

Abstract
Accurate and rapid identification of perturbed pathways through the analysis of genome-wide expression profiles facilitates the generation of biological hypotheses. We propose a statistical framework for determining whether a specified group of genes for a pathway has a coordinated association with a phenotype of interest. Several issues on proper hypothesis-testing procedures are clarified. In particular, it is shown that the differences in the correlation structure of each set of genes can lead to a biased comparison among gene sets unless a normalization procedure is applied. We propose statistical tests for two important but different aspects of association for each group of genes. This approach has more statistical power than currently available methods and can result in the discovery of statistically significant pathways that are not detected by other methods. This method is applied to data sets involving diabetes, inflammatory myopathies, and Alzheimer's disease, using gene sets we compiled from various public databases. In the case of inflammatory myopathies, we have correctly identified the known cytotoxic T lymphocyte-mediated autoimmunity in inclusion body myositis. Furthermore, we predicted the presence of dendritic cells in inclusion body myositis and of an IFN-α/β response in dermatomyositis, neither of which was previously described. These predictions have been subsequently corroborated by immunohistochemistry.
Keywords: microarrays, gene ontology, normalization, correlated data, inflammatory myopathies
Extracting clear and coherent hypotheses from genome-wide expression data remains an important challenge. Much of the initial work has focused on the development of techniques for accurate identification of differentially expressed genes and their statistical significance in a variety of experimental designs (1). However, the main difficulty in analysis lies not in the identification of differentially expressed genes but in their interpretation. Attempting to understand individual genes on a list of significant genes is demanding and laborious. Also, a comparison of gene lists from random subsets of a data set in simulation studies clearly shows that the gene list based on a small number of samples can be highly variable and that studying each gene separately may be ineffective in many cases (2, 3). The problem is compounded when the pathway of interest involves moderate effects that are not captured by the genes near the top of the list. Therefore, recent efforts have focused on the discovery of biological pathways rather than individual gene function, with the development of methods that are robust to the inaccuracies of specific gene estimates and provide a more expansive view of the underlying processes.
In the most common approach, genes are first ordered according to their evidence for differential expression, by one of many statistical methods available. Then, a short list of specified length containing the top genes is examined against each of the predefined sets of genes representing different pathways, to determine whether any set is overrepresented in the short list compared with the whole list. Suppose there are B0 differentially expressed genes from the total of B genes, and m0 genes of the pathway that involves m genes are among the differentially expressed genes. To examine the evidence of association in this case, Fisher's exact test based on the hypergeometric distribution or its large-sample approximation χ2 test is typically used. Given its simplicity, numerous software and web sites provide this capability, most often by using Gene Ontology as the source of gene sets. Examples include genmapp (4), chipinfo (5), gominer (6), onto-tools (7), and funcassociate (8).
This approach is reasonable but has at least three shortcomings, some of which are pointed out in ref. 9. First, only the most significant portion of the gene list is used to compute the statistic, treating the less-relevant genes as irrelevant. Second, the order of genes on the significant gene list is not taken into consideration. Simply counting the number of gene set members contained in the short list leads to loss of information, especially if the list is long and the difference between the more significant and the less significant is substantial. Third, the correlation structure of gene sets is not considered at all. This last issue is perhaps not as conspicuous as the first two, but it is an important aspect to consider in assessing statistical significance. We discuss this issue extensively in the present work.
An alternative and more successful technique should consider the distribution of pathway genes in the entire list of genes (9-12) as well as adjust for the correlation structure. In the innovative Gene Set Enrichment Analysis (GSEA) method (13), the following steps are applied: (i) all genes are ranked by using a signal-to-noise ratio; (ii) for each gene set, the distribution of gene ranks from the gene set is compared against the distribution for the rest of the genes by using the enrichment score (ES) based on a one-sided Kolmogorov-Smirnov statistic; (iii) class labels are permuted to generate a null distribution of ES; and (iv) statistical significance of the observed score is assessed for the top-ranking gene set by comparison with the null distribution of maximum scores from each permutation. By considering the distribution of the gene ranks belonging to each gene set over the entire list, this method is a clear improvement over previous ones. However, the effect of the gene-set size and the influence of other gene sets not under consideration can be counterintuitive in some instances (14). Its normalization and permutation procedures also may lead to inaccurate assessment of statistical significance.
A successful approach for finding significant pathways depends on two components: (i) an accurate and powerful statistical method to discover significant patterns for a group of genes and (ii) a comprehensive and well-characterized pathway information mapped to microarray probes. In this work, we introduce a previously undescribed statistical framework (Fig. 1). We present two related hypotheses that test for complementary aspects of the gene sets and develop a testing procedure for each case. In particular, we point out that a normalization step is necessary to account for the different correlation structure of gene sets before they can be compared. The proposed approach includes a correct estimate of the statistical significance for each group of genes in addition to correct rank order, with proper adjustment for multiple testing based on q values (15). The advantages of the method are demon-strated on data sets from studies on diabetes, inflammatory myopathies, and Alzheimer's disease (AD). The examples are carried out by using >600 gene sets we have collected from pathway databases (Biocarta, KEGG, and BioCyc) and pathway-specific microarray annotations (www.superarray.com), as well as >5,000 gene sets from Gene Ontology.
