

Abstract
Two-way communication between neurons and nonneural cells called glia is essential for axonal conduction, synaptic transmission, and information processing and thus is required for normal functioning of the nervous system during development and throughout adult life. The signals between neurons and glia include ion fluxes, neurotransmitters, cell adhesion molecules, and specialized signaling molecules released from synaptic and nonsynaptic regions of the neuron. In contrast to the serial flow of information along chains of neurons, glia communicate with other glial cells through intracellular waves of calcium and via intercellular diffusion of chemical messengers. By releasing neurotransmitters and other extracellular signaling molecules, glia can affect neuronal excitability and synaptic transmission and perhaps coordinate activity across networks of neurons.
Historically, neuroscientists suspected that nonneural cells called glial cells might contribute to information processing in the brain. However, the supporting evidence was comparatively meager because glia have been studied with tools used to probe the electrical excitability of neurons. Although many of the same voltage-sensitive ion channels and neurotransmitter receptors of neurons are found in glia (1), glial cells lack the membrane properties required to fire action potentials. Nevertheless, these ion channels and electrogenic membrane transporters allow glia to sense indirectly the level of neuronal activity by monitoring activity-dependent changes in the chemical environment shared by these two cell types. Advanced imaging methods, which allow observation of changes in intracellular and extracellular signaling molecules in real time, show that glia communicate with one another and with neurons primarily through chemical signals rather than electrical signals (see Movie S1). Many of these signaling systems overlap with the neurotransmitter signaling systems of neurons, but some are specialized for glial-glial and neuron-glial communication.
This expanded relationship between neurons and glia is challenging traditional neurobiology. Contrary to dogma, some neurons in the central nervous system (CNS) use rapid neurotransmission not only at synapses with other neurons, but also at synapses with glia as well. Furthermore, neural activity releases chemical messengers not only at synaptic junctions, but also in extrasynaptic regions of neurons. This suggests functions for neuron-glial communication beyond those associated with synaptic transmission. For example, glia can regulate synapse formation, can control synaptic strength, and may participate in information processing by coordinating activity among sets of neurons. Conversely, neural impulse activity regulates a wide range of glial activities, including their proliferation, differentiation, and myelination.
Glial Cells
There are three categories of glia: Schwann cells and oligodendrocytes—the myelin-forming cells of the peripheral nervous system (PNS) and CNS, respectively, that wrap layers of myelin membrane around axons to insulate them for impulse conduction—and astrocytes, which are closely associated with neurons in the brain but do not form myelin (Fig. 1). Astrocytes ensheath synaptic junctions, associate with nodes of Ranvier, and respond to disease and injury by clearing cellular debris, secreting trophic factors and forming scars. The name refers to their stellate morphology observed in histological preparations, but their morphology varies widely (Fig. 1B). Some astrocytes span the entire width of the brain radially from the hollow ventricles to the pial surface, providing scaffolding along which neurons migrate during fetal development. Others stretch from blood capillaries to neurons, transporting ions and other substances to sustain neurons and to regulate the extracellular environment. So intimate is the association between astrocytes and neurons that monitoring activity of these nonneuronal cells is a reliable surrogate for measuring neural activity. The marvelous imaging techniques that provide a window into brain function for both basic research and medical diagnosis actually rely on responses of astrocytes to the changing metabolic demands of neurons (2, 3). The prevailing view, based on histology, has been that the stellate processes of astrocytes form a tightly intermingled web throughout the brain, but now it seems that these cells are much larger than previously thought, and the processes of adjacent astrocytes do not overlap extensively (4). Thus, parts of the brain (such as the hippocampus where memories are formed) are divided by astrocytes into separate compartments, each one the sole domain of an individual astrocyte. The functional significance of this structural organization is completely unknown. In the CNS, oligodendroglia (Fig. 1C) extend multiple processes to myelinate several axons at a time. In the PNS, a single highly versatile cell, the Schwann cell, performs all of the functions of CNS astrocytes and oligodendrocytes (Fig. 1D): forming myelin (Fig. 1A), ensheathing synaptic junctions, and bundling small-diameter axons together.
Fig. 1.
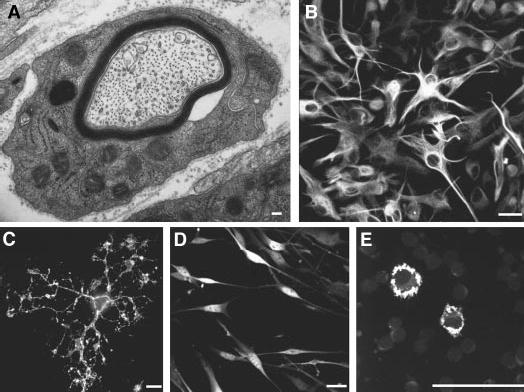
Major types of glial cells in the nervous system. (A) An electron micrograph of a mouse sensory axon in the process of becoming myelinated by a Schwann cell. Note the multiple layers of dark myelin membrane that the Schwann cell is wrapping around the nerve axon to insulate it for rapid long-distance conduction of neural impulses. (B) Astrocytes do not form myelin, but they form networks of communicating cells within the CNS, and they interact with neurons to support and modulate many of their functions. (C) Olidgodendrocytes form myelin around CNS axons with multiple cellular extensions from the cell body. (D) Schwann cells form myelin around PNS axons and ensheath multiple small unmyelinated axons into bundles. (E) Microglia enter the CNS early in development from embryonic cells of nonectodermal origin and they respond to brain injury and disease. These cells were grown in cell culture and labeled by fluorescence immunocytochemistry for specific proteins expressed by each cell type (GFAP, O4, S100, OX-42, in B to E, respectively). Scale bars, 100 nm (A), 25 μm (B, C).
Microglia make up a fourth category of nonneuronal cells in the brain (Fig. 1E). In contrast to macroglia, which derive embryologically from ectodermal precursors within the nervous system, microglia derive from bone marrow monocyte precursors (5). Like their counterparts in the hematopoietic system, microglia respond to injury or disease by engulfing cellular debris and triggering inflammatory responses. New findings suggests that microglia can respond to neural impulse activity, and thereby mediate neuroimmune interactions, for example in chronic pain conditions (6).
Glial Regulation of Synaptic Transmission
Fluorescent imaging of intracellular Ca2+ fluxes (Fig. 2 and Movie S1) reveals how neurons and glia communicate. In the brain, Ca2+ responses are seen in hippocampal astrocytes when axons are stimulated to fire action potentials, (7, 8). In the PNS, stimulation of motor axons causes Ca2+ responses in terminal Schwann cells, which are specialized glia that ensheath the synaptic junction between motor nerve endings and muscles (9–11).
