

Abstract
Repetitive elements in the human genome, once considered ‘junk DNA’, are now known to adopt more than a dozen alternative (that is, non-B) DNA structures, such as self-annealed hairpins, left-handed Z-DNA, three-stranded triplexes (H-DNA) or four-stranded guanine quadruplex structures (G4 DNA). These dynamic conformations can act as functional genomic elements involved in DNA replication and transcription, chromatin organization and genome stability. In addition, recent studies have revealed a role for these alternative structures in triggering error-generating DNA repair processes, thereby actively enabling genome plasticity. As a driving force for genetic variation, non-B DNA structures thus contribute to both disease aetiology and evolution.
Introduction
Sequencing of the human genome revealed that more than 50% is composed of repetitive elements1. Initially thought of as mere by-products in genetic evolutionary trajectories, we now know that many repetitive sequences have important biological functions, such as the regulation of chromatin structure, gene expression, DNA replication and genomic rearrangement2,3. A crucial feature of some repetitive sequences is the potential to fold into alternative, non-canonical DNA structures4,5 that differ from the right-handed DNA double helix, referred to as the canonical B-form or B-DNA structure, described by Watson, Crick, Wilkins and Franklin in 1953. Since then, more than 15 types of DNA structure that differ from canonical B-DNA have been reported6,7, with an estimated 13% of the human genome containing sequences that support such structures8. In addition to the primary sequence, the formation of non-B DNA structures is dictated by many cellular factors such as chromatin structure, DNA negative supercoiling stress and DNA binding proteins. Thus, depending on the conditions, rapid transitions from B-DNA to non-B DNA can occur, making this a highly dynamic process9. As a consequence, non-B DNA structures range from small single-stranded loop-outs of a few nucleotides formed by simple tandem repeats10 to more complex structures such as hairpin or cruciform DNA, Z-DNA, H-DNA and G quadruplexes (G4 DNA), which can contain hundreds of nucleotides (Fig. 1).
Fig. 1 |. Schematic of non-B DNA structures.
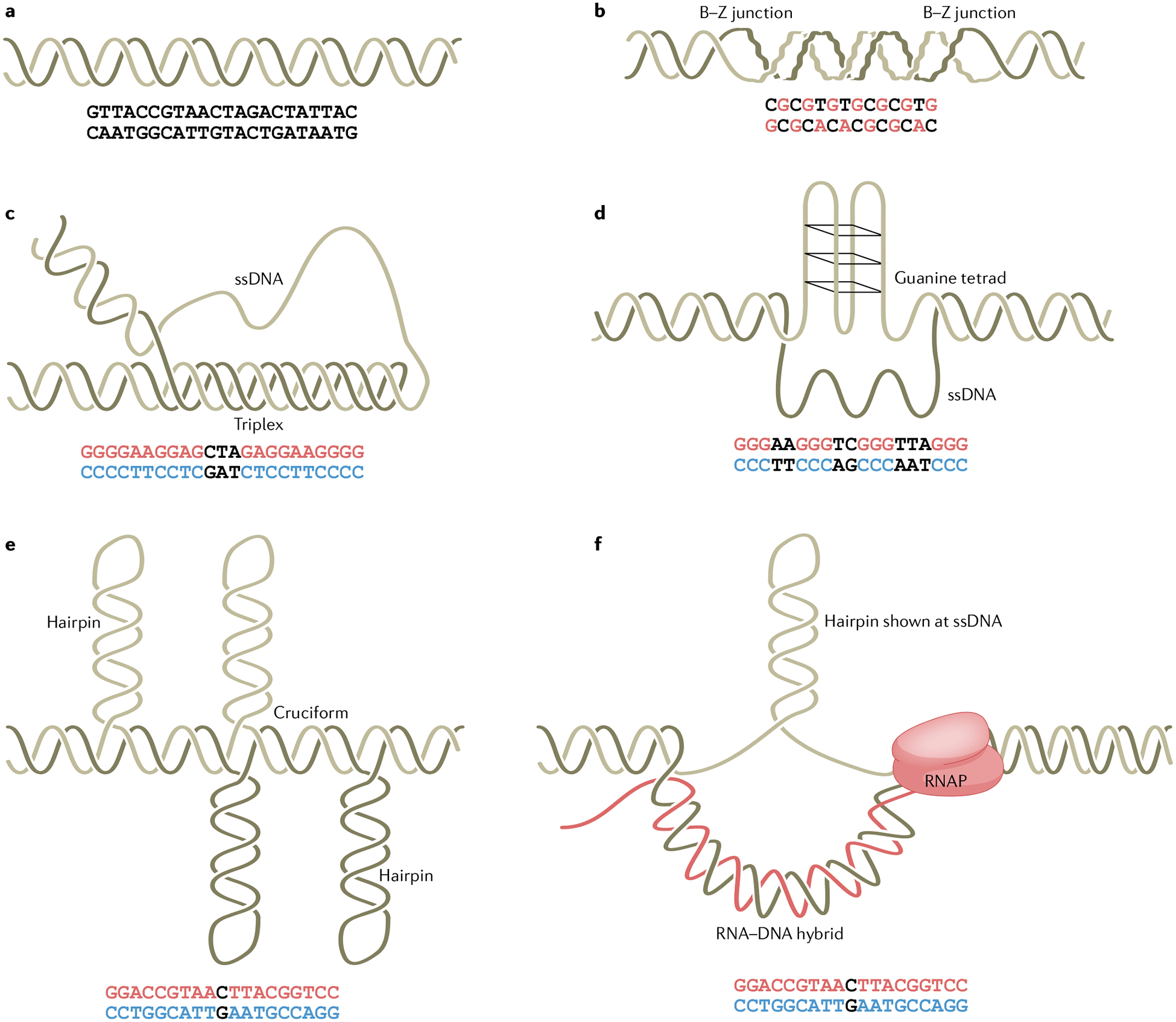
a, Canonical B-form DNA. b, Z-DNA forms at alternating purine–pyrimidine sequences, where the syn-formation purines and anti-conformation pyrimidines twist the backbone into a zigzag shape310. c, H-DNA forms at polypurine or polypyrimidine sequences that contain a mirror repeat, where half of the repeat in single-stranded form folds back into the major groove of the DNA duplex to form a triplex structure via Hoogsteen hydrogen bonding311,312. H-DNA can exist in various isomers depending on strand orientation and whether the purine-rich or pyrimidine-rich strand is used as the third strand. d, G quadruplexes form at sequences containing four runs of three or more guanines. Four guanine bases associate through Hoogsteen hydrogen bonding (guanine tetrad), and three continuous guanine tetrads stack to form a G quadruplex (G4 DNA)88,313. e, Cruciform or hairpin structures form at inverted-repeat sequences167,314,315, whereby two symmetrical arms self-anneal to form a duplex stem. f, R-loops contain a nascent RNA strand annealed to the DNA template strand316, leaving the non-template strand unpaired, which can adopt a stable structure, such as a hairpin or G4 DNA. The red/blue letters in the sequences represent the bases involved in the non-B conformation. RNAP, RNA polymerase; ssDNA, single-stranded DNA.
The co-localization of non-B DNA structures with functional genomic loci and genetic instability hotspots has suggested a role for non-B DNA in important physiological and pathophysiological events, including the regulation of transcription, DNA replication, DNA recombination and genome integrity. For example, non-B DNA structures can not only regulate the initiation of transcription11–13 and replication14–16 but also act as impediments to the transcription and replication complexes, leading to replication stalling, template slippage and/or replication fork collapse and DNA breakage17. Indeed, the dysregulation of DNA replication at non-B DNA structures is a major driving force of repeat expansion events18, which occur at different stages of development in different cell types and have been associated with human disease19. Since the initial discoveries connecting expansions of trinucleotide CGG repeats with fragile X syndrome20,21 and CAG repeat expansion with spinal and bulbar muscular atrophy22 more than 30 years ago, expansions of non-B DNA structure-forming mono-, di-, tri-, tetra-, penta- and hexa-nucleotide repeats have been implicated in more than 50 neurodevelopmental, neuromuscular and neurodegenerative disorders, among many other diseases23–29. Although the detailed mechanisms by which different repeats (different in both repeat unit and number) are involved in disease-related gene dysregulation and/or genetic instability may differ, expanded long repeats tend to form more stable structures and thus increase the risk of further instability events, a feature referred to as ‘dynamic mutation’30. An important recent discovery in the field is that non-B DNA structures can be recognized by DNA repair proteins, triggering error-generating repair processes, resulting in replication-independent genetic instability and variation31,32. This structure-specific repair processing mechanism can contribute to the DNA repeat-related mutations that occur in many diseases33–36.
In this Review, we discuss the key types of non-B DNA structure with a focus on their roles in genetic instability and disease aetiology. We begin by highlighting the dynamic nature of non-B DNA structures and the conditions that favour non-B DNA formation, before reviewing how non-B DNA structures influence cellular processes such as transcription, replication, recombination and DNA damage and repair. We discuss replication-dependent and replication-independent mechanisms of non-B DNA-induced mutagenesis, before concluding with a brief discussion of non-B DNA sequences in human disease.
Dynamic non-B DNA in the human genome
The nucleotide sequence dictates the potential for the formation and stability of a particular non-B DNA structure (Fig. 1). Hence, many sequence-based computer algorithms are now available to search for potential non-B DNA-forming sequences in genomes. Some examples include palindrome, detectIR37, QGRS Mapper38, G4Hunter39 and DNA Structure Search40 (see Related links) and others that search for inverted repeats, G4 DNA-forming, H-DNA-forming and Z-DNA-forming sequences41–44. Recently, deep learning or feature representative machine learning approaches that use large datasets to identify sequence features have been used to search for potential Z-DNA and G4 DNA-forming sequences45–47. Combining DNA sequence features with other biological factors could provide more accurate prediction power. However, many challenges remain in detecting and characterizing non-B DNA structures in genomes of living cells and organisms. Taking into consideration primary sequence analysis, RNA polymerase II (RNAPII) binding sites, and permanganate and S1 nuclease footprinting data, putative non-B DNA sites have been mapped at high resolution in mammalian genomes. The results showed that the promoter regions of oncogenes contained significantly more non-B DNA structures than other regions, even after excluding the non-B motifs that overlapped with transcription bubbles (RNAPII chromatin immunoprecipitation followed by sequencing (ChIP–seq) peaks)48. Although the reason for such enrichment is not clear, plausible explanations include the altered activities at these genes and/or that non-B DNA-induced genetic instability facilitates the formation of oncogenes from proto-oncogenes.
Genomic DNA is nevertheless largely maintained in the B-form, as this is the most energetically stable structure even when the sequences meet the pattern requirements for non-B DNA formation (Fig. 1). The transition from the canonical B-form DNA to a non-B DNA structure requires energy49. Therefore, non-B DNA formation is dependent not only on primary sequence features, but also on conditions induced by genomic activities. For example, when a DNA duplex is separated into single strands during transcription, replication or DNA repair processes, the B-DNA to non-B DNA transition can be facilitated by the negative supercoiling and open chromosome structure that occur during these processes (Fig. 2). Thus, the potential for repetitive elements to adopt non-B DNA structures, the type of non-B DNA formed and the location are determined by several factors, including negative supercoiling50, the presence of specific binding proteins51,52, open nucleosome and chromatin conformations53, and intracellular microenvironments such as pH54 and salt concentration55. For Z-DNA formation in the mouse prefrontal cortex, it was found that negative supercoiling levels and the presence of the Z-DNA-specific binding protein ADAR1 were the most important factors9.
Fig. 2 |. Dynamic non-B DNA structure induced by transcription.
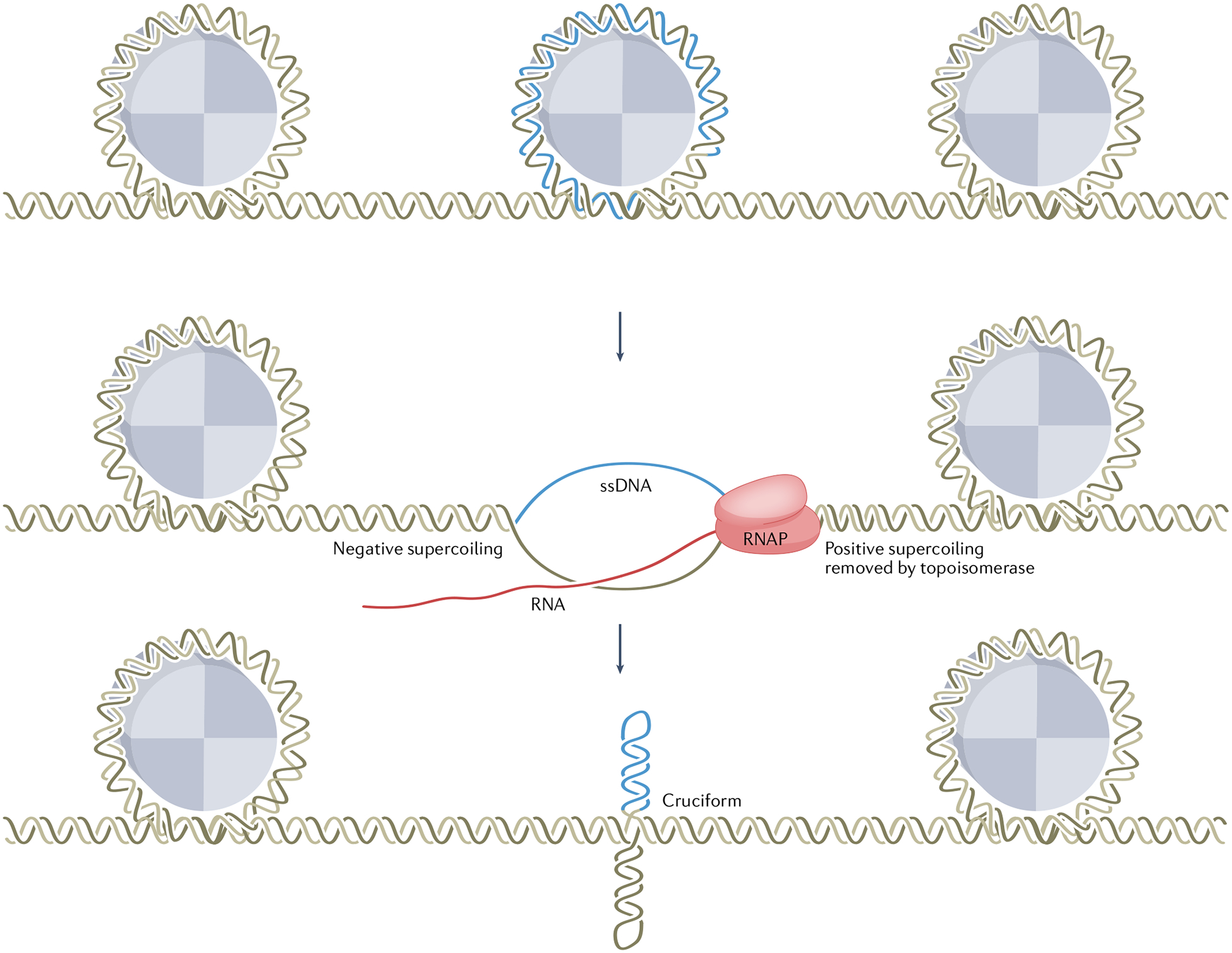
The inverted-repeat sequence (blue) is maintained in B-DNA form on histones and is unwrapped during transcription. The progressing transcription machinery unwinds DNA from the nucleosome structure and creates positive supercoiling in front (removed by topoisomerases) and negative supercoiling behind, which facilitates non-B DNA structure formation (shown in the schematic as a cruciform). RNAP, RNA polymerase; ssDNA, single-stranded DNA.
Depending on the nuclear environment, the same sequence has the potential to adopt different structures. For example, the purine-rich strand from an H-DNA-forming sequence can fold back to form a triplex structure at neutral pH in the presence of bivalent cations such as Mg2+ (Fig. 1c). However, under acidic conditions, the cytosines can be protonated, and the pyrimidine strand can serve as the third strand in the triplex structure56,57. As another example, high concentrations of aluminium maltolate can convert a CCG(12) repeat sequence in the FMR1 gene — involved in fragile X syndrome — into Z-DNA, as evidenced by circular dichroism spectra analyses58. Molecular dynamics simulations also predicted a stable Z-DNA structure at CCG repeats with alternately extruded Gs that favour syn conformations followed by symmetrically extruded junctions between adjacent Z-DNA formations59 (Fig. 1b). However, a similar CCG repeat was found to adopt a G-quadruplex structure in the presence of high concentrations of sodium chloride60 (Fig. 1d). Moreover, a recent single-molecule study found that a TG(11) duplex opened an unpaired bubble under low stretching tension and unwinding torsion. However, with increased negative supercoiling tension, the TG(11) repeat formed a Z-DNA structure61. These examples show that different environments can support the formation of different non-B DNA structures at the same or similar sequences. Thus, cellular and genomic metabolic conditions should be considered when studying DNA structure-induced cellular activities.
Chromatin conformation
DNA–histone interactions maintain B-DNA formation, such that an important initiating step for non-B DNA formation is nucleosome disassembly during DNA metabolic processes. A study using Fourier transform infrared spectroscopy found that histone acetylation could lead to open chromatin structures and a concomitant increase in Z-DNA formation in trichostatin A-treated HeLa cells62. Using the same technology, and confirmed by ChIP, the authors found that Z-DNA formation was increased in cells that overexpressed BRG1 or BRM ATPases, key components of the mammalian SWI/SNF chromatin remodelling complex. As expected, Z-DNA formation was reduced when BRG1 and BRM were depleted by short interfering RNA63.
Negative supercoiling
Genomic DNA is wrapped on histone cores in a left-handed toroidal manner, and the helical tension in the linker regions is released by topoisomerases. Thus, unless the DNA has been unwrapped from histones to allow for negative supercoiling, the energy to facilitate non-B DNA structure formation may not be available. A recent study mapped DNA supercoiling regions and non-B DNA structures in the genome of Caenorhabditis elegans embryos and found that 400 bp regions around transcription start sites had significantly increased negative supercoiling and non-B formation (Z-DNA and cruciform structures)64. Supercoiling induced by transcription through the MYC gene can stimulate Z-DNA formation in the promoter region, as shown by Z-DNA-specific antibody binding in permeabilized mammalian cell nuclei65–67. Approximately 1.5 kb upstream of the MYC promoter P1, a region known as the far upstream element (FUSE), lies a well-characterized supercoiling responsive region that can adopt non-B DNA structures when the gene is expressed68. Using an immunofluorescence labelling method, cruciform structures that had strong levels of negative supercoiling induced by active transcription were detected in growing mouse oocytes, but not in fully matured oocytes where transcription was not active69. Furthermore, the inhibition of transcription with α-amanitin treatment in growing oocytes significantly reduced cruciform DNA foci. When mouse genomic DNA was fragmented, circularized by ligase and negatively supercoiled at a near-physiological level, the single-stranded DNA (ssDNA) regions exposed by non-B DNA structures on the artificially supercoiled naked circular DNA resembled the ssDNA regions detected in vivo, demonstrating the contribution of negative supercoiling to non-B DNA conformation in vivo48.
Non-B DNA binding proteins
Various proteins have been identified that bind to non-B DNA and alter their stability. A family of Z-DNA binding proteins that share a common Z-DNA binding domain (ZDBD) have been described70, such as Z-DNA binding protein 1 (ZBP1), a pathogen-sensing protein that regulates cell death and inflammation71,72; PKZ, a PKR-like protein kinase that has a role in host responses to viruses73; and poxvirus virulence factor E3L and ORF112, which are crucial for viral pathogenesis74,75. The ZDBD of the ADAR1 protein binds to Z-DNA with high affinity and can convert even a short TA(3) repeat into a Z-DNA structure, which cannot form in the absence of ADAR1 (ref.52). Many chromosomal architectural proteins such as histones H1 and H5 and the high mobility group (HMG) proteins bind preferentially to cruciform structures (reviewed in ref.76), and the HMGB1 protein has high affinity for triplex DNA structures in collaboration with the nucleotide excision repair (NER) protein complex XPA–RPA77. Many proteins are known to interact with G4 DNA, such as POT1, RPA and BRCA1 (reviewed in ref.78). A recent study that used a cell-permeable G4 DNA ligand and crosslinking of the G4 DNA-interacting proteins identified hundreds of putative G4 DNA-associated proteins with known functions in transcription regulation, mRNA processing, cell cycle regulation and DNA damage and repair processes53. The presence and the local concentrations of these proteins in vivo could modulate the formation and activities of non-B DNA structures.