Fig. 1.

Outline of the methodology. An extensive collection of pathway information is assembled from various databases; a statistical test is applied to find relationships between the expression levels and the phenotype, and then two different testing procedures are used to find statistically significant pathways. Proper adjustments for correlation structure and multiple testing are critical.
Methods
Hypothesis Testing Framework. The overall objective of the analysis is to test whether a group of genes has a coordinated association with a phenotype of interest. In terms of formal statistical language, there are two ways to formulate the null hypothesis. Q1 and Q2 are related but not equivalent. When there is a significant proportion of genes associated with the phenotype of interest, a gene set would contain genes with association, even if the gene set is purely a random subset from the entire gene list. A less obvious but important fact, however, is that even if all the genes of the entire list are not associated with the phenotype of interest, the observed association of genes within a gene set could have a different distribution compared with that of the genes outside the gene set because of the special correlation structure among the genes in a given gene set.
Hypothesis Q1: The genes in a gene set show the same pattern of associations with the phenotype compared with the rest of the genes.
Hypothesis Q2: The gene set does not contain any genes whose expression levels are associated with the phenotype of interest.
We give a simple example to illustrate this second point. Assume that we only have three genes whose expression levels are independent with the phenotype of interest. Suppose the expression levels of the first two genes are positively correlated with a high-correlation coefficient, e.g., 0.95, and the expression level of the third gene is independent with that of the first two genes. Moreover, we assume that the test statistics, e.g., t statistics, for testing association between three genes and the phenotype, ti, i = 1, 2, 3, have the same marginal distribution. Let ri, i = 1, 2, 3 be the ranks of ti, i = 1, 2, 3. If we consider a gene set that consists of the first two genes, then the ranks of the genes in this gene set {r1, r2} can take six possible combinations: {1, 2}, {2, 1}, {1, 3}, {3, 1}, {2, 3}, and {3, 2}. However, because t1 and t2 are highly correlated, we would expect that r1 and r2 are more likely to be close to each other than otherwise. Therefore, the probability of observing {r1, r2} = {1, 3} would be smaller than that of {r1, r2} = {1, 2}, which suggests that {t1, t2} is not a random sample from {t1, t2, t3}.
An essential difference between Q1 and Q2 is that Q1 compares the association strength for genes in a gene set with the association strength for genes outside the gene set, whereas Q2 only focuses on the associations of genes within the gene set. The disadvantage of Q1 is that a gene set without any gene associated with the phenotype may be identified as demonstrating a special pattern of the associations, and the identified gene set list is often much longer than that according to Q2. The results from Q1, therefore, should be interpreted with the awareness that some gene sets are statistically significant because of correlation structure among expression profiles of its member genes. As a ranking criterion, Q2 also has its own limitation: When there is a significant proportion of genes associated with the phenotype of interest, large gene sets corresponding to irrelevant pathways could contain many genes associated with the phenotype by chance and be ranked highly according to Q2.
Mathematical descriptions of the hypothesis-testing procedures are presented in Appendix. Here, we describe the ideas briefly. The two statistics we introduce for Q1 and Q2 are Tk and Ek, respectively, for the kth gene set. Large magnitude indicates high significance, and the sign indicates the direction of change in expression. To obtain Tk, a measure of association ti is first computed between each gene i and the phenotype of interest. Then, for the kth set, these association measures of genes in that set are summed. To get statistical significance of the statistic, it is compared against the distribution under the null hypothesis, obtained by permuting the association measures. For Ek, the procedure is similar, but the null distribution is generated by permuting the phenotypes across samples. If the data are represented by a matrix where the rows are genes and columns are samples, the permutations of the null distributions of Tk and Ek correspond to permuting rows and columns, respectively. Because ti values are correlated, a special weighting function may also be used in the summation. The details are discussed in Appendix.
The original GSEA procedure generates the null distribution of the ES by permuting the phenotype (group labels) with Q2 as the implicit null hypothesis, although the claimed null hypothesis is that the differences between two states of genes in the gene set are randomly distributed in the list of all differences. As discussed before, Q1 and Q2 answer related but different questions. The fact that it generates the null distribution of the test statistic under hypothesis Q2 while using the Kolmogorov-Smirnov statistic to test hypothesis Q1 results in loss of power. Furthermore, to test Q2, it seems counterintuitive to use the expression levels of genes outside the gene set as done in GSEA. All of the genes of interest are within the gene set, and the test result should not be influenced by genes outside the set.
