

Abstract
Structural flexibility is an essential attribute, without which few proteins could carry out their biological functions. Much information about protein flexibility has come from x-ray crystallography, in the form of atomic mean-square displacements (AMSDs) or B factors. Profiles showing the AMSD variation along the polypeptide chain are usually interpreted in dynamical terms but are ultimately governed by the local features of a highly complex energy landscape. Here, we bypass this complexity by showing that the AMSD profile is essentially determined by spatial variations in local packing density. On the basis of elementary statistical mechanics and generic features of atomic distributions in proteins, we predict a direct inverse proportionality between the AMSD and the contact density, i.e., the number of noncovalent neighbor atoms within a local region of ∼1.5 nm3 volume. Testing this local density model against a set of high-quality crystal structures of 38 nonhomologous proteins, we find that it accurately and consistently reproduces the prominent peaks in the AMSD profile and even captures minor features, such as the periodic AMSD variation within α helices. The predicted rigidifying effect of crystal contacts also agrees with experimental data. With regard to accuracy and computational efficiency, the model is clearly superior to its predecessors. The quantitative link between flexibility and packing density found here implies that AMSDs provide little independent information beyond that contained in the mean atomic coordinates.
To date, x-ray crystallography has provided nearly 12,000 atomic-level models of protein structure (see http://www.rcsb.org/pdb/). The primary data, structure factors of Bragg reflections, result from diffraction of x-rays by the atoms in a single-crystal comprising some 1015 protein molecules. At any instant, the members of this molecular ensemble are continuously, but nonuniformly, distributed in conformational space. The structure factors yield a set of mean atomic positions r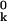 ≡ 〈rk〉 that define the “ground-state” protein structure, or, if resolution permits, a small number of substantially populated low-energy conformational substates. For each atom thus located, one also obtains a Debye–Waller factor, i.e., the spatial Fourier transform of the probability distribution function (PDF) Fk(uk) for displacements uk ≡ rk − r
≡ 〈rk〉 that define the “ground-state” protein structure, or, if resolution permits, a small number of substantially populated low-energy conformational substates. For each atom thus located, one also obtains a Debye–Waller factor, i.e., the spatial Fourier transform of the probability distribution function (PDF) Fk(uk) for displacements uk ≡ rk − r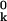 of atom k away from its mean position (1). For diffraction data of ultra-high resolution, Fk(uk) is usually modeled as a trivariate Gaussian function, parametrized in terms of the six independent elements of the atomic covariance matrix 〈uk u
of atom k away from its mean position (1). For diffraction data of ultra-high resolution, Fk(uk) is usually modeled as a trivariate Gaussian function, parametrized in terms of the six independent elements of the atomic covariance matrix 〈uk u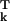 〉 (2). More commonly, one adopts a univariant Gaussian function, fully characterized by the (isotropic) mean-square displacement 〈uk⋅uk〉 ≡ σk, or the B factor Bk ≡ 8π2σk/3.
〉 (2). More commonly, one adopts a univariant Gaussian function, fully characterized by the (isotropic) mean-square displacement 〈uk⋅uk〉 ≡ σk, or the B factor Bk ≡ 8π2σk/3.
Like the mean atomic positions, the set of atomic mean square displacements (AMSDs) {σk}, k = 1, 2, … , N, is an intrinsic property of the protein (in its crystal environment), providing a spatially resolved measure of the small-amplitude pliability or flexibility of the ground-state protein conformation (3). Although Bragg diffraction data contain no information about the rate or mechanism of conformational motion, AMSDs are often discussed and interpreted in dynamical terms (3–5). Indeed, the terms flexibility, dynamics, and mobility are often used synonymously in this context. In principle, AMSDs can be calculated and the associated motions identified by molecular dynamics simulations based on semiempirical atomic force-field models (6–9). In practice, the agreement between simulated and experimental AMSDs is modest (7–9), even when the rigidifying effect of crystal contacts is taken into account (7, 9). Calculated AMSDs tend to increase with the length of the analyzed trajectory as slower motions of larger amplitude are sampled and do not converge even in nanosecond-length simulations (8, 9). Among the slower motions are dihedral barrier crossings between distinct conformational substates, such as alternative side-chain conformations. The displacement distribution Fk(uk) for atoms undergoing such motions is multimodal and hence not well approximated by a Gaussian function (10, 11). Therefore, ultra-high-resolution diffraction data are usually modeled with several residues in alternative conformations, each with its own set of AMSDs. In either case, conformational substates complicate the comparison with simulated AMSDs.
Disregarding minor quantum effects, AMSDs are static equilibrium properties, completely determined by the interactions within the system. In other words, AMSDs cannot depend on any kinetic parameters, such as libration frequencies, substate interconversion rates, or solvent viscosity. Consequently, AMSDs can be predicted without invoking motion. Moreover, this should be far less challenging than predicting the mean atomic positions (the folding problem), because AMSDs are governed by local features of the energy landscape near the global minimum. It has long been recognized that AMSDs correlate with structural features such as solvent exposure, packing density, and secondary structure (5, 12, 13). However, such observations have been of a qualitative nature and have not been pursued in a systematic way.
The aim of the present work is to explore the hypothesis that AMSDs can be predicted solely on the basis of packing density. This hypothesis is motivated by the following considerations. On average, protein interiors are as densely packed as crystalline solids (14–17). Most atoms therefore cannot be displaced much without also displacing some of their nonbonded neighbors. Yet, the local density, averaged over volume elements of 0.1–1 nm3, varies substantially within a protein (14, 17, 18). Equivalently stated, the distribution of voids (cavities and subatomic interstices) is highly inhomogeneous. Presumably, low-density regions can accommodate a variety of alternative packings or conformations, whereas high-density regions might be realized only for a few closely similar conformations. AMSDs should then be anticorrelated with local packing density.
