

Abstract
A positional cloning study of type 2 diabetes in Mexican Americans identified a region, termed “NIDDM1,” on chromosome 2q37 with significant linkage evidence. Haplotype combinations at the calpain-10 gene (CAPN10) within this region were shown to increase diabetes risk in several populations. On the basis of the thrifty genotype hypothesis, variants that increase susceptibility to type 2 diabetes under modern lifestyle conditions provided a survival advantage in past environments by increasing the efficiency of energy use and storage. Here, our goal is to make inferences about the evolutionary forces shaping variation in genes in the NIDDM1 region and to investigate the population genetics models that may underlie the thrifty genotype hypothesis. To this end, we surveyed sequence variation in CAPN10 and in an adjacent gene, G-protein–coupled receptor 35 (GPR35), in four population samples from different ethnic groups. These data revealed two distinct deviations from the standard neutral model in CAPN10, whereas GPR35 variation was largely consistent with neutrality. CAPN10 showed a significant deficit of variation in the haplotype class defined by the derived allele at SNP44, a polymorphism that is significantly associated with diabetes in meta-analysis studies. This suggests that this haplotype class was quickly driven to high frequency by positive natural selection. Interestingly, the derived allele at SNP44 is protective against diabetes. CAPN10 also showed a local excess of polymorphism and linkage disequilibrium decay in intron 13. Simulations show that this pattern may be explained by long-standing balancing selection that maintains multiple selected alleles. Alternatively, it is possible that the local mutation and recombination rates changed since the divergence of human and chimpanzee; this scenario does not require the action of natural selection on intron 13 variation.
Introduction
A genomewide scan in Mexican Americans identified a region on chromosome 2 (NIDDM1) that showed significant linkage to type 2 diabetes (MIM 125853) (Hanis et al. 1996). Subsequently, a combined linkage and case-control study of this region, using samples from the same population of Mexican Americans, identified a combination of haplotypes defined by three intronic variants at the calpain-10 locus (CAPN10) that was significantly associated with increased susceptibility to type 2 diabetes (Horikawa et al. 2000). Although CAPN10 variation was significantly associated with both disease susceptibility and the evidence of linkage, variation in the neighboring gene, GPR35, showed evidence of association with disease but not linkage. Subsequent studies of CAPN10 variation and type 2 diabetes and diabetes-related phenotypes have supported an association in some—but not all—populations studied (Weedon et al. 2003; Cox et al. 2004; Song et al. 2004 and references therein). The interpretation of these results rests on our understanding of the landscape of variation at this locus and how that variation differs across populations.
In 1962, Neel put forth a compelling hypothesis to explain the epidemiology of diabetes; this hypothesis remains a significant influence on our understanding of the evolutionary history of type 2 diabetes and other metabolic syndromes (Neel 1962; Weiss et al. 1984). The thrifty genotype hypothesis posits that variation that increases susceptibility to type 2 diabetes under modern lifestyle conditions provided an advantage in past environments by increasing the efficiency of energy use and storage. The thrifty genotype hypothesis has been modified and updated over the years in response to advances in understanding the disease pathophysiology and the role of specific environmental factors (Miller and Colagiuri 1994; Neel et al. 1998). However, it remains a hypothetical model that is mainly based on physiological and epidemiological considerations; no explicit population genetics model has been formulated within the context of the thrifty genotype hypothesis.
Indeed, population genetics studies of diabetes candidate genes, such as CAPN10 and GPR35, may inform the development of such models. For example, one population genetics scenario might envision that new thrifty variants arose by mutation in ancient human populations and were driven up in frequency—but not to fixation—by positive selection; these derived alleles, which may retain a signature of that selection in linked neutral variation, now result in increased risk of diabetes. In an alternative hypothetical scenario, the thrifty variants arose by mutation and were fixed as the result of more ancient adaptations; more specifically, their fixation was older than the average age of neutral human polymorphisms. Within this framework, the thrifty alleles are hypothesized to be ancestral and to have been maintained by purifying selection in ancient human populations. With the shift in lifestyle, these ancestral alleles are no longer advantageous and confer risk of diabetes (Sharma 1998). Concurrently, the derived alleles that used to be less efficient and, hence, slightly deleterious have become protective against diabetes. These protective alleles may have evolved neutrally or adaptively, depending on the fitness effects of diabetes and other pleiotropic phenotypes. The mechanism relating environmental changes and physiological adaptations is a topic of speculation. Regardless of the mechanism, the selective pressures acting on variants related to energy metabolism are likely to have fluctuated over time in response to changes in climate and diet, as well as changes in the physiological demands for energy.
The hypothesis that genes involved in type 2 diabetes evolved under changing selective pressures and may carry the signature of natural selection motivated us to describe patterns of sequence variation at CAPN10 and GPR35 in human populations. An initial population genetics study reported an unusually large difference in allele frequencies between African and non-African populations for the CAPN10 variants shown to influence risk of type 2 diabetes (Horikawa et al. 2000; Fullerton et al. 2002); it was proposed that this pattern reflects the impact of population-specific selective pressures.
Here, we follow up on this initial observation by conducting a full resequencing survey of CAPN10 and GPR35, and we use additional aspects of genetic variation to make inferences about the evolution of these genes. The effects of evolutionary forces on patterns of variation are complex; each aspect of variation is shaped by the stochastic effects of drift over time, the demographic history of the population, and natural selection acting on specific loci. As a result, observations made at any one locus cannot disentangle the effects of demography from the effects of natural selection (Hamblin et al. 2002; Akey et al. 2004; Hammer et al. 2004; Stajich and Hahn 2005). To characterize the effects of demography on patterns of sequence variation, we previously resequenced 50 unlinked noncoding regions in three population samples (Hausa of Cameroon, Italians, and Chinese) (Frisse et al. 2001; L.M.F. and A.D., unpublished data). This multilocus data set represents an empirical null distribution that captures the effects of each population’s unique demographic history and therefore can point toward unusual observations that may represent the signature of natural selection. Furthermore, these data showed that the sample from Cameroon fits the expectations of a model of long-term constant population size and random mating, but the same model did not fit the non-African samples for multiple aspects of the data (Frisse et al. 2001; Pluzhnikov et al. 2002); these results are in agreement with other multilocus studies of human populations (Akey et al. 2004; Hammer et al. 2004; Stajich and Hahn 2005). Thus, in addition to an empirical comparison, we use the multilocus data set to estimate the parameters of the neutral equilibrium model for the Hausa (i.e., the population mutation rate, θ [= 4Neμ], and recombination rate, ρ [= 4Ner]), to run coalescent simulations; these simulations are aimed at assessing the fit of the CAPN10 data to the model. Here, we investigate the same population samples, as well as a sample of a native Mexican population (Mazatecans), to show that patterns of variation at the CAPN10 gene do not fit the expectations of the standard neutral model.
Material and Methods
DNA Samples
Sequence variation was surveyed in DNA samples from four human populations: 16 Hausa from Yaoundé (Cameroon), 16 Han Chinese from Taiwan, 16 individuals from central Italy, and 16 Mazatecans from San Lorenzo Cuaunecuiltitla, Oaxaca (Mexico). This study was approved by the institutional review board of the University of Chicago. The CAPN10 and GPR35 genes were sequenced in one common chimpanzee; CAPN10 was also sequenced in one orangutan.
PCR Amplification
Primers for amplification and sequencing were designed using the program PRIMER, version 3. All primers were designed on the basis of GenBank sequence entry AF158748.3. All nucleotide positions in the present study, for both human and nonhuman primate data, refer to this entry. With few exceptions, PCR primers were designed to amplify a 600–800-bp fragment with ⩾200 bp overlap between amplicons. Amplification primers were used for sequencing. Additional sequencing primers were designed, adjacent to insertion/deletion polymorphisms, for nearly complete coverage in both orientations.
