

Abstract
Congenital muscular dystrophy (CMD) is characterized by severe muscle wasting, premature death in early childhood, and lack of effective treatment. Most of the CMD cases are caused by genetic mutations of laminin-α2, which is essential for the structural integrity of muscle extracellular matrix. Here, we report that somatic gene delivery of a structurally unrelated protein, a miniature version of agrin, functionally compensates for laminin-α2 deficiency in the murine models of CMD. Adeno-associated virus-mediated overexpression of miniagrin restored the structural integrity of myofiber basal lamina, inhibited interstitial fibrosis, and ameliorated dystrophic pathology. Furthermore, systemic gene delivery of miniagrin into multiple vital muscles significantly improved whole body growth and motility and quadrupled the lifespan (50% survival) of the dystrophic mice. Thus, our study demonstrated the efficacy of somatic gene therapy in a mouse model of CMD.
Keywords: adeno-associated virus, integrity, myofiber basal laming, gene therapy
Congenital muscular dystrophies (CMDs) are a group of autosomal recessive inherited disorders, manifesting severe and progressive muscle-wasting and weakness, which start at or shortly after birth and often lead to death in early childhood (1–4). There is no effective treatment currently available for this lethal disease (4). Most of the cases of laminin-α2-deficient CMD are associated with genetic mutations in the laminin-α2 gene (5, 6). The disease was also previously termed merosin-deficient CMD and more recently MDC1A (1). A number of mutations that result in the absence of laminin-α2 or a truncated form of laminin-α2 have been identified in human patients (1). However, in the Japanese Fukuyama-type CMD and the Finnish muscle-eye-brain disease, the reduction in laminin-α2 expression is secondary to mutations in other genes (7, 8).
Laminins are large heterotrimeric extracellular glycoproteins, consisting of α, β, and γ chains that play a key role in the integrity of the extracellular basement membrane, specifically the basal lamina of striated muscle. More than 11 laminin variants have been identified (9, 10). The predominant extrasynaptic laminin is laminin-2 (α2/β1/γ1), and synaptic basal lamina may contain laminin-4, -9, and -11 (α2/β2/γ1, α4/β2/γ1, and α5/β2/γ1, respectively) (11). Originally isolated from the muscle, laminin-α2 is also found in the basement membrane of Schwann cells, blood vessels of the brain (12, 13), and other tissues (14). The major role of laminin-2 in the muscle is to interconnect the myofiber extracellular basal lamina with the plasma membrane, mainly through dystroglycans (5, 15). The latter, in turn, binds to an intracellular protein dystrophin, which further connects the plasma membrane to the cortical cytoskeletal structures of the myofibers. This interlinked protein network protects the integrity of the muscle cell structure during repeated cycles of contraction and relaxation. In laminin-α2-deficient individuals, the basal lamina is corrupted and the transmembrane cytoskeletal structure described above is lost, which leads to a classic dystrophic pathology. Naturally, the deficiency of laminin-α2 is partially compensated by the up-regulation of laminin-α4. However, the latter fails to bind α-dystroglycan with significant affinity (16–18) and therefore does not fully compensate for the absence of laminin-α2.
Agrin, a basement membrane-associated heparan sulfate proteoglycan, functions to induce the formation of postsynaptic apparatus at the neuromuscular junctions. A naturally occurring, alternatively spliced agrin mRNA generates a muscle isoform, which is produced at very low levels in skeletal muscles (17, 19). This protein has no defined functions either in muscle fibers or at the neuromuscular junctions. However, it does bind via its carboxyl-terminal domain to α-dystroglycan of the dystrophin-associated protein complex with high affinity (20–22). In addition, its amino-terminal domain (NtA) binds to a number of laminin variants, including laminin-8, which contains the α4 chain that is up-regulated in the α2 chain–deficient muscle (16, 18). Thus, the agrin muscle isoform, if produced in sufficient quantity, could conceptually act as a crosslink between the α4-containing laminin and the dystroglycans, and therefore, restore the basal lamina and ameliorate the dystrophic pathology (17).
Recently, Ruegg and colleagues (17) demonstrated that overexpression of a novel miniature version of chick agrin (miniagrin), containing conserved regions of both N and C termini and deletions in the central region, rendered significant therapeutic benefits in a laminin-α2 knockout mouse model of CMD. Those investigators used a transgenic model to deliver the miniagrin, an alternative transgene product, to the laminin-α2 knockout mouse. Although therapeutic efficacy is an important finding, the transgenic approach is not clinically viable. This limitation could be potentially overcome by somatic gene transfer, which, however, has hurdles in delivering the therapeutic gene into every single muscle cell and is unable to render therapeutic benefits at the early embryonic stage. To investigate the potential of somatic gene therapy, we chose adeno-associated virus (AAV) vectors as the gene delivery system, mainly because of AAV's proven record as the most efficient vector for long-term gene transfer in both normal and dystrophic muscles (23–32). Recent development in AAV vector technology demonstrated feasibility of achieving systemic muscle gene delivery (33, 34). Here, we show that overexpression of a mouse miniagrin gene by AAV vectors in two different mouse models of laminin-α2-deficient CMD ameliorated muscle pathology, decreased fibrosis, and restored the structure of the muscle myofiber basal lamina. Moreover, the vector-treated dystrophic mice obtained significant improvement in body growth, locomotor functions, and lifespan.