Fig. 2.
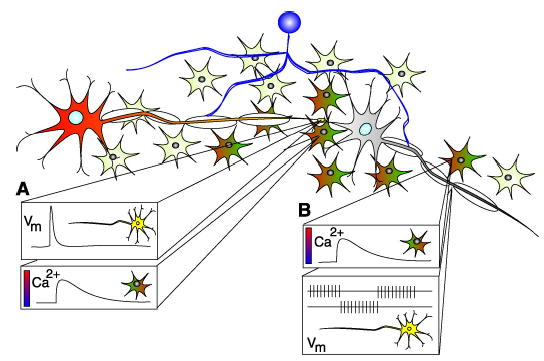
Calcium imaging reveals communication between neurons and glia. (A) Molecules released during synaptic transmission bind receptors on glia that cause increases in intracellular Ca2+ (rainbow colored cells), which are propagated as waves through glial networks. (B) Increases or decreases in axonal firing may coincide with the passage of a glial Ca2+ wave. Oligodendrocytes (purple) myelinate CNS axons. vm, membrane voltage.
Terminal Schwann cells not only monitor synaptic activity by detecting neuron-glial signaling molecules coreleased from the neuron with neurotransmitter, they control the strength of synaptic transmission by regulating the release of neurotransmitter from the nerve terminal. The process is initiated when G protein–coupled receptors on the Schwann cells become activated by neurotransmitter, extracellular ATP, or adenosine, which are released from the nerve terminal. This functional interaction between the synapse and synaptic glia was shown by injecting agents into terminal Schwann cells to modulate specific intracellular signaling pathways, while monitoring synaptic strength by electrophysiological recording (fig. S1). Stimulating guanosine 5′-triphosphate (GTP)–binding protein activity in terminal Schwann cells reduced synaptic strength (12) because of a decrease in neurotransmitter release from the presynaptic nerve terminal. In addition, inhibiting GTP-binding protein activity in the Schwann cell prevented the decrease in synaptic strength that normally accompanies prolonged trains of action potentials (12). This well studied response, termed synaptic depression, was thought to result from mechanisms operating entirely within the nerve terminal itself. But increasing the Ca2+ concentration in the terminal Schwann cell (by flash photolysis of caged Ca2+ or by injecting inositol trisphosphate) caused an increase in synaptic strength that was sustained for more than 30 min (13). Thus, signaling pathways activated by a rise in cytoplasmic Ca2+ in the Schwann cell may increase synaptic strength, possibly through release of prostaglandins (13). In contrast, signaling pathways linked to G-protein activation affect synaptic depression, possibly through release of nitric oxide (14). Together, these signaling networks in the terminal Schwann cell integrate the activity of the synapse and balance the strength of the connection.
Striking parallels are seen in the CNS, where astrocytes ensheath synaptic junctions in the brain and also use purinergic receptors for neuron-glial signaling. A rise in cytoplasmic Ca2+ in an astrocyte is associated with changes in synaptic strength in adjacent synapses in culture (15, 16) or in the intact retina (17). Interestingly, sudden increases or decreases in spike firing are both observed after a transient rise in astrocytic cytoplasmic Ca2+. Whether the response to astrocyte activation is inhibitory or excitatory may be determined by differential effects on neurotransmitter release or uptake (18) or by astrocytes activating an intervening inhibitory neuron in the synaptic circuit (19). In each case, imaging techniques reveal that the rise in cytoplasmic Ca2+ concentration in astrocytes is accompanied by an increase in extracellular concentration of ATP (20–22).
Astrocyte-Astrocyte Communication
Like neurons, astrocytes are functionally coupled over large areas of the brain. However, in addition to specialized cell contact–mediated communication via gap junctions—intercellular channels that allow ions and small molecules to pass between adjoining cells—astrocytes also communicate by extracellular signaling molecules that are released and propagated among networks of astrocytes in a chain reaction (fig. S2). This astrocyte-astrocyte signaling engages with chemical signaling at neuronal synapses. Fluorescent imaging methods show that a large number of substances, including the excitatory neurotransmitter glutamate, cause cytoplasmic Ca2+ changes in astrocytes (23, 24 ). Such methods also reveal ATP secretion from astrocytes by an unknown mechanism (20), as well as the accompanying calcium-dependent secretion of glutamate (25). Extracellular ATP spreads to neighboring astrocytes and activates membrane receptors (P2Y receptors) generating an increase in intracellular Ca2+, which spreads through neighboring astrocytes like ripples on a pond (21). Through this mechanism, synaptic signals could then resonate broadly through the brain through glial cells, and this could, in theory, modulate the efficacy of nearby synapses.
Terminal Schwann cells affect synaptic strength by regulating neurotransmitter release from the presynaptic neuron, but astrocytes can influence synaptic strength by their own actions. Imaging shows that the release of excitatory neurotransmitter glutamate from astrocytes, which accompanies the increase in cytoplasmic Ca2+, augments neurotransmitter released by the neuron (16). How can neurotransmitter release from astrocytes be reconciled with the well-established role of these cells in clearing neurotransmitter from the synaptic cleft (26)? One possibility is that these functions are either segregated in time or confined to specific subcellular regions of the astrocyte, thus subserving the appropriate function at different places. This implies substantial involvement of astrocytes with information processing in the brain.
Both Ca2+ wave propagation and coupling of astrocytes at gap junctions can be regulated in response to neuronal activity. Phosphorylation by calmodulin kinase regulates gap junctions, increasing coupling in response to elevated extracellular K+ and promoting clearance of K+ from the extracellular space after increased neuronal activity (27 ). High levels of extracellular K+ also decrease propagation of the intercellular Ca2+ wave (apparently by depleting intracellular Ca2+ stores), which reduces glutamate release and prevents overstimulation of synapses (28).
