

Summary
The effects of genetic variation on complex traits act mainly through changes in gene regulation. Although many genetic variants have been linked to target genes in cis, the trans-regulatory cascade mediating their effects remains largely uncharacterized. Mapping trans-regulators based on natural genetic variation has been challenging due to small effects, but experimental perturbations offer a complementary approach. Using CRISPR, we knocked out 84 genes in primary CD4+ T cells, targeting inborn error of immunity (IEI) disease transcription factors (TFs) and TFs without immune disease association. We developed a novel gene network inference method called linear latent causal Bayes (LLCB) to estimate the network from perturbation data and observed 211 regulatory connections between genes. We characterized programs affected by the TFs, which we associated with immune genome-wide association study (GWAS) genes, finding that JAK-STAT family members are regulated by KMT2A, an epigenetic regulator. These analyses reveal the trans-regulatory cascades linking GWAS genes to signaling pathways.
Keywords: T cells, gene regulatory networks, inbornb errors of immunity, CRISPR, GWAS, RNA-seq, immunology, network inference
Graphical abstract
Highlights
-
•
Linear latent causal Bayes (LLCB) enables gene network inference from CRISPR knockouts
-
•
Inborn error of immunity (IEI) transcription factors function upstream of immune GWASs
-
•
IEI transcription factors and background factors form largely interconnected networks
-
•
Epigenetic modifier KMT2A is a critical regulator of JAK-STAT family expression
Although many connections between genetic variation and immune disease genes have been discovered in cis, the regulatory cascade of these variants remains relatively unknown. To discover these cascades, Weinstock et al. performed arrayed gene knockouts of inborn error of immunity (IEI) disease transcription factors (TFs) and matched background TFs in CD4+ T cells and developed a novel network inference method to reconstruct the gene regulatory network. This network revealed highly interconnected signaling between IEI and background TFs and distinguished functional modules of factors, defining trans-regulatory cascades that control critical immune genes.
Introduction
A primary mission of human genetics is to discover genetic variation that is associated with disease. Genome-wide association studies (GWASs) have identified thousands of variant-disease pairs in recent years, spanning disease, behavioral, and molecular phenotypes. Functional analyses of GWAS loci have revealed that most GWAS single-nucleotide polymorphisms (SNPs) are non-coding, demonstrating that the effects of genetic variation on complex traits largely manifest through regulatory variation.1,2 However, the identification of the molecular consequences of non-coding SNPs has proven challenging. Recent efforts have cataloged expression quantitative trait loci (eQTLs) across diverse tissues and contexts.3,4,5,6 These eQTL studies have been very successful in identifying genetic variation that associates with expression variation of nearby genes in cis. However, except for a small number of examples, the trans-regulatory cascade beyond the associated locus of these cis-acting genetic variants remains largely unknown. Recent analyses of the genetic architecture of complex traits have shown that the bulk (60%–90%) of expression heritability is mediated through a constellation of trans effects, which typically have small effects individually but a large contribution in aggregate.7,8,9 These trans effects are difficult to discover with natural genetic variation because their effect sizes are small and may only exist in contexts that are missed in bulk tissue steady-state models of gene expression.10,11,12,13 Thus, alternative approaches are needed to map the trans-regulatory effects of cis-acting eQTLs.
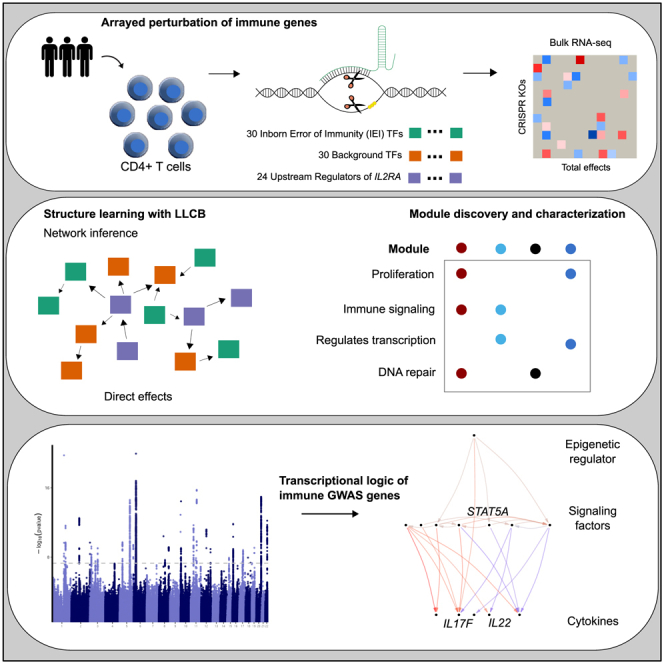
We previously mapped the trans-regulators of key autoimmune disease genes, including IL2RA, IL2, and CTLA4, in primary human CD4+ T cells using CRISPR knockouts (KOs).14,15 In contrast to natural genetic variation, experimental perturbations enable the manipulation of gene expression in ways that are unlikely to be permitted by natural selection.16 We therefore sought to apply this approach to inborn error of immunity (IEI) genes, which are associated with monogenic immune disease spanning regulation and function.17 Although hundreds of these genes have been reported, the transcriptional consequences of their loss of function remain largely uncharacterized. We selected 30 IEI transcription factors (TFs) for CRISPR ablation in human CD4+ T cells to both characterize their function and construct a regulatory network. CD4+ T cells have previously been implicated as a causal cell type in the pathology of many autoimmune traits, including rheumatoid arthritis, multiple sclerosis, and type 1 diabetes, among others.18,19,20 To enable characterization of the properties of the IEI TFs as a whole, we selected 30 background TFs that are matched to the IEI genes in terms of the constraint metric pLI (probability of loss-of-function intolerance21) and expression level in CD4+ T cells but have not been implicated in GWASs of immune phenotypes. We also included 24 upstream regulators of IL2RA, which we had previously perturbed using the same protocol,14 because these genes are likely enriched for master regulators of CD4+ gene regulatory networks (GRNs). In total, we perturbed 84 genes from three gene sets, which we used to construct a high-fidelity gene network relevant to immune disease.
Building on recent advances in the causal inference literature,22,23 we developed a novel statistical method for estimating causal GRNs from perturbation data. In contrast to differential expression or correlation analyses, incorporating causal inference approaches enables the estimation of both direct and indirect regulatory effects, where edges are interpreted as direct effects. We emphasize that in this work, the term “direct effect” is used to convey that the effect of one gene on another is adjusted for confounding pathways among other perturbed genes rather than acting as a claim of physical interaction. Direct effects are useful because they facilitate a coherent interpretation of gene networks as directed probabilistic graphical models. Our approach differs from many other gene networks in two key ways: (1) because our network is derived from experimental perturbations, the edges are much more likely to be causal than the edges in a network estimated from observational co-expression data, where the constituent variation is often of an unknown genesis, and (2) our method enables the estimation of possibly cyclic graphs rather than the common restriction to directed acyclic graphs (DAGs).22,24,25,26 Human genetics has identified several examples of cyclic regulatory behavior,27 so the restriction of GRNs to DAGs represents an artificial constraint that we circumvent with appropriate statistical technology.
We report the causal, cyclic GRN derived from applying our novel statistical method to the 84 CRISPR KOs. Because this method is a Bayesian modification of the linear latent causal (LLC) algorithm, we refer to our method as LLC Bayes (LLCB). Using our network, we systematically characterized the properties of genes that distinguish background TFs from IEI TFs and the IL2RA regulators. We show that although IEI TFs and IL2RA regulators are much more likely to have outgoing connections than background TFs, all the genes form a highly interconnected network rather than distinct communities of disease and background genes. Across the entire network, we found that IEI TFs and IL2RA regulators are more likely to disrupt immune-specific signaling pathways than background TFs. We then identified nine coherent gene programs among the 84 KOs and their downstream genes, which we characterized using enrichment analyses to identify points of functional convergence in T cell biology. In addition to downstream characterization, we used GWAS summary statistic heritability analyses to estimate the contribution of gene-program-linked SNPs to immune trait heritability. This profiling highlighted the importance of a module comprised of key JAK-STAT-IL-2 (interleukin-2) signaling regulators and KMT2A, a global epigenetic regulator that we observed to be upstream of classic IL-2 signaling TFs and receptors, including IRF4, STAT5B, and IL2RA.
In summary, we perturbed a diverse set of genes to characterize the immune regulatory landscape and develop novel statistical methodology to characterize the CD4+ T cell network centered around immune disease genes. Our network reveals the trans-regulatory cascade of these gene programs and elucidates the transcriptional logic of immune GWAS loci.
Results
Perturbation of IEI TFs and matched background TFs
To construct a network enriched for genes relevant to immune disease in CD4+ T cells we perturbed 30 TFs from the IEI genes implicated in Mendelian forms of immune disease.17 We also included 30 background TFs that were not annotated for immune function but were matched on gene constraint and expression to the IEI TFs in order to characterize the properties that distinguish IEI TFs. Lastly, to expand the breadth of our network, we integrated data from 24 previously mapped IL-2RA regulators.14 (STAR Methods; Figure 1). We used CRISPR Cas9 ribonucleoproteins (RNPs) to perform arrayed perturbations in CD4+ T cells from three donors as described in Freimer et al.14 We validated the efficiency of our CRISPR editing by genotyping the 60 additional targeted loci, which indicated a high editing efficiency (Figures S1A and S1B; Table S11). Using bulk RNA sequencing (RNA-seq), we detected ∼13,000 genes that were expressed highly enough for analysis (STAR Methods). As our data were generated in two batches, we performed stringent quality control of the RNA-seq data. We performed alignment and gene count quantification using one pipeline on the 84 samples and performed principal-component analysis (PCA) of the normalized expression data. Pathway enrichment analysis revealed that the first four PCs were associated with very broad biological phenomena, including cell cycle regulation and ribosome activity. Because the PCs also captured batch effects, we included the first four PCs as covariates in downstream analyses. Regressing out PCs has previously been shown to improve the inference of gene networks.28
Figure 1.

Study overview
Schematic describing the three gene sets that were perturbed with CRISPR knockouts and modeling of the gene network, network inference analyses, and gene module identification and integration with immune GWAS data.
