

Abstract
Kinetic parameters of human immunodeficiency virus type 1 (HIV-1) and hepatitis C virus (HCV) infections have been estimated from plasma virus levels following perturbation of the chronically infected (quasi-) steady state. We extend previous models by also considering the large pool of virus localized in the lymphoid tissue (LT) compartment. The results indicate that the fastest time scale of HIV-1 plasma load decay during therapy probably reflects the clearance rate of LT virus and not, as previously supposed, the clearance rate of virus in plasma. This resolves the discrepancy between the clearance rate estimates during therapy and those based on plasma apheresis experiments. In the extended models plasma apheresis measurements are indeed expected to reflect the plasma decay rate. We can reconcile all current HIV-1 estimates with this model when, on average, the clearance rate of virus in plasma is approximately 20 day−1, that of LT virus is approximately 3 day−1, and the death rate of virus-producing cells is approximately 0.5 day−1. The fast clearance in the LT compartment increases current estimates for total daily virus production. Because HCV is produced in the liver, we let virus be produced into the blood compartment of our model. The results suggest that extending current HCV models with an LT compartment is not likely to affect current estimates for kinetic parameters and virus production. Estimates for treatment efficacy might be affected, however.
Our understanding of the dynamics of human immunodeficiency virus type 1 (HIV-1) infection relies largely on the analysis of changes in the viral load in plasma after initiation of treatment with potent antiretroviral drugs. Quantitative mathematical models have been fitted to clinical data to provide estimates for the virion clearance rate, the average life span of productively infected cells, the viral generation time, and the total daily production of virus (14, 15, 20, 21). These studies have identified two time scales during the first 1 to 2 weeks of potent antiviral therapy. The slower of the two was supposed to reflect the turnover of virus-producing cells, with an average death rate (δ) of about 0.5 day−1. For the faster decay of virus particles a “clearance” rate (c) of about 3 day−1 was found. The estimation of c from the total virus decline was later suggested to be unreliable (7), but an independent measurement of the decline of plasma infectivity (21) supported the estimate of ≃3 day−1.
A recent plasma apheresis study yielded significantly faster estimates for the HIV-1 virus clearance (22). Large quantities of plasma were removed from infected patients to create an additional clearance term for the virus. During the 1 to 2 h of apheresis, the virus level in plasma dropped significantly and rapidly returned to baseline levels after the apheresis was stopped. A mathematical model was fitted to the data to obtain estimates for the natural clarance rate c. The estimates for four patients ranged from 9 to 36 day−1, with an average of approximately 23 day−1. Although the early estimates of approximately 3 day−1 were established as lower bounds, we show below that the large discrepancy in the estimates for c can be due to the compartmentalization of the virus pool. The estimates for the turnover rate, δ, of virus-producing cells have also been published as lower bounds (21) and have also been questioned (5). In HIV-1-infected patients, no correlation between δ and the magnitude of the cytotoxic response was found (17). This resulted in a debate on the importance of the cytotoxic immune response (4) and in more sophisticated immune control models (13).
It has long been known that the major site of HIV-1 infection is the lymphoid tissue (LT) (3, 19). The trafficking of lymphocytes between the plasma and the LT compartment has been shown to be of potential importance in HIV-1 infection (12, 23). Here we extend this approach to the distribution of virus. A series of studies have confirmed that most of the virus, i.e., on the order of 1010 virions in a typical patient, resides in the LT compartment (1, 6, 18, 24). Most of the virus is bound to the surfaces of follicular dendritic cells (FDCs). Surprisingly, virus in the LT was found to decline at about the same rate as free virus in plasma during therapy (1): during the first two days of therapy, virus in LT declined at a rate of about 0.4 day−1. Later the decay slows down, which may be due to multivalent binding (9). The early parallel declines of virus in LT and plasma suggest that on this time scale of a few days there is a (quasi-) steady state between the two virus compartments that is maintained by an exchange of viral particles. The decay of virus in LT introduces a third time scale, which is intermediate to those for c and δ and which hence influences the established estimates.
Mathematical analysis of plasma apheresis (22) and antiviral treatment data (14) has also been carried out for hepatitis C virus (HCV). To explain the rapid drop of the viral load in plasma in about 1 day, which is followed by a slower second-phase decay, Neumann et al. suggested that alpha interferon (IFN-α) blocks the production of new HCV virus particles (14). The first-phase decay was dominated by a c of approximately 6 days−1, and the slow second-phase decay, with 0.01 day−1 < δ < 0.4 day−1 was conjectured to reflect the death rate of virus-producing cells.
The HCV estimates from the plasma apheresis study are in much better agreement with the treatment estimates: in two patients virion clearance rates were estimated to be 5.5 and 9.9 day−1. Below we suggest that the reason for this difference between HIV-1 and HCV is the different localization of virus production. Whereas HIV-1 is mostly produced in the LT, HCV is produced in the liver, which connects to the blood plasma compartment. By implementing this difference in the models we are able to show that the impact of an LT virus reservoir on turnover parameter estimates is likely to be much smaller for HCV than for HIV-1. Total virus production estimates are also more reliable.
