

Abstract

There is an urgent need for the development of safe and effective modalities for the treatment of diseases owing to drug resistance, undesired side effects, and poor clinical outcomes. Combining cell-targeting and efficient cell-killing properties, peptide–drug conjugates (PDCs) have demonstrated superior efficacy compared with peptides and payloads alone. However, innovative molecular designs of PDCs are essential for further improving targeting precision, protease resistance and stability, cell permeability, and overall treatment efficacy. Several strategies have been developed to address these challenges, such as multivalency approaches, bispecific targeting, and long-acting PDCs. Other novel strategies, including overcoming biological barriers, conjugating novel functional payloads, and targeting macropinocytosis, have also shown promise. This perspective compiles the most recent strategies for enhancing PDC treatment efficacy, highlights key advancements in PDC, and provides insights on future directions for the development of novel PDCs.
Significance
Latest innovations in PDC emphasizing strategies that improve treatment efficacy.
Methods to enhance cell targeting, circulatory stability, and overcoming biological barriers of PDCs.
Conjugating novel functional payload in the augment of PDC bioactivity is summarized.
1. Introduction
Treatment methods for diseases have evolved from traditional small-molecule drugs to antibody–drug conjugates (ADCs) and peptide–drug conjugates (PDCs).1 Each of these drug types have distinct advantages and disadvantages. Small-molecule drugs are highly stable, easily penetrate cell membranes, and can be administered orally, making them the most prevalent drugs worldwide. However, their inability to distinguish normal cells from diseased cells often leads to high toxicity and limited therapeutic efficacy.2 Monoclonal antibodies (mAbs), known for their high target specificity and affinity, are widely used to treat inflammation, autoimmune diseases, and cancer. By conjugating mAbs with small molecular payloads, ADCs selectively deliver these payloads to diseased cells, considerably enhancing their treatment efficacy. However, ADCs face some limitations such as long circulation times in blood (causing off-target toxicity), poor solid tumor penetration, high development cost, premature payload release, and potential immunogenicity. In addition, the presence of many lysine residues in antibodies often results in heterogeneous ADC products, necessitating site-specific conjugation technologies to produce homogeneous ADCs. Solid tumors, characterized by a high concentration of dense connective tissue and extracellular matrix, further hamper effective drug penetration and weaken treatment efficacy. This unique microenvironment of solid cancers presents additional challenges, underscoring the urgent need for novel drugs or strategies to overcome these limitations. PDCs emerged as a promising approach to address these challenges of ADCs while retaining targeting specificity and affinity.3
PDCs share a structural similarity with ADCs, comprising a peptide, linker, and active payload (Figure 1). Compared with ADCs, PDCs offer many advantages, such as small molecular weight, enhanced penetration capability, low-cost synthesis, and minimal immunogenicity.4 PDCs have demonstrated as an efficient treatment modality after ADCs for
Figure 1.
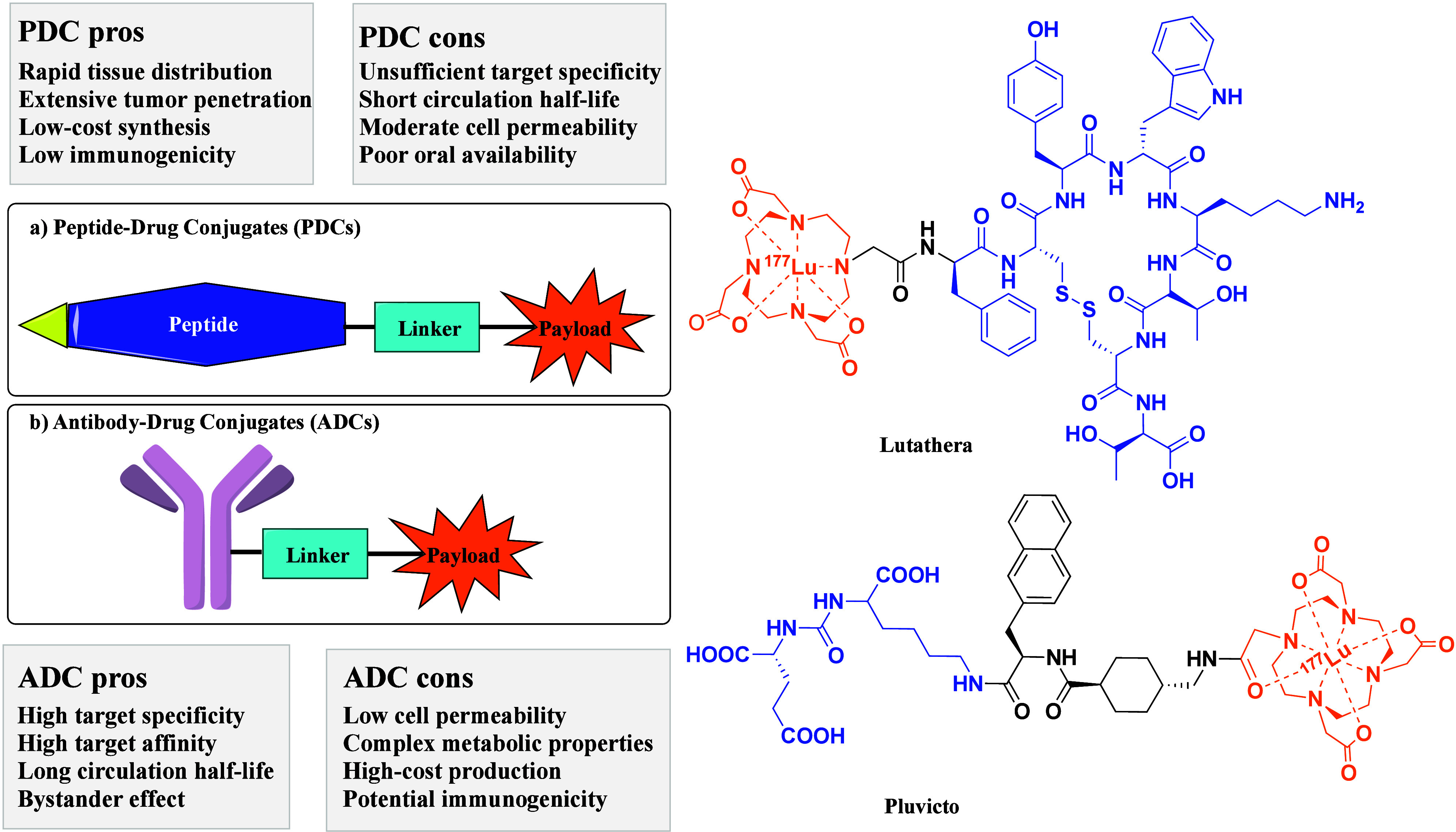
Schematic structure of a peptide–drug conjugate (PDC) and chemical structures of FDA-approved PDCs. (1) Lutathera features a cell-targeting peptide (CTP) derived from the tyrosine-containing somatostatin analogue Tyr3-octreotate (TATE), an amide bond linker, and a payload comprising the macrocyclic chelating agent tetraazacyclododecane-tetraacetic acid (DOTA) bound to the beta-emitting radionuclide lutetium-177 (177Lu). (2) Pluvicto contains a CTP with the PSMA-binding motif Glu–NH–CO–NH–Lys, a linker incorporating 2-naphthyl-l-alanine and tranexamic acid, and the same payload as the Lutathera. (3) The CTP is shown in blue, the payload in orange, and the linker in black.
drug-resistant infections and complex disease states, including autoimmune diseases, metabolic disorders, and cancer. Their small size facilitates rapid distribution in tissues and extensive tumor penetration, enabling rapid delivery of payload to tumors. Currently, two PDC drugs (Figure 1) are approved for marketing by the U.S. Food and Drug Administration (FDA). 177Lu-DOTA-TATE (Lutathera) was approved in 2018 as the first generation of PDCs for treating somatostatin receptor-positive gastroenteropancreatic neuroendocrine tumors (GEP-NETs).5 Recent Phase III clinical trials reveal that first-line Lutathera combined with long-acting octreotide substantially extended median progression-free survival (PFS, by 14 months) in patients with grade 2 or 3 advanced GEP-NETs.6 In March 2022, Pluvicto was approved by the FDA for treating prostate-specific membrane antigen (PSMA)-positive metastatic castration-resistant prostate cancer. Pluvicto combines a PSMA-specific peptidomimetic with a therapeutical radionuclide to selectively deliver ionizing radiation to tumor cells.
Designed to deliver highly potent payloads into targeted cells, PDCs have demonstrated treatment efficiency in diseases except for tumors. Various PDCs are developed to treat viral and bacterial infections.7 A novel PDC contains a kidney-targeting peptide G3-C12 (ANTPCGPYTHDCPVKR), a cleavable disulfide linker, and captopril payload showed profound angiotensin-converting enzyme inhibition activity in chronic kidney disease.8 The primary mechanism of PDCs involves cell-targeting peptides (CTPs) binding to membrane receptors, mediating endocytosis or internalization of PDCs (Figure 2). Small-molecule payloads can also mediate endocytosis of PDCs by binding to its receptor. Endocytosis is an energy-dependent process requiring ATP consumption and vesicle formation to translocate PDCs.9 Studies describe at least four endocytosis mechanisms: macropinocytosis, clathrin-dependent, caveolin-dependent, and clathrin/caveolin-independent pathways.10 Cell-penetrating peptides (CPPs) in PDCs may directly translocate through the lipid bilayer of the membrane in an energy-independent mechanism, interacting with the lipid bilayer of the plasma membrane to change membrane dynamics at the site of contact and facilitate cellular entry of PDCs.11 Once inside, PDCs are transported to endosomes or lysosomes, where low acidity and high enzyme concentrations enable the release of peptides and payloads into the cytoplasm, nucleus, or other organelles to perform their functions (Figure 2).
Figure 2.

Mechanism of action of PDCs. PDCs can enter cells via endocytosis mediated by CTPs or small-molecule payloads. Alternatively, they may cross the membrane through CPPs. The low acidity and high enzyme concentrations in endosomes or lysosomes facilitate the release of payloads into the cytoplasm, nucleus, or other organelles to exert their functions.
2. PDC Components and Conjugate Strategies
The identification of peptides or small-molecule payloads toward a particular target or disease is a logical starting point for the design of PDCs. Effective delivery of payloads to the target site is a challenging task. The peptide part of PDCs mainly drives target specificity, enabling site-specific payload delivery while reducing their toxicity. Over the past decade, innovations in PDC peptides (Figure 3), such as novel CTPs, CPPs, self-assembling peptides (SAPs), and stimuli-responsive peptides (SRPs), have tremendously improved the therapeutic efficacy of payloads.
Figure 3.
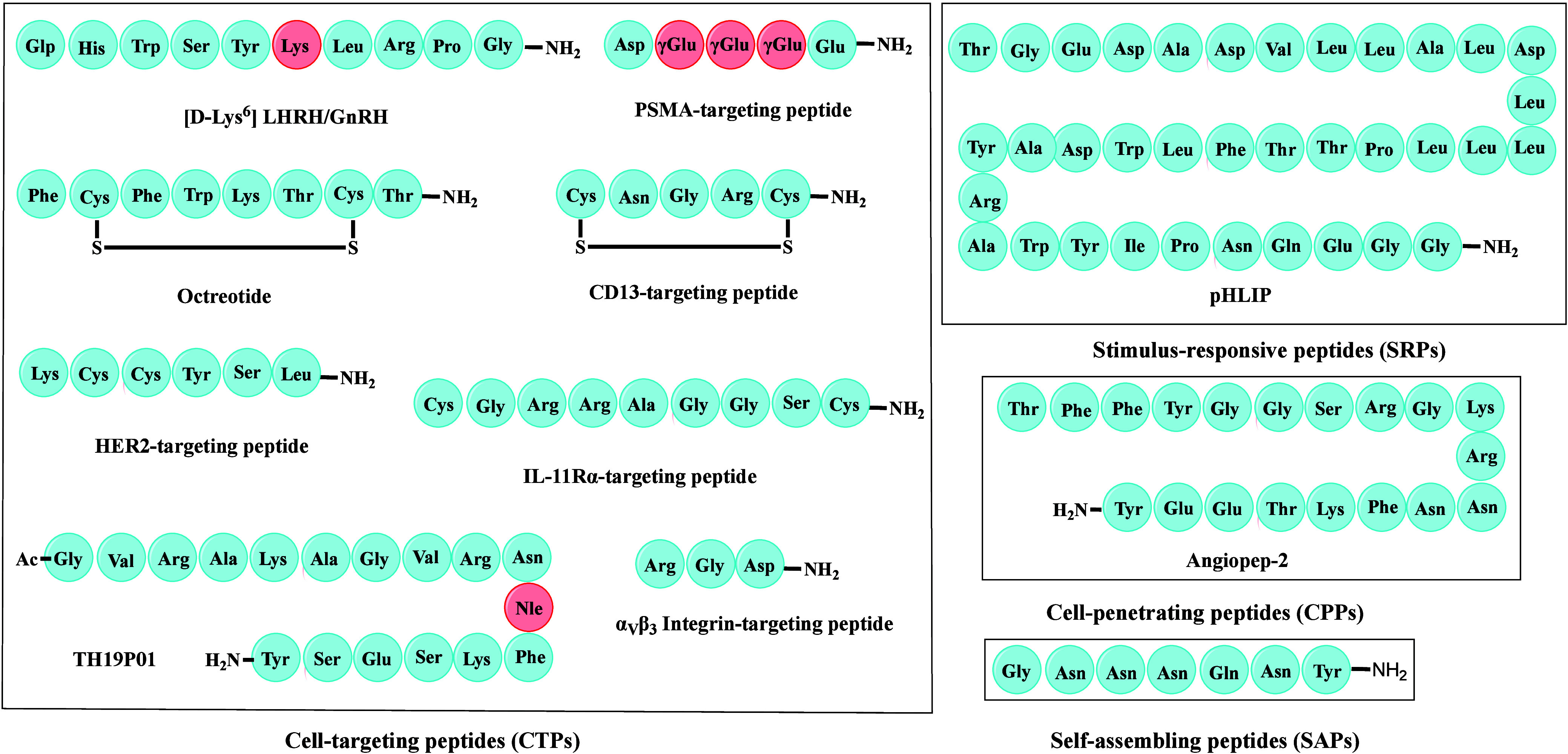
Representative peptides utilized in PDCs. The amino acid sequences of some typical CTPs are shown in the left box, where the amino acid in red color is a non-natural amino acid. The boxes on the right are examples of SRP, CPP, and SAP, respectively.
Most peptide drugs are hormone mimics or derived from natural products.12 The design of potent and selective peptides that target receptor ligand-binding domains remains challenging. Various encoding and display technologies are used to identify high-potency peptide sequences for target proteins.13 Nucleotide-encoded mass library screening techniques14 advance non-natural moieties or nonproteinogenic amino acids to address challenging inaccessible drug targets, including phage display screening, split-intein circular ligation of peptides and proteins, mRNA display, and the one-bead-one-compound or split-and-pool strategies. Peptides screened through these methods usually have high affinity for target proteins.
The linker plays an important role in PDC design, as it strongly impacts the safety and potency of the PDC. Two main categories of linkers have been developed based on their cleavability (Figure 4). Cleavable linkers are either sensitive to pH, glutathione, or proteases such as cathepsin B for dipeptide linkers. Noncleavable linkers, such as thiazole, oxime, or thioether, enhance PDC circulatory stability in humans.
Figure 4.
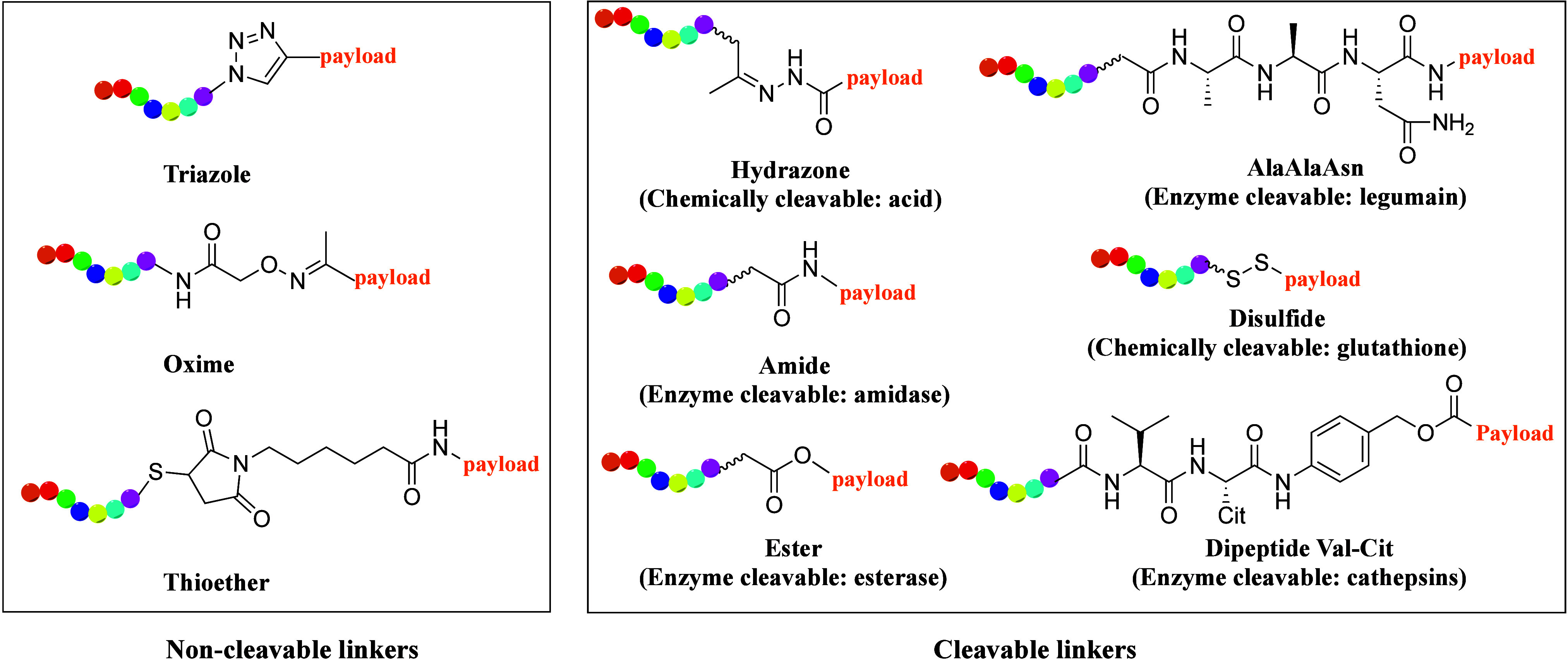
Chemical structures of different linkers in PDCs. The peptide is shown in rainbow color and the linker in black.