The surveyed region of CAPN10 contains a polymorphic 30-bp tandem repeat around nucleotides 21500–21800 that we were unable to characterize by PCR-based sequencing of diploid samples. PCR primers were designed on both sides of the repeat. Repeat size was determined by size fractionation of the PCR product on a 3.5% agarose gel. This approach necessarily precludes the identification of any SNP variation nested within the tandem repeat array.
Sequencing
PCR products were prepared for sequence analysis by treatment with a combination of shrimp alkaline phosphatase and exonuclease I (United States Biochemical). Dye-terminator sequencing was performed with the ABI BigDye terminator cycle sequencing kit, version 3, and analysis was performed on an ABI 3100 or ABI 3700 automated sequencer. All sequences were assembled and analyzed using the Phred-Phrap-Consed package (Nickerson et al. 1997). All putative polymorphisms and software-derived genotype calls were visually inspected (often by multiple independent operators) and were individually confirmed using Consed.
Statistical Analysis
Summary statistics were calculated for each gene by use of the Web application SLIDER (see the SLIDER Web site). Statistics were calculated for the entire length of the gene and on a sliding window of 1,000 bp, with a step size of 100 bp. For three statistics calculated on a sliding window (π, Tajima’s D, and ρH01), the maximum value observed in a single window (referred to as “max”), the ratio of the maximum value to the median value for all windows (referred to as “max/med”), and the maximum area above the median across all windows (referred to as “max area above med”) were calculated. These measures attempted to capture as many features of the peak as possible: its absolute magnitude, its magnitude relative to the rest of the surveyed region, and its size in terms of height and width of the region showing an elevation relative to the rest of the region. Haplotypes were inferred for each population sample by use of PHASE, version 2.02 (Stephens et al. 2001b; Stephens and Donnelly 2003). Estimates of the population recombination rate parameter 4Ner were obtained from diploid data by a composite likelihood method, with the use of the Web application MAXDIP, and are denoted by “ρH01” (Hudson 2001) (see the MAXDIP Web site). The Web application RECSLIDER was used to calculate ρH01 on a sliding window of 20 segregating sites, with a step size of 1 segregating site (Wall et al. 2003) (see the RECSLIDER Web site).
The haplotype test asks if a subset of haplotypes at a particular frequency contains fewer segregating sites than expected by simulating samples under neutrality, examining all subsets at the given frequency in each simulated sample, and determining how often a subset of haplotypes at the same frequency containing the same number or fewer segregating sites can be found (Hudson et al. 1994). This test was performed (separately for each population) using the program PSUBS, using recombination and mutation parameters estimated for each population to simulate 106 replicates under neutrality. We initially performed the test using the best reconstruction of haplotypes for each individual in the sample, based on the program PHASE. To assess whether the test results were robust to misspecification of haplotype phase, the list of possible haplotype pairs for each individual and the probabilities associated with each pair (generated by PHASE) were used to reassign haplotypes. After all individuals were assigned a pair of haplotypes, the number of segregating sites in the subset was recalculated and the test was performed again. This procedure was repeated 106 times for each population.
Coalescent Simulations
The significance of sliding-window observations was estimated by coalescent simulations with recombination. One thousand samples were simulated under a neutral model, using the program MS (Hudson 2002). For the neutral model, the recombination rate parameter used in the simulations was estimated for the Hausa at CAPN10, with the ratio of gene conversion to crossing over fixed at 2 and an average conversion tract length of 500 bp (Frisse et al. 2001). Likewise, the mutation rate parameter was estimated from π in the Hausa at CAPN10. The Hausa were used as a basis for comparison because evidence from unlinked noncoding regions suggests that they best fit an equilibrium model of demography, whereas the Italians and Chinese do not (Frisse et al. 2001; Pluzhnikov et al. 2002). A sliding-window analysis was performed on each simulated replicate, precisely mirroring the analyses performed on the CAPN10 data (described in the “Statistical Analysis” section): for each of the summary statistics (π, Tajima’s D, and ρH01), the maximum window value (max), the ratio of the maximum value to the median across all windows (max/med), and the maximum area above the median across all windows (max area above med) were calculated. The probability of observing a value as high or higher than that observed at CAPN10 was estimated on the basis of the simulated replicates. This probability can be converted to a two-tailed P value by use of the expression 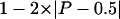 , where P is the above-estimated probability.
, where P is the above-estimated probability.
Samples were simulated under the model of biallelic balancing selection described by Hudson and Kaplan (1988), using a modification of the program MS and population parameters estimated from the Hausa data. Each simulated sequence was 10 kb long, with the selected site in the center. The allele frequency at the selected site was varied from 0.1 to 0.5. Multiallelic balancing selection was simulated using software by Mikkel Schierup that simulates sequences with recombination under the model described in the study by Schierup et al. (2001). Under this model, selection acts at a single genomic location to preserve M (the number of alleles at the selected site) at equal frequencies in the population. Over time, these allelic classes are lost and replaced (“turnover”), in accordance with a process described by Takahata (1990). At a turnover event, one class is lost and another is created, keeping M constant. The class that was newly created by mutation rises to equilibrium frequency approximately instantaneously. Each simulated sequence was 5 kb long, with a single site under selection placed at one end. One thousand replicates of 32 sequences were simulated for each of 30 combinations of the turnover rate (Q, where 2Ne/Q is the mean time between turnover events) and the number of allelic classes (M), with Q ranging from 0.1 to 1.3 and M ranging from 3 to 30. To assess the overall fit of the data to the model, a combined statistic was also calculated by summing the logs of the two-tailed P values for each statistic calculated from the Hausa sample; a P value for this combined statistic was then calculated as the proportion of simulated samples for which the combined statistic took a value that was greater than or equal to the value calculated for the Hausa.
Results
Whole-Gene Analyses
Summary statistics representing several aspects of variation were calculated for each gene as a whole, in each population sample (table 1). The total surveyed sequence length for CAPN10 and GPR35 was 33,465 bp and 2,310 bp, respectively, and included the coding regions, the introns, the UTRs, and ∼1 kb upstream of the transcription start site. The average sequence divergence between human and chimpanzee for CAPN10 and GPR35 was 1.77% and 1.92%, respectively—somewhat higher than that observed at other loci (Ebersberger et al. 2002). Average sequence divergence between human and orangutan was 3.67% for CAPN10, which is also higher than that observed at other loci across the genome (Nachman et al. 1998; Chen and Li 2001).
Table 1.
Summary Statistics of Polymorphism
|
CAPN10 |
GPR35 |
|||||||||
| Population | Sa | θWb | πc | ρH01d | Tajima’s D | Sa | θWb | πc | ρH01d | Tajima’s D |
| Hausa | 203 | 14.8 | 10.8 | 12.4 | −1.08 | 10 | 11.7 | 11.2 | 40.1 | −.14 |
| Chinese | 148 | 10.8 | 12.0 | 1.3 | .35 | 6 | 7.0 | 8.7 | 11.9 | .70 |
| Italians | 163 | 11.9 | 11.8 | 1.9 | −.08 | 16 | 18.7 | 16.0 | 9.9 | −.47 |
| Mazatecans | 115 | 8.4 | 7.5 | .5 | −.44 | 6 | 7.0 | 8.1 | 12.8 | .45 |
Number of polymorphic sites.
Watterson’s estimator of θ (= 4Nμ) per bp (× 10−4) (Watterson 1975).
Nucleotide diversity per bp (× 10−4).
