

Abstract
Immune checkpoint inhibitor-associated myocarditis is the most lethal side effect of immune checkpoint blockade. Myocarditis leads to persistently increased mortality and lacks effective treatments. The development of patient-relevant disease models may enable disease prediction, increased understanding of disease pathophysiology, and the development of effective treatment strategies. Here, we report a new method to model immune checkpoint inhibitor-associated myocarditis in vitro via a co-culture of activated primary human immune cells, human induced pluripotent stem cell-derived cardiomyocytes, and FDA-approved immune checkpoint inhibitors to recapitulate myocarditis in vitro. Significant cardiomyocyte necrosis, arrhythmia development, and sarcomere destruction occur, replicating clinical findings from myocarditis. This tissue culture myocarditis phenotype may rely on an induced pluripotent stem cell-derived cardiomyocyte antigen-specific CD8+ T cell response. The administration of dexamethasone rescued cardiomyocyte viability, morphology, and electrophysiology and suppressed inflammatory cytokine production. In conclusion, we detail how this platform can effectively model and provide critical information about the morphological and electrophysiological changes induced by immune checkpoint inhibitor-associated myocarditis. We have also validated the ability of this platform to screen potential medications to treat immune checkpoint inhibitor-associated myocarditis. This work establishes a robust, scalable model for identifying new therapies and risk factors, which is valuable in delineating the nature of interactions between the immune system and the heart during myocarditis.
Keywords: Immune checkpoint inhibitors, Myocarditis, Modeling, Personalized medicine, Human induced pluripotent stem cells, Cardiomyocytes
Graphical abstract
Immune checkpoint inhibitor associated myocarditis may be modeled in vitro using hiPSC derived cardiomyocytes, primary human leukocytes, and clinical immune checkpoint inhibitors.
Highlights
-
•
Immune checkpoint inhibitor-associated myocarditis is severe and poorly understood.
-
•
Novel model recapitulates myocarditis using primary human immune cells and hiPSC-CMs.
-
•
Confirms a role for cardiac-specific T cells in the development of ICI myocarditis
-
•
Future application for patient-specific disease modeling and therapeutic screening
1. Introduction
Myocarditis results from the pathological interaction of cardiac and immune cells, including CD4+, CD8+ T cells, and macrophages. Myocarditis most frequently occurs after viral infections, though it also commonly follows autoimmune diseases, vaccines, and cancer therapies[1]. While patients often recover following an uncomplicated disease course, they remain at chronically elevated risk of additional cardiac disease and death[2,3]. Insufficient understanding of myocarditis has led to ineffective treatment options and uncertainty about the development of chronic complications such as dilated cardiomyopathy[4]. Immune checkpoint inhibitor-associated myocarditis (ICI myocarditis) is a recently recognized form of myocarditis closely associated with the administration of ICI cancer therapy. ICI therapy blocks receptor-ligand interactions that typically maintain peripheral immune tolerance via immune checkpoints such as PD-1 and CTLA-4. This enables potent anti-tumor immune responses for cancer treatment, an approach that has extraordinary benefits for the patients who respond to ICI therapy[5]. ICI myocarditis is severe, presenting with gross myocardial necrosis, malignant arrhythmia, and hemodynamic instability. ICI myocarditis has an estimated 40 % mortality rate and occurs in 1–2 % of patients treated with ICI[6,7]. The increasing use of ICI in cancer treatment may increase the number of ICI myocarditis cases.
ICI myocarditis risk factors are largely unknown, as are the underlying mechanisms that drive disease development[8]. Myocarditis treatment goals include hemodynamic support and pain relief. Steroid immunosuppression is used in non-viral cases of myocarditis, including ICI myocarditis[1,9]. It is unclear if steroid-based immune suppression is beneficial for these patients, and there are no evidence-based therapies for ICI myocarditis. It is not known if patients may be treated again safely with ICI after developing ICI myocarditis[10]. Patients are empirically treated with corticosteroid-based immune suppression despite lacking evidence of clinical efficacy for ICI myocarditis[11,12]. The small patient population has hindered obtaining better clinical evidence, making traditional randomized controlled trials difficult. The need for personalized disease modeling platforms capable of predicting elevated risk of ICI myocarditis in patients is essential to address these medical needs effectively.
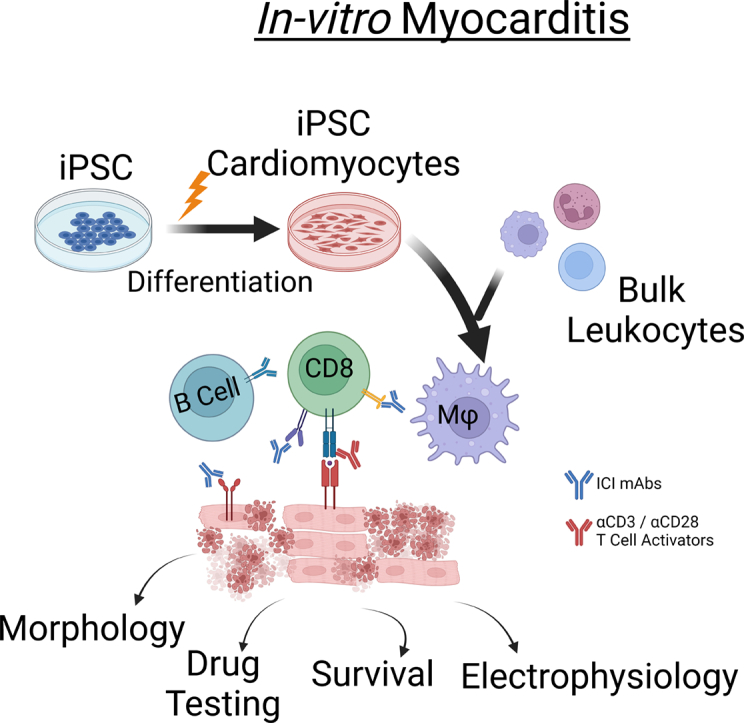
No scalable humanized model systems exist to characterize the interaction between human immune cells and cardiomyocytes in vitro[13]. Murine myocarditis models have proven invaluable in describing myocarditis pathophysiology but do not fully address the patient-specific cardiac and immune factors that may contribute to the disease[14,15]. Animal models are not scalable for large-scale therapeutic screening. There is a critical need for a platform that captures patient-specific risk factors, is scalable, and can provide prognostic and therapeutic information. The recent development of stem cell-derived cardiomyocytes has enabled personalized cardiac disease modeling in vitro. Primary human immune cells have long been used for in vitro disease modeling. Currently, there is no existing model for culturing stem cell-derived cardiomyocytes and immune cells in vitro to simulate inflammatory cardiac disease. This how-to article describes methods to 1) isolate primary human peripheral blood mononuclear cells (hPBMCs), 2) generate large numbers of human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs), and 3) generate and characterize a tissue culture-based myocarditis model. We introduce methodologies and provide example data from healthy donor samples treated with ICI biosimilars. This guide serves as a starting point for investigators interested in human in vitro disease modeling utilizing primary hPBMCs and hiPSC-CMs. The described methods will promote future studies that provide a comprehensive personalized disease modeling and therapeutic screening platform for myocarditis.
2. Material and methods
2.1. hiPSC culture and hiPSC-CM differentiation
Human iPSCs (ATCC ACS-1021, Thermo Fisher A18945) are cultured on Matrigel (Gibco A1413302) coated plates using StemFlex stem cell maintenance media (Gibco A3349401) (Supplemental Fig. 1A). Freshly thawed hiPSCs were cultured with ROCK inhibitor overnight to promote adhesion, after which media was replaced by fresh StemFlex medium daily until 50–60 % confluence was obtained. Cells were passaged using Versene (Gibco 15040066) 3–4 times before seeding for differentiation. Two-6-well plates were seeded for differentiation with 350,000 hiPSCs in ROCK inhibitor (Gibco A2644501) for each well. StemFlex medium was changed daily until hiPSC confluence was 80 %–90 %. hiPSC confluence that is either too high (>95 %) or too low (<50 %) may lead to poor differentiation efficiency. Differentiation was induced by the addition of 4 mL RPMI-1640 (Gibco 11875–119) containing B27 supplement without insulin (Gibco A1895601) and 4–6 μM CHIR99021 (Selleckchem S2924), a GSK3β inhibitor. This was considered differentiation Day 0 (Fig. 1A). Optimal concentrations of CHIR99021 vary by hiPSC line and should be determined by titration. GSK3β inhibition by CHIR99021 promotes mesoderm differentiation[16]. Day 2: Medium was replaced with 4 mL of RPMI-1640 supplemented with B27 without insulin for 24 h. Day 3: Medium was replaced with 4 mL of RPMI 1640 supplemented with 5 μM IWR (Selleckchem S7086), a Wnt inhibitor, and B27 without insulin for 48 h[16]. Day 5: Medium was replaced with RPMI 1640 supplemented with B27 without insulin for 48 h. Day 7: Media was replaced with 4 mL of RPMI 1640 supplemented with B27 supplement containing insulin (Gibco 17504–044) for 48 h. Beating hiPSC-CMs may be visible by phase contrast microscopy between days 7–11. Throughout the differentiation, substantial cell death and debris are present. This is normal, but the debris should be removed by gently swirling the plate and completely aspirating the spent culture media[17]. To obtain a high purity of hiPSC-CMs we performed metabolic purification using glucose deficient medium. hiPSC-CMs may utilize metabolic pathways other than glycolysis, while non-cardiomyocytes resulting from hiPSC differentiation typically cannot. Day 9: Medium was replaced with 4 mL of RPMI 1640 without glucose (Gibco 11897–020), supplemented with B27 containing insulin for 48 h. Day 11: Medium was replaced with RPMI 1640 supplemented with B27 containing insulin. Day 12: Successfully differentiated hiPSC-CMs have visually observable beating hiPSC-CMs. Wells with >80 % beating hiPSC-CMs visible on day 12 may be expanded or purified again using glucose starvation for an additional four days[[17], [18], [19]] (Supplemental Fig. 1B, Supplemental Video 1).
Fig. 1.

Setup for tissue culture myocarditis setup and model phenotype.
A) Schematic for hiPSC-CM differentiation and expansion.
B) Tissue Culture Myocarditis Setup.
C) Normalized hiPSC-CM survival at 72 h. Ratio represents the number of hPBMCs relative to the number of hiPSC-CMs. T cell activation mAbs (αCD3 & αCD28) dosages are indicated by ng/mL. Technical replicates indicated by individual dots on graph, n = 4 technical replicates per condition. Isotype treated conditions are shown in blue. ICI biosimilar treated conditions are shown in red.
D) Viability assay demonstrating impaired iPSC-CMs survival after 48–96 h. co-culture with activated hPBMCs and checkpoint inhibitors. Technical replicates indicated by individual dots on graph: CM Control (n = 8), quiescent PBMC (n = 9), Day 1 (n = 10), Day 2 (n = 6), Day 3 (n = 10), Day 4 (n = 6).