The final conjugation step is crucial in PDC synthesis. Amide or ester formation has proven useful and efficient for linking peptides to payloads or linkers. Recently, click reactions have gained prominence owing to their high efficiency, rapidity, and mild reaction conditions (Figure 5).15 Among these, azide–alkyne Huisgen cycloaddition is widely used, forming triazole units in both aqueous and organic solvents.16 This reaction has been a useful tool in the synthesis of ADCs or PDCs, especially multimeric PDCs.17 To address safety concerns associated with metal catalysts in Huisgen cycloaddition, strain-promoted azide–alkyne cycloaddition (SPAAC) was developed, eliminating the need for metal catalysts.18 However, the high reactivity of SPAAC can lead to poor regioselectivity and chemoselectivity. Staudinger ligation,19 which forms amide bonds between phosphine compounds and azides in aqueous solutions without metal catalysts, offers an alternative. Traditional coupling of primary or secondary amines with carboxylic acids also facilitates amide formation. A novel PDC was synthesized via azide–alkyne click reaction by conjugating an αVβ6 integrin-recognizing small cyclopeptide, c(AmpLRGDL), with the tyrosine kinase inhibitor (nintedanib) for idiopathic pulmonary fibrosis treatment.20 Another example involves an EphA2-targeting PDC generated by coupling an EphA2-targeting peptide to an azido-hexanoyl paclitaxel (PTX) group via azide–alkyne click reactions.21
Figure 5.

Schematic representation of the most common conjugate reactions used in PDCs. The peptide moieties are highlighted in rainbow color.
Cysteine is often present in peptide sequences and can be used for thiol–alkene conjugation. This thiol-mediated Michael addition usually needs the alkene to be linked to an electron-withdrawing group and is effective in both organic and aqueous solvents. Another conjugation strategy is the condensation of a carbonyl group with an oxyimino group to form oximes or with hydrazines to yield hydrazone compounds. Esters and carbamates are often utilized in PDC synthesis. For example, the hydrophobic drug PTX was successfully conjugated with a CPP via ester linkage with succinic acid as a pH-cleavable linker.22 The ester linkage remains stable until the PDC is internalized into cancer cells, where the acidic environment induces hydrolysis of the ester bond, releasing the PTX payload.
In brief, synthesizing PDCs can be challenging owing to the choice of chemistry for the final ligation. The selection of the conjugation reaction should be consciously evaluated in every aspect based on the PDC structure.
We will not elaborate on the components of PDCs, as several excellent reviews already cover the topics of peptides,23 linkers,24 and payloads25,26 of PDCs. Herein, we present an outline of the latest cancer-targeting PDCs in development, their applications in clinical trials, and recent strategies aimed at enhancing the treatment efficacy of PDCs.
3. Strategies for Improving PDC Therapeutic Efficacy
PDCs have great therapeutics potential if they exhibit improved cell-targeting ability, circulatory stability, and permeability. However, few PDCs can be delivered orally and reach a systemic concentration sufficient for treatment. Therefore, efficient intracellular trafficking and targeted delivery to the site of action are essential to overcoming the current drawbacks of PDCs in disease therapy. To address these issues, researchers have employed various strategies to enhance targeting ability, circulatory stability, toxicity management, and blood–brain barrier (BBB) penetration, aiming to optimize pharmacokinetic properties and therapeutic efficacy (Figure 6).
Figure 6.
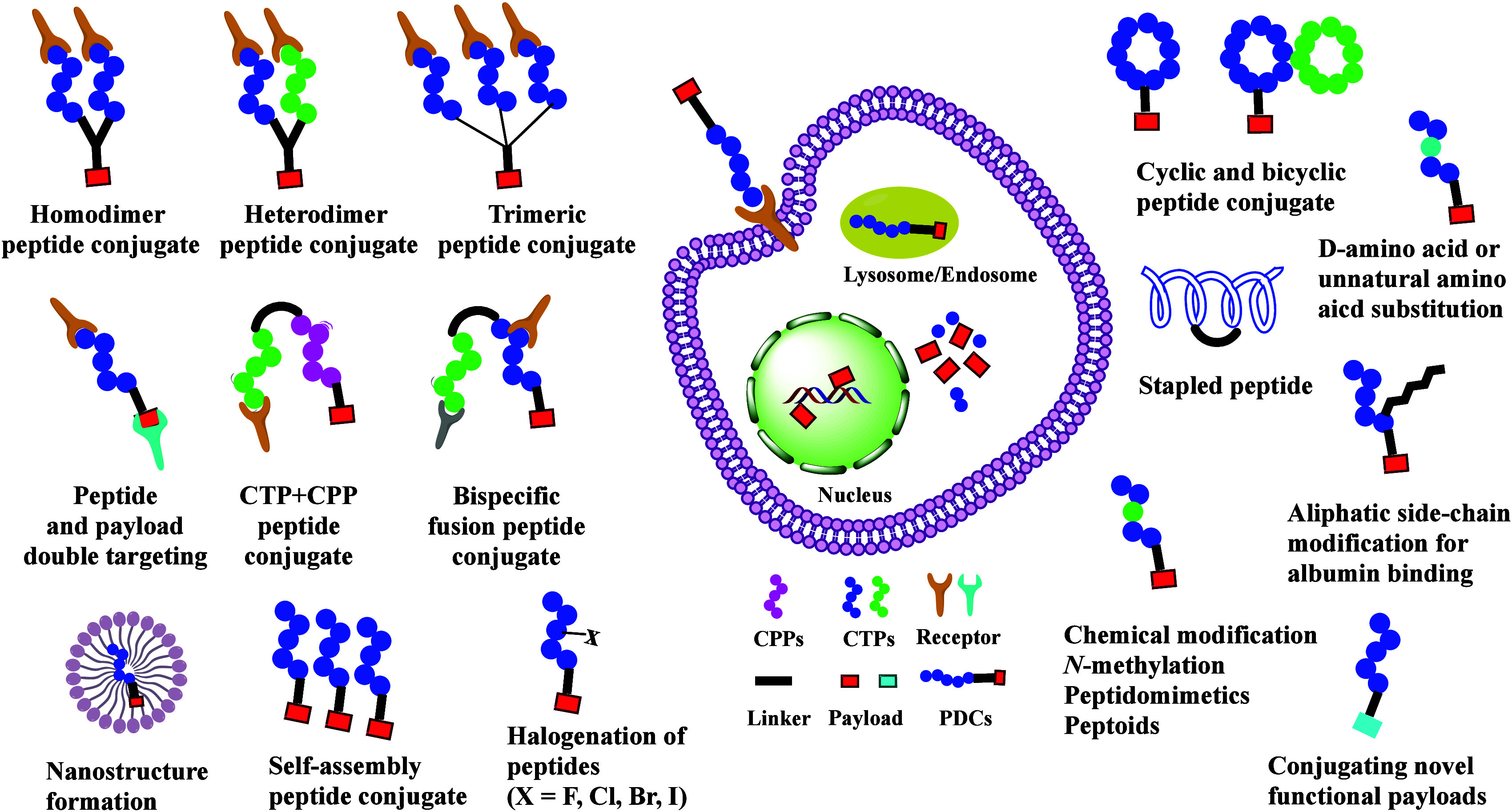
Overview of the current challenges and optimizations of PDCs.
3.1. Improving the Cell Targeting of PDCs
3.1.1. Multivalency Strategy
CTPs are peptides with moderate target specificity and affinity, allowing them to efficiently deliver conjugate payloads to cells that overexpress specific targets. To enhance the cell-targeting ability and binding affinity of CTPs with corresponding targets, the multivalency of dimeric or trimeric peptide strategy was developed in PDCs to improve treatment efficacy (Figure 6).27 Arginine–glycine–aspartic acid (RGD) peptides, known for targeting the αVβ3 integrin receptor, have garnered immense attention in tumor therapy. Dimeric RGD peptides show a 10-fold higher tumor affinity than the monomer and better retention in the tumor.28 Further studies found that three and four copies of RGD peptides showed better activity compared to derivatives with one to 16 clustered RGD motifs.29 To improve the antitumor efficacy of PTX through tumor targeting, dimeric and tetrameric cRGD–PTX conjugates were prepared. The dimeric PDCs showed similar antitumor activity to PTX but with stronger binding to the αvβ3 receptor.17 Binding tests with purified integrin αvβ3 receptor showed that IC50 values decreased as the number of ligand moieties increased, reaching a plateau with the trimeric conjugate.30
The ephrin receptor family, the largest receptor tyrosine kinase family, plays a key role in cancer. Ephrin type-A receptor 2 (EphA2) is overexpressed in many solid tumors, contributing to oncogenesis, tumor-associated angiogenesis, and metastasis. MEDI-547, an ADC composed of a human anti-EphA2 monoclonal antibody linked to the monomethylauristatin F payload, showed substantial antitumor activity in mouse models.31 However, a Phase I study was terminated because of drug-related adverse effects, possibly resulting from insufficient subcellular internalization of the ADC.32 An interesting feature of EphA2 ligands is that their agonistic activities can be enhanced by properly clustering the targeting agent, achieved by synthesizing dimers spaced by the appropriate linker.33 The dimeric peptide showed approximately 13-fold increased binding affinity for EphA2 compared to the monomeric peptide. Based on these results, novel dimeric EphA2-targeting PDCs were designed, proving highly effective at targeting circulating tumor cells and inhibiting lung metastasis in breast cancer21 and pancreatic cancer models.34 Recent time-resolved, live-cell fluorescence spectroscopy revealed that ligand-free EphA2 assembles into multimers through two types of intermolecular interactions in the ectodomain.35 Additionally, a homodimer cyclic peptide conjugated with the photosensitizer indocyanine green showed better photothermal therapy efficacy compared to the monomeric PDC in breast cancer.36
Human epidermal growth factor receptor 2 (HER2) is a receptor tyrosine-protein kinase, and its overexpression is associated with the development of several cancers, including breast, gastric, esophageal, ovarian, and endometrial cancer. A novel PDC containing a HER2-targeting homodimer cyclic peptide (RNWELRLK-PEG4)2 showed enhanced binding affinity, improved antitumor effects, and reduced systemic toxicities in HER2-positive breast cancer.37 These results highlight multivalency as an effective strategy to strengthen ligand–target interactions and enhance cell-targeting ability of PDCs.
3.1.2. Bispecific Targeting
Epidermal growth factor receptor (EGFR) is associated with cell proliferation and migration and is abnormally overexpressed in many solid tumors. A novel PDC was designed to simultaneously target both the extracellular and intracellular domains of EGFR. This PDC consists of an EGFR extracellular targeting peptide (sequence: LARLLT) and an EGFR intracellular targeting payload, gefitinib, which showed better binding affinity and antitumor activity than gefitinib alone in lung cancer cells.38 Another bispecific fusion peptide was designed to bind both HER2 domains II and IV. Compared to peptides targeting a single antigen domain, bispecific peptides targeting different domains of the same protein show enhanced efficiency in targeting and binding. Conjugating this fusion peptide with camptothecin resulted in enhanced antitumor activity in a HER2-positive breast cancer mouse model.39
3.1.3. Halogenation of Peptides
Halogenation is an emerging method to modulate the pharmacological activities of peptides.40 It has been empirically discovered over time that the insertion of halogens (fluorine, chlorine, bromine, or iodine) at specific positions in a drug molecule can improve its pharmacological properties and bioactivity.41 Late-stage halogenation of peptides can be achieved through chemical catalysts42 or biocatalytic methods using enzymes.43,44 Halogenation is generally associated with enhanced target selectivity and reduced off-target toxicity.45 The introduction of halotryptophans in RGD peptides increased their affinity for integrin αvβ3 and enhanced selectivity over integrin α5β1.46 A cyptophycin derivative with a chlorine substituent shows 8-fold increased activity against human cervical carcinoma cells.47 Substitution with chlorine or bromine in peptoids enhanced antimicrobial activity while reducing cytotoxicity of normal cells, whereas fluorination had minimal effect.48 Similarly, bromonation of antimicrobial peptides exhibits a 2-fold improvement in antimicrobial activity.49 These results indicate that the halogenation of peptides may impact target affinity and treatment efficacy, warranting further investigation in PDC development.
3.1.4. Self-Assembling Peptides (SAPs), Stimuli-Responsive Peptides (SRPs) and Nano Structure Formation
PDCs containing hydrophilic peptide segments and hydrophobic drugs can self-assemble into various nanostructures, such as nanotubes, nanofibers, hydrogels, and supramolecular filaments. Conjugating small molecular hydrophobic units with short peptide segments promotes new self-assembling features and functional properties.50 A balanced hydrophobic–hydrophilic ratio is crucial for the efficient translocation of the conjugate into cells and its optimal bioactivity.
The amino acid type and sequence of peptides form the basis of their assembly ability.51 The structure of SAPs includes the primary amino acid sequence, resulting in secondary structures such as helical, β-sheet, trans, or disordered conformations under specific stimuli. Peptides, composed of amino acids with active amino and hydroxyl groups, are particularly advantageous as SRPs. Specific stimuli for SRPs include pH, temperature, redox potential, light, and enzymes. Notably, pH-responsive peptides show great promise for tumor targeting owing to their ability to respond to pH changes. Various innovative pH-responsive peptides and polypeptides52 have emerged, such as pH-low insertion peptides (pHLIPs).53 pHLIPs are 36-residue polypeptides containing the C-helix sequence of the integral membrane protein bacteriorhodopsin.54 As a pH-sensitive peptide, pHLIP forms an α helix in the low-pH tumor microenvironment and inserts the C-terminus with the payload across the cell membrane.55 The insertion reaction proceeds rapidly at room temperature and is fully reversible. CBX-12 conjugates pHLIP with topoisomerase I inhibitors such as exatecan. The combination of CBX-12 with ceralasertib markedly suppressed tumor growth in mouse xenografts.56 CBX-12 combined with anti-PD-1 delayed tumor growth, induced a complete response, and promoted long-term antitumor immunity in a mouse model.57 A Phase II study of CBX-12 is currently underway in patients with platinum-resistant or refractory ovarian cancer (NCT06315491). Ye and co-workers developed a pH-responsive PDC to treat severe infectious diseases and combat antimicrobial resistance.
A novel PDC, SAP-CPT,58 consists of an RGD targeting peptide, a self-assembling peptide (GNNNQNY), and the cytotoxic payload camptothecin. SAP-CPT drug targets the tumor-overexpressed receptor integrin αVβ3 and assembles in situ to form nanoscale oligomers. This self-assembly strategy enhances endocytosis and improves tumor-killing effects in breast and bladder xenograft mice models. Compared to normal cells, mitochondria in cancer cells show profound differences, including lower mitochondrial membrane potential and increased reactive oxygen species (ROS) generation. A novel ROS-responsive PDC contains a mitochondria-targeted peptide (KLAK) with a ROS-cleavable thioketal linkage, enabling self-assembly into nanoparticles under physiological conditions. When the nanoparticles accumulate in tumors and enter cancer cells, they actively target the mitochondria, where they disassemble to release the camptothecin payload, effectively destroying cancer cells.59
Another novel PDC (PDC-DOX2), in which two doxorubicin (DOX) molecules are conjugated onto a short peptide with self-assembly function, was designed and synthesized. Hyaluronic acid (HA) was coated on PDC-DOX2 micelles to form an HA-shelled, PDC-cored nanomedicine.60 Anionic HA, overexpressed by many tumors, serves as an active tumor-targeting ligand with excellent biocompatibility and biodegradability.61 HA-modified nanocarriers have proven effective in targeting CD44-overexpressing tumor cells.62 HA can actively enhance the targeting effects of PDC-DOX2 by interacting with overexpressed CD44 receptors in cancer cells.60 Another strategy involving polypeptide–polyethylene glycol derivatives (PPDs) regulates PDC structure and properties to construct intravenous nanomedicine.63 PPD molecules act as targeting carriers, facilitating the delivery of the encapsulated PDCs into the hepatocyte mitochondria and enhancing antitumor efficiency. PEGylated PDCs can form self-assembled nanoparticles, passively targeting tumor sites via the enhanced permeability and retention effect.64 Overall, the nanostructure of PDCs enhances drug delivery potential by forming self-assembled particles capable of targeted delivery of drugs through active and passive targeting.
3.2. Overcome Biological Barriers via Cell-Penetrating Peptides (CPPs)
The development of efficient transcellular peptides capable of overcoming biological barriers, such as the BBB or epithelial barriers composed of multiple layers of diverse cells, remains a major challenge. CPPs are small molecules, typically 5 to 30 amino acids in length, known for their ability to cross cell membranes without requiring a receptor.65 While the exact mechanisms are still debated, most CPPs enter cells via endocytosis. When conjugated with a payload, CPPs act as carriers, facilitating the delivery of payloads, including drugs, nucleic acids, and probes, across biological barriers.
CPPs are generally classified as cationic, hydrophobic, or amphipathic based on their physicochemical properties. Early examples, such as the HIV-1 transcription activator protein (TAT) and the RGD sequence, have demonstrated the ability to deliver various cargo molecules. TAT and penetratin are cationic CPPs, rich in both lysine and arginine. Poly arginine peptides enter cells more effectively than equal-length poly lysine, although poly arginine peptides shorter than six amino acids are ineffective.66 Furthermore, CPPs rich in arginine residues have shown renal toxicity in rodent models, limiting their application.67 Improving the cell-penetrating ability of CPPs remains an active and attractive research field.
Cyclizing CPPs limits the formation of secondary structures, reducing the free energy required for binding the peptides to membranes and enhancing their cell penetration.68 Recently, a hydrophilic cell-penetrating cyclic peptide (EPP6) with no positive charge demonstrated prominent transcytosis abilities.69 Another novel CPP, peptide–bismuth bicycles, increased cellular uptake by more than 1 order of magnitude compared to linear peptide.70 Cyclization also increased the bactericidal activity of arginine-rich cationic CPPs compared with their linear counterparts.71 Statistically designed transcellular peptides conjugated with 6-paradol also exhibited improved cell permeability and superior therapeutic efficacy in human keratinocyte cells and mouse models of psoriasis.72 However, most CPPs lack cell specificity, limiting their application in drug delivery. To address this challenge, labile linkages can be introduced to prodrugs for stimuli-triggered drug release. Another prodrug strategy involves developing an activable CPP, where the cell-penetrating function of CPPs is masked by an anionic peptide with a cleavable linker. Upon reaching tumor tissue, proteolysis of the linker activates the cell-penetrating function of CPP. An activable CPP, conjugated with antitumor drug DOX and shielded by 2,3-dimethylmaleic anhydride, formed a novel prodrug for tumor-targeted drug delivery, exhibiting remarkable tumor growth inhibition.73 Further studies are needed to assess the toxicity of these PDCs.
The BBB is a major hurdle for central nervous system drug delivery. Several pathways enable molecules to cross the BBB, such as passive diffusion, carrier-mediated transcytosis, receptor-mediated transcytosis, adsorptive-mediated transcytosis, and cell-mediated transcytosis.74 A distinctive characteristic of the BBB is the expression of several receptors, including those associated with angiogenesis, such as vascular endothelial growth factor receptors (VEGFRs) and EGFRs, as well as transferrin receptors (TfRs), integrin receptors, and low-density lipoprotein receptors.
