

Abstract
Site-selective functionalizations of C–H bonds are often achieved with a directing group that leads to five- or six-membered metallacyclic intermediates. Analogous reactions that occur through four-membered metallacycles are rare. We report a challenging palladium-catalyzed oxidation of primary C–H bonds β to nitrogen in an imine of an aliphatic amine, a process that occurs through a four-membered palladacyclc intermediate. The success of the reaction relies on the identification, by H/D exchange, of a simple directing group (salicylaldehyde) capable of inducing the formation of this small ring. To gain insight into the steps of the catalytic cycle of this unusual oxidation reaction, a series of mechanistic experiments and density functional theory (DFT) calculations were conducted. The experimental studies showed that cleavage of the C–H bond is rate-limiting and formation of the strained four-membered palladacycle is thermodynamically uphill. DFT calculations corroborated these conclusions and suggested that the presence of an intramolecular hydrogen bond between the oxygen of the directing group and hydroxyl group of the ligating acetic acid is crucial for stabilization of the palladacyclic intermediate.
Graphical Abstract:
INTRODUCTION
Aliphatic amines are important organic compounds that are widely found in natural products, pharmaceuticals, and agrochemicals.1 Therefore, the development of synthetic methods that enable efficient synthesis or functionalization of aliphatic amines has been a long-standing goal.2 The functionalization of C(sp3)–H bonds in alkylamines3 and their derivatives4,3h,5 by transition-metal complexes has been pursued because it can transform ubiquitous inert C(sp3)–H bonds of these molecules to valuable functional groups that are usually inaccessible or difficult to introduce by traditional methods.
Cyclometalation is a crucial step in the functionalization of C–H bonds at specific sites by transition-metal catalysts.6 A wide range of directing groups have been developed to promote cyclometalation to form stable five- or six-membered metallacyclic intermediates (Scheme 1a, top left). Such cyclometalation with alkylamines and their derivatives has led to the functionalization at C–H bonds γ- or δ- to the nitrogen atom.6c In contrast, reactions that occur by C–H bond activation to form four-membered metallacycles are rare because formation of a strained-ring intermediate is kinetically and thermodynamically less favorable (Scheme 1a, top right). Thus, functionalization of an alkylamine derivative at the C–H bond β to nitrogen is challenging,3a,c–g,7,5l,8 and the corresponding four-membered metallacycles are only known in specific cases.3a,c For example, Gaunt and co-workers reported a seminal palladium-catalyzed, β-C(sp3)–H activation of alkylamines directed by the amine nitrogen to form strained aziridine derivatives; however, the amines were limited to sterically hindered, secondary amines (Scheme 1a, bottom). 3a
Scheme 1.

Transition-Metal-Catalyzed C–H Functionalization of Aliphatic Amines
To achieve the functionalization of β-C(sp3)–H bonds in alkylamine derivatives, other more circuitous pathways have been developed, but none of these involve four-membered metallacyclic intermediates.5l,8,9 For example, Dong and co-workers reported palladium-catalyzed acetoxylations of β-C(sp3)–H bonds of alkylamines to form 1,2-amino alcohol derivatives by converting the alkylamines to hydrazone derivatives, but this method is limited to secondary carbinamines, requires three steps to install the directing group on the nitrogen of the amine, and requires two steps to remove the directing group after the C–H acetoxylation (Scheme 1b).5l Our group recently developed an iridium-catalyzed intramolecular silylation of the β-C(sp3)–H bonds of secondary amines.8 In combination with oxidation of the C–Si bond, this reaction creates an alternative approach to form 1,2-amino alcohols (Scheme 1b), but preparation of the hydrosilane was required and the functional group tolerance of the synthesis of this silane with LiAlH4 would limit the application of this process. Thus, functionalizations that occur through four-membered metallacycles and direct oxidations of the β-C(sp3)–H bonds of alkylamine derivatives are needed.
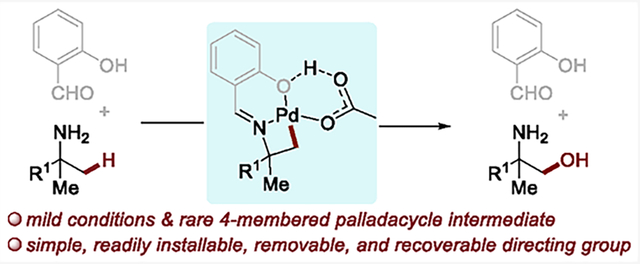
Here, we report an imine-directed acetoxylation of the β-C(sp3)–H bonds of tertiary carbinamines under mild reaction conditions by the formation of a four-membered palladacyclic intermediate (Scheme 1c). The appropriate imine (from salicylaldehyde)5b,g was revealed by initial studies on H/D exchange, and this directing group is inexpensive, readily installed, removed and recovered. Mechanistic studies show C–H bond cleavage is rate-limiting, and formation of the strained four-membered pallacacycle is thermodynamically uphill.
RESULTS AND DISCUSSION
Preliminary Studies on the Undirected C–H Oxidation of tert-Butyl Amine.
tert-Butyl amine, the simplest tertiary carbinamine, was selected as the model substrate to investigate the β-C(sp3)–H oxidation of primary alkylamines because of the utility of the corresponding oxidation product, 2-amino-2-methylpropanol (AMP).10 Preliminary investigations focused on reported conditions for platinum- and palladium-catalyzed C(sp3)–H oxidation of free amines,11,12 but no product derived from C–H bond functionalization was observed (eq 1). The lack of reactivity of tert-butyl amine in
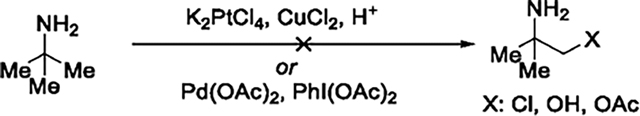 |
(1) |
the presence of the platinum catalyst in acid could be explained by the combination of steric hindrance and electron-poor property of the C–H bond of the protonated amine.11 The lack of reactivity in the presence of the palladium catalyst under more neutral conditions can be attributed to coordination of tert-butyl amine, leading to deactivation of the catalyst.7
Identification of a Suitable Directing Group.
