gene (trpRS1) from Streptomyces coelicolor is inducible by indolmycin (502), and amplification of the IleRS gene as a result of secondary mutations provides adaptation to mupirocin resistance in S. enterica (503). Altogether, these phenomena explain the recent interest to better understand the biology and pharmacology of natural compounds interacting with aaRSs (e.g., the mechanism of mupirocin resistance [504]) and to search for antibiotic analogs, as is the case for mupirocin (499).
Synthetic Inhibitors
A huge number of synthetic inhibitors targeting the complete set of aaRSs were discovered by high-throughput screening of chemical libraries or were rationally designed as substrate analogs, primarily for pharmacological reasons, but also for crystallization/crystallographic purposes (481, 482, 484, 486). The refined understanding of aaRS structures supports efforts for renewed rational approaches to discover new inhibitors of pathogenic aaRSs. Thus, in view of new antituberculosis drugs, the use of the adenosine-binding site for inhibitor binding to M. smegmatis MetRS was evaluated and a potential binding site for a specific allosteric inhibitor was identified (158).
Stable nonhydrolyzable analogs of aminoacyl-adenylates (aa-AMP), such as aminoalkyl-adenylates (aa-ol-AMP) or aminoacylsulfamoyl-adenosines (aa-AMS), have been widely used in structural and mechanistic studies of aaRSs (481, 505, 506, 507) and for solving most of the crystallographic structures discussed in this review. The β-ketophosphonates are other synthetic adenylate analogs that were used, e.g., to inhibit bacterial GlnRSs and GluRSs (508). Potential inhibitors docking the aminoacyl-adenylate binding sites in aaRSs were discovered by virtual or library high-throughput screening. Among them, several molecules with only partial or apparently unrelated structures with aaRS substrates were found to selectively target, e.g., S. aureus MetRS (509, 510, 511), Staphylococcus epidermidis TrpRS (512), E. coli, S. aureus and P. aeruginosa TyrRSs (513, 514, 515), S. aureus ProRS (516), mycobacterial AspRSs (517), S. aureus LysRS (516), and bacterial PheRSs (518).
The boron-containing antifungal agent AN2690 is of particular interest because it inhibits posttransfer editing of LeuRS. A crystal structure of T. thermophilus LeuRS shows binding of an AMP-AN2690 adduct in the editing site (2v0c) and reveals the inhibition mechanism (156). This mechanism requires boron and the oxaborole ring of AN2690 and implies binding of the inhibitor to the cis-diol of 3′-terminal ribose of tRNA via its boron atom with the consequence of tRNA being trapped in the editing site (156). Interestingly, a derivative of AN2690 binding to the freestanding editing domain of S. pneumoniae LeuRS (4k47, 4k48) inhibits LeuRS activity, thus being a potent antipneumococcal agent (519). Another organoboron LeuRS inhibitor targets clinical anaerobic Bacteria, such as bacteroidetes and clostridia (520). These results open up opportunities for the development of new aaRS inhibitors targeting posttransfer editing sites of aaRSs via boron chemistry. Likewise, bacterial supramolecular particles containing aaRSs, such as transamidosomes (the particles where glutamyl-tRNAGln or asparaginyl-tRNAAsn is formed in organisms lacking GlnRS or AsnRS; see “Alternative functions of bacterial aminoacyl-tRNA synthetases,” below, for details), are other attractive targets for specific inhibition of pathogens. Thus, among a series of new chloramphenicol analogs, one compound was identified as being a strong competitive inhibitor of the amidation of aspartyl-tRNAAsn by the GatCAB amidotransferase in the asparagine transamidosome of H. pylori (521).
Engineering of aaRSs, Toward Orthogonality, and Design of Chimeras
In the early 1980s, the TyrRS from B. stearothermophilus was one of the first proteins to be engineered by site-directed mutagenesis (522, 523, 524). Today, engineering of aaRSs is routine practice, both for structural and functional studies (see, e.g., reference 256 and the many other examples given in this review). Remarkably, in the past decade, this engineering has moved in important new directions with the design of orthogonal aaRSs, more precisely of aaRS/tRNA pairs that are orthogonal to all endogenous pairs in a given organism, notably in Bacteria. (An orthogonal aaRS:tRNA pair consists of variant aaRS and tRNA molecules engineered in such a way that, first, the orthogonal aaRS becomes specific to an unnatural amino acid and would solely charge it on the orthogonal tRNA partner, and, second, the orthogonal tRNA would not be charged by a natural amino acid by any other aaRSs present in the protein synthesis system for which the orthogonal aaRS:tRNA pair was designed.) In addition, emerging trends aim to design aaRS chimeras.
Availability of orthogonal pairs enabling specific tRNA aminoacylation by unnatural amino acids is the prerequisite for ribosome-dependent synthesis of alloproteins, i.e., proteins containing chemically or spectroscopically active or structurally diverse amino acid analogs. To date, orthogonal aaRS/tRNA pairs have been used both in vitro and in vivo to incorporate ~200 unnatural amino acids into proteins in either E. coli (notably ~55 unnatural amino acids incorporated in vivo), yeast or mammalian cells (470, 525, 526). The method relies on orthogonal aaRSs able to charge stop codon (amber, ochre, or opal) reading suppressor tRNAs or frameshift suppressor tRNAs. Knowledge of identity rules, aaRS structure, and natural orthogonal aaRS/tRNA pairs (see above and, e.g., references 46, 527, and 528) was essential for the rational choice of the aaRSs to be engineered. Because of kingdom-specific distinctions in tRNA aminoacylation systems, expression of alloproteins in E. coli cells essentially relies on engineered aaRS/tRNA pairs of heterologous origin (e.g., M. jannaschii TyrRS/tRNATyr or S. cerevisiae AspRS/tRNAAsp, GlnRS/tRNAGln, TyrRS/tRNATyr, or PheRS/tRNAPhe pairs). Interestingly, based on a GlnRS/tRNAGln pair, it was possible to generate a complete set of orthogonal pairs active for reading amber, ochre, and opal codons (529). Orthogonality is reached by adapting the active site of the aaRSs to the desired new amino acid analog either by structure-based mutagenesis or by selection of the desired variants from structure-based libraries. Thus, 22 unnatural amino acids could be incorporated into proteins by evolving the orthogonal E. coli TyrRS/tRNATyr and LeuRS/tRNALeu pairs (470). Noticeably, mutagenesis in editing domains of aaRSs could become useful to improve orthogonality (248). Altogether, the methods based on aaRS orthogonality have great potential, either in biotechnology and molecular medicine, besides being a powerful source of basic information on genetic code and aaRS evolution and the mechanism of tRNA recognition and aminoacylation.
In another perspective, and given the modular structure of aaRSs, a logical follow-up would be to fabricate aaRS chimeras encompassing structural elements from aaRSs of different specificities and/or of different phylogenetic origins. This idea has been explored based on conformational similarities in the catalytic domain (i.e., Rossmann fold) and the nearby SC-fold (i.e., a β-α-α-β-α fold that properly orients tRNA on the ACB domain) of E. coli MetRS (1qqt) and GlnRS (1exd). Thus, MetRS chimeras were designed with portions of the structurally similar SC-fold, but with highly degenerated sequence, of GlnRS motif or with alanine residues. The chimerical variants retained significant tRNA methionylation activity and indicated that the structural integrity of the SC-fold contributes more to tRNA aminoacylation than does amino acid identity (530). It can be anticipated that such studies will help to reveal new structure/function relationships between aaRSs and to get deeper insight into the evolutionary processes that shaped the modularity of these enzymes.
REGULATION OF AMINOACYL-tRNA SYNTHETASE GENE EXPRESSION IN E. COLI
General Organization and Genes
The 20 aaRSs from E. coli are encoded by 23 genes. Most (17 of 20) of the aaRSs are encoded by a single gene; the three exceptions are PheRS, GlyRS, and LysRS. The heteromultimeric PheRS and GlyRS are encoded by two different genes corresponding to their two different subunits. LysRS is encoded by two genes for two homologous but different proteins, one being constitutively synthesized (the housekeeping enzyme) and the other being under multiple regulations (see below). The E. coli genome also carries a number of genes paralogous to aaRS genes, such as genX (yjeA) that encodes a protein homologous to the catalytic core of LysRS (85, 531, 532) and yadB that encodes a protein homologous to the catalytic domain of GluRS (533, 534). The properties of these shorter aaRS versions are described in “Paralogs of bacterial aminoacyl-tRNA synthetases,” below.
The aaRS genes of E. coli are generally scattered throughout the whole chromosome, with the exception of the genes for the heteromultimeric enzymes mentioned above, which are grouped. Also, the genes for ThrRS (thrS) and PheRS (pheST) are grouped at 38 min (Table 6) and only separated by three genes, and those for AspRS (aspS) and ArgRS (argS) are located at 42 min and separated by nine genes. All the available bacterial genome sequences show that aaRS genes are generally scattered, even if, as for E. coli, some of these genes may be clustered. The clustering is higher for B. subtilis, where about half of the aaRS genes are situated between 323° and 268° (0/360° is the replication origin).
Table 6.
Properties of the E. coli aminoacyl-tRNA synthetase genes
| aaRS gene and map position a | Coordinates b | Promoter c | Regulation d |
|---|---|---|---|
| alaS 60.72 (−) | 2,817,403 ← 2,820,033 UG: recX (−) DG: csrA (−) p | alaSp TIS: 2820112; 79 nt from TIC (535) | Transcriptional autoregulation (536) |
| argS 42.2 (+) | 1,958,086 → 1,959,819 UG: yecM (−) DG: yecT (+) | argSp TIS: 195828; 58 nt from TIC ρ-independent terminator (537) | |
| asnS 21.27 (−) | 986,808 ← 988,208 UG: pcnB (−) t DG: ompF (−) p | asnSp TIS: 988267; 59 nt from TIC (538) | |
| aspS 41.96 (−) | 1,946,774 ← 1,948,546 UG: yecD (+) DG: nudB (−) p | aspSp TIS: 1948641; 95 nt from TIC aspSp1: 1948725; 179 nt from TIC (539) | |
| cysS 11.94 (+) | 553,834 → 555,219 UG: ppiB (−) DG: ybcI (−) | cysSp TIS: 553800 (+); 34 nt from TIC Putative ρ-independent terminator (540) | |
| glnS 15.2 (−) | 705,316 → 706,980 UG: nagE (−) t DG: ybfM (−) | glnSp2 TIS: 705222; 94 from TIC (541) glnSp1 TIS: 705283; 33 from TIC ρ-independent terminator (542) | Possible regulation by incompletely characterized loci: glnU and glnR (543); see also 544 for comments. glnSp1: −10 carries a Dam methylation site. Expression of glnS is decreased by methylation (545). glnSp2: σ70 (D) and σ32 (H) activated (541). Possibly under director indirect control of OmpR (546) |
| gltX 54.26 (−) | 2,517,279 ← 2,518,694 UG: valU (+) DG: yfeD (+) | gltXp1 TIS: 2518806: 112 nt from TIC gltXp2: 2518798; 104 nt from TIC gltXp3 TIS: 2518752; 58 nt from TIC (547) | Repressed by Fis (483) |
| glyQ 80.23 (−) glyS 80.19 (−) | 3,722,430 ← 3,723,341 3,720,351 ← 3,722,420 UG: ysaB (−) DG: sokA (+) | glyQS operon Putative glyQp TIS: 3723377; 36 nt from TIC ρ-independent terminator (549) | |
| hisS 56.84 (−) | 2,637,323 ← 2,638,597 UG: sroE (−) DG: yfgM (−) p | hisSp TIS: 2638664; 67 nt from TIC (550) | |
| ileS 0.48 (+) | 22,391 → 25,207 UG: ribF (+) p DG: lspA (+) p | ribFp: 21383; 1008 nt from TIC ileSp1: 21833; 558 nt from TIC ileSp2: 22034; 357 nt from TIC (551) ileSp3: 22229; 162 nt from TIC (541) | ileSp3: σ32 (H) activated (541) |
| leuS 14.47 (−) | 671,424 ← 674,006 UG: ybeL (+) p DG: lptE (−) | No experimental evidence, but putative promoter and terminator proposed (552) | Possible regulation by incompletely characterized mutants leuX and leuY (553, 554) and/or leuR (555); see also reference 544 for comments |
| lysS 65.34 (−) | 3,031,679 ← 3,033,196 UG: prfB (−) p DG: idi (+) | xerD-dsbC-recJ-prfB-lysS operon xerDp: 3037873; 108 nt from TIC (xerD) (541) dsbCp2: 3037655; 811 nt from TIC (dsbC) (556) (dsbCp): 3036856; 12 nt from TIC (dsbC) (557) prfBp: 3034342–47; 38–43 nt from TIC (prfB) (558) | xerDp: σ32 (H) (556) and σ24 (E)-activated promoter (541, 557) dsbCp2: σ24 (E)-dependent promoter (556) dsbCp: CpxR and σ24 (E) activated promoter (557) |
| lysU 93.78 (−) | 4,351,223 ← 4,352,740 UG: yjdL (−) DG: yjdO (+) | lysUp1: 4352828; 88 nt from TIC lysUp2: 4352820; 80 nt from TIC (559) | lysUp1: repressed by Lrp (560). Under control of rlu and lrp; see text |
| metG 47.25 (+) | 2,192,322 → 2,194,355 UG: mrp (−) DG: molR (+) p | metGp0+ 2192081; 241 nt from TIC metGp+: 2192290;32 nt from TIC (561) | Possible autoregulation by a tRNA-like structure in the mrp gene (561) |
| pheM (leader peptide) 38.74 (−) pheS (α subunit) 38.71 (−) pheT (β subunit) 38.66 (−) | 1,797,250 ← 1,797,294 1,795,983 ← 1,796,966 1,793,581 ← 1,795,968 UG: rplT (−) t DG: ihfA (−) p | pheST operon pheMp: 1797325 (−); 31 nt from pheM (562, 563) | Regulated by phenylalanyl-tRNAPhe levels (leader peptide-dependent attenuation) (562, 564). Possibly regulated by oxygen availability (Fnr and ArcA) via readthrough from rplT (565, 566). Regulated by IHF and SOS (567); see text |
| proS 4.68 (−) | 217,057 ← 218,775 UG: yaeB (−) DG: yaeF (−) | proSp: 218837; 62 nt from TIC putative ρ-independent terminator. Gene called DrpA (568) | |
| serS 20.23 (+) | 938,651 → 939,943 UG: rarA (+) DG: dmsA (+) p | serSp: 938589; 62 nt from TIC (569) | Possible regulation by incompletely characterized locus serR (555); see also reference 544 for comments |
| thrS 38.77 (−) | 1,798,666 ← 1,800,594 UG: arpB (+) DG: infC (−) p | thrS-infC operon thrSp: 1800757 (−); 163 nt from TIC (570) | Translational autoregulation (205, 563), see text. NsrR binding site in regulatory regions (571) |
| trpS 75.67 (−) | 3,510,656 ← 3,511,660 UG: rpe, gph (−) p DG: yhfZ (−) | rpe-gph- trpS operon rpep: 3513175; 94 from TIC of rpe (572) trpSp: 3511719; 59 from TIC of trpS (573) | Not under autogenous miaAor trpR control (573) |
| tyrS 36.94 (−) | 1,713,972 ← 1,715,246 UG: pdxH (−) p DG: pdxY (−) p | pdxH-tyrS-pdxY operon tyrSp2: 1715307 (−); 61 from TIC (538) tyrSp1: 1715293 (−); 47 from TIC (574) | |
| valS 96.54 (−) | 4,479,005 ← 4,481,860 UG: holC (−) p DG: yjgN (+) | valSp1: 4481950; 90 nt from TIC valSp2: 4481942; 82 nt from TIC; ρ-independent terminator (575) |
Listed are the names of the aaRS genes and the positions in centisomes. The direction of transcription is with increasing (+) or decreasing (−) centisomes.
Listed are the coordinates of the different genes of the E. coli K12 MG1655 strain (http://ecocyc.org/) and the name of the upstream gene (UG) and downstream gene (DG), with the direction of transcription followed by either p, indicating that the gene has his own promoter, or by t, indicating that the gene is followed by a (putative) ρ-independent terminator. The information comes from EcoCyc (http://ecocyc.org/) or regulon database (http://regulondb.ccg.unam.mx/index.jsp) (538).
Listed are the coordinates of the transcription initiation site (TIS) and the distance in nucleotides between the TIS and the A of the AUG of the translation initiation codon (TIC). The gene is followed by a sequence resembling a ρ-independent terminator when indicated.
Given is the type of regulation, if known.
Inspection of bacterial genomes, other than E. coli, also tells us that the two genes corresponding to the two subunits of PheRS (pheS and pheT) are mostly adjacent in Proteobacteria, the phylum to which E. coli belongs, but may be found separated by a single open reading frame, as for Pasteurella multocida (a γ-Proteobacteria as E. coli), or by several open reading frames, as for Neisseria gonorrhoeae and Rhodospirillum centenum (both β-Proteobacteria). In more distant phyla, such as Cyanobacteria, pheS can be found separately from pheT. For even more distant phyla, such as Aquifica, the two PheRS genes can be found grouped (T. maritima) or separately (D. radiodurans and A. aeolicus). The situation is similar for GlyRS, although its two genes were already found separately in the phylogenetic tree close to E. coli, as, e.g., in some γ-Proteobacteria (Francisella tularensis). Concerning the genes of different aaRSs that are grouped such as thrS and pheST (separated by three genes in E. coli), genomic sequences indicate that they remain grouped in γ-Proteobacteria, but also sometimes in much more distant phyla, such as some Firmicutes (Clostridium acetobutylicum), in a way not obviously related to the phylogenetic tree. The looser association of aspS and argS that are separated by nine genes in E. coli is generally conserved in Enterobacteria but not further in γ-Proteobacteria.
In E. coli, unlike the rRNA operons that are divergently transcribed from oriC, there is no correlation between the orientation of the transcription of the different aaRS genes and the direction of replication. Moreover, no correlation is found between the position of the aaRS genes and the tRNA genes that are also scattered over the chromosome, with the exception of the gene encoding GluRS (gltX), which is adjacent to three genes encoding tRNAVal that are transcribed divergently from the aaRS gene (576). Although aaRSs have a key role linking amino acid biosynthesis to translation, cotranscription of aaRS genes with biosynthetic genes is not found in E. coli, whereas such examples have been described in B. subtilis, for instance, with the gltX-cysE-cysS operon (577).
All aaRS genes from E. coli have been experimentally found to be essential (578, 579) (https://www.genome.wisc.edu/resources/essential.htm), with the exception of lysS and lysU (see below), which may replace each other. The deletion of lysU shows no clearly identifiable phenotype (85), whereas that of lysS causes cold sensitivity mainly because lysU is hardly expressed at temperatures below 37°C (559) (see below).
Putative σ70-promoters have been assigned to almost all E. coli aaRSs (Table 6). Although these enzymes are quite abundant and their codon content is that of relatively highly expressed genes, their promoters have sequences that are often quite far from the classical σ70-consensus. In some cases several promoters are being used to express a single aaRS gene with one of the promoters activated by an alternative σ-factor (Table 6).
Heterodimeric Enzymes and Multifunctional Operons
PheRS and GlyRS are the only aaRSs in E. coli composed of two different subunits. In both cases, the genes for the two different subunits are not only grouped on the E. coli chromosome, but are also expressed as operons where the promoter proximal gene codes for the smaller subunit. In the case of GlyRS, the genes for the two subunits, glyQ and glyS, are located at 80 min, and the evidence that they are cotranscribed is based on the polar effects of Tn5 insertions in glyQ, the promoter proximal gene, on the expression of glyS, the promoter distal gene (580). The intergenic distance between the promoter proximal glyQ gene and the distal glyS gene is only nine nucleotides. In the case of PheRS, the genes of the two subunits, pheS and pheT, are located at 38 min and are also cotranscribed (562, 581). The distance between the two genes of the operon is 14 base pairs, similar to the intercistronic distance in the glyQS operon. The pheST operon is controlled by attenuation and is part of a region, which includes the structural gene for ThrRS (see below).
Some aaRS genes are expressed from larger transcription units with apparently unrelated genes. For example, the gene for IleRS (ileS) is located at 0.48 min and is part of an operon composed of five cistrons: the first, ribF, encodes the flavokinase and FAD synthetase; the second, ileS, encodes IleRS; the third, lspA, encodes the prolipoprotein signal peptidase; and the two last cistrons, fkpB and ispH, encode a peptidylprolyl isomerase and a 1-hydroxy-2-methyl-2-(E)-butenyl-4-diphosphate reductase (http://biocyc.org/ECOLI/). The whole operon is expressed from a promoter upstream of ribF (ribFp). Three other promoters are located within ribF (ileSp1, ileSp2, ileSp3), and the last one is located within ileS upstream of lspA (lspAp) (551) (http://biocyc.org/ECOLI/). The characterized terminator of this operon is located after the ispH gene.
Another example is the constitutively synthesized gene for LysRS (the housekeeping enzyme), lysS, that is expressed from an operon located at 65 min of which the first cistron is the gene pfrB encoding the peptide-chain-release factor 2 (RF2), and the second cistron is the lysS gene originally called herC (558). The gene lysS might also be expressed from longer transcripts carrying recJ (involved in recombination) and dsbC (a subunit of the protein disulfide isomerase) sequences upstream of pfrB (http://biocyc.org/ECOLI/). The expression of prfB is autoregulated by a posttranscriptional mechanism involving a +1 frameshifting at a naturally occurring UGA codon early in the gene (582). The expression of lysS, located immediately downstream, might thus be influenced by the translation of prfB. Still another example is trpS encoding TrpRS, which is expressed as a distal gene of a complex operon (572) made of seven genes expressed from seven promoters (http://biocyc.org/ECOLI/).
A striking example of a larger transcription unit is that carrying the gene for ThrRS (thrS), which is located at 39 min and belongs to a region of six genes transcribed in the same direction and all involved in translation. The thrS gene is followed by infC (translation initiation factor 3), rpmI (ribosomal protein L35), rplT (ribosomal protein L20), and the genes pheS and pheT (the two subunits of PheRS). The gene thrS is expressed from the thrSp promoter and makes two detectable mRNAs: the shorter covers thrS and infC and stops at terminator t1, and the longer traverses t1 and also covers the next two genes, rpmI and rplT, and stops at terminator t2 (583). These results indicate that thrS is always expressed with infC and sometimes with infC, rpmI, and rplT. The expression of thrS is negatively autoregulated at the translational level (205). However, neither the expression of infC nor that of the other next genes is noticeably affected by this regulation, since these genes are mainly expressed from their own promoters. Transcripts that carry thrS and the very distal genes pheS or pheT were not detected. The latter genes are expressed from their own promoter, pheMp, but may also be expressed from upstream promoters since the terminator t2, located after rplT and upstream of pheM, the leader peptide of the pheST operon, is very leaky in vivo (584).
With reference to the association between aaRS genes with unrelated genes that exist in E. coli, the gene for ThrRS, thrS, is found associated with infC and rpmI-rplT, in almost all γ-Proteobacteria and β-Proteobacteria and more rarely so in α-Proteobacteria. This association persists in some species belonging to quite distant phyla such as Bacteroidetes or Chlorobium and even Firmicutes (C. acetobutylicum). Other associations, such as ribF and lspA with ileS, are found in E. coli and most γ-Proteobacteria and β-Proteobacteria and less so with α-Proteobacteria and seldom with more distant phyla. The prfB-lysS association is even less conserved.
In conclusion, it appears that in E. coli some aaRS genes are expressed as multicistronic operons carrying apparently unrelated genes. However, the significance of cotranscription of other genes with aaRS genes is unknown. In the case of the highly regulated thrS-pheS-pheT region, the existence of long transcripts cannot, at the present time, be associated with any cross-regulation (e.g., ThrRS regulating the expression of the downstream gene infC or ribosomal protein L20, the regulator of rpmI and rplT, regulating the expression of the downstream pheST genes).
Regulation Strategies
Specific regulations
Specific regulations deal with the modulation of the expression of individual aaRSs in response to growth medium and/or genetic changes. The study of several amino acid biosynthetic operons in E. coli and Salmonella enterica serovar Typhimurium showed that aaRSs could be involved in the regulation of these operons (585). Since the expression of the biosynthetic operons is generally induced under cognate amino acid starvation conditions, the question was raised of whether or not the expression of the genes for aaRSs themselves is controlled in a similar way in E. coli. To test this hypothesis, many experiments were performed to study the effect of amino acid shortage on the expression of cognate aaRSs (reviewed in reference 544). Although different studies gave contradictory results, it seems that many aaRSs are transiently derepressed under cognate amino acid shortage but much less than the corresponding biosynthetic operons under the same experimental conditions (586, 587). In addition to these physiological studies, some more detailed molecular analyses have been performed to identify the cis-acting signals and the trans-acting genes that are involved in aaRS expression regulation. Some of these results have been reviewed previously (544, 588) and are summarized in Table 6; a few are described below.
In vitro studies of the expression of the alaS gene encoding AlaRS indicated that this enzyme was able to inhibit its own transcription in the presence of alanine (536). In addition, the enzyme was shown to bind to the −10 region of its own promoter, protecting two palindromic sequences from DNase I digestion. Unfortunately, no experiments are available to confirm that this apparently unique regulatory mechanism among E. coli aaRSs occurs in vivo.
In E. coli, LysRS is encoded by two different genes, lysS and lysU, located at 65 and 94 min, respectively (Table 6). This example is the only one in which two different aaRSs are able to activate the same tRNA isoacceptor family in E. coli. The first gene, lysS, corresponds to the housekeeping aaRS and is constitutively expressed from one or several σ70-dependent promoters but might also be expressed from σ24 and σ32-dependent promoters (Table 6). The second gene, lysU, corresponds to a form that is not constitutively expressed but is induced by various stimuli such as high temperature shifts, external pH changes, anaerobiosis, or the presence of specific metabolites in the growth medium (559, 589, 590). The expression of lysU is controlled by several genes such as lrp, rpoH, and rlu, which code, respectively, for the leucine response protein, the heat shock σ-factor, and an uncharacterized regulatory locus, all described below.
The expression of the lysU gene is induced by heat shock but only in the presence of a wild-type copy of htpR (or rpoH), the structural gene for the heat shock σ-factor (591). The role of htpR in lysU expression is unclear since induction of htpR synthesis, without temperature shift, does not cause lysU derepression (592). A second regulatory locus for lysU was found by genetic means: second-site suppression analysis indicated that lysU expression is under the control of a locus named rlu (regulation of lysU), located at 49.5 min on the E. coli map (593). The third regulator of lysU expression is the lrp gene, which encodes the regulatory protein of the Leu-Lrp regulon and causes repression or activation of many genes in E. coli (594, 595). The direct involvement of lrp in lysU expression was shown by DNase footprinting of Lrp protein to DNA upstream of lysU (560, 596).
Although a great deal of information is available about the control of lysU expression, many points are still unclear. How are the effects of the different stimuli (temperature, pH, and metabolites) related? What is the role of rlu? What is the indirect mechanism by which htpR controls lysU expression? What is the effect of prfB (encoding RF2) autoregulation on lysU expression? Although sensible reasons have been proposed to explain the existence of a second gene for LysRS (85), the raison d’être of this second inducible gene remains hypothetical.
The two subunits of PheRS, a heterodimeric aaRS of the α2β2-type, are expressed from an operon (see above) whose expression is mainly due to a promoter located far upstream (~400 base pairs) of pheS, the first structural gene of the operon (562). Nucleotide sequencing and transcription studies (562) showed the presence of a ρ-independent transcription terminator between this promoter and the structural genes of the operon. Elements similar to those found upstream of amino acid biosynthetic operons controlled by attenuation were found between the promoter and the terminator of the pheST operon. These elements are a small 14-amino-acid-long open reading frame with five phenylalanine codons called the leader peptide and sequences that allow the formation of alternative structures on the transcript. These control elements were shown to be responsible for the induction under conditions of phenylalanyl-tRNAPhe starvation (564). The model for pheST expression regulation, similar to that described for the histidine and tryptophan biosynthetic genes (585), is as follows. Under conditions of phenylalanine starvation, the ribosome pauses in the leader peptide at the phenylalanine codons, which allows the downstream mRNA to fold into a structure, called the antiterminator, which is incompatible with the formation of the terminator located downstream. The absence of transcription termination allows RNA polymerase to reach the structural genes of the operon. On the contrary, if phenylalanine is synthesized at adequate levels, the leader peptide is translated normally and the antiterminator structure cannot form, allowing the terminator structure to form, which decreases transcription of the structural genes. A strong indication that the expression of the pheST operon is controlled by a leader peptide-dependent attenuation mechanism comes from the finding that the operon is induced in the presence of a mutation in the miaA gene, formerly called trpX, which codes for Δ2-isopentenylpyrophosphate transferase, a tRNA modification enzyme in E. coli (e.g., for tRNATrp and tRNAPhe modification). A mutation in this gene also causes induction of the biosynthetic trp and pheA operons, which are controlled by the same kind of leader peptide-dependent attenuation mechanism. In the presence of the miaA mutation, translation of the leader peptide is slowed down, favoring antiterminator structure formation downstream on the mRNA and thus increased expression of the structural genes. The involvement of specific attenuator-like mRNA structures in control of pheST expression was shown by detailed mutational analysis of the leader region (584, 597). The pheST operon is also under himA and SOS control (567). The former control may be direct because sequences resembling the IHF binding site consensus are present at the pheST promoter. SOS control is probably indirect since no consensus LexA binding site was found next to this promoter. Global transcription studies (565, 566) indicate that the expression of rplT, the gene encoding ribosomal protein L20 located immediately upstream of the pheST operon, is decreased during anaerobic growth and increased in Fnr- and ArcA-deficient strains (both are global transcriptional regulators involved in the response to oxygen). Since the terminator between rplT and pheST is weak (see above), pheST expression could also be regulated by aerobiosis.
Many Enterobacteria show, after the rplT transcription terminator and upstream of pheST structural genes, features corresponding to a leader peptide-dependent attenuation described above. Inspection of the regions upstream of pheS in other γ-Proteobacteria belonging to orders different from Enterobacteria, such as Alteromonadales (Shewanella putrefaciens), Pasteurellaceae (H. influenzae), Pseudomonadaceae (Pseudomonas aeruginosa), and Vibrionaceae (Vibrio cholerae), do not show features corresponding to leader peptide-dependent attenuation. Despite the limited occurrence of this type of regulation for the PheRS genes, leader peptide-dependent attenuation has been found to regulate the transcription of at least one other aaRS in Bacteria. For instance, the auxiliary TrpRS encoded by the trpS1 gene of S. coelicolor has been shown to be inducible by several antibiotics using the same type of mechanism (500).
The most complete work about regulation of aaRS gene expression in E. coli has been performed with ThrRS, which follows a mechanism completely different from PheRS. ThrRS negatively regulates the expression of its own gene (thrS) at the translational level (563). The protein binds to a site, the operator, positioned in the leader of its own mRNA upstream of the Shine-Dalgarno sequence and inhibits translational initiation by competing with the binding of the 30S ribosomal subunit (598). The RNA operator is composed of four structural domains with tRNAThr anticodon-like sequences present in the loops of two of the four domains. The role of these two anticodon-like sequences is somewhat different since mutations in the main anticodon-like domain (that closest to the translation initiation site) abolish control, whereas mutations in the other have only a limited effect. The fact that these sequences are recognized as anticodon-like sequences is highlighted by biochemical and genetic data (205). In particular, the first base of the anticodon-like sequences can be changed without major effect on control, whereas any change in the second or third base abolishes control. This pattern of mRNA recognition is reminiscent of that of the tRNAThr isoacceptors for which the first base can either be C, G, or U, but neither the second nor the third base can be changed without abolishing aminoacylation (599). Perhaps the most convincing data indicating that the RNA operator carries a tRNA-like structure comes from an experiment showing that, if its identity is switched from threonine to methionine, the gene thrS becomes regulated by MetRS instead of ThrRS levels in the cell. Indeed, the replacement of the anticodon-like CGU sequence in the first (and main) anticodon-like domain of operator by CAU, the tRNAMet anticodon, abolishes the negative feedback control by ThrRS and establishes control by MetRS both in vitro (600) and in vivo (601).
ThrRS is a homodimeric enzyme, with each subunit composed of three different domains. The N-terminal domain gives the enzyme a winged shape, while the catalytic and the C-terminal domain form the core of the protein (145) (see “Structure of aminoacyl-tRNA synthetases,” above, for more details on structure). The crystal structures of ThrRS complexed with two tRNAThr2 molecules (1qf6) (145) and to the main anticodon-like domain of mRNAThrRS (1kog) (206) provide a detailed picture of how this aaRS performs two distinct functions, aminoacylation and control. In aminoacylation, each tRNA interacts mainly with one subunit of the enzyme with the anticodon loop of tRNAThr recognized by the C-terminal domain of the protein and the acceptor arm sequestered between the catalytic and the N-terminal domains. In the regulatory complex, the two anticodon-like domains occupy positions equivalent to those of the anticodon arm of tRNAThr with the two apical loops of the operator in interaction with the two C-terminal domains of the ThrRS dimer. Remarkably, the same amino acids interact with the CGU anticodon sequence of the tRNA and with the analogous residues in the main anticodon-like sequence of the operator. In agreement with the structure, changes of amino acids in the C-terminal domain of ThrRS affect both aminoacylation and regulation (602). In conclusion, the conjunction of genetic, biochemical, and structural data definitively proves the existence of mimicry in the way ThrRS recognizes the anticodon arm of tRNA and the two analogous regions of the operator on its own mRNA. Other tRNA mimics have been described (see above), and their role in gene regulation has been discussed elsewhere (603).
In addition to the enterobacterial order, the entire thrS operator with its four domains is found in several other γ-proteobacterial orders such as the Alteromonadales (Shewanella putrefaciens) and Pseudomonadaceae (P. aeruginosa, putida, and syringae groups). In more distant γ-proteobacterial orders, such as the Pasteurellaceae, either the entire operator (H. influenzae, Pasteurella multocida) or a partial operator carrying only the translation initiation proximal part (domains 1 and 2) of the operator (Haemophilus ducreyi) is found. In the case of V. cholerae belonging to the order of Vibrionaceae, only a partial (domains 1 and 2) operator is found (206).
It should be noted that the regulation of aaRS in E. coli and related Bacteria is very different from that of Firmicutes. In B. subtilis, the derepression levels after cognate amino acid starvation of several aaRS genes resemble those obtained with biosynthetic genes (83, 604, 605). Also, the control regions of most B. subtilis aaRS genes show common characteristics, something that is not found in E. coli. These common features are located downstream of the transcription initiation site and include a 14-base-pair-long conserved sequence (the T-box) located upstream of a transcription termination site (83, 605, 606). These regulatory regions are generally composed of three helical domains, a pseudoknot element preceding the T-box and mutually exclusive secondary structures, one of them being the terminator. A key element of this regulation is a triplet, called the “specifier codon,” which is placed in the first helical domain (607). The “specifier codon” always corresponds to the amino acid specificity of the downstream aaRS gene. Under amino acid starvation conditions, the level of aminoacylation of the cognate tRNA decreases. This starvation allows the cognate uncharged tRNA to bind to the nascent mRNA in such a way that the tRNA anticodon recognizes the “specifier codon” and the 3′ CCA uncharged end of the tRNA attaches to a downstream sequence of the mRNA (608). This tRNA attachment favors the formation of an antiterminator structure excluding the formation of the terminator. This allows transcription beyond the terminator into the structural aaRS gene. Interestingly, these regulatory elements can also be found upstream of amino acid biosynthetic and uptake genes in bacilli (609). The presence of such regulatory elements is exactly what could be expected from the apparently interconnected role of aaRSs and biosynthetic genes. At the present time, more than 1000 T-box elements have been annotated in all groups of Gram-positive Bacteria, as well as in some other phyla such as Deinococcus and Thermus and in a few Gram-negative phyla such as Geobacter and Chloroflexus (608, 610, 611). Several variations in this mechanism were recently described, notably the discovery in organisms of the phylum Actinobacteria of a novel group of T-box leader sequences of IleRS genes (ileS) lacking conserved elements essential for interaction with tRNA in other T-boxes. In these organisms, regulation requires tRNAIle-dependent structural rearrangements in the T-box riboswitches (612). Another peculiarity occurs in Clostridium acetobutylicum, where efficient antitermination of the AlaRS gene (alaS) is explained by correlated sequence variations in tRNAAla and alaS T-box leader RNA (613). Lastly, a novel regulatory mechanism is found in B. subtilis strains lacking a D-aminoacyl-tRNA deacylase. This mechanism regulates expression of the two TyrRSs (TyrS and TyrZ) present in these strains via a T-box riboswitch and a MarR (Multiple antibiotic resistance Regulator) factor. This is accounted by differentially organized tyrS and tyrZ operons (with a T-box in both operons and a MarR sequence in the tyrZ operon) and their growth-dependent expression (606). Remarkably, tyrZ is mainly expressed when B. subtilis needs D-tyrosine under stationary phase growth and biofilm formation, and when TyrS is inactivated. Since TyrZ is less efficient than TyrS and more selective for L-tyrosine over D-tyrosine it is concluded that these features may provide the mechanism preventing misincorporation of D-tyrosine in proteins (606).
Global regulations
The bacterial cell becomes larger and contains more DNA, RNA, and protein when cellular growth rate increases. The relative increase in RNA is larger than that of proteins that, in turn, is larger than that of DNA (614). In addition, the proportion of stable RNA (ribosomal RNA and tRNA) to total RNA increases with growth rate. The number of ribosomes per cell increases from ~7,000 to ~70,000 when the doubling time varies from 100 to 24 min (614). Also, the number of tRNA molecules per cell increases from ~63,000 to ~700,000 when the cellular doubling time varies to the same extent (614).
Quantitative 2D gel analysis of cells grown at different growth rates shows that the cellular concentration of the majority of the aaRSs analyzed increases with growth rate (615). This increase is, if normalized to the total cellular proteins, between 1.5- and 3-fold. In the case of AlaRS and LysRS expressed from the regulated gene, lysU, the cellular concentration actually decreases with growth rate. The average increase with growth rate in the concentration of housekeeping aaRSs is 1.7-fold if normalized to total cellular proteins, which is a limited increase compared with that of ribosomes under the same growth conditions.
Although the molecular mechanisms of growth rate-dependent regulation are unknown for most aaRS genes (see discussion in reference 544), precise information is available for ThrRS. As explained above, the synthesis of this enzyme is negatively autoregulated at the level of translation because of its ability to bind to its RNA operator on its own mRNA causing translation inhibition. Interestingly, operator mutations that abolish autoregulation also abolish growth rate-dependent regulation (570) indicating a clear relationship between the specific and the more global regulation for this aaRS. A simple explanation for these results is that, at high growth rates when the tRNAThr concentration is higher, the majority of ThrRS is involved in aminoacylation and less is available to bind to its mRNA, increasing its translation. The involvement of tRNAThr is indicated by the fact that, if tRNAThr concentration is increased by the presence of a multicopy plasmid carrying the gene for a tRNAThr isoacceptor, the expression of thrS is induced independently of growth rate (570).