The angiopep family of peptides, derived from the Kunitz domain of human aprotinin, includes angiopep-2, which exhibits great transcytosis and BBB penetration.75 Angiopep-2 (sequence: TFFYGGSRGKRNNFKTEEY) is a 19-amino-acid oligopeptide that binds to the low-density lipoprotein receptor-related protein-1 (LRP1), facilitating brain penetration. ANG1005 (also known as GRN1005 or PTX trevatide) is a novel PDC consisting of three PTX molecules covalently linked to angiopep-2 via a succinyl linker. This ester linker in the conjugate is enzyme cleavable, such as esterases in lysosomes, releasing PTX. ANG1005 has been shown to cross the BBB via receptor-mediated transcytosis through LRP1 to penetrate malignant cells.76 Phase I studies of ANG1005 showed activity in recurrent malignant glioma with toxicity similar to PTX.77 A multicenter Phase II study of ANG1005 also demonstrated notable systemic treatment effects and prolonged overall survival (OS) in all patients with breast cancer and recurrent brain metastases.78 A randomized open-label, multicenter pivotal Phase III study of ANG1005 is ongoing, comparing it with the physician’s best choice in HER2-negative breast cancer patients with newly diagnosed leptomeningeal carcinomatosis and previously treated brain metastases (NCT03613181). Another novel PDC was developed by conjugating the TfR-targeting peptide (sequence: HAIYPRH) with the payload SN-38 via a cathepsin B cleavable linker. This PDC successfully delivers SN-38 across the BBB and selectively into glioblastoma cells, triggering a cytotoxic response.79
3.3. CPP and CTP Combination Strategy
CPPs enable efficient intracellular trafficking but lack targeting ability. Therefore, the conjugation of CPPs and CTPs provides a novel, efficient method for PDC design, enhancing drug delivery efficacy (Figure 6).80 A chemically modified polysaccharide was conjugated with integrin-target receptor tripeptide (RGD) and l-arginine as a cell-penetrating amino acid for synergistic targeting and enhanced internalization by cancer cells. This PDC showed a remarkable reduction in cytotoxicity toward normal cells while enhancing efficacy against cancer cells.81 Combining the integrin αVβ3 targeting peptide, CPPs, and the payload daunorubicin, novel PDCs demonstrated highly pronounced antitumor effects in cancer cells.82 The cellular uptake of internalization is mainly mediated by the CPP part, while the targeting unit directs the conjugates selectively to αvβ3 integrin-expressing cells. The CTP–CPP–PTX conjugate exhibited less cytotoxicity to normal cells and enhanced antitumor efficacy.83 Another strategy conjugates integrin-targeting peptide (RGDC) with cyclic CPPs via a disulfide bond and the payload cabazitaxel via an ester bond. This PDC reaches the tumor site, where the disulfide bond is cleaved by glutathione, allowing the cyclic CPPs to facilitate payload entry and release via esterase or acidic pH. This conjugation strategy has been shown to enhance antitumor activity and targeting in prostate and breast cancer.84
These examples highlight the synergistic effects of CPPs and CTPs in the design of PDCs to improve their treatment efficacy.
3.4. Improving the Circulatory Stability of PDCs
Proteolytic susceptibility and fast renal clearance have long posed major challenges in the development of peptide drugs. PDCs, with their low molecular weights, are vulnerable to peptidase hydrolysis, resulting in short half-lives in the blood. In most cases, a short plasma half-life is insufficient to deliver a sufficient drug load to targeted organs and tissues, and circulatory instability and rapid renal clearance persist in clinical use. However, considerably rapid tissue distribution may compensate for the short half-life in blood. Several strategies have been employed to address these challenges, including cyclization, albumin binding, and amino acid substitution or peptide modifications (Figure 6). A novel antibody-assisted PDC has also shown an extended circulation half-life and improved treatment efficacy in a glioblastoma mouse model.85
3.4.1. Cyclic, Bicyclic, and Stapled Peptide Formation
Cyclic peptides are more structurally and metabolically stable than linear peptides.86 Cyclization stabilizes peptides against proteolysis by concealing the mobile ends of peptides in the space, preventing exopeptidases from cleaving them. Studies have revealed that cyclization improves the stability, target affinity, and biological activity of linear peptides. For example, cyclic RGD was found to be 30-fold more stable than linear RGD in a neutral pH 7 solution.87 The increased stability of cyclic RGD is attributed to decreased structural flexibility and salt bridge formation between the side chains of the Arg and Asp residues.88 Consequently, cyclization improves the binding affinity of the peptide to targeted receptors. Cyclic-RGDKLAK exhibited selectively and specifically higher affinity for αvβ3-integrin in glioblastoma cells compared to liner analogues.89 Additionally, blockade of the interaction between programmed cell death ligand-1 (PD-L1) and its receptor PD-1 has shown great success in cancer immunotherapy. Cyclic anti-PD-L1 peptides, discovered through macrocyclization scanning, demonstrated improved blocking activity, serum stability, and excellent in vivo antitumor activity compared with their linear counterpart.90
Stapled peptides are a class of cyclic peptides modified by the insertion of aliphatic bridges to increase proteolytic stability and membrane permeability.91 The stapling strategy is a common macrocyclization strategy that promotes the α-helical conformation of peptides. Various stapled peptides have been designed as therapeutic drug candidates against different diseases, including cancer, infectious diseases, inflammation, and diabetes.92 For instance, stapling a tumor-suppressing p53-derived peptide improved its α-helical content, proteolytic stability, and binding affinity.93 A novel stapled peptide, SP9, can block mucus secretion and mucus accumulation in mouse airways. Four non-natural amino acids were incorporated into the SP9 sequence, and two hydrocarbon staples were formed using Grubbs’ catalyst. Compared to the nonstapled peptide, SP9 showed increased helical content, serum stability, and treatment efficacy in mice.94
Bicyclic peptides, a new class of cyclic peptides, have constrained conformations. Compared to monocyclic peptides, bicyclic peptides bind their targets with higher affinity and are more resistant to proteolytic degradation.95 Bicyclic peptides have been developed as therapeutics in PDCs against a wide range of diseases. Recently, metal bismuth was reported as a stable, nontoxic, and selective linker for the bicyclization of peptides by reacting with cysteine residues. Peptide–bismuth bicycles form instantaneously at physiological pH and remain stable in aqueous solution for weeks. Bicyclic peptides are up to 130 times more active and 19 times more proteolytically stable than their linear analogues without bismuth.96
Several bicyclic peptide–conjugated PDCs have shown positive results in preclinical studies and clinical trials. BT8009 consists of a nectin-4-binding bicyclic peptide, a cleavable Val–Cit linker, and the cell-penetrant toxin monomethylauristatin E (MMAE).97 The Val–Cit dipeptide linker is cleaved by the enzyme cathepsin B, which is abundant in lysosomes of tumor cells. BT8009 shows remarkable antitumor activity in preclinical tumor models across various cancer indications and is well tolerated in preclinical safety studies.98 Clinical trials of BT8009 are ongoing for advanced or metastatic solid cancers (Table 1). BT5528 is another novel bicycle toxin conjugate comprising a bicyclic peptide targeting the EphA2 tumor antigen linked to a payload MMAE. A Phase I dose-escalation study of BT5528 demonstrated good safety and antitumor activity (NCT04180371).99
Table 1. Summary of Representative PDCs in Clinical Trials.
| drug (manufacturer) | target | indication | peptide | linker | payload | phase | ClinicalTrials | recruitment status |
|---|---|---|---|---|---|---|---|---|
| AEZS-108 (AEterna Zentaris) | LHRH-R | endometrial cancer | [d-Lys6] LHRH | amide | doxorubicin | phase III | NCT01767155 | completed |
| EP-100 (Esperance Pharmaceuticals Inc.) | LHRH-R | ovarian cancer | LHRH | amide | lytic peptide | phase II | NCT01485848 | completed |
| ANG1005 (Angiochem, Inc.) | LRP1 | breast cancer with brain metastases, leptomeningeal | Angiopep-2 | succinic acid ester | paclitaxel | phase III | NCT03613181 | active, not recruiting |
| NGR-hTNF (AGC Biologics SpA) | CD13 neovasculature | malignant pleural mesothelioma | CNGRC | amide | hTNF | phase III | NCT01098266 | completed |
| G-202 (GenSpera, Inc.) | PSMA | Solid tumors, HCC | DγEγEγEE | amide | Thapsigargin | phase II | NCT01777594 | completed |
| BT8009 (BicycleTx Limited) | Nectin-4 | advanced solid tumor | bicyclic peptide | Val-Cit | MMAE | phase I/II | NCT04561362 | recruiting |
| urothelial Cancer | phase II/III | NCT06225596 | recruiting | |||||
| PEN-221 (Tarveda Therapeutics) | SSTR2 | advanced neuroendocrine and small cell lung cancer | octreotide | disulfide | DM1 | phase I/II | NCT02936323 | completed |
| CBX-12 (Cybrexa Therapeutics) | low pH and TOP1 | platinum resistant or refractory ovarian cancer | pHLIP | disulfide | Exatecan | phase II | NCT06315491 | recruiting |
| BMTP-11 (M.D. Anderson Cancer Center) | IL-11Rα | osteosarcoma and prostate cancer with bone metastasis | CGRRAGGSC | amide | peptidomimetic D(KLAKLAK)2 | phase I | NCT00872157 | completed |
| TH1902 (Theratechnologies) | SORT1 | advanced solid tumor | TH19P01 | succinic acid ester | docetaxel | phase I | NCT04706962 | recruiting |
| BPP-PTX | ACE | TNBC | EWPRPQIPP | succinic acid | paclitaxel | preclinical | preclinical | preclinical |
| KCC-TGX | HER2 | prostate cancer | KCCYSL | self-cycling | TGX-D1 | preclinical | preclinical | preclinical |
| SAP-CPT | integrin αVβ3 | breast and bladder cancer | RGD | self-assembly | camptothecin | preclinical | preclinical | preclinical |
Knottin peptides100 and multicyclic peptides,101 constrained by covalent disulfide cross-links, are emerging as promising candidates for drug discovery. A de novo disulfide-directed method has been developed to design and discover diverse multicyclic peptides with potent protein-binding capabilities.102 Knottin peptides, typically 30–50 amino acids in length, exhibit high thermal and proteolytic stability and potential for oral administration.103 A novel knottin peptide–drug conjugate (KDC) was designed to selectively deliver gemcitabine to malignant cells expressing integrins. This KDC binds to tumor cells with low-nanomolar affinity, is internalized via an integrin-mediated process, and releases its payload intracellularly.104 These results make multicyclic peptides an attractive molecular modality for the development of therapeutic agents.
3.4.2. Albumin-Binding Strategy
Low molecular weight (MW) of PDCs leads to their rapid removal from the bloodstream through renal clearance. One approach to overcome this issue is to increase their MW by conjugating them to albumin. Albumin, the most abundant plasma protein, is present in different body compartments, including the lymphatic, interstitial fluid, and blood. Human serum albumin (HSA) plays key roles in maintaining osmotic pressure and transporting exogenous drugs and endogenous small molecules such as bilirubin, metals, hormones, and fatty acids. With a serum half-life of ∼19 days in humans,105 HSA has prompted the development of several albumin-binding moieties to improve peptide stability and prolong their action time (Figure 7).106 For example, 4-(p-iodophenyl)butyric acid derivatives form stable, noncovalent binding interactions of variable affinity with both mouse and HSA.107 The addition of 4-(p-iodophenyl)butyric acid motifs to [177Lu]Lu-DOTA-TATE enhanced its mean residence time in blood.108 Cannabidiol also binds to HSA and γ-globulin in an exothermic manner with high binding affinity.109 Additionally, a series of truncated Evans Blue (tEB) derivatives was developed, with N(tEB)2 able to dimerize albumin to sandwich peptide therapeutics to protect them from proteolysis.110 PDCs incorporating four residues of either cationic lysine or anionic glutamate showed that cationic lysine possesses a high albumin-binding capacity and bioactivity.111
Figure 7.
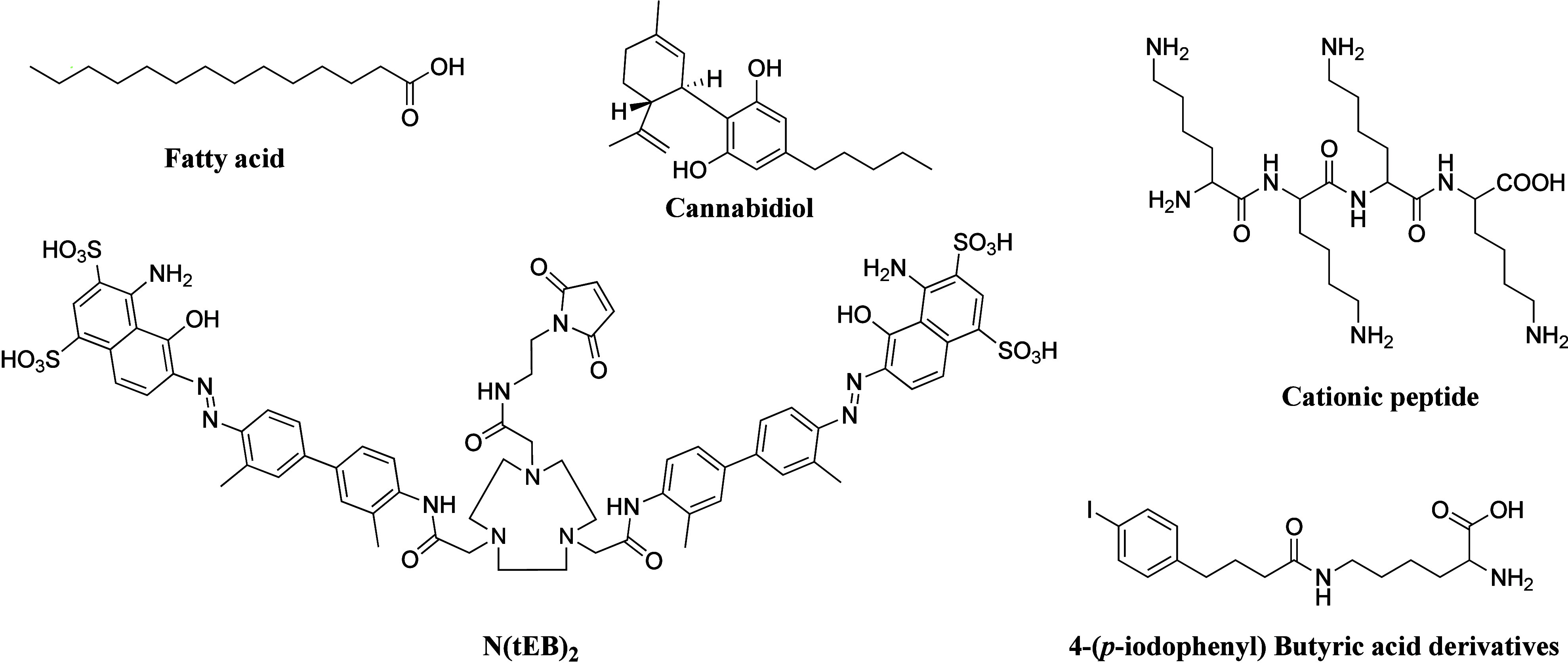
Chemical structures of small molecular albumin-binding moieties.
The albumin-binding strategy to prolong the action profile of insulins through acylation with fatty acids (Figure 8) was proposed and demonstrated in vivo approximately 30 years ago.112,113 Two long-acting insulin drugs, detemir and degludec, have since been approved by the FDA. In detemir, the last residue (threonine amino acid) is removed, and a fatty acid (meristic acid) is covalently bonded to the B29 lysine. In degludec, a fatty acid (hex-decanedioic acid) is bonded to B29 via glutamic acid as a linker. Both markedly prolong the average timespan of action.114 Insulin icodec, an ultralong-acting drug with a plasma half-life of 196 h in humans,115 is a once-weekly insulin analogue conjugated with a fatty diacid acylation moiety (1,18-octadecanedioic acid) and a linker as the albumin-binding motif. Icodec insulin was approved for the treatment of type 2 diabetes in Europe in March 2024.
Figure 8.
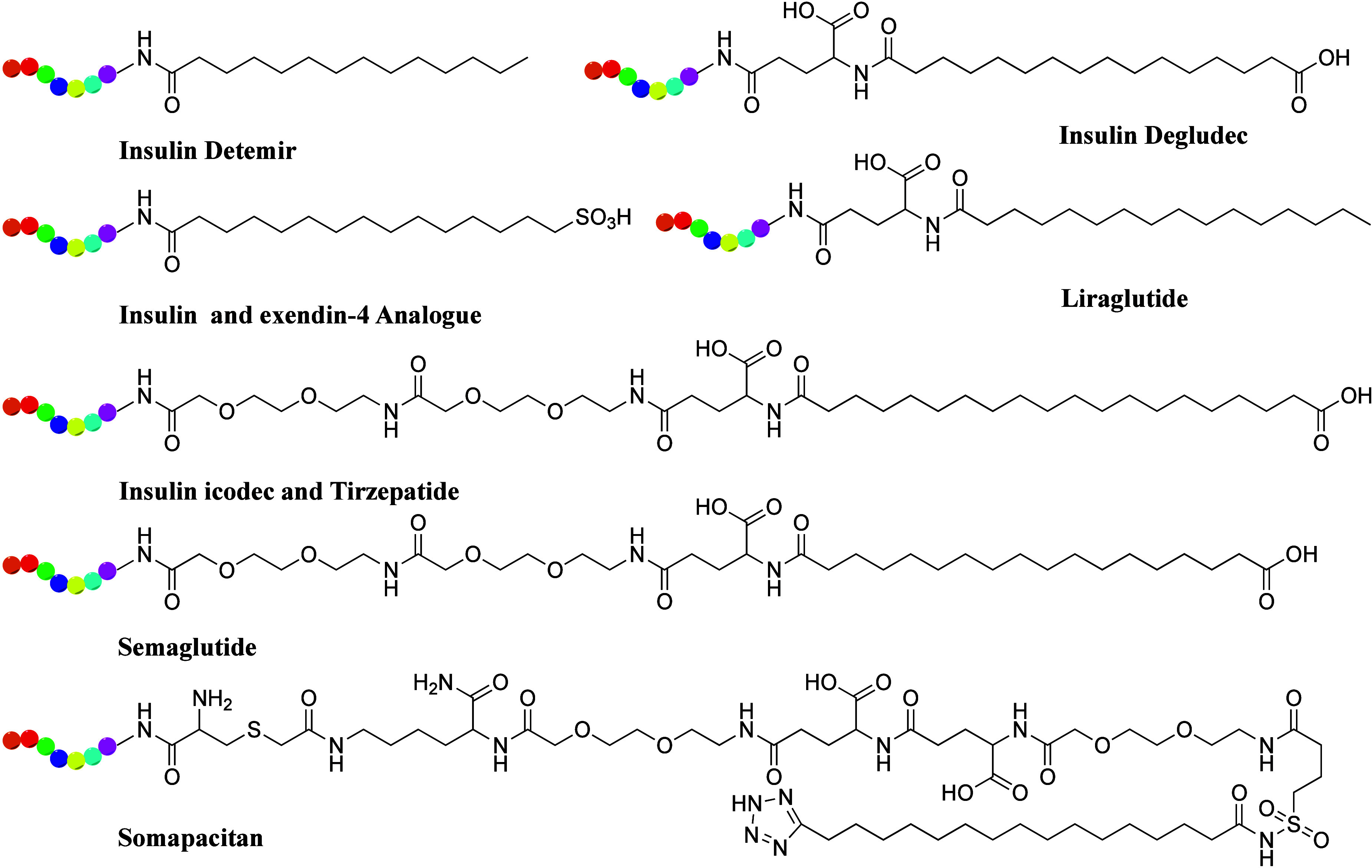
Chemical structures of albumin-binding fatty acids (in black color) used in long-acting peptide drugs and PDCs.