Next, we developed a statistical method to estimate the GRN among the 84 genes. We extended the LLC method introduced by Hyttinen et al.23 by recasting the statistical estimand in a Bayesian framework, which enabled the incorporation of prior knowledge about the properties of biological networks. Briefly, LLC proceeds in three steps. First, the total effect of a given perturbation of gene Xi on another gene, Xj, is estimated on all observed (non-perturbed) genes. These total effects are estimated pairwise between all perturbed genes and all observed genes . Second, a system of equations that relates to the direct effects, β, using trek rules is constructed. Third, this system of equations is solved to deconvolve into β. The conditions that permit the identifiability of β for the LLC method include a collection of single gene perturbations among all nodes in the graph, which corresponds to our experimental design, indicating that we have a sufficient number of perturbations to identify β. Because most of the 84 genes are TFs, the elements of β are likely to be greatly enriched for physical binding interactions and other mechanisms of direct transcriptional regulation. However, β may also capture post-transcriptional regulation mechanisms that manifest as statistical direct effects on expression. In this experiment, we are unable to account for the effects of genes that were not perturbed, suggesting that some effects of unmeasured genes may be attributed to direct effects among the 84 perturbed genes.
We extended the LLC framework in two ways (STAR Methods). First, we regressed out the first four expression PCs from the variance-stabilizing transform29 normalized expression data. Second, we estimated β in a Bayesian framework where we incorporated a graph prior, . We included a penalty on the sum of the L1 norms of the columns of β, which penalizes the number of incoming connections to a given gene. We included this penalty because it is known that the distribution of outgoing connections from a gene has more dispersion than the distribution of incoming connections. Following recent advances in differentiable DAG search,22,30,31 we also included a Gaussian prior over the norm of the spectral radius of β, which enables indirect tuning of the degree to which β contains cycles. We performed inference using pathfinder, a recently developed approach to inferring posteriors using pseudo-Hessian optimizers applied to a variational inference objective.32 We chose a variational inference approach rather than Markov chain Monte Carlo (MCMC) because MCMC approaches have been shown to be computationally very intensive when sampling over large discrete graph structures.25,26,33,34 We termed this statistical method LLCB. We validated LLCB theoretically using simulations of cyclic GRNs (STAR Methods).
Network inference from LLCB reveals that the gene groups are highly interconnected
We then used LLCB to estimate the causal CD4+ GRN among the 84 genes (Figures 2A and 2B). We identified 350, 211, and 151 total edges (out of 6,972 possible) when thresholding at 0.020, 0.025, and 0.030, respectively (Figures 2A–2C; Table S1). We reported the network after thresholding on β because filtering on local false sign rates (LFSR)35 resulted in very dense networks (67% network density at LFSR < 5 × 10−3), reflecting the challenges in the estimation of uncertainty in graph structures.
Figure 2.

The gene network of the 84 perturbed genes
(A) Estimate of the directed network that describes how the 84 perturbed genes interact. The radius of each point is proportional to the degree of that gene. Arrows are used to indicate directionality of the edges, such that an arrow pointing into a gene indicates that it is being regulated by another gene. For emphasis, the opacity of the edges from or to inborn error of immunity transcription factors is increased, and all other edges are displayed with greater transparency. Positive values in the color scale indicate that the parent gene is a positive regulator of the child gene.
(B) A sub-network centered around STAT1.
(C) A scatterplot of the indegree and outdegree of each of the 84 genes.
(D) Association analyses between gene properties and their indegree, outdegree, and total degree.
To assess whether edges in this network estimate could be validated through orthogonal approaches, we compared our network estimate to three other estimates of the same network constructed from different sources. First, we constructed a GRN using ATAC-seq data that we previously generated for the 24 IL-2RA regulators, permitting validation of a subset of the network. We gathered all possible enhancers of the 84 genes in CD4+ T cells using the predicted enhancer-gene pairs from the activity-by-contact (ABC)36 model and cross-referenced the enhancer-gene pairs with differentially accessible chromatin (DAC) that we previously identified. We defined the children of a gene i based on those genes that had ABC enhancers that intersected with the DACs from the KO of gene i, and we refer to this network as the ABC-DAC GRN (Table S2). We observed a strong enrichment (∼4×) of edges in the LLCB estimate for the same edges in the ABC-DAC GRN, and this enrichment was robust to different thresholds (Figure S2). Second, we used an external estimate of the T cell regulatory network reported in Green et al.,37 which was estimated using curated pathway information and co-expression data. We similarly observed an enrichment of our edges in this external network (Figure S3). Finally, we performed a similar analysis using the ABC model paired with CD4+ chromatin immunoprecipitation (ChIP)-seq data, where we also observed a strong enrichment of our edges (Figure S4; STAR Methods). Collectively, these three validations, derived from orthogonal data sources and modalities, show that our network estimate is replicable and reflective of biological properties.
We then asked whether the topological properties of genes distinguished the three gene groups. We computed the number of outgoing edges (“outdegree”), the number of incoming edges (“indegree”), and the total number of edges connected to a node (“total degree”) for each node, and we observed that the IEI TFs and IL2RA regulators were strongly enriched for outdegree, and the control TFs were relatively depleted (Figure 2C). We observed no outgoing connections and many incoming connections for the receptor IL2RA, which is expected for a non-TF gene. This result was likely facilitated by our inclusion of the downstream effectors of IL2RA signaling within the graph, such that downstream effects were more likely to be attributed to these genes, such as STAT5A/B and JAK3, rather than IL2RA itself. To identify the properties of genes that associated with their centrality in the graph, we performed negative binomial regressions for three measures of node centrality, including gene group status, gene expression at baseline, and gene constraint as covariates. We defined gene constraint using the quantity Shet, estimated using a recently developed empirical Bayes approach called GeneBayes.38 Shet is defined as the degree of selection acting against heterozygous loss-of-function variants in a given gene and is more predictive of functional and clinical importance than related measures, including pLI and LOEUF (loss-of-function observed/expected upper bound fraction).21 We observed that even after adjusting for Shet and expression, IL2RA regulators and IEI TFs were strongly enriched for outgoing connections relative to control TFs but were not enriched for incoming connections (Figure 2D). Taken together, these data suggest that constraint is much more strongly associated with the number of outgoing connections from a gene than the number of incoming connections and that IEI regulators exhibit more outgoing connections than control genes despite being matched for constraint.
We asked whether edges were enriched between genes that were members of the same gene group. To generate a null distribution, we permuted the edges of the network 2,000 times while preserving the gene degree distributions (STAR Methods). Of the edges in the unpermuted network, 37% had the same parent and child node gene group. Of the permuted networks, 8% had more edges within groups than in the original (unpermuted) network, indicating that the three gene groups do not cluster distinctly in the unpermuted network (Figure S5).
We then estimated indirect effects between pairs of genes, defined as the difference between the total effects and the direct effects . The indirect effects can be interpreted as the sum of all effects of gene i on gene j that are not mediated through the direct effect and thus may include proximal indirect effects comprised of short (<3 genes involved) paths between the two genes or potentially distal effects from long, possibly cyclic paths. These indirect effects may include instances of both transcriptional regulation and post-transcriptional indirect effects. We observed that the bulk of variation in total effects (R2 = 99%) is explained by direct effects (Figure S6), suggesting that direct effects between two genes are much larger than indirect effects. This observation is consistent with the intuition that indirect effects, which are defined as the product of several direct coefficients, are likely to be small unless all of the direct effects along the path are very large. Indeed, if all direct effects are less than 1.0 in magnitude, then the product is guaranteed to be no larger than the smallest direct effect included in the path. We observed that the largest indirect effects were mediated by length-2 cycles with two large direct effects (Figure S7). For example, we observed that KLF2 and MYB regulate each other in a length-2 negative feedback loop, which may help prevent aberrant proliferation.
trans-eQTL-derived networks have limited overlap with the perturbation-derived network
To compare our network estimate to one constructed from natural genetic variation, we first obtained the unfiltered trans-eQTL summary statistics from Yazar et al.,6 which contains the largest catalog of CD4+ eQTLs mapped to date. We observed that only 24 of the 84 perturbed genes had at least one cis-eQTL (q value < 0.01). Among these genes, 10 were background TFs, 8 were IEI TFs, and 6 were IL2RA regulators. We did not observe a significant association between the gene group and whether the gene had a cis-eQTL. The 24 genes with cis-eQTLs were much less constrained than the 60 without (difference in mean Shet = −0.07, 95% confidence interval [CI]: (−0.15, 0.01)), corroborating our prior observations that eQTL discovery is biased toward genes tolerant to loss-of-function variation.16 Notably, the average Shet for the perturbed genes is greater than the average for all genes (difference in mean Shet = 0.12, 95% CI: (0.10, 0.15)), which may limit overlap with eQTL-derived networks. None of the 24 cis-eQTL genes had a trans-eQTL, even at liberal significance thresholds (q value < 0.30), indicating that this eQTL catalog was incapable of recapitulating any of the edges in our GRN. To evaluate whether the absence of trans-eQTLs among the 24 genes was the result of trans-eQTL network sparsity, we tabulated the number of trans-eGenes in CD4 naive and effector cells at q values <0.30, resulting in 12,185 trans-eGenes out of 16,025 tested genes. This implies that the probability of observing 24 randomly selected genes with no trans-eQTLs is 1.3 × 10−15, indicating that trans-eQTL sparsity alone cannot explain this observation. Collectively, these observations indicate that these TFs are strongly depleted of trans-eQTLs, potentially due to selective constraint, suggesting that mapping trans-regulators of highly constrained TFs with natural genetic variation is very underpowered at current sample sizes.
Immune GWAS genes are enriched for regulation from IEI TFs and IL2RA regulators
Next, we expanded our network analyses to include all 12,803 other genes that were expressed highly enough for analysis (STAR Methods), which we refer to as non-perturbed genes. We estimated the effects of the 84 perturbed genes on the non-perturbed genes using two methods. First, we used a traditional differential expression approach using DESeq2,29 where we regressed the normalized expression of each gene against a design matrix that included an indicator for the perturbation status of the sample, the donor identity, and the first four expression PCs. Next, we used mashr39 to perform statistical shrinkage of the differential expression estimates. We refer to these results as DEG-mashr estimates. To model the effects of multiple upstream TFs at the same time, we developed a novel statistical estimator of the bipartite graph (BG), which models the effects of the 84 perturbed genes on the 12,803 non-perturbed genes jointly in a single linear model. In contrast to a differential expression approach, the BG model is less likely to detect redundant causal pathways. We term this approach the BG model (Figure 3A; STAR Methods).