MATERIALS AND METHODS
The models.
The models consider virus-producing infected cells, I, free plasma virus, VP, and virus in the lymphoid tissue, VL. To set the pretreatment steady state, we write a simplified term allowing for limited target cell availability (see Appendix). Thus, in the pretreatment situation new infections occur at a rate (β) given by β = βmaxV/(h + V), where βmax is the maximum value of β and h is a saturation parameter. Because we set βmax = 0 during highly active antiretroviral therapy (HAART), most results do not depend on the form of this equation. Altenatively, we could have allowed for a density-dependent δ to model an immune response.
Virus is produced at rate p, with units of per infected cell per day, and is cleared from the blood at rate c and from the LT at rate cL. Assuming a constant immune response on the time scale of treatment, the death rate of infected cells, δ, is a fixed parameter. Between the two compartments there is an exchange of virus at rates i, for influx into the blood, and e, for efflux from the blood into the LT. Most of the virus in the LT is in fact bound onto FDCs. Because the kinetics of virus association and dissociation seems to be fast (9), we treat LT virus as a single population. Our LT clearance rate cL should therefore be interpreted as an average over the bound and the free virus in the LT. In the Appendix we briefly discuss how this simplification can be derived.
In the paper we consider “typical” patients with most of the virus in the LT, i.e., VL ≫ VP, and “late-stage” patients with equal amounts of virus in the LT and in the plasma. This difference is probably caused by the destruction of the FDC network during disease progression, which reduces the amount of virus that can be bound to FDCs (11). The LT virus population in our model is a mixture of FDC-bound virus and free virus in the LT. Virus bound to FDCs with multiple bonds is probably protected from fast clearance and has a longer residence time in the LT than free virus (9). Destruction of the FDC network during disease progression is therefore expected to increase cL and the rate of transport (i) of our mixed-virus population in the LT to the plasma. In this paper we vary both parameters to (i) model late-stage patients with high i and (ii) study the effect of the unknown cL in the LT. Since the steady-state plasma viral load is proportional to the amount of virus released from the LT into the plasma (see below), increasing i decreased the VL/VP ratio.
In the HIV model virus-producing CD4+ T cells reside in the LT. Conversely, in the HCV model, where the infected cells are mostly hepatocytes, virus is released into the blood plasma compartment. Thus for the HIV-1 model we write
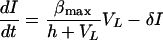 |
1 |
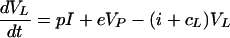 |
2 |
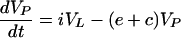 |
3 |
whereas in the HCV model produced virus enters the blood so we write
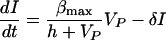 |
4 |
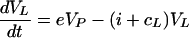 |
5 |
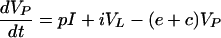 |
6 |
Due to the density-dependent infection term, the nontrivial equilibrium of both models is always stable.
We make a clear distinction between “true” virus clearance, cL and c, and the “transport” terms, i and e: the “total clearance” rates are defined by ĉ ≡ e + c and cL̂ ≡ i + cL. As there is no known biological mechanism that would transport large amounts of virus from the blood to the LT, e should be small. The flux of virus from the LT to the blood probably follows the flow of lymph to the blood. Later we will show that the plasma apheresis experiments suggest that this process is also likely to be slow, hence i should also be small. Because this flux is not passive diffusion, we write the explicit flux parameters e and i and formulate our system in terms of total body counts of productively infected cells, plasma virions, and LT virions. By appropriate scaling one can, however, rewrite the model in terms of concentrations and conventional diffusion-like transport terms.
During a treatment blocking new infections, we set β = 0. As this makes the model linear, one may solve the eigenvalues of the full system (equations 1 to 3) to find that λ1 = −δ and
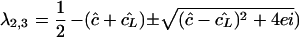 |
7 |
We have argued above, and suggest below, that transport parameters e and i are probably small. If the product ei is sufficiently small, i.e., if 4ei is ≪(ĉ − cL̂ )2, one obtains the simple eigenvalue structure λ1 = −δ, λ2 = cL̂ , and λ3 = ĉ, such that the solution becomes
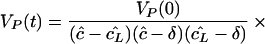 |
8 |
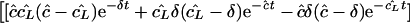 |
The kinetics of the viral load in the plasma, VP, thus has three time scales, set by δ, ĉ, and cL̂
Plasma apheresis.
The clearance rate of plasma virus has been studied with plasma apheresis experiments (22). During plasma apheresis the removal of plasma increases the clearance of free plasma virus. The changes in plasma virus concentration were modeled by the equation
 |
9 |
where P is the total input of virus into the blood and c′ is the sum of the natural clarance rate c and the rate of removal by apheresis, ɛ. The input P represents virus production and was considered to be constant during the 1 to 2 h of apheresis. Estimates for c and P were obtained by a nonlinear fitting procedure of the plasma virus concentrations before, during, and after apheresis. In total less than 108 virus particles were removed during an experiment.