DNA helicases
DNA helicases are responsible for unwinding duplex DNA during replication, transcription and repair and have a key role in genome maintenance79. Helicases have gained increasing attention recently regarding their roles in non-B DNA and disease (Table 1). In general, helicases have the capacity to unwind non-B DNA structures, such that their deficiency can increase the risk of non-B DNA-related diseases, but this conclusion should not be oversimplified. For example, deficiency of yeast Sgs1 helicase, a homologue of the bacterial RecQ helicase, resulted in an accumulation of cruciform-shaped replication intermediates80. However, Sgs1 deficiency reduced the expansion of GAA repeats in yeast81. It was hypothesized that Sgs1 unwinds nascent strands from their templates when replication forks are stalled by non-B DNA structures, with the shorter Okazaki fragments annealing to the longer nascent leading strand, resulting in extra repeat units (that is, repeat expansion)81. Thus, the effects of helicases on non-B DNA processing seem to be more complicated than simply resolving structures to maintain genome stability.
Table 1 |.
DNA helicases and non-B DNA structures
| Helicase | Cellular function | Reported types of non-B DNA processed | Results from helicase deficiency | Ref. |
|---|---|---|---|---|
| Superfamily 1 DNA helicases | ||||
| PIF1 | Unwinding replication barriers, assisting fork progression | G4 | Genetic instability, increased risk of cancer | 289 |
| RRM3 | Unwinding replication barriers, assisting fork progression | G4 | Genetic instability, increased risk of cancer | 290 |
| DNA2 | Telomere maintenance; helicase and G4 nuclease | G4 | Cell senescence, telomere replication defects, genetic instability, increased risk of cancer | 148 |
| Srs2 | Post-replication repair to UV, ionizing radiation or MMS lesions | Cruciform at inverted repeats, hairpin at triplet repeats | UV sensitivity, genetic instability | 291 |
| UvrD | Nucleotide excision repair, mismatch repair, HR | G4, cruciform/Holliday junctions, triplet repeats | UV sensitivity, genetic instability | 292 |
| Rep | DNA replication | G4 | Slow progression of chromosomal replication forks | 293 |
| RecBCD | Helicase and nuclease activities, DSB repair by HR | Cruciform at inverted repeat, hairpin at triplet repeat | Genetic instability | 294 |
| Superfamily 2 DNA helicases | ||||
| RECQ1 (also known as RECQL and RECQL1) | DNA repair, cell cycle and growth, telomere maintenance, transcription | Cruciform/Holliday junctions, very weak on G4 | Genetic instability and increased risk of cancer | 295 |
| RECQ2 (also known as BLM) | DNA replication, immunoglobulin class-switch recombination | G4, triplex H-DNA, direct repeats including triplet repeats | Hereditary Bloom syndrome: primordial dwarfism, genetic instability, increased risk of cancer | 296 |
| Him-6 | BLM homologue in Caenorhabditis elegans | G4 | Genetic instability | 297 |
| RECQ3 (also known as WRN) | Telomere maintenance | G4, hairpin at triplet repeat, triplex H-DNA, Z-DNA | Hereditary Werner syndrome, premature ageing, increased risk of cancer, cell senescence | 298 |
| Sgs1 | Yeast homologue of human BLM and WRN. Forms a complex with Top3 and Rmi1. DNA replication, regulating HR | Cruciform/palindrome, G4, hairpin at triplet repeat | Genetic instability | 291 |
| RECQ4 (also known as RTS) | ATPase activity and single-strand annealing activity, replisome assembly | Holliday junctions, G4 | Hereditary Rothmund–Thomson, RAPADILINO and Baller–Gerold syndromes: skin, hair, skeletal and dental abnormalities, increased risk of cancer | 299 |
| RECQ5 | DNA replication, transcription, repair, suppressing sister chromatid exchanges during HR | G4, an order of magnitude weaker than BLM and WRN | Genetic instability | 300 |
| RTEL1 | Telomere maintenance and HR regulation | G4, hairpin at triplet repeat | Hoyeraal–Hreidarsson syndrome, pulmonary fibrosis and/or bone marrow failure, telomere-related 3 (PFBMFT3), dyskeratosis congenita, autosomal recessive 5 (DKCB5) | 301 |
| DHX36 | RNA and DNA helicase activity, transcription and translation regulation, genetic stability, telomere maintenance | G4 | Genetic instability and increased risk of cancer | 302 |
| DHX9 | DNA replication, transcription, translation, microRNA biogenesis, genetic stability | Triplex H-DNA, Z-DNA | Genetic instability and increased risk of cancer | 303 |
| FANCJ | DNA repair, HR, replication fork progress during replication stress | G4, hairpin at triplet repeat | Hereditary breast and ovarian cancer, Fanconi anaemia, bone marrow failure | 304 |
| Dog-1 | FANCJ homologue in C. elegans | G4 | Genetic instability | 305 |
| XPB/XPD | TFIIH components with functions in nucleotide excision repair | G4, triplex H-DNA | Xeroderma pigmentosum (UV sensitivity, cancer); trichothiodystrophy, Cockayne syndrome (development) | 306 |
| DDX5 | DNA and RNA helicase activity, transcriptional regulation, splicing | G4 | Aberrantly expressed in many tumours | 307 |
| DDX11 (also known as CHLR1), CHL1 in yeast | Chromosome segregation, cell cycle progression, sister chromatid cohesion, putative RNA helicase, translation initiation, splicing | G4, triplex H-DNA | Warsaw breakage syndrome | 308 |
| ATRX | Chromosome alignment and meiotic spindle organization, recombination pathway selection | G4 | α-Thalassaemia with mental retardation | 309 |
DSB, double-strand break; HR, homologous recombination; MMS, methyl methanesulfonate.
Non-B DNA induced by DNA damage or repair
DNA damage and repair can affect non-B DNA structure formation by altering local topological conditions. Some types of DNA lesion or repair intermediate can alter the energetics of structural transitions of DNA, affect protein–DNA interactions and modulate nucleosome and chromosome conformations, eventually leading to non-B DNA structure formation. For example, DNA double-strand break (DSB) processing near short inverted repeats can stimulate the formation of hairpin structures, likely by creating ssDNA that enables self-annealing82.
Abasic (AP) sites generated during the repair of 8-oxo-7,8-dihydroguanine (8-oxo-G) by OGG1 can destabilize duplex DNA and provide thermodynamic energy for the transition from G-rich duplex DNA to a more stable G4 DNA structure83, or a cruciform structure when the processing occurs within an inverted-repeat region84. The distribution of AP sites, OGG1 and AP endonuclease 1 (APE1) binding sites in lung cancer genomic DNA determined by ChIP–seq exhibited genome-wide correlation with G4-forming motifs, particularly in promoter and gene regulatory regions85. Furthermore, binding of APE1 to AP sites within G4-forming motifs in the MYC promoter stimulated the formation of G4 DNA in vitro85. Another study reported that an AP site located in the centre spacer region between two symmetrical arms of an inverted repeat could destabilize B-DNA formation and increase the formation of hairpin structures86.
A tetrahydrofuran abasic site analogue within a GAA repeat was processed by base excision repair, during which the repeats on the template strand could form a loop of approximately eight TTC repeat units in vitro, which recruited DNA polymerase-β (Polβ) for bypass, resulting in large deletions87.
In summary, non-B DNA conformations are highly dynamic in living cells. Although the primary sequence is crucial for structural transitions, other cellular activities such as transcription, replication and DNA repair can have an impact on non-B DNA formation via the modulation of nucleosome or chromatin structures, alterations in DNA supercoiling levels and/or DNA binding proteins.
Biological functions of non-B DNA
Repetitive sequences capable of adopting non-B DNA structures are enriched at highly conserved regions with biological functions, such as promoters and replication origins. Although co-localization does not necessarily relate to function of non-B DNA in these processes, numerous studies have suggested that non-B DNA structures can contribute to several important biological functions88,89. Non-B DNA structure formation can change the local topology of genomic regions, thereby influencing interactions of DNA metabolic processes, protein binding and chromatin structures. The important biological roles of non-B DNA-forming sequences, perhaps leading to positive selection pressure during evolution, may explain the abundance and conservation of these unstable elements across genomes (Box 1).
Box 1.
Non-B DNA as a driver of evolution
Non-B DNA-forming sequences have been found to co-localize with evolutionarily active regions3,318. Transposable elements comprise a large fraction of many eukaryotic genomes and most contain terminal inverted repeats319 and direct repeats that can stimulate double-strand breaks (DSBs). A comparative autosomal map covering >90% of the mouse and human genomes revealed that the breakpoint regions of intrachromosomal rearrangements contained a high density of repetitive sequences320. Although it is not easy to distinguish which occurred first, a mutagenic non-B DNA-forming sequence or an active evolution hotspot over time, unstable non-B DNA is considered as a driving force for genetic variations and evolution321–323.
Insects contain a xenobiotic-metabolizing P450 gene that can detoxify xenobiotics, and its expression is regulated by a G4 motif in the promoter region that is thought to be acquired from an HzIS1–3 transposon324. Bacterial transposon Tn7, which encodes a TnsC protein that can bind to triple-helical DNA, created selective insertion of Tn7 adjacent to a H-DNA-forming sequence in an in vitro transposition assay325. It will be interesting to see whether similar H-DNA-directed transposon mobility and genome evolution also occurs in mammalian genomes.
The genome of muntjac deer has undergone drastic evolutionary changes with a dramatic reduction in the number of chromosomes from 2n = 70 in the ancestral karyotype to 2n = 6 in female and 7 in male Muntiacus muntjak vaginalis. Analysis of the fusion sites revealed repetitive elements that may have stimulated DSB formation and mediated recurrent fusions between different chromosomes326. Non-B DNA structures formed in simple sequence repeats (SSRs), including microsatellites, also stimulate replication slippage, crossover and/or gene conversion events327. SSRs are very abundant in penaeid shrimp and the distribution is highly associated with transposable element expansion and intrachromosomal rearrangements328. Compared with Fenneropenaeus chinensis, which lives exclusively in salt water, Litopenaeus vannamei, which is capable of surviving in a large range of salinities, showed significant alterations of SSRs within introns or untranslated regions (UTRs) of differentially expressed genes related to amino acid and lipid metabolism involved in osmoregulation, suggesting a regulatory role of these repetitive elements in adaptive evolution in these species328.
A more direct connection between non-B DNA structure and evolution comes from comparing marine stickleback fish, which have developed a robust pelvic apparatus, and many independently derived freshwater populations that have adaptively lost pelvic hind fins over the past ~15,000 years. This repeated pelvic loss maps to recurrent deletions of a pelvic enhancer (Pel) of the homeodomain transcription factor gene Pitx1 (ref.329). The Pel sequence from marine populations contains a long Z-DNA-forming GT repeat that was shown to stimulate the formation of DSBs and large deletions (>100 bp) in a repeat length- and orientation-dependent fashion in yeast and on mutation reporters in mammalian cells mirroring the situation in stickleback fish. Similar repeats in human genomes were also mapped with aphidicolin-sensitive breakage sites, suggesting that non-B DNA structure-induced genetic instability is a common process that has contributed to genetic evolution330.
The male-specific region of the human Y chromosome occupies ~95% of the chromosome, and eight nearly identical palindromic sequences, the result of duplication events, contain many of the testis-specific genes. The variations on palindromic sequences in existing human populations suggest frequent recurrent arm-to-arm gene conversion events in testis gene families331.
Notably, non-B DNA-forming sequences are not always deleterious. Many non-B DNA-forming sequences are associated with distinct genomic features that are evolutionarily conserved, such as regulatory elements in promoters5,88. Motif-containing elements for the formation of G4 DNA, triplexes and hairpins increased rapidly in eumetazoan genomes during evolution and seemed to be under positive selective pressure, suggesting that the conservation of non-B DNA-forming sequences may be beneficial during evolution89.
Non-B DNA in chromatin organization
Eukaryotic DNA is packaged into nucleosomes and then higher tertiary DNA structures in vivo. When B-DNA is wrapped around histone cores, the minor groove of the helix aligns and interacts with the positively charged arginines on the histones90. These electrostatic interactions are important for maintaining nucleosome structure and B-form DNA. Non-B DNA conformations change the orientation of DNA strands and the shape of the grooves and interrupt the DNA–histone arrangement and therefore alter nucleosome structures.
Non-B DNA affects local chromatin organization.
Some repetitive satellites serve as signalling sequences for nucleosome assembly, whereas other repeats, such as Z-DNA-forming CG or CGG repeats59 or H-DNA-forming GA repeats, are resistant to placement within the nucleosome structure91 (reviewed in ref.92). GAA repeats were more refractory to nucleosome assembly in supercoiled plasmid DNA when H-DNA formation was supported, yet the same repeats can be packaged into nucleosomes when in B-form DNA93. In Saccharomyces cerevisiae, most short inverted-repeat sequences were found in regions with low nucleosome occupancy94. G4 DNA sequences are enriched in nucleosome-depleted regions in both human cells and C. elegans95. Furthermore, a G4 DNA-stabilizing ligand created open nucleosome structures for RNAPII binding even in compacted chromatin regions96.
There are reports that suggest that non-B DNA-forming repeats facilitate the formation of nucleosome structures. CTG repeats (as short as six repeat units) are enough to facilitate nucleosome assembly, although expansion to 62 repeats — considered to be more prone to forming a hairpin structure — did not further affect the assembly of histone octamers97. Note that most studies were performed in vitro and details on the DNA conformation adopted at repetitive non-B-forming sequences are lacking owing to the difficulties in determining nucleosome and non-B DNA structures simultaneously.
Non-B DNA may affect distal chromatin organization.
Disrupting the formation of a key nucleosome in the β-globin gene by altering the underlying positioning sequence was shown to affect adjacent nucleosomes98. Although this has not been verified as a universal mechanism throughout the entire genome in all species, this discovery suggests the interesting possibility that changing the position of one nucleosome by non-B DNA structure formation could affect distant regions.
G4 DNA motifs are significantly enriched at distal interchromosomal interaction sites99 and can recruit the chromosomal architectural protein RIF1, which brings multiple G4 DNA-forming sequences together at different regions to create local chromosomal compartments via chromatin looping at the nuclear lamina100. The ssDNAs from the tips of hairpins can interact with each other to form ‘kissing’ complexes, similar to the NMR solution structure of a kissing complex formed between deoxyoligoribonucleotides corresponding to the dimerization initiation site SL1 of HIV-1Lai RNA101. This loop–loop interaction is important for tertiary and topology structure maintenance and provides a basis for molecular recognition102. Some long potential G4 DNA-forming sequences that contain multiple G4 elements were identified in antibody switch regions, where the single-stranded loops of neighbouring G4 structures were frequently complementary and base paired with each other, perhaps contributing to chromosomal rearrangements in cancer103. In addition, ssDNA regions from two H-DNA structures formed at long GAA repeats on plasmids were shown to interact to form a dumbbell-shaped complex referred to as ‘sticky’ DNA in bacterial cells104,105. Thus, non-B DNA-mediated interactions may bring distal elements together, contributing to 3D genome organization and stimulating crosstalk between chromosomal territories and DNA elements that regulate gene or chromosomal functions.
Taken together, the formation of both non-B DNA structures and nucleosome structures is dynamic, and often competitive. DNA in nucleosomes is typically maintained in the B-form structure by DNA–histone interactions, and non-B DNA structures, once formed, are often more refractory to nucleosome assembly than B-DNA. The impact of non-B DNA on chromosomal structure, particularly on long-range chromosomal architecture, may have important biological and pathological functions that remain to be discovered.
The impact of non-B DNA on transcription
Non-B DNA affects transcription initiation.
A recent bioinformatics study investigated the distribution of non-B DNA-forming sequences in 15 species and found that promoter regions contain a unique pattern of non-B DNA positioning: G4 DNA and Z-DNA are the most enriched of the non-B types and are frequently found in core promoter regions in nearly all species106. Direct repeats are enriched in the immediate (50–100 bp) upstream region of core promoters, and mirror repeats are often located (100–300 bp) upstream of the core. It will be interesting to determine whether these repeat patterns underlie as yet unknown mechanisms of transcription regulation.
In the yeast genome, G4 DNA-forming sequences are enriched at promoters approximately sixfold over random distribution107. Inverted repeats, which can form cruciform structures (Fig. 1), are substantially enriched in regions adjacent to stop codons, at the end of genes, near start codons, 5′-untranslated regions (UTRs) and promoter regions108. Their conservation and enrichment within or surrounding these key elements suggests a role for non-B DNA in transcription regulation. In promoters, non-B DNA formation could provide an open chromatin structure for transcription initiation complex formation or block or recruit transcription factors and thus affect transcription initiation109 (Fig. 3).
Fig. 3 |. Biological functions of non-B DNA.
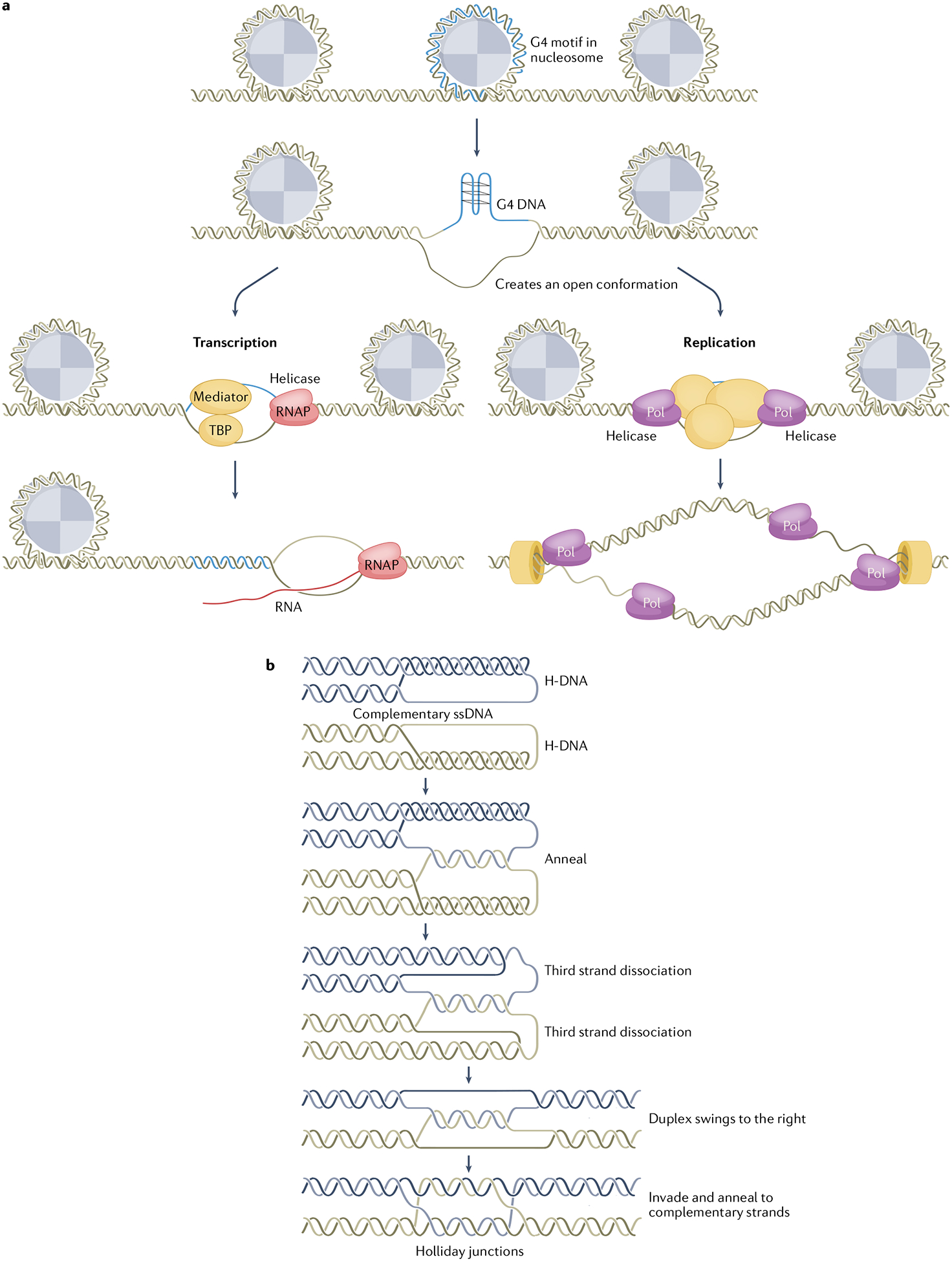
a, Non-B DNA can facilitate the initiation of transcription and replication. Non-B DNA formation (shown in the schematic as G quadruplexes (G4 DNA)) unwinds DNA from nucleosomes and creates an open structure that facilitates the assembly of transcription (left) and replication (right) complexes. b, Non-B DNA can stimulate homologous recombination (HR). There are multiple pathways by which non-B DNA can directly or indirectly stimulate HR. Shown in the schematic is a unique structural alteration between two H-DNA isomers containing complementary single-stranded DNA (ssDNA) regions. With the presence of a nick on either strand or with the assistance of a topoisomerase, the two strands could wind around each other to form Watson–Crick base pairs. Owing to the dynamic nature of H-DNA in vivo, the third strand in both H-DNA structures could disassociate from the duplex and anneal to each other to form a double Holliday junction structure and thereby stimulate HR317. RNAP, RNA polymerase; TBP, TATA-box-binding protein.