Figure 3.

The landscape of downstream effects
(A) The statistical model used to relate the 84 perturbed genes to the expressed genes.
(B) The distribution of the number of downstream effects for each of the 84 genes, stratified by gene group. Genes that are outliers with respect to their gene group distribution are labeled.
(C) The distribution of indegree for each of the non-perturbed genes. Outlier genes are labeled.
(D) Association between the properties of downstream genes and the gene set of the upstream regulators. Coefficients are estimated with negative binomial regressions of the gene-set-specific indegree. Downstream gene annotations are indicated on the y axis, and the facets are used to indicate the gene set of the upstream regulator. ∗The 306 denotes the total number of RNA-seq observations, which includes 84 genes perturbed in three donors and 54 samples from control guides.
Among the non-perturbed genes, 7,299 (57%) had an incoming edge from at least one KO. Among the non-perturbed genes with at least one incoming edge, the median number of incoming edges was 5. The median number of downstream effects from the BG model was 251.5, ranging from 52 (EGR3) to 2,634 (MED12). Estimates from both the DEG-mashr and BG approaches (Tables S3, S4, and S5) revealed the striking enrichment of IL2RA regulators among the genes with the largest number of downstream connections (Figure 3B). We observed that MED12 and CBFB regulated more genes than any canonical T cell TF. MED12 is a subunit of the mediator complex, which transmits signals from enhancer-bound TFs to RNA polymerase II bound at the promoter.40,41 Despite its large effects, MED12 has never been reported in any autoimmune GWAS, nor does it have a known cis-eQTLs in CD4+ T cells,6 underscoring the value of perturbations for characterizing its function.
To our surprise, we also observed that three of the background TFs (DR1, YBX1, and BPTF) regulated more genes than any of the IEI TFs. The widespread effects of these three background TFs highlight the value of large-scale searches for upstream regulators, even in cell types with well-annotated signaling pathways. Consistent with their large effects, these three TFs were highly constrained (Shet estimates of 0.38, 0.17, and 0.30 for DR1, YBX1, and BPTF). Although BPTF had no outgoing connections to the other 83 knocked out genes, it had an incoming connection from STAT1, suggesting that it may partially mediate the effect of STAT1 on downstream genes. Among the 7,299 downstream genes with at least one incoming connection, there were 10 genes with at least 26 incoming connections (Figure 3C), including genes involved in the DNA damage response (ZMAT3), cell cycle regulation (CCND2), granzymes (GZMA, GZMB), and a T cell costimulatory receptor (CD2).
Next, we asked which properties of the 12,803 non-perturbed genes were associated with regulation from the three gene groups. We performed a series of negative binomial regressions of the incoming connections to non-perturbed genes, including six gene annotations as covariates (Figure 3D). We observed that non-perturbed autoimmune GWAS genes were much more likely to be enriched for regulation from IEI TFs (∼20% enrichment) and IL2RA regulators (∼30% enrichment). Shet was negatively associated with incoming connections in three of the four regressions, consistent with our prior observation that gene constraint is more strongly associated with the number of outgoing connections from a gene than the number of incoming connections to the gene. We also observed that eQTL trans-eGenes were strongly enriched for incoming connections in each regression, suggesting that trans-eGenes reside in the periphery of the network with many incoming connections. Using GTEx, we also identified genes that were only expressed in whole blood and asked whether regulation of blood-specific genes varied by the three gene groups. We observed that blood-specific genes were much more likely to be regulated by IEI TFs (∼20% enrichment) and IL2RA regulators (∼40% enrichment) than background TFs. Collectively, these observations highlight that although background TFs have similar graph centrality to IEI TFs, they are much less likely to disrupt cell-type-specific transcriptional pathways.
Gene modules link groups of genes to a shared function
Next, we asked whether there were groups of the 84 perturbed genes with similar effects on downstream pathways among the 12,803 non-perturbed genes. Hierarchical clustering of the DEG-mashr results revealed the presence of nine gene modules (Figure 4), which we also grouped into a coarser set of super-modules. We remark that although the perturbed genes within each of these modules are mutually exclusive, the non-perturbed genes may overlap. To identify pathways that were regulated by these gene modules, we performed systematic enrichment analyses using KEGG genetic, signaling, and immune pathways42 (Figure 5A; Figures S8–S10; Table S6).
Figure 4.

The discovery of gene modules
Hierarchical clustering is used to identify clusters of shared downstream effects. The upstream gene members within each module are labeled in the left-handed margin of the plot, and the gene group of each gene is indicated by the text color. The total number of genes in the module, including both upstream and downstream effects, is included under the list of genes.
Figure 5.

Gene module characterization
(A) Enrichment analyses of KEGG genetic, immune, and signaling pathways for each of the 84 perturbed genes, stratified by gene module. The JAK-STAT pathway is highlighted with a dashed red box. The color bar maximum is set to 4.
(B) The JAK-STAT sub-network, which is organized such that cytokine genes are at the bottom and upstream regulators are at the top.
(C) Effects of KOs in the gene modules on a proliferation assay. Each point represents an individual gene perturbation sample plotted as the log2 fold change sample count as compared to AAVS1 KO control samples from the same donor (∗p < 0.05 and ∗∗∗∗p < 0.001; n = 3 donors per KO, the number of KOs per cluster is reflected in Figure 4).
The perturbed genes in module 1 included 14 IEI TFs, 19 background TFs, and two IL2RA regulators (RELA and YY1). The perturbed genes in modules 1–2 were primarily IEI and background TFs, and modules 3–4 were primarily IL2RA regulators. We observed that module 1A was enriched for the disruption of mitogen-activated protein kinase (MAPK) and p53 signaling. Module 1B included T-bet (TBX21), a TF that is required for interferon-gamma production and the T helper type 1 cell (Th1) phenotype,43 and three members of the Rel family (NFAT5, RELB, and REL), subunits of nuclear factor κB (NF-κB), a TF complex that plays a role in T cell activation.44 Surprisingly, this cluster also included four background TFs without any annotated immune function (ZNF329, ZNF791, ZBTB14, and ZKSCAN1). ZBTB7B has been observed to be required for CD4+ commitment and interacts with NF-κB,45 but many other members of the ZBTB family, including ZBTB14, remain relatively uncharacterized. The high proportion of shared effects between ZBTB14, T-bet, and the Rel family proteins suggests that ZBTB14 may have similar function to ZBTB7B.
Genes in super-module 2 were enriched for effects on cell cycle regulation and apoptosis. Modules 3–4 were much more strongly enriched for IL2RA regulators than clusters 1–2. Consistent with their annotation, every gene in modules 3–4 had downstream effects on the JAK-STAT and chemokine signaling pathways. Surprisingly, KMT2A, a methylation writer, clustered in the same module as JAK3, STAT5A, STAT5B, IRF4, and IL2RA. Although translocations of KMT2A have been shown to cause lymphoid malignancy,46 it has no annotated function in non-mutated cells in the JAK-STAT pathway.47 We then examined the structure of module 4 (Figure 5B), observing that KMT2A is upstream of IRF4, STAT5A, and IL2RA and directly regulates several downstream effector cytokines through pathways not mediated by the other perturbed genes.
Several modules were strongly enriched for cell cycle and proliferation pathways. To determine if there was a uniform effect on in vitro expansion within any of the modules, we quantified the number of live cells per KO compared to control cells where the guide RNA targeted the safe-harbor locus AAVS1 from the respective donor. Nearly all members of module 2A, which was enriched for cell cycle effects, showed a mean increase in cell counts across three donors as the result of the perturbation. Collectively, the module had a 1.14-fold increase in live cells when knocked out compared to the controls, suggesting that genes in 2A function as proliferation repressors (Figures 5C; Table S12). Concordant with these observations, a recent report described the proliferation-promoting effects of disruption of a module 2A member, TET2, in chimeric antigen receptor (CAR)-T cells.48 Our analyses suggest that other members of 2A may have similar properties to TET2 and thus may represent a group of genes that could be perturbed to alter engineered T cell function. Several upstream members of 2A upregulated three of four CDKN genes, which inhibit cyclin-dependent kinases and potentially lead to reduced cycling (Figure S11). Thus, our inference of gene modules recapitulates known regulators of immune signaling pathways and identifies novel members of these modules.
Heritability analyses link gene modules to immune disease risk
We then asked whether SNPs that were linked to the nine gene modules were enriched for the heritability of autoimmune traits. We included GWAS summary statistics for 10 phenotypes from a combination of Finngen and disease-specific consortia.49,50 After linking SNPs to each of the nine modules using the ABC method,36 we used linkage disequilibrium (LD) score regression2,51 to estimate the contribution of these SNPs to the heritability to eight autoimmune traits and two allergy traits (STAR Methods; Table S7). As a reference point, we also included a group of SNPs linked to genes that were not regulated by any of the 84 genes, which we term module 0. To adjust for confounding genomic annotations, we included the LD score baseline model. We observed that module 4 SNPs were potent contributors generally, as half of the traits analyzed were enriched (Figures 6A and S12; Table S8). Across the traits, there was substantial heterogeneity in the effects of modules. For example, only 4A and 2B SNPs were associated with psoriasis heritability, while 1A, 2B, 3A, 3B, and 4A all contributed to rheumatoid arthritis heritability. In the baseline module 0, only multiple sclerosis was enriched. Remarkably, module 1B contributed little to heritability enrichment of any trait despite including TBX21.
Figure 6.

Autoimmune and allergy association of SNPs linked with the gene modules
(A) Estimated coefficients from LD score regression are plotted for each gene module and phenotype. Module 0 is defined as genes that were not included in any module but are still expressed in CD4+ T cells.
(B) Exemplar analysis annotating the fine-mapped genes from a Finngen dermatitis GWAS based on their presence in module 2A.
We also observed that module 2A, which was strongly enriched for effects on cell cycle regulation pathways, was enriched for the regulation of atopic dermatitis GWAS genes. Next, we annotated the fine-mapped signals from the dermatitis GWAS. Of the 44 credible sets, 34 were linked to genes. Of these 34 hits, four were regulated by module 2A TFs, including SATB1, IL22, LTK, and EZH1 (Figure 6B). Given the putative effects of module 2A on cell proliferation, we then cross-referenced these four genes with cell proliferation annotation pathways. LTK is a receptor with tyrosine kinase activity and may contribute to proliferation through activation of the phosphatidylinositol 3-kinase (PI3K) signaling pathway.52 Similarly, IL22 has also been reported to regulate PI3K signaling.53 Taken together, these analyses highlight the value of unbiased module discovery for identifying specific pathways that contribute to trait heritability. We illustrate how module 2A TFs regulate a subset of dermatitis GWAS genes that have been implicated in PI3K signaling, a common proliferation pathway.
