

Abstract
The rise of antimicrobial resistance represents a significant global health threat, driven by the diminishing efficacy of existing antibiotics, a lack of novel antibacterials entering the market, and an over- or misuse of existing antibiotics, which accelerates the evolution of resistant bacterial strains. This review focuses on innovative therapies by highlighting 19 novel antibacterials in clinical development as of June 2024. These selected compounds are characterized by new chemical scaffolds, novel molecular targets, and/or unique mechanisms of action, which render their potential to break antimicrobial resistance particularly high. A detailed analysis of the scientific foundations behind each of these compounds is provided, including their pharmacodynamic profiles, current development state, and potential for overcoming existing limitations in antibiotic therapy. By presenting this subset of chemically novel antibacterials, the review highlights the ability to innovate in antibiotic drug development to counteract bacterial resistance and improve treatment outcomes.
Key Points
| The antibacterial pipeline is dominated by improved derivatives of known antibiotic classes, but it also contains novel chemical scaffolds addressing novel mechanisms of action, which are the focus of this review. |
| Antibiotics with novel chemical scaffolds address new targets for outer membrane biosynthesis, protein secretion, membrane integrity, or virulence factors. In addition, existing targets such as gyrase, RNA polymerase or β-lactamases are addressed with new chemistry. |
| The antibacterial pipeline shows the ability to innovate, but it is too small overall. |
Introduction
The discovery and widespread utilization of antibiotics—compounds that eradicate bacteria or inhibit their growth—heralded the advent of modern medicine. Antibiotics are indispensable not only for treating existing infections but also for enabling modern medical procedures. For example, they play a critical role in managing neutropenia [1], frequently induced by chemotherapy [2], and are essential for infection prophylaxis following invasive surgeries like Caesarean section [3] as well as during immunosuppressive therapy after organ or bone marrow transplantation [4].
Increasing antimicrobial resistance (AMR), arising from the survival of pathogenic microorganisms in the presence of antimicrobials, represents one of the most significant global health challenges of the 21st century. In 2021 alone, despite a notable decrease in non-COVID-related infections during the coronavirus pandemic, there were an estimated 1.14 million global deaths directly attributable to bacterial drug resistance, and this burden is estimated to increase to 1.91 million deaths by 2050 [5]. The 2022 World Health Organization (WHO) “Global Antimicrobial Resistance and Use Surveillance System (GLASS)” report disclosed alarming levels of resistance worldwide that substantiate the need for concerted action [6]. The rise of AMR is further exacerbated by the misuse of antibiotics in agriculture (e.g., as feed additives for infection prevention or for growth promotion), and aqua- or crop culture [7] inducing an increasing prevalence of resistant pathogens in these environments. Beyond its direct health implications, AMR poses a severe threat to the global economy with wide-ranging effects on international trade and overall productivity [8].
To effectively address AMR, an integrated approach has been proclaimed that encompasses, among other elements, improved surveillance to monitor resistance, the development of new vaccines, the implementation of rapid diagnostics to reduce unnecessary antibiotic use, and continued research and development efforts for novel antimicrobials [9]. To streamline these efforts, the WHO has categorized bacterial pathogens into critical, high and medium priority for the year 2024 [10]. The group of critical pathogens, including Carbapenem-resistant Acinetobacter baumannii (CRAB) as well as carbapenem-resistant (CRE) and third-generation cephalosporin-resistant (3GCRE) Enterobacterales exemplify the urgent need for novel therapeutic strategies as they exhibit resistance to last-resort antibiotics, leaving limited treatment options. Investment in the development of innovative and effective antibacterial agents is crucial to outpace the rapid evolution of resistant pathogens [10]. However, the pharmaceutical industry has mostly abandoned antibiotic development, because its economic profitability is unclear or non-existent given the current market environment [9].
To fill the void, several funding initiatives have been established, including Combating Antibiotic-Resistant Bacteria Biopharmaceutical Accelerator (CARB-X) in Boston [11], the INCubator for Antibacterial Therapies in Europe (INCATE) [12], the Replenishing and Enabling the Pipeline for Anti-Infective Resistance (REPAIR) Impact Fund established by the Novo Holdings in Denmark [13], the AMR Action Fund [14, 15] and the Global Antibiotic Research & Development Partnership (GARDP). Despite all these efforts, the WHO concludes in its recent reports that antibacterial agents in the clinical pipeline combined with those approved between 2018–2023 are still insufficient to combat the threat of the emergence and spread of drug-resistant infections [8, 10].
This review aims to shed light on small-molecule antibacterial agents currently in clinical development to stimulate future research. A tabular overview of the complete pipeline (Table 1) was compiled using existing reviews [16, 17], the Global AMR R&D Hub's Dynamic Dashboard funded by the German Federal Ministry of Health (cut-off date July 2022) [18], and the WHO's report "2023 Antibacterial agents in clinical and preclinical development: an overview and analysis" (cut-off date December 2023) [8], followed by a supplementary search for Phase I-III studies started, first posted, updated, or completed between January 2022 and June 10th 2024 on ClinicalTrials.gov with the keywords “infection” or “bacterial infection”. Rather than commenting on all pipeline entries, we focus this review on particularly innovative new antibacterials represented by compounds with novel chemical scaffolds. The presence of a novel scaffold is not a sufficient criterion for efficacy against a pathogen that has developed resistance to approved antibiotics or even for achieving clinically relevant innovation. However, we hypothesize that the new chemical classes have more likelihood of distinct pre-existing therapeutic potential for overcoming resistance than the new derivatives of existing chemical classes. This is because the latter, despite all newly acquired advantages, are unlikely to completely overcome resistance mechanisms or fundamentally alter the properties of the drug class. We have selected 19 antibacterials with new chemical scaffolds, a new target, and/or a novel mechanism of action (MoA) and discuss their scientific background and current development state. The presented examples do not cover all novel concepts and have been selected based on the authors' discretion. Compounds were excluded from this analysis that exclusively targeted Clostridium difficile as well as Mycobacteria including Mycobacterium tuberculosis, or non-traditional approaches such as antibodies, other proteins, live biotherapeutic products (LBPs) such as bacteriophages, microbiome-modulating or immune-modulating agents.
Table 1.
Global pipeline of small-molecule antibiotics with reported clinical trials
| Name | Cmpd. class | Developer/sponsor | Mechanism (target) | Indication: trial number (phase) | References |
|---|---|---|---|---|---|
| Phase III | |||||
| Benapenem | Carbapenem | Sihuan Pharmaceutical Holdings Group Ltd. | Cell wall (PBP) | cUTI or AP: NCT04505683 (II/III) | [234] |
| Epetraborole (GSK2251052, AN3365, BRII-658) | Oxaborole | AN2 Therapeutics, Brii Biosciences | Protein synthesis (leucyl-tRNA synthetase, LeuRS) | MAC Lung Disease: NCT05327803 (II/III) | [235] |
| Funobactam (XNW4107) + imipenem + cilastin | DBO BLI + carbapenem + degradation inhibitor | Evopoint Biosciences | Cell wall (PBP) |
cUTI: NCT05204368 (III) HABP/VABP: NCT05204563 (III) |
[236] |
| Gepotidacin (GSK2140944) | Triazaacenaphthylene | GSK | DNA organization (DNA gyrase, GyrA) |
uUTI: NCT04020341 (III), NCT05630833 (III), NCT04010539 (III), NCT04187144 (III) Gonorrhea: NCT04010539 (III) |
[34] |
| Nacubactam (OP0595, FPI-1459, RG6080, RO7079901) + cefepime or aztreonam (OP0595) | DBO BLI + cephalosporin or monobactam | Meiji Seika Pharma Co. | Cell wall (PBP) | cUTI, AP, HABP, VABP, cIAI: NCT05905055 (III), NCT05887908 (III) | [237] |
| Nafithromycin (WCK 4873) | Macrolide (Ketolide) | Wockhardt Limited | Protein synthesis (50S ribosomal subunit) | CABP: CTRI/2019/11/021964 (III) | [238] |
| Reltecimod (AB103) | Peptide mimetic | Atox Bio | Immunomodulating (T-lymphocyte receptor mimetic) | Necrotizing Soft-Tissue Infections: NCT02469857 (III) | [239] |
| Rifasutenizol (TNP-2198) | Rifamycin-nitroimidazole hybrid | TenNor Therapeutics | DNA-dependent RNA synthesis (RNA polymerase) + Formation of reactive radicals | H. pylori infections: NCT05857163 (III) | [240] |
| Solithromycin (T-4288) | Macrolide (Ketolide) | FUJIFILM Toyama Chemical Co. | Protein synthesis (50S ribosomal subunit) |
CABP: NCT01968733 (III), NCT01756339 (III), NCT02605122 (II/III) Gonorrhea: NCT02210325 (III) |
[241] |
| Sudapyridine (WX-081) | Diarylquinoline | Shanghai Jiatan Pharmatech Co. | ATP production (mycobacterial ATP synthase) | TB: NCT05824871 (III), NCT04608955 (II) | [242] |
|
Sulopenem (CP-65,207) Sulopenem etzadroxil (PF-03709270) (+ probenecid) |
Penem | Iterum Therapeutics | Cell wall (PBP) |
cIAI: NCT03358576 (III) cUTI: NCT03357614 (III) uUTI: NCT03354598 (III), NCT05584657 (III) |
[243] |
| Taniborbactam (VNRX-5133) + cefepime | Bicyclic boronate BLI + cephalosporin | VenatoRx Pharmaceuticals | Cell wall (PBP) |
cUTI: NCT03840148 (III) VABP, HABP: NCT06168734 (III) |
[244] |
| Zoliflodacin (ETX0914, AZD0914) | Spiropyrimidinetrione | Innoviva; Global Antibiotics Research and Development Partnership | DNA organization (DNA gyrase, GyrB) | Gonorrhea: NCT03959527 (III) | [245] |
| Phase II | |||||
| ACG-701 (ARV-1801, sodium fusidate, fusidic acid) + ceftazidime or meropenem | Steroid antibiotic | Aceragen | Protein synthesis (elongation factor G) | Melioidosis: NCT05105035 (II) | [246] |
| Afabicin (Debio 1450, AFN-1270) | Benzofuran naphthyridine | Debiopharm | Cell wall / fatty acid biosynthesis (Enoyl-acyl carrier protein reductase, FabI) |
S. aureus Bone or Joint Infection: NCT03723551 (II) Staphylococcal ABSSSI: NCT02426918 (II) |
[247] |
| Alpibectir (BVL-GSK098, GSK3729098) + ethionamide or isoniazid | Amido piperidine spiroisoxazoline | BioVersys AG, GSK | Cell wall / fatty acid biosynthesis (transcriptional regulators of Mtb; NAD+) | Pulmonary TB: NCT05473195 (II) | [248] |
| Brilacidin (PMX-30063) | Aryl amide foldamer | Innovation Pharmaceuticals | Cell wall (Host defense protein mimetic) |
ABSSSI: NCT02052388 (II) ABSSSI: NCT01211470 (II) |
[135] |
| BTZ-043 (+ bedaquiline + delamanid) or (+ GSK3036656 + bedaquiline) or (+ delamanid + bedaquiline) | Benzothiazinone | Ludwig-Maximilians-Universität in Munich | Cell wall (DprE1) | TB: NCT05926466 (II), NCT06114628 (II), NCT05382312 (II), NCT05807399 (II), NCT04044001 (I/II) | [249] |
| CRS3123 (REP3123) | Diaryldiamine | Crestone | Protein synthesis (Methionyl-tRNA synthetase) | CDI: NCT04781387 (II) | [250] |
| Cannabidiol (BTX 1801, BTX 1503, BTX 1204) | Cannabinoid | Botanix Pharmaceuticals | Cell wall |
Nasal S. aureus decolonization: ACTRN12620000456954 (II) Acne: NCT03573518 (II) Atopic Dermatitis: NCT03824405 (II) |
[251] |
|
Delpazolid (RMX2001, LCB01-0371) + bedaquiline + delamanid + moxifloxacin; delpazolid + vancomycin |
Oxazolidinone | LigaChem Biosciences | Protein synthesis (50S ribosomal subunit) |
Pulmonary TB: NCT04550832 (II), NCT02836483 (II) MRSA bacteraemia: NCT05225558 (II) |
[252] |
| Dovramilast (CC-11050, AMR-634) + isoniazid + rifampicin + pyrazinamide + ethambutol | 3-Oxo-1H-isoindol-4-yl | Medicines Development for Global Health | Anti-inflammatory (PDE4) |
Leprosy: NCT03807362 (II) TB: NCT02968927 (II) |
[253] |
| Exeporfinium chloride (XF-73) | Porphyrin | Destiny Pharma | Cell wall | Post-Op nasal S. aureus decolonization: NCT03915470 (II) | [254] |
| Finafloxacin | Fluoroquinolone | MerLion Pharmaceuticals | DNA organization (DNA gyrase, GyrA) |
cUTI and AP: NCT01928433 (II) uUTI: NCT00722735 (II) H. pylori infections: NCT00723502 (II) |
[255] |
|
Fobrepodacin (SPR720, pVXc-486) SPR719 (active metabolite) |
Aminobenzimidazole | Spero Therapeutics | DNA organization (DNA gyrase, GyrB) | MAC Lung Disease: NCT05496374 (II), NCT04553406 (II) | [256] |
| Ftortiazinon (fluorothiazinone, CL-55) + cefepime | Thyazinone + cephalosporin | Gamaleya Research Institute of Epidemiology and Microbiology | Bacterial virulence (Bacterial type III secretion system, T3SS) + Cell wall (PBP) |
Nosocomial G-ve: NCT06135350 (II) cUTI: NCT03638830 (II) |
[194] |
| Gallium citrate (AR501) | Organic metal salt | Aridis Pharmaceuticals | Disruption of bacterial iron metabolism | Pseudomonal cystic fibrosis: NCT03669614 (I/II) | [257] |
| Gallium nitrate (Ganite) | Inorganic metal salt | University of Washington | Disruption of bacterial iron metabolism | Pseudomonal cystic fibrosis: NCT02354859 (II) | [258] |
| Ganfeborole (GSK3036656, GSK656, GSK070) | Oxaborole | GSK | Protein synthesis (leucyl-tRNA synthetase, LeuRS) |
TB: NCT05382312 (II), NCT03557281 (II), NCT06114628 (II) MAC Lung Disease: NCT05327803 (II/III) |
[259] |
| Ibezapolstat (ACX-362E) | Dichlorobenzyl guanine | Acurx Pharmaceuticals | DNA synthesis (DNA polymerase IIIC) | CDI: NCT04247542 (II) | [260] |
| JDB0131 besylate | Undisclosed | Beijing Chest Hospital | – | Pulmonary TB: NCT06224036 (II) | – |
| MGB-BP-3 | Distamycin | MGB Biopharma | DNA (Minor groove binding) |
CDAD: NCT03824795 (II) CDI: NCT02518607 (I) |
[261] |
| Niclosamide (ATx201) | Salicylanilide | UNION therapeutics | Bacterial growth (Proton motive force, PMF) |
Atopic Dermatitis: NCT04339985 (II), NCT03009734 (I/II), NCT03304470 Impetigo: NCT03429595 (II) |
[262] |
| Nilofabicin (CG-549, CG400549) | Pyridone | CrystalGenomics | Cell wall / fatty acid biosynthesis (Enoyl-acyl carrier protein reductase, FabI) | ABSSSI: NCT01593761 (II) | [263] |
| OMN6 | Antimicrobial cyclic peptide (40 amino acids) | Omnix Medical | Cell wall / Bacterial membrane | HABP, VABP: NCT06087536 (II), CS0379-200475 Omnix (I) | [132] |
| Peceleganan (PL-5, V681) | Cationic peptide | Jiangsu ProteLight Pharmaceutical & Biotechnology Co. | Cell wall / Bacterial membrane | Wound infections: ChiCTR2000033334 (II) | [114] |
| Pravibismane (MBN-101, BisEDT) | Bismuth thiol | Microbion Corporation | Cellular bioenergetics via membrane potential |
DFI: NCT05174806 (II) Orthopedic-implant infection: NCT02436876 (II) |
[264] |
| Pyrifazimine (TBI-166) | Riminophenazine | Institute of Materia Medica, Chinese Academy of Medical Sciences, Peking Union Medical College | Bacterial proliferation (DNA) | TB: NCT04670120 (II) | [265] |
| Quabodepistat (opc-167832) (+ delamanid + bedaquiline + sutezolid + pretomanid) | Carbostyril Derivative | Otsuka Pharmaceutical Development and Commercialization | Cell wall (DprE1) | Pulmonary TB: NCT05971602 (II), NCT05221502 (II), NCT03678688 (I/II) | [266] |
| Recce 327 (R327) | Acrolein polymer | Recce Pharmaceuticals | Cellular bioenergetics via membrane potential and/or ATP synthesis |
Burn wound infections: ACTRN12621000412831 (I/II) DFI: ACTRN12623000056695 (I/II) UTI: ACTRN12623000448640 (I/II) Serious bacterial infections: ACTRN12621001313820 (I) |
[267] |
| Rifabutin (BV100) | Rifamycin | BioVersys AG | DNA-dependent RNA synthesis (RNA polymerase) | VABP: NCT05685615 | [268] |
| Sanfetrinem cilexetil (GV-104326) | Trinem | GSK | Cell wall (PBP) | Pulmonary TB: NCT05388448 (II) | [269] |
| Sutezolid (PF-2341272, PNU-100480) (+ bedaquiline + delamanid + moxifloxacin) (+ bedaquiline + pretomanid) | Oxazolidinone | TB Alliance | Protein synthesis (50S ribosomal subunit) | TB: NCT05686356 (II/III), NCT01225640 (II), NCT03959566 (II) | [270] |
| TBA-7371 | Azaindole | TB Alliance | Cell wall (DprE1) | TB: NCT04176250 (II) | [271] |
| TBAJ-876 | Diarylquinoline | TB Alliance | ATP production (mycobacterial ATP synthase) | TB: NCT06058299 (II) | [272] |
| Telacebec (Q-203) | Imidazopyridine amide | Qurient Co., Infectex | Cellular energy production (Respiratory cytochrome bc1 complex) | Pulmonary TB: NCT03563599 (II) | [273] |
| TNP-2092 (CBR-2092) | Rifamycin-quinolizinone hybrid | TenNor Therapeutics | DNA-dependent RNA synthesis; DNA organization (RNA polymerase; DNA gyrase, GyrA; Topoisomerase IV, ParC) |
ABSSSI: NCT03964493 (II) PJI: NCT04294862 (I) |
[274] |
| Voxvoganan (LTX-109) | Antimicrobial peptide | Pharma Holdings AS | Cell wall / Bacterial membrane | Nasal S. aureus decolonization: NCT05889351 (II), NCT04767321 (I/II) | [110] |
| Phase I | |||||
| ALS-4 | Undisclosed | Aptorum Therapeutics | Bacterial virulence (4,4ʹ-diapophytoene desaturase, CrtN) | NCT05274802 (I) | [275] |
| ANT3310 + meropenem | DBO BLI + carbapenem | Antabio | Cell wall (PBP) | NCT05905913 (I) | [276] |
| Apramycin (EBL-1003) | Aminoglycoside | Juvabis | Protein synthesis (30S ribosomal subunit) | NCT04105205 (I), NCT05590728 (I) | [277] |
| Avibactam (NXL104) + Meropenem (combination: TQD3606) | DBO BLI + carbapenem | Chia Tai Tianqing Pharmaceutical Group Co. | Cell wall (PBP) | NCT05340530 (I) | [278] |
