

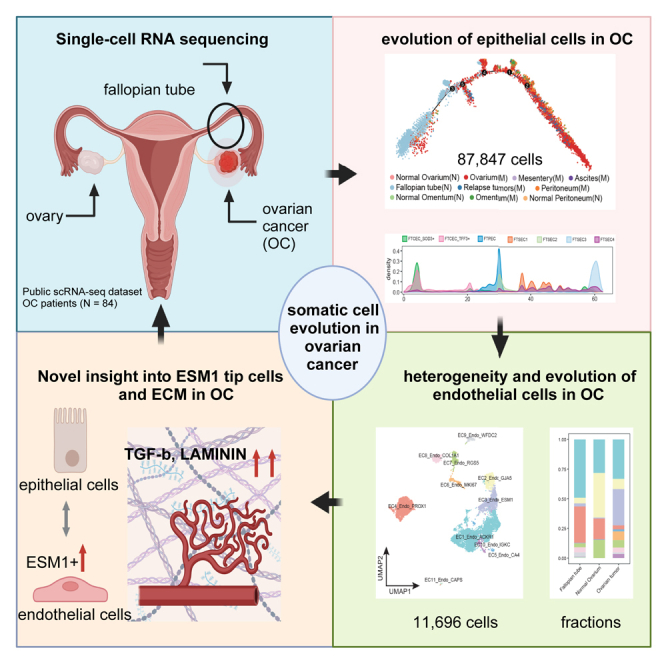
Summary
Tumor neovascularization mediated by endothelial cells (ECs) is essential for ovarian cancer (OC) progression, but interactions between epithelial cells and ECs are not well understood. Here, we analyze single-cell transcriptome of 87,847 epithelial cells and 11,696 ECs from fallopian tubes, primary and metastatic ovarian tumors. Cell differentiation trajectory analysis reveals that fallopian tube cells exhibit a potential development trend toward primary OC epithelial cells. We identify a sub-population of fallopian tube epithelial cells (FTSEC3), which highly express tumor cell markers and are enriched in vascular endothelial growth factor production. Two neovascularization-related EC phenotypes (MKI67+ proliferating ECs and ESM1+ tip cells) are specially found in ovarium tumors, which exhibit strong interactions with FTSEC3. We validate that genetic disruption of LAMININ and TGF-β with CRISPR in ECs inhibits sprouting angiogenesis. In summary, this study reveals a reciprocal evolution and interaction between epithelial and ECs in OC development and progression.
Subject areas: Biological sciences, Cancer, Omics
Graphical abstract
Highlights
-
•
Analysis of 87,847 epithelial cells and 11,696 endothelial cells in ovarian cancer
-
•
Fallopian tube secretory epithelial cells exhibit early ovarian tumor signatures
-
•
Endothelial cells in ovarian cancer are heterogeneous in functional phenotypes
-
•
ESM1+ endothelial tip cells are associated with tumor progression and prognosis
Biological sciences; Cancer; Omics
Introduction
Ovarian cancer (OC) is one of the most common forms of reproductive tumor and leading-cause of cancer death in women. A widely accepted hypothesis proposes that OC (epithelial) cells originate from the fallopian tube epithelial cells. Triggered by a continuous inflammatory environment, fallopian tube epithelial cells undergo malignant transformations, leading to their migration onto the ovarian surface and subsequent formation of malignant tumors. Hence, the fallopian tube is recognized as a potential origin for OC.1 The epithelial-mesenchymal transition (EMT) state is recognized as a crucial process necessary for the metastasis of cancer stem cells (CSCs).2 With the rapid development of single-cell RNA sequencing (scRNA-seq) technologies,3 single-cell studies focusing on epithelial cells from normal fallopian tube have revealed the presence of normal epithelial cells under partially EMT state.4,5,6,7 Recent studies have shown that the EMT status of fallopian tube epithelial cells may be closely associated with disease conditions and OC risk. Furthermore, multiple EMT states of fallopian tube epithelium contribute to the phenotypic heterogeneity of OC.5 Additionally, in individuals at high risk for OC, they were found to exhibit the presence of fallopian tube secretory epithelial cells expressing EMT markers, which were recognized as progenitor cells for OC.7 Furthermore, the transition process from normal fallopian tube ciliated epithelial cells to secretory epithelial cells may represent an early step in tumorigenesis.6 These findings provide important clues for further investigating the mechanisms underlying the development and progression of OC from fallopian tube. However, further interpretation is still required to comprehend the relationship between EMT state cells and tumor cells. Therefore, we aim to integrate the vast amount of single-cell transcriptome data from normal fallopian tube epithelial cells and tumor cells to gain more insights into somatic evolution in the development of OC. Through subpopulation analysis, we seek to elucidate the potential sub clusters of fallopian tube epithelial cells that may serve as the origin of OC.
Another important cell type in the development and progression of OC is vascular endothelial cells, which play essential roles in shaping the tumor microenvironment and serve as crucial contributors to tumor progression through neoangiogenesis. Endothelial cells are functionally heterogeneous. Previously, we have applied scRNA-seq to unravel the functionally diverse endothelial cell phenotypes in several malignant tumors.8,9,10 Angiogenic tip cells possess the ability to modulate the metabolism of tumor cells by regulating glycolysis and oxidative phosphorylation. These tip cells are predominantly found in tumor tissues, including colorectal cancer (CRC), gastric cancer (GC), lung cancer (LC), OC, pancreatic ductal adenocarcinoma (PDAC), and renal cell carcinoma (RCC), while they are nearly absent in normal tissues.11 Tumor endothelial cells exhibit immunomodulatory functions, which can influence tumor progression by decreasing antigen presentation and regulating immune cell recruitment. In OC, activated endothelial cells (VCAM1+) may contribute to lymphocyte infiltration and participate in antigen processing and presentation, thereby enhancing chemotherapy sensitivity.12 Interaction between extracellular matrix (ECM) remodeling and tumor endothelial cells can regulate tumor cell adhesion and migration.13 Lymphatic endothelial cells have been shown to promote lymph node metastasis in high-grade serous tubo-ovarian cancer.14
Here, we conducted an extensive analysis of 87,847 single-cell transcriptome of epithelial cells and 11,696 endothelial cells from OC to investigate the origin of this malignancy. We identified a secretory fallopian tube epithelial cells sub-population (FTSEC3), which are positioned at the late differentiation trajectory of fallopian tube epithelial cells, express ovarium tumor epithelial cell signatures. In addition, we deciphered the EC heterogeneity in OC and identified ESM1+ endothelial tip cells, which interact with FTSEC3 and drive the OV evolution. Through single-cell transcriptome-based pan-cancer ligand-receptor analysis, we further discovered and validated that the ECM remodeling pathways LAMININ and TGF-β are involving in modulating vascular neoangiogenesis, adding more biological and cellular insights into the somatic cell evolution and tumorigenesis of OV.
Results
Heterogeneity and evolution of epithelial cells in OC
To provide deeper insights into the heterogeneity and evolution of OC cells, we conducted comprehensive single-cell transcriptome analysis of epithelial cells, which were generated using the 10X Genomics technology.5,6,14,15,16,17,18,19,20,21,22,23,24,25 In total, we obtained 87,847 epithelial cells from both normal tissues and various sites of ovarian tumor tissues, including malignant ascites, and ovarian tumors located on the surface of the ovaries, omentum, peritoneum, and mesentery. Normal tissues included the fallopian tubes, ovaries, peritoneum, and omentum (Table S1).
To study the potential evolution trends of OC cells, we performed pseudotime trajectory analysis of epithelial cells derived from different tissue group. Pseudotime analysis revealed that epithelial cells follow one differentiation trajectory (Figures 1A and S1A). The cells from different tissue sources were differentially distributed along the trajectory. Cells derived from normal fallopian tube and ovarium were located at the beginning of the trajectory, while cells from malignant tissues were positioned at the later trajectory as compared to cells from normal tissues. Cell from relapse tumors were positioned at the terminal of trajectory, illustrating the epithelial cell evolution in OC at the single-cell levels (Figures 1B and S1B). Compared with cells derive from normal ovarium, fallopian tube cells exhibit continuous distribution at pseudotime trajectory. Besides, the ovarium tumor epithelial cells and normal fallopian tube epithelial cells partially co-localize at an earlier position along the pseudo time trajectory (Figure 1B), implying the presence of fallopian tube epithelial cell subtypes with a developmental degree resembling early ovary tumor epithelial cells.
Figure 1.

Heterogeneity and evolution of epithelial cells in OC
(A) Pseudo-time analysis of OC epithelial cells inferred by Monocle2. Each data point corresponds to a single cell. The colors of the points represent different tissue types. M: malignant, N: normal.
(B) The cell proportion of epithelial cells of ovarium tumor, normal fallopian tube and ovarium on the pseudo-time trajectory.
(C) UMAP plot with cells colored according to tissue origin.
(D) UMAP plot with epithelial cells colored according to molecular clusters.
(E) Pseudo-time analysis of OC epithelial cells inferred by Monocle2. Each data point corresponds to a single cell. The colors of the data points represent OC epithelial cell subtypes.
(F) Proportion of cells in cluster Epi1, Epi2, Epi3 on the pseudo-time trajectory.