E) Immunocytochemistry staining of cTnT(red) and DAPI(blue) showing perturbations to sarcomere structure induced by activated hPBMCs and exacerbated by ICI treatment at 72 h.
F) Proportion of intracellular area containing cTnT obtained by particle analysis. CTRL (n = 17), PBMC (n = 21), Isotype (n = 22), Checkpoint (n = 28) individually gated hiPSC-CMs. Normalized to untreated control hiPSC-CMs.
*P < .05, **P < .01, ***P < .001, ****P < .0001 by unpaired t-test with Welch's correction for unequal variances for (B), Dunnett's multiple comparisons test for (C), and Tukey's multiple comparisons test for (E).
Error bars indicates mean ± s.d.
Experiments repeated >10 (A, B, C, D, E, F) times. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
2.2. Expansion of CMS
Following successful differentiation, cardiomyocytes may be expanded using a combination of sparse passaging and continued Wnt inhibition. On day 12, purified hiPSC-CMs were passaged to Matrigel-coated T75 tissue culture flasks. Adherent hiPSC-CMs were passaged using TrypLE Express (Gibco 12,605–010). A wash buffer of 80 % PBS and 20 % heat-inactivated FBS was used to quench the TypeLE Express enzyme. hiPSC-CMs were resuspended in a recovery medium (RPMI 1640, 5 % FBS, 2 % B27 supplement, and ROCK inhibitor). After 24 h, the culture medium was replaced with RPMI 1640 supplemented with B27 and 4 μM CHIR99021. This combination of sparse cell density and continued Wnt suppression by CHIR 99021 prevents the maturation of hiPSC-CMs, allowing them to retain significant proliferative capacity[17,18]. Media was replaced three times a week until hiPSC-CMs approached 90 % confluence. Cells were split in the same manner into 3-T185 flasks. hiPSC-CMs may be expanded again into 6–8 T185 flasks and then into 12–20 T185 flasks once 80–90 % confluence has been reached. Each passage may take 3–7 days to reach confluence. At this point, hiPSC-CMs may be replated for experimental use. Critical note: continued Wnt suppression by CHIR99021 prolongs hiPSC-CM proliferative capacity by maintaining hiPSC-CM immaturity. CHIR99021 must be removed to allow expanded hiPSC-CMs to mature to a similar level as non-expanded hiPSC-CMs[17]. Following differentiation and expansion, hiPSC-CMs were seeded into plates for further experimentation. hiPSC-CMs were allowed to mature for a minimum of ten days following removal of ROCK inhibitor (see section 2.1) before experiments were performed. Following this protocol, similar expressions of sarcomere maturity genes, similar sarcomere morphology, electrophysiology, force generation, and metabolism between expanded and non-expanded hiPSC-CMs have been demonstrated[17]. For additional information regarding hiPSC-CM differentiation, refer to the references cited in 2.1, 2.2.
2.3. hPBMC isolation
Healthy donor buffy coats were obtained from a local blood bank. hPBMCs were isolated from peripheral blood using Lymphoprep density gradient centrifugation (StemCell 07851). Donor whole blood was diluted with an equal volume of PBS. Lymphoprep, a comparable Ficoll-Paque alternative, was added to a conical tube according to manufacturer guidelines. Diluted whole blood was layered on top of the Lymphoprep. Critical note: do not mix blood samples with the Lymphoprep during sample layering or transport to the centrifuge. To minimize mixing from centrifugation, samples were centrifuged at 800 xg for 30 min at room temperature with a slow start and the centrifuge brake turned off. hPBMCs were present at the interface layer of the plasma and Lymphoprep. Collect hPBMCs with a transfer pipette and wash four times with PBS to remove Lymphoprep. Centrifuge at 400 xg for 10 min. The remaining red blood cells were removed using ACK lysis buffer (Gibco A1049201) at room temperature. Lysis was quenched after 5 min. by the addition of PBS. hPBMCs were resuspended in PBS and filtered through a 40 μM cell strainer to eliminate aggregated cells. RBC lysis may be repeated as necessary to remove remaining RBCs. Isolated hPBMCs should be kept on ice and may be used immediately or cryopreserved for later use. Before use, we performed an unstained flow cytometric evaluation to ensure sufficient hPBMC purity. Forward scatter (FSC) and side scatter (SSC) evaluation can quickly determine the percentage of granulocytes in the sample. Granulocytes have significantly greater SSC than PBMC populations, including lymphocytes (low FSC, low SSC) and myeloid cells (intermediate FSC and SSC) (Supplemental Fig. 1C). Care must be taken to minimize granulocyte contamination as these cells have short lifespans and may alter hPBMC behavior in vitro[20]. If cryopreserved hPBMCs are used, we typically allow thawed hPBMCs to recover on ice for three or more hours before use, though this has not been shown to significantly impact hPBMC function in vitro[21]. We filtered thawed hPBMCs through a 40 μM strainer to ensure no cellular aggregates were present before use. hPBMCs were analyzed on a BD LSR-II and sorted on a BD FACSFusion cytometer. For leukocyte depletion studies, populations of interest were stained, and PE-positive populations were depleted from bulk hPBMCs. The negatively selected hBPMCs were used for downstream assays. CD8+ T cells were stained and sorted from bulk hPBMCs using the BD FACSFusion cytometer.
2.4. Quantitating hiPSC-CM viability
Co-cultured hiPSC-CMs and hPBMCs were rinsed with PBS to remove suspension hPBMCs and necrotic hiPSC-CM debris. The remaining cells were stained with αCD45 (BioLegend 982,316) for 30 min, washed twice, and fixed in 4 % PFA for 30 min at room temperature. Cells were stained with 6.7 nM phalloidin (PerkinElmer CP25681) and 600 nM DAPI (BioLegend 422,801) for 2 h in PBS + 0.02 % Triton-X-100, then washed two times with PBS before imaging via automated confocal microscopy using an InCell Analyzer 6000 or Molecular Devices ImageXpress imaging platform using a 4× NA = 0.20 objective (Nikon). Four non-overlapping centrally located fields covering the entire well area were collected (Supplemental Fig. 1D). Images were analyzed using a custom script developed using Pipeline Pilot 2022 Server Edition (Dassault Systemes BioVia, San Diego). This method first subtracted the image background using the mode pixel intensity followed by segmenting nuclei using a thresholding and watershed-based approach applied to the DAPI channel. Intensity measurements were then collected under the nuclear mask for each fluorescently labeled channel, and GFP-positive PBMCs were digitally filtered out. The count of remaining cardiomyocytes was then used for downstream analysis. hiPSC-CM viability was normalized to untreated hiPSC-CM control wells. Workflows generated using Pipeline Pilot will be made available upon request and will require an active server license.
2.5. Western blot
Samples were lysed in RIPA II Cell lysis buffer (GenDepot R2400–010) containing 1.0 mM Pefabloc (Sigma-Aldrich 11429868001) and protease inhibitor cocktail (Sigma 11697498001). Samples were washed with lysis buffer and boiled in NuPAGE LDS Sample Buffer (Invitrogen NP0007) containing 10 % 2-mercaptoethanol (Sigma-Aldrich M6250). Samples were then separated in a 4–12 % gradient SDS-PAGE gel (GenScript M00654), transferred to a PVDF membrane (Thermo Scientific 88518), blocked with EveryBlot Blocking Buffer (BioRad 12010020), incubated with primary antibodies against Sox2 (Invitrogen 48-1400), OCT4 (Invitrogen PA5-27438), cTnT (Abcam ab45932), αactinin (sigma A7811), NANOG (Abcam ab21624), vimentin (Cell Signaling 5741p), GAPDH (Cell Signaling 2118s) overnight at 4 °C, washed 4 times, and stained with secondary antibodies (GenDepot SA002, GenDepot SA001). Membranes were imaged using the KindleBio KwikQuant Digital Western Blot Detection System. GenDepot West-Q Pico Dura ECL Solution (GenDepot W3653) was used. Images were processed using Kindle Biosciences KwikQuant Image Analyzer 5.9.
2.6. Immunocytochemistry
hiPSC-CMs were rinsed with PBS and fixed using 4 % PFA with 1 % BSA for 15 min at room temperature, permeabilized in 0.2 % Triton-X-100 in PBS for 10 min at room temperature and blocked in 10 % goat serum in PBS for 30 min at room temperature. Primary antibodies against cTnT (Abcam ab45932) and α-actinin (Sigma-Aldrich A7811) were incubated overnight at 4C without shaking. Samples were washed three times with room temperature PBS. Goat anti-mouse (Invitrogen A-11001) and goat anti-rabbit (Invitrogen A-11012) secondary antibodies were incubated with samples in 1 % BSA in PBS for 1 h. at room temperature. Samples were washed three times with PBS, and coverslips were mounted with DAPI containing Vectashield (Vector Laboratories H-1500-10). The samples were analyzed using a W1-Yokogawa/Nikon Live cell Imaging Spinning Disk Confocal microscope with a 60×, 1.4 NA, oil immersion lens using identical image acquisition settings. Images were processed using the Nikon NIS-Elements Advanced Research package. Particle analysis was performed in FIJI version 1.54f to obtain hiPSC-CM sarcomere coverage. Measured values were normalized to untreated control hiPSC-CMs.
2.7. Transwell assay
hiPSC-CMs were cultured on sterilized Matrigel (150 μg/mL) coated 0.13-.16 mm thick, 12 mm diameter glass coverslips (Ted Pella 26,020). Coated coverslips were cultured in 0.4 μM pore polycarbonate membrane transwell containing 24-well plates (Corning 3396). hPBMCs were added to the transwell chamber, with hiPSC-CMs cultured on the Matrigel-coated coverslips below. hPBMCs were cultured at a ratio of 10:1 with the hiPSC-CMs, with αCD3 and αCD28 mAbs at a final concentration of 100 ng/mL for 72 h. ICI mAbs were added at 200 nM concentration. Brightfield images were obtained on a Leica DMi4000b using a 5× NA = 0.12 objective. Coverslips were then removed from the transwell plate and stained using the immunocytochemistry protocol previously described in section 2.6. This technique was used only for the first part of section 4.1. Thereafter, where no transwell assay is described in the results, no transwell was used.
2.8. ELISA assays
Classical inflammatory cytokines were measured using a bead-based multiplex ELISA kit. Cytokine-containing medium was collected from hPBMCs after 72 h of activation and culture with hiPSC-CMs. We used the BioLegend LEGENDPlex Human Inflammation Panel 1 kit (Cat: 740809) to measure thirteen cytokines (IL-1β, IFN-α2, IFNγ, TNF-α, MCP-1, IL-6, CXCL7, IL-10, IL-12p70, IL-17A, IL-18, IL-23, and IL-33). ELISA beads were evaluated on a BD Biosciences LSRII analytical flow cytometer according to assay instructions. 10,000 events were collected and analyzed with the included analysis software from BioLegend using 4-parameter logistic regression analysis.