| Avibactam tomilopil (PF-07338233, ARX-1796, AV-006, AVP, AVI-ARX) + ceftibuten (PF-06264006, CTB) | DBO BLI + cephalosporin | Pfizer | Cell wall (PBP) |
cUTI: NCT05554237 (I) NCT03931876 (I) |
[160] |
| BWC0977 | Oxazolidinone containing NBTI | Bugworks | DNA organization (DNA gyrase, GyrA; Topoisomerase IV) | NCT05088421 (I) | [279] |
| EQ-778 | Undisclosed | Vedic Lifesciences | – | RTI: NCT05835375 | – |
| ETX0282 + cefpodoxime proxetil (combination: ETX0282CPDP) | DBO BLI + cephalosporin | Entasis Therapeutics | Cell wall (PBP) | NCT03491748 (I) | [280] |
| GSK2556286 (GSK286) | Uracil aryloxypiperidine | GSK | Bacterial growth (Adenylyl cyclase Rv1625c) | TB: NCT04472897 (I) | [281] |
| GSK3882347 | Mannose derivative | GSK, Fimbrion Therapeutics | Bacterial virulence (Type 1 fimbrin D-mannose specific adhesin, FimH) |
uUTI: NCT05138822 (I) NCT04488770 (I) |
[214] [216] |
| KSP-1007 + meropenem | Bicyclic boronate BLI + carbapenem | Sumitomo Dainippon Pharma | Cell wall (PBP) | NCT05226923 (I) | [186] |
| Ledaborbactam etzadroxil (VNRX-7145) + ceftibuten | Bicyclic boronate BLI + cephalosporin | VenatoRx Pharmaceuticals | Cell wall (PBP) | NCT05527834 (I), NCT05488678 (I), NCT04243863 (I), NCT04877379 (I) | [166] |
| Macozinone (PBTZ-169) | Benzothiazinone | Innovative Medicines for Tuberculosis Foundation, Nearmedic Plus | Cell wall (DprE1) | TB: NCT03776500 (I), NCT03423030 (I), NCT03036163 (I) | [282] |
| MK-7762 (TBD09) | Oxazolidinone | Bill & Melinda Gates Medical Research Institute | Protein synthesis (50S ribosomal subunit) | TB: NCT05824091 (I) | [283] |
| MRX-8 | Polymyxin | MicuRx Pharmaceuticals | Cell wall / Bacterial membrane | NCT04649541 (I) | [284] |
| Murepavadin (POL7080, RG7929) | Protegrin I | Spexis | Cell wall (LPS-assembly protein LptD) | Pseudomonal cystic fibrosis (I) | [66] |
| Nacubactam (OP0595, FPI-1459, RG6080, RO7079901) + meropenem | DBO BLI + carbapenem | Meiji Seika, Roche | Cell wall (PBP) | NCT02134834 (I), NCT02975388 (I), NCT03182504 (I), NCT02972255 (I), NCT03174795 (I) | [237] |
| PL-18 | Cationic peptide | Jiangsu ProteLight Pharmaceutical and Biotechnology | Cell wall / Bacterial membrane | Bacterial vaginosis: NCT05340790 (I) | [114] |
| PLG0206 (WLBU2) | Cationic peptide | Peptilogics | Cell wall / Bacterial membrane | PJI: NCT05137314 (I) | [118] |
| QPX9003 (F365, BRII-693) | Polymyxin | Brii Biosciences | Cell wall / Bacterial membrane | NCT04808414 (I) | [285] |
| RG6436 (GDC-0829) | Arylomycin | Genentech | Cell wall (Type I signal peptidase LepB) | ISRCTN18049481 (I) | – |
| SZEY-2108 | Monobactam | Suzhou Erye Pharmaceutical Co. | Cell wall (PBP) | CRE: NCT06055777 (I) | – |
| TBAJ-587 | Diarylquinoline | TB Alliance | ATP production (mycobacterial ATP synthase) | TB: NCT04890535 (I) | [286] |
| TBI-223 | Oxazolidinone | TB Alliance | Protein synthesis (50S ribosomal subunit) | TB: NCT03758612 (I), NCT04865536 (I) | [287] |
| TXA709 | Benzamide | TAXIS Pharmaceuticals | Cell wall (Cell division protein FtsZ) | S. aureus | [95] |
| Upleganan (SPR206) | Polymyxin | Spero Therapeutics | Cell wall / Bacterial membrane | NCT03792308 (I), NCT04868292 (I), NCT04865393 (I) | [288] |
| Ursodeoxycholic acid (ursodiol) | Steroid | Nottingham University Hospitals NHS Trust | – | CDI: NCT05526807 (I) | – |
| Xeruborbactam (QPX7728) or prodrug (QPX7831) (+ QPX2014) (+ ceftibuten/QPX2015) | Bicyclic boronate BLI + (undisclosed) BL | Qpex Biopharma | Cell wall (PBP) | NCT04380207 (I), NCT05072444 (I), NCT04578873 (I), NCT03939429 (I) | [179] |
|
Zidebactam (WCK 5107) + cefepime (combination: WCK 5222, FEP-ZID) zidebactam + ertapenem (WCK 6777) |
DBO BLI + cephalosporin or carbapenem | Wockhardt | Cell wall (PBP) | cUTI and AP: NCT05645757 (I) | [289] |
| Zifanocycline (KBP-7072) | Tetracycline | KBP BioSciences Pharmaceutical Technical Co. Ltd. | Protein synthesis (30S ribosomal subunit) | NCT02454361(I), NCT02654626 (I), NCT04532957 (I), NCT05507463 (I) | [290] |
| Zosurabalpin (RG6006, RO7223280, Abx MCP) | Macrocyclic peptide | Roche | Cell wall (LptB2FGC complex) | NCT05614895 (I), NCT04605718 (I), BP43532 | [62] |
Compounds categorized by the WHO as “agents not under active development” are not listed [8]
ABSSSI acute bacterial skin and skin structure infections, ADC antibody drug conjugate, AP acute pyelonephritis, BL β-lactam, BLI β-lactamase inhibitor, CABP community-acquired bacterial pneumonia, CDAD Clostridium difficile-associated diarrhea, CDI Clostridium difficile infections, cIAI complicated intra-abdominal infections, CRE carbapenem-resistant Enterobacteriaceae, cUTI complicated urinary tract infections, DBO diazabicyclooctane, DFI diabetic foot infections, G-ve Gram-negative, HABP hospital-acquired bacterial pneumonia, MAC Mycobacterium avium complex, NBTI novel bacterial topoisomerase inhibitor, PBP penicillin binding protein, PJI prosthetic joint infections, RTI respiratory tract infection, TB tuberculosis, UTI urinary tract infections, uUTI uncomplicated urinary tract infections, VABP ventilator-associated bacterial pneumonia
Antibiotics with New Chemical Scaffolds
DNA Gyrase and Topoisomerase IV Inhibitors
Type IIa topoisomerases, including DNA gyrase and topoisomerase IV (TI IV), play critical roles in the control of bacterial DNA topology (3D configuration). The DNA gyrase introduces negative supercoils into DNA [19], facilitating essential processes such as replication and transcription, whereas TI IV primarily resolves DNA entanglements [20] and decatenates replicated chromosomes [21, 22]. Both enzymes create transient double-strand breaks in the DNA helix, allowing the passage of another DNA segment before resealing the breaks, and have a heterotetramer structure: DNA gyrase comprises two GyrA and two GyrB subunits [23], while TI IV consists of two ParC and two ParE subunits [24]. Novobiocin, the sole approved aminocoumarin antibiotic, binds to the GyrB (gyrase) and ParE (TI IV) subunits [25, 26]. It was indicated for the treatment of serious Staphylococcus aureus infections. However, the oral formulation of the drug has been discontinued from the market due to lack of effectiveness and safety concerns [27]. In contrast, the quinolone antibiotics, dual gyrase and topoisomerase inhibitors, span a major class of antibiotics that has been widely used over decades, until safety concerns have limited their use [28]. Two quinolones bind to the GyrA (gyrase) or ParC (TI IV) subunits in the presence of DNA, forming a ternary complex immediately after the DNA double strand break was introduced, thereby inhibiting DNA relegation. The inhibition ultimately results in irreversible double-strand breaks, leading to DNA fragmentation, eventually causing cell death [29, 30]. The ‘Novel Bacterial Topoisomerase Inhibitors’ (NBTI) represent a new, chemically heterogeneous class following a similar MoA, yet with distinct binding sites (Fig. 1) [31].
Fig. 1.
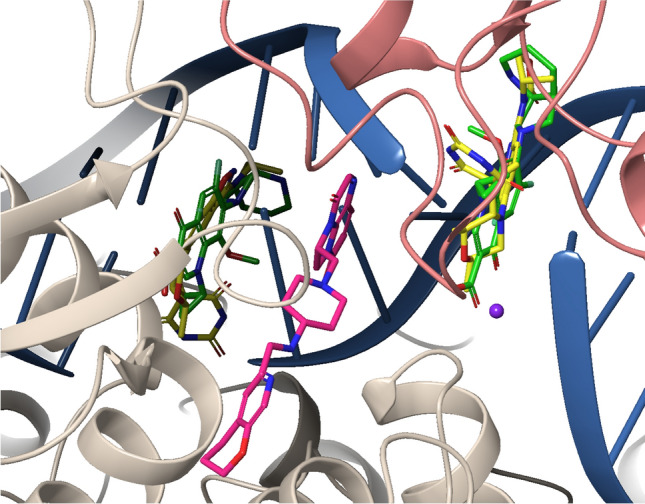
Overlay of the crystal structures of moxifloxacin (green), zoliflodacin (yellow) and gepotidacin (pink) in a ternary complex with S. aureus DNA gyrase (GyrA: beige; GyrB: coral) and doubly nicked DNA (blue) with magnesium (purple) (PDB: 5CDQ, 8BP2, and 6QTK) [32–34]
Gepotidacin (GSK2140944)
Gepotidacin is a triazaacenaphthylene (Fig. 2) developed by GlaxoSmithKline [35]. Although gepotidacin binds to the same subunits (GyrA and ParC) as the quinolones, it occupies a different binding pocket (Fig. 1), thereby conferring a lower potential for cross-resistance [36, 37]. Nevertheless, the simultaneous binding of quinolones and gepotidacin to the complex is not possible [30, 34]. Within the topoisomerase cleavage-complex, a gepotidacin molecule interacts with the DNA between the two staggered cleavage sites of the topoisomerase, inducing single-strand breaks that culminate in a bactericidal effect [30]. The in vitro activity spectrum includes Gram-positives such as methicillin-susceptible S. aureus (MSSA) and methicillin-resistant S. aureus (MRSA), Streptococcus pneumoniae, S. pyrogenes and Gram-negatives such as Haemophilus influenzae, Moraxella catarrhalis, Escherichia coli and Neisseria gonorrhoeae with MIC90 values ranging from ≤ 0.06–2 μg/mL [38, 39]. In Phase III studies for uncomplicated urinary tract infections (uUTIs), the compound showed non-inferiority (NCT04020341) or superiority (NCT04187144) after 1.5 g peroral (po) twice daily administration against nitrofurantoin 100 mg po twice daily [40]. Additionally, non-inferiority was shown in a Phase III study for 3 g gepotidacin administered po twice daily compared to ceftriaxone 500 mg intramuscular (IM) plus azithromycin 1 g po as comparative therapy (NCT04010539) in patients with uncomplicated urogenital gonorrhea [41].
Fig. 2.
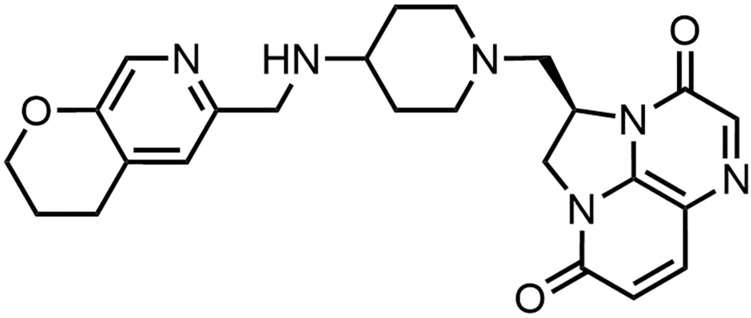
Structure of gepotidacin
Zoliflodacin (ETX0914, AZD0914)
Zoliflodacin is another member of the NBTIs featuring a spiropyrimidinetrione core (Fig. 3), commonly known as barbituric acid [42]. It was originally developed by AstraZeneca and its spin-off Entasis, which was acquired by Innoviva Specialty Therapeutics in 2022 [43]. Although the compound shares a similar binding pocket with the quinolone antibiotics (Fig. 1), zoliflodacin does not interact with GyrA but with the GyrB subunits. As a result, it is less susceptible to GyrA mutations that lead to quinolone resistance [33, 44]. The in vitro spectrum includes S. aureus (MSSA and MRSA), Enterococcus faecalis, Streptococci including S. pneumoniae, N. gonorrhoeae, Helicobacter pylori and atypical bacteria with MIC90 values ranging from 0.125–1 μg/mL, while the activity against E. faecium shows high variability with MIC values between ≤ 0.06-128 μg/mL [44–46]. In partnership with GARDP, zoliflodacin is currently under development against uncomplicated gonorrhea infections [47]. Similar to gepotidacin, a non-inferiority Phase III study on uncomplicated urogenital gonorrhae patients with 3 g zoliflodacin po compared to ceftriaxone 500 mg IM plus azithromycin 1 g po in the comparator arm was successful (NCT03959527).
Fig. 3.
Structure of zoliflodacin
BWC0977
BWC0977 is an oxazolidinone NBTI (Fig. 4) under development by Bugworks with support of GARDP [48]; a backup program is supported by CARB-X [49]. The compound shows very high in vitro activity against all members of the ESKAPE panel (E. faecium, S. aureus, E. coli, Klebsiella pneumoniae, A. baumannii, Pseudomonas aeruginosa and Enterobacter cloacae) with MIC90 values ranging from ≤ 0.5–2 μg/mL as well as N. gonorrhoeae, independent of their resistance against marketed drug classes [50, 51]. Thus, in contrast to the NBTIs mentioned above, BWC0977 has potential for a broad-spectrum antibiotic. The data also indicate an overlapping binding pocket with gepotidacin and the same MoA [30, 52, 53]. First Phase I safety and tolerability studies of intravenous (IV) infusions have been completed (NCT05088421).
Fig. 4.

Structure of BWC0977
RNA Polymerase Inhibitor
DSTA4637S
DSTA4637S is the first antibody-antibiotic conjugate (AAC) in clinical development. The conjugate comprises the small molecule rifamycin analog dmDNA31 (named ‘rifalogue’) that is coupled to the monoclonal immunoglobulin G1 (IgG1) antibody MSTA3852A [54] via a cathepsin-cleavable valine-citrulline linker (Fig. 5) [55]. Isolated from human blood and engineered by the THIOMAB™ [56] technology, the antibody targets β-O-linked N-acetylglucosamine modifications on bacterial wall-teichoic acids (WTAs) [54]. The AAC is developed for the treatment of bacteremia caused by S. aureus within host phagocytic cells, such as neutrophils and macrophages [54, 57, 58]. While the Fab region of the antibody directs binding of the conjugate to the cell wall of S. aureus, the Fc region mediates uptake of the opsonized bacteria into immune cells, where the active agent rifalogue is enzymatically released in the phagolysosomes and thus eliminates bacteria intracellularly. Advantages of this approach, apart from an increase in intracellular efficacy and minimization of toxicity through targeted drug delivery, are the extension of the half-life from 3–4 h (free rifalogue) to four days (conjugate) in mice [59].
Fig. 5.

Structure of DSTA4637S
Safety, pharmacokinetics (PK) and tolerability (immunogenicity) were determined in a first single-ascending dose (5–150 mg/kg) Phase I study (NCT02596399) in 20 healthy volunteers [60]. Besides one infusion-related moderate adverse event, no serious side effects were observed and no significant changes in laboratory parameters or vital signs occurred. Anti-drug antibodies (ADAs) induced by DSTA4637S conjugate were not observed. In a multiple-ascending dose Phase Ib study (NCT03162250), patients who tested positive for S. aureus were administered a low, intermediate or high dose of DSTA4637S via IV infusion, followed by up to five additional doses every seven days in combination with anti-staphylococcal standard of care antibiotics. Since this study, development has been suspended [8].
Small Molecules and Peptides Affecting Cell Wall Integrity
Zosurabalpin (Abx MCP, RG6006, RO7223280)
The outer membrane (OM) of Gram-negative bacteria is difficult to penetrate for xenobiotics due to its specific structure, consisting of an asymmetric bilayer with phospholipids in the inner leaflet and lipopolysaccharides (LPS) in the outer leaflet. The latter are transported to the cell surface by a multiprotein complex (LPS transporter), which extends from the inner membrane through the periplasm to the OM (Fig. 6). Zosurabalpin belongs to a novel class of synthetic cyclic tripeptides with a non-peptidic tether [61, 62]. By blocking LptB2FGC, the inner membrane component of the LPS transporter, zosurabalpin traps an LPS-bound conformation (Fig. 6c) [61]. This ternary complex disrupts LPS transport to the OM, leading to toxic accumulation of LPS biosynthetic intermediates with relatively slow in vitro killing kinetics (≥ 12 h) in Acinetobacter spp., including carbapenem-resistant Acinetobacter baumannii-calcoaceticus (ABC) complex [63, 64]. The selectivity for Acinetobacter spp. (MICs: ≤ 0.06–1 mg/L) is attributed to the significant variation in the protein sequences of the target LptFG across different Gram-negative species. Zosurabalpin's zwitterionic character, imparted by the carboxylic acid, reduced the formation of aggregated low-density lipoprotein (LDL)/high-density lipoprotein (HDL) vesicles and enhanced the tolerability in rats significantly, compared to more basic progenitors [62]. In addition to in vivo activity in a neutropenic mouse thigh infection model and in an immunocompetent mouse sepsis model, a > 5 log reduction in colony-forming units (CFUs) at a total daily dose of 360 mg/kg could be demonstrated in a murine pneumonia model with a pan-drug–resistant contemporary clinical isolate (A. baumannii ACC01073). In two Phase I studies in healthy volunteers (NCT04605718, ISRCTN37043682), a single IV infusion of 10–2000 mg was found to be safe and well tolerated [65]. Moreover, approximately dose-proportional plasma exposure (Cmax and AUCinf) up to 1000 mg and more than dose-proportional plasma exposure at > 1000 mg was observed.
Fig. 6.
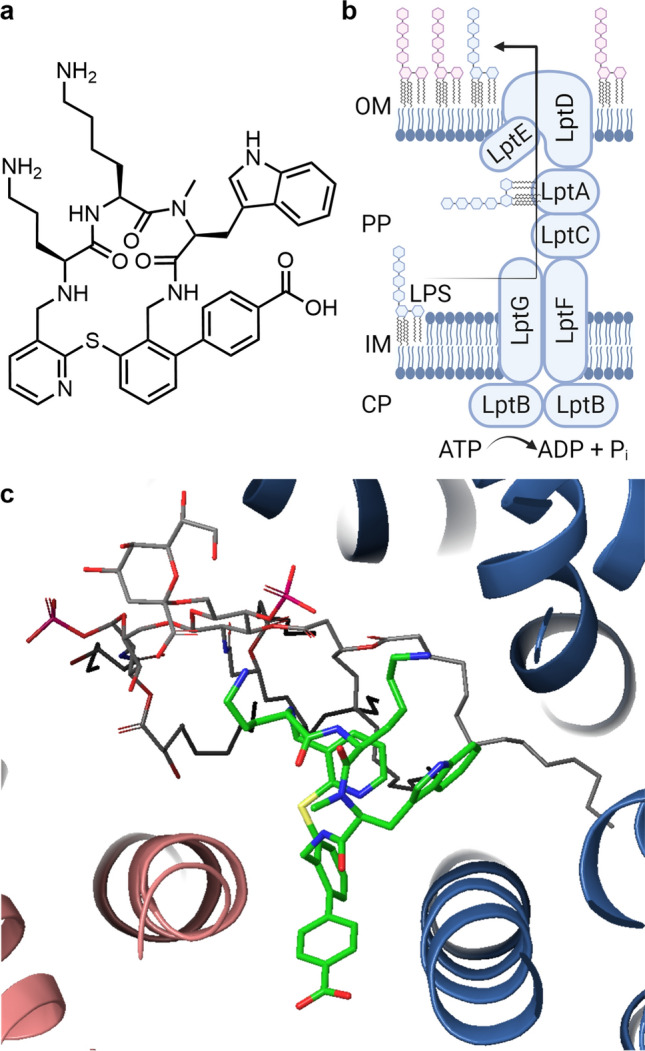
a Structure of zosurabalpin. b Schematic structure of the lipopolysaccharide (LPS) transporter. Adapted from [61]. c Cryo-EM structure of zosurabalpin (green) with Acinetobacter baylyi LptB2FG (F: blue; G: red) and Acinetobacter LPS (grey) (PDB: 8FRN) [61]
Murepavadin (POL7080, RG7929)
Murepavadin is a synthetic, cyclic 14-amino-acid cationic peptide (Fig. 7) based on the membranolytic host-defense peptide Protegin I, whose d-proline-l-proline scaffold facilitates the stabilization of a β-hairpin conformation [66]. By binding to the OM-embedded β-barrel protein LptD of the LPS transporter (Fig. 6b), which resides in a complex with the lipoprotein LptE [67], murepavadin inhibits the transport of LPS to their final location on the cell surface of Pseudomonas spp. in the nanomolar range (MIC90 = 0.25 mg/L) [68–70]. This leads to changes in the OM [71] and ultimately to bacterial cell death. Photolabeling experiments indicated a binding site at the N-terminal periplasmic segment of LptD, which contains an additional 100 amino acids in Pseudomonas spp., thereby explaining murepavadin's high selectivity for these bacteria over other Gram-negative species [68, 71]. Following the observation of elevated serum creatinine levels [72], indicative of higher-than-anticipated incidences of acute kidney injury post-IV murepavadin administration [73], Phase III trials NCT03409679 and NCT03582007 for the treatment of ventilator-associated bacterial pneumonia (VABP)/hospital-acquired bacterial pneumonia were terminated, despite the absence of increased mortality in the murepavadin arm relative to the control arm [74]. To mitigate those nephrotoxic side effects [75, 76], an inhalation of single doses ranging from 12.5–300 mg in healthy volunteers was explored in a first-in-human Phase I study (POL7080-201-01) [71, 77]. At the highest dose level, a systemic bioavailability of just 5% was observed, while a murepavadin concentration that inhibits the growth of 90% of P. aeruginosa isolates (MIC90) obtained from people with cystic fibrosis (CF) was still detected in the epithelial lining fluid after 24 hours [78]. In a future Phase II clinical trial, Spexis intends to investigate the treatment of P. aeruginosa infections in patients with CF and non-CF bronchiectasis. A detailed review about murepavadin was published in 2018 [79].
Fig. 7.