Glucagon-like peptide 1 (GLP-1) is a proglucagon-derived peptide secreted from intestinal L-cells to lower postprandial glycemic excursion by stimulating insulin secretion and inhibiting glucagon secretion. The blood half-life of bioactive GLP-1 is less than 2 min owing to inactivation by the protease enzyme dipeptidyl peptidase-4 (DPPIV) and rapid renal clearance. In 2003, a modified GLP-1 (CJC-1131) exhibited a prolonged duration of action and long serum half-life in vivo by substituting l-Ala8 with d-Ala8 at position 2 and adding a Lys37 to the C-terminus with selective attachment of an albumin-binding residue. Novel fatty acid chain-modified GLP-1 analogues were designed and synthesized, showing enhanced stability and prolonged activity. These analogues introduced cysteine into the parent peptide, with a half-life closely related to the length of the aliphatic chain.116 Semaglutide, first approved by the FDA in 2017 for type 2 diabetes and in 2021 for chronic weight management in adults with obesity or overweight, features a long stearic (C18) fatty diacid on the side chain of Lys26, which increases albumin-binding affinity and considerably extends circulating half-life. Tirzepatide, a dual glucose-dependent insulinotropic polypeptide and GLP-1 receptor agonist, includes a C20 fatty acid moiety similar to semaglutide and icodec insulin (Figure 8). Several peptide candidates for cancer immunotherapy have been designed, showing improved stability and activity via fatty acid modification.117 Recently, novel long-acting PDCs were developed by conjugating peptides with fatty acid side chains for albumin binding, demonstrating enhanced stability in plasma and prolonged duration of antitumor action in mouse models.118
Although macromolecules such as polyethylene glycol (PEG), polysialic acids, and hydroxyethyl starch can be attached to a PDC to reduce renal clearance, the resulting polymerized PDC may lower tumor penetration ability and limit treatment efficacy. A novel strategy involves using polymer–drug conjugates to target tumor-associated myeloid cells, converting cold tumors to hot tumors and improving cancer immunotherapy.119
3.4.3. Amino Acid Substitution and Chemical Modification
To enhance the bioactivity and stability of peptides, researchers have explored various chemical modification techniques, including substitution with d-amino acids or unnatural amino acids, N-methylation, and the formation of peptidomimetics or peptoids.
d-Amino acids render peptides much more resistant to protease than their l-amino acid counterparts.120 Proteolytic susceptibility of peptides is primarily owing to the presence of l-chiral amino acids in their sequences. These poly-L sequences are recognized by various receptors and enzymes, triggering receptor-mediated endocytosis and proteolytic cleavage in the endosome or lysosome, respectively. d- and l-peptides bind to cellular heparan sulfates with similar propensity and affinities but differ in their capacity to trigger their endocytic uptake.121 Pentapeptides with the sequence Lys–Arg–Tyr–Arg–Arg (KRYRR), composed entirely of both d- and l-amino acids, were evaluated, and d-amino acid peptide enhanced cellular retention and tumor xenograft targeting.122 Increased stability in serum has been observed not only in peptides composed entirely of d-amino acids but also in those with partial d-amino acid substitutions at the termini in small peptides.123 Both the all-d-amino acid derivative and partial d-lysine substitution enhanced the stability of the antimicrobial peptide polybia-CP while maintaining its parental peptide activity.124d-Amino acids were also introduced at the N- and C-terminus of LFP-6 to obtain a proteolysis-resistant peptide, which retained equivalent blocking activity compared to the parental LFP-6 peptide.125 However, it is important to consider that d-amino acid substitution may affect peptide activity owing to changes in three-dimensional conformation.
The stability of peptides can be improved by altering single or multiple amino acids. The isoelectric point and net charge of peptides are important factors in their systemic circulatory half-lives and stability. Renal clearance occurs because the glomerular basement membrane in the kidneys, constructed of anionic carbohydrate moieties, acts as a strong barrier to the filtration of anionic molecules into the urine.126 In insulin glargine, glycine replaces asparagine at position 21 of the A chain, and two arginine amino acids are added at the C-terminus of the B chain, greatly improving the time of action.127 These alterations shift the isoelectric point closer to neutral, making insulin glargine highly soluble in acidic environments but less soluble at physiological pH, thereby prolonging bioavailability. These changes also favor the formation of insulin hexamers, further delaying absorption into the bloodstream. Therefore, the impact of the three-dimensional structure must also be considered during amino acid substitution.
Another strategy to improve peptide stability is to decrease the substrate recognition and binding affinity of proteolytic enzymes by introducing steric hindrance through amino acid substitutions. As previously mentioned, native GLP-1 is unsuitable for therapeutic applications owing to its short half-life caused by DPPIV degradation, with the N-terminal dipeptide serving as the proteolytic cleavage site for DPPIV.128 A single amino acid substitution at position 8 (alanine to glycine) in the GLP-1 sequence improves its stability against DPPIV.129 Amino-isobutyric acid (Aib), an α,α-disubstituted amino acid, is incorporated into peptide structures to stabilize their secondary structure elements, such as α-helices and β-sheets.130 Even a single Aib residue in heptapeptide sequences induces helical structure.131 Aib and 2′-naphthylalanine substitution in Ipamorelin improve its terminal half-life to 2 h in humans.132 The GLP-1 analogue semaglutide, with two amino acid substitutions compared to human GLP-1 (Aib8, Arg34) and a side chain modification at lysine 26, substantially prolongs its half-life to 165 h in humans through these substitutions combined with albumin binding.133
In addition to d-amino acid or other amino acid substitutions, other strategies to improve the stability of peptides include chemical modifications such as N-methylation134,135 and the development of peptidomimetics or peptoids.136,137 Peptide glycosylation has also been used to improve the stability of peptides.138N-methylation of the amide bond in peptides forms an organized hydrogen bonding network, thereby improving the stability of peptides. A notable example of peptidomimetics is the clinical trial candidate BMTP-11, which consists of a functional IL-11Rα-binding peptide (cyclic CGRRAGGSC) fused to the apoptosis-inducing peptidomimetic D(KLAKLAK)2, showing translational potential against malignant diseases.139 This peptidomimetic motif induces cell death via mitochondrial membrane disruption upon cell internalization.140 The IL-11 receptor is an established molecular target in osteosarcoma and secondary bone metastases from solid tumors, such as prostate cancer. IL-11 receptor is also a suitable cell surface target for human leukemia and lymphoma. BMTP-11 demonstrated promising antitumor activity in preclinical mouse models of human osteosarcoma141 and lung cancer.142 A Phase I clinical trial of BMTP-11 was completed in patients with castrate-resistant prostate cancer and osteoblastic bone metastases.143 Peptoids, peptidomimetics that contain N-substituted glycines, provide improved proteolytic stability, decreased immunogenicity, and improved cellular permeability.144 A novel conjugate of manganese tetraphenylporphyrin with a mitochondria-targeting peptoid boosted ROS production and induced cytotoxicity in cancer cells, sparing normal fibroblasts.145 Peptoid–peptide hydrogels also showed promising results in long-acting drug delivery systems.146
3.5. Conjugating Novel Functional Payloads
Owing to biodistribution, uptake, and loss of conjugation in circulation, only a small fraction of PDC payloads reach their intracellular target. Thus, the payload must be potent enough to eradicate target cells even at low accumulated concentrations. PDC payloads are typically much more toxic than conventional chemotherapies. Compared to ADCs, PDCs benefit from the good permeability and low MW of peptides, allowing for higher IC50 values for the payloads used. PDCs also offer a wider range of payload options than ADCs (Figure 9), including radionuclides such as 177Lu, 111In, and 90Y.
Figure 9.
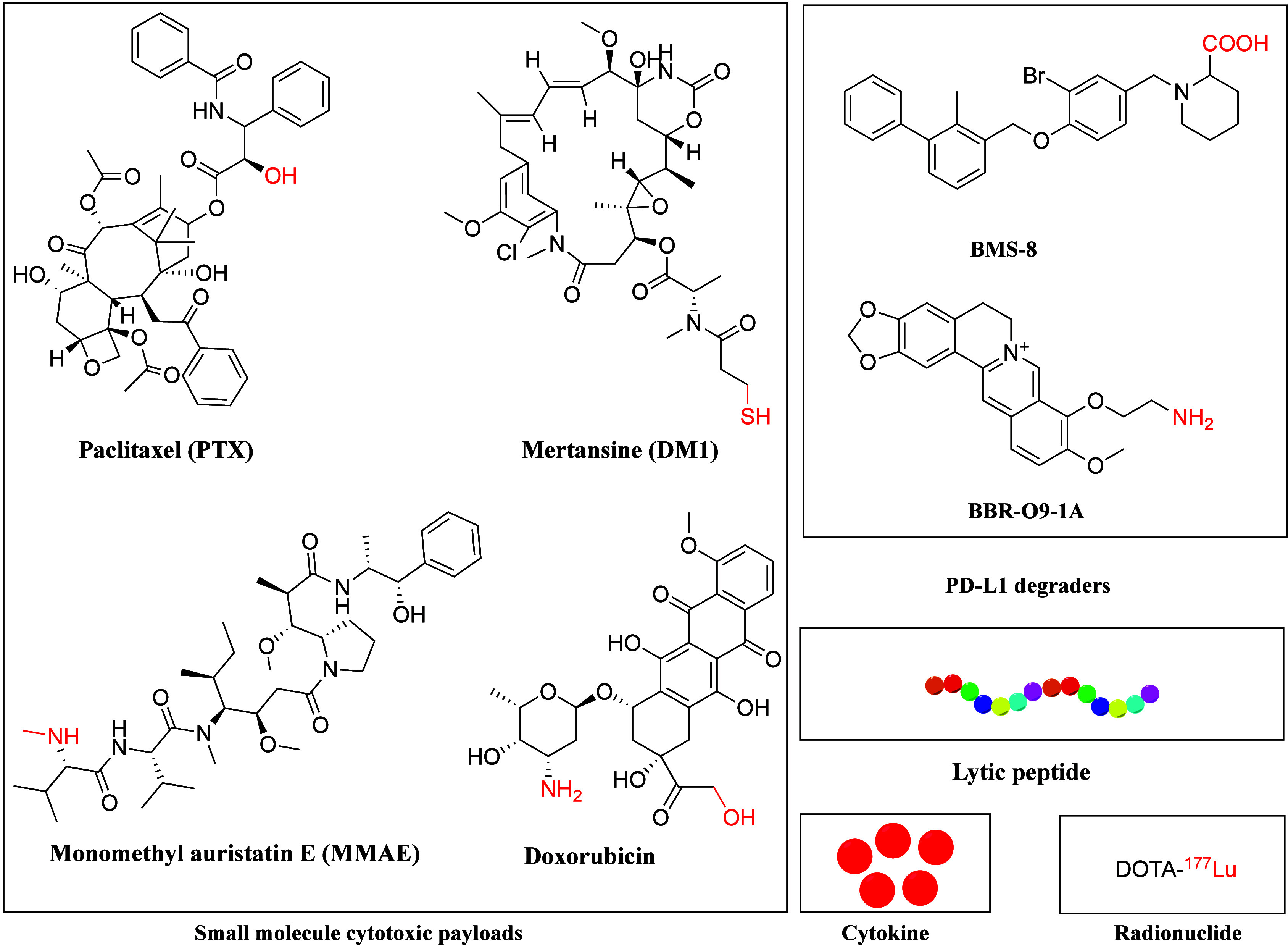
Chemical structures of representative payloads of PDCs. The payload and linker conjugating functional groups are highlighted in red.
In addition to the potency of payload, drug loading markedly impacts the release rate of the free drug, intracellular accumulation, and ultimately, the bioactivity of the PDCs. High drug loading is often desirable for low-potency drugs and may also be beneficial for the treatment of drug-resistant tumors. While high drug loading improves intracellular accumulation, it can slow the release rate of the free drug from the conjugate. Conjugates with balanced cellular uptake and drug release rates are most efficient in killing cancer cells.147
Traditional cytotoxic payloads for PDCs include microtubule-targeting drugs like PTX, antimitotic agents such as MMAE and maytansine derivatives (DM1), DNA-damaging agents like pyrrolobenzodiazepine dimers, and topoisomerase I inhibitors such as SN-38. Recently, novel payloads have emerged, such as cytokines, lytic peptides, toxic proteins, proteolysis-targeting chimeras, oligonucleotides, and PD-L1 degraders (Figure 9).
PD-L1 is an essential immune checkpoint receptor exploited by cancer cells to evade immune surveillance. Increasing evidence suggests that degradation of PD-L1 improves the effectiveness of cancer immunotherapy.148,149 By conjugating the PD-L1 degrader BMS-8 with cRGD, BMS-L1-RGD exhibited improved PD-L1 degradation and tumor suppression effects with minimal side effects in a melanoma mouse model.150 Another novel PDC (PMT-O9-1A) was synthesized by conjugating an anti-PD-L1 peptide with the PD-L1 degrader palmatine via a disulfide linker. PMT-O9-1A substantially mitigated PD-L1 expression and improved its treatment efficacy through both external PD-L1 blockade and internal PD-L1 degradation mechanisms in lung cancer.151
Lytic peptides, usually composed of a 15-amino acid sequence containing leucine and lysine, eliminate cancer cells by disrupting the phospholipid bilayer of cell membranes. However, their application is limited by nonspecific toxicity. By conjugating an EGFR-targeting peptide or interleukin-4 receptor–targeting peptide with a lytic peptide via a three-glycine linker, these novel hybrid peptides displayed remarkable antitumor activity.152,153 QR-KLU is a novel PDC with a VEGFR-targeting peptide conjugated to a lytic peptide. QR-KLU showed strong antitumor effects154 and enhanced the efficacy of anti-PD-1 antibodies with good safety in hepatocellular carcinoma (HCC).155
EP-100 consists of a hormone-based peptide linked to a cationic α-helical lytic peptide (ClIP-71) without a linker. The α-helical domain of ClIP-71 interacts with negatively charged membranes, causing cell death through membrane lysis.156 EP-100 was well tolerated in patients with advanced, LHRH-receptor-expressing solid tumors.157 Even though EP-100 combined with PTX showed a similar ORR to PTX alone in patients with refractory or recurrent ovarian cancer, a subset of patients with liver metastases appeared to benefit from the combination.158 A mechanism study revealed that EP-100 increased PD-L1 levels and enhanced immunotherapy, particularly in combination with an anti-PD-L1 antibody in ovarian cancer.159
Tumor necrosis factor alpha (TNF-α) is a cytokine that initiates apoptosis and regulates cell survival and proliferation. NGR-hTNF comprises a CD13-directed tumor-homing peptide (sequence: CNGRC) fused to human TNF-α. CD13, also called aminopeptidase N, is overexpressed on tumor blood vessels. Neovasculature plays a crucial role in the development and metastasis of tumors, making targeting neovasculature a promising drug delivery method.160 Effective drug delivery and penetration into neoplastic cells distant from tumor vessels are critical for successful chemotherapy in solid tumors. Malignant pleural mesothelioma (MPM) is an aggressive cancer with highly vascularized tumors, poor prognosis, and limited treatment options after first-line chemotherapy failure, with no standard second-line therapy. A Phase Ib study of NGR-hTNF combined with doxorubicin showed promising activity in patients with advanced solid tumors.161 The NGR peptide targets tumor neovasculature, while hTNF alters endothelial barrier function and tumor interstitial pressure, enhancing vascular permeability and penetration of doxorubicin in tumors. NGR-hTNF was well tolerated, with no grade 3 to 4 toxicities in metastatic colorectal cancer (CRC),162 advanced HCC,163 and MPM164 in Phase II studies. In combination with capecitabine, oxaliplatin, or doxorubicin, NGR-hTNF showed manageable toxicity and promising antitumor activity in patients with advanced CRC,165 relapsed ovarian cancer,166 small cell lung cancer (SCLC),167 and other refractory solid tumors.168 A randomized, double-blind Phase III study (NGR015) comparing NGR-hTNF plus best investigator’s choice (BIC) versus placebo plus BIC in previously treated patients with advanced MPM (NCT01098266) showed no prominent difference in OS between groups. However, a noticeable interaction between a short treatment-free interval and improved OS and PFS was observed.169
4. PDC Clinical Applications
In addition to the marketed Lutathera and Pluvicto, diverse PDC drugs are under active investigation in preclinical or clinical trials (Table 1).
Luteinizing hormone-releasing hormone (LHRH, also GnRH) is a hormone-based peptide released by the hypothalamus. LHRH receptor is overexpressed in ovarian cancer, endometrium cancer (EC), prostate cancer, breast cancer, etc. AEZS-108 (zoptarelin doxorubicin, AN-152, and ZEN-008) is composed of doxorubicin chemically linked to the carrier LHRH agonist, targeting the LHRH receptor. The amide linker can be selectively cleaved by amidases in the lysosomes. The ovary and endometrium are hormone-dependent organs. There is no clear standard of care for advanced or recurrent EC following platinum-based therapy. As binding sites are present on tumors in higher concentrations than on most normal tissues, these receptors represent a specific target for AEZS-108.170 A multicenter Phase II trial demonstrated that AEZS-108 has excellent activity and low toxicity in women with advanced or recurrent LHRH receptor-positive EC.171 Another Phase II trial of AEZS-108 showed an acceptable safety profile and anticancer activity in castration- and taxane-resistant prostate cancer.172 However, a Phase III randomized controlled study (NCT01767155) found that AEZS-108 did not improve OS and PFS compared with doxorubicin or dostarlimab as second-line therapy for locally advanced, recurrent, or metastatic EC.173 Recently, another group proposed that the lack of efficacy was owing to linker instability and payload potency. After replacing the linker and payload, a novel PDC (d-Cys6-LHRH vedotin) showed markedly superior bioactivity and selectivity over AEZS-108 (GI50 4 vs 453 nM) in ovarian cancers.174
G-202 (mipsagargin) contains a peptide (DγEγEγEE) targeting PSMA linked to the small-molecule payload thapsigargin. A Phase II, multicenter, single-arm study of G-202 showed antitumor activity and prolonged disease stabilization as second-line therapy following sorafenib in adult patients with progressive advanced HCC.175 High expression of sortilin (SORT1) was found in most clinically annotated breast cancer, ovarian, and endometrial tumors. Somatostatin is a hormone that inhibits the release of other hormones and regulates digestion and cell growth. Somatostatin receptor 2 (SSTR2) is frequently overexpressed in several types of solid tumors. To overcome the limitations of natural somatostatin, several somatostatin analogues have been synthesized, such as Lanreotide, Vapreotide, and Octreotide. PEN-221 is a conjugate consisting of the microtubule-targeting agent DM1 linked to the C-terminal side chain of octreotide, targeting SSTR2. The reducible disulfide linker is chemically cleavable, and the excessive reductive environment in tumor cells facilitates the release of the free drug at the cancer site. The intracellular glutathione concentration is much higher than the extracellular concentration in cancer cells. PEN-221 showed antitumor activity in xenograft mouse models and in patients with SSTR2-positive SCLC.176
TH1902 (Sudocetaxel Zendusortide) is a newly developed SORT1-targeted peptide-docetaxel conjugate, showing superior anticancer activity compared to unconjugated docetaxel in SORT1-positive ovarian and triple-negative breast cancer xenograft mouse models.177 A recent mechanism study found that TH1902 triggers the cGAS/STING pathway, enhancing anti-PD-L1 immune-mediated tumor cell killing.178 This is the first evidence that TH1902 exerts its antitumor activity, in part, through modulation of the immune tumor microenvironment.
