

Abstract
ATM, the gene that is mutated in ataxia-telangiectasia, is associated with cerebellar degeneration, abnormal proliferation of small blood vessels, and cancer. These clinically important manifestations have stimulated interest in defining the sequence variation in the ATM gene. Therefore, we undertook a comprehensive survey of sequence variation in ATM in diverse human populations. The protein-encoding exons of the gene (9,168 bp) and the adjacent intron and untranslated sequences (14,661 bp) were analyzed in 93 individuals from seven major human populations. In addition, the coding sequence was analyzed in one chimpanzee, one gorilla, one orangutan, and one Old World monkey. In human ATM, 88 variant sites were discovered by denaturing high-performance liquid chromatography, which is 96%–100% sensitive for detection of DNA sequence variation. ATM was compared to 14 other autosomal genes for nucleotide diversity. The noncoding regions of ATM had diversity values comparable to other genes, but the coding regions had very low diversity, especially in the last 29% of the protein sequence. A test of the neutral evolution hypothesis, through use of the Hudson/Kreitman/Aguadé statistic, revealed that this region of the human ATM gene was significantly constrained relative to that of the orangutan, the Old World monkey, and the mouse, but not relative to that of the chimpanzee or the gorilla. ATM displayed extensive linkage disequilibrium, consistent with suppression of meiotic recombination at this locus. Seven haplotypes were defined. Two haplotypes accounted for 82% of all chromosomes analyzed in all major populations; two others carrying the same D126E missense polymorphism accounted for 33% of chromosomes in Africa but were never observed outside of Africa. The high frequency of this polymorphism may be due either to a population expansion within Africa or to selective pressure.
Introduction
The human ATM gene is an important case study for mutation analysis. ATM is the gene mutated in the human recessive disease ataxia-telangiectasia (A-T [MIM 208900]) (Gatti 2001). In addition to the characteristic ataxia and oculocutanaceous telangiectasias that gave the disease its name, patients with A-T have a 100-fold increased risk for cancer (Morrell et al. 1986). Furthermore, obligate heterozygous carriers may have a sixfold relative risk for cancer, implicating ATM as a possible tumor-suppressor gene (Morrell et al. 1990). This has been corroborated by a recent Dutch study showing an increased risk of breast cancer in ATM heterozygotes (Broeks et al. 2000). Other breast cancer studies, however, have been at odds with these results (Vorechovsky et al. 1996b; FitzGerald et al. 1997; Bay et al. 1998; Chen et al. 1998; Bebb et al. 1999; Janin et al. 1999; Olsen et al. 2001).
The conflicting estimates of ATM mutation frequencies in patients with breast cancer may be due to the different patient populations that have been studied or to the insensitivity of available mutation detection methods. Indeed, there are many technical challenges involved in analysis of the ATM gene. The 13-kb mRNA is assembled from 66 exons distributed across a genomic region of 150 kb. The coding sequence alone encompasses 9,168 bp (Uziel et al. 1996; Platzer et al. 1997). Gel-based strategies for finding sequence variation in this gene—such as mismatch cleavage analysis, dideoxy sequencing, the protein truncation test, or single-strand conformation polymorphism analysis—are cumbersome and/or expensive. As a result, analyses of samples from patients with A-T detected only 60%–80% of the expected alleles (Gilad et al. 1996; Telatar et al. 1996; Vorechovsky et al. 1996a; Concannon and Gatti 1997; Broeks et al. 1998; Ejima and Sasaki 1998; Sasaki et al. 1998; Castellví-Bel et al. 1999; Gatti et al. 1999; Sandoval et al. 1999; Teraoka et al. 1999).
Denaturing high-performance liquid chromatography (DHPLC) is a novel, non–gel-based method that is very sensitive for detection of DNA sequence variation. Detection of sequence variation by DHPLC is based on differences in retention of perfectly matched homoduplexes and heteroduplexes containing one or more mismatched base pairs (Oefner and Underhill 1995, 1998). The choice of temperature during DHPLC is critical for achieving maximum detection sensitivity, which is 96%–100%. Blinded analyses have shown that DHPLC is superior to conventional methods (O'Donovan et al. 1998; Choy et al. 1999; Jones et al. 1999; Wagner et al. 1999b).
This article presents the results of the application of DHPLC to an analysis of global genetic variation in ATM. The sequence diversity of human ATM is compared with orthologs in four primate species, as well as with 14 other human autosomal genes. The implications of these findings for the identification of neutral sequence variation are discussed.