Structure of murepavadin
Afabicin (Debio 1450, AFN-1270)
The benzofuran napthyridinone afabicin is a prodrug of afabicin desphosphono (Debio 1452, AFN-1252), an enoyl-acyl carrier protein (ACP) reductase (FabI) inhibitor developed by Debiopharm (Fig. 8) [81]. Debiopharm acquired Affinium Pharmaceuticals in the year 2014, who had owned the license for the lead compound, originally discovered by GSK [82, 83]. The target FabI catalyzes the final step of the bacterial fatty acid biosynthesis cycle, the reduction of the double bond in the enoyl-ACP intermediate [84]. Inhibition of FabI disrupts this process, preventing the bacteria from synthesizing fatty acids for membrane assembly (Fig. 9) [85].
Fig. 8.
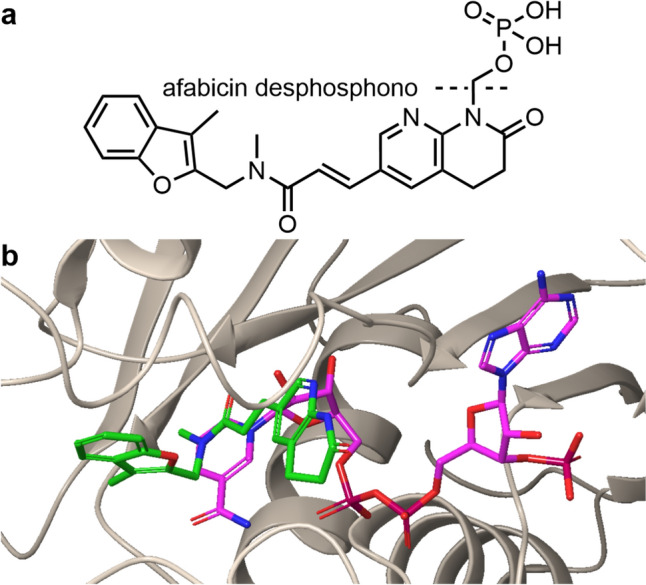
a Structure of afabicin. The cleavage site to give the active agent afabicin dephospho is indicated by a dotted line. b Crystal structure of afabicin (green) with S. aureus FabI (beige) and the unnatural substrate 3´NADPH (pink) (PDB: 4FS4). Besides interactions with the target, the carbonyl of afabicin desphospho cis-amide can interact with NADPH [80]
Fig. 9.
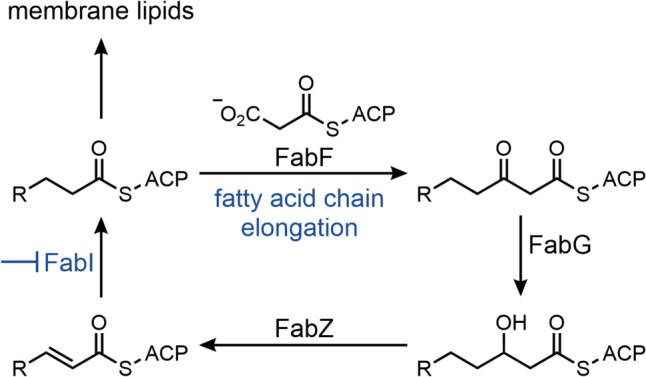
Bacterial fatty acid biosynthesis and the inhibition of the FabI-catalyzed step by afabicin desphospho. Adapted from [85]
This mechanism is particularly effective for species such as Staphylococci (MIC90 = 0.008–0.06 mg/L) [86], which have not developed salvage pathways. After oral or IV administration of afabicin, dephosphorylation and cleavage of the resulting hemiaminal lead to the formation of the active species, showing selective and potent antibacterial activity against Staphylococcus spp., including MRSA [87]. In a Phase II study against acute bacterial skin and skin structure infections (ABSSSI) (NCT02426918), IV-to-oral afabicin (low-dose: 80 mg IV followed by 120 mg po twice a day and high-dose: 160 mg IV followed by 240 mg po twice a day) showed non-inferiority to a combination of vancomycin (1 g or 15 mg/kg IV) followed by linezolid (600 mg po twice a day). Most frequent treatment-emergent adverse events were mild headache and nausea [87]. In another, ongoing Phase II trial, afabicin is evaluated using an IV-to-oral switch strategy for the treatment of S. aureus bone or joint infections (NCT03723551).
TXA709
The cell division protein filamenting temperature-sensitive mutant Z (FtsZ) is a new, potentially broad-spectrum target for the inhibition of bacterial cytokinesis, the process by which a bacterial cell divides into two daughter cells (Fig. 10) [90]. Its structure and function are comparable to that of mammalian β-tubulin [91]. Filamenting temperature-sensitive mutant Z forms a ring-like, inner cytoplasmic membrane anchored structure in the center of the cell via GTP-dependent self-polymerization. This “Z-ring“ serves as a scaffold for recruiting and organizing other components of the divisome complex for septum assembly and guides cell division (Fig. 10). Filamenting temperature-sensitive mutant Z is a key component for most Gram-negatives and -positives as it is highly conserved and ubiquitous across most bacteria [92, 93].
Fig. 10.
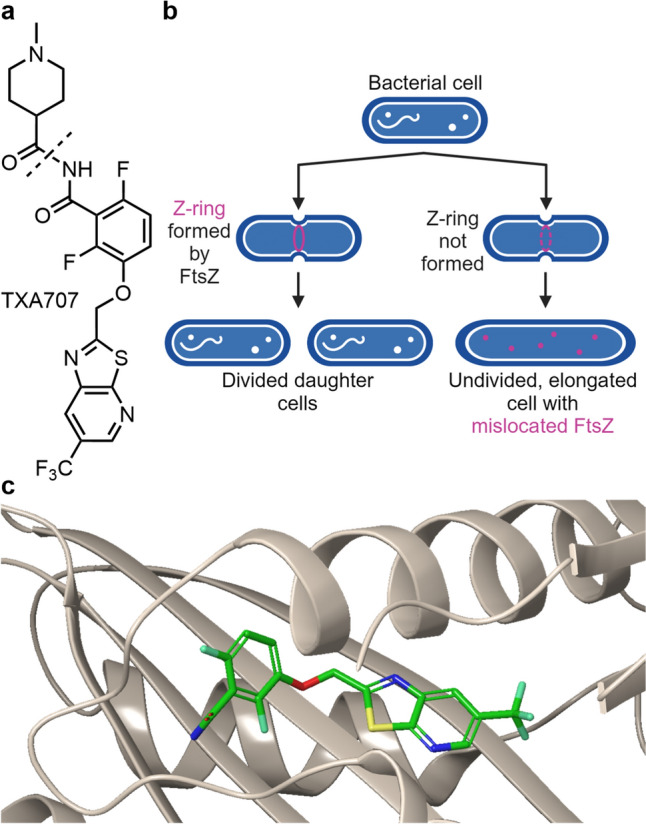
a Structure of TXA709. The cleavage site to give the active agent TXA707 is indicated by a dotted line. b TXA707’s mechanism of action. Adapted from [88]. c Crystal structure of TXA 707 binding to an inter-subdomain pocket of Staphylococcus aureus FtsZ (PDB: 5XDT) [89]
The imide TXA709 (Fig. 10) is an anti-MRSA prodrug [94] of benzamide TXA707, an inhibitor of FtsZ, developed by TAXIS Pharmaceuticals [95]. The imide TXA709 is based on Prolysis’s compound PC190723 [91, 96], a potent FtsZ-inhibitor with bactericidal activity against Staphylococcus spp. (incl. MRSA), yet limited metabolic stability. In contrast, the imide prodrug TXA709 possesses enhanced oral bioavailability and superior in vivo efficacy against MRSA. The analogues also exhibited bactericidal activity against S. aureus strains (MIC ≤ 1 μg/mL) resistant to the current standard of care drugs vancomycin and linezolid [90]. TXA709 was the first FtsZ-inhibitor entering human clinical trials and serves as the lead compound for further development targeting bacteria such as E. coli and K. pneumoniae. Synergistic effects with other antibiotics such as β-lactams have been described [88, 90].
RG6436 (GDC-0829)
Bacterial type I signal peptidases (SPase I enzymes) are essential, membrane-bound proteases that cleave membrane-bound preproteins, thus releasing mature proteins into the periplasm or the outside of the cell [97]. Their inhibition leads to an accumulation of the preproteins in the membrane, which ultimately results in cell death (Fig. 11) [98]. Natural arylomycins are macrocyclic lipopeptides, that inhibit the active side of SPases on the cell surface of Gram-positive bacteria, while not being able to reach the inner membrane-bound SPases of Gram-negative bacteria [99]. The structure optimization of arylomycins resulted in G0775 (Fig. 11), a compound with improved penetration of the OM through a porin-independent mechanism, yet insufficient physicochemical properties. In vitro tests showed activity against ESKAPE-pathogens and improved potency against Gram-positive MRSA [100]. The remarkable widening of the bacterial spectrum illustrates the power of medicinal chemistry in antibiotic hit optimization, and it positions the macrocycles as a promising new class of antibiotics with novel MoA.
Fig. 11.

a Structure of G0775. b Mechanism of action of optimized arylomycin with activity against Gram-negative bacteria. CP cytoplasm, IM inner membrane, OM outer membrane, PP periplasm. Adapted from [101]
RG6436 is an arylomycin with an undisclosed structure inhibiting the SPase LepB in Gram-negative bacteria. Since it was acquired from Enterprise Therapeutics, the development is led by the Roche subsidiary Genentech [102, 103]. In vitro data showed a broad activity against Gram-negative bacteria, including Enterobacterales—most importantly E. coli, K. pneumoniae, E. cloacae—as well as A. baumannii, P. aeruginosa and Serratia marcescens (MIC90 = 0.5–2 μg/mL) [98]. RG6436 is currently under development for the treatment of complicated urinary tract infections (cUTIs) and is being investigated in a Phase I clinical trial on healthy volunteers (ISRCTN18049481) [102].
Small Molecules and Peptides Acting as Membrane Disruptors
Antimicrobial peptides (AMPs), which are integral components of the innate immune system and thus also referred to as host defense peptides (HDPs), are found across a broad spectrum of species [104]. Most AMPs have an amphiphilic structure [105]. Positively charged groups preferably bind to negatively charged bacterial membrane components [104] over mostly zwitterionic eukaryotic membranes [106], while the hydrophobic side of AMPs mediates their integration into the membrane, resulting in membrane disruption and subsequent cell lysis [105]. However, the split between antibiotic activity and eukaryotic cytotoxicity seen in cellular assays often did not translate in sufficient therapeutic windows in vivo following systemic use and hemolysis has been an often-encountered safety issue [107]. Therefore, no earlier-generation AMPs have reached drug approval. On the other hand, bacterial resistance development to AMPs is very difficult due to the non-proteogenic lipid targets. We therefore present a selection of next-generation AMPs in the current development pipeline.
Voxvoganan (LTX-109)
Staphylococcus aureus infections are associated with prolonged hospital stays, high mortality rates, and medical costs [108]. Nasal decolonization protocols for S. aureus have been implemented to mitigate the risk of severe infections in surgical, dialysis, and immunocompromised patients, as well as to prevent transmission to non-carriers [109]. Due to rising resistance to the widely established decolonization regimen with the polyketide mupirocin, new agents are currently under development. Voxvoganan is a synthetic cationic tripeptide, characterized by a preferred amphipathic conformation and a C-terminal 2-phenylethylamine cap (Fig. 12) [110], which impeded proteolytic degradation [111]. Irrespective of the reduced susceptibility of S. aureus strains to β-lactams, vancomycin, daptomycin, linezolid, clindamycin, trimethoprim-sulfamethoxazole, and mupirocin, voxvoganan maintained a consistent minimum inhibitory concentration (MIC) range of 2–4 µg/mL and a rapid dose-dependent bactericidal effect within 0.5–4 h in vitro [112]. Additionally, it possessed a longer post-antibiotic effect than mupirocin [113] and exhibited a low tendency for resistance development [112]. Presumably owing to its hemolytic activity, evidenced by 50% lysis of erythrocytes at 175 µg/mL [110], voxvoganan’s clinical investigation is currently confined to topical application. In persistent nasal MRSA/MSSA carriers, notable nasal decolonization efficacy was achieved within two days using a 2% or 5% voxvoganan hydrogel applied to the anterior nares thrice daily, complemented by a standard local hygiene regimen encompassing body and hair cleansing, with low systemic bioavailability and no safety concerns over the 9-week follow-up period (NCT01158235) [109]. However, recurrence of the bacteria after five days following completion of the three-day treatment was noted in all participants except one.
Fig. 12.
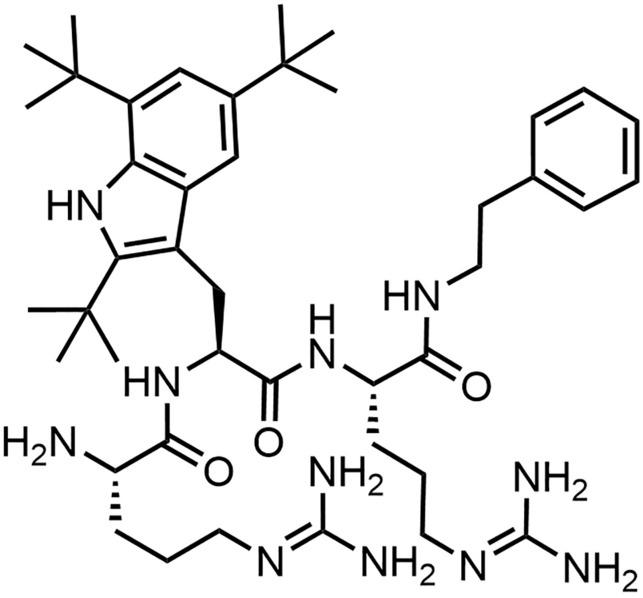
Structure of voxvoganan
PL-18
PL-18 is a 15mer, all-d-amino-acid, α-helical, cationic, amphipathic peptide derived from the N-terminus of ribosomal protein L1 (RpL1) of H. pylori (Fig. 13). The peptide is further stabilized by an N-terminal acetyl residue and C-terminal primary amide [114]. With MIC values between 1–4 µM, PL-18 demonstrated a broad spectrum of activity against Gram-negative and Gram-positive bacteria, notwithstanding H. pylori's intrinsic resistance to the N-terminal region of the protein [115]. Presumably due to the demonstrated hemolytic activity of the enantiomer HPRP-A1 [116, 117], PL-18 is currently only being clinically investigated as a topical application in the form of a 1–15 mg suppository against bacterial vaginosis (NCT05340790).
Fig. 13.

Structure of PL-18
PLG0206 (WLBU2)
PLG0206 is a de novo designed, 24-amino-acid, α-helical, cationic, amphipathic peptide containing valine and tryptophan residues in the hydrophobic face and exclusively arginine residues on the hydrophilic side (Fig. 14) [118, 119]. Interaction of PLG0206 with the bacterial cell membrane led to local stiffening and changes in membrane thickness [120, 121]. Those effects caused a spacing mismatch in lipid headgroups and reduced the energy barrier for ion flux across the membrane, resulting in a bactericidal effect within minutes [122]. PLG0206 exhibited broad-spectrum activity against Gram-negative and -positive bacteria, including > 1200 multidrug-resistant (MDR) ESKAPEE clinical isolates (MIC90 = 0.25–16 μg/mL) [123], while having little to no cytotoxic effects on human primary cells at bactericidal concentrations [124]. Serial passage experiments with P. aeruginosa strains have shown a development of resistance only after 25 days [119]. The low tendency for resistance development was corroborated through single-step spontaneous mutation frequency assays [123]. Of particular value is the strong PLG0206 anti-biofilm activity [122, 123, 125] against S. aureus, the most common pathogen responsible for persistent infections that develop around joint prostheses, such as hip and knee replacements. Five-year mortality for such periprosthetic joint infection (PJI) is about 25% [126]. After explanted components of total knee arthroplasties (TKA) from patients with chronic PJI were treated ex vivo with a 1 mg/mL PLG0206 solution for 15 minutes, rinsed and sonicated in 1% Tween 20, 10 of 17 prostheses became culture-negative (NCT05137314) [127, 128]. The observation that infected prostheses exposed to PLG0206 showed a mean 4-log10 reduction in CFU compared to the untreated control suggests the development of PLG0206 as a local irrigation solution directly in the wound cavity for patients with PJI after total knee/hip arthroplasty. In a further Phase I study, the IV administration of PLG0206 as a single dose between 0.05–1 mg/kg appeared to be safe and well tolerated as long as the drug concentration and infusion rate remained below 0.5 mg/mL and 25 mg/h, respectively [129, 130]. Detailed reviews of the preclinical and clinical development of PLG0206 have recently been published [124, 131].
Fig. 14.

Amino acid sequence of PLG0206
OMN6
Cecropins are 29–42 amino acids long, cationic, linear AMPs found in the orders of Diptera, commonly known as flies, and Lepidoptera, commonly known as butterflies and moths [132]. Their membrane-disrupting properties are attributed to the secondary structure consisting of two amphipathic α-helices connected by a hinge. OMN6 is a synthetic 40mer, all-l-amino-acid peptide based on cecropin A, cyclized via a disulfide bridge between engineered Cys2 and Cys40 residues to increase stability and decrease proteolytic degradation (Fig. 15). OMN6 exhibited activity against Gram-negative bacteria, in particular against 401 A. baumannii strains, with a very narrow MIC range of 4–8 µg/mL, despite partial colistin resistance [133]. In vitro serial passaging studies with four strains of the ABC complex demonstrated unchanged MIC values after 20 passages (20 days), and thus no development of resistance [132]. Up to a concentration of 868 μg/mL, OMN6 had no effect on the survival rate of HEK293T cells or the hemolysis of erythrocytes. Since OMN6 showed increased in vitro activity in the presence of bovine pulmonary surfactant [133], it might be suitable for the treatment of lung infections. In a corresponding mouse model with the MDR A. baumannii strain ACC0000535, three IV injections of 21, 35, or 49 mg/kg OMN6 at 1-hour intervals starting 2 hours post-infection resulted in a reduction in bacterial load of > 4.4 log CFU/lungs compared to the vehicle-treated group after 24 hours. For a first-in-human Phase I study (CS0379-200475 Omnix), healthy volunteers received doses of 7.5–300 mg OMN6 spread across up to three 3-hour infusions, interspersed with a 5-hour wash-out period [134]. All treatments were well tolerated and adverse events were mild and transient. A half-life of 0.9 hours was determined for single infusions of 80 or 100 mg OMN6.
Fig. 15.
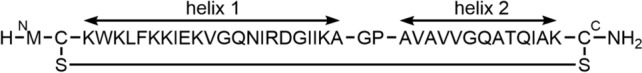
Structure of OMN6
Brilacidin (PMX-30063)
Researchers at the University of Pennsylvania found small, synthetic, non-peptidic HDP mimetics against a variety of multi-drug–resistant Gram-negative and -positive bacteria [135, 136]. Their spin-off PolyMedix was founded in 2002. The most promising compounds had an arylamide backbone equipped with multiple charged and hydrophobic elements, resulting in an overall amphiphilic structure with a molecular size < 1000 Da [135]. Further structure optimization for selectivity against S. aureus led to the discovery of brilacidin. The arylamide foldamer shows a planar structure with four positively charged guanadine- and pyrrolidine-groups as well as two hydrophobic trifluoromethane elements (Fig. 16) [135, 137].
Fig. 16.

Structure of brilacidin
Brilacidin was active against a broad range of both Gram-negative and -positive bacteria including MRSA with MIC90 values ranging from 0.25–64 μg/mL [138]. It has been tested in two randomized Phase II trials for the treatment of ABSSSI (NCT02052388 and NCT01211470) [139, 140]. In the latter, a single dose of 0.6 mg/kg IV brilacidin, the lowest dosage tested in the trial, was well tolerated and showed an efficacy similar to 7-day treatment with daptomycin. Although Innovation Pharmaceuticals Inc., formerly known as Cellceutic, who acquired PolyMedix in 2013, announced plans in 2015 to advance to Phase III ABSSSI trials, we are not aware that such a trial has commenced to date [141].
Brilacidin also shows antifungal [142] and antiviral [136, 143, 144] properties and has been identified as a potential drug against the malaria-causing parasite Plasmodium falciparum [145]. In more recent studies, Innovation Pharmaceuticals Inc. investigated the efficacy of a brilacidin oral rinse (NCT02324335) and in COVID-19 patients (NCT04784897).