(G) Heat maps show the GSVA scores of cancer-related function gene sets for module1 and module3.
(H) The differentially expressed genes (rows) along the pseudo-time (columns) in OC epithelial cells.
(I) GO enrichment result of pseudo-time variable gene clusters.
To reduce the noises from epithelial cells of other tissues, we selected epithelial cells derived from normal fallopian tubes and ovarian tumors for further analysis. The epithelial cells from normal fallopian tubes and ovarian tumors were separated into 12 distinct clusters (Epi0-Epi11) with strong transcriptional signatures of tissue origin (Figures 1C and 1D). Cluster Epi2 represents a mixed cell population that includes epithelial cells from both normal fallopian tube and ovarium tumor. In the UMAP plot, Cluster Epi3, Epi2, and Epi1 exhibit a linear distribution, suggesting a potential evolutionary relationship among these three epithelial cell clusters (Figure 1D). Pseudotime trajectory analysis suggested that Epi2 cells were situated between Epi3 (early) and Epi1 (late) (Figures 1E, 1F, and S1C). Additionally, the Epi2 cluster exhibits functional characteristics of both an inflammation gene module (module3) and an invasion and EMT gene module (module1), indicating that Epi2 might undergo malignant transformation from normal tubal epithelial cells under the influence of chronic inflammation (Figures 1G and S1D; Table S2). The co-appearance of tumor cells and normal cells over the pseudotime trajectory coincides with the location of cluster Epi2 (Figures 1B–1F), implying that the fallopian tube epithelial cells presented in cluster Epi2 shares similar transcriptomic signatures with OC epithelial cells.
To decipher the functional alterations during the process of epithelial cell transition, we analyzed the variable genes (differentially expressed) in cluster Epi3, Epi2, and Epi1 along the pseudotime progression (Figure 1H). The significantly altered genes along the pseudotime trajectory were categorized into five distinct gene clusters, among which gene cluster 2 consisted of genes highly expressed in early trajectory (Epi3 and partially Epi2). These genes are functionally associated with cilium organization. Genes within cluster 4 were highly expressed in Epi2, with functions enriched in blood vessel development. On the other hand, genes within clusters 1, 3, and 5 were highly expressed in Epi1, and enriched in functions related to tumor migration and basal metabolism, suggesting after malignant transition, cancer cell clusters play a role in promoting tumor survival and invasion (Figure 1I and Table S3). Our analysis of epithelial cell evolution suggests that fallopian tube epithelial cell is involved in OC development.
FTSEC3 resembles OC epithelial cell signatures and functions
Fallopian tube epithelium is highly heterogeneous across patients. To identify the specific epithelial cell subtype resembling OC signatures, we used the consensus non-negative matrix factorization (cNMF) method26 to reduce dimension and cluster the cells. Fallopian tube epithelial cells were divided into 8 clusters based on 8 gene modules (Figures 2A and S2A). Based on the expression of functional marker genes in fallopian tube epithelial cells and the contribution of specific gene modules, we identified three distinct cell types: fallopian tube ciliated epithelial cells (FTCEC) (FTE2 and FTE5) expressing FOXJ1, CCDC17, and CCDC78; fallopian tube secretory epithelial cells (FTSEC) (FTE0-1, 3–4, and 6) expressing KRT7 and PAX8; and fallopian tube peg epithelial cells (FTPEC) (FTE7, CD44+PAX8-) (Figures 2B–2D and S2B). In addition to the known CD44 and RUNX3,5,27 we also found that FTPEC, located in the basal layer of the fallopian tube epithelium, specifically expressed marker genes such as SRGN (Figure S2C and Table S4).
Figure 2.

FTSEC3 resembles OC epithelial cell signatures and functions
(A) UMAP plot of fallopian tube epithelial cells sub clusters.
(B) Expression of classic marker genes in secretory epithelial cells and ciliated epithelial cells within the fallopian tube epithelial subgroups.
(C) Heatmap of top-ranking marker genes expression in different fallopian tube epithelial cell sub clusters. In this and all further heatmaps depicting marker genes, colors indicate row-wise scaled gene expression, with a mean of 0 and an SD of 1 (Z scores).
(D) Sankey diagram to visualize fallopian tube epithelial cell subtype projection. The number represents the number of cells (k = ∗1000).
(E) Pseudo-time analysis of fallopian tube epithelial cells inferred by Monocle2. The colors of the data points represent pseudo time (top). Position of FTSEC3 along the fallopian tube epithelial cell pseudotime trajectory (bottom).
(F) Bubble plots showing the expression of selected marker genes in fallopian tube epithelial cell sub cluster.
(G) The differentially expressed genes (rows) along the pseudo-time (columns) in fallopian tube epithelial cells.
(H) GO enrichment result of pseudo-time variable gene cluster 4.
To provide deeper insights into the functional characteristics of the fallopian tube epithelial cell subtypes, we performed gene ontology (GO) enrichment analysis based on the gene modules and cell markers (Figures S2D and S2E). The enrichment results reveal that the two subgroups of ciliated cells: FTCEC1 (TFF3+) and FTCEC2 (SOD1+) share similar enrichment in cilium functions, but FTCEC2 exhibits marker genes for both FTCECs and FTSECs (Figure S2B). In FTPEC, the majority of differentially expressed genes (DEGs) are immune-related genes. The enriched signaling pathways and dominant gene modules of these DEGs further support this finding (Figures S2D and S2E; Tables S5 and S6). The subgroups of FTSECs show comparable tumor signaling patterns (Figure S2F and Table S7), however, they display notable distinctions in terms of DEGs and corresponding gene modules. The functional characteristics of FTSEC1 and FTSEC2 predominantly involve terms of cytoplasmic translation and protein secretion. On the other hand, FTSEC3 and FTSEC4 are enriched in functions associated with angiogenesis, ECM synthesis and promoting cell migration. FTSEC3 and FTSEC4 demonstrate functions more closely linked to tumor-related processes compared to the other two FTSEC subgroups. Based on these findings, we speculate that these two cell types represent distinct subtypes of epithelial cells that may be involved in OC development.
To validate this, we performed pseudotime analysis and confirmed temporal ordering of these cell types along the trajectory. The FTCECs are located at the early stage of the pseudotime trajectory, FTPECs occupy the intermediate position of the trajectory, and FTSECs are positioned relatively toward the end of the trajectory (Figures 2E and S2G). FTSEC3 and FTSEC4 are indeed positioned toward the very end of the pseudotime trajectory. Notably, FTSEC3 exhibits higher expression of CSC marker genes (VEGFA, CD44, CD24, and ALDH1A1) compared to other FTSEC sub clusters23,25 (Figure 2F), indicating that the presence of FTSEC3 is associated with OC development. Several sex hormone receptors (AR, ESR1, and PGR) (Figure 2F), which aligns with the functional characteristics associated with FTSEC3, were highly expressed in FTSEC3. We analyzed the expression of signatures genes along the pseudotime trajectory and revealed that genes (cluster 4) predominantly expressed in FTSEC3 were located at the end of the pseudotime trajectory (Figure 2G). The genes within cluster 4 exhibit significant enrichment in cytokine production, cell migration, epithelial cell morphogenesis, and protein translation (Figure 2H and Table S8). Furthermore, it is noteworthy to mention that genes in the late stage of pseudotime exhibit an enrichment of vascular endothelial growth factor (VEGF) related signaling pathways (Figures 2H and S2E), is consistent with the functional characteristics of FTSEC3, implying FTSEC3 may potentially contribute to tumor progression, through its regulation of tumor angiogenesis.
Endothelial cell heterogeneity and evolution in OC
Tumor endothelial cells are highly heterogeneous and play essential role in tumor progression.8,28 Our previous analyses suggested that fallopian tube epithelial cells (FTSEC3) positively regulate VEGF production, which reciprocally promotes tumor neoangiogenesis and OC progression. To unravel the endothelial cell heterogeneity, evolution, interaction with epithelial cells, and roles in OC progression, we carried out a comprehensive investigation focusing on endothelial cells in OC. Initially, we performed a rigorous annotation of 11,696 OC endothelial cells and identified 11 distinct endothelial cell phenotypes (subtypes) (Figures 3A, S3A, and S3B). Based on the expression profiles of classical endothelial cell marker genes, we have identified distinct subtypes of endothelial cells, including typical vascular bed endothelial cells, such as arterial endothelial cells (EC2, positively expressing GJA5+), venous endothelial cells (EC1, ACKR1+), capillary endothelial cells (EC5, CA4+), and lymphatic endothelial cells (EC4, PROX1+) (Figures S3A and S3B, Table S9). We also identify several functionally distinct endothelial cell subtypes based on expression of marker genes, GO functional enrichment analysis and gene set enrichment analysis (Figures 3B and S3C). These include the tip cells (EC3, ESM1+), proliferating endothelial cells (EC6, MKI67+), immune activated endothelial cells (EC10, IGKC+), pericytes (not endothelial cells but annotated as EC7 to be consistent, RGS5+), and an intermediate phenotype of endothelial cells transitioning into mesenchymal cells (fibroblasts)29 (EC8, COL1A1+) (Figure S3D and Table S10). Two endothelial cells phenotypes: one (EC9) exhibiting proangiogenic (WFDC2) and immune suppressive signatures (leukocyte peptidase inhibitor SLPI) and the other (EC11) expressing strong ciliated cell markers (CAPS and TPPP3) were mainly found detected in normal fallopian tubes.