Although 108 particles correspond to a significant fraction of the total plasma virus pool, VP, it is less than 1% of the total virus load, VP + VL. Thus, the mere fact that one finds an observable decline in the plasma virus load by removing less than 1% of the total body virus suggests that the exchange between the LT compartment and the blood cannot be too rapid, i.e., transport rates i and e have to be sufficiently small. In our two-compartment model the input term in equation 9 becomes P = iVL and the clearance term becomes c′ = c + e + ɛ. Because typically VL is ≫VP, it is reasonable to assume that P (=iVL) hardly varies during the apheresis. Thus equation 9 applies equally well to our two-compartment model for HIV-1, with the only change being that the estimated clearance rate of 23 day−1 (22) should reflect the total clearance, ĉ = c + e.
For HCV, the input of free plasma virus is production plus influx from the LT (see equation 6), i.e., P = pI + iVL, which can again be treated as constant during plasma apheresis. The clearance rate in equation 9 remains c′ = c + e + ɛ. Thus, the plasma apheresis estimates for the total clearance, ĉ = c + e, are 5.5 and 9.9 day−1 (22). These estimates are close to those obtained from treatment of HCV-infected patients, which suggested a ĉ of 6 day−1 (14).
Because the transport rate of virus from the plasma to the LT is probably small and because the plasma apheresis experiments suggest a limited exchange on a time scale of hours, we will assume that the efflux rate, e, is small, i.e., we set e = 1 day−1 for both viruses. This seems a conservative choice because the rapid total clearance rates measured during the plasma apheresis experiments would largely reflect the true clearance, c (22). Accordingly, we set c at 24 day−1 for HIV-1 and 7 day−1 for HCV. For robustness all simulations were checked for a c of 5 day−1 and an e of 20 day−1 for HIV-1 and for a c of 2 day−1 and an e of 6 day−1 for HCV.
RESULTS
Antiviral treatment: HIV-1. (i) The 1- to 2-week time scale of total virus decline.
Combination therapy of HIV-1 infection with protease and reverse transcriptase inhibitors blocks new infections and renders newly produced virus noninfectious. As a consequence, the population of virus-producing cells declines exponentially at rate δ (10). After a fast initial transient dominated by the fast clearance of plasma virus, the decline of the plasma virus load was believed to approach the same slope, δ (21). In models allowing for additional virus compartments, such as LT, this need not remain true. Assuming for simplicity a 100% drug efficacy and considering the total virus load (i.e., infectious plus noninfectious virus), we model similar treatment by setting the maximum infection rate βmax = 0.
To study how the different compartments affect the estimate of δ, we first ignore the fast transient by a quasi-steady-state (QSS) assumption for the plasma virus, VP = iVL/ĉ. Solving the QSS model for βmax = 0, one obtains for the LT virus
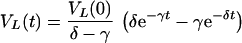 |
10 |
where
 |
11 |
Because of the QSS assumption the plasma virus load remains proportional to equation 10. The two different true clearance rates, cL and c, of virus turnover are combined into a composite decay rate, γ. Note that for the supposedly small value of e, γ ≃ cL̂ After a fast initial transient, the behavior of the system will be dominated by the slowest exponent in equation 10, i.e., by γ or δ. Thus, the observed slope at which virus declines in the blood will not reflect the death rate, δ, of productively infected cells whenever γ is <δ. This we call the “masking” of infected-cell turnover rates. In equations 11 γ can only be smaller than δ under the following condition: cL + ci/(e + c) ≃ cL + i = cL̂ < δ. To remain consistent with the data (10, 21), the slower of the two exponents should be about 0.5 day−1. We therefore set δ = 1 day−1 and we restrict ourselves to values of cL̂ ≥0.5 day−1. When the FDC network is degraded during disease progression, one may expect the flow rate, i, of virus from the LT to the blood to increase because a smaller fraction of the LT virus is bound to the FDCs (11). In terms of equations 11, disease progression is thus expected to increase γ and the VP/VL ratio.
We studied the full model numerically to find parameter values that are consistent with the treatment and the plasma apheresis estimates and to investigate when we find masking (i.e., γ < δ). In all numerical examples we found similar behavior for the QSS model and the full system, confirming the validity of equation 10. Typically, in asymptomatic patients one finds that most of the total body virus is confined to the LT (6, 18). A patient with a QSS I of 108 virus-producing cells and VP of 108 plasma virus particles might harbor as many as 5 × 1010 virus particles in the LT (6). The first 10 day of the treatment of such a patient is depicted in Fig. 1a, where we first set 0.5 day−1 for the decay rate of LT virus. All other parameter values are set such as to obtain the required initial steady state (Fig. 1). Because VL is ≫VP, we find that γ is <δ in this example, and the observed decline of the plasma virus load is dominated by the decay rate, cL̂, of LT virus and not by the death rate, δ, of virus-producing cells. This provides an example of the masking of δ.