Considering that the spread of extracellular ATP from astrocyte to astrocyte is a critical component in communication among these glial cells, it will be essential to determine the mechanism for the nonvesicular release of ATP. The mechanism for glutamate release from astrocytes is different from the mechanism for ATP release (these two extracellular signaling molecules act synergistically). Glutamate release depends on a rise in Ca2+ in the astrocytes, but ATP release does not. Astrocytes have many of the same synaptic vesicle proteins associated with neurotransmitter release from neurons, and when these molecules are disrupted by specific toxins, the release of glutamate from astrocytes is inhibited (29). ATP release may involve the ATP-binding cassette transporters (30), the cystic fibrosis transmembrane conductance regulator or other stretch-activated Cl− channel (31), or gap junctions unpaired with adjacent cells (hemijunctions) (32, 33). Recent experiments reveal that ATP release may be associated with transient opening of a nonselective membrane channel, and that simple diffusion of extracellular ATP may regulate expansion of Ca2+ waves through astrocytes (34 ).
Glial Regulation of Synaptic Strength
Glutamate release from astrocytes may involve membrane channels or transporters or extracellular synthesis from secreted precursors such as N-acetylaspartylglutamate (35). Also, substances other than glutamate and ATP, for example, the inhibitory neurotransmitter γ-aminobutyric acid (GABA) (36 ), peptides (37 ), or growth factors (38), may participate in neuron-glial signaling. For example, amino acids in the mammalian body are typically in the levorotatory optical isomer, but d-serine is found in the brain in astrocytes that ensheath synapses containing N-methyl-d-aspartate (NMDA) glutamate receptors, which are involved in synaptic plasticity and memory. d-Serine released by these astrocytes may stimulate NMDA receptors on the postsynaptic membrane of neurons by activating the “glycine” site on the NMDA receptor (39) (Fig. 3). Although glycine was presumed to be the endogenous coagonist acting together with glutamate to activate the NMDA receptor, d-serine—synthesized from l-serine by a serine racemase in astrocytes (40)—may be the endogenous ligand for this modulatory site on the NMDA receptors of postsynaptic neurons (41).
Fig. 3.
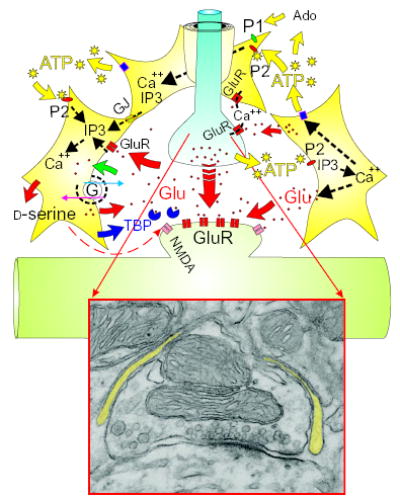
Synaptic astrocytes (yellow) regulate synaptic transmission by responding to signaling molecules, such as ATP and glutamate, released from the presynaptic neuron during synaptic transmission. Astrocytes communicate with adjacent astrocytes via gap junctions (GJ) and with distant astrocytes via extracellular ATP. The rise in Ca2+ causes release of glutamate from astrocytes, and ATP is released via an unknown mechanism, which propagates ATP signaling to adjacent cells. Astrocytes may also regulate synaptic transmission by uptake of glutamate from the synaptic cleft via membrane transporters (green arrow) or the release of glutamate upon reversal of the transporter induced by elevated intracellular Na+ (red arrow). Other substances, such as d-serine, strengthen synaptic transmission by coactivating NMDA receptors in the postsynaptic membrane, or reduce synaptic transmission by secreting transmitter-binding proteins ( TBP). (Inset) An electron micrograph of a synapse surrounded by an astrocyte (yellow) from the spinal cord of rat (Courtesy of M. H. Ellisman, National Center for Microscopy and Imaging Research, University of California, San Diego). GluR, glutamate receptor; Ado, adenosine; IP3, inositol trisphosphate; P1, adenosine receptor; P2, ATP receptor.
The traditional functions of glia in regulating the ionic extracellular environment, clearing neurotransmitter from the synaptic cleft (26) and responding to the metabolic demands of synaptic transmission (3), provide abundant possibilities for regulating synaptic strength in either direction. For instance, a newly described mechanism for modulating synaptic transmission involves secretion by synaptic glia of a neurotransmitter-binding protein into the synaptic cleft of CNS neurons of the mollusc Lymnaea stagnalis. This protein resembles the acetylcholine neurotransmitter receptor, but lacks the ion channel. When secreted by glia in response to neuronal firing, it binds to neurotransmitter released from the presynaptic membrane and suppresses transmission (42) (Fig. 3).
Glial Participation in Information Processing
There are curious differences in the responses of astrocytes in culture and in brain slices. In the hippocampus, local glutamate application via flash photolysis with an ultraviolet laser caused Ca2+ responses in only a few neighboring astrocytes (43), rather than the chain reaction that spreads widely through cultured astrocytes (24). This raises the intriguing possibility that there may be preferred astrocytic circuits in the brain. These circuits could result from differences among astrocytes that become lost in cell culture or result from preferred physical connections among astrocytes that would limit information flow to particular networks of these cells.
Could astrocytic circuits process information in parallel with neuronal circuits? By manipulating the principal excitatory neurotransmitter in the brain, astrocytes would seem equipped with the power to regulate synaptic strength in either direction. Recently, withdrawal of astrocytic processes from neurons in the hypothalamus has been shown to control glutamate clearance and synaptic efficacy (44). The ability to control glutamate levels in the synapse, together with the Ca2+-dependent mechanisms of long-distance communication, may enable astrocytes to participate in information processing by bridging the “hardwired” lines of neuronal contacts. If this is the case, how plastic are these astrocytic circuits, and how might glia contribute to neuronal plasticity?
Synapse Formation and Remodeling
Sensory input remodels neuronal topography and the distribution of synaptic connections during development. The instructive influence of glia on migration and morphology of neurons during brain morphogenesis, mediated by changes in cell adhesion molecules and secretion of neurotrophic factors, suggests that glia may participate in this activity-dependent remodeling. In ol-factory glomeruli (45), and in the visual relay nucleus (lateral geniculate nucleus) (46 ), glial boundaries precede formation of neuronal topography, and abnormal sensory input interferes with the formation of appropriate glial boundaries. This implies that afferent neural activity influences glia, which subsequently interferes with the glial instructive influence on neuronal organization and synapse formation (47 ).