Several aaRS genes carry a recognition sequence (GATC) for the Dam methylase in their regulatory regions. This is the case for trpS (at the 5′ end of the −35 box), glnS (over the −10 region), and possibly for cysS (two sequences upstream of the −35 box). The expression of a few E. coli genes is influenced by Dam methylation (616). The expression of glnS was shown to be under Dam methylase control since glnS-lacZ translational or transcriptional fusions are derepressed in a strain mutated in the dam gene (545). The expression of trpS was also reported to be derepressed in a dam strain (617).
At high growth rate, there are ~7 × 105 tRNAs/cell and protein synthesis rates are of approximately 5 × 105 amino acids/s/cell (614). These numbers mean that, on average, an aminoacylated tRNA is discharged and recharged about every second. This, in turn, means that, as soon as the supply of an amino acid is restricted, the concentration of the corresponding aminoacylated tRNA is immediately limiting (544). As a consequence, stable RNA synthesis is immediately stopped in E. coli. The effect of amino acid starvation on stable RNA synthesis is called the stringent response and depends on the relA and spoT genes that allow pppGpp and ppGpp synthesis. The synthesis of individual proteins is differentially affected by amino acid starvation. A study of the effect on aaRS synthesis of a decrease of aminoacylated tRNA indicates that most of the aaRSs are not under stringent control, i.e., less affected by amino acid starvation than, for instance, ribosomal proteins, which are known to be under stringent control. Among the nine aaRSs that were studied, ArgRS and maybe ValRS could be under weak stringent control (618).
ALTERNATIVE FUNCTIONS OF BACTERIAL AMINOACYL-tRNA SYNTHETASES
Indirect Pathways of Specific tRNA Aminoacylation for Ribosome-Mediated Translation
Besides direct charging of the amino acid onto its cognate tRNA(s) by an aaRS, there are also indirect pathways with tRNA-dependent modification of a noncognate amino acid mischarged to tRNA (619). Three such cases occur in Bacteria and concern formation of asparaginyl-tRNAAsn, glutaminyl-tRNAGln, and selenocysteinyl-tRNASec (with Sec for selenocysteine, the 21st amino acid in ribosome-mediated protein synthesis). A fourth case, tRNA-dependent biosynthesis of cysteine from O-phosphoserine in methanogens (107), is archaea-specific and will not be further discussed here.
ND-GluRSs and ND-AspRS for tRNA-dependent glutamine and asparagine biosynthesis
In those Bacteria (also in most Archaea and in some organelles) deprived of AsnRS or GlnRS, asparagine or glutamine can be synthesized by tRNA-dependent mechanisms after the mischarging of tRNAAsn or tRNAGln by ND-AspRS or ND-GluRS (ND means Non-Discriminating for efficient tRNA charging by these ND-aaRSs of both their cognate tRNAs and a related noncognate tRNA) followed by amidation of mischarged aspartate or glutamate by a tRNA-dependent amidotransferase (AdT) (7, 72). AdT enzymes designated by the generic name Gat (for Glutamine amidotransferase) are of different oligomeric organizations (heterotrimeric GatCAB in Bacteria and heterodimeric GatDE in Archaea) with structurally different subunits that amidate both tRNA-bound aspartate and glutamate. Surprisingly, in the H. pylori system, AspRS was shown to increase the affinity of aspartyl-tRNAAsn for the GatCAB amidotransferase (620), suggesting a coupling between aaRS and amidotransferase activities (see below, for structural validation). Note that the GatE subunit contains a subdomain that is included in the insertion domain of bacterial AspRSs making a structure-function link between aaRSs and tRNA-dependent amidotransferases. Also of interest is the presence in a few Bacteria of ND-AspRSs of archaeal-type coexisting with D-AspRSs (e.g., in T. thermophilus and D. radiodurans) (621, 622), making an evolutionary link between Bacteria and Archaea. In Bacteria, however, ND-AspRSs are mostly of bacterial-type (e.g., in P. aeruginosa and H. pylori) (221, 623). Simultaneous presence of the two GluRS forms (D and ND) in Bacteria is also documented in H. pylori and Acidithiobacillus ferrooxidans and, intriguingly, the ND-version does solely mischarge tRNAGln and has lost the ability to aminoacylate tRNAGlu (77, 78, 79). Altogether, the phylogenetic distribution of ND-aaRSs is intricate and not well understood, but likely recapitulates part of the evolutionary history of the class Ib GluRS/GlnRS and class IIb AspRS/AsnRS couples. At the tRNA level, the identity sets of the tRNAGlu/tRNAGln and tRNAAsp/tRNAAsn couples are related with participation of discriminator N73 and anticodon N34N35N36 residues (188). Finally, it should also be emphasized that both direct and indiect pathways for the synthesis of asparaginyl-tRNAAsn can be operational in the same organism, e.g., in T. thermophilus (624), in Synechococcus elongatus, and probably in other Cyanobacteria (239).
Based on these considerations, it is anticipated that the relaxed specificity of ND-AspRSs and ND-GluRSs relies on altered recognition of the tRNA identity determinants because of correlated architectural idiosyncrasies within the ND-aaRSs. In the AspRS case, the ND-enzyme has to efficiently recognize both tRNAAsp and tRNAAsn, two tRNAs sharing the same major identity determinants with the sole exception of the third anticodon determinant C36 in tRNAAsp replaced by U36 in tRNAAsn (Figure 4). Thus, the structural understanding appears straightforward and relies primarily on peculiarities in the OB-fold ACB domain of ND-AspRSs. In the particular case of archaeal-type T. thermophilus ND-AspRS (1n9w), this domain is lacking a helix inserted between two β-strands of the OB-fold and contains a peculiar L1 loop differing from the large loops interacting with tRNAAsp identity determinant C36 in conventional D-AspRSs (161). More generally, these features are accompanied by additional idiosyncrasies in archaeal-type ND-AspRSs (625), but remain more elusive in bacterial-type ND-AspRSs (220, 221). Although of the same nature, the recognition of tRNAGln and tRNAGlu by ND-GluRSs appears more intricate, especially since ND-GluRS has lost the ability to aminoacylate cognate tRNAGlu (77, 78) and since the nonconservation of the glutamine G73 identity determinant that can be A73 in tRNAGlu2, as in H. pylori and in Archaea (19). A structural basis accounting for the relaxed specificity of ND-GluRSs is brought by the crystal structure of the enzyme from the thermophilic cyanobacterium Thermosynechococcus elongatus (2cfo) (626). As expected, it shares the overall architecture of a D-GluRS (that of T. thermophilus [142]) and shows a sievelike recognition of the third anticodon base N36 (C36 in tRNAGlu and G36 in tRNAGln). However, while in D-GluRS from T. thermophilus, Arg358 recognizes C36, it is Gly366 that fulfills this role in ND-GluRSs. This amino acid change, conserved in most other ND-GluRSs, triggers relaxation, because the smaller glycine allows both small C36 and bulkier G36 to be tolerated by the protein (626). In agreement, homologous Arg350 from the ACB region in H. pylori D-GluRS rejects tRNAGln for glutaminylation (77).
For reasons of functional efficiency, specificity, and evolution, one can conjecture that biosynthesis of asparaginyl-tRNAAsn or glutaminyl-tRNAGln is coupled within supramolecular ternary aaRS:tRNA:AdT complexes in organisms utilizing the indirect tRNA-dependent pathway of asparagine or glutamine synthesis (Figure 4). Experimental support for this idea comes from the analysis of the crystal structure of archaeal Methanothermobacter thermautotrophicus GatDE complexed to tRNAGln (2d6f) (627). This structure visualizes a 40-Å-long channel for ammonia transport connecting the active sites in GatD and GatE and reveals the interactions that the accepting branch and the D-loop of tRNAGln make with GatE (627). In addition, the structure allows straightforward docking of GluRS for forming the aaRS:tRNA:AdT complex. Existence of such a ternary complex, called a transamidosome, is known for the asparagine (628) and glutamine (186) systems (Figure 4). Thus, the asparagine transamidosome assembles T. thermophilus ND-AspRS with cognate mischarged tRNAAsp and AdT (GatCAB) and remains stable during the aspartate to asparagine transformation process. By channeling the mischarged aspartyl-tRNAAsn intermediate between the ND-AspRS and AdT active sites, it prevents toxic interactions of the mischarged tRNA with elongation factor. Specificity of aspartyl-tRNAAsn amidation is conferred by the U1–A72 pair of tRNAAsn and prevented on aspartyl-tRNAAsp by its G1–C72 pair and U20a in the D-loop that act as antideterminants (189). This scheme finds robust experimental support from crystallographic and solution data on the archael-type T. thermophilus asparagine transamidosome that is a particle formed by 14 macromolecular units, namely, two GatCAB trimers, two ND-AspRS dimers, and four asparaginyl-tRNAAsp molecules (3kfu) (187).
The distinct structure of the asparagine transamidosome from Pseudomonas aeruginosa (4wj3) (629) is representative of Bacteria, while that from T. thermophilus is representative of Archaea. In the Thermus archaeal-type structure, the Yqey domain of GatB contacts specifically the D-loop of tRNAAsn (Yqey refers to the name of the Yqey genes that code for a family of bacterial and yeast proteins of unknown function containing a multihelical domain consisting of two different α-helical bundles; because of the structural similarity of Yqey with the C-terminal domain of GatB—and with the mobile C-terminal extension in D. radiodurans GlnRS [363]—Yqey was renamed GatB/Yqey motif in the SCOP-hierarchy [http://supfam.org/SUPERFAMILY]). The larger D-loop structure of tRNAAsp prevents recognition by the Yqey domain of GatB and thereby contributes to the rejection of tRNAAsp from the transamidosome. In the bacterial-type P. aeruginosa asparagine transamidosome, the bacteria-specific GAD domain of ND-AspRS provokes a new architecture of the complex and both tRNAAsn molecules in the transamidosome simultaneously serve as substrates and scaffolds for the complex assembly (4wj3). This architecture rationalizes an elevated dynamic and a greater turnover of ND-AspRS within bacterial-type transamidosomes (629).
Since the same combination of nucleotides also determines specific tRNAGln-dependent formation of glutamine (189), it can be suggested that the glutamine transamidosome would operate as the asparagine transamidosome. If so, one is faced with the problem of a steric hindrance, since both class I ND-GluRS and GatCAB would recognize the acceptor stem of tRNAGln by the minor groove side, implying that the transamidosome could not be formed. The crystal structure of the glutamine transamidosome from T. maritima shows how the dilemma is solved (3al0) (630). In contrast to the asparagine transamidosome where the accepting stem of tRNAAsp is contacted simultaneously by AspRS and GatCAB, it is the anticodon loop and the outer corner of the L-shape of tRNAGln that are simultaneously contacted by GluRS and GatCAB. This binding mode of tRNAGln provides flexibility for alternative conformations that can flip from a productive glutamylation/nonproductive amidation state to a productive amidation/nonproductive glutamylation state, thereby evading the steric hindrance dilemma (630). Interestingly, a dynamic glutamine transamidosome was also characterized in the mesophilic Bacteria H. pylori, of which the assembly is consistent with the crystal structure of the T. maritima homolog (631). Hence, two strategies for asparaginylation are possible: either tRNAAsn binds GatCAB first, allowing aminoacylation and immediate transamidation once ND-AspRS joins the complex, or tRNAAsn is bound by ND-AspRS which releases the Asp-tRNAAsn product much slower than the cognate Asp-tRNAAsp (632). Formation of the H. pylori Asn-transamidosome is tRNA-independent and requires Hp0100, a protein component that stabilizes the dynamic transamidosome and additionally enhances the capacity of AdT to convert Asp-tRNAAsn into Asn-tRNAAsn (633).
Peculiar properties of SerRS in selenocysteine biology
Selenocysteine, isosteric to cysteine with the sulfur atom replaced by selenium, is the 21st amino acid in ribosome-mediated protein synthesis. Specific incorporation of selenocysteine is coded by UGA stop codons (under the control of SECIS elements [i.e., SElenoCysteine Insertion Sequences] within mRNAs coding for selenocysteine-containing proteins) that are read by an atypical tRNA called tRNASec. Synthesis of selenocysteinyl-tRNASec occurs by an indirect pathway comprising tRNASec serylation by SerRS followed by conversion of serine to selenocysteine by a selenophosphate synthetase (634, 635). In E. coli, serylation efficiency of tRNASec is only ~1% that of the five canonical serine isoacceptors (636). This weak catalytic efficiency is explained by the atypical cloverleaf structure of tRNASec that differs from tRNASer by extended accepting- and D-arms as well as by a short D-loop with the consequence of an altered 3D-fold (637) that is explicitly seen in the crystal structure of human tRNASec (3a3a), particularly an absence of tertiary interactions between the D-stem and the long extra arm (638). A selenophosphate synthetase can than catalyze the tRNA-dependent conversion of serine to selenocysteine in a mechanism deciphered on the basis of the crystal structure of A. aeolicus selenophosphate synthetase (2zod) (630). In contrast to the situation of tRNA-dependent amidation, the serine to selenocysteine conversion in Bacteria likely proceeds in uncoupled reactions since tRNASec encompasses antideterminants in its acceptor arm (C7–G66/G49•U65/C50–G64 box) that prevent interaction with elongation factor EF-Tu, but not with SelB, the specialized factor in Bacteria replacing EF-Tu in the selenocysteine pathway (639).
Aminoacylation of tmRNAs by AlaRSs
The transfer messenger RNA (tmRNA) system performs translational quality control in Bacteria and some organelles in a process called trans-translation (640). This system rescues stalled ribosomes and consists of AlaRS, EF-Tu/GTP, the SmpB factor (for Small protein B), and tmRNA. E. coli tmRNA is a chimera of ~360 nucleotides with modular and phylogenetically conserved structure (641). This chimera encompasses a 3′-tRNAAla-like domain connected via a pseudoknot-containing linker to a 5′-mRNA-like domain that encodes a short peptide used as a degradation signal. For rescuing stalled ribosomes alanyl-tmRNA enters the A site, thus allowing the incompletely synthesized protein to be transpeptidated on tmRNA and protein synthesis to be resumed on the mRNA-like moiety of the tmRNA. The tagged aberrant protein is then degraded by cellular proteases. The prerequisite is alanylation of tmRNA by AlaRS (642). Alanylation occurs because of the presence in the tRNA-like domain of tmRNA of residues homologous to the major G3•U70 and A73 alanine identity determinants and of the modular arrangement of tmRNA allowing interaction with AlaRS. After alanylation, the charged tmRNA is complexed to EF-Tu (643) and is then ready for entering the ribosome. In this process, the SmpB factor (1k8h) (644) has a crucial role that is not entirely elucidated. It is a RNA-binding protein containing an OB-fold (a common structural motif present in many proteins with Oligonucleotide/oligosaccharide Binding capacity [218]) that interacts with tmRNA (2czj,1p6v) (645, 646) at each step of the rescue system, in particular, for increasing the alanylation capacity of AlaRS (640). Interestingly, the OB-fold of the SmpB factor resembles ribosomal protein S17, initiation factor IF1, and the N-terminal ACB domain of class IIb aaRSs (644).
BACTERIUM-LIKE AMINOACYL-tRNA SYNTHETASES
Organellar aaRSs
Mitochondria and chloroplasts are believed to be endosymbionts originating from α-Proteobacteria and Cyanobacteria, which opens the question of the evolutionary origin of their aaRSs, since many of them (not all, e.g., human mt-GlyRS and mt-LysRS are eukaryal [647]) are considered to be bacterium-like (648). Organellar aaRSs are nuclear encoded and have intricate phylogenetic origins with gene duplication and horizontal gene transfer from bacterial endosymbionts to the Eukarya (649). This nuclear origin implies that their gene products are targeted toward the organelles. In plants, as documented for Arabidopsis thaliana, the same bacterium-like gene product is dual-targeted by different N-terminal signatures for delivery in either mitochondria or chloroplasts (650, 651). It is noteworthy that mt-aaRSs seem to exhibit a reduced catalytic efficiency for tRNA aminoacylation in comparison with that of the bacterial or cytosolic homologs, as explicitly shown, e.g., for several yeast (104) and mammalian mt-aaRSs (647, 652).
It is not within the scope of this essay to cover in depth the field of organellar aaRSs. However, peculiar structural and functional features of a few typical mt-aaRSs (mt-AspRS, mt-LeuRS, mt-PheRS, mt-ThrRS, and mt-TyrRS) should be pointed out. Thus, mt-PheRSs present atypical monomeric structures idiosyncratic to mitochondria as first observed for the S. cerevisiae mt-PheRS (103, 104) and confirmed by crystallography for the human enzyme (3cmq) (103), in sharp contrast with bacterial and other cytosolic PheRSs that are (αβ)2 tetramers. On the other hand, S. cerevisiae mt-ThrRS recognizes two tRNAThr isoacceptors with distinct anticodon loops. Remarkably, the crystal structure of the complex with tRNAThr2 shows contacts with only the second and third anticodon base in contrast with the orthologous bacterial ThrRS that reads the entire anticodon sequence (4yye) (653). As to human mt-AspRS, functional studies have shown a greater sensitivity to adenylate analogs than bacterial AspRSs (one adenylate analog provides ~500-fold stronger competitive inhibition). Although the catalytic site of this mt-AspRS is almost identical to that of E. coli AspRS (654), it distinguishes by thermodynamic properties (655).
Furthermore, this mt-AspRS is not sensitive to identity position 73 and recognizes a minimalist identity set in cognate tRNAAsp restricted to the GUC anticodon. This result is in contrast to bacterial AspRSs, where G73 is a universally conserved major aspartate determinant (656). The reason for the relaxed specificity lies in two amino acid replacements in motif 2 within the AspRS catalytic site allowing accommodation of any base in the G73 binding pocket. Likewise, human and other mammalian mt-TyrRSs disobey the tyrosine identity rules (290) and rely more on A73 for specifying tyrosine than on the otherwise universal N1–N72 identity pair. This result is in agreement with the crystal structure of human mt-TyrRS (2pid) (657). These restrictions in aminoacylation identity sets could result from reverse evolution of identity elements in mt-tRNAs because of the rapid mutagenetic rate of the mitochondrial genomes (656, 658). Another example of an uncommon property comes from the editing capacity of Neurospora crassa mt-LeuRS. In fact, this yeast enzyme is competent for posttransfer editing like other LeuRSs. However, mutants with altered LeuRS editing capacity do not dramatically affect yeast viability and mitochondrial function in the presence of high levels of nonleucine amino acids, in contrast to E. coli, in which the editing-defective mutations limit cell viability. It is possible that yeast mitochondria have evolved to tolerate lower fidelity in protein synthesis or have developed alternate correction mechanisms (659). On the other hand, this LeuRS, besides being an actor in mt-protein synthesis, acts in intron RNA splicing, an activity requiring the C terminus of the protein (660, 661). Likewise, another N. crassa mt-aaRS, namely TyrRS, is a splicing factor and, as shown by a cocrystal structure (2rkj), binds the intronic RNA across the two subunits of the TyrRS but at a side opposite from that which binds tRNATyr (662).
aaRSs and aaRS-Like Proteins in Cell-Wall Biogenesis and in Other Secondary Metabolic Pathways
It has been known since the 1960s that an atypical tRNAGly species from S. aureus can be aminoacylated by canonical GlyRS and participates in cell-wall peptidoglycan synthesis (663, 664). Mirroring this finding, atypical aaRSs involved in nonribosomal peptide synthesis pathways were identified more recently in various Bacteria. Thus, a CysRS paralog is involved in mycothiol (1-D-myo-inosityl-2-[N-acetyl-L-cysteinyl]amido-2-deoxy-α-D-glucopyranoside) biosynthesis in M. smegmatis (3c8z) (665) and LysRS and AlaRS paralogs in P. aeruginosa or Clostridium perfringens participate in tRNA-dependent phospholipid aminoacylation (666, 667, 668). Another atypical aaRS-like protein, named MurM (Muropeptide factor M), present in Streptococcus pneumoniae attaches l-serine or l-alanine (provided by seryl-tRNAAla and alanyl-tRNAAla) to the stem peptide lysine of Lipid II in cell-wall peptidoglycans (669).
Furthermore, peptide bond-forming systems that depend on tRNA and aaRS-like proteins (CDPSs or CycloDiPeptide Synthases) were identified in several Bacteria, such as B. subtilis, Streptomyces noursei, Mycobacterium tuberculosis (670), and Bacillus licheniformis (671). In addition, paralogous versions of MetRS are widespread in Bacteria (672). Remarkably, the CDPS from M. tuberculosis that uses tyrosyl-tRNATyr as substrate to catalyze the formation of cyclo(L-Tyr–L-Tyr), demonstrates a structural resemblance with the catalytic domain of class Ic TyrRS (2x9q) (673). Likewise, AlbC from S. noursei (the protein coded by the third gene “C” of the antibacterial peptide Albonoursin biosynthesis operon acting at the first step of the biosynthetic pathway [674]) is one other member of the CDPS family and is similar to the catalytic domain of class Ic aaRSs (3oqv). Unprecedented, however, this monomeric protein uses phenylalanyl-tRNAPhe and leucyl-tRNALeu to synthesize cyclo(L-Phe–L-Leu) (675) instead of tyrosyl-tRNATyr that is typically formed by a dimeric TyrRS. In the case of S. noursei AlbC, the two charged tRNA substrates are accommodated in different binding sites and not all leucyl-tRNALeu isoacceptors are used as second substrate (676). The same mimicry with class Ic aaRSs exists in a CDPS from B. licheniformis that catalyzes cyclodileucine formation. The crystal structure of this CDPS (3oqh) (designated YvmC-Blic according to the name of the third gene “C” in the gene cluster yvm of the cyclodileucine biosynthetic pathway and its origin from B. licheniformis) suggests that all CDPS enzymes share a common aaRS-like architecture and a catalytic mechanism involving the formation of an enzyme-bound intermediate (671).
Other paralogous aaRSs were identified in a few peculiar Bacteria, where they have key roles in unexpected metabolic pathways. An example is a SerRS paralog from Streptomyces viridifaciens that provides seryl-tRNASer in the biosynthetic pathway of valinimycin (a cyclic decapeptide antibiotic, so named because it contains valine residues) (677). Another example comes from the radiation-resistant bacterium D. radiodurans that codes for an auxiliary TrpRS that contains an N-terminal extension similar to that of proteins involved in stress responses (678, 679). This TrpRS-2 is induced during responses to radiation damage and binds stoichiometrically to nitric oxide synthase, and, furthermore, it efficiently charges tRNATrp with 4-nitrotryptophan and 5-hydroxytryptophan (84, 680). The crystal structures of this TrpRS, either the apo-form (2a4m) or ATP-bound (1yid) and tryptophan-bound (1yi8) versions show overall structure similarity to standard bacterial TrpRSs but reveal also several idiosyncrasies, such as smaller amplitude motions of the helical ACB domain upon substrate binding and atypical tryptophan recognition. Interestingly, the loop conformations of the ACB domain resemble more those of human TrpRS than those of B. stearothermophilus TrpRS, indicating different modes of tRNA recognition in TrpRSs (678). The biological function of D. radiodurans TrpRS-2, also found in six other bacterial genomes (678), remains elusive. The 4-nitrotryptophanyl-tRNATrp is probably not used in protein synthesis but rather participates in the biosynthesis of metabolites such as thaxtomin, a dipeptidic phytotoxin containing a nitrotryptophan moiety produced by Streptomyces species (681). This unusual activity of D. radiodurans TrpRS-2 is a nice example of an additional aaRS function that provides a link between tRNA and nitric oxide metabolism (84).
The existence of other bacterium-like aaRSs remains an open question since genomes have not been systematically screened for such proteins. From considerations on aaRS evolution, gene transfer processes, and metabolomics, it can be anticipated that such proteins exist and wait to be identified.
PARALOGS OF BACTERIAL AMINOACYL-tRNA SYNTHETASES
The modular architecture of aaRSs recapitulates how addition of novel functional domains to the primordial catalytic cores led to their modern structures. This modularity implies, first, that isolated domains within aaRSs can be produced as stable proteins that may have conserved functionality (this has been amply documented, especially for editing domains; see above) and, second, that paralogs of aaRS domains may be present in extant organisms (371, 682). On the other hand, because aaRSs are among the oldest proteins, one would expect that memory of structural elements present in aaRSs is imprinted in contemporary proteins that participate in functions differing from tRNA aminoacylation in protein synthesis (371, 682, 683, 684). Note that several such proteins were discovered in the frame of structural genomics programs aiming to understand proteins of unknown function, as was, e.g., the case of the YbaK (a trans-editing protein related with the editing domain of ProRS) and YadB (a paralog of the catalytic domain of GluRS) proteins (452, 533).
Mimicry of Catalytic Domains
At present, paralogs of the catalytic domain of 10 aaRS specificities (namely, 5 class I mimics [CysRS, GluRS, MetRS, TrpRS, and TyrRS] and 5 class II mimics [AlaRS, AspRS, HisRS, LysRS, and SerRS]) have been characterized in Bacteria. These proteins participate in a variety of functions that are seemingly unrelated, but that all necessitate activation of amino acids. For the paralogs participating in nonribosomal peptide synthesis (AlaRS, CysRS, LysRS, MetRS, and TyrRS), in antibiotic biogenesis (SerRS), and in tRNA mischarging by two tryptophan analogs (TrpRS), see “aaRSs and aaRS-like proteins in cell-wall biogenesis and in other secondary metabolic pathways,” above.
Catalytic site mimicry of class I aaRSs
Bacterial YadB proteins are paralogs of the catalytic domain of GluRSs. These proteins were identified in various bacterial phyla, including proteobacterial, deinococcal, and cyanobacterial genera (239, 685). The E. coli protein has 34% sequence identity with E. coli GluRS, and its crystal structure (1nzj, 2zlz) (533, 686) shows the canonical fold of the GluRS catalytic domain (180). This protein activates glutamate and charges activated glutamate on tRNA (534), but, in contrast to canonical GluRSs, the activation step is tRNA independent, probably because of the replacement of a critical tryptophan in GluRS by a leucine (Leu194) in YadB, thereby allowing proper binding of ATP without tRNA-aided adaptation of the catalytic pocket (533). The accepting tRNA, however, is not tRNAGlu but instead bacterial tRNAAsp where glutamylation occurs on a cis-hydroxyl group of a ribose mimic at position 34 of its anticodon, namely, the dihydroxycyclopentane ring of the Q-base (or queuosine, a 7-deazaG-derivative) forming a labile ester linkage (685, 687). Thus, YadB is a tRNA modification enzyme (renamed Glu-Q-RS for Glutamyl-Q-tRNAAsp-Synthetase) and a new member of the group of enzymes that incorporate amino acid moieties in anticodon loops, such as lysine or threonine moieties, respectively, on C34 or A37 of certain tRNAs (319, 320, 688). It is likely that the function of Glu-Q-RSs is reminiscent of the early evolution of tRNA aminoacylation systems when primordial tRNA was restricted to minihelices. Although Glu-Q-RS and GluRS differ widely in function, they share strong structural resemblance within their catalytic core and quasi-equivalence of their zinc-binding sites (a SWIM domain [Cys98Cys100Cys125His127] in E. coli GluRS slightly altered [Cys101Cys103Cys119Tyr115] in E. coli Glu-Q-RS with His replaced by Tyr). While zinc is crucial for the positioning of tRNAGlu on GluRS, it contributes to enhanced protein solubility and decreased aggregation in Glu-Q-RS (689).
Other proteins related to the catalytic domain from class I aaRSs, in particular, that of TyrRSs, but containing only altered HIGH signature motifs (the HIGH sequence is one of the class I signature involved in ATP recognition), are the Glycerol 3-phosphate Cytidyl-Transferases (GCTs) and the ATPsulfurylases (ATPS). These enzymes catalyze reactions that are very different from tRNA aminoacylation (GCTs participate in the biosynthesis of lipids and intricate carbohydrates and use CTP instead ATP to activate their alcoholic or sugar substrates, and ATPSs catalyze the first step of the incorporation of inorganic sulfate into the precursors of cysteine and methionine). In addition they present various types of oligomeric structures in sharp contrast to monomeric class I aaRSs. Despite these peculiarities, both GCT and ATPS families recognize bound CTP or ATP via interactions with the conserved HxGH signature motif, as is the case of ATP recognition by class I aaRSs. Crystal structures of B. subtilis GCT (1coz) (690) and bacterium-like S. cerevisiae ATPS (1g8h) (691) support these assumptions. In summary, aaRSs, GCTs, and ATPSs utilize the same active site and catalytic strategy with virtually identical positions of their HxGH signature, suggesting that the three families diverged from a common ancestor (682).
Knowledge of the structure of class I aaRSs and the plausible assumption that primordial proteins had simplified structures provided the conceptual background for the design of a minimal TrpRS as a prototype of class I minimalist/ancestral aaRSs. The resulting 130-residue protein (embedding the HIGH and KMSKS signatures and a short inserted CP1 domain) retains substantial tryptophan-dependent PPi exchange activity (28, 198). The nucleotide sequence of this minimalist catalytic domain based on the gene of B. stearothermophilus TrpRS (named TrpRS urzyme [198]) shows significant antisense complementarity with the sequence of class II E. coli HisRS. It is most interesting that an artificial construct of the anticipated HisRS urzyme shows catalytic activity for histidine activation (692). Altogether, this antisense complementarity gives support to the appealing hypothesis that class I and class II aaRSs arose opposite one another on the same ancestral gene (in other words, that primordial class I aaRSs would be complementary replicas of class II aaRSs) (31, 693, 694).
Catalytic site mimicry of class II aaRSs
An E. coli protein similar to an isolated catalytic domain of AspRS is an asparagine-biosynthesizing enzyme (11as) (695). Such asparagine synthetases (AsnA) present in Bacteria and Archaea have a reaction mechanism implying formation of an aspartyl-adenylate intermediate in agreement with the conserved architecture of the catalytic sites in AsnA and AspRS (695). It is likely that these proteins have evolved from a common AspRS ancestor even though their sequence similarities are small (696). However, in contrast with YadB (see above), AsnA proteins have lost the capacity to transfer the activated amino acid to tRNA.
Various α- and γ-Proteobacteria, such as E. coli and S. enterica, contain a gene (called genX, poxA, or yjeA) coding for the catalytic domain of class II LysRSs (86, 531, 532). In S. enterica, mutants of poxA are associated with multiple phenotypes, including decreased growth rate, hypersensitivity to herbicides and amino acid analogs, and decreased pathogenicity (86). Bacterial PoxA proteins (also referred to as YjeA and GenX) are paralogs of class IIb LysRSs (3g1z, 3a5y). Importantly and unexpectedly, they specifically aminoacylate bacterial EF-P with lysine via a lysyl-adenylate-dependent reaction, but are unable to aminoacylate tRNALys despite a great resemblance with the catalytic core of canonical LysRSs (229, 697). Lysylation occurs on a highly conserved Lys34 of EF-P and is followed by isomerization of Lys34-α-Lysine catalyzed by YjeK that leads to Lys34-β-Lysine. This pathway is analogous to the two-step modification of the eukaryal EF-P homolog, eIF5A that generates hypusine (a polyamide-derived unusual amino acid only found in Eukarya and a few Archaea) (698). Interestingly, the crystal structures of the free (with bound AMP [3g1z] or adenylate [3a5y]) and of the EF-P:adenylate bound (3a5z) paralog, together with a structure-based sequence comparison of the paralogs with the catalytic domain of bacterial LysRSs account well for the lysylation of EF-P at a position mimicking the tRNA-accepting end and for the lack of lysylation of tRNA as the result of amino acid replacements in the catalytic domain (229, 697). The Lys34 modification of EF-P is essential for cell survival and likely plays a regulatory role in translation (229, 697, 699).
HisZ is based on the catalytic domain of HisRS and is widely represented in Bacteria (but not in E. coli) and Archaea. This HisRS paralog lacks aminoacylation activity, but, in contrast, is the allosteric regulator of ATP PhosphoRibosyl-Transferase (ATP-PRT), the first enzyme of the histidine biosynthesis pathway where it joins ATP and 5-PhosphoRibosyl-1-PyroPhosphate (PRPP) (700). Structural and functional studies shed light on the evolution and activity of this enzyme of hetero-octameric organization with four HisG catalytic subunits and four HisZ regulatory subunits. Structures of ATP-PRTs under two functional states (with or without PRPP), respectively, from Lactococcus lactis (1z7m, 1z7n) (701) and of the HisG/HisZ complex from T. maritima (1usy) (702) reveal a phosphate ion located in the HisG/HisZ interface and a total of eight histidine binding sites located within these interfaces in a region highly conserved between HisZ and HisRS. Steady-state kinetics indicates that only the complete octameric complex is active and noncompetitively inhibited by the pathway product histidine (626). Crystal structures of E. coli ATP-PRT have been solved in complex with the inhibitor AMP and the product PR-ATP (PhosphoRibosyl-ATP, with the PR-moiety linked to adenosine N2 atom) (1h3d, 1q1k) (703). They clearly identify AMP in the PRPP-binding site, with the adenosine ring occupying the ATP-binding site. Comparison with two structures of the ATP-phosphoribosyltransferase from M. tuberculosis (apo-form [1nh7] and enzyme in complex with inhibitor histidine and AMP [1nh8] [704]) indicates that histidine is solely responsible for the large conformational changes observed between the hexameric forms of the enzyme (703).
BirA is a biotin ligase with regulatory properties of its own gene that comprises a domain having striking structural similarity with the catalytic domain of SerRSs (705) and more generally of the catalytic domain of class II aaRSs. Note also the similarity of BirA with domain B8 of the large and noncatalytic β-subunit of PheRSs (236). While the crystal structure of E. coli BirA (1hxd) (706) clearly shows a biotin adenylation site resembling the catalytic domain of class II aaRSs, it reveals also the absence of structural features associated with amino acid binding and monomer dimerization, thus explaining the monomeric state of BirA proteins in contrast to class II aaRSs that are dimers or multiples of dimers.
Intriguing is the discovery in various bacterial species of a novel group of enzymes that are similar to class II aaRSs and transfer activated amino acids to the phosphopantetheine group of small Carrier Proteins, and therefore were named aa:CP ligases. Some of them are truncated SerRSs homologous to the catalytic domain of the atypical SerRSs found in methanogenic Archaea, others derive from the catalytic domain of AlaRS and GlyRS (707, 708). These proteins have relaxed amino acid specificity and lack tRNA aminoacylation activity. In contrast, they transfer activated amino acids to the phosphopantetheine prosthetic group of putative carrier proteins. A representative member from the bacterium Bradyrhizobium japonicum (a microsymbiotic nitrogen-fixing Proteobacteria in legume-root nodules) has been functionally and crystallographically characterized. It activates preferentially glycine instead of serine and deviates slightly from the canonical SerRS catalytic domain, although it presents the characteristic signature motifs of class II aaRSs and the canonical Zn-binding site of SerRSs (3mf2, 3mey, 3mf1) (707). A series of crystal structures of B. japonicum Gly:CP ligase in functional complexes with the carrier protein from B. japonicum (4h2s, 4h2t, 4h2u, 4h2v) and Agrobacterium tumefaciens (4h2w, 4h2x, 4h2y) show a fundamentally different binding of the carrier protein compared to the tRNA binding of the structurally related aaRSs (708). These structures reveal how a conserved class II aaRS catalytic core can adapt to another function through minor structural alterations (708).
Editing site mimicry of class II aaRSs
Ybak, ProX, and AlaX factors are discussed in “Error correction” under “Aminoacylation of tRNA,” above.
Mimicry of tRNA Binding Domains
The OB-fold with β-barrel architecture is a widespread RNA binding motif, notably in aaRSs, where it is present under various versions in both class I and class II enzymes. For example, it was found in bacterial and eukaryal MetRSs and in class IIb aaRSs, where it is the canonical ACB domain. OB-folds are also present in aaRS-related yeast Arc1p and mammalian p43 proteins (Arc1p, a cofactor for MetRS and GluRS, is homologous to the auxiliary p43 protein of the multi-aaRS MARS complex [709]) and in EMAP II cytokines whose structure is conserved in the B2 domain of the β-subunit of heterotetrameric PheRSs. The structure of the isolated OB domain of E. coli LysRS (1krs, 1krt), either free or in complex with polyU, was solved by NMR (710). It is identical to that of native E. coli LysRS (224) and other class IIb aaRSs and shows striking similarity to the nuclear ribonucleoprotein U1A (711). Interestingly, the complex with polyU identified the amino acids important for binding the UUU anticodon of tRNALys (710).
Trbp111 and its ortholog CsaA are other proteins related to aaRSs that are present in various Bacteria and Archaea. Trbp111 is a dimeric protein (2x111 amino acids) discovered in the hyperthermophilic bacterium A. aeolicus. As pinpointed by its name, Trbp111 is a tRNA-binding protein interacting with any kind of tRNA (712). Its crystal structure and that of the E. coli ortholog show a classical OB-fold in the core of the monomer (1pyb, 3ers) (713). Docking and solution data are consistent with a 2:1 Trbp111:tRNA complex with tRNA recognition occurring through its T-stem opposite to the concave site recognized by the aaRSs (713, 714). Given this functional property, it is likely that the Trbp111 domain in MetRS (see “Structure of aminoacyl-tRNA synthetases,” below) acts in cis to increase the affinity of cognate tRNAMet.
Bacterial CsaA secretion chaperones are dimeric proteins (2x ~110 amino acids) with export-related activity that share sequence and conformational homology with Trbp111 and the C-terminal domain of archaeal MetRSs (715). Several crystal structures are available, notably from T. thermophilus (1gd7) (715) and B. subtilis (2nzh) (715) that show an OB-fold core with N- and C-terminal α-helical extensions involved in dimer formation. On the basis of the structural similarity with Trbp111, it has been proposed that CsaA proteins possess a tRNA-binding ability (716) that still remains elusive.
Given the ancestry of tRNA, it was not unreasonable to conjecture that many constitutive domains or subdomains of extant proteins including aaRSs, but also freestanding proteins recognizing tRNA (or tRNA domains) unrelated with the functioning of tRNAs in ribosome-mediated protein biosynthesis, should be present in proteomes. The few examples discussed above show the correctness of the anticipation. It is likely that many other examples will become available in the near future.