FIG. 1.

Viral load during HAART as obtained by equations 1 to 3. (a and c) Typical situation with a large LT virus compartment (VL ≫ VP) for slow and rapid LT clearance rates, respectively. (b) Late-stage patient, with equal amounts of virus in the LT and blood compartment and a slow LT clearance rate. In panel a there is masking of δ, whereas in panels b and c the plasma viral load is coming down at rate δ. Parameters: c = 24 day−1, e = 1 day−1, δ = 1 day−1, and h = 109. We set βmax = 1.02 × 108 to attain the stable pretreatment equilibrium and set βmax = 0 at day 0. Other parameters are as follows: i, 0.05 (a and c) and 25 day−1 (b); cL, 0.5 (a and b) and 5 day−1 (c); p, 274 (a and b) and 2,524 particles cell−1 day−1 (c). Initial steady state, VL = 5 × 1010 particles, Vp = 108 particles, I = 108 cells (a and c) and VL = 1.4 × 108 particles, VP = 1.4 × 108 particles, I = 1.3 × 107 cells (b).
Because patients in a more-advanced stage of the disease can have a much smaller compartment of LT virus (11), our next example is a case with equal amounts of virus in the blood and in the LT. To achieve VL = VP, we increased the influx rate, i, of virions from LT to blood 500-fold. All other parameter values were unchanged. The time course of treatment is depicted in Fig. 1b. The decline of virus compartments approaches that for the productively infected cells (δ) (the graphs of VL and VP coincide with the graph depicting δ). In this case γ is >δ, and indeed there is no masking. Because c is >δ, such a situation is expected when a sufficiently large fraction of the total-body virus is located in the blood (see equations 11).
The turnover rate of LT virus is unknown and need not be a simple exponential decline (9). Equation 10 only shows that the slower of the γ and δ timescales is reflected in the parallel declines of plasma and LT virus. Figure 1c depicts the same system also for a 10-fold-higher clearance of LT virus. To allow for the same initial steady state, we compensated by changing the production rate accordingly (Fig. 1c). Because γ is >δ in Fig. 1c, δ masks cL, and a realistic δ of 1 day−1 is observed. The latter scenario implies a rapid clearance of virus from the LT, i.e., implies that cL̂ is >δ.
Summarizing, for cases representing typical asymptomatic patients where VL is ≫VP, there are two parameter settings that remain consistent with the previous observations (10, 21, 22). First, the turnover of LT virus may be slow, i.e., cL̂ ≃ 0.5 day−1, and would be expected to be responsible for the observed decline of virus in the blood. Second, the turnover of LT virus may be fast, i.e., cL̂ > δ, such that the observed decline of virus in the blood reflects the turnover, δ, of productively infected cells. In the first scenario LT virus turnover masks δ, whereas in the second δ masks LT virus turnover. Finally, because the masking in the slow-turnover scenario depends on the VL/VP ratio, it may depend on disease stage. Such a dependence is not expected for the rapid-turnover scenario.
(ii) The rapid time scale of infectious virus.
During complete protease inhibitor treatment the infectious virus titer was expected to fall with a slope of c (21). Data from a single late-stage patient revealed that this slope was approximately 3 day−1 (21). For the same patient δ was estimated to be 0.53 day−1. Considering the infectious virus titer only, we modeled complete protease inhibitor treatment by setting p = 0. Assuming, as above, that the product of the transport parameters, ei, is sufficiently small, we obtain
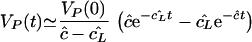 |
12 |
with cL̂ and ĉ time scales. Since, the turnover rate, cL̂ , of the LT virus has the slower time scale, it is expected to dominate the dynamics. In Fig. 2 we consider the full model with p = 0 for same three cases as in Fig. 1. Indeed we see that the decay rate of LT virus dominates, and that it masks clearance rate ĉ in the blood, in Fig. 2a and c, representing typical patients with VL ≫ VP (i.e., small i). The observed slope of 3 day−1 (21) could therefore correspond to the total clearance, cL̂ , of LT virus. To model the late-stage patient of Fig. 2b, with VL = VP, we increased the rate of transport, i, of virus from the LT to the plasma. Increasing i increases cL̂ , such that ĉ may come to represent the slower time scale. Indeed in Fig. 2b we observe a rapid slope of approximately 24 day−1.
FIG. 2.

Blocking viral production in HIV-1 infection. The plasma virus titer follows the decay rate of LT virus if the LT reservoir is large (a and c) and follows the decay rate of plasma virus if it is small (b). The parameters and initial steady states are the same as in the corresponding panels of Fig. 1. Treatment is modeled by setting p = 0.
(iii) Interpretation.
