

Abstract
The scaffold protein AMBRA1, which participates in the autophagy pathway, also promotes CD4+ T cell differentiation to Tregs independent of autophagy through its interactor PP2A. Here we have investigated the role of AMBRA1 in CD8+ T cell differentiation to cytotoxic T cells (CTL). AMBRA1 depletion in CD8+ T cells was associated with impaired expression of the transcription factors RUNX3 and T-BET that drive CTL differentiation and resulted in impaired acquisition of cytotoxic potential. These effects were recapitulated by pharmacological inhibition of the AMBRA1 activator ULK1 or its interactor PP2A. Based on the ability of PP2A to activate TFEB, we hypothesized a role for TFEB in the CTL differentiation program regulated by AMBRA1. We show that TFEB modulates RUNX3 and T-BET expression and the generation of killing-competent CTLs, and that AMBRA1 depletion, or ULK1 or PP2A inhibition, suppresses TFEB activity. These data highlight a role for AMBRA1, ULK1 and PP2A in CTL generation, mediated by TFEB, which we identify as a new pioneering transcription factor in the CTL differentiation program.
Keywords: AMBRA1, Cytotoxic T cell, Lytic granule /, PP2A / ULK1
Subject terms: Immunology, Cell biology
Introduction
Autophagy is a fundamental process that ensures the lysosome-mediated degradation of damaged or old cytosolic components and organelles, while providing building blocks and energy for the maintenance of cellular homeostasis1. However, a variegated role of autophagy beyond this basic function has emerged in recent years2. T lymphocytes are no exception3,4. Autophagy is required for thymocyte development, promoting thymocyte transition from the double negative to the double positive stage and regulating positive and negative selection in response to MHCII-bound self antigens5,6. Autophagy also participates in the activation and proliferation of peripheral naive T cells and impinges on the function of both CD4+ and CD8+ effectors, promoting Th1, Th2 and Treg survival as well as cytotoxic T lymphocyte (CTL) function and memory CD8+ T cell formation7–13.
Autophagy is initiated by the ULK complex, which is required for the activation of the class III PI3-K complex (PI3KIII). By locally producing PI3P, PI3KIII marks the sites of autophagosome nucleation, allowing for the recruitment of the autophagy machinery. PI3KIII consists of the lipid kinase VPS34, the membrane protein VPS15, ATG14 and BECLIN 1. The latter is essential for the activity and localization of PI3KIII, and interactions of BECLIN 1 with alternative molecular partners modulate the function of the complex14. A key interactor of BECLIN 1 is AMBRA1, a WD40 domain-containing scaffold protein that, when phosphorylated by the ULK complex component ULK1, promotes the detachment of PI3KIII from the cytoskeleton and its translocation to the sites of autophagosome formation15. AMBRA1 has been shown to be upregulated in activated T cells and to promote Treg differentiation and maintenance independently of its pro-autophagic function by interacting with the phosphatase PP2A, which stabilizes the transcription factor FOXO3, leading to the expression of the master Treg transcription factor FOXP316. Additionally, AMBRA1 has been reported to tune down alloreactive T cell responses in the mouse in the context of graft-vs-host disease17.
Here we have investigated the role of AMBRA1 in the differentiation of human CD8+ T cells to cytotoxic effectors. We show that AMBRA1 is required for the activation of the gene expression program that drives the differentiation of activated CD8+ T cells to killing-component CTLs. This function, which we found to be mediated by the transcription factor TFEB, is shared by the AMBRA1 activator ULK1 and its interactor PP2A. Our data identify a new role for AMBRA1 and the components of its interaction network in CTL differentiation and cytotoxicity.
Results
AMBRA1 expression is upregulated during CD8+ T cell differentiation to CTLs
AMBRA1 has been reported to increase in activated CD4+ T cells, with the highest levels in in vitro polarized Tregs16. To understand whether AMBRA1 is modulated during CD8+ T cell differentiation to CTLs, CD8+ T cells purified from peripheral blood from healthy donors were stimulated for different times with beads coated with anti-CD3 and anti-CD28 antibodies in the presence of IL-2 (Fig. 1A). Under these conditions CD8+ T cells differentiate to fully functional cytotoxic effectors by day 518. RT-qPCR analysis showed that the levels of AMBRA1 mRNA increased at days 2 and 5, with a drop at day 7 (Fig. 1B). This was paralleled by an increase in the protein level at all times of activation tested (Fig. 1C).
Fig. 1.

AMBRA1 expression is upregulated during CD8+ T cell differentiation to CTLs. (A) Workflow of in vitro CD8+ T cell differentiation starting from human CD8+ T cells purified from buffy coats of healthy donors. Freshly isolated CD8+ T cells (day 0) were activated with anti-CD3/CD28 coated beads in the presence of IL-2 and expanded for 5–7 days to generate CTLs. (B) RT-qPCR analysis of AMBRA1 mRNA and (C) immunoblot analysis of AMBRA1 protein levels in CD8+ T cells collected at days 0, 2, 5 and 7 after stimulation. 18S was used for normalization in RT-qPCR analysis. Actin was used as loading control. The migration of molecular mass markers is indicated. The histograms show the quantification of AMBRA1 expression during CTL differentiation (ndonor
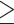 3, one sample t test, day 0 value = 1). (D) RT-qPCR analysis of human TBET and RUNX3 mRNA in CD8+ T cells at days 0, 2, 5 and 7 after stimulation. 18S was used for normalization (ndonor = 3, one-way ANOVA test, day 0 value = 1). Data are shown as mean fold ± SD, day 0 value = 1. * P ≤ 0.05; ** P ≤ 0.01; *** P ≤ 0.001; **** P ≤ 0.0001.
3, one sample t test, day 0 value = 1). (D) RT-qPCR analysis of human TBET and RUNX3 mRNA in CD8+ T cells at days 0, 2, 5 and 7 after stimulation. 18S was used for normalization (ndonor = 3, one-way ANOVA test, day 0 value = 1). Data are shown as mean fold ± SD, day 0 value = 1. * P ≤ 0.05; ** P ≤ 0.01; *** P ≤ 0.001; **** P ≤ 0.0001.
To investigate the impact of IL-2 in the modulation of AMBRA1 expression during the expansion and differentiation phase, we first activated freshly purified CD8+ T cells with anti-CD3/CD28-coated beads, and then cultured the cells in presence or absence of IL-2. Exogenously added IL-2 increased AMBRA1 levels (Fig.S1A), which were paralleled by enhanced upregulation of the surface levels of the high-affinity IL-2 receptor (IL-2R) component CD25 (Fig.S1B), thus indicating that both TCR/CD28 co-stimulation and IL-2R signaling concur to promote AMBRA1 expression.
The acquisition of killing potential by effector CD8+ T cells requires the expression of a battery of cytotoxic molecules, including the serine proteases granzyme (GZM) A and B, the pore-forming proteins perforin (PRF) and granulysin (GNLY), and the scaffolding proteoglycan serglycin (SRGN), which are stored in specialized secretory lysosomes known as lytic granules (LG)19. This differentiation program is coordinated by a network of transcription factors of which RUNX3 and T-BET are key components20–24. As expected, RT-qPCR analysis of RUNX3 and T-BET mRNA showed an upregulation following CD8+ T cell activation (Fig. 1D), with an expression profile paralleling the one observed for AMBRA1.