Studies in culture show that astrocytes play a powerful part in regulating the formation and maintenance of synaptic connections between neurons during development. The number of synapses a neuron forms may not be entirely intrinsic to the neuron; it may be influenced by extrinsic factors, notably continuous signaling from astrocytes. When astrocytes were excluded from retinal ganglion neuron cultures, electrophysiological recordings revealed little synaptic activity. Adding astrocytes back to the cultures increased synaptic strength through a presynaptic effect causing more neurotransmitter to be released (48). Fluorescent imaging revealed that more synaptic connections were formed in astrocyte cultures, and individual connections were more active in releasing and recycling synaptic vesicles (49). Analysis of astrocyte-conditioned medium and functional studies revealed that apoE/cholesterol particles secreted from astrocytes become internalized by neurons, which leads to increased cholesterol in neuronal membranes, where it promotes an increase in synapse number (50). The extent to which synaptogenesis depends on cholesterol as a limiting membrane component or cholesterol-mediated intracellular signaling remains to be determined.
Synapses on Glia?
There are presumed to be two general mechanisms enabling glia to respond electrically to the neurotransmitter glutamate released by the presynaptic neuron: activation of various glutamate receptors on glia (51) or electrogenic transporter currents (26) that depolarize the glial membrane during the process of removing glutamate from the extracellular space. Patch electrode recordings from glial cells in the hippocampus, for example, show depolarizing responses when axons are stimulated electrically to release glutamate (52). This shows that, in violation of the usual serial flow of information along chains of neurons, the influence of neurotransmitters extends well beyond the synaptic cleft to include perisynaptic astrocytes and possibly nearby synapses. In performing electrophysiological recordings in glial cells of the hippocampus, occasional currents with extremely fast kinetics, peaking within 250 μs, have been observed. This is too fast to be accounted for by spillover of neurotransmitter from the synaptic cleft. Immunologically and morphologically, these glial cells are similar to cultured oligodendrocyte precursor (O2A) glia. Glia with these characteristics mature into oligodendrocytes, which form myelin around CNS axons (Fig. 1C). Electron microscopic analysis revealed ultrastructural specializations characteristic of synaptic contacts between axons of the Schaeffer collaterals and these oligodendrocyte precursor cells. Recordings from these glia demonstrate that, like neuronal synapses, they generate spontaneous and evoked excitatory synaptic currents, through activation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors (53), and inhibitory synaptic currents, through activation of GABAA receptors.
There are several possible functions for this neuron-glial synapse: It may permit glia to have a neuromodulatory role, perhaps integrating activity across groups of axons, or regulating myelination in accordance with functional activity in axons. Whatever the purpose, these cells and neuron-glial synapses are not limited to a transient period in development; they are present in the adult brain as well. Still to be determined is whether release of glutamate from astrocytes might activate oligodendrocyte precursor cells, whether the O2A-like cells have the biochemical release apparatus for secreting neurotransmitter, or whether these glia might release other neuromodulatory substances (such as trophic factors) in response to electrical activity. Already, these new findings are undermining traditional defining features of neurons, astrocytes, and oligodendrocytes.
Genesis of Neurons and Glia
Neurons and macroglia were once thought to derive from different embryonic stem cells, but we now know that both can be derived from the same stem-cell precursors. Their fate is determined in part by exposure to growth factors and by developmental programs that ultimately restrict the precursors to either glial or neuronal fates. Neural stem cells persisting in the adult nervous system participate in plasticity and regeneration, but they have the immunocytochemical markers of glia (54). Examination of neurogenesis early in development using green fluorescent protein–tagged radial glia, reveals that not only do these glia provide the scaffolding upon which newly-formed neurons migrate into the cortex, they can generate neurons by asymmetrical cell division (55). The single transmembrane Notch receptor is a major determinant of whether precursor cells develop into neurons or glia. Activation of Notch signaling promotes radial glial identity and maintains the proliferative state (56). Similarly, formation of Muller glia by progenitor cells in the retina (57), Schwann cells in the PNS (58), and astrocytes in the adult hippocampus (59) are stimulated by Notch activation. Whereas Notch signaling is necessary for glial specification, neuron-glial interactions (and interactions with target cells) are critical in specifying final cell fate through cell contact–mediated and soluble extrinsic signals (60). Research on secretion of neurotrophins has shown (61) that secretion of many soluble factors is modulated by neural impulse activity.
Neuron-Glial Communication in Nonsynaptic Regions
Communication between neurons and glia is not limited to the synapse. High-frequency axonal firing causes phosphorylation of myelin basic protein (MBP), a component of myelin in the white matter tract of the hippocampus. This activity-dependent axon-oligodendrocyte signaling is mediated by nitric oxide released from axons, which stimulates phosphorylation of MBP in oligodendrocytes by a mitogen-activated protein kinase (MAP-K)–dependent mechanism (62).
Studies in the PNS and CNS indicate that neural impulse activity in fetal and early post-natal life can influence development of myelinating glia (63). This communication is mediated by ionic changes in the extracellular space accompanying neural impulse activity, as well as nonsynaptic release of neurotransmitter, growth factors, or specialized axon-glial signaling molecules. Blocking K+ channels in oligodendrocyte precursor cells, with depolarizing agents or specific ion channel toxins, prevents differentiation and proliferation of these glia (64), suggesting that release of K+ by axonal firing could help coordinate development of glia with functional activity in the brain.
Similarly in the PNS, axonal firing inhibits Schwann cell proliferation and differentiation during development. However, the responsible axon-glial signaling molecule was found to be extracellular ATP released from nonsynaptic regions of axons and activating P2Y receptors on Schwann cells. Imaging techniques have revealed that action potentials cause release of ATP from axons and an increase in intracellular Ca2+ in the Schwann cells situated along the axons (65). This rise in Ca2+-activated signaling pathways controls transcription of genes that are involved in regulating proliferation and differentiation of Schwann cells. This process of axon-glial communication may help to terminate early developmental events, such as Schwann cell division, at the point when the nervous system becomes functionally active. Also, arresting differentiation of Schwann cells could help to prevent their premature differentiation into an unmyelinating phenotype.
Effects of Impulse Activity on Myelination
Rapid impulse conduction depends on specialized glia that form the insulating wraps of myelin around axons (Fig. 1A). The signals regulating the development and maintenance of this exquisite neuron-glial organization have been a matter of intense research. The close association between myelination and axonal conduction suggests that impulse activity might have an influence on formation of myelin, but experimental evidence has yielded contradictory results (66). In the PNS, evidence for two activity-dependent mechanisms affecting myelination have been identified: changes in the molecular composition of the axonal membrane and soluble molecules released by axonal firing that activate membrane receptors on myelinating glia. In culture, axonal firing at a low frequency down-regulates expression of a cell adhesion molecule (L1-CAM) on axons (67), a molecule essential for initiating myelination by Schwann cells (68). Interestingly, higher frequency stimulation had no effect on L1-CAM expression. In coculture with Schwann cells, axons stimulated at low frequency (0.1 Hz) developed fewer myelinated profiles (69). Earlier in development, electrical stimulation of mouse dorsal root ganglion (DRG) axons in culture blocked myelination by a different mechanism that prevented differentiation of Schwann cells into a promyelinating phenotype. This effect was traced to the release of extracellular ATP from axons firing neural impulses, which activated purinergic receptors on Schwann cells that arrested development at an immature stage (65).