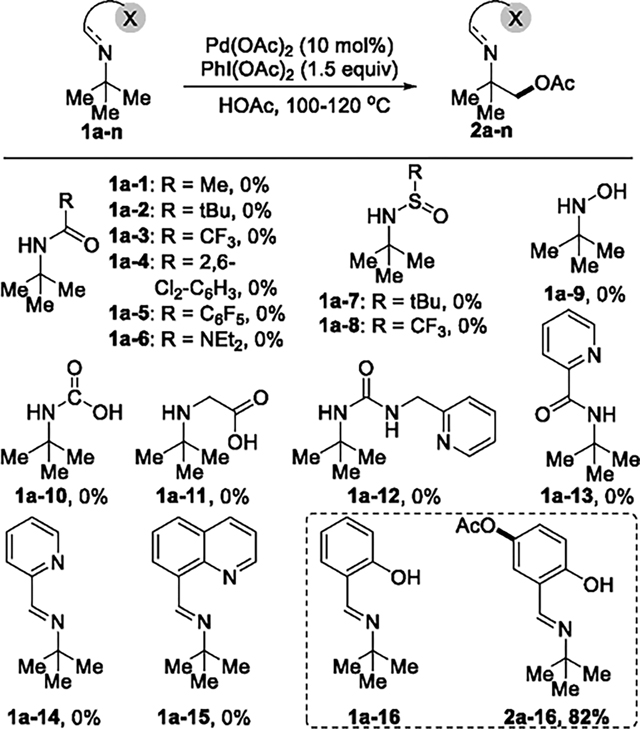
Further studies involving directing groups6c on nitrogen led to functionalization of the β-C–H bond, but obtaining good yields of the oxidized product required substantial investigation. A series of auxiliary groups were installed on the nitrogen of tert-butyl amine to form the corresponding amine derivatives 1a-1 to 1a-16 (Table 1). Under commonly employed conditions for the acetoxylation of C(sp3)–H bonds (10 mol % Pd(OAc)2, 1.2 equiv of PhI(OAc)2, in AcOH at 100–120 °C),4b,c,13 none of the reactions of 1a-1 to 1a-15 gave any detectable product from acetoxylation; the reaction with 1a-16 gave the product from acetoxylation at the C(sp2)–H bond para to the hydroxyl group of the imine to form 2a-16 in 82% yield without evidence for reaction at the C(sp3)–H bond.
Table 1.
Pd-Catalyzed C−H Acetoxylation of Amine Derivatives
|
We considered two possible origins of the lack of reactivity of amine derivatives 1a-1 to 1a-15 and the lack of reactivity of 1a-16 at the alkyl C–H bond based on the typical mechanism for palladium-catalyzed directed C–H oxidation (Figure 1). First, cleavage of the C(sp3)–H bond might not occur because the barrier to formation of a four-membered palladacycle B is too high. Second, C(sp3)–H cleavage could occur, but the transition-state energy for subsequent oxidation of the Pd(II) intermediate B to the Pd(IV) intermediate C is too high or formation of the palladacycle C is too endothermic.
Figure 1.

Proposed catalytic cycle for the β-C–H functionalization
To distinguish between these possibilities, a set of H/D-exchange experiments was conducted in the absence of oxidant (Scheme 2). The combination of tert-butyl amine derivatives 1a-1 to 1a-16, Pd(OAc)2 (10 mol %), and AcOD (2.0 equiv) was heated in CHCl3 at 80 °C for 12 h. No detectable incorporation of deuterium into 1a-1 to 1a-15 was observed by 2H NMR spectroscopy, showing that cleavage of the C–H bond in these substrates does not occur. In contrast, H/D exchange with 1a-16 occurred at the C(sp3)–H bonds; 9.3% of the hydrogen atoms in the tert-butyl group of 1a-16 were deuterated, indicating that the C(sp3)–H bond activation occurred with the imine derived from salicaldehyde. These data indicate that cleavage of the C(sp3)–H bond in 1a-16 occurs under more neutral conditions without activation of the aryl C–H bond that underwent oxidation in neat acetic acid. Yu recently investigated salicylaldehyde and its analogues systematically as a transient directing group for the arylation of the γ-C–H bonds in alkylamines.5g,k However, the functionalization of the β-C–H bond in alkylamines with these types of directing groups or others (e.g., picolinic acid, 8-quinoline-carbaldehyde, etc.) has not been observed.
Scheme 2.

H/D Exchange Experiments
Development of the β-C(sp3)–H Bond Acetoxylation.
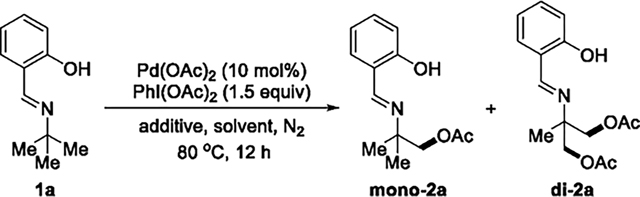
Having shown that the C(sp3)–H bond of the salicylaldimine 1a-16 (hereafter abbreviated as 1a) undergoes cleavage, we considered that solvent and acidity could influence the selectivity for functionalization of the sp2 versus sp3 C–H bonds (compare results in Table 1 and Scheme 2) and enable oxidation of the Pd–C bond of palladacycle B. Indeed, in the absence of acetic acid in nonpolar solvents, the product from acetoxylation of the C(sp3)–H bond was obtained (Table 2). Among the reactions conducted in nonpolar solvents, the reaction in benzene gave a 4:1 mixture of acetoxylated products mono2a and di2a in a combined 51% yield (entry 3). Reactions conducted with a series of added bases showed that weak bases had little effect on the yield (entries 3, 11–14). Reactions with some of the ligands14 currently used for palladium-catalyzed oxidation showed that the reaction with N-(tert-butoxycarbonyl)-l-alanine (N-Boc-Ala) occurred in a slightly higher 58% yield than that without ligand (51%) (entry 15; see the Supporting Information for more details). In this reaction 10–20% of the reactant remained, leading to a mass balance of 70–80%; no side product formed in greater than 5% yield.
Table 2.