Subjects, Material, and Methods
Subjects and DNA Samples
DNA was obtained from chimpanzee, gorilla, orangutan, and macaque (Old World monkey) blood samples, and from 93 human lymphoblastoid cell lines derived from individuals enrolled according to approved human subject protocols. These individuals were believed to have no obvious genetic disease, although no medical records are available to confirm this. More than 60% of the samples came from males. In detail, the individuals included 18 from Africa (4 Biaka pygmies, 3 Mbuti Zaire pygmies, 4 Lisongo, 1 Omega San, 1 Namibian San, 1 Tsumkwe San, 1 Mandenka, 2 Ethiopian Jews, and 1 Sudanese), 9 from the Middle East (2 Druze, 2 Bedouin, 2 Palestinian, 2 Sephardim, and 1 Iranian), 12 from the Indian peninsula (1 Pathan, 2 Sindhi, 2 Brushaski, 2 Baluchi, 1 Brahui, 1 Makran, 1 Hazara, 1 Kalash, and 1 Tamil), 20 from Asia (3 Cambodians, 1 Hmong, 1 Atayal, 1 Ami, 8 Han Chinese, 4 Japanese, 1 Korean, and 1 Yakut), 16 from Europe (2 French, 2 European Americans from Utah, 1 Amish, 2 Germans, 2 Italian, 1 Dane, 2 Ashkenazi, 1 Adygei, 1 Georgian, 1 Caucasian, and 1 Russian), 8 from Oceania (2 New Guinean, 2 Melanesian, 2 Australian Aborigines, 1 Micronesian, and 1 Samoan), and 10 American Indians (1 Karitiana, 1 Surui, 2 Mayans, 2 Colombian Indians, 1 Quechua, 1 Muskogee, 1 Pima, and 1 Navaho).
PCR primers designed from the human ATM sequence were used to amplify the homologous regions of ATM for sequencing in four primate species: chimpanzee, gorilla, orangutan, and macaque monkey. The orangutan and macaque samples were obtained from Coriell Cell Repositories. Some exons from the primate species did not readily amplify with the human primers. The proportion of the ATM coding sequence amplified in each species is listed in the Results section. The chimpanzee sample was also analyzed by DHPLC for all ATM exon regions except exons 14 and 35. The PCR products from the chimpanzee template were sequenced even when the DHPLC profile was homozygous.
DNA Extraction
Total genomic DNA was prepared through use of a QIAamp DNA Mini Kit (Qiagen). DNA samples were diluted with TE buffer (10 mM Tris, 1 mM EDTA) to a working concentration of 30–50 ng/μl and were stored in an 96-well microtiter plate format. For PCR, 2 μl of the DNA stocks were dispensed into 96-well microtiter plates by a Hydra96 robot (Robbins Scientific) and were dried for 10–20 min by a SpeedVac Concentrator (Model SC210A or AES1010, Savant Instruments).
PCR Conditions
All 62 coding exons (9,168 bp) of ATM, as well as 14,661 bp of noncoding exon sequence (9,110 bp of intron sequence adjacent to coding exons and 5,551 bp of 5′ and 3′ noncoding exon sequence), were PCR amplified from genomic DNA through use of the 71 primer pairs listed in table 1. All primers were complementary to intron sequences near the exon boundaries except those in the 5′ and 3′ untranslated sequence that were amplified in overlapping segments. The exons were numbered according to established convention (Platzer et al. 1997). PCR primers used for amplification of regions containing ATM exons 10, 12, 19, 20, 23–37, 40–44, 55, 56, 57, 58, and 63 have been published elsewhere (Hacia et al. 1998). Primers for the remaining exon regions were selected from the published genomic sequence for ATM (GenBank accession number HSU82828), using the computer program Oligo 4.0 or Oligo 6.4 (Molecular Biology Insights). Amplicons were optimized for DHPLC by the choice of primer pairs that minimized the temperature difference in melting domains across the region and that maximized the length screened, to as many as 550 bp. The exons were amplified individually, except in four cases, in which two nearby exons could be amplified together.
Table 1.