CONCLUSIONS AND PERSPECTIVES
The Present Status
The overview on aaRSs published in 1996 in the last printed EcoSal corpus already covered a large body of structural and functional results that beautifully and convincingly demonstrated the primordial role of aaRSs in translation (10). At that time, aaRS understanding became textbook knowledge and many scientists outside the field claimed that no paradigm shift would occur anymore with aaRSs so that only modest progress could be expected. However, since 1996, the science of aaRSs has made significant advances and has undergone great mutations. The insight moved from essentially reductionist and classical genetics viewpoints toward more integrated cellular biology and physiology perspectives. Contrary to the common belief prophesying modest incremental progress, one has observed an abundance of new findings that amplified in the past decade. A number of anticipated new aaRS properties received robust experimental support, and new atypical and often unexpected structural and functional properties were discovered. On the other hand, unexplored topics became rejuvenated, as is the case of the biology of ApnA compounds produced and consumed by aaRSs (e.g., references 717 and 718). At present, aaRS research is invading all biology. Three issues deserve special comments. They are as follows.
(i) The remarkable development of aaRS structural biology. Indeed, a plethora of new X-ray structures (and some NMR structures) became accessible, particularly in the past 10 years (~20 structures known in 1996 corresponding to 6 amino acid specificities and 3 complexes with tRNA, compared with ~600 structures presently in the PDB). This trend was especially prominent in Bacteria, not only for canonical aaRSs but also for the wealth of paralogs discovered in an increasing number of sequenced genomes. The fact that structures of the same aaRS could be visualized under different conformational states and that structures from different organisms could be compared completely renewed our understanding of aaRS function and evolution. For several aaRSs, the mechanism of tRNA aminoacylation has reached a high degree of sophistication (18) (see details in “Aminoacylation of tRNA,” above). In this respect, the example of the TrpRS from B. stearothermophilus is emblematic, since a precise structural and temporal description of the steps leading to tryptophan activation could be derived on the basis of a large panel of crystal structures (11 PDB entries in 2015 for the protein in complex with different small ligand associations) completed by molecular dynamics simulations and kinetic data on mutant proteins (328, 354, 365). Likewise, structural studies focusing on tRNA aminoacylation gave clues for more precise descriptions of the amino acid transfer on the 3′-terminal –CCAOH of tRNA (18) and, importantly, for explaining posttransfer editing, i.e., the aaRS-catalyzed hydrolysis of mischarged amino acids (e.g., references 248, 455, and 719). Also remarkable is the finding of dynamic coupling between editing and synthetic sites in class IIa bacterial-like ProRS (720). On the other hand, exploration of genomes demonstrated the universal presence of aaRS paralogs in living organisms and structural genomics established definitively the evolutionary origin of the modular architecture of aaRSs.
(ii) The discovery of a broad variety of new functions for canonical aaRSs and their paralogs. The new functions concern not only the translation machinery itself (e.g., for atypical tRNA or protein aminoacylation, for clearing errors) but also diverse metabolic pathways a priori unrelated to translation (e.g., for nonribosomal peptide bond synthesis, for atypical ester bond formations using activated amino acids) (see details and examples in the three preceding sections). In most cases, these new functions rely on subtle modifications of the catalytic sites of canonical aaRSs. This large diversity of functions illustrates how the invention of the two types of aaRS catalytic cores (class I and class II specific) and their tinkering during evolution was beneficial for life and how translation is intermingled with other cellular processes.
(iii) Progress in aaRS gene regulation. Various experimental evidence has indicated that quite a few E. coli aaRS genes may be inducible under cognate amino acid starvation conditions but at a level much lower than the corresponding amino acid biosynthetic operons under the same experimental conditions (reviewed in reference 479). In E. coli, the molecular mechanisms underlying these specific regulations are understood in only a limited number of cases and seem to vary from one aaRS to the other. For instance, the operon encoding PheRS is regulated by a leader peptide-dependent transcriptional attenuation mechanism similar to that of the E. coli trp operon, whereas the gene for ThrRS has been shown to be autoregulated at the level of translation in a way similar to that of ribosomal protein genes. In E. coli, a majority of the aaRS genes also seem to be under more global regulation, such as growth rate-dependent control. The situation is very different in B. subtilis and many other Gram-positive Bacteria, in which the induction levels after cognate amino acid starvation of several aaRS genes resemble those obtained with biosynthetic genes. Also, the control mechanism, common to most B. subtilis aaRS genes, is very different from those of E. coli aaRS genes.
In summary, the advances in aaRS structural biology, enzymology, and genomics, together with the deeper knowledge on aaRS genes, led to a more complete biology of bacterial aaRSs. These advances were also beneficial for a better understanding of the archaeal and eukaryal aaRSs, especially the human aaRSs in both their cytosolic and mitochondrial versions. Moreover, progress in the aaRS field was paralleled by equally important progress in the tRNA field (e.g., references 264, 268, 721, and 722), both being the result of intensive interdisciplinary research efforts. From the viewpoint of applications, it is remarkable to realize how engineered aaRSs and aaRS domains are becoming important actors in biotechnology (e.g., as tools for the fabrication of proteins having incorporated nonnatural amino acids with focused chemical or biophysical/spectroscopic properties) and molecular medicine (e.g., for the search of new antipathogen drugs or for therapeutic use).
The Future of aaRS Research
It can be anticipated that the perspectives in the aaRS science will be flourishing, because they are founded on a state-of-the-art, robust theoretical, experimental, and methodological background. Interdisciplinary approaches will remain fundamental and will continue to combine reductionist and integrated approaches. Deciphering the systems biology of aaRSs will be one of the next challenges. The mechanisms underlying the expression and the regulation of aaRS genes in Bacteria are far from being understood in depth, and many questions remain unanswered, among others: Why is there such heterogeneity in E. coli control mechanisms? Why is the situation so different in Enterobacteria and Firmicutes? Why are the induction levels of aaRS genes so weak under starvation conditions in E. coli? Why is there a need for clearly inducible aaRSs in B. subtilis? Is there a general mechanism for global controls in Enterobacteria? What about global controls of aaRS synthesis in Firmicutes? If such controls exist, are they related, or not (723), to the mechanism of induction under starvation conditions? Likewise, understanding the biology of the aaRSs under normal and stress conditions presents other challenges. Finding answers implies understanding aaRS properties (e.g., enzymology, structural plasticity, supramolecular complexes, and characterization of partners, crowding—the effect of related or unrelated macromolecules on aaRS properties in crude mixtures/media—channeling, trafficking, regulation, degradation) under a variety of cellular conditions. Continuing to explore genomes will certainly reveal aaRSs and aaRS paralogs with new idiosyncratic properties and divulge novel connections with metabolic pathways. Moreover, deep sequencing of genomes will also enable establishment of the polymorphic sequence variations in specific aaRSs of given organisms and thus allow us to distinguish toxic from polymorphic mutations.
Finally, and because of their ancient origin, aaRS sequences will remain markers of choice for refined structure-based taxonomies (14, 68) worth being compared with phenotype-based taxonomies. Studies of aaRS phylogenies will provide renewed understanding of the origin and evolutionary history of life on Earth. Despite countless speculations many questions remain essentially open. Thus, one would like to know the molecular identity of the progenitor of the first aaRS urzyme in a prebiotic world (RNA, proteic, or mixed?). How did this molecule acquire specificity? What is the connection between the proteic urzyme and ribozyme catalysts for RNA acylation? Why are there two classes of aaRSs? Was primordial life possible with aaRSs from only one class? Was class II before class I, or vice versa, or did the two classes emerge together? Class II aaRSs could have been first since they are specific for amino acids with broader chemical diversity than class I aaRSs and, therefore, better suited for catalysis and have kept memory of ancestral capacity to aminoacylate tRNA minihelices, but other arguments would favor the opposite possibility. Answering these questions will be challenging, but new experimental data are expected to insightfully enrich the ongoing debates on the emergence of aaRSs in life (2, 14, 27, 30, 34, 37, 198, 705, 724, 725, 726, 727).

ACKNOWLEDGMENTS
Because of space limitations, all contributors could not be cited, and we apologize for this. We thank Catherine Florentz, Jacques Lapointe, and Eric Westhof for comments and helpful suggestions. We are also indebted to Claude Sauter for help in figure preparation and for advice.
This work received support from Centre National de la Recherche Scientifique (CNRS), Université de Strasbourg, ACI BCMS, ANR-07-BLAN-0351-03, and ANR-09-BLAN-0091-01.
No potential conflicts of interest relevant to this review were reported.
REFERENCES
- 1.de Duve C. 1988. The second genetic code. Nature 333:117–118. [PubMed] 10.1038/333117a0 [DOI] [PubMed] [Google Scholar]
- 2.Schimmel P, Giegé R, Moras D, Yokoyama S. 1993. An operational RNA code for amino acids and possible relationship to genetic code. Proc Natl Acad Sci USA 90:8763–8768. [PubMed] 10.1073/pnas.90.19.8763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bullwinkle TJ, Ibba M. 2014. Emergence and evolution. Top Curr Chem 344:43–87. [PubMed] 10.1007/128_2013_423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carter CW Jr. 1993. Cognition, mechanism, and evolutionary relationships in aminoacyl-tRNA synthetases. Annu Rev Biochem 62:715–748. [PubMed] 10.1146/annurev.bi.62.070193.003435 [DOI] [PubMed] [Google Scholar]
- 5.Giegé R. 2006. The early history of tRNA recognition by aminoacyl-tRNA synthetases. J Biosci 31:477–488. [PubMed] 10.1007/BF02705187 [DOI] [PubMed] [Google Scholar]
- 6.Giegé R, Eriani G. 2014. Transfer RNA recognition and aminoacylation by synthetases, 21 pages. Encyclopedia of Life Sciences (ELS). John Wiley & Sons, Ltd, Chichester. [Google Scholar]
- 7.Ibba M, Söll D. 2000. Aminoacyl-tRNA synthesis. Annu Rev Biochem 69:617–650. [PubMed] 10.1146/annurev.biochem.69.1.617 [DOI] [PubMed] [Google Scholar]
- 8.Lapointe J, Giegé R. 1991. Transfer RNAs and aminoacyl-tRNA synthetases, p 35–69. In Trachsel H (ed), Translation in Eukaryotes. CRC Presss Inc. Boca Raton, FL. [Google Scholar]
- 9.Martinis SA, Plateau P, Cavarelli J, Florentz C. 1999. Aminoacyl-tRNA synthetases: A family of expanding functions. EMBO J 18:4591–4596. [PubMed] 10.1093/emboj/18.17.4591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martinis SA, Schimmel P. 1996. Aminoacyl-tRNA synthetases: General features and relationships, p 887–901. In Neidhardt FC, Curtiss R III, Ingraham JL, Lin ECC, Low KB, Magasanik b, Reznikoff WS, Riley M, Schaechter M, and Umbarger EH (ed), Escherichia coli and Salmonella: Cellular and Molecular Biology, 2nd ed., ASM Press, Washington, DC. [Google Scholar]
- 11.Meinnel T, Mechulam Y, Blanquet S. 1995. Aminoacyl-tRNA synthetases: Occurrence, structure, and function, p 251–290. In Söll D and RajBhandary UL (ed), tRNA: Structure, Biosynthesis, and Function. American Society for Microbiology, Washington, DC. [Google Scholar]
- 12.Schimmel P. 1987. Aminoacyl-tRNA synthetases: General scheme of structure-function relationships in the polypeptides and recognition of transfer RNAs. Annu Rev Biochem 56:125–158. [PubMed] 10.1146/annurev.bi.56.070187.001013 [DOI] [PubMed] [Google Scholar]
- 13.Söll D, Schimmel PR. 1974. Aminoacyl-tRNA synthetases, p 489–538. In Boyer P (ed), The Enzymes, vol. 10. Academic Press, New York, NY. 10.1016/s1874-6047(08)60147-x [DOI] [Google Scholar]
- 14.Woese CR, Olsen GJ, Ibba M, Söll, D. 2000. Aminoacyl-tRNA synthetases, the genetic code, and the evolutionary process. Microbiol Mol Biol Rev 64:202–236. [PubMed] 10.1128/MMBR.64.1.202-236.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo M, Yang X-L, Schimmel, P. 2010. New functions of aminoacyl-tRNA synthetases beyond translation. Nat Rev Mol Cell Biol 11:668–674. [PubMed] 10.1038/nrm2956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo M, Schimmel P. 2013. Essential nontranslational functions of tRNA synthetases. Nat Chem Biol 9:145–153. [PubMed] 10.1038/nchembio.1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pang YL, Poruri K., Martinis SA. 2014. tRNA synthetase: tRNA aminoacylation and beyond. Wiley Interdiscip Rev RNA 5:461–480. [PubMed] 10.1002/wrna.1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ibba M, Francklyn C, Cusack S (ed). 2005. The Aminoacyl-tRNA Synthetases, Landes Bioscience, Georgetown, TX. [Google Scholar]
- 19.Jühling F, Mörl M, Hartmann R, Sprinzl M, Stadler PF, Pütz J. 2009. Compilation of tRNA sequences and tRNA genes. Nucleic Acids Res 37:D159–D162. [PubMed] 10.1093/nar/gkn772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goodsell DS, Dutta S, Zardecki C, Voigt M, Berman HM, Burley SK. 2015. The RCSB PDB “Molecule of the month”: Inspiring a molecular view of biology. PLoS Biol 13:e1002140. [PubMed] 10.1371/journal.pbio.1002140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prilusky J, Hodis E, Canner D, Decatur WA, Oberholser K, Martz E, Berchanski A, Harel M, Sussman JL. 2011. Proteopedia: A status report on the collaborative, 3D web-encyclopedia of proteins and other biomolecules. J Struct Biol 175:244–252. [PubMed] 10.1016/j.jsb.2011.04.011 [DOI] [PubMed] [Google Scholar]
- 22.Davie EW, Königsberger VV, Lipmann F. 1956. The isolation of a tryptophan-activating enzyme from pancreas. Arch Biochem Biophys 65:21–36. 10.1016/0003-9861(56)90173-4 [DOI] [PubMed] [Google Scholar]
- 23.Berg P. 1961. Specificity in protein synthesis. Annu Rev Biochem 30:293–322. 10.1146/annurev.bi.30.070161.001453 [DOI] [Google Scholar]
- 24.Berg P, Offengand EJ. 1958. An enzymatic mechanism for linking amino acids to RNA. Proc Natl Acad Sci USA 44:78–86. [PubMed] 10.1073/pnas.44.2.78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cusack S, Berthet-Colominas C, Härtlein M, Nassar N, Leberman R. 1990. A second class of synthetase structure revealed by X-ray analysis of Escherichia coli seryl-tRNA synthetase at 2.5Å. Nature 347:249–255. [PubMed] 10.1038/347249a0 [DOI] [PubMed] [Google Scholar]
- 26.Eriani G, Delarue M, Poch O, Gangloff J, Moras D. 1990. Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs. Nature 347:203–206. [PubMed] 10.1038/347203a0 [DOI] [PubMed] [Google Scholar]
- 27.Carter CW Jr, Duax WL. 2002. Did tRNA synthetase classes arise on opposite strands of the same gene? Mol Cell 10:705–708. [PubMed] 10.1016/S1097-2765(02)00688-3 [DOI] [PubMed] [Google Scholar]
- 28.Pham Y, Li L, Kim A, Erdogan O, Weinreb V, Butterfoss GL, Kuhlman B, Carter CW Jr. 2007. A minimal TrpRS catalytic domain supports sense/antisense ancestry of class I and II aminoacyl-tRNA synthetases. Mol Cell 25:851–862. [PubMed] 10.1016/j.molcel.2007.02.010 [DOI] [PubMed] [Google Scholar]
- 29.Ribas de Pouplana L, Schimmel P. 2001. Two classes of tRNA synthetases suggested by sterically compatible dockings on tRNA acceptor stem. Cell 104:191–193. [PubMed] 10.1016/S0092-8674(01)00204-5 [DOI] [PubMed] [Google Scholar]
- 30.Rodin AS, Szathmary E, Rodin SN. 2009. One ancestor for two codes viewed from the perspective of two complementary modes of tRNA aminoacylation. Biol Direct 4:4. [PubMed] 10.1186/1745-6150-4-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rodin SN, Rodin AS. 2006. Partitioning of aminoacyl-tRNA synthetases in two classes could have been encoded in a strand-symmetric RNA world. DNA Cell Biol 25:617–626. [PubMed] 10.1089/dna.2006.25.617 [DOI] [PubMed] [Google Scholar]
- 32.Smith TF, Hartman H. 2015. The evolution of class II aminoacyl-tRNA synthetases and the first code. FEBS Lett 589:3499–3507. [PubMed] 10.1016/j.febslet.2015.10.006 [DOI] [PubMed] [Google Scholar]
- 33.Williams TA, Wolfe KH, Fares MA. 2009. No rosetta stone for a sense-antisense origin of aminoacyl tRNA synthetase classes. Mol Biol Evol 26:445–450. [PubMed] 10.1093/molbev/msn267 [DOI] [PubMed] [Google Scholar]
- 34.Martinez-Rodriguez L, Erdogan O, Jimenez-Rodriguez M, Gonzalez-Rivera K, Williams T, Li L, Weinreb V, Collier M, Chandrasekaran SN, Ambroggio X, Kuhlman B, Carter CW Jr. 2015. Functional class I and II amino acid activating enzymes can be coded by opposite strands of the same gene. J Biol Chem 290:19710–19725. [PubMed] 10.1074/jbc.M115.642876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Normanly J, Ogden RC, Horvath SJ, Abelson J. 1986. Changing the identity of a transfer RNA. Nature 321:213–219. [PubMed] 10.1038/321213a0 [DOI] [PubMed] [Google Scholar]
- 36.Shimura Y, Ozeki H. 1973. Genetic study on transfer RNA. Adv Biophys 4:191–226. [PubMed] [PubMed] [Google Scholar]
- 37.Giegé R, Puglisi JD, Florentz C. 1993. tRNA structure and aminoacylation efficiency. Prog Nucleic Acid Res Mol Biol 45:129–206. [PubMed] 10.1016/S0079-6603(08)60869-7 [DOI] [PubMed] [Google Scholar]
- 38.Jakubowski H, Goldman E. 1992. Editing of errors in selection of amino acids for protein synthesis. Microbiol Rev 56:412–429. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wolf YI, Aravind L, Grishin NV Koonin EV. 1999. Evolution of aminoacyl-tRNA synthetases – analysis of unique domain architectures and phylogenetic trees reveals a complex history of horizontal gene transfer events. Genome Res 9:689–710. [PubMed] [PubMed] [Google Scholar]
- 40.O’Donoghue P, Luthey-Schulten Z. 2005. Evolutionary profiles derived from the QR factorization of multiple structural alignments gives an economy of information. J Mol Biol 346:875–894. [PubMed] 10.1016/j.jmb.2004.11.053 [DOI] [PubMed] [Google Scholar]
- 41.Sissler M, Lorber B, Messmer M, Schaller A, Pütz J, Florentz C. 2008. Handling mammalian mitochondrial tRNAs and aminoacyl-tRNA synthetases for functional and structural characterization. Methods 44:176–189. [PubMed] 10.1016/j.ymeth.2007.11.002 [DOI] [PubMed] [Google Scholar]
- 42.Baouz S, Schmitter J-M, Chenoune L, Beauvallet C, Blanquet S, Woisard A, Hountondji C. 2009. Primary structure revision and active site mapping of E. coli isoleucyl-tRNA synthetase by means of Maldi mass spectrometry. Open Biochem J 3:26–38. [PubMed] 10.2174/1874091X00903010026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kern D, Giegé R, Ebel J-P. 1981. Glycyl-tRNA synthetase from baker’s yeast. Interconversion between active and inactive forms of the enzyme. Biochemistry 20:122–131. [PubMed] 10.1021/bi00504a021 [DOI] [PubMed] [Google Scholar]
- 44.Paravisi S, Fumagalli G, Riva M, Morandi P, Morosi R, Konarev PV, Petoukhov MV, Bernier S, Chênevert R, Svergun DI, Curti B, Vanoni MA. 2009. Kinetic and mechanistic characterization of Mycobacterium tuberculosis glutamyl-tRNA synthetase and determination of its oligomeric structure in solution. FEBS J 276:1398–1417. [PubMed] 10.1111/j.1742-4658.2009.06880.x [DOI] [PubMed] [Google Scholar]
- 45.Terada T, Nureki O, Ishitani R, Ambrogelly A, Ibba M, Söll D, Yokoyama S. 2002. Functional convergence of two lysyl-tRNA synthetases with unrelated topologies. Nat Struct Biol 9:257–262. [PubMed] 10.1038/nsb777 [DOI] [PubMed] [Google Scholar]
- 46.NozawaK, O’Donoghue P, Gundllapalli S, Araiso Y, Ishitani R, Umehara T, Söll D, Nureki O. 2009. Pyrrolysyl-tRNA synthetase-tRNAPyl structure reveals the molecular basis of orthogonality. Nature 457:1163–1167. [PubMed] 10.1038/nature07611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yanagisawa T, Ishii R, Fukunaga R, Kobayashi T, Sakamoto K, Yokoyama S. 2008. Crystallographic studies on multiple conformational states of active-site loops in pyrrolysyl-tRNA synthetase. J Mol Biol 378:634–652. [PubMed] 10.1016/j.jmb.2008.02.045 [DOI] [PubMed] [Google Scholar]
- 48.Fukunaga R, Yokoyama S. 2007. Structural insights into the second step of RNA-dependent cysteine biosynthesis in archaea: Crystal structure of Sep-tRNA:Cys-tRNA synthase from Archaeoglobus fulgidus. J Mol Biol 370:128–141. [PubMed] 10.1016/j.jmb.2007.04.050 [DOI] [PubMed] [Google Scholar]
- 49.Kamtekar S, Hohn MJ, Park HS, Schnitzbauer M, Sauerwald A, Söll D, Steitz TA. 2007. Toward understanding phosphoseryl-tRNACys formation: The crystal structure of Methanococcus maripaludis phosphoseryl-tRNA synthetase. Proc Natl Acad Sci USA 104:2620–2625. [PubMed] 10.1073/pnas.0611504104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jasin M, Regan L, Schimmel P. 1983. Modular arrangement of functional domains along the sequence of an aminoacyl tRNA synthetase. Nature 306:441–447. [PubMed] 10.1038/306441a0 [DOI] [PubMed] [Google Scholar]
- 51.Giegé R, Touzé E, Lorber B, Théobald-Dietrich A, Sauter C. 2008. Crystallogenesis trends of free and liganded aminoacyl-tRNA synthetases. Cryst Growth Des 8:4297–4306. 10.1021/cg8007766 [DOI] [Google Scholar]
- 52.Winter GP, Hartley BS. 1977. The amino acid sequence of tryptophanyl-tRNA synthetase from Bacillus stearothermophilus. FEBS Lett 80:340–342. [PubMed] 10.1016/0014-5793(77)80471-7 [DOI] [PubMed] [Google Scholar]
- 53.Putney SD, Royal NJ, DeVegvar HN, Herlihy WC, Biemann K, Schimmel P. 1981. Primary structure of a large aminoacyl-tRNA synthetase. Science 213:1497–1501. [PubMed] 10.1126/science.7025207 [DOI] [PubMed] [Google Scholar]
- 54.Perna NT, Plunkett G 3rd, Burland V, Mau B, Glasner JD, Rose DJ, Mayhew GF, Evans PS, Gregor J, Kirkpatrick HA, Posfai G, Hackett J, Klink S, Boutin A, Shao Y, Miller L, Grotbeck EJ, Davis NW, Lim A, Dimalanta ET, Potamousis KD, Apodaca J, Anantharaman TS, Lin J, Yen G, Schwartz DC, Welch RA, Blattner FR. 2001. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature 409:529–533. [PubMed] 10.1038/35054089 [DOI] [PubMed] [Google Scholar]
- 55.Welch RA, Burland V, Plunkett G 3rd, Redford P, Roesch P, Rasko D, Buckles EL, Liou SR, Boutin A, Hackett J, Stroud D, Mayhew GF, Rose DJ, Zhou S, Schwartz DC, Perna NT, Mobley HL, Donnenberg MS, Blattner FR. 2002. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc Natl Acad Sci USA 99:17020–17024. [PubMed] 10.1073/pnas.252529799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Legendre M, Arslan D, Abergel C, Claverie J-M. 2012. Genomics of Megavirus and the elusive fourth domain of life. Commun Integr Biol 5:102–106. [PubMed] 10.4161/cib.18624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Raoult D, Audic S, Robert C, Abergel C, Renesto P, Ogata H, La Scola B, Suzan M, Claverie J-M. 2004. The 1.2-megabase genome sequence of Mimivirus. Science 306:1344–1350. [PubMed] 10.1126/science.1101485 [DOI] [PubMed] [Google Scholar]
- 58.Ruff M, Krishnaswamy S, Boeglin M, Poterszman A, Mitschler A, Podjarny A, Rees B, Thierry J-C, Moras D. 1991. Class II aminoacyl transfer RNA synthetases: crystal structure of yeast aspartyl-tRNA synthetase complexed with tRNAAsp. Science 252:1682–1689. [PubMed] 10.1126/science.2047877 [DOI] [PubMed] [Google Scholar]
- 59.Cusack S. 1993. Aminoacyl-tRNA synthetases. Curr Opin Struct Biol 3:39–44. 10.1016/0959-440X(93)90199-U [DOI] [PubMed] [Google Scholar]
- 60.Moras D. 1992. Aminoacyl-tRNA synthetases. Curr Opin Struct Biol 2:138–142. 10.1016/0959-440X(92)90189-E [DOI] [Google Scholar]
- 61.Schimmel P. 1991. Classes of aminoacyl-tRNA synthetases and the establishment of the genetic code. Trends Biochem Sci 16:1–2. 10.1016/0968-0004(91)90002-D [DOI] [PubMed] [Google Scholar]
- 62.Rossmann MG, Moras D, Olsen KW. 1974. Chemical and biological evolution of a nucleotide-binding protein. Nature 250:194–199. [PubMed] 10.1038/250194a0 [DOI] [PubMed] [Google Scholar]
- 63.Shen N, Guo L, Yang B, Jin Y, Ding J. 2006. Structure of human tryptophanyl-tRNA synthetase in complex with tRNATrp reveals the molecular basis of tRNA recognition and specificity. Nucleic Acids Res 34:3246–3258. [PubMed] 10.1093/nar/gkl441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yaremchuk A, Kriklivyi I, Tukalo M, Cusack S. 2002. Class I tyrosyl-tRNA synthetase has a class II mode of cognate tRNA recognition. EMBO J 21:3829–3840. [PubMed] 10.1093/emboj/cdf373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chinault AC, Tan KH, Hassur SM, Hecht SM. 1977. Initial position of aminoacylation of individual Escherichia coli, yeast, and calf liver transfer RNAs. Biochemistry 16:766–776. [PubMed] 10.1021/bi00623a031 [DOI] [PubMed] [Google Scholar]
- 66.Fraser TH, Rich A. 1975. Amino acids are not all initially attached to the same position on transfer RNA molecules. Proc Natl Acad Sci USA 72:3044–3048. [PubMed] 10.1073/pnas.72.8.3044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sprinzl M, Cramer M. 1975. Site of aminoacylation of tRNAs from Escherichia coli with respect to the 2′- or 3′-hydroxyl group of the terminal adenosine. Proc Natl Acad Sci USA 72:3049–3053. [PubMed] 10.1073/pnas.72.8.3049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.O’Donoghue P, Luthey-Schulten Z. 2003. On the evolution of structure in aminoacyl-tRNA synthetases. Microbiol Mol Bio. Rev 67:550–573. 10.1128/MMBR.67.4.550-573.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cammer S, Carter CW Jr. 2010. Six Rossmannoid folds, including the class I aminoacyl-tRNA synthetases, share a partial core with the anti-codon-binding domain of a class II aminoacyl-tRNA synthetase. Bioinformatics 26:709–714. [PubMed] 10.1093/bioinformatics/btq039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lapointe J, Duplain L, Proulx M. 1986. A single glutamyl-tRNA synthetase aminoacylates tRNAGlu and tRNAGln in Bacillus subtilis and efficiently misacylates Escherichia coli tRNAGln1 in vitro. J Bacteriol 165:88–93. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wilcox M, Nirenberg M. 1968. Transfer RNA as a cofactor coupling amino acid synthesis with that of protein. Proc Natl Acad Sci USA 61:229–236. [PubMed] 10.1073/pnas.61.1.229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ibba M, Söll, D. 2001. The renaissance of aminoacyl-tRNA synthesis. EMBO Rep 2:382–387. [PubMed] 10.1093/embo-reports/kve095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chang KM, Hendrickson TL. 2009. Recognition of tRNAGln by Helicobacter pylori GluRS2 – a tRNAGln-specific glutamyl-tRNA synthetase. Nucleic Acids Res 37:6942–6949. [PubMed] 10.1093/nar/gkp754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nehmé B, Ganga MA, Lonvaud-Funel A. 2006. The arginine deiminase locus of Oenococcus oeni includes a putative arginyl-tRNA synthetase ArgS2 at its 3′-end. Appl Microbiol Biotechnol 70:590–597. [PubMed] 10.1007/s00253-005-0095-6 [DOI] [PubMed] [Google Scholar]
- 75.Becker HD, Roy H, Moulinier L, Mazauric M-H, Keith G, Kern D. 2000. Thermus thermophilus contains an eubacterial and an archaebacterial aspartyl-tRNA synthetase. Biochemistry 39:3216–3230. [PubMed] 10.1021/bi992573y [DOI] [PubMed] [Google Scholar]
- 76.Sareen D, Steffek M, Newton GL, Fahey RC. 2002. ATP-dependent L-cysteine:1D-myo-inosityl 2-amino-2-deoxy-alpha-D-glucopyranoside ligase, mycothiol biosynthesis enzyme MshC, is related to class I cysteinyl-tRNA synthetases. Biochemistry 41:6885–6890. [PubMed] 10.1021/bi012212u [DOI] [PubMed] [Google Scholar]
- 77.Lee J, Hendrickson TL 2004. Divergent anticodon recognition in contrasting glutamyl-tRNA synthetases. J Mol Biol 344:1167–1174. 10.1016/j.jmb.2004.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Salazar, JC, Ahel I, Orellana O, Tumbula-Hansen D, Krieger R, Daniels L, Söll D. 2003. Coevolution of an aminoacyl-tRNA synthetase with its tRNA substrates. Proc Natl Acad Sci USA 100:13863–13868. [PubMed] 10.1073/pnas.1936123100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Skouloubris S, Ribas de Pouplana L, De Reuse H, Hendrickson TL. 2003. A noncognate aminoacyl-tRNA synthetase that may resolve a missing link in protein evolution. Proc Natl Acad Sci USA 100:11297–11302. [PubMed] 10.1073/pnas.1932482100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gilbart J, Perry CR, Slocombe B. 1993. High-level mupirocin resistance in Staphylococcus aureus: Evidence for two distinct isoleucyl-tRNA synthetases. Antimicrob Agents Chemother 37:32–38. [PubMed] 10.1128/AAC.37.1.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lévêque F, Plateau P, Dessen P, Blanquet S. 1990. Homology of lysS and lysYU, the two Escherichia coli genes encoding distinct lysysl-tRNA synthetas species. Nucleic Acids Res 18:305–312. [PubMed] 10.1093/nar/18.2.305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zeng Y, Roy H, Patil PB, Ibba M, Chen S. 2009. Characterization of two seryl-tRNA synthetases in albomycin-producing Streptomyces sp. ATCC 700974. Antimicrob Agents Chemother 53:4619–4627. [PubMed] 10.1128/AAC.00782-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Henkin TM, Glass BL, Grundy FJ. 1992. Analysis of the Bacillus subtilis tyrS gene: Conservation of a regulatory sequence in multiple tRNA synthetase genes. J Bacteriol 174:1299–1306. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Buddha MR, Crane BR. 2005. Structure and activity of an aminoacyl-tRNA synthetase that charges tRNA with nitro-tryptophan. Nat Struct Mol Biol 12:274–275. [PubMed] 10.1038/nsmb907 [DOI] [PubMed] [Google Scholar]
- 85.Brevet A, Chen J, Lévêque F, Blanquet S, Plateau P. 1995. Comparison of the enzymatic properties of the two Escherichia coli lysyl-tRNA synthetase species. J Biol Chem 270:14439–14444. [PubMed] 10.1074/jbc.270.24.14439 [DOI] [PubMed] [Google Scholar]
- 86.Kaniga K, Compton MS, Curtiss R 3rd, Sundaram P. 1998. Molecular and functional characterization of Salmonella enterica serovar typhimurium poxA gene: Effect on attenuation of virulence and protection. Infect Immun 66:5599–5606. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lenhard B, Orellana O, Ibba M, Weygand-Durasevic I. 1999. tRNA recognition and evolution of determinants in seryl-tRNA synthesis. Nucleic Acids Res 27:721–729. [PubMed] 10.1093/nar/27.3.721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Guo M, Yang, XL. 2014. Architecture and metamorphosis. Top Curr Chem 344:89–118. [PubMed] 10.1007/128_2013_424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Frugier M, Moulinier L, Giegé R. 2000. A domain in the N-terminal extension of class IIb eukaryotic aminoacyl-tRNA synthetases is important for tRNA binding. EMBO J 19:2371–2380. [PubMed] 10.1093/emboj/19.10.2371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Francin M, Kaminska M, Kerjan P, Mirande M. 2002. The N-domain of mammalian lysyl-tRNA synthetase is a functional tRNA binding domain. J Biol Chem 277:1762–1769. [PubMed] 10.1074/jbc.M109759200 [DOI] [PubMed] [Google Scholar]
- 91.Ray PS, Sullivan JC, Jia J, Francis J, Finnerty JR, Fox PL. 2011. Evolution of function of a fused metazoan tRNA synthetase. Mol Biol Evol 28:437–447. [PubMed] 10.1093/molbev/msq246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Antonellis A, Green ED. 2008. The role of aminoacyl-tRNA synthetases in genetic diseases. Annu Rev Genomics Hum Genet 9:87–107. [PubMed] 10.1146/annurev.genom.9.081307.164204 [DOI] [PubMed] [Google Scholar]
- 93.Park SG, Schimmel P, Kim S. 2008. Aminoacyl tRNA synthetases and their connections to disease. Proc Natl Acad Sci USA 105:11043–11049. [PubMed] 10.1073/pnas.0802862105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yao P, Fox PL. 2013. Aminoacyl-tRNA synthetases in medicine and disease. EMBO Mol Med 5:332–343. [PubMed] 10.1002/emmm.201100626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu J, Shue E, Ewalt KL, Schimmel P. 2004. A new gamma-interferon-inducible promoter and splice variants of an anti-angiogenic human tRNA synthetase. Nucleic Acids Res 32:719–727. [PubMed] 10.1093/nar/gkh240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yu Y, Liu Y, Shen N, Xu X, Xu F, Jia J, Jin Y, Arnold E, Ding J. 2004. Crystal structure of human tryptophanyl-tRNA synthetase catalytic fragment: Insights into substrate recognition, tRNA binding, and angiogenesis activity. J Biol Chem 279:8378–8388. [PubMed] 10.1074/jbc.M311284200 [DOI] [PubMed] [Google Scholar]
- 97.Greenberg Y, King M. Kiosses WB, Ewalt K, Yang X, Schimmel P, Reader JS, Tzima E. 2008. The novel fragment of tyrosyl-tRNA synthetase, mini-TyrRS, is secreted to induce an angiogenic response in endothelial cells. FASEB J 22:1597–1605. [PubMed] 10.1096/fj.07-9973com [DOI] [PubMed] [Google Scholar]
- 98.Arif A, Jia J, Mukhopadhyay R, Willard B, Kinter M, Fox PL. 2009. Two-site phosphorylation of EPRS coordinates multimodal regulation of noncanonical translational control activity. Mol Cell 35:164–180. [PubMed] 10.1016/j.molcel.2009.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kaminska M, Havrylenko S, Decottignies P, Le Maréchal P, Negrutskii B, Mirande M. 2009. Dynamic organization of aminoacyl-tRNA synthetase complexes in the cytoplasm of human cells. J Biol Chem 284:13746–13754. [PubMed] 10.1074/jbc.M900480200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Robinson JC, Kerjan P, Mirande M. 2000. Macromolecular assemblage of aminoacyl-tRNA synthetases: quantitative analysis of protein-protein interactions and mechanism of complex assembly. J Mol Biol 304:983–994. [PubMed] 10.1006/jmbi.2000.4242 [DOI] [PubMed] [Google Scholar]
- 101.Kyriacou SV, Deutscher MP. 2008. An important role for the multienzyme aminoacyl-tRNA synthetase complex in mammalian translation and cell growth. Mol Cell 29:419–427. [PubMed] 10.1016/j.molcel.2007.11.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Park SG, Choi EC, Kim S. 2010. Aminoacyl-tRNA synthetase-interacting multifunctional proteins (AIMPs): A triad for cellular homeostasis. IUBMB Life 62:296–302. [PubMed] [DOI] [PubMed] [Google Scholar]
- 103.KlipcanL, Levin I, Kessler N, Moor N, Finarov I, Safro M. 2008. The tRNA-induced conformational activation of human mitochondrial phenylalanyl-tRNA synthetase. Structure 16:1095–1104. [PubMed] 10.1016/j.str.2008.03.020 [DOI] [PubMed] [Google Scholar]
- 104.Sanni A,Walter P, Boulanger Y, Ebel J-P, Fasiolo F. 1991. Evolution of aminoacyl-tRNA synthetase quaternary structure and activity: Saccharomyces cerevisiae mitochondrial phenylalanyl-tRNA synthetase. Proc Natl Acad Sci USA 88:838–8391. 10.1073/pnas.88.19.8387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bour T, Akaddar A, Lorber B, Blais S, Balg C, Candolfi E, Frugier M. 2009. Plasmodial aspartyl-tRNA synthetases and peculiarities in Plasmodium falciparum. J Biol Chem 284:18893–18903. [PubMed] 10.1074/jbc.M109.015297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Merritt EA,Arakaki TL, Larson ET, Kelley A, Mueller N, Napuli AJ, Zhang L, De Titta G, Luft J, Verlinde CL, Fan E, Zucker F, Buckner FS, Van Voorhis WC, Hol WG. 2010. Crystal structure of the aspartyl-tRNA synthetase from Entamoeba histolytica. Mol Biochem Parasitol 169:95–100. [PubMed] 10.1016/j.molbiopara.2009.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Merritt EA, Arakaki TL, Gillespie JR, Larson ET, Kelley A, Mueller N, Napuli AJ, Kim J, Zhang L, Verlinde CL, Fan E, Zucker F, Buckner FS, van Voorhis WC, Hol WG. 2010. Crystal structures of trypanosomal histidyl-tRNA synthetase illuminate differences between eukaryotic and prokaryotic homologs. J Mol Biol 397:481–494. [PubMed] 10.1016/j.jmb.2010.01.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.SauerwaldA, Zhu W, Major TA, Roy H, Palioura S, Jahn D, Whitman WB, Yates JR 3rd, Ibba M, Söll D. 2005. RNA-dependent cysteine biosynthesis in archaea. Science 307:1969–1972. [PubMed] 10.1126/science.1108329 [DOI] [PubMed] [Google Scholar]
- 109.Gaston MA, Jiang R, Krzycki JA. 2011. Functional context, biosynthesis, and genetic encoding of pyrrolysine. Curr Opin Microbiol 14:342–349. [PubMed] 10.1016/j.mib.2011.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.PolycarpoC, Ambrogelly A, Bérubé A, Winbush SM, McCloskey JA, Crain PF, Wood JL, Söll D. 2004. An aminoacyl-tRNA synthetase that specifically activates pyrrolysine. Proc Natl Acad Sci USA 101:12450–12454. [PubMed] 10.1073/pnas.0405362101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hauenstein SI, Hou Y-M, Perona JJ. 2008. The homotetrameric phosphoseryl-tRNA synthetase from Methanosarcina mazei exhibits half-of-the-sites activity. J Biol Chem 283:21997–22006. [PubMed] 10.1074/jbc.M801838200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Fukunaga R, Yokoyama S. 2007. Structural insights into the first step of RNA-dependent cysteine biosynthesis in archaea. Nat Struct Mol Biol 14:272–279. [PubMed] 10.1038/nsmb1219 [DOI] [PubMed] [Google Scholar]
- 113.O’Donoghue P, Sethi A, Woese CR, Luthey-Schulten ZA. 2005. The evolutionary history of Cys-tRNACys formation. Proc Natl Acad Sci USA 102:19003–19008. [PubMed] 10.1073/pnas.0509617102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Englert M, Moses, S., Hohn, M., Ling, J., O’Donoghue, P., Söll D. 2013. Aminoacylation of tRNA 2′- or 3′-hydroxyl by phosphoseryl- and pyrrolysyl-tRNA synthetases. FEBS J 587:3360–3364. [PubMed] 10.1016/j.febslet.2013.08.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Imanaka T, Lee S, Takagi M, Fujiwara S. 1995. Aspartyl-tRNA synthetase of the hyperthermophilic archaeon Pyrococcus sp. KOD1 has a chimerical structure of eukaryotic and bacterial enzymes. Gene 164:153–156. [PubMed] 10.1016/0378-1119(95)00491-N [DOI] [PubMed] [Google Scholar]
- 116.Korencic D, Ahel I, Schelert J, Sacher M, Ruan B, Stathopoulos C, Blum P, Ibba M, Söll D. 2004. A freestanding proofreading domain is required for protein synthesis quality control in Archaea. Proc Natl Acad Sci USA 101:10260–10265. [PubMed] 10.1073/pnas.0403926101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shimizu S, Juan EC, Sato Y, Miyashita Y-i, Hoque MM, Suzuki K, Sagara T, Tsunoda M, Sekiguchi T, Dock-Bregeon A-C, Moras D, Takénaka A. 2009. Two complementary enzymes for threonylation of tRNA in Crenarchaeota: Crystal structure of Aeropyrum pernix threonyl-tRNA synthetase lacking a cis-editing domain. J Mol Biol 394:286–296. [PubMed] 10.1016/j.jmb.2009.09.018 [DOI] [PubMed] [Google Scholar]