Although these arguments are intuitively appealing, the functional form an AMSD–density relationship is not obvious. We show here that a simple inverse proportionality, σk ∝ n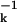 , emerges from a series of crude but well-defined approximations. As the measure of local packing, we use the contact density nk, i.e., the number of nonhydrogen atoms within a spherical region of ∼1.5 nm3 volume centered on atom k. We then test the predictive power of this simple relation on a set of 38 nonhomologous protein crystal structures of exceptionally high quality. We find that the simple inverse relationship faithfully reproduces the variation of backbone as well as side-chain AMSDs along the polypeptide chain. Because the AMSD profile can be predicted with good accuracy from the contact density, it does not furnish much independent information beyond that already contained in the mean coordinates. This finding has implications for how we think about AMSDs. For example, the use of AMSDs to infer likely pathways for ligand access to internal sites (3, 4, 19), sometimes called thermal motion paths (19), essentially amounts to an identification of contiguous regions of low packing density.
, emerges from a series of crude but well-defined approximations. As the measure of local packing, we use the contact density nk, i.e., the number of nonhydrogen atoms within a spherical region of ∼1.5 nm3 volume centered on atom k. We then test the predictive power of this simple relation on a set of 38 nonhomologous protein crystal structures of exceptionally high quality. We find that the simple inverse relationship faithfully reproduces the variation of backbone as well as side-chain AMSDs along the polypeptide chain. Because the AMSD profile can be predicted with good accuracy from the contact density, it does not furnish much independent information beyond that already contained in the mean coordinates. This finding has implications for how we think about AMSDs. For example, the use of AMSDs to infer likely pathways for ligand access to internal sites (3, 4, 19), sometimes called thermal motion paths (19), essentially amounts to an identification of contiguous regions of low packing density.
The present local density approach to AMSDs is superficially related to the use of effective harmonic potentials with distance-dependent force constants to predict AMSDs and large-scale conformational transitions (20–23). However, the underlying physical models are qualitatively different. Moreover, the present approach is both more accurate and more computationally efficient.
Methods
Statistical–Mechanical Basis.
The isotropic AMSD, σk, of atom k is defined by
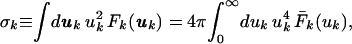 |
1 |
where F̄k(uk) is the orientational average of the displacement PDF Fk(uk). The potential of mean force (POMF) wk associated with Fk(uk) may be defined through (24)
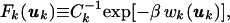 |
2 |
where β = (kBT)−1, and Ck is a constant that normalizes Fk(uk) to unity. Expanding wk around the mean position of atom k, we obtain (in matrix notation)
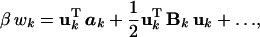 |
3 |
where the Cartesian components of the vector ak and the tensor Bk are first and second derivatives, respectively, of βwk with respect to the Cartesian components of the displacement vector uk, evaluated at the mean position r (or uk = 0).
(or uk = 0).
The isotropic PDF F̄k(uk) in Eq. 1 involves the orientational average of the Boltzmann factor in Eq. 2. We approximate this by the Boltzmann factor of the orientationally averaged POMF:
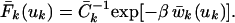 |
4 |
When we take the isotropic average of wk in Eq. 3, all terms containing odd powers of uk vanish, so that
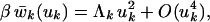 |
5 |
with Λk = Tr Bk/3. For sufficiently small displacements, we can neglect terms of fourth and higher order in Eq. 5. The orientationally averaged POMF then becomes harmonic, as generally assumed in the interpretation of diffraction data (1, 2), and a combination of Eqs. 1, 4, and 5 yields σk = 3/(2Λk).
To relate Λk to the local density, we make a bold assumption: when atom k is displaced from its mean position, all other atoms remain at their mean positions. The N-particle problem then becomes a one-particle problem, and the POMF wk reduces to the sum of pair interactions νki(rk − r ) of atom k with every other atom i, each confined to its mean position r
) of atom k with every other atom i, each confined to its mean position r . The pair interaction νki depends on the atomic configuration in a complicated way. In several recent treatments of protein conformational dynamics and flexibility, harmonic pair interactions have been postulated (20–23). Although νki may be approximately harmonic near its minimum (for the isolated atom pair), it is certainly not harmonic at the separations of the vast majority of atom pairs in the mean configuration of the protein, where the second derivatives in Λk are to be evaluated. In fact, most atoms i in the protein hardly interact at all with the reference atom k and, therefore, do not contribute significantly to Λk. In evaluating Λk, we therefore need to consider only those nk atoms i whose mean positions are within some cutoff distance RC of atom k, i.e., for which r
. The pair interaction νki depends on the atomic configuration in a complicated way. In several recent treatments of protein conformational dynamics and flexibility, harmonic pair interactions have been postulated (20–23). Although νki may be approximately harmonic near its minimum (for the isolated atom pair), it is certainly not harmonic at the separations of the vast majority of atom pairs in the mean configuration of the protein, where the second derivatives in Λk are to be evaluated. In fact, most atoms i in the protein hardly interact at all with the reference atom k and, therefore, do not contribute significantly to Λk. In evaluating Λk, we therefore need to consider only those nk atoms i whose mean positions are within some cutoff distance RC of atom k, i.e., for which r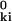 ≡ |r
≡ |r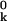 − r
− r | ≤ RC. We can then express Λk as a sum of contributions from these nk atoms, or as Λk = nk λk, where λk is the mean of the nk atomic contributions. We shall now argue that the dependence of Λk on k derives mainly from local density (nk) variations.
| ≤ RC. We can then express Λk as a sum of contributions from these nk atoms, or as Λk = nk λk, where λk is the mean of the nk atomic contributions. We shall now argue that the dependence of Λk on k derives mainly from local density (nk) variations.