IFNγ concentrations after treatment with dexamethasone were measured using a human IFNγ ELISA kit (Invitrogen 88-7316-22). Standard curves were fitted using 4-parameter logistical regression to calculate sample concentration. IFNγ suppression was accomplished using an αIFNγ antibody (BioLegend 502501).
2.9. Multi-electrode array (MEA)
20,000 hiPSC-CMs were seeded over the electrode-containing region in Axion Biosystems 24 or 48 well MEA plates (Axion Biosystems M384-tMEA-24 W, M768-tMEA-48 W) in RPMI supplemented with B27 supplement (Gibco 17,504,044), 10 % FBS, and 10 μM ROCK inhibitor Y-27632 2HCl (SelleckChem S1049). Media was replaced after 24 h with RPMI supplemented with B27. After seven or more days of recovery, baseline cardiomyocyte electrophysiology was measured on the Axion Maestro Pro. The Maestro Pro culture chamber maintained a constant temperature of 37 °C and 5 % CO2. Voltage data was collected from all plate electrodes simultaneously, with FPD readings taken at 1 Hz to 2 kHz and LEAP readings at 0.01 Hz to 2 kHz. Local extracellular action potential induction was performed using AxIS Navigator. Field potential, contractility, and action potential readings were taken on the Maestro Pro. The threshold level for field potential duration was set to 100 μV with a recognized beat period range between 250 msec and 15 s. Polynomial regression modeling was used to identify hiPSC-CM repolarization T waves, with a post-spike detection holdoff of 120 msec and a pre-spike detection holdoff of 75 msec. A maximum post-spike search duration of 1.2 s and a 10-beat running beat average were used to assess field potentials. A maximum propagation delay of 40 msec with 25 % of electrodes detecting a field potential was required for field potential labeling. Identical settings were used for contractility and LEAP data analysis. A minimum threshold of 20 kΩ was used to confirm hiPSC-CM viability in the MEA tissue culture plate. hiPSC-CM electrophysiology data was processed using Axion Biosystems' AxIS Navigator, Cardiac Analysis Tool, and Plotting tool according to manufacturer instructions.
2.10. Statistical analysis and reproducibility
Data was plotted, and statistical analysis was performed using GraphPad Prism version 10.0.2. All data are presented as mean ± s.d. with P-values. The minimum sample size for each experiment was an N = 3 minimum technical replicates. Two disparate hiPSC lines and eight healthy PBMC donors were used. Reagents, cell lines, and software will be made available upon request in compliance with the NIH Grants Policy Statement 8.2, the Sharing of Research Resources[22].
3. Results
Primary human cardiomyocytes are challenging to obtain and have a short lifespan in a tissue culture setting[23]. Access to fresh human cardiac tissue is not possible or practical on a large scale, limiting the use of primary human cardiomyocytes in the preclinical research setting. Recent developments in the production of hiPSCs and their subsequent differentiation into stem cell-derived cardiomyocytes enable large-scale use of hiPSC-CMs for in vitro disease modeling[24,25]. This may partially address the lack of access to primary cardiomyocytes by providing a scalable alternative to model human disease[26]. Many hiPSC-CM differentiation methods are available[[27], [28], [29], [30]]. A widely used method relies on monolayer hiPSC culture and utilizes sequential GSK3β and Wnt signal pathway modulation to induce cardiomyocyte differentiation[27]. Differentiated hiPSC-CMs may also be temporarily retained in a proliferative stage through continued Wnt modulation[17,18]. Removal of Wnt/Gsk3β pathway modulators after cardiomyocyte proliferation allows continued maturation of hiPSC-CM sarcomere, electrophysiology, and force generation[18]. To streamline and standardize the generation of large numbers of hiPSC-CMs, we use a combination of monolayer hiPSC culture followed by a standardized expansion method to ensure reproducibility.
3.1. Evaluating hiPSC-CM differentiation quality evaluation
Following expansion, cardiomyocyte differentiation quality is assessed before experimentation using a combination of flow cytometry, Western blot, and immunocytochemistry. Our approach follows well-established methods widely available in the published literature [[16], [17], [18], [19],[24], [25], [26], [27], [28], [29], [30]]. HiPSC-CMs should demonstrate expected morphology, express canonical cardiomyocyte proteins, and be pure and free from contaminating non-differentiated hiPSCs or fibroblasts. The absence of non-differentiated hiPSCs and fibroblasts is confirmed by Western blot. Stem cell markers should not be present in differentiated hiPSC-CM cultures. Successful differentiations used for modeling myocarditis should be free of significant fibroblast contamination. We used Western blot for cardiac α-actinin and cTnT to show cardiac marker expressions in the hiPSC-CMs. Canonical hiPSC markers, OCT4, SOX2, and NANOG, were only present in lysate collected from hiPSCs but not in the cell lysate collected from differentiated cardiomyocytes. Vimentin, a fibroblast marker, was not detected in lysate from differentiated cardiomyocytes or hiPSCs (Supplemental Fig. 2A). hiPSC-CM purity was quantified by flow cytometry staining for αMyHC (Supplemental Fig. 2B). Differentiations contaminated by significant numbers of fibroblasts or yielding poor hiPSC-CM purity (< 90 %) should not be used. Immunocytochemistry was used to evaluate sarcomere structure and morphology for differentiated hiPSC-CMs. For successful differentiations, intracellular staining for cTnT and/or α-actinin should reveal expected striated sarcomere morphology (Supplemental Fig. 2C). These results demonstrate highly pure hiPSC-CMs free from significant contamination from competing cell types, including undifferentiated hiPSCs or fibroblasts. In summary, we have used Western blot to demonstrate successful expression of cardiac lineage markers as well as the absence of iPSC and fibroblast markers. We used flow cytometry to demonstrate the expression of cTnT, a cardiac-specific marker, in >90 % of cells evaluated. We used imaging to evaluate differentiated hiPSC-CM sarcomere structure. These results suggest a successful differentiation with a highly pure population of hiPSC-CMs.
3.2. Myocarditis model setup
Our tissue culture myocarditis model is comparable to assays used in modeling anti-tumor T cell responses that co-culture tumor cell lines and human immune cells[31]. We set up our myocarditis assay by combining a culture of activated hPBMCs with hiPSC-CMs. First, adherent hiPSC-CMs are plated and matured for a minimum of ten days. Then, hPBMCs, which are suspension cells, were added with other compounds to our previously plated hIPSC-CMs to initiate the myocarditis model (Fig. 1B). We have found that hPBMCs must be activated during this assay. T cells used in vitro typically require either activating stimuli or cytokines. Non-stimulated T cells do not proliferate or respond well to antigens in vitro, particularly in the absence of a costimulatory signal to complement the so-called “signal 1” through the T cell receptor[32]. This can be accomplished through different approaches, including monoclonal antibodies that stimulate the T cell receptor, T cell co-receptor, or cytokines that support T cell activation against antigens presented in their target cells. We compared methods to stimulate hPBMCs and found that T cell activation with αCD3/28 stimulating antibodies (mAbs)is required to induce loss of hiPSC-CM viability in vitro. Quiescent hPBMCs do not induce hiPSC-CM necrosis (Supplemental Fig. 3A). hPBMCs treated with either LPS or cytokines (IL-2, IL-7, and IL-15) did not induce hiPSC-CM necrosis (Supplemental Fig. 3B).
Using a combination of αCD3 and αCD28 stimulating mAbs to activate T cells within the hPBMCs, we systematically evaluated both ratios of hPBMCs to hiPSC-CMs and concentrations of stimulating antibodies (Fig. 1C). We found that a ratio of three hPBMCs to[18,18,28,29] one hiPSC-CMs with αCD3/CD28 mAbs concentrations between 10 and 30 ng/mL allows this model to be used for 72–96 h. Unless stated otherwise, all experiments described hereafter use these optimized conditions, the 3:1 ratio of hPBMCs to hiPSC-CMs with 30 ng/mL of stimulating anti-αCD3/CD28 monoclonal antibodies (mAbs). ICI biosimilars, if present, were used at 200 nM. A combination of αPD1/αPD-L1(BioXcell SIM0010 or BioXcell SIM0003) and αCTLA-4 (BioXcell SIM0004) antibodies were used. hiPSC-CM viability was normalized to untreated negative control hiPSC-CMs. There was no difference in cardiomyocyte viability between negative control hiPSC-CMs or treatment control hiPSC-CMs (hiPSC-CMs treated with isotype mAbs, αCD3 and αCD28, ICI biosimilars, or quiescent hPBMCs) (Supplemental Fig. 3A). Treatment control hiPSC-CMs cultured with quiescent hPBMCs with or without ICI biosimilars do not have significantly different viability (105 % for isotype treated and 103 % for ICI biosimilar treated). Activated hPBMCs alone reduce hiPSC-CM viability to 86 % of control at 48 h., 28 % at 72 h., and 19 % at 96 h (Fig. 1D). Adding ICI biosimilars to activated hPBMCs further significantly decreases hiPSC-CM viability to 60 %, 18 %, and 8 % of controls in hiPSC-CM viability at 48, 72, and 96 h respectively (Fig. 1D). hiPSC-CM viability at 24 h was not significantly different compared to untreated hiPSC-CMs. We observed the largest decrease in hiPSC-CM viability occurs between 48 and 72 h. Based on these findings, we characterize the impact of both activation and ICI biosimilar administration of hiPSC-CM morphology and electrophysiology and validate potential therapeutic responses over 48–72 h.
3.3. Myocarditis-mediated sarcomere disruption
Aggregation and disarray of sarcomere structural components, including cTnT, occurs during cardiomyocyte injury and often leads to loss of characteristic striations[33,34]. Sarcomere disruption was visible on imaging from our viability assay (Supplemental Fig. 3C). To better evaluate this finding, we performed immunocytochemistry staining for cTnT across the same range of conditions used in our viability assay (Supplemental Fig. 4). cTnT imaging demonstrates disruption to the hiPSC-CM cytoskeleton when cultured with activated hPBMCs and was exacerbated by the addition of ICI biosimilars (Fig. 1E). Sarcomere coverage, the intracellular area positive for cTnT, was not significantly different between control groups (negative control hiPSC-CMs and treatment control hiPSC-CMs cultured with quiescent hPBMCs, and ICI biosimilars). iPSC-CMs cultured with activated hPBMCs treated with isotype control antibodies induced cTnT aggregation and significantly reduced hiPSC-CM sarcomere coverage to 0.68. ICI treatment of activated hPBMCs further reduced sarcomere coverage to 0.50 (Fig. 1F).