Although Z-DNA-forming sequences are essential for transcription initiation in viruses, a Z-DNA-forming sequence in the promoter region of the rat Ncl gene, which encodes nucleolin, has been shown to inhibit promoter activity, such that its deletion increased transcription by ~50%25. Interestingly, in this study the effect of Z-DNA on promoter activity was neither location nor orientation dependent; relocating the repeat 458 bp from the promoter or cloning it in the opposite direction did not change its inhibitory effects25. A plausible explanation is that local negative supercoiling stress is essential for transcription initiation because it not only initiates melting of the DNA duplex, which is an energy-consuming step110,111, but also facilitates the interaction of transcription factors with promoters112. However, formation of non-B DNA also requires negative supercoiling and relaxes the local supercoiling level once formed. One left-handed helical turn of Z-DNA can relieve 1.8 helical turns of negative superhelical twisting on the B-DNA helix113. Therefore, non-B DNA formation could affect gene expression by altering the local DNA topological tension, independently of its (short-range) location and orientation.
G4 DNA has been shown to either enhance or suppress gene expression (reviewed in ref.114). Many of the published reports on this topic were based on the overlap of computational predictions of G4 DNA-forming sequences and gene functional analyses. A recent study that used an antibody-based G4 ChIP–seq approach identified ~10,000 G4 DNA structures in human chromatin, predominantly in the promoters and 5′-UTRs of highly transcribed genes115. However, these data reveal only a correlation between non-B DNA and transcription regulation rather than providing direct evidence for cause and effect. In another study, G4 DNA-stabilizing ligands were used to observe transcriptional alterations associated with increased G4 DNA structures in human cells. A database of transcriptome alterations induced by seven distinct G4 DNA ligands, including 25,228 genes, was recently published116. Again, although such correlations are informative, the alterations in transcription may be due to other cellular responses induced by the ligands and not necessarily a direct consequence of G4 DNA formation. Thus, studies that demonstrate direct evidence of DNA structure-associated transcription regulation are warranted. However, given the dynamic nature of non-B DNA structures, it is challenging to determine DNA structure in living cells in real time; studies that combine bioinformatic approaches, small-molecule compounds that regulate non-B DNA conformation, and genetic and molecular studies with engineered non-B DNA conformations in the same model systems can provide convincing evidence for the effects of non-B DNA in vivo.
Non-B DNA affects transcription elongation.
Transcription complexes use DNA helicases and transcription elongation factors to unwind or bypass barriers during elongation, including those imposed by non-B DNA. However, if a non-B DNA conformation is stable enough to resist helicase activity, or is stabilized by structure-specific proteins, it can act as a barrier to transcription and reduce the processivity and fidelity of RNA polymerases117–119. In yeast, Spt4/5, individually or cooperatively with Elf1, interacted with RNAPII to facilitate transcription elongation and increased the run-off transcripts through CTG(40) repeats in the B-form in an in vitro assay; however, when the CTG repeat formed a stem–loop structure, the presence of Spt4/5 in fact enhanced transcription pausing in front of the stem–loop120.
Interestingly, RNA polymerases not only pause in front of non-B DNA conformations during extension but can also stall after passing through the non-B DNA-forming sequences. For example, transcription by T7 RNA polymerase or RNAPII was paused at H-DNA-, Z-DNA- and G4 DNA-forming sequences within and downstream of the non-B DNA sequences in a length- and supercoiling-dependent manner in in vitro multiple-round transcription assays117,118,121. It is plausible that the negative supercoiling generated behind the progressing polymerase, the non-template ssDNA and/or the nascent RNA stimulated a structural complex in this area that impeded the progression of the RNA polymerase complexes121.
In contrast to H-DNA, Z-DNA or cruciform structures, in which both strands are involved in the conformation, G4 DNA forms on the G-rich strand of a duplex. Therefore, the location of G4 DNA on the template versus the non-template strand during transcription results in different effects. For example, G4 DNA in the template strand upstream of the start codon in the Renilla luciferase gene substantially inhibited transcription, but showed no effect when located in the non-template strand122.
Non-B DNA affects RNA splicing.
A genome-wide screen and statistical analyses suggested strong associations between non-B DNA structures, including G4 DNA, cruciform DNA, triplex DNA, slipped DNA and Z-DNA, and exon skipping in both human and mouse genomes123. Although the mechanisms are still unclear, it is possible that non-B DNA-induced stalling of RNA polymerase complexes and the delay in elongation could facilitate the recruitment of splicing factors and the recognition of splice sites. It has also been proposed that non-B DNA on the template strand enables transcription initiation from non-contiguous regions, producing alternative RNA isomers124,125. However, to date, the experimental evidence is scarce; therefore, further studies on the roles of non-B DNA structures in regulating alternative splicing are warranted and would provide timely and important advances for the field.
Although many details still remain to be elucidated, it is clear that the effects of non-B DNA on transcription are associated with functional genomic regions and higher-order chromosome conformations, such that they cannot be considered simply as ‘activators’ or ‘repressors’126. With such a wide variety of effects, the specific manipulation of DNA structure formation could serve as a unique type of ‘epigenetic’ regulation of gene expression and subsequent cellular activities127.
The impact of non-B DNA on replication
Non-B DNA affects replication initiation.
Non-B DNA-forming sequences have emerged as key controllers of genome replication at the level of both replication origin firing and replication fork progression. Genome-wide studies have revealed significant enrichment of non-B DNA-forming sequences, including G4 DNA, H-DNA, Z-DNA and cruciform- or hairpin-forming inverted repeats, at replication origins14–16. These sequences are important for prokaryotic DNA replication128, viral replication in eukaryotic cells129,130 and the replication of eukaryotic genomes131,132. For example, it was recently found that direct, inverted and mirror repeats as well as Z-DNA- and G4 DNA-forming sequences are associated with origins of replication that are consistent with the position and firing of the origins. However, the regulation of replication origins is complex and involves many different factors. Interestingly, many of these factors, such as base composition, DNA accessibility and chromatin structure, are often overlain by or cluster with non-B DNA-forming sequences in human genomes133. Formation of non-B DNA structures at or near replication origins can alter local topological conditions that affect melting of the DNA duplex and loading of replication factors, and can recruit structure-specific binding proteins for replication machinery assembly134,135 (Fig. 3). A G4 DNA-forming sequence from the βA origin in the chicken DT40 cell line was able to initiate replication when it was cloned into a region that lacked strong initiation sites, and a G>A point mutation that destabilized the G4 DNA structure reduced origin efficiency136. Cruciform-binding proteins that belong to the 14-3-3 protein family form dimers and bind to cruciform structures at the four-way junctions. Deleting the DNA binding domain reduced the cruciform-binding activity and suppressed the replication of plasmids that contained a yeast replication origin in yeast cells137.
The human origin recognition complex (ORC) binds preferentially to replication origins at G-rich ssDNA that can form G quadruplex structures to facilitate the initiation of DNA replication in eukaryotic cells138. A recent study directly explored the functions of a G4 DNA-forming sequence from an origin G-rich repeated element (OGRE) on different types of replication origin139. Deletion of the OGRE-G4 sequence substantially decreased the corresponding origin activity, whereas adding the OGRE-G4 element created a new replication origin. G4 DNA stabilizer binding to G4 DNA in intergenic regions tended to activate new origins or enhance existing origin activities. By contrast, G4 DNA ligand binding reduced firing efficiency of origins that were clustered and located in active promoters, likely owing to the G4 DNA-induced reduction in transcription, thereby attenuating the stimulating effects of transcription on replication origin firing139. Thus, non-B DNA can affect replication initiation even when located hundreds of base pairs from the initiation sites.
Non-B DNA affects replication elongation.
After initiation and priming, replication forks progress, and DNA polymerases act on both the leading and lagging strands. Non-B DNA on the template strand can impose a barrier for many DNA polymerases35,128,140,141, which can reduce their fidelity, stall replication and cause replication fork collapse, resulting in DNA strand breaks142,143. Topoisomerases and helicases are actively involved in replication and can unwind some non-B DNA structures in front of replication forks144–146. The template DNA enters and is pulled through a tunnel formed at the zinc finger region of the N-tier ring and C-tier motor domain of human replicative helicase complexes. DNA in a non-B conformation is generally much bulkier than the tunnel and cannot pass through147. Although the detailed thermal energy characteristics of helicase activities in the context of non-B DNA structures are not fully determined, at least some types of non-B DNA can resist helicase unwinding148,149.
Direct evidence of replication fork stalling at non-B DNA structures in vivo is challenging because both the non-B DNA structure and replication stalling are transient, and a single paused replication fork at a specific non-B DNA region is difficult to distinguish from a normal progressing fork. 2D gel electrophoresis has been used to successfully determine replication stalling at specific non-B regions40,150. However, it requires a substantial number of forks to stall at the same location simultaneously, making this technology more appropriate for bacterial and/or yeast genomes. DNA fibre analysis has also been used to monitor replication rates; however, this technique detects large regions and therefore must be combined with a targeted technique, such as fluorescence in situ hybridization (FISH), to identify non-B DNA regions in the genome151.
The impact of non-B DNA on recombination
Non-B DNA-forming sequences are enriched at recombination hotspots, implying a link between non-B DNA and homologous recombination. For example, a 1,000 bp motif in the I–B and I–J subregions within the mouse Eβ gene in the major histocompatibility complex (MHC) contains several types of repeat such as AGGC and GC/GT-rich repeats that can adopt non-B DNA structures, including Z-DNA. This short region has been estimated to account for ~2% of the recombination events in the entire genome152. Unequal sister chromatid exchange between the Cγ2a and Cγ2b heavy chain genes in MPC-11 mouse myeloma cells occurs at a GA repeat that can adopt an H-DNA structure, followed by a GT repeat that can form Z-DNA153. G4 DNA is abundant in immunoglobulin switch (S) regions and may contribute to class-switch recombination (CSR) by stalling transcription, leading to the nicking of DNA strands154. In human bladder carcinoma EJ cells, a Z-DNA-forming GT(30) repeat was found to increase recombination between non-replicating plasmids up to 20-fold. Both gene conversion and reciprocal exchange events were found 237–1,269 bp from the Z-DNA-forming sequence155.
Certainly, there are different mechanisms involved in non-B DNA-induced recombination. Some non-B DNA structures are composed of self-folding formations on one strand and create long ssDNA on the complementary strand, such as H-DNA and G4 DNA structures. The exposed ssDNA could potentially invade a homologous duplex and form a structure similar to a D-loop, which is known to induce homologous recombination156 (Fig. 3). Guanosines in Z-DNA are in the syn position and are exposed, and the N7 and C8 of guanosines in Z-DNA are ‘stickier’ and can interact with other DNA molecules157. The left-handed helix of Z-DNA has been shown to facilitate the formation of paranemic joints during synapsis between two topo domains containing homologous sequences158. In addition, non-B DNA structures can stall DNA replication forks and generate DNA nicks and breaks that could stimulate recombination.
A distinct mechanism of non-B DNA-stimulated recombination in immune cells includes the AID protein, which belongs to the APOBEC cytidine deaminase family, and involves G4 DNA structures on the non-template DNA strand of the Sμ and Sγ regions. AID is expressed in B cells and can deaminate deoxycytidine, converting it into deoxyuridine within single-stranded regions159. As AID targets ssDNA, many different types of non-B DNA structure that expose ssDNA regions could potentially serve as targets. In mice, MSH2–MSH6 can bind to both G4 DNA formed within transcribed S regions and G–U mismatches to facilitate DNA synapsis and recombination160.
Interestingly, when G4 DNA-forming sequences in human or mouse S regions were replaced with chicken or Xenopus laevis Sμ sequences, which are rich in palindromic and stem–loop structures, CSR was still functional in murine B lymphoma cells although it was less efficient. The Xenopus Sμ sequence is in fact AT-rich, rather than G-rich, and it supported CSR in an orientation-independent manner. By contrast, a non-palindromic G-rich sequence was not able to activate CSR161. These results suggest that common features of non-B DNA structures, such as exposure of ssDNA or transcription modulation, rather than the primary sequences, are important for modulating recombination and immune reactions, providing a potential therapeutic target to manipulate CSR and immunoglobulin secretion levels in autoimmune or immunoallergic disorders.
Non-B DNA-induced mutations
Many non-B DNA-forming sequences have been shown to stimulate genetic instability in various organisms162,163. DNA replication has long been considered a major process implicated in non-B DNA-induced mutagenesis10,28. Hairpin-forming triplet repeats are often more mutagenic in highly proliferative tissues and rapidly dividing cells than in differentiated non-replicating cells18,164,165. Consistent with this finding, the signature mutations stimulated by triplet repeats are expansions or contractions of repeat units, which are likely the result of slippage errors during DNA replication18,166 (Fig. 4). Using a forward mutagenesis assay, different forms of non-B DNA structure, including H-DNA, Z-DNA and cruciform DNA, were found to induce point mutations, DNA single-strand breaks and DSBs, and large-scale deletions in replication-deficient HeLa cell extracts167–169. Therefore, there are evidently multiple mechanisms involved in the mutagenic processing of non-B DNA structures that depend on several factors, including the type of DNA structure, topological conditions and genomic processes such as transcription, replication and DNA repair. In this section, we summarize the replication-dependent and replication-independent mechanisms of non-B DNA structure-induced mutagenesis.
Fig. 4 |. Replication-associated genetic instability induced by non-B DNA.
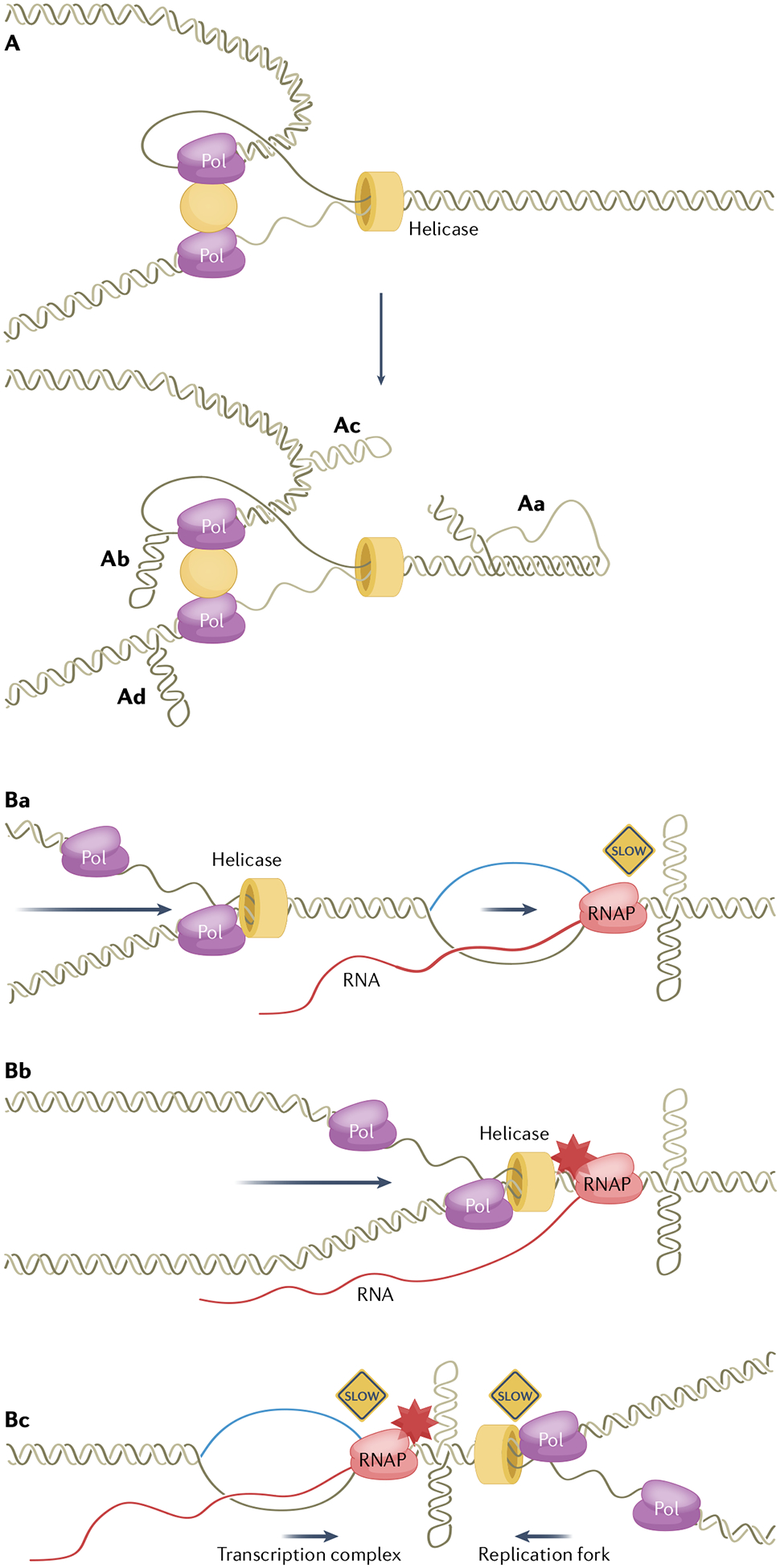
A, Non-B DNA formed at a progressing replication fork. A progressing DNA replication fork is depicted on the top. Aa, A non-B DNA structure (shown in the schematic as H-DNA) in front of a replication fork slows or stalls replication, which gives rise to further structural alterations on the replication complex. Ab, A hairpin structure formed on the template of a lagging strand can lead to replication stalling or repeat contraction (repeat template skipping). Ac, Ad, Hairpin structures formed on the nascent strands on the leading and lagging strands can lead to repeat expansion (via nascent strand self-folding and misalignment). B, Non-B DNA-induced transcription and replication collisions. Ba, Transcription and replication forks in the same direction. Bb, Non-B DNA (shown as a cruciform structure) slows or stalls transcription elongation and leads to a co-directional collision. Bc, Non-B DNA slows or stalls replication or transcription and disrupts the coordination, leading to headon collisions. Collisions in either direction can lead to replication stress and genetic instability. RNAP, RNA polymerase; Pol, DNA polymerase.
Replication-dependent mutations
Both the stability and topological features of non-B DNA on the template strand can affect the processivity and fidelity of DNA polymerases170,171. New advances in nucleic acid sequencing, such as single-molecule real-time (SMRT) sequencing technology have made it possible to determine the processivity and fidelity of DNA polymerases at the nucleotide level8. Studies using such techniques have revealed that many types of non-B DNA motif, such as G4 DNA-forming sequences, Z-DNA-forming GC repeats and hairpin/cruciform-forming AT and CAG repeats, increased sequencing errors that were positively associated with the reduced kinetics of the DNA polymerases8. In a high-throughput primer extension assay at 20,000 different sequences, T7 DNA polymerase was found to be significantly stalled at G4 DNA, hairpins and loop structures, even after controlling for GC context. Its fidelity was also reduced, supporting a plausible mechanism for non-B DNA-induced DNA polymerization difficulties and errors that lead to genetic instability172.
Hairpin or G4 DNA structures formed at CGG repeats in the FMR1 gene impeded all three eukaryotic replicative B-family DNA polymerases; Polα, Polδ and Polε173. An AT(24) repeat and a run of 19–28 As from the common fragile site FRA16D were capable of stalling both Polα and Polδ174. In addition, H-DNA-forming GA or GGAA repeats resulted in Polα pausing, which was more pronounced when the polypurine sequence served as the template140. Polδ can be stalled on the G-rich template of telomeric TTAGGG repeats even in the presence of proliferating cell nuclear antigen (PCNA) and replication factor C (RFC). In addition, a G4 DNA stabilizer, BRACO-19, further inhibited Polδ stalling within G-rich regions175. The proofreading activity of the B-family polymerases helps to reduce misalignment-based replicative errors176,177, and deficiency of Pols α, δ and ε enhanced expansion of the GAA triplet repeat in yeast178. Still, the B-family polymerases created ~1,000-fold more misalignment-based insertion or deletion events on GT(10) or CA(10) repeat templates than in adjacent non-repetitive sequences179, demonstrating the impact of non-B DNA structure-forming repeats in this process.