β-Lactamase Inhibitors
β-Lactamase inhibitors (BLIs) prevent the hydrolytic inactivation of β-lactam motives found in penicillins, cephalosporines, penems, and monobactams [146]. The targeted β-lactamases are predominantly responsible for the resistance of Gram-negative pathogens towards those β-lactam antibiotics. They are categorized into Ambler classes A, B, C and D based on amino acid sequence homology [147, 148], but other classification systems, such as the one of Bush-Jacoby-Medeiros, that groups by substrate specificity and response to inhibitors, also exist [149–151]. The Ambler classes A, C and D comprise serine β-lactamases (SBLs), which catalyze the ring-opening hydrolysis of β-lactam substrates via a serine residue at the active site, resulting in the formation of an acylated-enzyme intermediate (Fig. 17) [152]. Numerous inhibitors targeting SBLs are known to react with the serine residue in a reversible or irreversible fashion [153, 154]. The Ambler class B comprises metallo-β-lactamases (MBLs) that hydrolyse β-lactams through the attack of water facilitated by one or more zinc cations. Aztreonam, a monobactam antibiotic, was generally unaffected by MBLs, yet hydrolyzed by extended-spectrum β-lactamases [155]. The recently introduced cefiderocol showed reduced susceptibility in vitro [156], yet all other β-lactam antibiotics were vulnerable to MBLs [157]. The first generation of BLIs, such as clavulanic acid [158] or tazobactam (YTR 830) [159], were β-lactams themselves. In the past decade, a second chemical class named diazabicyclooctane (DBO) with an activated urea moiety, was introduced to the market with launched products such as avibactam [160], durlobactam (ETX2514) [161] or relebactam (MK-7655) [162], and several analogs in clinical development (Table 1). In this review, we would like to highlight a third class named after its unusual reactivity center, the boronates [163]. Taniborbactam, xeruborbactam, and KSP-1007 are currently in clinical trials as the first broad-spectrum BLIs, targeting both SBLs and MBLs. Given that in 2022 approximately 50% of all antibiotics prescribed in outpatient settings in the USA were β-lactams, this new class of BLIs holds particular significance [164]. The dual MoA for taniborbactam and xeruborbactam is shown in Figure 17. While the boron atom in the boronic acid is a strong electrophile that blocks the active serine in SBLs, also the active Zn2+ ions of MBLs could be bound by the same inhibitor. This remarkable achievement cannot be generalized for all bicyclic boronate BLIs; for example ledaborbactam lacks activity against MBLs [165, 166].
Fig. 17.

a Penicillin hydrolysis by serine β-lactamases (top) and a di-zinc B1 metallo-β-lactamase (bottom). BA base for acylation step, BD base for the deacylation step. Adapted from [152, 167]. b Dual mechanism of action of some bicyclic boronate β-lactamase inhibitors based on the crystal structures of taniborbactam (PDB: 6SP6 and 6SP7) and xeruborbactam (PDB: 6V1J and 6V1P) [168, 169]
Taniborbactam (VNRX-5133)
Taniborbactam is a bicyclic boronate BLI (Fig. 18) developed by Venatorx Pharmaceutical [170]. In 2020, GARDP supported the development of the drug and in early 2024, the Menarini group acquired the exclusive rights for commercialization of a cefepime/taniborbactam (FEP/TAN) combination in 96 countries [170]. According to in vitro data, taniborbactam was highly active against SBLs and most MBLs. Yet, weaker effects were found against certain MBLs, such as IMP-1 [169, 171]. In a Phase III study, the combination FEP/TAN showed non-inferiority in adults with cUTI while having a lower adverse effect frequency compared to meropenem (NCT03840148) [172]. Superiority was demonstrated in pre-specified secondary parameters, specifically microbiologic and clinical success during late follow-up, 28–35 days after therapy initiation. An additional Phase III study against HABP/VABP is planned (NCT06168734) [173]. A detailed review about FEP/TAN was published recently [174].
Fig. 18.
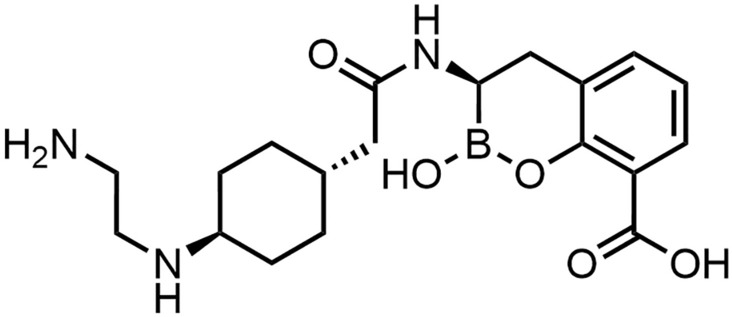
Structure of taniborbactam
Xeruborbactam (QPX7728)
Xeruborbactam (Fig. 19) was developed by Qpex Biopharma, which was acquired by Shionogi in 2023 [175]. Compared to taniborbactam, xeruborbactam showed a broader spectrum against both SBLs and MBLs in vitro [176–178], which is particularly remarkable given the very small size (molecular weight: 222 g/mol) of the compound. Additionally, at concentrations (MIC90 = 16 to > 64 μg/mL) generally higher than those required for β-lactamase inhibition, an intrinsic antibacterial effect was observed [179]. Xeruborbactam and its orally available isobutyryloxymethyl ester prodrug QPX7831 underwent Phase I studies alone and in combination with QPX2014, a compound with an undisclosed structure (NCT04380207, NCT04578873) [180–182]. The steady-state PK of xeruborbactam (500 mg initial and 250 mg q8h or 1000 mg initial and 500 mg q8h) showed a half-life of 31 h and 24.9 h and a low volume of distribution at steady-state of 15.5 L and 17.8 L [181]. More recently, clinical trials have been initiated to evaluate PK and safety of the combination of QPX7831 (prodrug) and the orally applicable cephalosporin ceftibuten (QPX2015) in healthy volunteers (NCT06079775) [183].
Fig. 19.

Structure of xeruborbactam
KSP-1007
KSP-1007 (Fig. 20) was discovered by Sumitomo Pharma and is developed in collaboration with the Kitsato Institute in Japan [184]. In combination with meropenem, Sumitomo Pharma proposed an indication against cUTI, complicated intra-abdominal infections as well as HABP/VABP [185]. In vitro studies demonstrated that, when combined with meropenem, there was a significant antimicrobial activity against meropenem-susceptible and non-susceptible Enterobacterales. Lower in vitro activity was found against A. baumannii and P. aeruginosa. In a direct comparison with taniborbactam, the in vitro studies hint towards a better activity of KSP-1007 against MBL-producing strains, yet conclusive evidence about the superiority over taniborbactam and xeruborbactam is missing [186]. During single and multiple ascending IV dose Phase I studies, both with and without meropenem, a half-life of 2–4 hours was determined (NCT05226923). At the highest dose of 1500 mg KSP-1007 combined with 2 g of meropenem every 8 hours for 5 days, the most common adverse events were nausea, vomiting and transient increases of serum creatinine [187].
Fig. 20.

Structure of KSP-1007
Anti-virulence Therapeutics
Anti-virulence therapeutics represent a new approach for the prevention and treatment of bacterial infections. In contrast to a bactericidal (killing) or bacteriostatic (growth-limiting) antibiotic, these novel substances limit the disease-causing potential of bacteria or severity of an infection [188]. A variety of virulence mechanisms involved in the colonization, invasion and persistence in a susceptible host have been addressed by such virulence inhibitors, including bacterial adhesion to host cells, the pili- or flagella-mediated motility, biofilm formation, the assembly of secretion systems, communication through quorum sensing, immune evasion, as well as the secretion of toxins and destructive enzymes [188–190]. Although the bacterial load is not reduced directly, blocking virulence facilitates the elimination of the pathogen by the host's natural immune response. As the viability of the bacteria remains unaffected, it is believed that the reduced selection pressure will slow down the development of resistances [188]. A second major advantage is that, since virulence factors are mainly associated with pathogenic bacteria, an anti-virulence treatment preserves the integrity of the host microbiome [189]. This prevents the colonization of pathogenic bacteria in the gastrointestinal tract, thereby reducing the risk of infection and associated health complications, including antibiotic-associated diarrhea. Unlike antibiotics, anti-virulence treatment also allows for uncomplicated preventive use in high-risk patients (e.g., immunocompromised patients) [190]. Furthermore, it is anticipated that combination therapy [190] with low-dose antibiotics will result in a more effective treatment with fewer side effects. On the other hand, the clinical translation of anti-virulence approaches is challenging, because it poses special requirements on biomarker-driven patient selection and the establishment of pharmacodynamic markers for dose-finding. If virulence blockers are to be used as an adjunctive therapy on top of standard antibiotics, a clinical trial design that demonstrates superiority is required [190]. In spite of these challenges, monoclonal antibodies that neutralize toxins have been launched (against Bacillus anthracis, C. difficile) or are in clinical development (against S. aureus, [191, 192]). The first small molecules that entered clinical trials are highlighted below.
Fluorothiazinone (Ftortiazinon, CL-55)
Fluorothiazinone (Fig. 21), developed by the Gamaleya Research Institute of Epidemiology and Microbiology, is a thiadiazinone-based inhibitor of the type 3 secretion system (T3SS), one of the main pathogenicity factors of many Gram-negative bacteria [193, 194]. This multiprotein complex consists of more than 20 different proteins, spans the bacterial cell wall and has a structure reminiscent of a needle with a syringe mechanism [195]. Direct secretion of linearly unrolled effector proteins from the bacterial cell into the cytoplasm of eukaryotic cells facilitates bacterial colonization of these cells. In addition, this secretion system enables the pathogen to evade the host's immune response or even to kill host cells by altering signaling pathways. In numerous murine infection models against urogenital Chlamydia trachomatis serovar D [196], intraperitoneal (IP) injected [197] or orally administered Salmonella enterica serovar Typhimurium [198], septicemic A. baumannii [199], uropathogenic E. coli [200], as well as pulmonary K. pneumoniae [201] and P. aeruginosa clinical isolates [202, 203], it has been shown that fluorothyazinone, given as prevention or treatment, led to a decrease in the bacterial load. Subsequently, fewer pathomorphological changes in organs, decreased modulation of the local immune response, inhibition of biofilm formation as well as improved survival rates of mice, without affecting bacterial growth in vitro, were demonstrated. Neither a single IP dose of 5000 mg/kg in mice and rats, nor 50 mg/kg po for 14 days in rats and rabbits were toxic to the animals [204]. However, a 10-fold increase of the dose in the latter experiment led to reversible pathological findings in rat thymi. Furthermore, no carcinogenic or mutagenic effects (Ames test) or increased levels of chromosomal aberrations in bone marrow cells (comet assay) were observed in vitro [204]. This favorable safety profile is particularly important for a prophylactic setting.
Fig. 21.
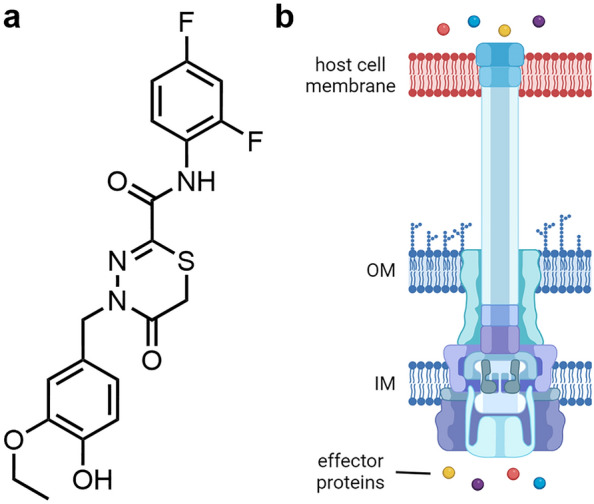
a Structure of fluorothiazinone. b Schematic structure of a bacterial type 3 secretion system injecting effector proteins from the bacterial cytoplasm into the host cell. IM inner membrane, OM outer membrane. Adapted from [193]
Two Phase II studies assessed PK, safety, and efficacy. In NCT03638830, the treatment of hospitalized patients with cUTI caused by P. aeruginosa in combination with the fourth-generation cephalosporin cefepime was investigated. The results have not yet been published. In NCT06135350, the prophylaxis of ventilator-associated pneumonia caused by Gram-negative bacteria is being investigated with a monotherapy of up to 2400 mg/day fluorothiazinone over a period of up to 14 days. The study is expected to be completed by the end of 2024.
GSK3882347
Uropathogenic E. coli (UPEC) bacteria form reservoirs in the host gut, allowing them to repeatedly colonize the periurethral area or the vagina through fecal transmission [205]. Followed by an ascent in the urethra, they reach the urinary tract, where they cause UTIs. In this process, appendages, such as type 1 pili, facilitate adhesion to human uroplakins, proteins found in the cell membrane of the urothelium (epithelium of the urinary tract). FimH, a lectin (carbohydrate-binding protein) at the tip of the type 1 pilus of UPEC, binds to those mannosylated proteins, enabling colonization of the bladder and invasion of urothelial cells, where the pathogen replicates intracellularly [206]. GSK3882347 is an orally available [207] mannose-based (mannoside) [208] FimH antagonist [209], co-developed by Fimbrion Therapeutics and GSK with CARB-X support. It prevented E. coli from binding to the bladder wall, thus allowing the body to naturally eliminate the pathogen [207].
Mannosides like M4284 (Fig. 22) also reduce the intestinal colonization of different UPEC isolates in the feces, cecum, and colon of mice, without notably disrupting the gut microbial communities [205]. This dual MoA could significantly reduce the rate of (recurrent) UTIs. A single and multiple ascending dose Phase I study of GSK3882347 has been completed (NCT04488770). Although the exact structure of GSK3882347 has not been reported, detailed studies on O- [205, 206, 210–214] and C-mannosides [215, 216] have been published.
Fig. 22.

a Structure of M4284. b Schematic structure of the type 1 pilus of uropathogenic E. coli, displaying FimH at the tip. Adapted from [217]. c Cellular mechanism of action of FimH antagonists. Adapted from [218]
Conclusion
A previous pipeline analysis conducted 10–15 years ago detected and warned about a steady decline of antibiotic approvals over time [219, 220]. However, compared to an all-time low with 7 launches in the years 2000–2009, the following decade again saw an increase with 15 antibiotic drug launches [221]. And yet, the AMR problem has not been alleviated. First, the availability of novel antibiotics for patients has remained limited because of economic constraints that hinder a launch in low-, middle-, and even in a large portion of high-income countries [222]. Second, the licensed drugs were dominantly derivatives of established chemical classes, that hardly met the four innovation criteria established by the WHO – namely, the introduction of a new chemical class, a novel target, a new mode of action, and a lack of cross-resistance to existing antibiotics [8]. The current development pipeline is also dominated by derivatives of established chemical classes, which carry smaller development risks, but rarely overcome the existing resistance mechanisms completely. Despite these concerns, research and development activities strengthened by large, global consortia and public-private partnerships start yielding in innovative and unprecedented solutions against AMR. This includes, on the one hand, a broad range of new drug formats such as engineered phages [223], LBPs, monoclonal antibodies, RNA-targeting drugs, or new vaccination approaches [8]. On the other hand, small molecule antibiotics addressing new targets with new chemical structures have entered development. These were the main focus of this review.
Targeted therapies against specific bacterial species, like zosurabalpin for Acinetobacter spp., murepavadin for Pseudomonas spp., and afabicin for Staphylococcus ssp. mitigate unwanted selection pressure on other pathogenic but non-disease–causing bacteria and thus the development of resistance in the normal flora. The former two examples exemplify first successes in targeting outer membrane biosynthesis of Gram-negative bacteria. However, the effective use of such narrow spectrum agents necessitates the prior identification of the causative pathogen and the exclusion of a polybacterial, mixed infection. Therefore, it is essential to assure the availability of appropriate diagnostic tools, which often require a co-development alongside the selective therapeutic.
Antibody–antibiotic conjugates represent an innovative new approach, enabling the selective delivery of otherwise too toxic substances or compounds with poor pharmacokinetics to the site of infection [224]. Furthermore, as exemplified by DSTA4637S, targeted eradication of intracellular pathogens is achievable through an appropriate linker chemistry [54]. Given the successful use of antibody-drug conjugates in cancer treatment, this format allows a re-evaluation of previously unsuccessful small molecule antibiotic projects by considering their development as conjugates [225–227]. On the other hand, the costs of development but also production of AACs exceed those of standard antibiotics significantly.
The low propensity towards resistance development, coupled with the broad-spectrum activity against resistant pathogens, are attractive features of peptidic membrane disruptors that mimic a part of the innate immune responses against pathogens, even though their in vitro activity is often lower (MIC values between 1–4 µM) compared to conventional antibiotics. But their biggest challenge is to define a sufficient therapeutic window by limiting eukaryotic cell toxicity. Some development programs therefore focus on a non-systemic application; in addition, the switch from peptides to peptidomimetic structures (e.g., as in voxvoganan or brilacidin) help to fine-tune stability and toxicity properties.
The high number of β-lactam antibiotics in combination with a BLI in the development pipeline is striking. Given their broad spectrum of activity, bactericidal effect, low toxicity, good tissue penetration, inexpensive production, and extensive clinical experience spanning over 50 years, it is anticipated that these agents will continue to play a critical role in therapeutic practice globally [228]. We would like to highlight two developments: the optimization of BLIs targeting all four Ambler classes was enabled by chemical innovations, in particular through the switch to boronate as a chemical motif that binds to both serine nucleophiles as well as ionic zinc lewis acids. Second, the diazabicyclooctanes ETX0462 [229] and EBL-1463, [49] as well as other compounds [230], demonstrate that it is possible to evolve a conventional BLI to a dual-action inhibitor targeting additionally penicillin-binding proteins (PBPs). Inhibiting PBPs with non–β-lactam pharmacophores that are not affected by the widespread β-lactamases may open up a sustained exploration of this mechanism.
Anti-virulence therapeutics offer a promising new strategy to limit the damage caused by virulence factors on host cells that is orthogonal to direct-acting antibiotics. These therapeutics may render bacterial eradication by the host immune system more effective, when used synergistically with standard-of-care antibiotics, which needs to be demonstrated clinically through superiority trials. Alternatively, their prophylactic use is conceivable, in particular when antibiotics are not indicated, requiring careful patient stratification strategies. While the first successes have been achieved with toxin-neutralizing monoclonal antibodies, the fact that small molecules have also entered clinical development indicates that virulence blockers indeed represent a viable, complementary approach to fight AMR [188].
Counteracting AMR with novel antibiotics is possible and ongoing. Some resistance mechanisms could be alleviated through derivatization of established compound classes [231]. But novel chemical scaffolds – as illustrated in this review – are of particular interest for addressing AMR, due to their new modes of action and/or distinct binding sites compared to clinically used drug classes. The research and preclinical development pipeline demonstrates that antibiotic research does not suffer from a lack of ideas or innovative concepts. However, the overall scale in terms of number of projects is regarded as too small. For comparison, > 2000 oncology clinical trials started in 2023 with novel modalities [232]. With a Phase I to approval success rate for antibiotics of about 16%, attrition is high in particular for new, initially non-validated targets and new chemical scaffolds with unknown human side effects [233]. Because the clinical translation of antibiotics is both challenging and costly, in particular the unclear economic incentives are seen as major hurdles on the way to medicines against drug-resistant infections that are broadly available for patients. This calls for further action, in science and beyond.
Declarations
Conflict of interest
Alexandra Jana Kohnhäuser is employee of IPG DXTRA (Germany) GmbH, working for dna communications, but the work on this review was neither work-related, nor did she write about content connected to her work-related clients. Dominik Heimann, Daniel Kohnhäuser, Alexandra Jana Kohnhäuser and Mark Brönstrup have declared no conflicts of interest that may be relevant to the contents of this review.
Ethics approval
Not applicable.
Consent for publication
Not applicable.
Consent to participate
Not applicable.
Availability of data and materials
Not applicable.
Code availability
Not applicable.
Funding
Open Access funding enabled and organized by Projekt DEAL. Deutsches Zentrum für Infektionsforschung (TTU09.712).
Authors’ contribution
All authors: Design and concept of the entire manuscript. DH: Draft of the introduction, conclusion and chapters about DSTA4637S, zosurabalpin, murepavadin, voxvoganan, PL-18, PLG0206, OMN6 and anti-virulence therapeutics; creation of all figures and the global pipeline table. DK: Draft of chapters about DNA gyrase and topoisomerase IV inhibitors as well as β-lactamase inhibitors; creation of the global pipeline table. AJK: Draft of chapters about afabicin, TXA709, RG6436 and brilacidin. MB: Writing—review and editing, supervision.
References
- 1.Dickter J, Logan C, Taplitz R. Neutropenia and antibiotics: when, what, how and why? Curr Opin Infect Dis. 2023;36:218–27. 10.1097/QCO.0000000000000932. [DOI] [PubMed] [Google Scholar]
- 2.Cossey J, Cote MCB. Evaluation and management of febrile neutropenia in patients with cancer. JAAPA. 2024;37:16–20. 10.1097/01.jaa.0000000000000054. [DOI] [PubMed] [Google Scholar]
- 3.Nabhan AF, Allam NE, Hamed Abdel-Aziz Salama M. Routes of administration of antibiotic prophylaxis for preventing infection after caesarean section. Cochrane Database Syst Rev. 2016;2016:011876. 10.1002/14651858.cd011876.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Subramanian AK. Antimicrobial prophylaxis regimens following transplantation. Curr Opin Infect Dis. 2011;24:344–9. 10.1097/qco.0b013e328348b379. [DOI] [PubMed] [Google Scholar]
- 5.GBD 2021 Antimicrobial resistance collaborators. Global burden of bacterial antimicrobial resistance 1990–2021: a systematic analysis with forecasts to 2050. Lancet. 2024;404:1199–226. 10.1016/S0140-6736(24)01867-1. [DOI] [PMC free article] [PubMed]
- 6.Global antimicrobial resistance and use surveillance system (GLASS) report: 2022: Bacterial Pathogens of Public Health Importance, to Guide Research, Development, and Strategies to Prevent and Control Antimicrobial Resistance. 1st ed. Geneva: World Health Organization; 2022.