Polymorphism levels are summarized by the estimator of the population mutation rate parameter θW (Watterson 1975), based on the number of polymorphic sites, as well as by nucleotide diversity (π). At CAPN10, the Hausa sample shows the highest polymorphism levels in terms of θW, but not in terms of nucleotide diversity. At GPR35, the Italian sample shows the highest polymorphism levels for both summaries of the data. African populations have been found to harbor more variation than non-African populations at most loci across the genome (Cann et al. 1987; Hammer 1995; Harding et al. 1997; Zietkiewicz et al. 1998), as well as in our data set of noncoding regions, but there is great variation from one locus to another. None of the 50 noncoding regions shows a difference in θW between Hausa and Italians that is greater than that observed at GPR35; the same holds for the difference between Italians and Chinese. This raises the possibility that GPR35 did not evolve neutrally in the Italians and that the high polymorphism levels reflect the action of natural selection. This hypothesis was investigated by means of a test of neutrality that is based on polymorphism and divergence levels (Hudson et al. 1987) and that was performed on the Italian data for GPR35 and the 50 noncoding regions: no significant departure was observed. A test of neutrality that is based on the ratio between synonymous and nonsynonymous changes in polymorphism and divergence data and that was performed for each population, compared with the chimpanzee and orangutan divergence, did not detect a significant departure (McDonald and Kreitman 1991). Consistent with previous data from other Native American populations (Mulligan et al. 2004 and references therein), the Mazatecan sample harbors low levels of variation.
The spectrum of allele frequencies is summarized by the Tajima’s D statistic, which is expected to be ∼0 under the neutral equilibrium model (Tajima 1989). A negative value indicates an excess of rare variants, whereas a positive value indicates an excess of intermediate frequency variants. Certain demographic or selective departures from neutral equilibrium predict skews in the frequency spectrum. The Tajima’s D value for the Hausa at CAPN10 is significantly negative as assessed by simulations of the equilibrium model (P=.01) but is well within the range of values observed at the noncoding regions. This is consistent with a model of recent population growth from a population at equilibrium (Pluzhnikov et al. 2002). All other values of Tajima’s D are not unusual.
Estimates of the population recombination rate parameter provide an assessment of linkage disequilibrium (LD) across a region that is less sensitive to sample size and allele frequency than other commonly used summary statistics, such as D′ and r2 (Pritchard and Przeworski 2001). A composite maximum-likelihood estimator, ρH01, was calculated for each population under a model of recombination that includes both crossover and gene conversion events. The Hausa show the highest value of ρH01, corresponding to the lowest levels of LD. This is consistent with the decay of LD over distances that are shorter among African populations than among non-African populations, an observation made for many loci across the genome (Reich et al. 2001; Stephens et al. 2001a; Crawford et al. 2004b; McVean et al. 2004) and in our data from noncoding regions (Frisse et al. 2001).
Testing for Selection on the Haplotype Class Defined by SNP44
Meta-analyses of published studies supported a significant association of the C allele at SNP44 (position 22751) with increased risk of type 2 diabetes (Weedon et al. 2003; Song et al. 2004). Although it is located in intron 3, only 11 bp from SNP43 (which shows linkage and association with diabetes in Mexican Americans), SNP44 is not in perfect LD with the SNPs that define the risk haplotype: SNP43 (position 22762), Indel19 (position 25830), and SNP63 (position 34288) (table 2) (see also Horikawa et al. [2000] and Fullerton et al. [2002]).
Table 2.
Pairwise LD (r2) between SNP44 and Other SNPs
|
r2 for Data Set |
|||||
| SNP | Position(bp) | Hausa | Italian | Chinese | Mazatecan |
| SNP134 | 17749 | 1 | 1 | 1 | 1 |
| SNP135 | 17841 | 1 | 1 | 1 | 1 |
| SNP43 | 22762 | .0025 | .0476 | .0290 | .0472 |
| SNP19 | 25830 | .0051 | .2088 | .4667 | .0616 |
| SNP110 | 27713 | 1 | .6191 | 1 | .3118 |
| SNP63 | 34288 | .0816 | .0114 | .0074 | .0147 |
The risk allele at SNP44 (i.e., C) is inferred to be ancestral on the basis of the chimpanzee and orangutan sequences. Thus, this variant may fit one of the population genetics models for the thrifty genotype hypothesis proposed in the “Introduction” section (i.e., one in which alleles increasing risk to type 2 diabetes are ancestral and were maintained by purifying selection in ancient human populations). Interestingly, the derived (T) allele appears at a high frequency (78%–97%) on a haplotype background with little variation in all four population samples. A large subset of haplotypes containing little variation is the signature expected if positive selection recently and quickly drove variant(s) on the haplotype to high frequency.
To test whether the haplotypes bearing the derived allele at SNP44 carried less variation than expected under neutrality, we used the haplotype test described by Hudson et al. (1994). PHASE was used to infer haplotypes in each population for a 6-kb region that centered on SNP44; using the haplotypes inferred by PHASE as the best reconstruction, the test was significant for all populations (Hausa: P=.0007; Chinese: P=.0277; Italians: P=.0328; and Mazatecans: P=.0034). To determine whether the test results were robust to misspecification of haplotype assignment, we generated 106 haplotype samples in which phase was assigned on the basis of the uncertainty estimated by PHASE (see the “Material and Methods” section) and reran the test for each sample. The proportion of runs with a P value <.05 was 99.95% for the Hausa, 68.82% for the Chinese, 51.41% for the Italians, and 88.69% for the Mazatecans. Thus, at least the Hausa harbor a clear signal of natural selection on the haplotype class defined by the derived allele at SNP44; given the substantial proportion of haplotype reconstructions yielding a significant haplotype test result, it seems plausible that the non-African samples also retain evidence of selection. In the Hausa and the Chinese, SNP44 is in perfect LD with an amino acid polymorphism, Thr504Ala (SNP110, position 22713) (table 2). We performed the haplotype test again on a 10-kb region centered on both SNPs by defining the haplotype subset on the basis of the derived alleles at both SNPs. Using the haplotypes inferred by PHASE as the best reconstruction, the test was significant in the Hausa (P=.0002) but not in the Chinese.
Sliding-Window Analyses of CAPN10
A signature of natural selection, if present, may extend over a relatively short distance. Thus, such a signal could be diluted and could become undetectable when summaries of the data are calculated over a large segment, as is the case here for CAPN10. To reveal patterns of variation on a finer scale and to ask whether other signatures of natural selection (in addition to that around SNP44) are present in this gene, each summary statistic was also investigated using a sliding-window analysis.
Values of Tajima’s D showed stochastic variation around the mean but no unexpected extremes in any of the populations (data not shown). However, levels of polymorphism, assessed by either π or θW, varied dramatically across the gene, with a prominent peak in intron 13 (fig. 1). Although levels of interspecies divergence showed random variation across the surveyed region, there is no corresponding peak in sequence divergence from either chimpanzee or orangutan (fig. A1 [online only]), as would be expected for a long-standing local increase in mutation rate. The peak in polymorphism overlaps two of the variants that define the diabetes susceptibility haplotype in Mexican Americans (SNP30 and SNP63) (fig. 1). In the Hausa, the peak in polymorphism level also corresponds to a peak in LD decay, as estimated by ρH01; this peak is either much lower or absent in the Italians, Chinese, and Mazatecans (fig. 1).
Figure 1.

Sliding-window plots of π (blue line) and ρH01 (red line) for the CAPN10 gene. The size of the window is 20 polymorphic sites, and the step size is 1 polymorphic site. Each summary statistic is calculated per bp by normalizing the summary by the varying length in bp of each window. The black triangles along the horizontal axes indicate the position of the SNPs included in the diabetes risk haplotype, as defined by Horikawa et al. (2000) (ordered 5′→3′: SNP43, SNP56, SNP59, Indel19, SNP48, SNP30, SNP63, and SNP65). Nucleotide positions are expressed in bp.
In the analyses that follow, we investigate the processes underlying this striking pattern of variation by performing coalescent simulations of the standard neutral model and two models of long-standing selection. Determining whether an observation made through a sliding-window analysis represents a significant departure from a null model presents a difficult multiple-testing problem: the window is slid across a contiguous region, so that not only do the windows overlap but the sites within even distant windows may also be in LD. Testing observed data against an empirical distribution drawn from simulated samples analyzed in the same manner circumvents this problem.