Consider the radial distribution function gk(r), which, when multiplied by 4π r2dr, gives the number nk(r) of nonhydrogen atoms in a spherical shell of thickness dr at a distance r from reference atom k. We compute this quantity by summing over the isotropic displacement PDFs F̄i(ui) for all other atoms i and taking the isotropic average over the orientation of the vector r:
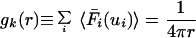 |
6 |
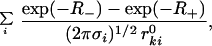 |
where R± = (r ± r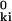 )2/(2σi). This result is obtained by inserting the Gaussian PDF F̄i(ui) (see Eqs. 1, 4, and 5) and noting that u
)2/(2σi). This result is obtained by inserting the Gaussian PDF F̄i(ui) (see Eqs. 1, 4, and 5) and noting that u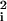 = r2 + (r
= r2 + (r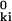 )2 − 2 r r
)2 − 2 r r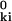 cos θ, where θ is the angle between r and r
cos θ, where θ is the angle between r and r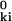 . Fig. 1 shows gk(r) for each α carbon in parvalbumin; similar results are obtained for other proteins. The first peak in gk(r), with maximum at 1.5 Å and extending to about 3.5 Å, corresponds to the covalent neighbors: 6–10 atoms linked to the reference α carbon by 1–3 bonds. Except for a few α carbons near the chain termini, gk(r) exhibits a second peak, with maximum near 5 Å, produced by a much larger number of atoms. Although close in space, most of these atoms are many bonds away from the reference α carbon and therefore have predominantly noncovalent interactions with it. We refer to them as noncovalent neighbors.
. Fig. 1 shows gk(r) for each α carbon in parvalbumin; similar results are obtained for other proteins. The first peak in gk(r), with maximum at 1.5 Å and extending to about 3.5 Å, corresponds to the covalent neighbors: 6–10 atoms linked to the reference α carbon by 1–3 bonds. Except for a few α carbons near the chain termini, gk(r) exhibits a second peak, with maximum near 5 Å, produced by a much larger number of atoms. Although close in space, most of these atoms are many bonds away from the reference α carbon and therefore have predominantly noncovalent interactions with it. We refer to them as noncovalent neighbors.
Figure 1.

Radial distribution of nonhydrogen atoms around each α carbon in parvalbumin (2pvb), computed from Eq. 6. The thick black curve is the average of the 107 gray curves.
By far the largest contribution to Λk comes from the covalent neighbors. Because displacements of these atoms are highly correlated with displacement of the reference α carbon, the rigid-environment approximation (ri = r ) is strongly violated. But because the covalent neighbors are distributed in much the same way around all α carbons (see Fig. 1), they hardly contribute to the AMSD variation that we seek to model. We therefore ignore the covalent neighbors and attribute Λk entirely to the noncovalent neighbors. We must then also reinterpret the pair potential νki as the interaction of atom i with the cluster comprising atom k and its 6–10 covalent neighbors.
) is strongly violated. But because the covalent neighbors are distributed in much the same way around all α carbons (see Fig. 1), they hardly contribute to the AMSD variation that we seek to model. We therefore ignore the covalent neighbors and attribute Λk entirely to the noncovalent neighbors. We must then also reinterpret the pair potential νki as the interaction of atom i with the cluster comprising atom k and its 6–10 covalent neighbors.
The mean contribution λk from the noncovalent neighbors depends mainly on their mean positions. Because the position of the second peak in gk(r) varies relatively little (see Fig. 1), whereas nk varies by a factor 5 (see below), we attribute the variation of Λk with k entirely to nk, writing Λk = nk λ. We then arrive at the desired result
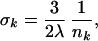 |
7 |
predicting that the AMSD is inversely proportional to the contact density nk, i.e., the number of noncovalent neighbors. The set of approximations leading to Eq. 7 will be referred to as the local density model (LDM). The LDM can predict the AMSD variation within a protein but, because the parameter λ is undetermined, it cannot yield the mean AMSD. In comparing LDM predictions with experimental results, we therefore scale the calculated AMSDs such that 〈σ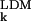 〉 = 〈σ
〉 = 〈σ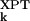 〉. This scaling takes care of the temperature dependence of the AMSDs; if the (renormalized) pair interactions are temperature independent, it follows from the foregoing that λ ∝ T−1.
〉. This scaling takes care of the temperature dependence of the AMSDs; if the (renormalized) pair interactions are temperature independent, it follows from the foregoing that λ ∝ T−1.
Selection of Proteins.
To test the hypothesis that AMSDs scale with local density according to Eq. 7, we use a set of 38 crystal structures taken from the current (Feb. 25, 2001) pdb select list of structures with less than 25% sequence identity between any two proteins (25). From this list, we selected the structures of highest resolution (≤1.30 Å) and best quality (R factor < 0.16), retaining only single-chain proteins with more than 50 residues. Because experimental AMSDs depend to some extent on the refinement method, we included only structures refined with the program shelxl (26), the most widely used protocol for ultra-high-resolution data. Although nearly all of the selected structures were refined with anisotropic Debye–Waller factors, we use only the isotropic AMSDs. (Relevant characteristics of the analyzed protein structures are collected in Table 2, which is published as supporting information on the PNAS web site, www.pnas.org).