5. Conclusions and Prospectives
PDCs possess unique characteristics that make them highly desirable as pharmaceutical agents. Despite their considerable success, their widespread clinical use remains limited, partly owing to their insufficient cell-targeting ability and susceptibility to enzymatic degradation. Cell-targeting ability can be considerably improved through multivalency, bispecific, or halogenation strategies. Stability and circulation half-life can be improved by cyclic, bicyclic, albumin-binding, d-amino acids or unnatural amino acid substitution, N-methylation, peptidomimetics, or peptoid formation strategies. However, each strategy has limitations, and no single approach can achieve all the desirable characteristics of an ideal PDC. While d-amino acid substitution improves the proteolytic stability of peptides, it may also compromise the biological activity of modified PDCs. The development of efficient CPPs that overcome biological barriers has been demonstrated to improve the treatment efficacy of PDCs. Combining CTPs with CPPs or SAPs with SRPs has also substantially improved the bioactivity of PDCs.
In recent years, continuous research has focused on developing novel treatment strategies and combinations to improve outcomes for various diseases. Bacteria loaded with aptamer–drug conjugates (ApDC) have considerably enhanced the treatment efficacy in pancreatic cancer.179 This synergistic strategy combines the penetrative capability of bacteria with the targeting and toxic effects of ApDC by conjugating the ApDC to bacteria through straightforward, one-step click chemistry. Bacteria prolong the serum stability of ApDC for up to 48 h, resulting in increased drug concentration at tumor sites. By utilizing the characteristics of tumors and the different mechanisms of drug entry into cells, novel PDCs can be developed to improve therapeutic efficacy and reduce drug resistance. The Kirsten rat sarcoma viral oncogene homologue (KRAS) mutation remains nontargetable owing to the lack of conventional drug-binding sites. Drugs bound to albumin can exploit the macropinocytic features of KRAS mutant cancer to enable pan-KRAS tumor targeting. A novel macropinocytosis-targeting PDC has been developed to treat KRAS mutant cancers, showing escalated apoptosis and enhanced payload accumulation within tumors.180 Recently, covalent peptide with unnatural amino acid that can target noncysteine residues (such as histidine181) and bind to flat protein surfaces of undruggable targets.182 PDC containing the covalent peptide have the potential to benefit for the therapeutics effects in cancer. In summary, the field of PDCs is rapidly expanding in the pharmaceutical industry. The key roles of peptide design, linker chemistry, and novel payloads in PDCs in enhancing treatment efficacy make this area worth exploring as our understanding of cell biology grows.
Acknowledgments
This work was supported by National Natural Science Foundation of China (no. 82473266, no. 82160129), Cuiying Scientific and Technological Innovation Program of Lanzhou University Second Hospital (no. CY2021-MS-B09 and no. CY2023-ZD-01), Key Talents Project of Gansu Province (no. 2019RCXM020), Science and Technology Project of Chengguan District of Lanzhou City (no. 2020JSCX0073), Gansu Provincial Department of Education Project (no. 2022B-034), Gansu Provincial Science and Technology Major Project (no. 24ZDFA011), Gansu Provincial Innovation Fund for Small and Medium-Sized Technology-based Firms (no. 24CXGA107).
Glossary
Abbreviations Used
- ADC
antibody–drug conjugate
- Aib
amino-isobutyric acid
- ApDC
aptamer–drug conjugate
- BIC
best investigator’s choice
- CPP
cell-penetrating peptide
- CRC
colorectal cancer
- CTP
cell-targeting peptide
- DM1
maytansine derivatives
- DOTA
tetraazacyclododecane-tetraacetic acid
- DOX
doxorubicin
- EC
endometrium cancer
- EphA2
ephrin type-A receptor 2
- GEP-NETs
gastroenteropancreatic neuroendocrine tumors
- HA
hyaluronic acid
- HCC
hepatocellular carcinoma
- HER2
human epidermal growth factor receptor 2
- KDC
knottin peptide–drug conjugate
- KRAS
Kirsten rat sarcoma viral oncogene homologue
- LRP1
low-density lipoprotein receptor-related protein-1
- 177Lu
lutetium-177
- mAb
monoclonal antibody
- MMAE
monomethylauristatin E
- MPM
malignant pleural mesothelioma
- OS
overall survival
- PDC
peptide–drug conjugate
- PD-L1
programmed cell death ligand-1
- PFS
progression-free survival
- pHLIP
pH-low insertion peptides
- PPD
polypeptide–polyethylene glycol derivative
- PSMA
prostate-specific membrane antigen
- PTX
paclitaxel
- RGD
arginine–glycine–aspartic acid
- SAP
self-assembling peptide
- SCLC
small cell lung cancer
- SORT1
sortilin
- SPAAC
strain-promoted azide–alkyne cycloaddition
- SRP
stimuli-responsive peptide
- SSTR2
somatostatin receptor 2
- TAT
transcription activator protein
- TATE
tyrosine-containing somatostatin analog Tyr3-octreotate
- tEB
truncated Evans Blue
- TfR
transferrin receptor
Biographies
Jiahui Ma is a graduate student in the Second Clinical Medical School at Lanzhou University. She obtained her Bachelor’s degree in Radiology at the Medical School of Suzhou University. Her research focuses on the bioactivity of PDC drugs.
Xuedan Wang obtained her Bachelor’s degree in Pharmaceutical Engineering at School of Life Sciences and Engineering, Lanzhou University of Technology. She is a graduate student at the Lanzhou University of Technology and also training at the Shenzhen DIVBIO Pharmaceutical. Her research focuses on the structural design and synthesis of PDC drugs.
Yonghua Hu is a Ph.D. student in the Second Clinical Medical School at Lanzhou University. She obtained her Bachelor’s and Master degree in the Medical School at Lanzhou University. She is an Associate Professor at the Gansu University of Chinese Medicine. Her research focuses on the traditional Chinese medicine and pharmaceutical sciences.
Jianping Ma received her Bachelor’s degree, Master degree, and Ph.D. in Organic Chemistry from the Lanzhou University. She is a Professor of Pharmaceutical Sciences at the Lanzhou University of Technology. Her research focuses on the natural product and pharmaceutical engineering.
Yaping Ma obtained his Ph.D. degree in Organic Chemistry at Chinese Academy of Sciences. He is currently the CEO of Shenzhen DIVBIO Pharmaceutical. He served successively as Vice President and Chief Technology Officer of Chengdu DiAo Pharmaceutical Group, Hainan Zhonghe Group, and Shenzhen Hanyu Pharmaceutical from 2006 to 2016. He is the technical leader in the development of a series of peptide drugs and the organizer for the formation of key technologies.
Hao Chen is Chief Surgeon and Professor in the tumor center of Lanzhou University Second Hospital. He obtained his Ph.D. degree from the University of Florida. He worked as research assistant professor and associated professor at the Vascular Surgery Department of the University of Florida after three years postdoc training there. He mainly engaged in clinical, teaching, and scientific research of digestive system tumors.
Zhijian Han is an Associate Professor of Pharmaceutical Sciences at the Second Hospital of Lanzhou University. He holds a Ph.D. in Biochemistry and Master’s degree in Organic Chemistry from the Lanzhou University. As a group leader, he worked in the Basilea Pharmaceutia for seven years, where he was responsible for the synthesis and scale-up of small molecular lead compounds. He also worked as a visiting scholar at the University of California Riverside. His research interests include small molecules and PDCs against tumors.
Author Contributions
⊥ Jiahui Ma, Xuedan Wang, and Yonghua Hu contributed equally.
The authors declare no competing financial interest.
References
- Zhang B.; Wang M.; Sun L.; Liu J.; Yin L.; Xia M.; Zhang L.; Liu X.; Cheng Y. Recent Advances in Targeted Cancer Therapy: Are PDCs the Next Generation of ADCs?. J. Med. Chem. 2024, 67, 11469–11487. 10.1021/acs.jmedchem.4c00106. [DOI] [PubMed] [Google Scholar]
- Fu C.; Yu L.; Miao Y.; Liu X.; Yu Z.; Wei M. Peptide-drug conjugates (PDCs): a novel trend of research and development on targeted therapy, hype or hope?. Acta Pharm. Sin B 2023, 13, 498–516. 10.1016/j.apsb.2022.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi S. F. A.; Zhang L.; Zhang H.; Fang Q. Peptide-Drug Conjugates: Design, Chemistry, and Drug Delivery System as a Novel Cancer Theranostic. ACS Pharmacol Transl Sci. 2024, 7, 309–334. 10.1021/acsptsci.3c00269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong L.; Zhao H.; Liu Y.; Wu H.; Liu C.; Chang S.; Chen L.; Jin M.; Wang Q.; Gao Z.; Huang W. Research advances in peptide–drug conjugates. Acta Pharm. Sin B 2023, 13, 3659–3677. 10.1016/j.apsb.2023.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S.; Al-Toubah T.; El-Haddad G.; Strosberg J. (177)Lu-DOTATATE for the treatment of gastroenteropancreatic neuroendocrine tumors. Expert Rev. Gastroenterol Hepatol 2019, 13, 1023–1031. 10.1080/17474124.2019.1685381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S.; Halperin D.; Myrehaug S.; Herrmann K.; Pavel M.; Kunz P. L.; Chasen B.; Tafuto S.; Lastoria S.; Capdevila J.; García-Burillo A.; Oh D. Y.; Yoo C.; Halfdanarson T. R.; Falk S.; Folitar I.; Zhang Y.; Aimone P.; de Herder W. W.; Ferone D. [(177)Lu]Lu-DOTA-TATE plus long-acting octreotide versus high-dose long-acting octreotide for the treatment of newly diagnosed, advanced grade 2–3, well-differentiated, gastroenteropancreatic neuroendocrine tumours (NETTER-2): an open-label, randomised, phase 3 study. Lancet 2024, 403, 2807–2817. 10.1016/S0140-6736(24)00701-3. [DOI] [PubMed] [Google Scholar]
- Dean T. T.; Jelú-Reyes J.; Allen A. C.; Moore T. W. Peptide-Drug Conjugates: An Emerging Direction for the Next Generation of Peptide Therapeutics. J. Med. Chem. 2024, 67, 1641–1661. 10.1021/acs.jmedchem.3c01835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng Q.; Sun X.; Gong T.; Zhang Z. R. Peptide-drug conjugate linked via a disulfide bond for kidney targeted drug delivery. Bioconjug Chem. 2012, 23, 1200–10. 10.1021/bc300020f. [DOI] [PubMed] [Google Scholar]
- Doherty G. J.; McMahon H. T. Mechanisms of endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. 10.1146/annurev.biochem.78.081307.110540. [DOI] [PubMed] [Google Scholar]
- Kaksonen M.; Roux A. Mechanisms of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2018, 19, 313–326. 10.1038/nrm.2017.132. [DOI] [PubMed] [Google Scholar]
- Allolio C.; Magarkar A.; Jurkiewicz P.; Baxová K.; Javanainen M.; Mason P. E.; Šachl R.; Cebecauer M.; Hof M.; Horinek D.; Heinz V.; Rachel R.; Ziegler C. M.; Schröfel A.; Jungwirth P. Arginine-rich cell-penetrating peptides induce membrane multilamellarity and subsequently enter via formation of a fusion pore. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 11923–11928. 10.1073/pnas.1811520115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Wang N.; Zhang W.; Cheng X.; Yan Z.; Shao G.; Wang X.; Wang R.; Fu C. Therapeutic peptides: current applications and future directions. Signal Transduct Target Ther 2022, 7, 48. 10.1038/s41392-022-00904-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y.; Feng R.; Zhang X.; Wang Z. L.; Xiong F.; Zhang S.; Zhong Z. F.; Yu H.; Zhang Q. W.; Zhang Z.; Wang Y.; Li G. Encoding and display technologies for combinatorial libraries in drug discovery: The coming of age from biology to therapy. Acta Pharm. Sin B 2024, 14, 3362–3384. 10.1016/j.apsb.2024.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colas K.; Bindl D.; Suga H. Selection of Nucleotide-Encoded Mass Libraries of Macrocyclic Peptides for Inaccessible Drug Targets. Chem. Rev. 2024, 124, 12213–12241. 10.1021/acs.chemrev.4c00422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W.; Becker M. L. ″Click″ reactions: a versatile toolbox for the synthesis of peptide-conjugates. Chem. Soc. Rev. 2014, 43, 7013–39. 10.1039/C4CS00139G. [DOI] [PubMed] [Google Scholar]
- Taiariol L.; Chaix C.; Farre C.; Moreau E. Click and Bioorthogonal Chemistry: The Future of Active Targeting of Nanoparticles for Nanomedicines?. Chem. Rev. 2022, 122, 340–384. 10.1021/acs.chemrev.1c00484. [DOI] [PubMed] [Google Scholar]
- Bianchi A.; Arosio D.; Perego P.; De Cesare M.; Carenini N.; Zaffaroni N.; De Matteo M.; Manzoni L. Design, synthesis and biological evaluation of novel dimeric and tetrameric cRGD-paclitaxel conjugates for integrin-assisted drug delivery. Org. Biomol Chem. 2015, 13, 7530–41. 10.1039/C5OB00497G. [DOI] [PubMed] [Google Scholar]
- Luu T.; Gristwood K.; Knight J. C.; Jörg M. Click Chemistry: Reaction Rates and Their Suitability for Biomedical Applications. Bioconjug Chem. 2024, 35, 715–731. 10.1021/acs.bioconjchem.4c00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednarek C.; Wehl I.; Jung N.; Schepers U.; Bräse S. The Staudinger Ligation. Chem. Rev. 2020, 120, 4301–4354. 10.1021/acs.chemrev.9b00665. [DOI] [PubMed] [Google Scholar]
- Bugatti K.; Andreucci E.; Monaco N.; Battistini L.; Peppicelli S.; Ruzzolini J.; Curti C.; Zanardi F.; Bianchini F.; Sartori A. Nintedanib-Containing Dual Conjugates Targeting α(V)β(6) Integrin and Tyrosine Kinase Receptors as Potential Antifibrotic Agents. ACS Omega 2022, 7, 17658–17669. 10.1021/acsomega.2c00535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salem A. F.; Wang S.; Billet S.; Chen J. F.; Udompholkul P.; Gambini L.; Baggio C.; Tseng H. R.; Posadas E. M.; Bhowmick N. A.; Pellecchia M. Reduction of Circulating Cancer Cells and Metastases in Breast-Cancer Models by a Potent EphA2-Agonistic Peptide-Drug Conjugate. J. Med. Chem. 2018, 61, 2052–2061. 10.1021/acs.jmedchem.7b01837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi S. F. A.; Abbas N.; Zhang H.; Fang Q. Identification of a pH-Responsive Peptide-Paclitaxel Conjugate as a Novel Drug with Improved Therapeutic Potential. J. Med. Chem. 2023, 66, 8324–8337. 10.1021/acs.jmedchem.3c00382. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Li Y.; Huang W.; Shi W.; Qian H. Source and exploration of the peptides used to construct peptide-drug conjugates. Eur. J. Med. Chem. 2021, 224, 113712. 10.1016/j.ejmech.2021.113712. [DOI] [PubMed] [Google Scholar]
- Alas M.; Saghaeidehkordi A.; Kaur K. Peptide-Drug Conjugates with Different Linkers for Cancer Therapy. J. Med. Chem. 2021, 64, 216–232. 10.1021/acs.jmedchem.0c01530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Li H.; Gou L.; Li W.; Wang Y. Antibody-drug conjugates: Recent advances in payloads. Acta Pharm. Sin B 2023, 13, 4025–4059. 10.1016/j.apsb.2023.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi M.; Zhu J.; Zhang F.; Shen H.; Chen J.; Xiao Z.; Huangfu Y.; Wu C.; Sun H.; Xia G. Antibody-drug conjugates for targeted cancer therapy: Recent advances in potential payloads. Eur. J. Med. Chem. 2024, 276, 116709. 10.1016/j.ejmech.2024.116709. [DOI] [PubMed] [Google Scholar]
- Cossu J.; Thoreau F.; Boturyn D. Multimeric RGD-Based Strategies for Selective Drug Delivery to Tumor Tissues. Pharmaceutics 2023, 15, 525. 10.3390/pharmaceutics15020525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen M.; Oyen W. J.; Massuger L. F.; Frielink C.; Dijkgraaf I.; Edwards D. S.; Radjopadhye M.; Corstens F. H.; Boerman O. C. Comparison of a monomeric and dimeric radiolabeled RGD-peptide for tumor targeting. Cancer Biother Radiopharm 2002, 17, 641–6. 10.1089/108497802320970244. [DOI] [PubMed] [Google Scholar]
- Garanger E.; Boturyn D.; Coll J. L.; Favrot M. C.; Dumy P. Multivalent RGD synthetic peptides as potent alphaVbeta3 integrin ligands. Org. Biomol Chem. 2006, 4, 1958–65. 10.1039/B517706E. [DOI] [PubMed] [Google Scholar]