4. Model validation – ICI myocarditis
4.1. CD8+ T cells contribute to hiPSC-CM necrosis
In murine ICI myocarditis models, intramyocardial CD8+ T cells cause cardiomyocyte necrosis. It is hypothesized that these cells are potentially autoreactive[14,35]. We performed a transwell assay to evaluate the contribution of specific leukocyte populations to myocarditis development in our model. The transwell insert physically separates one population of cells from the others. hPBMCs are placed in the transwell insert, and hiPSC-CMs are cultured on Matrigel-coated glass coverslips for future confocal imaging. This allows culture media containing nutrients and soluble factors, such as cytokines, to freely diffuse throughout the well without direct contact between hPBMCs and hiPSC-CMs (Fig. 2A). No changes to hiPSC-CM sarcomere morphology were observed under any condition despite increased ratios of hPBMCs to hiPSC-CMs (10: 1) and increased stimulating antibodies (100 ng/mL αCD3/αCD28 mAbs) (Fig. 2A). Sarcomere coverage was not different from control groups (Fig. 2B). The data suggest direct cell contact is required for the development of our myocarditis phenotype, which was unknown before this experiment. This is consistent with the known function of CD8+ T lymphocytes [32,36]. Having demonstrated that direct contact is required for our myocarditis phenotype we did not use the transwell assay further.
Fig. 2.

Mechanisms of tissue culture myocarditis.
A) Graphical schematic of transwell assay setup conditions. Phase contrast microscopy of quiescent or activated hPBMCs after 72 h. (top) and ICC staining for hiPSC-CMs (bottom). cTnT(Red), α-actinin(green), DAPI (blue). hPBMC were cultured in a transwell insert separating them from hiPSC-CMs in the bottom of the well. Conditions included are indicated by a (+) in the underlying table. Control CMs contains hiPSC-CMs cultured in isolation.
B) hiPSC-CM sarcomere coverage obtained by particle analysis. Normalized to untreated hiPSC-CM control. Isotype (n = 18), Checkpoint (n = 20), Activated Isotype (n = 25), Activated Checkpoint (n = 18), technical replicates are individually gated cells from immunocytochemistry images.
C) Viability assay using FACS sorted hPBMCs at 72 h. Depleted populations are indicated by (−). hiPSC-CM viability normalized to untreated control hiPSC-CM condition. CD8+ and CD3+ T cells are required for myocarditis development. Technical replicates indicated on graph, n = 12 for all conditions.
D) Immunocytochemistry staining for cTnT (red) and DAPI (blue) to assess sarcomere structure at 72 h. Myocarditis was induced after specific cellular populations were stained and depleted from bulk hPBMCs. Depleted cells are indicated by (−). T cells (+) were obtained by depleting all other cell types. Sarcomere disarray is visible when CD8+ or CD3+ T cells are present.
*P < .05, ***P < .001, ****P < .0001 by one-way ANOVA with Tukey's correction for multiple comparisons for (B) and (C).
Experiments repeated 2 (A, B) and 3 (C, D) times. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
To determine which specific hPBMC populations are required for myocarditis development in vitro, we used FACS to deplete specific hPBMC populations, including CD8+ cytotoxic T cells, CD3+ T cells, CD19+ B cells, and CD56+ natural killer cells (Supplemental Fig. 5A). These populations were chosen because their requirement for direct interaction with target cells is well described in the literature. Except for the population-specific depletion of hPBMCs, we set up this myocarditis assay as described in section 3.2. There was no difference in hiPSC-CM viability among controls (negative control hiPSC-CMs or treatment control hiPSC-CMs cultured with quiescent hPBMCs ± isotype/ICI biosimilar mAbs). Activated hPBMCs significantly reduced hiPSC-CM viability to 73 % of controls. Depletion of CD3+ T cells eliminates the anti-hiPSC-CM activity of bulk hPBMCs, leading to preserved hiPSC-CM viability (99 %). hiPSC-CM cultured with activated purified CD8+ cells have significantly lower viability (65 %) compared to bulk-activated hPBMCs (73 %) (Fig. 2C). We confirmed this finding through sarcomere analysis of cTnT-stained hiPSC-CMs cultured with sorted hPBMCs. hPBMCs depleted of either CD8+ T cells or CD3+ T cells (of which CD8+ T cells are a part) show markedly reduced capacity for inducing hiPSC-CM sarcomere disruption compared to bulk hPBMCs (Fig. 2D). Enriched T cells were obtained by depleting hPBMCs of CD14+, CD16+, CD19+, and CD56+ cells. These enriched negatively selected T cells demonstrate a similar capacity for hiPSC-CM sarcomere destruction compared to bulk hPBMC populations (Fig. 2D). Depleting natural killer cells did not lead to improved hiPSC-CM sarcomere structure. These results suggest a requirement for CD8+ T cells in the pathophysiology of our tissue culture-based ICI myocarditis model.
We sought to evaluate the relationship between the proportion of CD8+ T lymphocytes and the severity of myocarditis in the tissue culture setting. We measured the percentage of bulk hPBMCs from eight donors that stained positive for CD8 via flow cytometry and performed myocarditis modeling as previously described, using un-sorted, activated hPBMCs cultured with previously plated hiPSC-CMs (Supplemental Fig. 5B). Linear regression analysis demonstrated a weak but significant correlation between the number of CD8+ T lymphocytes and hiPSC-CM killing by activated hPBMCs, R2 = 0.2855, p < 0001 (Supplemental Fig. 5C, D). Our findings suggest that the proportion of CD8+ T lymphocytes among hPBMCs is not the major factor contributing to myocarditis severity. Therefore, additional factors may be considered, including the potential contribution of pre-existing hiPSC-CM-reactive CD8+ lymphocytes.
4.2. Evaluating hiPSC-CM electrophysiology
A frequent cause of death in ICI myocarditis is malignant arrhythmias such as AV block or ventricular fibrillation[[37], [38], [39]]. We used MEA technology to characterize the electrophysiological changes present in our tissue culture-based myocarditis model. Beat period, the time interval between hiPSC-CM beats, was unchanged in control conditions, including negative control hiPSC-CMs and treatment control hiPSC-CMs cultured with quiescent hPBMCs ±isotype / ICI biosimilar mAbs (beat period 1.7–2.8 s). Beat periods for hiPSC-CMs cultured with activated hPBMCs did not differ from controls at baseline. At time points 18 h. and longer, the beat period for hiPSC-CMs cultured with activated hPBMCs treated with isotype antibodies was significantly longer than that of controls. ICI biosimilar-treated conditions containing activated hPBMCs had significantly longer beat periods than isotype-treated conditions. At 18 h. the beat period was 3.05 s with isotype treated active hPBMCs and 3.62 s for ICI biosimilar treated activated hPBMCs. At 44 h., hiPSC-CMs cultured with isotype-treated activated hPBMCs had a beat period of 19.6 s. ICI biosimilar treated activated hPBMCs caused hiPSC-Cm beat periods to increase to 22.2 s (Fig. 3A).
Fig. 3.

Electrophysiology of tissue culture myocarditis.
A) hiPSC-CM beat period measured by MEA. Control CMs (n = 8), Isotype (n = 8), Checkpoint (n = 8), Activated Isotype (n = 12), Activated Checkpoint (n = 12). Individual replicates are averaged values obtained from individual wells containing 16 microelectrodes. Timepoints are indicated on the graph.
B) Beat period irregularity of hiPSC-CMs under indicated conditions. # = P < .05 at 26, 32, and 44 h. between activated hPBMCs treated with isotype or ICI, and between ICI and steroid treated activated hPBMCs, as indicated on the graph. Statistics from Tukey's multiple comparisons test. Technical replicates are indicated on graph Control CMs (n = 8), Isotype (n = 8), Checkpoint (n = 8), Activated Isotype (n = 12), Activated Checkpoint (n = 12). Individual replicates are averaged values obtained from individual wells containing 16 microelectrodes. Timepoints are indicated on the graph.
C) Representative beat period Poincare plots demonstrating hiPSC-CM beat period irregularity.
*P < .05, **P < .01, ***P < .001, ****P < .0001 by Tukey's multiple comparisons test for (A), (B).
Experiments performed 4 (A, B, C) times.
hiPSC-CM beat period irregularity is defined as the beat period standard deviation divided by the mean beat period duration. Beat period irregularity was unchanged among the negative control hiPSC-CMs and treatment control hiPSC-CMs cultured with quiescent hPBMCs. Beat period irregularity was significantly increased in conditions with activated hPBMCs and further increased by the addition of ICI biosimilars. In isotype-treated conditions, the beat period irregularity was 6.5 %, 20.6 %., 35.6 %., and 56 % at 18, 26, 32, and 44 h, respectively. Beat period irregularity was significantly greater with ICI biosimilar treatment than in the isotype comparison group at these time points (9.7 %, 32.1 %, 57 %, and 77.7 % at 18, 26, 32, and 44 h, respectively) (Fig. 3B, C). Following 48 h., the beat period for myocarditis conditions was outside the analysis parameters for the multi-electrode array analysis. There were no significant changes to hiPSC-CM contractility under any condition relative to untreated control hiPSC-CMs (Fig. 4A).
Fig. 4.

Electrophysiology of tissue culture myocarditis.
A) hiPSC-CM beat amplitude measured by MEA. Timepoints are indicated on the graph.
B) hiPSC-CM 90th percentile action potential duration measured by MEA. Timepoints are indicated on the graph.
*P < .05, **P < .01, ***P < .001, ****P < .0001 by Tukey's multiple comparisons test for (A) and Brown-Forsythe ANOVA for (B).
Experiments performed 4 (A, B) times.
Altered cardiomyocyte action potential generation can lead to arrhythmia. To evaluate the contribution of altered action potentials to the beat period irregularity we observed, we evaluated hiPSC-CM local extracellular action potentials (LEAP). We measured LEAP at 24 h to avoid the significant hiPSC-CM cytotoxicity occurring at later time points. Prolonged hiPSC-CM beat periods and significant beat period irregularity are present before 24 h. (Fig. 3A, B). We observed a substantial increase in the 90th percentile of hiPSC-CM action potential duration (APD90) across all conditions containing hPBMCs regardless of activation. APD 90 was identical for conditions cultured with both quiescent hPBMCs (0.63 s) and activated hPBMCs (0.63 s) cultured with ICI biosimilars (Fig. 4B). Our previous experiments have not identified significant changes to hiPSC-CM viability, sarcomere structure, or hiPSC-CM beat metrics when hiPSC-CMs are cultured with quiescent hPBMCs. Significant changes in hiPSC-CM viability, altered sarcomere structure, and arrhythmia only occur when hiPSC-CMs are cultured with activated hPBMCs. Thus, it is possible that the increased number of cells caused by including hPBMCs results in prolonged APD90 measurements. This may suggest that the observed beat period irregularity is not caused by altered hiPSC-CM action potentials.
4.3. Cytokine production and impact on electrophysiology
We found significant increases in classical inflammatory cytokines in our model. Multiplex ELISA demonstrates increased IFNα2, IFNγ, TNFα, MCP1, IL6, IL8, IL-10, IL12-p70, IL17a, IL18, IL23, IL33, and IL1β under conditions where hiPSC-CMs were cultured with activated hPBMCs and ICI biosimilars (Supplemental Fig. 6A). Cytokines that are known to be increased in ICI myocarditis patients include IFNγ, IL-8, IL-6, MCP1, IL-10, IL-18, and IL-1, though the specific cytokine response in ICI myocarditis is not fully characterized [[40], [41], [42]]. To evaluate the contribution of activated hPBMCs-derived cytokines on the electrophysiology of hiPSC-CMs, we cultured previously untreated hiPSC-CMs with conditioned medium collected from myocarditis cultures containing activated hPBMCs and ICI biosimilars. The conditioned medium was centrifuged at >10,000 ×g for 15 min. and filtered through a 20 μM cell strainer to ensure no residual hiPSC-CMs or hPBMCs were present. HiPSC-CMs exposed to conditioned media did not develop significant changes in either the beat period or beat period irregularity. The hiPSC-CM beat period was 1.7 s before and 1.7 s after exposure to conditioned media (Supplemental Fig. 6B). Beat period irregularity was 1.4 % before and 1.1 % after exposure to conditioned medium (Supplemental Fig. 6C).