- 118.Hussain T, Kruparani SP, Pal B, Dock-Bregeon A-C, Dwivedi S, Shekar MR, Sureshbabu K, Sankaranarayanan R. 2006. Post-transfer editing mechanism of a D-aminoacyl-tRNA deacylase-like domain in threonyl-tRNA synthetase from archaea. EMBO J 25:4152–4162. [PubMed] 10.1038/sj.emboj.7601278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sasaki HM, Sekine S, Sengoku T, Fukunaga R, Hattori M, Utsunomiya Y, Kuroishi C, Kuramitsu S, Shirouzu M, Yokoyama S. 2006. Structural and mutational studies of the amino acid-editing domain from archaeal/eukaryal phenylalanyl-tRNA synthetase. Proc Natl Acad Sci USA 103:14744–14749. [PubMed] 10.1073/pnas.0603182103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Waller J-P, Risler J, Monteilhet C, Zelwer C. 1971. Crystallisation of trypsin-modified methionyl-tRNA synthetase from Escherichia coli. FEBS Lett 16:186–188. [PubMed] 10.1016/0014-5793(71)80128-X [DOI] [PubMed] [Google Scholar]
- 121.Crépin T, Schmitt E, Mechulam Y, Sampson PB, Vaughan MD, Honek JF, Blanquet S. 2003. Use of analogues of methionine and methionyl adenylate to sample conformational changes during catalysis in Escherichia coli methionyl-tRNA synthetase. J Mol Biol 332:59–72. [PubMed] 10.1016/S0022-2836(03)00917-3 [DOI] [PubMed] [Google Scholar]
- 122.Mechulam Y, Schmitt E, Maveyraud L, Zelwer C, Nureki O, Yokoyama S, Konno M, Blanquet S. 1999. Crystal structure of Escherichia coli methionyl-tRNA synthetase highlights species-specific features. J Mol Biol 294:1287–1297. [PubMed] 10.1006/jmbi.1999.3339 [DOI] [PubMed] [Google Scholar]
- 123.Serre L, Verdon G, Choinowski T, Hervouet N, Risler J-L, Zelwer C. 2001. How methionyl-tRNA synthetase creates its amino acid recognition pocket upon L-methionine binding. J Mol Biol 306:863–876. [PubMed] 10.1006/jmbi.2001.4408 [DOI] [PubMed] [Google Scholar]
- 124.Sugiura I, Nureki O, Ugaji-Yoshikawa Y, Kuwabara S, Shimada A, Tateno M, Lorber B, Giegé R, Moras D, Yokoyama S, Konno M. 2000. The 2.0Å crystal structure of Thermus thermophilus methionyl-tRNA synthetase reveals two RNA-binding modules. Structure 8:197–208. 10.1016/S0969-2126(00)00095-2 [DOI] [PubMed] [Google Scholar]
- 125.Giegé R, Lorber B, Ebel J-P, Moras D, Thierry J-C. 1980 Crystallization of the complex formed between yeast aspartyl tRNA and its specific aminoacyl tRNA synthetase. C R Acad Sci Paris D 291:393–396. [PubMed] [PubMed] [Google Scholar]
- 126.Lorber B, Giegé R, Ebel J-P, Berthet C, Thierry J-C, Moras D. 1983. Crystallization of a tRNA-aminoacyl-tRNA synthetase complex. Characterization and first crystallographic data. J Biol Chem 258:8429–8435. [PubMed] [PubMed] [Google Scholar]
- 127.Florentz C, Kern D, Giegé R. 1990. Stimulatory effect of ammonium sulfate at high concentrations on the aminoacylation of tRNA and tRNA-like molecules. FEBS Lett 261:335–338. [PubMed] 10.1016/0014-5793(90)80585-7 [DOI] [PubMed] [Google Scholar]
- 128.Giegé R, Lorber B, Ebel J-P, Moras D, Thierry J-C, Jacrot B, Zaccaï G. 1982. Formation of a catalytically active complex between tRNAAsp and aspartyl-tRNA synthetase from yeast in high concentrations of ammonium sulphate. Biochimie 64:357–362. [PubMed] 10.1016/S0300-9084(82)80440-9 [DOI] [PubMed] [Google Scholar]
- 129.Bonnet J, Ebel J-P. 1975. Influence of various factors on the recognition specificity of tRNAs by yeast valyl-tRNA synthetase. Eur J Biochem 58:193–201. [PubMed] 10.1111/j.1432-1033.1975.tb02364.x [DOI] [PubMed] [Google Scholar]
- 130.Smith DW. 1969. The effect of salt solutions on the acceptance of amino acids by transfer ribonucleic acid. J Biol Chem 244:896–901. [PubMed] [PubMed] [Google Scholar]
- 131.Perona JJ, Swanson R, Steitz TA, Söll D. 1988. Overproduction and purification of Escherichia coli tRNAGln2 and its use in crystallization of glutaminyl-tRNA synthetase-tRNAGln complex. J Mol Biol 202:121–126. [PubMed] 10.1016/0022-2836(88)90524-4 [DOI] [PubMed] [Google Scholar]
- 132.Yaremchuk AD, Tukalo MA, Krikliviy I, Malchenko N, Biou V, Berthet-Colominas C, Cusack S. 1992. A new crystal form of the complex between seryl-tRNA synthetase and tRNASer from Thermus thermophilus that diffracts to 2.8Å resolution. FEBS Lett 310:157–161. [PubMed] 10.1016/0014-5793(92)81319-H [DOI] [PubMed] [Google Scholar]
- 133.Reshetnikova L, Khodyreva S, Lavrik O, Ankilova V, Frolow F, Safro M. 1993. Crystals of the phenylalanyl-tRNA synthetase from Thermus thermophilus HB8 complexed with tRNAPhe. J Mol Biol 231:927–929. [PubMed] 10.1006/jmbi.1993.1338 [DOI] [PubMed] [Google Scholar]
- 134.Swairjo MA, Schimmel PR. 2005. Breaking sieve for steric exclusion of a noncognate amino acid from active site of a tRNA synthetase. Proc Natl Acad Sci USA 102:988–993. [PubMed] 10.1073/pnas.0409024102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Naganuma M, Sekine S, Fukunaga R, Yokoyama S. 2009. Unique protein architecture of alanyl-tRNA synthetase for aminoacylation, editing, and dimerization. Proc Natl Acad Sci USA 106:8489–8494. [PubMed] 10.1073/pnas.0901572106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ibba M, Bono JL, Rosa PA, Söll D. 1997. Archaeal-type lysyl-tRNA synthetase in the Lyme disease spirochete Borrelia burgdorferi. Proc Natl Acad Sci USA 94:14383–14388. [PubMed] 10.1073/pnas.94.26.14383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tukalo M, Yaremchuk A, Fukunaga R, Yokoyama S, Cusack S. 2005. The crystal structure of leucyl-tRNA synthetase complexed with tRNALeu in the post-transfer-editing conformation. Nat Struct Mol Biol 12:923–930. [PubMed] 10.1038/nsmb986 [DOI] [PubMed] [Google Scholar]
- 138.Silvian LF, Wang J, Steitz, TA. 1999. Insights into editing from an ile-tRNA synthetase structure with tRNAIle and mupirocin. Science 285:1074–1077. [PubMed] 10.1126/science.285.5430.1074 [DOI] [PubMed] [Google Scholar]
- 139.Fukai S, Nureki O, Sekine S, Shimada A, Tao J, Vassylyev DG, Yokoyama S. 2000. Structural basis for double-sieve discrimination of L-valine from L-isoleucine and L-threonine by the complex of tRNAVal and valyl-tRNA synthetase. Cell 103:793–803. 10.1016/S0092-8674(00)00182-3 [DOI] [PubMed] [Google Scholar]
- 140.Shimada A, Nureki O, Goto M, Takahashi S, Yokoyama S. 2001. Structural and mutational studies of the recognition of the arginine tRNA-specific major identity element, A20, by arginyl-tRNA synthetase. Proc Natl Acad Sci USA 98:13537–13542. [PubMed] 10.1073/pnas.231267998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hauenstein S, Zhang C-M, Hou Y-M, Perona JJ. 2004. Shape-selective RNA recognition by cysteinyl-tRNA synthetase. Nat Struct Mol Biol 11:1134–1141. [PubMed] 10.1038/nsmb849 [DOI] [PubMed] [Google Scholar]
- 142.Sekine S, Shichiri M, Bernier S, Chênevert R, Lapointe J, Yokoyama S. 2006. Structural bases of transfer RNA-dependent amino acid recognition and activation by glutamyl-tRNA synthetase. Structure 14:1791–1799. [PubMed] 10.1016/j.str.2006.10.005 [DOI] [PubMed] [Google Scholar]
- 143.Bullock TL, Uter N, Nissan TA, Perona JJ. 2003. Amino acid discrimination by a class I aminoacyl-tRNA synthetase specified by negative determinants. J Mol Biol 328:395–408. [PubMed] 10.1016/S0022-2836(03)00305-X [DOI] [PubMed] [Google Scholar]
- 144.Kobayashi T, Takimura T, Sekine R, Kelly VP, Kamata K, Sakamoto K, Nishimura S, Yokoyama S. 2005. Structural snapshots of the KMSKS loop rearrangement for amino acid activation by bacterial tyrosyl-tRNA synthetase. J Mol Biol 346:105–117. [PubMed] 10.1016/j.jmb.2004.11.034 [DOI] [PubMed] [Google Scholar]
- 145.Sankaranarayanan R, Dock-Bregeon A-C, Romby P, Caillet J, Springer M, Rees B, Ehresmann C, Ehresmann B, Moras D. 1999. The structure of threonyl-tRNA synthetase-tRNAThr complex enlightens its repressor activity and reveals an essential zinc ion in the active site. Cell 97:371–381. [PubMed] 10.1016/S0092-8674(00)80746-1 [DOI] [PubMed] [Google Scholar]
- 146.Yaremchuk A, Tukalo M, Grotli M, Cusack S. 2001. A succession of substrate induced conformational changes ensures the amino acid specificity of Thermus thermophilus prolyl-tRNA synthetase: Comparison with histidyl-tRNA synthetase. J Mol Biol 309:989–1002. [PubMed] 10.1006/jmbi.2001.4712 [DOI] [PubMed] [Google Scholar]
- 147.Arnez JG, Augustine JG, Moras D, Francklyn CS. 1997. The first step of aminoacylation at the atomic level in histidyl-tRNA synthetase. Proc Natl Acad Sci USA 94:7144–7149. [PubMed] 10.1073/pnas.94.14.7144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Logan D, Mazauric M-H, Kern D, Moras D. 1995. Crystal structure of glycyl-tRNA synthetase from Thermus thermophilus. EMBO J 14:4156–4167. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Eiler S, Dock-Bregeon A-C, Moulinier L, Thierry J-C, Moras D. 1999. Synthesis of aspartyl-tRNAAsp in Escherichia coli - a snapshot of the second step. EMBO J 18:6532–6541. [PubMed] 10.1093/emboj/18.22.6532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Berthet-Colominas C, Seignovert L, Härtlein M, Grotli M, Cusack S, Leberman R. 1998. The crystal structure of asparaginyl-tRNA synthetase from Thermus thermophilus and its complexes with ATP and asparaginyl-adenylate: The mechanism of discrimination between asparagine and aspartic acid. EMBO J 17:2947–2960. [PubMed] 10.1093/emboj/17.10.2947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Desogus G, Todone F, Brick P, Onesti S. 2000. Active site of lysyl-tRNA synthetase: Structural studies of the adenylation reaction. Biochemistry 39:8418–8425. [PubMed] 10.1021/bi0006722 [DOI] [PubMed] [Google Scholar]
- 152.Moor N, Kotik-Kogan O, Tworowski D, Sukhanova M, Safro M. 2006. The crystal structure of the ternary complex of phenylalanyl-tRNA synthetase with tRNAPhe and a phenylalanyl-adenylate analogue reveals a conformational switch of the CCA end. Biochemistry 45:10572–10583. [PubMed] 10.1021/bi060491l [DOI] [PubMed] [Google Scholar]
- 153.Chopra S, Palencia A, Virus C, Tripathy A, Temple BR, Velazquez-Campoy A, Cusack S, Reader JS. 2013. Plant tumour biocontrol agent employs a tRNA-dependent mechanism to inhibit leucyl-tRNA synthetase. Nat Commun 4:1417. [PubMed] 10.1038/ncomms2421 [DOI] [PubMed] [Google Scholar]
- 154.Hernandez V, Crépin T, Palencia A, Cusack S, Akama T, Baker SJ, Bu W, Feng L, Freund YR, Liu L, Meewan M, Mohan M, Mao W, Rock FL, Sexton H, Sheoran A, Zhang Y, Zhang YK, Zhou Y, Nieman JA, Anugula MR, Keramane el M, Savariraj K, Reddy DS, Sharma R, Subedi R, Singh R, O’Leary A, Simon NL, De Marsh PL, Mushtaq S, Warner M, Livermore DM, Alley MR, Plattner JJ. 2013. Discovery of a novel class of boron-based antibacterials with activity against gram-negative bacteria. Antimicrob Agents Chemother 57:1394–1403. [PubMed] 10.1128/AAC.02058-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Li L, Palencia A, Lukk T, Li Z, Luthey-Schulten ZA, Cusack S, Martinis SA, Boniecki MT. 2013. Leucyl-tRNA synthetase editing domain functions as a molecular rheostat to control codon ambiguity in Mycoplasma pathogens. Proc Natl Acad Sci USA 110:3817–3822. [PubMed] 10.1073/pnas.1218374110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Rock FL, Mao W, Yaremchuk A, Tukalo M, Crépin T, Zhou H, Zhang Y-K, Hernandez V, Akama T, Baker SJ, Plattner JJ, Shapiro L, Martinis SA, Benkovic SJ, Cusack S, Alley MR. 2007. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science 316:1759–1761. [PubMed] 10.1126/science.1142189 [DOI] [PubMed] [Google Scholar]
- 157.Nakama T, Nureki O, Yokoyama S. 2001. Structural basis for the recognition of isoleucyl-adenylate and an antibiotic, mupirocin, by isoleucyl-tRNA synthetase. J Biol Chem 276:47387–47393. [PubMed] 10.1074/jbc.M109089200 [DOI] [PubMed] [Google Scholar]
- 158.Ingvarsson H Unge T. 2010. Flexibility and communication within the structure of the Mycobacterium smegmatis methionyl-tRNA synthetase. FEBS J 277:3947–3962. [PubMed] 10.1111/j.1742-4658.2010.07784.x [DOI] [PubMed] [Google Scholar]
- 159.Teng M, Hilgers M, Cunningham M, Borchardt A, Locke J, Abraham S, Haley G, Kwan B, Hall C, Hough G, Finn J, Shaw K. 2013. Identification of bacteria selective threonyl-tRNA synthetase (ThrRS) substrate inhibitors by structure-based design. J Med Chem 56:1748–1760. [PubMed] 10.1021/jm301756m [DOI] [PubMed] [Google Scholar]
- 160.Qiu X, Janson CA, Smith WW, Green SM, McDevitt P, Johanson K, Carter P, Hibbs M, Lewis C, Chalker A, Fosberry A, Lalonde J, Berge J, Brown P, Houge-Frydrych CS, Jarvest RL. 2001. Crystal structure of Staphylococcus aureus tyrosyl-tRNA synthetase in complex with a class of potent and specific inhibitors. Protein Sci 10:2008–2016. [PubMed] 10.1110/ps.18001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Charron C, Roy H, Blaise M, Giegé R, Kern D. 2003. Non-discriminating and discriminating aspartyl-tRNA synthetases differ in the anticodon-binding domain. EMBO J 22:1632–1643. [PubMed] 10.1093/emboj/cdg148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Schmitt E, Moulinier L, Fujiwara S, Imanaka T, Thierry J-C, Moras D. 1998. Crystal structure of aspartyl-tRNA synthetase from Pyrococcus kodakaraensis KOD: Archeon specificity and catalytic mechanism of adenylate formation. EMBO J 17:5227–5237. [PubMed] 10.1093/emboj/17.17.5227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Delarue M, Poterszman A, Nikonov S, Garber M, Moras D, Thierry J-C. 1994. Crystal structure of a prokaryotic aspartyl-tRNA synthetase. EMBO J 13:3219–3229. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ng JD, Sauter C, Lorber B, Kirkland N, Arnez J, Giegé R. 2002. Comparative analysis of space-grown and earth-grown crystals of an aminoacyl-tRNA synthetase: Space-grown crystals are more useful for structural determination. Acta Crystallgr D 58:645–652. 10.1107/S0907444902003177 [DOI] [PubMed] [Google Scholar]
- 165.Rees B, Webster G, Delarue M, Boeglin M, Moras D. 2000. Aspartyl-tRNA synthetase from Escherichia coli: Flexibility and adaptability to the substrates. J Mol Biol 299:1157–1164. [PubMed] 10.1006/jmbi.2000.3792 [DOI] [PubMed] [Google Scholar]
- 166.Sauter C, Lorber E, Cavarelli J, Moras D, Giegé R. 2000. The free yeast aspartyl-tRNA synthetase differs from the tRNAAsp-complexed enzyme by structural changes in the catalytic site, hinge region, and anticodon-binding domain. J Mol Biol 299:1313–1324. [PubMed] 10.1006/jmbi.2000.3791 [DOI] [PubMed] [Google Scholar]
- 167.Schmitt E, Tanrikulu IC, Yoo TH, Panvert M, Tirrell DA, Mechulam Y. 2009. Switching from an induced fit to a lock and key mechanism in an aminoacyl-tRNA synthetase with modified specificity. J Mol Biol 394:843–851. [PubMed] 10.1016/j.jmb.2009.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Cassio D, Waller J-P. 1971. Modification of methionyl-tRNA synthetase by proteolytic cleavage and properties of the trypsin-modified enzyme. Eur J Biochem 20:283–300. [PubMed] 10.1111/j.1432-1033.1971.tb01393.x [DOI] [PubMed] [Google Scholar]
- 169.Fourmy D, Dardel F, Blanquet S. 1993. Methionyl-tRNA synthetase zinc binding domain. Three-dimensional structure and homology with rubredoxin and gag retroviral proteins. J Mol Biol 231:1078–1089. [PubMed] 10.1006/jmbi.1993.1353 [DOI] [PubMed] [Google Scholar]
- 170.Cusack S, Yaremchuk A, Tukalo M. 2000. The 2Å crystal structure of leucyl-tRNA synthetase and its complex with a leucyl-adenylate analogue. EMBO J 19:2351–2361. [PubMed] 10.1093/emboj/19.10.2351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Vu MT, Martinis SA. 2007. A unique insert of leucyl-tRNA synthetase is required for aminoacylation and not amino acid editing. Biochemistry 46:5170–5176. [PubMed] 10.1021/bi062078j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hsu JL, Martinis SA. 2008. A flexible peptide tether controls accessibility of a unique C-terminal RNA-binding domain in leucyl-tRNA synthetases. J Mol Biol 376:482–491. [PubMed] 10.1016/j.jmb.2007.11.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Xu M-G, Chen J-F, Martin F, Zhao M-W, Eriani G, Wang E-D. 2002. Leucyl-tRNA synthetase consisting of two subunits from hyperthermophilic bacteria Aquifex aeolicus. J Biol Chem 277:41590–41596. [PubMed] 10.1074/jbc.M205126200 [DOI] [PubMed] [Google Scholar]
- 174.Yamane T, Hopfield JJ. 1977. Experimental evidence for kinetic proofreading in the aminoacylation of tRNA by synthetase. Proc Natl Acad Sci USA 74:2246–2250. [PubMed] 10.1073/pnas.74.6.2246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Newberry KJ, Hou Y-M, Perona JJ. 2002. Structural origins of amino acid selection without editing by cysteinyl-tRNA synthetase. EMBO J 21:2778–2787. [PubMed] 10.1093/emboj/21.11.2778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Fersht AR, Dingwall C. 1979. Cysteinyl-tRNA synthetase from Escherichia coli does not need an editing mechanism to reject serine and alanine. High binding energy of small groups in specific molecular interactions. Biochemistry 18:1245–1249. [PubMed] 10.1021/bi00574a020 [DOI] [PubMed] [Google Scholar]
- 177.Cavarelli J, Delagoutte B, Eriani G, Gangloff J, Moras D. 1998. L-arginine recognition by yeast arginyl-tRNA synthetase. EMBO J 17:5438–5448. [PubMed] 10.1093/emboj/17.18.5438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Bi, K, Zheng, Y, Gao, F, Dong, J, Wang, J, Wang, Y, Gong, W. 2014. Crystal structure of E. coli arginyl-tRNA synthetase and ligand binding studies revealed key residues in arginine recognition. Protein Cell 5:151–159. [PubMed] 10.1007/s13238-013-0012-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Rould MA, Perona JJ, Söll D, Steitz TA. 1989. Structure of E. coli glutaminyl-tRNA synthetase complexed with tRNAGln and ATP at 2.8Å resolution. Science 246:1135–1142. [PubMed] 10.1126/science.2479982 [DOI] [PubMed] [Google Scholar]
- 180.Nureki O, Vassylyev DG, Katayanagi K, Shimizu T, Sekine S-i, Kigawa T, Miyazawa T, Yokoyama S, Morikawa K. 1995. Architectures of class-defining and specific domains of glutamyl-tRNA synthetase. Science 267:1958–1965. [PubMed] 10.1126/science.7701318 [DOI] [PubMed] [Google Scholar]
- 181.Makarova KS, Aravind L, Koonin EV. 2002. SWIM, a novel Zn-chelating domain present in bacteria, archaea and eukaryotes. Trends Biochem Sci 27:384–386. [PubMed] 10.1016/S0968-0004(02)02140-0 [DOI] [PubMed] [Google Scholar]
- 182.Banerjee R, Dubois DY, Gauthier J, Lin S-X, Roy S, Lapointe J. 2004. The zinc-binding site of a class I aminoacyl-tRNA synthetase is a SWIM domain that modulates amino acid binding via the tRNA acceptor arm. Eur J Biochem 271:724–733. [PubMed] 10.1111/j.1432-1033.2003.03976.x [DOI] [PubMed] [Google Scholar]
- 183.Perona JJ, Rould MA, Steitz TA, Rissler J-L, Zelwer C, Brunie S. 1991. Structural similarities in glutaminyl- and methionyl-tRNA synthetases suggest a common overall orientation of tRNA binding. Proc Natl Acad Sci USA 88:2903–2907. [PubMed] 10.1073/pnas.88.7.2903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Siatecka M, Rozek M, Barciszewski J, Mirande M. 1998. Modular evolution of the Glx-tRNA synthetase family – rooting of the evolutionary tree between the bacteria and archaea/eukarya branches. Eur J Biochem 256:80–87. [PubMed] 10.1046/j.1432-1327.1998.2560080.x [DOI] [PubMed] [Google Scholar]
- 185.Sekine S, Nureki O, Shimada A, Vassylyev D, Yokoyama S. 2001. Structural basis for anticodon recognition by discriminating glutamyl-tRNA synthetase. Nat Struct Mol Biol 8:203–206. [PubMed] 10.1038/84927 [DOI] [PubMed] [Google Scholar]
- 186.Ito T, Yokoyama S. 2010. Two enzymes bound to one tRNA assume alternative conformations for consecutive reactions. Nature 467:612–616. [PubMed] 10.1038/nature09411 [DOI] [PubMed] [Google Scholar]
- 187.Blaise M, Bailly M, Fréchin M, Behrens MA, Fischer F, Oliveira CL, Becker HD, Pedersen JS, Thirup S, Kern D. 2010. Crystal structure of a transfer-ribonucleoprotein particle that promotes asparagine formation. EMBO J 29:3118–3129. [PubMed] 10.1038/emboj.2010.192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Giegé R, Sissler M, Florentz C. 1998. Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res 26:5017–5035. [PubMed] 10.1093/nar/26.22.5017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Bailly M, Giannouli S, Blaise M, Stathopoulos C, Kern D, Becker HD. 2006. A single tRNA base pair mediates bacterial tRNA-dependent biosynthesis of asparagine. Nucleic Acids Res 34:6083–6094. [PubMed] 10.1093/nar/gkl622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ibba M, Losey HC, Kawarabayasi Y, Kikuchi H, Bunjun S, Söll D. 1999. Substrate recognition by class I lysyl-tRNA synthetases: A molecular basis for gene displacement. Proc Natl Acad Sci USA 96:418–423. [PubMed] 10.1073/pnas.96.2.418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Doublié S, Bricogne G, Gilmore C, Carter CW Jr. 1995. Tryptophanyl-tRNA synthetase crystal structure reveals an unexpected homology to tyrosyl-tRNA synthetase. Structure 3:17–31. [PubMed] 10.1016/S0969-2126(01)00132-0 [DOI] [PubMed] [Google Scholar]
- 192.Tzima E, Schimmel P. 2006. Inhibition of tumor angiogenesis by a natural fragment of a tRNA synthetase. Trends Biochem Sci 31:7–10. [PubMed] 10.1016/j.tibs.2005.11.002 [DOI] [PubMed] [Google Scholar]
- 193.Bedouelle H, Winter G. 1986. A model of synthetase/transfer RNA interaction as deduced by protein engineering. Nature 320:371–373. [PubMed] 10.1038/320371a0 [DOI] [PubMed] [Google Scholar]
- 194.Bonnefond L, Giegé R, Rudinger-Thirion J. 2005. Evolution of the tRNATyr/TyrRS aminoacylation systems. Biochimie 87:873–883. [PubMed] 10.1016/j.biochi.2005.03.008 [DOI] [PubMed] [Google Scholar]
- 195.Ilyin VA, Temple B, Hu M, Li G, Yin Y, Vachette P, Carter CW Jr. 2000. 2.9Å crystal structure of ligand-free tryptophanyl-tRNA synthetase: Domain movements fragment the adenine nucleotide binding site. Protein Sci 9:218–231. [PubMed] 10.1110/ps.9.2.218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Brick P, Bhat TN, Blow DM. 1989. Structure of tyrosyl-tRNA synthetase refined at 2.3Å resolution. Interaction of the enzyme with the tyrosyl adenylate intermediate. J Mol Biol 208:83–98. 10.1016/0022-2836(89)90090-9 [DOI] [PubMed] [Google Scholar]
- 197.Guijarro JI, Pintar A, Prochnicka-Chalufour A, Guez V, Gilquin B, Bedouelle H, Delepierre M. 2002. Structure and dynamics of the anticodon arm binding domain of Bacillus stearothermophilus tyrosyl-tRNA synthetase. Structure 10:311–317. [PubMed] 10.1016/S0969-2126(02)00699-8 [DOI] [PubMed] [Google Scholar]
- 198.Pham Y, Kuhlman B, Butterfoss GL, Hu H, Weinreb V, Carter CW Jr. 2010. Tryptophanyl-tRNA synthetase Urzyme: A model to recapitulate molecular evolution and investigate intramolecular complementation. J Biol Chem 285:38590–38601. [PubMed] 10.1074/jbc.M110.136911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Retailleau P, Huang X, Yin Y, Hu M, Weinreb V, Vachette P, Vonrhein C, Bricogne G, Roversi P, Ilyin V, Carter CW Jr. 2003. Interconversion of ATP binding and conformational free energies by tryptophanyl-tRNA synthetase: structures of ATP bound to open and closed, pre-transition-state conformations. J Mol Biol 325:39–63. [PubMed] 10.1016/S0022-2836(02)01156-7 [DOI] [PubMed] [Google Scholar]
- 200.Retailleau P, Weinreb V, Hu M, Carter CW Jr. 2007. Crystal structure of tryptophanyl-tRNA synthetase complexed with adenosine-5′ tetraphosphate: Evidence for distributed use of catalytic binding energy in amino acid activation by class I aminoacyl-tRNA synthetases. J Mol Biol 369:108–128. [PubMed] 10.1016/j.jmb.2007.01.091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Han GW, Yang X-L, McMullan D, Chong YE, Krishna SS, Rife CL, Weekes D, Brittain SM, Abdubek P, Ambing E, Astakhova T, Axelrod HL, Carlton D, Caruthers J, Chiu H-J, Clayton T, Duan L, Feuerhelm J, Grant JC, Grzechnik SK, Jaroszewski L, Jin KK, Klock HE, Knuth MW, Kumar A, Marciano D, Miller MD, Morse AT, Nigoghossian E, Okach L, Paulsen J, Reyes R, van den Bedem H, White A, Wolf G, Xu Q, Hodgson KO, Wooley J, Deacon AM, Godzik A, Lesley SA, Elsliger MA, Schimmel P, Wilson IA. 2010. Structure of a tryptophanyl-tRNA synthetase containing an iron-sulfur cluster. Acta Crystallgr F 66:1326–1334. [PubMed] 10.1107/S1744309110037619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Pierrel F, Douki T, Fontecave M, Atta M. 2004. MiaB protein is a bifunctional radical-S-adenosylmethionine enzyme involved in thiolation and methylation of tRNA. J Biol Chem 279:47555–47563. [PubMed] 10.1074/jbc.M408562200 [DOI] [PubMed] [Google Scholar]
- 203.Biou V, Yaremchuk A, Tukalo M, Cusack S. 1994. The 2.9Å crystal structure of T. thermophilus seryl-tRNA synthetase complexed with tRNASer. Science 263:1404–1410. [PubMed] 10.1126/science.8128220 [DOI] [PubMed] [Google Scholar]
- 204.Gruic-Sovulj I, Rokov-Plavec J, Weygand-Durasevic I. 2007. Hydrolysis of non-cognate aminoacyl-adenylates by a class II aminoacyl-tRNA synthetase lacking an editing domain. FEBS Lett 581:5110–5114. [PubMed] 10.1016/j.febslet.2007.09.058 [DOI] [PubMed] [Google Scholar]
- 205.Romby P, Springer M. 2003. Bacterial translational control at atomic resolution. Trends Genet 19:155–161. [PubMed] 10.1016/S0168-9525(03)00020-9 [DOI] [PubMed] [Google Scholar]
- 206.Torres-Larios A, Dock-Bregeon A-C, Romby P, Rees B, Sankaranarayanan R, Caillet J, Springer M, Ehresmann C, Ehresmann B, Moras D. 2002. Structural basis of translational control by Escherichia coli threonyl-tRNA synthetase. Nat Struct Biol 9:343–347. [PubMed] 10.1038/nsb789 [DOI] [PubMed] [Google Scholar]
- 207.Beebe K, Ribas de Pouplana L, Schimmel P. 2003. Elucidation of tRNA-dependent editing by a class II tRNA synthetase and significance for cell viability. EMBO J 22:668–675. [PubMed] 10.1093/emboj/cdg065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Fukunaga R, Yokoyama S. 2007. Structure of the AlaX-M trans-editing enzyme from Pyrococcus horikoshii. Acta Crystallgr D 63:390–400. [PubMed] 10.1107/S090744490605640X [DOI] [PubMed] [Google Scholar]
- 209.Dock-Bregeon A-C, Rees B, Torres-Larios A, Bey G, Caillet J, Moras D. 2004. Achieving error-free translation; the mechanism of proofreading of threonyl-tRNA synthetase at atomic resolution. Mol Cell 16:375–386. 10.1016/S1097-2765(04)00591-X [DOI] [PubMed] [Google Scholar]
- 210.Torres-Larios A, Sankaranarayanan R, Rees B, Dock-Bregeon A-C, Moras D. 2003. Conformational movements and cooperativity upon amino acid, ATP and tRNA binding in threonyl-tRNA synthetase. J Mol Biol 331:201–211. [PubMed] 10.1016/S0022-2836(03)00719-8 [DOI] [PubMed] [Google Scholar]
- 211.Crépin T, Yaremchuk A, Tukalo M, Cusack S. 2006. Structures of two bacterial prolyl-tRNA synthetases with and without a cis-editing domain. Structure 14:1511–1525. [PubMed] 10.1016/j.str.2006.08.007 [DOI] [PubMed] [Google Scholar]
- 212.Yaremchuk A, Cusack S, Tukalo M. 2000. Crystal structure of a eukaryote/archaeon-like prolyl-tRNA synthetase and its complex with tRNAPro(CGG). EMBO J 19:4745–4758. [PubMed] 10.1093/emboj/19.17.4745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Arnez JG, Harris DC, Mitschler A, Rees B, Francklyn CS, Moras D. 1995. Crystal structure of histidyl-tRNA synthetase from Escherichia coli complexed with histidyl-adenylate. EMBO J 14:4143–4155. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Aberg A, Yaremchuk A, Tukalo M, Rasmussen B, Cusack S. 1997. Crystal structure analysis of the activation of histidine by Thermus thermophilus histidyl-tRNA synthetase. Biochemistry 36:3084–3094 [PubMed] 10.1021/bi9618373 [DOI] [PubMed] [Google Scholar]
- 215.Qiu X, Janson CA, Blackburn MN, Chhohan IK, Hibbs M, Abdel-Meguid SS. 1999. Cooperative structural dynamics and a novel fidelity mechanism in histidyl-tRNA synthetases. Biochemistry 38:12296–12304. [PubMed] 10.1021/bi990482v [DOI] [PubMed] [Google Scholar]
- 216.Guth E, Farris M, Bovee M, Francklyn CS. 2009. Asymmetric amino acid activation by class II histidyl-tRNA synthetase from Escherichia coli. J Biol Chem 284:20753–20762. [PubMed] 10.1074/jbc.M109.021311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Kaur G, Subramanian S. 2015. The insertion domain 1 of class IIA dimeric glycyl-tRNA synthetase is a rubredoxin-like zinc ribbon. J Struct Biol 190:38–46. [PubMed] 10.1016/j.jsb.2015.02.004 [DOI] [PubMed] [Google Scholar]
- 218.Murzin AG. 1993. OB (oligonucleotide/oligosaccharide binding)-binding fold: Common structural and functional solution for non-homologous sequences. EMBO J 12:861–867. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Commans S, Lazard M, Delort F, Blanquet S, Plateau P. 1998. tRNA anticodon recognition and specification within subclass IIb aminoacyl-tRNA synthetases. J Mol Biol 278:801–813. [PubMed] 10.1006/jmbi.1998.1711 [DOI] [PubMed] [Google Scholar]
- 220.Bernard D, Akochy PM, Beaulieu D, Lapointe J, Roy PH. 2006. Two residues in the anticodon recognition domain of the aspartyl-tRNA synthetase from Pseudomonas aeroginosa are individually implicated in the recognition of tRNAAsn. J Bacteriol 188:269–274. [PubMed] 10.1128/JB.188.1.269-274.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Chuawong P, Hendrickson TL. 2006. The nondiscriminating aspartyl-tRNA synthetase from Helicobacter pylori: Anticodon-binding domain mutations that impact tRNA specificity and heterologous toxicity. Biochemistry 45:8079–8087. [PubMed] 10.1021/bi060189c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Archontis G, Simonson T, Moras D, Karplus M. 1998. Specific amino acid recognition by aspartyl-tRNA synthetase studied by free energy simulations. J Mol Biol 275:823–846. [PubMed] 10.1006/jmbi.1997.1470 [DOI] [PubMed] [Google Scholar]
- 223.Iwasaki W, Sekine S, Kuroishi C, Kuramitsu S, Shirouzu M, Yokoyama S. 2006. Structural basis of the water-assisted asparagine recognition by asparaginyl-tRNA synthetase. J Mol Biol 360:329–342. [PubMed] 10.1016/j.jmb.2006.04.068 [DOI] [PubMed] [Google Scholar]
- 224.Onesti S, Miller AD, Brick P. 1995. The crystal structure of the lysyl-tRNA synthetase (LysU) from Escherichia coli. Structure 3:163–176. [PubMed] 10.1016/S0969-2126(01)00147-2 [DOI] [PubMed] [Google Scholar]
- 225.Brevet A, Chen J, Lévêque F, Plateau P, Blanquet S. 1989. In vivo synthesis of adenylylated bis(5′-nucleosidyl) tetraphosphates (Ap4N) by Escherichia coli aminoacyl-tRNA synthetases. Proc Natl Acad Sci USA 86:8275–8279. [PubMed] 10.1073/pnas.86.21.8275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Kisselev LL, Justesen J, Wolfson AD, Frolova LY. 1998. Diadenosine oligophosphates (Ap(n)A), a novel class of signalling molecules? FEBS Lett 427:157–163. [PubMed] 10.1016/S0014-5793(98)00420-7 [DOI] [PubMed] [Google Scholar]
- 227.Cusack S, Yaremchuk A, Tukalo M. 1996. The crystal structures of T. thermophilus lysyl-tRNA synthetase complexed with E. coli tRNALys and a T. thermophilus tRNALys transcript: Anticodon recognition and conformational changes upon binding of a lysyl-adenylate analogue. EMBO J 15:6321–6334. [PubMed] [PMC free article] [PubMed] [Google Scholar]
- 228.Sakurama H, Takita T, Mikami B, Itoh T, Yasukawa K, Inouye K. 2009. Two crystal structures of lysyl-tRNA synthetase from Bacillus stearothermophilus in complex with lysyladenylate-like compounds: Insights into the irreversible formation of the enzyme-bound adenylate of L-lysine hydroxamate. J Biochem 154:555–563. [PubMed] 10.1093/jb/mvp014 [DOI] [PubMed] [Google Scholar]
- 229.Navarre WW, Zou SB, Roy H, Xie JL, Savchenko A, Singer A, Edvokimova E, Prost LR, Kumar R, Ibba, M, Fang FC. 2010. PoxA, YjeK, and elongation factor P coordinately modulate virulence and drug resistance in Salmonella enterica. Mol Cell 39:209–221. [PubMed] 10.1016/j.molcel.2010.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Swairjo MA, Otero FJ, Yang X-L, Lovato MA, Skene RJ, McRee DE, Ribas de Pouplana L, Schimmel P. 2004. Alanyl-tRNA synthetase crystal structure and design for acceptor-stem recognition. Mol Cell 13:829–841. [PubMed] 10.1016/S1097-2765(04)00126-1 [DOI] [PubMed] [Google Scholar]
- 231.Fishman R, Ankilova V, Moor N, Safro M. 2001. Structure at 2.6Å resolution of phenylalanyl-tRNA synthetase complexed with phenylalanyl-adenylate in the presence of manganese. Acta Crystallgr D 57:1534–1544. [PubMed] 10.1107/S090744490101321X [DOI] [PubMed] [Google Scholar]
- 232.Goldgur Y, Mosyak L, Reshetnikova L, Ankilova V, Lavrik O, Khodyreva S, Safro M. 1997. The crystal structure of phenylalanyl-tRNA synthetase from Thermus thermophilus complexed with cognate tRNAPhe. Structure 5:59–68. [PubMed] 10.1016/S0969-2126(97)00166-4 [DOI] [PubMed] [Google Scholar]
- 233.Kotik-Kogan O, Moor N, Tworowski D, Safro M. 2005. Structural basis for discrimination of L-phenylalanine from L-tyrosine by phenylalanyl-tRNA synthetase. Structure 13:1799–1807. [PubMed] 10.1016/j.str.2005.08.013 [DOI] [PubMed] [Google Scholar]
- 234.Mosyak L, Reshetnikova L, Goldgur Y, Delarue M, Safro MG. 1995. Structure of phenylalanyl-tRNA synthetase from Thermus thermophilus. Nat Struct Biol 2:537–547. [PubMed] 10.1038/nsb0795-537 [DOI] [PubMed] [Google Scholar]
- 235.Mermershtain I, Finarov I, Klipcan L, Kessler N, Rozenberg H, Safro MG. 2011. Idiosyncrasy and identity in the prokaryotic phe-system: Crystal structure of E. coli phenylalanyl-tRNA synthetase complexed with phenylalanine and AMP. Protein Sci 20:160–167. [PubMed] 10.1002/pro.549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Safro M, Mosyak L. 1995. Structural similarities in the noncatalytic domains of phenylalanyl-tRNA and biotin synthetases. Protein Sci 4:2429–2432. [PubMed] 10.1002/pro.5560041122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Evdokimov AG, Mekel M, Hutchings K, Narasimhan L, Holler T, McGrath T, Beattie B, Fauman E, Yan C, Heaslet H, Walter R, Finzel B, Ohren J, McConnell P, Braden T, Sun F, Spessard C, Banotai C, Al-Kassim L, Ma W, Wengender P, Kole D, Garceau N, Toogood P, Liu J. 2008. Rational protein engineering in action: The first crystal structure of a phenylalanine tRNA synthetase from Staphylococcus haemolyticus. J Struct Biol 162:152–169. [PubMed] 10.1016/j.jsb.2007.11.002 [DOI] [PubMed] [Google Scholar]
- 238.Abibi A, Ferguson AD, Fleming PR, Gao N, Hajec LI, Hu J, Laganas VA, McKinney DC, McLeod SM, Prince DB, Shapiro AB, Buurman ET. 2014. The role of a novel auxiliary pocket in bacterial phenylalanyl-tRNA synthetase druggability. J Biol Chem 289:21651–22162. [PubMed] 10.1074/jbc.M114.574061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Luque I, Riera-Alberola M, Andujar A, Ochoa de Alda JA. 2008. Intraphylum diversity and complex evolution of cyanobacterial aminoacyl-tRNA synthetases. Mol Biol Evol 25:2369–2389. [PubMed] 10.1093/molbev/msn197 [DOI] [PubMed] [Google Scholar]
- 240.First EA. 2005. Catalysis of the tRNA aminoacylation reaction, p 328–352. In Ibba M, Francklyn C, and Cusack S (ed), The Aminoacyl-tRNA Synthetases. Landes Bioscience, Georgetown, TX. [Google Scholar]
- 241.Airas K. 1996. Differences in the magnesium dependences of the class I and class II aminoacyl-tRNA synthetases from Escherichia coli. Eur J Biochem 240:223–231. [PubMed] 10.1111/j.1432-1033.1996.0223h.x [DOI] [PubMed] [Google Scholar]
- 242.Perona JJ, Rould MA, Steitz TA. 1993. Structural basis for transfer RNA aminoacylation by glutaminyl-tRNA synthetase. Biochemistry 32:8758–8771. [PubMed] 10.1021/bi00085a006 [DOI] [PubMed] [Google Scholar]
- 243.Weinreb V, Li L, Carter Jr CW. 2012. A master switch couples Mg2+-assisted catalysis to domain motion in B. stearothermophilus tryptophanyl-tRNA synthetase. Structure 20:128–138. [PubMed] 10.1016/j.str.2011.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Rould MA, Perona JJ, Steitz TA. 1991. Structural basis of anticodon loop recognition by glutaminyl-tRNA synthetase. Nature 352:213–218. [PubMed] 10.1038/352213a0 [DOI] [PubMed] [Google Scholar]
- 245.Briand C, Poterszman A, Eiler S, Webster G, Thierry JC, Moras D. 2000. An intermediate step in the recognition of tRNAAsp by aspartyl-tRNA synthetase. J Mol Biol 299:1051–1060. [PubMed] 10.1006/jmbi.2000.3819 [DOI] [PubMed] [Google Scholar]
- 246.Pauling L. 1957. The probability of errors in the process of synthesis of protein molecules, p 597–602. Festschrift Prof. Dr. Arthur Stoll zum siebzigsten Geburtstag, 8 Januar 1957. Birkhäuser Verlag, Basel, Switzerland. [Google Scholar]
- 247.Geslain R, Ribas de Pouplana L. 2004. Regulation of RNA function by aminoacylation and editing? Trends Genet 20:604–610. [PubMed] 10.1016/j.tig.2004.09.012 [DOI] [PubMed] [Google Scholar]
- 248.Mascarenhas AP, An S, Rosen AE, Martinis SA, Musier-Forsyth K. 2009. Fidelity mechanisms of aminoacyl-tRNA synthetases, p 155–203. In Köhrer C and RajBhandary UL (ed), Protein Engineering, vol. 22. Springer, Berlin, Germany. 10.1007/978-3-540-70941-1_6 [DOI] [Google Scholar]
- 249.Thompson D, Lazennec C, Plateau P, Simonson T. 2007. Ammonium scanning in an enzyme active site. The chiral specificity of aspartyl-tRNA synthetase. J Biol Chem 282:30856–30868. [PubMed] 10.1074/jbc.M704788200 [DOI] [PubMed] [Google Scholar]
- 250.Banik SD, Nandi N. 2010. Aminoacylation reaction in the histidyl-tRNA synthetase: Fidelity mechanism of the activation step. J Phys Chem B 114:2301–2311. [PubMed] 10.1021/jp910730s [DOI] [PubMed] [Google Scholar]
- 251.Thompson D, Lazennec C, Plateau P, Simonson T. 2008. Probing electrostatic interactions and ligand binding in aspartyl-tRNA synthetase through site-directed mutagenesis and computer simulations. Proteins 71:1450–1460. [PubMed] 10.1002/prot.21834 [DOI] [PubMed] [Google Scholar]
- 252.Stephen P, Lin S-X, Giegé R. 2016. Interplay between catalysts and substrates for activity of class Ib aminoacyl-tRNA synthetases and implications for pharmacology. Curr Top Med Chem 16:616–633. [PubMed] 10.2174/1568026615666150819110018 [DOI] [PubMed] [Google Scholar]
- 253.Banerjee R, Chen S, Dare K, Gilreath M, Praetorius-Ibba M, Raina M, Reynolds NM, Rogers T, Roy H, Yadavalli SS, Ibba M. 2010. tRNAs: Cellular barcodes for amino acids. FEBS Lett 584:387–395. [PubMed] 10.1016/j.febslet.2009.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Uter NT, Gruic-Sovulj I, Perona JJ. 2005. Amino acid-dependent transfer RNA affinity in a class I aminoacyl-tRNA synthetase. J Biol Chem 280:23966–23977. [PubMed] 10.1074/jbc.M414259200 [DOI] [PubMed] [Google Scholar]
- 255.Uter NT, Perona JJ. 2006. Active-site assembly in glutaminyl-tRNA synthetase by tRNA-mediated induced fit. Biochemistry 45:6858–6865. [PubMed] 10.1021/bi052606b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Bullock TL, Rodriguez-Hernandez A, Corigliano EM, Perona JJ. 2008. A rationally engineered misacylating aminoacyl-tRNA synthetase. Proc Natl Acad Sci USA 105:7428–7433. [PubMed] 10.1073/pnas.0711812105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Delagoutte B, Moras D, Cavarelli J. 2000. tRNA aminoacylation by arginyl-tRNA synthetase: Induced conformations during substrates binding. EMBO J 19:5599–5610. [PubMed] 10.1093/emboj/19.21.5599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Zhou M, Ye S, Stephen P, Zhang R-G, Wang E-D, Giegé R, Lin S-X. 2016. Structures of Escherichia coli arginyl-tRNA synthetase mediated by arginine and tRNAArg binding. in preparation.