The dynamics of plasma virus is characterized by three time scales, each of which can be estimated from existing data sets. Plasma apheresis (22) provides an estimate of approximately 23 day−1 for the total clearance of plasma virus. For patients with a large LT virus pool, the observed decline of infectious virus at rates of at least 3 day−1 (21) should reflect the slower of clearance rates ĉ, and cL̂ , which is then necessarily the latter. Total plasma virus and LT virus decline at a rate of 0.5 day−1 (10), which in typical patients with VL ≫ VP should be dominated by the slower of cL̂ and δ, which would then be δ. Hence the model is consistent with current data when ĉ is ≃23 per day, cL̂ is ≃3 per day, and δ is ≃0.5 per day.
HCV.
The biphasic decline of plasma virus titers during IFN-α treatment of HCV infection suggests that the inhibition of virus production is incomplete and that the second-phase slope reflects the decline of residual production by the death rate, δ, of virus-producing cells (14). This was modeled by decreasing virus production by factor ɛ. Because we cannot set β = βmax = 0 during IFN-α treatment, we fixed the infection rate, β, at its pretreatment value to keep it constant on the short time scale of interest. For the plasma viral load we obtain
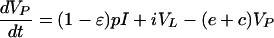 |
13 |
For HCV the typical total blood pool of virus is on the order of 1010 particles and the size of the LT virus compartment is unknown. We investigated two scenarios, one with a large LT virus compartment and one with an LT virus compartment equal to the blood compartment. Obviously, when the LT virus compartment is sufficiently small, it will fail to affect the behavior of the new model. In “Plasma apheresis” we suggested setting ĉ = 8 day−1; hence we set e = 1 day−1 and c = 7 day−1. The fast initial drop of viremia does not allow for a much smaller true clearance rate, c (not shown), which further confirms the assumption of a small efflux, e. We set δ = 0.5 day−1 and VP(0) = 1010 particles, while the remaining free parameters, i.e., p, β, and i, were set so as to obtain the required pretreatment steady state. Figure 3 shows different drug efficacies (i.e., ɛ = 0.99 and 0.80 [14]), depicting the slopes of the three decay rates and the total body count of plasma and LT virus. For comparison with the model of Neumann et al. (14), Fig. 3 also depicts the solution of their one-compartment model.
FIG. 3.

HCV treatment dynamics where treatment blocks 99 (a and c) or 80% (b and d) of virus production. The initial decline reflects ĉ, the total viral clearance in the blood. In panels a and b the LT virus compartment is large and the second-phase decline of plasma virus (heavy solid line) follows the total plasma LT clearance rate, cL̂. In panels c and d the plasma and the LT virus compartments are of equal size, and the rate of the second-phase decline is between cL̂ and δ. Symbols are as in Fig. 1. Heavy dash-dotted line, behavior of the original one-compartment model (14). Parameters: c, 7 day−1, e, 1 day−1; δ, 0.5 day−1; cL, 0.05 day−1; i, 0.05 (a and b) and 0.95 day−1 (c and d). The pretreatment plasma viral load is identical in all panels (VP = 1010 particles), which was achieved by setting β and p. The initial total body counts of LT virus are 1011 particles (a and b) and 1010 particles (c and d).
For Fig. 3a and b where VL(0) = 10VP(0), we have to allow for a small decay rate of LT virus, e.g., cL = 0.05 day−1. This constraint stems from the pretreatment ratio of the two virus pools, i.e., VL/VP = e/cL̂. As in the original model (14), the decline of plasma virus is biphasic (Fig. 3). The two models agree on the rapid first phase, which fits well to a slope of ĉ. Thus, no masking is expected for the decay rate of plasma virus. The models differ with respect to the second-phase decay, however. When the LT virus compartment is large, the kinetics of the two-compartment model is significantly slower than that of the earlier model. In this case a slow decay of LT virus may mask δ. The masked death rate could be even higher than the 0.5 day−1 in our example. Thus, if the two-compartment model reflects reality better, the death rate of virus-producing cells could be much larger than the previous estimates (14). Additionally, the depth of the drop in the virus load during the rapid first phase is significantly smaller in the two-compartment model than it is in the previous model. Since this initial drop of viremia was used to assess the efficacy of the IFN-α treatment, the drug efficacy, ɛ, could also be an underestimate. Note that this effect is strongest at high efficacy (cf. Fig. 3a and b).
Figure 3c and d depict the situation with equally large virus compartments. Although for both phases the slopes are in a fairly good agreement with those of the original model, the second-phase decline of plasma viremia reflects an intermediate rate between δ and cL̂. In the two-compartment model, the initial drop of plasma viremia remains small, however. To obtain equal pool sizes, we increased the influx, i, of virus from the LT compartment into the blood. This situation also allows us to study higher LT virus decay rates. Increasing the decay rate to 0.5 day−1 had little effect on the difference in the initial drop in the viremia (not shown). For even smaller LT compartment sizes, the masking effect of cL disappears altogether.
Summarizing, the two models always agree on the estimates for ĉ during the initial phase. They differ in the estimation of treatment efficacy. Whenever the LT compartment is sufficiently large, they also differ in the estimate for δ. This interpretation is consistent with the observation that the HCV estimates for ĉ from the IFN-α treatment and the plasma apheresis experiments were in good agreement (14, 22).