Simulations of the Standard Neutral Model
A great deal of stochastic variation in fine-scale patterns of sequence variation is expected even under the neutral model with uniform recombination and mutation rate. To assess the probability that the intron 13 data were simply due to chance, we ran coalescent simulations under the standard neutral model to generate samples that were analyzed using the same sliding-window method that was used on the CAPN10 data. The patterns of variation were summarized by three sliding-window statistics: (1) the maximum value observed in a window (max); (2) the ratio of the maximum value to the median across all windows (max/med); and (3) the maximum area above the median across all windows (max area above med). Each measure was calculated from the Hausa data for the CAPN10 region and was compared with the same measures calculated for each of 1,000 samples simulated under a neutral equilibrium model. The Hausa data were used because previous analyses of noncoding region variation indicated that this population sample conforms to the assumptions of the demographic model under which the neutral samples were simulated, whereas the Chinese and Italian samples do not (Frisse et al. 2001; Pluzhnikov et al. 2002). The same analysis was performed for ρH01 and Tajima’s D. The area above the median for nucleotide diversity is significantly larger than that expected under neutrality, as is the ratio of the maximum ρH01 to the median (table 3). These results, taken together, suggest that the standard neutral model cannot explain the Hausa data for CAPN10.
Table 3.
Estimated Probability of Observing a Value as High as or Higher than That Observed at CAPN10 for the Hausa, under the Standard Neutral Model
|
P Value, by Summary Statistic |
|||
| Sliding-WindowStatistica | πb | ρH01c | Tajima’s D |
| max | .259 | .457 | .850 |
| max/med | .239 | .008 | .924 |
| max area above med | .006 | .381 | .131 |
See the “Statistical Analysis” section.
Nucleotide diversity per bp (× 10−4).
Simulations of the Biallelic Model of Balancing Selection
Long-standing balancing selection may preserve a polymorphism at the site under selection longer than would be expected under neutrality. As a result, neutral mutations at linked sites are not fixed or lost as quickly as expected if all variation in the region were neutral, and a signature of increased polymorphism develops over time. In intron 13, we observed a peak of polymorphism relative to divergence overlapping variants that have been associated with disease susceptibility; this pattern was shown to be unlikely under neutrality and is similar to that expected under long-standing balancing selection. However, little is known about the expected frequency spectrum and LD under long-standing balancing selection.
Thus, to test whether balancing selection could indeed explain the polymorphism levels, as well as other aspects of sequence variation data in intron 13, we simulated samples under the simplest model of balancing selection. Under this model, selection acts at a single site to maintain two alleles at a stable equilibrium frequency, which was varied in our simulations over a range from 0.1 to 0.5. Furthermore, we considered the same three aspects of the data (i.e., polymorphism levels, allele-frequency spectrum, and LD) that we had examined in the population data.
The median nucleotide diversity across simulated samples is elevated above the 97.5th percentile from neutral simulations for a narrow region (∼1 kb) centered on the selected site (fig. 2). This signal is consistent across equilibrium allele frequencies and increases with increasing equilibrium frequency. Most—but not all—models resulted in a shift in allele-frequency spectrum (fig. 2). For an equilibrium allele frequency of 0.1, the median Tajima’s D value was indistinguishable from the value expected under neutrality. The median Tajima’s D value is elevated, but it is below the neutral 97.5th percentile under a model with an equilibrium allele frequency of 0.2; a significant skew toward alleles of intermediate frequency is expected only if the equilibrium frequency is ⩾0.3. For all equilibrium frequencies, median ρH01 is reduced near the selected site, although it is well within expectations under the neutral model, indicating an increase of LD in an ∼2-kb region surrounding the selected site. LD near the selected site increases as equilibrium allele frequency decreases.
Figure 2.

Sliding-window plots of π, ρH01, and Tajima’s D for samples of 32 sequences generated by simulations of a biallelic model of balancing selection over a range of equilibrium allele frequencies of 0.1–0.5. Each summary is calculated as the median across 1,000 simulated samples for each value of equilibrium frequency. Nucleotide positions are expressed in bp.
These results indicate that, under a simple biallelic model, the signature of selection in polymorphism levels, frequency spectrum, and LD is tightly localized around the selected site and is detectable by use of a sliding-window analysis for most equilibrium allele frequencies. However, the simulations also show that this selection model is not compatible with CAPN10 for any equilibrium allele frequency. As shown in table 4, the ratio of the maximum ρH01 to the median is significantly higher than expected at all equilibrium allele frequencies, Tajima’s D value is lower than expected for allele frequencies ⩾0.3, and polymorphism levels are lower than expected for allele frequencies ⩾0.4.
Table 4.
Estimated Probability of Observing a Value as High as or Higher than That Observed at CAPN10 for the Hausa, under the Biallelic Balancing Selection Model
|
P Value, by Summary Statistic |
|||||||||
| πa |
ρH01b |
Tajima’s D |
|||||||
| EquilibriumFrequency | max | max/med | max area above med | max | max/med | max area above med | max | max/med | max area above med |
| .1 | .742 | .564 | .137 | .504 | .021c | .365 | .874 | .934 | .201 |
| .2 | .943 | .834 | .421 | .504 | .020c | .374 | .880 | .924 | .245 |
| .3 | .967 | .902 | .606 | .504 | .020c | .375 | .985c | .927 | .317 |
| .4 | .980 | .946 | .708 | .528 | .020c | .366 | .996c | .903 | .465 |
| .5 | .985c | .953 | .744 | .526 | .022c | .381 | .993c | .898 | .511 |
Simulations of a Multiallelic Model of Balancing Selection
Two of the features of the CAPN10 data that are inconsistent with the predictions of a simple biallelic model suggest that a more complex selection hypothesis may be more plausible. Haplotype analyses of the ∼5 kb surrounding the peak of polymorphism reveal five distinct deep lineages (V. J. Clark and A. Di Rienzo, unpublished results), instead of two, as a biallelic model would predict. Also, the value of Tajima’s D at the peak of polymorphism is not strikingly positive, as expected under the biallelic model of balancing selection for most equilibrium frequencies. These observations led us to explore the model of multiallelic balancing selection previously developed to investigate the major histocompatibility complex (MHC) in mammals and self-incompatibility genes in plants, as described by Schierup et al. (2001). Because more than two alleles are maintained in the population under this model, the skew toward intermediate frequency variants may be less marked than under the biallelic model and, hence, may be consistent with the intron 13 data. To investigate whether this more complex model of balancing selection could explain the intron 13 data, samples were simulated using 30 combinations of the parameters Q and M, and the probability of the Hausa data for each summary statistic was estimated (table A1 [online only]). As expected, median nucleotide diversity was elevated near the selected site for all combinations of the parameters; the observed levels of polymorphism at CAPN10 were consistent with high turnover rates (Q=1 [i.e., a mean time between turnover events of 500,000 years, under the assumptions of Ne=10,000 and 25 years per generation) and low numbers of allelic classes (M=5–6). On average, the value of Tajima’s D was slightly higher near the selected site for models with few allelic classes; as the number of allelic classes was increased, the median Tajima’s D value near the selected site became closer to zero (fig. A2 [online only]); the observed value of Tajima’s D was consistent with the model for all combinations of parameter values considered. Remarkably, the median ρH01 was elevated around the selected site, which is the opposite of the trend for the biallelic model and is consistent with our observations. To assess the overall fit of the data over the two-dimensional parameter space, we calculated a composite statistic combining the P values of the three aspects of variation for each sliding-window statistic. This was compared with the distribution of the same statistic in simulated samples to estimate the probability of a departure as large as or larger than observed (see the “Material and Methods” section). The results indicate that the Hausa data are compatible with the multiallelic balancing selection model for a portion of the parameter space (shown as the unshaded area in fig. 3).
Figure 3.

Contour plot of the probability of the Hausa data for CAPN10 over a grid of values of the parameters Q and M, under the multiallelic model of balancing selection. The shaded area indicates the portion of the parameter space that is not compatible with the data. The P values are for the statistic combining the probability of three aspects of the data (i.e., π, ρH01, and Tajima’s D) and are calculated by comparison with simulated samples (see the “Material and Methods” section).