In contrast to the PNS, electrical activity appears to stimulate myelination in the CNS, as shown by using pharmacological agents to increase or decrease action potential firing (70). The mechanism is unknown, but identifying the axon-glial signaling molecule that stimulates myelination could offer new approaches for treating demyelinating diseases such as multiple sclerosis (MS). The reason for the discrepancy between the effects of impulse activity on myelination in the PNS and CNS is puzzling. New research on DRG neurons, which have axons in both the CNS and PNS, suggests that action potentials may be communicated to myelinating glia via different purinergic signaling molecules in the CNS and PNS (71). ATP and related molecules released from axons by electrical activity reportedly inhibit proliferation of both oligodendrocyte progenitor cells and Schwann cells, but had opposite effects on differentiation of these two types of glia (72). Acting through distinct receptors on oligodendrocytes and Schwann cells, the effects of impulse activity on myelination may be independently regulated along central and peripheral axons.
Node of Ranvier
Involvement of glia at the synapse is only now widely appreciated, yet neuron-glial interactions at the node of Ranvier have long been recognized as essential for rapid impulse conduction. Nodes of Ranvier, situated at regular intervals along axons, act as repeating amplifiers to propagate the neural impulse over long distances (Fig. 4). Voltage-gated Na+ channels are enriched at the nodes of Ranvier, in the exposed areas of axons where Na+ ions cross and depolarize the membrane between segments of compact myelin. In the juxtaparanodal region of the axon, the delayed rectifier K+ channels are highly concentrated to allow K+ ions to exit the axon and to restore the resting membrane potential after the impulse (73). The physiological importance of this arrangement is clear, but how do Na+ channels become concentrated at the node? The magnitude of this problem is greatly multiplied, considering that there are 10 genes encoding Na+ channel α subunits, and that multiple Na+ channel isoforms are differentially expressed and localized in neurons (74). The expression of different Na+ channel isoforms changes during development, in association with disease, and in response to chronic pain or alterations in neuronal activity (75).
Fig. 4.

Nodal glia play an important role in the formation, organization, and maintenance of myelinated axons. (A) An electron micrograph of a longitudinal section through the Node of Ranvier in the spinal dorsal root of rat, showing the intricate association between myelinating glia and axons (courtesy of M. H. Ellisman, National Center for Microscopy and Imaging Research, University of California, San Diego). (B) Three specific domains are defined by axon-glial interactions at the node of Ranvier: the Na+ channel-enriched node of Ranvier, the adjacent paranode (PN), the juxtaparanodal region (JP), which contains delayed rectifier K+ channels, and the internode (IN). This axon domain organization is regulated by soluble signals from myelinating glia as well as direct contact and interactions between proteins expressed on the surface of axons and glia. At the paranode, the transmembrane protein Caspr is found on the axon surface in association with the GPI anchored cell adhesion molecule, contactin (Cont). This molecular complex interacts with the glial cell adhesion molecule, neurofascin 155 (NF) and anchors the intercellular junction to the axonal cytoskeleton through the actin associated protein 4.1B, which binds to the cytoplasmic domain of Caspr. (*) Demyelination can lead to axon degeneration, indicating the necessity of continual communication between the axon and myelinating glia for maintaining axonal integrity (87, 95). KCh, K+ channel, NaCh, Na+ channel.
The chicken-and-egg problem of whether the axon determines nodal specialization and directs the glia to form nodal structure, or whether the glia direct the axonal membrane to aggregate Na+ channels to the nodal region and K+ channels to the paranodal region, is still controversial. There is evidence in favor of both mechanisms, but it is clear that no matter which cell has the first word, a continuing dialogue between nodal glia and axons is essential for the formation and maintenance of the node of Ranvier. After disease caused Schwann cells to demyelinate, sodium channels were found uniformly distributed along the axon. However, as the Schwann cells reassociated with the axon, Na+ channels became clustered in the axonal membrane at both ends of the Schwann cell, and were swept along as the Schwann cell enlarged and expanded along the axon (76). Ultimately, the Na+ channels became concentrated at the points where two Schwann cells came together to form a node of Ranvier. In the CNS, Na+ channels are also clustered adjacent to myelinating oligodendrocytes (77).
This compelling evidence that direct contact with nodal glia imposes molecular organization on the axon is apparently at odds with other observations in myelin-deficient dystrophic mice (78), which show clusters of Na+ channels spaced along the axon at roughly appropriate internodal distances, despite the absence of myelin. Although this suggests that there is some axonal specification of Na+ channel clustering, an alternative interpretation is that instructive soluble signals from nearby myelinating glia may be involved. Clustering of Na+ channels can be induced along retinal ganglion axons by conditioned medium collected from mature oligodendroglial (but not astrocyte) cultures (79). Although the soluble signal is still not known, this clustering is regulated by electrical activity and requires an intact cytoskeleton (80). The Na+ channel clusters are spaced at intervals 100 times the axon caliber, suggesting that the precise localization of Na+ channels may be intrinsic to the axon. The most parsimonious conclusion to this controversy is that although there may be differences in the PNS and CNS, there are several mechanisms involved in nodal organization of Na+ channels, including glial-derived soluble factors, glial contact, and axonal specification (Fig. 4).
New research has identified protein complexes in the nodal and paranodal regions that regulate the localization, formation, and stabilization of axonal domains and glial specializations at the node. On either side of the node of Ranvier, a series of paranodal loops of noncompact myelin closely appose the axon to form specialized septate-like junctions (Fig. 4). Recent evidence suggests that these paranodal junctions are a site of axon-glial communication regulating axon domain organization. The axonal membrane protein contactin binds to contactin-associated protein (Caspr) through cis interactions (81), and the Caspr-contactin protein complex is necessary for proper nodal domain organization and ion channel localization (Fig. 4B). In the absence of Caspr, the paranodal loops are disrupted, there is a reduction of contactin on the surface of the axon, K+ channels are displaced, and nerve conduction velocity is reduced (82). Similarly, mice that lack contactin exhibit a strikingly similar phenotype (83). Interaction between contactin and the extracellular domain of Caspr is required for the proper transport of Caspr out of the neuronal cell body, and the Caspr/contactin complex is stabilized at the paranode by binding the intracellular region of Caspr to the axonal cytoskeleton through the cytoskeleton-associated protein 4.1B (84). This complex spans the extracellular cleft between axon and glial cell by binding the 155-kD isoform of neurofascin (NF155) (85), a cell adhesion molecule expressed on the glial surface on the opposing paranodal loop (86) (Fig. 4B).