Figure 3.

Endothelial cell heterogeneity and evolution in OC
(A) UMAP plot of endothelial cell sub clusters.
(B) Bubble plot showing differentially enriched GO terms in each endothelial cell sub cluster.
(C) UMAP plots of the distribution of endothelial cell sub cluster in three tissue types.
(D) Bar plot shows the proportion of different endothelial cell sub clusters in each tissue.
(E) The fractions of four endothelial cell sub clusters between fallopian tube, normal ovarium and OC tissues in OC bulk RNA-seq (GSE223426). Comparisons of cellular proportions between fallopian tube and ovarian tumor tissues for each endothelial cell sub cluster were performed using Student’s t test.
∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001, ∗∗∗∗, p < 0.0001. Data are represented as median with 25% and 75% percentile.
We further quantified the proportions of endothelial cell subtypes in the fallopian tube, normal ovarium, and OC tissue. Our results showed that angiogenic tip cells (EC3), proliferating endothelial cells (EC6), and immune activated endothelial cells (EC10) were profoundly detected in OC tissues. The mucosal ciliated endothelial cells were only detected in the fallopian tubes (Figures 3C and 3D). We confirmed our finding of increase in tip and proliferating endothelial cells by deconvolved bulk transcriptome data for normal fallopian tubes, normal ovarium, and OC tumor (Figure 3E).
Endothelial tip cells exhibit strong TGF-β signaling and are associated with poor outcome
Since tip cells are leading the tumor neoangiogenesis, we first sought to validate the finding of increase ESM1+ tip cell in OC. We measured ESM1 expression at the RNA level in 40 OC samples and 10 normal ovary control. Indeed, the ESM1 expression was significantly increased in OC compared to control (p value = 0.0001, un-pair t test, Figure 4A). We further confirmed that expression of ESM1 was specific to endothelial cells based on immunofluorescent assay (Figure 4B). To obtain deeper insights into tip cell functions, we performed GO enrichment analysis and found that endothelial tip cells were significantly enriched in terms related to epithelial and endothelial cell migration (Figure 4C), suggesting that the endothelial tip cells promote the epithelial cell migration and angiogenesis, thereby playing a critical role in the progression of OC. We further analyzed the DEGs in tip cells as compared to other subtypes of endothelial cells (Figure 4D and Table S11), as well as 14 signaling pathways in the endothelial cell subtypes. Our results showed that in addition to VEGF signaling, tip cells also exhibited high TGF-β signaling (Figure 4E). The TGF-β pathway has been reported to enhance cell invasion and migration and induce EMT in various cancers.30,31,32,33,34 We analyzed the intersection of the TGF-β signaling pathway genes and the DEGs related to epithelial cell migration, and identified 5 key genes including PTK2, ACVRL1, CDH5, TGFB1, and ITGB1 (Figure 4F). All these genes have been reported promoting cancer cell migration and metastasis.34,35,36,37,38 Notably, we observed high expression levels of these five genes within the endothelial tip cells (Figure 4G), as well as proliferating endothelial cells. To identify the regulons in OC endothelial tip cells, we performed gene regulatory network analysis and identified the specific expression of the transcriptional repressor Blimp-1 (PRDM1) (Figure S4A). Blimp-1 plays a crucial role in TGF-β1-induced EMT by inhibiting BMP-5 in breast cancer.39 Furthermore, we examined the association between tip cell signatures and the long-term prognosis of OC patients using data from The Cancer Genome Atlas (TCGA). We found that patients with high expression of the top 10 tip cell signature genes had a significantly lower overall survival rate (Wald test, p value = 0.022) (Figure 4H and Table S12), further confirming their functions in tumor angiogenesis and progression. We also fit a multivariate Cox-PH regression model, after adjusting for other covariates in the Cox-PH model, EC3_Endo_ESM1 remained significantly correlated with the survival rate of OC patients (Wald test, p value <0.001) (Figure S4B). In summary, our study highlights the important role of endothelial tip cells in OC progression, in part through the TGF-β pathway, providing important insights into the underlying molecular mechanisms.
Figure 4.

Endothelial tip cells exhibit strong TGF-β signaling and are associated with worse prognosis outcome
(A) Quantification of ESM1 expression in 40 OC samples and 10 control specimens using quantitative PCR. Values were represented as the mean ± SD of minus delta CT value between ESM1 and ACTIN. p value = 0.0001, un-pair t test.
(B) Representative immunofluorescence staining images from three OC patient tumor tissues with antibodies against human CD31 and ESM1.
(C) Bubble diagram representation of top 10 significantly enriched GO terms for EC3_Endo_ESM1.
(D) Volcano plot of DEGs in EC3_Endo_ESM1 compared with other endothelial cell sub clusters. The epithelial cell migration genes are labeled.
(E) Mean pathway activity scores of endothelial cell sub cluster.
(F) The Venn diagram shows the intersection of TGF-β signaling pathway genes and DEGs related to epithelial cell migration.
(G) Gene expression levels of TGFB1, PTK2, ITGB1, ACVRL1 and CDH5 in fallopian tube and OC tissues at bulk transcriptome.
(H) Survival analysis with Cox-PH regression model of patients with TCGA-OV grouped by the top10 marker gene of EC3_Endo_ESM1. p values were calculated by Wald test. p value = 0.022.
Endothelial tip cells interact with FTSEC3 in promoting OC progression
To further investigate the specific interaction between tip endothelial cells and FTSECs, which resemble ovarian tumor epithelial phenotype, we conducted cell-cell communication between tip cells and FTSEC3 using ligand-receptor pairs analysis. Firstly, we studied the output signaling pathways from endothelial tip cells (EC3) and found that FTSEC3 is the epithelial cell type receiving the most signals (Figure 5A). Among the various ligand-receptor pairs identified, the LAMININ, EGF, EPHA, HSPG, SEMA4, and TRAIL signaling pathways exhibited the highest signal strength in FTSEC3. We further calculated the specific strengths of communication between these signaling pathways and their corresponding ligand-receptor pairs (Figures 5A and S5A). Notably, FTSEC3 displayed a higher specificity in receiving HSPG and TRAIL signals. Notably, both HSPG and TRAIL have been reported to promote tumor cell migration and EMT,40,41,42 suggesting that endothelial tip cells promotes the EMT of FTSEC3 and migration.
Figure 5.

Cell-cell interactions between tip endothelial cells and FTSEC3
(A) EC3_Endo_ESM1 output signal pathway intensity. Left side indicates outgoing signaling patterns, right side represents incoming signaling patterns. Color represents relative cell communication strength, with darker colors indicating greater intensity.
(B) UMAP plot showing pan-cancer endothelial cells clusters by color.
(C) The violin plots show the expression of indicated genes in tip endothelial cells of 5 different cancer types.
(D) The circle plot shows the TGF-β signal pathway network. Colors represent different clusters of epithelial and endothelial cells. The thickness of the lines indicates the strength of cell communication. Thicker lines indicate greater communication intensity.
(E) The violin diagram shows the distribution of ligand-receptor gene expression in the TGF-β signaling pathway in different epithelial and endothelial clusters.
(F) Quantification of CRISPR knockout (KO) efficiency (indel %) in HUVECs (mean ± SD, n = 5).
(G) Quantification of spheroid-based in vitro sprouting angiogenesis (n = 10 spheroid per group, mean ± SD). ∗, adjusted p value <0.05; ∗∗∗, adjusted p value <0.001; ∗∗∗∗, adjusted p value <0.0001, one-way ANOVA.
Conserved endothelial signaling pathways across tumor types
To investigate the specificity of these pathways in promoting OC cell migration and EMT, we identified endothelial tip cells within a pan-cancer cohort of endothelial cells43 (Figures 5B and S5C). Subsequently, we examined the expression patterns of the ligand genes associated with these pathways in five different types of cancers, including BC, GC, ICC, LC, and PDAC (Figure 5C). Our findings revealed that EPHA, HSPG, TGF β, and LAMININ signaling pathways were conserved across all five types of cancers, while SEMA4 is specific in OC and TRAIL signaling was minimal in BCs. In addition, our analysis revealed the presence of the TGF-β pathway, which interestingly, was exclusively received by FTSEC3 among the various fallopian tube epithelial subtypes (Figures 5A and 5D). To gain further insights into this interaction, we examined the expression patterns of TGF-β receptor genes and found that the receptor genes TGFBR1 and TGFBR2 were specifically expressed in FTSEC3 (Figure 5E). At the protein level, we also observed the expression of TGFB1 in vascular endothelium and TGFBR1 in fallopian tube epithelium (Figure S5D). We explored the signaling pathway of FTSEC3 as the signal sender and identified that the strongest signal received by tip cells via VEGF signaling pathway (Figure S5E). To further validate the effect of TGF-β, and LAMININ pathway on EC angiogenesis, we applied CRISPR-Cas9 to KO the LAMA4, TGFB1, and TGFBR1 genes in cultured primary human umbilical vein endothelial cells (HUVECs). We obtained over 70% gene KO efficiency for all three genes (Figure 5F). To study the effect on angiogenesis, we performed spheroid-based in vitro angiogenesis and validated that the genetic disruption of LAMA4, TGFB1, and TGFBR1 significantly impairs the angiogenic capacity of HUVECs (Figure 5G). These results collectively suggest that endothelial tip cells (EC3) and FTSEC3 exhibit strong signaling communications, in part through the ECM modeling pathway, which reciprocally promote OC development and progression.