The transcriptional logic linking the JAK-STAT module to immune GWAS genes
Given the substantial contribution of module 4 to autoimmune and allergy phenotype heritability and its large effects on T cell differentiation, we integrated ChIP-seq and ATAC readouts to elucidate the regulatory structure of module 4 (STAR Methods). We observed that KMT2A was a positive regulator of IL-17F and IL-21 expression, two Th17-secreted factors (Figure 7A). Notably, IL-17F had a striking decrease in expression (−5.9 log2 fold change) upon KMT2A KO. We also observed concordant decreases in chromatin accessibility near (5.7 and 40 kb upstream of the transcription start site [TSS]) IL-17F and IL-21 upon KO of KMT2A via ATAC-seq. We then intersected the DAC regions from the KMT2A KO condition with each of the KOs within module 4 and observed that STAT5B shared several differentially expressed sites, including possible distal enhancer regions upstream of IL-17F (Figure S13). An additional Th17-secreted factor, IL-22, also had a shared region between the two conditions, although the transcript was only differentially expressed in the STAT5B KO. The STAT5B KO also abrogated chromatin accessibility 5.7 kb upstream of the IL-17F promoter, in a region bound by STAT5B in CD4+ T cell ChIP-seq (Figure 7B). Because KMT2A is a methyltransferase that deposits activating methylation marks on H3K4, we then asked whether H3K4me3 was present at the same locus in activated Th17s, finding broad H3K4me3 within the region (Figure 7B). These observations led us to suggest the following mechanism for the regulatory logic of module 4: KMT2A, a global epigenetic regulator of transcription, collaborates with downstream factors, including members of JAK-STAT, to positively regulate IL-17F by modulating a putative IL-17F-specific enhancer.
Figure 7.

The transcriptional logic linking module 4 to GWAS loci
(A) The sub-network of module 4 and Th17 cytokines.
(B) Locus plot including tracks describing the functional characteristics of the region. Each track is constructed from publicly available ChIP-seq data (STAR Methods) or ATAC-seq data from Freimer et al.14 Gray boxes indicate significantly different regions between the respective KO and AAVS1 control KO ATAC data (adjusted p value [padj] < 0.05, n = 3 donors per KO). The y axis displays normalized counts.
These observations suggest that cis-regulatory elements near KMT2A may harbor autoimmune risk variants. To assess this hypothesis, we examined recent biobank GWASs in the UK Biobank (UKB),54,55 Finngen,49 and Biobank Japan56 (BBJ) for variants associated with autoimmune phenotypes near KMT2A. The A allele of rs45480496, a common variant (minor-allele frequency [MAF] of 21% in TOPMed57) 36 kb from the TSS of KMT2A, is suggestively associated with autoimmune disease (“diseases marked as autoimmune origin,” odds ratio [OR] = 1.04, p = 2 × 10−7) in Finngen and was also reported as a suggestive hit in a BBJ-UKB meta-analysis58 (“autoimmune multi-trait,” OR = 1.08, p = 2 × 10−6). A meta-analysis of these two signals results in genome-wide significance (p = 2 × 10−12; Figure S14) for this variant. We then looked for functional evidence linking rs45480496 to KMT2A. Although rs45480496 has not yet been reported as an eQTL for KMT2A, lookup of the SNP in a promoter Hi-C capture in immune cells59 revealed that it resides in a regulatory element that interacts with the promoter of KMT2A in megakaryocytes, naive CD4s and CD8s, and effector CD4s and CD8s. Concordant with these observations, lookup of rs45480496 in RegulomeDB60 indicated that it is in an active enhancer in Th17s. The haplotype that rs45480496 tags also intersects with a predicted KMT2A enhancer in CD4+ T cells from the ABC model.36 Although the variant-to-gene predictions from OpenTargets61 suggest that other causal genes are possible in this locus, we remark that these predictions are made without knowledge of the causal cell type for a given phenotype. Collectively, these data report a novel risk locus for autoimmune traits upstream of KMT2A within a putative KMT2A enhancer.
Discussion
Human genetics has been remarkably productive in discovering complex-trait-associated SNPs. There are now several resources to map the effects of these SNPs to molecular phenotypes in cis; however, the development of maps of the regulatory cascades of these SNPs has progressed much more slowly. Enabled by recent innovations in large-scale perturbation technologies, we are now able to systematically perturb large numbers of genes in primary human cell contexts. These perturbations complement natural genetic variation approaches to mapping trans-regulators, as they facilitate the examination of biological variance that is unlikely to be observed in healthy cells. After network inference with LLCB, we observed 211 trans-regulatory causal connections in our upstream GRN, none of which were reported in the largest catalog of CD4+ eQTLs performed to date.6
To infer the gene network, we developed LLCB, which builds upon recent advances in the structure learning literature to estimate a graph with edge weights that are interpretable as direct effects. This stands in contrast to the majority of effect estimates reported in the functional genomics literature, which primarily report estimates from differential expression analyses performed separately in each perturbed gene. These estimates confer results that are difficult to interpret because they do not attempt to adjust for confounding pathways in the GRN, which are known to be highly abundant in biological networks. We use LLCB to estimate the topology and effect size of these confounding pathways. We found that direct effects were generally much larger than indirect effects in magnitude and that the largest indirect effects were mediated by local feedback cycles.
Using experimental perturbations, we investigated the properties of IEI TFs, which are infrequently mutated in natural genetic variation. We performed a series of systematic analyses that delineate the commonalities and differences among the IEI TFs, background TFs, and IL2RA regulators. Consistent with our previous report,14 we found that the IL2RA regulators were potent regulators of downstream effects. Both the IEI TFs and IL2RA regulators were enriched for being upstream and much more likely than background TFs to disrupt autoimmune GWAS loci and whole-blood-specific genes even after adjustment for gene constraint. We also observed that the topology of the regulatory network is strongly associated with selective constraint. Shet was among the best predictors of the topological properties of the perturbed genes: Shet was strongly associated with the number of outgoing connections of a gene but not the number of incoming connections. This is reflected in the dense downstream network identified for the IL2RA regulators with overall high levels of constraint compared to the other TF groups. Overall, the difference in enrichment based on Shet suggests that the centrality of genes is best expressed as a multi-dimensional construct. This further highlights the value of estimating GRNs with directed edges, as opposed to estimating undirected graphs from observational co-expression data, as the richer graphical structure enables much more granular topological analyses.
Utilizing the novel connections in the GRN, we report several observations that improve annotation of canonical immune pathways. We observed that three of the background TFs (DR1, BPTF, and YBX1) regulated more downstream genes than any of the 30 IEI TFs, including TBX21, a master regulator of Th1 differentiation. After identifying gene modules and their downstream pathways, we observed multiple novel members of canonical gene modules, including KMT2A, in the JAK-STAT pathway. We observed that KMT2A, a methyltransferase that deposits activating methylation marks, modulated the expression of canonical IL-2 signaling TFs. KMT2A collaborated with these TFs to upregulate IL-17F, a pro-inflammatory cytokine that is secreted by Th17s, indicating that KMT2A is an underappreciated regulator of the IL2-JAK-STAT axis and Th17 regulation. Meta-analysis of biobank autoimmune GWASs revealed a novel risk locus in a Th17 enhancer upstream of KMT2A. Given the success of JAK inhibitors in treating a subset of patients with autoimmunity,62,63 our pathway analysis could offer an expanded set of candidate drug targets.
Limitations of the study
Although we have demonstrated that our regulatory network is useful for the discovery of novel immune pathway biology and that it is validated by orthogonal data modalities, our study is not without limitations. While CD4+ T cells play a role in many immune pathologies, the construction of networks in more cell types and cellular contexts would undoubtedly result in increased discovery, as would the inclusion of additional perturbations. The restriction to transcriptional regulation also inhibits the interrogation of post-translational regulation, which makes the interpretation of edges from genes where post-translational regulation is important challenging. For example, STAT proteins, which are known to be sensitive to phosphorylation, may regulate more genes than is estimated in our transcriptional network. The use of a bulk expression readout, although more sensitive to genes with low expression than single-cell assay transcriptome analysis, also precludes the analysis of more granular cell types and contexts, which are easier to assess in parallel using single-cell profiling methodologies, including Perturb-seq and single-cell eQTL studies.
Conclusion
In conclusion, we describe the GRN of key CD4+ T cell regulators. This network enabled both the broad characterization of the properties of immune disease genes and the discovery of novel regulatory connections between TFs and signaling pathways that modulate immune disease genes. We anticipate that our approach can be applied in other cell types and contexts to generate maps of the molecular consequences of regulatory variation of disease genes.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jonathan K. Pritchard (pritch@stanford.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
Bulk RNA-seq data have been deposited at GEO: GSE271788 and GSE171737 and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Genotyping data and cell counts are available in Tables S1, S2, S3, S4, S5, S6, S7, S8, S9, S10, S11, and S12.
-
•
All original code has been deposited at GitHub and is publicly available as of the date of publication. Code used to generate the main figures can be found here: 10.5281/zenodo.12807946. LLCB code is available at 10.5281/zenodo.12807979 and https://github.com/weinstockj/LLCB.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Acknowledgments
We thank Romain Lopez, Matt Aguirre, Eric Kernfeld, and Prashanthi Ravichandran for several stimulating conversations about gene networks and structure learning. We also thank members of the Pritchard, Battle, and Marson labs for feedback and insightful discussions. This work was supported by NHGRI 2R01HG008140. Additionally, M.M.A. is a National Science Foundation Graduate Research Fellow supported under grant no. 2038436. M.O. was supported by the Astellas Foundation for Research on Metabolic Disorder and Chugai Foundation for Innovative Drug Discovery Science (C-FINDs). A.M. received funding from the Simons Foundation, a Lloyd J. Old STAR Award (Cancer Research Institute), the Parker Institute for Cancer Immunotherapy, and the Innovative Genomics Institute. A.B. was supported by NIGMS R35GM139580.