- Raposo Moreira Dias A.; Pina A.; Dal Corso A.; Arosio D.; Belvisi L.; Pignataro L.; Caruso M.; Gennari C. Multivalency Increases the Binding Strength of RGD Peptidomimetic-Paclitaxel Conjugates to Integrin α(V) β(3). Chemistry 2017, 23, 14410–14415. 10.1002/chem.201703093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. W.; Stone R. L.; Lee S. J.; Nam E. J.; Roh J. W.; Nick A. M.; Han H. D.; Shahzad M. M.; Kim H. S.; Mangala L. S.; Jennings N. B.; Mao S.; Gooya J.; Jackson D.; Coleman R. L.; Sood A. K. EphA2 targeted chemotherapy using an antibody drug conjugate in endometrial carcinoma. Clin. Cancer Res. 2010, 16, 2562–70. 10.1158/1078-0432.CCR-10-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annunziata C. M.; Kohn E. C.; LoRusso P.; Houston N. D.; Coleman R. L.; Buzoianu M.; Robbie G.; Lechleider R. Phase 1, open-label study of MEDI-547 in patients with relapsed or refractory solid tumors. Invest New Drugs 2013, 31, 77–84. 10.1007/s10637-012-9801-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggineni S.; Mitra S.; Lamberto I.; Han X.; Xu Y.; An J.; Pasquale E. B.; Huang Z. Design and Synthesis of Potent Bivalent Peptide Agonists Targeting the EphA2 Receptor. ACS Med. Chem. Lett. 2013, 4, 344–8. 10.1021/ml3004523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baggio C.; Udompholkul P.; Gambini L.; Pellecchia M. Targefrin: A Potent Agent Targeting the Ligand Binding Domain of EphA2. J. Med. Chem. 2022, 65, 15443–15456. 10.1021/acs.jmedchem.2c01391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X.; Lingerak R.; Herting C. J.; Ge Y.; Kim S.; Toth P.; Wang W.; Brown B. P.; Meiler J.; Sossey-Alaoui K.; Buck M.; Himanen J.; Hambardzumyan D.; Nikolov D. B.; Smith A. W.; Wang B. Time-resolved live-cell spectroscopy reveals EphA2 multimeric assembly. Science 2023, 382, 1042–1050. 10.1126/science.adg5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Zhang D.; Li J.; Li F.; Wei R.; Jiang G.; Xu H.; Wang X.; Zhou Y.; Xi L. A novel ICG-labeled cyclic TMTP1 peptide dimer for sensitive tumor imaging and enhanced photothermal therapy in vivo. Eur. J. Med. Chem. 2022, 227, 113935. 10.1016/j.ejmech.2021.113935. [DOI] [PubMed] [Google Scholar]
- Liu S.; Tian Y.; Jiang S.; Wang Z. A Novel Homodimer Peptide-Drug Conjugate Improves the Efficacy of HER2-Positive Breast Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 4590. 10.3390/ijms24054590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan X.; Le M.; Wang H.; Huo B.; Yu T.; Huang P.; Luan T.; Wen S. Design and synthesis of peptide-drug conjugates to double target EGFR. Phytochemistry Letters 2023, 56, 30–37. 10.1016/j.phytol.2023.06.003. [DOI] [Google Scholar]
- Zhou J.; Bian X.; Kan Z.; Cai Z.; Jiang Y.; Wang Z.; Li Y.; Shi W.; Qian H. In Silico Exploration and Biological Evaluation of Bispecific Peptides Derived from Anti-HER2 Antibodies and Peptide-Camptothecin Conjugates for HER2-Positive Breast Cancer. J. Med. Chem. 2022, 65, 15123–15139. 10.1021/acs.jmedchem.2c00968. [DOI] [PubMed] [Google Scholar]
- Wang W.; Song S.; Jiao N. Late-Stage Halogenation of Complex Substrates with Readily Available Halogenating Reagents. Acc. Chem. Res. 2024, 57, 3161–3181. 10.1021/acs.accounts.4c00501. [DOI] [PubMed] [Google Scholar]
- Sun H.; Keefer C. E.; Scott D. O. Systematic and pairwise analysis of the effects of aromatic halogenation and trifluoromethyl substitution on human liver microsomal clearance. Drug Metab Lett. 2011, 5, 232. 10.2174/187231211798472575. [DOI] [PubMed] [Google Scholar]
- Mondal H.; Patra S.; Saha S.; Nayak T.; Sengupta U.; Sudan Maji M. Late-Stage Halogenation of Peptides, Drugs and (Hetero)aromatic Compounds with a Nucleophilic Hydrazide Catalyst. Angew. Chem., Int. Ed. Engl. 2023, 62, e202312597 10.1002/anie.202312597. [DOI] [PubMed] [Google Scholar]
- Schnepel C.; Moritzer A. C.; Gäfe S.; Montua N.; Minges H.; Nieß A.; Niemann H. H.; Sewald N. Enzymatic Late-Stage Halogenation of Peptides. Chembiochem 2023, 24, e202200569 10.1002/cbic.202200569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sana B.; Ke D.; Li E. H. Y.; Ho T.; Seayad J.; Duong H. A.; Ghadessy F. J. Halogenation of Peptides and Proteins Using Engineered Tryptophan Halogenase Enzymes. Biomolecules 2022, 12, 1841. 10.3390/biom12121841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig A. J.; Ermolovich Y.; Cameron A.; Rodler A.; Wang H.; Hawkes J. A.; Hubert M.; Björkling F.; Molchanova N.; Brimble M. A.; Moodie L. W. K.; Svenson J. Antimicrobial Peptides Incorporating Halogenated Marine-Derived Amino Acid Substituents. ACS Med. Chem. Lett. 2023, 14, 802–809. 10.1021/acsmedchemlett.3c00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemker I.; Schröder D. C.; Feiner R. C.; Müller K. M.; Marion A.; Sewald N. Tuning the Biological Activity of RGD Peptides with Halotryptophans†. J. Med. Chem. 2021, 64, 586–601. 10.1021/acs.jmedchem.0c01536. [DOI] [PubMed] [Google Scholar]
- Weiss C.; Figueras E.; Borbely A. N.; Sewald N. Cryptophycins: cytotoxic cyclodepsipeptides with potential for tumor targeting. J. Pept Sci. 2017, 23, 514–531. 10.1002/psc.3015. [DOI] [PubMed] [Google Scholar]
- Molchanova N.; Nielsen J. E.; So̷rensen K. B.; Prabhala B. K.; Hansen P. R.; Lund R.; Barron A. E.; Jenssen H. Halogenation as a tool to tune antimicrobial activity of peptoids. Sci. Rep 2020, 10, 14805. 10.1038/s41598-020-71771-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L.; Kuipers O. P.; Broos J. Facile Halogenation of Antimicrobial Peptides As Demonstrated by Producing Bromotryptophan-Labeled Nisin Variants with Enhanced Antimicrobial Activity. J. Nat. Prod 2024, 87, 1548–1555. 10.1021/acs.jnatprod.4c00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheetham A. G.; Zhang P.; Lin Y. A.; Lock L. L.; Cui H. Supramolecular nanostructures formed by anticancer drug assembly. J. Am. Chem. Soc. 2013, 135, 2907–10. 10.1021/ja3115983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y.; Zhang Z.; Cao Y.; Yu Z. Stimulus-Responsive Amino Acids Behind In Situ Assembled Bioactive Peptide Materials. Chembiochem 2023, 24, e202200497 10.1002/cbic.202200497. [DOI] [PubMed] [Google Scholar]
- Hou M.; Liu S. Recent Progress of pH-Responsive Peptides, Polypeptides, and Their Supramolecular Assemblies for Biomedical Applications. Biomacromolecules 2024, 25, 5402–5416. 10.1021/acs.biomac.4c00688. [DOI] [PubMed] [Google Scholar]
- Wyatt L. C.; Moshnikova A.; Crawford T.; Engelman D. M.; Andreev O. A.; Reshetnyak Y. K. Peptides of pHLIP family for targeted intracellular and extracellular delivery of cargo molecules to tumors. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E2811. 10.1073/pnas.1715350115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt J. F.; Rath P.; Rothschild K. J.; Engelman D. M. Spontaneous, pH-dependent membrane insertion of a transbilayer alpha-helix. Biochemistry 1997, 36, 15177–92. 10.1021/bi970147b. [DOI] [PubMed] [Google Scholar]
- Reshetnyak Y. K.; Andreev O. A.; Engelman D. M. Aiming the magic bullet: targeted delivery of imaging and therapeutic agents to solid tumors by pHLIP peptides. Front Pharmacol 2024, 15, 1355893. 10.3389/fphar.2024.1355893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo U.; Murai Y.; Agama K. K.; Sun Y.; Saha L. K.; Yang X.; Arakawa Y.; Gayle S.; Jones K.; Paralkar V.; Sundaram R. K.; Van Doorn J.; Vasquez J. C.; Bindra R. S.; Choi W. S.; Pommier Y. TOP1-DNA Trapping by Exatecan and Combination Therapy with ATR Inhibitor. Mol. Cancer Ther 2022, 21, 1090–1102. 10.1158/1535-7163.MCT-21-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayle S.; Paradis T.; Jones K.; Vasquez J.; Paralkar V. M. Antigen-independent tumor targeting by CBX-12 (alphalex()-exatecan) induces long-term antitumor immunity. Immunotherapy 2022, 14, 1467–1480. 10.2217/imt-2022-0121. [DOI] [PubMed] [Google Scholar]
- Wang M. D.; Hou D. Y.; Lv G. T.; Li R. X.; Hu X. J.; Wang Z. J.; Zhang N. Y.; Yi L.; Xu W. H.; Wang H. Targeted in situ self-assembly augments peptide drug conjugate cell-entry efficiency. Biomaterials 2021, 278, 121139. 10.1016/j.biomaterials.2021.121139. [DOI] [PubMed] [Google Scholar]
- Ji L.; Li Z.-X.; Cheng D.-B.; Zhang X.-H.; Liu J.; Yang Z.; Qiao Z.-Y.; Wang H. Reactive Oxygen Species-Responsive Peptide-Drug Conjugate for Mitochondria-Specific Chemotherapy. ChemNanoMat 2022, 8, e202100532 10.1002/cnma.202100532. [DOI] [Google Scholar]
- Wang J.; Qian Y.; Xu L.; Shao Y.; Zhang H.; Shi F.; Chen J.; Cui S.; Chen X.; Zhu D.; Hu R.; Chen Z. Hyaluronic acid-shelled, peptide drug conjugate-cored nanomedicine for the treatment of hepatocellular carcinoma. Mater. Sci. Eng. C Mater. Biol. Appl. 2020, 117, 111261. 10.1016/j.msec.2020.111261. [DOI] [PubMed] [Google Scholar]
- Bokatyi A. N.; Dubashynskaya N. V.; Skorik Y. A. Chemical modification of hyaluronic acid as a strategy for the development of advanced drug delivery systems. Carbohydr. Polym. 2024, 337, 122145. 10.1016/j.carbpol.2024.122145. [DOI] [PubMed] [Google Scholar]
- Song M.; Liang Y.; Li K.; Zhang J.; Zhang N.; Tian B.; Han J. Hyaluronic acid modified liposomes for targeted delivery of doxorubicin and paclitaxel to CD44 overexpressing tumor cells with improved dual-drugs synergistic effect. Journal of Drug Delivery Science and Technology 2019, 53, 101179. 10.1016/j.jddst.2019.101179. [DOI] [Google Scholar]
- Chen R.; Wang J.; Qian C.; Ji Y.; Zhu C.; Wu L.; Li W.; Bi X.; Wang Y.; Cao G.; Chen Z. From Nanofibers to Nanorods: Nanostructure of Peptide-Drug Conjugates Regulated by Polypeptide-PEG Derivative and Enhanced Antitumor Effect. Adv. Funct. Mater. 2019, 29, 1806058. 10.1002/adfm.201806058. [DOI] [Google Scholar]
- Hao T.; Fu Y.; Yang Y.; Yang S.; Liu J.; Tang J.; Ridwan K. A.; Teng Y.; Liu Z.; Li J.; Guo N.; Yu P. Tumor vasculature-targeting PEGylated peptide-drug conjugate prodrug nanoparticles improve chemotherapy and prevent tumor metastasis. Eur. J. Med. Chem. 2021, 219, 113430. 10.1016/j.ejmech.2021.113430. [DOI] [PubMed] [Google Scholar]
- Lopuszynski J.; Wang J.; Zahid M. Beyond Transduction: Anti-Inflammatory Effects of Cell Penetrating Peptides. Molecules 2024, 29, 4088. 10.3390/molecules29174088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell D. J.; Kim D. T.; Steinman L.; Fathman C. G.; Rothbard J. B. Polyarginine enters cells more efficiently than other polycationic homopolymers. J. Pept Res. 2000, 56, 318–25. 10.1034/j.1399-3011.2000.00723.x. [DOI] [PubMed] [Google Scholar]
- Amantana A.; Moulton H. M.; Cate M. L.; Reddy M. T.; Whitehead T.; Hassinger J. N.; Youngblood D. S.; Iversen P. L. Pharmacokinetics, biodistribution, stability and toxicity of a cell-penetrating peptide-morpholino oligomer conjugate. Bioconjug Chem. 2007, 18, 1325–31. 10.1021/bc070060v. [DOI] [PubMed] [Google Scholar]
- Andreev K.; Martynowycz M. W.; Ivankin A.; Huang M. L.; Kuzmenko I.; Meron M.; Lin B.; Kirshenbaum K.; Gidalevitz D. Cyclization Improves Membrane Permeation by Antimicrobial Peptoids. Langmuir 2016, 32, 12905–12913. 10.1021/acs.langmuir.6b03477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Li Z.; Aispuro D.; Guevara N.; Van Valkenburgh J.; Chen B.; Zhou X.; McCarroll M. N.; Ji F.; Cong X.; Sarkar P.; Chaudhuri R.; Guo Z.; Perkins N. P.; Shao S.; Sello J. K.; Chen K.; Xue M. Hydroxyl-Rich Hydrophilic Endocytosis-Promoting Peptide with No Positive Charge. J. Am. Chem. Soc. 2022, 144, 20288–20297. 10.1021/jacs.2c07420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss S.; Adair L. D.; Achazi K.; Kim H.; Bergemann S.; Bartenschlager R.; New E. J.; Rademann J.; Nitsche C. Cell-Penetrating Peptide-Bismuth Bicycles. Angew. Chem., Int. Ed. Engl. 2024, 63, e202318615 10.1002/anie.202318615. [DOI] [PubMed] [Google Scholar]
- John C. M.; Otala S. A.; Jarvis G. A. Cyclization increases bactericidal activity of arginine-rich cationic cell-penetrating peptide for Neisseria gonorrhoeae. Microbiol Spectr 2024, 12, e0099724 10.1128/spectrum.00997-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae D. H.; Bae H.; Yu H. S.; Dorjsembe B.; No Y. H.; Kim T.; Kim N. H.; Kim J. W.; Kim J.; Lee B. S.; Kim Y. J.; Park S.; Khaleel Z. H.; Sa D. H.; Lee E. C.; Lee J.; Ham J.; Kim J. C.; Kim Y. H. Peptide-Drug Conjugate with Statistically Designed Transcellular Peptide for Psoriasis-Like Inflammation. Adv. Healthc Mater. 2024, 13, e2303480 10.1002/adhm.202303480. [DOI] [PubMed] [Google Scholar]
- Cheng H.; Zhu J. Y.; Xu X. D.; Qiu W. X.; Lei Q.; Han K.; Cheng Y. J.; Zhang X. Z. Activable Cell-Penetrating Peptide Conjugated Prodrug for Tumor Targeted Drug Delivery. ACS Appl. Mater. Interfaces 2015, 7, 16061–9. 10.1021/acsami.5b04517. [DOI] [PubMed] [Google Scholar]
- Zhou X.; Smith Q. R.; Liu X. Brain penetrating peptides and peptide-drug conjugates to overcome the blood-brain barrier and target CNS diseases. Wiley Interdiscip Rev. Nanomed Nanobiotechnol 2021, 13, e1695 10.1002/wnan.1695. [DOI] [PubMed] [Google Scholar]
- Demeule M.; Régina A.; Ché C.; Poirier J.; Nguyen T.; Gabathuler R.; Castaigne J. P.; Béliveau R. Identification and design of peptides as a new drug delivery system for the brain. J. Pharmacol Exp Ther 2008, 324, 1064–72. 10.1124/jpet.107.131318. [DOI] [PubMed] [Google Scholar]
- Régina A.; Demeule M.; Ché C.; Lavallée I.; Poirier J.; Gabathuler R.; Béliveau R.; Castaigne J. P. Antitumour activity of ANG1005, a conjugate between paclitaxel and the new brain delivery vector Angiopep-2. Br. J. Pharmacol. 2008, 155, 185–97. 10.1038/bjp.2008.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drappatz J.; Brenner A.; Wong E. T.; Eichler A.; Schiff D.; Groves M. D.; Mikkelsen T.; Rosenfeld S.; Sarantopoulos J.; Meyers C. A.; Fielding R. M.; Elian K.; Wang X.; Lawrence B.; Shing M.; Kelsey S.; Castaigne J. P.; Wen P. Y. Phase I study of GRN1005 in recurrent malignant glioma. Clin. Cancer Res. 2013, 19, 1567–76. 10.1158/1078-0432.CCR-12-2481. [DOI] [PubMed] [Google Scholar]
- Kumthekar P.; Tang S. C.; Brenner A. J.; Kesari S.; Piccioni D. E.; Anders C.; Carrillo J.; Chalasani P.; Kabos P.; Puhalla S.; Tkaczuk K.; Garcia A. A.; Ahluwalia M. S.; Wefel J. S.; Lakhani N.; Ibrahim N. ANG1005, a Brain-Penetrating Peptide-Drug Conjugate, Shows Activity in Patients with Breast Cancer with Leptomeningeal Carcinomatosis and Recurrent Brain Metastases. Clin. Cancer Res. 2020, 26, 2789–2799. 10.1158/1078-0432.CCR-19-3258. [DOI] [PubMed] [Google Scholar]
- Bataille Backer P.; Adekiya T. A.; Kim Y.; Reid T. R.; Thomas M.; Adesina S. K. Development of a Targeted SN-38-Conjugate for the Treatment of Glioblastoma. ACS Omega 2024, 9, 2615–2628. 10.1021/acsomega.3c07486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dissanayake S.; Denny W. A.; Gamage S.; Sarojini V. Recent developments in anticancer drug delivery using cell penetrating and tumor targeting peptides. J. Controlled Release 2017, 250, 62–76. 10.1016/j.jconrel.2017.02.006. [DOI] [PubMed] [Google Scholar]
- Mansur A. A. P.; Carvalho S. M.; Lobato Z. I. P.; Leite M. F.; Cunha A. D. S. Jr.; Mansur H. S. Design and Development of Polysaccharide-Doxorubicin-Peptide Bioconjugates for Dual Synergistic Effects of Integrin-Targeted and Cell-Penetrating Peptides for Cancer Chemotherapy. Bioconjug Chem. 2018, 29, 1973–2000. 10.1021/acs.bioconjchem.8b00208. [DOI] [PubMed] [Google Scholar]
- Feni L.; Parente S.; Robert C.; Gazzola S.; Arosio D.; Piarulli U.; Neundorf I. Kiss and Run: Promoting Effective and Targeted Cellular Uptake of a Drug Delivery Vehicle Composed of an Integrin-Targeting Diketopiperazine Peptidomimetic and a Cell-Penetrating Peptide. Bioconjug Chem. 2019, 30, 2011–2022. 10.1021/acs.bioconjchem.9b00292. [DOI] [PubMed] [Google Scholar]
- Deng X.; Mai R.; Zhang C.; Yu D.; Ren Y.; Li G.; Cheng B.; Li L.; Yu Z.; Chen J. Discovery of novel cell-penetrating and tumor-targeting peptide-drug conjugate (PDC) for programmable delivery of paclitaxel and cancer treatment. Eur. J. Med. Chem. 2021, 213, 113050. 10.1016/j.ejmech.2020.113050. [DOI] [PubMed] [Google Scholar]