Discussion
In conclusion, we have provided a deep investigation of epithelial cells and endothelial cell evolution in OC based on single-cell transcriptome analysis. A number of single-cell RNA sequencing studies have been conducted on fallopian tube epithelial cells,4,5,6,7 which have unveiled the existence of diverse functional subtypes within normal fallopian tube epithelial cells. These distinct functional subtypes are hypothesized to potentially serve as the origin cells for OC. The reduction in the abundance of ciliated cells significantly related with OC risk and disease progression.44,45,46 The process of reciprocal transformation between ciliated and secretory cells may be one of the potential causes in FTCEC loss.45,47 In line with these, our analysis identified SOD3+ FTCEC2, which expresses both markers of ciliated cell and secretory cells (Figure S2B). The SOD3+ FTCEC2 cells were located at both early and late stages of pseudo-time trajectory, suggesting that SOD3+ FTCEC cells may be undergoing a transition to secretory cells. Nevertheless, the mechanisms of ciliated cell loss and their relationship with tumor progression requires further investigation.
It has been suggested that fallopian tube secretory cells, especially those under an EMT state, may represent a potential cellular origin for OC.4,5,6,7,45 Gene modules associated with EMT functions were found to be enriched in FTSEC3. Furthermore, FTSEC3 expresses multiple tumor stem cell marker genes23,25 and sexual hormone receptor genes,48 lacking the expression of stromal fibroblast transcription factor PRRX1 gene.5 The evolution of fallopian tube epithelial cells may be associated with the progression of OC, with FTSEC3 representing the terminal stage of fallopian tube epithelium evolution. Additionally, the DEGs of FTSEC3 are also enriched in VEGF-related signaling pathways, indicating that FTSEC3 plays an important role in tumor angiogenesis via VEGF signaling. However, it should be noted that a limitation of our study is the lack of paired fallopian tube and tumor tissues from the same patients, as well as the absence of follow-up information on the fallopian tube donors. Therefore, it is currently not possible to establish a direct cause-and-effect relationship between fallopian tube epithelial cells in an EMT state and the transformation into cancer cells. Additional research and analysis are necessary to provide more comprehensive insights. Unfortunately, most of the tumor cell data in Epi2 did not include information on the International Federation of Gynecology and Obstetrics (FIGO) stage, making it impossible to determine the potential connection between OC precursor cells and cancer stage.
Vascular endothelial cells play a crucial role in the development and progression of tumors. It has been reported that tip and proliferating endothelial cells are detectable only in tumor tissues.8 Our findings indicate that a certain population of tip endothelial cells also exists in normal fallopian tube tissues, which might play a role in fallopian tube epithelial cells migration and OC formation. Furthermore, our results suggest that tip endothelial cells are highly likely to facilitate the process of EMT in fallopian tube epithelial cells through LAMININ and TGF-β signaling. This molecular cascade ultimately enhances the invasive and migratory capacities of the fallopian tube epithelium, thereby contributing to the onset and progression of OC.
Through cell-cell communication analysis between endothelial cells and fallopian tube epithelial cells, we discovered that FTSEC3 is the epithelial cell subtype receiving the most output signals from tip endothelial cells. Previous studies have also indicated that the interaction between secretory epithelial cells and endothelial cells in the fallopian tubes is more prominent compared to that with ciliated epithelial cells.48 Moreover, we found that HSPG and TRAIL are more specifically received by FTSEC3, suggesting that tip endothelial cells might regulate the EMT of OC-initiating cells, FTSEC3, through the ligand-receptor pairs of HSPG and TRAIL, thereby promoting the occurrence of OC. Meanwhile, FTSEC3 regulates tip endothelial cells through the VEGF ligand-receptor pair to promote angiogenesis. Tip endothelial cells respond to various angiogenic signals, guiding and navigating vascular sprouting, and providing the necessary nutrients and oxygen for tumor growth.13 Finally, we identified tip endothelial cells in the pan-cancer dataset and examined the expression of ligand genes involved in the EMT signaling and ECM modeling pathways. And our findings provide evidence of the widespread presence of the EPHA, HSPG, TRAIL, TGF-β, LAMININ pathways in cancer.
In summary, our research has identified FTSEC3 cells are positioned at the late differentiation trajectory of fallopian tube epithelial cells, express ovarium tumor epithelial cell signatures, and are enriched in VEGF production. Additionally, ESM1+ tip endothelial cells play a significant role in promoting the occurrence of OC. Therefore, understanding and intervening in the functions and regulatory mechanisms of tip endothelial cells can enhance our understanding of tumor growth and metastasis mechanisms, and provide new targets and strategies for cancer therapy.
Limitations of the study
We integrated single-cell transcriptome data that had been conducted over 14 studies. Although we have used bioinformatic tools to carefully correct any batch effect, the integrated nature of these single-cell transcriptome data from 84 subjects remains as a potential caveat of the work. Endothelial cells are functionally heterogeneous. Due to the technical difficulties in establishing endothelial cell cultures from OC tissues, we have used normal human umbilical vein endothelial cells (HUVECs) to validate the effect of LAMININ and TGF-β connectomes on cancer endothelial cell functions, which represents another limitation of the study.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Yonglun Luo (alun@biomed.au.dk).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
Data that support the findings of this study are available in the key resources table and from the lead contact author upon reasonable request.
-
•
Code for analyzing the single-cell data are available from the GitHub depository https://github.com/lianglangchao/ovarian_cancer_llc_V2.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Acknowledgments
We thank the China National GeneBank Database for providing computational resource for the storage, processing, and analysis of the scRNA-seq data. We thank Dr. Jianqing Zhu for heading the ethical approval application for the validation patient samples (IRB-2023-978). This project is supported by the Qingdao-Europe Advanced Institute for Life Sciences. Y.L. is supported by Danish Research Council (9041-00317B), European Union’s Horizon 2020 research and innovation program under grant agreement No 899417 (Y.L.), the Novo Nordisk Foundation (NNF21OC0068988; NNF21OC0071031), M-ERA.Net and the Innovation Fund Denmark (9355 PIECRISCI), and the Lundbeck Foundation Ascending Investigator award (R396-2022-350).
Author contributions
Conceptualization: L.Liang., C.C. and Y.L.; methodology: L.Liang., C.C., A.L., A.S.N., S.K., C.B.B., and W.Z.; validation: A.L., A.S.N., S.K., C.B.B., and W.Z.; formal analysis: L.Liang. and C.C.; investigation: L.Liang., C.C., A.L., A.S.N., S.K., C.B.B., W.Z., H.L., and L.Lin.; resources: L.Lin., J.W., and Y.L.; writing—original draft: L.Liang. and C.C.; writing—review and editing: all authors; visualization: L.Liang., C.C., and Y.L.; supervision: J.W. and Y.L.; project administration: Y.L.; funding acquisition: Y.L.