Normalization for Comparing Gene Set Scores. After the test statistics Tk and Ek are computed for testing hypotheses Q1 and Q2, respectively, we rank the K gene sets in order of their significance and control for the inflated Type I error due to multiple comparisons of gene sets. It is tempting to use a permutation-type procedure as in Significance Analysis of Microarrays (16), where a regularized t statistic is computed for each gene, and its significance is determined by how each observed order statistic compares with the mean of the same order statistic in permuted samples. The difficulty, however, is that unlike in the Significance Analysis of Microarrays procedure for regular microarray data, the null distributions of the test statistics (Tk or Ek) for different gene sets are not the same. It is therefore unfair to rank the gene sets simply by the observed raw test statistics. For example, when we test hypothesis Q1, the null distribution of T1,..., TK could be very different because of different gene set sizes and correlation structure. This effect is a subtle but critical issue.
This phenomenon is observed, for example, in the application of GSEA to a diabetes data set (13). Focusing on the difference between the 17 normal glucose tolerance and 18 Type 2 diabetes mellitus subjects, we repeated the analysis with the same preprocessing steps and the same gene sets. When we simulated the null distribution of raw ES for each gene set by permuting the group labels with 1,000 permutations, we indeed found that the null distributions are markedly different, with their standard deviations (SDs) ranging from 25 to 110. This result implies that the same ES for different gene sets could suggest evidence of different strengths for association of interest and that the raw ES across different gene sets should not be compared directly. The marked difference in distributions is due to the complex correlation structure of genes within different gene sets. The highest ranked gene set representing oxidative phosphorylation, for example, contains genes that are tightly regulated and hence highly correlated. When we plot the SDs of the approximated null distribution for ES vs. the observed ES (Fig. 2), we see a clear positive correlation. In fact, the three highest gene sets by ES are the same three with the largest variance in the null distribution.
Fig. 2.

A scatterplot of the SDs of null distributions for the ES vs. the observed ES for the diabetes data. Each point represents a gene set. The Pearson correlation coefficient is 0.55. Without proper normalization among different gene sets, a high score may be due to its wide null distribution, which depends on the size and correlation structure of the gene set.
To remedy this problem, we suggest a simple standardization that results in normalized statistics NTk and NEk, which have the same null distributions for all gene sets. We then can rank the gene sets by NTk, and a resampling procedure similar to the one in Significance Analysis of Microarrays can be carried out to approximate the joint null distribution of ordered {NT1,..., NTK}. See Appendix for a mathematical description.
Results
Example I: Reanalysis of Diabetes Data. We carry out a more comprehensive analysis of the data set described above to illustrate the properties of the method, especially in relation to GSEA. To be sure that the differences observed are only due to the main feature of the algorithm, we make a couple of adjustments. First, the original GSEA was a one-sided approach to identify gene sets containing down-regulated genes in Type 2 diabetes mellitus subjects, but we implement a two-sided test. Second, we use the t statistic as the difference metric for individual genes.
For the measure of significance, we estimated the q value from the permutation procedure for each gene set, resulting in the identification of eight gene sets significant at the 0.05 level. As discussed in Appendix, the q value is the counterpart of the p value in the multiple testing scenario. Some of the test statistics and their corresponding ranks for 26 gene sets, including the five top-ranked gene sets for each procedure, are listed in Table 1. One statistic shown is the normalized ES (NES), applying the normalization step described above to ES. Interestingly, the oxidative phosphorylation gene set is still ranked first, but the rest of the rankings for ES and NES are substantially different, confirming the necessity of standardization. Overall, even when the various test statistics produced similar rankings, only some of them yielded statistically significant findings. In particular, we find that none of the NES based q values are <0.1. This loss in power is not surprising: In GSEA we tested the hypothesis Q2, from which we generated the null distribution, by using the Kolmogorov-Smirnov test, which is designed to test Q1 and hence is less powerful.
Table 1. Analysis of diabetes data.
| Gene set | Set size | ES | NES | NTk | 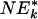 |
|---|---|---|---|---|---|
| OXPHOS (oxidative phosphorylation)† | 106 | 1 | 1 | 1 | 4 |
| human-mitoDB† | 440 | 2 | 22 | 2 | 11 |
| Mitochondria† | 450 | 3 | 25 | 3 | 7 |
| c18-U133 (muscle development‡)† | 202 | 4 | 18 | 4 | 19 |
| c20-U13 (macromolecule metabolism‡)† | 216 | 5 | 26.5 | 6 | 36 |
| MAP00190 (oxidative phosphorylation)† | 53 | 6 | 21 | 5 | 13 |
| MAP00910 (nitrogen metabolism)†§ | 19 | 11 | 2 | 8 | 2 |
| MAP00330 (arginine and proline metabolism) | 41 | 12 | 3 | 31 | 14 |
| MAP00500 (starch and sucrose metabolism) | 13 | 13 | 5 | 20 | 5 |
| c22-U133 (cell growth and maintenance‡)†§ | 84 | 18 | 24 | 7 | 1 |
| MAP00631 (1,2-Dichloroethane degradation)¶ | 8 | 31 | 4 | 66 | 67 |
| MAP00960 (alkaloid biosynthesis II) | 3 | 48 | 16 | 21 | 11 |
| c29-U133 (musculoskeletal movement‡)§ | 102 | 82 | 60 | 82 | 3 |
The five top-ranked sets for each statistic are shown, ordered by the ES rank. ES and normalized ES give substantially different ranks.
q value is <0.05 according to NTk.