- 7.Stockwell VO, Duffy B. Use of antibiotics in plant agriculture. Rev Sci Tech. 2012;31:199–210. 10.20506/rst.31.1.2104. [DOI] [PubMed] [Google Scholar]
- 8.2023 Antibacterial agents in clinical and preclinical development: an overview and analysis. 1st ed. Geneva: World Health Organization; 2024.
- 9.O’Neill J. Tackling drug-resistant infections globally: final report and recommendations. 2016. https://amr-review.org/sites/default/files/160525_Final%20paper_with%20cover.pdf. Accessed 6 Sept 2024.
- 10.WHO Bacterial Priority Pathogens List 2024: Bacterial Pathogens of Public Health Importance, to Guide Research, Development, and Strategies to Prevent and Control Antimicrobial Resistance. 1st ed. Geneva: World Health Organization; 2024.
- 11.Alm RA, Gallant K. Innovation in antimicrobial resistance: the CARB-X perspective. ACS Infect Dis. 2020;6:1317–22. 10.1021/acsinfecdis.0c00026. [DOI] [PubMed] [Google Scholar]
- 12.Alt S, Haggstrom D, Kessmann H, Kloss F, Schneider CE, Jäger T, et al. INCATE: a partnership to boost the antibiotic pipeline. Nat Rev Drug Discov. 2022;21:621–2. 10.1038/d41573-022-00138-7. [DOI] [PubMed] [Google Scholar]
- 13.Engel A. Fostering antibiotic development through impact funding. ACS Infect Dis. 2020;6:1311–2. 10.1021/acsinfecdis.0c00069. [DOI] [PubMed] [Google Scholar]
- 14.McCall B. New fund stimulates the ailing antibiotic pipeline. Lancet Infect Dis. 2020;20:1017. 10.1016/S1473-3099(20)30629-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mullard A. Pharmaceutical firms commit US$1 billion to antibiotic development. Nat Rev Drug Discov. 2020;19:575–6. 10.1038/d41573-020-00143-8. [DOI] [PubMed] [Google Scholar]
- 16.Butler MS, Henderson IR, Capon RJ, Blaskovich MAT. Antibiotics in the clinical pipeline as of December 2022. J Antibiot (Tokyo). 2023;76:431–73. 10.1038/s41429-023-00629-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Butler MS, Vollmer W, Goodall ECA, Capon RJ, Henderson IR, Blaskovich MAT. A review of antibacterial candidates with new modes of action. ACS Infect Dis. 2024. 10.1021/acsinfecdis.4c00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Global AMR R&D Hub. Dynamic Dashboard: Antibacterials In Clinical Development. 2022. https://dashboard.globalamrhub.org/reports/pipelines/pipelines. Accessed 6 Sept 2024.
- 19.Higgins NP. Gyrase. In: Brenner's Encyclopedia of Genetics. Elsevier; 2013. p. 374–377. 10.1016/B978-0-12-374984-0.00670-7.
- 20.Stone MD, Bryant Z, Crisona NJ, Smith SB, Vologodskii A, Bustamante C, Cozzarelli NR. Chirality sensing by Escherichia coli topoisomerase IV and the mechanism of type II topoisomerases. Proc Natl Acad Sci USA. 2003;100:8654–9. 10.1073/pnas.1133178100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Helgesen E, Sætre F, Skarstad K. Topoisomerase IV tracks behind the replication fork and the SeqA complex during DNA replication in Escherichia coli. Sci Rep. 2021;11:474. 10.1038/s41598-020-80043-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang SC, Shapiro L. The topoisomerase IV ParC subunit colocalizes with the Caulobacter replisome and is required for polar localization of replication origins. Proc Natl Acad Sci USA. 2004;101:9251–6. 10.1073/pnas.0402567101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spencer AC, Panda SS. DNA gyrase as a target for quinolones. Biomedicines. 2023. 10.3390/biomedicines11020371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Veselkov DA, Laponogov I, Pan X-S, Selvarajah J, Skamrova GB, Branstrom A, et al. Structure of a quinolone-stabilized cleavage complex of topoisomerase IV from Klebsiella pneumoniae and comparison with a related Streptococcus pneumoniae complex. Acta Crystallogr D Struct Biol. 2016;72:488–96. 10.1107/S2059798316001212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bellon S, Parsons JD, Wei Y, Hayakawa K, Swenson LL, Charifson PS, et al. Crystal structures of Escherichia coli topoisomerase IV ParE subunit (24 and 43 kilodaltons): a single residue dictates differences in novobiocin potency against topoisomerase IV and DNA gyrase. Antimicrob Agents Chemother. 2004;48:1856–64. 10.1128/aac.48.5.1856-1864.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maxwell A, Lawson DM. The ATP-binding site of type II topoisomerases as a target for antibacterial drugs. Curr Top Med Chem. 2003;3:283–303. 10.2174/1568026033452500. [DOI] [PubMed] [Google Scholar]
- 27.Federal Register. Determination That ALBAMYCIN (Novobiocin Sodium) Capsule, 250 Milligrams, Was Withdrawn From Sale for Reasons of Safety or Effectiveness. 2011. https://www.federalregister.gov/documents/2011/01/19/2011-1000/determination-that-albamycin-novobiocin-sodium-capsule-250-milligrams-was-withdrawn-from-sale-for. Accessed 6 Sept 2024.
- 28.Collin F, Karkare S, Maxwell A. Exploiting bacterial DNA gyrase as a drug target: current state and perspectives. Appl Microbiol Biotechnol. 2011;92:479–97. 10.1007/s00253-011-3557-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anderson VE, Osheroff N. Type II topoisomerases as targets for quinolone antibacterials: turning Dr. Jekyll into Mr. Hyde. Curr Pharm Des. 2001;7:337–53. 10.2174/1381612013398013. [DOI] [PubMed]
- 30.Oviatt AA, Gibson EG, Huang J, Mattern K, Neuman KC, Chan PF, Osheroff N. Interactions between gepotidacin and Escherichia coli gyrase and topoisomerase IV: genetic and biochemical evidence for well-balanced dual-targeting. ACS Infect Dis. 2024;10:1137–51. 10.1021/acsinfecdis.3c00346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kokot M, Weiss M, Zdovc I, Hrast M, Anderluh M, Minovski N. Structurally optimized potent dual-targeting NBTI antibacterials with an enhanced bifurcated halogen-bonding propensity. ACS Med Chem Lett. 2021;12:1478–85. 10.1021/acsmedchemlett.1c00345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chan PF, Srikannathasan V, Huang J, Cui H, Fosberry AP, Gu M, et al. Structural basis of DNA gyrase inhibition by antibacterial QPT-1, anticancer drug etoposide and moxifloxacin. Nat Commun. 2015;6:10048. 10.1038/ncomms10048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morgan H, Lipka-Lloyd M, Warren AJ, Hughes N, Holmes J, Burton NP, et al. A 2.8 Å structure of zoliflodacin in a DNA cleavage complex with Staphylococcus aureus DNA gyrase. Int J Mol Sci. 2023. 10.3390/ijms24021634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gibson EG, Bax B, Chan PF, Osheroff N. Mechanistic and structural basis for the actions of the antibacterial gepotidacin against staphylococcus aureus gyrase. ACS Infect Dis. 2019;5:570–81. 10.1021/acsinfecdis.8b00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Watkins RR, Thapaliya D, Lemonovich TL, Bonomo RA. Gepotidacin: a novel, oral, “first-in-class” triazaacenaphthylene antibiotic for the treatment of uncomplicated urinary tract infections and urogenital gonorrhoea. J Antimicrob Chemother. 2023;78:1137–42. 10.1093/jac/dkad060. [DOI] [PubMed] [Google Scholar]
- 36.Bax BD, Chan PF, Eggleston DS, Fosberry A, Gentry DR, Gorrec F, et al. Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature. 2010;466:935–40. 10.1038/nature09197. [DOI] [PubMed] [Google Scholar]
- 37.Vanden Broeck A, Lotz C, Ortiz J, Lamour V. Cryo-EM structure of the complete E. coli DNA gyrase nucleoprotein complex. Nat Commun. 2019;10:4935. 10.1038/s41467-019-12914-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Biedenbach DJ, Bouchillon SK, Hackel M, Miller LA, Scangarella-Oman NE, Jakielaszek C, Sahm DF. In vitro activity of gepotidacin, a novel triazaacenaphthylene bacterial topoisomerase inhibitor, against a broad spectrum of bacterial pathogens. Antimicrob Agents Chemother. 2016;60:1918–23. 10.1128/aac.02820-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scangarella-Oman NE, Dixon P, Koeth LM, DiFranco-Fisher J, Miller LA. Analysis of antimicrobial susceptibility testing methods and variables and in vitro activity of gepotidacin against urogenital Neisseria gonorrhoeae in men. Diagn Microbiol Infect Dis. 2021;101: 115484. 10.1016/j.diagmicrobio.2021.115484. [DOI] [PubMed] [Google Scholar]
- 40.Wagenlehner F, Perry CR, Hooton TM, Scangarella-Oman NE, Millns H, Powell M, et al. Oral gepotidacin versus nitrofurantoin in patients with uncomplicated urinary tract infection (EAGLE-2 and EAGLE-3): two randomised, controlled, double-blind, double-dummy, phase 3, non-inferiority trials. Lancet. 2024;403:741–55. 10.1016/S0140-6736(23)02196-7. [DOI] [PubMed] [Google Scholar]
- 41.Perry CR, Scangarella-Oman NE, Millns H, Flight W, Gatsi S, Jakielaszek C, et al. Efficacy and safety of gepotidacin as treatment of uncomplicated urogenital gonorrhea (EAGLE-1): design of a randomized, comparator-controlled, phase 3 study. Infect Dis Ther. 2023;12:2307–20. 10.1007/s40121-023-00862-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shi C, Zhang Y, Wang T, Lu W, Zhang S, Guo B, et al. Design, synthesis, and biological evaluation of novel DNA gyrase-inhibiting spiropyrimidinetriones as potent antibiotics for treatment of infections caused by multidrug-resistant gram-positive bacteria. J Med Chem. 2019;62:2950–73. 10.1021/acs.jmedchem.8b01750. [DOI] [PubMed] [Google Scholar]
- 43.Innoviva. Innoviva to Acquire Entasis Therapeutics. 2022. https://investor.inva.com/news-releases/news-release-details/innoviva-acquire-entasis-therapeutics. Accessed 6 Sept 2024.
- 44.Bradford PA, Miller AA, O’Donnell J, Mueller JP. Zoliflodacin: an oral spiropyrimidinetrione antibiotic for the treatment of neisseria gonorrheae, including multi-drug-resistant isolates. ACS Infect Dis. 2020;6:1332–45. 10.1021/acsinfecdis.0c00021. [DOI] [PubMed] [Google Scholar]
- 45.Biedenbach DJ, Huband MD, Hackel M, de Jonge BLM, Sahm DF, Bradford PA. In vitro activity of AZD0914, a novel bacterial DNA gyrase/topoisomerase IV inhibitor, against clinically relevant gram-positive and fastidious gram-negative pathogens. Antimicrob Agents Chemother. 2015;59:6053–63. 10.1128/aac.01016-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu J, Jia J, Shi T, Bai Y, Huang Y, Zeng L, Bi H. Evaluating the potency of zoliflodacin against Helicobacter pylori: In vitro activity and conserved GyrB target. Helicobacter. 2024;29: e13075. 10.1111/hel.13075. [DOI] [PubMed] [Google Scholar]
- 47.Innoviva. Innoviva Reports First Quarter 2024 Financial Results; Highlights Recent Company Progress. 2024. https://investor.inva.com/news-releases/news-release-details/innoviva-reports-first-quarter-2024-financial-results-highlights. Accessed 6 Sept 2024.
- 48.Global Antibiotic Research and Development Partnership (GARDP). Novel broad-spectrum antibiotic compound prioritized for development in fight against AMR. 2024. https://gardp.org/novel-broad-spectrum-antibiotic-compound-prioritized-for-development-in-fight-against-amr/. Accessed 6 Sept 2024.
- 49.Carb-X. Pipeline. 2024. https://carb-x.org/wp-content/uploads/2024/06/CARB-X-Pipeline-2024.5.13.pdf. Accessed 6 Sept 2024.
- 50.Huband MD, Lindley JM, Watters AA, Becker HK, Ramachandran V, Balasubramanian V, Flamm RK. In Vitro Activity of BWC0977 (a Novel Bacterial Topoisomerase Inhibitor) and Comparators against Recent Clinical and Molecularly Characterized Enterobacteriaceae and Non-Fermenter Isolates from the United States and Europe. ECCMID Amsterdam Poster #P1845. 2019. https://bugworksresearch.com/wp-content/uploads/2021/11/b-20190415_ECCMID_P1847.pdf. Accessed 6 Sept 2024.
- 51.Peer Mohamed SH, Bharatham N, Katagihallimath N, Sharma S, Nandishaiah R, Ramachandran V, Venkataraman B. Bugworks Research India Pvt Ltd. Preparation of heterocyclic derivatives for use as antibacterial agents. 2018.
- 52.Nandishaiah R, Katagihallimath N, Bharatham N, Datta S, Hameed S. Molecular interactions of BWC0977; a dual inhibitor of DNA gyrase and Topoisomerase IV. ECCMID Copenhagen Poster #2170. 2023. https://bugworksresearch.com/wp-content/uploads/2024/07/P2170_Radha.pdf. Accessed 6 Sept 2024.
- 53.Hameed S, Sharma S, Nandishaiah R, Katagihallimath N, Bharatham N, Shanbhag A, et al. BWC0977, a novel dual target topoisomerase inhibitor: antimicrobial potency, spectrum and mechanism of action. ECCMID Amsterdam Poster #P1844. 2019. https://bugworksresearch.com/wp-content/uploads/2021/11/e-20190415_ECCMID_P1844.pdf. Accessed 6 Sept 2024.
- 54.Lehar SM, Pillow T, Xu M, Staben L, Kajihara KK, Vandlen R, et al. Novel antibody-antibiotic conjugate eliminates intracellular S. aureus. Nature. 2015;527:323–8. 10.1038/nature16057. [DOI] [PubMed] [Google Scholar]
- 55.Poreba M. Protease-activated prodrugs: strategies, challenges, and future directions. FEBS J. 2020;287:1936–69. 10.1111/febs.15227. [DOI] [PubMed] [Google Scholar]
- 56.Zhou Q. Site-specific antibody conjugation for ADC and beyond. Biomedicines. 2017. 10.3390/biomedicines5040064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shopsin B, Kaveri SV, Bayry J. Tackling difficult Staphylococcus aureus infections: antibodies show the way. Cell Host Microbe. 2016;20:555–7. 10.1016/j.chom.2016.10.018. [DOI] [PubMed] [Google Scholar]
- 58.Mariathasan S, Tan M-W. Antibody-antibiotic conjugates: a novel therapeutic platform against bacterial infections. Trends Mol Med. 2017;23:135–49. 10.1016/j.molmed.2016.12.008. [DOI] [PubMed] [Google Scholar]
- 59.Zhou C, Lehar S, Gutierrez J, Rosenberger CM, Ljumanovic N, Dinoso J, et al. Pharmacokinetics and pharmacodynamics of DSTA4637A: a novel THIOMAB™ antibody antibiotic conjugate against Staphylococcus aureus in mice. MAbs. 2016;8:1612–9. 10.1080/19420862.2016.1229722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Peck M, Rothenberg ME, Deng R, Lewin-Koh N, She G, Kamath AV, et al. A phase 1, randomized, single-ascending-dose study to investigate the safety, tolerability, and pharmacokinetics of DSTA4637S, an anti-Staphylococcus aureus thiomab antibody-antibiotic conjugate, in healthy volunteers. Antimicrob Agents Chemother. 2019. 10.1128/aac.02588-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pahil KS, Gilman MSA, Baidin V, Clairfeuille T, Mattei P, Bieniossek C, et al. A new antibiotic traps lipopolysaccharide in its intermembrane transporter. Nature. 2024;625:572–7. 10.1038/s41586-023-06799-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zampaloni C, Mattei P, Bleicher K, Winther L, Thäte C, Bucher C, et al. A novel antibiotic class targeting the lipopolysaccharide transporter. Nature. 2024;625:566–71. 10.1038/s41586-023-06873-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Erbetti I, Ferrari L, Ortombina A, Savoia P, Felici A, Bissantz C, Zampaloni C. 2109. In vitro and in vivo killing kinetics of zosurabalpin (RG6006) against Acinetobacter baumannii. Open Forum Infect Dis. 2023. 10.1093/ofid/ofad500.1733. [Google Scholar]
- 64.Hawser S, Kothari N, Valmont T, Louvel S, Zampaloni C. 2131. Activity of the novel antibiotic zosurabalpin (RG6006) against clinical Acinetobacter isolates from China. Open Forum Infect Dis. 2023. 10.1093/ofid/ofad500.1754. [Google Scholar]
- 65.Guenther A, Millar L, Messer A, Giraudon M, Patel K, Deurloo EJ, et al. 2126. Safety, Tolerability, and Pharmacokinetics (PK) in Healthy Participants Following Single Dose Administration of Zosurabalpin, a Novel Pathogen-Specific Antibiotic for the Treatment of Serious Acinetobacter Infections. Open Forum Infect Dis 2023. 10.1093/ofid/ofad500.1749.
- 66.Srinivas N, Jetter P, Ueberbacher BJ, Werneburg M, Zerbe K, Steinmann J, et al. Peptidomimetic antibiotics target outer-membrane biogenesis in Pseudomonas aeruginosa. Science. 2010;327:1010–3. 10.1126/science.1182749. [DOI] [PubMed] [Google Scholar]
- 67.Botos I, Noinaj N, Buchanan SK. Insertion of proteins and lipopolysaccharide into the bacterial outer membrane. Philos Trans R Soc Lond B Biol Sci. 2017. 10.1098/rstb.2016.0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Andolina G, Bencze L-C, Zerbe K, Müller M, Steinmann J, Kocherla H, et al. A peptidomimetic antibiotic interacts with the periplasmic domain of LptD from Pseudomonas aeruginosa. ACS Chem Biol. 2018;13:666–75. 10.1021/acschembio.7b00822. [DOI] [PubMed] [Google Scholar]
- 69.Werneburg M, Zerbe K, Juhas M, Bigler L, Stalder U, Kaech A, et al. Inhibition of lipopolysaccharide transport to the outer membrane in Pseudomonas aeruginosa by peptidomimetic antibiotics. ChemBioChem. 2012;13:1767–75. 10.1002/cbic.201200276. [DOI] [PubMed] [Google Scholar]
- 70.Sader HS, Flamm RK, Dale GE, Rhomberg PR, Castanheira M. Murepavadin activity tested against contemporary (2016–17) clinical isolates of XDR Pseudomonas aeruginosa. J Antimicrob Chemother. 2018;73:2400–4. 10.1093/jac/dky227. [DOI] [PubMed] [Google Scholar]
- 71.Díez-Aguilar M, Hernández-García M, Morosini M-I, Fluit A, Tunney MM, Huertas N, et al. Murepavadin antimicrobial activity against and resistance development in cystic fibrosis Pseudomonas aeruginosa isolates. J Antimicrob Chemother. 2021;76:984–92. 10.1093/jac/dkaa529. [DOI] [PubMed] [Google Scholar]
- 72.Spexis AG. Polyphor closes the Phase III PRISM studies of murepavadin intravenous formulation and evaluates further product improvement options. 2019. https://spexisbio.com/news-adhoc/news-detail/?newsid=1800485. Accessed 6 Sept 2024.
- 73.Spexis AG. Polyphor temporarily halts enrollment in the Phase III studies of murepavadin for the treatment of patients with nosocomial pneumonia. 2019. https://spexisbio.com/news/corporate-news-details/?newsid=1775911. Accessed 6 Sept 2024.
- 74.Prasad NK, Seiple IB, Cirz RT, Rosenberg OS. Leaks in the pipeline: a failure analysis of gram-negative antibiotic development from 2010 to 2020. Antimicrob Agents Chemother. 2022;66: e0005422. 10.1128/aac.00054-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Spexis AG. Spexis reports solid safety and pharmacokinetics results from first-in-human study with inhaled murepavadin, a novel macrocycle compound. 2023. https://spexisbio.com/news-adhoc/news-detail/?newsid=2419969. Accessed 6 Sept 2024.
- 76.Wang Y, Chang RYK, Britton WJ, Chan H-K. Advances in the development of antimicrobial peptides and proteins for inhaled therapy. Adv Drug Deliv Rev. 2022;180: 114066. 10.1016/j.addr.2021.114066. [DOI] [PubMed] [Google Scholar]
- 77.Ekkelenkamp MB, Cantón R, Díez-Aguilar M, Tunney MM, Gilpin DF, Bernardini F, et al. Susceptibility of Pseudomonas aeruginosa recovered from cystic fibrosis patients to murepavadin and 13 comparator antibiotics. Antimicrob Agents Chemother. 2020. 10.1128/AAC.01541-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Spexis AG. Spexis provides business update and announces financial results for the full year 2022. 2023. https://spexisbio.com/news-adhoc/news-detail/?newsid=2522559. Accessed 6 Sept 2024.