4.4. Potential as a therapeutic screening platform
To validate the drug responsiveness of our myocarditis in a dish platform and demonstrate its potential as a therapeutic testing platform, we evaluated two potential therapeutic compounds chosen from the scientific literature: corticosteroid treatment and an IFNγ neutralizing antibody[43,44]. hiPSC-CM viability was not different among controls (negative control hiPSC-CMs and treatment control hiPSC-CMs cultured with quiescent hPBMCs). hiPSC-CM viability with activated hPBMCs and ICI biosimilar was significantly reduced to 73 %. Dexamethasone treatment prevented the development of myocarditis and maintained 100 % hiPSC-CM viability (Fig. 5A). Dexamethasone treatment also prevented damage to the hiPSC-CM sarcomere structure at 72 h (Fig. 5B). Beat period prolongation and beat period irregularity were also controlled by 500 nM dexamethasone treatment at 72 h (Fig. 5C-E). Dexamethasone is a well-described glucocorticoid receptor agonist that has been widely used since 1957 as an immunosuppressant. No beneficial effect on hiPSC-CM viability was detected from the IFNγ neutralizing mAbs (Fig. 5A).
Fig. 5.

Treatment validation for tissue culture myocarditis platform.
A) hiPSC-CM survival modulation by steroid treatment or IFN suppression at 72 h. n = 12 technical replicates per condition.
B) ICC staining for α-actinin (green), cTnT (red), and DAPI (blue) after steroid treatment for 72 h.
C) Beat period after steroid treatment at 72 h. n = 8 or 12 technical replicates per condition.
D) Beat period irregularity after steroid treatment at 72 h. n = 8 or 12 technical replicates per condition.
E) Beat period Poincare after steroid treatment.
***P < .001, ****P < .0001 from One Way ANOVA with Dunnett's T3 multiple comparisons test for (A), one way ANOVA for (C) and (D).
Experiments performed 2 (A, B, C, D, E) times. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
4.5. Antigen specificity
Recent murine models suggest autoreactive intramyocardial T cells specific for cardiac α-myosin heavy chain (αMyHC) may contribute to the development of ICI myocarditis[15]. Similar cells are present in the circulation of affected patients and healthy donors, raising additional questions about their roles in the underlying disease pathophysiology[35,45]. To define the cardiomyocyte antigen-specific response in this model, we pre-exposed hPBMCs to hiPSC-CM lysate in the presence of IL-2 and IL-15 to support the expansion of hiPSC-CM-specific T lymphocytes. These antigen-expanded hPBMCs (exPBMCs) were compared to both hPBMCs cultured with IL-2 and IL-15 and non-expanded hPBMCs from the same donor. exPBMCs demonstrate a significantly increased ability to induce hiPSC-CM death, resulting in a relative viability of 36 % for isotype and 31 % for ICI biosimilar treatment without stimulating αCD3/CD28 mAbs. Non-antigen-exposed quiescent hPBMCs did not reduce hiPSC-CM viability. Additional activation of exPBMCs with αCD3/CD28 further reduced the relative viability of hiPSC-CMs to 16.7 % (Fig. 6A). Considering these findings and our observation that the percentage of CD8 T cells did not fully explain myocarditis severity among our donors, this may suggest a role for hiPSC-CM antigen-specific T cells in developing myocarditis in our tissue culture model.
Fig. 6.

αhiPSC-CM specific hPBMCs contribute to myocarditis phenotype.
A) Viability assay utilizing hiPSC-CM antigen exPBMCs at 72 h. See methods for exPBMC culture protocol details. ExPBMCs demonstrate increased cell mediated cytotoxicity compared to non-exPBMCs. Technical replicates indicated on graph, CM Control (n = 8), Quiescent PBMC Isotype (n = 9), Quiescent PBMC Checkpoint (n = 10), hPBMCs + IL2/15 (n = 12), Ex-PBMC Isotype (n = 12), Ex-PBMC Checkpoint (n = 12), Activated Ex-PBMC (n = 12).
B) Phalloidin stain of murine colon cancer CT26 line (i and ii) and HEK293T cell line (iii and iv). Control lines cultured in isolation (i and iii) as well as cell lines cultured with activated hPBMCs and ICI mAbs (ii and iv) are shown. Imaging performed at 72 h. Experiment performed once.
Tukey's multiple comparisons test for (A). Experiments performed twice.
To address the potential impact of non-specific immune-mediated cytotoxicity resulting from activation of T lymphocytes via αCD3/CD28 stimulation, we utilized two unrelated non-cardiac cell lines, the murine colon cancer CT26 and HEK293T cell line, in place of hiPSC-CMs. These were cultured with hPBMCs under the same conditions as our myocarditis assay, a 3:1 ratio with 30 ng/mL αCD3/28 mAbs, and 200 nM ICI biosimilar treatment. No apparent CD26 or HEK293T cell coverage loss was exhibited compared to the untreated control cell lines that were not cultured with hPBMCs after 72 h (Fig. 6B).
5. Discussion
ICI myocarditis is a devastating complication of ICI treatment. There are critical unmet needs for evidence-based treatment strategies, a deeper understanding of disease pathophysiology, and patient-specific disease modeling platforms that recapitulate genetic and non-genetic risk factors for ICI myocarditis. Cardiac biopsies are invasive and suffer from low sensitivity, severely limiting the study of myocarditis using human samples. This necessitates using disease models to discover disease pathophysiology and perform preclinical testing of potential treatment strategies. In vivo myocarditis models utilize immunization with specific peptide fragments from αMyHC, viral infection, or deletion of immune checkpoints for modeling myocarditis or ICI myocarditis[46]. Murine models excel at describing disease pathophysiology but are poorly suited for patient-specific disease modeling and large-scale therapeutic screening. The availability of both hiPSC-CMs and primary human PBMCs may address these two needs. We have described a new method to model myocarditis in vitro by co-culturing primary human leukocytes and human-derived iPSC-CMs. hiPSC-CMs partially address the difficulty in obtaining human cardiomyocyte samples. Primary hPBMCs are easily obtained with minimal invasiveness and carry patient-specific genetic and non-genetic immune response factors that may impact ICI myocarditis pathophysiology and treatment response. We have validated this method using samples from multiple healthy blood donors and hiPSC lines. Additionally, we have demonstrated a similar phenotype using either hPBMCs or purified T lymphocytes. Our data suggests that additional factors beyond the number of CD8+ T lymphocytes may be responsible for the phenotype severity observed using this model. Our findings also suggest a role for cardiac-specific T lymphocytes in the development of our tissue culture myocarditis phenotype, seemingly in agreement with findings from murine ICI myocarditis models[15,47]. This raises interesting questions as deficient central tolerance to major cardiac antigens, including αMyHC, is well described in the literature, leading to frequent cardiac-reactive lymphocytes in the general population[[47], [48], [49], [50]].
A critical first step of this model is the ability to culture hiPSCs and differentiate them into hiPSC-CMs successfully. As previously noted, continued Wnt suppression by CHIR-99021 treatment maintains an immature differentiation state for hiPSC-CMs to preserve their proliferative capacity. This causes reduced contractile force, potentially caused by decreased expression of the ryanodine receptor 2, myosin light chain, and troponin I genes[51]. However, if previously expanded hiPSC-CMs are cultured without CHIR99021, expression of these genes returns to normal expression levels and is comparable with non-expanded hiPSC-CMs[18]. Expanded hiPSC-CMs must be cultured without CHIR to allow continued maturation to a normal hiPSC-CM morphology[18]. If hiPSC-CMs are produced in-house, differentiation purity should be assessed to ensure that high-purity hiPSC-CMs are used. We observed identical phenotypes using multiple hiPSC-CM lines. This may indicate that commercially produced hiPSC-CMs may be used in place of hiPSC-CMs differentiated “in-house.” We have not assessed the impact of additional cardiac cell types in this model. Adding cardiac fibroblasts or endothelial cells to this model may change the results and is an interesting experiment for future study.
Alternative methods may be used to quantify hiPSC-CM viability in vitro, but few are appropriate in culture protocols with multiple cellular populations. When selecting analysis methods, care must be taken to ensure that a specific count of hiPSC-CMs is obtained. Alternatives such as the Chromium-51 release assay or reporter-based viability assays may work, but consideration must be given to potential immune responses to reporter proteins[52,53]. Similar challenges exist when measuring hiPSC-CM beat and contractility measurements. hiPSC-CM analysis platforms frequently rely on Ca2+ staining to quantify hiPSC-CM beating. Substantial numbers of hPBMCs prevent adequate visualization of hiPSC-CMs using these platforms. Additionally, the cytosolic Ca2+ influx contributing to leukocyte activation may interfere with Ca2+ imaging[54,55].
We chose to activate hPBMCs using αCD3 and αCD28 stimulating mAbs. T cells require activation before proliferation, cytolytic activity, and cytokine production[56]. Various methods have been developed to activate T cells in vitro. Immune-oncology assays frequently use T cells activated by α-CD3 and α-CD28 mAbs. This well-established methodology partially mimics endogenous T cell activation by stimulating both the T cell receptor and coreceptor[57,58]. Several potential mechanisms may lead to T cell activation in ICI myocarditis. Tumor antigens displayed on MHC may lead to T cell activation via the CD3 and CD28 receptors, and necrotic regions may activate leukocytes via pattern recognition receptors[[59], [60], [61]]. ICI therapy does not directly lead to the activation of cytotoxic T lymphocytes in vivo. It simply removes an inhibitory signal that some tumors use to prevent anti-tumor immunity[62]. We have shown that T cells activated by αCD3/CD28 mAbs demonstrate potent anti-hiPSC-CM activity in vitro that is enhanced by treatment with ICI biosimilars. In our hands, non-activated hPBMCs do not result in hiPSC-CM death over 96 h (Supplemental Fig. 3A). hPBMCs without T cell receptor activation also do not result in hiPSC-CM death (Supplemental Fig. 3B). PBMCs exposed to hiPSC-CM lysate for prolonged periods also demonstrate significant anti-hiPSC-CM activity in vitro without the need for non-specific activation with αCD3/28 mAbs. A similar impact on hiPSC-CM viability was present using both methods. This may suggest a similar mechanism underlying both methods of activating T cells (Fig. 1, Fig. 4).