If the replicative Polδ and Polε are stalled for an extended period of time, DNA polymerases from other families with lower fidelity can be recruited to take over the synthesis through non-B DNA regions to complete genome replication. Such DNA polymerases include the DNA repair X-family members Polβ180 and Polλ181, the Y-family translesion synthesis polymerases Polκ, Polη182–188 and REV1 (refs.189–191), and the A-family translesion polymerase PolQ (Polθ)192,193. Depletion of Polκ and Polη sensitized human cells to the G4 DNA stabilizer telomestatin and led to more DSBs in transgenic HeLa cells that harboured multiple copies of G-rich sequences from the human MYC promoter, which contains multiple G4 DNA, Z-DNA and H-DNA motifs. Furthermore, there were more DSBs in transgenic HeLa cells that contained either the BCL2 gene major break region (Mbr), which contains an H-DNA-forming sequence, or the H-DNA-forming sequences from Kaposi’s sarcoma-associated herpesvirus (KSHV)183. Polκ and Polη facilitated replication through a CTG(100) repeat or a polypurine–polypyrimidine sequence from the PKD1 gene that can adopt H-DNA or G4 DNA structures and attenuated the formation of DSBs185. These results suggest that the less stringent repair and translesion bypass DNA polymerases can facilitate DNA synthesis through non-B DNA-forming regions. However, these lower-fidelity polymerases can lead to base misincorporations and misalignments, resulting in various mutations194. Replication stalling at G4 DNA caused DSBs, and their repair required PolQ, which led to small deletions, in a mechanism that differs from non-homologous end-joining or homologous recombination192. Thus, recruitment of error-prone polymerases to bypass non-B DNA-induced impediments to replication seems to be a double-edged sword for the maintenance of genomic integrity and stability.
Replication fork collapse and DSB formation can occur if the non-B DNA-induced impediment is not unwound or bypassed35,128 (Fig. 4). Using a unique exogenous G-rich sequence with two distinct G4 DNA structure folding possibilities that stall replication forks at different positions, a study in C. elegans revealed that G4 DNA was stable enough to be maintained at the same location during proliferation and stimulated deletions in daughter cells similar to those in parental cells195. Whether or not this persistency occurs in other organisms and/or is unique to G4 DNA remains to be determined.
Expansion of simple repeats in genomic DNA occurs at various stages of development in different cell types and is associated with more than 30 hereditary human diseases. The dysregulation of DNA replication at non-B DNA formed within these repeats is a major driving force of expansion events10,28. Large expansions of CTG/CAG triplet repeats occurred more frequently when the CAG repeats were used as the template during lagging strand replication196, with the nascent CTG strands more likely to form stable hairpin structures than the CAG repeats10,28. A CAG repeat at the 3′ end of an Okazaki initiation zone resulted in expansion, yet it caused contraction events when located at the 5′ end197. In the yeast URA3 reporter gene, long GAA repeats tended to gain a relatively narrow range of 44–63 extra triplets within the length of an Okazaki fragment178. Together, these data demonstrate replication-dependent mechanisms of non-B DNA-induced mutagenesis.
However, a CTG(55) repeat in a transgenic mouse genome surrounded by its native 45 kb genomic segment of the human DMPK gene, which encodes dystrophia myotonica protein kinase, showed expansion with age in the heart, gastrocnemius, liver, pancreas and kidney, with no obvious relationship to cellular proliferation rates198. CTG repeats are also highly unstable in tissues with low levels of proliferation, such as the basal ganglia, cerebral cortex and frontal cortex199,200. Cells derived from various tissues of transgenic mice carrying long CTG(162) repeats exhibited different levels of expansion events, with the highest levels found in the kidney and lower levels in the lung; thus, there was no simple correlation between repeat instability and cell proliferation19. In addition, several cell types in the cerebellum and hippocampus, such as Purkinje cells, showed high levels of CTG repeat expansion, and granule cells were relatively more stable in spinocerebellar ataxias200, suggesting a genomic DNA replication-independent mechanism of DNA structure-induced mutagenesis (see below).
Non-B induced replication–transcription collision
Replication and transcription can occur on the same DNA strand simultaneously in both prokaryotic and eukaryotic genomes. In prokaryotic genomes, the transcriptional templates of highly expressed genes are predominantly on the leading strand during replication201, such that replication and transcription move in the same direction. Eukaryotic genomes are more complicated, as there are tens of thousands of replication origins202, with no substantial preference of positioning genes on replication leading versus lagging strands. As a result, both transcription and replication can initiate at multiple sites and move in different directions on the same chromosome, which increases the risk of ‘head-on’ collisions. Formation of non-B DNA on the templates that are shared by both transcription and replication complexes can enhance the potential of such collisions17 (Fig. 4).
Non-B DNA-forming sequences such as CGG repeats, which can adopt Z-DNA, hairpin or cruciform structures, or cruciform-forming AT-rich palindromes are enriched in fragile sites203. These sites are generally replicated more slowly and at later stages compared with other regions, and are associated with chromosome breakage and disease development204–206. Many common fragile sites nestle in or overlap with large genes that are actively transcribed207,208. Both the large size of the genes and non-B DNA-forming sequences can drastically slow the RNA polymerase complexes, and transcription can take longer than a cell cycle209. Therefore, active transcription and replication must occur together within these fragile sites, increasing the risk of collision. Replication–transcription collision in the genome of actively dividing Bacillus subtilis bacterial cells resulted in duplications, deletions and base substitutions not only at the sites of collision but also in adjacent areas210. Because transcription requires NTPs for RNA synthesis and the progressing transcription complex recruits UTP, thus increasing the local UTP concentration near the stalled DNA polymerases, misincorporation-incorporation of UTP at the template of A could occur when the collision is resolved and replication is restarted211.
Notably, it is common that cancer cells have replication stresses that lead to slower replication fork progression and dysregulated firing of replication origins212. As a result, the inappropriately timed and prolonged replication and overactivated transcription could result in an increased risk of collisions in cancer cells, particularly at non-B DNA-containing regions, which may contribute to oncogene-induced DNA damage and genomic instability in cancer.
In addition to increasing the risk of replication–transcription collision, non-B DNA such as G4 DNA or hairpin structures could also form within the transcription bubble on the non-template ssDNA region. It is reasonable to speculate that, together with an RNA–DNA hybrid formed on the template strand, known as an R-loop (Fig. 1f), non-B DNA formation on the non-template strand could render the stalled R-loop more difficult to resolve before restarting of the replication fork.
Structure-specific repair cleavage models
Some DNA repair proteins screen genomes by sensing distortions of the DNA double helix induced by lesions, which is the initial signal to recognize DNA damage213, followed by recruitment of repair enzymes (for example, nucleases) to the sites of damage to remove the lesions and restore genome integrity. Non-B DNA structures induce distortions in the DNA, affecting protein binding and chromosome organization, similar to some types of DNA damage. As a result, non-B DNA can stimulate DNA damage responses and may be recognized and cleaved by structure-specific DNA repair proteins (Fig. 5). For example, long tracts of CAG(175) repeats on plasmids stimulated expression of the sfiA (sulA) gene, an inhibitor of septum formation induced early in the SOS response214. In comparison with an SOS-defective strain, cells with activated SOS responses increased the supercoiling density of the plasmid, which in turn stimulated non-B DNA formation and deletion frequencies at the CAG repeats214. Long CAG repeats in a yeast artificial chromosome (YAC) also stimulated a DNA damage checkpoint response34,215. A plasmid containing a 2.5 kb H-DNA-forming polypurine–polypyrimidine tract from intron 21 of the unstable human PKD1 gene was recognized by the NER proteins UvrB and UvrC, induced an SOS response in bacteria and delayed growth of the transformed cells216. These findings suggest that non-B DNA structures may be recognized as ‘damage’ to stimulate cellular DNA damage responses. If the DNA structure-specific repair cleavage is subsequently processed in an error-free fashion, the primary sequence will be retained, which could result again in non-B DNA formation, triggering further recognition and cleavage until mutations occur to remove or prevent non-B DNA formation. Thus, structure-specific DNA repair cleavage models provide a plausible explanation for replication-independent non-B DNA-induced mutagenesis in cells, particularly in those with low proliferation rates.
Fig. 5 |. Structure-specific cleavage modulates non-B DNA structure-induced genetic instability.
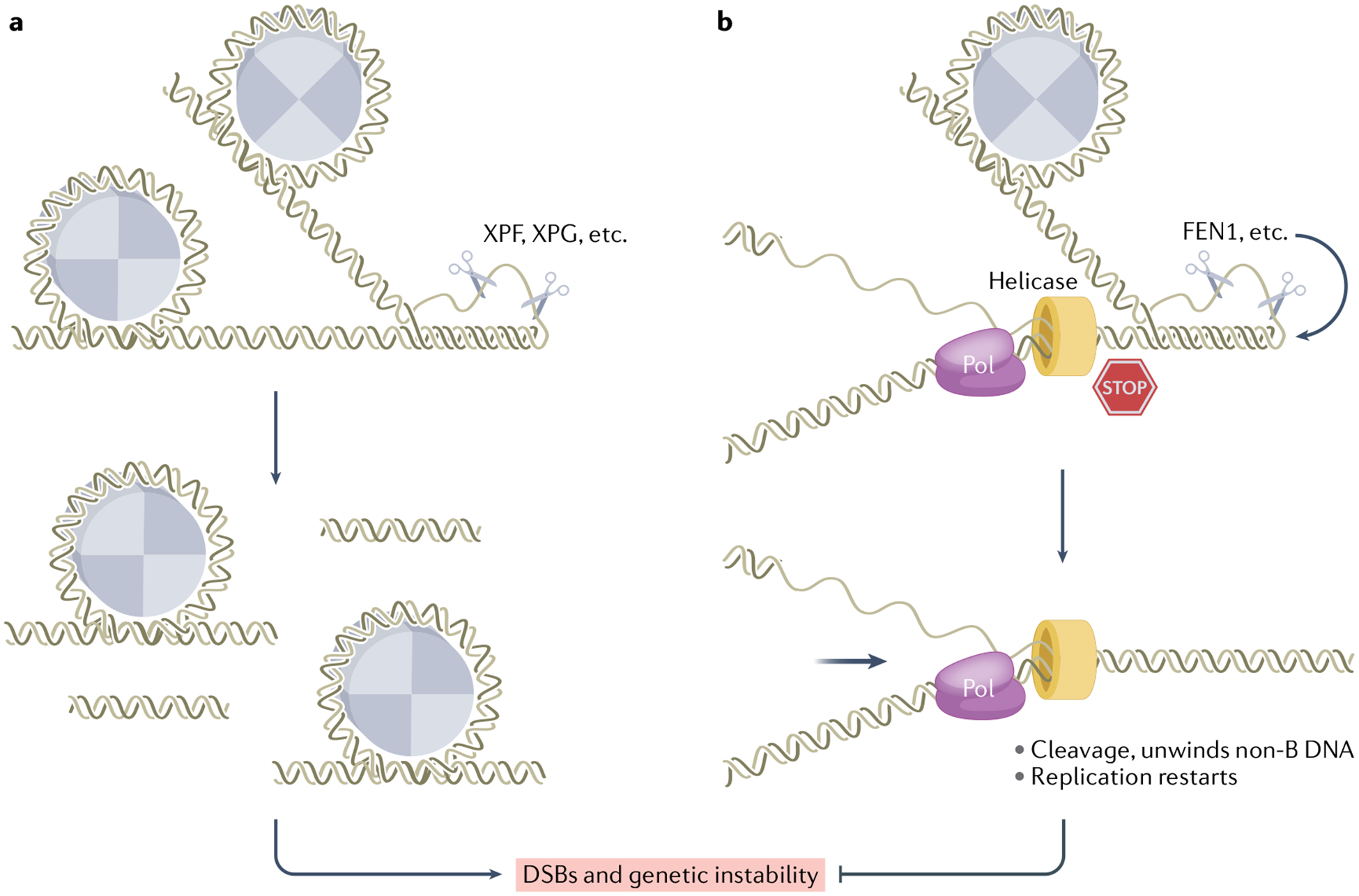
a, Structure-specific cleavage of non-B DNA leads to genetic instability. A non-B DNA structure (shown in the schematic as H-DNA) causes helical distortions and creates an open structure for recruiting DNA repair nucleases. DNA structure-specific cleavage generates breaks within or surrounding the non-B DNA structure, followed by error-free or error-generating repair. This ‘structure forming–repair’ cycle can occur repeatedly until a mutation interrupts the formation of non-B DNA or a deletion removes the non-B DNA-forming sequence. b, A non-B DNA structure (shown in the schematic as H-DNA) is formed in front of a progressing replication fork and stalls DNA replication, increasing the chance for fork collapse and double-strand break (DSB) formation. Structure-specific cleavage of the non-B DNA structure creates a nick or DSB, which unwinds the non-B DNA conformation and reduces structure-induced genetic instability by allowing continuous replication.
Mismatch repair proteins.
Mismatch repair (MMR) proteins, particularly the MSH2–MSH3 complex (MutSβ), sense heteroduplex DNA that contains small loops generated by slippage events at microsatellite repeats (reviewed in ref.33). Single-strand loop-outs are common in many types of non-B DNA structure, such as the simple repeat-induced slippage loops, the unpaired tips of hairpin structures and the unpaired junctions of B-DNA to non-B DNA transitions. Thus, it is reasonable to speculate that MMR proteins recognize and process non-B DNA structures that contain small loops. Indeed, MSH2–MSH3 was found to process short loops within CTG trinucleotide repeats, resulting in repeat unit number alterations, but was not required for processing larger CTG trinucleotide repeat loops (more than five CTG repeats)217–219. Consistent with this result, long CTG(20) loop-outs were processed in cell extracts from multiple different human cells, including neuronal cells, independently of MSH2, MSH3, MSH6, MLH1, PMS2 or PMS1 (ref.220), despite the potential A–A or T–T mismatches formed in the loop-outs. Interestingly, the MSH2–MSH3 complex has a higher binding affinity for small loop-outs than for mismatched base pairs221, and A–A mismatches in fact reduced the binding affinity of MSH2–MSH3 for the hairpins formed at CTG or CAG repeats. The ATPase activity of MSH2–MSH3 was also reduced on the small CA(4) loop compared with perfect base-paired hairpins of the same length222; therefore, the mismatches do not seem to be the main recruiter of MSH2–MSH3 in this case. Similarly, hairpins with 3–17 bp stems and 6–8 nt tips219 or longer perfect inverted repeats217,218 were typically processed independently of MMR proteins. A reasonable hypothesis is that the MSH2–MSH3 complex binds to the junctions of hairpins and can also interact with the tips; yet the tips of longer hairpins are too distant from the junctions and the binding repair complex. However, direct evidence is still lacking. Note that a long CAG repeat could form either a large hairpin or multiple small loops, and the effects of MSH2–MSH3 could be very different depending on the size of each loop. This may explain the different effects of MSH2–MSH3 on CAG repeats in different species and under different experimental conditions. As an example, deficiency of MSH2 (ref.223) or MSH3 (ref.224) reduced CAG expansions and resulted in more contractions in the genomes of mice225 but reduced contractions at long repeats in genomic DNA from human cells226. Therefore, although there is strong evidence to support a role for MMR in the mutagenic processing of hairpin structures, factors such as nucleosome and chromosome structures, transcription and replication activities, and the presence of DNA binding proteins could affect the stability and/or MMR-associated processing of non-B DNA structures.
The MSH2–MSH3 complex has been shown to bind to intermolecular triplex DNA structures with high affinity together with a NER damage or distortion recognition complex, XPA–RPA or XPC–RAD23B227. GAA repeats from the FXN gene, involved in Friedreich’s ataxia, can form loop-outs owing to slippage events or H-DNA structures228, and GAA(120–340) repeats were shown to impede DNA replication forks, resulting in chromosomal breakage and gross chromosomal rearrangements in yeast229. Deficiency of the MMR proteins MSH2, MSH3, MLH1 or PMS1, but not MSH6, suppressed DSBs and reduced large deletions in yeast, suggesting a role for MMR in creating DSBs at GAA repeats, although the MMR deficiency increased the small deletions within these repeats, consistent with the canonical MMR activity229. In addition, the MMR proteins MSH2, MSH3, MLH1 and PMS1 have been shown to be involved in stimulating the formation of DSBs at H-DNA formed at GAA(100) repeats in non-dividing yeast cells, where the DSBs were processed by Exo1 and re-joined by non-homologous end-joining activity, leading to large deletions230.
The human MSH2–MSH6 complex was found to bind to G4 DNA as visualized by electron microscopy and could bind a G4 DNA-forming oligonucleotide as assessed by slower migration in gel mobility-shift assays160. Surprisingly, bacterial MutS binds to G4 DNA with a higher affinity than to G–T mismatches, a canonical MMR substrate. However, adding ATP to the reaction failed to release MutS from G4 DNA as it does on duplex DNA, suggesting a G4 DNA-specific interaction231. In addition, when MutS and MutL were bound to G4 DNA, the hydrolysis activity of MutH was increased by about threefold over that of MutH alone. Interestingly, the binding of MutS to G4 DNA did not seem to require its mismatch discrimination function, as the specific binding was maintained after a highly conserved crucial residue for heteroduplex recognition and mismatch correction was mutated232. Thus, it is possible that MutS has a unique G4-DNA binding motif, leading to repair activity that differs from canonical mismatch-direct MMR. The MSH2–MSH3 complex in conjunction with Polβ facilitated synthesis through GAA and CAG repeats containing abasic sites in vitro. The interaction of MSH2–MSH3 with Polβ increased the potential for flap formation and repeat expansion, rather than contraction events that occurred when Polβ acted on the repeats alone233. Consistent with MMR proteins functioning outside of their canonical roles on non-B DNA, we recently discovered that the MSH2–MSH3 complex, in conjunction with the NER complex Rad10–Rad1 (ERCC1–XPF), was required for Z-DNA-induced genetic instability in yeast and human cells. MSH2–MSH3 associated with Z-DNA, as evidenced by ChIP assays. However, instead of recruiting downstream MMR proteins, the NER complex ERCC1–XPF was recruited to the MSH2–MSH3-bound Z-DNA-containing region. Cleavage of Z-DNA by ERCC1–XPF led to DSBs and genetic instability in yeast and mammalian cells31,234.
Nucleotide excision repair proteins.
Interestingly, another non-B DNA structure, H-DNA, is also cleaved by ERCC1–XPF32, similarly to Z-DNA. However, in contrast to Z-DNA, the functional NER pathway was involved in a distinct mechanism of H-DNA-induced genetic instability, independently of MMR proteins. Deficiency of the NER nucleases ERCC1–XPF and XPG, or the central NER scaffold molecule XPA, reduced H-DNA-induced mutations in yeast and human cells, independently of the DNA replication status. Both ERCC1–XPF and XPG were able to cleave H-DNA in vitro, and ERCC1–XPF binding to H-DNA was reduced in XPA-deficient cells32. These results suggest that NER is responsible for a replication-independent, structure-specific cleavage model of H-DNA-induced mutagenesis. NER proteins monitor helical distortions in the DNA helix induced by bulky DNA adducts235; therefore, H-DNA might be recognized as ‘damage’ owing to its associated helical distortions. In fact, the NER mechanism was also required for intermolecular triplex structure-induced mutagenesis in mammalian cells236, and purified human recombinant XPA–RPA237 and XPC–RAD23B238 were shown to bind to intermolecular triplex DNA structures in vitro with high affinity and specificity.
Purified UvrA binds to supercoiled CAG repeats with higher affinity (by two orders of magnitude) than to linear CAG repeats, suggesting a role for DNA structure in protein–DNA binding interactions. Moreover, deficiency of UvrA reduced the deletion events at CAG repeats in Escherichia coli239, as did its damage recognition partner UvrB240,241. UvrD helicase deficiency increased deletion events at long CAG repeats as expected, perhaps owing to increased or stabilized non-B DNA formation in the helicase-deficient bacterial cells and/or its role in MMR242. Surprisingly, deficiency in the endonuclease UvrC also enhanced CAG-repeat-induced deletions, perhaps because other enzymes can cleave the UvrAB-bound CAG repeats240. The NER proteins XPA, XPC, ERCC1 and XPG, and the MMR proteins MSH2 and MSH3 have also been implicated in contraction of CAG repeats in human cells in a transcription-dependent fashion226,243. Why so many proteins are required for CAG instability and how they coordinate with each other in recognizing and processing these structures remains to be fully elucidated.
Other DNA repair proteins.
In addition to MMR and NER proteins, many other enzymes have been found to have activities on non-B DNA. For example, in yeast, Mre11 can bind to long (>160 bp) palindromic DNA sequences244,245. Purified Mre11 exhibited DNA structure-specific endonuclease activity at hairpin and cruciform structures, and cleaved the DNA at the 5′-junction at the loop of a hairpin, and the junction of a 3′-end flap structure244. The MRN (Mre11–Rad50–Nbs1) complex also interacts with BRCA1, which contains a four-way branched DNA structure binding domain246 and can facilitate the recruitment of the MRN complex to cruciforms247. Sae2 functions together with the MRX complex in yeast to initiate DNA end resection for DSB repair and to process hairpin or cruciform structures248. The Mre11 protein can bind to G4 DNA with higher affinity than to B-DNA and cleave it in a Mn2+-dependent manner249. MRX generates DSBs at cruciform structures at an early stage during pre-meiotic replication, and the meiotic recombination protein Rec12 creates DSBs at a later stage250. Meiosis-specific endonuclease Spo11 also cleaved hairpin structures and was involved in CAG repeat expansions and deletions in yeast251.