- 259.Lam SSM, Schimmel PR. 1975. Equilibrium measurements of cognate and noncognate interactions between aminoacyl transfer RNA synthetases and transfer RNA. Biochemistry 14:2775–2780. [PubMed] 10.1021/bi00683a034 [DOI] [PubMed] [Google Scholar]
- 260.Ebel J-P, Giegé R, Bonnet J, Kern D, Befort N, Bollack C, Fasiolo F, Gangloff J, Dirheimer G. 1973. Factors determining the specificity of the tRNA aminoacylation reaction. Biochimie 55:547–557. [PubMed] 10.1016/S0300-9084(73)80415-8 [DOI] [PubMed] [Google Scholar]
- 261.Beuning PJ, Musier-Forsyth K. 1999. Transfer RNA recognition by aminoacyl-tRNA synthetases. Biopolymers 52:1–28. [PubMed] [DOI] [PubMed] [Google Scholar]
- 262.Giegé R, Frugier M. 2003. Transfer RNA structure and identity, p 1–24. In Lapointe J and Brakier-Gringas L (ed), Translation Mechanisms. Landes Biociences, Georgetown, TX. [Google Scholar]
- 263.McClain WH. 2005. tRNA aminoacylation: the crucial roles of tRNA deformabilty and backbone-mediated interactions by synthetase, p 266–270. In Ibba M, Francklyn C, and Cusack S (ed), The Aminoacyl-tRNA Synthetases. Landes Bioscience, Georgetown, TX. [Google Scholar]
- 264.Giegé R. 2008. Toward a more complete view of tRNA biology. Nat Struct Mol Biol 15:1007–1014. [PubMed] 10.1038/nsmb.1498 [DOI] [PubMed] [Google Scholar]
- 265.Frugier M, Helm M, Felden B, Giegé R, Florentz C. 1998. Sequences outside recognition sets are not neutral for tRNA aminoacylation: Evidence for non-permissive combinations of nucleotides in the acceptor stem of yeast tRNAPhe. J Biol Chem 273:11605–11610. [PubMed] 10.1074/jbc.273.19.11605 [DOI] [PubMed] [Google Scholar]
- 266.Perona JJ, Hou Y-M. 2007. Indirect readout of tRNA for aminoacylation. Biochemistry 46:10419–10432. [PubMed] 10.1021/bi7014647 [DOI] [PubMed] [Google Scholar]
- 267.Choi H, Kay G, Schneider JA, Otten S, McClain WH. 2003. Recognition of acceptor-stem structure of tRNAAsp by Escherichia coli aspartyl-tRNA synthetase. RNA 9:386–393. [PubMed] 10.1261/rna.2139703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Giegé R, Jühling F, Pütz J, Stadler P, Sauter C, Florentz C. 2012. Structure of transfer RNAs: Similarity and variability. Wiley Interdiscip Rev RNA 3:37–61. [PubMed] 10.1002/wrna.103 [DOI] [PubMed] [Google Scholar]
- 269.Salinas-Giegé T, Giegé R., Giegé P. 2015. tRNA biology in mitochondria. Int J Mol Sci 16:4518–4559. [PubMed] 10.3390/ijms16034518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Musier-Forsyth K, Schimmel P. 1999. Atomic determinants for aminoacylation of RNA minihelices and relationship to genetic code. Acc Chem Res 32:368–375. 10.1021/ar970148w [DOI] [Google Scholar]
- 271.Musier-Forsyth K, Schimmel P. 1992. Functional contacts of a transfer RNA synthetase with 2′-hydroxyl groups in the RNA minor groove. Nature 357:513–515. [PubMed] 10.1038/357513a0 [DOI] [PubMed] [Google Scholar]
- 272.Aphasizhev R, Théobald-Dietrich A, Kostyuk D, Kochetkov SN, Kisselev L, Giegé R, Fasiolo F. 1997. Structure and aminoacylation capacities of tRNA transcripts containing deoxyribonucleotides. RNA 3:893–904. [PubMed] [PMC free article] [PubMed] [Google Scholar]
- 273.Giegé R, Lapointe J. 2009. Transfer RNA aminoacylation and modified nucleosides, p 475–492. In Grosjean H (ed), DNA and RNA Modification Enzymes: Structure, Mechanism, Function and Evolution. Landes Bioscience, Georgetown, TX. [Google Scholar]
- 274.Muramatsu T, Nishikawa K, Nemoto F, Kuchino Y, Nishimura S, Miyazawa T, Yokoyama S. 1988. Codon and amino-acid specificities of a transfer RNA are both converted by a single post-transcriptional modification. Nature 336:179–181. [PubMed] 10.1038/336179a0 [DOI] [PubMed] [Google Scholar]
- 275.Pütz J, Florentz C, Benseler F, Giegé R. 1994. A single methyl group prevents the mischarging of a tRNA. Nat Struct Biol 1:580–582. [PubMed] 10.1038/nsb0994-580 [DOI] [PubMed] [Google Scholar]
- 276.Giegé R, Rees B. 2005. Aspartyl-tRNA synthetases, p 210–226. In Ibba M, Francklyn C, and Cusack S (ed), The Aminoacyl-tRNA Synthetases. Landes Bioscience, Georgetown, TX. [Google Scholar]
- 277.Freyhult E, Cui Y, Nilsson O, Ardell DH. 2007. New computational methods reveal tRNA identity element divergence between Proteobacteria and Cyanobacteria. Biochimie 89:1276–1288. [PubMed] 10.1016/j.biochi.2007.07.013 [DOI] [PubMed] [Google Scholar]
- 278.Perret V, Florentz C, Puglisi JD, Giegé R. 1992. Effect of conformational features on the aminoacylation of tRNAs and consequences on the permutation of tRNA specificities. J Mol Biol 226:323–333. [PubMed] 10.1016/0022-2836(92)90950-O [DOI] [PubMed] [Google Scholar]
- 279.McClain WH, Nicholas HBJ. 1987. Differences between transfer RNA molecules. J Mol Biol 194:635–642. [PubMed] 10.1016/0022-2836(87)90240-3 [DOI] [PubMed] [Google Scholar]
- 280.Ardell DH, Andersson SG. 2006. TFAM detects co-evolution of tRNA identity rules with lateral transfer of histidyl-tRNA synthetase. Nucleic Acids Res 34:893–904. [PubMed] 10.1093/nar/gkj449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Freyhult E, Moulton V, Ardell DH. 2006. Visualizing bacterial tRNA identity determinants and antideterminants using function logos and inverse function logos. Nucleic Acids Res 34:905–916. [PubMed] 10.1093/nar/gkj478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Jako E, Ittzes P, Szenes A, Kun A, Szathmary E, Pal G. 2007. In silico detection of tRNA sequence features characteristic to aminoacyl-tRNA synthetase class membership. Nucleic Acids Res 35:5593–5609. [PubMed] 10.1093/nar/gkm598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Korencic D, Söll D, Ambrogelly A. 2002. A one-step method for in vitro production of tRNA transcripts. Nucleic Acids Res 30:e105. [PubMed] 10.1093/nar/gnf104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Beuning PJ, Gulotta M, Musier-Forsyth K. 1997. Atomic group “mutagenesis” reveals major groove fine interactions of a tRNA synthetase with an RNA helix. J Am Chem Soc 119:8397–8402. 10.1021/ja971020c [DOI] [Google Scholar]
- 285.Martinis SA, Schimmel P. 1995. Small RNA oligonucleotide substrates for specific aminoacylations, p 349–370, In Söll D and RajBhandary UL (ed), tRNA: Structure, Biosynthesis, and Function. American Society for Microbiology, Washington, DC. [Google Scholar]
- 286.Normanly J, Abelson J. 1989. tRNA identity. Annu Rev Biochem 58:1029–1049. [PubMed] 10.1146/annurev.bi.58.070189.005121 [DOI] [PubMed] [Google Scholar]
- 287.McClain WH. 1993. Transfer RNA identity. FASEB J 7:72–78. [PubMed] [DOI] [PubMed] [Google Scholar]
- 288.McClain WH. 1993. Rules that govern tRNA identity in protein synthesis. J Mol Biol 234:257–280. [PubMed] 10.1006/jmbi.1993.1582 [DOI] [PubMed] [Google Scholar]
- 289.Kobayashi T, Nureki O, Ishitani R, Yaremchuk A, Tukalo M, Cusack S, Sakamoto K, Yokoyama S. 2003. Structural basis for orthogonal tRNA specificities of tyrosyl-tRNA synthetases for genetic code expansion. Nat Struct Biol 10:425–432. [PubMed] 10.1038/nsb934 [DOI] [PubMed] [Google Scholar]
- 290.Bonnefond L, Frugier M, Giegé R, Rudinger-Thirion J. 2005. Human mitochondrial TyrRS disobeys the tyrosine idenity rules. RNA 11:558–562. [PubMed] 10.1261/rna.7246805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Moulinier L, Eiler S, Eriani G, Gangloff J, Thierry J-C, Gabriel K, McClain WH, Moras D. 2001. The structure of an AspRS-tRNAAsp complex reveals a tRNA-dependant control mechanism. EMBO J 20:5290–5301. [PubMed] 10.1093/emboj/20.18.5290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Nameki N, Tamura K, Himeno H, Asahara H, Hasegawa T, Shimuzu M. 1992. Escherichia coli tRNAAsp recognition mechanism differing from that of the yeast system. Biochem Biophys Res Commun 189:856–862. [PubMed] 10.1016/0006-291X(92)92282-3 [DOI] [PubMed] [Google Scholar]
- 293.Hou Y-M, Westhof E, Giegé R. 1993. An unusual RNA tertiary interaction has a role for the specific aminoacylation of a transfer RNA. Proc Natl Acad Sci USA 90:6776–6780. 10.1073/pnas.90.14.6776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Hou Y-M, Motegi H, Lipman RSA, Hamann CS, Shiba K. 1999. Conservation of a tRNA core for aminoacylation. Nucleic Acids Res 27:4743–4750. [PubMed] 10.1093/nar/27.24.4743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Tocchini-Valentini G, Saks ME, Abelson J. 2000. tRNA leucine identity and recognition sets. J Mol Biol 298:779–793. [PubMed] 10.1006/jmbi.2000.3694 [DOI] [PubMed] [Google Scholar]
- 296.Burke B, Yang F, Chen F, Stehlin C, Chan B, Musier-Forsyth K. 2000. Evolutionary coadaptation of the motif 2-acceptor stem interaction in the class II prolyl-tRNA synthetase system. Biochemistry 39:15540–15547. [PubMed] 10.1021/bi001835p [DOI] [PubMed] [Google Scholar]
- 297.Larkin DC, Williams AM, Martinis SA, Fox GE. 2002. Identification of essential domains for Escherichia coli tRNALeu aminoacylation and amino acid editing using minimalist RNA molecules. Nucleic Acids Res 30:2103–2113. [PubMed] 10.1093/nar/30.10.2103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Fukunaga J, Ohno S, Nishikawa K, Yokogawa T. 2006. A base pair at the bottom of the anticodon stem is reciprocally preferred for discrimination of cognate tRNAs by Escherichia coli lysyl- and glutaminyl-tRNA synthetases. Nucleic Acids Res 34:3181–3188. [PubMed] 10.1093/nar/gkl414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Wong F-C, Beuning PJ, Silvers C, Musier-Forsyth K. 2003. An isolated class II aminoacyl-tRNA synthetase insertion domain is functional in amino acid editing. J Biol Chem 278:52857–52864. [PubMed] 10.1074/jbc.M309627200 [DOI] [PubMed] [Google Scholar]
- 300.Zhao M-W, Zhu B, Hao R, Xu M-G, Eriani G, Wang E-D. 2005. Leucyl-tRNA synthetase from the ancestral bacterium Aquifex aeolicus contains relics of synthetase evolution. EMBO J 24:1430–1439. [PubMed] 10.1038/sj.emboj.7600618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Tamura K, Himeno H, Asahara H, Hasegawa T, Shimizu M. 1992. In vitro study of E. coli tRNAArg and tRNALys identity elements. Nucleic Acids Res 20:2335–2339. [PubMed] 10.1093/nar/20.9.2335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Schmitt E, Meinnel T, Panvert M, Mechulam Y, Blanquet S. 1993. Two acidic residues of Escherichia coli methionyl-tRNA synthetase act as negative discriminants towards the binding of non-cognate tRNA anticodons. J Mol Biol 233:615–628. [PubMed] 10.1006/jmbi.1993.1540 [DOI] [PubMed] [Google Scholar]
- 303.Bedouelle H, Guez-Ivanier V, Nageotte R. 1993. Discrimination between transfer-RNAs by tyrosyl-tRNA synthetase. Biochimie 75:1099–1108. [PubMed] 10.1016/0300-9084(93)90009-H [DOI] [PubMed] [Google Scholar]
- 304.Nureki O, Niimi T, Muramatsu T, Kanno H, Kohno T, Florentz C, Giegé R, Yokoyama S. 1994. Molecular recognition of the identity-determinant set of isoleucine transfer RNA from Escherichia coli. J Mol Biol 236:710–724. [PubMed] 10.1006/jmbi.1994.1184 [DOI] [PubMed] [Google Scholar]
- 305.Madore E, Florentz C, Giegé R, Sekine S, Yokoyama S, Lapointe J. 1999. Effect of modified nucleotides on Escherichia coli tRNAGlu structure and on its aminoacylation by glutamyl-tRNA synthetase – Predominant and distinct roles of the mnm5 and s2 modifications of U34. Eur J Biochem 266:1128–1135. [PubMed] 10.1046/j.1432-1327.1999.00965.x [DOI] [PubMed] [Google Scholar]
- 306.Beebe K, Merriman E, Schimmel P. 2003. Structure-specific tRNA determinants for editing a mischarged amino acid. J Biol Chem 278:45056–45061. [PubMed] 10.1074/jbc.M307080200 [DOI] [PubMed] [Google Scholar]
- 307.Zhu B, Zhao M-W, Eriani G, Wang E-D. 2007. A present-day aminoacyl-tRNA synthetase with ancestral editing properties. RNA 13:15–21. [PubMed] 10.1261/rna.228707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Francklyn C, Schimmel P. 1989. Aminoacylation of RNA minihelices with alanine. Nature 337:478–481. [PubMed] 10.1038/337478a0 [DOI] [PubMed] [Google Scholar]
- 309.Musier-Forsyth K, Usman N, Scaringe S, Doudna J, Green R, Schimmel P. 1991. Specificity for aminoacylation of an RNA helix: An unpaired, exocyclic amino group in the minor groove. Science 253:784–786. [PubMed] 10.1126/science.1876835 [DOI] [PubMed] [Google Scholar]
- 310.Fischer AE, Beuning PJ, Musier-Forsyth K. 1999. Identification of discriminator base atomic groups that modulate the alanine aminoacylation reaction. J Biol Chem 274:37093–37096. [PubMed] 10.1074/jbc.274.52.37093 [DOI] [PubMed] [Google Scholar]
- 311.Martinis SA, Schimmel P. 1992. Enzymatic aminoacylation of sequence-specific RNA minihelices and hybrid duplexes with methionine. Proc Natl Acad Sci USA 89:65–69. [PubMed] 10.1073/pnas.89.1.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Quinn CL, Tao N, Schimmel P. 1995. Species-specific microhelix aminoacylation by a eukaryotic pathogen tRNA synthetase dependent on a single base pair. Biochemistry 34:12489–12495. [PubMed] 10.1021/bi00039a001 [DOI] [PubMed] [Google Scholar]
- 313.Liu H, Yap L-P, Musier-Forsyth K. 1996. Single atomic group in RNA helix needed for positive and negative tRNA synthetase discrimination. J Am Chem Soc 118:2523–2524. 10.1021/ja954062e [DOI] [Google Scholar]
- 314.Saks ME, Sampson JR. 1996. Variant minihelix RNAs reveal sequence-specific recognition of the helical tRNASer acceptor stem by E. coli seryl-tRNA synthetase. EMBO J 15:2843–2849. [PubMed] [PMC free article] [PubMed] [Google Scholar]
- 315.Frugier M, Florentz C, Giegé R. 1992. Anticodon-independent aminoacylation of an RNA minihelix with valine. Proc Natl Acad Sci USA 89:3990–3994. [PubMed] 10.1073/pnas.89.9.3990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Frugier M, Florentz C, Giegé R. 1994. Efficient aminoacylation of resected RNA helices by class II aspartyl-tRNA synthetase dependent on a single nucleotide. EMBO J 13:2218–2226. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Rosen AE, Musier-Forsyth K. 2004. Recognition of G-1:C73 atomic groups by Escherichia coli histidyl-tRNA synthetase. J Am Chem Soc 126:64–65. [PubMed] 10.1021/ja0381609 [DOI] [PubMed] [Google Scholar]
- 318.Gustilo EM, Dubois DY, Lapointe J, Agris PF. 2007. E. coli glutamyl-tRNA synthetase is inhibited by anticodon stem-loop domains and a minihelix. RNA Biol 4:85–92. [PubMed] 10.4161/rna.4.2.4736 [DOI] [PubMed] [Google Scholar]
- 319.Grosjean H, de Crécy-Lagard V, Björk GR. 2004. Aminoacylation of the anticodon stem by a tRNA-synthetase paralog: Relic of an ancient code? Trends Biochem Sci 29:519–522. [PubMed] 10.1016/j.tibs.2004.08.005 [DOI] [PubMed] [Google Scholar]
- 320.Ibba M, Francklyn C. 2004. Turning tRNA upside down: When aminoacylation is not a prerequisite to protein synthesis. Proc Natl Acad Sci USA 101:7493–7494. [PubMed] 10.1073/pnas.0402276101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Di Giulio M. 2004. The origin of the tRNA molecule: Implications for the origin of protein synthesis. J Theor Biol 226:89–93. [PubMed] 10.1016/j.jtbi.2003.07.001 [DOI] [PubMed] [Google Scholar]
- 322.Dubois DY, Blais SP, Huot JL, Lapointe J. 2009. A C-truncated glutamyl-tRNA synthetase specific for tRNAGlu is stimulated by its free complementary distal domain: Mechanistic and evolutionary implications. Biochemistry 48:6012–6021. [PubMed] 10.1021/bi801690f [DOI] [PubMed] [Google Scholar]
- 323.Carter CW Jr, Wolfenden R. 2015. tRNA acceptor stem and anticodon bases form independent codes related to protein folding. Proc Natl Acad Sci USA 112:7489–7494. [PubMed] 10.1073/pnas.1507569112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.First EA. 1998. Catalysis of tRNA aminoacylation by class I and class II aminoacyl-tRNA synthetases, p 573–607. In Sinnott M (ed), Comprehensive Biological Catalysis, vol. 1. Academic Press, New-York, NY. [Google Scholar]
- 325.Perona JJ, Gruic-Sovulj I. 2014. Synthetic and editing mechanisms of aminoacyl-tRNA synthetases. Top Curr Chem 344:1–41. [PubMed] 10.1007/128_2013_456 [DOI] [PubMed] [Google Scholar]
- 326.Onesti S, Desogus G, Brevet A, Chen J, Plateau P, Blanquet S, Brick P. 2000. Structural studies of lysyl-tRNA synthetase: Conformational changes induced by substrate binding. Biochemistry 39:12853–12861. [PubMed] 10.1021/bi001487r [DOI] [PubMed] [Google Scholar]
- 327.Kapustina M, Weinreb V, Li L, Kuhlman B, Carter CW Jr. 2007. A conformational transition state accompanies tryptophan activation by B. stearothermophilus tryptophanyl-tRNA synthetase. Structure 15:1272–1284. [PubMed] 10.1016/j.str.2007.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Laowanapiban P, Kapustina M, Vonrhein C, Delarue M, Koehl P, Carter CW Jr. 2009. Independent saturation of three TrpRS subsites generates a partially assembled state similar to those observed in molecular simulations. Proc Natl Acad Sci USA 106:1790–1795. [PubMed] 10.1073/pnas.0812752106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Weinreb V, Li L, Campbell CL, Kaguni LS, Carter CW Jr. 2009. Mg2+-assisted catalysis by B. stearothermophilus TrpRS is promoted by allosteric effects. Structure 17:952–964. [PubMed] 10.1016/j.str.2009.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Cramer F, Freist W. 1993. Aminoacyl-tRNA synthetases: The division into two classes predicted by the chemistry of substrates and enzymes. Angew Chem Int Ed Engl 32:190–200. 10.1002/anie.199301901 [DOI] [Google Scholar]
- 331.Sprinzl M. 2006. Chemistry of aminoacylation and peptide bond formation on the 3′-terminus of tRNA. J Biosci 31:489–496. [PubMed] 10.1007/BF02705188 [DOI] [PubMed] [Google Scholar]
- 332.Förster C, Limmer S, Zeidler W, Sprinzl M. 1994. Effector region or the translation elongation factor EF-Tu-GTP complex stabilizes an orthoester acid intermediate structure of aminoacyl-tRNA in a ternary complex. Proc Natl Acad Sci USA 92:4254–4257. 10.1073/pnas.91.10.4254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Ward WH, Fersht AR. 1988. Tyrosyl-tRNA synthetase acts as an asymmetric dimer in charging tRNA. A rationale for half-of-the-sites activity. Biochemistry 27:5525–5530. [PubMed] 10.1021/bi00415a021 [DOI] [PubMed] [Google Scholar]
- 334.Kern D, Lorber B, Boulanger Y, Giegé R. 1985. A peculiar property of aspartyl-tRNA synthetase from bakers yeast: Chemical modification of the protein by the enzymatically synthesized aminoacyl adenylate. Biochemistry 24:1321–1332. 10.1021/bi00327a009 [DOI] [Google Scholar]
- 335.Guth E, Connolly SH, Bovee M, Francklyn CS. 2005. A substrate-assisted concerted mechanism for aminoacylation by a class II aminoacyl-tRNA synthetase. Biochemistry 44:3785–3794. [PubMed] 10.1021/bi047923h [DOI] [PubMed] [Google Scholar]
- 336.Minajigi A, Francklyn CS. 2010. Aminoacyl transfer rate dictates choice of editing pathway in threonyl-tRNA synthetase. J Biol Chem 285:23810–23817. [PubMed] 10.1074/jbc.M110.105320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Guth EC, Francklyn CS. 2007. Kinetic discrimination of tRNA identity by the conserved motif 2 loop of a class II aminoacyl-tRNA synthetase. Mol Cell 25:531–542. [PubMed] 10.1016/j.molcel.2007.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Hughes SJ, Tanner JA, Miller AD, Gould IR. 2006. Molecular dynamics simulations of LysRS: An asymmetric state. Proteins 62:649–662. [PubMed] 10.1002/prot.20609 [DOI] [PubMed] [Google Scholar]
- 339.Freist W, Logan DT, Gauss DH. 1996. Glycyl-tRNA synthetases. Biol Chem 377:343–356. [PubMed] [PubMed] [Google Scholar]
- 340.Bovee ML, Yan W, Sproat BS, Francklyn CS. 1999. tRNA discrimination at the binding step by a class II aminoacyl-tRNA synthetase. Biochemistry 38:13725–13735. [PubMed] 10.1021/bi991182g [DOI] [PubMed] [Google Scholar]
- 341.Ibba M, Sever S, Praetorius-Ibba M, Söll D. 1999. Transfer RNA identity contributes to transition state stabilization during aminoacyl-tRNA synthesis. Nucleic Acids Res 27:3631–3637. [PubMed] 10.1093/nar/27.18.3631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Pütz J, Puglisi JD, Florentz C, Giegé R. 1993. Additive, cooperative and anti-cooperative effects between identity nucleotides of a tRNA. EMBO J 12:2949–2957. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Uter NT, Perona JJ. 2004. Long-range intramolecular signaling in a tRNA synthetase complex revealed by pre-steady-state kinetics. Proc Natl Acad Sci USA 101:14396–14401. [PubMed] 10.1073/pnas.0404017101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Bonnet J, Ebel J-P. 1972. Interpretation of incomplete reactions in tRNA aminoacylation. Aminoacylation of yeast tRNAValII with yeast valyl-tRNA synthetase. Eur J Biochem 31:335–344. [PubMed] 10.1111/j.1432-1033.1972.tb02538.x [DOI] [PubMed] [Google Scholar]
- 345.Schimmel PR, Söll D. 1979. Aminoacyl-tRNA synthetases: General features and recognition of transfer RNAs. Annu Rev Biochem 48:601–648. [PubMed] 10.1146/annurev.bi.48.070179.003125 [DOI] [PubMed] [Google Scholar]
- 346.Black Pyrkosz A, Eargle J, Sethi A, Luthey-Schulten Z. 2010. Exit strategies for charged tRNA from GluRS. J Mol Biol 397:1350–1371. [PubMed] 10.1016/j.jmb.2010.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Banerjee P, Warf MB, Alexander R. 2009. Effect of a domain-spanning disulfide on aminoacyl-tRNA synthetase activity. Biochemistry 48:10113–10119. [PubMed] 10.1021/bi9012275 [DOI] [PubMed] [Google Scholar]
- 348.Cui Q, Karplus M. 2008. Allostery and cooperativity revisited. Protein Sci 17:1295–1307. [PubMed] 10.1110/ps.03259908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Zhang CM, Hou Y-M. 2005. Domain-domain communication for tRNA aminoacylation: The importance of covalent connectivity. Biochemistry 44:7240–7249. [PubMed] 10.1021/bi050285y [DOI] [PubMed] [Google Scholar]
- 350.Alexander RW, Schimmel P. 2001. Domain-domain communication in aminoacyl-tRNA synthetases. Prog Nucleic Acid Res Mol Biol 69:317–349. [PubMed] 10.1016/S0079-6603(01)69050-0 [DOI] [PubMed] [Google Scholar]
- 351.Li R, Macnamara LM, Leuchter JD, Alexander RW, Cho SS. 2015. MD Simulations of tRNA and aminoacyl-tRNA synthetases: Dynamics, folding, binding, and allostery. Int J Mol Sci 16:15872–15902. [PubMed] 10.3390/ijms160715872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Budiman ME, Knaggs MH, Fetrow JS, Alexander RW. 2007. Using molecular dynamics to map interaction networks in an aminoacyl-tRNA synthetase. Proteins 68:670–689. [PubMed] 10.1002/prot.21426 [DOI] [PubMed] [Google Scholar]
- 353.Bushnell EAC, Huang W, Llano J, Gauld JW. 2012. Molecular dynamics investigation into substrate binding of the catalytic base in the mechanism of threonyl-tRNA synthetase. J Phys Chem B 116:5205–5212. [PubMed] 10.1021/jp302556e [DOI] [PubMed] [Google Scholar]
- 354.Weinreb V, Li L, Chandrasekaran SN, Koehl P, Delarue M, Carter Jr CW. 2014. Enhanced amino acid selection in fully evolved tryptophanyl-tRNA synthetase, relative to its urzyme, requires domain motion sensed by the D1 switch, a remote dynamic packing motif. J Biol Chem 289:4367–4376. [PubMed] 10.1074/jbc.M113.538660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Ghosh A, Vishveshwara S. 2007. A study of communication pathways in methionyl-tRNA synthetase by molecular dynamics simulations and structure network analysis. Proc Natl Acad Sci USA 104:15711–15716. [PubMed] 10.1073/pnas.0704459104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Ghosh A, Vishveshwara S. 2008. Variations in clique and community patterns in protein structures during allosteric communication: Investigation of dynamically equilibrated structures of methionyl tRNA synthetase complexes. Biochemistry 47:11398–11407. [PubMed] 10.1021/bi8007559 [DOI] [PubMed] [Google Scholar]
- 357.Sethi A, Eargle J, Black AA, Luthey-Schulten Z. 2009. Dynamical networks in tRNA:protein complexes. Proc Natl Acad Sci USA 106:6620–6625. [PubMed] 10.1073/pnas.0810961106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Fukunaga R, Yokoyama S. 2005. Crystal structure of leucyl-tRNA synthetase from the archaeon Pyrococcus horikoshii reveals a novel editing domain orientation. J Mol Biol 346:57–71. [PubMed] 10.1016/j.jmb.2004.11.060 [DOI] [PubMed] [Google Scholar]
- 359.Sekine S-i, Nureki O, Dubois DY, Bernier S, Chênevert R, Lapointe J, Vassylyev DG, Yokoyama S. 2003. ATP binding by glutamyl-tRNA synthetase is switched to the productive mode by tRNA binding. EMBO J 22:676–688. [PubMed] 10.1093/emboj/cdg053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Rodriguez-Hernandez A, Perona JJ. 2011. Heat maps for intramolecular communication in an RNP enzyme encoding glutamine. Structure 19:386–396. [PubMed] 10.1016/j.str.2010.12.017 [DOI] [PubMed] [Google Scholar]
- 361.Weimer KM, Shane BL, Brunetto M, Bhattacharyya S, Hati S. 2009. Evolutionary basis for the coupled-domain motions in Thermus thermophilus leucyl-tRNA synthetase. J Biol Chem 284:10088–10099. [PubMed] 10.1074/jbc.M807361200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Johnson JM, Sanford BL, Strom AM, Tadayon SN, Lehman BP, Zirbes AM, Bhattacharyya S, Musier-Forsyth K, Hati S. 2013. Multiple pathways promote dynamical coupling between catalytic domains in Escherichia coli prolyl-tRNA synthetase. Biochemistry 52:4399–4412. [PubMed] 10.1021/bi400079h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Deniziak M, Sauter C, Becker HD, Paulus CA, Giegé R, Kern D. 2007. Deinococcus glutaminyl-tRNA synthetase is a chimer between proteins from an ancient and the modern pathways of aminoacyl-tRNA formation. Nucleic Acids Res 35:1421–1431. [PubMed] 10.1093/nar/gkl1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Touzé E, Lorber B, Deniziak M, Becker HD, Kern D, Giegé R, Sauter C. 2007. Disorder can exist inside well-diffracting crystals. Cryst Growth Des 7:2195–2197. 10.1021/cg7007057 [DOI] [Google Scholar]
- 365.Kapustina M, Carter CW Jr. 2006. Computational studies of tryptophanyl-tRNA synthetase: Activation of ATP by induced-fit. J Mol Biol 362:1159–1180. [PubMed] 10.1016/j.jmb.2006.06.078 [DOI] [PubMed] [Google Scholar]
- 366.Bhattacharyya M, Ghosh A, Hansia P, Vishveshwara S. 2010. Allostery and conformational free energy changes in human tryptophanyl-tRNA synthetase from essential dynamics and structure networks. Proteins 78:506–517. [PubMed] 10.1002/prot.22573 [DOI] [PubMed] [Google Scholar]
- 367.Strom AM, Fehling SC, Bhattacharyya S, Hati S. 2014. Probing the global and local dynamics of aminoacyl-tRNA synthetases using all-atom and coarse-grained simulations. J Mol Model 20:2245. [PubMed] 10.1007/s00894-014-2245-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Guillon J-M, Mechulam Y, Blanquet S, Fayat G. 1993. Importance of formylability and anticodon stem sequence to give a tRNAMet an initiator identity in Escherichia coli. J Bacteriol 175:4507–4514. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.LaRiviere FJ, Wolfson A.D, Uhlenbeck, OC. 2001. Uniform binding of aminoacyl-tRNAs to elongation factor Tu by thermodynamic compensation. Science 294:165–168. [PubMed] 10.1126/science.1064242 [DOI] [PubMed] [Google Scholar]
- 370.Ogle JM, Murphy FV, Tarry MJ, Ramakrishnan V. 2002. Selection of tRNA by the ribosome requires a transition from an open to a closed form. Cell 111:721–732. [PubMed] 10.1016/S0092-8674(02)01086-3 [DOI] [PubMed] [Google Scholar]
- 371.Francklyn C. 2003. tRNA synthetase paralogs: Evolutionary links in the transition from tRNA-dependent amino acid biosynthesis to de novo biosynthesis. Proc Natl Acad Sci USA 100:9650–9652. [PubMed] 10.1073/pnas.1934245100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Calendar R, Berg P. 1967. D-Tyrosyl RNA: Formation, hydrolysis and utilization for protein synthesis. J Mol Biol 26:39–54. [PubMed] 10.1016/0022-2836(67)90259-8 [DOI] [PubMed] [Google Scholar]
- 373.Ferri-Fioni ML, Schmitt E. Soutourina J, Plateau P, Mechulam Y, Blanquet S. 2001. Structure of crystalline D-Tyr-tRNATyr deacylase. A representative of a new class of tRNA-dependent hydrolases. J Biol Chem 276:47285–47290. [PubMed] 10.1074/jbc.M106550200 [DOI] [PubMed] [Google Scholar]
- 374.Soutourina J, Plateau P, Blanquet S. 2000. Metabolism of D-aminoacyl-tRNAs in Escherichia coli and Saccharomyces cerevisiae cells. J Biol Chem 275:32535–32542. [PubMed] 10.1074/jbc.M005166200 [DOI] [PubMed] [Google Scholar]
- 375.Soutourina O, Soutourina J, Blanquet S, Plateau P. 2004. Formation of D-tyrosyl-tRNATyr accounts for the toxicity of D-tyrosine toward Escherichia coli. J Biol Chem 279:42560–42565. [PubMed] 10.1074/jbc.M402931200 [DOI] [PubMed] [Google Scholar]
- 376.Wydau S, van der Rest G, Aubard C, Plateau P, Blanquet S. 2009. Widespread distribution of cell defense against D-aminoacyl-tRNAs. J Biol Chem 284:14096–14104. [PubMed] 10.1074/jbc.M808173200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Jakubowski H. 1999. Misacylation of tRNALys with noncognate amino acids by lysyl-tRNA synthetase. Biochemistry 38:8088–8093. [PubMed] 10.1021/bi990629i [DOI] [PubMed] [Google Scholar]
- 378.Hendrickson WA, Horton JR, LeMaster DM. 1990. Selenomethionyl proteins produced for analysis by multiwavelength anomalous diffraction (MAD): A vehicle for direct determination of the three-dimensional structure. EMBO J 9:1665–1672. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Strub M-P, Hoh F, Sanchez J-F, Strub JM, Böck A, Aumelas A, Dumas C. 2003. Selenomethionine and selenocysteine double labeling strategy for crystallographic phasing. Structure 11:1359–1367. [PubMed] 10.1016/j.str.2003.09.014 [DOI] [PubMed] [Google Scholar]
- 380.Sankaranarayanan R, Dock-Bregeon A-C, Rees B, Bovee M, Caillet J, Romby P, Francklyn CS, Moras D. 2000. Zinc ion mediated amino acid discrimination by threonyl-tRNA synthetase. Nat Struct Biol 7:461–465. [PubMed] 10.1038/75856 [DOI] [PubMed] [Google Scholar]
- 381.Bacher JM, de Crécy-Lagard V, Schimmel PR. 2005. Inhibited cell growth and protein functional changes from an editing-defective tRNA synthetase. Proc. Natl. Acad. Sci. USA 102:1697–1701. [PubMed] 10.1073/pnas.0409064102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Nangle LA, de Crécy-Lagard V, Döring V, Schimmel P. 2002. Genetic code ambiguity: Cell viability related to severity of editing defects in mutant tRNA synthetases. J Biol Chem 277:45729–45733. [PubMed] 10.1074/jbc.M208093200 [DOI] [PubMed] [Google Scholar]
- 383.Lee JW, Beebe K, Nangle LA, Jang J, Longo-Guess CM, Cook SA, Davisson MT, Sundberg JP, Schimmel P, Ackerman SL. 2006. Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature 443:50–55. [PubMed] 10.1038/nature05096 [DOI] [PubMed] [Google Scholar]
- 384.Nangle LA, Motta C.M, Schimmel P. 2006. Global effects of mistranslation from an editing defect in mammalian cells. Chem Biol 13:1091–1100. [PubMed] 10.1016/j.chembiol.2006.08.011 [DOI] [PubMed] [Google Scholar]