Materials and Methods
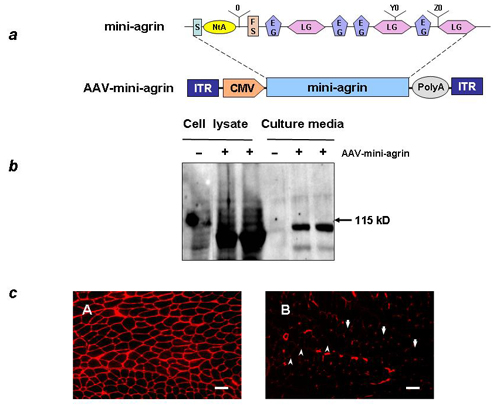
Construction of Miniagrin Gene and AAV Vector Production. The mouse miniagrin cDNA (GenBank accession no. AY914875) was generated by RT-PCR and a series of cloning processes. Briefly, the total RNA from the kidney, which also expresses the muscle-form agrin, was used as a template for miniagrin cDNA synthesis with the ThermoScript RT-PCR System (Invitrogen). A signal peptide sequence that was deduced from the mouse agrin genomic DNA (35) was added in front of the N-terminus RT-PCR product to increase protein secretion levels. The primers used for amplification of the C terminus were 5′-cccccaaagtcctgtgattcc-3′ and 5′-tcagagagtggggcagggtc-3′. The primers used for the N terminus were 5′-aaaggcaaagatgtggtgg-3′ and 5′-acatggcccttggcggagtag-3′. The miniagrin cDNA was then cloned into an AAV vector plasmid under the transcriptional control of CMV promoter (Fig. 7a, which is published as supporting information on the PNAS web site) (36).
The recombinant viral vector stocks were produced according to the three-plasmid cotransfection method (37). The viral particles were purified twice through CsCl density gradient ultracentrifugation by using the previously published protocol (38). The vector titers of viral particle numbers were determined by the DNA dot blot method and were in the range of 2 × 1012 to 5 × 1012 vector genomes per ml.
Mice and Vector Administration. All experiments involving animals were approved by the University of Pittsburgh Animal Care and Use Committee. The spontaneous mutant dy/dy mice were purchased from The Jackson Laboratory. The experimental null mutant dyw/dyw mice were kindly provided by E. Engvall (The Burnham Institute, La Jolla, CA) (6, 39). The AAV serotype 2 miniagrin vector was injected into three sites (tibialis anterior, gastrocneminus, and thigh) of the hind leg muscle of 3-week-old dy/dy mice with 30 μl at each site. The AAV serotype 1 miniagrin vector was delivered into the neonates (3–5 days old) of dyw/dyw mice by i.p. injection with 100 μl per mouse.
Immunofluorescent (IF) Staining. Goat anti-type III collagen antibody was purchased from Southern Biotechnology Associates (catalogue no. 1330-01). Anti-rat-agrin antibody was bought from R & D Systems (catalogue no. AF550). Rabbit anti-neurofilament antibody (catalogue no. AB1981) and rat-anti-laminin β2(γ1) chain (catalogue no. MAB1914) were obtained from Chemicon. IF staining of muscle cryo-thin sections was performed as described (26, 40).
Results
Construction and Characterization of Mouse Miniagrin Gene. The spontaneous dy/dy mouse (41, 42) and the laminin-α2 knockout dyw/dyw mouse (6, 43) represent currently available CMD mouse models. To avoid potential host immune responses to the commonly used chick agrin, we wanted to use mouse agrin for the somatic gene therapy experiments. However, the published mouse agrin N-terminal coding sequence lacks a definitive signal peptide for efficient secretion. To resolve this problem, we cloned and constructed mouse miniagrin cDNA by RT-PCR and added a signal peptide sequence that was deduced from the mouse agrin genomic DNA (35). Similar to the chick miniagrin reported by Ruegg and colleagues (17), the mouse miniagrin gene construct consists of the following elements: a signal peptide sequence, the N-terminal region for laminin binding, the first follistatin-like domain, and the C-terminus region for dystroglycan binding (Fig. 7a). The mouse miniagrin gene was subcloned into the AAV vector under the control of a CMV promoter to assure strong and constitutive expression in the muscle and heart. Before it was used for the in vivo gene transfer experiments, we confirmed by transfection in vitro that the miniagrin could be synthesized and properly secreted in cell culture as shown by Western analysis (Fig. 7b). We further tested the miniagrin gene expression in vivo in normal muscle by direct intramuscular injection of the AAV vector. IF staining with an anti-agrin antibody revealed both overexpression and appropriate localization of the miniagrin at the muscle basal lamina 4 weeks after gene delivery. The untreated normal muscle as a control only showed punctuated IF staining at the blood vessels and the neuromuscular junctions where the nonmuscle isoforms were located (19) (Fig. 7c).