A brief summary; many categories of genes are present in these clusters.
q value is <0.05 according to 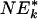 , a variation of NEk that is used for increased power [see NEk (λk) in Supporting Text].
, a variation of NEk that is used for increased power [see NEk (λk) in Supporting Text].
MAP00631/MAP00053 (Ascorbate and aldarate metabolism) are identical gene sets after filtering.
The final interpretation requires consideration of both NTk and NEk. As described earlier, the correlation structure in gene sets can give false positives in NTk; conversely, NEk can be influenced by the gene set size. Therefore, the gene sets that rank high in both categories are the best candidates. It appears that all procedures point to gene set OXPHOS (oxidative phosphorylation), whose members tend to be expressed relatively higher in normal glucose tolerance subjects. This finding is consistent with the previous conclusion (13) and also is supported by another gene set (MAP00190) and the mitochondria gene sets. Of particular interest are two gene sets that are significant by NTk and NEk criteria (q value < 0.01), even though their rankings by ES are not as high. MAP00910 group has 19 genes related to amino acid metabolism that are up-regulated in Type 2 diabetes mellitus patients, which has been repeatedly reported (17). c22-U133 is more difficult to interpret because it refers to a cluster in ref. 18 consisting of heterogeneous set of genes. It contains many mitochondrial genes as well as those related to protein and carbohydrate metabolism and transcription. Further investigation is needed to identify targets of interest.
Example II: Inflammatory Myopathies. We examined 49 muscle samples consisting of 23 from patients with inclusion body myositis (IBM), 13 from patients with dermatomyositis, and 13 from normal subjects (NORM). After a global normalization, we eliminated those genes whose expression levels were below the trimmed mean of the sample in all patients, which resulted in 10,526 genes. We used 926 gene sets whose size is between 20 and 500 based on these genes; to compare the gene expression levels between NORM and dermatomyositis, we used the t test for individual genes. We identified 272 and 829 gene sets with q value < 0.01 according to NTk and NEk, respectively. Similarly, we compared the expression levels between NORM and IBM and identified 269 and 378 gene sets with q value of <0.01 according to NTk and NEk, respectively. The gene sets are ordered by their average ranks for NTk and NEk.
The pathogenesis of dermatomyositis has been modeled as a humorally mediated disorder initiated by autoantibody-mediated muscle capillary destruction and ischemia of muscle (19). Our analysis (Table 2) indicates a major disturbance of transcription of interferon-α/β-inducible genes, not predicted by the current model of this disease. The same finding also was made based on separate analyses of this data set, and subsequent immunohistochemical studies of muscle tissue confirmed a key role in the pathogenesis of proteins encoded by these genes (20).
Table 2. Analysis of inflammatory myopathy data.
| Gene set category | Pathway | Set size | NTk | Rank | NEk | Rank |
|---|---|---|---|---|---|---|
| Normal (NORM) vs. Dermatomyositis | ||||||
| Customarray†‡ | Interferon α/β response | 107 | −10.61 | 8 | −4.71 | 4 |
| GO:0019883†‡ | Antigen presentation, endogenous antigen | 25 | −7.56 | 38 | −4.95 | 2 |
| GO:0030106†‡ | MHC class I receptor activity | 23 | −7.30 | 43 | −5.05 | 1 |
| GO:0019885†‡ | Antigen processing, endogenous antigen via MHC class I | 29 | −7.12 | 45 | −4.80 | 3 |
| GO:0019882†‡ | Antigen presentation | 49 | −5.88 | 70 | −4.09 | 8 |
| GO:0045298†‡§ | Tubulin | 21 | −5.55 | 77 | −4.54 | 5 |
| GO:0030705†‡§ | Cytoskeleton-dependent intracellular transport | 31 | −4.96 | 85 | −4.25 | 7 |
| GO:0005874†‡ | Microtubule | 32 | −4.80 | 89 | −4.31 | 6 |
| GO:0015075† | Ion transporter activity | 219 | 11.09 | 4 | −1.11 | 790.5 |
| GO:0008324† | Cation transporter activity | 190 | 11.12 | 3 | −0.92 | 806 |
| GO:0006091† | Energy pathways | 217 | 12.95 | 2 | −0.90 | 808 |
| GO:0015399† | Primary active transporter activity | 175 | 10.24 | 10 | −1.00 | 803 |
| GO:0015980† | Energy derivation by oxidation of organic compounds | 140 | 10.71 | 7 | −0.88 | 809 |
| GO:0015077† | Monovalent inorganic cation transporter activity | 144 | 10.51 | 9 | −0.74 | 816 |
| KEGG† | Ribosome | 153 | 13.09 | 1 | −0.46 | 825 |
| GO:0015078† | Hydrogen ion transporter activity | 140 | 10.89 | 6 | −0.62 | 820 |
| KEGG† | Oxidative-phosphorylation | 130 | 10.94 | 5 | −0.52 | 822 |
| NORM vs. IBM | ||||||