Evaluation of the Conditions for the Acetoxylation of Compound 1aa
| ||||
|---|---|---|---|---|
| entry | additive | solvent | 2a% | mono:di |
| 1 | NaOAc (1.5 equiv) | CHCl3 | 48% | 2.2:1 |
| 2 | NaOAc (1.5 equiv) | m-xylene | 34% | 8:1 |
| 3 | NaOAc (1.5 equiv) | benzene | 51% | 4:1 |
| 4 | NaOAc (1.5 equiv) | toluene | 34% | 10:1 |
| 5 | NaOAc (1.5 equiv) | THF | 7% | >20:1 |
| 6 | NaOAc (1.5 equiv) | dioxane | 14% | >20:1 |
| 7 | NaOAc (1.5 equiv) | EA | 28% | 8:1 |
| 8 | NaOAc (1.5 equiv) | MeCN | 25% | 7:1 |
| 9 | NaOAc (1.5 equiv) | DMF | 4% | >20:1 |
| 10 | NaOAc (1.5 equiv) | DMPU | 0% | |
| 11 | Na2HPO4 (1.5 equiv) | benzene | 48% | 3.8:1 |
| 12 | KH2PO4 (1.5 equiv) | benzene | 49% | 4:1 |
| 13 | PhSO3Na (1.5 equiv) | benzene | 51% | 4:1 |
| 14 | — | benzene | 48% | 4:1 |
| 15 | N-Boc-Ala (10 mol %) | benzene | 58% | 4:1 |
Conditions: Reaction was conducted with 1a (0.1 mmol), Pd(OAc)2 (10 mmol %), and Phl(OAc)2 (1.5 equiv) in solvent (1.0 mL) at 80 °C under N2; the yields and ratio of mono2a vs di2a were determined by gas chromatography (GC) with dodacane as the internal standard.
Abbreviations: ethyl acetate, EA; dimethylformamide, DMF; N,N’-dimethylpropyleneurea, DMPU.
Scope of the β-C(sp3)–H Bond Acetoxylation.
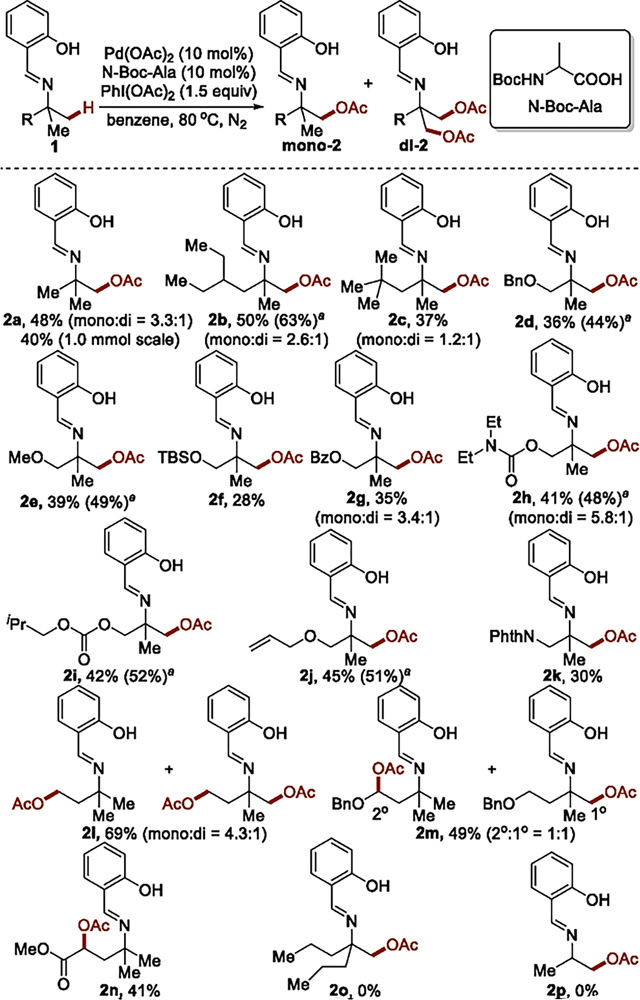
The scope of the acetoxylation of amines under the developed conditions via imine 1a is shown in Table 3. Reactions of imines from tertiary carbinamines containing a range of substitution patterns and functional groups on the amine afforded isolated products from acetoxylation of the β-C(sp3)–H bonds in moderate yields. In most cases, conversion of the starting substrate was greater than 80%; the mass balance consisted of multiple products from side reactions. The acetoxylation of the β-C(sp3)–H bond of imines 1a–1c bearing a series of alkyl substituents afforded the corresponding products (2a–2c). The ratio of monoacetoxylation product to diacetoxylation product was higher for the examples containing a smaller alkyl substituent. The acetoxylation also occurred in the presence of a variety of functional groups (1d–1k), including benzyl and alkyl ethers (1d and 1e), a silyl ether (1f), an ester (1g), a carbamate (1h), a carbonate (1i), an alkene (1j), and an amide (1k). The yield from acetoxylation on a larger scale was similar to that on smaller scale (Table 3, substrate 1a).
Table 3.
Scope of the β-Acetoxylation of Amine Derivativesa
|
The yields refer to isolated yields, and the yields in parentheses refer to yields based on recovered starting material.
The regioselectivity and yield for the acetoxylation depended on the architecture of the alkyl group. Acetoxylation of 1l, which contains both γ- and β-methyl C–H bonds, occurred predominantly at the γ-methyl C–H bonds through a more favorable five-membered palladacycle intermediate. The site of acetoxlylation of 1m and 1n, which contain both secondary γ-C(sp3)–H bonds and unactivated primary β-C(sp3)–H bonds, depended on the nature of the substituents on the secondary carbon. Reaction of 1m containing a benzyloxy substituent at the γ methylene position gave a 1:1 mixture of products from reaction at the primary β-C(sp3)–H bond and secondary γ-C(sp3)–H bond, while reaction with substrate 1n containing an alkoxycarbonyl group at the γ-methylene position gave the product from acetoxylation at the acidic secondary γ-C(sp3)–H bond exclusively.15 Imine 1o containing a single β-methyl group and imine 1p derived from a secondary, rather than tertiary, carbinamine did not react.