Local Gene Delivery Inhibits Muscle Pathology and Fibrosis. We first evaluated the therapeutic effects of the miniagrin gene by local intramuscular gene delivery into the hind leg muscles of the spontaneous mutant dy/dy mice. These commercially available mice show typical pathological signs of CMD, including wide variation in myofiber sizes, severe interstitial fibrosis, myofiber necrosis, and atrophy (4). AAV serotype 2 vector carrying the mouse miniagrin gene was injected into the gastrocnemius, tibialis anterior, and quadricep muscles of homozygous dy/dy mice at 3 weeks of age, at which time the diseased homozygous mice could be identified from their healthy littermates. Two months after vector injection, muscles from AAV-miniagrin–treated dy/dy mice and the littermate controls were collected for evaluation of miniagrin expression and histological examination. Agrin IF staining and hematoxylin/eosin (H&E) staining on the age-matched, untreated dystrophic muscles showed absence of sarcolemmal agrin, myofiber degeneration, mononuclear cell infiltration, heterogeneous myofiber sizes, and pronounced fibrosis (Fig. 1 Left). In contrast, the AAV-agrin vector-treated muscle showed efficient miniagrin expression as well as muscle morphology improvement including more uniform myofiber sizes, less mononuclear cell infiltration, and minimal fibrosis (Fig. 1 Middle), very similar to the histology of the WT control muscle (Fig. 1 Right). We further confirmed the inhibition of fibrosis in the vector-treated dy/dy mice by both IF staining for collagen III (44, 45) and Masson's Trichrome staining for general collagen depositions (Fig. 1). These results clearly demonstrated that overexpression of the miniagrin protein in the dystrophic muscle improved muscle pathology and inhibited collagen deposition in the dy/dy mice.
Fig. 1.

Local overexpression of miniagrin gene and amelioration of histopathology in the dystrophic muscle of dy/dy mice. Homozygous dy/dy mice were injected in quadriceps with AAV2-miniagrin vector at the age of 3 weeks. At 2 months after vector injection, cryo-thin sections of quadriceps muscle from untreated homozygous mice dy/dy (Left), AAV-treated homozygous mice (dy/dy + AAV) (Center), and littermate WT control mice (Right) were subjected to IF staining against agrin, collagen type III, H & E staining, and Masson's Trichrome staining. In Trichrome staining, collagen was stained as a blue color. Note that miniagrin overexpression greatly improved dystrophic muscle histology and inhibited fibrosis. (Scale bars: 100 μm.)
Restoration of Myofiber Basal Lamina Structure Integrity. A complete or partial loss of the myofiber basal lamina is one of the pathological causes of CMD at the cellular level. The deficiency is secondary to the primary loss of laminin-α2 chain, which is essential for the formation of a laminin-based primary scaffold to protect the integrity of the basal lamina (11). We further tested whether the overexpression of miniagrin protein was able to restore the disintegrated basal lamina. Electron microscopy was used for ultrastructure examination of the tibialis anterior muscle myofibers. The treated mice were killed 2 months after vector administration, and age-matched controls were used. Whereas the untreated dystrophic myofibers showed an absence of intact basal lamina, the AAV-miniagrin-treated dystrophic myofibers showed a continuous and dense basal lamina structure, immediately outside the myofiber plasma membrane, equivalent to that of the WT muscles (Fig. 2). This result indicated that somatic gene transfer of the miniagrin could effectively restore the integrity of the basal lamina of the laminin-α2-deficient dystrophic muscle, to an extent similar to that of the miniagrin transgenic mice (17).
Fig. 2.

Electron microscopy on restoration of ultra structure of the muscle cell basement membrane after miniagrin overexpression. The tibialis anterior muscles from 3-month-old untreated dy/dy mice (Left), AAV vector-treated dy/dy mice (Center), and the control WT mice (Right) were examined by electron microscopy. Shown are cross sections of a myofiber from the mice. The black arrows highlight the extracellular basement membrane/basal lamina structures, and the hollow arrowheads point to the dense plasma membrane. (Magnification: ×15,000.)
Systemic Gene Delivery Improves Multiple Vital Muscles and Heart. The success of local AAV-miniagrin delivery prompted us to further examine whether systemic delivery of the vector into large groups of vital muscles could be achieved and the disease phenotypes ameliorated. Recently, we found that i.p. injection of AAV vectors into neonatal mice could render systemic gene transfer in multiple muscles (34). However, the homozygous dy/dy mice that were used earlier in this study for local treatment could not be screened by DNA analysis because the mutation site was unknown. The disease phenotypes would not be evident until 3–4 weeks after birth, which makes it difficult for early diagnosis and treatment at the neonatal age. For this reason, we chose to use the genetically more defined laminin-α2 knockout dyw/dyw model, because rapid PCR screening could be used for identification of homozygous (dyw/dyw) neonates. Furthermore, the dyw/dyw mice manifest very severe clinical phenotypes and rarely live beyond 2 months of age (6, 17, 39). The mice are also physically passive and much smaller and thinner than their WT and heterozygote littermates, therefore serving as a clinically relevant model for the evaluation of therapeutic efficacies.