Declaration of interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-CD31/PECAM-1 antibody (H-3) | Santa Cruz Biotechnology | sc-376764 |
| anti-ESM1 | Abcam | ab235966 |
| goat anti-mouse Alexa Fluor 488 | Invitrogen | A-11001 |
| donkey anti-rabbit Alexa Fluor 647 | Invitrogen | A-31573 |
| Chemicals, peptides, and recombinant proteins | ||
| M199 medium | Gibco | Cat#22350-029 |
| L-glutamine | Gibco | Cat#35050-061 |
| Endothelial Cell Growth Supplement (ECGS)/Heparin | PromoCell | Cat#C-30120 |
| fetal bovine serum | Sigma | Cat#F7524 |
| SpCas9 | IDT | Cat#1081059 |
| Opti-MEM | Thermo Scientific | Cat#31985062 |
| AccuPrime PFX Reaction Mix | Invitrogen | Cat#92008 |
| methylcellulose | Merck | Cat#M05129 |
| Collagen-type I solution | Merck | Cat#08-115 |
| 4% paraformaldehyde | AH Diagnostics | Cat#SC-281692 |
| Critical commercial assays | ||
| NucleoSpin Gel and PCR Clean-up kit | Macherey | Cat#2006/001 |
| Mix2Seq Kit | Eurofins Genomics | N/A |
| Experimental models: Cell lines | ||
| Human umbilical vein endothelial cells (HUVECs) | Isolated from human umbilical vein, this study | N/A |
| Oligonucleotides | ||
| LAMA4 gRNA (spacer sequences): TGACATTGAAGGGAGCTCAG | Synthego, US | N/A |
| TGFB1 gRNA (spacer sequences): GGCACCTCCCCCTGGCTCG | Synthego, US | N/A |
| TGFBR1 gRNA (spacer sequences): TTGACTTAATTCCTCGAGAT | Synthego, US | N/A |
| Software and algorithms | ||
| Seurat | https://satijalab.org/seurat/ | version 4.1.0 |
| cNMF | https://github.com/dylkot/cNMF | version 1.3.5 |
| Monocle2 | https://cole-trapnell-lab.github.io/monocle-release/ | version 2.22.0 |
| harmony | https://github.com/immunogenomics/harmony | version 0.1.0 |
| org.Hs.eg.db | https://bioconductor.org/packages/release/data/annotation/html/org.Hs.eg.db.html | version 3.16.0 |
| clusterProfiler | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html | version 4.6.0 |
| ggplot2 | https://ggplot2.tidyverse.org/ | version 3.3.5 |
| GSVA | https://bioconductor.org/packages/release/bioc/html/GSVA.html | version 1.46.0 |
| pheatmap | https://www.rdocumentation.org/packages/pheatmap/versions/1.0.12/topics/pheatmap | version 1.0.12 |
| PROGENy | https://github.com/saezlab/progeny | version 1.16.0 |
| pyscenic | https://github.com/aertslab/pySCENIC | version 0.11.2 |
| CellChat | https://github.com/sqjin/CellChat | version 1.5.0 |
| CIBERSORTx | https://cibersortx.stanford.edu/ | version 0.1.0 |
| ImageJ | https://imagej.net/ij/ | version 2.9.0 |
| Code availability | https://github.com/lianglangchao/ovarian_cancer_llc_V2 | N/A |
| Ovarian cancer single cell RNA expression database | https://dreamapp.biomed.au.dk/OvaryCancer_DB/ | N/A |
| Integrated OC scRNA-seq data | https://lambrechtslab.sites.vib.be/en/pan-cancer-blueprint-tumor-microenvironment-0 | N/A |
| Integrated OC scRNA-seq data | https://lambrechtslab.sites.vib.be/en/high-grade-serous-tubo-ovarian-cancer-refined-single-cell-rna-sequencing-specific-cell-subtypes | N/A |
| Integrated OC scRNA-seq data | https://github.com/vicDRC/CCR_CD39study/tree/master/data | N/A |
| Integrated OC scRNA-seq data | Gene Expression Omnibus (GEO) |
GSE140819 GSE173682 GSE165897 GSE178101 GSE151214 GSE150443 GSE151316 GSE184880 GSE160755 GSE193371 GSE194105 GSE154600 GSE130000 GSE158937 |
| Other | ||
| Nikon laser confocal microscope | Japan | A1 HD25 |
Experimental model and study participant details
Cell lines
Primary human umbilical vein endothelial cells (HUVEC, less than passage 3) were used for CRISPR KO experiment. HUVECs were authenticated by evaluating the expression of CD31.
Human samples
Three achieved ovarian tumor tissues (age 52; 56; 69) were used for immunofluorescence staining of ESM1 and CD31 (ethical approval number of the sample achieve: IRB-2023-978).
Method details
Data collection and processing
All published human OC scRNA-seq data generated using the 10X genomics platforms were selected for our analysis. We filtered the data according to the quality control criteria described in the original study. If the specific quality control conditions were not mentioned by the original article, cells with the number of expressed genes <500 (low quality) or >7000 (doublets), or MT% transcript > 25 % (dead cells), were removed. For more details regarding the data source, patient clinical information, and data processing, please refer to our previous study.49
Consensus Non-negative matrix factorization (cNMF) analysis for single cell data matrix
The cNMF analysis was performed with the following parameters: numiter=200, numhvgenes=3000, and components=np.arange(2,30). From the resulting decomposition of 8 gene modules out of the range of 2 to 30, we proceeded with dimensionality reduction and clustering. The “RunUMAP” function from the Seurat package was used for dimensionality reduction, with the reduction parameter set to 'nmf' and dims parameter set to 1:8. To identify cell clusters, the “FindNeighbors” and “FindClusters” functions were applied with default parameters and a resolution parameter of 0.1.
Pseudotime analysis
Monocle2 (version 2.22.0) was used to explore the underlying changes in epithelial cell function and identify potential lineage differentiation.50 Original UMI count, gene and cell matrix were used as input using the “newCellDataSet” function to create a Monocle object with lowerDetectionLimit = 0.1, expressionFamily = negbinomial.size(). “reduceDimension” was then used to reduce the data dimension with method = “DDRTree” and max_components = 2. We performed “diferentialGeneTest” to identify genes that differed significantly over time.
Endothelial cell single cell data integration and clustering
We extracted the cluster of endothelial cells from the total cells. After applying the criteria: positive for the endothelial cell marker PECAM1 < 0 and negative for immune cell marker PTPRC=0, we obtained a total of 11,696 endothelial cells. Subsequently, we applied the “NormalizeData” and “ScaleData” functions on the Seurat object to perform data normalization and scaling. The “FindVariableFeatures” function was used to identify variable genes. We then performed Principal Component Analysis (PCA) using the “RunPCA” function.51 After PCA, we used R package harmony (version 0.1.0) to remove batch effects between samples, using parameters assay.use = “RNA”, max.iter.harmony = 15.52 Finally, we performed clustering using the “FindNeighbors” and “FindClusters” functions with a resolution of 0.8. We utilized 30 principal components for the analysis. Subsequently, we applied Uniform Manifold Approximation and Projection (UMAP) to visualize the cellular landscape.
Gene ontology (GO) enrichment analysis
We employed the R package org.Hs.eg.db (version 3.16.0) for the conversion of symbols and ENTREZIDs. The subsequent GO enrichment analysis was conducted utilizing the "enrichGO" function of clusterProfiler (version 4.6.0) with the parameters ont="BP" and pvalueCutoff=0.05.53 The results were visualized using ggplot2 (version 3.3.5).
Identification of differentially expressed genes (DEGs)
To identify markers (DEGs) for identity classes, we utilized the “FindMarkers” function from the R package Seurat. The analysis was conducted with the following parameters: logfc.threshold = 0.25, min.pct = 0.25, only.pos = T, and return.thresh = 0.05.
Gene Set Variation Analysis (GSVA)
We used the GSVA package (version 1.46.0) for gene set enrichment analysis.54 Specifically, we used single sample gene set enrichment analysis (ssGSEA) to identify enrichment pathways for 50 hallmark gene sets in the Molecular Signature Database (MSigDB) of each endothelial cell phenotype. The pheatmap (version 1.0.12) was used for data visualization.
Inference of tumor-related signaling pathway activity
We used the PROGENy package (version 1.16.0) to infer the activity of tumor-associated signaling pathways.55 Inference of pathway activity is based on the gene set of the first 500 most responsive genes after the corresponding pathway perturbation. Subsequently, we computed the Progeny activity scores and added them to our Seurat object. The Progeny scores were then summarized by cell population, and the results were visualized using pheatmap (version 1.0.12).
Gene regulatory network analysis
SCENIC (version 0.11.2) was used to construct regulatory networks from scRNA-seq data of endothelial cell sub cluster.56 To identify specific TF regulons for endothelial cell sub cluster, SCENIC analysis was performed on all endothelial cells. We used the pyscenic (version 0.11.2) and hg38__refseq-r80__10kb_up_and_down_tss.mc9nr.genes_vs_motifs.rankings.feather databases for GRNboost, RcisTarget, and AUCell. The input matrix was the raw expression matrix that was from Seurat object.
Survival analysis
The Cancer Genome Atlas (TCGA) ovarian cancer data (TCGA, Nature 2011), which consists of clinical information and mRNA expression matrix, were downloaded from the cBioPortal database (http://www.cbioportal.org/). We calculated the average gene expression levels of the top 10 genes for EC3_Endo_ESM1 and determined the optimal cutpoint of this variable using “surv_cutpoint” function. Following this, we conducted univariable Cox-PH survival analysis on seven factors: Fraction Genome Altered, Neoplasm Histologic Grade, Mutation Count, Platinum Status, TMB (nonsynonymous), Tumor Stage 2009, and EC3_Endo_ESM1 (Table S12), identifying 5 factors significantly associated with survival. Subsequently, we used these five significant factors to fit a multivariate Cox-PH regression model. Finally, we visualized the results using “ggsurvplot” and “ggforest” functions.
Cell-cell communication analysis
Intercellular interaction analysis was performed using CellChat (version 1.5.0) based on the expression of known ligand-receptor pairs in different cell clusters.57 The normalized scRNA-seq gene expression matrix was used as the input data. The reference database used for ligand-receptor pairs was “CellChatDB.human”. The “computeCommunProb” was used to infer the probability and strength of intercellular communication. After that, we used “netAnalysis_computeCentrality” to calculate and visualize the network centrality scores. Lastly, we used “netAnalysis_signalingRole_network”, “netVisual_bubble” and “netVisual_aggregate” for visualization.
Cell composition deconvolution
Cell composition deconvolution was performed using CIBERSORTx.58 Firstly, a feature gene expression matrix was constructed based on the marker gene of endothelial cell subtype from the scRNA-seq dataset. Secondly, org.Hs.eg.db was used to convert ENTREZID of bulk RNA-seq data to gene names. Subsequently, CIBERSORT was employed to estimate the cellular proportions of bulk RNA-seq samples based on bulk RNA-seq dataset, with parameters set as “perm = 1000” and “QN = T”.