- 79.Martin-Loeches I, Dale GE, Torres A. Murepavadin: a new antibiotic class in the pipeline. Expert Rev Anti Infect Ther. 2018;16:259–68. 10.1080/14787210.2018.1441024. [DOI] [PubMed] [Google Scholar]
- 80.Kaplan N, Albert M, Awrey D, Bardouniotis E, Berman J, Clarke T, et al. Mode of action, in vitro activity, and in vivo efficacy of AFN-1252, a selective antistaphylococcal FabI inhibitor. Antimicrob Agents Chemother. 2012;56:5865–74. 10.1128/aac.01411-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Menetrey A, Janin A, Pullman J, Overcash JS, Haouala A, Leylavergne F, et al. Bone and joint tissue penetration of the staphylococcus-selective antibiotic afabicin in patients undergoing elective hip replacement surgery. Antimicrob Agents Chemother. 2019. 10.1128/AAC.01669-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Payne DJ, Miller WH, Berry V, Brosky J, Burgess WJ, Chen E, et al. Discovery of a novel and potent class of FabI-directed antibacterial agents. Antimicrob Agents Chemother. 2002;46:3118–24. 10.1128/AAC.46.10.3118-3124.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Debiopharm. Debiopharm Group™ to Acquire Affinium’s Antibiotic Clinical Assets and Platform to Identify and Develop Targeted Antibiotics. 2014. https://www.debiopharm.com/drug-development/press-releases/debiopharm-group-to-acquire-affiniums-antibiotic-clinical-assets-and-platform-to-identify-and-develop-targeted-antibiotics/. Accessed 6 Sept 2024.
- 84.Heath RJ, Rock CO. Enoyl-acyl carrier protein reductase (fabI) plays a determinant role in completing cycles of fatty acid elongation in Escherichia coli. J Biol Chem. 1995;270:26538–42. 10.1074/jbc.270.44.26538. [DOI] [PubMed] [Google Scholar]
- 85.Zhang Y-M, Rock CO. Membrane lipid homeostasis in bacteria. Nat Rev Microbiol. 2008;6:222–33. 10.1038/nrmicro1839. [DOI] [PubMed] [Google Scholar]
- 86.Hawser S, Gueny M, Rochat I, Morrissey I, Magnet S, Dieppois G. Activity of Debio 1452 against Staphylococcus spp. collected in 2015 / 2016. ECCMID Poster P1824. 2018. https://www.ihma.com/app/uploads/Debiopharm_IHMA_IHMA_ECCMID_2018_FINAL.pdf. Accessed 26 Nov 2024.
- 87.Wittke F, Vincent C, Chen J, Heller B, Kabler H, Overcash JS, et al. Afabicin, a first-in-class antistaphylococcal antibiotic, in the treatment of acute bacterial skin and skin structure infections: clinical noninferiority to vancomycin/linezolid. Antimicrob Agents Chemother. 2020. 10.1128/AAC.00250-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rosado-Lugo JD, Sun Y, Banerjee A, Cao Y, Datta P, Zhang Y, et al. Evaluation of 2,6-difluoro-3-(oxazol-2-ylmethoxy)benzamide chemotypes as Gram-negative FtsZ inhibitors. J Antibiot (Tokyo). 2022;75:385–95. 10.1038/s41429-022-00531-9. [DOI] [PubMed] [Google Scholar]
- 89.Fujita J, Maeda Y, Mizohata E, Inoue T, Kaul M, Parhi AK, et al. Structural flexibility of an inhibitor overcomes drug resistance mutations in Staphylococcus aureus FtsZ. ACS Chem Biol. 2017;12:1947–55. 10.1021/acschembio.7b00323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kaul M, Mark L, Parhi AK, LaVoie EJ, Pilch DS. Combining the FtsZ-targeting prodrug TXA709 and the cephalosporin cefdinir confers synergy and reduces the frequency of resistance in methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2016;60:4290–6. 10.1128/AAC.00613-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Haydon DJ, Bennett JM, Brown D, Collins I, Galbraith G, Lancett P, et al. Creating an antibacterial with in vivo efficacy: synthesis and characterization of potent inhibitors of the bacterial cell division protein FtsZ with improved pharmaceutical properties. J Med Chem. 2010;53:3927–36. 10.1021/jm9016366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Monteiro JM, Pereira AR, Reichmann NT, Saraiva BM, Fernandes PB, Veiga H, et al. Peptidoglycan synthesis drives an FtsZ-treadmilling-independent step of cytokinesis. Nature. 2018;554:528–32. 10.1038/nature25506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schäper S, Brito AD, Saraiva BM, Squyres GR, Holmes MJ, Garner EC, et al. Cell constriction requires processive septal peptidoglycan synthase movement independent of FtsZ treadmilling in Staphylococcus aureus. Nat Microbiol. 2024;9:1049–63. 10.1038/s41564-024-01629-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kaul M, Mark L, Zhang Y, Parhi AK, LaVoie EJ, Pilch DS. Pharmacokinetics and in vivo antistaphylococcal efficacy of TXY541, a 1-methylpiperidine-4-carboxamide prodrug of PC190723. Biochem Pharmacol. 2013;86:1699–707. 10.1016/j.bcp.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 95.Kaul M, Mark L, Zhang Y, Parhi AK, Lyu YL, Pawlak J, et al. TXA709, an FtsZ-targeting benzamide prodrug with improved pharmacokinetics and enhanced in vivo efficacy against methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2015;59:4845–55. 10.1128/aac.00708-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Haydon DJ, Stokes NR, Ure R, Galbraith G, Bennett JM, Brown DR, et al. An inhibitor of FtsZ with potent and selective anti-staphylococcal activity. Science. 2008;321:1673–5. 10.1126/science.1159961. [DOI] [PubMed] [Google Scholar]
- 97.Smith PA, Romesberg FE. Mechanism of action of the arylomycin antibiotics and effects of signal peptidase I inhibition. Antimicrob Agents Chemother. 2012;56:5054–60. 10.1128/AAC.00785-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dieppois G, Smith P, Man Wah Tan. In vitro activity of two novel antibiotics, GDC-5780 and GDC-0829, against a global panel of gram-negative priority pathogens. ECCMID Barcelona Poster BES1603. 2024. https://medically.roche.com/content/dam/pdmahub/restricted/infectious-disease/escmid-2024/ESCMID-2024-poster-dieppois-in-vitro-activity.pdf. Accessed 6 Sept 2024.
- 99.Koehler MFT, Chen Y-C, Chen Y, Chen Y, Crawford JJ, Durk MR, et al. Lipid tales: optimizing arylomycin membrane anchors. ACS Med Chem Lett. 2023;14:1524–30. 10.1021/acsmedchemlett.3c00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Smith PA, Koehler MFT, Girgis HS, Yan D, Chen Y, Chen Y, et al. Optimized arylomycins are a new class of Gram-negative antibiotics. Nature. 2018;561:189–94. 10.1038/s41586-018-0483-6. [DOI] [PubMed] [Google Scholar]
- 101.Xu M, La H, Smith P, Ma F, Girgis H, Weiss W, et al. Determination of the PK/PD driver for the LepB inhibitor, GDC-0829, a novel broad-spectrum antibiotic against gram-negative bacterial infections. ECCMID Barcelona Poster #2315. 2024. https://medically.roche.com/content/dam/pdmahub/restricted/infectious-disease/escmid-2024/ESCMID-2024-poster-xu-determination-of-the-PK-PD.pdf. Accessed 6 Sept 2024.
- 102.Genentech. Pipeline. 2024. https://www.gene.com/medical-professionals/pipeline. Accessed 6 Sept 2024.
- 103.Roche. Roche Group development pipeline. 2024. https://assets.roche.com/f/176343/x/a282874c3a/pharmaq124.pdf. Accessed 6 Sept 2024.
- 104.Mookherjee N, Anderson MA, Haagsman HP, Davidson DJ. Antimicrobial host defence peptides: functions and clinical potential. Nat Rev Drug Discov. 2020;19:311–32. 10.1038/s41573-019-0058-8. [DOI] [PubMed] [Google Scholar]
- 105.Fjell CD, Hiss JA, Hancock REW, Schneider G. Designing antimicrobial peptides: form follows function. Nat Rev Drug Discov. 2011;11:37–51. 10.1038/nrd3591. [DOI] [PubMed] [Google Scholar]
- 106.Choi S, Isaacs A, Clements D, Liu D, Kim H, Scott RW, et al. De novo design and in vivo activity of conformationally restrained antimicrobial arylamide foldamers. Proc Natl Acad Sci USA. 2009;106:6968–73. 10.1073/pnas.0811818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Xuan J, Feng W, Wang J, Wang R, Zhang B, Bo L, et al. Antimicrobial peptides for combating drug-resistant bacterial infections. Drug Resist Updat. 2023;68: 100954. 10.1016/j.drup.2023.100954. [DOI] [PubMed] [Google Scholar]
- 108.Sakr A, Brégeon F, Rolain J-M, Blin O. Staphylococcus aureus nasal decolonization strategies: a review. Expert Rev Anti Infect Ther. 2019;17:327–40. 10.1080/14787210.2019.1604220. [DOI] [PubMed] [Google Scholar]
- 109.Nilsson AC, Janson H, Wold H, Fugelli A, Andersson K, Håkangård C, et al. LTX-109 is a novel agent for nasal decolonization of methicillin-resistant and -sensitive Staphylococcus aureus. Antimicrob Agents Chemother. 2015;59:145–51. 10.1128/aac.03513-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Isaksson J, Brandsdal BO, Engqvist M, Flaten GE, Svendsen JSM, Stensen W. A synthetic antimicrobial peptidomimetic (LTX 109): stereochemical impact on membrane disruption. J Med Chem. 2011;54:5786–95. 10.1021/jm200450h. [DOI] [PubMed] [Google Scholar]
- 111.Svenson J, Stensen W, Brandsdal B-O, Haug BE, Monrad J, Svendsen JS. Antimicrobial peptides with stability toward tryptic degradation. Biochemistry. 2008;47:3777–88. 10.1021/bi7019904. [DOI] [PubMed] [Google Scholar]
- 112.Saravolatz LD, Pawlak J, Johnson L, Bonilla H, Fakih MG, Fugelli A, Olsen WM. In vitro activities of LTX-109, a synthetic antimicrobial peptide, against methicillin-resistant, vancomycin-intermediate, vancomycin-resistant, daptomycin-nonsusceptible, and linezolid-nonsusceptible Staphylococcus aureus. Antimicrob Agents Chemother. 2012;56:4478–82. 10.1128/aac.00194-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Saravolatz LD, Pawlak J, Martin H, Saravolatz S, Johnson L, Wold H, et al. Postantibiotic effect and postantibiotic sub-MIC effect of LTX-109 and mupirocin on Staphylococcus aureus blood isolates. Lett Appl Microbiol. 2017;65:410–3. 10.1111/lam.12792. [DOI] [PubMed] [Google Scholar]
- 114.Feng Q, Huang Y, Chen M, Li G, Chen Y. Functional synergy of α-helical antimicrobial peptides and traditional antibiotics against Gram-negative and Gram-positive bacteria in vitro and in vivo. Eur J Clin Microbiol Infect Dis. 2015;34:197–204. 10.1007/s10096-014-2219-3. [DOI] [PubMed] [Google Scholar]
- 115.Pütsep K, Brändén CI, Boman HG, Normark S. Antibacterial peptide from H. pylori. Nature. 1999;398:671–2. 10.1038/19439. [DOI] [PubMed] [Google Scholar]
- 116.Zhao L, Huang Y, Gao S, Cui Y, He D, Wang L, Chen Y. Comparison on effect of hydrophobicity on the antibacterial and antifungal activities of α-helical antimicrobial peptides. Sci China Chem. 2013;56:1307–14. 10.1007/s11426-013-4884-y. [Google Scholar]
- 117.Mai X, Huang J, Tan J, Huang Y, Chen Y. Effects and mechanisms of the secondary structure on the antimicrobial activity and specificity of antimicrobial peptides. J Pept Sci. 2015;21:561–8. 10.1002/psc.2767. [DOI] [PubMed] [Google Scholar]
- 118.Deslouches B, Phadke SM, Lazarevic V, Cascio M, Islam K, Montelaro RC, Mietzner TA. De novo generation of cationic antimicrobial peptides: influence of length and tryptophan substitution on antimicrobial activity. Antimicrob Agents Chemother. 2005;49:316–22. 10.1128/aac.49.1.316-322.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Deslouches B, Steckbeck JD, Craigo JK, Doi Y, Burns JL, Montelaro RC. Engineered cationic antimicrobial peptides to overcome multidrug resistance by ESKAPE pathogens. Antimicrob Agents Chemother. 2015;59:1329–33. 10.1128/AAC.03937-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kumagai A, Dupuy FG, Arsov Z, Elhady Y, Moody D, Ernst RK, et al. Elastic behavior of model membranes with antimicrobial peptides depends on lipid specificity and d-enantiomers. Soft Matter. 2019;15:1860–8. 10.1039/c8sm02180e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Heinrich F, Salyapongse A, Kumagai A, Dupuy FG, Shukla K, Penk A, et al. Synergistic biophysical techniques reveal structural mechanisms of engineered cationic antimicrobial peptides in lipid model membranes. Chemistry. 2020;26:6247–56. 10.1002/chem.202000212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mandell JB, Deslouches B, Montelaro RC, Shanks RMQ, Doi Y, Urish KL. Elimination of antibiotic resistant surgical implant biofilms using an engineered cationic amphipathic peptide WLBU2. Sci Rep. 2017;7:18098. 10.1038/s41598-017-17780-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Huang DB, Brothers KM, Mandell JB, Taguchi M, Alexander PG, Parker DM, et al. Engineered peptide PLG0206 overcomes limitations of a challenging antimicrobial drug class. PLoS ONE. 2022;17: e0274815. 10.1371/journal.pone.0274815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Huang D, Pachuda N, Sauer JM, Dobbins D, Steckbeck J. The engineered antibiotic peptide PLG0206 Eliminates biofilms and is a potential treatment for periprosthetic joint infections. Antibiotics (Basel). 2021. 10.3390/antibiotics11010041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Masihzadeh S, Amin M, Farshadzadeh Z. In vitro and in vivo antibiofilm activity of the synthetic antimicrobial peptide WLBU2 against multiple drug resistant Pseudomonas aeruginosa strains. BMC Microbiol. 2023;23:131. 10.1186/s12866-023-02886-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zmistowski B, Karam JA, Durinka JB, Casper DS, Parvizi J. Periprosthetic joint infection increases the risk of one-year mortality. J Bone Joint Surg Am. 2013;95:2177–84. 10.2106/JBJS.L.00789. [DOI] [PubMed] [Google Scholar]
- 127.Huang D, Parker DM, Mandell JB, Brothers KM, Gish CG, Koch JA, et al. Prospective activity of PLG0206, an engineered antimicrobial peptide, on chronic periprosthetic joint infection total knee arthroplasty components ex vivo: the knee explant analysis (KnEA) study. Microbiol Spectr. 2021;9: e0187921. 10.1128/spectrum.01879-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Huang D, Parker D, Pachuda N, Dobbins D, Steckbeck J, Urish K. 1040. Knee Explant Analysis (KnEA) Using PLG0206 in Periprosthetic Joint Infection (KnEA Study). Open Forum Infect Dis. 2021;8:S610-S611. 10.1093/ofid/ofab466.1234.
- 129.Huang D, Dobbins D, Ghahramani P, Friedland I, Steckbeck J. A phase 1 study of the safety, tolerability, and pharmacokinetics of single ascending doses of a first-in-human engineered cationic peptide, PLG0206, intravenously administered in healthy subjects. Antimicrob Agents Chemother. 2022;66: e0144121. 10.1128/AAC.01441-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Huang D, Dobbins D, Ghahramani P, Steckbeck J. 1098. A phase 1 safety and tolerability of single ascending doses of a novel engineered cationic peptide, PLG0206, in healthy subjects. Open Forum Infect Dis. 2021;8:S641. 10.1093/ofid/ofab466.1292.
- 131.Elsalem L, Khasawneh A, Al SS. WLBU2 antimicrobial peptide as a potential therapeutic for treatment of resistant bacterial. Turk J Pharm Sci. 2022;19:110–6. 10.4274/tjps.galenos.2020.43078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mandel S, Michaeli J, Nur N, Erbetti I, Zazoun J, Ferrari L, et al. OMN6 a novel bioengineered peptide for the treatment of multidrug resistant Gram negative bacteria. Sci Rep. 2021;11:6603. 10.1038/s41598-021-86155-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Michaeli J, Mandel S, Maximov S, Zazoun J, Savoia P, Kothari N, et al. In vitro and in vivo antimicrobial activity of the novel peptide OMN6 against multidrug-resistant Acinetobacter baumannii. Antibiotics (Basel). 2022. 10.3390/antibiotics11091201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Omnix Medical. Safety and pharmacokinetic results from the first-in-human study with intravenously administered OMN6, a novel antimicrobial peptide for Acinetobacter baumannii (AB), conducted in healthy volunteers. 2024. https://omnixmedical.com/2024/05/10/safety_and_pharmacokinetic_results_from_the_first_in_human_study/. Accessed 6 Sept 2024.
- 135.Mensa B, Howell GL, Scott R, DeGrado WF. Comparative mechanistic studies of brilacidin, daptomycin, and the antimicrobial peptide LL16. Antimicrob Agents Chemother. 2014;58:5136–45. 10.1128/aac.02955-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bakovic A, Risner K, Bhalla N, Alem F, Chang TL, Weston WK, et al. Brilacidin demonstrates inhibition of SARS-CoV-2 in cell culture. Viruses. 2021;13:271. 10.3390/v13020271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kratochvil HT, Newberry RW, Mensa B, Mravic M, DeGrado WF. Spiers memorial lecture: analysis and de novo design of membrane-interactive peptides. Faraday Discuss. 2021;232:9–48. 10.1039/D1FD00061F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kowalski RP, Romanowski EG, Yates KA, Mah FS. An independent evaluation of a novel peptide mimetic, Brilacidin (PMX30063), for ocular anti-infective. J Ocul Pharmacol Ther. 2016;32:23–7. 10.1089/jop.2015.0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Jorgensen DM, Scott RW, O'Riordan WA, Tack KJ. A randomized, double-blind study comparing single-dose and short-course brilacidin to daptomycin in the treatment of acute bacterial skin and skin structure infections (ABSSSI). ECCMID Copenhagen oral presentation O195. 2015. https://static1.squarespace.com/static/5715352e20c647639137f992/t/583f8286d2b85731b8713a36/1480557192947/A-Randomized-Double-Blind-Study-Comparing-Single-Dose-and-Short-Course-Brilacidin-to-Daptomycin-in-the-Treatment-of-Acute-Bacterial-Skin-Skin-Structure-Infections-ABSSSI1.pdf. Accessed 6 Sept 2024.
- 140.Scott RW, Sonis ST, Kroczak B, Freeman KB, DeGrado WF, Holden SA, et al. Brilacidin, host defence peptide mimetic, one of a new class of immunomodulatory agents that can target multiple disease indications. ECCMID Copenhagen Poster EV0201. 2015. https://static1.squarespace.com/static/5715352e20c647639137f992/t/583f83821b631be3d85bc071/1480557442978/ECCMID-2015-OM-poster.pdf. Accessed 6 Sept 2024.
- 141.Innovation Pharmaceuticals. Cellceutix to Start Brilacidin Phase 3 Program in ABSSSI. 2015. https://www.ipharminc.com/press-release/2016/11/16/cellceutix-to-start-brilacidin-phase-3-program-in-absssi. Accessed 6 Sept 2024.