Installation, Removal, and Recovery of the Directing Group.
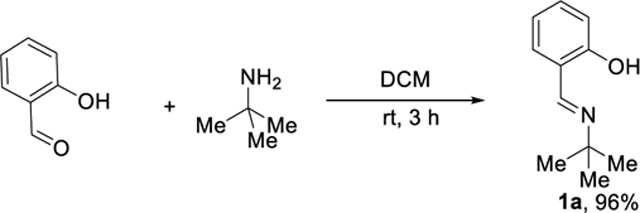
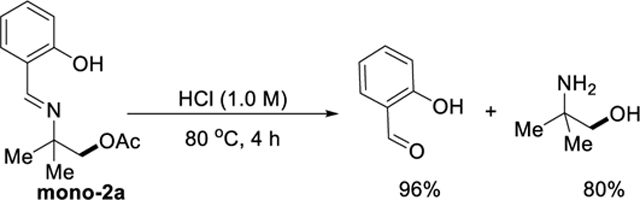
Installation and removal of the directing group are efficient and operationally simple, and the directing group is recoverable. The imine 1a was obtained almost quantitatively (96%) by condensation of tert-butyl amine with salicylaldehyde in DCM at room temperature (eq 2). To demonstrate the ability to remove and recover the directing group, mono2a was subjected to a 1.0 M aqueous solution of HCl at 80 °C (eq 3). The salicylaldehyde was removed and recovered in 96% yield, based on the amount of mono2a. After basification of the aqueous solution, followed by extraction by organic solvent (DCM), the free amino alcohol was obtained in 80% yield.
Mechanistic Investigation.
To determine whether cleavage of the C–H bond or oxidation of the Pd–C bond is rate-limiting and to gain insight into the thermodynamics for
 |
(2) |
 |
(3) |
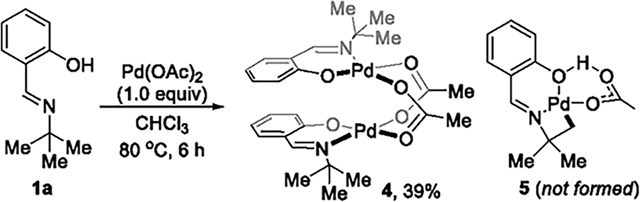
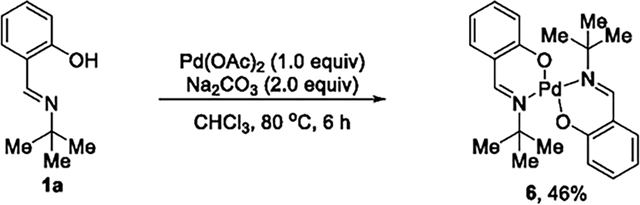
formation of the four-membered palladacycle, we conducted synthetic organometallic studies of cyclopalladated species. Reaction of imine 1a with 1.0 equiv of Pd(OAc)2 at 80 °C formed a dinuclear bis-μ-acetatopalladium complex 4 in 39% yield. No cyclopalladated complex 5 formed (eq 4). The same
 |
(4) |
 |
(5) |
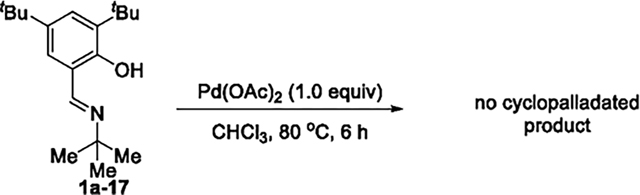 |
(6) |
reaction in the presence of base (e.g., Na2CO3) to promote the C–H bond cleavage formed the palladium complex 6 containing two deprotonated salicaldimines, but, again, no cyclopalladated species formed (eq 5). Stoichiometric reactions were also conducted with 1a-17 that contains a bulky tert-butyl substituent at the ortho position of the hydroxyl group on the aryl group, but the cyclopalladated species was again absent (eq 6). To potentially stabilize the four-membered palladium intermediate, external ligands, such as triphenyl phosphine and pyridine, were added to the reaction of 1a with Pd(OAc)2, but no palladacycle formed from cleavage of the C(sp3)–H bond. Consistent with these synthetic studies, no palladacyclic intermediate was observed during the catalytic reactions (eqs 4–6), as judged by analysis of the crude reaction mixture by 1H NMR spectroscopy. This set of mechanistic experiments suggests that the formation of the four-membered palladacycle is unfavorable and could lie uphill from the salicylaldimine complex 4.
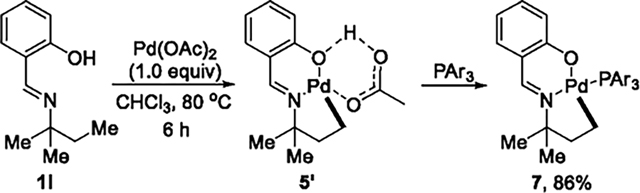
To test if a salicylaldimine-ligated palladium is capable of forming a metallacycle by cleavage of a C(sp3)–H bond, we allowed Pd(OAc)2 to react with substrate 1l that could form a five-membered palladacycle under the same conditions as the reaction with 1a. Indeed, the five-membered palladium complex 5′ formed in quantitative yield, as determined by 1H NMR spectroscopy (eq 7). Although complex 5′ is stable in solution, it decomposed during attempts to purify it by various methods. Thus, an external ligand (3,5-bis(trifluoromethyl)-
 |
(7) |
phenyl)phosphine was added to the crude reaction mixture of 5′ to form a more stable complex 7, which was isolated by silica gel column chromatography in 86% yield. The structures of palladium complexes 4, 6, and 7 were confirmed by X-ray crystallography (Figure 2).
Figure 2.

Solid-state structures of 4, 5, and 7 determined by single crystal X-ray diffraction.