Discussion
Our analysis of two genes, CAPN10 and GPR35, within a positional candidate region for type 2 diabetes identified several interesting patterns that may result from the action of positive natural selection. First, in CAPN10, the haplotype class defined by the derived allele at SNP44, a polymorphism associated with increased diabetes risk, has a significant deficit of polymorphism, as expected if recent positive selection rapidly drove this haplotype class to high frequency. Second, a region of markedly high polymorphism and decay of LD was identified in intron 13 of CAPN10, which is consistent with a model of balancing selection involving multiple alleles. Although additional studies will be necessary to assess whether these findings support the thrifty genotype hypothesis, it is likely that selective pressures acting on this genomic region have changed over the time scale of human evolution and have shaped its patterns of variation.
Of the two potential signatures of selection mentioned above, the low variability associated with the haplotypes carrying the derived allele at SNP44 may be most easily reconciled with selection on variation affecting diabetes risk. Recent meta-analyses have confirmed a role for this variant—or one in perfect LD with it—in diabetes risk. A meta-analysis of >7,000 cases and controls supported an association of the ancestral (C) allele at SNP44 with increased risk of type 2 diabetes, with an odds ratio (OR) of 1.17 and a P value of .0007 (Weedon et al. 2003). A separate meta-analysis of CAPN10 variation and type 2 diabetes reported a significant undertransmission of the derived (T) allele at SNP44 to affected offspring in the pooled sample from three family-based studies, with a pooled OR of 0.66 and a P value of .004 (Song et al. 2004). Functional studies also suggest a role for SNP44. On the basis of in vitro assays, it was proposed that SNP44 is located within an enhancer element and influences its activity (Horikawa et al. 2000). Although binding assays of nuclear extracts in HepG2 cells showed that the SNP44 polymorphism did not affect binding, a reporter gene assay showed that SNP44, in addition to SNP43, may modulate transcription of CAPN10. These results are compatible with the notion that SNP44 itself was the target of positive selection and one of the causative disease variants; however, the possibility that the signature of selection and the disease-association signal are both due to a polymorphic site in strong LD with SNP44 cannot be excluded. In fact, our data show that the derived allele at SNP44 is part of a long-range haplotype containing several other variants, including the derived allele at Thr504Ala (SNP110), an amino acid replacement in domain III of calpain-10. It is possible that Thr504Ala, alone or in combination with SNP44, was the target of positive natural selection and drove the association in disease-mapping studies. However, Thr504Ala is not the only polymorphism in strong LD with SNP44 that might be functional. For example, in all four population samples, SNP44 is in perfect LD with two polymorphisms in the 5′ UTR, SNP134 (position 17749) and SNP135 (position 17841) (table 2). Notably, at these two sites, it is the ancestral allele that is associated with the derived allele at SNP44. Given the rapid breakdown of LD in intron 13, it is unlikely that the target of selection resides in the unsurveyed region 3′ to CAPN10. Independent data show that LD breaks down (r2⩽0.04) in populations from the major ethnic groups between SNP131 (position 4061), in the RNPEPL1 gene 5′ to CAPN10, and SNP66 (position 10676) (L. del Bosque-Plata and G. Hayes, unpublished data); this suggests that at least part of the RNPEPL1 gene can be excluded as the target of selection.
It was noted elsewhere that several of the variants (i.e., Indel19 and SNP63) that make up the risk haplotypes had unusually large differences in allele frequencies between Africans and non-Africans, as compared with a set of likely neutral loci (Fullerton et al. 2002). This finding was interpreted as possibly resulting from population-specific selective pressures on these SNPs. However, the evidence of a partial selective sweep that emerges from the present survey suggests an alternative explanation. Theoretical work showed that, if a selective sweep occurs in geographically subdivided populations and migration rates are low relative to the selection coefficient, then linked neutral alleles may exhibit unusually large differences in allele frequencies across subpopulations (Slatkin and Wiehe 1998). Thus, if the selective event that drove up the frequency of the SNP44-haplotype class occurred in African and non-African populations after their separation, then the observed degree of differentiation of allele frequencies at linked SNPs would not be unexpected and would not reflect a selective advantage of the highly differentiated SNPs. A haplotype test performed on segments centered on either Indel19 or SNP63 in each population did not yield a significant result; this is consistent with the idea that these SNPs were not the target of directional selection.
The possibility of an ancestral allele, such as would be the case for SNP44 or Thr504Ala, that increases risk of diabetes is particularly interesting in light of other findings about risk variants for common diseases. The common polymorphism Pro12Ala at the PPARG gene was shown to increase risk of type 2 diabetes, with an OR of 1.25 (Altshuler et al. 2000). On the basis of an alignment with the orthologous chimpanzee sequence, we determined that the risk allele (Pro12) is ancestral, whereas the protective and less common allele is derived. If the parallel with SNP44 (or Thr504Ala) holds, then one might expect a signature of positive selection around the derived Ala12 allele. However, given its relatively low frequency (∼15%), the power to detect the signature of selection, if present, may be low. The ɛ4 allele at the APOE gene is another pertinent example: this allele is defined by the presence of the ancestral allele at two common amino acid polymorphisms and was shown to increase risk of coronary artery disease (Davignon et al. 1988; de Knijff et al. 1994; Stengard et al. 1995) and Alzheimer disease (Corder et al. 1993; Strittmatter et al. 1993). Interestingly, an analysis of polymorphism data in human populations showed that the haplotype class defined by the derived allele ɛ3—the most common in all populations—was associated with an excess of low-frequency variants, which was interpreted as evidence of the action of positive selection on this haplotype (Fullerton et al. 2000). The hypothesis that the ɛ3 allele increased in frequency as a result of positive selection had been independently proposed on the basis of its distribution across populations with different subsistence strategies, as well as on the basis of functional considerations (Corbo and Scacchi 1999). Within the same context, it was postulated that the ancestral ɛ4 allele was a “thrifty” allele in ancient human populations and that it had become deleterious under more recent environmental conditions.
Thus, SNP44 and Thr504Ala may be additional examples of polymorphisms in which the ancestral allele increases risk of common metabolic syndromes and which probably became deleterious during the relatively recent history of human populations. The causes of such a deleterious effect are unclear. One might speculate that the disease phenotype is deleterious, per se, or that the thrifty alleles have pleiotropic effects with negative fitness consequences (e.g., variation in birth weight). Overall, these results suggest that one of the population genetics models associated with the thrifty genotype hypothesis involves ancestral risk alleles that were beneficial in ancient human populations but that became detrimental as a result of recent lifestyle changes; these changes led, in turn, to the rapid rise in frequency of the derived alleles and the resulting signature of positive selection.
The second deviation from the standard neutral model is the one observed in intron 13 of CAPN10. This signal consists of a significantly large peak in polymorphism that overlaps, in the Hausa, with a significant peak in the decay of LD. One possible source of deviation from the null model is the action of natural selection. We performed coalescent simulations to explore a biallelic model of long-standing balancing selection, and, when we rejected it, we turned to a more complex model in which selection maintains more than two alleles. Under the multiallelic balancing selection model, the data are consistent with a limited range of combinations of parameter values. In this model, which was originally developed to explain variation at the MHC, a fixed number of alleles is maintained through a process of turnover, whereby selected alleles are lost by chance and are replaced by new selected alleles (Takahata 1990). Although this long-standing diversifying selection model is not easily reconciled with diabetes susceptibility, it is possible that other selective models (e.g., long-term fluctuations of environmental conditions and selective pressures [Gillespie 1991]) result in patterns of variation that are similar to those predicted by the model of Takahata (Takahata 1990; Hedrick 2002) and may be more relevant to the evolution of genes involved in energy metabolism.