Cruciform DNA shares some structural similarities with Holliday junctions and, as expected, the Yen1/GEN1 (refs.252,253) resolvases, SLX1/SLX4 (refs.254,255) and Mus81–Eme1 (Mms4 in budding yeast) resolvases256 were all reported to cleave cruciform structures257,258. These enzymes were also recruited to and cleaved many common fragile site sequences that contained repetitive sequences, leading to DSBs and subsequent genetic instability in mammalian cells259. Recently, a YAC reporter system was used to investigate the genes involved in processing long inverted repeats ranging from 320 bp to 2.7 kb, and identified many endonucleases including the MRX–Sae2 complex, Mus81–Mms4 resolvase, and replication and DSB repairrelated proteins such as Rfa2 (a subunit of heterotrimeric RPA) to be involved. Surprisingly, MUS81, YEN1, SLX4, RAD1 and MLH1 did not significantly affect the DSBs and gross chromosomal rearrangements at these long inverted repeats260. Whether these discrepancies are due to the different reporter systems and/or the different hairpin or cruciform substrates used remains to be determined.
Repair proteins that suppress non-B DNA-induced mutation.
Notably, not all nucleases increase non-B DNA-induced genetic instability. For example, the FANCI-associated nuclease 1 (FAN1), involved in DNA inter-strand crosslink repair, has been shown to bind to MLH1 and inhibit its interaction with MSH3, thereby reducing MMR-promoted CAG repeat expansion in human cells261. FAN1 can dimerize and bind to trinucleotide repeats and cleave slipped CAG or CTG repeats near the junctions262. In addition to its endonuclease activity, FAN1 shows 5′ to 3′ exonuclease activity on hairpin loops containing A–A and T–T mismatches, but is paused by perfectly paired hairpins. Thus, it is plausible that the nuclease activities of FAN1 on CAG repeats represent a counterforce against repeat expansion. In support of this idea, reduced exonuclease activity of FAN1 in individuals with autism was found to be associated with CGG expansions262.
Interestingly, we found that flap structure-specific endonuclease 1 (FEN1) can cleave H-DNA structures in vitro and attenuates H-DNA-induced mutagenesis in eukaryotic cells31,32. FEN1 deficiency increased H-DNA-induced mutagenesis approximately fivefold only in replicationcompetent human and yeast cells. A possible explanation for this finding is that FEN1, as a replication assistant, cleaves H-DNA and diminishes the structural impediment ahead of replication forks, allowing for continuous replication to maintain genetic stability31,32 (Fig. 5).
This section summarizes mechanisms of non-B DNA-induced mutagenesis, and the roles of non-B DNA on replication, transcription–replication collisions, stimulation of structure-specific cleavage and the impact of alternative structures on DNA damage and repair. Of note, although many types of non-B DNA share some structural and functional features, and many enzymes show similar activities towards different types of non-B DNA conformation, each structure has distinct features in terms of how it is recognized and processed. For example, the hairpin structure formed at CAG repeats affects the function of MSH2–MSH3 on DNA mismatches222, whereas G4 DNA does not affect MMR activity on a G–T mismatch in close proximity232. Instead, G4 DNA has been shown to prevent the recognition and excision of 8-oxoG by NEIL1, NEIL3 and OGG1 (ref.263). We also present evidence for the distinct processing of Z-DNA and H-DNA, with both being cleaved by ERCC1–XPF, albeit via different mechanisms31,32. Therefore, individual types of non-B DNA structure should be investigated separately for their distinct mechanisms of mutagenic processing and associated biological outcomes.
Non-B DNA and human disease
DNA is no longer considered passive with regard to incurring damage or mutagenesis. Instead, the DNA itself, in the presence or absence of exogenous damage, can act as a causative factor for genetic instability, leading to DSBs, deletions, recombination and/or large genomic rearrangements that are associated with disease aetiology. Expansions of non-B DNA structure-forming repeats have been implicated in many diseases via various mechanisms, depending on the sequence, length and/or the location of repeats in the affected genes (reviewed in refs.26,28). For example, expansion of CAG repeats within coding regions of genes leads to stretches of polyglutamine (polyQ) in the resulting proteins, contributing to more than 20 neurodegenerative and neuromuscular diseases, including Huntington disease, spinal and bulbar muscular atrophy, dentatorubral-pallidoluysian atrophy and several types of spinocerebellar ataxia (reviewed in ref.26). CGG expansions in the non-coding 5′-UTR of FMR1 inhibit transcription and contribute to fragile X syndrome264. The repeats can also stall replication265 and recruit structure-specific ‘repair’ cleavage enzymes, leading to chromosomal abnormalities266. The CGG repeats in the FMR1 RNA can also affect the translation of other RNAs within the same RNA granules267. Expansion of CTG repeats in the 3′-UTR of the DMPK gene leads to long CUG runs in the RNA that can interfere with the developmentally regulated alternative splicing of pre-mRNAs, resulting in toxicity associated with myotonic dystrophy type 1 (ref.268).
In addition to disorders caused by repetitive DNA itself, many types of non-B DNA structure are involved in human diseases associated with genetic instability, such as cancer (reviewed in ref.32). Using a bioinformatic approach, it was found that ~33% of missense mutations and ~37% of microdeletions in the Human Gene Mutation Database (HGMD)269 occurred within non-B DNA-forming repeats, which is significantly higher than by chance alone270. Potential non-B DNA-forming sequences were also found to have higher substitution frequencies in the UCSC Genome Browser and the Simons Genome Diversity datasets162. From an analysis of 1,809 whole-genome sequences from ten cancer types, more mutations were found near or within non-B DNA regions in general, particularly at the single-stranded spacer regions between symmetrical arms of direct and inverted repeats or H-DNA mirror repeats163. Non-B DNA-forming sequences are also enriched at chromosomal breakpoints identified in many diseases, including translocation-related cancers, such as lymphomas and leukaemias271,272. For example, the human MYC promoter region contains multiple overlapping non-B DNA-forming sequences with the capacity to adopt H-DNA, G4 DNA and Z-DNA within a small region (~400 bp) surrounding the P1 promoter, which is one of the major breakpoint cluster regions in MYC-related translocations in lymphomas40. H-DNA has been implicated in translocation events within the BCL2 major breakpoint region in follicular lymphomas273. G4 DNA is associated with mutations in ataxias and fragile X syndrome274. In a dataset from the International Cancer Genome Consortium Data Portal, comprising 2,234 samples from ten cancer types, stem–loop structures fit the blood, brain, liver and prostate cancer breakpoint hotspot profiles, and G4 DNA-forming sequences co-localized with mutation hotspots in bone, breast, ovarian, pancreatic and skin cancers at levels much higher than by chance alone275. In 200–400 bp windows flanking breakpoints, characterized in 19,947 translocations in human cancer genomes, non-B DNA-forming sequences were significantly enriched, including simple AT repeats, GAA or GAAA repeats and other repetitive sequences that have the propensity to adopt H-DNA, Z-DNA, G4 DNA and cruciform or hairpin structures32,272. A very recent study analysed ~630,000 cancer breakpoints using machine learning tools to search for genetic features associated with genome breakage. It was found that the cancer-associated breakpoint hotspots were predictable, and that transcription and the presence of non-B DNA motifs were predominant factors responsible for the development of the hotspots276.
Although non-B DNA-forming sequences have been implicated in the development of many diseases, it is likely that many of the non-B DNA-induced mutations are silent and do not exhibit an obvious phenotype. For example, long palindromic AT-rich repeats on chromosomes 11q23 and 22q11 have been shown to stimulate a high frequency (~10–5) of de novo t(11;22) translocations in sperm cells from healthy men277. However, most cells that carry these mutations do not show a phenotype and/or are lost in screening systems, whereas other cells with non-B DNA-induced mutations in crucial genomic regions could trigger apoptosis and would no longer be detectable in the population. Therefore, non-B DNA-induced mutations in human genomes are likely much more frequent than estimated.
Conclusions and perspectives
Although our knowledge of non-B DNA structures, their functions in the genome and contributions to evolution and disease has advanced substantially over the past few decades, there is still much to be discovered. Thus, many challenges remain to be addressed in future studies, some of which are listed below.
To date, it remains a challenge to directly detect non-B DNA structures in genomes of living cells, in part because their formation in vivo is dynamic and transient. The methods to detect non-B DNA in cells thus far depend on indirect measurements and/or the inclusion of steps that can alter non-B DNA formation, such as cell fixation, protein removal, changes in pH and salt concentration or the addition of probes or antibodies that can induce structure formation, and/or stabilize or destabilize existing non-B DNA structures. Some recent progress has been made in this respect. For example, a small-molecule fluorophore was conjugated with a G4 DNA ligand, pyridostatin, to form a fluorescent G4 DNA probe278. The small molecular weight and polarity of the probe molecule enabled penetration of the cell membrane, and the low cell toxicity fluorescent probe was used for real-time detection of single G4 DNA structures in living cells. This small-molecule probebased procedure is less toxic to host cells than other methods and is compatible with studies in living cells, and as such better reflects the physiological situation in vivo. Note that owing to the dynamic nature of non-B DNA conformations, using a DNA structure-specific probe will inevitably affect the balance of the B-DNA to non-B DNA structure formation transition and equilibrium. Thus, more sensitive and less invasive methods that can directly detect non-B DNA without interfering with DNA structural transitions in cells would substantially advance the field.
Our understanding of free energy alterations, the kinetics of B-DNA to non-B DNA transitions, the interactions and bonds between the atoms involved in non-B DNA formation and stabilization are still limited, particularly measurements under physiological conditions. As a result, although many computer programs for predicting non-B DNA conformations in genomes are available, they are empirically derived. Thus, a better understanding of these most fundamental and basic factors in non-B DNA structure formation is crucial for future progress in the field.
On the basis of decades of research, we now know that non-B DNA-forming sequences have important roles in various biological processes and can stimulate genetic instability, implicating them in evolution and disease aetiology. Therefore, strategies to modulate these structures using small molecules or probes are warranted owing to their ability to provide spatial and temporal tools to interrogate DNA structures and their potential as therapeutic agents279,280. Much progress has been made towards this goal by employing in silico simulation and high-throughput methods to identify small-molecule ligands to modulate non-B DNA formation and/or stability. However, small ligands can interact with genomic and mitochondrial DNA and affect DNA structure throughout the genome and may have off-target effects, potentially limiting their therapeutic efficacy. Thus, the development of approaches to increase specificity by combining ligands with sequence-specific targeting is warranted. Such approaches may include the use of non-B DNA-specific probes, antisense oligonucleotides, intermolecular triplex-forming oligonucleotides or peptide nucleic acids281.
In a recent study, the structure-specific DNA ligand naphthyridine-azaquinolone (NA) was used to treat cells from patients with Huntington disease or a Huntington disease mouse model that harbours a transgene of HTT (huntingtin) exon 1 containing CAG repeats282. NA, which binds specifically to slipped CAG DNA intermediates and thus affects expanded CAG repeats, induced repeat contractions in both cultured human cells and medium spiny neurons of the mouse striatum. NA injection reduced mutant HTT protein aggregates in mice, a biomarker of Huntington disease pathogenesis, suggesting a promising approach to reduce the pathogenic repeat length with non-B DNA structure-specific DNA ligands282. This type of compound that can specifically interact with pathology-related non-B DNA conformations is needed to advance the effort towards developing DNA-targeted therapeutics.
Using the G4 DNA-specific binding domain of RNA helicase associated with AU-rich element (RHAU, also known as DHX36) fused with a cleavage domain of the Fok1 nuclease, a G-quadruplex-specific DNA endonuclease was constructed to cleave double-stranded DNA adjacent to G4 DNA-forming sequences283. Such enzymes represent powerful tools for DNA structure research. If they can be delivered in vivo and their expression controlled, they may provide another approach for targeted genome modification. Although not necessarily a DNA structure-specific approach, CRISPR–Cas9 techniques have also been applied to delete core G4 DNA sequences within a microRNA cluster in rat, which led to increased microRNA levels in the heart, contributing to cardiac contractile dysfunction284. In future studies, similar strategies could be employed to modulate the structure and function of other non-B DNA-forming sequences in vivo.
Taking advantage of sequence-specific DNA triplex formation, short triplex-forming oligonucleotides have been developed that can form intermolecular triplexes that share structural similarities with intramolecular H-DNA. Binding of triplex-forming oligonucleotides to target duplexes to form stable triplex structures can inhibit DNA replication and gene expression, and stimulate site-specific mutations in genomes281. Chemotherapeutic DNA-damaging agents can also be used in conjunction with or covalently linked to triplex-forming oligonucleotides to direct site-specific damage in genomes to enhance the efficacy of cancer chemotherapy285,286.
In summary, because non-B DNA structures have important roles in various biological and pathological processes and provide unique structural features for recognition and binding, they represent potential druggable targets for use as both research tools and therapeutics. Although limitations still exist in this type of approach, such as specificity for the targeted non-B DNA versus B-DNA regions, potential off-target effects and efficacy based on targeting multiple similar structures, progress in this area is promising. For example, a small-molecule G4 DNA-specific ligand, CX-5461, is currently being evaluated in an open-label, multi-centre phase Ib study as a potential targeted cancer therapeutic for BRCA1/BRCA2 homologous recombination-deficient tumours287,288. Nonetheless, until direct and conclusive methods are available to detect and specifically target non-B DNA in vivo, caution is warranted when studying such complex and dynamic non-B DNA structures in the genome.
Acknowledgements
The authors thank A. Wang for support with artwork. The author’s work is supported by the NIH (NIH/NCI CA093729 to K.M.V.).
Glossary
- Circular dichroism
Absorption spectroscopy method to detect the differential absorption of leftand right-handed light spectra for rapid evaluation of the secondary structures of macromolecules such as protein and DNA
- DNA helicases
A class of motor proteins that move along DNA and transiently separate duplexes into two single strands using energy from ATP hydrolysis
- Fourier transform infrared spectroscopy
A spectroscopy method that simultaneously collects the absorption, emission and photoconductivity of a wide spectral range at high resolution to measure the intensity and wavelength of light required to vibrate molecules in a sample
- Holliday junctions
Branched DNA structures containing four arms covalently linked together that serve as key intermediates in many meiotic and mitotic homologous recombination events
- Negative supercoiling
A segment of underwound DNA in which the two strands wind around the helical axis less than 360° every 10.5 bp and retain twist strain (free energy)
- Okazaki fragments
Short fragments of DNA produced by discontinuous replication on the lagging strand during DNA replication. Because the template for lagging strand synthesis is exposed in the 5′–3′ direction at the progressing replication fork, the nascent strand is composed of sequential Okazaki fragments created by DNA polymerase working backwards from the replication fork
- Satellites
A subfraction of genomic DNA consisting of short repetitive nucleotide sequences that are repeated a large number of times. These non-coding repeats are important for centromere and heterochromatin construction and separate from the rest of the genomic DNA on a density gradient because of their higher content of AT base pairs
- SOS response
A complex global response to DNA damage identified in bacteria that includes activation of multiple factors, leading to the stalling of cell division and alteration of DNA replication, recombination and repair to promote genome integrity and cell survival, at the cost of increased mutagenesis
- Stretching tension
When both ends of a segment of DNA are anchored (for example, by proteins) and the DNA is pulled mechanically, it carries stretching tension coupled with twisting torsion along the helix and can be elongated by up to 70% without disrupting base pairs
- Topoisomerases
A class of enzymes that are able to cleave one or both strands of DNA to release topological stress on DNA duplex, and to link or unlink, knot or unknot associated DNA molecules
- Translesion synthesis polymerases
Polymerases that can catalyse DNA polymerization at damaged templates during replication and/or repair, although often with lower fidelty than replicative polymerases
- Unequal sister chromatid exchange
A mitotic crossover event that leads to the exchange of genetic material between homologous chromosomes and is also a major repair pathway for double-strand breaks
Related links
detectIR: https://sourceforge.net/projects/detectir/
DNA Structure Search: http://utw10685.utweb.utexas.edu/nonbdna/
G4Hunter: http://bioinformatics.ibp.cz
palindrome: http://emboss.bioinformatics.nl/cgi-bin/emboss/palindrome
QGRS Mapper: https://bioinformatics.ramapo.edu/QGRS/index.php
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Nurk S et al. The complete sequence of a human genome. Science 376, 44–53 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]; The most recent and compete human genome sequencing assembly reveals more repetitive elements in the human genome than researchers have previously estimated, which could potentially support non-B DNA formation.
- 2.Plohl M, Luchetti A, Mestrovic N & Mantovani B Satellite DNAs between selfishness and functionality: structure, genomics and evolution of tandem repeats in centromeric (hetero)chromatin. Gene 409, 72–82 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Thakur J, Packiaraj J & Henikoff S Sequence, chromatin and evolution of satellite DNA. Int. J. Mol. Sci 22, 4309 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Herbert A ALU non-B-DNA conformations, flipons, binary codes and evolution. R. Soc. Open. Sci 7, 200222 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kasinathan S & Henikoff S Non-B-form DNA is enriched at centromeres. Mol. Biol. Evol 35, 949–962 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang G & Vasquez KM Impact of alternative DNA structures on DNA damage, DNA repair, and genetic instability. DNA Repair 19, 143–151 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi J & Majima T Conformational changes of non-B DNA. Chem. Soc. Rev 40, 5893–5909 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Guiblet WM et al. Long-read sequencing technology indicates genome-wide effects of non-B DNA on polymerization speed and error rate. Genome Res. 28, 1767–1778 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marshall PR et al. Dynamic regulation of Z-DNA in the mouse prefrontal cortex by the RNA-editing enzyme Adar1 is required for fear extinction. Nat. Neurosci 23, 718–729 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; The ADAR1 protein binds to Z-DNA in the mouse prefrontal cortex during fear extinction learning and supresses or reduces Z-DNA formation, which is suggested to be required for memory flexibility.
- 10.Mirkin SM Expandable DNA repeats and human disease. Nature 447, 932–940 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Praseuth D, Guieysse AL & Helene C Triple helix formation and the antigene strategy for sequence-specific control of gene expression. Biochim. Biophys. Acta 1489, 181–206 (1999). [DOI] [PubMed] [Google Scholar]
- 12.Huppert JL & Balasubramanian S G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 35, 406–413 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marsico G et al. Whole genome experimental maps of DNA G-quadruplexes in multiple species. Nucleic Acids Res. 47, 3862–3874 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Valton AL & Prioleau MN G-quadruplexes in DNA replication: a problem or a necessity? Trends Genet. 32, 697–706 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Wang G & Vasquez KM Effects of replication and transcription on DNA structure-related genetic instability. Genes 8, 17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prioleau MN G-quadruplexes and DNA replication origins. Adv. Exp. Med. Biol 1042, 273–286 (2017). [DOI] [PubMed] [Google Scholar]
- 17.St Germain C, Zhao H & Barlow JH Transcription-replication collisions — a series of unfortunate events. Biomolecules 11, 1249 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu G, Chen X, Bissler JJ, Sinden RR & Leffak M Replication-dependent instability at (CTG) x (CAG) repeat hairpins in human cells. Nat. Chem. Biol 6, 652–659 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gomes-Pereira M, Fortune MT & Monckton DG Mouse tissue culture models of unstable triplet repeats: in vitro selection for larger alleles, mutational expansion bias and tissue specificity, but no association with cell division rates. Hum. Mol. Genet 10, 845–854 (2001). [DOI] [PubMed] [Google Scholar]
- 20.Fu YH et al. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell 67, 1047–1058 (1991). [DOI] [PubMed] [Google Scholar]
- 21.Kremer EJ et al. Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n. Science 252, 1711–1714 (1991). [DOI] [PubMed] [Google Scholar]
- 22.La Spada AR, Wilson EM, Lubahn DB, Harding AE & Fischbeck KH Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 352, 77–79 (1991). [DOI] [PubMed] [Google Scholar]
- 23.Catasus L et al. Frameshift mutations at coding mononucleotide repeat microsatellites in endometrial carcinoma with microsatellite instability. Cancer 88, 2290–2297 (2000). [PubMed] [Google Scholar]
- 24.Georgakopoulos-Soares I et al. Transcription-coupled repair and mismatch repair contribute towards preserving genome integrity at mononucleotide repeat tracts. Nat. Commun 11, 1980 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; Using bioinformatic approaches, this study reports transcription-associated asymmetrical distribution of repetitive elements, insertions and deletions at repeats in human cancer genomes, with involvement of DNA repair pathways.