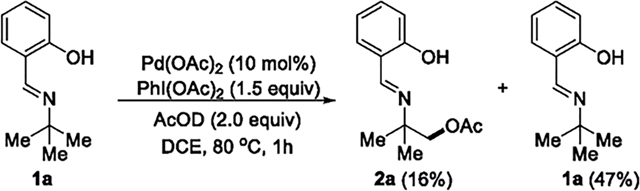
To probe whether the β-C(sp3)–H bond was cleaved reversibly under the conditions of the acetoxylation, we ran the catalytic reaction of 1a in the presence of 2.0 equiv of AcOD and 1.5 equiv of PhI(OAc)2 to partial conversion. After 1 h at 80 °C (eq 8), 1H NMR analysis of the crude reaction mixture
 |
(8) |
showed that 47% of the starting material 1a remained (16% of the acetoxylated product 2a formed), and no deuterium had been incorporated into the unreacted 1a, as determined by 2H NMR spectroscopy. These data indicate that the C–H cleavage is irreversible and, therefore, that oxidation of the metallacyclic intermediate is more rapid than reversion to the acyclic precursor.
To gauge the rate of the oxidation of a palladacycle derived from the salicylaldimines, we studied the oxidation of the five-membered metallacycle 5′ generated in solution (eq 9). The
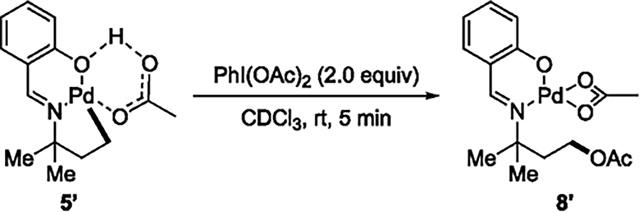 |
(9) |
reaction of 5′ with 2 equiv of PhI(OAc)2 occurred to completion at room temperature in less than 5 min to form the C-acetoxylated complex in 46–50% yield. These data are consistent with rapid oxidation of the four-membered palladacycle at 80 °C that is required for irreversible formation of the metallacycle.
Density Functional Theory (DFT) Calculations of the Catalytic Acetoxylation.
To gain insight into the thermodynamics for formation of a four-membered metallacycle, we performed DFT calculations on the catalytic acetoxylation process. These data are summarized in Figure 3. We determined the barrier and thermodynamics for cleavage of the β-C–H bond of the bound salicaldimine by a concerted metalation–deprotonation process involving the coordinated acetate in the monomeric complexes 9 and 9′ derived from the isolated dimeric complexes 4 and 4′. Quasi-harmonic Gibbs free energies were calculated of the observed dimeric salicaldimine palladium complexes 4 and 4′, the monomeric 9 and 9′ that would undergo C–H bond cleavage, the transition states for C–H activation by 9 and 9′ (TS(9–10) and TS(9′–10′)) to form palladacycles 10 and 10′ containing a bound acetic acid, and palladacycles 5 and 5′, which are isomers of 10 and 10′, in which the carboxylic acid is engaged in an intramolecular hydrogen bond. These calculations showed that formation of the four-membered metallacycle is endergonic but formation of the five-membered metallacycle is exergonic.
Figure 3.

Quasi-harmonic Gibbs free energy (T = 353.15 K) profile of Pd-catalyzed α-acetoxylation of primary amines. Calculations were performed at the B3LYP-d3/LANL2DZ+6–31G(d)/SMD(benzene)//M06-d3/SDD+6–311+G(d,p)/SMD(benzene) level of theory.
The free energy barriers for C–H activation within complexes 9 and 9′ to form the four-membered and five-membered palladacycles 10 and 10′ are 22.5 and 17.9 kcal/mol, respectively. The computed thermodynamics for the formation of the metallacycle depended strongly on the presence or absence of an intramolecular hydrogen bond involving the coordinated acetic acid. The computed energy of the four-membered palladacycle 5 containing the hydrogen bond is lower in energy than that of palladacycle 10 lacking the hydrogen bond by 13.8 kcal/mol. The computed difference in energy between the analogous structures 5′ and 10′ containing the hydrogen bond is 14.2 kcal/mol. Thus, the formation of the four-membered palladacycle 5 is computed to be thermodynamically uphill of the monomeric salicaldimine complex 9 by 1.3 kcal/mol and uphill of the isolated dimeric analogue 4 by 10.6 kcal/mol, whereas formation of the five-membered pallacycle 5′ is computed to be downhill of monomeric 9′ by −8.8 kcal/mol and nearly thermoneutral with the dimeric complex 4’. These results corroborate our experimental observation of the formation of five-membered palladacycle 5′ from 4′ but our detection of the formation of the four-membered ring palladacycle 5 only indirectly by H/D exchange experiments in the absence of oxidant (see Scheme 2).
DFT calculations on the remainder of the reaction pathway are included in Figure 3. Oxidation of both palladacycles 5 and 5′ by PhI(OAc)2 was computed to be thermodynamically downhill by ~20 kcal/mol. The resulting Pd(IV) intermediates 11 and 11′ are computed to undergo nearly thermoneutral dissociation of HOAc to form complexes 12 and 12′, which subsequently undergo reductive elimination to form acetoxylated products 8 and 8′ in which the functionalized salicaldimine remains bound to palladium.16 The free energy barriers for reductive elimination from intermediates 12 and 12′ were calculated to be 12.2 and 15.4 kcal/mol, respectively. A comparison of these barriers to the 22.5 and 17.9 kcal/mol barriers for C–H bond cleavage imply that C–H activation is irreversible and turnover-limiting for the catalytic acetoxylation process. This conclusion is consistent with the lack of incorporation of deuterium into the reactant under the conditions of the catalytic process.
CONCLUSION
In conclusion, we developed a palladium-catalyzed, directed β-C(sp3)–H acetoxylation of tertiary carbinamines, which likely proceeds through a rare four-membered palladacycle intermediate. Identification of the appropriate group on the nitrogen of the amine to enable C–H activation resulted from studies on the H/D exchange. The directing group is inexpensive and readily installed, removed, and recovered. Mechanistic studies revealed that the C(sp3)–H bond is cleaved irreversibly and is the rate-determining step of the reaction. DFT calculations were consistent with experimental results in which the four-membered palladacycle was not observed during the reaction but five-membered palladacycles formed readily. The DFT studies suggest that formation of the four-membered palladacycle is thermodynamically uphill, but the formation of the corresponding five-membered palladacycle is downhill. The combination of directing group on nitrogen to facilitate formation of a four-membered metallacycle and oxidant that reacts rapidly with the unstable metallacycle enables this functionalization of the C–H bond of an amine to occur, and these principles should facilitate the discovery of additional reactions through such strained intermediates.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by BASF through the California Research Alliance (CARA) and Computation Facility at UC Berkeley for DFT calculations. We gratefully acknowledge Dr. Hasan Celik for assistance with NMR experiments and Dr. Nicholas Settineri for X-ray crystallography. Instruments in the CoC-NMR are supported in part by NIH S10OD024998. A.B. thanks the Swiss National Science Foundation for a postdoctoral fellowship. B.S. thanks Dr. Yumeng Xi for helpful discussion.