Assessment of LDM Predictions.
Two different indicators, a merit function and a measure of association, are used here to quantitatively assess the agreement between calculated (σ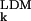 ) and experimental (σ
) and experimental (σ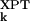 ) AMSDs. For most proteins, the σk distribution is highly skewed, with a sharp cutoff on the low-σk side, corresponding to atoms in densely packed core regions, and a pronounced tail on the high-σk side, corresponding to atoms in flexible loops or chain termini. The conventional merit function, the mean-square deviation, and the usual measure of association, Pearson's linear correlation coefficient, are unsuitable here because they can be dominated by a few outliers (27). We therefore use more robust indicators. As merit function, we use the relative mean absolute deviation, i.e., 〈|σ
) AMSDs. For most proteins, the σk distribution is highly skewed, with a sharp cutoff on the low-σk side, corresponding to atoms in densely packed core regions, and a pronounced tail on the high-σk side, corresponding to atoms in flexible loops or chain termini. The conventional merit function, the mean-square deviation, and the usual measure of association, Pearson's linear correlation coefficient, are unsuitable here because they can be dominated by a few outliers (27). We therefore use more robust indicators. As merit function, we use the relative mean absolute deviation, i.e., 〈|σ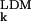 − σ
− σ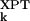 |〉 divided by 〈|σ
|〉 divided by 〈|σ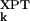 − 〈σ
− 〈σ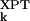 〉|〉. Normalization by the experimental σk variation allows us to compare Δ values from protein structures determined at ambient and cryogenic temperatures. As a measure of association, we use the Spearman rank-order correlation coefficient, ρ, which is based on the rank order of σk rather than its actual value (27). In contrast to Pearson's coefficient, the nonparametric correlation coefficient ρ can be meaningfully compared among different protein structures.
〉|〉. Normalization by the experimental σk variation allows us to compare Δ values from protein structures determined at ambient and cryogenic temperatures. As a measure of association, we use the Spearman rank-order correlation coefficient, ρ, which is based on the rank order of σk rather than its actual value (27). In contrast to Pearson's coefficient, the nonparametric correlation coefficient ρ can be meaningfully compared among different protein structures.
The Contact Density.
Although the LDM can be used to predict AMSDs for any atom type, we shall mainly discuss α carbons here. The contact density nk is then the number of nonhydrogen atoms within a distance RC from the reference α carbon k. The cutoff radius RC should be chosen to include most of the second peak in gk(r). For the calculations reported here, we have fixed RC to the radial distance, R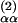 , of the second minimum in the Cα–Cα radial density, 4π r2gαα(r). For our data set, R
, of the second minimum in the Cα–Cα radial density, 4π r2gαα(r). For our data set, R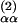 has a mean of 7.35 Å and a standard deviation of only 0.18 Å. Virtually identical results are obtained for any RC value in the range 7–10 Å (see Fig. 6, which is published as supporting information on the PNAS web site).
has a mean of 7.35 Å and a standard deviation of only 0.18 Å. Virtually identical results are obtained for any RC value in the range 7–10 Å (see Fig. 6, which is published as supporting information on the PNAS web site).
The contact density can be obtained simply by counting the number of nonhydrogen atoms whose mean positions are within RC of atom k. However, we can compensate to some extent for the shortcomings of the rigid-environment approximation by taking into account thermal displacements of neighbor atoms in the calculation of nk. We thus obtain nk by integrating the radial density nk(r) = 4π r2gk(r), with gk(r) given by Eq. 6, from r = 0 to r = RC. Because this requires knowledge of the AMSD variation that we want to predict, we perform a self-consistent calculation starting from a flat AMSD profile. When integrating nk(r), we should also use a lower cutoff of about 3.5 Å to exclude the covalent neighbors. However, because nk is heavily dominated by noncovalent neighbors, a lower cutoff has little effect.
The Cα contact density distribution P(n), calculated in this way with RC = 7.35 Å, is shown in Fig. 2 for the entire data set. For each of the 38 proteins, the contact density spans essentially the whole range, n ≈ 20–100, of the cumulative distribution. The small-n flank of P(n) is caused by α carbons in exposed termini and loops. On the other flank, P(n) drops sharply as the close-packing limit of n ≈ 100 is approached. At intermediate densities, P(n) exhibits a broad plateau for n ≈ 60–90. The mean contact density for all 6,231 α carbons is 67.5.
Figure 2.

Normalized contact density distribution (binned to integral n values) for all 6,231 α carbons in the set of 38 proteins (black) and for the uniform-sphere model described in the text (gray). For the latter case, the n = 100 bar has been truncated at about 25% of its real height, P(100) = 0.196.
Some α carbons have low contact density simply because they are near the surface of the protein. To examine this geometric contribution to the contact density, we calculated P(n) for a set of 38 uniformly packed spherical “proteins” with the equivalent-sphere radii of the real proteins. The resulting P(n) has two striking features (see Fig. 2). First, it is dominated by a large peak (truncated in Fig. 2) from atoms that are further than RC from the surface. These “core” atoms all have the same contact density, which we have set to 100. Second, P(n) decreases with n over a wide range, because the number of atoms in a spherical shell decreases towards the center. Because P(n) for real proteins displays neither of these features, we conclude that it mainly reflects local variations in packing density (including the detailed shape of the surface).