AMBRA1 is required for CD8+ T cell differentiation to CTLs
Since AMBRA1 is able to indirectly modulate gene expression16,25,26, we hypothesized a functional link between AMBRA1 and CTL-promoting transcription factors during effector CD8+ T cell differentiation. To verify this hypothesis, we depleted AMBRA1 from day 2-activated CD8+ T cells by RNA interference (RNAi) and analyzed CTLs 48 h later (Fig. 2A). AMBRA1 depletion did not affect either CTL expansion (Fig.S2A) or CD25 expression (Fig.S2B). Additionally, AMBRA1 deficiency did not impact IL-2R signaling, as assessed by immunoblot analysis of STAT3 phosphorylation (Fig.S2C).
Fig. 2.

AMBRA1 is required for CD8+ T cell differentiation to CTLs. (A) Immunoblot analysis of human AMBRA1 levels in control (ctr, scramble RNAi) and AMBRA1 KD CTLs (KD). Actin and ß-tubulin were used as loading controls. The migration of molecular mass markers is indicated. The histogram shows the quantification of AMBRA1 expression in CTLs normalized to actin or ß-tubulin (n = 3, one sample t test, ctr value = 1). (B) RT-qPCR analysis of human RUNX3, T-BET and EOMES mRNA in control and AMBRA1 KD CTLs. 18S was used for normalization (ndonor = 3, one sample t test, ctr value = 1). (C) RT-qPCR analysis of the GZMA, GZMB, PRF, GNLY and SRGN mRNA and (D) Immunoblot analysis of the LG components GZMB and PRF in control and AMBRA1 KD CTLs. Actin was used as loading control. The migration of molecular mass markers is indicated. The histogram shows the quantification of GZMB and PRF expression in AMBRA1 KD CTLs related to control CTLs (n = 3, one sample t test, ctr value = 1). (E) Real-time calcein release-based killing assay. Control or AMBRA1 KD CTLs were co-cultured with sAg-loaded Raji B cells at the target:effector (T:E) ratios indicated. The graph shows the kinetics of target cell killing quantified by measuring calcein fluorescence every 10 min for 4 h. The histogram shows the quantification of the percentage of target cell death at the endpoint (4 h) of independent experiments carried out on CTLs from ndonor = 3, performed in duplicate (one-way ANOVA test). Data are shown as mean fold ± SD. * P ≤ 0.05; ** P ≤ 0.01; *** P ≤ 0.001; **** P ≤ 0.0001.
We next measured the impact of AMBRA1 depletion on the transcription factors RUNX3, T-BET and EOMES expression. AMBRA1 knocked-down (KD) CTLs showed a reduction in the levels of RUNX3, T-BET and EOMES mRNA compared to control CD8+ T cells nucleofected with scrambled siRNAs (Fig. 2B). Consistent with the crucial role of these transcription factors in CTL differentiation to cytotoxic effectors, expression of GZMA, GZMB, PRF and GNLY, but not SRGN, was impaired in AMBRA1 KD CTLs (Fig. 2C,D).
To investigate whether these defects impinge on the killing ability of CTLs, we mixed control or AMBRA1 KD CTLs with Raji cells used as targets. CTL activation was elicited by pulsing Raji cells with a mix of staphylococcal superantigens (SAg) that trigger the polyclonal activation of T cells with unknown TCR specificity27. Raji cells were loaded with calcein-AM, a probe that becomes fluorescent following cleavage by intracellular esterases, and killing was monitored at different times as a drop in fluorescence28. In agreement with the defect in the expression of the LG-associated cytotoxic effectors, AMBRA1 depletion resulted in impaired target cell killing at all effector:target cell ratios analyzed (Fig. 2E). Hence, AMBRA1 participates in the differentiation program of CTLs, leading to acquisition of killing activity by promoting the expression of key transcription factors that regulate the biogenesis of LGs.
The AMBRA1 activator ULK1 and its interactor PP2A are required for CTL differentiation and function
The pro-autophagic function of AMBRA1 is regulated by ULK1-mediated phosphorylation15. To understand whether AMBRA1 may function downstream of ULK1 in promoting CTL differentiation, we treated 24 h-activated CD8+ T cells with the ULK1 inhibitor SBI-020696529 and tested gene expression and CTL function 48 h later. The effectiveness of the inhibitor concentration used for these experiments was confirmed by immunoblot analysis of LC3, which showed a decrease in the ratio of the LC3II, the autophagosome-associated form of LC3, versus the cytosolic one, namely LC3I (Fig.S3A). Consistent with a previous report29, no effect was observed on critical pathways involving AKT or ERK1/2 (Fig.S3B). Cell viability was not affected under these conditions (Fig.S3C). SBI-0206965 treatment led to a strong inhibition of RUNX3 and T-BET expression in CTLs (Fig. 3A), concomitant with an impaired ability to upregulate the expression of cytotoxic effectors of LGs, including GZMA, GZMB, GNLY and SRGL, but not PRF (Fig. 3B,C). These defects led to a reduced killing ability of CTLs, as assessed through calcein-based cytotoxicity assays using SAg-pulsed Raji cells as targets (Fig. 3D).
Fig. 3.

The AMBRA1 activator ULK1 is required for CTL differentiation and function. (A) RT-qPCR analysis of human RUNX3 and T-BET mRNA in CTLs treated with SBI-0206965 or vehicle (DMSO).18S was used for normalization. Data are shown as mean fold ± SD of treated vs untreated CTLs (ndonor = 3, one sample t test, vehicle value in CTLs set as 1). (B) RT-qPCR analysis of the GZMA, GZMB, PRF, GNLY and SRGN mRNA in CTLs treated with SBI-0206965 or vehicle. Data are shown as mean fold ± SD of treated vs untreated CTLs. 18S was used for normalization (ndonor = 3, one sample t test, vehicle value = 1). (C) Immunoblot analysis of the LG components GZMB and PRF in CTLs treated with SBI-0206965 or vehicle. Actin was used as loading control. The migration of molecular mass markers is indicated. The histogram shows the quantification of GZMB and PRF protein expression in SBI-0206965-treated CTLs related to untreated CTLs. Data are shown as mean fold ± SD (ndonor = 3, one sample t test, vehicle value = 1). (D) Real-time calcein release-based killing assay. CTLs either untreated or treated with SBI-0206965 were co-cultured with sAg-loaded Raji B cells at the target:effector ratios indicated. The graph shows the kinetics of target cell killing quantified by measuring calcein fluorescence every 10 min for 4 h. The histogram shows the quantification of the percentage of target cell death at the endpoint (4 h) of independent experiments carried out on CTLs from ndonor = 3, performed in duplicate. Data are shown as mean fold ± SD (n = 3, one-way ANOVA test). ** P ≤ 0.01; *** P ≤ 0.001; **** P ≤ 0.0001.
The phosphatase PP2A has been identified as an interactor of AMBRA1 that promotes the autophagy-independent CD4+ T cell polarization to Tregs through the FOXO3-mediated expression of FOXP316. To assess the potential role of PP2A in CTL differentiation we treated 24 h-activated CD8+ T cells with the PP2A inhibitor LB-10030. CTL treatment with LB-100 resulted in an increase in FOXO3A phosphorylation on S253, which is modulated by AMBRA1/PP2A, confirming the inhibition of PP2A at the concentration used (Fig.S3D). No effect on cell viability was observed (Fig.S3C). Similar to SBI-0206965, LB-100 treatment recapitulated the effects of AMBRA1 deficiency, leading to impaired expression of CTL-relevant genes (Fig. 4A-C) and CTL-mediated killing (Fig. 4D). An exception was PFR expression which, at variance with SBI-0206965 treated CTLs, was downregulated in LB-100-treated CTLs (Fig. 4B,C). Together, these data indicate that AMBRA1, as well as two proteins belonging to its interaction network, regulate CD8+ T cell differentiation to killing-competent CTLs.