Footnotes
The authors declare the following competing financial interest(s): A patent has been filed on this process.
ASSOCIATED CONTENT
SI Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c01629.
Experimental procedures, characterization of new compounds, X-ray data, and computational details (PDF)
X-ray crystallographic data for compounds 4 (CIF)
X-ray crystallographic data for compounds 7 (CIF)
X-ray crystallographic data for compounds 6 (CIF)
Contributor Information
Bo Su, Department of Chemistry, University of California, Berkeley, Berkeley, California 94720, United States.
Ala Bunescu, Department of Chemistry, University of California, Berkeley, Berkeley, California 94720, United States.
Yehao Qiu, Department of Chemistry, University of California, Berkeley, Berkeley, California 94720, United States.
Stephan J. Zuend, BASF Corporation, Fremont, California 94538, United States
Martin Ernst, BASF SE, 67056 Ludwigshafen, Germany.
John F. Hartwig, Department of Chemistry, University of California, Berkeley, Berkeley, California 94720, United States
REFERENCES
- (1).Kittakoop P; Mahidol C; Ruchirawat S Alkaloids as Important Scaffolds in Therapeutic Drugs for the Treatments of Cancer, Tuberculosis, and Smoking Cessation. Curr. Top. Med. Chem. 2013, 14, 239. [DOI] [PubMed] [Google Scholar]
- (2).(a) Hager A; Vrielink N; Hager D; Lefranc J; Trauner D Synthetic approaches towards alkaloids bearing α-tertiary amines. Nat. Prod. Rep. 2016, 33, 491. [DOI] [PubMed] [Google Scholar]; (b) Salvatore RN; Yoon CH; Jung KW Synthesis of secondary amines. Tetrahedron 2001, 57, 7785. [Google Scholar]
- (3).(a) McNally A; Haffemayer B; Collins BSL; Gaunt MJ Palladium-catalysed C-H activation of aliphatic amines to give strained nitrogen heterocycles. Nature 2014, 510, 129. [DOI] [PubMed] [Google Scholar]; (b) Calleja J; Pla D; Gorman TW; Domingo V; Haffemayer B; Gaunt MJ A steric tethering approach enables palladium-catalysed C–H activation of primary amino alcohols. Nat. Chem. 2015, 7, 1009. [DOI] [PubMed] [Google Scholar]; (c) He C; Gaunt MJ Ligand-Enabled Catalytic C-H Arylation of Aliphatic Amines by a Four-Membered-Ring Cyclopalladation Pathway. Angew. Chem., Int. Ed. 2015, 54, 15840. [DOI] [PubMed] [Google Scholar]; (d) Willcox D; Chappell BGN; Hogg KF; Calleja J; Smalley AP; Gaunt MJ A general catalytic β-C–H carbonylation of aliphatic amines to β-lactams. Science 2016, 354, 851. [DOI] [PubMed] [Google Scholar]; (e) Zakrzewski J; Smalley AP; Kabeshov MA; Gaunt MJ; Lapkin AA Continuous-Flow Synthesis and Derivatization of Aziridines through Palladium-Catalyzed C(sp3)-H Activation. Angew. Chem., Int. Ed. 2016, 55, 8878. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Cabrera-Pardo JR; Trowbridge A; Nappi M; Ozaki K; Gaunt MJ Selective Palladium(II)-Catalyzed Carbonylation of Methylene β-C-H Bonds in Aliphatic Amines. Angew. Chem., Int. Ed. 2017, 56, 11958. [DOI] [PubMed] [Google Scholar]; (g) Smalley AP; Cuthbertson JD; Gaunt MJ Palladium-Catalyzed Enantioselective C–H Activation of Aliphatic Amines Using Chiral Anionic BINOL-Phosphoric Acid Ligands. J. Am. Chem. Soc. 2017, 139, 1412. [DOI] [PubMed] [Google Scholar]; (h) He C; Whitehurst WG; Gaunt MJ Palladium-Catalyzed C(sp3)–H Bond Functionalization of Aliphatic Amines. Chem. 2019, 5, 1031. [Google Scholar]
- (4).(a) Wang D-H; Hao X-S; Wu D-F; Yu J-Q Palladium-Catalyzed Oxidation of Boc-Protected N-Methylamines with IOAc as the Oxidant: A Boc-Directed sp3 C-H Bond Activation. Org. Lett. 2006, 8, 3387. [DOI] [PubMed] [Google Scholar]; (b) Neufeldt SR; Sanford MS O-Acetyl Oximes as Transformable Directing Groups for Pd-Catalyzed C-H Bond Functionalization. Org. Lett. 2010, 12, 532. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Desai LV; Hull KL; Sanford MS Palladium-Catalyzed Oxygenation of Unactivated sp3 C-H Bonds. J. Am. Chem. Soc. 2004, 126, 9542. [DOI] [PubMed] [Google Scholar]; (d) Zaitsev VG; Shabashov D; Daugulis O Highly Regioselective Arylation of sp3 C-H Bonds Catalyzed by Palladium Acetate. J. Am. Chem. Soc. 2005, 127, 13154. [DOI] [PubMed] [Google Scholar]; (e) He G; Chen G A Practical Strategy for the Structural Diversification of Aliphatic Scaffolds through the Palladium-Catalyzed Picolinamide-Directed Remote Functionalization of Unactivated C(sp3)-H Bonds. Angew. Chem., Int. Ed. 2011, 50, 5192. [DOI] [PubMed] [Google Scholar]; (f) He G; Zhao Y; Zhang S; Lu C; Chen G Highly Efficient Syntheses of Azetidines, Pyrrolidines, and Indolines via Palladium Catalyzed Intramolecular Amination of C(sp3)–H and C(sp2)–H Bonds at γ and δ Positions. J. Am. Chem. Soc. 2012, 134, 3. [DOI] [PubMed] [Google Scholar]; (g) Nadres ET; Daugulis