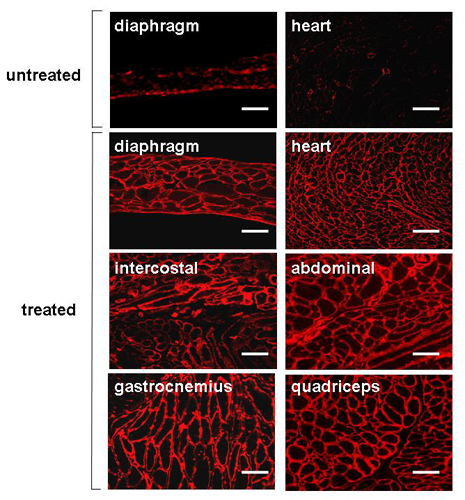
AAV serotype 1 vector carrying the miniagrin gene was delivered into the dyw/dyw neonates at 3–5 days of age by i.p. injection. At 4 months of age the animals were killed and IF staining of agrin was performed to confirm gene transfer and expression in the major groups of muscles. Indeed, overexpression of miniagrin protein was observed in many muscle groups, including the diaphragm, intercostal, abdominal, and limb muscles, as well as the heart (Figs. 3a and 4, and Fig. 8, which is published as supporting information on the PNAS web site). Systemic miniagrin gene delivery, similar to local intramuscular delivery, also ameliorated pathological phenotypes of the dystrophic skeletal muscles as shown by H&E staining (Fig. 3a). The therapeutic effect of miniagrin expression was also reflected by the increased muscle fiber sizes. Whereas the untreated dystrophic mice showed consistently smaller and more variable myofiber diameters (17, 46) caused by muscle atrophy, the AAV-miniagrin–treated dyw/dyw mice displayed myofibers with larger diameters (Fig. 3b). Furthermore, overexpression of miniagrin in heart muscle as a result of systemic gene delivery also prevented dystrophic pathology such as fibrosis. The hearts of the untreated dyw/dyw mice displayed more cell nuclei, which were likely to be the fibroblast infiltration and more collagen deposition as shown by anticollagen staining (Fig. 4 Left). By contrast, the hearts of AAV vector-treated dyw/dyw mice showed normal cell nuclei density and normal collagen deposition (Fig. 4 Middle), which were essentially indistinguishable from the hearts of their WT littermates (Fig. 4 Right).
Fig. 3.

Overexpression of the miniagrin protein and the improvement of muscle histology in skeletal muscle after systemic AAV-miniagrin gene delivery. The AAV1 vector was injected i.p. in dyw/dyw neonates, and the mice were killed at 4 months of age. (a) Agrin staining of diaphragm (first row) and quadriceps (third row) showed overexpression of miniagrin protein after treatment, whereas H & E staining (second and fourth rows, respectively) displayed improved muscle histology. Untreated homozygous mice dy/dy (Left), AAV-treated homozygous mice (dy/dy + AAV) (Center), and littermate WT control mice (Right). (Scale bars: 100 μm.) (b) The untreated dystrophic mice (empty columns) displayed consistently smaller and more variable myofiber diameters. The AAV-miniagrin-treated dyw/dyw mice (filled columns) displayed myofibers larger than the untreated mice but smaller than the WT control (hatched columns). In detail, 8- to 10-μm cryo-thin sections of the quadriceps muscles were subjected to IF staining against the dystrophin to display circumferences of the myofiber. Pictures were taken and the radius of the myofiber was analyzed by using metamorph software with a minimum of 300 myofibers from each mouse and three mice for each group. Student's t test was used; *, P < 0.05. Standard error is labeled as a bar.
Fig. 4.

Overexpression of miniagrin in heart after systemic delivery and improvement of cardiac histology. The AAV1 vector was injected i.p. in dyw/dyw neonates, and the mice [untreated homozygous mice dy/dy (Left), AAV-treated homozygous mice (dy/dy + AAV) (Center), and littermate WT control mice (Right)] were killed at 2 ½ months of age. Cryo-thin sections of heart muscle were subjected to IF staining of agrin (a–c), agrin (red) counterstained with cell nuclei (green) (d–f), and collagen III (g–i). (Insets) Enlargement of a representative area of each panel to show more details. (Scale bars: 100 μm.)
Improvement of Growth Rate, Locomotive Activity, and Lifespan. In addition to the improvement of histopathological phenotypes, the general health of the dyw/dyw mice after systemic gene therapy with the AAV vector was greatly improved when compared with that of their untreated littermates. The treated mice grew faster and were larger than their untreated dystrophic littermates. However, the therapy was not sufficient to ameliorate all symptoms and the treated animals still lagged behind the WT and heterozygote littermates, as shown by the growth curve (Fig. 5a) and appearance (Fig. 5b). At the age of 6 weeks, the average body weight of the treated dyw/dyw mice was 12.32 ± 1.29 g, with 80% increase over the untreated counterparts of 6.87 ± 2.34 g. Furthermore, the AAV-treated mice were physically more active than the untreated ones and showed highly improved locomotor function when tested by the rotarod method. The AAV vector-treated dyw/dyw mice could stay on the rotarod for 160 ± 84 s, whereas most of the untreated dystrophic mice could barely stay on the rotarod (Fig. 5c). Finally, the lifespan of the vector–treated mice was also significantly increased. The 50% survival time of the untreated dyw/dyw mice was ≈4 weeks, but the vector-treated mice survival times were quadrupled to >17 weeks. Whereas all of the untreated mice (n = 18) had died by 13 weeks after birth, only one of the seven treated mice had died by that time, and the longest lifespan was 35 weeks (Fig. 5d). These results demonstrated that systemic somatic gene delivery of the miniagrin gene could improve both longevity and general health of the CMD mice.
Fig. 5.