| GO:0030106†‡ | MHC class I receptor actitivy | 23 | −14.69 | 3 | −8.59 | 1 |
| GO:0019882†‡ | Antigen presentation | 49 | −16.12 | 1 | −7.07 | 4 |
| GO:0019883†‡ | Antigen presentation, endogenous antigen | 25 | −14.2 | 4 | −8.30 | 2 |
| GO:0030333†‡ | Antigen processing | 52 | −15.68 | 2 | −6.82 | 5 |
| GO:0019885†‡ | Antigen processing, endogenous antigen via MHC class I | 29 | −13.84 | 5 | −7.76 | 3 |
| Customarray†‡ | Interferon α/β response | 107 | −12.77 | 7 | −5.07 | 9 |
| Biocarta†‡ | CTL-mediated immune response against target cells | 23 | −8.90 | 10 | −6.08 | 6 |
| GO:0045012†‡ | MHC class II receptor activity | 23 | −8.42 | 12 | −5.48 | 7 |
| Customarray†‡ | Dendritic antigen presenting cell | 158 | −13.33 | 6 | −4.50 | 14 |
| GO:0019886†‡§ | Antigen processing, exogenous antigen via MHC class II | 22 | −7.95 | 17 | −5.37 | 8 |
| GO:0003735† | Structural constituent of ribosome | 211 | 9.01 | 9 | −0.53 | 797 |
| KEGG† | Ribosome | 153 | 9.40 | 8 | 0.06 | 840.5 |
q value is <0.01 according to NTk.
q value is <0.01 according to NEk.
GO:0045298/GO:0046785 (microtubule polymerization), GO:0030705/GO:0007018 (microtubule-based movement), and GO:0019886/GO:0019884 (antigen presentation, exogenous antigen) are identical gene sets after filtering.
IBM has been modeled as having cytotoxic T lymphocyte (CTL)-mediated destruction of MHC class I antigen expressing myofibers (19). Our analysis predicts that this model is largely correct, with enrichment of genes encoding MHC class I antigen presentation and the category “CTL-mediated immune response against target cells.” Additionally, the analysis predicts the presence of dendritic cells in IBM muscle, which was not previously described. These results have been subsequently corroborated by immunohistochemistry, which showed a substantial number of dendritic cells infiltrating IBM muscle. The details of this observation will be reported separately (S.A.G., unpublished data). We emphasize that this finding is not evident from the list of differentially expressed genes but was found by the proposed method.
Example III: AD Data. Recently, the pathogenesis of AD was studied based on gene expression of hippocampal specimens, with 22 AD subjects and 9 controls (21). For all subjects, Mini Mental Status Examination (MMSE) scores as well as neurofibrillary tangle scores were obtained. We applied our method to this data set, concentrating on the MMSE score as the phenotype and excluding the control group. In the two previous examples, the phenotype was a class label, and the t statistic was used to measure the strength of association between the expression values and the phenotype. In this example, because the MMSE score is a continuous variable, Fisher's z, 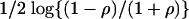 , is used as a metric for association, where ρ is the Pearson correlation coefficient between expression level and MMSE. The same filtering procedure used in the previous example was applied.
, is used as a metric for association, where ρ is the Pearson correlation coefficient between expression level and MMSE. The same filtering procedure used in the previous example was applied.
Strikingly, according to our analysis, MMSE score showed a strong positive correlation with calcium ion transport and calcium channel activity. More than 80% of the genes in each group were in fact up-regulated in the subjects with high MMSE scores. Destabilization of calcium signaling has been shown to be central to the pathogenesis of AD (22). Calcium ion transport group also was found to be significant by the authors of ref. 21, but only by marginal significance (p value = 0.0482). The second most significant group was “signal transduction in cancer,” which contains numerous genes related to apoptosis, such as TP53, BAX, BCL2, AKT, and TNF. This group showed negative correlation with MMSE score or, equivalently, positive correlation with severity of AD. The third category “Tumor metastasis” also contained a number of genes implicated in AD including matrix metalloproteinase 2, 3, and 9 (MMP9), MAP2K4, TGFB1, and ERBB2; previously, MMP9 concentration was found to be elevated in cerebrospinal fluid of AD patients (23). Others on the list included several classes of genes related to energy metabolism including mitochondrial electron transport pathways, which showed positive correlation with MMSE score. This finding supports the mitochondria-mediated pathophysiology of AD (24). The table containing the top 10 gene sets in NTk and NEk (ordered by their average ranks) and more information on the three data sets in this section can be found in Supporting Text and Tables 3 and 4, which are published as supporting information on the PNAS web site.