- Park S. E.; El-Sayed N. S.; Shamloo K.; Lohan S.; Kumar S.; Sajid M. I.; Tiwari R. K. Targeted Delivery of Cabazitaxel Using Cyclic Cell-Penetrating Peptide and Biomarkers of Extracellular Matrix for Prostate and Breast Cancer Therapy. Bioconjug Chem. 2021, 32, 1898–1914. 10.1021/acs.bioconjchem.1c00319. [DOI] [PubMed] [Google Scholar]
- Kim H.; Hwang D.; Choi M.; Lee S.; Kang S.; Lee Y.; Kim S.; Chung J.; Jon S. Antibody-Assisted Delivery of a Peptide-Drug Conjugate for Targeted Cancer Therapy. Mol. Pharmaceutics 2019, 16, 165–172. 10.1021/acs.molpharmaceut.8b00924. [DOI] [PubMed] [Google Scholar]
- Deyle K.; Kong X. D.; Heinis C. Phage Selection of Cyclic Peptides for Application in Research and Drug Development. Acc. Chem. Res. 2017, 50, 1866–1874. 10.1021/acs.accounts.7b00184. [DOI] [PubMed] [Google Scholar]
- Bogdanowich-Knipp S. J.; Chakrabarti S.; Williams T. D.; Dillman R. K.; Siahaan T. J. Solution stability of linear vs. cyclic RGD peptides. J. Pept Res. 1999, 53, 530–41. 10.1034/j.1399-3011.1999.00052.x. [DOI] [PubMed] [Google Scholar]
- Bogdanowich-Knipp S. J.; Jois D. S.; Siahaan T. J. The effect of conformation on the solution stability of linear vs. cyclic RGD peptides. J. Pept Res. 1999, 53, 523–9. 10.1034/j.1399-3011.1999.00055.x. [DOI] [PubMed] [Google Scholar]
- Rizvi S. F. A.; Mu S.; Wang Y.; Li S.; Zhang H. Fluorescent RGD-based pro-apoptotic peptide conjugates as mitochondria-targeting probes for enhanced anticancer activities. Biomed Pharmacother 2020, 127, 110179. 10.1016/j.biopha.2020.110179. [DOI] [PubMed] [Google Scholar]
- Fetse J.; Zhao Z.; Liu H.; Mamani U. F.; Mustafa B.; Adhikary P.; Ibrahim M.; Liu Y.; Patel P.; Nakhjiri M.; Alahmari M.; Li G.; Cheng K. Discovery of Cyclic Peptide Inhibitors Targeting PD-L1 for Cancer Immunotherapy. J. Med. Chem. 2022, 65, 12002–12013. 10.1021/acs.jmedchem.2c00539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moiola M.; Memeo M. G.; Quadrelli P. Stapled Peptides-A Useful Improvement for Peptide-Based Drugs. Molecules 2019, 24, 3654. 10.3390/molecules24203654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Wu M.; Fu Y.; Xue J.; Yuan F.; Qu T.; Rissanou A. N.; Wang Y.; Li X.; Hu H. Therapeutic stapled peptides: Efficacy and molecular targets. Pharmacol. Res. 2024, 203, 107137. 10.1016/j.phrs.2024.107137. [DOI] [PubMed] [Google Scholar]
- Bernal F.; Tyler A. F.; Korsmeyer S. J.; Walensky L. D.; Verdine G. L. Reactivation of the p53 tumor suppressor pathway by a stapled p53 peptide. J. Am. Chem. Soc. 2007, 129, 2456–7. 10.1021/ja0693587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y.; Fois G.; Flores J. R.; Tuvim M. J.; Zhou Q.; Yang K.; Leitz J.; Peters J.; Zhang Y.; Pfuetzner R. A.; Esquivies L.; Jones P.; Frick M.; Dickey B. F.; Brunger A. T. Inhibition of calcium-triggered secretion by hydrocarbon-stapled peptides. Nature 2022, 603, 949–956. 10.1038/s41586-022-04543-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes C. A.; Pei D. Bicyclic Peptides as Next-Generation Therapeutics. Chemistry 2017, 23, 12690–12703. 10.1002/chem.201702117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss S.; Rademann J.; Nitsche C. Peptide-Bismuth Bicycles: In Situ Access to Stable Constrained Peptides with Superior Bioactivity. Angew. Chem., Int. Ed. Engl. 2022, 61, e202113857 10.1002/anie.202113857. [DOI] [PubMed] [Google Scholar]
- Mudd G. E.; Scott H.; Chen L.; van Rietschoten K.; Ivanova-Berndt G.; Dzionek K.; Brown A.; Watcham S.; White L.; Park P. U.; Jeffrey P.; Rigby M.; Beswick P. Discovery of BT8009: A Nectin-4 Targeting Bicycle Toxin Conjugate for the Treatment of Cancer. J. Med. Chem. 2022, 65, 14337–14347. 10.1021/acs.jmedchem.2c00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigby M.; Bennett G.; Chen L.; Mudd G. E.; Harrison H.; Beswick P. J.; Van Rietschoten K.; Watcham S. M.; Scott H. S.; Brown A. N.; Park P. U.; Campbell C.; Haines E.; Lahdenranta J.; Skynner M. J.; Jeffrey P.; Keen N.; Lee K. BT8009; A Nectin-4 Targeting Bicycle Toxin Conjugate for Treatment of Solid Tumors. Mol. Cancer Ther 2022, 21, 1747–1756. 10.1158/1535-7163.MCT-21-0875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashir B.; Wang J. S.; Falchook G.; Fontana E.; Arkenau H. T.; Carter L.; Galot R.; Basu B.; Greystoke A.; Subbiah V.; Richardson D. L.; Orr H.; Bennett G.; Sharma R.; Xu H.; Paganoni P.; Xu C.; Campbell C.; McKean M. Results From First-in-Human Phase I Dose-Escalation Study of a Novel Bicycle Toxin Conjugate Targeting EphA2 (BT5528) in Patients With Advanced Solid Tumors. J. Clin Oncol 2024, 42, 3443–3452. 10.1200/JCO.23.01107. [DOI] [PubMed] [Google Scholar]
- Kim J. W.; Cochran F. V.; Cochran J. R. A chemically cross-linked knottin dimer binds integrins with picomolar affinity and inhibits tumor cell migration and proliferation. J. Am. Chem. Soc. 2015, 137, 6–9. 10.1021/ja508416e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Z.; Kong C.; Fan S.; Wu C. Triscysteine disulfide-directing motifs enabling design and discovery of multicyclic peptide binders. Nat. Commun. 2024, 15, 7799. 10.1038/s41467-024-51723-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S.; Fan S.; Xiao S.; Li J.; Zhang S.; Wu Y.; Kong C.; Zhuang J.; Liu H.; Zhao Y.; Wu C. Disulfide-Directed Multicyclic Peptide Libraries for the Discovery of Peptide Ligands and Drugs. J. Am. Chem. Soc. 2023, 145, 1964–1972. 10.1021/jacs.2c12462. [DOI] [PubMed] [Google Scholar]
- Werle M.; Schmitz T.; Huang H. L.; Wentzel A.; Kolmar H.; Bernkop-Schnürch A. The potential of cystine-knot microproteins as novel pharmacophoric scaffolds in oral peptide drug delivery. J. Drug Target 2006, 14, 137–46. 10.1080/10611860600648254. [DOI] [PubMed] [Google Scholar]
- Cox N.; Kintzing J. R.; Smith M.; Grant G. A.; Cochran J. R. Integrin-Targeting Knottin Peptide-Drug Conjugates Are Potent Inhibitors of Tumor Cell Proliferation. Angew. Chem., Int. Ed. Engl. 2016, 55, 9894–7. 10.1002/anie.201603488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeken W. L.; Volwiler W.; Goldsworthy P. D.; Garby L. E.; Reynolds W. E.; Stogsdill R.; Stemler R. S. Studies of I-131-albumin catabolism and distribution in normal young male adults. J. Clin Invest 1962, 41, 1312–33. 10.1172/JCI104594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullah A.; Shin G.; Lim S. I. Human serum albumin binders: A piggyback ride for long-acting therapeutics. Drug Discov Today 2023, 28, 103738. 10.1016/j.drudis.2023.103738. [DOI] [PubMed] [Google Scholar]
- Dumelin C. E.; Trüssel S.; Buller F.; Trachsel E.; Bootz F.; Zhang Y.; Mannocci L.; Beck S. C.; Drumea-Mirancea M.; Seeliger M. W.; Baltes C.; Müggler T.; Kranz F.; Rudin M.; Melkko S.; Scheuermann J.; Neri D. A portable albumin binder from a DNA-encoded chemical library. Angew. Chem., Int. Ed. Engl. 2008, 47, 3196–201. 10.1002/anie.200704936. [DOI] [PubMed] [Google Scholar]
- Rousseau E.; Lau J.; Zhang Z.; Uribe C. F.; Kuo H. T.; Zhang C.; Zeisler J.; Colpo N.; Lin K. S.; Bénard F. Effects of adding an albumin binder chain on [(177)Lu]Lu-DOTATATE. Nucl. Med. Biol. 2018, 66, 10–17. 10.1016/j.nucmedbio.2018.08.001. [DOI] [PubMed] [Google Scholar]
- Liu C.; Cai A.; Li H.; Deng N.; Cho B. P.; Seeram N. P.; Ma H. Characterization of molecular interactions between cannabidiol and human plasma proteins (serum albumin and γ-globulin) by surface plasmon resonance, microcalorimetry, and molecular docking. J. Pharm. Biomed Anal 2022, 214, 114750. 10.1016/j.jpba.2022.114750. [DOI] [PubMed] [Google Scholar]
- Tian R.; Zhu S.; Zeng Q.; Lang L.; Ma Y.; Kiesewetter D. O.; Liu Y.; Fu X.; Lau J.; Zhu G.; Jacobson O.; Wang Z.; Dai Y.; Yu G.; Brooks B. R.; Liu G.; Niu G.; Chen X. An Albumin Sandwich Enhances in Vivo Circulation and Stability of Metabolically Labile Peptides. Bioconjug Chem. 2019, 30, 1711–1723. 10.1021/acs.bioconjchem.9b00258. [DOI] [PubMed] [Google Scholar]
- Lau C. Y. J.; Benne N.; Lou B.; Zharkova O.; Ting H. J.; Ter Braake D.; van Kronenburg N.; Fens M. H.; Broere F.; Hennink W. E.; Wang J. W.; Mastrobattista E. Modulating albumin-mediated transport of peptide-drug conjugates for antigen-specific Treg induction. J. Controlled Release 2022, 348, 938–950. 10.1016/j.jconrel.2022.06.025. [DOI] [PubMed] [Google Scholar]
- Kurtzhals P.; Havelund S.; Jonassen I.; Kiehr B.; Larsen U. D.; Ribel U.; Markussen J. Albumin binding of insulins acylated with fatty acids: characterization of the ligand-protein interaction and correlation between binding affinity and timing of the insulin effect in vivo. Biochem. J. 1995, 312, 725. 10.1042/bj3120725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markussen J.; Havelund S.; Kurtzhals P.; Andersen A. S.; Halstro̷m J.; Hasselager E.; Larsen U. D.; Ribel U.; Schäffer L.; Vad K.; Jonassen I. Soluble, fatty acid acylated insulins bind to albumin and show protracted action in pigs. Diabetologia 1996, 39, 281. 10.1007/BF00418343. [DOI] [PubMed] [Google Scholar]
- Aktas G.; Taslamacioglu Duman T. Current usage of long-acting insulin analogs in patients with type 2 diabetes mellitus. Expert Rev. Endocrinol Metab 2024, 19, 155–161. 10.1080/17446651.2024.2320631. [DOI] [PubMed] [Google Scholar]
- Kjeldsen T. B.; Hubálek F.; Hjo̷rringgaard C. U.; Tagmose T. M.; Nishimura E.; Stidsen C. E.; Porsgaard T.; Fledelius C.; Refsgaard H. H. F.; Gram-Nielsen S.; Naver H.; Pridal L.; Hoeg-Jensen T.; Jeppesen C. B.; Manfè V.; Ludvigsen S.; Lautrup-Larsen I.; Madsen P. Molecular Engineering of Insulin Icodec, the First Acylated Insulin Analog for Once-Weekly Administration in Humans. J. Med. Chem. 2021, 64, 8942–8950. 10.1021/acs.jmedchem.1c00257. [DOI] [PubMed] [Google Scholar]
- Han J.; Huang X.; Sun L.; Li Z.; Qian H.; Huang W. Novel fatty chain-modified glucagon-like peptide-1 conjugates with enhanced stability and prolonged in vivo activity. Biochem. Pharmacol. 2013, 86, 297–308. 10.1016/j.bcp.2013.05.012. [DOI] [PubMed] [Google Scholar]
- Niu X.; Wu M.; Li G.; Zhou X.; Cao W.; Zhai W.; Wu A.; Zhou X.; Jin S.; Chen G.; Li Y.; Du J.; Wu Y.; Qiu L.; Zhao W.; Gao Y. Identification and optimization of peptide inhibitors to block VISTA/PSGL-1 interaction for cancer immunotherapy. Acta Pharm. Sin B 2023, 13, 4511–4522. 10.1016/j.apsb.2023.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Fan Y.; Liu S.; Guan Y.; Wan J.; Ren Q.; Wang J.; Zhong L.; Hu Z.; Shi W.; Qian H. Development of Peptide Paratope Mimics Derived from the Anti-ROR1 Antibody and Long-Acting Peptide-Drug Conjugates for Targeted Cancer Therapy. J. Med. Chem. 2024, 67, 10967–10985. 10.1021/acs.jmedchem.4c00511. [DOI] [PubMed] [Google Scholar]
- Sheehy T. L.; Kwiatkowski A. J.; Arora K.; Kimmel B. R.; Schulman J. A.; Gibson-Corley K. N.; Wilson J. T. STING-Activating Polymer-Drug Conjugates for Cancer Immunotherapy. ACS Cent Sci. 2024, 10, 1765–1781. 10.1021/acscentsci.4c00579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmquist A.; Langel U. In vitro uptake and stability study of pVEC and its all-D analog. Biol. Chem. 2003, 384, 387. 10.1515/BC.2003.044. [DOI] [PubMed] [Google Scholar]
- Verdurmen W. P.; Bovee-Geurts P. H.; Wadhwani P.; Ulrich A. S.; Hällbrink M.; van Kuppevelt T. H.; Brock R. Preferential uptake of L- versus D-amino acid cell-penetrating peptides in a cell type-dependent manner. Chem. Biol. 2011, 18, 1000–10. 10.1016/j.chembiol.2011.06.006. [DOI] [PubMed] [Google Scholar]
- Foulon C. F.; Reist C. J.; Bigner D. D.; Zalutsky M. R. Radioiodination via D-amino acid peptide enhances cellular retention and tumor xenograft targeting of an internalizing anti-epidermal growth factor receptor variant III monoclonal antibody. Cancer Res. 2000, 60, 4453. [PubMed] [Google Scholar]
- Tugyi R.; Uray K.; Iván D.; Fellinger E.; Perkins A.; Hudecz F. Partial D-amino acid substitution: Improved enzymatic stability and preserved Ab recognition of a MUC2 epitope peptide. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 413–8. 10.1073/pnas.0407677102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia F.; Wang J.; Peng J.; Zhao P.; Kong Z.; Wang K.; Yan W.; Wang R. D-amino acid substitution enhances the stability of antimicrobial peptide polybia-CP. Acta Biochim Biophys Sin (Shanghai) 2017, 49, 916–925. 10.1093/abbs/gmx091. [DOI] [PubMed] [Google Scholar]
- Qian Y.; Sun Y.; Shi P.; Zhou X.; Zhang Q.; Dong Q.; Jin S.; Qiu L.; Niu X.; Zhou X.; Zhao W.; Wu Y.; Zhai W.; Gao Y. Development of LAG-3/FGL1 blocking peptide and combination with radiotherapy for cancer immunotherapy. Acta Pharm. Sin B 2024, 14, 1150–1165. 10.1016/j.apsb.2023.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy K. J.; Wassenhove-McCarthy D. J. The glomerular basement membrane as a model system to study the bioactivity of heparan sulfate glycosaminoglycans. Microsc Microanal 2012, 18, 3–21. 10.1017/S1431927611012682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender K. D. Basal insulin: physiology, pharmacology, and clinical implications. Postgrad Med. 2011, 123, 17–26. 10.3810/pgm.2011.07.2300. [DOI] [PubMed] [Google Scholar]
- de Meester I.; Lambeir A. M.; Proost P.; Scharpé S. Dipeptidyl peptidase IV substrates. An update on in vitro peptide hydrolysis by human DPPIV. Adv. Exp. Med. Biol. 2004, 524, 3–17. 10.1007/0-306-47920-6_1. [DOI] [PubMed] [Google Scholar]
- Matthews J. E.; Stewart M. W.; De Boever E. H.; Dobbins R. L.; Hodge R. J.; Walker S. E.; Holland M. C.; Bush M. A. Pharmacodynamics, pharmacokinetics, safety, and tolerability of albiglutide, a long-acting glucagon-like peptide-1 mimetic, in patients with type 2 diabetes. J. Clin Endocrinol Metab 2008, 93, 4810–7. 10.1210/jc.2008-1518. [DOI] [PubMed] [Google Scholar]
- Haldar D.; Drew M. G. B.; Banerjee A. α-Aminoisobutyric acid modified protected analogues of β-amyloid residue 17–20: a change from sheet to helix. Tetrahedron 2006, 62, 6370–6378. 10.1016/j.tet.2006.04.036. [DOI] [Google Scholar]
- Mahalakshmi R.; Balaram P. Non-protein amino acids in the design of secondary structure scaffolds. Methods Mol. Biol. 2006, 340, 71–94. 10.1385/1-59745-116-9:71. [DOI] [PubMed] [Google Scholar]
- Raun K.; Hansen B. S.; Johansen N. L.; Tho̷gersen H.; Madsen K.; Ankersen M.; Andersen P. H. Ipamorelin, the first selective growth hormone secretagogue. Eur. J. Endocrinol. 1998, 139, 552. 10.1530/eje.0.1390552. [DOI] [PubMed] [Google Scholar]
- Lau J.; Bloch P.; Schäffer L.; Pettersson I.; Spetzler J.; Kofoed J.; Madsen K.; Knudsen L. B.; McGuire J.; Steensgaard D. B.; Strauss H. M.; Gram D. X.; Knudsen S. M.; Nielsen F. S.; Thygesen P.; Reedtz-Runge S.; Kruse T. Discovery of the Once-Weekly Glucagon-Like Peptide-1 (GLP-1) Analogue Semaglutide. J. Med. Chem. 2015, 58, 7370–80. 10.1021/acs.jmedchem.5b00726. [DOI] [PubMed] [Google Scholar]
- Li X.; Wang N.; Liu Y.; Li W.; Bai X.; Liu P.; He C. Y. Backbone N-methylation of peptides: Advances in synthesis and applications in pharmaceutical drug development. Bioorg Chem. 2023, 141, 106892. 10.1016/j.bioorg.2023.106892. [DOI] [PubMed] [Google Scholar]
- Chatterjee J.; Gilon C.; Hoffman A.; Kessler H. N-methylation of peptides: a new perspective in medicinal chemistry. Acc. Chem. Res. 2008, 41, 1331–42. 10.1021/ar8000603. [DOI] [PubMed] [Google Scholar]
- Li Petri G.; Di Martino S.; De Rosa M. Peptidomimetics: An Overview of Recent Medicinal Chemistry Efforts toward the Discovery of Novel Small Molecule Inhibitors. J. Med. Chem. 2022, 65, 7438–7475. 10.1021/acs.jmedchem.2c00123. [DOI] [PubMed] [Google Scholar]
- Lenci E.; Trabocchi A. Peptidomimetic toolbox for drug discovery. Chem. Soc. Rev. 2020, 49, 3262–3277. 10.1039/D0CS00102C. [DOI] [PubMed] [Google Scholar]
- Rodriguez M. C.; Cudic M. Optimization of physicochemical and pharmacological properties of peptide drugs by glycosylation. Methods Mol. Biol. 2013, 1081, 107–36. 10.1007/978-1-62703-652-8_8. [DOI] [PubMed] [Google Scholar]