Evaluating the plausibility of this selective scenario is challenging because of the uncertainties concerning the biological role of calpain-10 and the significance of its variants with regard to gene function and disease susceptibility. However, from a strictly evolutionary standpoint, our findings could be consistent with the “carnivore connection” hypothesis (Miller and Colagiuri 1994; Colagiuri and Brand Miller 2002), which postulates a critical role for the quantity of dietary protein and carbohydrate in the evolution of insulin resistance. More specifically, it was proposed that insulin resistance offered survival and reproductive advantages during the Ice Ages, which started ∼2.5 million years ago (mya) and dominated most of human evolution, when a shift to a high-protein, low-carbohydrate diet occurred as a consequence of climatic changes. Notably, when we apply a molecular clock calculation (eq. 3 in Thomson et al. [2000]) to estimate the average time since the most recent common ancestor of the population samples for the 1-kb segment centered on the peak of polymorphism, we obtain age estimates of 2.1–2.8 mya and 2.4–3.1 mya, depending on whether the chimpanzee or the orangutan sequences were used as an outgroup species (we assumed a divergence time of 6 mya and 14 mya for human and chimpanzee and for human and orangutan, respectively). This suggests that, if indeed a balanced polymorphism exists in this location, its age would be consistent with the onset of new selective pressures associated with the Ice Ages. Given the likely role of calpains in insulin secretion and action (Sreenan et al. 2001), these results offer a new working hypothesis for the evolution of intron 13 variation and its role in human metabolic adaptations.
However, natural selection is not the only possible cause for the pattern observed in intron 13. One possibility is that the elevation in polymorphism levels is the result of a complex history of population structure. It can be speculated that ancient admixture in sub-Saharan Africa prior to the dispersal into Europe and Asia could have led to the patches of high diversity observed in intron 13; however, one might also expect an excess of LD, which is contrary to our observations. Additional data from unlinked genomic regions surveyed in multiple human populations will allow a more accurate reconstruction of the history of population structure and will offer the opportunity to reevaluate our findings.
Nonuniform rates of recombination or mutation are also possible violations of the assumptions of the standard neutral model. More specifically, a hotspot of both recombination and mutation, with all variation being selectively neutral, may explain the data. However, the absence of a peak of interspecies sequence divergence corresponding to the peak of polymorphism implies that, if a mutational hotspot is present, this must have arisen after the divergence of human and chimpanzee. The coincidence of high π and ρH01 in intron 13 may reflect a relationship between mutation and recombination. Evidence that mutation and recombination may be related processes on a genomewide scale comes from the observation that regions of high recombination in humans tend to have slightly higher levels of divergence between human and mouse (Lercher and Hurst 2002), human and baboon, and human and macaque (Hellmann et al. 2003). On a finer scale, the recombinational hotspot in the human TAP2 gene, which was confirmed by sperm typing (Jeffreys et al. 2000), is associated with an excess of polymorphism relative to divergence (Jeffreys et al. 2000) and rapid decay of LD (Cullen et al. 1995; Jeffreys et al. 2001), just like intron 13 of CAPN10. Interestingly, a recent analysis of LD in the orthologous region of TAP2 in chimpanzees indicates that the recombination hotspot is absent in this species (Ptak et al. 2004). Thus, the rate of both mutation and recombination changed dramatically at the TAP2 gene since the divergence of human and chimpanzee. However, the β-globin recombination hotspot has no corresponding peak of polymorphism or divergence and there is no evidence that the hotspot exists in the orthologous region of macaques and, perhaps, chimpanzees (Wall et al. 2003). Importantly, a recent large-scale study of sequence variation in two population samples detected numerous regions of rapid LD decay, consistent with the presence of recombinational hotspots; however, no association between levels of polymorphism and LD decay was detected in these regions (Crawford et al. 2004a). Thus, an increase in polymorphism levels is unlikely to be a universal feature of human recombination hotspots. Sperm typing and LD analysis in chimpanzees are currently being performed to investigate this alternative neutral explanation for the observations at intron 13 of CAPN10.
The GPR35 gene was included in our survey because of its location immediately 3′ to CAPN10 and because it showed evidence of association with type 2 diabetes. Also, its expression in tissues including pancreatic islets and skeletal muscle is consistent with a possible role in type 2 diabetes. Our results showed unusually high polymorphism levels in the Italian sample, relative to the Hausa and Chinese samples; this was determined by comparison with the data from 50 noncoding regions obtained in the same population samples. However, a test of the standard neutral model based on polymorphism and divergence—using the same data—did not detect a significant departure in the Italians. Thus, although these results remain unusual and deserve further attention, they do not constitute strong evidence of positive selection.
Elucidating the evolutionary models of common human diseases is crucial for defining powerful approaches to mapping susceptibility variants. In particular, models in which selective pressures on disease risk variants changed during human evolution are likely to apply broadly to diseases of modernization (e.g., obesity, hypertension, and asthma), which represent a major health burden in industrialized countries. The analysis of candidate genes, such as CAPN10 and GPR35, is a step toward outlining the relevant models for these diseases and developing further working hypotheses. Further studies of CAPN10 and GPR35, as well as other candidate genes, will be necessary to develop a comprehensive understanding of the evolution of common-disease susceptibility.
Acknowledgments
We are grateful to C. Spencer for pointing out to us that the multiallelic balancing selection model could generate patterns of variation that are similar to those observed at CAPN10 and for sharing simulation results with us. We thank N. Cox, for helpful comments and discussions throughout this project; G. Bell, for comments on the manuscript; and M. Hammond, for technical help. This work was supported by the National Institutes of Health (NIH) (grants DK56670 and DK55889). L.M.F. was supported by a National Research Service Award postdoctoral fellowship (HG00219). J.V. was supported by an NIH training grant (GM07197).
Appendix A: Supplemental Material
Figure A1.

Sliding-window analysis of polymorphism levels in the Hausa and sequence divergence of human versus chimpanzee and human versus orangutan. The window size was 1,000 bp, and the window step was 100 bp. Nucleotide positions are expressed in bp.
Figure A2.

Aspects of variation under a multiallelic model of balancing selection. For each statistic calculated for a 1,000-bp segment adjacent to the site under selection, the median across 1,000 simulated samples is plotted over a grid of the parameter values (Q and M) used in the simulation.
Table A1.
Estimated Probability of Observing a Value as High as or Higher than That Observed at CAPN10 for the Hausa, under the Multiallelic Balancing Selection Model
| Combinations of Parameter Values | P Value, by Summary Statistica | |||
| Q | M | π | Tajima’s D | ρH01 |
| 1.3 | 3 | .122 | .838 | .070 |
| 1.3 | 5 | .682 | .976b | .109 |
| 1.3 | 6 | .877 | .988b | .213 |
| 1.3 | 10 | 1b | .988b | .988b |
| 1.3 | 15 | 1b | .995b | .819 |
| 1.3 | 30 | 1b | .999b | .975b |
| 1 | 3 | .099 | .849 | .160 |
| 1 | 5 | .660 | .969 | .209 |
| 1 | 6 | .856 | .983b | .334 |
| 1 | 10 | .999b | .990b | .771 |
| 1 | 15 | 1b | .993b | .912 |
| 1 | 30 | 1b | .999b | .993b |
| .7 | 3 | .262 | .870 | .065 |
| .7 | 5 | .863 | .987b | .151 |
| .7 | 6 | .975b | .992b | .269 |
| .7 | 10 | 1b | .998b | .702 |
| .7 | 15 | 1b | .998b | .866 |
| .7 | 30 | 1b | .999b | .995b |
| .4 | 3 | .436 | .933 | .060 |
| .4 | 5 | .940 | .995b | .205 |
| .4 | 6 | .993b | .997b | .326 |
| .4 | 10 | 1b | .996b | .762 |
| .4 | 15 | 1b | 1b | .928 |
| .4 | 30 | 1b | .999b | 1b |
| .1 | 3 | .543 | .970 | .168 |
| .1 | 5 | .985b | .997b | .475 |
| .1 | 6 | .999b | .996b | .701 |
| .1 | 10 | 1b | 1b | .949 |
| .1 | 15 | 1b | .998b | .992b |
| .1 | 30 | 1b | .999b | 1b |
For each summary statistic, the P value is the probability of the value calculated for the first window away from the selected site. Nucleotide diversity (π) and Tajima’s D values were calculated for a window of 1 kb, whereas ρH01 value was calculated for a window of 20 polymorphic sites.