- 385.Nangle LA, Zhang W, Xie W, Yang X-L, Schimmel P. 2007. Charcot-Marie-Tooth disease-associated mutant tRNA synthetases linked to altered dimer interface and neurite distribution defect. Proc Natl Acad Sci USA 104:11239–11244. [PubMed] 10.1073/pnas.0705055104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Igloi GL, Schiefermayr E. 2009. Amino acid discrimination by arginyl-tRNA synthetases as revealed by an examination of natural specificity variants. FEBS J 276:1307–1318. [PubMed] 10.1111/j.1742-4658.2009.06866.x [DOI] [PubMed] [Google Scholar]
- 387.Zhang CM, Perona JJ, Hou Y-M. 2003. Amino acid discrimination by a highly differentiated metal center of an aminoacyl-tRNA synthetase. Biochemistry 42:10931–10937. [PubMed] 10.1021/bi034812u [DOI] [PubMed] [Google Scholar]
- 388.Swanson R, Hoben P, Sumner-Smith M, Uemura H, Watson L, Söll D. 1988. Accuracy of in vivo aminoacylation requires proper balance of tRNA and aminoacyl-tRNA synthetase. Science 242:1548–1551. [PubMed] 10.1126/science.3144042 [DOI] [PubMed] [Google Scholar]
- 389.Lim K, Tempczyk A, Bonander N, Toedt J, Howard A, Eisenstein E, Herzberg O. 2003. A catalytic mechanism for D-Tyr-tRNATyr deacylase based on the crystal structure of Hemophilus influenzae HI0670. J Biol Chem 278:13496–13502. [PubMed] 10.1074/jbc.M213150200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Bhatt TK, Yogavel M, Wydau S, Berwal R, Sharma A. 2010. Ligand-bound structures provide atomic snapshots for the catalytic mechanism of D-amino acid deacylase. J Biol Chem 285:5917–5930. [PubMed] 10.1074/jbc.M109.038562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Beebe K, Merriman E, Ribas de Pouplana L, Schimmel P. 2004. A domain for editing by an archaebacterial tRNA synthetase. Proc. Natl. Acad. Sci. USA 101:5958–5963. [PubMed] 10.1073/pnas.0401530101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Ahmad S, Muthukumar S, Kuncha SK, Routh SB, Yerabham AS, Hussain T, Kamarthapu V, Kruparani SP, Sankaranarayanan R. 2015. Specificity and catalysis hardwired at the RNA-protein interface in a translational proofreading enzyme. Nat Commun 6:7552. [PubMed] 10.1038/ncomms8552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Sherman JM, Rogers MJ, Söll D. 1992. Competition of aminoacyl-tRNA synthetases for tRNA ensures the accuracy of aminoacylation. Nucleic Acids Res 20:2847–2852. [PubMed] 10.1093/nar/20.11.2847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Bonnet J, Giegé R, Ebel J-P. 1972. Lack of specificity in the aminoacyl-tRNA synthetase-catalysed deacylation of aminoacyl-tRNA. FEBS Lett 27:139–144. [PubMed] 10.1016/0014-5793(72)80427-7 [DOI] [PubMed] [Google Scholar]
- 395.Eldred EW, Schimmel PR. 1972. Rapid deacylation by isoleucyl transfer ribonucleic acid synthetase of isoleucine-specific transfer ribonucleic acid aminoacylated with valine. J Biol Chem 247:2961–2964. [PubMed] [PubMed] [Google Scholar]
- 396.Yarus M. 1972. Phenylalanyl-tRNA synthetase and isoleucyl-tRNAPhe: A possible verification mechanism for aminoacyl-tRNA. Proc Natl Acad Sci USA 69:1915–1919. [PubMed] 10.1073/pnas.69.7.1915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Igloi GL, von der Haar F, Cramer F. 1978. Aminoacyl-tRNA synntheses from yeast: Generality of chemical proofreading in the prevention of misaminoacylation of tRNA. Biochemistry 17:3459–3468. [PubMed] 10.1021/bi00610a006 [DOI] [PubMed] [Google Scholar]
- 398.Fersht AR, Dingwall C. 1979. Evidence for the double-sieve editing mechanism in protein synthesis. Steric exclusion of isoleucine by valyl-tRNA synthetase. Biochemistry 18:2627–2631. [PubMed] 10.1021/bi00579a030 [DOI] [PubMed] [Google Scholar]
- 399.Nureki O, Vassylyev DG, Tateno M, Shimida A, Nakama T, Fukai S, Konno M, Hendrickson T, Schimmel P, Yokoyama S. 1998. Enzyme structure with two catalytic sites for double-sieve selection of substrate. Science 280:578–582. [PubMed] 10.1126/science.280.5363.578 [DOI] [PubMed] [Google Scholar]
- 400.Bishop AC, Nomanbhoy TK, Schimmel P. 2002. Blocking site-to-site translocation of a misactivated amino acid by mutation of a class I tRNA synthetase. Proc Natl Acad Sci USA 99:585–590. [PubMed] 10.1073/pnas.012611299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Bishop AC, Beebe K, Schimmel PR. 2003. Interstice mutations that block site-to-site translocation of a misactivated amino acid bound to a class I tRNA synthetase. Proc Natl Acad Sci USA 100:490–494. [PubMed] 10.1073/pnas.0237335100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Ahel I, Korencic D, Ibba M, Söll D. 2003. Trans-editing of mischarged tRNAs. Proc Natl Acad Sci USA 100:15422–15427. [PubMed] 10.1073/pnas.2136934100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.An S, Musier-Forsyth K. 2004. Trans-editing of Cys-tRNAPro by Haemophilus influenzae YbaK protein. J Biol Chem 279:42359–42362. [PubMed] 10.1074/jbc.C400304200 [DOI] [PubMed] [Google Scholar]
- 404.Hendrickson TL, Schimmel P. 2003. Transfer RNA-depedent amino acid discrimination by aminoacyl-tRNA synthetases, p 34–64. In Lapointe J and Brakier-Gringas L (ed), Translation Mechanisms. Landes Biociences, Georgetown, TX. [Google Scholar]
- 405.Ling J, Reynolds N, Ibba M. 2009. Aminoacyl-tRNA synthesis and translational quality control. Annu Rev Microbiol 63:61–78. [PubMed] 10.1146/annurev.micro.091208.073210 [DOI] [PubMed] [Google Scholar]
- 406.Schimmel P. 2008. An editing activity that prevents mistranslation and connection to disease. J Biol Chem 283:28777–28782. [PubMed] 10.1074/jbc.X800007200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Gruic-Sovulj I, Uter N, Bullock T, Perona JJ. 2005. tRNA-dependent aminoacyl-adenylate hydrolysis by a nonediting class I aminoacyl-tRNA synthetase. J Biol Chem 280:23978–23986. (check) [PubMed] 10.1074/jbc.M414260200 [DOI] [PubMed] [Google Scholar]
- 408.Baldwin AN, Berg P. 1966. Transfer ribonucleic acid-induced hydrolysis of valyladenylate bound to isoleucyl ribonucleic acid synthetase. J Biol Chem 241:839–845. [PubMed] [PubMed] [Google Scholar]
- 409.Nomanbhoy, TK, Hendrickson TL, Schimmel P. 1999. Transfer RNA-dependent translocation of misactivated amino acids to prevent errors in protein synthesis. Mol Cell 4:519–528. [PubMed] 10.1016/S1097-2765(00)80203-8 [DOI] [PubMed] [Google Scholar]
- 410.Lin L, Hale SP, Schimmel P. 1996. Aminoacylation error correction. Nature 384:33–34. [PubMed] 10.1038/384033b0 [DOI] [PubMed] [Google Scholar]
- 411.Tan M, Zhu B, Zhou X-L, He R, Chen X, Eriani G, Wang E-D. 2010. tRNA-dependent pre-transfer editing by prokaryotic leucyl-tRNA synthetase. J Biol Chem 285:3235–3244. [PubMed] 10.1074/jbc.M109.060616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Tardif KD, Horowitz J. 2002. Transfer RNA determinants for translational editing by Escherichia coli valyl-tRNA synthetase. Nucleic Acids Res 30:2538–2545. [PubMed] 10.1093/nar/30.11.2538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Williams AM, Martinis SA. 2006. Mutational unmasking of a tRNA-dependent pathway for preventing genetic code ambiguity. Proc Natl Acad Sci USA 103:3586–3591. [PubMed] 10.1073/pnas.0507362103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Boniecki MT, Vu MT, Betha AK, Martinis SA. 2008. CP1-dependent partitioning of pretransfer and posttransfer editing in leucyl-tRNA synthetase. Proc Natl Acad Sci USA 105:19223–19228. [PubMed] 10.1073/pnas.0809336105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Zhu B, Yao P, Tan M, Eriani G, Wang E-D. 2009. tRNA-independent pretransfer editing by class I leucyl-tRNA synthetase. J Biol Chem 284:3418–3424. [PubMed] 10.1074/jbc.M806717200 [DOI] [PubMed] [Google Scholar]
- 416.Li L, Palencia A, Lukk T, Li Z, Luthey-Schulten ZA, Cusack S, Martinis SA, Boniecki MT. 2013. Leucyl-tRNA synthetase editing domain functions as a molecular rheostat to control codon ambiguity in Mycoplasma pathogens. Proc Natl Acad Sci USA 110:3817–3822. [PubMed] 10.1073/pnas.1218374110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Bharatham N, Bharatham K, Lee Y, Woo Lee K. 2009. Molecular dynamics simulation study of valyl-tRNA synthetase with its pre- and post-transfer editing substrates. Biophys Chem 143:34–43. [PubMed] 10.1016/j.bpc.2009.03.009 [DOI] [PubMed] [Google Scholar]
- 418.Dulic M, Cvetesic N, Perona JJ, Gruic-Sovulj I. 2010. Partitioning of tRNA-dependent editing between pre- and post-transfer pathways in class I aminoacyl-tRNA synthetases. J Biol Chem 285:23799–23809. [PubMed] 10.1074/jbc.M110.133553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Cvetesic N, Perona JJ, Gruic-Sovulj I. 2012. Kinetic partitioning between synthetic and editing pathways in class I aminoacyl-tRNA synthetases occurs at both pre-transfer and post-transfer hydrolytic steps. J Biol Chem 287:25381–25394. [PubMed] 10.1074/jbc.M112.372151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Fortowsky GB, Simard DJ, Aboelnga MM, Gauld JW. 2015. Substrate-assisted and enzymatic pretransfer editing of nonstandard amino acids by methionyl-tRNA synthetase. Biochemistry 54:5757–5765. [PubMed] 10.1021/acs.biochem.5b00588 [DOI] [PubMed] [Google Scholar]
- 421.Beuning PJ, Musier-Forsyth K. 2000. Hydrolytic editing by a class II aminoacyl-tRNA synthetase. Proc Natl Acad Sci USA 97:8916–8920. [PubMed] 10.1073/pnas.97.16.8916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Splan KE, Ignatov ME, Musier-Forsyth K. 2008. Transfer RNA modulates the editing mechanism used by class II prolyl-tRNA synthetase. J Biol Chem 283:7128–7134. [PubMed] 10.1074/jbc.M709902200 [DOI] [PubMed] [Google Scholar]
- 423.Dock-Bregeon A-C, Sankaranarayanan R, Romby P, Caillet J, Springer M, Rees B, Francklyn CS, Ehresmann C, Moras D. 2000. Transfer RNA-mediated editing in threonyl-tRNA synthetase. The class II solution to the double discrimination problem. Cell 103:877–884. 10.1016/s0092-8674(00)00191-4 [DOI] [PubMed] [Google Scholar]
- 424.Budisa N, Steipe B, Demange P, Eckerskorn C, Kellermann J, Huber R. 1995. High-level biosynthetic substitution of methionine in proteins by its analogs 2-aminohexanoic acid, selenomethionine, telluromethionine and ethionine in Escherichia coli. Eur J Biochem 230:788–796. [PubMed] 10.1111/j.1432-1033.1995.tb20622.x [DOI] [PubMed] [Google Scholar]
- 425.Lin S-X, Baltzinger M, Remy P. 1983. Fast kinetic study of yeast phenylalanyl-tRNA synthetase: An efficient discrimination between tyrosine and phenylalanine at the level of the aminoacyladenylate-enzyme complex. Biochemistry 22:681–689. [PubMed] 10.1021/bi00272a024 [DOI] [PubMed] [Google Scholar]
- 426.Ibba M, Kast P, Hennecke H. 1994. Substrate specificity is determined by amino acid binding pocket size in Escherichia coli phenylalanyl-tRNA synthetase. Biochemistry 33:7107–7112. [PubMed] 10.1021/bi00189a013 [DOI] [PubMed] [Google Scholar]
- 427.Zhai Y, Nawaz MH, Lee KW, Kirkbride E, Briggs JM, Martinis SA. 2007. Modulation of substrate specificity within the amino acid editing site of leucyl-tRNA synthetase. Biochemistry 46:3331–3337. [PubMed] 10.1021/bi061778l [DOI] [PubMed] [Google Scholar]
- 428.Mascarenhas AP, Martinis SA. 2008. Functional segregation of a predicted “hinge” site within the beta-strand linkers of Escherichia coli leucyl-tRNA synthetase. Biochemistry 47:4808–4816. [PubMed] 10.1021/bi702494q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Karkhanis VA, Mascarenhas AP, Martinis SA. 2007. Amino acid toxicities of Escherichia coli that are prevented by leucyl-tRNA synthetase amino acid editing. J Bacteriol 189:8765–8768. [PubMed] 10.1128/JB.01215-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Lincecum TL Jr, Tukalo M, Yaremchuk A, Mursinna RS, Williams AM, Sproat BS, Van Den Eynde W, Link A, Van Calenbergh S, Grotli M, Martinis SA, Cusack S. 2003. Structural and mechanistic basis of pre- and posttransfer editing by leucyl-tRNA synthetase. Mol Cell 11:951–963. [PubMed] 10.1016/S1097-2765(03)00098-4 [DOI] [PubMed] [Google Scholar]
- 431.Liu RJ, Tan M, Du DH, Xu BS, Eriani G, Wang E-D. 2011. Peripheral insertion modulates editing activity of the isolated CP1 domain of leucyl-tRNA synthetase. Biochem J 440:217–227. [PubMed] 10.1042/BJ20111177 [DOI] [PubMed] [Google Scholar]
- 432.Hu Q-H, Huang Q, Wang E-D. 2013. Crucial role of the C-terminal domain of Mycobacterium tuberculosis leucyl-tRNA synthetase in aminoacylation and editing. Nucleic Acids Res 41:1859–1872. [PubMed] 10.1093/nar/gks1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Palencia A, Crépin T, Vu MT, Linecum TL Jr, Martinis, SA, Cusak S. 2012. Structural dynamics of the aminoacylation and proofreading functional cycle of bacterial leucyl-tRNA synthetase. Nat Struct Mol Biol 19:677–684. [PubMed] 10.1038/nsmb.2317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Zhou X-L, Zhu B, Wang E-D. 2008. The CP2 domain of leucyl-tRNA synthetase is crucial for amino acid activation and post-transfer editing. J Biol Chem 283:36608–36616. [PubMed] 10.1074/jbc.M806745200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Wong FC, Beuning PJ, Nagan M, Shiba K, Musier-Forsyth K. 2002. Functional role of the prokaryotic proline-tRNA synthetase insertion domain in amino acid editing. Biochemistry 41:7108–7115. [PubMed] 10.1021/bi012178j [DOI] [PubMed] [Google Scholar]
- 436.Guo M, Chong YE, Beebe K, Shapiro R, Yang X-L, Schimmel P. 2009. The C-Ala domain brings together editing and aminoacylation functions on one tRNA. Science 325:744–747. [PubMed] 10.1126/science.1174343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Ling J, Yadavalli SS, Ibba M. 2007. Phenylalanyl-tRNA synthetase editing defects result in efficient mistranslation of phenylalanine codons as tyrosine. RNA 13:1881–1886. [PubMed] 10.1261/rna.684107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Ling J, Roy H, Ibba M. 2007. Mechanism of tRNA-dependent editing in translational quality control. Proc Natl Acad Sci USA 104:72–77. [PubMed] 10.1073/pnas.0606272104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Roy H, Ibba M. 2006. Phenylalanyl-tRNA synthetase contains a dispensable RNA-binding domain that contributes to the editing of noncognate aminoacyl-tRNA. Biochemistry 45:9156–9162. [PubMed] 10.1021/bi060549w [DOI] [PubMed] [Google Scholar]
- 440.Roy H, Ling J, Irnov M, Ibba M. 2004. Post-transfer editing in vitro and in vivo by the beta subunit of phenylalanyl-tRNA synthetase. EMBO J 23:4639–4648. [PubMed] 10.1038/sj.emboj.7600474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Tworowski D, Klipcan L, Peretz M, Moor N, Safro MG. 2015. Universal pathway for posttransfer editing reactions: Insights from the crystal structure of TtPheRS with puromycin. Proc Natl Acad Sci USA 112:3967–3972. [PubMed] 10.1073/pnas.1414852112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Cvetesic N, Bilus M, Gruic-Sovuly. 2015. The tRNA A76 hydroxyl groups control partitioning of the tRNA-dependent pre- and post-transfer editing pathways in class I tRNA synthetase. J Biol Chem 290:13981–13991. [PubMed] 10.1074/jbc.M115.648568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Hellmann RA, Martinis SA. 2009. Defects in transient tRNA translocation bypass tRNA synthetase quality control mechanisms. J Biol Chem 284:11478–11484. [PubMed] 10.1074/jbc.M807395200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Tan M, Zhu B, Liu RJ, Chen X, Zhou XL, Wang E-D. 2013. Interdomain communication modulates the tRNA-dependent pre-transfer editing of leucyl-tRNA synthetase. Biochem J 449:123–131. [PubMed] 10.1042/BJ20121258 [DOI] [PubMed] [Google Scholar]
- 445.Seiradake E, Mao W, Hernandez V, Baker SJ, Plattner JJ, Alley MR, Cusack S. 2009. Crystal structures of the human and fungal cytosolic leucyl-tRNA synthetase editing domains: A structural basis for the rational design of antifungal benzoxaboroles. J Mol Biol 390:196–207. [PubMed] 10.1016/j.jmb.2009.04.073 [DOI] [PubMed] [Google Scholar]
- 446.Liu Y, Liao J, Zhu B, Wang E-D, Ding J. 2006. Crystal structures of the editing domain of Escherichia coli leucyl-tRNA synthetase and its complexes with Met and Ile reveal a lock-and-key mechanism for amino acid discrimination. Biochem J 394:399–407. [PubMed] 10.1042/BJ20051249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Fukunaga R, Fukai S, Ishitani R, Nureki O, Yokoyama S. 2004. Crystal structures of the CP1 domain from Thermus thermophilus isoleucyl-tRNA synthetase and its complex with L-valine. J Biol Chem 279:8396–8402. [PubMed] 10.1074/jbc.M312830200 [DOI] [PubMed] [Google Scholar]
- 448.Fukunaga R, Yokoyama S. 2006. Structural basis for substrate recognition by the editing domain of isoleucyl-tRNA synthetase. J Mol Biol 359:901–912. [PubMed] 10.1016/j.jmb.2006.04.025 [DOI] [PubMed] [Google Scholar]
- 449.Chen J-F, Guo N-N, Li T, Wang E-D, Wang Y-L. 2000. CP1 domain in Escherichia coli leucyl-tRNA synthetase is crucial for its editing function. Biochemistry 39:6726–6731. [PubMed] 10.1021/bi000108r [DOI] [PubMed] [Google Scholar]
- 450.Betha AK, Williams AM, Martinis SA. 2007. Isolated CP1 domain of Escherichia coli leucyl-tRNA synthetase is dependent on flanking hinge motifs for amino acid editing activity. Biochemistry 46:6258–6267. [PubMed] 10.1021/bi061965j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451.Beebe K, Mock M, Merriman E, Schimmel P. 2008. Distinct domains of tRNA synthetase recognize the same base pair. Nature 451:90–93. [PubMed] 10.1038/nature06454 [DOI] [PubMed] [Google Scholar]
- 452.Zhang H, Huang K, Li Z, Banerjei L, Fisher KE, Grishin NV, Eisenstein E, Herzberg O. 2000. Crystal structure of YbaK protein from Haemophilus influenzae (HI1434) at 1.8Å resolution: Functional implications. Proteins 40:86–97. [DOI] [PubMed] [Google Scholar]
- 453.An S, Musier-Forsyth K. 2005. Cys-tRNAPro editing by Haemophilus influenzae YbaK via a novel synthetase•YbaK•tRNA ternary complex. J Biol Chem 280:34465–34472. [PubMed] 10.1074/jbc.M507550200 [DOI] [PubMed] [Google Scholar]
- 454.Ruan B, Söll D. 2005. The bacterial YbaK protein is a Cys-tRNAPro and Cys-tRNACys deacylase. J Biol Chem 280:25887–25891. [PubMed] 10.1074/jbc.M502174200 [DOI] [PubMed] [Google Scholar]
- 455.Vargas-Rodriguez O, Musier-Forsyth K. 2013. Exclusive use of trans-editing domains prevents proline mistranslation. J Biol Chem 288:14391–14399. [PubMed] 10.1074/jbc.M113.467795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Das M, Vargas-Rodriguez O, Goto Y, Suga H, Musier-Forsyth K. 2014. Distinct tRNA recognition strategies used by a homologous family of editing domains prevent mistranslation. Nucleic Acids Res 42:3943–3953. [PubMed] 10.1093/nar/gkt1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Liu Z, Vargas-Rodriguez O, Goto Y, Novoa EM, Ribas de Pouplana L, Suga H, Musier-Forsyth K. 2015. Homologous trans-editing factors with broad tRNA specificity prevent mistranslation caused by serine/threonine misactivation. Proc Natl Acad Sci USA 112:6027–6032. [PubMed] 10.1073/pnas.1423664112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Ishijima J, Uchida Y, Kuroishi C, Tuzuki C, Takahashi N, Okazaki N, Yutani K, Miyano M. 2006. Crystal structure of alanyl-tRNA synthetase editing-domain homolog (PH0574) from a hyperthermophile, Pyrococcus horikoshii OT3 at 1.45Å resolution. Proteins 62:1133–1137. [PubMed] 10.1002/prot.20760 [DOI] [PubMed] [Google Scholar]
- 459.Sokabe M, Okada A, Yao M, Nakashima T, Tanaka I. 2005. Molecular basis of alanine discrimination in editing site. Proc Natl Acad Sci USA 102:11669–11674. [PubMed] 10.1073/pnas.0502119102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Chong YE, Yang X-L, Schimmel P. 2008. Natural homolog of tRNA synthetase editing domain rescues conditional lethality caused by mistranslation. J Biol Chem 283:30073–30078. [PubMed] 10.1074/jbc.M805943200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Pasman Z, Robey-Bond SM, Mirando AC, Smith GJ, Lague AH, Francklyn CS. 2011. Substrate specificity and catalysis by the editing active site of alanyl-tRNA synthetase from Escherichia coli. Biochemistry 50:1474–1482. [PubMed] 10.1021/bi1013535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Novoa EM, Vargas-Rodriguez O, Lange S, Goto Y, Suga H, Musier-Forsyth K, Ribas de Pouplana L. 2015. Ancestral AlaX editing enzymes for control of genetic code fidelity are not tRNA specific. J Biol Chem 290:10495–10503. [PubMed] 10.1074/jbc.M115.640060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Hale SP, Auld DS, Schmidt E, Schimmel P. 1997. Discrete determinants in transfer RNA for editing and aminoacylation. Science 276:1250–1252. [PubMed] 10.1126/science.276.5316.1250 [DOI] [PubMed] [Google Scholar]
- 464.Yao P, Zhu B, Jaeger S, Eriani G, Wang E-D. 2008. Recognition of tRNALeu by Aquifex aeolicus leucyl-tRNA synthetase during the aminoacylation and editing steps. Nucleic Acids Res 36:2728–2738. [PubMed] 10.1093/nar/gkn028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Tardif KD, Horowitz J. 2004. Functional group recognition at the aminoacylation and editing sites of E. coli valyl-tRNA synthetase. RNA 10:493–503. [PubMed] 10.1261/rna.5166704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Mursinna RS, Lee KW, Briggs JM, Martinis SA. 2004. Molecular dissection of a critical specificity determinant within the amino acid editing domain of leucyl-tRNA synthetase. Biochemistry 43:155–165. [PubMed] 10.1021/bi034919h [DOI] [PubMed] [Google Scholar]
- 467.Bartholow TG, Sanford BL, Cao B, Schmit HL, Johnson JM, Meitzner J, Bhattacharyya S, Musier-Forsyth K, Hati S. 2014. Strictly conserved lysine of prolyl-tRNA synthetase editing domain facilitates binding and positioning of misacylated tRNAPro. Biochemistry 53:1059–1068. [PubMed] 10.1021/bi401279r [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Dulic M, Perona JJ, Gruic-Sovulj I. 2014. Determinants for tRNA-dependent pretransfer editing in the synthetic site of isoleucyl-tRNA synthetase. Biochemistry 53:6189–6198. [PubMed] 10.1021/bi5007699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Ling J, So BR, Yadavalli SS, Roy H, Shoji S, Fredrick K, Musier-Forsyth K, Ibba M. 2009. Resampling and editing of mischarged tRNA prior to translation elongation. Mol Cell 33:654–660. [PubMed] 10.1016/j.molcel.2009.01.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Liu CC, Schultz PG. 2010. Adding new chemistries to the genetic code. Annu Rev Biochem 79:413–444. [PubMed] 10.1146/annurev.biochem.052308.105824 [DOI] [PubMed] [Google Scholar]
- 471.Crépin T, Schmitt E, Blanquet S, Mechulam Y. 2002. Structure and function of the C-terminal domain of methionyl-tRNA synthetase. Biochemistry 41:13003–13011. [PubMed] 10.1021/bi026343m [DOI] [PubMed] [Google Scholar]
- 472.Frugier M, Giegé R, Schimmel P. 2003. RNA recognition by designed peptide fusion creates “artificial” tRNA synthetase. Proc Natl Acad Sci USA 100:7471–7475. [PubMed] 10.1073/pnas.1332771100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Frugier M, Schimmel P. 1997. Subtle atomic group discrimination in the RNA minor groove. Proc Natl Acad Sci USA 94:11291–11294. [PubMed] 10.1073/pnas.94.21.11291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Ibba M, Söll D. 1999. Quality control mechanisms during translation. Science 286:1893–1897. [PubMed] 10.1126/science.286.5446.1893 [DOI] [PubMed] [Google Scholar]
- 475.Jakubowski H. 2012. Quality control in tRNA charging. Wiley Interdiscip Rev RNA 3:295–310. [PubMed] 10.1002/wrna.122 [DOI] [PubMed] [Google Scholar]
- 476.Rodnina MV. 2012. Quality control of mRNA decoding on the bacterial ribosome. Adv Protein Chem Struct Biol 86:95–128. [PubMed] 10.1016/B978-0-12-386497-0.00003-7 [DOI] [PubMed] [Google Scholar]
- 477.Yadavalli SS, Ibba M. 2012. Quality control in aminoacyl-tRNA synthesis its role in translational fidelity. Adv Protein Chem Struct Biol 86:1–43. [PubMed] 10.1016/B978-0-12-386497-0.00001-3 [DOI] [PubMed] [Google Scholar]
- 478.Kumar S, Das M, Hadad CM, Musier-Forsyth K. 2013. Aminoacyl-tRNA substrate and enzyme backbone atoms contribute to translational quality control by YbaK. J Phys Chem B 117:4521–4527. [PubMed] 10.1021/jp308628y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Cvetesic N, Palencia A, Halasz I, Cusack S, Gruic-Sovulj I. 2014. The physiological target for LeuRS translational quality control is norvaline. EMBO J 33:1639–1653. [PubMed] 10.15252/embj.201488199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Ataide SF, Ibba M. 2006. Small molecules: Big players in the evolution of protein synthesis. ACS Chem Biol 1:285–297. [PubMed] 10.1021/cb600200k [DOI] [PubMed] [Google Scholar]
- 481.Chênevert R, Bernier S, Lapointe J. 2003. Inhibitors of aminoacyl-tRNA synthetases as antibiotics and tools for structural and mechanistic studies, p 416–428. In Lapointe J and Brakier-Gringas L (ed), Translation Mechanisms. Landes Sciences, Georgetown, TX. [Google Scholar]
- 482.Finn J, Tao J. 2005. Aminoacyl-tRNA synthetases as anti-infective drug targets, p 405–413. In Ibba M, Francklyn C, and Cusack S (ed), The Aminoacyl-tRNA Synthetases. Landes Bioscience, Georgetown, TX. [Google Scholar]
- 483.Kim S, Lee C, Choi EC, Choi SY. 2003. Aminoacyl-tRNA synthetases and their inhibitors as a novel family of antibiotics. Appl Microbiol Biotechnol 61:278–288. [PubMed] 10.1007/s00253-003-1243-5 [DOI] [PubMed] [Google Scholar]
- 484.Ochsner UA, Jarvis TC. 2014. Aminoacyl-tRNA synthetase inhibitors, p 387–410. In Gualerzi CO, Brandi L, Fabbretti A, Pon CL (ed), Antibiotics: Targets, Mechanisms, and Resistance. Wiley-VCH Verlag GmbH & Co. KgaA. [Google Scholar]
- 485.Tao J, Schimmel P. 2000. Inhibitors of aminoacyl-tRNA synthetases as novel anti-infectives. Exp Opin Invest Drugs 9:1767–1775. [PubMed] 10.1517/13543784.9.8.1767 [DOI] [PubMed] [Google Scholar]
- 486.Zhao Y, Meng Q, Bai L, Zhou H. 2014. In silico discovery of aminoacyl-tRNA synthetase inhibitors. Int J Mol Sci 15:1358–1373. [PubMed] 10.3390/ijms15011358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Brown MJ, Carter PS, Fenwick AS, Fosberry AP, Hamprecht DW, Hibbs MJ, Jarvest RL, Mensah L, Milner PH, O’Hanlon PJ, Pope AJ, Richardson CM, West A, Witty DR. 2002. The antimicrobial natural product chuangxinmycin and some synthetic analogues are potent and selective inhibitors of bacterial tryptophanyl-tRNA synthetase. Bioorg Med Chem Lett 12:3171–3174. [PubMed] 10.1016/S0960-894X(02)00604-2 [DOI] [PubMed] [Google Scholar]
- 488.Bullard JM. 2015. High throughput screen identifies natural product inhibitor of phenylalanyl-tRNA synthetase from Pseudomonas aeruginosa and Streptococcus pneumoniae. Curr Drug Discov Technol 11:279–292. 10.2174/1570163812666150120154701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Zeng Y, Kulkarni A, Yang Z, Patil PB, Zhou W, Chi X, Van Lanen S, Chen S. 2012. Biosynthesis of albomycin delta2 provides a template for assembling siderophore and aminoacyl-tRNA synthetase inhibitor conjugates. ACS Chem Biol 7:1565–1575. [PubMed] 10.1021/cb300173x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Metlitskaya A, Kazakov T, Kommer A, Pavlova O, Praetorius-Ibba M, Ibba M, Krasheninnikov I, Kolb V, Khmel I, Severinov K. 2006. Aspartyl-tRNA synthetase is the target of peptide nucleotide antibiotic microcin C. J Biol Chem 281:18033–18042. [PubMed] 10.1074/jbc.M513174200 [DOI] [PubMed] [Google Scholar]
- 491.Battenberg OA, Yang Y, Verhelst SH, Sieber SA. 2013. Target profiling of 4-hydroxyderricin in S. aureus reveals seryl-tRNA synthetase binding and inhibition by covalent modification. Mol Biosyst 9:343–351. [PubMed] 10.1039/c2mb25446h [DOI] [PubMed] [Google Scholar]
- 492.Perl TM, Cullen JJ, Wenzel RP, Zimmerman MB, Pfaller MA, Sheppard D, Twombley J, French PP, Herwaldt LA. 2002. Intranasal mupirocin to prevent postoperative Staphylococcus aureus infections. N Engl J Med 346:1871–1877. [PubMed] 10.1056/NEJMoa003069 [DOI] [PubMed] [Google Scholar]
- 493.Walker ES, Levy F, Shorman M, David G, Abdalla J, Sarubbi FA. 2004. A decline in mupirocin resistance in methicillin-resistant Staphylococcus aureus accompanied administrative control of prescriptions. J Clin Microbiol 42:2792–2795. [PubMed] 10.1128/JCM.42.6.2792-2795.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494.Stefanska AL, Coates NJ, Mensah LM, Pope AJ, Ready SJ, Warr SR. 2000. SB-219383, a novel tyrosyl-tRNA synthetase inhibitor from a Micromonospora sp. I. Fermentation, isolation and properties. J Antibiot 53:345–350. [PubMed] 10.7164/antibiotics.53.345 [DOI] [PubMed] [Google Scholar]
- 495.Vondenhoff GH, Pugach K, Gadakh B, Carlier L, Rozenski J, Froeyen M, Severinov K, Van Aerschot A. 2013. N-alkylated aminoacyl sulfamoyladenosines as potential inhibitors of aminoacylation reactions and microcin C analogues containing D-amino acids. PLoS One 8:e79234. [PubMed] 10.1371/journal.pone.0079234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496.Brown MJ, Mensah LM, Doyle ML, Broom NJ, Osbourne N, Forrest AK, Richardson CM, O’Hanlon PJ, Pope AJ. 2000. Rational design of femtomolar inhibitors of isoleucyl-tRNA synthetase from a binding model for pseudomonic acid-A. Biochemistry 39:6003–6011. [PubMed] 10.1021/bi000148v [DOI] [PubMed] [Google Scholar]
- 497.Cooper SM, Laosripaiboon W, Rahman AS, Hothersall J, El-Sayed AK, Winfield C, Crosby J, Cox RJ, Simpson TJ, Thomas CM. 2005. Shift to pseudomonic acid B production in P. fluorescens NCIMB10586 by mutation of mupirocin tailoring genes mupO, mupU, mupV, and macpE. Chem Biol 12:825–833. [PubMed] 10.1016/j.chembiol.2005.05.015 [DOI] [PubMed] [Google Scholar]
- 498.Kitabatake M, Ali K, Demain A, Sakamoto K, Yokoyama S, Söll D. 2002. Indolmycin resistance of Streptomyces coelicolor A3(2) by induced expression of one of its two tryptophanyl-tRNA synthetases. J Biol Chem 277:23882–23887. [PubMed] 10.1074/jbc.M202639200 [DOI] [PubMed] [Google Scholar]
- 499.Thomas CM, Hothersall J, Willis CL, Simpson TJ. 2010. Resistance to and synthesis of the antibiotic mupirocin. Nat Rev Microbiol 8:281–289. [PubMed] 10.1038/nrmicro2278 [DOI] [PubMed] [Google Scholar]