Human clinical specimens used for validation experiments
Forty primary ovarian tumors and ten normal ovary tissues as described previously59 were used for quantification of ESM1 expression at the RNA levels. Three ovarian tumor tissues were collected for multiple immunofluorescence staining. Informed consent was obtained from all patients for the collection and use of clinical samples, and the study was approved by the Scientific Ethics Committee of Zhejiang Cancer Hospital (IRB-2023-978).
RT-qPCR
TRIzol reagent was utilized for total RNA extraction. To assess mRNA levels, cDNA synthesis was performed utilizing Hiscript II Q RT SuperMix for qPCR (+gDNA wiper) (Vazyme Biotech, Nanjing, China), followed by PCR amplification using ChamQ Universal SYBR qPCR Master Mix (Vazyme). β-actin mRNA served as a reference. The qPCR primer sequences are listed in Table S13.
Immunofluorescence
Paraffin sections (4 μm) from clinical specimens underwent a series of dewaxing and hydration steps. Antigen retrieval was then executed using 0.01M citrate buffer (pH 6.0), employing microwave treatment for 25 min, followed by gradual cooling to room temperature and thorough washing with PBST. Tissue permeabilization was achieved by treating the sections with 0.1% Triton X-100 for 10 min. Subsequently, endogenous peroxidase activity and non-specific antigens were blocked, followed by incubation with the primary antibodies anti-CD31/PECAM-1 antibody (H-3) (1:500; sc-376764, Santa Cruz Biotechnology) and anti-ESM1 (1:500, ab235966, Abcam), overnight at 4°C. After washing, the sections were incubated with the appropriate Alexa Fluor-conjugated secondary antibodies [goat anti-mouse Alexa Fluor 488 (1:2000; A-11001, Invitrogen) and donkey anti-rabbit Alexa Fluor 647 (1:20000; A-31573, Invitrogen)] at room temperature for 40 min. Finally, the sections were incubated with DAPI for 10 minutes before image acquisition using a Nikon laser confocal microscope (A1 HD25, Japan).
Human umbilical vein endothelial cells (HUVEC)
Primary HUVECs were cultured in M199 medium (Gibco, #22350-029) supplemented with 2 mM L-glutamine (Gibco, #35050-061), 0.4% Endothelial Cell Growth Supplement (ECGS)/Heparin (PromoCell, #C-30120), and 20% fetal bovine serum (FBS) (Sigma, #F7524).
CRISPR-Cas9 based gene editing of HUVECs
CRISPRon60 was used to design and evaluate the guide RNAs for LAMA4, TGFB1, and TGFBR1 knockout (KO). The designed gRNAs were then purchased from Synthego, US. For nucleofection, ribonucleoprotein (RNP) complex required was prepared by mixing 2.4 μl nuclease free water with 1.2 μl of gRNA (3.2 μg/μl) and 1.2 μl of SpCas9 (10 μg/μl) (IDT, #1081059), and placed in room temperature between 10 to 60 minutes. HUVEC cells (2 × 105) were resuspended in 36.2 μl Opti-MEM (Thermo Scientific, #31985062) and 4.8 μl RNP complex for the gene KO. Nucleofection was performed using the 4D-Nucleofector X Unit (Lonza, CH) under the program termed CM138. Immediately after nucleofection, 150 μL prewarmed M199 medium with supplements was added to each well in the nucelocuvette. The nucleofected HUVECs were then seeded in the 0.1% gelatin coated 24-well plate with a total volume of 500 μL culturing medium and incubated for 24 h in 5% CO2 at 37°C. CRISPR KO cells were harvested for KO efficiency quantification and sprouting angiogenesis 48 hours after nucleofection.
KO efficiency quantification with ICE assay
PCR was carried out using the AccuPrime PFX Reaction Mix (Invitrogen, Waltham, MA, USA, #92008) and gene specific primers (Table S14). The PCR product was purified using a NucleoSpin Gel and PCR Clean-up kit (Macherey, #2006/001) for Sanger sequencing at Eurofins Genomics with a Mix2Seq Kit (Eurofins Genomics, Germany). The Sanger sequencing data were analyzed with the web tool “ICE”, a KO analysis tool by Synthego, to calculate the overall CRISPR gene editing efficiency.
Spheroid assay
5 × 105 HUVECs cells were resuspended in 12.5 ml of EGM2 medium with 20% methylcellulose (Merck, #M05129) solution. The well-mixed cell suspension was transferred to multichannel pipette reservoir. Cell suspensions were evenly transferred to non-adherent plastic dishes, with 25 μl per droplet. The plates with individual cell droplets were cultured upside down and incubated for 24 hours at 5% CO2 at 37°C. Evenly form and rounded spheroids were observed on the succeeding day. 10 ml of DPBS with 10% FBS solution was spread on the non-adherent plate to harvest spheroids in a 50 ml conical tube and sedimented by centrifuging at 300 g for 5minutes and then at 500 g for 3 minutes. The centrifuge was set to no brake mode for this sedimentation process. The supernatant was carefully aspirated, and the pellet obtained was gently loosened by inverting the tubes. The pellet was then overlayed with polymerization solution (made on ice) constituting of 375 μl of Collagen-type I solution (Merck, #08-115), 475 μl of methyl cellulose with 40% FBS, 150 μl of Sodium Bicarbonate (15.6 mg/ml), and 10 μl of 1 M Sodium Hydroxide respectively per unit volume of polymerization reagent required. The required volume of polymerization solution was calculated as 1 ml per 60 spheroids. 500 μl of the polymerization solution containing spheroids was transferred to 24 well plates and incubated with an incubator for polymerization. After 20 minutes, we added 500 μl of EGM2 media and incubate it at 37°C and 5% CO2. Sprouts were visible within 6 hours and the media was removed and replaced with 500 μl 4% paraformaldehyde (AH Diagnostics, #SC-281692) solution for fixation. Sprouts were imaged using ZOE fluorescent Cell Imager system. The images were then analyzed using Fiji (ImageJ) v. 2.9.0 analysis tool and sprout length were quantified. The graph obtained were plotted using the Graphpad Prism 9 software.
Quantification and statistical analysis
Statistical analyses were conducted using the ggpubr R package (version 0.4.0) and Graphpad Prism 9. The significance of the ratio of endothelial cell subtypes was evaluated by Student's t test. Statistical significance was defined as p < 0.05 (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; ns, not significant). To determine the statistical significance of differences between survival curves in Cox-PH regression model, we performed Wald test using the implementation provided in the survival package. One-way ANOVA with multiple comparison was used for statistical analysis of sprouting between different cell lines. Pooled data are represented as mean +/- SD. Box plot data are presented as median with 25% and 75% percentile.
Additional resources
The integrated ovarian cancer single cell expression database can be accessed through this url: https://dreamapp.biomed.au.dk/OvaryCancer_DB/.
Published: November 19, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.111417.
Contributor Information
Jianmin Wu, Email: jianminwu81@foxmail.com.
Yonglun Luo, Email: alun@biomed.au.dk.