The contact density used in the following to predict AMSDs includes probability density from all nonhydrogen atoms within a sphere of radius RC, whether or not these atoms belong to the same protein molecule as the reference atom k. In other words, nk may contain contributions from atoms in neighboring protein molecules in the crystal. The contact density may also contain contributions from cofactors, such as heme groups, iron–sulphur clusters, and specifically bound metal ions and, in a few cases, from internally bound substrates. On the other hand, nk does not include any contributions from water molecules or cosolvents. (In a separate calculation on two BPTI structures, inclusion of the four internal water molecules was found to slightly improve the agreement between predicted and experimental AMSDs.) Most of the crystal structures in our data set contain multiple conformations of several residues, particularly for side-chains. For such residues, the dominant conformation and its associated AMSDs were used for the analysis.
Results
We consider first backbone flexibility, comparing predicted and experimental AMSD profiles for the α carbons along the polypeptide chain. Fig. 3 shows such profiles for Serratia marcescens endonuclease (241 residues). Predicted AMSDs were calculated self-consistently by using Eqs. 6 and 7 with RC = 7.32 Å. The LDM reproduces all prominent peaks in the AMSD profile and even captures minor features, such as the periodic AMSD variation often seen in α helices. The prediction quality indicators are Δ = 0.54 and ρ = 0.82. (For a compilation of quality indicators for all protein structures, see Table 3, which is published as supporting information on the PNAS web site.)
Figure 3.

Experimental (dots) and calculated (curve) AMSD profiles for the α carbons in S. marcescens endonuclease (1ql0). Predicted AMSDs are based on contact densities including all nonhydrogen protein atoms in the crystal. Experimental points are color coded according to secondary structure: α helix (blue), β strand (red), and turn (orange).
The LDM yields consistently accurate predictions of AMSD profiles; for our set of 38 protein structures, Δ = 0.72 ± 0.11 and ρ = 0.70 ± 0.09 (mean ± one standard deviation). For 82% of these structures, Δ < 0.78 and ρ > 0.60. The indicators Δ and ρ do not correlate with protein size or with secondary structure content (see Fig. 7, which is published as supporting information on the PNAS web site).
Among the analyzed structures, 31 were determined at cryogenic temperatures (85–120 K) and only 7 at ambient temperatures (287–300 K). Although proteins are less flexible at low temperature, any static lattice disorder should be unaffected by cryogenic quenching. The effect of lattice disorder might be modeled by fitting the two parameters in σk = σ0 + c/nk to the experimental AMSDs. Because the relation between σk and 1/nk remains linear, the correlation coefficient is not affected. Although the ambient-temperature structures have larger mean AMSD than the cryo-structures (0.54 versus 0.43 Å2), the agreement between predicted and experimental AMSD profiles is hardly better for the room-temperature structures: Δ = 0.70 ± 0.11 versus 0.73 ± 0.11 and ρ = 0.70 ± 0.12 versus 0.70 ± 0.08. This near-invariance suggests that lattice disorder does not contribute significantly to our data set, in accordance with the expectation that crystals diffracting to atomic resolution should exhibit little mosaicity (3).
As seen from Table 1, the self-consistent calculation of the contact density, according to Eq. 6, leads to a small improvement compared to a fixed-atom calculation. When the contact density includes only atoms in the same protein molecule as the reference α carbons, the predicted AMSD profile often exhibits peaks that are absent in the experimental profile. These spurious peaks tend to coincide with loop- and turn regions in intimate contact with adjacent protein molecules. In the LDM, the effect of such crystal contacts can be taken into account by including all nonhydrogen atoms in neighboring proteins that are within RC of any of the reference α carbons. This inclusion markedly improves the agreement with experiment (see Table 1, rows b and d), particularly for small proteins, which have a larger fraction of their residues involved in crystal contacts. Fig. 4 illustrates the effect of crystal contacts for Bacillus caldolyticus cold-shock protein (66 residues, RC = 7.71 Å). The agreement with experiment is clearly better when the contact density includes contributions from crystal neighbors (Δ = 0.77, ρ = 0.71) rather than just atoms in the reference molecule (Δ = 1.36, ρ = 0.58). The strongest crystal interaction, clearly manifested in the AMSD profile, involves a loop region (G21, E22, G23) in close contact with the N terminus (M1, Q2, R3) of a symmetry-related molecule.
Table 1.
Indicators for model predictions of Cα AMSDs for full protein set
| Model | Density* | 〈Δ〉† | Range of Δ | 〈ρ〉† | Range of ρ | |
|---|---|---|---|---|---|---|
| a | LDM | all/ref/fix | 0.89 ± 0.27 | 0.63–2.19 | 0.62 ± 0.09 | 0.41–0.80 |
| b | LDM | all/ref/scd | 0.86 ± 0.26 | 0.62–2.09 | 0.64 ± 0.09 | 0.43–0.81 |
| c | LDM | all/xtl/fix | 0.75 ± 0.12 | 0.52–1.21 | 0.67 ± 0.09 | 0.45–0.83 |
| d | LDM | all/xtl/scd | 0.72 ± 0.11 | 0.53–1.13 | 0.70 ± 0.09 | 0.49–0.85 |
| e | P-GNM | Cα/ref/fix | 1.08 ± 0.42 | 0.65–3.06 | 0.58 ± 0.17 | 0.05–0.84 |
| f | LDM | Cα/ref/fix | 1.02 ± 0.32 | 0.74–2.58 | 0.51 ± 0.11 | 0.20–0.70 |
| g | LDM | Cα/ref/scd | 0.97 ± 0.29 | 0.68–2.32 | 0.58 ± 0.08 | 0.42–0.75 |
In all calculations, the cutoff radius RC was set equal to the distance, R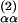 , of the second minimum in the Cα–Cα radial density.