Discussion
The proposed method allows for the detection of subtle processes that are not likely to be revealed by examining a small list of highly significant genes. No technique for identifying differentially expressed genes can circumvent the problem of small sample sizes that arises in nearly all microarray studies, but by examining the pattern for a group of genes, it is possible to mitigate the effect of errors on individual gene estimates. By applying proper normalization and considering both Q1 and Q2, the proposed approach selects those gene sets that are likely to be relevant, with good statistical power. Valid statistical tests have been previously described (10, 25), but we emphasize the need to test both aspects: a gene set with tightly correlated genes, but otherwise unimportant, can appear significant if only Q1 is tested; when a high fraction of genes are associated with the phenotype, a large gene set can appear significant by chance if only Q2 is tested. We have found that ordering by the average rank of the two statistics can be a useful heuristic in ranking gene sets.
For this approach to be successful, a large collection of carefully curated information on pathways must be available. Although Gene Ontology has been a useful source in this regard, pathways involving multiple processes and functions are not well represented. In this work, we have collected several hundred pathways from public databases and demonstrated their utility. In the future, a coordinated effort to define the pathways and to map the gene identifiers of each pathway to the target sequence IDs of each array type will be essential. Defining the relationships among the gene sets them-selves and organizing them also would facilitate interpretation, especially given the hierarchical structure in Gene Ontology.
We have considered three examples in which the phenotype of interest was a class label or a continuous variable, but the same approach also can be used for more complicated phenotypes. For example, for multiclass comparisons, we can use the F-statistic from ANOVA-type comparisons. If the phenotype is the right censored survival time, the standardized log-rank test statistic can be used.
Supplementary Material
Acknowledgments
We thank Profs. L. J. Wei and Atul Butte for reading the manuscript. This work was supported by grants from the National Institute of General Medical Sciences (to P.J.P.); the Muscular Dystrophy Association and the National Institute of Neurological Disorders and Stroke (to S.A.G.); and Specialized Centers of Clinically Oriented Research in Pediatric Heart Development and Disease (to S.W.K.). Inflammatory myopathy data were generated by the Children's Hospital Gene Expression Core (NS40828).
Appendix
Testing Procedure for Q1. For this hypothesis, we can test whether the observed associations of genes in a gene set is a random sample from the background distribution of all observed associations. Because we have multiple gene sets under consideration, once we perform a statistical test for a specific gene set, we can rank the gene sets according to the strength of the statistical evidence against the null hypothesis Q1.
We first introduce some notations. Let the indices i and j denote the ith gene and jth sample, with i = 1,..., B and j = 1,..., n for B genes and n subjects. We assume that the phenotype of interest is measured by {z1,..., zn} for the n subjects, with the resulting association measure ti between the ith gene and the phenotype of interest.
We also assume that Gki, k = 1,..., K and i = 1,..., B, indexes the corresponding K gene sets of interest, i.e., Gki = 1ifthe kth gene set contains ith gene and 0 otherwise. With the prespecified gene set information, the data can be represented as the following matrix:
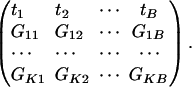 |
If we view Gi1,..., GiB and t1,..., tB as B independent and identically distributed copies of random variable Gi and t, respectively, then testing whether the observed associations of genes in the kth gene set is a random sample from the background distribution is equivalent to testing the independence between Gi and t. Various statistical tests can be used here.
To detect possibly moderate but coordinated associations for genes in a gene set, the specific alternative is likely to be a location (mean) shift from the background distribution. Therefore, a test against the omnibus alternative (any type of deviation from the reference distribution) such as the Kolmogorov-Smirnov test, which is used in GSEA, is not the most appropriate candidate in terms of power. Instead, we suggest the t test or the Wilcoxon rank test, both of which are more powerful for detecting location difference between two distributions. If we use the t test, the test statistics for kth gene set can be written as
 |
where  , the number of genes in the kth gene set. Under the null hypothesis, Tk should be centered at
, the number of genes in the kth gene set. Under the null hypothesis, Tk should be centered at  . Because different gene sets may or may not share the same genes, Tk, k = 1,..., K are dependent. Their null distributions can be generated by permuting {t1,..., tB}. To be more specific, under the null hypothesis that t is independent with (G1,..., GK), the null distribution of (T1,..., TK) can be approximated by the empirical distribution of (
. Because different gene sets may or may not share the same genes, Tk, k = 1,..., K are dependent. Their null distributions can be generated by permuting {t1,..., tB}. To be more specific, under the null hypothesis that t is independent with (G1,..., GK), the null distribution of (T1,..., TK) can be approximated by the empirical distribution of ( ), where
), where
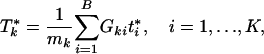 |
and {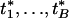 } is permuted {t1,..., tB}.