O Heterocycle Synthesis via Direct C-H/N-H Coupling. J. Am. Chem. Soc. 2012, 134, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Zhang S-Y; He G; Zhao Y; Wright K; Nack WA; Chen G Efficient Alkyl Ether Synthesis via Palladium-Catalyzed, Picolinamide-Directed Alkoxylation of Unactivated C(sp3)–H and C(sp2)–H Bonds at Remote Positions. J. Am. Chem. Soc. 2012, 134, 7313. [DOI] [PubMed] [Google Scholar]; (i) Zhang S-Y; He G; Nack WA; Zhao Y; Li Q; Chen G Palladium-Catalyzed Picolinamide-Directed Alkylation of Unactivated C(sp3)–H Bonds with Alkyl Iodides. J. Am. Chem. Soc. 2013, 135, 2124. [DOI] [PubMed] [Google Scholar]; (j) Fan M; Ma D Palladium-Catalyzed Direct Functionalization of 2-Aminobutanoic Acid Derivatives: Application of a Convenient and Versatile Auxiliary. Angew. Chem., Int. Ed. 2013, 52, 12152. [DOI] [PubMed] [Google Scholar]; (k) Ye X; He Z; Ahmed T; Weise K; Akhmedov NG; Petersen JL; Shi X 1,2,3-Triazoles as versatile directing group for selective sp2 and sp3 C–H activation: cyclization vs substitution. Chem. Sci. 2013, 4, 3712. [Google Scholar]; (l) Ling P-X; Fang S-L; Yin X-S; Chen K; Sun B-Z; Shi B-F Palladium-Catalyzed Arylation of Unactivated γ-Methylene C(sp3)-H and δ-C-H Bonds with an Oxazoline-Carboxylate Auxiliary. Chem. - Eur. J. 2015, 21, 17503. [DOI] [PubMed] [Google Scholar]; (m) Zhang L-S; Chen G; Wang X; Guo Q-Y; Zhang X-S; Pan F; Chen K; Shi Z-J Direct Borylation of Primary C-H Bonds in Functionalized Molecules by Palladium Catalysis. Angew. Chem., Int. Ed. 2014, 53, 3899. [DOI] [PubMed] [Google Scholar]; (n) Chan KSL; Wasa M; Chu L; Laforteza BN; Miura M; Yu J-Q Ligand-enabled cross-coupling of C(sp3)–H bonds with arylboron reagents via Pd(II)/Pd(0) catalysis. Nat. Chem. 2014, 6, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Topczewski JJ; Cabrera PJ; Saper NI; Sanford MS Palladium-catalysed transannular C–H functionalization of alicyclic amines. Nature 2016, 531, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen Y-Q; Wang Z; Wu Y; Wisniewski SR; Qiao JX; Ewing WR; Eastgate MD; Yu J-Q Overcoming the Limitations of γ- and δ-C–H Arylation of Amines through Ligand Development. J. Am. Chem. Soc. 2018, 140, 17884. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhan B-B; Li Y; Xu J-W; Nie X-L; Fan J; Jin L; Shi B-F Site-Selective δ-C(sp3)-H Alkylation of Amino Acids and Peptides with Maleimides via a Six-Membered Palladacycle. Angew. Chem., Int. Ed. 2018, 57, 5858. [DOI] [PubMed] [Google Scholar]; (d) Shao Q; Wu Q-F; He J; Yu J-Q Enantioselective γ-C(sp3)–H Activation of Alkyl Amines via Pd(II)/Pd(0) Catalysis. J. Am. Chem. Soc. 2018, 140, 5322. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Kapoor M; Liu D; Young MC Carbon Dioxide-Mediated C(sp3)–H Arylation of Amine Substrates. J. Am. Chem. Soc. 2018, 140, 6818. [DOI] [PubMed] [Google Scholar]; (f) Yang Q-L; Li Y-Q; Ma C; Fang P; Zhang X-J; Mei T-S Palladium-Catalyzed C(sp3)-H Oxygenation via Electrochemical Oxidation. J. Am. Chem. Soc. 2017, 139, 3293. [DOI] [PubMed] [Google Scholar]; (g) Yada A; Liao W; Sato Y; Murakami M Buttressing Salicylaldehydes: A Multipurpose Directing Group for C(sp3)-H Bond Activation. Angew. Chem., Int. Ed. 2017, 56, 1073. [DOI] [PubMed] [Google Scholar]; (h) Liu Y; Ge H Site-selective C–H arylation of primary aliphatic amines enabled by a catalytic transient directing group. Nat. Chem. 2017, 9, 26. [Google Scholar]; (i) Yang K; Li Q; Liu Y; Li G; Ge H Catalytic C–H Arylation of Aliphatic Aldehydes Enabled by a Transient Ligand. J. Am. Chem. Soc. 2016, 138, 12775. [DOI] [PubMed] [Google Scholar]; (j) Xu Y; Young MC; Wang C; Magness DM; Dong G Catalytic C(sp3)-H Arylation of Free Primary Amines with an exo Directing Group Generated In Situ. Angew. Chem., Int. Ed. 2016, 55, 9084. [DOI] [PubMed] [Google Scholar]; (k) Wu Y; Chen Y-Q; Liu T; Eastgate MD; Yu J-Q Pd-Catalyzed γ-C(sp3)–H Arylation of Free Amines Using a Transient Directing Group. J. Am. Chem. Soc. 2016, 138, 14554. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Huang Z; Wang C; Dong G A Hydrazone-Based exo-Directing-Group Strategy for β-C-H Oxidation of Aliphatic Amines. Angew. Chem., Int. Ed. 2016, 55, 5299. [DOI] [PubMed] [Google Scholar]; (m) Li B; Li X; Han B; Chen Z; Zhang X; He G; Chen G Construction of Natural-Product-Like Cyclophane-Braced Peptide Macrocycles via sp3 C–H Arylation. J. Am. Chem. Soc. 2019, 141, 9401. [DOI] [PubMed] [Google Scholar]; (n) Wang H; Tong H-R; He G; Chen G An Enantioselective Bidentate Auxiliary Directed Palladium-Catalyzed Benzylic C-H Arylation of Amines Using a BINOL Phosphate Ligand. Angew. Chem., Int. Ed. 2016, 55, 15387. [DOI] [PubMed] [Google Scholar]