Author contributions
Formal analysis, J.S.W., M.A.A., and M.O.; investigation, J.S.W. and M.M.A.; supervision, J.K.P., A.B., and A.M.; funding acquisition, J.K.P., A.B., and A.M.; experimental work, M.A.A. and J.W.F.; writing – original draft preparation, J.S.W. and M.M.A.; writing – review & editing, J.K.P., A.B., A.M., M.O., and J.W.F.
Declaration of interests
A.M. is a cofounder of Site Tx, Arsenal Biosciences, Spotlight Therapeutics, and Survey Genomics; serves on the boards of directors at Site Tx, Spotlight Therapeutics, and Survey Genomics; is a member of the scientific advisory boards of Site Tx, Arsenal Biosciences, Cellanome, Spotlight Therapeutics, Survey Genomics, NewLimit, Amgen, and Tenaya; owns stock in Arsenal Biosciences, Site Tx, Cellanome, Spotlight Therapeutics, NewLimit, Survey Genomics, Tenaya, and Lightcast; and has received fees from Site Tx, Arsenal Biosciences, Cellanome, Spotlight Therapeutics, NewLimit, Gilead, Pfizer, 23andMe, PACT Pharma, Juno Therapeutics, Tenaya, Lightcast, Trizell, Vertex, Merck, Amgen, Genentech, GLG, ClearView Healthcare, AlphaSights, Rupert Case Management, Bernstein, and ALDA. A.M. is an investor in and informal advisor to Offline Ventures and a client of EPIQ. The Marson laboratory has received research support from the Parker Institute for Cancer Immunotherapy, the Emerson Collective, Arc Institute, Juno Therapeutics, Epinomics, Sanofi, GlaxoSmithKline, Gilead, and Anthem and reagents from Genscript and Illumina. J.W.F. was a consultant for NewLimit, is an employee of Genentech, and has equity in Roche. A.B. is a stockholder in Alphabet, Inc., and a consultant for Third Rock Ventures. J.S.W. was a consultant to Spiral Genetics.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Human Peripheral Blood Leukopaks | STEMCELL Technologies | 70500 |
| Chemicals, peptides, and recombinant proteins | ||
| Cas9 protein (MacroLab, Berkeley | – | – |
| Lonza P3 buffer | Lonza, catalog no. | V4XP-3032 |
| EasySep™ Human CD4+CD127lowCD25+ Regulatory T cell Isolation Kit | STEMCELL Technologies | 18063 |
| ImmunoCult™ Human CD3/CD28/CD2 T cell Activator | STEMCELL Technologies | 10990 |
| Critical commercial assays | ||
| RNA lysis buffer (Zymo, #R1060-1-100). | Zymo | R1060-1-100 |
| Zymo-Quick RNA micro prep kit (#R1051) | Zymo | R1051 |
| Turbo-DNAse (Fisher Scientific, AM2238) | Zymo | AM2238 |
| RNA Clean & Concentrator-5 kit (Zymo, #R1016) | Zymo | R1016 |
| QuantSeq FWD kit (Lexogen) | Lexogen | K0152x96-2-0162 |
| Deposited data | ||
| RNAseq data for IEI and background TFs | GEO | GSE271788 |
| RNAseq data from Freimer et al.14 | GEO | GSE171737 |
| Oligonucleotides | ||
| Custom crRNAs (Dharmacon) for CRISPR KO | – | See Table S9 |
| Primers for genotyping | – | See Table S10 |
| Dharmacon Edit-R CRISPR-Cas9 Synthetic tracrRNA | Dharmacon | U-002005-20 |
| Single-stranded donor oligonucleotides (ssODN): TTAGCTCTGTTTACG TCCCAGCGGGCATGAGAGTAACA AGAGGGTGTGGTAATATTAC GGTACCGAGCACTATCGATACAAT ATGTGTCATACGGACACG |
IDT custom oligo | N/A |
| Software and algorithms | ||
| Figure generation | Github | https://github.com/weinstockj/RNAseq-perturbation-CD4-pipeline |
| LLCB code | Github | https://github.com/weinstockj/LLCB |
| Other | ||
| 96-well electroporation cuvette plate | Lonza | VVPA-1002 |
Experimental model and study participant details
Primary human T cells were isolated from blood samples procured from STEMCELL technologies. Healthy male and female donors were utilized without selection for age, weight or for ethnicity.
Method details
Cell isolation and expansion
Primary CD25−CD4+ effector T cells were isolated from fresh Human Peripheral Blood Leukopaks (STEMCELL Technologies, #70500) from healthy donors, after institutional review board–approved informed written consent (STEMCELL Technologies). Peripheral blood mononuclear cells (PBMCs) were washed twice with a 1X volume of EasySep buffer (DPBS, 2% fetal Bovine Serum (FBS), 1mM pH 8.0 EDTA). The washed PBMCs were resuspended at 200E6 cells/mL in EasySep buffer and isolated with the EasySep Human CD4+CD127lowCD25+ Regulatory T cell Isolation Kit (STEMCELL Technologies, #18063), according to the manufacturer’s protocol. Cells were seeded at 1x106 cells/mL in complete RPMI-1640 supplemented with 10% FCS, 2 mM L-Glutamine (Fisher Scientific #25030081), 10 mM HEPES (Sigma, #H0887-100ML), 1X MEM Non-essential Amino Acids (Fisher, #11140050), 1 mM Sodium Pyruvate (Fisher Scientific #11360070), 100 U/mL Penicillin-Streptomycin (Sigma, #P4333-100ML), and 50 U/mL IL-2 (Amerisource Bergen, #10101641) and stimulated with 6.25 μL/mL ImmunoCult Human CD3/CD28/CD2 T cell Activator (STEMCELL Technologies, #10990). Cells were cultured at 37°C with 5% CO2. Following activation and electroporation, cells were split 1:2 every other day to maintain an approximate density of 1x106 cells/mL.
Cas9 RNP preparation and delivery
Custom crRNAs (Dharmacon) and Dharmacon Edit-R CRISPR-Cas9 Synthetic tracrRNA (Dharmacon, #U-002005-20) were resuspended in Nuclease Free Duplex Buffer (IDT, #11-01-03-01) at 160uM stock concentration. In a 96 well plate, each crRNA was combined with tracrRNA at a 1:1 M ratio and incubated at 37°C for 30 min. Custom crRNA sequences are included in Table S9. Single-stranded donor oligonucleotides (ssODN; sequence: TTAGCTCTGTTTACGTCCCAGCGGGCATGAGAGTAACAAGAGGGTGTGGTAATATTACGGTACCGAGCACTATCGATACAATATGTGTCATACGGACACG, 100uM stock) was added to the complex at a 1:1 M ratio and incubated at 37°C for 5 min. Finally, Cas9 protein (MacroLab, Berkeley, 40 μM stock) was added at a 1:2 M ratio and incubated at 37°C for 15 min. The resulting RNPs were frozen at −80°C until the day of electroporation. 48 h following effector T cell activation, the cells were pelleted at 100x g for 10 min and resuspended in room temperature Lonza P3 buffer (Lonza, catalog no. V4XP-3032) at 1.5x106 cells per 20 μl P3. The cells were combined with 5 μl aliquots of the thawed RNPs, transferred to a 96-well electroporation cuvette plate (Lonza, #VVPA-1002) and nucleofected with pulse code EH-115. Immediately following electroporation, the cells were gently resuspended in 90 μl warmed complete RPMI with IL-2 and incubated at 37°C for 15 min. After recovery, the cells were cultured in 96 well plates at 1x106 cells/mL for the duration of the experiment. To prevent edge effects, the guides were randomly distributed across each plate and the first and last column of each plate was excluded, being filled instead with PBS to prevent evaporation.
RNA isolation and library preparation
8 days after T cell isolation and activation, the cells were pelleted and resuspended at 1x106 cells per 300 μl of RNA lysis buffer (Zymo, #R1060-1-100). Cells were pipette mixed and frozen at −80 until RNA isolation was performed. RNA was isolated using the Zymo-Quick RNA micro prep kit (#R1051) according to the manufacturer’s protocol with the following modifications: After thawing the samples, each tube was vortexed vigorously to ensure complete lysis prior to loading into the extraction columns. In lieu of the kit provided DNAse, RNA was eluted from the isolation column after the recommended washes and digested with Turbo-DNAse (Fisher Scientific, #AM2238) at 37°C for 20 min. Following digestion, RNA was purified using the RNA Clean & Concentrator-5 kit (Zymo, #R1016) according to the manufacturer’s protocol. The resulting purified RNA was submitted to the UC Davis DNA Technologies and Expression Analysis Core to generate 3′ Tag-seq libraries with unique molecular indices (UMIs). Barcoded sequencing libraries were prepared using the QuantSeq FWD kit (Lexogen) for multiplexed sequencing on an Hiseq 4000 (Illumina).
Genotyping of arrayed KOs
On the day of cell collection for RNAseq, genomic DNA was isolated using DNA QuickExtract (Lucigen, #QE09050) according to the manufacturer’s protocol. Primers were designed to flank each sgRNA cut site (Table S10). Amplicons of the region were generated by adding 1.25 μL each of forward and reverse primer at 10uM to 5 μL of sample in QuickExtract, 12.5 μL of NEBNext Ultra II Q5 master mix (NEB, Cat #M0544L), and H2O to a total 25 μL reaction volume. Touchdown PCR was used with the following cycling conditions: 98°C for 3 min, 15 cycles of 94°C for 20 s followed by 65°C–57.5°C for 20 s (0.5°C incremental decreases per cycle), and 72°C for 1 min, and a subsequent 20 cycles at 94°C for 20 s, 58°C for 20 s and 72°C for 1 min, and a final 10 min extension at 72°C. Amplicons were diluted 1:200 and Illumina sequencing adapters were then added in a second PCR reaction. Indexing reactions included 1 μL of the diluted PCR1 sample, 2.5 μL of each the forward and reverse Illumina TruSeq indexing primers at 10 μM each, 12.5 μL of NEB Q5 master mix, and H2O to a total 25 μL reaction volume. The following PCR cycling conditions were used: 98°C for 30 s, followed by 98°C for 10 s, 60°C for 30 s, and 72°C for 30 s for 12 cycles, and a final extension period at 72°C for 2 min. Samples were pooled at an equivolume ratio and SPRI purified prior to sequencing on an Illumina MiSeq with PE 150 reads. Analysis with performed with CRISPResso253 (v2.2.7) CRISPRessoBatch --skip_failed --n_processes 4 --exclude_bp_from_left 0 --exclude_bp_from_right 0 --plot_window_size 10.