Neuron-Glial Communication in Injury and Disease
When myelination and nodal specializations go awry through injury, disease or birth defects, serious medical problems result owing to erratic impulse conduction. A two-way communication between axons and myelinating glia is important for maintenance of both glia and axons. Although myelin forms normally in mice lacking the myelin cell adhesion molecule MAG (87) or the myelin membrane proteolipid PLP (88), the axons ultimately degenerate. Whether this is a secondary effect due to the disruption in electrical conduction caused by the myelin abnormality or some other mechanism, the resulting axonal deficits demonstrate the necessity for continual glial-neuron communication. MS, which results from the loss of myelin in the CNS, causes a wide range of functional impairments in patients because of abnormal conduction of action potentials. However, brain scan and autopsy material from chronic MS patients show extensive loss of axons and neurons in some patients (89). Thus, MS should be regarded in part as a neurodegenerative disease (not just a glial disease) resulting from disruption of the normal neuron-glial communication in myelinated fibers. Better methods for treating such diseases may develop from studying the ways glia signal to axons, and by taking aggressive action to prevent demyelination caused by inflammatory responses or other brain insults.
Neuron-Glial Communication in Axon Outgrowth and Regeneration
During development, glia have a powerful role in setting up the basic scaffolding of the brain. By interacting with specific cell adhesion molecules on the glial membrane, neurons migrate along appropriate glial processes and extend axons and dendrites using glia as “guide posts” to form proper synaptic connections (90). However, after axons become injured, these same guidance molecules can act to impede the regenerative process by blocking the outgrowth of axons, as in the case of the myelin protein Nogo, which hinders axon regeneration in the CNS (91). Interestingly, signaling between neurons and glia via cell adhesion molecules can be context-dependent, and this property may be exploited for medical benefit. For instance, a cell adhesion molecule expressed on oligodendrocytes, MAG, promotes neurite outgrowth from embryonic neurons, but blocks axon outgrowth from postnatal neurons (92). This shift in response is associated with changes in adenosine 3′,5′-monophosphate (cAMP) levels that can be stimulated by exposure to neurotrophins, such as brain-derived neurotrophic factor and glial cell line– derived neurotrophic factor. If cAMP is elevated in neurons before exposure to MAG, the inhibitory effect on neurite outgrowth is blocked (93). This can be explained by studies showing that Gi proteins are activated by MAG and act to inhibit cAMP (94 ).
Future Directions
Many aspects of neuron-glial communication remain controversial. For example, how ATP, glutamate and other chemical messengers are released from glia in response to physiological stimuli is still hotly debated. An imposing array of chemical messengers stimulate Ca2+ responses in glia, and suggest multiple mechanisms of neuron-glial and glial-glial communication, which may operate in parallel. Moreover, in glia, information is integrated and transmitted by intracellular Ca2+, but the ubiquitous involvement of Ca2+ as a second messenger makes it difficult to distinguish direct effects from indirect effects. Our understanding of the diversity of glia is too simplistic. There are likely to be important developmental, physiological, and anatomical differences among glial cells, which make it difficult to uncover the mechanisms of communication and interactions with neurons at this early phase of research. The similarities and differences between neuron-glial communication in cell culture and in situ is an important example. As in any emerging field, this reflects a situation in which information is currently insufficient to encompass the true complexity of the phenomena. Future research on glial diversity and the mechanisms of cell-cell communication will lead to a greater understanding of the ways in which these nonneuronal cells participate in information processing in the nervous system.
Footnotes
Supporting Online Material www.sciencemag.org/cgi/content/full/298/5593/556/DC1, Figs. S1 and S2, Movie S1
References
- 1.Verhratsky A, Kettenmann H. Trends Neurosci. 1996;19:346. doi: 10.1016/0166-2236(96)10048-5. [DOI] [PubMed] [Google Scholar]
- 2.Logothetis NK, Pauls J, Augat M, Trinath T, Oeltermann A. Nature. 2001;412:150. doi: 10.1038/35084005. [DOI] [PubMed] [Google Scholar]
- 3.Shulman RG, Hyder F, Rothman DL. Proc Natl Acad Sci USA. 2001;98:6417. doi: 10.1073/pnas.101129298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bushong EA, Martone ME, Jones YZ, Ellisman MH. J Neurosci. 2002;22:183. doi: 10.1523/JNEUROSCI.22-01-00183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaur C, Hao AJ, Wu CH, Ling EA. Microsc Res Tech. 2001;54:2. doi: 10.1002/jemt.1114. [DOI] [PubMed] [Google Scholar]
- 6.Watkinis LR, Milligan EE, Maier SF. Trends Neurosci. 2001;24:4505. doi: 10.1016/s0166-2236(00)01854-3. [DOI] [PubMed] [Google Scholar]
- 7.Dani JW, Chernjavsky A, Smith SJ. Neuron. 1992;8:429. doi: 10.1016/0896-6273(92)90271-e. [DOI] [PubMed] [Google Scholar]