} is permuted {t1,..., tB}.
This procedure is therefore different from GSEA in two respects. First, we favor the t test in view of the analysis objective. More importantly, permuting the phenotype {z1,..., zn} does not give the correct null distribution for Q1, and therefore we propose to permute the association metric {t1,..., tB}.
Testing Procedure for Q2. We propose a test for Q2 based on expression levels of genes within the gene set. One simple test statistic is the average of association metric ti of genes in the gene set
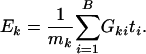 |
It is important to note that although the formula for Ek is the same as that of Tk for testing Q1, their probability interpretations and hence their testing procedures are quite different. In Tk, ti is deterministic and the gene set structure is random; in Ek, the opposite is true.
Because ti approximately follows N(0, 1) when the expression is independent of phenotype, it is tempting to conduct a test by using the approximation 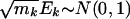 , under hypothesis Q2. However, the approximation is not valid even under the null hypothesis because of the potential correlations among ti. Because the genes in the same gene set are functionally related, their expression levels and association metric ti are likely to be dependent. Therefore, permutation methods should be used to approximate the null distribution of (E1,..., EK), where phenotypes {z1,..., zn} are permuted, as was done in the original GSEA.
, under hypothesis Q2. However, the approximation is not valid even under the null hypothesis because of the potential correlations among ti. Because the genes in the same gene set are functionally related, their expression levels and association metric ti are likely to be dependent. Therefore, permutation methods should be used to approximate the null distribution of (E1,..., EK), where phenotypes {z1,..., zn} are permuted, as was done in the original GSEA.
The power of the test against a certain alternative could be improved by using a more general linear combination of the form 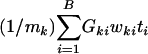 , where wki are appropriate weights used to combine mk test statistics while accounting for the correlations in ti. This procedure is described in Supporting Text.
, where wki are appropriate weights used to combine mk test statistics while accounting for the correlations in ti. This procedure is described in Supporting Text.
Standardization of Gene Set Scores. Assuming F1(·),..., FK(·) are the estimated null distributions of T1,..., TK by permutation, we first find the corresponding transformations 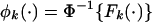 , k = 1,..., K, where Φ(·) is the cumulative distribution function for standard normal. This transformation results in the null distribution of NTk = φk(Tk) being N(0, 1) for all k.
, k = 1,..., K, where Φ(·) is the cumulative distribution function for standard normal. This transformation results in the null distribution of NTk = φk(Tk) being N(0, 1) for all k.
Multiple Testing Adjustment. Because the testing procedure is carried out for hundreds of gene sets, it is critical to apply a proper adjustment to control for type I error. In our analysis, we use the q value, which is a counterpart of the p value in the context of false discovery rate, to assess the statistical significance of associations for each gene set (15). Family-wise error rates, such as those obtained by the Bonferroni or the Westfall-Young method, are too stringent. A simple version of the q value for the kth gene set is
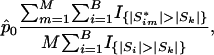 |
where p̂0 is an estimated upper bound for the proportion of null hypotheses, Si is the observed test statistic for the ith gene set, 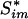 is the permuted test statistic for the ith gene set in the mth permuted sample, m = 1,..., M, and I{·} is the indicator function giving 1 if the argument is true and 0 otherwise.
is the permuted test statistic for the ith gene set in the mth permuted sample, m = 1,..., M, and I{·} is the indicator function giving 1 if the argument is true and 0 otherwise.
There are different ways to estimate p̂0 in the literature, most involving a subjectively chosen smoothing parameter. Here, we adopt a previously undescribed, objective approach. We first compute {p1,..., pB}, p values testing the association for all gene sets. Because the marginal density function f0(p) for p values is nonincreasing, 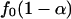 is an upper bound for the proportion of null hypotheses, and its estimator can be used to replace p̂0. We propose to estimate f0(p) by the left derivative of the smallest concave function greater than the empirical distribution function of {p1,..., pB} (least concave majorant). This value is the nonparametric maximum likelihood estimator (NPMLE) of f0(p). Therefore p̂0 can be replaced by
is an upper bound for the proportion of null hypotheses, and its estimator can be used to replace p̂0. We propose to estimate f0(p) by the left derivative of the smallest concave function greater than the empirical distribution function of {p1,..., pB} (least concave majorant). This value is the nonparametric maximum likelihood estimator (NPMLE) of f0(p). Therefore p̂0 can be replaced by 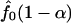 for a prespecified small α, e.g., 0.05, where f0(p) is the aforementioned NPMLE of f0(p).
for a prespecified small α, e.g., 0.05, where f0(p) is the aforementioned NPMLE of f0(p).
Abbreviations: GSEA, Gene Set Enrichment Analysis; ES, enrichment score; NES, normalized ES; IBM, inclusion body myositis; AD, Alzheimer's disease; MMSE, Mini Mental Status Examination.