Significant at the 5% level.
Electronic-Database Information
The accession number and URLs for data presented herein are as follows:
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for human reference sequence [accession number AF158748.3])
- MAXDIP, http://genapps.uchicago.edu/maxdip/index.html
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for type 2 diabetes)
- RECSLIDER, http://genapps.uchicago.edu/recslider1/index.html
- SLIDER, http://genapps.uchicago.edu/slider/index.html
References
- Akey JM, Eberle MA, Rieder MJ, Carlson CS, Shriver MD, Nickerson DA, Kruglyak L (2004) Population history and natural selection shape patterns of genetic variation in 132 genes. PLoS Biol 2:E286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altshuler D, Hirschhorn JN, Klannemark M, Lindgren CM, Vohl MC, Nemesh J, Lane CR, Schaffner SF, Bolk S, Brewer C, Tuomi T, Gaudet D, Hudson TJ, Daly M, Groop L, Lander ES (2000) The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet 26:76–80 10.1038/79216 [DOI] [PubMed] [Google Scholar]
- Cann RL, Stoneking M, Wilson AC (1987) Mitochondrial DNA and human evolution. Nature 325:31–36 10.1038/325031a0 [DOI] [PubMed] [Google Scholar]
- Chen FC, Li WH (2001) Genomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees. Am J Hum Genet 68:444–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colagiuri S, Brand Miller J (2002) The “carnivore connection”—evolutionary aspects of insulin resistance. Eur J Clin Nutr Suppl 56:S30–S35 10.1038/sj.ejcn.1601351 [DOI] [PubMed] [Google Scholar]
- Corbo RM, Scacchi R (1999) Apolipoprotein E (APOE) allele distribution in the world: is APOE*4 a “thrifty” allele? Ann Hum Genet 63:301–310 10.1046/j.1469-1809.1999.6340301.x [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261:921–923 [DOI] [PubMed] [Google Scholar]
- Cox NJ, Hayes MG, Roe CA, Tsuchiya T, Bell GI (2004) Linkage of calpain 10 to type 2 diabetes: the biological rationale. Diabetes Suppl 53:S19–S25 [DOI] [PubMed] [Google Scholar]
- Crawford DC, Bhangale T, Li N, Hellenthal G, Rieder MJ, Nickerson DA, Stephens M (2004a) Evidence for substantial fine-scale variation in recombination rates across the human genome. Nat Genet 36:700–706 10.1038/ng1376 [DOI] [PubMed] [Google Scholar]
- Crawford DC, Carlson CS, Rieder MJ, Carrington DP, Yi Q, Smith JD, Eberle MA, Kruglyak L, Nickerson DA (2004b) Haplotype diversity across 100 candidate genes for inflammation, lipid metabolism, and blood pressure regulation in two populations. Am J Hum Genet 74:610–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen M, Erlich H, Klitz W, Carrington M (1995) Molecular mapping of a recombination hotspot located in the second intron of the human TAP2 locus. Am J Hum Genet 56:1350–1358 [PMC free article] [PubMed] [Google Scholar]
- Davignon J, Gregg RE, Sing CF (1988) Apolipoprotein E polymorphism and atherosclerosis. Arteriosclerosis 8:1–21 [DOI] [PubMed] [Google Scholar]
- de Knijff P, van den Maagdenberg AM, Frants RR, Havekes LM (1994) Genetic heterogeneity of apolipoprotein E and its influence on plasma lipid and lipoprotein levels. Hum Mutat 4:178–194 [DOI] [PubMed] [Google Scholar]
- Ebersberger I, Metzler D, Schwarz C, Paabo S (2002) Genomewide comparison of DNA sequences between humans and chimpanzees. Am J Hum Genet 70:1490–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisse L, Hudson RR, Bartoszewicz A, Wall JD, Donfack J, Di Rienzo A (2001) Gene conversion and different population histories may explain the contrast between polymorphism and linkage disequilibrium levels. Am J Hum Genet 69:831–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullerton SM, Bartoszewicz A, Ybazeta G, Horikawa Y, Bell GI, Kidd KK, Cox NJ, Hudson RR, Di Rienzo A (2002) Geographic and haplotype structure of candidate type 2 diabetes susceptibility variants at the calpain-10 locus. Am J Hum Genet 70:1096–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullerton SM, Clark AG, Weiss KM, Nickerson DA, Taylor SL, Stengard JH, Salomaa V, Vartiainen E, Perola M, Boerwinkle E, Sing CF (2000) Apolipoprotein E variation at the sequence haplotype level: implications for the origin and maintenance of a major human polymorphism. Am J Hum Genet 67:881–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie JH (1991) The causes of molecular evolution. Oxford University Press, New York, pp 142–230 [Google Scholar]
- Hamblin MT, Thompson EE, Di Rienzo A (2002) Complex signatures of natural selection at the Duffy blood group locus. Am J Hum Genet 70:369–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer MF (1995) A recent common ancestry for human Y chromosomes. Nature 378:376–378 10.1038/378376a0 [DOI] [PubMed] [Google Scholar]
- Hammer MF, Garrigan D, Wood E, Wilder JA, Mobasher Z, Bigham A, Krenz JG, Nachman MW (2004) Heterogeneous patterns of variation among multiple human X-linked loci: the possible role of diversity-reducing selection in non-Africans. Genetics 167:1841–1853 10.1534/genetics.103.025361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanis CL, Boerwinkle E, Chakraborty R, Ellsworth DL, Concannon P, Stirling B, Morrison VA, et al (1996) A genome-wide search for human non-insulin-dependent (type 2) diabetes genes reveals a major susceptibility locus on chromosome 2. Nat Genet 13:161–166 10.1038/ng0696-161 [DOI] [PubMed] [Google Scholar]
- Harding RM, Fullerton SM, Griffiths RC, Bond J, Cox MJ, Schneider JA, Moulin DS, Clegg JB (1997) Archaic African and Asian lineages in the genetic ancestry of modern humans. Am J Hum Genet 60:772–789 [PMC free article] [PubMed] [Google Scholar]
- Hedrick PW (2002) Pathogen resistance and genetic variation at MHC loci. Evolution Int J Org Evolution 56:1902–1908 [DOI] [PubMed] [Google Scholar]
- Hellmann I, Ebersberger I, Ptak SE, Paabo S, Przeworski M (2003) A neutral explanation for the correlation of diversity with recombination rates in humans. Am J Hum Genet 72:1527–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horikawa Y, Oda N, Cox NJ, Li X, Orho-Melander M, Hara M, Hinokio Y, Lindner TH, Mashima H, Schwarz PE, del Bosque-Plata L, Oda Y, Yoshiuchi I, Colilla S, Polonsky KS, Wei S, Concannon P, Iwasaki N, Schulze J, Baier LJ, Bogardus C, Groop L, Boerwinkle E, Hanis CL, Bell GI (2000) Genetic variation in the gene encoding calpain-10 is associated with type 2 diabetes mellitus. Nat Genet 26:163–175 10.1038/79876 [DOI] [PubMed] [Google Scholar]
- Hudson RR (2001) Two-locus sampling distributions and their application. Genetics 159:1805–1817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— (2002) Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18:337–338 10.1093/bioinformatics/18.2.337 [DOI] [PubMed] [Google Scholar]
- Hudson RR, Bailey K, Skarecky D, Kwiatowski J, Ayala FJ (1994) Evidence for positive selection in the superoxide dismutase (Sod) region of Drosophila melanogaster. Genetics 136:1329–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR, Kaplan NL (1988) The coalescent process in models with selection and recombination. Genetics 120:831–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR, Kreitman M, Aguade M (1987) A test of neutral molecular evolution based on nucleotide data. Genetics 116:153–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffreys AJ, Kauppi L, Neumann R (2001) Intensely punctate meiotic recombination in the class II region of the major histocompatibility complex. Nat Genet 29:217–222 10.1038/ng1001-217 [DOI] [PubMed] [Google Scholar]