- 25.Rothenburg S, Koch-Nolte F, Rich A & Haag F A polymorphic dinucleotide repeat in the rat nucleolin gene forms Z-DNA and inhibits promoter activity. Proc. Natl Acad. Sci. USA 98, 8985–8990 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malik I, Kelley CP, Wang ET & Todd PK Molecular mechanisms underlying nucleotide repeat expansion disorders. Nat. Rev. Mol. Cell Biol 22, 589–607 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paulson HL & Fischbeck KH Trinucleotide repeats in neurogenetic disorders. Annu. Rev. Neurosci 19, 79–107 (1996). [DOI] [PubMed] [Google Scholar]
- 28.McMurray CT Mechanisms of trinucleotide repeat instability during human development. Nat. Rev. Genet 11, 786–799 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones L, Houlden H & Tabrizi SJ DNA repair in the trinucleotide repeat disorders. Lancet Neurol. 16, 88–96 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Cleary JD & Pearson CE Replication fork dynamics and dynamic mutations: the fork-shift model of repeat instability. Trends Genet. 21, 272–280 (2005). [DOI] [PubMed] [Google Scholar]
- 31.McKinney JA et al. Distinct DNA repair pathways cause genomic instability at alternative DNA structures. Nat. Commun 11, 236 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports that the MMR protein complex MSH2–MSH3 binds to Z-DNA and recruits the NER nuclease ERCC1–XPF to the site, resulting in structure-specific cleavage and DSBs at Z-DNA regardless of DNA replication status.
- 32.Zhao J et al. Distinct mechanisms of nuclease-directed DNA-structure-induced genetic instability in cancer genomes. Cell Rep. 22, 1200–1210 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iyer RR, Pluciennik A, Napierala M & Wells RD DNA triplet repeat expansion and mismatch repair. Annu. Rev. Biochem 84, 199–226 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sundararajan R & Freudenreich CH Expanded CAG/CTG repeat DNA induces a checkpoint response that impacts cell proliferation in Saccharomyces cerevisiae. PLoS Genet. 7, e1001339 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; Long CAG/CTG repeats trigger an MRX-dependent DNA damage checkpoint response in budding yeast, which affects the cell cycle, leading to repeat-dependent S-phase delays and G2/M arrests, which results in morphological abnormalities.
- 35.Voineagu I, Surka CF, Shishkin AA, Krasilnikova MM & Mirkin SM Replisome stalling and stabilization at CGG repeats, which are responsible for chromosomal fragility. Nat. Struct. Mol. Biol 16, 226–228 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Orr HT & Zoghbi HY Trinucleotide repeat disorders. Annu. Rev. Neurosci 30, 575–621 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Ye C, Ji G, Li L & Liang C detectIR: a novel program for detecting perfect and imperfect inverted repeats using complex numbers and vector calculation. PLoS ONE 9, e113349 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kikin O, D’Antonio L & Bagga PS QGRS Mapper: a web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res. 34, W676–W682 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brazda V et al. G4Hunter web application: a web server for G-quadruplex prediction. Bioinformatics 35, 3493–3495 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang G, Gaddis S & Vasquez KM Methods to detect replication-dependent and replication-independent DNA structure-induced genetic instability. Methods 64, 67–72 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barshai M, Aubert A & Orenstein Y G4detector: convolutional neural network to predict DNA G-quadruplexes. IEEE/ACM Trans. Comput. Biol. Bioinform 19, 1946–1955 (2022). [DOI] [PubMed] [Google Scholar]
- 42.Jenjaroenpun P & Kuznetsov VA TTS mapping: integrative WEB tool for analysis of triplex formation target DNA sequences, G-quadruplets and non-protein coding regulatory DNA elements in the human genome. BMC Genomics 10, S9 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cer RZ et al. Non-B DB v2.0: a database of predicted non-B DNA-forming motifs and its associated tools. Nucleic Acids Res. 41, D94–D100 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang G, Zhao J & Vasquez KM Detection of cis- and trans-acting factors in DNA structure-induced genetic instability using in silico and cellular approaches. Front. Genet 7, 135 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beknazarov N, Jin S & Poptsova M Deep learning approach for predicting functional Z-DNA regions using omics data. Sci. Rep 10, 19134 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rocher V, Genais M, Nassereddine E & Mourad R DeepG4: a deep learning approach to predict cell-type specific active G-quadruplex regions. PLoS Comput. Biol 17, e1009308 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bian Y et al. Insights into the kinetic partitioning folding dynamics of the human telomeric G-quadruplex from molecular simulations and machine learning. J. Chem. Theory Comput 16, 5936–5947 (2020). [DOI] [PubMed] [Google Scholar]
- 48.Kouzine F et al. Permanganate/S1 nuclease footprinting reveals non-B DNA structures with regulatory potential across a mammalian genome. Cell Syst. 4, 344–356 e347 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abeysinghe SS, Chuzhanova N, Krawczak M, Ball EV & Cooper DN Translocation and gross deletion breakpoints in human inherited disease and cancer I: nucleotide composition and recombination-associated motifs. Hum. Mutat 22, 229–244 (2003). [DOI] [PubMed] [Google Scholar]
- 50.Rahmouni AR & Wells RD Stabilization of Z DNA in vivo by localized supercoiling. Science 246, 358–363 (1989). [DOI] [PubMed] [Google Scholar]
- 51.Koeris M, Funke L, Shrestha J, Rich A & Maas S Modulation of ADAR1 editing activity by Z-RNA in vitro. Nucleic Acids Res. 33, 5362–5370 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Herbert A et al. The Zalpha domain from human ADAR1 binds to the Z-DNA conformer of many different sequences. Nucleic Acids Res. 26, 3486–3493 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang X, Spiegel J, Martinez Cuesta S, Adhikari S & Balasubramanian S Chemical profiling of DNA G-quadruplex-interacting proteins in live cells. Nat. Chem 13, 626–633 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; Hundreds of putative G4 DNA binding proteins from various functional classes are identified using G4-ligand probes crosslinked to G4 binding proteins in situ in living cells, suggesting complex and active DNA structure-related metabolism in vivo.
- 54.Zheng LL et al. pH-responsive DNA motif: from rational design to analytical applications. Front. Chem 9, 732770 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Son H, Bae S & Lee S A thermodynamic understanding of the salt-induced B-to-Z transition of DNA containing BZ junctions. Biochem. Biophys. Res. Commun 583, 142–145 (2021). [DOI] [PubMed] [Google Scholar]
- 56.Potaman VN, Ussery DW & Sinden RR Formation of a combined H-DNA/open TATA box structure in the promoter sequence of the human Na,K-ATPase alpha2 gene. J. Biol. Chem 271, 13441–13447 (1996). [DOI] [PubMed] [Google Scholar]
- 57.Htun H & Dahlberg JE Topology and formation of triple-stranded H-DNA. Science 243, 1571–1576 (1989). [DOI] [PubMed] [Google Scholar]
- 58.Latha KS, Anitha S, Rao KS & Viswamitra MA Molecular understanding of aluminum-induced topological changes in (CCG)12 triplet repeats: relevance to neurological disorders. Biochim. Biophys. Acta 1588, 56–64 (2002). [DOI] [PubMed] [Google Scholar]
- 59.Fakharzadeh A, Zhang J, Roland C & Sagui C Novel eGZ-motif formed by regularly extruded guanine bases in a left-handed Z-DNA helix as a major motif behind CGG trinucleotide repeats. Nucleic Acids Res. 50, 4860–4876 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ajjugal Y, Kolimi N & Rathinavelan T Secondary structural choice of DNA and RNA associated with CGG/CCG trinucleotide repeat expansion rationalizes the RNA misprocessing in FXTAS. Sci. Rep 11, 8163 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim SH, Jung HJ, Lee IB, Lee NK & Hong SC Sequence-dependent cost for Z-form shapes the torsion-driven B-Z transition via close interplay of Z-DNA and DNA bubble. Nucleic Acids Res. 49, 3651–3660 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang F, Huang Q, Yan J & Chen Z Histone acetylation induced transformation of B-DNA to Z-DNA in cells probed through FT-IR spectroscopy. Anal. Chem 88, 4179–4182 (2016). [DOI] [PubMed] [Google Scholar]
- 63.Li Y et al. Remodeling chromatin induces Z-DNA conformation detected through Fourier transform infrared spectroscopy. Anal. Chem 92, 14452–14458 (2020). [DOI] [PubMed] [Google Scholar]
- 64.Krassovsky K, Ghosh RP & Meyer BJ Genome-wide profiling reveals functional interplay of DNA sequence composition, transcriptional activity, and nucleosome positioning in driving DNA supercoiling and helix destabilization in C. elegans. Genome Res. 31, 1187–1202 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; DNA supercoiling regions and non-B DNA structures in the genome of C. elegans embryos are mapped and found to co-localize at functional regions in the genome, such as transcription start sites.
- 65.Wittig B, Dorbic T & Rich A Transcription is associated with Z-DNA formation in metabolically active permeabilized mammalian cell nuclei. Proc. Natl Acad. Sci. USA 88, 2259–2263 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wolfl S, Wittig B & Rich A Identification of transcriptionally induced Z-DNA segments in the human c-myc gene. Biochim. Biophys. Acta 1264, 294–302 (1995). [DOI] [PubMed] [Google Scholar]
- 67.Wittig B, Wolfl S, Dorbic T, Vahrson W & Rich A Transcription of human c-myc in permeabilized nuclei is associated with formation of Z-DNA in three discrete regions of the gene. EMBO J. 11, 4653–4663 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Michelotti GA et al. Multiple single-stranded cis elements are associated with activated chromatin of the human c-myc gene in vivo. Mol. Cell. Biol 16, 2656–2669 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Feng X, Xie FY, Ou XH & Ma JY Cruciform DNA in mouse growing oocytes: its dynamics and its relationship with DNA transcription. PLoS ONE 15, e0240844 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schwartz T, Behlke J, Lowenhaupt K, Heinemann U & Rich A Structure of the DLM-1-Z-DNA complex reveals a conserved family of Z-DNA-binding proteins. Nat. Struct. Biol 8, 761–765 (2001). [DOI] [PubMed] [Google Scholar]
- 71.Baik JY et al. ZBP1 not RIPK1 mediates tumor necroptosis in breast cancer. Nat. Commun 12, 2666 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ha SC et al. The crystal structure of the second Z-DNA binding domain of human DAI (ZBP1) in complex with Z-DNA reveals an unusual binding mode to Z-DNA. Proc. Natl Acad. Sci. USA 105, 20671–20676 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rothenburg S et al. A PKR-like eukaryotic initiation factor 2alpha kinase from zebrafish contains Z-DNA binding domains instead of dsRNA binding domains. Proc. Natl Acad. Sci. USA 102, 1602–1607 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim YG, Lowenhaupt K, Oh DB, Kim KK & Rich A Evidence that vaccinia virulence factor E3L binds to Z-DNA in vivo: implications for development of a therapy for poxvirus infection. Proc. Natl Acad. Sci. USA 101, 1514–1518 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kus K et al. The structure of the Cyprinid herpesvirus 3 ORF112-Zalpha.Z-DNA complex reveals a mechanism of nucleic acids recognition conserved with E3L, a poxvirus inhibitor of interferon response. J. Biol. Chem 290, 30713–30725 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brazda V, Laister RC, Jagelska EB & Arrowsmith C Cruciform structures are a common DNA feature important for regulating biological processes. BMC Mol. Biol 12, 33 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Reddy MC, Christensen J & Vasquez KM Interplay between human high mobility group protein 1 and replication protein A on psoralen-cross-linked DNA. Biochemistry 44, 4188–4195 (2005). [DOI] [PubMed] [Google Scholar]
- 78.Meier-Stephenson V G4-quadruplex-binding proteins: review and insights into selectivity. Biophys. Rev 14, 635–654 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chu WK & Hickson ID RecQ helicases: multifunctional genome caretakers. Nat. Rev. Cancer 9, 644–654 (2009). [DOI] [PubMed] [Google Scholar]
- 80.Liberi G et al. Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev. 19, 339–350 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shishkin AA et al. Large-scale expansions of Friedreich’s ataxia GAA repeats in yeast. Mol. Cell 35, 82–92 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Butler DK, Yasuda LE & Yao MC Induction of large DNA palindrome formation in yeast: implications for gene amplification and genome stability in eukaryotes. Cell 87, 1115–1122 (1996). [DOI] [PubMed] [Google Scholar]
- 83.Fleming AM, Ding Y & Burrows CJ Oxidative DNA damage is epigenetic by regulating gene transcription via base excision repair. Proc. Natl Acad. Sci. USA 114, 2604–2609 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fleming AM, Zhu J, Jara-Espejo M & Burrows CJ Cruciform DNA sequences in gene promoters can impact transcription upon oxidative modification of 2′-deoxyguanosine. Biochemistry 59, 2616–2626 (2020). [DOI] [PubMed] [Google Scholar]
- 85.Roychoudhury S et al. Endogenous oxidized DNA bases and APE1 regulate the formation of G-quadruplex structures in the genome. Proc. Natl Acad. Sci. USA 117, 11409–11420 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Volker J, Plum GE, Klump HH & Breslauer KJ DNA repair and DNA triplet repeat expansion: the impact of abasic lesions on triplet repeat DNA energetics. J. Am. Chem. Soc 131, 9354–9360 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lai Y et al. Base excision repair of chemotherapeutically-induced alkylated DNA damage predominantly causes contractions of expanded GAA repeats associated with Friedreich’s ataxia. PLoS ONE 9, e93464 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bochman ML, Paeschke K & Zakian VA DNA secondary structures: stability and function of G-quadruplex structures. Nat. Rev. Genet 13, 770–780 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Smith SS Evolutionary expansion of structurally complex DNA sequences. Cancer Genomics Proteomics 7, 207–215 (2010). [PubMed] [Google Scholar]
- 90.Luger K, Mader AW, Richmond RK, Sargent DF & Richmond TJ Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389, 251–260 (1997). [DOI] [PubMed] [Google Scholar]
- 91.Godde JS, Kass SU, Hirst MC & Wolffe AP Nucleosome assembly on methylated CGG triplet repeats in the fragile X mental retardation gene 1 promoter. J. Biol. Chem 271, 24325–24328 (1996). [DOI] [PubMed] [Google Scholar]
- 92.Linxweller W & Horz W Reconstitution experiments show that sequence-specific histone-DNA interactions are the basis for nucleosome phasing on mouse satellite DNA. Cell 42, 281–290 (1985). [DOI] [PubMed] [Google Scholar]
- 93.Ruan H & Wang YH Friedreich’s ataxia GAA.TTC duplex and GAA.GAA.TTC triplex structures exclude nucleosome assembly. J. Mol. Biol 383, 292–300 (2008). [DOI] [PubMed] [Google Scholar]
- 94.Miura O, Ogake T, Yoneyama H, Kikuchi Y & Ohyama T A strong structural correlation between short inverted repeat sequences and the polyadenylation signal in yeast and nucleosome exclusion by these inverted repeats. Curr. Genet 65, 575–590 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wong HM & Huppert JL Stable G-quadruplexes are found outside nucleosome-bound regions. Mol. Biosyst 5, 1713–1719 (2009). [DOI] [PubMed] [Google Scholar]
- 96.Shen J et al. Promoter G-quadruplex folding precedes transcription and is controlled by chromatin. Genome Biol. 22, 143 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Godde JS & Wolffe AP Nucleosome assembly on CTG triplet repeats. J. Biol. Chem 271, 15222–15229 (1996). [DOI] [PubMed] [Google Scholar]
- 98.Wada-Kiyama Y & Kiyama R Conservation and periodicity of DNA bend sites in the human beta-globin gene locus. J. Biol. Chem 270, 12439–12445 (1995). [DOI] [PubMed] [Google Scholar]
- 99.Hou Y et al. Integrative characterization of G-quadruplexes in the three-dimensional chromatin structure. Epigenetics 14, 894–911 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kanoh Y et al. Rif1 binds to G quadruplexes and suppresses replication over long distances. Nat. Struct. Mol. Biol 22, 889–897 (2015). [DOI] [PubMed] [Google Scholar]; Rif1 is able to bind to G4 DNA motifs at selected intergenic regions in the fission yeast genome and create local chromatin structures that suppress late-firing of dormant origins located up to 50 kb from these regions.
- 101.Barbault F, Huynh-Dinh T, Paoletti J & Lanceloti G A new peculiar DNA structure: NMR solution structure of a DNA kissing complex. J. Biomol. Struct. Dyn 19, 649–658 (2002). [DOI] [PubMed] [Google Scholar]
- 102.Xu X & Chen SJ Topological constraints of RNA pseudoknotted and loop-kissing motifs: applications to three-dimensional structure prediction. Nucleic Acids Res. 48, 6503–6512 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Williams JD et al. Characterization of long G4-rich enhancer-associated genomic regions engaging in a novel loop:loop ‘G4 Kissing’ interaction. Nucleic Acids Res. 48, 5907–5925 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Son LS, Bacolla A & Wells RD Sticky DNA: in vivo formation in E. coli and in vitro association of long GAA*TTC tracts to generate two independent supercoiled domains. J. Mol. Biol 360, 267–284 (2006). [DOI] [PubMed] [Google Scholar]
- 105.Vetcher AA, Napierala M & Wells RD Sticky DNA: effect of the polypurine. polypyrimidine sequence. J. Biol. Chem 277, 39228–39234 (2002). [DOI] [PubMed] [Google Scholar]
- 106.Vanaja A & Yella VR Delineation of the DNA structural features of eukaryotic core promoter classes. ACS Omega 7, 5657–5669 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hershman SG et al. Genomic distribution and functional analyses of potential G-quadruplex-forming sequences in Saccharomyces cerevisiae. Nucleic Acids Res. 36, 144–156 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Miura O, Ogake T & Ohyama T Requirement or exclusion of inverted repeat sequences with cruciform-forming potential in Escherichia coli revealed by genome-wide analyses. Curr. Genet 64, 945–958 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Du X et al. The genome-wide distribution of non-B DNA motifs is shaped by operon structure and suggests the transcriptional importance of non-B DNA structures in Escherichia coli. Nucleic Acids Res. 41, 5965–5977 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Drew HR, Weeks JR & Travers AA Negative supercoiling induces spontaneous unwinding of a bacterial promoter. EMBO J. 4, 1025–1032 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang JC & Lynch AS Transcription and DNA supercoiling. Curr. Opin. Genet. Dev 3, 764–768 (1993). [DOI] [PubMed] [Google Scholar]
- 112.Mizutani M, Ohta T, Watanabe H, Handa H & Hirose S Negative supercoiling of DNA facilitates an interaction between transcription factor IID and the fibroin gene promoter. Proc. Natl Acad. Sci. USA 88, 718–722 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Aboul-ela F, Bowater RP & Lilley DM Competing B-Z and helix-coil conformational transitions in supercoiled plasmid DNA. J. Biol. Chem 267, 1776–1785 (1992). [PubMed] [Google Scholar]
- 114.Varshney D, Spiegel J, Zyner K, Tannahill D & Balasubramanian S The regulation and functions of DNA and RNA G-quadruplexes. Nat. Rev. Mol. Cell Biol 21, 459–474 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lago S et al. Promoter G-quadruplexes and transcription factors cooperate to shape the cell type-specific transcriptome. Nat. Commun 12, 3885 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Revikumar A et al. Multiple G-quadruplex binding ligand induced transcriptomic map of cancer cell lines. J. Cell Commun. Signal 16, 129–135 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ditlevson JV et al. Inhibitory effect of a short Z-DNA forming sequence on transcription elongation by T7 RNA polymerase. Nucleic Acids Res. 36, 3163–3170 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Belotserkovskii BP et al. A triplex-forming sequence from the human c-MYC promoter interferes with DNA transcription. J. Biol. Chem 282, 32433–32441 (2007). [DOI] [PubMed] [Google Scholar]
- 119.Pandey S et al. Transcription blockage by stable H-DNA analogs in vitro. Nucleic Acids Res. 43, 6994–7004 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Xu J, Chong J & Wang D Opposite roles of transcription elongation factors Spt4/5 and Elf1 in RNA polymerase II transcription through B-form versus non-B DNA structures. Nucleic Acids Res. 49, 4944–4953 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Belotserkovskii BP et al. Mechanisms and implications of transcription blockage by guanine-rich DNA sequences. Proc. Natl Acad. Sci. USA 107, 12816–12821 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Agarwal T, Roy S, Kumar S, Chakraborty TK & Maiti S In the sense of transcription regulation by G-quadruplexes: asymmetric effects in sense and antisense strands. Biochemistry 53, 3711–3718 (2014). [DOI] [PubMed] [Google Scholar]
- 123.Tsai ZT, Chu WY, Cheng JH & Tsai HK Associations between intronic non-B DNA structures and exon skipping. Nucleic Acids Res. 42, 739–747 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Darnell JE Jr. Implications of RNA-RNA splicing in evolution of eukaryotic cells. Science 202, 1257–1260 (1978). [DOI] [PubMed] [Google Scholar]
- 125.Nieto Moreno N, Giono LE, Cambindo Botto AE, Munoz MJ & Kornblihtt AR Chromatin, DNA structure and alternative splicing. FEBS Lett. 589, 3370–3378 (2015). [DOI] [PubMed] [Google Scholar]
- 126.Dai X & Rothman-Denes LB DNA structure and transcription. Curr. Opin. Microbiol 2, 126–130 (1999). [DOI] [PubMed] [Google Scholar]
- 127.Kim N The Interplay between G-quadruplex and transcription. Curr. Med. Chem 26, 2898–2917 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Samadashwily GM, Raca G & Mirkin SM Trinucleotide repeats affect DNA replication in vivo. Nat. Genet 17, 298–304 (1997). [DOI] [PubMed] [Google Scholar]
- 129.Kim YS & Kang HS Sequence-specific functions of the early palindrome domain within the SV40 core origin of replication. Nucleic Acids Res. 17, 9279–9289 (1989). [PMC free article] [PubMed] [Google Scholar]
- 130.Lin S & Kowalski D DNA helical instability facilitates initiation at the SV40 replication origin. J. Mol. Biol 235, 496–507 (1994). [DOI] [PubMed] [Google Scholar]
- 131.Pearson CE, Zorbas H, Price GB & Zannis-Hadjopoulos M Inverted repeats, stem-loops, and cruciforms: significance for initiation of DNA replication. J. Cell. Biochem 63, 1–22 (1996). [DOI] [PubMed] [Google Scholar]
- 132.Lerner LK & Sale JE Replication of G quadruplex DNA. Genes 10, 95 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Guilbaud G et al. Determination of human DNA replication origin position and efficiency reveals principles of initiation zone organisation. Nucleic Acids Res. 50, 7436–7450 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bartholdy B, Mukhopadhyay R, Lajugie J, Aladjem MI & Bouhassira EE Allele-specific analysis of DNA replication origins in mammalian cells. Nat. Commun 6, 7051 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Schneider TD Strong minor groove base conservation in sequence logos implies DNA distortion or base flipping during replication and transcription initiation. Nucleic Acids Res. 29, 4881–4891 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Valton AL et al. G4 motifs affect origin positioning and efficiency in two vertebrate replicators. EMBO J. 33, 732–746 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Yahyaoui W, Callejo M, Price GB & Zannis-Hadjopoulos M Deletion of the cruciform binding domain in CBP/14-3-3 displays reduced origin binding and initiation of DNA replication in budding yeast. BMC Mol. Biol 8, 27 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hoshina S et al. Human origin recognition complex binds preferentially to G-quadruplex-preferable RNA and single-stranded DNA. J. Biol. Chem 288, 30161–30171 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Prorok P et al. Involvement of G-quadruplex regions in mammalian replication origin activity. Nat. Commun 10, 3274 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; G4 DNA-forming sequences in the OGRE are required for the activity of several types of replication origin; adding G4 DNA-stabilizing ligands affects origin activities accordingly, suggesting a role for G4 DNA in replication regulation.