- 8.Porter JY, McCarthy KD. J Neurosci. 1996;16:5073. doi: 10.1523/JNEUROSCI.16-16-05073.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reist NE, Smith SJ. Proc Natl Acad Sci USA. 1992;89:7625. doi: 10.1073/pnas.89.16.7625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jahromi BS, Robitaille R, Charlton MP. Neuron. 1992;8:1069. doi: 10.1016/0896-6273(92)90128-z. [DOI] [PubMed] [Google Scholar]
- 11.Rochon D, Rousse L, Robitaille R. J Neurosci. 2001;21:3819. doi: 10.1523/JNEUROSCI.21-11-03819.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robitaille R. Neuron. 1998;21:847. doi: 10.1016/s0896-6273(00)80600-5. [DOI] [PubMed] [Google Scholar]
- 13.Castonguay A, Robitaille R. J Neurosci. 2001;21:1911. doi: 10.1523/JNEUROSCI.21-06-01911.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomas S, Robitaille R. J Neurosci. 2001;21:1087. doi: 10.1523/JNEUROSCI.21-04-01087.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Araque A, et al. J Neurosci. 1998;18:6822. doi: 10.1523/JNEUROSCI.18-17-06822.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parpura V, Haydon PG. Proc Natl Acad Sci USA. 2000;97:8629. doi: 10.1073/pnas.97.15.8629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Newman EA, Zahs KR. J Neurosci. 1998;18:4022. doi: 10.1523/JNEUROSCI.18-11-04022.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barbour B, Brew H, Attwell D. Nature. 1988;335:433. doi: 10.1038/335433a0. [DOI] [PubMed] [Google Scholar]
- 19.Kang J, Jiang L, Goldman SA, Nedergaard M. Nature Neurosci. 1998;1:683. doi: 10.1038/3684. [DOI] [PubMed] [Google Scholar]
- 20.Wang Z, Haydon PG, Yeung ES. Anal Chem. 2000;72:2001. doi: 10.1021/ac9912146. [DOI] [PubMed] [Google Scholar]
- 21.Guthrie PB. J Neurosci. 1999;19:520. doi: 10.1523/JNEUROSCI.19-02-00520.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newman EA. J Neurosci. 2001;21:2215. doi: 10.1523/JNEUROSCI.21-07-02215.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Finkbeiner SM. Glia. 1993;9:83. doi: 10.1002/glia.440090202. [DOI] [PubMed] [Google Scholar]
- 24.Cornell-Bell AH, Finkbeiner SM, Cooper MS, Smith SJ. Science. 1990;247:470. doi: 10.1126/science.1967852. [DOI] [PubMed] [Google Scholar]
- 25.Innocenti B, Papura V, Haydon PG. J Neurosci. 2000;20:1800. doi: 10.1523/JNEUROSCI.20-05-01800.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bergles DE, Jahr CE. J Neurosci. 1998;18:7709. doi: 10.1523/JNEUROSCI.18-19-07709.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pina-Benabou MH, Srinivas M, Spray DC, Scemes E. J Neurosci. 2001;21:6635. doi: 10.1523/JNEUROSCI.21-17-06635.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.E. Scemes, Am. Soc. Cell. Biol. Abstr. (2001).
- 29.Araque A, Li N, Doyle RT, Haydon PG. J Neurosci. 2000;20:666. doi: 10.1523/JNEUROSCI.20-02-00666.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abraham EH. Proc Natl Acad Sci USA. 1993;90:312. doi: 10.1073/pnas.90.1.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Queiroz G, et al. Neuroscience. 1999;91:1171. doi: 10.1016/s0306-4522(98)00644-7. [DOI] [PubMed] [Google Scholar]
- 32.Contreras JE. Proc Natl Acad Sci USA. 2002;99:495. [Google Scholar]
- 33.Cotrina ML. Proc Natl Acad Sci USA. 1998;95:15735. doi: 10.1073/pnas.95.26.15735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arcuino G, et al. Proc Natl Acad Sci USA. 2002;99:9840. doi: 10.1073/pnas.152588599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Urazaev AK. Neuroscience. 2001;106:237. doi: 10.1016/s0306-4522(01)00270-6. [DOI] [PubMed] [Google Scholar]
- 36.Liu QY, Schaffner AE, Chang YH, Maric D, Barker JL. J Neurophysiol. 2000;84:1392. doi: 10.1152/jn.2000.84.3.1392. [DOI] [PubMed] [Google Scholar]
- 37.Blondel O, et al. J Neurosci. 2000;20:8012. doi: 10.1523/JNEUROSCI.20-21-08012.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beattie EC. Science. 2002;295:2282. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- 39.Baranano DE, Ferris CD, Snyder SH. Trends Neurosci. 2001;24:99. doi: 10.1016/s0166-2236(00)01716-1. [DOI] [PubMed] [Google Scholar]
- 40.Wolosker H, Blackshaw S, Snyder SH. Proc Natl Acad Sci USA. 1999;96:13409. [Google Scholar]
- 41.Mothet JP. Proc Natl Acad Sci USA. 2000;97:4926. doi: 10.1073/pnas.97.9.4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smit AB. Nature. 2001;411:261. doi: 10.1038/35077000. [DOI] [PubMed] [Google Scholar]
- 43.P. G. Haydon et al.: Soc. Neurosci. Abstr. (2001).
- 44.Oliet SH, Piet R, Poulain DA. Science. 2001;292:923. doi: 10.1126/science.1059162. [DOI] [PubMed] [Google Scholar]
- 45.Rossler W, Oland LA, Higgins MR, Hildebrand JG, Tolbert LP. J Neurosci. 1999;19:9865. doi: 10.1523/JNEUROSCI.19-22-09865.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hutchins JB, Casagrande VA. J Comp Neurol. 1990;298:113. doi: 10.1002/cne.902980109. [DOI] [PubMed] [Google Scholar]
- 47.Muller CM. Int Rev Neurobiol. 1992;34:215. doi: 10.1016/s0074-7742(08)60099-9. [DOI] [PubMed] [Google Scholar]
- 48.Pfrieger FW, Barres BA. Science. 1997;277:1684. doi: 10.1126/science.277.5332.1684. [DOI] [PubMed] [Google Scholar]
- 49.Ullian EM, Sapperstein SK, Christopherson KS, Barres BA. Science. 2001;291:657. doi: 10.1126/science.291.5504.657. [DOI] [PubMed] [Google Scholar]
- 50.Mauch DH. Science. 2001;294:1354. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- 51.Gallo V, Ghiani CA. Trends Pharmacol. 2000;21:252. doi: 10.1016/s0165-6147(00)01494-2. [DOI] [PubMed] [Google Scholar]