- 142.Larwood DJ, Stevens DA. Antifungal activity of brilacidin, a nonpeptide host defense molecule. Antibiotics (Basel). 2024;13:405. 10.3390/antibiotics13050405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Anderson CA, Barrera MD, Boghdeh NA, Smith M, Alem F, Narayanan A. Brilacidin as a broad-spectrum inhibitor of enveloped, acutely infectious viruses. Microorganisms. 2024;12:54. 10.3390/microorganisms12010054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hu Y, Jo H, DeGrado WF, Wang J. Brilacidin, a COVID-19 drug candidate, demonstrates broad-spectrum antiviral activity against human coronaviruses OC43, 229E, and NL63 through targeting both the virus and the host cell. J Med Virol. 2022;94:2188–200. 10.1002/jmv.27616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Agrawal P, Kumari S, Mohmmed A, Malhotra P, Sharma U, Sahal D. Identification of novel, potent, and selective compounds against malaria using glideosomal-associated protein 50 as a drug target. ACS Omega. 2023;8:38506–23. 10.1021/acsomega.3c05323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Bush K, Bradford PA. Interplay between β-lactamases and new β-lactamase inhibitors. Nat Rev Microbiol. 2019;17:295–306. 10.1038/s41579-019-0159-8. [DOI] [PubMed] [Google Scholar]
- 147.Bush K. Past and present perspectives on β-lactamases. Antimicrob Agents Chemother. 2018. 10.1128/aac.01076-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ambler RP. The structure of beta-lactamases. Philos Trans R Soc Lond B Biol Sci. 1980;289:321–31. 10.1098/rstb.1980.0049. [DOI] [PubMed] [Google Scholar]
- 149.Bush K, Jacoby GA. Updated functional classification of beta-lactamases. Antimicrob Agents Chemother. 2010;54:969–76. 10.1128/aac.01009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bush K, Jacoby GA, Medeiros AA. A functional classification scheme for beta-lactamases and its correlation with molecular structure. Antimicrob Agents Chemother. 1995;39:1211–33. 10.1128/aac.39.6.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sawa T, Kooguchi K, Moriyama K. Molecular diversity of extended-spectrum β-lactamases and carbapenemases, and antimicrobial resistance. J Intensive Care. 2020;8:13. 10.1186/s40560-020-0429-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.He Y, Lei J, Pan X, Huang X, Zhao Y. The hydrolytic water molecule of Class A β-lactamase relies on the acyl-enzyme intermediate ES* for proper coordination and catalysis. Sci Rep. 2020;10:10205. 10.1038/s41598-020-66431-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Li R, Chen X, Zhou C, Dai Q-Q, Yang L. Recent advances in β-lactamase inhibitor chemotypes and inhibition modes. Eur J Med Chem. 2022;242: 114677. 10.1016/j.ejmech.2022.114677. [DOI] [PubMed] [Google Scholar]
- 154.Docquier J-D, Mangani S. An update on β-lactamase inhibitor discovery and development. Drug Resist Update. 2018;36:13–29. 10.1016/j.drup.2017.11.002. [DOI] [PubMed] [Google Scholar]
- 155.Morroni G, Bressan R, Fioriti S, D’Achille G, Mingoia M, Cirioni O, et al. Antimicrobial activity of aztreonam in combination with old and new β-lactamase inhibitors against MBL and ESBL co-producing gram-negative clinical isolates: possible options for the treatment of complicated infections. Antibiotics (Basel). 2021. 10.3390/antibiotics10111341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ito-Horiyama T, Ishii Y, Ito A, Sato T, Nakamura R, Fukuhara N, et al. Stability of novel siderophore cephalosporin S-649266 against clinically relevant carbapenemases. Antimicrob Agents Chemother. 2016;60:4384–6. 10.1128/aac.03098-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Boyd SE, Livermore DM, Hooper DC, Hope WW. Metallo-β-lactamases: structure, function, epidemiology, treatment options, and the development pipeline. Antimicrob Agents Chemother. 2020. 10.1128/aac.00397-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Reading C, Cole M. Clavulanic acid: a beta-lactamase-inhiting beta-lactam from Streptomyces clavuligerus. Antimicrob Agents Chemother. 1977;11:852–7. 10.1128/aac.11.5.852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Aronoff SC, Jacobs MR, Johenning S, Yamabe S. Comparative activities of the beta-lactamase inhibitors YTR 830, sodium clavulanate, and sulbactam combined with amoxicillin or ampicillin. Antimicrob Agents Chemother. 1984;26:580–2. 10.1128/aac.26.4.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Negatu DA, Del González RR, Cacho-Izquierdo M, Barros-Aguirre D, Lelievre J, Rullas J, et al. Activity of oral tebipenem-avibactam in a mouse model of mycobacterium abscessus lung infection. Antimicrob Agents Chemother. 2023;67: e0145922. 10.1128/aac.01459-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Durand-Réville TF, Guler S, Comita-Prevoir J, Chen B, Bifulco N, Huynh H, et al. ETX2514 is a broad-spectrum β-lactamase inhibitor for the treatment of drug-resistant Gram-negative bacteria including Acinetobacter baumannii. Nat Microbiol. 2017;2:17104. 10.1038/nmicrobiol.2017.104. [DOI] [PubMed] [Google Scholar]
- 162.Mangion IK, Ruck RT, Rivera N, Huffman MA, Shevlin M. A concise synthesis of a β-lactamase inhibitor. Org Lett. 2011;13:5480–3. 10.1021/ol202195n. [DOI] [PubMed] [Google Scholar]
- 163.Yang Y, Yan Y-H, Schofield CJ, McNally A, Zong Z, Li G-B. Metallo-β-lactamase-mediated antimicrobial resistance and progress in inhibitor discovery. Trends Microbiol. 2023;31:735–48. 10.1016/j.tim.2023.01.013. [DOI] [PubMed] [Google Scholar]
- 164.Centers for Disease Control and Prevention (CDC). Antibiotic use and stewardship: all antibiotic classes. 2022. https://arpsp.cdc.gov/profile/antibiotic-use/all-classes?class-select-community=class212,class213,class214,class216,class215,class208,class210,class204,class205&tabsection-724=0. Accessed 6 Sept 2024.
- 165.Trout RE, Zulli A, Mesaros E, Jackson RW, Boyd S, Liu B, et al. Discovery of VNRX-7145 (VNRX-5236 Etzadroxil): an orally bioavailable β-lactamase inhibitor for enterobacterales expressing ambler class A, C, and D enzymes. J Med Chem. 2021;64:10155–66. 10.1021/acs.jmedchem.1c00437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Chatwin CL, Hamrick JC, Trout REL, Myers CL, Cusick SM, Weiss WJ, et al. Microbiological characterization of VNRX-5236, a broad-spectrum β-lactamase inhibitor for rescue of the orally bioavailable cephalosporin ceftibuten as a carbapenem-sparing agent against strains of enterobacterales expressing extended-spectrum β-lactamases and serine carbapenemases. Antimicrob Agents Chemother. 2021;65: e0055221. 10.1128/aac.00552-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Karsisiotis AI, Damblon CF, Roberts GCK. A variety of roles for versatile zinc in metallo-β-lactamases. Metallomics. 2014;6:1181–97. 10.1039/C4MT00066H. [DOI] [PubMed] [Google Scholar]
- 168.Hecker SJ, Reddy KR, Lomovskaya O, Griffith DC, Rubio-Aparicio D, Nelson K, et al. Discovery of cyclic boronic acid QPX7728, an ultrabroad-spectrum inhibitor of serine and metallo-β-lactamases. J Med Chem. 2020;63:7491–507. 10.1021/acs.jmedchem.9b01976. [DOI] [PubMed] [Google Scholar]
- 169.Liu B, Trout REL, Chu G-H, McGarry D, Jackson RW, Hamrick JC, et al. Discovery of taniborbactam (VNRX-5133): a broad-spectrum serine- and metallo-β-lactamase inhibitor for carbapenem-resistant bacterial infections. J Med Chem. 2020;63:2789–801. 10.1021/acs.jmedchem.9b01518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Menarini Group. Venatorx Pharmaceuticals and Menarini Group Enter Commercial Agreement for Cefepime-Taniborbactam in 96 Countries. 2024. https://www.menarini.com/en-us/news/news-detail/venatorx-pharmaceuticals-and-menarini-group-enter-commercial-agreement-for-cefepime-taniborbactam-in-96-countries. Accessed 6 Sept 2024.
- 171.Le Terrier C, Nordmann P, Freret C, Seigneur M, Poirel L. Impact of acquired broad spectrum β-lactamases on susceptibility to novel combinations made of β-lactams (aztreonam, cefepime, meropenem, and imipenem) and novel β-lactamase inhibitors in Escherichia coli and Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2023;67: e0033923. 10.1128/aac.00339-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wagenlehner FM, Gasink LB, McGovern PC, Moeck G, McLeroth P, Dorr M, et al. Cefepime-taniborbactam in complicated urinary tract infection. N Engl J Med. 2024;390:611–22. 10.1056/NEJMoa2304748. [DOI] [PubMed] [Google Scholar]
- 173.Venatorx Pharmaceuticals. Pipeline: Cefepime-Taniborbactam. 2024. https://venatorx.com/pipeline/cefepime-taniborbactam/. Accessed 6 Sept 2024.
- 174.Zhanel GG, Mansour C, Mikolayanko S, Lawrence CK, Zelenitsky S, Ramirez D, et al. Cefepime-taniborbactam: a novel cephalosporin/β-lactamase inhibitor combination. Drugs. 2024. 10.1007/s40265-024-02082-9. [DOI] [PubMed] [Google Scholar]
- 175.Shionogi. Shionogi Further Extends Infectious Disease Innovation Platform with Planned Acquisition of Qpex Biopharma, Inc. 2023. https://www.shionogi.com/global/en/news/2023/6/20230626_1.html. Accessed 6 Sept 2024.
- 176.Lomovskaya O, Tsivkovski R, Totrov M, Dressel D, Castanheira M, Dudley M. New boronate drugs and evolving NDM-mediated beta-lactam resistance. Antimicrob Agents Chemother. 2023;67: e0057923. 10.1128/aac.00579-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lomovskaya O, Tsivkovski R, Sun D, Reddy R, Totrov M, Hecker S, et al. QPX7728, an ultra-broad-spectrum B-lactamase inhibitor for intravenous and oral therapy: overview of biochemical and microbiological characteristics. Front Microbiol. 2021;12: 697180. 10.3389/fmicb.2021.697180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Lomovskaya O, Castanheira M, Lindley J, Rubio-Aparicio D, Nelson K, Tsivkovski R, et al. In vitro potency of xeruborbactam in combination with multiple β-lactam antibiotics in comparison with other β-lactam/β-lactamase inhibitor (BLI) combinations against carbapenem-resistant and extended-spectrum β-lactamase-producing Enterobacterales. Antimicrob Agents Chemother. 2023;67: e0044023. 10.1128/aac.00440-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Sun D, Tsivkovski R, Pogliano J, Tsunemoto H, Nelson K, Rubio-Aparicio D, Lomovskaya O. Intrinsic antibacterial activity of xeruborbactam in vitro: assessing spectrum and mode of action. Antimicrob Agents Chemother. 2022;66: e0087922. 10.1128/aac.00879-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Griffith D, Roberts J, Wallis S, Hernandez-Mitre MP, Morgan E, Dudley M, Loutit J. 218. A phase 1 study of the single-dose safety, tolerability, and pharmacokinetics of the beta-lactamase inhibitor xeruborbactam administered as the isobutyryloxymethyl oral prodrug to healthy adult subjects. Open Forum Infect Dis 2022. 10.1093/ofid/ofac492.296.
- 181.Griffith D, Roberts J, Wallis S, Hernandez-Mitre MP, Morgan E, Gehrke S, et al. 216. A phase 1 study of the safety, tolerability, and pharmacokinetics of multiple doses of the beta-lactamase inhibitor xeruborbactam alone and in combination meropenem in healthy adult subjects. Open Forum Infect Dis 2022. 10.1093/ofid/ofac492.294.
- 182.Reddy KR, Parkinson J, Sabet M, Tarazi Z, Boyer SH, Lomovskaya O, et al. Selection of QPX7831, an orally bioavailable prodrug of boronic acid β-lactamase inhibitor QPX7728. J Med Chem. 2021;64:17523–9. 10.1021/acs.jmedchem.1c01722. [DOI] [PubMed] [Google Scholar]
- 183.Hernández-Mitre MP, Wallis SC, Morgan EE, Dudley MN, Loutit JS, Griffith DC, Roberts JA. A phase I, randomized, double-blind, placebo-controlled, ascending single- and multiple-dose study of the pharmacokinetics, safety, and tolerability of oral ceftibuten in healthy adult subjects. Antimicrob Agents Chemother. 2024;68: e0109923. 10.1128/aac.01099-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Sumitomo Pharma. Joint Research between The Kitasato Institute and Sumitomo Pharma:Drug Combination of Meropenem and KSP-1007 for Treatment of CarbapenemResistant Bacteria Infections Designated as Qualified Infectious Disease Product (QIDP)/Fast Track. 2022. https://www.sumitomo-pharma.com/news/assets/pdf/ene20220929.pdf. Accessed 6 Sept 2024.
- 185.Sumitomo Pharma. Development Pipeline. 2024. https://www.sumitomo-pharma.com/rd/pipeline_new-medicine/pipeline.html. Accessed 6 Sept 2024.
- 186.Takemoto K, Nakayama R, Fujimoto K, Suzuki Y, Takarabe Y, Honsho M, et al. In vitro and in vivo activities of KSP-1007, a broad-spectrum inhibitor of serine- and metallo-β-lactamases, in combination with meropenem against carbapenem-resistant Gram-negative bacteria. Antimicrob Agents Chemother. 2024:e0160223. 10.1128/aac.01602-23. [DOI] [PMC free article] [PubMed]
- 187.Naik H, Shen S, Bordelon N, Meyer J, Tomita Y, Azoulay S. AAR-FRIDAY-519. Safety, Tolerability, And Pharmacokinetics Of KSP-1007 After Single And Multiple Ascending Doses Alone Or In Combination With Meropenem In Healthy Subjects. 2023. https://www.abstractsonline.com/pp8/#!/10789/presentation/6827. Accessed 6 Sept 2024.
- 188.Rasko DA, Sperandio V. Anti-virulence strategies to combat bacteria-mediated disease. Nat Rev Drug Discov. 2010;9:117–28. 10.1038/nrd3013. [DOI] [PubMed] [Google Scholar]
- 189.Dickey SW, Cheung GYC, Otto M. Different drugs for bad bugs: antivirulence strategies in the age of antibiotic resistance. Nat Rev Drug Discov. 2017;16:457–71. 10.1038/nrd.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Theuretzbacher U, Piddock LJV. Non-traditional antibacterial therapeutic options and challenges. Cell Host Microbe. 2019;26:61–72. 10.1016/j.chom.2019.06.004. [DOI] [PubMed] [Google Scholar]
- 191.François B, Mercier E, Gonzalez C, Asehnoune K, Nseir S, Fiancette M, et al. Safety and tolerability of a single administration of AR-301, a human monoclonal antibody, in ICU patients with severe pneumonia caused by Staphylococcus aureus: first-in-human trial. Intensive Care Med. 2018;44:1787–96. 10.1007/s00134-018-5229-2. [DOI] [PubMed] [Google Scholar]
- 192.François B, Jafri HS, Chastre J, Sánchez-García M, Eggimann P, Dequin P-F, et al. Efficacy and safety of suvratoxumab for prevention of Staphylococcus aureus ventilator-associated pneumonia (SAATELLITE): a multicentre, randomised, double-blind, placebo-controlled, parallel-group, phase 2 pilot trial. Lancet Infect Dis. 2021;21:1313–23. 10.1016/s1473-3099(20)30995-6. [DOI] [PubMed] [Google Scholar]
- 193.Deng W, Marshall NC, Rowland JL, McCoy JM, Worrall LJ, Santos AS, et al. Assembly, structure, function and regulation of type III secretion systems. Nat Rev Microbiol. 2017;15:323–37. 10.1038/nrmicro.2017.20. [DOI] [PubMed] [Google Scholar]
- 194.Zigangirova NA, Zayakin ES, Kapotina LN, Kost EA, Didenko LV, Davydova DY, et al. Development of chlamydial Type III secretion system inhibitors for suppression of acute and chronic forms of chlamydial infection. Acta Naturae. 2012;4:87–97. [PMC free article] [PubMed] [Google Scholar]
- 195.Lv C, Li Y, Wei Y, Wang J, Yu H, Gao F, et al. Research progress on small molecular inhibitors of the type 3 secretion system. Molecules. 2022. 10.3390/molecules27238348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Koroleva EA, Kobets NV, Zayakin ES, Luyksaar SI, Shabalina LA, Zigangirova NA. Small molecule inhibitor of type three secretion suppresses acute and chronic Chlamydia trachomatis infection in a novel urogenital Chlamydia model. Biomed Res Int. 2015;2015: 484853. 10.1155/2015/484853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Nesterenko LN, Zigangirova NA, Zayakin ES, Luyksaar SI, Kobets NV, Balunets DV, et al. A small-molecule compound belonging to a class of 2,4-disubstituted 1,3,4-thiadiazine-5-ones suppresses Salmonella infection in vivo. J Antibiot (Tokyo). 2016;69:422–7. 10.1038/ja.2015.131. [DOI] [PubMed] [Google Scholar]
- 198.Zigangirova NA, Nesterenko LN, Sheremet AB, Soloveva AV, Luyksaar SI, Zayakin ES, et al. Fluorothiazinon, a small-molecular inhibitor of T3SS, suppresses salmonella oral infection in mice. J Antibiot (Tokyo). 2021;74:244–54. 10.1038/s41429-020-00396-w. [DOI] [PubMed] [Google Scholar]
- 199.Bondareva NE, Soloveva AV, Sheremet AB, Koroleva EA, Kapotina LN, Morgunova EY, et al. Preventative treatment with Fluorothiazinon suppressed Acinetobacter baumannii-associated septicemia in mice. J Antibiot (Tokyo). 2022;75:155–63. 10.1038/s41429-022-00504-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Koroleva EA, Soloveva AV, Morgunova EY, Kapotina LN, Luyksaar SI, Luyksaar SV, et al. Fluorothiazinon inhibits the virulence factors of uropathogenic Escherichia coli involved in the development of urinary tract infection. J Antibiot (Tokyo). 2023;76:279–90. 10.1038/s41429-023-00602-5. [DOI] [PubMed] [Google Scholar]
- 201.Tsarenko SV, Zigangirova NA, Soloveva AV, Bondareva NE, Koroleva EA, Sheremet AB, et al. A novel antivirulent compound fluorothiazinone inhibits Klebsiella pneumoniae biofilm in vitro and suppresses model pneumonia. J Antibiot (Tokyo). 2023;76:397–405. 10.1038/s41429-023-00621-2. [DOI] [PubMed] [Google Scholar]
- 202.Sheremet AB, Zigangirova NA, Zayakin ES, Luyksaar SI, Kapotina LN, Nesterenko LN, et al. Small molecule inhibitor of type three secretion system belonging to a class 2,4-disubstituted-4H-1,3,4-thiadiazine-5-ones improves survival and decreases bacterial loads in an airway Pseudomonas aeruginosa infection in mice. Biomed Res Int. 2018;2018:5810767. 10.1155/2018/5810767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Savitskii MV, Moskaleva NE, Brito A, Zigangirova NA, Soloveva AV, Sheremet AB, et al. Pharmacokinetics, quorum-sensing signal molecules and tryptophan-related metabolomics of the novel anti-virulence drug Fluorothiazinon in a .-induced pneumonia murine model. J Pharm Biomed Anal. 2023;236: 115739. 10.1016/j.jpba.2023.115739. [DOI] [PubMed] [Google Scholar]
- 204.Zigangirova NA, Kost EA, Didenko LV, Kapotina LN, Zayakin ES, Luyksaar SI, et al. A small-molecule compound belonging to a class of 2,4-disubstituted 1,3,4-thiadiazine-5-ones inhibits intracellular growth and persistence of Chlamydia trachomatis. J Med Microbiol. 2016;65:91–8. 10.1099/jmm.0.000189. [DOI] [PubMed] [Google Scholar]
- 205.Spaulding CN, Klein RD, Ruer S, Kau AL, Schreiber HL, Cusumano ZT, et al. Selective depletion of uropathogenic E. coli from the gut by a FimH antagonist. Nature. 2017;546:528–32. 10.1038/nature22972. [DOI] [PMC free article] [PubMed]
- 206.Han Z, Pinkner JS, Ford B, Obermann R, Nolan W, Wildman SA, et al. Structure-based drug design and optimization of mannoside bacterial FimH antagonists. J Med Chem. 2010;53:4779–92. 10.1021/jm100438s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Carb-X. GSK3882347 – FiMH antagonist. 2020. https://carb-x.org/gallery/glaxosmithkline/. Accessed 6 Sept 2024.
- 208.Fimbrion Therapeutics. CARB-X AWARDS FUNDING TO GSK FOR THE EARLY CLINICAL DEVELOPMENT OF A NOVEL THERAPY FOR URINARY TRACT INFECTIONS. 2020. https://www.fimbrion.com/carb-x. Accessed 6 Sept 2024.
- 209.GSK. Pipeline. 2024. https://www.gsk.com/en-gb/innovation/pipeline/. Accessed 6 Sept 2024.
- 210.Cusumano CK, Pinkner JS, Han Z, Greene SE, Ford BA, Crowley JR, et al. Treatment and prevention of urinary tract infection with orally active FimH inhibitors. Sci Transl Med. 2011;3:109ra115. 10.1126/scitranslmed.3003021. [DOI] [PMC free article] [PubMed]
- 211.Han Z, Pinkner JS, Ford B, Chorell E, Crowley JM, Cusumano CK, et al. Lead optimization studies on FimH antagonists: discovery of potent and orally bioavailable ortho-substituted biphenyl mannosides. J Med Chem. 2012;55:3945–59. 10.1021/jm300165m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Totsika M, Kostakioti M, Hannan TJ, Upton M, Beatson SA, Janetka JW, et al. A FimH inhibitor prevents acute bladder infection and treats chronic cystitis caused by multidrug-resistant uropathogenic Escherichia coli ST131. J Infect Dis. 2013;208:921–8. 10.1093/infdis/jit245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Jarvis C, Han Z, Kalas V, Klein R, Pinkner JS, Ford B, et al. Antivirulence isoquinolone mannosides: optimization of the biaryl aglycone for fimh lectin binding affinity and efficacy in the treatment of chronic UTI. ChemMedChem. 2016;11:367–73. 10.1002/cmdc.201600006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Bishop MJ, Janetka JW, McGrane LK, Widdowson KL. GlaxoSmithKline, Fimbrion Therapeutics. Substituted biphenyl or phenylheteroaryl-mannosides as antagonists of FimH. 2020. WO2020254369.