Cell proliferation quantification
One replica plate of cells from each donor was run on the Attune NxT Flow Cytometer (Thermo Fisher) within 24 h of cell lysis for RNA extraction. The lymphocyte count was collected for each well using an equi-volume amount of sample. Counts were normalized to the mean AAVS1 lymphocyte count for the respective donor and experiment. Samples with a total Lymphocyte_count <1000 were excluded from the analysis, removing one donor sample from SP110, EPAS1, and ZBTB14 KOs.
Quantification and statistical analysis
RNA-seq alignment and gene count quantification
Adapters were trimmed from fastq files with cutadapt.64 Low-quality bases from reads were trimmed using the Phred algorithm implemented in seqtk (https://github.com/lh3/seqtk). Reads were then aligned with STAR65 and mapped to GRCh38. Gene counts from deduplicated reads were quantified using featureCounts.66 Sample quality control reports were generated with Fastqc (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), rseqc,67 and Multiqc.68
Gene filtering and PCA analysis
Genes were first filtered to those with at least 10 counts in at least five samples. PCA was then performed on the variance stabilizing transformed29 (vst) counts of the 500 most variable genes. Three outlier samples were excluded and then the above process was repeated. The PCs were then assessed for association with batch effects and very broad cellular pathways. PCs 1–2 associated with batch effects, and PCs 3–4 were associated with cell cycle state, suggesting that PCs 1–4 should be included as covariates or otherwise adjusted for in downstream analysis.
Differential expression analysis
Differential expression analysis was performed using DESeq2,29 including donor identity, PCs 1–4, and the KO as predictors of the response. Donor identity and PCs 1–4 were included as covariates to mitigate their confounding effects on gene expression. We emphasize that the statistical estimand in this analysis the total effect of the perturbation of a given gene on the readout gene. This effect may include several indirect paths between the perturbed gene and the readout gene.
We used mashr39 to perform shrinkage of the effect sizes of the differentially expressed genes. This yielded an estimate of the local false-sign rate (LFSR), which is the posterior probability that the true effect has a different sign (positive or negative) than the sign that is most compatible with the posterior distribution. We used a threshold of LFSR <5 x 10−3 as a significance threshold.
LLCB
We formulate the GRN as a graph , where the nodes are each a vector of the vst normalized gene expression values. We restrict this analysis to the 84 KO’d genes reflecting the importance of satisfying the identifiability condition described in Hyttinen et al. is the adjacency matrix describing the direct linear effects between genes, where the rows encode the parent genes and the columns encode the children genes. We then construct a covariate matrix where the columns indicate covariates to regress out. We then orthogonalize based on with the transformation , where . We add back in the column means to roughly preserve the original scale of . In , we include the donor identity and first four PCs as covariates.
We define as the indices indicating the samples in which was intervened upon and we set . We define as the indices in which safe-harbor AAVS1 control samples were used. For all we recode for all . This reflects our belief that the CRISPR KOs were effectively forcing the normalized functional transcript abundance to 0, i.e., we assume perfect interventions.
We then estimate in two steps: 1. Estimate the total effects between every pair of genes ; 2. Estimate from using a modification of the LLC algorithm.23 To estimate , we first center and scale based on its mean and standard deviation in the control samples . Then, we use OLS to estimate the total effect of on , limiting the samples used to , such that we exclude all instances in which the child node has been KO’d. This analysis results in the matrix of estimated total effects, . We emphasize that these coefficients are on a correlation scale because of the standardization procedure.
We assume asymptotic stability23 over the true , which is equivalent to assuming that the largest eigenvalue is less than 1. Because we know is asymptotically stable, the following decomposition of true effects into direct effects is coherent:
This relationship indicates that total effect of a gene on gene is the sum of all possible paths between them, where the value of an individual path is defined by the product of direct effects along that path.
To estimate from , we use the LLC procedure Algorithm 1:
Algorithm 1.

This procedure results in matrices of size and column vectors . We then concatenate vertically into a column vector of length and we form the block matrix
We then define the likelihood as the probability of conditional on and parameters and . and represent a system of linear equations relating the total effects to the direct effects . For a given gene we define the set of rows in corresponding to experimental observations where we perturb a putative parent of and record the effect on as . For each row of and where the gene is the child node, we specific the likelihood as:
Including a child node specific dispersion parameter allows for heterogeneity in the residual variance across the genes.
Because we have prior knowledge of what realistic gene networks look like, we specify the prior in three parts as follows:
where is defined as the spectral radius of , i.e., the maximum eigenvalue of . We estimate the maximum eigenvalue of using power iteration. We incorporate a prior on the spectral radius because it is an upper bound over the NOTEARS DAG penalty,22 which is a differentiable penalty that enables DAG search in a continuous optimization framework. Importantly, we encode this prior as a “soft-constraint” with the Gaussian density to weakly penalize the divergence of from the space of DAGs while still allowing for cyclic elements.
Over the columns of , i.e., we place a sparsity inducing L1 prior:
The purpose of this term in the prior is to reflect our belief that the indegree of a gene should be relatively small; we know that genes are not directly regulated by hundreds of TFs. In contrast, a given TF may regulate hundreds of downstream genes, so we do not penalize the rows of . Overall – this prior encodes the following three prior beliefs: 1. The effects should be somewhat small on a partial correlation scale; 2. The maximum eigenvalue should not be very large to penalize graphs with many cycles; 3. The indegree for each gene should be relatively small, while the outdegree should not be penalized.
On the dispersion terms, , we place a prior. We estimate total dispersion terms because there may heterogeneity in the residual variance of the total effects across the KO’d genes.
Causal network posterior inference
We use pathfinder32 to estimate the posterior. Briefly, pathfinder is a variational inference algorithm that optimizes the joint log probability of the model using L-BFGS, i.e., the maximum a posteriori objective. Along this optimization trajectory, it constructs a surrogate posterior at each point using the estimate of the hessian from L-BFGS as the precision of the surrogate posterior. Then, at each point, the evidence lower bound (ELBO) is evaluated. The variational approximation resulting in the largest ELBO is then returned as the posterior estimate. We compute seven runs of this optimization procedure in parallel, and then use importance resampling to combine the fits. We initialize based on the component-wise sum of the MLE estimate of and a vector of Gaussian noise i.e., .
Causal network posterior uncertainty quantification
We compute a pseudo-posterior inclusion probability (PIP) we defined as . We set We also computed local-false sign rates (LFSR) estimates: . We note that these summary statistics, although likely proportional to the ‘true’ values, are likely somewhat uncalibrated given that a) we do not model the underling discrete graph structure separately from the parameters and b) calibrated inference in a network setting has been shown empirically to be extremely challenging.
Simulation of a cyclic network in a steady state
We start by simulating a given expression vector of genes as . Then, for a given adjacency matrix we model the effect of a perturbation on the gene as setting , i.e., we remove the incoming edges to this node and set the value of this node to 0. We denote this perturbed adjacency matrix as . We then sample the “steady-state” limit as We assessed the performance of our algorithm on a corresponding to a cyclic network (Supplemental Methods 1).
ABC-DAC GRN
We extracted the CD4+ enhancer to gene predictions from the ABC model36 and we intersected them with the differential ATAC peaks from Freimer et al., which were generated on samples where the 24 IL2RA regulators were KO’d. For the gene we included as an edge in this graph if one its differential ATAC peaks intersected with an ABC enhancer for gene , suggesting that perturbation of gene was perturbing a cis regulatory element for gene . We then calculated the enrichment of these edges among those detected in the IL2RA regulator sub-network of causal network estimate.
HBase validation network
We downloaded the HumanBase37 predicted “T-Lymphocyte” network from https://hb.flatironinstitute.org/download. We downloaded the version of the network with only the top edges included. We then estimated enrichment in the same manner as with the ABC-GRN network.
ABC-ChIP GRN
We downloaded ChIP-seq tracks for nine factors (NFKB1, ETS1, FOXP3, REL, RELA, STAT5B, IRF4, TBX21, YY1) encoded by the KO’d genes where the data were available in human CD4+ cells from the ChIP-seq atlas. We then intersected these peaks with enhancer to gene predictions from the ABC model. For the gene we included as an edge in this graph if one its ChIP peaks intersected with an ABC enhancer for gene . We refer to the network defined from these edges as the ABC-ChIP GRN.
Comparison to CD4+Trans-eQTLs
Full trans-eQTL summary statistics were obtained from Yazar et al.6 from correspondence with the authors. For each of the 84 perturbed genes, we then intersected those with the cis-eQTL summary statistics in CD4+ effector and naive cells, finding that only 24 of the 84 genes had at least one cis-eQTL. Among these 24, we then searched for a trans effect of these cis-eQTLs at various q-value thresholds (5%, 10%, 20%, 30%) of the trans-eQTL summary statistics; we included a range of liberal q-value thresholds in order to construct trans-eQTL derived networks at a permissive range of network densities. We defined an edge in the trans-eQTL network as a cis-eQTL for one of the perturbed genes that also was a trans-eQTL for another of the perturbed genes.
Bipartite graph model of downstream gene expression
We refer to a “downstream” gene as those that were measured among the 12,803 genes that were highly expressed but not among the perturbed genes. We form a matrix with 12,803 columns containing the vst normalized gene expression data. We define a matrix with the expression values of the 84 perturbed genes. We applied the same normalization data procedure as in our causal network estimation such that both and are vst transformed data that is orthogonal to covariates (donor identity, PCs 1–4). We specified the following likelihood for the measurement of the downstream gene:
Over the we place the following prior , where is defined as the asymptotic steady state covariance implied by our point estimate from the causal network model, i.e., . This prior encodes the belief that similar effects among the 84 genes in the causal network will increase the likelihood of similar downstream effects. Because we used a conjugate prior the posterior has an analytic form
where and .
We set 0 in practice, although in principle empirical Bayesian approaches or other criteria could be used to set this hyperparameter. We estimate the residual variance parameter using maximum likelihood and we use LFSR as our variable selection criteria.
Pathway analysis
Downstream enriched pathways were identified for each perturbation using pathfindR (v1.6.4).69 For each upstream gene perturbed, outgoing edges within the BG model were used as input for pathfinder, with a significance threshold of LFSR <5 x 10−3. Gene sets were limited to KEGG,42 Reactome,47 and GO-BP52 and the minimum gene set size and enrichment threshold were set to 10 and 0.05 respectively. Pathways were prioritized for visualization based on the number of genes within the module with enrichment for the pathway, median fold enrichment across all members of the module, and relevance to T cell biology.