DISCUSSION
During asymptomatic HIV-1 infection the LT virus compartment is known to be large. We have investigated how this LT compartment may affect current procedures for estimating the virus clearance rate, c, and the average lifetime of productively infected cells, 1/δ. We have found good agreement between all previous studies when the total clearance rate in the blood, ĉ, is ≃23 day−1, the total clearance rate in the LT, cL̂ (=cL + i) is ≃3 day−1, and δ is ≃0.5 day−1. As virus infusion experiments suggest a relatively slow efflux from the blood, the true clearance rate might be close to the above estimate, i.e., c ≃ 20 day−1. The dominance of the LT virus compartment in turn requires a small influx rate, which implies that cL is ≃3 day−1. Our results suggest that the LT total clearance rate, cL̂ , and the total plasma clearance rate, ĉ, can be estimated from the deline of infectious virus (21) and by plasma apheresis (22), respectively. The death rate of virus-producing cells, δ, can indeed (10, 21) be obtained from the decline of total plasma or LT virus.
Importantly, when the LT virus compartment is large, the influx rate, i, has to be small and the decline of infectious virus follows the LT clearance rate, cL (which was found to be 3 per day in the single studied case [21]). This implies high estimates for the total daily production of HIV-1 particles. An infection involving a total body burden of 5 × 1010 particles would require a production of at least 1.5 × 1011 particles per day. Late-stage patients, having a smaller LT virus compartment (11), would require a lower daily production.
We let virus production in HCV infection take place in the blood compartment because the liver releases virus into the blood. As yet, there are no good estimates available for the LT virus compartment in HCV infection. Irrespective of its size, however, allowing for a LT virus compartment hardly affects current estimates for the total viral clearance rate, ĉ. Indeed, the HCV estimates for ĉ obtained from plasma apheresis (22) are in good agreement with those obtained from IFN-α treatment (14). Because HCV production takes place in the blood compartment, we can only have a large LT virus compartment in our model by allowing for a very slow clearance rate, cL, in the LT and a slow influx into blood. If our HIV-1 estimate of cL̂ ≃ 3 day−1 also holds for HCV, we would expect much a smaller pool of LT virus in hepatitis infections. This implies that current estimates of δ (14) are not likely to be confounded by the LT virus compartment and that the high plasma viral loads associated with hepatitis B virus and HCV (14, 16) need not reflect a much higher total body viral burden or production than that estimated for HIV-1. The LT virus compartment may nevertheless remain responsible for an underestimation of the efficacy of IFN-α treatment.
The current model allowing for three time scales is still lacking the possibly important slow time scale of virus attached to the FDC network by multiple bonds (8, 9). Our QSS assumption for virus bound to the FDC network is expected to be valid during the first few day of treatment only. To reliably study the later stages, one would have to merge the current model with that of Hlavacek et al. (8, 9). Their work incorporates multiply bound virus particles but neglects the breakdown of FDC-associated virus and does not account for the observed rapid initial decline of plasma infectivity.
Another simplification of our model is the omission of virus production by infected cells in the blood compartment. In our typical patient, about 2.5 × 109 virions must enter the plasma per day to balance the rate of loss observed in the plasma apheresis experiments. With the most likely parameter setting of Fig. 1c, this could be achieved by production from about 106 productively infected cells, i.e., 1% of the number present in LT. This is close to the ratio of total CD4 T cells in blood to CD4 T cells in LT. Even though infection is likely to induce cells to home to lymph nodes (2), the contribution of blood resident cells cannot be excluded completely. However, the results of the present work are not affected by this possibility. The behavior of the large LT virus pool during the first day of HAART is hardly affected by the plasma pool. Hence our first result remains valid: the 0.5-per-day decay rate of LT virus is the slower of δ and cL̂. Nor is the decline of plasma infectivity affected, as the production of infectious virions is blocked both in the plasma and in LT. This decline of 3 per day therefore also remains the slower of ĉ and ĉL. As the estimates obtained by plasma apheresis reflect ĉ, irrespective of the source term of plasma virus, our interpretation remains valid for all three time scales. Virus production in the blood might only affect the short initial transient of total plasma virus, which is hard to analyze due to the confounding effect of pharmaceutical delay. In all, production of virus in the blood cannot be excluded by our analysis but does not affect our results.
To summarize, the presence of an LT virus compartment in current models of viral infection may confound parameter estimation. Current data (1, 21) suggest that the average total clearance rate cL̂ , of virus in the LT compartment is relatively fast. We can reconcile most data when the following relation exists: ĉ > cL̂ ≃ 3 > δ. Such a fast LT clearance implies that current estimates of ĉ from plasma apheresis and of δ from antiviral treatment are hardly confounded by the LT virus compartment and that virus production during HIV-1 infection is even higher than was estimated previously. Finally, an LT clearance rate of approximately 3 day−1 would reconcile the difference between the estimates for the plasma virus clearance as obtained by plasma apheresis and HAART.