- 215.Mydock-McGrane L, Cusumano Z, Han Z, Binkley J, Kostakioti M, Hannan T, et al. Antivirulence C-mannosides as antibiotic-sparing, oral therapeutics for urinary tract infections. J Med Chem. 2016;59:9390–408. 10.1021/acs.jmedchem.6b00948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Bishop MJ, Colandrea VJ, Li Y, Stewart EL, Strambeanu I, Widdowson KL, et al. GlaxoSmithKline, Fimbrion Therapeutics. C-mannoside compounds useful for the treatment of urinary tract infections. 2019. WO2020012336.
- 217.Busch A, Waksman G. Chaperone-usher pathways: diversity and pilus assembly mechanism. Philos Trans R Soc Lond B Biol Sci. 2012;367:1112–22. 10.1098/rstb.2011.0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Abgottspon D, Ernst B. In vivo evaluation of FimH antagonists - a novel class of antimicrobials for the treatment of urinary tract infection. Chimia (Aarau). 2012;66:166–9. 10.2533/chimia.2012.166. [DOI] [PubMed] [Google Scholar]
- 219.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, et al. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48:1–12. 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 220.Cooper MA, Shlaes D. Fix the antibiotics pipeline. Nature. 2011;472:32. 10.1038/472032a. [DOI] [PubMed] [Google Scholar]
- 221.McKenna M. The antibiotic paradox: why companies can’t afford to create life-saving drugs. Nature. 2020;584:338–41. 10.1038/d41586-020-02418-x. [DOI] [PubMed] [Google Scholar]
- 222.Outterson K, Orubu ESF, Rex J, Årdal C, Zaman MH. Patient access in 14 high-income countries to new antibacterials approved by the US Food and Drug Administration, European Medicines Agency, Japanese Pharmaceuticals and Medical Devices Agency, or Health Canada, 2010–2020. Clin Infect Dis. 2022;74:1183–90. 10.1093/cid/ciab612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Strathdee SA, Hatfull GF, Mutalik VK, Schooley RT. Phage therapy: from biological mechanisms to future directions. Cell. 2023;186:17–31. 10.1016/j.cell.2022.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Bargh JD, Isidro-Llobet A, Parker JS, Spring DR. Cleavable linkers in antibody-drug conjugates. Chem Soc Rev. 2019;48:4361–74. 10.1039/c8cs00676h. [DOI] [PubMed] [Google Scholar]
- 225.O’Leary MK, Ahmed A, Alabi CA. Development of host-cleavable antibody-bactericide conjugates against extracellular pathogens. ACS Infect Dis. 2023;9:322–9. 10.1021/acsinfecdis.2c00492. [DOI] [PubMed] [Google Scholar]
- 226.Tvilum A, Johansen MI, Glud LN, Ivarsen DM, Khamas AB, Carmali S, et al. Antibody-drug conjugates to treat bacterial biofilms via targeting and extracellular drug release. Adv Sci (Weinh). 2023;10: e2301340. 10.1002/advs.202301340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Johnson K, Delaney JC, Guillard T, Reffuveille F, Varin-Simon J, Li K, et al. Development of an antibody fused with an antimicrobial peptide targeting Pseudomonas aeruginosa: a new approach to prevent and treat bacterial infections. PLoS Pathog. 2023;19: e1011612. 10.1371/journal.ppat.1011612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Lima LM, Da Silva BNM, Barbosa G, Barreiro EJ. β-Lactam antibiotics: an overview from a medicinal chemistry perspective. Eur J Med Chem. 2020;208: 112829. 10.1016/j.ejmech.2020.112829. [DOI] [PubMed] [Google Scholar]
- 229.Durand-Reville TF, Miller AA, O’Donnell JP, Wu X, Sylvester MA, Guler S, et al. Rational design of a new antibiotic class for drug-resistant infections. Nature. 2021;597:698–702. 10.1038/s41586-021-03899-0. [DOI] [PubMed] [Google Scholar]
- 230.Bertonha AF, Silva CCL, Shirakawa KT, Trindade DM, Dessen A. Penicillin-binding protein (PBP) inhibitor development: a 10-year chemical perspective. Exp Biol Med (Maywood). 2023;248:1657–70. 10.1177/15353702231208407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Chellat MF, Raguž L, Riedl R. Targeting antibiotic resistance. Angew Chem Int Ed Engl. 2016;55:6600–26. 10.1002/anie.201506818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.IQVIA. Global Oncology Trends 2024: Outlook to 2028. 2024. https://www.iqvia.com/insights/the-iqvia-institute/reports-and-publications/reports/global-oncology-trends-2024. Accessed 6 Sept 2024.
- 233.Thomas D, Wessel C. The State of Innovation in Antibacterial Therapeutics. 2022. https://www.bio.org/sites/default/files/2022-04/BIO-Antibacterial-Report-2022.pdf. Accessed 6 Sept 2024.
- 234.Zhao C-Y, Lv Y, Zhu Y, Wei M-J, Liu M-Y, Ji X-W, et al. A first-in-human safety, tolerability, and pharmacokinetics study of benapenem in healthy Chinese volunteers. Antimicrob Agents Chemother. 2019. 10.1128/aac.02188-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Hernandez V, Crépin T, Palencia A, Cusack S, Akama T, Baker SJ, et al. Discovery of a novel class of boron-based antibacterials with activity against gram-negative bacteria. Antimicrob Agents Chemother. 2013;57:1394–403. 10.1128/aac.02058-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Wu Y, Huang S, Chen X, Hu Y, Liu X. Suzhou Sinovent Pharma. β-Lactamase inhibitor and use thereof. 2019. WO2019144912A1.
- 237.Morinaka A, Tsutsumi Y, Yamada M, Suzuki K, Watanabe T, Abe T, et al. OP0595, a new diazabicyclooctane: mode of action as a serine β-lactamase inhibitor, antibiotic and β-lactam “enhancer.” J Antimicrob Chemother. 2015;70:2779–86. 10.1093/jac/dkv166. [DOI] [PubMed] [Google Scholar]
- 238.Hackel MA, Karlowsky JA, Dressel D, Sahm DF. Determination of disk diffusion and mic quality control ranges for nafithromycin (WCK 4873), a new lactone-ketolide. J Clin Microbiol. 2017;55:3021–7. 10.1128/jcm.00972-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Ramachandran G, Tulapurkar ME, Harris KM, Arad G, Shirvan A, Shemesh R, et al. A peptide antagonist of CD28 signaling attenuates toxic shock and necrotizing soft-tissue infection induced by Streptococcus pyogenes. J Infect Dis. 2013;207:1869–77. 10.1093/infdis/jit104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Surur AS, Sun D. Macrocycle-antibiotic hybrids: a path to clinical candidates. Front Chem. 2021;9: 659845. 10.3389/fchem.2021.659845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Donald BJ, Surani S, Deol HS, Mbadugha UJ, Udeani G. Spotlight on solithromycin in the treatment of community-acquired bacterial pneumonia: design, development, and potential place in therapy. Drug Des Dev Ther. 2017;11:3559–66. 10.2147/DDDT.S119545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Yao R, Wang B, Fu L, Li L, You K, Li Y-G, Lu Y. Sudapyridine (WX-081), a novel compound against Mycobacterium tuberculosis. Microbiol Spectr. 2022;10: e0247721. 10.1128/spectrum.02477-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Gootz T, Retsema J, Girard A, Hamanaka E, Anderson M, Sokolowski S. In vitro activity of CP-65,207, a new penem antimicrobial agent, in comparison with those of other agents. Antimicrob Agents Chemother. 1989;33:1160–6. 10.1128/aac.33.8.1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Krajnc A, Brem J, Hinchliffe P, Calvopiña K, Panduwawala TD, Lang PA, et al. Bicyclic boronate VNRX-5133 inhibits metallo- and serine-β-lactamases. J Med Chem. 2019;62:8544–56. 10.1021/acs.jmedchem.9b00911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Jacobsson S, Golparian D, Alm RA, Huband M, Mueller J, Jensen JS, et al. High in vitro activity of the novel spiropyrimidinetrione AZD0914, a DNA gyrase inhibitor, against multidrug-resistant Neisseria gonorrhoeae isolates suggests a new effective option for oral treatment of gonorrhea. Antimicrob Agents Chemother. 2014;58:5585–8. 10.1128/aac.03090-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Aceragen. Our Pipeline. 2024. https://www.aceragen.com/our-pipeline/. Accessed 6 Sept 2024.
- 247.Saxena D, Kaul G, Dasgupta A, Chopra S. Afabicin. Enoyl-(acyl-carrier-protein) reductase FabI inhibitor, Antibacterial drug. Drugs Future. 2021;46:5. 10.1358/dof.2021.46.1.3179432.
- 248.Weston DJ, Thomas S, Boyle GW, Pieren M. Alpibectir: early qualitative and quantitative metabolic profiling from a first-time-in-human study by combining 19F-NMR (nuclear magnetic resonance), 1H-NMR, and high-resolution mass spectrometric analyses. Drug Metab Dispos. 2024;52:858–74. 10.1124/dmd.124.001562. [DOI] [PubMed] [Google Scholar]
- 249.Makarov V, Manina G, Mikusova K, Möllmann U, Ryabova O, Saint-Joanis B, et al. Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science. 2009;324:801–4. 10.1126/science.1171583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Critchley IA, Green LS, Young CL, Bullard JM, Evans RJ, Price M, et al. Spectrum of activity and mode of action of REP3123, a new antibiotic to treat Clostridium difficile infections. J Antimicrob Chemother. 2009;63:954–63. 10.1093/jac/dkp041. [DOI] [PubMed] [Google Scholar]
- 251.Blaskovich MAT, Kavanagh AM, Elliott AG, Zhang B, Ramu S, Amado M, et al. The antimicrobial potential of cannabidiol. Commun Biol. 2021;4:7. 10.1038/s42003-020-01530-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Kim TS, Choe JH, Kim YJ, Yang C-S, Kwon H-J, Jeong J, et al. Activity of LCB01-0371, a novel oxazolidinone, against Mycobacterium abscessus. Antimicrob Agents Chemother. 2017. 10.1128/aac.02752-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Subbian S, Tsenova L, Holloway J, Peixoto B, O’Brien P, Dartois V, et al. Adjunctive phosphodiesterase-4 inhibitor therapy improves antibiotic response to pulmonary tuberculosis in a rabbit model. EBioMedicine. 2016;4:104–14. 10.1016/j.ebiom.2016.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Ooi N, Miller K, Hobbs J, Rhys-Williams W, Love W, Chopra I. XF-73, a novel antistaphylococcal membrane-active agent with rapid bactericidal activity. J Antimicrob Chemother. 2009;64:735–40. 10.1093/jac/dkp299. [DOI] [PubMed] [Google Scholar]
- 255.Higgins PG, Stubbings W, Wisplinghoff H, Seifert H. Activity of the investigational fluoroquinolone finafloxacin against ciprofloxacin-sensitive and -resistant Acinetobacter baumannii isolates. Antimicrob Agents Chemother. 2010;54:1613–5. 10.1128/aac.01637-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Stokes SS, Vemula R, Pucci MJ. Advancement of GyrB inhibitors for treatment of infections caused by mycobacterium tuberculosis and non-tuberculous mycobacteria. ACS Infect Dis. 2020;6:1323–31. 10.1021/acsinfecdis.0c00025. [DOI] [PubMed] [Google Scholar]
- 257.Bonchi C, Imperi F, Minandri F, Visca P, Frangipani E. Repurposing of gallium-based drugs for antibacterial therapy. BioFactors. 2014;40:303–12. 10.1002/biof.1159. [DOI] [PubMed] [Google Scholar]
- 258.Goss CH, Kaneko Y, Khuu L, Anderson GD, Ravishankar S, Aitken ML, et al. Gallium disrupts bacterial iron metabolism and has therapeutic effects in mice and humans with lung infections. Sci Transl Med. 2018. 10.1126/scitranslmed.aat7520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Li X, Hernandez V, Rock FL, Choi W, Mak YSL, Mohan M, et al. Discovery of a Potent and Specific M. tuberculosis Leucyl-tRNA Synthetase Inhibitor: (S)-3-(Aminomethyl)-4-chloro-7-(2-hydroxyethoxy)benzoc1,2oxaborol-1(3H)-ol (GSK656). J Med Chem. 2017;60:8011–26. 10.1021/acs.jmedchem.7b00631. [DOI] [PubMed]
- 260.van Eijk E, Boekhoud IM, Kuijper EJ, Bos-Sanders IMJG, Wright G, Smits WK. Genome location dictates the transcriptional response to PolC inhibition in Clostridium difficile. Antimicrob Agents Chemother. 2019. 10.1128/aac.01363-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Hind C, Clifford M, Woolley C, Harmer J, McGee LMC, Tyson-Hirst I, et al. Insights into the spectrum of activity and mechanism of action of MGB-BP-3. ACS Infect Dis. 2022;8:2552–63. 10.1021/acsinfecdis.2c00445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Weiss A, Delavenne E, Matias C, Lagler H, Simon D, Li P, et al. Topical niclosamide (ATx201) reduces Staphylococcus aureus colonization and increases Shannon diversity of the skin microbiome in atopic dermatitis patients in a randomized, double-blind, placebo-controlled Phase 2 trial. Clin Transl Med. 2022;12: e790. 10.1002/ctm2.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Yum JH, Kim CK, Yong D, Lee K, Chong Y, Kim CM, et al. In vitro activities of CG400549, a novel FabI inhibitor, against recently isolated clinical staphylococcal strains in Korea. Antimicrob Agents Chemother. 2007;51:2591–3. 10.1128/aac.01562-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Folsom JP, Baker B, Stewart PS. In vitro efficacy of bismuth thiols against biofilms formed by bacteria isolated from human chronic wounds. J Appl Microbiol. 2011;111:989–96. 10.1111/j.1365-2672.2011.05110.x. [DOI] [PubMed] [Google Scholar]
- 265.Xu J, Wang B, Fu L, Zhu H, Guo S, Huang H, et al. In vitro and in vivo activities of the riminophenazine TBI-166 against Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2019. 10.1128/aac.02155-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Hariguchi N, Chen X, Hayashi Y, Kawano Y, Fujiwara M, Matsuba M, et al. OPC-167832, a novel carbostyril derivative with potent antituberculosis activity as a DprE1 inhibitor. Antimicrob Agents Chemother. 2020. 10.1128/aac.02020-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Recce Pharmaceuticals. New class of synthetic antibiotics set to tackle superbugs. Biopharm. Deal. 2019. https://www.nature.com/articles/d43747-020-00804-y.pdf. Accessed 6 Sept 2024.
- 268.Trebosc V, Kemmer C, Lociuro S, Gitzinger M, Dale GE. Rifabutin for infusion (BV100) for the treatment of severe carbapenem-resistant Acinetobacter baumannii infections. Drug Discov Today. 2021;26:2099–104. 10.1016/j.drudis.2021.07.001. [DOI] [PubMed] [Google Scholar]
- 269.Iavarone L, Bottacini M, Pugnaghi F, Morandini C, Grossi P. Sanfetrinem and sanfetrinem-cilexetil: disposition in rat and dog. Xenobiotica. 1997;27:693–709. 10.1080/004982597240280. [DOI] [PubMed] [Google Scholar]
- 270.Sbardella G, Mai A, Artico M, Loddo R, Setzu MG, La Colla P. Synthesis and in vitro antimycobacterial activity of novel 3-(1H-pyrrol-1-yl)-2-oxazolidinone analogues of PNU-100480. Bioorg Med Chem Lett. 2004;14:1537–41. 10.1016/j.bmcl.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 271.Shirude PS, Shandil R, Sadler C, Naik M, Hosagrahara V, Hameed S, et al. Azaindoles: noncovalent DprE1 inhibitors from scaffold morphing efforts, kill Mycobacterium tuberculosis and are efficacious in vivo. J Med Chem. 2013;56:9701–8. 10.1021/jm401382v. [DOI] [PubMed] [Google Scholar]
- 272.Sarathy JP, Ragunathan P, Shin J, Cooper CB, Upton AM, Grüber G, Dick T. TBAJ-876 retains Bedaquiline’s activity against subunits c and ε of mycobacterium tuberculosis F-ATP synthase. Antimicrob Agents Chemother. 2019. 10.1128/aac.01191-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Pethe K, Bifani P, Jang J, Kang S, Park S, Ahn S, et al. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat Med. 2013;19:1157–60. 10.1038/nm.3262. [DOI] [PubMed] [Google Scholar]
- 274.Ma Z, Lynch AS. Development of a dual-acting antibacterial agent (TNP-2092) for the treatment of persistent bacterial infections. J Med Chem. 2016;59:6645–57. 10.1021/acs.jmedchem.6b00485. [DOI] [PubMed] [Google Scholar]
- 275.Kao YTR, Gao P, Li X, Liu M. Versitech. Compounds affecting pigment production and methods for treatment of bacterial diseases. 2020. US11052078B2.
- 276.Davies DT, Leiris S, Zalacain M, Sprynski N, Castandet J, Bousquet J, et al. Discovery of ANT3310, a novel broad-spectrum serine β-lactamase inhibitor of the diazabicyclooctane class, which strongly potentiates meropenem activity against carbapenem-resistant enterobacterales and Acinetobacter baumannii. J Med Chem. 2020;63:15802–20. 10.1021/acs.jmedchem.0c01535. [DOI] [PubMed] [Google Scholar]
- 277.O’Connor S, Lam LK, Jones ND, Chaney MO. Apramycin, a unique aminocyclitol antibiotic. J Org Chem. 1976;41:2087–92. 10.1021/jo00874a003. [DOI] [PubMed] [Google Scholar]
- 278.Stachyra T, Levasseur P, Péchereau M-C, Girard A-M, Claudon M, Miossec C, Black MT. In vitro activity of the {beta}-lactamase inhibitor NXL104 against KPC-2 carbapenemase and Enterobacteriaceae expressing KPC carbapenemases. J Antimicrob Chemother. 2009;64:326–9. 10.1093/jac/dkp197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Desai J, Sachchidamand S, Kumar S, Sharma R. Novel bacterial topoisomerase inhibitors (NBTIs)—a comprehensive review. Eur J Med Chem Rep. 2021;3:100017. 10.1016/j.ejmcr.2021.100017. [Google Scholar]
- 280.Durand-Réville TF, Comita-Prevoir J, Zhang J, Wu X, May-Dracka TL, Romero JAC, et al. Discovery of an orally available diazabicyclooctane inhibitor (ETX0282) of class A, C, and D serine β-lactamases. J Med Chem. 2020;63:12511–25. 10.1021/acs.jmedchem.0c00579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Nuermberger EL, Martínez-Martínez MS, Sanz O, Urones B, Esquivias J, Soni H, et al. GSK2556286 is a novel antitubercular drug candidate effective in vivo with the potential to shorten tuberculosis treatment. Antimicrob Agents Chemother. 2022;66: e0013222. 10.1128/aac.00132-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Makarov V, Lechartier B, Zhang M, Neres J, van der Sar AM, Raadsen SA, et al. Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol Med. 2014;6:372–83. 10.1002/emmm.201303575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Crowley BM, Nantermet P, Olsen DB, Suzuki T, Yang L, You L. Merck Sharp & Dohme. Oxazolidinone compound and methods of use thereof as an antibacterial agent. 2021. WO2021188606.
- 284.Qu X, Guo C, Liu S, Li X, Xi L, Liu X, Zhang J. Pharmacokinetics and nephrotoxicity of polymyxin MRX-8 in rats: a novel agent against resistant gram-negative bacteria. Antibiotics (Basel). 2024. 10.3390/antibiotics13040354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Roberts KD, Zhu Y, Azad MAK, Han M-L, Wang J, Wang L, et al. A synthetic lipopeptide targeting top-priority multidrug-resistant Gram-negative pathogens. Nat Commun. 2022;13:1625. 10.1038/s41467-022-29234-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Sutherland HS, Tong AST, Choi PJ, Blaser A, Conole D, Franzblau SG, et al. 3,5-Dialkoxypyridine analogues of bedaquiline are potent antituberculosis agents with minimal inhibition of the hERG channel. Bioorg Med Chem. 2019;27:1292–307. 10.1016/j.bmc.2019.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Foti C, Piperno A, Scala A, Giuffrè O. Oxazolidinone antibiotics: chemical, biological and analytical aspects. Molecules. 2021. 10.3390/molecules26144280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Brown P, Abbott E, Abdulle O, Boakes S, Coleman S, Divall N, et al. Design of next generation polymyxins with lower toxicity: the discovery of SPR206. ACS Infect Dis. 2019;5:1645–56. 10.1021/acsinfecdis.9b00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Livermore DM, Mushtaq S, Warner M, Vickers A, Woodford N. In vitro activity of cefepime/zidebactam (WCK 5222) against Gram-negative bacteria. J Antimicrob Chemother. 2017;72:1373–85. 10.1093/jac/dkw593. [DOI] [PubMed] [Google Scholar]
- 290.Huband MD, Mendes RE, Pfaller MA, Lindley JM, Strand GJ, Benn VJ, et al. In vitro activity of KBP-7072, a novel third-generation tetracycline, against 531 recent geographically diverse and molecularly characterized acinetobacter baumannii species complex isolates. Antimicrob Agents Chemother. 2020. 10.1128/aac.02375-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.