, of the second minimum in the Cα–Cα radial density.
Contact density is based on nonhydrogen atoms (all) or Cα atoms (Cα) in reference molecule (ref) or entire crystal (xtl) and is calculated with fixed (fix) or self-consistently distributed (scd) atoms.
Mean value ± one standard deviation.
Figure 4.

Experimental (dots) and calculated (curves) AMSD profiles for the α carbons in B. caldolyticus cold-shock protein (1c9o). Predicted AMSDs are based on contact densities including all nonhydrogen protein atoms in the crystal (thick black curve) or only atoms in the same protein molecule as the reference α carbons (thin blue curve). Experimental points are color coded according to secondary structure: α helix (blue), β strand (red), and turn (orange).
The LDM is not limited to α carbons but can be applied to any or all atom types. Fig. 5 shows the AMSD profile for all 460 nonhydrogen atoms in a Kunitz-type domain from collagen (58 residues, RC = 7.40 Å). The LDM correctly predicts that side-chain atoms are more flexible than adjacent backbone atoms and, in most cases, also reproduces the relative flexibility of different side-chains. The overall agreement with experiment is comparable to that found for α carbons only. Fig. 5 also shows, in several instances, that the LDM correctly identifies side-chains with reduced flexibility because of crystal contacts.
Figure 5.

AMSD profile for all nonhydrogen atoms in the Kunitz-type domain (C5) from the α-3 chain of human type VI collagen (2knt). Circles represent experimental backbone (filled) and side-chain (open) AMSDs, and curves represent predicted AMSDs on the basis of contact densities including all nonhydrogen protein atoms in the crystal (thick black curve, Δ = 0.63, ρ = 0.72) or only atoms in the same protein molecule as the reference atoms (thin blue curve, Δ = 0.64, ρ = 0.76). Experimental points for atoms in disulfide Cys residues are colored orange.
Discussion
Interactions, Dynamics, and Flexibility.
Considering its extreme simplicity, the LDM is remarkably successful. Its central idea, that AMSD profiles are governed by spatial variations in packing density, might seem to ignore all interactions apart from excluded volume. In particular, the LDM does not recognize covalent bonds or hydrogen bonds explicitly. However, all types of interactions, specific as well as nonspecific, are implicitly manifested in the LDM via their effect on the local density. Elements of regular secondary structure, such as α helices and β sheets, not only are extensively hydrogen-bonded but also are densely packed. Disulfide bridges not only impose connectivity constraints on conformational motions, but, by forcing backbone segments together, also increase the local atomic density. Thus, for example, the LDM accurately predicts the AMSDs of all six disulfide cysteine residues in the Kunitz-type domain C5 (see Fig. 5).
Protein conformational flexibility may be thought of and rationalized in different ways. The most widely adopted viewpoint is to interpret AMSDs in terms of conformational motion. Ultimately, however, both flexibility and dynamics are determined by interactions. It is therefore possible, in principle, to predict AMSDs from detailed interaction models. This approach is computationally demanding and has met with limited success so far, partly because interactions in proteins are extremely complex and not yet fully understood. By relating flexibility directly to local density, the LDM offers a conceptual shortcut that bypasses the intricacies of detailed interaction models.
As demonstrated here, variations in small-amplitude structural flexibility within native proteins are largely governed by spatial inhomogeneities in packing density. By unifying these aspects of protein structure, the LDM contributes to our understanding of the physical properties of proteins. On the other hand, the success of the LDM implies that (isotropic) crystallographic B factors supply very little independent information not already present in the mean atomic coordinates. It should be possible to improve the accuracy of LDM predictions by including ordered water molecules buried in internal cavities or trapped at crystal contacts, by using weight factors for different (united) atom types, or by optimizing the cutoff radius for each protein structure. Yet, truly quantitative accuracy cannot be expected from such a simple model. Further insight about the determinants of structural flexibility might come from a systematic study of those instances where the LDM predictions are least accurate. In some cases, such discrepancies might be traced to unresolved conformational substates; in other cases, they might reflect deficiencies in the model.
Other AMSD Correlations.
Protein flexibility correlates with a variety of physical properties, such as solvent exposure, distance from center-of-mass, and secondary structure (3–5, 12, 28, 29). The most frequently invoked correlation is that with solvent-accessible surface area (SASA) (5, 12, 29). The simplest linear relationship between AMSD (σk) and SASA (ak) of atom k is σk = σ0 + cak. Like the uniform-sphere model discussed above, this model predicts that all buried atoms have the same AMSD, σ0. Although the SASA model identifies many of the prominent peaks in the AMSD profile, arising from exposed loop- and turn residues, the predicted AMSD variation is far too small, and most of the fine structure is lost. To quantitatively assess the SASA model, we adjusted the two parameters σ0 and c by minimizing the mean absolute deviation between predicted and experimental α carbon AMSDs, using for ak the SASA (1.4-Å probe radius) for the backbone atoms of residue k. This yields rather poor agreement for our data set. For example, for protein G Igg-binding domain III (2igd, 61 residues), the SASA model yields Δ = 0.85 and ρ = 0.49, whereas the LDM yields Δ = 0.53 and ρ = 0.83; and for S. marcescens endonuclease (1ql0, 241 residues), the SASA model yields Δ = 0.82 and ρ = 0.54, whereas the LDM yields Δ = 0.54 and ρ = 0.82.