References
- 1.Speed, T., ed. (2003) Statistical Analysis of Gene Expression Microarray Data (Chapman & Hall/CRC, Boca Raton, FL).
- 2.Pavlidis, P., Li, Q. & Noble, W. S. (2003) Bioinformatics 19, 1620-1627. [DOI] [PubMed] [Google Scholar]
- 3.Kim, R. D. & Park, P. J. (2004) Genome Biol. 5, R70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dahlquist, K. D., Salomonis, N., Vranizan, K., Lawlor, S. C. & Conklin, B. R. (2002) Nat. Genet. 31, 19-20. [DOI] [PubMed] [Google Scholar]
- 5.Zhong, S., Li, C. & Wong, W. H. (2003) Nucleic Acids Res. 31, 3483-3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zeeberg, B. R., Feng, W., Wang, G., Wang, M. D., Fojo, A. T., Sunshine, M., Narasimhan, S., Kane, D. W., Reinhold, W. C., Lababidi, S., et al. (2003) Genome Biol. 4, R28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Draghici, S., Khatri, P., Bhavsar, P., Shah, A., Krawetz, S. A. & Tainsky, M. A. (2003) Nucleic Acids Res. 31, 3775-3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berriz, G. F., King, O. D., Bryant, B., Sander, C. & Roth, F. P. (2003) Bioinformatics 19, 2502-2504. [DOI] [PubMed] [Google Scholar]
- 9.Pavlidis, P., Qin, J., Arango, V., Mann, J. J. & Sibille, E. (2004) Neurochem. Res. 29, 1213-1222. [DOI] [PubMed] [Google Scholar]
- 10.Pavlidis, P., Lewis, D. P. & Noble, W. S. (2002) Pac. Symp. Biocomput., 474-485. [PubMed]
- 11.Breitling, R., Amtmann, A. & Herzyk, P. (2004) BMC Bioinformatics 5, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rahnenführer, J., Domingues, F. S., Maydt, J. & Lengauer, T. (2004) Stat. Applications Genet. Mol. Biol. 3, 16. [DOI] [PubMed] [Google Scholar]
- 13.Mootha, V. K., Lindgren, C. M., Eriksson, K., Subramanian, A., Sihag, S., Lehar, J., Puigserver, P., Carlsson, E., Ridderstrale, M., Laurila, E., et al. (2003) Nat. Genet. 34, 267-273. [DOI] [PubMed] [Google Scholar]
- 14.Damian, D. & Gorfine, M. (2004) Nat. Genet. 36, 663 (lett.). [DOI] [PubMed] [Google Scholar]
- 15.Storey, J. D. & Tibshirani, R. (2003) Proc. Natl. Acad. Sci. USA 100, 9440-9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tusher, V. G., Tibshirani, R. & Chu, G. (2001) Proc. Natl. Acad. Sci. USA 98, 5116-5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halvatsiotis, P., Short, K. R., Bigelow, M. & Nair, K. S. (2002) Diabetes 51, 2395-2404. [DOI] [PubMed] [Google Scholar]
- 18.Su, A. I., Cooke, M. P., Ching, K. A., Hakak, Y., Walker, J. R., Wiltshire, T., Orth, A. P., Vega, R. G., Sapinoso, L. M., Moqrich, A., et al. (2002) Proc. Natl. Acad. Sci. USA 99, 4465-4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dalakas, M. C. & Hohlfeld, R. (2003) Lancet 362, 971-982. [DOI] [PubMed] [Google Scholar]
- 20.Greenberg, S. A., Pinkus, J. L., Pinkus, G. S., Burleson, T., Sanoudou, D., Tawil, R., Barohn, R. J., Saperstein, D. S., Briemberg, H. R., Ericsson, M., et al. (2005) Ann. Neurol. 57, 664-678. [DOI] [PubMed] [Google Scholar]
- 21.Blalock, E. M., Geddes, J. W., Chen, K. C., Porter, N. M., Markesbery, W. R. & Landfield, P. W. (2004) Proc. Natl. Acad. Sci. USA 101, 2173-2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.LaFerla, F. M. (2002) Nat. Rev. Neurosci. 3, 862-872. [DOI] [PubMed] [Google Scholar]
- 23.Adair, J. C., Charlie, J., Dencoff, J. E., Kaye, J. A., Quinn, J. F., Camicioli, R. M., Stetler-Stevenson, W. G. & Rosenberg, G. A. (2004) Stroke 35, e159-e162. [DOI] [PubMed] [Google Scholar]
- 24.Melov, S. (2004) Trends Neurosci. 27, 601-606. [DOI] [PubMed] [Google Scholar]
- 25.Simon, R. & Lam, A. P. (2005) BRB Array-Tools User's Guide, Version 3.3 (National Cancer Institute Biometric Research Branch, Bethesda, MD), Technical Report 28.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.