- 140.Hile SE & Eckert KA Positive correlation between DNA polymerase alpha-primase pausing and mutagenesis within polypyrimidine/polypurine microsatellite sequences. J. Mol. Biol 335, 745–759 (2004). [DOI] [PubMed] [Google Scholar]
- 141.Anand RP et al. Overcoming natural replication barriers: differential helicase requirements. Nucleic Acids Res. 40, 1091–1105 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wang Q et al. G-quadruplex formation at the 3′ end of telomere DNA inhibits its extension by telomerase, polymerase and unwinding by helicase. Nucleic Acids Res. 39, 6229–6237 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Paeschke K, Capra JA & Zakian VA DNA replication through G-quadruplex motifs is promoted by the Saccharomyces cerevisiae Pif1 DNA helicase. Cell 145, 678–691 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kopel V, Pozner A, Baran N & Manor H Unwinding of the third strand of a DNA triple helix, a novel activity of the SV40 large T-antigen helicase. Nucleic Acids Res. 24, 330–335 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Yangyuoru PM, Bradburn DA, Liu Z, Xiao TS & Russell R The G-quadruplex (G4) resolvase DHX36 efficiently and specifically disrupts DNA G4s via a translocation-based helicase mechanism. J. Biol. Chem 293, 1924–1932 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Le TT et al. Synergistic coordination of chromatin torsional mechanics and topoisomerase activity. Cell 179, 619–631.e15 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yuan Z et al. DNA unwinding mechanism of a eukaryotic replicative CMG helicase. Nat. Commun 11, 688 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Masuda-Sasa T, Polaczek P, Peng XP, Chen L & Campbell JL Processing of G4 DNA by DNA2 helicase/nuclease and replication protein A (RPA) provides insights into the mechanism of DNA2/RPA substrate recognition. J. Biol. Chem 283, 24359–24373 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Peleg M, Kopel V, Borowiec JA & Manor H Formation of DNA triple helices inhibits DNA unwinding by the SV40 large T-antigen helicase. Nucleic Acids Res. 23, 1292–1299 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lopes J et al. G-quadruplex-induced instability during leading-strand replication. EMBO J. 30, 4033–4046 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dovrat D et al. A live-cell imaging approach for measuring DNA replication rates. Cell Rep. 24, 252–258 (2018). [DOI] [PubMed] [Google Scholar]
- 152.Kobori JA, Strauss E, Minard K & Hood L Molecular analysis of the hotspot of recombination in the murine major histocompatibility complex. Science 234, 173–179 (1986). [DOI] [PubMed] [Google Scholar]
- 153.Weinreb A, Collier DA, Birshtein BK & Wells RD Left-handed Z-DNA and intramolecular triplex formation at the site of an unequal sister chromatid exchange. J. Biol. Chem 265, 1352–1359 (1990). [PubMed] [Google Scholar]
- 154.Vallur AC & Maizels N Activities of human exonuclease 1 that promote cleavage of transcribed immunoglobulin switch regions. Proc. Natl Acad. Sci. USA 105, 16508–16512 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wahls WP, Wallace LJ & Moore PD The Z-DNA motif d(TG)30 promotes reception of information during gene conversion events while stimulating homologous recombination in human cells in culture. Mol. Cell. Biol 10, 785–793 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Roy U & Greene EC Demystifying the D-loop during DNA recombination. Nature 586, 677–678 (2020). [DOI] [PubMed] [Google Scholar]
- 157.Haniford DB & Pulleyblank DE The in-vivo occurrence of Z DNA. J. Biomol. Struct. Dyn 1, 593–609 (1983). [DOI] [PubMed] [Google Scholar]
- 158.Blaho JA & Wells RD Left-handed Z-DNA and genetic recombination. Prog. Nucleic Acid Res. Mol. Biol 37, 107–126 (1989). [DOI] [PubMed] [Google Scholar]
- 159.Xu Z, Zan H, Pone EJ, Mai T & Casali P Immunoglobulin class-switch DNA recombination: induction, targeting and beyond. Nat. Rev. Immunol 12, 517–531 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Larson ED, Duquette ML, Cummings WJ, Streiff RJ & Maizels N MutSalpha binds to and promotes synapsis of transcriptionally activated immunoglobulin switch regions. Curr. Biol 15, 470–474 (2005). [DOI] [PubMed] [Google Scholar]
- 161.Tashiro J, Kinoshita K & Honjo T Palindromic but not G-rich sequences are targets of class switch recombination. Int. Immunol 13, 495–505 (2001). [DOI] [PubMed] [Google Scholar]
- 162.Guiblet WM et al. Non-B DNA: a major contributor to small- and large-scale variation in nucleotide substitution frequencies across the genome. Nucleic Acids Res. 49, 1497–1516 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Georgakopoulos-Soares I, Morganella S, Jain N, Hemberg M & Nik-Zainal S Noncanonical secondary structures arising from non-B DNA motifs are determinants of mutagenesis. Genome Res. 28, 1264–1271 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Thornton CA, Johnson K & Moxley RT III Myotonic dystrophy patients have larger CTG expansions in skeletal muscle than in leukocytes. Ann. Neurol 35, 104–107 (1994). [DOI] [PubMed] [Google Scholar]
- 165.Zatz M et al. Analysis of the CTG repeat in skeletal muscle of young and adult myotonic dystrophy patients: when does the expansion occur? Hum. Mol. Genet 4, 401–406 (1995). [DOI] [PubMed] [Google Scholar]
- 166.Rider SD Jr et al. Stable G-quadruplex DNA structures promote replication-dependent genome instability. J. Biol. Chem 298, 101947 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lu S et al. Short inverted repeats are hotspots for genetic instability: relevance to cancer genomes. Cell Rep. 10, 1674–1680 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Wang G, Christensen LA & Vasquez KM Z-DNA-forming sequences generate large-scale deletions in mammalian cells. Proc. Natl Acad. Sci. USA 103, 2677–2682 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Wang G & Vasquez KM Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells. Proc. Natl Acad. Sci. USA 101, 13448–13453 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Takahashi S, Brazier JA & Sugimoto N Topological impact of noncanonical DNA structures on Klenow fragment of DNA polymerase. Proc. Natl Acad. Sci. USA 114, 9605–9610 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Burrow AA, Marullo A, Holder LR & Wang YH Secondary structure formation and DNA instability at fragile site FRA16B. Nucleic Acids Res. 38, 2865–2877 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Murat P, Guilbaud G & Sale JE DNA polymerase stalling at structured DNA constrains the expansion of short tandem repeats. Genome Biol. 21, 209 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kamath-Loeb AS, Loeb LA, Johansson E, Burgers PM & Fry M Interactions between the Werner syndrome helicase and DNA polymerase delta specifically facilitate copying of tetraplex and hairpin structures of the d(CGG)n trinucleotide repeat sequence. J. Biol. Chem 276, 16439–16446 (2001). [DOI] [PubMed] [Google Scholar]
- 174.Shah SN, Opresko PL, Meng X, Lee MY & Eckert KA DNA structure and the Werner protein modulate human DNA polymerase delta-dependent replication dynamics within the common fragile site FRA16D. Nucleic Acids Res. 38, 1149–1162 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lormand JD et al. DNA polymerase delta stalls on telomeric lagging strand templates independently from G-quadruplex formation. Nucleic Acids Res. 41, 10323–10333 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Iyer RR, Pluciennik A, Rosche WA, Sinden RR & Wells RD DNA polymerase III proofreading mutants enhance the expansion and deletion of triplet repeat sequences in Escherichia coli. J. Biol. Chem 275, 2174–2184 (2000). [DOI] [PubMed] [Google Scholar]
- 177.Teng FY et al. Escherichia coli DNA polymerase I can disrupt G-quadruplex structures during DNA replication. FEBS J. 284, 4051–4065 (2017). [DOI] [PubMed] [Google Scholar]
- 178.Shah KA et al. Role of DNA polymerases in repeat-mediated genome instability. Cell Rep. 2, 1088–1095 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Abdulovic AL, Hile SE, Kunkel TA & Eckert KA The in vitro fidelity of yeast DNA polymerase delta and polymerase epsilon holoenzymes during dinucleotide microsatellite DNA synthesis. DNA Repair 10, 497–505 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Guo J, Gu L, Leffak M & Li GM MutSbeta promotes trinucleotide repeat expansion by recruiting DNA polymerase beta to nascent (CAG)n or (CTG)n hairpins for error-prone DNA synthesis. Cell Res. 26, 775–786 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Meyer D et al. Cooperation between non-essential DNA polymerases contributes to genome stability in Saccharomyces cerevisiae. DNA Repair 76, 40–49 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Eddy S et al. Human translesion polymerase kappa exhibits enhanced activity and reduced fidelity two nucleotides from G-quadruplex DNA. Biochemistry 55, 5218–5229 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Betous R et al. Role of TLS DNA polymerases eta and kappa in processing naturally occurring structured DNA in human cells. Mol. Carcinog 48, 369–378 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Eddy S et al. Evidence for the kinetic partitioning of polymerase activity on G-quadruplex DNA. Biochemistry 54, 3218–3230 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Gadgil RY et al. Replication stress at microsatellites causes DNA double-strand breaks and break-induced replication. J. Biol. Chem 295, 15378–15397 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Stern HR, Sefcikova J, Chaparro VE & Beuning PJ Mammalian DNA polymerase kappa activity and specificity. Molecules 24, 2805 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Walsh E, Wang X, Lee MY & Eckert KA Mechanism of replicative DNA polymerase delta pausing and a potential role for DNA polymerase kappa in common fragile site replication. J. Mol. Biol 425, 232–243 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Twayana S et al. Translesion polymerase eta both facilitates DNA replication and promotes increased human genetic variation at common fragile sites. Proc. Natl Acad. Sci. USA 118, e2106477118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Ketkar A et al. Human Rev1 relies on insert-2 to promote selective binding and accurate replication of stabilized G-quadruplex motifs. Nucleic Acids Res. 49, 2065–2084 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Northam MR et al. DNA polymerases zeta and Rev1 mediate error-prone bypass of non-B DNA structures. Nucleic Acids Res. 42, 290–306 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Sarkies P, Reams C, Simpson LJ & Sale JE Epigenetic instability due to defective replication of structured DNA. Mol. Cell 40, 703–713 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Koole W et al. A Polymerase Theta-dependent repair pathway suppresses extensive genomic instability at endogenous G4 DNA sites. Nat. Commun 5, 3216 (2014). [DOI] [PubMed] [Google Scholar]
- 193.Chan KY, Li X, Ortega J, Gu L & Li GM DNA polymerase theta promotes CAG*CTG repeat expansions in Huntington’s disease via insertion sequences of its catalytic domain. J. Biol. Chem 297, 101144 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Boyer AS, Grgurevic S, Cazaux C & Hoffmann JS The human specialized DNA polymerases and non-B DNA: vital relationships to preserve genome integrity. J. Mol. Biol 425, 4767–4781 (2013). [DOI] [PubMed] [Google Scholar]
- 195.Lemmens B, van Schendel R & Tijsterman M Mutagenic consequences of a single G-quadruplex demonstrate mitotic inheritance of DNA replication fork barriers. Nat. Commun 6, 8909 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Kang S, Jaworski A, Ohshima K & Wells RD Expansion and deletion of CTG repeats from human disease genes are determined by the direction of replication in E. coli. Nat. Genet 10, 213–218 (1995). [DOI] [PubMed] [Google Scholar]
- 197.Cleary JD, Nichol K, Wang YH & Pearson CE Evidence of cis-acting factors in replication-mediated trinucleotide repeat instability in primate cells. Nat. Genet 31, 37–46 (2002). [DOI] [PubMed] [Google Scholar]
- 198.Lia AS et al. Somatic instability of the CTG repeat in mice transgenic for the myotonic dystrophy region is age dependent but not correlated to the relative intertissue transcription levels and proliferative capacities. Hum. Mol. Genet 7, 1285–1291 (1998). [DOI] [PubMed] [Google Scholar]
- 199.Telenius H et al. Somatic and gonadal mosaicism of the Huntington disease gene CAG repeat in brain and sperm. Nat. Genet 6, 409–414 (1994). [DOI] [PubMed] [Google Scholar]
- 200.Hashida H, Goto J, Kurisaki H, Mizusawa H & Kanazawa I Brain regional differences in the expansion of a CAG repeat in the spinocerebellar ataxias: dentatorubral-pallidoluysian atrophy, Machado-Joseph disease, and spinocerebellar ataxia type 1. Ann. Neurol 41, 505–511 (1997). [DOI] [PubMed] [Google Scholar]
- 201.Liu T, Luo H & Gao F Position preference of essential genes in prokaryotic operons. PLoS ONE 16, e0250380 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Prioleau MN & MacAlpine DM DNA replication origins-where do we begin? Genes Dev. 30, 1683–1697 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Thys RG, Lehman CE, Pierce LC & Wang YH DNA secondary structure at chromosomal fragile sites in human disease. Curr. Genomics 16, 60–70 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Arlt MF, Durkin SG, Ragland RL & Glover TW Common fragile sites as targets for chromosome rearrangements. DNA Repair 5, 1126–1135 (2006). [DOI] [PubMed] [Google Scholar]
- 205.Sutherland GR Rare fragile sites. Cytogenet. Genome Res 100, 77–84 (2003). [DOI] [PubMed] [Google Scholar]
- 206.Glover TW Instability at chromosomal fragile sites. Recent Results Cancer Res. 154, 185–199 (1998). [DOI] [PubMed] [Google Scholar]
- 207.Brison O et al. Transcription-mediated organization of the replication initiation program across large genes sets common fragile sites genome-wide. Nat. Commun 10, 5693 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Smith DI, Zhu Y, McAvoy S & Kuhn R Common fragile sites, extremely large genes, neural development and cancer. Cancer Lett. 232, 48–57 (2006). [DOI] [PubMed] [Google Scholar]
- 209.Helmrich A, Ballarino M & Tora L Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 44, 966–977 (2011). [DOI] [PubMed] [Google Scholar]
- 210.Sankar TS, Wastuwidyaningtyas BD, Dong Y, Lewis SA & Wang JD The nature of mutations induced by replication–transcription collisions. Nature 535, 178–181 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; Replication–transcription collisions in the genome of actively dividing bacterial cells result in duplications and deletions at sites of replication stalling where replication forks enter a transcription unit, resulting in T>C base substitutions on the non-template strand, not only at the sites of collision but also in adjacent areas.
- 211.Kim N & Jinks-Robertson S dUTP incorporation into genomic DNA is linked to transcription in yeast. Nature 459, 1150–1153 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Macheret M & Halazonetis TD DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol 10, 425–448 (2015). [DOI] [PubMed] [Google Scholar]
- 213.Buschta-Hedayat N, Buterin T, Hess MT, Missura M & Naegeli H Recognition of nonhybridizing base pairs during nucleotide excision repair of DNA. Proc. Natl Acad. Sci. USA 96, 6090–6095 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Majchrzak M, Bowater RP, Staczek P & Parniewski P SOS repair and DNA supercoiling influence the genetic stability of DNA triplet repeats in Escherichia coli. J. Mol. Biol 364, 612–624 (2006). [DOI] [PubMed] [Google Scholar]
- 215.Lahiri M, Gustafson TL, Majors ER & Freudenreich CH Expanded CAG repeats activate the DNA damage checkpoint pathway. Mol. Cell 15, 287–293 (2004). [DOI] [PubMed] [Google Scholar]
- 216.Bacolla A, Jaworski A, Connors TD & Wells RD Pkd1 unusual DNA conformations are recognized by nucleotide excision repair. J. Biol. Chem 276, 18597–18604 (2001). [DOI] [PubMed] [Google Scholar]
- 217.Tran H, Degtyareva N, Gordenin D & Resnick MA Altered replication and inverted repeats induce mismatch repair-independent recombination between highly diverged DNAs in yeast. Mol. Cell. Biol 17, 1027–1036 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Nag DK & Kurst AA 140-bp-long palindromic sequence induces double-strand breaks during meiosis in the yeast Saccharomyces cerevisiae. Genetics 146, 835–847 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Bill CA, Taghian DG, Duran WA & Nickoloff JA Repair bias of large loop mismatches during recombination in mammalian cells depends on loop length and structure. Mutat. Res 485, 255–265 (2001). [DOI] [PubMed] [Google Scholar]
- 220.Panigrahi GB, Lau R, Montgomery SE, Leonard MR & Pearson CE Slipped (CTG)*(CAG) repeats can be correctly repaired, escape repair or undergo error-prone repair. Nat. Struct. Mol. Biol 12, 654–662 (2005). [DOI] [PubMed] [Google Scholar]
- 221.Palombo F et al. hMutSbeta, a heterodimer of hMSH2 and hMSH3, binds to insertion/deletion loops in DNA. Curr. Biol 6, 1181–1184 (1996). [DOI] [PubMed] [Google Scholar]
- 222.Owen BA et al. CAG)(n)-hairpin DNA binds to Msh2–Msh3 and changes properties of mismatch recognition. Nat. Struct. Biol 12, 663–670 (2005). [DOI] [PubMed] [Google Scholar]
- 223.Manley K, Shirley TL, Flaherty L & Messer A Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice. Nat. Genet 23, 471–473 (1999). [DOI] [PubMed] [Google Scholar]
- 224.van den Broek WJ et al. Somatic expansion behaviour of the (CTG)n repeat in myotonic dystrophy knock-in mice is differentially affected by Msh3 and Msh6 mismatch-repair proteins. Hum. Mol. Genet 11, 191–198 (2002). [DOI] [PubMed] [Google Scholar]
- 225.Savouret C et al. CTG repeat instability and size variation timing in DNA repair-deficient mice. EMBO J. 22, 2264–2273 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Lin Y, Dion V & Wilson JH Transcription promotes contraction of CAG repeat tracts in human cells. Nat. Struct. Mol. Biol 13, 179–180 (2006). [DOI] [PubMed] [Google Scholar]
- 227.Zhao J, Jain A, Iyer RR, Modrich PL & Vasquez KM Mismatch repair and nucleotide excision repair proteins cooperate in the recognition of DNA interstrand crosslinks. Nucleic Acids Res. 37, 4420–4429 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Potaman VN et al. Length-dependent structure formation in Friedreich ataxia (GAA) n*(TTC)n repeats at neutral pH. Nucleic Acids Res. 32, 1224–1231 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Kim HM et al. Chromosome fragility at GAA tracts in yeast depends on repeat orientation and requires mismatch repair. EMBO J. 27, 2896–2906 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Neil AJ et al. Replication-independent instability of Friedreich’s ataxia GAA repeats during chronological aging. Proc. Natl Acad. Sci. USA 118, e2013080118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study finds that MMR proteins are involved in generating DBSs at long GAA(100) repeats that can form H-DNA in non-dividing cells, resulting in large-scale deletions, including the repeat and adjacent regions, and mediated by error-prone non-homologous end-joining or gene conversions via ectopic homologous recombination.