Another approach for predicting α carbon AMSDs in proteins (21) has been inspired by a simple model of rubber elasticity (30, 31). In this Gaussian phantom network model (GNM), an elastomer material is modeled as a network of noninteracting polymer segments, where any two connected junctions are subject to a restoring force proportional to their separation and with a force constant inversely proportional to the segment contour length (30). This force, which increases with separation, is entirely generated by the configurational entropy of the polymer segment; all mechanical interactions, even excluded volume, are neglected. To prevent the model network from collapsing, the mean positions of the junctions are taken to be fixed by external forces. The GNM leads to a harmonic POMF wk(uk) and, consequently, to a Gaussian displacement PDF Fk(uk) (30, 31).
In the protein version of the GNM, here denoted as P-GNM, the α carbons are regarded as junctions in a virtual network characterized by pairwise interactions of the form νki = (γ/2) ɛki |rki − r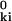 |2 = (γ/2) ɛki |uk − ui|2, where ɛki = 1 if r
|2 = (γ/2) ɛki |uk − ui|2, where ɛki = 1 if r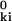 ≤ RC and ɛki = 0 otherwise. In contrast to the entropic interaction in the original GNM, this pair potential is postulated without an underlying physical model. This fundamental difference between the two models reflects the fact that the junctions are physically linked in the real network (GNM) but not in the virtual network (P-GNM). The real interaction between α carbons is negligibly weak and monotonically decaying (as r
≤ RC and ɛki = 0 otherwise. In contrast to the entropic interaction in the original GNM, this pair potential is postulated without an underlying physical model. This fundamental difference between the two models reflects the fact that the junctions are physically linked in the real network (GNM) but not in the virtual network (P-GNM). The real interaction between α carbons is negligibly weak and monotonically decaying (as r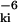 ) at most separations of interest here.
) at most separations of interest here.
As in the original GNM, the postulated harmonic form of the pair interaction leads to a Gaussian displacement PDF with the AMSD given by
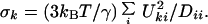 |
8 |
Here, D is the diagonal eigenvalue matrix, and U is the orthogonal eigenvector matrix that diagonalizes the symmetric matrix Γ according to UT Γ U = D. The so-called Kirchoff adjacency matrix Γ (32) has off-diagonal elements Γki = −ɛki, whereas its diagonal elements are the Cα–Cα contact densities, Γkk = ∑i≠kɛki = nk. When expressed in terms of the atomic displacements uk, the partition function diverges, because the adjacency matrix is singular (rank N − 1) in the P-GNM. Therefore, the inverse of Γ does not exist. In Eq. 8, the zero eigenvalue, corresponding to a uniform displacement of all atoms, is omitted from the sum. The adjacency matrix is dominated by its diagonal elements, giving the number of α carbons within RC (averaging 8.3 ± 0.6 for our data set). If all off-diagonal elements (−1 or 0) in Γ are set to zero, one obtains σk = 3 kBT/(γ nk), which coincides with the LDM result in Eq. 7, with the correspondence λ ↔ γ/(2 kBT).
We have tested the P-GNM on our set of 38 high-quality protein structures. As seen from Table 1 (rows d and e), the LDM predicts α carbon AMSDs considerably more accurately than the P-GNM. It is also of interest to compare P-GNM and LDM predictions when both models are based on α carbons only. As seen from Table 1 (rows e and f), the off-diagonal elements of Γ do not substantially improve the AMSDs. (This is also the case when both models are based on all nonhydrogen atoms.) Furthermore, when the contact density is calculated self-consistently, the LDM performs slightly better (and more consistently) than the P-GNM, even when only α carbons are included in the contact density (rows e and g).
A major weakness of the P-GNM is its obscure physical basis. Although the mathematical formalism conforms closely to the original GNM, the underlying physics is quite different. For example, in the original GNM, γ is proportional to T, making the AMSDs independent of temperature, in contrast to what is observed for proteins (33). The P-GNM approach has been justified a posteriori through its agreement with experimental AMSDs (34, 35). We believe that the reason for the relative success of the P-GNM can be found in the physical justification given here for the LDM, to which it reduces after a numerical approximation (neglect of off-diagonal Γ elements). The LDM is not only more accurate than the P-GNM; it is also more computationally efficient. Because it does not involve any matrix diagonalization, the LDM can readily be used to predict AMSDs for all nonhydrogen atoms. To the extent that NMR derived second-rank orientational order parameters for peptide N—H bonds correlate with (crystal-contact-corrected) x-ray-derived AMSDs (29, 35–37), they can also be predicted by the LDM.
Supplementary Material
Acknowledgments
This work was supported by the Swedish Research Council.