Fig. 4.

The AMBRA1 interactor PP2A is required for CTL differentiation and function. (A) RT-qPCR analysis of RUNX3 and T-BET mRNA in CTLs treated with LB-100 or vehicle (DMSO). 18S was used for normalization. Data are shown as mean fold ± SD (ndonor = 3, one sample t test, vehicle value in CTLs set as 1). (B) RT-qPCR analysis of GZMA, GZMB, PRF, GNLY and SRGN mRNA in CTLs treated with LB-100 inhibitor or vehicle. Data are shown as mean fold ± SD (treated vs untreated CTLs). 18S was used for normalization (n 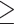 3, one sample t test, vehicle value = 1). (C) Immunoblot analysis of the LG components GZMB and PRF in CTLs treated with LB-100 inhibitor or vehicle. Actin was used as loading control. The migration of molecular mass markers is indicated. The histogram shows the quantification of GZMB and PRF protein expression in LB-100-treated CTLs related to untreated CTLs. Data are shown as mean fold ± SD (ndonor = 3, one sample t test, vehicle value = 1). (D) Real-time calcein release-based killing assay. CTLs either untreated or treated with LB-100 were co-cultured with sAg-loaded Raji B cells at the target:effector ratios indicated. The graph shows the kinetics of target cell killing quantified by measuring calcein fluorescence every 10 min for 4 h. The histogram shows the quantification of the percentage of target cell death at the endpoint (4 h) of independent experiments carried out on CTLs from ndonor = 3, performed in duplicate. Data are shown as mean fold ± SD (n = 3, one-way ANOVA test). * P ≤ 0.05; ** P ≤ 0.01; *** P ≤ 0.001; **** P ≤ 0.0001.
3, one sample t test, vehicle value = 1). (C) Immunoblot analysis of the LG components GZMB and PRF in CTLs treated with LB-100 inhibitor or vehicle. Actin was used as loading control. The migration of molecular mass markers is indicated. The histogram shows the quantification of GZMB and PRF protein expression in LB-100-treated CTLs related to untreated CTLs. Data are shown as mean fold ± SD (ndonor = 3, one sample t test, vehicle value = 1). (D) Real-time calcein release-based killing assay. CTLs either untreated or treated with LB-100 were co-cultured with sAg-loaded Raji B cells at the target:effector ratios indicated. The graph shows the kinetics of target cell killing quantified by measuring calcein fluorescence every 10 min for 4 h. The histogram shows the quantification of the percentage of target cell death at the endpoint (4 h) of independent experiments carried out on CTLs from ndonor = 3, performed in duplicate. Data are shown as mean fold ± SD (n = 3, one-way ANOVA test). * P ≤ 0.05; ** P ≤ 0.01; *** P ≤ 0.001; **** P ≤ 0.0001.
The transcription factor TFEB regulates the expression of CTL-specific pioneer transcription factors downstream of ULK1, AMBRA1 and PP2A
The transcription factor TFEB, which coordinates the transcriptional programs of lysosome biogenesis and autophagy31, has been recently implicated in the regulation of CD4+ T cell differentiation to Tregs32. Interestingly, TFEB expression was upregulated during CD8+ T cell differentiation to killing-competent CTLs, as assessed by RT-qPCR and immunoblot analysis (Fig. 5A,B). To address the potential role of TFEB in the transcriptional program that coordinates CD8+ T cell differentiation to CTLs, we looked for putative TFEB binding sites in the gene promoters of RUNX3 and T-BET through an in silico analysis of the ~ 2 kb region upstream of the respective transcription start sites, using the JASPAR software (http://jaspar.genereg.net/). We identified 3 and 2 high-score (≥ 8) TFEB binding sites on the T-BET and RUNX3 promoter, respectively (Table 1). To validate the TFEB binding sites identified in the RUNX3 and T-BET promoters by the in silico analysis, chromatin was extracted from day-2 CTLs and processed for immunoprecipitation using an anti-TFEB antibody or non-specific IgG. PCR amplification of the DNA recovered from the anti-TFEB immunoprecipitates using specific primer pairs showed binding to the RUNX3 and T-BET promoters (Fig. 5C). As expected, TFEB was found to bind to the promoter of gene encoding cathepsin D (CTSD), used as a positive control.
Fig. 5.

TFEB regulates expression of the CTL-specific pioneer transcription factors RUNX3 and T-BET. (A) RT-qPCR analysis of TFEB mRNA and (B) immunoblot analysis of TFEB protein in CD8+ T cells at days 0, 2, 5 and 7. 18S was used for normalization in RT-qPCR analysis. Actin was used as loading control. The migration of molecular mass markers is indicated. The histograms show the quantification of TFEB levels during CTL differentiation related to CD8+ T cells at day 0. Data are shown as mean fold ± SD (ndonor = 3, one-way ANOVA test, day 0 value = 1). (C) ChIP assays of nuclear extracts of CD8+ T cells at day 2 of differentiation using either anti-TFEB or control unspecific rabbit IgG antibodies. Selected regions of the RUNX3 and TBET promoters containing putative binding sites for TFEB were amplified by qRT-PCR. Data are show as fold enrichment (the percentage of input DNA of TFEB-Ab IP samples vs ctrl IgG-Ab samples (ndonor = 3, one sample t test). (D) Immunoblot analysis of human TFEB protein in control (scramble RNAi) and TFEB KD CTLs. Actin was used as loading control. Molecular weights are indicated at the left side of the representative immunoblot image. The histogram shows the quantification of TFEB expression in KD CTLs vs control CTLs. Data are shown as mean fold ± SD (ndonor = 3, one sample t test, ctr value = 1). (E) RT-qPCR analysis of human RUNX3 and T-BET mRNA in control and TFEB KD CTLs.18S was used for normalization. Data are shown as mean fold ± SD (ndonor = 3, one sample t test, ctr value = 1). * P ≤ 0.05; ** P ≤ 0.01; **** P ≤ 0.0001.
Table 1.
Putative TFEB binding sites: in silico analysis of the ~ 2 kb region upstream of the respective transcription start sites using the JASPAR software.
| Gene | Matrix ID | Matrix name | Max score | N. putative sites |
|---|---|---|---|---|
| RUNX3 | MA0692.1 | TFEB | 13.1 | 10 |
| T-BET | MA0692.1 | TFEB | 12.0 | 8 |
These data suggest that TFEB may regulate CTL differentiation by driving the transcription of critical pioneer transcription factors. To assess whether TFEB is indeed required for the expression of RUNX3 and T-BET during CTL differentiation, we depleted day 2-activated CD8+ T cells of TFEB by RNAi, using scrambled siRNAs as control (Fig. 5D). TFEB depletion resulted in a significant drop in the levels of RUNX3 and T-BET transcripts as compared to controls (Fig. 5E), indicating that TFEB is required for the differentiation program of CTLs. Of note, TFEB shares similar roles in lysosome biogenesis with the transcription factor binding to IGHM enhancer 3 (TFE3), suggesting the possibility that the reduction in TFEB expression might be related to a concomitant increase in TFE3 levels33. We thus evaluated the expression of TFE3 transcripts in TFEB KD CTLs. We found that TFEB deficiency does not modify the mRNA levels of TFE3 in CTLs (ctr = 1; TFEB KD = 0.97 ± 0.28), supporting the notion that the observed effects on CTL differentiation are specifically related to TFEB depletion.