- Karjalainen K.; Jaalouk D. E.; Bueso-Ramos C.; Bover L.; Sun Y.; Kuniyasu A.; Driessen W. H.; Cardó-Vila M.; Rietz C.; Zurita A. J.; O’Brien S.; Kantarjian H. M.; Cortes J. E.; Calin G. A.; Koivunen E.; Arap W.; Pasqualini R. Targeting IL11 Receptor in Leukemia and Lymphoma: A Functional Ligand-Directed Study and Hematopathology Analysis of Patient-Derived Specimens. Clin. Cancer Res. 2015, 21, 3041–51. 10.1158/1078-0432.CCR-13-3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellerby H. M.; Arap W.; Ellerby L. M.; Kain R.; Andrusiak R.; Rio G. D.; Krajewski S.; Lombardo C. R.; Rao R.; Ruoslahti E.; Bredesen D. E.; Pasqualini R. Anti-cancer activity of targeted pro-apoptotic peptides. Nat. Med. 1999, 5, 1032–8. 10.1038/12469. [DOI] [PubMed] [Google Scholar]
- Lewis V. O.; Devarajan E.; Cardó-Vila M.; Thomas D. G.; Kleinerman E. S.; Marchiò S.; Sidman R. L.; Pasqualini R.; Arap W. BMTP-11 is active in preclinical models of human osteosarcoma and a candidate targeted drug for clinical translation. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 8065–8070. 10.1073/pnas.1704173114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardó-Vila M.; Marchiò S.; Sato M.; Staquicini F. I.; Smith T. L.; Bronk J. K.; Yin G.; Zurita A. J.; Sun M.; Behrens C.; Sidman R. L.; Lee J. J.; Hong W. K.; Wistuba I. I.; Arap W.; Pasqualini R. Interleukin-11 Receptor Is a Candidate Target for Ligand-Directed Therapy in Lung Cancer: Analysis of Clinical Samples and BMTP-11 Preclinical Activity. Am. J. Pathol. 2016, 186, 2162–2170. 10.1016/j.ajpath.2016.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualini R.; Millikan R. E.; Christianson D. R.; Cardó-Vila M.; Driessen W. H.; Giordano R. J.; Hajitou A.; Hoang A. G.; Wen S.; Barnhart K. F.; Baze W. B.; Marcott V. D.; Hawke D. H.; Do K. A.; Navone N. M.; Efstathiou E.; Troncoso P.; Lobb R. R.; Logothetis C. J.; Arap W. Targeting the interleukin-11 receptor α in metastatic prostate cancer: A first-in-man study. Cancer 2015, 121, 2411–21. 10.1002/cncr.29344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapperton A. M.; Babi J.; Tran H. A Field Guide to Optimizing Peptoid Synthesis. ACS Polym. Au 2022, 2, 417–429. 10.1021/acspolymersau.2c00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun M.; Vijayan V.; Shin S.; Nam H. Y.; Song D.; Choi J.; Vasvani S.; Cho S. K.; Park I. K.; Seo J. A bleomycin-mimicking manganese-porphyrin-conjugated mitochondria-targeting peptoid for cancer therapy. Bioorg. Med. Chem. 2025, 117, 118023. 10.1016/j.bmc.2024.118023. [DOI] [PubMed] [Google Scholar]
- Coulter S. M.; Pentlavalli S.; An Y.; Vora L. K.; Cross E. R.; Moore J. V.; Sun H.; Schweins R.; McCarthy H. O.; Laverty G. In Situ Forming, Enzyme-Responsive Peptoid-Peptide Hydrogels: An Advanced Long-Acting Injectable Drug Delivery System. J. Am. Chem. Soc. 2024, 146, 21401–21416. 10.1021/jacs.4c03751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z.; Zhang P.; Cheetham A. G.; Moon J. H.; Moxley J. W. Jr.; Lin Y. A.; Cui H. Controlled release of free doxorubicin from peptide-drug conjugates by drug loading. J. Controlled Release 2014, 191, 123–30. 10.1016/j.jconrel.2014.05.051. [DOI] [PubMed] [Google Scholar]
- Gou Q.; Dong C.; Xu H.; Khan B.; Jin J.; Liu Q.; Shi J.; Hou Y. PD-L1 degradation pathway and immunotherapy for cancer. Cell Death Dis 2020, 11, 955. 10.1038/s41419-020-03140-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.; Yu X.; Yuan Z.; Li L.; Yin P. New horizons in the mechanisms and therapeutic strategies for PD-L1 protein degradation in cancer. Biochim Biophys Acta Rev. Cancer 2024, 1879, 189152. 10.1016/j.bbcan.2024.189152. [DOI] [PubMed] [Google Scholar]
- Zheng J.; He W.; Li J.; Feng X.; Li Y.; Cheng B.; Zhou Y.; Li M.; Liu K.; Shao X.; Zhang J.; Li H.; Chen L.; Fang L. Bifunctional Compounds as Molecular Degraders for Integrin-Facilitated Targeted Protein Degradation. J. Am. Chem. Soc. 2022, 144, 21831–21836. 10.1021/jacs.2c08367. [DOI] [PubMed] [Google Scholar]
- Zeng Z.; Yang Z.; Li C.; Liu S.; Wei W.; Zhou Y.; Wang S.; Sui M.; Li M.; Lin S.; Cheng Y.; Hou P. Advancing Cancer Immunotherapy through Engineering New PD-L1 Degraders: A Comprehensive Study from Small Molecules to PD-L1-Specific Peptide-Drug Conjugates. J. Med. Chem. 2024, 67, 19216–19233. 10.1021/acs.jmedchem.4c01652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno M.; Horibe T.; Haramoto M.; Yano Y.; Ohara K.; Nakajima O.; Matsuzaki K.; Kawakami K. A novel hybrid peptide targeting EGFR-expressing cancers. Eur. J. Cancer 2011, 47, 773–83. 10.1016/j.ejca.2010.10.021. [DOI] [PubMed] [Google Scholar]
- Yang L.; Horibe T.; Kohno M.; Haramoto M.; Ohara K.; Puri R. K.; Kawakami K. Targeting interleukin-4 receptor α with hybrid peptide for effective cancer therapy. Mol. Cancer Ther 2012, 11, 235–43. 10.1158/1535-7163.MCT-11-0363. [DOI] [PubMed] [Google Scholar]
- Wang D.; Liu J.; Li T.; Wang Y.; Liu X.; Bai Y.; Wang C.; Ju S.; Huang S.; Yang C.; Zhou C.; Zhang Y.; Xiong B. A VEGFR targeting peptide-drug conjugate (PDC) suppresses tumor angiogenesis in a TACE model for hepatocellular carcinoma therapy. Cell Death Discov 2022, 8, 411. 10.1038/s41420-022-01198-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Bai Y.; Liu X.; Zhou B.; Sun P.; Wang Y.; Ju S.; Zhou C.; Wang C.; Yao W.; Yang H.; Jiang X.; Yang L.; Wang D.; Zheng C. Enhanced efficacy of combined VEGFR peptide-drug conjugate and anti-PD-1 antibody in treating hepatocellular carcinoma. Sci. Rep 2024, 14, 21728. 10.1038/s41598-024-72907-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuschner C.; Hansel W. Membrane disrupting lytic peptides for cancer treatments. Curr. Pharm. Des 2004, 10, 2299–310. 10.2174/1381612043383971. [DOI] [PubMed] [Google Scholar]
- Curtis K. K.; Sarantopoulos J.; Northfelt D. W.; Weiss G. J.; Barnhart K. M.; Whisnant J. K.; Leuschner C.; Alila H.; Borad M. J.; Ramanathan R. K. Novel LHRH-receptor-targeted cytolytic peptide, EP-100: first-in-human phase I study in patients with advanced LHRH-receptor-expressing solid tumors. Cancer Chemother Pharmacol 2014, 73, 931–41. 10.1007/s00280-014-2424-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelariu-Raicu A.; Nick A.; Urban R.; Gordinier M.; Leuschner C.; Bavisotto L.; Molin G. Z. D.; Whisnant J. K.; Coleman R. L. A multicenter open-label randomized phase II trial of paclitaxel plus EP-100, a novel LHRH receptor-targeted, membrane-disrupting peptide, versus paclitaxel alone for refractory or recurrent ovarian cancer. Gynecol Oncol 2021, 160, 418–426. 10.1016/j.ygyno.2020.11.013. [DOI] [PubMed] [Google Scholar]
- Kim M. S.; Ma S.; Chelariu-Raicu A.; Leuschner C.; Alila H. W.; Lee S.; Coleman R. L.; Sood A. K. Enhanced Immunotherapy with LHRH-R Targeted Lytic Peptide in Ovarian Cancer. Mol. Cancer Ther 2020, 19, 2396–2406. 10.1158/1535-7163.MCT-20-0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentinis B.; Porcellini S.; Asperti C.; Cota M.; Zhou D.; Di Matteo P.; Garau G.; Zucchelli C.; Avanzi N. R.; Rizzardi G. P.; Degano M.; Musco G.; Traversari C. Mechanism of Action of the Tumor Vessel Targeting Agent NGR-hTNF: Role of Both NGR Peptide and hTNF in Cell Binding and Signaling. Int. J. Mol. Sci. 2019, 20, 4511. 10.3390/ijms20184511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorc V.; Santoro A.; Bennicelli E.; Punt C. J.; Citterio G.; Timmer-Bonte J. N.; Caligaris Cappio F.; Lambiase A.; Bordignon C.; van Herpen C. M. Phase Ib study of NGR-hTNF, a selective vascular targeting agent, administered at low doses in combination with doxorubicin to patients with advanced solid tumours. Br. J. Cancer 2009, 101, 219–24. 10.1038/sj.bjc.6605162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro A.; Rimassa L.; Sobrero A. F.; Citterio G.; Sclafani F.; Carnaghi C.; Pessino A.; Caprioni F.; Andretta V.; Tronconi M. C.; Finocchiaro G.; Rossoni G.; Zanoni A.; Miggiano C.; Rizzardi G. P.; Traversari C.; Caligaris-Cappio F.; Lambiase A.; Bordignon C. Phase II study of NGR-hTNF, a selective vascular targeting agent, in patients with metastatic colorectal cancer after failure of standard therapy. Eur. J. Cancer 2010, 46, 2746–52. 10.1016/j.ejca.2010.07.012. [DOI] [PubMed] [Google Scholar]
- Santoro A.; Pressiani T.; Citterio G.; Rossoni G.; Donadoni G.; Pozzi F.; Rimassa L.; Personeni N.; Bozzarelli S.; Rossoni G.; Colombi S.; De Braud F. G.; Caligaris-Cappio F.; Lambiase A.; Bordignon C. Activity and safety of NGR-hTNF, a selective vascular-targeting agent, in previously treated patients with advanced hepatocellular carcinoma. Br. J. Cancer 2010, 103, 837–44. 10.1038/sj.bjc.6605858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorc V.; Zucali P. A.; Santoro A.; Ceresoli G. L.; Citterio G.; De Pas T. M.; Zilembo N.; De Vincenzo F.; Simonelli M.; Rossoni G.; Spreafico A.; Grazia Viganò M.; Fontana F.; De Braud F. G.; Bajetta E.; Caligaris-Cappio F.; Bruzzi P.; Lambiase A.; Bordignon C. Phase II study of asparagine-glycine-arginine-human tumor necrosis factor alpha, a selective vascular targeting agent, in previously treated patients with malignant pleural mesothelioma. J. Clin Oncol 2010, 28, 2604–11. 10.1200/JCO.2009.27.3649. [DOI] [PubMed] [Google Scholar]
- Mammoliti S.; Andretta V.; Bennicelli E.; Caprioni F.; Comandini D.; Fornarini G.; Guglielmi A.; Pessino A.; Sciallero S.; Sobrero A. F.; Mazzola G.; Lambiase A.; Bordignon C. Two doses of NGR-hTNF in combination with capecitabine plus oxaliplatin in colorectal cancer patients failing standard therapies. Ann. Oncol 2011, 22, 973–978. 10.1093/annonc/mdq436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorusso D.; Scambia G.; Amadio G.; di Legge A.; Pietragalla A.; De Vincenzo R.; Masciullo V.; Di Stefano M.; Mangili G.; Citterio G.; Mantori M.; Lambiase A.; Bordignon C. Phase II study of NGR-hTNF in combination with doxorubicin in relapsed ovarian cancer patients. Br. J. Cancer 2012, 107, 37–42. 10.1038/bjc.2012.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorc V.; Cavina R.; Novello S.; Grossi F.; Lazzari C.; Capelletto E.; Genova C.; Salini G.; Lambiase A.; Santoro A. NGR-hTNF and Doxorubicin as Second-Line Treatment of Patients with Small Cell Lung Cancer. Oncologist 2018, 23, 1133–e112. 10.1634/theoncologist.2018-0292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorc V.; De Braud F. G.; De Pas T. M.; Scalamogna R.; Citterio G.; Milani A.; Boselli S.; Catania C.; Donadoni G.; Rossoni G.; Ghio D.; Spitaleri G.; Ammannati C.; Colombi S.; Caligaris-Cappio F.; Lambiase A.; Bordignon C. Phase I study of NGR-hTNF, a selective vascular targeting agent, in combination with cisplatin in refractory solid tumors. Clin. Cancer Res. 2011, 17, 1964–72. 10.1158/1078-0432.CCR-10-1376. [DOI] [PubMed] [Google Scholar]
- Gregorc V.; Gaafar R. M.; Favaretto A.; Grossi F.; Jassem J.; Polychronis A.; Bidoli P.; Tiseo M.; Shah R.; Taylor P.; Novello S.; Muzio A.; Bearz A.; Greillier L.; Fontana F.; Salini G.; Lambiase A.; O’Brien M. NGR-hTNF in combination with best investigator choice in previously treated malignant pleural mesothelioma (NGR015): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol 2018, 19, 799–811. 10.1016/S1470-2045(18)30193-1. [DOI] [PubMed] [Google Scholar]
- Engel J. B.; Schally A. V.; Buchholz S.; Seitz S.; Emons G.; Ortmann O. Targeted chemotherapy of endometrial, ovarian and breast cancers with cytotoxic analogs of luteinizing hormone-releasing hormone (LHRH). Arch Gynecol Obstet 2012, 286, 437–42. 10.1007/s00404-012-2335-1. [DOI] [PubMed] [Google Scholar]
- Emons G.; Gorchev G.; Harter P.; Wimberger P.; Stähle A.; Hanker L.; Hilpert F.; Beckmann M. W.; Dall P.; Gruindker C.; Sindermann H.; Sehouli J. Efficacy and safety of AEZS-108 (LHRH agonist linked to doxorubicin) in women with advanced or recurrent endometrial cancer expressing LHRH receptors: a multicenter phase 2 trial (AGO-GYN5). Int. J. Gynecol Cancer 2014, 24, 260. 10.1097/IGC.0000000000000044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S. S.; Athreya K.; Liu S. V.; Schally A. V.; Tsao-Wei D.; Groshen S.; Quinn D. I.; Dorff T. B.; Xiong S.; Engel J.; Pinski J. A Phase II Trial of AEZS-108 in Castration- and Taxane-Resistant Prostate Cancer. Clin Genitourin Cancer 2017, 15, 742–749. 10.1016/j.clgc.2017.06.002. [DOI] [PubMed] [Google Scholar]
- Mathews C.; Lorusso D.; Coleman R. L.; Boklage S.; Garside J. An Indirect Comparison of the Efficacy and Safety of Dostarlimab and Doxorubicin for the Treatment of Advanced and Recurrent Endometrial Cancer. Oncologist 2022, 27, 1058–1066. 10.1093/oncolo/oyac188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vankadara S.; Ke Z.; Wang S.; Foo S. Y.; Gunaratne J.; Lee M. A.; Koh X.; Chia C. S. B. Cytotoxic activity and cell specificity of a novel LHRH peptide drug conjugate, D-Cys6-LHRH vedotin, against ovarian cancer cell lines. Chem. Biol. Drug Des 2024, 103, e14516 10.1111/cbdd.14516. [DOI] [PubMed] [Google Scholar]
- Mahalingam D.; Peguero J.; Cen P.; Arora S. P.; Sarantopoulos J.; Rowe J.; Allgood V.; Tubb B.; Campos L. A Phase II, Multicenter, Single-Arm Study of Mipsagargin (G-202) as a Second-Line Therapy Following Sorafenib for Adult Patients with Progressive Advanced Hepatocellular Carcinoma. Cancers (Basel) 2019, 11, 833. 10.3390/cancers11060833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen K. A.; White B. H.; Quinn J. M.; Kriksciukaite K.; Alargova R.; Au Yeung T. P.; Bazinet P.; Brockman A.; DuPont M. M.; Oller H.; Gifford J.; Lemelin C. A.; Lim Soo P.; Perino S.; Moreau B.; Sharma G.; Shinde R.; Sweryda-Krawiec B.; Bilodeau M. T.; Wooster R. Targeting the Somatostatin Receptor 2 with the Miniaturized Drug Conjugate, PEN-221: A Potent and Novel Therapeutic for the Treatment of Small Cell Lung Cancer. Mol. Cancer Ther 2019, 18, 1926–1936. 10.1158/1535-7163.MCT-19-0022. [DOI] [PubMed] [Google Scholar]
- Demeule M.; Charfi C.; Currie J. C.; Larocque A.; Zgheib A.; Kozelko S.; Béliveau R.; Marsolais C.; Annabi B. TH1902, a new docetaxel-peptide conjugate for the treatment of sortilin-positive triple-negative breast cancer. Cancer Sci. 2021, 112, 4317–4334. 10.1111/cas.15086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demeule M.; Currie J. C.; Charfi C.; Zgheib A.; Cousineau I.; Lullier V.; Béliveau R.; Marsolais C.; Annabi B. Sudocetaxel Zendusortide (TH1902) triggers the cGAS/STING pathway and potentiates anti-PD-L1 immune-mediated tumor cell killing. Front Immunol 2024, 15, 1355945. 10.3389/fimmu.2024.1355945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y.; Pan T.; Da W.; Liu Y.; Chen S.; Chen D.; Liu K.; Zheng Y.; Xie D.; Gao Y.; Xu H.; Sun Y.; Tan W. Aptamer-drug conjugates-loaded bacteria for pancreatic cancer synergistic therapy. Signal Transduct Target Ther 2024, 9, 272. 10.1038/s41392-024-01973-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. R.; Park S. J.; Cho Y. S.; Moyo M. K.; Choi J. U.; Lee N. K.; Chung S. W.; Kweon S.; Park J.; Kim B.; Ko Y. G.; Yeo J. H.; Lee J.; Kim S. Y.; Byun Y. Stimulating macropinocytosis of peptide-drug conjugates through DNA-dependent protein kinase inhibition for treating KRAS-mutant cancer. J. Controlled Release 2024, 372, 176–193. 10.1016/j.jconrel.2024.06.028. [DOI] [PubMed] [Google Scholar]
- Compain G.; Monsarrat C.; Blagojevic J.; Brillet K.; Dumas P.; Hammann P.; Kuhn L.; Martiel I.; Engilberge S.; Oliéric V.; Wolff P.; Burnouf D. Y.; Wagner J.; Guichard G. Peptide-Based Covalent Inhibitors Bearing Mild Electrophiles to Target a Conserved His Residue of the Bacterial Sliding Clamp. JACS Au 2024, 4, 432–440. 10.1021/jacsau.3c00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdan V. Y.; Klauser P. C.; Wang L. Covalent peptides and proteins for therapeutics. Bioorg. Med. Chem. 2021, 29, 115896. 10.1016/j.bmc.2020.115896. [DOI] [PubMed] [Google Scholar]