5.1. Summary of the required levels of expertise to perform analysis
The techniques used in performing this model include differentiation of hiPSC-CMs, isolation and stimulation of primary hPBMCs, confocal microscopy, flow cytometry, and multielectrode array.
-
•
hiPSC culture, hiPSC-CM differentiation, and expansion – basic tissue culture experience required. Care must be taken to properly passage the hiPSCs to prevent spontaneous differentiation. Differentiation is straightforward and requires basic experience in cell culture training.
-
•
Flow Cytometry – basic experience in flow cytometry and access to a flow cytometer is required to evaluate hiPSC-CM and isolate specific leukocyte populations.
-
•
Multi-electrode array – access to and expertise in multielectrode array platforms compatible with hiPSC-CM evaluation is required. Non-experts should consult with MEA operators before designing experiments to receive instrument-specific guidance for experimental setup and analysis.
-
•
Viability evaluation – expertise in confocal microscopy is required. We have used automated confocal microscopy platforms, including the GE InCell analyzer 6000 and Molecular Devices ImageXpress platforms, with analysis performed in Pipeline Pilot. Workflows generated using Pipeline Pilot will be made available upon request and will require an active server license.
5.2. Limitations
hiPSC-CMs are notably different than adult primary human cardiomyocytes. Primary human cardiomyocytes are functionally inaccessible for large-scale research. Primary animal cardiomyocytes are similarly difficult to obtain and have poor survival in vitro, significantly reducing their usefulness in disease modeling in vitro. HiPSC-CMs represent the most viable option for in vitro cardiac disease modeling experiments like our proposed model. To minimize the limitations of hiPSC-CMs, we have standardized our differentiation and expansion protocol, tested multiple hiPSC-CM lines, and allowed time after expansion for hiPSC-CMs to mature to a normal hiPSC-CM morphology[18]. We have not yet evaluated this protocol using hPBMCs or hiPSC-CMs obtained from ICI myocarditis patients. We do not know how such autologous disease modeling may differ from the results using commercially available cell lines. This is a future study we will perform. Our data demonstrates no significant CT26 or HEK293T cell coverage loss under identical conditions to our myocarditis assay. Despite this, there remains a possibility that allogenic interactions from MHC mismatch may contributed to our model phenotype[63]. We note that the Thermo Fisher hiPSC line is no longer sold. Alternatives include the episomal reprogramming kit used to create this cell line (Invitrogen A14703) that may be purchased. We have shown identical phenotypes between two disparate hiPSC-CM lines. Commercially produced hiPSC-CMs are broadly available, as are hiPSC lines that may be differentiated in-house.
5.3. Future studies
Future studies can apply the culture protocol described to investigate the pathophysiology and risk factors further and discover potential interventions for ICI myocarditis. The most exciting aspect of the human tissue culture model presented in this study is the opportunity for personalized disease modeling using autologous hiPSC-CMs and hPBMCs derived from the same donor. This approach will be used to determine both the clinical translational and prognostic relevance of this model and to assess the contribution of allogenic interactions resulting from MHC mismatch in this model. Murine immunologic disease models have complex and poorly understood genetic backgrounds and are not representative of human immunology and disease pathophysiology[64,65]. The possibility of autologous, patient-specific disease is one advantage of using human cells to model disease. It may partially address the shortcomings of disease modeling in mice to meet the critical needs and knowledge gaps of ICI myocarditis. For ICI myocarditis, we anticipate using this model to describe differences in the leukocyte-cardiomyocyte interaction among three groups: healthy donors, patients who experience myocarditis following ICI treatment, and patients who do not develop ICI myocarditis. This model is also valuable for large-scale therapeutic testing to identify novel medications that may effectively treat ICI myocarditis models in vivo.
In summary, we describe a novel cell culture method that combined primary human leukocytes with human induced pluripotent stem cell-derived cardiomyocytes to model myocarditis in vitro. We show how this model effectively replicates critical disease findings of ICI myocarditis, including cardiomyocyte necrosis, inflammatory mediated sarcomere disruption, and arrhythmia resulting from cardiomyocyte-specific immune responses. Finally, while the data presented here utilized commercially available cell lines and samples from healthy donors, we expect this approach to be valuable for samples from myocarditis patients.
The following are the supplementary data related to this article.
Video demonstrating spontaneous, non-paced beating of successfully differentiated hiPSC-CMs.
Validation of hiPSC-CM differentiation and hPBMC isolation. Brightfield images demonstrating visual iPSCs (A) and differentiated hiPSC-CMs (B). C) Representative SSC x FSC plot for unstained flow cytometric evaluation of hPBMCs. D) Representative Phalloidin (left), DAPI (middle) and αCD45 (right) stained images used for visual viability analysis pipeline. Graphical overview of hiPSC-CM viability analysis pipeline. Experiments repeated 10 times for (A, B, C, D).
hiPSC-CM Purity. A) Western blot for lineage specific markers for iPSCs (NANOG, OCT4, SOX2), day 14 post-differentiation hiPSC-CMs (cTnT, α-actinin), and iPSC-fibroblasts (vimentin) and GAPDH control. Lysate sample source indicated below Western blot. B) Flow cytometry staining for cTnT—PE on day 14 post-differentiated hiPSC-CMs. Unstained control (orange), isotype stained control (blue), stained hiPSC-CMs (red). C) Immunocytochemistry staining of day 24 post-differentiation hiPSC-CMs for cTnT (red), α-actinin (green), counterstained with DAPI (blue). Merged image in the left panel.
Myocarditis model setup. A) hiPSC-CM survival for indicated conditions including isotype mAbs, ICI biosimilars, αCD3/CD28, and quiescent hPBMCs at 72 h. B) hPBMCs without T cell activators do not induce hiPSC-CM death at 72 h following co-culture. C) Confocal images of phalloidin-stained hiPSC-CMs. Sarcomere structure for control hiPSC-CMs and hiPSC-CMs cultured with activated hPBMCs at 72 h. ns = not significant, Dunnett's multiple comparisons ANOVA for (A). One-way ANOVA for (B). Experiments repeated >10 times (A, C), 3 times (B).
Impact of tissue culture myocarditis on sarcomere morphology. Sarcomere analysis for hiPSC-CMs across a range of myocarditis conditions. ICC for cTnT (Red) AND DAPI (Blue) to assess the impact on sarcomere morphology induced by indicated ratios of hPBMCs and the concentration of αCD3/CD28 mAbs. A. CM (−) control. B. PBMC + Checkpoint. C. 3:1 15 ng Isotype. D. 3:1 15 ng Checkpoint. E. 3:1 30 ng Isotype. F. 3:1 30 ng Checkpoint. G. 4:1 15 ng Isotype. H. 4:1 15 ng Checkpoint. I. 4:1 30 ng Isotype. J. 4:1 30 ng Checkpoint. K. 5:1 15 ng Isotype. L. 5:1 15 ng Checkpoint. Ratio indicates PBMC numbers relative to hiPSC-CM numbers. Amount of αCD3/CD28 mAbs indicated by “ng”. ‘Isotype’ refers to isotype control mAbs, while ‘Checkpoint’ refers to ICI biosimilars. Experiments performed >10 times.
hPBMC FACS sorting and cytokine evaluation. A) Flow cytometric staining for PE stained hPBMC populations pre and post-FACS sorting. B) Table listing the percentage of CD8+ T lymphocytes among bulk hPBMCs for eight unique healthy donors. C) hiPSC-CM survival following myocarditis modeling with hPBMCs obtained from eight different donors. n = 8 technical replicates per condition. D) Simple linear regression and Pearson correlation analysis to evaluate the correlation between the proportion of a donors' CD8+ T cells relative to total hPBMC numbers and the survival of hiPSC-CMs following myocarditis in vitro. n = 8 technical replicates per condition. Experiments performed 4 (A), 1 (B, C, D) times.
Myocarditis-produced cytokines do not cause hiPSC-CM arrhythmias in vitro. A) Multiplex ELISA measuring 13 classical inflammatory cytokines and chemokines for hiPSC-CMs, quiescent hPBMCs cultured with hiPSC-CMs, and activated hPBMCs cultured with hiPSC-CMs after 72 h. Technical replicates (n = 3). B) hiPSC-CM beat period after treatment with conditioned medium containing inflammatory cytokines. (n = 8). C) hiPSC-CM beat period irregularity following treatment with conditioned medium (n = 8). ***P < .001 by Welch's unpaired t-test for (C) and (D). Experiments performed 2 (A, B, C) times.
CRediT authorship contribution statement
Garrett Jensen: Writing – review & editing, Writing – original draft, Visualization, Validation, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Xinjie Wang: Methodology, Investigation, Conceptualization. Jacob Kuempel: Investigation. Zhishi Chen: Resources, Methodology, Investigation, Conceptualization. Wei Yu: Resources, Methodology, Investigation, Conceptualization. Nicolas Palaskas: Supervision, Methodology, Investigation, Conceptualization. Mary Sobieski: Visualization, Resources. Nghi Nguyen: Visualization, Resources. Reid T. Powell: Visualization, Software, Methodology. Clifford Stephan: Visualization, Software, Resources. Weijia Luo: Writing – original draft, Supervision, Resources, Project administration, Methodology, Investigation, Funding acquisition, Formal analysis, Data curation, Conceptualization. Jiang Chang: Writing – review & editing, Writing – original draft, Supervision, Software, Resources, Project administration, Methodology, Investigation, Funding acquisition, Formal analysis, Data curation, Conceptualization.
Declaration of Generative AI and AI-assisted technologies in the writing process
The authors did not use generative AI or AI-assisted technologies in the development of this manuscript.
Declaration of competing interest
Authors report no competing interests to disclose.
Acknowledgements
National Institutes of Health grant NHLBI R01HL141215.
National Institutes of Health grant NHLBI R01HL150124.
National Institutes of Health grant NHLBI R01HL148133.
American Heart Association grant 23TPA1142716.
National Institutes of Health grant NHLBI R21HL157708.
Department of Defense Peer Reviewed Medical Research Program grant (PRMRP) Discovery AwardPR192609.
TAMU Alkek Early Career Investigator Fellowship Award (WL).