The activity of TFEB is regulated by its subcellular localization. Under homeostatic conditions, TFEB is phosphorylated by the serine-threonine kinases mTORC1 and ERK2, both of which prevent its nuclear translocation31,34. Dephosphorylation in response to a number of stimuli that signal the requirement for de novo lysosome biogenesis, such as cellular stress, starvation or lysosome dysfunction, relieves the inhibitory state of TFEB, allowing its localization to the nucleus to regulate the expression of target genes35,36. PP2A has been implicated in TFEB activation upon oxidative stress and PIKfyve inhibition36,37, suggesting a potential role for PP2A in fine-tuning the activity of TFEB and thus the transcription of TFEB target genes, including the newly identified RUNX3 and T-BET (Fig. 5C), in CTLs. To address this issue, we transfected CTLs with a construct encoding GFP-tagged TFEB and evaluated its subcellular localization by confocal microscopy. Nuclear TFEB-GFP could be observed in a major proportion of carrier-treated CTLs, consistent with the activation status. LB-100 treatment led to a significant reduction in the proportion of cells harboring nuclear TFEB-GFP (Fig. 6A), indicating that PP2A participates in the regulation of TFEB activity in CTLs. Interestingly, similar results were obtained when CTLs were treated with the ULK1 inhibitor SBI-0206965 (Fig. 6B). Consistent with published reports showing the inhibitory effects of ULK138 and PP2A39,40 on p70S6K activation, both SBI-0206965 and LB-100 enhanced the mTORC1 pathway, as assessed by immunoblot analysis of the mTORC1 target p70S6K (Fig.S4A).
Fig. 6.

TFEB activity during CTL differentiation is regulated by ULK1, AMBRA1 and PP2A. (A-C) Quantification of nuclear GFP-tagged TFEB in untreated and LB-100- (A) or SBI-0206965-treated CTLs (B) and control and AMBRA1 KD CTLs (C). The histograms show the quantification of the percentage of cells showing nuclear TFEB localization. Representative images (medial optical sections) are shown (20 cells/sample, ndonor = 3, Student’s t test). Scale bar: 5 μm. * P ≤ 0.05.
To directly address the role of AMBRA1 in TFEB activation we analysed GFP-tagged TFEB localization in AMBRA1 KD CTLs. AMBRA1 deficiency was associated with a decrease in TFEB nuclear translocation, and hence an impairment of its activity, compared to control CTLs (Fig. 6C). Consistent with the role of AMBRA1 in stabilizing ULK141, which is a negative regulator of mTORC1 activity42, phosphorylation of the mTORC1 target p70S6K was enhanced in AMBRA1 deficient CTLs (Fig.S4B). Collectively, the data provide evidence that AMBRA1, its upstream activator ULK1 and its interactor PP2A participate in the program of CD8+ T cell differentiation through the activation of TFEB, which regulates the expression of master transcription factors and lytic effectors to generate functional CTLs.
Discussion
Components of the pathways that regulate and execute autophagy have emerged in recent years as important players in cellular processes beyond degradation of damaged macromolecules and organelles, such as metabolism, cell differentiation and elimination of pathogens1,43,44. While autophagy has been implicated in CTL function and memory7,8,13, the underlying mechanisms remain to be defined. Here we show that the autophagy regulator AMBRA1 regulates the differentiation of CD8+ T cells to killing-competent CTLs by promoting the expression of the pioneer transcription factors RUNX3 and T-BET, both of which coordinate the expression of the cytotoxic effectors of LGs20–23. This function of AMBRA1 is recapitulated by its upstream regulator ULK1 and its interactor PP2A. We show that, in turn, RUNX3 and T-BET expression is regulated by the transcription factor TFEB, which is modulated by AMBRA1 as well as by ULK1 and PP2A. These results highlight a new role for AMBRA1, that appears mediated by TFEB, in CTL differentiation. They moreover identify the AMBRA1 network components ULK1 and PP2A as participants in the TFEB-dependent CTL differentiation program that may act in concert with AMBRA1 in this process (Fig. 7).
Fig. 7.

Schematic representation of the function of AMBRA1 in CTL differentiation. AMBRA1 promotes the expression of the key transcription factors RUNX3 and T-BET that drive the CTL differentiation program through TFEB activation. The AMBRA1 upstream regulator ULK1 and its interactor PP2A may act in concert with AMBRA1 in this process.
AMBRA1 has been previously demonstrated to regulate the differentiation of CD4+ T cells to Tregs as well as their maintenance independently of its pro-autophagic function. This activity of AMBRA1 is mediated by its interactor PP2A, which indirectly enhances the expression of the lineage-specific transcription factor FOXP3 by dephosphorylating and activating its transcriptional activator FOXO316. Intriguingly, AMBRA1 has been implicated in the metabolic reprogramming of activated mouse CD8+ T cells, restraining glycolysis and cell cycle progression45. Additionally, AMBRA1 has been shown to limit alloreactive responses of both CD4+ and CD8+ T cells in vivo as assessed in T cell-specific AMBRA1 KO mice in the context of graft-vs-host disease17. The enhanced responses of Ambra1-/- T cells were ascribed to an intrinsic hyperreactivity rather than the expected Treg defect, although the underlying mechanism was not addressed17. This role of AMBRA1 in tuning down T cell responses is in contrast with our identification of AMBRA1 as a positive regulator of human CTL differentiation and function. This may reflect species-specific differences, which may be amplified by differences between the cells used in each study, which come from very different physiological conditions (in vivo development of mouse Ambra1-/- T cells vs in vitro generation of human CTLs). A side-by-side comparison of the outcome of AMBRA1 depletion in human and mouse CD8+ T cells, using the same experimental settings, is needed to confirm this hypothesis.
ULK1 has been reported to participate in a metabolic AMPKα/ULK1/ATG7 axis that regulates CD8+ T cell memory generation in the presence of the mTORC1 inhibitor rapamycin46,47 or of the cytokines IL-748 or IL-1549, which trigger weak mTORC1-activating signals. However its specific role in this process has not been addressed as yet. Our data show that pharmacological inhibition of ULK1 in CD8+ T cells leads to their impaired differentiation to CTL effectors, highlighting this kinase not only as a putative regulator of CD8+ T cell memory generation but also as a molecular player in the generation of effector CD8+ T cells. The fact that ULK1 inhibition largely recapitulates the defects observed in AMBRA1 KD CD8+ T cells suggests that AMBRA1 may mediate the effects of ULK1 in these cells, however these data do not rule out the possibility that ULK1 may modulate CD8+ T differentiation through alternative effectors. The fact that, among the LG components analyzed, AMBRA1 deficiency does not impact on SRGN expression and, viceversa, ULK1 inhibition does not affect PRF expression, suggests that ULK1 may participate in CD8+ T differentiation independently of AMBRA1. Investigating the role of AMBRA1 in CD8+ T cell memory generation will help unravelling this question. Additionally, whether the ability of ULK1 to modulate CTL differentiation is related to its function as regulator of autophagy remains to be established.