LD score regression analyses
We first defined gene sets corresponding to each of the nine modules (1A-C, 2A-C, 3A-B, 4) and module 0, which we defined as the set of genes that were expressed highly enough for analysis but were not associated with any of the KO’d genes (at an LFSR threshold of 5 x 10−3). For each of these 10 gene sets, we then linked SNPs to these genes (S2G) using seven possible methods following Dey at al,70 including approaches that link SNPs based purely on physical distance to the nearest gene, fine-mapped eQTLs, promoter Hi-C capture, the ABC model, among others.
For each of the 10 phenotypes analyzed (Table S7) we obtained the GWAS summary statistics and performed LD score regression analysis. We included the LD score baseline model v2.1 in the regression. We used the publicly available European ancestry LD score estimates for the HapMap SNPs available from:
gs://broad-alkesgroup-public/LDSCORE/Dey_Enhancer_MasterReg/processed_data.
ATAC and ChIPseq data visualization
Bigwigs for each of the tracks were downloaded from ChIP-Atlas. ATAC bigwigs and differentially expressed regions were procured from Freimer et al. and a representative donor was used for visualization of each perturbation effect at the IL17F locus. Visualization was performed with rtracklayer (v1.52.1) and ggplot2 (v3.4.1). APRIS gene structure was used for gene annotation with gggenes (v0.5.0). Differentially accessible chromatin regions were defined in Freimer at al.,14 Supplementary Data 2.
Bigwig files were obtained for visualization from the following ChIP-Atlas sources: STAT5B KO ATAC- SRX10558086, KMT2A KO ATAC- SRX10558079, AAVS1 KO ATAC- SRX10558063 (all ATAC samples from CD4+ T cells treated with IL-2), H3K4me3 ChIP- activated Th17 ChIP71 (stimulated with anti-CD3/CD28 beads and IL-2)- SRX16500373 (GSM6376841), STAT5B ChIP72 (treated with IL-2)- SRX041293 (GSM671402).
Published: October 11, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2024.100671.
Contributor Information
Alexander Marson, Email: alex.marson@gladstone.ucsf.edu.
Alexis Battle, Email: ajbattle@jhu.edu.
Jonathan K. Pritchard, Email: pritch@stanford.edu.
Supplemental information
References
- 1.Maurano M.T., Humbert R., Rynes E., Thurman R.E., Haugen E., Wang H., Reynolds A.P., Sandstrom R., Qu H., Brody J., et al. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finucane H.K., Bulik-Sullivan B., Gusev A., Trynka G., Reshef Y., Loh P.-R., Anttila V., Xu H., Zang C., Farh K., et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 2015;47:1228–1235. doi: 10.1038/ng.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.GTEx Consortium. Laboratory Data Analysis &Coordinating Center LDACC—Analysis Working Group. Statistical Methods groups—Analysis Working Group. Enhancing GTEx eGTEx groups. NIH Common Fund. Biospecimen Collection Source Site—NDRI. Jo B., Mohammadi P., Park Y., Parsana P., et al. Genetic effects on gene expression across human tissues. Nature. 2017;550:204–213. doi: 10.1038/nature24277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318–1330. doi: 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Võsa U., Claringbould A., Westra H.-J., Bonder M.J., Deelen P., Zeng B., Kirsten H., Saha A., Kreuzhuber R., Yazar S., et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 2021;53:1300–1310. doi: 10.1038/s41588-021-00913-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yazar S., Alquicira-Hernandez J., Wing K., Senabouth A., Gordon M.G., Andersen S., Lu Q., Rowson A., Taylor T.R.P., Clarke L., et al. Single-cell eQTL mapping identifies cell type–specific genetic control of autoimmune disease. Science. 2022;376:eabf3041. doi: 10.1126/science.abf3041. [DOI] [PubMed] [Google Scholar]
- 7.Liu X., Li Y.I., Pritchard J.K. Trans Effects on Gene Expression Can Drive Omnigenic Inheritance. Cell. 2019;177:1022–1034.e6. doi: 10.1016/j.cell.2019.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boyle E.A., Li Y.I., Pritchard J.K. An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell. 2017;169:1177–1186. doi: 10.1016/j.cell.2017.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Price A.L., Helgason A., Thorleifsson G., McCarroll S.A., Kong A., Stefansson K. Single-Tissue and Cross-Tissue Heritability of Gene Expression Via Identity-by-Descent in Related or Unrelated Individuals. PLoS Genet. 2011;7:e1001317. doi: 10.1371/journal.pgen.1001317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Connally N.J., Nazeen S., Lee D., Shi H., Stamatoyannopoulos J., Chun S., Cotsapas C., Cassa C.A., Sunyaev S.R. The missing link between genetic association and regulatory function. Elife. 2022;11:e74970. doi: 10.7554/eLife.74970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elorbany R., Popp J.M., Rhodes K., Strober B.J., Barr K., Qi G., Gilad Y., Battle A. Single-cell sequencing reveals lineage-specific dynamic genetic regulation of gene expression during human cardiomyocyte differentiation. PLoS Genet. 2022;18:e1009666. doi: 10.1371/journal.pgen.1009666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Strober B.J., Elorbany R., Rhodes K., Krishnan N., Tayeb K., Battle A., Gilad Y. Dynamic genetic regulation of gene expression during cellular differentiation. Science. 2019;364:1287–1290. doi: 10.1126/science.aaw0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nathan A., Asgari S., Ishigaki K., Valencia C., Amariuta T., Luo Y., Beynor J.I., Baglaenko Y., Suliman S., Price A.L., et al. Single-cell eQTL models reveal dynamic T cell state dependence of disease loci. Nature. 2022;606:120–128. doi: 10.1038/s41586-022-04713-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Freimer J.W., Shaked O., Naqvi S., Sinnott-Armstrong N., Kathiria A., Garrido C.M., Chen A.F., Cortez J.T., Greenleaf W.J., Pritchard J.K., Marson A. Systematic discovery and perturbation of regulatory genes in human T cells reveals the architecture of immune networks. Nat. Genet. 2022;54:1133–1144. doi: 10.1038/s41588-022-01106-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mowery C.T., Freimer J.W., Chen Z., Casaní-Galdón S., Umhoefer J.M., Arce M.M., Gjoni K., Daniel B., Sandor K., Gowen B.G., et al. Systematic decoding of cis gene regulation defines context-dependent control of the multi-gene costimulatory receptor locus in human T cells. Nat. Genet. 2024;56:1156–1167. doi: 10.1038/s41588-024-01743-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mostafavi H., Spence J.P., Naqvi S., Pritchard J.K. Limited overlap of eQTLs and GWAS hits due to systematic differences in discovery. bioRxiv. 2022 doi: 10.1101/2022.05.07.491045. Preprint at. [DOI] [Google Scholar]
- 17.Bousfiha A., Moundir A., Tangye S.G., Picard C., Jeddane L., Al-Herz W., Rundles C.C., Franco J.L., Holland S.M., Klein C., et al. The 2022 Update of IUIS Phenotypical Classification for Human Inborn Errors of Immunity. J Clin Immunol. J. Clin. Immunol. 2022;42:1508–1520. doi: 10.1007/s10875-022-01352-z. [DOI] [PubMed] [Google Scholar]
- 18.Marrack P., Kappler J., Kotzin B.L. Autoimmune disease: why and where it occurs. Nat. Med. 2001;7:899–905. doi: 10.1038/90935. [DOI] [PubMed] [Google Scholar]
- 19.Attfield K.E., Jensen L.T., Kaufmann M., Friese M.A., Fugger L. The immunology of multiple sclerosis. Nat. Rev. Immunol. 2022;22:734–750. doi: 10.1038/s41577-022-00718-z. [DOI] [PubMed] [Google Scholar]
- 20.Sun L., Su Y., Jiao A., Wang X., Zhang B. T cells in health and disease. Signal Transduct. Targeted Ther. 2023;8:235–250. doi: 10.1038/s41392-023-01471-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P., et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng X., Aragam B., Ravikumar P., Xing E.P. DAGs with NO TEARS: Continuous Optimization for Structure Learning. arXiv. 2018 doi: 10.48550/arXiv.1803.01422. Preprint at. [DOI] [Google Scholar]
- 23.Hyttinen A., Eberhardt F., Hoyer P.O. Learning Linear Cyclic Causal Models with Latent Variables. J. Mach. Learn. Res. 2012;13:3387–3439. [Google Scholar]
- 24.Friedman N., Koller D. Being Bayesian About Network Structure. A Bayesian Approach to Structure Discovery in Bayesian Networks. Mach. Learn. 2003;50:95–125. doi: 10.1023/A:1020249912095. [DOI] [Google Scholar]
- 25.Battle A., Jonikas M.C., Walter P., Weissman J.S., Koller D. Automated identification of pathways from quantitative genetic interaction data. Mol. Syst. Biol. 2010;6:379. doi: 10.1038/msb.2010.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Agrawal R., Uhler C., Broderick T. Proceedings of the 35th International Conference on Machine Learning (PMLR) 2018. Minimal I-MAP MCMC for Scalable Structure Discovery in Causal DAG Models.https://proceedings.mlr.press/v80/agrawal18a/agrawal18a-supp.pdf [Google Scholar]
- 27.Harris S.L., Levine A.J. The p53 pathway: positive and negative feedback loops. Oncogene. 2005;24:2899–2908. doi: 10.1038/sj.onc.1208615. [DOI] [PubMed] [Google Scholar]
- 28.Parsana P., Ruberman C., Jaffe A.E., Schatz M.C., Battle A., Leek J.T. Addressing confounding artifacts in reconstruction of gene co-expression networks. Genome Biol. 2019;20 doi: 10.1186/s13059-019-1700-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15 doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lopez R., Hütter J.-C., Pritchard J.K., Regev A. Large-Scale Differentiable Causal Discovery of Factor Graphs. arXiv. 2022 doi: 10.48550/arXiv.2206.07824. Preprint at. [DOI] [Google Scholar]
- 31.Ng I., Ghassami A., Zhang K. On the Role of Sparsity and DAG Constraints for Learning Linear DAGs. arXiv. 2021 doi: 10.48550/arXiv.2006.10201. Preprint at. [DOI] [Google Scholar]
- 32.Zhang L., Carpenter B., Gelman A., Vehtari A. Pathfinder: Parallel quasi-Newton variational inference. arXiv. 2022 doi: 10.48550/arXiv.2108.03782. Preprint at. [DOI] [Google Scholar]
- 33.Su C., Borsuk M.E. Improving Structure MCMC for Bayesian Networks through Markov Blanket Resampling. J. Mach. Learn. Res. 2016;17:1–20. [Google Scholar]