Whole-body efficacies after systemic miniagrin gene delivery. (a) Growth curves of the untreated dyw/dyw mice (•), AAV-treated dyw/dyw mice (gray triangle), and WT littermate control mice (gray diamond). Most of the untreated dyw/dyw mice died by 9 weeks. (b) Whole-body photograph of the 1 ½-month-old WT control (Top), the AAV-treated (Middle), and untreated (Bottom) dyw/dyw mice. (c) Rotarod assays performed with 4-week-old mice. Animals were first trained three times to allow for accommodation to the task. After resting for at least 1 h, animals were tested on the machine. Each data point represents mean ± SD from four trials of each mouse (n = 4). (d) Survival rate of the mice. Fifty percent survival rate of the untreated dyw/dyw mice is 4 weeks (n = 18, •), the AAV-treated dyw/dyw mice is 17 weeks (n = 7, ▴), and the normal life span of the WT mice is >1 year (▪).
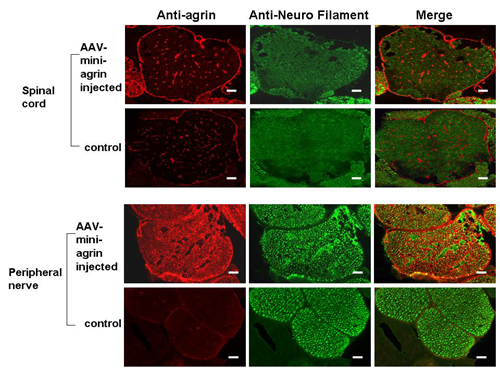
Expression of Miniagrin in Peripheral Nerve. The loss of laminin-α2 in endoneurial basal lamina is considered a direct cause of nerve deficiency in the CMD mice (6), because laminin-α2 normally is found in the Schwann cell basement membrane and plays a role in the ensheathment and myelination of the peripheral nerve (5). We wanted to see whether the miniagrin was expressed in the peripheral nerve such as the Schwann cells after systemic miniagrin gene delivery. Indeed, IF staining on thin sections of the spine revealed the extensive presence of miniagrin surrounding individual axon-Schwann cell units in the peripheral nerve of AAV-miniagrin–treated dyw/dyw mice, but not in the untreated control dyw/dyw mice (Fig. 6a and Fig. 9, which is published as supporting information on the PNAS web site). However, there was no significant presence of miniagrin within the spinal cord (Fig. 8) or the brain of the mice (data not shown). Although laminin-α2 is absent in dyw/dyw mice, laminin-γ1 is expectedly present in the basal lamina of the peripheral nerves (18). To further confirm the location of the miniagrin protein, we performed double-IF staining for both agrin and laminin-γ1 and analyzed it by confocal microscopy. As expected, colocalization of miniagrin protein and the laminin-γ1 chain was found in the peripheral nerve (Fig. 6b). In addition, costaining of neural filament and miniagrin (Fig. 6a) displayed identical patterns as costaining with laminin-γ1 (Fig. 6c). These results further indicated that the miniagrin was present in the basal lamina of the peripheral nerve cells. Hind limb paralysis was not prevented despite the expression of miniagrin in those cells. Our current analysis thus could not determine whether miniagrin offered any beneficial effects to the nervous system and to what extent.
Fig. 6.

Expression of miniagrin in the basal lamina of treated peripheral nerves. (a) AAV-miniagrin treatment resulted in overexpression of agrin protein in peripheral nerve. Intramuscular nerves from intercostals muscles of the AAV serotype 1 virus-treated dyw/dyw mice were stained with antibodies against agrin and neuron filament. Agrin was stained as red, and neuron filament that indicated large myelinated neuron axons was stained as green. In the AAV-miniagrin-injected mice, agrin protein surrounded individual axon-Schwann cell units in the peripheral nerve. Control nerve only displayed weak agrin expression around fascicles of nerve fibers. (b) Confocal analysis indicated that the miniagrin protein was positioned in the basal lamina of treated peripheral nerve (arrow area). Laminin-γ1 chain was expressed in both control and AAV-injected dyw/dyw mice, and the miniagrin protein (green) was colocalized with laminin-γ1 chain (red) in AAV-miniagrin-injected mice. (c) Costaining of laminin-γ1 chain and neuron filament. (Scale bars: 100 μm.)
Discussion
Laminin-α2-deficient CMD is one of the most severe muscular dystrophies with no effective therapy currently available. The aggressive pathology of CMD is often so severe that patients die at a very early age and are never able to walk (1, 4). Because of the early morbidity and mortality, a therapeutic strategy that improves both muscle histology and general health and lifespan is highly desirable. In this study, we present evidence of a successful gene therapy study to treat CMD in animal models by somatic gene delivery. We showed that the dystrophic muscle morphology was noticeably improved, myofiber basal lamina was restored, and fibrosis was inhibited as a result of either local intramuscular or whole-body systemic gene delivery of a novel miniagrin gene by the AAV vectors. More importantly, systemic gene delivery by a single i.p. injection of the AAV1 vector in CMD neonatal mice, without any pharmaceutical interventions (27, 33), transduced multiple vital muscle groups, improved whole-body growth and locomotive functions, and achieved life-saving effects by quadrupling the 50% survival time from 4 to 17 weeks. Thus, our study demonstrated the feasibility of AAV-mediated somatic gene therapy for CMD in an animal model.