As opposed to ULK1, the role of PP2A in T cells has been amply investigated, mainly in mice. Initially identified as a negative regulator of TCR signaling, PP2A has emerged as an important player in CD4+ T cells, inhibiting their differentiation to Th1 cells and promoting their differentiation to Th2, Th17, Treg and Th9 cells50. While some of these effects may be accounted for by the ability of PP2A to modulate mTORC1 activity51,52, mechanistic studies on Th17 cells have identified SMADs as substrates of PP2A that modulate binding of the Th17 lineage-specific transcription factor RORγT to the IL-17 gene promoter53. Of note, an enhancement in CD8+ T cell immunity has been observed in mice treated with a PP2A inhibitor in combination with PD1 blockade54. Here we show that, conversely, PP2A inhibition suppresses the generation of CTLs from human CD8+ T cells. Species-specific functions of PP2A may account for our finding, as suggested by the opposite outcome of PP2A inhibition on Th17 differentiation in mouse and human CD4+ T cells 53,55. The fact that PP2A has been identified as an interactor of AMBRA1 that modulates gene expression in CD4+ T cells16 suggests that it may act in concert with AMBRA1 in CD8+ T cell differentiation, however the data do not rule out the possibility that AMBRA1 and PP2A may participate in independent pathways that converge on this process.
The program of CD8+ T cell differentiation to effector or memory cells is orchestrated by a complex transcriptional network where pioneer transcription factors, that include RUNX3, T-BET, EOMES and GATA3, coordinate the acquisition of the specific functions that characterize these cells20–24,56,57. In effector CD8+ T cells, this involves the coordinated transcription of the genes that encode the cytotoxic machinery. Here we show that TFEB depletion impairs RUNX3 and T-BET expression, identifying TFEB as a novel transcriptional regulator of the CTL differentiation program. TFEB is the master regulator of the gene network that coordinates lysosome biogenesis, known as the CLEAR network31. Here we provide evidence for the existence of functional TFEB binding sites both on the RUNX3 and T-BET promoters. This suggests that TFEB may regulate the generation of killing-competent CTLs by promoting the expression of pioneer transcription factors. Interestingly, TFEB has been recently implicated in the expression of genes that regulate Treg differentiation in IL-2-treated CD4+ T cells58. Taken together with the data presented in the present report, these findings highlight TFEB as a new major player in T cell differentiation.
TFEB is retained in the cytosol by phosphorylation on negative regulatory serine residues by the serine-threonine kinases mTOR and ERK235. Additionally, PP2A has been implicated in the modulation of TFEB activity in response to oxidative stress, promoting its nuclear translocation through removal of the inhibitory phosphate on S14236. The enhancement in TFEB nuclear localization both in CTLs treated with the PP2A inhibitor and in AMBRA1-deficient CTLs suggests that PP2A may provide the functional link between AMBRA1 and TFEB. Of note, AMBRA1 has been shown not only to regulate FOXP3 expression through the PP2A-dependent activation of its transcription factor FOXO316, but also to control the levels of the transcription factor MYC by promoting its interaction with PP2A, which leads to its degradation59,60. MYC deficiency has been causally associated with impaired effector and memory T cell differentiation61. Hence AMBRA1 could represent a hub for the activation of key transcription factors that drive diverse T cell fates.
It is noteworthy that small molecule drugs that inhibit components of the regulatory network identified in this study, namely PP2A and ULK1, are being assessed as cancer therapeutics62,63. The PP2A inhibitor LB-100 has been shown to sensitize cancer cells to chemo- and radio-therapy by interfering with DNA repair, thus promoting cell apoptosis, and has completed a phase I trial for the treatment of solid tumors in combination with Decetaxel64,65. Of note, in addition to direct effects on cancer cells, a number of pre-clinical data reported on LB-100 having enhancing effects on both CD4+ and CD8+ T cell function, while suppressing Treg cell function30,66. A remarkable in vivo efficacy of LB-100 in combination with PD1 blockade or CAR T cell therapy has been demonstrated in mouse models of colorectal cancer, melanoma and glioblastoma54,67,68. Regarding ULK1 inhibitors, DCC-3116 has shown efficacy in mouse models of lung cancer and gastrointestinal stromal tumors69,70 and is currently in phase I/II clinical on advanced solid and metastatic tumors (https://clinicaltrials.gov/study/NCT04892017), however potential effects on immune cells have not been addressed. The implication of both PP2A and ULK1 in T cell differentiation and function46,47,50,53, for which we provide further insights in this report, highlights potential immunomodulatory effects that could be harnessed to enhance anti-tumor immunity or, in a different disease context, to mitigate autoimmunity.
Methods
Cells, T cell transfectants and antibodies
Peripheral blood samples were obtained from the blood banks of Siena University Hospital. All healthy donors, anonymized as per protocol approved by the local ethics committee of Siena University (Comitato Etico Regione Toscana Area Vasta Sud Est and Area Vasta Centro; 20759; Prof. Baldari), provided voluntary informed written consent to use their blood for research purposes. All procedures were performed according to the Declaration of Helsinki guidelines. Human CD8+ T cells were purified from peripheral blood of healthy donors by negative selection using RosetteSep™ Human CD8+ T cells Enrichment Cocktail (STEMCELL Technologies, #15063) followed by density gradient centrifugation on Lympholyte (Cedarlane Laboratories, #CL5026). Freshly purified CD8+ T cells were stimulated in the presence of anti-CD3/anti-CD28 coated Dynabeads (ThermoFisher Scientific, #111.13D) and grown at 37 °C, 5% CO2 with or without 50 U/ml recombinant human IL-2 (Miltenyi, #130–097-745) in RPMI-HEPES medium (Merck, #R7388) supplemented with 10% iron-enriched bovine calf serum (BCS; GE Healthcare HyClone, #SH30072.03) and 1X MEM nonessential amino acids (MEM NEAA; #11140050). After 48 h of activation, the beads were removed and CD8+ T cells (day 2) were expanded in complete medium in the presence of IL-2 (day 5–7). Cells were collected on day 0 (resting CD8+ T cells), 2, 5 and 7 for expression analysis. RNAi-mediated knockdown of AMBRA1 was performed by a specific silencing RNA oligonucleotide (siRNA) (Biofab) resuspended at the final concentration of 100 μM in 1 × Annealing buffer (60 mM KCl, 6 mM HEPES-pH 7.5, 0.2 mM MgCl2). Sense and antisense strand (ss: 5’-GAGUAGAACUGCCGGAUAG-3’, as: 5’-CUAUCCGGCAGUUCUACUC-3’) were annealed in 1 × Annealing buffer at 92 °C for 1 min and cooled slowly before use. Scrambled esiRNA control Renilla luciferase (Mission RLUC-Renilla luciferase-esiRNA, #EHURLUC) (Sigma-Aldrich, Milan, Italy) was used as negative control. CTLs were transiently transfected at day 2 by electroporation with either human AMBRA1-siRNA or negative control siRNA using the Human T cell Nucleofector Kit (Amaxa Biosystem, #VPA-1002) at the final concentration of 150 ng/106 cells. Cells were analysed 48 h post transfection.
CD8+ T cells were treated with either PP2A-inhibitor LB-100 (Selleckchem, #S7537) or ULK1-inhibitor SBI0206965 (Selleckchem, #S7885) 24 h after cell stimulation at the final concentration of 1 μM and 10 μM29,54, respectively. Control samples were treated with DMSO. Cell viability, drug activity and assays were analysed 48 h post-treatment.
Alternatively, for RNAi-mediated TFEB knockdown, CTLs were transiently transfected at day 2 by electroporation with either TFEB (#FE5L009798000010) specific siRNAs or negative control siRNA (#FE5D0018101020) (Dharmacon) using the Human T cell Nucleofector Kit at the final concentration of 150 ng/106 cells. Cells were analysed 24 h post transfection.