References
- 1.Ammirati E., Moslehi J.J. Diagnosis and treatment of acute myocarditis: a review. JAMA. 2023 Apr 4;329(13):1098–1113. doi: 10.1001/jama.2023.3371. [DOI] [PubMed] [Google Scholar]
- 2.Ghanizada M., Kristensen S.L., Bundgaard H., Rossing K., Sigvardt F., Madelaire C., et al. Long-term prognosis following hospitalization for acute myocarditis - a matched nationwide cohort study. Scand Cardiovasc J. 2021 Oct 1;55(5):264–269. doi: 10.1080/14017431.2021.1900596. [DOI] [PubMed] [Google Scholar]
- 3.Grun S., Schumm J., Greulich S., Wagner A., Schneider S., Bruder O., et al. Long-term follow-up of biopsy-proven viral myocarditis. J Am Coll Cardiol. 2012 May;59(18):1604–1615. doi: 10.1016/j.jacc.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 4.Tschöpe C., Ammirati E., Bozkurt B., Caforio A.L.P., Cooper L.T., Felix S.B., et al. Myocarditis and inflammatory cardiomyopathy: current evidence and future directions. Nat Rev Cardiol. 2021 Mar;18(3):169–193. doi: 10.1038/s41569-020-00435-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haslam A., Prasad V. Estimation of the percentage of US patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA Netw Open. 2019 May 3;2(5) doi: 10.1001/jamanetworkopen.2019.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang D.Y., Salem J.E., Cohen J.V., Chandra S., Menzer C., Ye F., et al. Fatal toxic effects associated with immune checkpoint inhibitors: a systematic review and meta-analysis. JAMA Oncol. 2018 01;4(12):1721–1728. doi: 10.1001/jamaoncol.2018.3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palaskas N., Lopez-Mattei J., Durand J.B., Iliescu C., Deswal A. Immune checkpoint inhibitor myocarditis: pathophysiological characteristics, diagnosis, and treatment. JAHA [Internet] 2020 Jan 21;9(2) doi: 10.1161/JAHA.119.013757. https://www.ahajournals.org/doi/10.1161/JAHA.119.013757 [cited 2021 Jan 8]. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yousif L.I., Screever E.M., Versluis D., Aboumsallem J.P., Nierkens S., Manintveld O.C., et al. Risk factors for immune checkpoint inhibitor-mediated cardiovascular toxicities. Curr Oncol Rep. 2023 Jul;25(7):753–763. doi: 10.1007/s11912-023-01414-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weaver J.M., Dodd K., Knight T., Chaudhri M., Khera R., Lilleker J.B., et al. Improved outcomes with early immunosuppression in patients with immune-checkpoint inhibitor induced myasthenia gravis, myocarditis and myositis: a case series. Support Care Cancer. 2023 Aug 12;31(9):518. doi: 10.1007/s00520-023-07987-x. [DOI] [PubMed] [Google Scholar]
- 10.Moslehi J., Salem J.E. Immune checkpoint inhibitor myocarditis treatment strategies and future directions. JACC CardioOncol. 2022 Dec;4(5):704–707. doi: 10.1016/j.jaccao.2022.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cautela J., Zeriouh S., Gaubert M., Bonello L., Laine M., Peyrol M., et al. Intensified immunosuppressive therapy in patients with immune checkpoint inhibitor-induced myocarditis. J Immunother Cancer. 2020 Dec;8(2) doi: 10.1136/jitc-2020-001887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang L., Zlotoff D.A., Awadalla M., Mahmood S.S., Nohria A., Hassan M.Z.O., et al. Major adverse cardiovascular events and the timing and dose of corticosteroids in immune checkpoint inhibitor-associated myocarditis. Circulation. 2020 Jun 16;141(24):2031–2034. doi: 10.1161/CIRCULATIONAHA.119.044703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lam C.K., Wu J.C. Clinical trial in a dish: using patient-derived induced pluripotent stem cells to identify risks of drug-induced cardiotoxicity. ATVB. 2021 Mar;41(3):1019–1031. doi: 10.1161/ATVBAHA.120.314695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wei S.C., Meijers W.C., Axelrod M.L., Anang N.A.A.S., Screever E.M., Wescott E.C., et al. A genetic mouse model recapitulates immune checkpoint inhibitor-associated myocarditis and supports a mechanism-based therapeutic intervention. Cancer Discov. 2021;11(3):614–625. doi: 10.1158/2159-8290.CD-20-0856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Won T., Kalinoski H.M., Wood M.K., Hughes D.M., Jaime C.M., Delgado P., et al. Cardiac myosin-specific autoimmune T cells contribute to immune-checkpoint-inhibitor-associated myocarditis. Cell Rep. 2022 Nov;41(6) doi: 10.1016/j.celrep.2022.111611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laco F., Woo T.L., Zhong Q., Szmyd R., Ting S., Khan F.J., et al. Unraveling the inconsistencies of cardiac differentiation efficiency induced by the GSK3β inhibitor CHIR99021 in human pluripotent stem cells. Stem Cell Reports. 2018 Jun;10(6):1851–1866. doi: 10.1016/j.stemcr.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maas R.G.C., Lee S., Harakalova M., Snijders Blok C.J.B., Goodyer W.R., Hjortnaes J., et al. Massive expansion and cryopreservation of functional human induced pluripotent stem cell-derived cardiomyocytes. STAR Protocols. 2021 Mar;2(1) doi: 10.1016/j.xpro.2021.100334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buikema J.W., Lee S., Goodyer W.R., Maas R.G., Chirikian O., Li G., et al. Wnt activation and reduced cell-cell contact synergistically induce massive expansion of functional human iPSC-derived cardiomyocytes. Cell Stem Cell. 2020 Jul 2;27(1):50–63.e5. doi: 10.1016/j.stem.2020.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thomas D., Cunningham N.J., Shenoy S., Wu J.C. Human-induced pluripotent stem cells in cardiovascular research: current approaches in cardiac differentiation, maturation strategies, and scalable production. Cardiovasc Res. 2022 Jan 7;118(1):20–36. doi: 10.1093/cvr/cvab115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brostjan C., Oehler R. The role of neutrophil death in chronic inflammation and cancer. Cell Death Dis. 2020 Apr 22;6(1):26. doi: 10.1038/s41420-020-0255-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baboo J., Kilbride P., Delahaye M., Milne S., Fonseca F., Blanco M., et al. The impact of varying cooling and thawing rates on the quality of cryopreserved human peripheral blood T cells. Sci Rep. 2019 Mar 4;9(1):3417. doi: 10.1038/s41598-019-39957-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.NIH Grants Policy Statement | grants.nih.gov [Internet]. [cited 2024 Apr 1]. Available from: https://grants.nih.gov/policy/nihgps/index.htm.
- 23.Zhou B., Shi X., Tang X., Zhao Q., Wang L., Yao F., et al. Functional isolation, culture and cryopreservation of adult human primary cardiomyocytes. Sig Transduct Target Ther. 2022 Jul 27;7(1):254. doi: 10.1038/s41392-022-01044-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karakikes I., Ameen M., Termglinchan V., Wu J.C. Human induced pluripotent stem cell–derived cardiomyocytes: insights into molecular, cellular, and functional phenotypes. Circ Res. 2015 Jun 19;117(1):80–88. doi: 10.1161/CIRCRESAHA.117.305365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharma A., McKeithan W.L., Serrano R., Kitani T., Burridge P.W., Del Álamo J.C., et al. Use of human induced pluripotent stem cell-derived cardiomyocytes to assess drug cardiotoxicity. Nat Protoc. 2018 Dec;13(12):3018–3041. doi: 10.1038/s41596-018-0076-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burridge P.W., Diecke S., Matsa E., Sharma A., Wu H., Wu J.C. Modeling cardiovascular diseases with patient-specific human pluripotent stem cell-derived cardiomyocytes. Methods Mol Biol. 2016;1353:119–130. doi: 10.1007/7651_2015_196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lyra-Leite D.M., Gutiérrez-Gutiérrez Ó., Wang M., Zhou Y., Cyganek L., Burridge P.W. A review of protocols for human iPSC culture, cardiac differentiation, subtype-specification, maturation, and direct reprogramming. STAR Protocols. 2022 Sep;3(3) doi: 10.1016/j.xpro.2022.101560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zwi L., Caspi O., Arbel G., Huber I., Gepstein A., Park I.H., et al. Cardiomyocyte differentiation of human induced pluripotent stem cells. Circulation. 2009 Oct 13;120(15):1513–1523. doi: 10.1161/CIRCULATIONAHA.109.868885. [DOI] [PubMed] [Google Scholar]
- 29.Batalov I., Feinberg A.W. Differentiation of cardiomyocytes from human pluripotent stem cells using monolayer culture. Biomark Insights. 2015;10(Suppl. 1):71–76. doi: 10.4137/BMI.S20050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hsueh Y.C., Pratt R.E., Dzau V.J., Hodgkinson C.P. Novel method of differentiating human induced pluripotent stem cells to mature cardiomyocytes via Sfrp2. Sci Rep. 2023 Mar 9;13(1):3920. doi: 10.1038/s41598-023-31144-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bonte S., De Munter S., Billiet L., Goetgeluk G., Ingels J., Jansen H., et al. In vitro OP9-DL1 co-culture and subsequent maturation in the presence of IL-21 generates tumor antigen-specific T cells with a favorable less-differentiated phenotype and enhanced functionality. OncoImmunology. 2021 Jan;10(1) doi: 10.1080/2162402X.2021.1954800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murphy K.M. Garland Science; 2012. Janeway’s immunobiology. [Google Scholar]
- 33.Kumar V., Abbas A.K., Aster J.C., editors. Robbins and Cotran pathologic basis of disease. 9th ed. Elsevier/Saunders; Philadelphia, PA: 2015. 1391 p. [Google Scholar]
- 34.Kang P.M., Haunstetter A., Aoki H., Usheva A., Izumo S. Morphological and molecular characterization of adult cardiomyocyte apoptosis during hypoxia and reoxygenation. Circ Res. 2000 Jul 21;87(2):118–125. doi: 10.1161/01.res.87.2.118. [DOI] [PubMed] [Google Scholar]
- 35.Balko J., Axelrod M., Meijers W., Screever E., Carroll M.G., Sun X., et al. Cytotoxic T cells specific for alpha-myosin drive immunotherapy related myocarditis [Internet] 2022. https://www.researchsquare.com/article/rs-1315661/v1 In Review. Feb [cited 2022 Aug 2]. Available from: [DOI] [PMC free article] [PubMed]
- 36.Punt J., Stranford S.A., Jones P.P., Owen J.A. 8th ed. Macmillan Learning; New York: 2019. Kuby immunology; p. 1. [Google Scholar]
- 37.Power J.R., Alexandre J., Choudhary A., Ozbay B., Hayek S.S., Asnani A., et al. Association of early electrical changes with cardiovascular outcomes in immune checkpoint inhibitor myocarditis. Arch Cardiovasc Dis. 2022 May;115(5):315–330. doi: 10.1016/j.acvd.2022.03.003. [DOI] [PubMed] [Google Scholar]
- 38.Song W., Zheng Y., Dong M., Zhong L., Bazoukis G., Perone F., et al. Electrocardiographic features of immune checkpoint inhibitor-associated myocarditis. Curr Probl Cardiol. 2023 Feb;48(2) doi: 10.1016/j.cpcardiol.2022.101478. [DOI] [PubMed] [Google Scholar]