Supplemental information
References
- 1.Kindelberger D.W., Lee Y., Miron A., Hirsch M.S., Feltmate C., Medeiros F., Callahan M.J., Garner E.O., Gordon R.W., Birch C., et al. Intraepithelial carcinoma of the fimbria and pelvic serous carcinoma: Evidence for a causal relationship. Am. J. Surg. Pathol. 2007;31:161–169. doi: 10.1097/01.pas.0000213335.40358.47. [DOI] [PubMed] [Google Scholar]
- 2.Li J., Condello S., Thomes-Pepin J., Ma X., Xia Y., Hurley T.D., Matei D., Cheng J.X. Lipid Desaturation Is a Metabolic Marker and Therapeutic Target of Ovarian Cancer Stem Cells. Cell Stem Cell. 2017;20:303–314.e5. doi: 10.1016/j.stem.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jovic D., Liang X., Zeng H., Lin L., Xu F., Luo Y. Single-cell RNA sequencing technologies and applications: A brief overview. Clin. Transl. Med. 2022;12:e694. doi: 10.1002/ctm2.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hu Z., Artibani M., Alsaadi A., Wietek N., Morotti M., Shi T., Zhong Z., Santana Gonzalez L., El-Sahhar S., Carrami E.M., et al. The Repertoire of Serous Ovarian Cancer Non-genetic Heterogeneity Revealed by Single-Cell Sequencing of Normal Fallopian Tube Epithelial Cells. Cancer Cell. 2020;37:226–242.e7. doi: 10.1016/j.ccell.2020.01.003. [DOI] [PubMed] [Google Scholar]
- 5.Ulrich N.D., Shen Y.C., Ma Q., Yang K., Hannum D.F., Jones A., Machlin J., Randolph J.F., Jr., Smith Y.R., Schon S.B., et al. Cellular heterogeneity of human fallopian tubes in normal and hydrosalpinx disease states identified using scRNA-seq. Dev. Cell. 2022;57:914–929.e7. doi: 10.1016/j.devcel.2022.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dinh H.Q., Lin X., Abbasi F., Nameki R., Haro M., Olingy C.E., Chang H., Hernandez L., Gayther S.A., Wright K.N., et al. Single-cell transcriptomics identifies gene expression networks driving differentiation and tumorigenesis in the human fallopian tube. Cell Rep. 2021;35 doi: 10.1016/j.celrep.2021.108978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu X., Lin W., Spirtos A., Wang Y., Chen H., Ye J., Parker J., Liu C.C., Wang Y., Quinn G., et al. Dissection of transcriptome dysregulation and immune characterization in women with germline BRCA1 mutation at single-cell resolution. BMC Med. 2022;20:283. doi: 10.1186/s12916-022-02489-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goveia J., Rohlenova K., Taverna F., Treps L., Conradi L.C., Pircher A., Geldhof V., de Rooij L.P.M.H., Kalucka J., Sokol L., et al. An Integrated Gene Expression Landscape Profiling Approach to Identify Lung Tumor Endothelial Cell Heterogeneity and Angiogenic Candidates. Cancer Cell. 2020;37:421–436. doi: 10.1016/j.ccell.2020.03.002. [DOI] [PubMed] [Google Scholar]
- 9.Teuwen L.-A., De Rooij L.P.M.H., Cuypers A., Rohlenova K., Dumas S.J., García-Caballero M., Meta E., Amersfoort J., Taverna F., Becker L.M., et al. Tumor vessel co-option probed by single-cell analysis. Cell Rep. 2021;35 doi: 10.1016/j.celrep.2021.109253. [DOI] [PubMed] [Google Scholar]
- 10.Rohlenova K., Goveia J., García-Caballero M., Subramanian A., Kalucka J., Treps L., Falkenberg K.D., de Rooij L.P.M.H., Zheng Y., Lin L., et al. Single-Cell RNA Sequencing Maps Endothelial Metabolic Plasticity in Pathological Angiogenesis. Cell Metabol. 2020;31:862–877.e14. doi: 10.1016/j.cmet.2020.03.009. [DOI] [PubMed] [Google Scholar]
- 11.Zhang J., Lu T., Lu S., Ma S., Han D., Zhang K., Xu C., Liu S., Gan L., Wu X., et al. Single-cell analysis of multiple cancer types reveals differences in endothelial cells between tumors and normal tissues. Comput. Struct. Biotechnol. J. 2023;21:665–676. doi: 10.1016/j.csbj.2022.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng X., Wang X., Cheng X., Liu Z., Yin Y., Li X., Huang Z., Wang Z., Guo W., Ginhoux F., et al. Single-cell analyses implicate ascites in remodeling the ecosystems of primary and metastatic tumors in ovarian cancer. Nat. Can. (Ott.) 2023;4:1138–1156. doi: 10.1038/s43018-023-00599-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeng Q., Mousa M., Nadukkandy A.S., Franssens L., Alnaqbi H., Alshamsi F.Y., Safar H.A., Carmeliet P. Understanding tumour endothelial cell heterogeneity and function from single-cell omics. Nat. Rev. Cancer. 2023;23:544–564. doi: 10.1038/s41568-023-00591-5. [DOI] [PubMed] [Google Scholar]
- 14.Olbrecht S., Busschaert P., Qian J., Vanderstichele A., Loverix L., Van Gorp T., Van Nieuwenhuysen E., Han S., Van den Broeck A., Coosemans A., et al. High-grade serous tubo-ovarian cancer refined with single-cell RNA sequencing: specific cell subtypes influence survival and determine molecular subtype classification. Genome Med. 2021;13:111. doi: 10.1186/s13073-021-00922-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Regner M.J., Wisniewska K., Garcia-Recio S., Thennavan A., Mendez-Giraldez R., Malladi V.S., Hawkins G., Parker J.S., Perou C.M., Bae-Jump V.L., Franco H.L. A multi-omic single-cell landscape of human gynecologic malignancies. Mol. Cell. 2021;81:4924–4941.e10. doi: 10.1016/j.molcel.2021.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Slyper M., Porter C.B.M., Ashenberg O., Waldman J., Drokhlyansky E., Wakiro I., Smillie C., Smith-Rosario G., Wu J., Dionne D., et al. A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat. Med. 2020;26:792–802. doi: 10.1038/s41591-020-0844-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang K., Erkan E.P., Jamalzadeh S., Dai J., Andersson N., Kaipio K., Lamminen T., Mansuri N., Huhtinen K., Carpén O., et al. Longitudinal single-cell RNA-seq analysis reveals stress-promoted chemoresistance in metastatic ovarian cancer. Sci. Adv. 2022;8 doi: 10.1126/sciadv.abm1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qian J., Olbrecht S., Boeckx B., Vos H., Laoui D., Etlioglu E., Wauters E., Pomella V., Verbandt S., Busschaert P., et al. A pan-cancer blueprint of the heterogeneous tumor microenvironment revealed by single-cell profiling. Cell Res. 2020;30:745–762. doi: 10.1038/s41422-020-0355-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu J., Fang Y., Chen K., Li S., Tang S., Ren Y., Cen Y., Fei W., Zhang B., Shen Y., Lu W. Single-Cell RNA Sequencing Reveals the Tissue Architecture in Human High-Grade Serous Ovarian Cancer. Clin. Cancer Res. 2022;28:3590–3602. doi: 10.1158/1078-0432.CCR-22-0296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laumont C.M., Wouters M.C.A., Smazynski J., Gierc N.S., Chavez E.A., Chong L.C., Thornton S., Milne K., Webb J.R., Steidl C., Nelson B.H. Single-cell Profiles and Prognostic Impact of Tumor-Infiltrating Lymphocytes Coexpressing CD39, CD103, and PD-1 in Ovarian Cancer. Clin. Cancer Res. 2021;27:4089–4100. doi: 10.1158/1078-0432.CCR-20-4394. [DOI] [PubMed] [Google Scholar]
- 21.Wan C., Keany M.P., Dong H., Al-Alem L.F., Pandya U.M., Lazo S., Boehnke K., Lynch K.N., Xu R., Zarrella D.T., et al. Enhanced Efficacy of Simultaneous PD-1 and PD-L1 Immune Checkpoint Blockade in High-Grade Serous Ovarian Cancer. Cancer Res. 2021;81:158–173. doi: 10.1158/0008-5472.CAN-20-1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anadon C.M., Yu X., Hänggi K., Biswas S., Chaurio R.A., Martin A., Payne K.K., Mandal G., Innamarato P., Harro C.M., et al. Ovarian cancer immunogenicity is governed by a narrow subset of progenitor tissue-resident memory T cells. Cancer Cell. 2022;40:545–557.e13. doi: 10.1016/j.ccell.2022.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geistlinger L., Oh S., Ramos M., Schiffer L., LaRue R.S., Henzler C.M., Munro S.A., Daughters C., Nelson A.C., Winterhoff B.J., et al. Multiomic Analysis of Subtype Evolution and Heterogeneity in High-Grade Serous Ovarian Carcinoma. Cancer Res. 2020;80:4335–4345. doi: 10.1158/0008-5472.CAN-20-0521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kan T., Zhang S., Zhou S., Zhang Y., Zhao Y., Gao Y., Zhang T., Gao F., Wang X., Zhao L., Yang M. Single-cell RNA-seq recognized the initiator of epithelial ovarian cancer recurrence. Oncogene. 2022;41:895–906. doi: 10.1038/s41388-021-02139-z. [DOI] [PubMed] [Google Scholar]