To test TFEB nuclear translocation CTLs were transiently co-transfected by electroporation with TFEB-GFP plasmid (kindly provided by D. Medina) (0.5 μg/106 cells) and either AMBRA1 or control siRNA (75 ng/106 cells) using the Human T cell Nucleofector Kit and cultured in complete medium added with 500 U/ml IL-2. Cells were analysed 24 h post transfection. In addition, CD8+ T cells either treated or untreated with inhibitors were transfected with TFEB-GFP plasmid (1 μg/106 cells) 24 h post treatment and analysed after additional 24 h.
Cells used in this work include the Burkitt lymphoma-derived B cell line Raji, grown at 37 °C and 5% CO2 in RPMI-1640 medium (Merck, #R8758) supplemented with 7.5% BCS. Primary antibodies used for immunoblotting and immunofluorescence are described in detail in Supplementary Table 1. Secondary horseradish peroxidase (HRP)-labelled antibodies were purchased from Jackson ImmunoResearch Laboratories (West Grove, Pennsylvania, USA) and Alexa Fluor 488-labeled secondary antibodies from ThermoFisher Scientific (Waltham, Massachusetts, USA).
RNA isolation, cDNA synthesis and quantitative real-time PCR (RT-qPCR)
Total RNA was isolated using RNeasy Mini Kit (Qiagen, #74136) according to the manufacturer instructions. Concentration of clean-up RNA was determined using QIAexpert spectrophotometer (Qiagen) and 500 ng of RNA was reverse transcribed to first-strand cDNAs using iScript cDNA Synthesis Kit (Bio-Rad, #1708891). Real-time quantitative PCR (RT-qPCR) was performed on 96-well optical PCR plates (Sarstedt) using SSo FastTM EvaGreenR SuperMix and a CFX96 Real-Time system (Bio-Rad Laboratories, Waltham, MA). The thermal cycling conditions denaturation for 3 min at 95 °C, denaturation in the subsequent 40 cycles was performed for 10 s at 95 °C, followed by 30 s of primer annealing at 60 °C. Results were processed and analyzed using CFX Manager Version 1.5 software (Bio-Rad). The transcript levels were determined on triplicate samples using the ΔΔCt method and normalized to 18S, used as housekeeping gene. Primers used for amplification are listed in Supplementary Table 2.
Immunofluorescence and image analysis
Cells were allowed to adhere for 15 min to poly-L-lysine (Merck, Darmstadt, Germany)-coated slides (ThermoFisher Scientific, Waltham, Massachusetts, USA) and then fixed in 4% paraformaldehyde (PFA) for 20 min at room temperature (RT). Following fixation, the samples were washed in PBS for 5 min and permeabilized with 0.1% Triton X-100 and 1% BSA PBS. Immunostaining was performed using primary anti-GFP antibody (Supplementary Table 2) 1 h at RT and goat anti-mouse AlexaFluor 488-labeled secondary antibody for 45 min at RT. Nuclei were counterstained with Hoechst 33342 dye (Thermo Fisher Scientific, #62249). Samples were observed using a super-resolution microscope (CSU-W1-SoRA Nikon), with 63x/1.49 oil objective. Denoise were performed using the software NIS Elements AR Nikon and applied to high-resolution images. The quantification of nuclear TFEB was reported as percentage of cells with nuclear TFEB calculated on TFEB-GFP expressing cells.
Cell activation, lysis and Immunoblotting
IL-2 receptor was triggered by incubating 5 × 105 CTLs with 500U/ml of recombinant human IL-2 (Miltenyi) for 15 min at 37 °C. Cells were washed twice with ice-cold PBS and lysed in lysis buffer (0.5% Triton X-100 in 20 mM Tris–HCl, pH 8.0, 150 mM NaCl) in the presence of protease inhibitors (Calbiochem, #539,134) and the phosphatase inhibitor sodium vanadate (Merck, #S6508). Protein extracts were quantified with the BCA Assay kit (EuroClone, #EMP014500) and denatured in 4X BoltTM LDS Sample Buffer (InvitrogenTM, ThermoFisher Scientific, #B0007) supplemented with 10X Bolt™ Sample Reducing Buffer (Invitrogen™, #B009) for 5 min at 100 °C.
Proteins were separated by SDS-PAGE on precast 4–12% Bis–Tris plus gels (ThermoFisher Scientific #04,125) with MOPS running buffer and transferred to nitrocellulose membrane (GE Healthcare, #9004–70-0). After 1 h blocking with 5% non-fat-dry milk in PBS containing 0.2% Tween-20 (Sigma-Aldrich, St. Louis, Missouri, USA), the membranes were incubated with primary antibodies (Supplementary Table 2) for 1 h at room temperature or overnight at 4 °C and further incubated with 20 ng/ml HRP-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, Pennsylvania, USA) for 45 min at room temperature. Stripping was carried out by using Re-Blot Plus Mild Antibody Stripping Solution, 10x (Merck Millipore, #2502). Labeled antibodies were detected using ECL kit (SuperSignal® West Pico Chemiluminescent Substrate, Thermo Scientific), and immunoblots were digitally acquired and densitometric levels were measured using Alliance Q9-Atom chemiluminescence imaging system (Uvitec).
All antibodies were initially tested on full-length filters to check that the electrophoretic mobility of the proteins against which they were raised corresponded to the expected molecular mass and that, even for those that did not have optimal performance, they gave a clear signal above background (Supplementary material 2). After that we cut the filters in order to have, on the same samples and on the same run, blots with different antibodies recognizing proteins with different molecular mass. This allowed for a more rigorous comparison of the relative intensity of each signal within samples. Full scans of the immunoblots shown in the figures are presented in Supplementary material 2.
Real-time calcein release-based killing assay
Raji B cells, used as target cells, were resuspended in serum-free AIM V medium (ThermoFisher Scientific, Waltham, Massachusetts, USA) with 10 mM HEPES and 500 nM calcein-AM (Invitrogen, #C1430) for 15 min at room temperature.
After staining, target cells were supplemented with fresh growth medium and loaded with 2 μg/ml Staphylococcal Superantigens A, B, and E (Toxin Technologies) for 1 h in serum-free AIM V medium (Gibco, #12,055–091) and thereafter settled in a 96-well black flat bottom plate (5 × 104/well). To evaluate killing ability of effector T cells, CTLs were added to target cells at different E:T ratio in duplicate (i.e., 3:1, 5:1, 10:1) in 200 μl AIMV medium and incubated at 37 °C for 4 h. Target cells alone and target cells lysed with 1% Triton X- 100 were used as control samples. The decrease in calcein fluorescence due to target cell lysis was measured at 485 nm excitation wavelength and 528 nm emission wavelength in the bottom reading mode by using a Synergy HTX multimode plate reader (BioTek). Cytotoxicity (% target cell lysis) was calculated using the following equation, based on the loss of calcein fluorescence in target cells: (Flive − γ × Fexp)/(Flive − Flyse) × 100, where Flive is the fluorescence of target cell alone, Fexp is the CTLs with target cells, and Flyse is the maximal target lysis. The γ value was measured at time zero: Flive (0)/Fexp (0). The experiments were performed in duplicate and the average was used as a dataset28,71.
Chromatin immunoprecipitation
Chip assay for the analysis of TFEB binding to the promoters of CTSD, T-BET and RUNX3 was carried out using MAGnify Chromatin Immunoprecipitation System (Thermo Fisher Scientific, #492024). 2 × 106 CD8+ T cells at day 2 of activation were crosslinked with 1% formaldehyde for 10 min at room temperature. Cells were lysed and sonicated 10 times for 20 s to obtain average DNA fragment sizes of 200–500 bases. Immunoprecipitations were carried out using 2 μg of either anti-TFEB (Cell Signaling, #37785) or control rabbit IgG. The immunoprecipitated DNA fragments were quantitated by qRT-PCR as described above. The primer sets used for the analyses are listed in Supplementary Table 2.