This report demonstrates somatic gene therapy for CMD, although transgenic mouse studies have been successfully performed (6, 17). The lack of results from gene or cell therapy for CMD might be partly caused by the severity of the disease itself and the lack of effective ways to deliver the therapeutic genes or cells to large groups of muscles to achieve significant therapeutic effects. Previously the AAV vectors have been well documented as the most efficient gene delivery system in striated muscle, primarily by local injections. The discovery of new AAV serotypes has made systemic muscle gene delivery possible (33, 34, 47–49). These new AAV vectors have a high infectivity in muscle and are capable of disseminating systemically (33, 34). We showed highly efficient miniagrin gene expression in muscle and heart as a result of systemic delivery by the AAV1 vector, which disseminated into and infected muscle more efficiently than the most commonly used AAV2 vector. However, AAV1 carrying reporter gene did not show high efficiency in heart and upper limb muscles (data not shown). Therefore, additional explanation for the widespread distribution of the miniagrin protein throughout the muscle and heart comes from the secretable nature of the miniagrin, which is able to reach and protect beyond the cells that produce it. As a result, it is unnecessary to transduce every muscle cell to achieve systemic efficacy. The long-term overexpression of the miniagrin gene is attributed to the low cellular immunity profile of AAV vectors (50) and the endogenous nature of the miniagrin, whose immunogenicity should be minimal, if any. Furthermore, overexpression of miniagrin apparently did not cause detectable toxicity in both normal and dystrophic mice as shown in a transgenic mouse study (17) and in our current somatic gene therapy study (data not shown). These combined advantages clearly support the use of AAV vectors and miniagrin for somatic gene therapy of CMD.
Despite the major improvement of muscle morphology and general health in the CMD mice, the efficacy of a somatic gene therapy regimen was still far from ideal. Neonatal treatment delayed the dystrophic phenotypes but failed to completely prevent them. As a result, in utero gene therapy may be required to achieve earlier and possibly better therapeutic effects, similar to those seen in the miniagrin transgenic CMD mice (17). Future development of new gene delivery systems, including novel AAV vectors, could also boost the efficiency of miniagrin gene expression. Because miniagrin is a structurally unrelated protein to laminin-α2, whose mutation is the underlining cause of CMD, a complete phenotypic rescue by the miniagrin may not be expected. Another major pathological feature, thus the progressive hind limb atrophy and paralysis, was still observed in the AAV-treated CMD mice, although it was delayed when compared with the untreated mice (data not shown). Noticeably, muscle-specific expression of miniagrin gene or even the laminin-α2 gene itself in the transgenic CMD mice failed to prevent the similar pathology in the hind limbs, suggesting that CMD is more than simply a muscle disease. Neurological dysfunction is apparently another contributing factor (6, 17). Indeed, in the normal peripheral nervous system, laminin-2 is expressed in the endoneurial basement membrane surrounding the myelin sheath of nerve fibers, where it binds to α-dystroglycan, which in turn is attached to the Schwann cell membrane by β-dystroglycan (51). Surprisingly, we observed expression of miniagrin protein in the basal lamina of the peripheral nerves in AAV-treated dyw/dyw mice but it did not prevent the neuromuscular pathology. As a result, overexpression of laminin-α2 itself in the endoneurial basement membrane may be required to alleviate the nerve defect of the CMD mice. Furthermore, therapeutic genes that promote muscle and nerve growth such as insulin-like growth factor (52, 53) and glial cell-derived nerve growth factor (53, 54) could also be used in conjunction with the miniagrin to further improve the structure as well as functions of the dystrophic muscles. Finally, new development in AAV vector technologies may offer novel vectors with even higher efficiencies than the currently used vectors for systemic muscle gene delivery.
Supplementary Material
Acknowledgments
We thank Dr. Eva Engvall for the CMD mice and Michael Xiao and Allison Sciullo for critical reading of the manuscript. C.Q. is a recipient of a Muscular Dystrophy Association fellowship. This work was supported in part by National Institutes of Health Paul Wellstone Muscular Dystrophy Cooperative Research Center Grant AR 50733 and National Institutes of Health Grant NS 46546.
Author contributions: C.Q., T.Z., Jianbin Li, S.W., X.Y., Juan Li, and X.X. designed research; C.Q., T.Z., Jianbin Li, R.D., S.W., X.Y., C.C., Juan Li, and X.X. performed research; C.Q., T.Z., Jianbin Li, R.D., S.W., X.Y., Juan Li, and X.X. contributed new reagents/analytic tools; C.Q., T.Z., Jianbin Li, R.D., S.W., X.Y., Juan Li, and X.X. analyzed data; and C.Q., T.Z., X.Y., Juan Li, and X.X. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: CMD, congenital muscular dystrophy; AAV, adeno-associated virus; IF, immunofluorescent; H&E, hematoxylin/eosin.
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. AY914875).