ACKNOWLEDGMENTS
V.M. was supported by the Hungarian Soros Fund and the Hungarian Scientific Research Fund (OTKA).
We thank Alan Perelson and Bill Hlavacek (Los Alamos) for extensive discussions and Béla Novák, Béla Győrffy, and Péter Simon (Budapest) for helpful comments.
Appendix
A standard equation for simple target cell dynamics is dT/dt = ς − δTT − β*TV, where susceptible target cells, T, arise at rate ς, die at rate δT, and are infected at rate β*. For the pretreatment steady state one sets dT/dt = 0 to obtain T(0) = 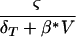 . Substituting this into infection term βTV, one obtains βmaxV/(h + V), where βmax = ς and h = δT/β*.
. Substituting this into infection term βTV, one obtains βmaxV/(h + V), where βmax = ς and h = δT/β*.
Virus in the LT is either free or bound to FDCs. As exchange with the blood involves free virus only and binding to FDCs is reversible, we write for the HIV-1 model VL = V*L + B and
 |
A1 |
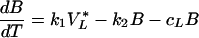 |
A2 |
where V*L is free virus, B is bound LT virus, i* is the influx rate of free LT virus into the blood, and k1 and k2 are association and dissociation rates, respectively. k2 for monovalent binding was estimated as 0.1 per s (9), which is orders of magnitude faster than the other processes in the system. As the association and dissociation of virus mostly involve virions attached to FDCs by one or a few ligands, one expects this fraction of bound virus to remain at QSS with free virus. Setting dB/dt = 0 one obtains B = k1V*L/(k2 + cL). Adding equation A2 to equation A1 one obtains equation 2 with i = i*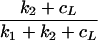 .
.
REFERENCES
- 1.Cavert W, Notermans D W, Staskus K, Wietgrefe S W, Zupancic M, Gebhard K, Henry K, Zhang Z Q, Mills R, McDade H, Goudsmit J, Danner S A, Haase A T. Kinetics of response in lymphoid tissues to antiretroviral therapy of HIV-1 infection. Science. 1997;276:960–964. doi: 10.1126/science.276.5314.960. [DOI] [PubMed] [Google Scholar]
- 2.Cloyd M W, Chen J J, Wang L. How does HIV cause AIDS? The homing theory. Mol Med Today. 2000;6:108–111. doi: 10.1016/s1357-4310(99)01663-9. [DOI] [PubMed] [Google Scholar]
- 3.Embretson J, Zupancic M, Ribas J L, Burke A, Racz P, Tenner-Racz K, Haase A T. Massive covert infection of helper T lymphocytes and macrophages by HIV during the incubation period of AIDS. Nature. 1993;362:359–362. doi: 10.1038/362359a0. [DOI] [PubMed] [Google Scholar]
- 4.Feinberg M B, McLean A R. AIDS: decline and fall of immune surveillance? Curr Biol. 1997;7:R136–R140. doi: 10.1016/s0960-9822(97)70072-1. [DOI] [PubMed] [Google Scholar]
- 5.Ferguson N M, DeWolf F, Ghani A C, Fraser C, Donnelly C A, Reiss P, Lange J M, Danner S A, Garnett G P, Goudsmit J, Anderson R M. Antigen-driven CD4+ T cell and HIV-1 dynamics: residual viral replication under highly active antiretroviral therapy. Proc Natl Acad Sci USA. 1999;96:15167–15172. doi: 10.1073/pnas.96.26.15167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haase A T, Henry K, Zupancic M, Sedgewick G, Faust R A, Melroe H, Cavert W, Gebhard K, Staskus K, Zhang Z Q, Dailey P J, Balfour H H, Jr, Erice A, Perelson A S. Quantitative image analysis of HIV-1 infection in lymphoid tissue. Science. 1996;274:985–989. doi: 10.1126/science.274.5289.985. [DOI] [PubMed] [Google Scholar]
- 7.Herz A V, Bonhoeffer S, Anderson R M, May R M, Nowak M A. Viral dynamics in vivo: limitations on estimates of intracellular delay and virus decay. Proc Natl Acad Sci USA. 1996;93:7247–7251. doi: 10.1073/pnas.93.14.7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hlavacek W S, Stilianakis N I, Notermans D W, Danner S A, Perelson A S. Influence of follicular dendritic cells on decay of HIV during antiretroviral therapy. Proc Natl Acad Sci USA. 2000;97:10966–10971. doi: 10.1073/pnas.190065897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hlavacek W S, Wofsy C, Perelson A S. Dissociation of HIV-1 from follicular dendritic cells during HAART: mathematical analysis. Proc Natl Acad Sci USA. 1999;96:14681–14686. doi: 10.1073/pnas.96.26.14681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ho D D, Neumann A U, Perelson A S, Chen W, Leonard J M, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 11.Hockett R D, Kilby J M, Derdeyn C A, Saag M S, Sillers M, Squires K, Chiz S, Nowak M A, Shaw G M, Bucy R P. Constant mean viral copy number per infected cell in tissues regardless of high, low, or undetectable plasma HIV RNA. J Exp Med. 1999;189:1545–1554. doi: 10.1084/jem.189.10.