- 231.Ehrat EA, Johnson BR, Williams JD, Borchert GM & Larson ED G-quadruplex recognition activities of E. coli MutS. BMC Mol. Biol 13, 23 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Pavlova AV et al. Responses of DNA mismatch repair proteins to a stable G-quadruplex embedded into a DNA duplex structure. Int. J. Mol. Sci 21, 8773 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Lai Y et al. Crosstalk between MSH2-MSH3 and polbeta promotes trinucleotide repeat expansion during base excision repair. Nat. Commun 7, 12465 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; The MMR protein complex MSH2–MSH3 is found to crosstalk with the BER machinery, stimulating the synthesis activity of DNA Polβ through triplet repeats and facilitating the formation of flap structures, which leads to repeat expansions.
- 234.McKinney JA, Wang G & Vasquez KM Distinct mechanisms of mutagenic processing of alternative DNA structures by repair proteins. Mol. Cell Oncol 7, 1743807 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Wood RD DNA damage recognition during nucleotide excision repair in mammalian cells. Biochimie 81, 39–44 (1999). [DOI] [PubMed] [Google Scholar]
- 236.Wang G, Seidman MM & Glazer PM Mutagenesis in mammalian cells induced by triple helix formation and transcription-coupled repair. Science 271, 802–805 (1996). [DOI] [PubMed] [Google Scholar]
- 237.Vasquez KM, Christensen J, Li L, Finch RA & Glazer PM Human XPA and RPA DNA repair proteins participate in specific recognition of triplex-induced helical distortions. Proc. Natl Acad. Sci. USA 99, 5848–5853 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Thoma BS, Wakasugi M, Christensen J, Reddy MC & Vasquez KM Human XPC-hHR23B interacts with XPA-RPA in the recognition of triplex-directed psoralen DNA interstrand crosslinks. Nucleic Acids Res. 33, 2993–3001 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Oussatcheva EA, Hashem VI, Zou Y, Sinden RR & Potaman VN Involvement of the nucleotide excision repair protein UvrA in instability of CAG*CTG repeat sequences in Escherichia coli. J. Biol. Chem 276, 30878–30884 (2001). [DOI] [PubMed] [Google Scholar]
- 240.Szwarocka ST, Staczek P & Parniewski P Chromosomal model for analysis of a long CTG/CAG tract stability in wild-type Escherichia coli and its nucleotide excision repair mutants. Can. J. Microbiol 53, 860–868 (2007). [DOI] [PubMed] [Google Scholar]
- 241.Parniewski P, Bacolla A, Jaworski A & Wells RD Nucleotide excision repair affects the stability of long transcribed (CTG*CAG) tracts in an orientation-dependent manner in Escherichia coli. Nucleic Acids Res. 27, 616–623 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Matson SW & Robertson AB The UvrD helicase and its modulation by the mismatch repair protein MutL. Nucleic Acids Res. 34, 4089–4097 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Lin Y & Wilson JH Transcription-induced CAG repeat contraction in human cells is mediated in part by transcription-coupled nucleotide excision repair. Mol. Cell. Biol 27, 6209–6217 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Trujillo KM & Sung P DNA structure-specific nuclease activities in the Saccharomyces cerevisiae Rad50*Mre11 complex. J. Biol. Chem 276, 35458–35464 (2001). [DOI] [PubMed] [Google Scholar]
- 245.Farah JA, Hartsuiker E, Mizuno K, Ohta K & Smith GRA 160-bp palindrome is a Rad50.Rad32-dependent mitotic recombination hotspot in Schizosaccharomyces pombe. Genetics 161, 461–468 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Paull TT, Cortez D, Bowers B, Elledge SJ & Gellert M Direct DNA binding by Brca1. Proc. Natl Acad. Sci. USA 98, 6086–6091 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.De la Torre C, Pincheira J & Lopez-Saez JF Human syndromes with genomic instability and multiprotein machines that repair DNA double-strand breaks. Histol. Histopathol 18, 225–243 (2003). [DOI] [PubMed] [Google Scholar]
- 248.Lengsfeld BM, Rattray AJ, Bhaskara V, Ghirlando R & Paull TT Sae2 is an endonuclease that processes hairpin DNA cooperatively with the Mre11/Rad50/Xrs2 complex. Mol. Cell 28, 638–651 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Ghosal G & Muniyappa K Saccharomyces cerevisiae Mre11 is a high-affinity G4 DNA-binding protein and a G-rich DNA-specific endonuclease: implications for replication of telomeric DNA. Nucleic Acids Res. 33, 4692–4703 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Farah JA, Cromie G, Steiner WW & Smith GR A novel recombination pathway initiated by the Mre11/Rad50/Nbs1 complex eliminates palindromes during meiosis in Schizosaccharomyces pombe. Genetics 169, 1261–1274 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Jankowski C & Nag DK Most meiotic CAG repeat tract-length alterations in yeast are SPO11 dependent. Mol. Genet. Genomics 267, 64–70 (2002). [DOI] [PubMed] [Google Scholar]
- 252.Mankouri HW, Ashton TM & Hickson ID Holliday junction-containing DNA structures persist in cells lacking Sgs1 or Top3 following exposure to DNA damage. Proc. Natl Acad. Sci. USA 108, 4944–4949 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Olmezer G et al. Replication intermediates that escape Dna2 activity are processed by Holliday junction resolvase Yen1. Nat. Commun 7, 13157 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Wyatt HD, Sarbajna S, Matos J & West SC Coordinated actions of SLX1-SLX4 and MUS81-EME1 for Holliday junction resolution in human cells. Mol. Cell 52, 234–247 (2013). [DOI] [PubMed] [Google Scholar]
- 255.Xu X et al. Structure specific DNA recognition by the SLX1-SLX4 endonuclease complex. Nucleic Acids Res. 49, 7740–7752 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Ashton TM, Mankouri HW, Heidenblut A, McHugh PJ & Hickson ID Pathways for Holliday junction processing during homologous recombination in Saccharomyces cerevisiae. Mol. Cell. Biol 31, 1921–1933 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Agostinho A et al. Combinatorial regulation of meiotic holliday junction resolution in C. elegans by HIM-6 (BLM) helicase, SLX-4, and the SLX-1, MUS-81 and XPF-1 nucleases. PLoS Genet. 9, e1003591 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Cote AG & Lewis SM Mus81-dependent double-strand DNA breaks at in vivo-generated cruciform structures in S. cerevisiae. Mol. Cell 31, 800–812 (2008). [DOI] [PubMed] [Google Scholar]
- 259.Minocherhomji S & Hickson ID Structure-specific endonucleases: guardians of fragile site stability. Trends Cell Biol. 24, 321–327 (2014). [DOI] [PubMed] [Google Scholar]
- 260.Ait Saada A et al. Structural parameters of palindromic repeats determine the specificity of nuclease attack of secondary structures. Nucleic Acids Res. 49, 3932–3947 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Goold R et al. FAN1 controls mismatch repair complex assembly via MLH1 retention to stabilize CAG repeat expansion in Huntington’s disease. Cell Rep. 36, 109649 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; The DNA-structure-specific nuclease FAN1 binds to the MMR protein MLH1 and suppresses its interaction with MSH3, thereby reducing MMR-promoted CAG repeat expansion in human cells.
- 262.Deshmukh AL et al. FAN1 exo- not endo-nuclease pausing on disease-associated slipped-DNA repeats: a mechanism of repeat instability. Cell Rep. 37, 110078 (2021). [DOI] [PubMed] [Google Scholar]
- 263.Zhou J, Fleming AM, Averill AM, Burrows CJ & Wallace SS The NEIL glycosylases remove oxidized guanine lesions from telomeric and promoter quadruplex DNA structures. Nucleic Acids Res. 43, 4039–4054 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Loomis EW, Sanz LA, Chedin F & Hagerman PJ Transcription-associated R-loop formation across the human FMR1 CGG-repeat region. PLoS Genet. 10, e1004294 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.de Graaff E et al. Hotspot for deletions in the CGG repeat region of FMR1 in fragile X patients. Hum. Mol. Genet 4, 45–49 (1995). [DOI] [PubMed] [Google Scholar]
- 266.Hayward BE & Usdin K Mechanisms of genome instability in the fragile X-related disorders. Genes 12, 1633 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Rovozzo R et al. CGG repeats in the 5′UTR of FMR1 RNA regulate translation of other RNAs localized in the same RNA granules. PLoS ONE 11, e0168204 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Liu G et al. Altered replication in human cells promotes DMPK (CTG)(n). (CAG)(n) repeat instability. Mol. Cell. Biol 32, 1618–1632 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Stenson PD et al. Human Gene Mutation Database (HGMD): 2003 update. Hum. Mutat 21, 577–581 (2003). [DOI] [PubMed] [Google Scholar]
- 270.Kamat MA, Bacolla A, Cooper DN & Chuzhanova N A role for non-B DNA forming sequences in mediating microlesions causing human inherited disease. Hum. Mutat 37, 65–73 (2016). [DOI] [PubMed] [Google Scholar]
- 271.Weckselblatt B & Rudd MK Human structural variation: mechanisms of chromosome rearrangements. Trends Genet. 31, 587–599 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Bacolla A, Tainer JA, Vasquez KM & Cooper DN Translocation and deletion breakpoints in cancer genomes are associated with potential non-B DNA-forming sequences. Nucleic Acids Res. 44, 5673–5688 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Raghavan SC, Swanson PC, Ma Y & Lieber MR Double-strand break formation by the RAG complex at the BCL-2 major breakpoint region and at other non-B DNA structures in vitro. Mol. Cell. Biol 25, 5904–5919 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Wu Y & Brosh RM Jr. G-quadruplex nucleic acids and human disease. FEBS J. 277, 3470–3488 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Cheloshkina K & Poptsova M Tissue-specific impact of stem-loops and quadruplexes on cancer breakpoints formation. BMC Cancer 19, 434 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Cheloshkina K & Poptsova M Comprehensive analysis of cancer breakpoints reveals signatures of genetic and epigenetic contribution to cancer genome rearrangements. PLoS Comput. Biol 17, e1008749 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Kurahashi H et al. Palindrome-mediated chromosomal translocations in humans. DNA Repair 5, 1136–1145 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Di Antonio M et al. Single-molecule visualization of DNA G-quadruplex formation in live cells. Nat. Chem 12, 832–837 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Del Mundo IMA, Vasquez KM & Wang G Modulation of DNA structure formation using small molecules. Biochim. Biophys. Acta 1866, 118539 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Du Y & Zhou X Targeting non-B-form DNA in living cells. Chem. Rec 13, 371–384 (2013). [DOI] [PubMed] [Google Scholar]
- 281.Vasquez KM, Narayanan L & Glazer PM Specific mutations induced by triplex-forming oligonucleotides in mice. Science 290, 530–533 (2000). [DOI] [PubMed] [Google Scholar]
- 282.Nakamori M et al. A slipped-CAG DNA-binding small molecule induces trinucleotide-repeat contractions in vivo. Nat. Genet 52, 146–159 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study uses a non-B DNA structure-specific ligand to induce repeat contractions at expanded CAG repeats in both cultured human cells and medium spiny neurons of the mouse striatum, suggesting a promising therapeutic approach to reduce pathogenic repeat length.
- 283.Dang DT, Nguyen LTA, Truong TTT, Nguyen HD & Phan AT Construction of a G-quadruplex-specific DNA endonuclease. Chem. Commun 57, 4568–4571 (2021). [DOI] [PubMed] [Google Scholar]
- 284.Zhu M et al. Novel roles of an intragenic G-quadruplex in controlling microRNA expression and cardiac function. Nucleic Acids Res. 49, 2522–2536 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Christensen LA, Finch RA, Booker AJ & Vasquez KM Targeting oncogenes to improve breast cancer chemotherapy. Cancer Res. 66, 4089–4094 (2006). [DOI] [PubMed] [Google Scholar]
- 286.Boulware SB et al. Triplex-forming oligonucleotides targeting c-MYC potentiate the anti-tumor activity of gemcitabine in a mouse model of human cancer. Mol. Carcinog 53, 744–752 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Xu H et al. CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat. Commun 8, 14432 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Bywater MJ et al. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell 22, 51–65 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Schuldt A DNA replication: Pif1 overcomes a quadruplex hurdle. Nat. Rev. Mol. Cell Biol 12, 402 (2011). [DOI] [PubMed] [Google Scholar]
- 290.Muellner J & Schmidt KH Yeast genome maintenance by the multifunctional PIF1 DNA helicase family. Genes 11, 224 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Kerrest A et al. SRS2 and SGS1 prevent chromosomal breaks and stabilize triplet repeats by restraining recombination. Nat. Struct. Mol. Biol 16, 159–167 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Saha T, Shukla K, Thakur RS, Desingu A & Nagaraju G Mycobacterium tuberculosis UvrD1 and UvrD2 helicases unwind G-quadruplex DNA. FEBS J. 286, 2062–2086 (2019). [DOI] [PubMed] [Google Scholar]
- 293.Paul T et al. E. coli Rep helicase and RecA recombinase unwind G4 DNA and are important for resistance to G4-stabilizing ligands. Nucleic Acids Res. 48, 6640–6653 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Eykelenboom JK, Blackwood JK, Okely E & Leach DR SbcCD causes a double-strand break at a DNA palindrome in the Escherichia coli chromosome. Mol. Cell 29, 644–651 (2008). [DOI] [PubMed] [Google Scholar]
- 295.Pike AC et al. Human RECQ1 helicase-driven DNA unwinding, annealing, and branch migration: insights from DNA complex structures. Proc. Natl Acad. Sci. USA 112, 4286–4291 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.van Wietmarschen N et al. BLM helicase suppresses recombination at G-quadruplex motifs in transcribed genes. Nat. Commun 9, 271 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Meier B et al. Protection of the C. elegans germ cell genome depends on diverse DNA repair pathways during normal proliferation. PLoS ONE 16, e0250291 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.van Wietmarschen N et al. Repeat expansions confer WRN dependence in microsatellite-unstable cancers. Nature 586, 292–298 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Keller H et al. The intrinsically disordered amino-terminal region of human RecQL4: multiple DNA-binding domains confer annealing, strand exchange and G4 DNA binding. Nucleic Acids Res. 42, 12614–12627 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Budhathoki JB et al. A comparative study of G-quadruplex unfolding and DNA reeling activities of human RECQ5 helicase. Biophys. J 110, 2585–2596 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Vannier JB, Pavicic-Kaltenbrunner V, Petalcorin MI, Ding H & Boulton SJ RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell 149, 795–806 (2012). [DOI] [PubMed] [Google Scholar]
- 302.Schult P & Paeschke K The DEAH helicase DHX36 and its role in G-quadruplex-dependent processes. Biol. Chem 402, 581–591 (2021). [DOI] [PubMed] [Google Scholar]
- 303.Jain A et al. DHX9 helicase is involved in preventing genomic instability induced by alternatively structured DNA in human cells. Nucleic Acids Res. 41, 10345–10357 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Wu Y, Shin-ya K & Brosh RM Jr. FANCJ helicase defective in Fanconia anemia and breast cancer unwinds G-quadruplex DNA to defend genomic stability. Mol. Cell. Biol 28, 4116–4128 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Tarailo-Graovac M et al. Spectrum of variations in dog-1/FANCJ and mdf-1/MAD1 defective Caenorhabditis elegans strains after long-term propagation. BMC Genomics 16, 210 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Kaushik Tiwari M & Rogers FA XPD-dependent activation of apoptosis in response to triplex-induced DNA damage. Nucleic Acids Res. 41, 8979–8994 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Wu G, Xing Z, Tran EJ & Yang D DDX5 helicase resolves G-quadruplex and is involved in MYC gene transcriptional activation. Proc. Natl Acad. Sci. USA 116, 20453–20461 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.van Schie JJM et al. Warsaw breakage syndrome associated DDX11 helicase resolves G-quadruplex structures to support sister chromatid cohesion. Nat. Commun 11, 4287 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Wang Y et al. G-quadruplex DNA drives genomic instability and represents a targetable molecular abnormality in ATRX-deficient malignant glioma. Nat. Commun 10, 943 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Rich A & Zhang S Timeline: Z-DNA: the long road to biological function. Nat. Rev. Genet 4, 566–572 (2003). [DOI] [PubMed] [Google Scholar]
- 311.Voloshin ON, Mirkin SM, Lyamichev VI, Belotserkovskii BP & Frank-Kamenetskii MD Chemical probing of homopurine-homopyrimidine mirror repeats in supercoiled DNA. Nature 333, 475–476 (1988). [DOI] [PubMed] [Google Scholar]
- 312.Frank-Kamenetskii MD & Mirkin SM Triplex DNA structures. Annu. Rev. Biochem 64, 65–95 (1995). [DOI] [PubMed] [Google Scholar]
- 313.Lane AN, Chaires JB, Gray RD & Trent JO Stability and kinetics of G-quadruplex structures. Nucleic Acids Res. 36, 5482–5515 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Voineagu I, Narayanan V, Lobachev KS & Mirkin SM Replication stalling at unstable inverted repeats: interplay between DNA hairpins and fork stabilizing proteins. Proc. Natl Acad. Sci. USA 105, 9936–9941 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Sinden RR, Zheng GX, Brankamp RG & Allen KN On the deletion of inverted repeated DNA in Escherichia coli: effects of length, thermal stability, and cruciform formation in vivo. Genetics 129, 991–1005 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Castillo-Guzman D & Chedin F Defining R-loop classes and their contributions to genome instability. DNA Repair 106, 103182 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Sinden RR DNA Structure and Function (Academic, 1994). [Google Scholar]
- 318.Achaz G, Coissac E, Netter P & Rocha EP Associations between inverted repeats and the structural evolution of bacterial genomes. Genetics 164, 1279–1289 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Feschotte C & Pritham EJ DNA transposons and the evolution of eukaryotic genomes. Annu. Rev. Genet 41, 331–368 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Schibler L et al. High-resolution comparative mapping among man, cattle and mouse suggests a role for repeat sequences in mammalian genome evolution. BMC Genomics 7, 194 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Moxon R, Bayliss C & Hood D Bacterial contingency loci: the role of simple sequence DNA repeats in bacterial adaptation. Annu. Rev. Genet 40, 307–333 (2006). [DOI] [PubMed] [Google Scholar]
- 322.Galen SC et al. Contribution of a mutational hot spot to hemoglobin adaptation in high-altitude Andean house wrens. Proc. Natl Acad. Sci. USA 112, 13958–13963 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Lavrov DV, Maikova OO, Pett W & Belikov SI Small inverted repeats drive mitochondrial genome evolution in Lake Baikal sponges. Gene 505, 91–99 (2012). [DOI] [PubMed] [Google Scholar]
- 324.Deng Z et al. A transposon-introduced G-quadruplex motif is selectively retained and constrained to downregulate CYP321A1. Insect Sci. 10.1111/1744-7917.13021 (2022). [DOI] [PubMed] [Google Scholar]
- 325.Rao JE & Craig NL Selective recognition of pyrimidine motif triplexes by a protein encoded by the bacterial transposon Tn7. J. Mol. Biol 307, 1161–1170 (2001). [DOI] [PubMed] [Google Scholar]
- 326.Yin Y et al. Molecular mechanisms and topological consequences of drastic chromosomal rearrangements of muntjac deer. Nat. Commun 12, 6858 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Ellegren H Microsatellites: simple sequences with complex evolution. Nat. Rev. Genet 5, 435–445 (2004). [DOI] [PubMed] [Google Scholar]
- 328.Yuan J et al. Simple sequence repeats drive genome plasticity and promote adaptive evolution in penaeid shrimp. Commun. Biol 4, 186 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Chan YF et al. Adaptive evolution of pelvic reduction in sticklebacks by recurrent deletion of a Pitx1 enhancer. Science 327, 302–305 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Xie KT et al. DNA fragility in the parallel evolution of pelvic reduction in stickleback fish. Science 363, 81–84 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; Structure-specific DSBs and large deletions caused by a Z-DNA-forming GT repeat in the Pel gene from marine stickleback fish populations have important roles in the evolutionary loss of pelvic hindfins in freshwater sticklebacks, suggesting that non-B DNA structure-induced genetic instability has contributed to evolution.
- 331.Rozen S et al. Abundant gene conversion between arms of palindromes in human and ape Y chromosomes. Nature 423, 873–876 (2003). [DOI] [PubMed] [Google Scholar]