References
- 1.Allamand, V. & Guicheney, P. (2002) Eur. J. Hum. Genet. 10, 91–94. [DOI] [PubMed] [Google Scholar]
- 2.Pegoraro, E., Fanin, M., Trevisan, C. P., Angelini, C. & Hoffman, E. P. (2000) Neurology 55, 1128–1134. [DOI] [PubMed] [Google Scholar]
- 3.D'Alessandro, M., Naom, I., Ferlini, A., Sewry, C., Dubowitz, V. & Muntoni, F. (1999) Hum. Genet. 105, 308–313. [DOI] [PubMed] [Google Scholar]
- 4.Emery, A. E. (2002) Lancet 359, 687–695. [DOI] [PubMed] [Google Scholar]
- 5.O'Brien, D. P., Johnson, G. C., Liu, L. A., Guo, L. T., Engvall, E., Powell, H. C. & Shelton, G. D. (2001) J. Neurol. Sci. 189, 37–43. [DOI] [PubMed] [Google Scholar]
- 6.Kuang, W., Xu, H., Vachon, P. H., Liu, L., Loechel, F., Wewer, U. M. & Engvall, E. (1998) J. Clin. Invest. 102, 844–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cormand, B., Avela, K., Pihko, H., Santavuori, P., Talim, B., Topaloglu, H., de la Chapelle, A. & Lehesjoki, A. E. (1999) Am. J. Hum. Genet. 64, 126–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kobayashi, K., Nakahori, Y., Miyake, M., Matsumura, K., Kondo-Iida, E., Nomura, Y., Segawa, M., Yoshioka, M., Saito, K., Osawa, M., et al. (1998) Nature 394, 388–392. [DOI] [PubMed] [Google Scholar]
- 9.Colognato, H. & Yurchenco, P. D. (2000) Dev. Dyn. 218, 213–234. [DOI] [PubMed] [Google Scholar]
- 10.Aumailley, M. & Smyth, N. (1998) J. Anat. 193, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanes, J. R. (2003) J. Biol. Chem. 278, 12601–12604. [DOI] [PubMed] [Google Scholar]
- 12.Leivo, I. & Engvall, E. (1988) Proc. Natl. Acad. Sci. USA 85, 1544–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Villanova, M., Malandrini, A., Toti, P., Salvestroni, R., Six, J., Martin, J. J. & Guazzi, G. C. (1996) J. Submicrosc. Cytol. Pathol. 28, 1–4. [PubMed] [Google Scholar]
- 14.Wewer, U. M. & Engvall, E. (1996) Neuromuscul. Disord. 6, 409–418. [DOI] [PubMed] [Google Scholar]
- 15.Pall, E. A., Bolton, K. M. & Ervasti, J. M. (1996) J. Biol. Chem. 271, 3817–3821. [DOI] [PubMed] [Google Scholar]
- 16.Ringelmann, B., Roder, C., Hallmann, R., Maley, M., Davies, M., Grounds, M. & Sorokin, L. (1999) Exp. Cell Res. 246, 165–182. [DOI] [PubMed] [Google Scholar]
- 17.Moll, J., Barzaghi, P., Lin, S., Bezakova, G., Lochmuller, H., Engvall, E., Muller, U. & Ruegg, M. A. (2001) Nature 413, 302–307. [DOI] [PubMed] [Google Scholar]
- 18.Patton, B. L., Miner, J. H., Chiu, A. Y. & Sanes, J. R. (1997) J. Cell Biol. 139, 1507–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gesemann, M., Brancaccio, A., Schumacher, B. & Ruegg, M. A. (1998) J. Biol. Chem. 273, 600–605. [DOI] [PubMed] [Google Scholar]
- 20.Ibraghimov-Beskrovnaya, O., Ervasti, J. M., Leveille, C. J., Slaughter, C. A., Sernett, S. W. & Campbell, K. P. (1992) Nature 355, 696–702. [DOI] [PubMed] [Google Scholar]
- 21.Gee, S. H., Montanaro, F., Lindenbaum, M. H. & Carbonetto, S. (1994) Cell 77, 675–686. [DOI] [PubMed] [Google Scholar]
- 22.Jacobson, C., Cote, P. D., Rossi, S. G., Rotundo, R. L. & Carbonetto, S. (2001) J. Cell Biol. 152, 435–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiao, X., Li, J. & Samulski, R. J. (1996) J. Virol. 70, 8098–8108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kessler, P. D., Podsakoff, G. M., Chen, X., McQuiston, S. A., Colosi, P. C., Matelis, L. A., Kurtzman, G. J. & Byrne, B. J. (1996) Proc. Natl. Acad. Sci. USA 93, 14082–14087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang, B., Li, J. & Xiao, X. (2000) Proc. Natl. Acad. Sci. USA 97, 13714–13719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xiao, X., Li, J., Tsao, Y. P., Dressman, D., Hoffman, E. P. & Watchko, J. F. (2000) J. Virol. 74, 1436–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greelish, J. P., Su, L. T., Lankford, E. B., Burkman, J. M., Chen, H., Konig, S. K., Mercier, I. M., Desjardins, P. R., Mitchell, M. A., Zheng, X. G., et al. (1999) Nat. Med. 5, 439–443. [DOI] [PubMed] [Google Scholar]
- 28.Cordier, L., Hack, A. A., Scott, M. O., Barton-Davis, E. R., Gao, G., Wilson, J. M., McNally, E. M. & Sweeney, H. L. (2000) Mol. Ther. 1, 119–129. [DOI] [PubMed] [Google Scholar]