Flow cytometry
Flow cytometry analysis of surface CD25 expression was carried out by incubating 2 × 105 CTLs for 30 min on ice with PE-labeled anti-hCD25 antibody (BioLegend, #302,606). Viable cells were identified by flow cytometry by adding 20 μg/ml propidium iodide (PI; #537059, Merck) to stain dead cells. Flow cytometry was perfomed using a Guava easyCyte Flow Cytometer and guavaSoft InCyte 2.7 software (Merck Millipore) using the appropriate lasers and emission filters. Data were analyzed using guavaSoft InCyte 2.7 (Merck Millipore).
Statistics and reproducibility
Each experiment was performed ≥ 3 independent times. One-way and two-way ANOVA tests with post-hoc Tukey correction and multiple comparisons were used to compare multiple groups. One-sample t tests were used where samples were compared to a known standard value. Statistical analyses were performed using GraphPad (Prism Software). A p-value < 0.05 was considered as statistically significant.
Supplementary Information
Acknowledgements
This research has received funding from Ministero dell’Istruzione, dell’Università e della Ricerca (Grant PRIN bando 2017-2017FS5SHL to CTB and FC; PRIN 2022 Bando PNRR P2022TJZYZ to VC; MIUR-Italy Departments of Excellence 2023-2027 to the Dept. of Science, Univ. Roma TRE), the European Commission (ERC_2021_SyG 951329—ATTACK) to CTB, AIRC (IG 2017-20148 to CTB; IG 2019-23543 to FC), and the Italian Ministry of Health (GR-2021-12372771 to VC). FC is also supported by Novo Nordisk (0070834), and KBVU (R231-A14034).
The authors wish to thank Annamaria Fusillo for technical assistance. The figure was generated with Biorender.com.
Author contributions
L.M., V.C., F.C., F.F. and C.T.B. contributed to the conception and design of the experiments. L.M, F.Z., M.G., L.M. and F.F. performed the experiments. L.M. and F.F. analyzed the data. L.P, C.U., G.M., V.C. and F.C. contributed reagents/materials/analysis tools. L.M., F.F. and C.T.B wrote the manuscript. All authors approved the manuscript for publication.
Data availability
Data generated in this study are available upon request from corresponding author FF.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Francesca Finetti and Cosima T. Baldari contributed equally to this work.
Contributor Information
Francesca Finetti, Email: finetti4@unisi.it.
Cosima T. Baldari, Email: cosima.baldari@unisi.it
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-82957-9.
References
- 1.Dikic, I. & Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol.19, 349–364 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Klionsky, D. J. et al. Autophagy in major human diseases. EMBO J40, 108863 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Metur, S. P. & Klionsky, D. J. Adaptive immunity at the crossroads of autophagy and metabolism. Cellular Mol. Immunol.10.1038/s41423-021-00662-3 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Macian, F. Autophagy in T cell function and aging. Front. Cell Dev. Biol.7, 213 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nedjic, J., Aichinger, M., Emmerich, J., Mizushima, N. & Klein, L. Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature455, 396–400 (2008). [DOI] [PubMed] [Google Scholar]
- 6.Kasai, M. et al. Autophagic compartments gain access to the MHC class II compartments in thymic epithelium. J. Immunol.183, 7278–7285 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Puleston, D. J. et al. Autophagy is a critical regulator of memory CD8+ T cell formation. Elife3, 1–21 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu, X. et al. Autophagy is essential for effector CD8 T cell survival and memory formation. Nat. Immunol.15, 1152 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zaffagnini, G. & Martens, S. Mechanisms of selective autophagy. J. Mol. Biol.10.1016/j.jmb.2016.02.004 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kovacs, J. R. et al. Autophagy promotes T-cell survival through degradation of proteins of the cell death machinery. Cell Death Differ19, 144–152 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kabat, A. M. et al. The autophagy gene Atg16l1 differentially regulates Treg and TH2 cells to control intestinal inflammation. Elife5, e12444 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wei, J. et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat. Immunol.17, 277–285 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carleton, G. & Lum, J. J. Autophagy metabolically suppresses CD8+ T cell antitumor immunity. Autophagy15, 1648–1649 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wirth, M., Joachim, J. & Tooze, S. A. Autophagosome formation—The role of ULK1 and Beclin1–PI3KC3 complexes in setting the stage. Semin. Cancer Biol.23, 301–309 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Cianfanelli, V., Nazio, F. & Cecconi, F. Connecting autophagy: AMBRA1 and its network of regulation. Mol. Cell Oncol.10.4161/23723548.2014.970059 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Becher, J. et al. AMBRA1 controls regulatory T-cell differentiation and homeostasis upstream of the FOXO3-FOXP3 axis. Dev. Cell47, 592-607.e6 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Masuhara, K. et al. AMBRA1 controls antigen-driven activation and proliferation of naive T cells. Int. Immunol.33, 107–118 (2021). [DOI] [PubMed] [Google Scholar]
- 18.Onnis, A. et al. SARS-CoV-2 Spike protein suppresses CTL-mediated killing by inhibiting immune synapse assembly. J. Exp. Med.10.1084/jem.20220906 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cassioli, C. & Baldari, C. T. The expanding arsenal of cytotoxic T cells. Front. Immunol.10.3389/fimmu.2022.883010 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cruz-Guilloty, F. et al. Runx3 and T-box proteins cooperate to establish the transcriptional program of effector CTLs. J. Exp. Med.206, 51–59 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pritchard, G. H. et al. Early T-bet promotes LFA1 upregulation required for CD8+ effector and memory T cell development. J. Exp. Med.10.1084/jem.20191287 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lotem, J. et al. Runx3-mediated transcriptional program in cytotoxic lymphocytes. PLoS One8, 80467 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fixemer, J. et al. Eomes cannot replace its paralog T-bet during expansion and differentiation of CD8 effector T cells. PLoS Pathog.10.1371/journal.ppat.1008870 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang, J. T., Wherry, E. J. & Goldrath, A. W. Molecular regulation of effector and memory T cell differentiation. Nat. Immunol.10.1038/ni.3031 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schoenherr, C. et al. The autophagy protein Ambra1 regulates gene expression by supporting novel transcriptional complexes. J. Biol. Chem.295, 12045 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yao, Y. & Lenardo, M. AMBRA1 control immune-specific gene expression in T lymphocytes. J. Immunol.10.4049/jimmunol.210.Supp.154.04 (2023).37358278 [Google Scholar]
- 27.Herrmann, T. & MacDonald, H. R. The CD8 T cell response to staphylococcal enterotoxins. Semin Immunol5, 33–39 (1993). [DOI] [PubMed] [Google Scholar]