- 25.Hippen A.A., Falco M.M., Weber L.M., Erkan E.P., Zhang K., Doherty J.A., Vähärautio A., Greene C.S., Hicks S.C. miQC: An adaptive probabilistic framework for quality control of single-cell RNA-sequencing data. PLoS Comput. Biol. 2021;17 doi: 10.1371/journal.pcbi.1009290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kotliar D., Veres A., Nagy M.A., Tabrizi S., Hodis E., Melton D.A., Sabeti P.C. Identifying gene expression programs of cell-type identity and cellular activity with single-cell RNA-Seq. Elife. 2019;8 doi: 10.7554/eLife.43803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paik D.Y., Janzen D.M., Schafenacker A.M., Velasco V.S., Shung M.S., Cheng D., Huang J., Witte O.N., Memarzadeh S. Stem-like epithelial cells are concentrated in the distal end of the fallopian tube: a site for injury and serous cancer initiation. Stem Cell. 2012;30:2487–2497. doi: 10.1002/stem.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Geldhof V., de Rooij L.P.M.H., Sokol L., Amersfoort J., De Schepper M., Rohlenova K., Hoste G., Vanderstichele A., Delsupehe A.M., Isnaldi E., et al. Single cell atlas identifies lipid-processing and immunomodulatory endothelial cells in healthy and malignant breast. Nat. Commun. 2022;13:5511. doi: 10.1038/s41467-022-33052-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang F., Ding P., Liang X., Ding X., Brandt C.B., Sjöstedt E., Zhu J., Bolund S., Zhang L., de Rooij L.P.M.H., et al. Endothelial cell heterogeneity and microglia regulons revealed by a pig cell landscape at single-cell level. Nat. Commun. 2022;13:3620. doi: 10.1038/s41467-022-31388-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu L., Li N., Zhang Q., Zhou J., Lin L., He X. Inhibition of ERK1/2 Signaling Impairs the Promoting Effects of TGF-β1 on Hepatocellular Carcinoma Cell Invasion and Epithelial-Mesenchymal Transition. Oncol. Res. 2017;25:1607–1616. doi: 10.3727/096504017X14938093512742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sritananuwat P., Sueangoen N., Thummarati P., Islam K., Suthiphongchai T. Blocking ERK1/2 signaling impairs TGF-β1 tumor promoting function but enhances its tumor suppressing role in intrahepatic cholangiocarcinoma cells. Cancer Cell Int. 2017;17:85. doi: 10.1186/s12935-017-0454-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muscella A., Vetrugno C., Cossa L.G., Marsigliante S. TGF-β1 activates RSC96 Schwann cells migration and invasion through MMP-2 and MMP-9 activities. J. Neurochem. 2020;153:525–538. doi: 10.1111/jnc.14913. [DOI] [PubMed] [Google Scholar]
- 33.Li T., Huang H., Shi G., Zhao L., Li T., Zhang Z., Liu R., Hu Y., Liu H., Yu J., Li G. TGF-β1-SOX9 axis-inducible COL10A1 promotes invasion and metastasis in gastric cancer via epithelial-to-mesenchymal transition. Cell Death Dis. 2018;9:849. doi: 10.1038/s41419-018-0877-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu Z., Shen M.X., Ma D.Z., Wang L.Y., Zha X.L. TGF-beta1-promoted epithelial-to-mesenchymal transformation and cell adhesion contribute to TGF-beta1-enhanced cell migration in SMMC-7721 cells. Cell Res. 2003;13:343–350. doi: 10.1038/sj.cr.7290179. [DOI] [PubMed] [Google Scholar]
- 35.Xie J., Guo T., Zhong Z., Wang N., Liang Y., Zeng W., Liu S., Chen Q., Tang X., Wu H., et al. ITGB1 Drives Hepatocellular Carcinoma Progression by Modulating Cell Cycle Process Through PXN/YWHAZ/AKT Pathways. Front. Cell Dev. Biol. 2021;9 doi: 10.3389/fcell.2021.711149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu H.-J., Hao M., Yeo S.K., Guan J.-L. FAK signaling in Cancer-Associated Fibroblasts promotes breast cancer cell migration and metastasis by exosomal miRNAs-mediated intercellular communication. Oncogene. 2020;39:2539–2549. doi: 10.1038/s41388-020-1162-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cunha S.I., Bocci M., Lövrot J., Eleftheriou N., Roswall P., Cordero E., Lindström L., Bartoschek M., Haller B.K., Pearsall R.S., et al. Endothelial ALK1 Is a Therapeutic Target to Block Metastatic Dissemination of Breast Cancer. Cancer Res. 2015;75:2445–2456. doi: 10.1158/0008-5472.CAN-14-3706. [DOI] [PubMed] [Google Scholar]
- 38.Fry S.A., Robertson C.E., Swann R., Dwek M.V. Cadherin-5: a biomarker for metastatic breast cancer with optimum efficacy in oestrogen receptor-positive breast cancers with vascular invasion. Br. J. Cancer. 2016;114:1019–1026. doi: 10.1038/bjc.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Romagnoli M., Belguise K., Yu Z., Wang X., Landesman-Bollag E., Seldin D.C., Chalbos D., Barillé-Nion S., Jézéquel P., Seldin M.L., Sonenshein G.E. Epithelial-to-mesenchymal transition induced by TGF-β1 is mediated by Blimp-1-dependent repression of BMP-5. Cancer Res. 2012;72:6268–6278. doi: 10.1158/0008-5472.CAN-12-2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang H., Qin G., Zhang C., Yang H., Liu J., Hu H., Wu P., Liu S., Yang L., Chen X., et al. TRAIL promotes epithelial-to-mesenchymal transition by inducing PD-L1 expression in esophageal squamous cell carcinomas. J. Exp. Clin. Cancer Res. 2021;40:209. doi: 10.1186/s13046-021-01972-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ishimura N., Isomoto H., Bronk S.F., Gores G.J. Trail induces cell migration and invasion in apoptosis-resistant cholangiocarcinoma cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2006;290:G129–G136. doi: 10.1152/ajpgi.00242.2005. [DOI] [PubMed] [Google Scholar]
- 42.Hua R., Yu J., Yan X., Ni Q., Zhi X., Li X., Jiang B., Zhu J. Syndecan-2 in colorectal cancer plays oncogenic role via epithelial-mesenchymal transition and MAPK pathway. Biomed. Pharmacother. 2020;121 doi: 10.1016/j.biopha.2019.109630. [DOI] [PubMed] [Google Scholar]
- 43.Luo H., Xia X., Huang L.B., An H., Cao M., Kim G.D., Chen H.N., Zhang W.H., Shu Y., Kong X., et al. Pan-cancer single-cell analysis reveals the heterogeneity and plasticity of cancer-associated fibroblasts in the tumor microenvironment. Nat. Commun. 2022;13:6619. doi: 10.1038/s41467-022-34395-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Richardson M.T., Recouvreux M.S., Karlan B.Y., Walts A.E., Orsulic S. Ciliated Cells in Ovarian Cancer Decrease with Increasing Tumor Grade and Disease Progression. Cells. 2022;11 doi: 10.3390/cells11244009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tao T., Lin W., Wang Y., Zhang J., Chambers S.K., Li B., Lea J., Wang Y., Wang Y., Zheng W. Loss of tubal ciliated cells as a risk for “ovarian” or pelvic serous carcinoma. Am. J. Cancer Res. 2020;10:3815–3827. [PMC free article] [PubMed] [Google Scholar]
- 46.Wu J., Raz Y., Recouvreux M.S., Diniz M.A., Lester J., Karlan B.Y., Walts A.E., Gertych A., Orsulic S. Focal Serous Tubal Intra-Epithelial Carcinoma Lesions Are Associated With Global Changes in the Fallopian Tube Epithelia and Stroma. Front. Oncol. 2022;12 doi: 10.3389/fonc.2022.853755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ghosh A., Syed S.M., Tanwar P.S. In vivo genetic cell lineage tracing reveals that oviductal secretory cells self-renew and give rise to ciliated cells. Development. 2017;144:3031–3041. doi: 10.1242/dev.149989. [DOI] [PubMed] [Google Scholar]
- 48.Lengyel E., Li Y., Weigert M., Zhu L., Eckart H., Javellana M., Ackroyd S., Xiao J., Olalekan S., Glass D., et al. A molecular atlas of the human postmenopausal fallopian tube and ovary from single-cell RNA and ATAC sequencing. Cell Rep. 2022;41 doi: 10.1016/j.celrep.2022.111838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chai C., Liang L., Mikkelsen N.S., Wang W., Zhao W., Sun C., Bak R.O., Li H., Lin L., Wang F., Luo Y. Single-cell transcriptome analysis of epithelial, immune, and stromal signatures and interactions in human ovarian cancer. Commun. Biol. 2024;7:131. doi: 10.1038/s42003-024-05826-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qiu X., Mao Q., Tang Y., Wang L., Chawla R., Pliner H.A., Trapnell C. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods. 2017;14:979–982. doi: 10.1038/nmeth.4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., 3rd, Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.R., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., Feng T., Zhou L., Tang W., Zhan L., et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation. 2021;2 doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hänzelmann S., Castelo R., Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schubert M., Klinger B., Klünemann M., Sieber A., Uhlitz F., Sauer S., Garnett M.J., Blüthgen N., Saez-Rodriguez J. Perturbation-response genes reveal signaling footprints in cancer gene expression. Nat. Commun. 2018;9:20. doi: 10.1038/s41467-017-02391-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.C., Geurts P., Aerts J., et al. SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jin S., Guerrero-Juarez C.F., Zhang L., Chang I., Ramos R., Kuan C.H., Myung P., Plikus M.V., Nie Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021;12:1088. doi: 10.1038/s41467-021-21246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Newman A.M., Steen C.B., Liu C.L., Gentles A.J., Chaudhuri A.A., Scherer F., Khodadoust M.S., Esfahani M.S., Luca B.A., Steiner D., et al. Determining cell-type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019;37:773–782. doi: 10.1038/s41587-019-0114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang Y., Li G., Mao F., Li X., Liu Q., Chen L., Lv L., Wang X., Wu J., Dai W., et al. Ras-induced Epigenetic Inactivation of the RRAD (Ras-related Associated with Diabetes) Gene Promotes Glucose Uptake in a Human Ovarian Cancer Model. J. Biol. Chem. 2014;289:14225–14238. doi: 10.1074/jbc.M113.527671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xiang X., Corsi G.I., Anthon C., Qu K., Pan X., Liang X., Han P., Dong Z., Liu L., Zhong J., et al. Enhancing CRISPR-Cas9 gRNA efficiency prediction by data integration and deep learning. Nat. Commun. 2021;12:3238. doi: 10.1038/s41467-021-23576-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data that support the findings of this study are available in the key resources table and from the lead contact author upon reasonable request.
-
•
Code for analyzing the single-cell data are available from the GitHub depository https://github.com/lianglangchao/ovarian_cancer_llc_V2.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.