- 52.Bergles DE, Jahr CE. Neuron. 1997;19:1297. doi: 10.1016/s0896-6273(00)80420-1. [DOI] [PubMed] [Google Scholar]
- 53.Bergles DE, Roberts JD, Somogyi P, Jahr CE. Nature. 2000;405:187. doi: 10.1038/35012083. [DOI] [PubMed] [Google Scholar]
- 54.Alvarez-Buylla A, Garcia-Verdugo JM, Tramontin AD. Nature Rev Neurosci. 2001;2:287. doi: 10.1038/35067582. [DOI] [PubMed] [Google Scholar]
- 55.Noctor SC, Flint AC, Weissman TA, Dammerman RS, Kriegstein AR. Nature. 2001;409:714. doi: 10.1038/35055553. [DOI] [PubMed] [Google Scholar]
- 56.Gaiano N, Nye JS, Fishell G. Neuron. 2000;361:258. doi: 10.1016/s0896-6273(00)81172-1. [DOI] [PubMed] [Google Scholar]
- 57.Furukawa T. Neuron. 2000;26:383. doi: 10.1016/s0896-6273(00)81171-x. [DOI] [PubMed] [Google Scholar]
- 58.Morrison SJ. Cell. 2000;101:499. doi: 10.1016/s0092-8674(00)80860-0. [DOI] [PubMed] [Google Scholar]
- 59.Tanigaki K, et al. Neuron. 2001;29:45. doi: 10.1016/s0896-6273(01)00179-9. [DOI] [PubMed] [Google Scholar]
- 60.Jessen K, Mirsky R. Ann NY Acad Sci. 1999;14:883. [PubMed] [Google Scholar]
- 61.Thoenen H. Science. 1995;270:593. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- 62.Atkins CM, Yon M, Groome NP, Sweatt JD. J Neurochem. 1999;73:1090. doi: 10.1046/j.1471-4159.1999.0731090.x. [DOI] [PubMed] [Google Scholar]
- 63.Barres BA, Raff M. Nature. 1993;361:258. doi: 10.1038/361258a0. [DOI] [PubMed] [Google Scholar]
- 64.Chittajallu R, et al. Proc Natl Acad Sci USA. 2002;9:2350. [Google Scholar]
- 65.Stevens B, Fields RD. Science. 2000;287:2267. doi: 10.1126/science.287.5461.2267. [DOI] [PubMed] [Google Scholar]
- 66.Zalc B, Fields RD. Neuroscientist. 2000;6:5. doi: 10.1177/107385840000600109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Itoh K, Stevens B, Schachner M, Fields RD. Science. 1995;270:1369. doi: 10.1126/science.270.5240.1369. [DOI] [PubMed] [Google Scholar]
- 68.Seilheimer B, Persohn E, Schachner M. J Cell Biol. 1989;109:3095. doi: 10.1083/jcb.109.6.3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stevens B, Tanner S, Fields RD. J Neurosci. 1998;18:9303. doi: 10.1523/JNEUROSCI.18-22-09303.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Demerens C, et al. Proc Natl Acad Sci USA. 1996;93:9887. [Google Scholar]
- 71.Fields RD, Stevens B. Trends Neurosci. 2000;23:625. doi: 10.1016/s0166-2236(00)01674-x. [DOI] [PubMed] [Google Scholar]
- 72.B. Stevens, S. Porta, L. Haak, V. Gallo, R. D. Fields, Soc. Neurosci. Abstr (2001).
- 73.Peles E, Salzer JL. Curr Opin Neurobiol. 2000;10:558. doi: 10.1016/s0959-4388(00)00122-7. [DOI] [PubMed] [Google Scholar]
- 74.Goldin AL. Annu Rev Physiol. 2001;63:871. doi: 10.1146/annurev.physiol.63.1.871. [DOI] [PubMed] [Google Scholar]
- 75.Waxman SG, Dib-Hajj S, Cummins TR, Black JA. Brain Res. 2000;886:5. doi: 10.1016/s0006-8993(00)02774-8. [DOI] [PubMed] [Google Scholar]
- 76.Dugandzija-Novakovic S, Koszowski AG, Levinson SR, Shrager P. J Neurosci. 1995;15:492. doi: 10.1523/JNEUROSCI.15-01-00492.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rasband MN. J Neurosci. 1999;19:7516. doi: 10.1523/JNEUROSCI.19-17-07516.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Deerinck TJ, Levinson SR, Bennett GV, Ellisman MH. J Neurosci. 1997;17:5080. doi: 10.1523/JNEUROSCI.17-13-05080.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kaplan MR. Nature. 1997;386:724. doi: 10.1038/386724a0. [DOI] [PubMed] [Google Scholar]
- 80.Kaplan M, et al. Neuron. 2001;30:105. doi: 10.1016/s0896-6273(01)00266-5. [DOI] [PubMed] [Google Scholar]
- 81.Peles E, et al. EMBO J. 1997;16:978. doi: 10.1093/emboj/16.5.978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Boyle ME. Neuron. 2001;30:385. doi: 10.1016/s0896-6273(01)00296-3. [DOI] [PubMed] [Google Scholar]
- 83.Bhat MA. Neuron. 2001;30:369. doi: 10.1016/s0896-6273(01)00294-x. [DOI] [PubMed] [Google Scholar]
- 84.Gollan LJ. Cell Biol. 2002;157:1247. doi: 10.1083/jcb.200203050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Charles P, et al. Curr Biol. 2002;12:217. doi: 10.1016/s0960-9822(01)00680-7. [DOI] [PubMed] [Google Scholar]
- 86.Tait S, et al. J Cell Biol. 2000;150:657. doi: 10.1083/jcb.150.3.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yin X, et al. J Neurosci. 1998;18:1953. doi: 10.1523/JNEUROSCI.18-06-01953.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Griffiths I, et al. Science. 1998;280:1610. doi: 10.1126/science.280.5369.1610. [DOI] [PubMed] [Google Scholar]
- 89.Bjartmar C, et al. Neurology. 2001;57:1248. doi: 10.1212/wnl.57.7.1248. [DOI] [PubMed] [Google Scholar]
- 90.Hidalgo A, Booth GE. Development. 2000;127:393. doi: 10.1242/dev.127.2.393. [DOI] [PubMed] [Google Scholar]
- 91.Chen MS. Nature. 2000;403:434. doi: 10.1038/35000219. [DOI] [PubMed] [Google Scholar]
- 92.Mukhopadhyay G, Doherty P, Walsh FS, Crocker PR, Filbin MT. Neuron. 1994;13:757. doi: 10.1016/0896-6273(94)90042-6. [DOI] [PubMed] [Google Scholar]
- 93.Cai D, Shen Y, De Bellard M, Tang S, Filbin MT. Neuron. 1999;22:89. doi: 10.1016/s0896-6273(00)80681-9. [DOI] [PubMed] [Google Scholar]
- 94.Cai D, et al. J Neurosci. 2001;21:4731. doi: 10.1523/JNEUROSCI.21-13-04731.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.de Waegh SM, Lee VM, Brady ST. Cell. 1992;68:461. doi: 10.1016/0092-8674(92)90183-d. [DOI] [PubMed] [Google Scholar]