- 29.Atlas, I. & Smolin, A. (1999) Eur. J. Obstet. Gynecol. Reprod. Biol. 87, 175–178. [DOI] [PubMed] [Google Scholar]
- 30.Dressman, D., Araishi, K., Imamura, M., Sasaoka, T., Liu, L. A., Engvall, E. & Hoffman, E. P. (2002) Hum. Gene. Ther. 13, 1631–1646. [DOI] [PubMed] [Google Scholar]
- 31.Hoshijima, M., Ikeda, Y., Iwanaga, Y., Minamisawa, S., Date, M. O., Gu, Y., Iwatate, M., Li, M., Wang, L., Wilson, J. M., et al. (2002) Nat. Med. 8, 864–871. [DOI] [PubMed] [Google Scholar]
- 32.High, K. A. (2004) Semin. Thromb. Hemostasis 30, 257–267. [DOI] [PubMed] [Google Scholar]
- 33.Gregorevic, P., Blankinship, M. J., Allen, J. M., Crawford, R. W., Meuse, L., Miller, D. G., Russell, D. W. & Chamberlain, J. S. (2004) Nat. Med. 10, 828–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang, Z., Zhu, T., Qiao, C., Zhou, L., Wang, B., Zhang, J., Chen, C., Li, J. & Xiao, X. (2005) Nat. Biotechnol. 23, 321–328. [DOI] [PubMed] [Google Scholar]
- 35.Shigemoto, K., Kubo, S., Maruyama, N., Yamada, S., Obata, K., Kikuchi, K. & Kondo, I. (2000) Biochim. Biophys. Acta 1494, 170–174. [DOI] [PubMed] [Google Scholar]
- 36.Li, J., Dressman, D., Tsao, Y. P., Sakamoto, A., Hoffman, E. P. & Xiao, X. (1999) Gene. Ther. 6, 74–82. [DOI] [PubMed] [Google Scholar]
- 37.Xiao, X., Li, J. & Samulski, R. J. (1998) J. Virol. 72, 2224–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Snyder, R., Xiao, X. & Samulski, R. J. (1996) in Current Protocols in Human Genetics, eds. Dracopoli, N., Haines, J., Krof, B., Moir, D., Morton, C., Seidman, C. & Smith, D. (Wiley, New York), pp. 12.1.1–24.
- 39.Guo, L. T., Zhang, X. U., Kuang, W., Xu, H., Liu, L. A., Vilquin, J. T., Miyagoe-Suzuki, Y., Takeda, S., Ruegg, M. A., Wewer, U. M. & Engvall, E. (2003) Neuromuscul. Disord. 13, 207–215. [DOI] [PubMed] [Google Scholar]
- 40.Watchko, J., O'Day, T., Wang, B., Zhou, L., Tang, Y., Li, J. & Xiao, X. (2002) Hum. Gene. Ther. 13, 1451–1460. [DOI] [PubMed] [Google Scholar]
- 41.Sunada, Y., Bernier, S. M., Kozak, C. A., Yamada, Y. & Campbell, K. P. (1994) J. Biol. Chem. 269, 13729–13732. [PubMed] [Google Scholar]
- 42.Xu, H., Christmas, P., Wu, X. R., Wewer, U. M. & Engvall, E. (1994) Proc. Natl. Acad. Sci. USA 91, 5572–5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gawlik, K., Miyagoe-Suzuki, Y., Ekblom, P., Takeda, S. & Durbeej, M. (2004) Hum. Mol. Genet. 13, 1775–1784. [DOI] [PubMed] [Google Scholar]
- 44.Stephens, H. R., Duance, V. C., Dunn, M. J., Bailey, A. J. & Dubowitz, V. (1982) J. Neurol. Sci. 53, 45–62. [DOI] [PubMed] [Google Scholar]
- 45.Morrison, J., Lu, Q. L., Pastoret, C., Partridge, T. & Bou-Gharios, G. (2000) Lab. Invest. 80, 881–891. [DOI] [PubMed] [Google Scholar]
- 46.Vachon, P. H., Loechel, F., Xu, H., Wewer, U. M. & Engvall, E. (1996) J. Cell Biol. 134, 1483–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xiao, W., Chirmule, N., Berta, S. C., McCullough, B., Gao, G. & Wilson, J. M. (1999) J. Virol. 73, 3994–4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gao, G. P., Alvira, M. R., Wang, L., Calcedo, R., Johnston, J. & Wilson, J. M. (2002) Proc. Natl. Acad. Sci. USA 99, 11854–11859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakai, H., Fuess, S., Storm, T. A., Muramatsu, S., Nara, Y. & Kay, M. A. (2005) J. Virol. 79, 214–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Monahan, P. E. & Samulski, R. J. (2000) Gene Ther. 7, 24–30. [DOI] [PubMed] [Google Scholar]
- 51.Moral-Naranjo, M. T., Cabezas-Herrera, J., Vidal, C. J. & Campoy, F. J. (2002) Neurosci. Lett. 331, 155–158. [DOI] [PubMed] [Google Scholar]
- 52.Barton, E. R., Morris, L., Musaro, A., Rosenthal, N. & Sweeney, H. L. (2002) J. Cell Biol. 157, 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kaspar, B. K., Llado, J., Sherkat, N., Rothstein, J. D. & Gage, F. H. (2003) Science 301, 839–842. [DOI] [PubMed] [Google Scholar]
- 54.Baron, W., Decker, L., Colognato, H. & ffrench-Constant, C. (2003) Curr. Biol. 13, 151–155. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}