- 39.Zlotoff D.A., Hassan M.Z.O., Zafar A., Alvi R.M., Awadalla M., Mahmood S.S., et al. Electrocardiographic features of immune checkpoint inhibitor associated myocarditis. J Immunother Cancer. 2021 Mar;9(3) doi: 10.1136/jitc-2020-002007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xie X., Wang L., Li Y., Xu Y., Wu J., Lin X., et al. Multi-organ immune-related adverse event is a risk factor of immune checkpoint inhibitor-associated myocarditis in cancer patients: a multi-center study. Front Immunol. 2022 Jul;18(13) doi: 10.3389/fimmu.2022.879900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsuruda T., Yoshikawa N., Kai M., Yamaguchi M., Toida R., Kodama T., et al. The cytokine expression in patients with cardiac complication after immune checkpoint inhibitor therapy. Intern Med. 2021 Feb 1;60(3):423–429. doi: 10.2169/internalmedicine.5317-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ammirati E., Bizzi E., Veronese G., Groh M., Van De Heyning C.M., Lehtonen J., et al. Immunomodulating therapies in acute myocarditis and recurrent/acute pericarditis. Front Med. 2022 Mar;7(9) doi: 10.3389/fmed.2022.838564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thuny F., Alexandre J., Salem J.E., Mirabel M., Dolladille C., Cohen-Solal A., et al. Management of immune checkpoint inhibitor–induced myocarditis: the French Working Group’s plea for a pragmatic approach. JACC: CardioOncology. 2021 Mar 1;3(1):157–161. doi: 10.1016/j.jaccao.2020.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma P., Liu J., Qin J., Lai L., Heo G.S., Luehmann H., et al. Expansion of pathogenic cardiac macrophages in immune checkpoint inhibitor myocarditis. Circulation. 2024;149(1):48–66. doi: 10.1161/CIRCULATIONAHA.122.062551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu H., Galdos F.X., Lee D., Waliany S., Huang Y.V., Ryan J., et al. Identification of pathogenic immune cell subsets associated with checkpoint inhibitor–induced myocarditis. Circulation. 2022 Jul 26;146(4):316–335. doi: 10.1161/CIRCULATIONAHA.121.056730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Błyszczuk P. Myocarditis in humans and in experimental animal models. Front Cardiovasc Med. 2019;6:64. doi: 10.3389/fcvm.2019.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Axelrod M.L., Meijers W.C., Screever E.M., Qin J., Carroll M.G., Sun X., et al. T cells specific for α-myosin drive immunotherapy-related myocarditis. Nature. 2022 Nov 24;611(7937):818–826. doi: 10.1038/s41586-022-05432-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lv H., Lipes M.A. Role of impaired central tolerance to α-myosin in inflammatory heart disease. Trends Cardiovasc Med. 2012 Jul;22(5):113–117. doi: 10.1016/j.tcm.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lv H., Havari E., Pinto S., Gottumukkala R.V.S.R.K., Cornivelli L., Raddassi K., et al. Impaired thymic tolerance to α-myosin directs autoimmunity to the heart in mice and humans. J Clin Invest. 2011 Apr 1;121(4):1561–1573. doi: 10.1172/JCI44583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pummerer C.L., Luze K., Grässl G., Bachmaier K., Offner F., Burrell S.K., et al. Identification of cardiac myosin peptides capable of inducing autoimmune myocarditis in BALB/c mice. J Clin Invest. 1996 May 1;97(9):2057–2062. doi: 10.1172/JCI118642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang H., Yang Y., Kiskin F.N., Shen M., Zhang J.Z. Recent advances in regulating the proliferation or maturation of human-induced pluripotent stem cell-derived cardiomyocytes. Stem Cell Res Ther. 2023 Aug 30;14(1):228. doi: 10.1186/s13287-023-03470-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baklaushev V.P., Kilpeläinen A., Petkov S., Abakumov M.A., Grinenko N.F., Yusubalieva G.M., et al. Luciferase expression allows bioluminescence imaging but imposes limitations on the Orthotopic mouse (4T1) model of breast cancer. Sci Rep. 2017 Aug 10;7(1):7715. doi: 10.1038/s41598-017-07851-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baumgaertner P., Speiser D., Romero P., Rufer N., Hebeisen M. Chromium-51 (51Cr) release assay to assess human T cells for functional avidity and tumor cell recognition. BIO-PROTOCOL [Internet] 2016;6(16) https://bio-protocol.org/e1906 [cited 2023 Dec 8]. Available from: [Google Scholar]
- 54.Fracchia K.M., Pai C.Y., Walsh C.M. Modulation of T cell metabolism and function through calcium signaling. Front Immunol. 2013 Oct;11(4):324. doi: 10.3389/fimmu.2013.00324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trebak M., Kinet J.P. Calcium signalling in T cells. Nat Rev Immunol. 2019 Mar;19(3):154–169. doi: 10.1038/s41577-018-0110-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mak T.W., Saunders M.E. The immune response [internet] Elsevier; 2006. T cell activation; pp. 373–401.https://linkinghub.elsevier.com/retrieve/pii/B9780120884513500168 [cited 2023 Dec 1]. Available from: [Google Scholar]
- 57.Dijkstra K.K., Cattaneo C.M., Weeber F., Chalabi M., Van De Haar J., Fanchi L.F., et al. Generation of tumor-reactive T cells by co-culture of peripheral blood lymphocytes and tumor organoids. Cell. 2018 Sep;174(6):1586–1598.e12. doi: 10.1016/j.cell.2018.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Olivo Pimentel V., Yaromina A., Marcus D., Dubois L.J., Lambin P. A novel co-culture assay to assess anti-tumor CD8+ T cell cytotoxicity via luminescence and multicolor flow cytometry. J Immunol Methods. 2020 Dec;487 doi: 10.1016/j.jim.2020.112899. [DOI] [PubMed] [Google Scholar]
- 59.Zeng Z., Chew H.Y., Cruz J.G., Leggatt G.R., Wells J.W. Investigating T cell immunity in cancer: achievements and prospects. IJMS. 2021 Mar 12;22(6):2907. doi: 10.3390/ijms22062907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Raskov H., Orhan A., Christensen J.P., Gögenur I. Cytotoxic CD8+ T cells in cancer and cancer immunotherapy. Br J Cancer. 2021 Jan 19;124(2):359–367. doi: 10.1038/s41416-020-01048-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Man S.M., Jenkins B.J. Context-dependent functions of pattern recognition receptors in cancer. Nat Rev Cancer. 2022 Jul;22(7):397–413. doi: 10.1038/s41568-022-00462-5. [DOI] [PubMed] [Google Scholar]
- 62.Wei S.C., Duffy C.R., Allison J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018 Sep 1;8(9):1069–1086. doi: 10.1158/2159-8290.CD-18-0367. [DOI] [PubMed] [Google Scholar]
- 63.Marino J., Paster J., Benichou G. Allorecognition by T lymphocytes and allograft rejection. Front Immunol [Internet] 2016 Dec 14;7 doi: 10.3389/fimmu.2016.00582. http://journal.frontiersin.org/article/10.3389/fimmu.2016.00582/full [cited 2023 Dec 11]. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mestas J., Hughes C.C.W. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004 Mar 1;172(5):2731–2738. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 65.Of men, not mice. Nat. Med. 2013;19(4):379. doi: 10.1038/nm.3163. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video demonstrating spontaneous, non-paced beating of successfully differentiated hiPSC-CMs.
Validation of hiPSC-CM differentiation and hPBMC isolation. Brightfield images demonstrating visual iPSCs (A) and differentiated hiPSC-CMs (B). C) Representative SSC x FSC plot for unstained flow cytometric evaluation of hPBMCs. D) Representative Phalloidin (left), DAPI (middle) and αCD45 (right) stained images used for visual viability analysis pipeline. Graphical overview of hiPSC-CM viability analysis pipeline. Experiments repeated 10 times for (A, B, C, D).
hiPSC-CM Purity. A) Western blot for lineage specific markers for iPSCs (NANOG, OCT4, SOX2), day 14 post-differentiation hiPSC-CMs (cTnT, α-actinin), and iPSC-fibroblasts (vimentin) and GAPDH control. Lysate sample source indicated below Western blot. B) Flow cytometry staining for cTnT—PE on day 14 post-differentiated hiPSC-CMs. Unstained control (orange), isotype stained control (blue), stained hiPSC-CMs (red). C) Immunocytochemistry staining of day 24 post-differentiation hiPSC-CMs for cTnT (red), α-actinin (green), counterstained with DAPI (blue). Merged image in the left panel.
Myocarditis model setup. A) hiPSC-CM survival for indicated conditions including isotype mAbs, ICI biosimilars, αCD3/CD28, and quiescent hPBMCs at 72 h. B) hPBMCs without T cell activators do not induce hiPSC-CM death at 72 h following co-culture. C) Confocal images of phalloidin-stained hiPSC-CMs. Sarcomere structure for control hiPSC-CMs and hiPSC-CMs cultured with activated hPBMCs at 72 h. ns = not significant, Dunnett's multiple comparisons ANOVA for (A). One-way ANOVA for (B). Experiments repeated >10 times (A, C), 3 times (B).
Impact of tissue culture myocarditis on sarcomere morphology. Sarcomere analysis for hiPSC-CMs across a range of myocarditis conditions. ICC for cTnT (Red) AND DAPI (Blue) to assess the impact on sarcomere morphology induced by indicated ratios of hPBMCs and the concentration of αCD3/CD28 mAbs. A. CM (−) control. B. PBMC + Checkpoint. C. 3:1 15 ng Isotype. D. 3:1 15 ng Checkpoint. E. 3:1 30 ng Isotype. F. 3:1 30 ng Checkpoint. G. 4:1 15 ng Isotype. H. 4:1 15 ng Checkpoint. I. 4:1 30 ng Isotype. J. 4:1 30 ng Checkpoint. K. 5:1 15 ng Isotype. L. 5:1 15 ng Checkpoint. Ratio indicates PBMC numbers relative to hiPSC-CM numbers. Amount of αCD3/CD28 mAbs indicated by “ng”. ‘Isotype’ refers to isotype control mAbs, while ‘Checkpoint’ refers to ICI biosimilars. Experiments performed >10 times.
hPBMC FACS sorting and cytokine evaluation. A) Flow cytometric staining for PE stained hPBMC populations pre and post-FACS sorting. B) Table listing the percentage of CD8+ T lymphocytes among bulk hPBMCs for eight unique healthy donors. C) hiPSC-CM survival following myocarditis modeling with hPBMCs obtained from eight different donors. n = 8 technical replicates per condition. D) Simple linear regression and Pearson correlation analysis to evaluate the correlation between the proportion of a donors' CD8+ T cells relative to total hPBMC numbers and the survival of hiPSC-CMs following myocarditis in vitro. n = 8 technical replicates per condition. Experiments performed 4 (A), 1 (B, C, D) times.
Myocarditis-produced cytokines do not cause hiPSC-CM arrhythmias in vitro. A) Multiplex ELISA measuring 13 classical inflammatory cytokines and chemokines for hiPSC-CMs, quiescent hPBMCs cultured with hiPSC-CMs, and activated hPBMCs cultured with hiPSC-CMs after 72 h. Technical replicates (n = 3). B) hiPSC-CM beat period after treatment with conditioned medium containing inflammatory cytokines. (n = 8). C) hiPSC-CM beat period irregularity following treatment with conditioned medium (n = 8). ***P < .001 by Welch's unpaired t-test for (C) and (D). Experiments performed 2 (A, B, C) times.