- 500.Vecchione JJ, Sello JK. 2010. Regulation of an auxiliary, antibiotic-resistant tryptophanyl-tRNA synthetase gene via ribosome-mediated transcriptional attenuation. J Bacteriol 192:3565–3573. [PubMed] 10.1128/JB.00290-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501.Antonio M, McFerran N, Pallen MJ. 2002. Mutations affecting the Rossman fold of isoleucyl-tRNA synthetase are correlated with low-level mupirocin resistance in Staphylococcus aureus. Antimicrob Agents Chemother 46:438–442. [PubMed] 10.1128/AAC.46.2.438-442.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Vecchione JJ, Sello JK. 2008. Characterization of an inducible, antibiotic-resistant aminoacyl-tRNA synthetase gene in Streptomyces coelicolor. J Bacteriol 190:6253–6257. [PubMed] 10.1128/JB.00737-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503.Paulander W, Andersson DI, Maisnier-Patin S. 2010. Amplification of the gene for isoleucyl-tRNA synthetase facilitates adaptation to the fitness cost of mupirocin resistance in Salmonella enterica. Genetics 185:305–312. [PubMed] 10.1534/genetics.109.113514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Seah C, Alexander DC, Louie L, Simor A, Low DE, Longtin J, Melano RG. 2012. MupB, a new high-level mupirocin resistance mechanism in Staphylococcus aureus. Antimicrob Agents Chemother 56:1916–1920. [PubMed] 10.1128/AAC.05325-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Forrest AK, Jarvest RL, Mensah LM, O’Hanlon PJ, Pope AJ, Sheppard RJ. 2000. Aminoalkyl-adenylate and aminoacyl-sulfamate intermediate analogues differing greatly in affinity for their cognate Staphylococcus aureus aminoacyl-tRNA synthetases. Bioorg Med Chem Lett 10:1871–1874. [PubMed] 10.1016/S0960-894X(00)00360-7 [DOI] [PubMed] [Google Scholar]
- 506.Bernier S, Akochy PM, Lapointe J, Chênevert R. 2005. Synthesis and aminoacyl-tRNA synthetase inhibitory activity of aspartyl adenylate analogs. Bioorg Med Chem 13:69–75. [PubMed] 10.1016/j.bmc.2004.09.055 [DOI] [PubMed] [Google Scholar]
- 507.Bernard D, Akochy PM, Bernier S, Fisette O, Brousseau OC, Chênevert R, Roy PH, Lapointe J. 2007. Inhibition by L-aspartol adenylate of a nondiscriminating aspartyl-tRNA synthetase reveals differences between the interactions of its active site with tRNAAsp and tRNAAsn. J Enzyme Inhib Med Chem 22:77–82. [PubMed] 10.1080/14756360600952316 [DOI] [PubMed] [Google Scholar]
- 508.Balg C, Blais SP, Bernier S, Huot JL, Couture M, Lapointe J, Chênevert R. 2007. Synthesis of beta-ketophosphonate analogs of glutamyl and glutaminyl adenylate, and selective inhibition of the corresponding bacterial aminoacyl-tRNA synthetases. Bioorg Med Chem 15:295–304. [PubMed] 10.1016/j.bmc.2006.09.056 [DOI] [PubMed] [Google Scholar]
- 509.Farhanullah, Kang T, Yoon E-J, Choi EC, Kim S, Lee J. 2009. 2-[2-Substituted-3-(3,4-dichlorobenzylamino) propylamino]-1H-quinolin-4-ones as Staphylococcus aureus methionyl-tRNA synthetase inhibitors. Eur J Med Chem 44:239–250. [PubMed] 10.1016/j.ejmech.2008.02.021 [DOI] [PubMed] [Google Scholar]
- 510.Finn J, Mattia K, Morytko M, Ram S, Yang Y, Wu X, Mak E, Gallant P, Keith D. 2003. Discovery of a potent and selective series of pyrazole bacterial methionyl-tRNA synthetase inhibitors. Bioorg Med Chem Lett 13:2231–2234. [PubMed] 10.1016/S0960-894X(03)00298-1 [DOI] [PubMed] [Google Scholar]
- 511.Green LS, Bullard JM, Ribble W, Dean F, Ayers DF, Ochsner UA, Janjic N, Jarvis TC. 2009. Inhibition of methionyl-tRNA synthetase by REP8839 and effects of resistance mutations on enzyme activity. Antimicrob Agents Chemother 53:86–94. [PubMed] 10.1128/AAC.00275-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 512.Wu Y, Yu K, Xu B, Chen L, Chen X, Mao J, Danchin A, Shen X, Qu D, Jiang H. 2007. Potent and selective inhibitors of Staphylococcus epidermidis tryptophanyl-tRNA synthetase. J Antimicrob Chemother 60:502–509. [PubMed] 10.1093/jac/dkm229 [DOI] [PubMed] [Google Scholar]
- 513.Wei W, Shi WK, Wang PF, Zeng XT, Li P, Zhang JR, Li Q, Tang ZP, Peng J, Wu LZ, Xie MQ, Liu C, Li XH, Wang YC, Xiao ZP, Zhu HL. 2015. Adenosine analogs as inhibitors of tyrosyl-tRNA synthetase: Design, synthesis and antibacterial evaluation. Bioorg Med Chem 23:6602–6611. [PubMed] 10.1016/j.bmc.2015.09.018 [DOI] [PubMed] [Google Scholar]
- 514.Xiao ZP, Wei W, Wang PF, Shi WK, Zhu N, Xie MQ, Sun YW, Li LX, Xie YX, Zhu LS, Tang N, Ouyang H, Li XH, Wang GC, Zhu HL. 2015. Synthesis and evaluation of new tyrosyl-tRNA synthetase inhibitors as antibacterial agents based on a N2-(arylacetyl)glycinanilide scaffold. Eur J Med Chem 102:631–638. [PubMed] 10.1016/j.ejmech.2015.08.025 [DOI] [PubMed] [Google Scholar]
- 515.Zhu N, Lin Y, Li D, Gao N, Liu C, You X, Jiang J, Jiang W, Si S. 2015. Identification of an anti-TB compound targeting the tyrosyl-tRNA synthetase. J Antimicrob Chemother 70:2287–2294. [PubMed] 10.1093/jac/dkv110 [DOI] [PubMed] [Google Scholar]
- 516.Montgomery JI, Smith JF, Tomaras AP, Zaniewski R, McPherson CJ, McAllister LA, Hartman-Neumann S, Arcari JT, Lescoe M, Gutierrez J, Yuan Y, Limberakis C, Miller AA. 2015. Discovery and characterization of a novel class of pyrazolopyrimidinedione tRNA synthesis inhibitors. J Antibiot (Tokyo) 68:361–367. [PubMed] 10.1038/ja.2014.163 [DOI] [PubMed] [Google Scholar]
- 517.Gurcha SS, Usha V, Cox JA, Futterer K, Abrahams KA, Bhatt A, Alderwick LJ, Reynolds RC, Loman NJ, Nataraj V, Alemparte C, Barros D, Lloyd AJ, Ballell L, Hobrath JV, Besra GS. 2014. Biochemical and structural characterization of mycobacterial aspartyl-tRNA synthetase AspS, a promising TB drug target. PLoS One 9:e113568. [PubMed] 10.1371/journal.pone.0113568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518.Montgomery JI, Toogood PL,Hutchings KM, Liu J, Narasimhan L, Braden T, Dermyer MR, Kulynych AD, Smith YD, Warmus JS, Taylor C. 2009. Discovery and SAR of benzyl phenyl ethers as inhibitors of bacterial phenylalanyl-tRNA synthetase. Bioorg Med Chem Lett 19:665–669. [PubMed] 10.1016/j.bmcl.2008.12.054 [DOI] [PubMed] [Google Scholar]
- 519.Hu Q-H, Liu R-J, Fang Z-P, Zhang J, Ding Y-Y, Tan M, Wang M, Pan W, Zhou H-C, Wang E-D. 2013. Discovery of a potent benzoxaborole-based anti-pneumococcal agent targeting leucyl-tRNA synthetase. Sci Rep 3:2475. [PubMed] 10.1038/srep02475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Goldstein EJ, Citron DM, Tyrrell KL, Merriam VC. 2013. Comparative in vitro activity of GSK2251052, a novel boron leucyl-tRNA synthetase inhibitor, against 916 anaerobic organisms. Antimicrob Agents Chemother 57:2401–2404. [PubMed] 10.1128/AAC.02580-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521.Balg C, Mieri MD, Huot JL, Blais SP, Lapointe J, Chênevert R. 2010. Inhibition of Helicobacter pylori aminoacyl-tRNA amidotransferase by chloramphenicol analogs. Bioorg Med Chem Lett 18:7868–7872. [PubMed] 10.1016/j.bmc.2010.09.045 [DOI] [PubMed] [Google Scholar]
- 522.Brown KA. 2011. A brief perspective of the determination of crystal structures of site-directed mutants of tyrosyl-tRNA synthetase. Protein Eng Des Sel 24:229–231. [PubMed] 10.1093/protein/gzq102 [DOI] [PubMed] [Google Scholar]
- 523.Leatherbarrow RJ, Fersht AR. 1986. Protein engineering. Protein Eng Des Sel 1:7–16. [PubMed] 10.1093/protein/1.1.7 [DOI] [PubMed] [Google Scholar]
- 524.Winter G, Fersht AR, Wilkinson AJ, Zoller M, Smith M. 1982. Redesigning enzyme using structure by site-directed mutagenesis: Tyrosyl-tRNA synthetase and ATP binding. Nature 299:756–758. [PubMed] 10.1038/299756a0 [DOI] [PubMed] [Google Scholar]
- 525.Köhrer C, RajBhandary UL (ed). 2009. Protein Engineering, vol. 22. Springer-Verlag, Berlin, Germany. [Google Scholar]
- 526.Young TS, Schultz PG. 2010. Beyond the canonical 20 amino acids: Expanding the genetic lexicon. J Biol Chem 285:11039–11044. [PubMed] 10.1074/jbc.R109.091306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527.Kobayashi T, Sakamoto K, Takimura T, Sekine R, Kelly VP, Kamata K, Nishimura S, Yokoyama S. 2005. Structural basis of nonnatural amino acid recognition by an engineered aminoacyl-tRNA synthetase for genetic code expansion. Proc Natl Acad Sci USA 102:1366–1371. [PubMed] 10.1073/pnas.0407039102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 528.Kobayashi T, Yanagisawa T, Sakamoto K, Yokoyama S. 2009. Recognition of non-alpha-amino substrates by pyrrolysyl-tRNA synthetase. J Mol Biol 385:1352–1360. [PubMed] 10.1016/j.jmb.2008.11.059 [DOI] [PubMed] [Google Scholar]
- 529.Köhrer C, Sullivan EL, RajBhandary UL. 2004 Complete set of orthogonal 21st aminoacyl-tRNA synthetase-amber, ochre and opal suppressor tRNA pairs: Concomitant suppression of three different termination codons in an mRNA in mammalian cells. Nucleic Acids Res 32:6200–6211. [PubMed] 10.1093/nar/gkh959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 530.Casina VC, Lobashevsky AA, McKinney WE, Brown CL, Alexander RW. 2011. Role for a conserved structural motif in assembly of a class I aminoacyl-tRNA synthetase active site domain. Biochemistry 50:763–769. [PubMed] 10.1021/bi101375d [DOI] [PubMed] [Google Scholar]
- 531.Ambrogelly A, O’Donoghue P, Söll D, Moses S. 2010. A bacterial ortholog of class II lysyl-tRNA synthetase activates lysine. FEBS Lett 584:3055–3060. [PubMed] 10.1016/j.febslet.2010.05.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532.Kong L, Fromant M, Blanquet S, Plateau P. 1991. Evidence for a new Escherichia coli protein resembling a lysyl-transfer RNA synthetase. Gene 108:163–164. [PubMed] 10.1016/0378-1119(91)90503-4 [DOI] [PubMed] [Google Scholar]
- 533.Campanacci V, Dubois DY, Becker HD, Kern D, Spinelli S, Valencia C, Pagot F, Salomoni A, Grisel S, Vincentelli R, Bignon C, Lapointe J, Giegé R, Cambillau C. 2004. The Escherichia coli YadB gene product reveals a novel aminoacyl-tRNA synthetase like activity. J Mol Biol 337:273–283. [PubMed] 10.1016/j.jmb.2004.01.027 [DOI] [PubMed] [Google Scholar]
- 534.Dubois DY, Blaise M, Becker HD, Campanacci V, Keith G, Giegé R, Cambillau C, Lapointe J, Kern D. 2004. An aminoacyl-tRNA synthetase-like protein encoded by the Escherichia coli yadB gene glutamylates specifically tRNAAsp. Proc Natl Acad Sci USA 101:7530–7535. [PubMed] 10.1073/pnas.0401634101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.Putney SD, Melendez DL, Schimmel PR. 1981. Cloning, partial sequencing, and in vitro transcription of the gene for alanine tRNA synthetase. J Biol Chem 256:205–211. [PubMed] [PubMed] [Google Scholar]
- 536.Putney SD, Schimmel P. 1981. An aminoacyl-tRNA synthetase bind to a specific DNA sequence and regulates its gene transcription. Nature 291:632–635. [PubMed] 10.1038/291632a0 [DOI] [PubMed] [Google Scholar]
- 537.Eriani G, Dirheimer G, Gangloff J. 1989. Isolation and characterization of the gene coding for Escherichia coli arginyl-tRNA synthetase. Nucleic Acids Res 17:5725–5736. [PubMed] 10.1093/nar/17.14.5725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538.Gama-Castro S, Jimenez-Jacinto V, Peralta-Gil M, Santos-Zavaleta A, Penaloza-Spinola MI, Contreras-Moreira B, Segura-Salazar J, Muniz-Rascado L, Martinez-Flores I, Salgado H, Bonavides-Martinez C, Abreu-Goodger C, Rodriguez-Penagos C, Miranda-Rios J, Morett E, Merino E, Huerta AM, Trevino-Quintanilla L, Collado-Vides J. 2008. RegulonDB (version 6.0): Gene regulation model of Escherichia coli K12 beyond transcription, active (experimental) annotated promoters and Textpresso navigation. Nucleic Acids Res 36:D120–D124. [PubMed] 10.1093/nar/gkm994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539.Eriani G, Dirheimer G, Gangloff J. 1990. Aspartyl-tRNA synthetase from Escherichia coli: Cloning and characterization of the gene, homologies of its translated amino acid sequence with asparaginyl- and lysyl-tRNA synthetases. Nucleic Acids Res 18:7109–7118. [PubMed] 10.1093/nar/18.23.7109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540.Eriani G, Dirheimer G, Gangloff J. 1991. Cysteinyl-tRNA synthetase: Determination of the last E. coli aminoacyl-tRNA synthetase primary structure. Nucleic Acids Res 19:265–269. [PubMed] 10.1093/nar/19.2.265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541.Nonaka G, Blankschien M, Herman C, Gross CA, Rhodius VA. 2006. Regulon and promoter analysis of the E. coli heat-shock factor, sigma32, reveals a multifaceted cellular response to heat stress. Genes Dev 20:1776–1789. [PubMed] 10.1101/gad.1428206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 542.Cheung AY, Söll D. 1984. In vivo and in vitro transcription of the Escherichia coli glutaminyl-tRNA synthetase gene. J Biol Chem 259:9953–9958. [PubMed] [PubMed] [Google Scholar]
- 543.Cheung A, Morgan S, Low KB, Söll D. 1979. Regulation of the biosynthesis of aminoacyl-transfer ribonucleic acid synthetases and of transfer ribonucleic acid in Escherichia coli. VI. Mutants with increased levels of glutaminyl-transfer ribonucleic acid synthetase and of glutamine transfer ribonucleic acid. J Bacteriol 139:176–184. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544.Putzer H, Grunberg-Manago M, Springer M. 1995. Bacterial aminoacyl-tRNA synthetases: Genes and regulation of expression, p 293–333. In Söll D and RajBhandary UL (ed), tRNA: Structure, Biosynthesis, and Function. American Society for Microbiology, Washington, DC. [Google Scholar]
- 545.Plumbridge J, Söll D. 1987. The effect of dam methylation on the expression of glnS in E. coli. Biochimie 69:539–541. [PubMed] 10.1016/0300-9084(87)90091-5 [DOI] [PubMed] [Google Scholar]
- 546.Brombacher E, Baratto A, Dorel C, Landini P. 2006. Gene expression regulation by the Curli activator CsgD protein: Modulation of cellulose biosynthesis and control of negative determinants for microbial adhesion. J Bacteriol 188:2027–2037. [PubMed] 10.1128/JB.188.6.2027-2037.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 547.Brun YV, Breton R, Lanouette P, Lapointe J. 1990. Precise mapping and comparison of two evolutionarily related regions of the Escherichia coli K12 chromosome. Evolution of valU and lysT from an ancestral tRNA operon. J Mol Biol 214:825–843. 10.1016/0022-2836(90)90339-N [DOI] [PubMed] [Google Scholar]
- 548.Champagne N, Lapointe J. 1998. Influence of FIS on the transcription from closely spaced and non-overlapping divergent promoters for an aminoacyl-tRNA synthetase gene (gltX) and a tRNA operon (valU) in Escherichia coli. Mol Microbiol 27:1141–1156. [PubMed] 10.1046/j.1365-2958.1998.00745.x [DOI] [PubMed] [Google Scholar]
- 549.Webster TA, Gibson BW, Keng T, Biemann K, Schimmel P. 1983. Primary structures of both subunits of Escherichia coli glycyl-tRNA synthetase. J Biol Chem 258:10637–10641. [PubMed] [PubMed] [Google Scholar]
- 550.Freedman R, Gibson B, Donovan D, Biemann K, Eisenbeis S, Parker J, Schimmel P. 1985. Primary structure of histidine-tRNA synthetase and characterization of hisS transcripts. J Biol Chem 260:10063–10068. [PubMed] [PubMed] [Google Scholar]
- 551.Miller KW, Bouvier J, Stragier P, Wu HC. 1987. Identification of the genes in the Escherichia coli ileS-lsp operon. Analysis of multiple polycistronic mRNAs made in vivo. J Biol Chem 262:7391–7397. [PubMed] [PubMed] [Google Scholar]
- 552.Härtlein M, Madern D. 1987. Molecular cloning and nucleotide sequence of the gene for Escherichia coli leucyl-tRNA synthetase. Nucleic Acids Res. 15:10199–10210. [PubMed] 10.1093/nar/15.24.10199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553.LaRossa R, Vogell G, Low KB, Söll D. 1977. Regulation of biosynthesis of aminoacyl-tRNA synthetases and of tRNA in Escherichia coli. II. Isolation of regulatory mutants affecting leucyl-tRNA synthetase levels. J Mol Biol 117:1033–1048. 10.1016/S0022-2836(77)80011-9 [DOI] [PubMed] [Google Scholar]
- 554.LaRossa RA, Mao J-i, Low KB, Söll D. 1977. Regulation of biosynthesis of aminoacyl-tRNA synthetases and of tRNA in Escherichia coli. III. Biochemical characterization of regulatory mutants affecting leucyl-tRNA synthetase levels. J Mol Biol 117:1049–1059. 10.1016/S0022-2836(77)80012-0 [DOI] [PubMed] [Google Scholar]
- 555.Theall G, Low KB, Söll D. 1979. Regulation of the biosynthesis of aminoacyl-tRNA synthetases and of tRNA in Escherichia coli. IV. Mutants with increased levels of leucyl- or seryl-tRNA synthetase. Mol Gen Genet 169:205–211. [PubMed] 10.1007/bf00271672 [DOI] [PubMed] [Google Scholar]
- 556.Rhodius VA, Suh WC, Nonaka G, West J, Gross CA. 2006. Conserved and variable functions of the sigmaE stress response in related genomes. PLoS Biol 4:e2. [PubMed] 10.1371/journal.pbio.0040002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557.Dartigalongue C, Missiakas D, Raina S. 2001. Characterization of the Escherichia coli sigma E regulon. J Biol Chem 276:20866–20875. [PubMed] 10.1074/jbc.M100464200 [DOI] [PubMed] [Google Scholar]
- 558.Kawakami K, Jönsson YH, Björk GR, Ikeda H, Nakamura Y. 1988. Chromosomal location and structure of the operon encoding peptide-chain-release factor 2 of Escherichia coli. Proc Natl Acad Sci USA 85:5620–5624. [PubMed] 10.1073/pnas.85.15.5620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 559.Lévêque F, Gazeau M, Fromant M, Blanquet S, Plateau P. 1991. Control of Escherichia coli lysyl-transfer-RNA synthetase expression by anaerobiosis. J Bacteriol 173:7903–7910. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560.Gazeau M, Delort F, Fromant M, Dessen P, Blanquet S, Plateau P. 1994. Structure-function relationship of the Lrp-binding region upstream of lysU in Escherichia coli. J Mol Biol 241:378–389. [PubMed] 10.1006/jmbi.1994.1514 [DOI] [PubMed] [Google Scholar]
- 561.Dardel F, Panvert M, Fayat G. 1990. Transcription and regulation of expression of the Escherichia coli methionyl-tRNA synthetase gene. Mol Gen Genet 223:121–133. [PubMed] 10.1007/BF00315804 [DOI] [PubMed] [Google Scholar]
- 562.Fayat G, Mayaux J-F, Sacerdot C, Fromant M, Springer M, Grunberg-Manago M, Blanquet S. 1983. Escherichia coli phenylalanyl-tRNA synthetase operon region: Evidence for an attenuation mechanism. Identification of the gene for the ribosomal protein L20. J Mol Biol 171:239–261. 10.1016/0022-2836(83)90092-X [DOI] [PubMed] [Google Scholar]
- 563.Springer M, Plumbridge JA, Butler JS, Graffe M, Dondon J, Mayaux J-F, Fayat G, Lestienne P, Blanquet S, Grunberg-Manago M. 1985. Autogenous control of Escherichia coli threonyl-tRNA synthetase expression in vivo. J Mol Biol 185:93–104. [PubMed] 10.1016/0022-2836(85)90185-8 [DOI] [PubMed] [Google Scholar]
- 564.Springer M., Trudel M, Plumbridge J, Fayat G, Mayaux J-F, Sacerdot C, Blanquet S, Grunberg-Manago M. 1983. Escherichia coli phenylalanyl-tRNA synthetase operon is controlled by attenuation in vivo. J Mol Biol 171:263–279. [PubMed] 10.1016/0022-2836(83)90093-1 [DOI] [PubMed] [Google Scholar]
- 565.Salmon K, Hung SP, Mekjian K, Baldi P, Hatfield GW, Gunsalus RP. 2003. Global gene expression profiling in Escherichia coli K12. The effects of oxygen availability and FNR. J Biol Chem 278:29837–29855. [PubMed] 10.1074/jbc.M213060200 [DOI] [PubMed] [Google Scholar]
- 566.Salmon KA, Hung SP, Steffen NR, Krupp R, Baldi P, Hatfield GW, Gunsalus RP. 2005. Global gene expression profiling in Escherichia coli K12: Effects of oxygen availability and ArcA. J Biol Chem 280:15084–15096. [PubMed] 10.1074/jbc.M414030200 [DOI] [PubMed] [Google Scholar]
- 567.Mechulam Y, Blanquet S, Fayat G. 1987. Dual level control of the Escherichia coli pheST-himA operon expression. J Mol Biol 197:453–470. [PubMed] 10.1016/0022-2836(87)90558-4 [DOI] [PubMed] [Google Scholar]
- 568.Zhou Z, Syvanen M. 1990. Identification and sequence of the drpA gene from Escherichia coli. J Bacteriol 172:281–286. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 569.Ferri ML, Vincent C, Leberman R, Härtlein M. 1997. Characterization of a temperature-sensitive Escherichia coli mutant and revertants with altered seryl-tRNA synthetase activity. J Bacteriol 179:2446–2448. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570.Comer MM, Dondon J, Graffe, M, Yarchuk O, Springer M. 1996. Growth rate-dependent control, feedback regulation and steady-state mRNA levels of the threonyl-tRNA synthetase gene of Escherichia coli.J Mol Biol 261:108–124. [PubMed] 10.1006/jmbi.1996.0445 [DOI] [PubMed] [Google Scholar]
- 571.Partridge JD, Bodenmiller DM, Humphrys MS, Spiro S. 2009. NsrR targets in the Escherichia coli genome: new insights into DNA sequence requirements for binding and a role for NsrR in the regulation of motility. Mol Microbiol 73:680–694. [PubMed] 10.1111/j.1365-2958.2009.06799.x [DOI] [PubMed] [Google Scholar]
- 572.Lyngstadaas A, Lobner-Olesen A, Grelland E, Boye E. 1999. The gene for 2-phosphoglycolate phosphatase (gph) in Escherichia coli is located in the same operon as dam and at least five other diverse genes. Biochim Biophys Acta 1472:376–384. [PubMed] 10.1016/S0304-4165(99)00146-4 [DOI] [PubMed] [Google Scholar]
- 573.Hall CV, Yanofsky C. 1982. Regulation of tryptophanyl-tRNA synthetase formation. J Bacteriol 151:918–923. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 574.Yang Y, Tsui HC, Man TK, Winkler ME. 1998. Identification and function of the pdxY gene, which encodes a novel pyridoxal kinase involved in the salvage pathway of pyridoxal 5′-phosphate biosynthesis in Escherichia coli K12. J Bacteriol 180:1814–1821. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575.Heck JD, Hatfield GW. 1988. Valyl-tRNA synthetase gene of Escherichia coli K12. Molecular genetic characterization. J Biol Chem 263:857–867. [PubMed] [PubMed] [Google Scholar]
- 576.Brun YV, Sanfacon H, Breton R, Lapointe J. 1990. Closely spaced and divergent promoters for an aminoacyl-tRNA synthetase gene and a tRNA operon in Escherichia coli. Transcriptional and post-transcriptional regulation of gltX, valU and alaW. J Mol Biol 214:845–864. [PubMed] 10.1016/0022-2836(90)90340-r [DOI] [PubMed] [Google Scholar]
- 577.Gagnon Y, Breton R, Putzer H, Pelchat M, Grunberg-Manago M, Lapointe J. 1994. Clustering and co-transcription of the Bacillus subtilis genes encoding the aminoacyl-tRNA synthetases specific for glutamate and for cysteine and the first enzyme for cysteine biosynthesis. J Biol Chem 269:7473–7482. [PubMed] [PubMed] [Google Scholar]
- 578.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol Syst Biol 2:2006 0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 579.Gerdes SY, Scholle MD, Campbell JW, Balazsi G, Ravasz E, Daugherty MD, Somera AL, Kyrpides NC, Anderson I, Gelfand MS, Bhattacharya A, Kapatral V, D’Souza M, Baev MV, Grechkin Y, Mseeh F, Fonstein MY, Overbeek R, Barabasi AL, Oltvai ZN, Osterman AL. 2003. Experimental determination and system level analysis of essential genes in Escherichia coli MG1655. J Bacteriol 185:5673–5684. [PubMed] 10.1128/JB.185.19.5673-5684.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 580.Keng T, Webster TA, Sauer RT, Schimmel P. 1982. Gene for Escherichia coli glycyl-tRNA synthetase has tandem subunit coding regions in the same reading frame. J Biol Chem 257:12503–12508. [PubMed] [PubMed] [Google Scholar]
- 581.Plumbridge JA, Springer M. 1980. Genes for the two subunits of phenylalanyl-tRNA synthesis of Escherichia coli are transcribed from the same promoter. J Mol Biol 144:595–600. [PubMed] 10.1016/0022-2836(80)90341-1 [DOI] [PubMed] [Google Scholar]
- 582.Craigen WJ, Cook RG, Tate WP, Caskey CT. 1985. Bacterial peptide chain release factors: Conserved primary structure and possible frameshift regulation of release factor 2. Proc Natl Acad Sci USA 82:3616–3620. [PubMed] 10.1073/pnas.82.11.3616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 583.Nogueira T, de Smit M, Graffe M, Springer M. 2001. The relationship between translational control and mRNA degradation for the Escherichia coli threonyl-tRNA synthetase gene. J Mol Biol 310:709–722. [PubMed] 10.1006/jmbi.2001.4796 [DOI] [PubMed] [Google Scholar]
- 584.Springer M, Mayaux J-F, Fayat G, Plumbridge JA, Graffe M, Blanquet S, Grunberg-Manago M. 1985. Attenuation control of the Escherichia coli phenylalanyl-tRNA synthetase operon. J Mol Biol 181:467–478. [PubMed] 10.1016/0022-2836(85)90420-6 [DOI] [PubMed] [Google Scholar]
- 585.Yanofsky C. 1981. Attenuation in the control of expression of bacterial operons. Nature 289:751–758. [PubMed] 10.1038/289751a0 [DOI] [PubMed] [Google Scholar]
- 586.Morgan SD, Söll D. 1978. Regulation of the biosynthesis of aminoacid: tRNA ligases and of tRNA. Prog Nucleic Acid Res Mol Biol 21:181–207. 10.1016/S0079-6603(08)60270-6 [DOI] [PubMed] [Google Scholar]
- 587.Neidhardt FC, Parker J, McKeever WG. 1975. Function and regulation of aminoacyl-tRNA synthetases in prokaryotic and eukaryotic cells. Annu Rev Microbiol 29:215–250. [PubMed] 10.1146/annurev.mi.29.100175.001243 [DOI] [PubMed] [Google Scholar]
- 588.Henkin TM. 2005. Regulation of aminoacyl-tRNA synthetase gene expression in bacteria, p 309–313. In Ibba M, Francklyn C, and Cusack S (ed), The Aminoacyl-tRNA Synthetases. Landes Bioscience, Georgetown, TX. [Google Scholar]
- 589.Hassani M, Pincus DH, Bennett GN, Hirshfield IN. 1992. Temperature-dependent induction of an acid-inducible stimulon of Escherichia coli in broth. Appl Environ Microbiol 58:2704–2707. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 590.Hickey EW, Hirshfield IN. 1990. Low-pH-induced effects on patterns of protein synthesis and on internal pH in Escherichia coli and Salmonella typhimurium. Appl Environ Microbiol 56:1038–1045. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 591.Neidhardt FC, VanBogelen RA. 1981. Positive regulatory gene for temperature-controlled proteins in Escherichia coli. Biochem Biophys Res Commun 100:894–900. [PubMed] 10.1016/S0006-291X(81)80257-4 [DOI] [PubMed] [Google Scholar]
- 592.VanBogelen RA, Acton MA, Neidhardt FC. 1987. Induction of the heat shock regulon does not produce thermotolerance in Escherichia coli. Genes Dev 1:525–531. [PubMed] 10.1101/gad.1.6.525 [DOI] [PubMed] [Google Scholar]
- 593.Hirshfield IN, Tenreiro R, Vanbogelen RA, Neidhardt FC. 1984. Escherichia coli K12 lysyl-tRNA synthetase mutant with a novel reversion pattern. J Bacteriol 158:615–620. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 594.Cho BK, Barrett CL, Knight EM, Park YS, Palsson BO. 2008. Genome-scale reconstruction of the Lrp regulatory network in Escherichia coli. Proc Natl Acad Sci USA 105:19462–19467. [PubMed] 10.1073/pnas.0807227105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 595.Brinkman AB, Ettema TJ, de Vos WM, van der Oost J. 2003. The Lrp family of transcriptional regulators. Mol Microbiol 48:287–294. [PubMed] 10.1046/j.1365-2958.2003.03442.x [DOI] [PubMed] [Google Scholar]
- 596.Lin R, Ernsting B, Hirshfield IN, Matthews RG, Neidhardt FC, Clark RL, Newman EB. 1992. The lrp gene product regulates expression of lysU in Escherichia coli K12. J Bacteriol 174:2779–2784. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 597.Mayaux J-F, Fayat G, Panvert M, Springer M, Grunberg-Manago M, Blanquet S. 1985. Control of phenylalanyl-tRNA synthetase genetic expression. J Mol Biol 184:31–44. [PubMed] 10.1016/0022-2836(85)90041-5 [DOI] [PubMed] [Google Scholar]
- 598.Moine H, Romby P, Springer M, Grunberg-Manago M, Ebel J-P, Ehresmann B, Ehresmann C. 1990. Escherichia coli threonyl-tRNA synthetase and tRNAThr modulate the binding of the ribosome to the translational initiation site of the thrS mRNA. J Mol Biol 216:299–310. [PubMed] 10.1016/S0022-2836(05)80321-3 [DOI] [PubMed] [Google Scholar]
- 599.Hasegawa T, Miyano M, Himeno H, Sano Y, Kimura K, Shimizu M. 1992. Identity determinants of E. coli threonine tRNA. Biochem Biophys Res Commun 184:478–484. [PubMed] 10.1016/0006-291X(92)91219-G [DOI] [PubMed] [Google Scholar]
- 600.Romby P, Brunel C, Caillet J, Springer M, Grunberg-Manago M, Westhof E, Ehresmann C, Ehresmann B. 1992. Molecular mimicry in translational control of E. coli threonyl-tRNA synthetase gene. Competitive inhibition in tRNA aminoacylation and operator-repressor recognition switch using tRNA identity rules. Nucleic Acids Res 20:5633–5640. [PubMed] 10.1093/nar/20.21.5633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 601.Graffe M, Dondon J, Caillet J, Romby P, Ehresmann C, Ehresmann B, Springer M. 1992. The specificity of translational control switched with transfer RNA identity rules. Science 255:994–996. [PubMed] 10.1126/science.1372129 [DOI] [PubMed] [Google Scholar]
- 602.Caillet J, Nogueira T, Masquida B, Winter F, Graffe M, Dock-Bregeon A-C, Torres-Larios A, Sankaranarayanan R, Westhof E, Ehresmann B, Ehresmann C, Romby P, Springer M. 2003. The modular structure of Escherichia coli threonyl-tRNA synthetase as both an enzyme and a regulator of gene expression. Mol Microbiol 47:961–974. [PubMed] 10.1046/j.1365-2958.2003.03364.x [DOI] [PubMed] [Google Scholar]
- 603.Ryckelynck M, Giegé R, Frugier M. 2005. tRNAs and tRNA mimics as cornerstones of aminoacyl-tRNA synthetase regulations. Biochimie 87:835–845. [PubMed] 10.1016/j.biochi.2005.02.014 [DOI] [PubMed] [Google Scholar]
- 604.Dale BA, Nester EW. 1971. Regulation of tyrosyl-transfer ribonucleic acid synthetase in Bacillus subtilis. J Bacteriol 108:586–588. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 605.Putzer H, Gendron N, Grunberg-Manago M. 1992. Co-ordinate expression of the two threonyl-tRNA synthetase genes in Bacillus subtilis: Control by transcriptional antitermination invoving a conserved regulatory sequence. EMBO J 11:3117–3127. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 606.Williams-Wagner RN, Grundy FJ, Raina M, Ibba M, Henkin TM. 2015. The Bacillus subtilis tyrZ gene encodes a highly selective tyrosyl-tRNA synthetase and is regulated by a MarR regulator and T box riboswitch. J Bacteriol 197:1624–1631. [PubMed] 10.1128/JB.00008-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 607.Grundy FJ, Henkin TM. 1993. tRNA as a positive regulator of transcription antitermination in B. subtilis. Cell 74:475–482. [PubMed] 10.1016/0092-8674(93)80049-K [DOI] [PubMed] [Google Scholar]
- 608.Green NJ, Grundy FJ, Henkin TM. 2010. The T box mechanism: tRNA as a regulatory molecule. FEBS Lett 584:318–324. [PubMed] 10.1016/j.febslet.2009.11.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 609.Grundy FJ, Henkin TM. 1994. Conservation of a transcription antitermination mechanism in aminoacyl-tRNA synthetase and amino acid biosynthesis genes in gram-positive bacteria. J Mol Biol 235:798–804. [PubMed] 10.1006/jmbi.1994.1038 [DOI] [PubMed] [Google Scholar]
- 610.Gutierrez-Preciado A, Henkin TM, Grundy FJ, Yanofsky C, Merino E. 2009. Biochemical features and functional implications of the RNA-based T-box regulatory mechanism. Microbiol Mol Biol Rev 73:36–61. [PubMed] 10.1128/MMBR.00026-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 611.Vitreschak AG, Mironov AA, Lyubetsky VA, Gelfand MS. 2008. Comparative genomic analysis of T-box regulatory systems in bacteria. RNA 14:717–735. [PubMed] 10.1261/rna.819308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 612.Sherwood AV, Grundy FJ, Henkin TM. 2015. T box riboswitches in Actinobacteria: Translational regulation via novel tRNA interactions. Proc Natl Acad Sci USA 112:1113–1118. [PubMed] 10.1073/pnas.1424175112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 613.Liu LC, Grundy FJ, Henkin TM. 2015. Non-conserved residues in Clostridium acetobutylicum tRNAAla contribute to tRNA tuning for efficient antitermination of the alaS T box riboswitch. Life (Basel) 5:1567–1582. 10.3390/life5041567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 614.Bremer H, Dennis PP. 1987. Modulation of chemical composition and other parameters of the cell at different exponential growth rates, p 1527–1542. In Neidhardt FC, Ingraham JL, Low KB, Magasanik B, Schaechter M, and Umbarger HE (ed), Escherichia coli and Salmonella: Cellular and Molecular Biology. American Society for Microbiology, Washington, DC. [Google Scholar]