- 34.Grzegorczyk M., Husmeier D. Improving the structure MCMC sampler for Bayesian networks by introducing a new edge reversal move. Mach. Learn. 2008;71:265–305. doi: 10.1007/s10994-008-5057-7. [DOI] [Google Scholar]
- 35.Stephens M. False discovery rates: a new deal. Biostatistics. 2017;18:275–294. doi: 10.1093/biostatistics/kxw041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fulco C.P., Nasser J., Jones T.R., Munson G., Bergman D.T., Subramanian V., Grossman S.R., Anyoha R., Doughty B.R., Patwardhan T.A., et al. Activity-by-contact model of enhancer–promoter regulation from thousands of CRISPR perturbations. Nat. Genet. 2019;51:1664–1669. doi: 10.1038/s41588-019-0538-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Greene C.S., Krishnan A., Wong A.K., Ricciotti E., Zelaya R.A., Himmelstein D.S., Zhang R., Hartmann B.M., Zaslavsky E., Sealfon S.C., et al. Understanding multicellular function and disease with human tissue-specific networks. Nat. Genet. 2015;47:569–576. doi: 10.1038/ng.3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zeng T., Spence J.P., Mostafavi H., Pritchard J.K. Bayesian estimation of gene constraint from an evolutionary model with gene features. bioRxiv. 2024 doi: 10.1101/2023.05.19.541520. Preprint at. [DOI] [PubMed] [Google Scholar]
- 39.Urbut S.M., Wang G., Carbonetto P., Stephens M. Flexible statistical methods for estimating and testing effects in genomic studies with multiple conditions. Nat. Genet. 2019;51:187–195. doi: 10.1038/s41588-018-0268-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Soutourina J. Transcription regulation by the Mediator complex. Nat. Rev. Mol. Cell Biol. 2018;19:262–274. doi: 10.1038/nrm.2017.115. [DOI] [PubMed] [Google Scholar]
- 41.Richter W.F., Nayak S., Iwasa J., Taatjes D.J. The Mediator complex as a master regulator of transcription by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2022;23:732–749. doi: 10.1038/s41580-022-00498-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kanehisa M., Furumichi M., Sato Y., Kawashima M., Ishiguro-Watanabe M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023;51:D587–D592. doi: 10.1093/nar/gkac963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu J., Yamane H., Paul W.E. Differentiation of Effector CD4 T Cell Populations. Annu. Rev. Immunol. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oh H., Ghosh S. NF-κB: Roles and Regulation In Different CD4+ T cell subsets. Immunol. Rev. 2013;252:41–51. doi: 10.1111/imr.12033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang L., Wildt K.F., Castro E., Xiong Y., Feigenbaum L., Tessarollo L., Bosselut R. The zinc finger transcription factor Zbtb7b represses CD8-lineage gene expression in peripheral CD4+ T cells. Immunity. 2008;29:876–887. doi: 10.1016/j.immuni.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baffa R., Negrini M., Schichman S.A., Huebner K., Croce C.M. Involvement of the ALL-1 gene in a solid tumor. Proc. Natl. Acad. Sci. USA. 1995;92:4922–4926. doi: 10.1073/pnas.92.11.4922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gillespie M., Jassal B., Stephan R., Milacic M., Rothfels K., Senff-Ribeiro A., Griss J., Sevilla C., Matthews L., Gong C., et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res. 2022;50:D687–D692. doi: 10.1093/nar/gkab1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jain N., Zhao Z., Feucht J., Koche R., Iyer A., Dobrin A., Mansilla-Soto J., Yang J., Zhan Y., Lopez M., et al. TET2 guards against unchecked BATF3-induced CAR T cell expansion. Nature. 2023;615:315–322. doi: 10.1038/s41586-022-05692-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kurki M.I., Karjalainen J., Palta P., Sipilä T.P., Kristiansson K., Donner K.M., Reeve M.P., Laivuori H., Aavikko M., Kaunisto M.A., et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature. 2023;613:508–518. doi: 10.1038/s41586-022-05473-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ishigaki K., Sakaue S., Terao C., Luo Y., Sonehara K., Yamaguchi K., Amariuta T., Too C.L., Laufer V.A., Scott I.C., et al. Multi-ancestry genome-wide association analyses identify novel genetic mechanisms in rheumatoid arthritis. Nat. Genet. 2022;54:1640–1651. doi: 10.1038/s41588-022-01213-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bulik-Sullivan B.K., Loh P.-R., Finucane H.K., Ripke S., Yang J., Schizophrenia Working Group of the Psychiatric Genomics Consortium. Patterson N., Daly M.J., Price A.L., Neale B.M. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 2015;47:291–295. doi: 10.1038/ng.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mitra A., Raychaudhuri S.K., Raychaudhuri S.P. IL-22 induced cell proliferation is regulated by PI3K/Akt/mTOR signaling cascade. Cytokine. 2012;60:38–42. doi: 10.1016/j.cyto.2012.06.316. [DOI] [PubMed] [Google Scholar]
- 54.Bycroft C., Freeman C., Petkova D., Band G., Elliott L.T., Sharp K., Motyer A., Vukcevic D., Delaneau O., O’Connell J., et al. Genome-wide genetic data on ∼500,000 UK Biobank participants. bioRxiv. 2017:166298. doi: 10.1101/166298. Preprint at. [DOI] [Google Scholar]
- 55.Zhou W., Nielsen J.B., Fritsche L.G., Dey R., Gabrielsen M.E., Wolford B.N., LeFaive J., VandeHaar P., Gagliano S.A., Gifford A., et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet. 2018;50:1335–1341. doi: 10.1038/s41588-018-0184-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nagai A., Hirata M., Kamatani Y., Muto K., Matsuda K., Kiyohara Y., Ninomiya T., Tamakoshi A., Yamagata Z., Mushiroda T., et al. Overview of the BioBank Japan Project: Study design and profile. J. Epidemiol. 2017;27:S2–S8. doi: 10.1016/j.je.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Taliun D., Harris D.N., Kessler M.D., Carlson J., Szpiech Z.A., Torres R., Taliun S.A.G., Corvelo A., Gogarten S.M., Kang H.M., et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature. 2021;590:290–299. doi: 10.1038/s41586-021-03205-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shirai Y., Nakanishi Y., Suzuki A., Konaka H., Nishikawa R., Sonehara K., Namba S., Tanaka H., Masuda T., Yaga M., et al. Multi-trait and cross-population genome-wide association studies across autoimmune and allergic diseases identify shared and distinct genetic component. Ann. Rheum. Dis. 2022;81:1301–1312. doi: 10.1136/annrheumdis-2022-222460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Javierre B.M., Burren O.S., Wilder S.P., Kreuzhuber R., Hill S.M., Sewitz S., Cairns J., Wingett S.W., Várnai C., Thiecke M.J., et al. Lineage-Specific Genome Architecture Links Enhancers and Non-coding Disease Variants to Target Gene Promoters. Cell. 2016;167:1369–1384.e19. doi: 10.1016/j.cell.2016.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boyle A.P., Hong E.L., Hariharan M., Cheng Y., Schaub M.A., Kasowski M., Karczewski K.J., Park J., Hitz B.C., Weng S., et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–1797. doi: 10.1101/gr.137323.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carvalho-Silva D., Pierleoni A., Pignatelli M., Ong C., Fumis L., Karamanis N., Carmona M., Faulconbridge A., Hercules A., McAuley E., et al. Open Targets Platform: new developments and updates two years on. Nucleic Acids Res. 2019;47:D1056–D1065. doi: 10.1093/nar/gky1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Benucci M., Bernardini P., Coccia C., De Luca R., Levani J., Economou A., Damiani A., Russo E., Amedei A., Guiducci S., et al. JAK inhibitors and autoimmune rheumatic diseases. Autoimmun. Rev. 2023;22 doi: 10.1016/j.autrev.2023.103276. [DOI] [PubMed] [Google Scholar]
- 63.Kotyla P., Gumkowska-Sroka O., Wnuk B., Kotyla K. Jak Inhibitors for Treatment of Autoimmune Diseases: Lessons from Systemic Sclerosis and Systemic Lupus Erythematosus. Pharmaceuticals. 2022;15 doi: 10.3390/ph15080936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. j. 2011;17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 65.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 67.Wang L., Wang S., Li W. RSeQC: quality control of RNA-seq experiments. Bioinformatics. 2012;28:2184–2185. doi: 10.1093/bioinformatics/bts356. [DOI] [PubMed] [Google Scholar]
- 68.Ewels P., Magnusson M., Lundin S., Käller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32:3047–3048. doi: 10.1093/bioinformatics/btw354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ulgen E., Ozisik O., Sezerman O.U. pathfindR: An R Package for Comprehensive Identification of Enriched Pathways in Omics Data Through Active Subnetworks. Front. Genet. 2019;10:858. doi: 10.3389/fgene.2019.00858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dey K.K., Gazal S., van de Geijn B., Kim S.S., Nasser J., Engreitz J.M., Price A.L. SNP-to-gene linking strategies reveal contributions of enhancer-related and candidate master-regulator genes to autoimmune disease. Cell Genomics. 2022;2:100145. doi: 10.1016/j.xgen.2022.100145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.The ENCODE Project Consortium An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liao W., Lin J.-X., Wang L., Li P., Leonard W.J. Modulation of cytokine receptors by IL-2 broadly regulates differentiation into helper T cell lineages. Nat. Immunol. 2011;12:551–559. doi: 10.1038/ni.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Bulk RNA-seq data have been deposited at GEO: GSE271788 and GSE171737 and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Genotyping data and cell counts are available in Tables S1, S2, S3, S4, S5, S6, S7, S8, S9, S10, S11, and S12.
-
•
All original code has been deposited at GitHub and is publicly available as of the date of publication. Code used to generate the main figures can be found here: 10.5281/zenodo.12807946. LLCB code is available at 10.5281/zenodo.12807979 and https://github.com/weinstockj/LLCB.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.