- Jeffreys AJ, Ritchie A, Neumann R (2000) High resolution analysis of haplotype diversity and meiotic crossover in the human TAP2 recombination hotspot. Hum Mol Genet 9:725–733 10.1093/hmg/9.5.725 [DOI] [PubMed] [Google Scholar]
- Lercher MJ, Hurst LD (2002) Human SNP variability and mutation rate are higher in regions of high recombination. Trends Genet 18:337–340 10.1016/S0168-9525(02)02669-0 [DOI] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M (1991) Adaptive protein evolution at the Adh locus in Drosophila. Nature 351:652–654 10.1038/351652a0 [DOI] [PubMed] [Google Scholar]
- McVean GA, Myers SR, Hunt S, Deloukas P, Bentley DR, Donnelly P (2004) The fine-scale structure of recombination rate variation in the human genome. Science 304:581–584 10.1126/science.1092500 [DOI] [PubMed] [Google Scholar]
- Miller JC, Colagiuri S (1994) The carnivore connection: dietary carbohydrate in the evolution of NIDDM. Diabetologia 37:1280–1286 10.1007/s001250050246 [DOI] [PubMed] [Google Scholar]
- Mulligan CJ, Hunley K, Cole S, Long JC (2004) Population genetics, history, and health patterns in Native Americans. Annu Rev Genomics Hum Genet 5:295–315 10.1146/annurev.genom.5.061903.175920 [DOI] [PubMed] [Google Scholar]
- Nachman MW, Bauer VL, Crowell SL, Aquadro CF (1998) DNA variability and recombination rates at X-linked loci in humans. Genetics 150:1133–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neel JV (1962) Diabetes mellitus: a “thrifty” genotype rendered detrimental by “progress”? Am J Hum Genet 14:353–362 [PMC free article] [PubMed] [Google Scholar]
- Neel JV, Weder AB, Julius S (1998) Type II diabetes, essential hypertension, and obesity as “syndromes of impaired genetic homeostasis”: the “thrifty genotype” hypothesis enters the 21st century. Perspect Biol Med 42:44–74 [DOI] [PubMed] [Google Scholar]
- Nickerson DA, Tobe VO, Taylor SL (1997) PolyPhred: automating the detection and genotyping of single nucleotide substitutions using fluorescence-based resequencing. Nucleic Acids Res 25:2745–2751 10.1093/nar/25.14.2745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluzhnikov A, Di Rienzo A, Hudson RR (2002) Inferences about human demography based on multilocus analyses of noncoding sequences. Genetics 161:1209–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Przeworski M (2001) Linkage disequilibrium in humans: models and data. Am J Hum Genet 69:1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptak SE, Roeder AD, Stephens M, Gilad Y, Paabo S, Przeworski M (2004) Absence of the TAP2 human recombination hotspot in chimpanzees. PLoS Biol 2:e155 (http://biology.plosjournals.org/plosonline/?request=get-document&doi=10.1371/journal.pbio.0020155) (electronically published June 15, 2004; accessed January 21, 2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich DE, Cargill M, Bolk S, Ireland J, Sabeti PC, Richter DJ, Lavery T, Kouyoumjian R, Farhadian SF, Ward R, Lander ES (2001) Linkage disequilibrium in the human genome. Nature 411:199–204 10.1038/35075590 [DOI] [PubMed] [Google Scholar]
- Schierup MH, Mikkelsen AM, Hein J (2001) Recombination, balancing selection and phylogenies in MHC and self-incompatibility genes. Genetics 159:1833–1844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma AM (1998) The thrifty-genotype hypothesis and its implications for the study of complex genetic disorders in man. J Mol Med 76:568–571 10.1007/s001090050251 [DOI] [PubMed] [Google Scholar]
- Slatkin M, Wiehe T (1998) Genetic hitch-hiking in a subdivided population. Genet Res 71:155–160 10.1017/S001667239800319X [DOI] [PubMed] [Google Scholar]
- Song Y, Niu T, Manson JE, Kwiatkowski DJ, Liu S (2004) Are variants in the CAPN10 gene related to risk of type 2 diabetes? a quantitative assessment of population and family-based association studies. Am J Hum Genet 74:208–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreenan SK, Zhou YP, Otani K, Hansen PA, Currie KP, Pan CY, Lee JP, Ostrega DM, Pugh W, Horikawa Y, Cox NJ, Hanis CL, Burant CF, Fox AP, Bell GI, Polonsky KS (2001) Calpains play a role in insulin secretion and action. Diabetes 50:2013–2020 [DOI] [PubMed] [Google Scholar]
- Stajich JE, Hahn MW (2005) Disentangling the effects of demography and selection in human history. Mol Biol Evol 22:63–73 10.1093/molbev/msh252 [DOI] [PubMed] [Google Scholar]
- Stengard JH, Zerba KE, Pekkanen J, Ehnholm C, Nissinen A, Sing CF (1995) Apolipoprotein E polymorphism predicts death from coronary heart disease in a longitudinal study of elderly Finnish men. Circulation 91:265–269 [DOI] [PubMed] [Google Scholar]
- Stephens JC, Schneider JA, Tanguay DA, Choi J, Acharya T, Stanley SE, Jiang R, et al (2001a) Haplotype variation and linkage disequilibrium in 313 human genes. Science 293:489–493 10.1126/science.1059431 [DOI] [PubMed] [Google Scholar]
- Stephens M, Donnelly P (2003) A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet 73:1162–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Smith NJ, Donnelly P (2001b) A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 68:978–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD (1993) Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA 90:1977–1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahata N (1990) A simple genealogical structure of strongly balanced allelic lines and trans-species evolution of polymorphism. Proc Natl Acad Sci USA 87:2419–2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson R, Pritchard JK, Shen P, Oefner PJ, Feldman MW (2000) Recent common ancestry of human Y chromosomes: evidence from DNA sequence data. Proc Natl Acad Sci USA 97:7360–7365 10.1073/pnas.97.13.7360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall JD, Frisse LA, Hudson RR, Di Rienzo A (2003) Comparative linkage-disequilibrium analysis of the β-globin hotspot in primates. Am J Hum Genet 73:1330–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson GA (1975) On the number of segregating sites in genetical models without recombination. Theor Popul Biol 7:256–276 10.1016/0040-5809(75)90020-9 [DOI] [PubMed] [Google Scholar]
- Weedon MN, Schwarz PE, Horikawa Y, Iwasaki N, Illig T, Holle R, Rathmann W, Selisko T, Schulze J, Owen KR, Evans J, Del Bosque-Plata L, Hitman G, Walker M, Levy JC, Sampson M, Bell GI, McCarthy MI, Hattersley AT, Frayling TM (2003) Meta-analysis and a large association study confirm a role for calpain-10 variation in type 2 diabetes susceptibility. Am J Hum Genet 73:1208–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss KM, Ferrell RE, Hanis CL (1984) A New World syndrome of metabolic diseases with a genetic and evolutionary basis. Yearb Phys Anthropol 27:153–178 [Google Scholar]
- Zietkiewicz E, Yotova V, Jarnik M, Korab-Laskowska M, Kidd KK, Modiano D, Scozzari R, Stoneking M, Tishkoff S, Batzer M, Labuda D (1998) Genetic structure of the ancestral population of modern humans. J Mol Evol 47:146–155 [DOI] [PubMed] [Google Scholar]