- 615.VanBogelen RA, Sankar P, Clark RL, Bogan JA, Neidhardt FC. 1992. The gene-protein database of Escherichia coli: edition 5. Electrophoresis 13:1014–1054. [PubMed] 10.1002/elps.11501301203 [DOI] [PubMed] [Google Scholar]
- 616.Barras F, Marinus MG. 1989. The great GATC: DNA methylation in E. coli. Trends Genet 5:139–143. [PubMed] 10.1016/0168-9525(89)90054-1 [DOI] [PubMed] [Google Scholar]
- 617.Yanofsky C. 1987. Operon-specific control by transcription attenuation. Trends Genet 3:356–360. 10.1016/0168-9525(87)90297-6 [DOI] [Google Scholar]
- 618.Blumenthal RM, Lemaux PG, Neidhardt FC, Dennis PP. 1976. The effects of the relA gene on the synthesis of aminoacyl-tRNA synthetases and other transcription and translation proteins in Escherichia coli A. Mol Gen Genet 149:291–296. [PubMed] 10.1007/BF00268530 [DOI] [PubMed] [Google Scholar]
- 619.Sheppard K, Yuan J, Hohn MJ, Jester B, Devine KM, Söll D. 2008. From one amino acid to another: tRNA-dependent amino acid biosynthesis. Nucleic Acids Res 36:1813–1825. [PubMed] 10.1093/nar/gkn015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 620.Huot JL, Balg C, Jahn D, Moser J, Emond A, Blais SP, Chênevert R, Lapointe J. 2007. Mechanism of a GatCAB amidotransferase: Aspartyl-tRNA synthetase increases its affinity for Asp-tRNAAsn and novel aminoacyl-tRNA analogues are competitive inhibitors. Biochemistry 46:13190–13198. [PubMed] 10.1021/bi700602n [DOI] [PubMed] [Google Scholar]
- 621.Becker HD, Reinbolt J, Kreutzer R, Giegé R, Kern D. 1997. Existence of two distinct aspartyl-tRNA synthetases in Thermus thermophilus. Structural and biochemical properties of the two enzymes. Biochemistry 36:8785–8797. [PubMed] 10.1021/bi970392v [DOI] [PubMed] [Google Scholar]
- 622.Curnow AW, Tumbula D, Pelaschier J, Min B, Söll D. 1998. Glutamyl-tRNAGln amidotransferase in Deinococcus radiodurans may be confined to asparagine biosynthesis. Proc Natl Acad Sci USA 95:12838–12843. [PubMed] 10.1073/pnas.95.22.12838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 623.Akochy PM, Bernard D, Roy PH, Lapointe J. 2004. Direct glutaminyl-tRNA biosynthesis and indirect asparaginyl-tRNA biosynthesis in Pseudomonas aeruginosa PAO1. J Bacteriol 186:767–776. [PubMed] 10.1128/JB.186.3.767-776.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 624.Becker HD, Kern D. 1998. Thermus thermophilus - a link in evolution of the tRNA-dependent amino acid amidation pathways. Proc Natl Acad Sci USA 95:12832–12837. 10.1073/pnas.95.22.12832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 625.Feng L, Yuan J, Toogood H, Tumbula-Hansen D, Söll D. 2005. Aspartyl-tRNA synthetase requires a conserved proline in the anticodon-binding loop for tRNAAsn recognition in vivo. J Biol Chem 280:20638–20641. [PubMed] 10.1074/jbc.M500874200 [DOI] [PubMed] [Google Scholar]
- 626.Schulze JO, Masoumi A, Nickel D, Jahn M, Jahn D, Schubert WD, Heinz DW. 2006. Crystal structure of a non-discriminating glutamyl-tRNA synthetase. J Mol Biol 361:888–897. [PubMed] 10.1016/j.jmb.2006.06.054 [DOI] [PubMed] [Google Scholar]
- 627.Oshikane H, Sheppard K, Fukai S. Nakamura Y, Ishitani R, Numata T, Sherrer RL, Feng L, Schmitt E, Panvert M, Blanquet S, Mechulam Y, Söll D, Nureki O. 2006. Structural basis of RNA-dependent recruitment of glutamine to the genetic code. Science 312:1950–1954. [PubMed] 10.1126/science.1128470 [DOI] [PubMed] [Google Scholar]
- 628.Bailly M, Blaise M, Lorber B, Becker HD, Kern D. 2007. The transamidosome: A dynamic ribonucleoprotein particle dedicated to prokaryotic tRNA-dependent asparagine biosynthesis. Mol Cell 28:228–239. [PubMed] 10.1016/j.molcel.2007.08.017 [DOI] [PubMed] [Google Scholar]
- 629.Suzuki T, Nakamura A, Kato K, Söll D, Tanaka I, Sheppard K, Yao M. 2015. Structure of the Pseudomonas aeruginosa transamidosome reveals unique aspects of bacterial tRNA-dependent asparagine biosynthesis. Proc Natl Acad Sci USA 112:382–387. [PubMed] 10.1073/pnas.1423314112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 630.Itoh Y, Sekine S, Matsumoto E, Akasaka R, Takemoto C, Shirouzu M, Yokoyama S. 2009. Structure of selenophosphate synthetase essential for selenium incorporation into proteins and RNAs. J Mol Biol 385:1456–1469. [PubMed] 10.1016/j.jmb.2008.08.042 [DOI] [PubMed] [Google Scholar]
- 631.Huot JH, Fischer F, Corbeil J, Madore E, Lorber B, Diss G,Hendrickson TL, Kern D, Lapointe J. 2011. Gln-tRNAGln synthesis in a dynamic transamidosome from Helicobacter pylori, where GluRS2 hydrolyzes excess Glu-tRNAGln. Nucleic Acids Res 39:9306–9315. [PubMed] 10.1093/nar/gkr619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 632.Silva GN, Fatma S, Floyd AM, Fischer F, Chuawong P, Cruz AN, Simari RM, Joshi N, Kern D, Hendrickson TL. 2013. A tRNA-independent mechanism for transamidosome assembly promotes aminoacyl-tRNA transamidation. J Biol Chem 288:3816–3822. [PubMed] 10.1074/jbc.M112.441394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 633.Fischer F, Huot JL, Lorber B, Diss G, Hendrickson TL, Becker HD, Lapointe J, Kern D. 2012. The asparagine-transamidosome from Helicobacter pylori: A dual-kinetic mode in non-discriminating aspartyl-tRNA synthetase safeguards the genetic code. Nucleic Acids Res 40:4965–4976. [PubMed] 10.1093/nar/gks167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 634.Böck A, Thanbichler M, Rother M, Resch A. 2005. Selenocysteine, p 320–327. In Ibba M, Francklyn C, and Cusack S (ed), The Aminoacyl-tRNA Synthetases. Landes Biosciences, Georgetown, TX. [Google Scholar]
- 635.Commans S, Böck A. 1999. Selenocysteine inserting tRNAs: An overview. FEMS Microbiol Rev 23:335–351. [PubMed] 10.1111/j.1574-6976.1999.tb00403.x [DOI] [PubMed] [Google Scholar]
- 636.Amberg R, Mizutani T, Wu X-Q, Gross HJ. 1996. Selenocysteine synthesis in mammalia: An identity switch from tRNASer to tRNASec. J Mol Biol 263:8–19. [PubMed] 10.1006/jmbi.1996.0552 [DOI] [PubMed] [Google Scholar]
- 637.Baron C, Westhof E, Böck A, Giegé R. 1993. Solution structure of selenocysteine inserting tRNASec from Escherichia coli. Comparison with canonical tRNASer. J Mol Biol 231:274–292. [PubMed] 10.1006/jmbi.1993.1282 [DOI] [PubMed] [Google Scholar]
- 638.Itoh Y, Chiba S, Sekine SI, Yokoyama S. 2009. Crystal structure of human selenocysteine tRNA. Nucleic Acids Res 37:6259–6268. [PubMed] 10.1093/nar/gkp648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 639.Rudinger J, Hillenbrandt R, Sprinzl M, Giegé R. 1996. Antideterminants present in minihelixSec hinder its recognition by prokaryotic elongation factor Tu. EMBO J 15:650–657. [PubMed] [PMC free article] [PubMed] [Google Scholar]
- 640.Moore SD, Sauer RT. 2007. The tmRNA system for translational surveillance and ribosome rescue. Annu Rev Biochem 76:101–124. [PubMed] 10.1146/annurev.biochem.75.103004.142733 [DOI] [PubMed] [Google Scholar]
- 641.Felden B, Hanawa K, Atkins JF, Himeno H, Muto A, Gesteland RF, McCloskey JA, Crain PF. 1998. Presence and location of modified nucleotides in Escherichia coli tmRNA: Structural mimicry with tRNA acceptor branches. EMBO J 17:3188–3196. [PubMed] 10.1093/emboj/17.11.3188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 642.Komine Y, Kitabatake M, Yokogawa T, Nishikava K, Inokuchi H. 1994. A tRNA-like structure is present in 10Sa RNA, a small stable RNA from Escherichia coli. Proc Natl Acad Sci USA 91:9223–9227. [PubMed] 10.1073/pnas.91.20.9223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 643.Rudinger-Thirion J, Giegé R, Felden B. 1999. Aminoacylated tmRNA from E. coli interacts with prokaryotic elongation factor Tu. RNA 5:989–992. [PubMed] 10.1017/S135583829999101X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 644.Dong G, Nowakowsky J, Hoffman DW. 2002. Structure of small protein B: The protein component of the tmRNA-SmpB system for ribosome rescue. EMBO J 12:1845–1854. [PubMed] 10.1093/emboj/21.7.1845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 645.Bessho Y, Shibata R, Sekine SI, Murayama K, Higashijima K, Hori-Takemoto C, Shirouzu M, Kuramitsu S, Yokoyama S. 2007. Structural basis for functional mimicry of long-variable-arm tRNA by transfer-messenger RNA. Proc Natl Acad Sci USA 104:8293–8298. [PubMed] 10.1073/pnas.0700402104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 646.Gutmann S, Haebel PW, Metzinger L, Sutter M, Felden B, Ban N. 2003. Crystal structure of the transfer-RNA domain of transfer-messenger RNA in complex with SmpB. Nature 424:699–703. [PubMed] 10.1038/nature01831 [DOI] [PubMed] [Google Scholar]
- 647.Bonnefond L, Fender A, Rudinger-Thirion J, Giegé R, Florentz C, Sissler M. 2005. Towards the full set of human mitochondrial aminoacyl-tRNA synthetases: Characterization of AspRS and TyrRS. Biochemistry 44:4805–4816. [PubMed] 10.1021/bi047527z [DOI] [PubMed] [Google Scholar]
- 648.Brindefalk B, Viklund J, Larsson D, Thollesson M, Andersson SG. 2007. Origin and evolution of the mitochondrial aminoacyl-tRNA synthetases. Mol Biol Evol 24:743–756. [PubMed] 10.1093/molbev/msl202 [DOI] [PubMed] [Google Scholar]
- 649.Diodato D, Ghezzi D, Tiranti V. 2014. The mitochondrial aminoacyl tRNA synthetases: Genes and syndromes. Int J Cell Biol 2014:787956. [PubMed] 10.1155/2014/787956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 650.Brandao MM, Silva-Filho MC. 2011. Evolutionary history of Arabidopsis thaliana aminoacyl-tRNA synthetase dual-targeted proteins. Mol Biol Evol 28:79–85. [PubMed] 10.1093/molbev/msq176 [DOI] [PubMed] [Google Scholar]
- 651.Duchêne A-M, Giritch A, Hoffmann B, Cognat V, Lancelin D, Peeters NM, Zaepfel M, Maréchal-Drouard L, Small ID. 2005. Dual targeting is the rule for organellar aminoacyl-tRNA synthetases in Arabidopsis thaliana. Proc Natl Acad Sci USA 102:16484–16489. [PubMed] 10.1073/pnas.0504682102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 652.Bullard JM, Cai YC, Spremulli LL. 2000. Expression and characterization of the human mitochondrial leucyl-tRNA synthetase. Biochim Biophys Acta 1490:245–258. [PubMed] 10.1016/S0167-4781(99)00240-7 [DOI] [PubMed] [Google Scholar]
- 653.Holman KM, Wu J, Ling J, Simonović M. 2016.The crystal structure of yeast mitochondrial ThrRS in complex with the canonical threonine tRNA. Nucleic Acids Res 44:1428–1439. [PubMed] 10.1093/nar/gkv1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 654.Messmer M, Blais S, Balg C, Chênevert R, Grenier L, Lagüe P, Sauter C, Sissler M, Giegé R, Lapointe J, Florentz C. 2009. Peculiar inhibition of human mitochondrial aspartyl-tRNA synthetase by adenylate analogs. Biochimie 91:596–603. [PubMed] 10.1016/j.biochi.2009.02.005 [DOI] [PubMed] [Google Scholar]
- 655.Neuenfeldt A, Lorber B, Ennifar E, Gaudry A, Sauter C, Sissler M, Florentz C. 2013. Thermodynamic properties distinguish human mitochondrial aspartyl-tRNA synthetase from bacterial homolog with same 3D architecture. Nucleic Acids Res 41:2698–2708. [PubMed] 10.1093/nar/gks1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 656.Fender A, Sauter C, Messmer M, Pütz J, Giegé R, Florentz C, Sissler M. 2006. Loss of a primordial identity element for a mammalian mitochondrial aminoacylation system. J Biol Chem 281:15980–15986. [PubMed] 10.1074/jbc.M511633200 [DOI] [PubMed] [Google Scholar]
- 657.Bonnefond L, Frugier M, Touzé E, Lorber B, Florentz C, Giegé R, Sauter C, Rudinger-Thirion J. 2007. Crystal structure of human mitochondrial tyrosyl-tRNA synthetase reveals common and idiosyncratic features. Structure 15:1505–1516. [PubMed] 10.1016/j.str.2007.09.018 [DOI] [PubMed] [Google Scholar]
- 658.Fender A, Gaudry A, Jühling F, Sissler M, Florentz C. 2012. Adaptation of aminoacylation identity rules to mammalian mitochondria. Biochimie 94:1090–1097. [PubMed] 10.1016/j.biochi.2012.02.030 [DOI] [PubMed] [Google Scholar]
- 659.Karkhanis VA, Boniecki MT, Poruri K, Martinis SA. 2006. A viable amino acid editing activity in the leucyl-tRNA synthetase CP1-splicing domain is not required in the yeast mitochondria. J Biol Chem 281:33217–33225. [PubMed] 10.1074/jbc.M607406200 [DOI] [PubMed] [Google Scholar]
- 660.Boniecki MT, Rho SB, Tukalo M, Hsu JL, Romero EP, Martinis SA. 2009. Leucyl-tRNA synthetase-dependent and -independent activation of a group I intron. J Biol Chem 284:26243–26250. [PubMed] 10.1074/jbc.M109.031179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 661.Hsu JL, Rho SB, Vannella KM, Martinis SA. 2006. Functional divergence of a unique C-terminal domain of leucyl-tRNA synthetase to accommodate its splicing and aminoacylation roles. J Biol Chem 281:23075–23082. [PubMed] 10.1074/jbc.M601606200 [DOI] [PubMed] [Google Scholar]
- 662.Paukstelis PJ, Chen JH, Chase E, Lambowitz AM, Golden BL. 2008. Structure of a tyrosyl-tRNA synthetase splicing factor bound to a group I intron RNA. Nature 451:94–97. [PubMed] 10.1038/nature06413 [DOI] [PubMed] [Google Scholar]
- 663.Francklyn CS, Minajigi A. 2010. tRNA as an active chemical scaffold for diverse chemical transformations. FEBS Lett 584:366–375. [PubMed] 10.1016/j.febslet.2009.11.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 664.Matsuhashi M, Dietrich CP, Strominger JL. 1965. Incorporation of glycine into the cell wall glycopeptide in Staphylococcus aureus: Role of sRNA and lipid intermediates. Proc Natl Acad Sci USA 54:587–594. [PubMed] 10.1073/pnas.54.2.587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 665.Tremblay LW, Fan F, Vetting MW, Blanchard JS. 2008. The 1.6Å crystal structure of Mycobacterium smegmatis MshC: The penultimate enzyme in the mycothiol biosynthetic pathway. Biochemistry 47:13326–13335. [PubMed] 10.1021/bi801708f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 666.Klein S, Lorenzo C, Hoffmann S, Walther JM, Storbeck S, Piekarski T, Tindall BJ, Wray V, Nimtz M, Moser J. 2009. Adaptation of Pseudomonas aeruginosa to various conditions includes tRNA-dependent formation of alanyl-phosphatidylglycerol. Mol Microbiol 71:551–565. [PubMed] 10.1111/j.1365-2958.2008.06562.x [DOI] [PubMed] [Google Scholar]
- 667.Roy H, Dare K, Ibba M. 2009. Adaptation of the bacterial membrane to changing environments using aminoacylated phospholipids. Mol Microbiol 71:547–550. [PubMed] 10.1111/j.1365-2958.2008.06563.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 668.Roy H, Ibba M. 2008. RNA-dependent lipid remodeling by bacterial multiple peptide resistance factors. Proc Natl Acad Sci USA 105:4667–4672. [PubMed] 10.1073/pnas.0800006105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 669.Shepherd J, Ibba M. 2013. Lipid II-independent trans editing of mischarged tRNAs by the penicillin resistance factor MurM. J Biol Chem 288:25915–22523. [PubMed] 10.1074/jbc.M113.479824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 670.Bonnefond L, Arai T, Sakaguchi Y, Suzuki T, Ishitani R, Nureki O. 2011. Structural basis for nonribosomal peptide synthesis by an aminoacyl-tRNA synthetase paralog. Proc Natl Acad Sci USA 108:3912–3917. [PubMed] 10.1073/pnas.1019480108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 671.Aravind L, de Souza RF, Iyer LM. 2010. Predicted class-I aminoacyl tRNA synthetase-like proteins in non-ribosomal peptide synthesis. Biol Direct 5:48. [PubMed] 10.1186/1745-6150-5-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 672.Vetting MW, Hegde SS, Blanchard JS. 2010. The structure and mechanism of the Mycobacterium tuberculosis cyclodityrosine synthetase. Nat Chem Biol 6:797–799. [PubMed] 10.1038/nchembio.440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 673.Gondry M, Sauguet L, Belin P, Thai R, Amouroux R, Tellier C, Tuphile K, Jacquet M, Braud S, Courcon M, Masson C, Dubois S, Lautru S, Lecoq A, Hashimoto S, Genet R, Pernodet J-L. 2009. Cyclodipeptide synthases are a family of tRNA-dependent peptide bond-forming enzymes. Nat Chem Biol 5:414–420. [PubMed] 10.1038/nchembio.175 [DOI] [PubMed] [Google Scholar]
- 674.Lautru S, Gondry M, Genet R, Pernodet, J-L. 2002. The albonoursin gene cluster of S. noursei biosynthesis of diketopiperazine metabolites independent of nonribosomal peptide synthetases. Chem Biol 9:1355–1364. 10.1016/S1074-5521(02)00285-5 [DOI] [PubMed] [Google Scholar]
- 675.Sauguet L, Moutiez M, Li Y, Belin P, Seguin J, Le Du M-H, Thai R, Masson C, Fonvielle M, Pernodet J-L, Charbonnier J-B, Gondry M. 2011. Cyclodipeptide synthases, a family of class-I aminoacyl-tRNA synthetase-like enzymes involved in non-ribosomal peptide synthesis. Nucleic Acids Res 39:4475–4489. [PubMed] 10.1093/nar/gkr027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 676.Moutiez M, Seguin J, Fonvielle M, Belin P, Jacques IB, Favry E, Arthur M, Gondry M. 2014. Specificity determinants for the two tRNA substrates of the cyclodipeptide synthase AlbC from Streptomyces noursei. Nucleic Acids Res 42:7247–7258. [PubMed] 10.1093/nar/gku348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 677.Garg RP, Qian XL, Alemany LB, Moran S, Parry RJ. 2008. Investigations of valanimycin biosynthesis: Elucidation of the role of seryl-tRNA. Proc Natl Acad Sci USA 105:6543–6547. [PubMed] 10.1073/pnas.0708957105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 678.Buddha MR, Crane BR. 2005. Structures of tryptophanyl-tRNA synthetase II from Deinococcus radiodurans bound to ATP and tryptophan. Insight into subunit cooperativity and domain motions linked to catalysis. J Biol Chem 280:31965–31973. [PubMed] 10.1074/jbc.M501568200 [DOI] [PubMed] [Google Scholar]
- 679.Mura C, Katz JE, Clarke SG, Eisenberg D. 2003. Structure and function of an archaeal homolog of survival protein E (SurEalpha): An acid phosphatase with purine nucleotide specificity. J Mol Biol 326:1559–1575. 10.1016/S0022-2836(03)00056-1 [DOI] [PubMed] [Google Scholar]
- 680.Buddha MR, Keery KM, Crane BR. 2004. An unusual tryptophanyl-tRNA synthetase interacts with nitric oxide synthase in Deinococcus radiodurans. Proc Natl Acad Sci USA 101:15881–15886. [PubMed] 10.1073/pnas.0405483101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 681.Kers JA, Wach MJ, Krasnoff SB, Widom J, Cameron KD, Bukhalid RA, Gibson DM, Crane BR, Loria R. 2004. Nitration of a peptide phytotoxin by bacterial nitric oxide synthase. Nature 429:79–82. [PubMed] 10.1038/nature02504 [DOI] [PubMed] [Google Scholar]
- 682.Francklyn C. 2005. tRNA synthetase-like proteins, p 285–297. In Ibba M, Francklyn C, and Cusack S (ed), The Aminoacyl-tRNA Synthetases. Landes Bioscience, Georgetown, TX. [Google Scholar]
- 683.Novoa EM, Castro de Moura M, Orozco M, Ribas de Pouplana L. 2010. A genomics method to identify pathogenicity-related proteins. Application to aminoacyl-tRNA synthetase-like proteins. FEBS Lett 584:460–466. [PubMed] 10.1016/j.febslet.2009.11.016 [DOI] [PubMed] [Google Scholar]
- 684.Schimmel P, Ribas de Pouplana L. 2000. Footprints of aminoacyl-tRNA synthetases are everywhere. Trends Biochem Sci 25:207–209. [PubMed] 10.1016/S0968-0004(00)01553-X [DOI] [PubMed] [Google Scholar]
- 685.Blaise M, Becker HD, Keith G, Cambillau C, Lapointe J, Giegé R, Kern D. 2004. A minimalist glutamyl-tRNA synthetase dedicated to aminoacylation of the tRNAAsp QUC anticodon. Nucleic Acids Res 32:2768–2775. [PubMed] 10.1093/nar/gkh608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 686.Blaise M, Olieric V, Sauter C, Lorber B, Roy B, Karmakar S, Banerjee R, Becker HD, Kern D. 2008. Crystal structure of glutamyl-queuosine tRNAAsp synthetase complexed with L-glutamate: Structural elements mediating tRNA-independent activation of glutamate and glutamylation of tRNAAsp anticodon. J Mol Biol 381:1224–1237. [PubMed] 10.1016/j.jmb.2008.06.053 [DOI] [PubMed] [Google Scholar]
- 687.Salazar JC, Ambrogelly A, Crain PF, McCloskey JA, Söll D. 2004. A truncated aminoacyl-tRNA synthetase modifies RNA. Proc Natl Acad Sci USA 101:7536–7541. [PubMed] 10.1073/pnas.0401982101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 688.Grosjean H (ed). 2009. DNA and RNA Modification Enzymes: Structure, Mechanisms, Function, and Evolution. Landes Bioscience, Austin, TX. [Google Scholar]
- 689.Ray S, Banerjee V, Blaise M, Banerjee B, Das KP, Kern D, Banerjee R. 2014. Critical role of zinc ion on E. coli glutamyl-Queuosine-tRNA synthetase (Glu-Q-RS) structure and function. Protein J. 33:143–149. [PubMed] 10.1007/s10930-014-9546-1 [DOI] [PubMed] [Google Scholar]
- 690.Weber CH, Park YS, Sanker S, Kent C, Ludwig ML (1999) A prototypical cytidylyltransferase: CTP:glycerol-3-phosphate cytidylyltransferase from Bacillus subtilis. Structure 7:1113–1124. [PubMed] 10.1016/S0969-2126(99)80178-6 [DOI] [PubMed] [Google Scholar]
- 691.Ullrich TC, Blaesse M, Huber R. 2001. Crystal structure of ATP sulfurylase from Saccharomyces cerevisiae, a key enzyme in sulfate activation. EMBO J 20:316–329. [PubMed] 10.1093/emboj/20.3.316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 692.Li L, Weinreb V, Francklyn C, Carter CW Jr. 2011. Histidyl-tRNA synthetase Urzymes: Class I and II aminoacyl-tRNA synthetase urzymes have comparable catalytic activities for cognate amino acid activation. J Biol Chem 286:10387–10395. [PubMed] 10.1074/jbc.M110.198929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 693.Rodin SN, Ohno S. 1995. Two types of aminoacyl-tRNA synthetases could be originally encoded by complementary strands of the same nucleic acid. Orig Life Evol Biosph 25:565–589. 10.1007/BF01582025 [DOI] [PubMed] [Google Scholar]
- 694.Chandrasekaran SN, Yardimci GG, Erdogan O, Roach J, Carter Jr CW. 2013. Statistical evaluation of the Rodin-Ohno hypothesis: Sense/antisense coding of ancestral class I and II aminoacyl-tRNA synthetases. Mol Biol Evol 30:1588–1604. [PubMed] 10.1093/molbev/mst070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 695.Nakatsu T, Kato H, Oda J. 1998. Crystal structure of asparagine synthetase reveals a close evolutionary relationship to class II aminoacyl-tRNA synthetase. Nat Struct Biol 5:15–19. [PubMed] 10.1038/nsb0198-15 [DOI] [PubMed] [Google Scholar]
- 696.Roy H, Becker HD, Reinbolt J, Kern D. 2003. When contemporary aminoacyl-tRNA synthetases invent their cognate amino acid metabolism. Proc Natl Acad Sci USA 100:9837–9842. [PubMed] 10.1073/pnas.1632156100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 697.Yanagisawa T, Sumida T, Ishii R, Takemoto C, Yokoyama S. 2010. A paralog of lysyl-tRNA synthetase aminoacylates a conserved lysine residue in translation elongation factor P. Nat Struct Mol Biol 17:1136–1143. [PubMed] 10.1038/nsmb.1889 [DOI] [PubMed] [Google Scholar]
- 698.Park MH. 2006. The post-translational synthesis of a polyamine-derived amino acid, hypusine, in the eukaryotic translation initiation factor 5A (eIF5A). J Biochem 139:161–169. [PubMed] 10.1093/jb/mvj034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 699.Bailly M, de Crécy-Lagard V. 2010. Predicting the pathway involved in post-translational modification of elongation factor P in a subset of bacterial species. Biol Direct 5:3. [PubMed] 10.1186/1745-6150-5-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 700.Sissler M, Delorme C, Bond J, Ehrlich SD, Renault P, Francklyn C. 1999. An aminoacyl-tRNA synthetase paralog with a catalytic role in histidine biosynthesis. Proc Natl Acad Sci USA 96:8985–8990. [PubMed] 10.1073/pnas.96.16.8985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 701.Champagne KS, Sissler M, Larrabee Y, Doublié S, Francklyn CS. 2005. Activation of the hetero-octameric ATP phosphoribosyl transferase through subunit interface rearrangement by a tRNA synthetase paralog. J Biol Chem 280:34096–34104. [PubMed] 10.1074/jbc.M505041200 [DOI] [PubMed] [Google Scholar]
- 702.Vega MC, Zou P, Fernandez FJ, Murphy GE, Sterner R, Popov A, Wilmanns, M. 2005. Regulation of the hetero-octameric ATP phosphoribosyl transferase complex from Thermotoga maritima by a tRNA synthetase-like subunit. Mol Microbiol 55:675–686. [PubMed] 10.1111/j.1365-2958.2004.04422.x [DOI] [PubMed] [Google Scholar]
- 703.Lohkamp B, McDermott G, Campbell SA, Coggins JR, Lapthorn AJ. 2004. The structure of Escherichia coli ATP-phosphoribosyltransferase: Identification of substrate binding sites and mode of AMP inhibition. J Mol Biol 336:131–144. [PubMed] 10.1016/j.jmb.2003.12.020 [DOI] [PubMed] [Google Scholar]
- 704.Cho Y, Sharma V, Sacchettini JC. 2003. Crystal structure of ATP phosphoribosyltransferase from Mycobacterium tuberculosis. J Biol Chem 278:8333–8339. [PubMed] 10.1074/jbc.M212124200 [DOI] [PubMed] [Google Scholar]
- 705.Artymiuk PJ, Rice DW, Poirrette AR, Willet P. 1994. A tale of two synthetases. Nat Struct Biol 1:758–760. [PubMed] 10.1038/nsb1194-758 [DOI] [PubMed] [Google Scholar]
- 706.Wilson KP, Shewchuk LM, Brennan RG, Otsuka AJ, Matthews BW. 1992. Escherichia coli biotin holoenzyme synthetase/bio repressor crystal structure delineates the biotin- and DNA-binding domains. Proc Natl Acad Sci USA 89:9257–9261. [PubMed] 10.1073/pnas.89.19.9257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 707.Mocibob M, Ivic N, Bilokapic S, Maier T, Luic M, Ban N, Weygand-Durasevic I. 2010. Homologs of aminoacyl-tRNA synthetases acylate carrier proteins and provide a link between ribosomal and nonribosomal peptide synthesis. Proc Natl Acad Sci USA 107:14585–14590. [PubMed] 10.1073/pnas.1007470107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 708.Mocibob M, Ivic N, Luic M, Weygand-Durasevic I. 2013. Adaptation of aminoacyl-tRNA synthetase catalytic core to carrier protein aminoacylation. Structure 21:614–626. [PubMed] 10.1016/j.str.2013.02.017 [DOI] [PubMed] [Google Scholar]
- 709.Simos G, Segref A, Fasiolo F, Hellmuth K, Shevchenko A, Mann M, Hurt EC. 1996. The yeast protein Arc1p binds to tRNA and functions as a cofactor for the methionyl- and glutamyl-tRNA synthetases. EMBO J 15:5437–5448. [PubMed] [PMC free article] [PubMed] [Google Scholar]
- 710.Commans S, Plateau P, Blanquet S, Dardel F. 1995. Solution structure of the anticodon-binding domain of Escherichia coli lysyl-tRNA synthetase and studies of its interaction with tRNALys. J Mol Biol 253:100–113. [PubMed] 10.1006/jmbi.1995.0539 [DOI] [PubMed] [Google Scholar]
- 711.Oubridge C, Ito N, Evans PR, Teo C-H, Nagai K. 1994. Crystal structure at 1.92Å resolution of the RNA-binding domaint of the U1A spliceosomal protein complexed with an RNA hairpin. Nature 372:432–438. [PubMed] 10.1038/372432a0 [DOI] [PubMed] [Google Scholar]
- 712.Morales AJ, Swairjo MA, Schimmel P. 1999. Structure-specific tRNA-binding protein from the extreme thermophile Aquifex aeolicus. EMBO J 18:3475–3483. [PubMed] 10.1093/emboj/18.12.3475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 713.Swairjo MA, Morales AJ, Wang C-C, Ortiz AR, Schimmel P. 2000. Crystal structure of Trbp111: A structure-specific tRNA-binding protein. EMBO J 19:6287–6298. [PubMed] 10.1093/emboj/19.23.6287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 714.Nomanbhoy T, Morales AJ, Abraham AT, Vörtler CS, Giegé R, Schimmel P. 2001. Simultaneous binding of two proteins to opposite sides of a single transfer RNA. Nat Struct Biol 8:344–348. [PubMed] 10.1038/86228 [DOI] [PubMed] [Google Scholar]
- 715.Shapova YA, Paetzel M. 2007. Crystallographic analysis of Bacillus subtilis CsaA. Acta Crystallgr D 63:478–485. [PubMed] 10.1107/S0907444907005045 [DOI] [PubMed] [Google Scholar]
- 716.Kawaguchi S, Müller J, Linde D, Kuramitsu S, Shibata T, Inoue Y, Vassylyev DG, Yokoyama S. 2001. The crystal structure of the ttCsaA protein: An export-related chaperone from Thermus thermophilus. EMBO J 20:562–569. [PubMed] 10.1093/emboj/20.3.562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 717.Lloyd AJ, Potter NJ, Fishwick CW, Roper DI, Dowson CG. 2013. Adenosine tetraphosphoadenosine drives a continuous ATP-release assay for aminoacyl-tRNA synthetases and other adenylate-forming enzymes. ACS Chem Biol 8:2157–2163. [PubMed] 10.1021/cb400248f [DOI] [PubMed] [Google Scholar]
- 718.Oka M, Takegawa K, Kimura Y. 2015. Enzymatic characterization of a class II lysyl-tRNA synthetase, LysS, from Myxococcus xanthus. Arch Biochem Biophys 579:33–39. [PubMed] 10.1016/j.abb.2015.05.014 [DOI] [PubMed] [Google Scholar]
- 719.Yan W, Ye Q, Tan M, Chen X, Eriani G, Wang E-D. 2015. Modulation of aminoacylation and editing properties of leucyl-tRNA synthetase by a conserved structural module. J Biol Chem 290:12256–12267. [PubMed] 10.1074/jbc.M115.639492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 720.Sanford B, Cao B, Johnson JM, Zimmerman K, Strom AM, Mueller RM, Bhattacharyya, S, Musier-Forsyth K, Hati S. 2012. Role of coupled dynamics in the catalytic activity of prokaryotic-like prolyl-tRNA synthetases. Biochemistry 51:2146–2156. [PubMed] 10.1021/bi300097g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 721.Phizicky EM, Hopper AK. 2010. tRNA biology charges to the front. Genes Dev 24:1832–1860. [PubMed] 10.1101/gad.1956510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 722.Santos MA, Orellana O, Ibba M. 2010. Transfer RNA’s latest port of call. RNA Biol 7:12–15. 10.4161/rna.7.5.13213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 723.Putzer H, Laalami S, Brakhage AA, Condon C, Grunberg-Manago M. 1995. Aminoacyl-tRNA synthetase gene regulation in Bacillus subtilis: Induction, repression and growth rate regulation. Mol Microbiol 16:709–718. [PubMed] 10.1111/j.1365-2958.1995.tb02432.x [DOI] [PubMed] [Google Scholar]
- 724.Aravind L, Anantharaman V, Koonin EV. 2002. Monophyly of class I aminoacyl tRNA synthetase, USPA, ETFP, photolyase, and PP-ATPase nucleotide-binding domains: Implications for protein evolution in the RNA. Proteins 48:1–14. [PubMed] 10.1002/prot.10064 [DOI] [PubMed] [Google Scholar]
- 725.Perona JJ, Hadd A. 2012. Structural diversity and protein engineering of the aminoacyl-tRNA synthetases. Biochemistry 51:8705–8729. [PubMed] 10.1021/bi301180x [DOI] [PubMed] [Google Scholar]
- 726.Xiao H, Murakami H, Suga H, Ferré-D’Amaré AR. 2008. Structural basis of specific tRNA aminoacylation by a small in vitro selected ribozyme. Nature 454:358–361. [PubMed] 10.1038/nature07033 [DOI] [PubMed] [Google Scholar]
- 727.Yarus M. 2002. Primordial genetics: Phenotype of the ribocyte. Annu Rev Genet 36:125–151. [PubMed] 10.1146/annurev.genet.36.031902.105056 [DOI] [PubMed] [Google Scholar]