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kirschner D, Webb G F, Cloyd M. Model of HIV-1 disease progression based on virus-induced lymph node homing and homing-induced apoptosis of CD4+ lymphocytes. J Acquir Immune Defic Syndr. 2000;24:352–362. doi: 10.1097/00126334-200008010-00010. [DOI] [PubMed] [Google Scholar]
- 13.Klenerman P, Phillips R E, Rinaldo C R, Wahl L M, Ogg G, May R M, McMichael A J, Nowak M A. Cytotoxic T lymphocytes and viral turnover in HIV type 1 infection. Proc Natl Acad Sci USA. 1996;93:15323–15328. doi: 10.1073/pnas.93.26.15323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neumann A U, Lam N P, Dahari H, Gretch D R, Wiley T E, Layden T J, Perelson A S. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science. 1998;282:103–107. doi: 10.1126/science.282.5386.103. [DOI] [PubMed] [Google Scholar]
- 15.Notermans D W, Goudsmit J, Danner S A, De Wolf F, Perelson A S, Mittler J. Rate of HIV-1 decline following antiretroviral therapy is related to viral load at baseline and drug regimen. AIDS. 1998;12:1483–1490. doi: 10.1097/00002030-199812000-00010. [DOI] [PubMed] [Google Scholar]
- 16.Nowak M A, Bonhoeffer S, Hill A M, Boehme R, Thomas H C, McDade H. Viral dynamics in hepatitis B virus infection. Proc Natl Acad Sci USA. 1996;93:4398–4402. doi: 10.1073/pnas.93.9.4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ogg G S, Jin X, Bonhoeffer S, Dunbar P R, Nowak M A, Monard S, Segal J P, Cao Y, Rowland-Jones S L, Cerundolo V, Hurley A, Markowitz M, Ho D D, Nixon D F, McMichael A J. Quantitation of HIV-1-specific cytotoxic T lymphocytes and plasma load of viral RNA. Science. 1998;279:2103–2106. doi: 10.1126/science.279.5359.2103. [DOI] [PubMed] [Google Scholar]
- 18.Pantaleo G, Cohen O J, Schacker T, Vaccarezza M, Graziosi C, Rizzardi G P, Kahn J, Fox C H, Schnittman S M, Schwartz D H, Corey L, Fauci A S. Evolutionary pattern of human immunodeficiency virus (HIV) replication and distribution in lymph nodes following primary infection: implications for antiviral therapy. Nat Med. 1998;4:341–345. doi: 10.1038/nm0398-341. [DOI] [PubMed] [Google Scholar]
- 19.Pantaleo G, Graziosi C, Demarest J F, Butini L, Montroni M, Fox C H, Orenstein J M, Kotler D P, Fauci A S. HIV infection is active and progressive in lymphoid tissue during the clinically latent stage of disease. Nature. 1993;362:355–358. doi: 10.1038/362355a0. [DOI] [PubMed] [Google Scholar]
- 20.Perelson A S, Essunger P, Cao Y, Vesanen M, Hurley A, Saksela K, Markowitz M, Ho D D. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature. 1997;387:188–191. doi: 10.1038/387188a0. [DOI] [PubMed] [Google Scholar]
- 21.Perelson A S, Neumann A U, Markowitz M, Leonard J M, Ho D D. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science. 1996;271:1582–1586. doi: 10.1126/science.271.5255.1582. [DOI] [PubMed] [Google Scholar]
- 22.Ramratnam B, Bonhoeffer S, Binley J, Hurley A, Zhang L, Mittler J E, Markowitz M, Moore J P, Perelson A S, Ho D D. Rapid production and clearance of HIV-1 and hepatitis C virus assessed by large volume plasma apheresis. Lancet. 1999;354:1782–1785. doi: 10.1016/S0140-6736(99)02035-8. [DOI] [PubMed] [Google Scholar]
- 23.Stekel D J. The role of inter-cellular adhesion in the recirculation of T lymphocytes. J Theor Biol. 1997;186:491–501. doi: 10.1006/jtbi.1997.0408. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Z Q, Schuler T, Cavert W, Notermans D W, Gebhard K, Henry K, Havlir D V, Gunthard H F, Wong J K, Little S, Feinberg M B, Polis M A, Schrager L K, Schacker T W, Richman D D, Corey L, Danner S A, Haase A T. Reversibility of the pathological changes in the follicular dendritic cell network with treatment of HIV-1 infection. Proc Natl Acad Sci USA. 1999;96:5169–5172. doi: 10.1073/pnas.96.9.5169. [DOI] [PMC free article] [PubMed] [Google Scholar]