- (6).(a) Kleiman JP; Dubeck M The Preparation of Cyclopentadienyl [o-(Phenylazo)Phenyl]Nickel. J. Am. Chem. Soc. 1963, 85, 1544. [Google Scholar]; (b) Dehand J; Pfeffer M Cyclometallated compounds. Coord. Chem. Rev. 1976, 18, 327. [Google Scholar]; (c) Sambiagio C; Schönbauer D; Blieck R; Dao-Huy T; Pototschnig G; Schaaf P; Wiesinger; Zia MF; Wencel-Delord J; Besset T; Maes BUW; Schnürch M A comprehensive overview of directing groups applied in metal-catalysed C–H functionalisation chemistry. Chem. Soc. Rev. 2018, 47, 6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Smalley AP; Gaunt MJ Mechanistic Insights into the Palladium-Catalyzed Aziridination of Aliphatic Amines by C–H Activation. J. Am. Chem. Soc. 2015, 137, 10632. [DOI] [PubMed] [Google Scholar]
- (8).Su B; Lee T; Hartwig JF Iridium-Catalyzed, β-Selective C(sp3)–H Silylation of Aliphatic Amines To Form Silapyrrolidines and 1,2-Amino Alcohols. J. Am. Chem. Soc. 2018, 140, 18032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Kim M; Mulcahy JV; Espino CG; Du Bois J Expanding the substrate scope for C-H amination reactions: Oxidative cyclization of urea and guanidine derivatives. Org. Lett. 2006, 8, 1073. [DOI] [PubMed] [Google Scholar]; (b) Olson DE; Du Bois J Catalytic C-H amination for the preparation of substituted 1,2-diamines. J. Am. Chem. Soc. 2008, 130, 11248. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Strambeanu II; White MC Catalyst-Controlled C-O versus C-N Allylic Functionalization of Terminal Olefins. J. Am. Chem. Soc. 2013, 135, 12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Calleja P; Ernst M; Hashmi ASK; Schaub T Ruthenium-Catalyzed Deaminative Hydrogenation of Amino Nitriles: Direct Access to 1,2-Amino Alcohols. Chem. - Eur. J. 2019, 25, 9498. [DOI] [PubMed] [Google Scholar]
- (11).Lee M; Sanford MS Platinum-Catalyzed, Terminal-Selective C(sp3)–H Oxidation of Aliphatic Amines. J. Am. Chem. Soc. 2015, 137, 12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Chen K; Wang D; Li Z-W; Liu Z; Pan F; Zhang Y-F; Shi Z-J Palladium catalyzed C(sp3)-H acetoxylation of aliphatic primary amines to r-amino alcohol derivatives. Org. Chem. Front. 2017, 4, 2097. [Google Scholar]
- (13).(a) Zhang J; Khaskin E; Anderson NP; Zavalij PY; Vedernikov AN Catalytic aerobic oxidation of substituted 8-methylquinolines in PdII-2,6-pyridinedicarboxylic acid systems. Chem. Commun. 2008, 3625. [DOI] [PubMed] [Google Scholar]; (b) Ren Z; Mo F; Dong G Catalytic Functionalization of Unactivated sp3 C–H Bonds via exo-Directing Groups: Synthesis of Chemically Differentiated 1,2-Diols. J. Am. Chem. Soc. 2012, 134, 16991. [DOI] [PubMed] [Google Scholar]; (c) Rit RK; Yadav MR; Sahoo AK Pd(II)-Catalyzed Primary-C(sp3)–H Acyloxylation at Room Temperature. Org. Lett. 2012, 14, 3724. [DOI] [PubMed] [Google Scholar]; (d) Li Q; Zhang SY; He G; Nack WA; Chen G Palladium-Catalyzed Picolinamide-Directed Acetoxylation of Unactivated γ-C(sp3)-H Bonds of Alkylamines. Adv. Synth. Catal. 2014, 356, 1544. [Google Scholar]
- (14).(a) Engle KM; Wang D-H; Yu J-Q Ligand-Accelerated C-H Activation Reactions: Evidence for a Switch of Mechanism. J. Am. Chem. Soc. 2010, 132, 14137. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Engle KM; Thuy-Boun PS; Dang M; Yu J-Q Ligand-Accelerated Cross-Coupling of C(sp2)–H Bonds with Arylboron Reagents. J. Am. Chem. Soc. 2011, 133, 18183. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wang D-H; Engle KM; Shi B-F; Yu J-Q Ligand-Enabled Reactivity and Selectivity in a Synthetically Versatile Aryl C-H Olefination. Science 2010, 327, 315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).(a) Aihara Y; Chatani N Nickel-Catalyzed Direct Alkylation of C–H Bonds in Benzamides and Acrylamides with Functionalized Alkyl Halides via Bidentate-Chelation Assistance. J. Am. Chem. Soc. 2013, 135, 5308. [DOI] [PubMed] [Google Scholar]; (b) Lu H; Hu Y; Jiang H; Wojtas L; Zhang XP Stereoselective Radical Amination of Electron-Deficient C(sp3)–H Bonds by Co(II)-Based Metalloradical Catalysis: Direct Synthesis of α-Amino Acid Derivatives via α-C–H Amination. Org. Lett. 2012, 14, 5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).For other possible reductive elimination pathways, see: Buettner CS; Willcox D; Chappell BGN; Gaunt MJ Mechanistic investigation into the C(sp3)–H acetoxylation of morpholinones. Chem. Sci. 2019, 10, 83. Nappi M; He C; Whitehurst WG; Chappell BGN; Gaunt MJ Selective Reductive Elimination at Alkyl Palladium(IV) by Dissociative Ligand Ionization: Catalytic C(sp3)-H Amination to Azetidines. Angew. Chem., Int. Ed. 2018, 57, 3178.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.