- 28.Kummerow, C. et al. A simple, economic, time-resolved killing assay. Eur. J. Immunol.44, 1870–1872 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Egan, D. F. et al. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol. Cell59, 285 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ronk, H., Rosenblum, J. S., Kung, T. & Zhuang, Z. Targeting PP2A for cancer therapeutic modulation. Cancer Biol. Med.19, 1428 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Settembre, C. et al. TFEB links autophagy to lysosomal biogenesis. Science332, 1429–1433 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xia, M. et al. Transcription factor EB coordinates environmental cues to regulate T regulatory cells’ mitochondrial fitness and function. Proc. Natl. Acad. Sci. U. S. A.10.1073/pnas.2205469119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pastore, N. et al. TFEB and TFE3 cooperate in the regulation of the innate immune response in activated macrophages. Autophagy12, 1240 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Settembre, C. et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J.31, 1095–1108 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Puertollano, R., Ferguson, S. M., Brugarolas, J. & Ballabio, A. The complex relationship between <scp>TFEB</scp> transcription factor phosphorylation and subcellular localization. EMBO J.37, 1–12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martina, J. A. & Puertollano, R. Protein phosphatase 2A stimulates activation of TFEB and TFE3 transcription factors in response to oxidative stress. J. Biol. Chem.293, 12525 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hasegawa, J. et al. PP2A-dependent TFEB activation is blocked by PIKfyve-induced mTORC1 activity. Mol. Biol. Cell10.1091/mbc.E21-06-0309 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee, S. B. et al. ATG1, an autophagy regulator, inhibits cell growth by negatively regulating S6 kinase. EMBO Rep.8, 360–365 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsumoto, N. et al. PP1C and PP2A are p70S6K phosphatases whose inhibition ameliorates HLD12-associated inhibition of oligodendroglial cell morphological differentiation. Biomedicines8, 89 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peterson, R. T., Desai, B. N., Hardwick, J. S. & Schreiber, S. L. Protein phosphatase 2A interacts with the 70-kDa S6 kinase and is activated by inhibition of FKBP12-rapamycin-associated protein. Proc. Natl. Acad. Sci. U. S. A.96, 4438–4442 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nazio, F. et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell Biol.15, 406–416 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Jung, C. H., Seo, M., Otto, N. M. & Kim, D. H. ULK1 inhibits the kinase activity of mTORC1 and cell proliferation. Autophagy7, 1212–1221 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dowling, S. D. & Macian, F. Autophagy and T cell metabolism. Cancer Lett.10.1016/j.canlet.2018.01.033 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bronietzki, A. W., Schuster, M. & Schmitz, I. Autophagy in T-cell development, activation and differentiation. Immunol. Cell Biol.93, 25–34 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Akatsuka, H. et al. AMBRA1 is involved in T cell receptor-mediated metabolic reprogramming through an ATG7-independent pathway. Biochem. Biophys. Res. Commun.491, 1098–1104 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Araki, K. et al. mTOR regulates memory CD8 T cell differentiation. Nature460, 108 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ara, A. et al. The energy sensor AMPKα1 is critical in rapamycin-inhibition of mTORC1-S6K-induced t-cell memory. Int. J. Mol. Sci.10.3390/ijms23010037 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu, A. et al. Prosurvival IL-7–stimulated weak strength of mTORC1-S6K controls T cell memory via transcriptional FOXO1–TCF1–Id3 and metabolic AMPKα1–ULK1–ATG7 pathways. J. Immunol.208, 155–168 (2022). [DOI] [PubMed] [Google Scholar]
- 49.Choi, Y. J. et al. CD5 suppresses IL-15–induced proliferation of human memory CD8+ T cells by inhibiting mTOR pathways. J. Immunol.209, 1108–1117 (2022). [DOI] [PubMed] [Google Scholar]
- 50.Khan, M. M., Kalim, U. U., Khan, M. H. & Lahesmaa, R. PP2A and its inhibitors in helper T-cell differentiation and autoimmunity. Front. Immunol.10.3389/fimmu.2021.786857 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Apostolidis, S. A. et al. Protein phosphatase 2A is requisite for the function of regulatory T cells. Nat. Immunol.17, 556 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jiang, Y. et al. Protein phosphatase 2A propels follicular T helper cell development in lupus. J. Autoimmun.136, 103028 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu, Q. et al. Phosphatase PP2A is essential for T H 17 differentiation. Proc. Natl. Acad. Sci. U. S. A.116, 982–987 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ho, W. S. et al. Pharmacologic inhibition of protein phosphatase-2A achieves durable immune-mediated antitumor activity when combined with PD-1 blockade. Nat. Commun.10.1038/s41467-018-04425-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khan, M. M. et al. CIP2A constrains Th17 differentiation by modulating STAT3 signaling. iScience23, 100947 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pearce, E. L. et al. Control of effector CD8+ T cell function by the transcription factor eomesodermin. Science1979(302), 1041–1043 (2003). [DOI] [PubMed] [Google Scholar]
- 57.Tindemans, I., Serafini, N., DiSanto, J. P. & Hendriks, R. W. GATA-3 function in innate and adaptive immunity. Immunity41, 191–206 (2014). [DOI] [PubMed] [Google Scholar]
- 58.Xia, M. et al. Transcription factor EB coordinates environmental cues to regulate T regulatory cells’ mitochondrial fitness and function. Proc. Natl. Acad. Sci. U. S. A.119, e2205469119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cianfanelli, V. et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-MYC dephosphorylation and degradation. Nat. Cell Biol.17, 20 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Capizzi, M., Strappazzon, F., Cianfanelli, V., Papaleo, E. & Cecconi, F. MIR7–3HG, a MYC-dependent modulator of cell proliferation, inhibits autophagy by a regulatory loop involving AMBRA1. Autophagy13, 554 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nozais, M. et al. MYC deficiency impairs the development of effector/memory T lymphocytes. iScience24, 102761 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bryant, J. P., Levy, A., Heiss, J. & Banasavadi-Siddegowda, Y. K. Review of PP2A tumor biology and antitumor effects of PP2A inhibitor LB100 in the nervous system. Cancers10.3390/cancers13123087 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Karmacharya, U. & Jung, J. W. Small molecule inhibitors for Unc-51-like autophagy-activating kinase targeting autophagy in cancer. Int. J. Mol. Sci.10.3390/ijms24020953 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chung, V. et al. Safety, tolerability, and preliminary activity of lb-100, an inhibitor of protein phosphatase 2a, in patients with relapsed solid tumors: an open-label, dose escalation, first-in-human. Phase I Trial. Clin. Cancer Res.23, 3277–3284 (2017). [DOI] [PubMed] [Google Scholar]
- 65.Hong, C. S. et al. LB100, a small molecule inhibitor of PP2A with potent chemo- and radio-sensitizing potential. Cancer Biol. Ther.16, 821–833 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Qi, Y., Li, L., Wei, Y. & Ma, F. PP2A as a potential therapeutic target for breast cancer: Current insights and future perspectives. Biomed. Pharmacother.10.1016/j.biopha.2024.116398 (2024). [DOI] [PubMed] [Google Scholar]
- 67.Maggio, D. et al. Inhibition of protein phosphatase-2A with LB-100 enhances antitumor immunity against glioblastoma. J. Neurooncol.148, 231–244 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cui, J. et al. Inhibition of PP2A with LB-100 enhances efficacy of CAR-T cell therapy against glioblastoma. Cancers (Basel)10.3390/cancers12010139 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ghazi, P. C. et al. Inhibition of ULK1/2 and KRASG12C controls tumor growth in preclinical models of lung cancer. Elife10.7554/eLife.96992 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bogdan, M. et al. DCC-3116, a first-in-class selective ULK1/2inhibitor of autophagy, in combination with the KIT inhibitor ripretinib induces complete regressions in GIST preclinical models. In: Proceedings of the American Association for Cancer Research Annual Meeting 2023; Apr 14–19; Orlando, FL. Philadelphia (PA): AACR; Cancer Res 2023;83
- 71.Chang, H. F. et al. Cytotoxic granule endocytosis depends on the Flower protein. J. Cell Biol.217, 667 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data generated in this study are available upon request from corresponding author FF.