Abbreviations
- AMSD
atomic mean-square displacement
- GNM
Gaussian network model
- P-GNM
protein version of GNM
- LDM
local density model
probability distribution function
- POMF
potential of mean force
- SASA
solvent accessible surface area
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Willis B T M, Pryor A W. Thermal Vibrations in Crystallography. Cambridge, U.K.: Cambridge Univ. Press; 1975. [Google Scholar]
- 2.Trueblood K N, Bürgi H-B, Burzlaff H, Dunitz J D, Gramaccioli C M, Schulz H H, Shmueli U, Abrahams S C. Acta Crystallogr A. 1996;52:770–781. [Google Scholar]
- 3.Ringe D, Petsko G A. Prog Biophys Mol Biol. 1985;45:197–235. doi: 10.1016/0079-6107(85)90002-1. [DOI] [PubMed] [Google Scholar]
- 4.Frauenfelder H, Petsko G A, Tsernoglou D. Nature (London) 1979;280:558–563. doi: 10.1038/280558a0. [DOI] [PubMed] [Google Scholar]
- 5.Artymiuk P J, Blake C C F, Grace D E P, Oatley S J, Phillips D C, Sternberg M J E. Nature (London) 1979;280:563–568. doi: 10.1038/280563a0. [DOI] [PubMed] [Google Scholar]
- 6.Swaminathan S, Ichiye T, van Gunsteren W, Karplus M. Biochemistry. 1982;21:5230–5241. doi: 10.1021/bi00264a019. [DOI] [PubMed] [Google Scholar]
- 7.York D M, Wlodawer A, Pedersen L G, Darden T A. Proc Natl Acad Sci USA. 1994;91:8715–8718. doi: 10.1073/pnas.91.18.8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hünenberger P H, Mark A E, van Gunsteren W F. J Mol Biol. 1995;252:492–503. doi: 10.1006/jmbi.1995.0514. [DOI] [PubMed] [Google Scholar]
- 9.Eastman P, Pellegrini M, Doniach S. J Chem Phys. 1999;110:10141–10152. [Google Scholar]
- 10.Rejto P A, Freer S T. Prog Biophys Mol Biol. 1997;66:167–196. doi: 10.1016/s0079-6107(97)85629-5. [DOI] [PubMed] [Google Scholar]
- 11.Garcia A E, Krumhansl J A, Frauenfelder H. Proteins. 1997;29:153–160. [PubMed] [Google Scholar]
- 12.Watenpaugh K D, Sieker L C, Jensen L H. J Mol Biol. 1980;138:615–633. doi: 10.1016/s0022-2836(80)80020-9. [DOI] [PubMed] [Google Scholar]
- 13.Bhaskaran R, Prabhakaran M, Jayaraman G, Yu C, Ponnuswamy P K. J Biomol Struct Dyn. 1996;13:627–639. doi: 10.1080/07391102.1996.10508875. [DOI] [PubMed] [Google Scholar]
- 14.Richards F M. J Mol Biol. 1974;82:1–14. doi: 10.1016/0022-2836(74)90570-1. [DOI] [PubMed] [Google Scholar]
- 15.Chothia C. Nature (London) 1975;254:304–308. doi: 10.1038/254304a0. [DOI] [PubMed] [Google Scholar]
- 16.Harpaz Y, Gerstein M, Chothia C. Structure (London) 1994;2:641–649. doi: 10.1016/s0969-2126(00)00065-4. [DOI] [PubMed] [Google Scholar]
- 17.Liang J, Dill K A. Biophys J. 2001;81:751–766. doi: 10.1016/S0006-3495(01)75739-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuntz I D, Crippen G M. Int J Pept Protein Res. 1979;13:223–228. doi: 10.1111/j.1399-3011.1979.tb01872.x. [DOI] [PubMed] [Google Scholar]
- 19.Carugo O, Argos P. Proteins. 1998;31:201–213. [PubMed] [Google Scholar]
- 20.Tirion M M. Phys Rev Lett. 1996;77:1905–1908. doi: 10.1103/PhysRevLett.77.1905. [DOI] [PubMed] [Google Scholar]
- 21.Bahar I, Atilgan A R, Erman B. Folding Des. 1997;2:173–181. doi: 10.1016/S1359-0278(97)00024-2. [DOI] [PubMed] [Google Scholar]
- 22.Hinsen K. Proteins. 1998;33:417–429. doi: 10.1002/(sici)1097-0134(19981115)33:3<417::aid-prot10>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 23.Tama Y, Sanejouand Y-H. Protein Eng. 2001;14:1–6. doi: 10.1093/protein/14.1.1. [DOI] [PubMed] [Google Scholar]
- 24.Hill T L. Statistical Mechanics. New York: McGraw–Hill; 1956. [Google Scholar]
- 25.Hobohm U, Sander C. Protein Sci. 1994;3:522–524. doi: 10.1002/pro.5560030317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sheldrick G M, Schneider T R. Methods Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- 27.Press W H, Teukolsky S A, Vetterling W T, Flannery B P. Numerical Recipes in C. 2nd Ed. Cambridge, U.K.: Cambridge Univ. Press; 1992. [Google Scholar]
- 28.Kuriyan J, Weis W I. Proc Natl Acad Sci USA. 1991;88:2773–2777. doi: 10.1073/pnas.88.7.2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buck M, Boyd J, Redfield C, MacKenzie D A, Jeenes D J, Archer D B, Dobson C M. Biochemistry. 1995;34:4041–4055. doi: 10.1021/bi00012a023. [DOI] [PubMed] [Google Scholar]
- 30.James H M. J Chem Phys. 1947;15:651–668. [Google Scholar]
- 31.Flory P J. Proc R Soc London Ser A. 1976;351:351–380. [Google Scholar]
- 32.Eichinger B E. Macromolecules. 1972;5:496–505. [Google Scholar]
- 33.Tilton R F, Dewan J C, Petsko G A. Biochemistry. 1992;31:2469–2481. doi: 10.1021/bi00124a006. [DOI] [PubMed] [Google Scholar]
- 34.Bahar I. Rev Chem Eng. 1999;15:319–347. [Google Scholar]
- 35.Haliloglu T, Bahar I. Proteins. 1999;37:654–667. doi: 10.1002/(sici)1097-0134(19991201)37:4<654::aid-prot15>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 36.Powers R, Clore G M, Garrett D S, Gronenborn A M. J Magn Reson B. 1993;101:325–327. [Google Scholar]
- 37.Goodman J L, Pagel M D, Stone M J. J Mol Biol. 2000;295:963–978. doi: 10.1006/jmbi.1999.3419. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}