Primers and DHPLC Conditions for Mutational Analysis of Human ATM
|
Primer |
Length of Sequence(bp) |
DHPLCConditions |
|||||
| ATMExon | Forward(5′→3′) | Reverse(5′→3′) | Coding | Noncodinga | Total | Temperature(°C) | Gradient(% ofBuffer B) |
| Promotor | GCCCAGAACCTCCGAATGACG | CGACTTAGCGTTTGCGGCTCG | 0 | 493 | 535 | 63 | 59–64 |
| 1A | AATGTTTTGGGGCAGTGTTT | AGGAAAGATGGAGTGAGGAGAG | 0 | 370 | 412 | 63 | 58–63 |
| 1B | GACTCCTCCCTCTCCTCACTC | CCATCTGGAAGGCTTCTACC | 0 | 515 | 556 | 60 | 60–65 |
| 1B, 2 | TGCAAACTCAGCCTGAGACT | CATGCCATTCTTTTCTAGTGC | 0 | 491 | 532 | 59 | 54–60 |
| 3 | TGAACCTTGAGTGGAACCTA | CTCTGAGGAAACAAAAACACA | 0 | 294 | 335 | 57 | 53–58 |
| 3 | TTGGCACCAGTTAGTTCAGA | CAGAAGATGCTCATTCACTGA | 0 | 260 | 300 | 54 | 55–60 |
| 4, 5 | TACACATTTTTTCACACCTCTTTC | AGGCATAATGATATATAGGAAGCA | 185 | 263 | 496 | 50, 55 | 60–66, 58–64 |
| 6 | ATTGGTCTTGTAGGAGTTAGGC | AAAAACTCACGCGACAGTAATC | 146 | 202 | 392 | 54 | 58–64 |
| 7 | TTGCCAATTTCTTCTCTACAAAAG | TTAAGGGTCAGTTCGATAACCATA | 165 | 241 | 454 | 55 | 54–60 |
| 8 | CTTTTTCTGTATGGGATTATGGA | TTCTGTTATGATGGATCAATGTTT | 166 | 177 | 390 | 50, 55 | 57–63, 51–56 |
| 9 | CAGCATACCACTTCATAACTG | TCATATCCTCCTAAAGAACAC | 239 | 131 | 412 | 54 | 56–61 |
| 10 | CTAGCAGTGTAAACAGAGTA | ATAGGCTTTTTGTGAGAACA | 164 | 121 | 325 | 54 | 55–60 |
| 11 | GCAACAACAGCGAAACTCTG | GAATGAGAAAATGGTAACACTT | 170 | 139 | 350 | 53, 56 | 56–61, 53–58 |
| 12 | TGTGATGGAATAGTTTTCAA | AGTAACAAACTATGAAAATGA | 372 | 98 | 511 | 57 | 56–62 |
| 13 | CAATAGCTTGCTTTTCACAAT | TGGCATCAAATAAGTGGAGAG | 195 | 92 | 335 | 57 | 54–59 |
| 14 | GATGAAAGCAATTTTAATCTAGGA | TCAGTTTTTCTCATTGGCACTT | 96 | 340 | 482 | 54 | 56–62 |
| 15 | CATATAAGGCAAAGCATTAG | AGTTTACCAAAGTTGAATCATA | 226 | 72 | 340 | 55 | 53–59 |
| 16 | GTTGTTTTTAGAGCTATCCAGGA | CTGCATCATGTACCCCAGAAC | 126 | 332 | 502 | 53, 57 | 58–63, 55–61 |
| 17 | CGTGGAACTTCTAAAAACATTTC | CAAAACAGTAACACCAACCAGT | 126 | 346 | 517 | 54, 59 | 59–65, 54–59 |
| 18 | AGCTCACTTATCTTTAGGGTCAA | GGCCTTAATTTCCACATTTGT | 90 | 329 | 463 | 54 | 57–62 |
| 19 | CTCCTGCAAGAAGCCATCT | AAGAAATCCCAAGTAGTAAAT | 172 | 38 | 250 | 57 | 53–58 |
| 20 | CTTTTTTTTGTGAAGAGGAGGA | TTTCAATTCTTCAAAGACACCA | 200 | 156 | 400 | 51, 58 | 58–64, 52–58 |
| 21 | TTTTTCCCTCCTACCATCTT | CTTAACAGAACACATCAGTTATT | 83 | 78 | 173 | 55 | 50–55 |
| 22 | TAAAATAACTGATGTGTTCTGTT | CAAAACTTGCATTCGTATC | 156 | 150 | 286 | 56 | 54–60 |
| 23, 24 | TAGCACAGAAAGACATATTGGAAG | TGTAAGACATTCTACTGCCATCTG | 207 | 236 | 491 | 55 | 56–61 |
| 25 | AGCTGCTGGTCTGAACCTCTTTA | TTGCTATGATTTGACCCATTGTG | 118 | 324 | 488 | 54, 57 | 56–60, 51–57 |
| 26 | TGGAGTTCAGTTGGGATTTTA | TTCACAGTGACCTAAGGAAGC | 174 | 87 | 303 | 51, 55 | 58–64, 56–61 |
| 27 | CTTAACACATTGACTTTTTGG | GTATGTGTGTTGCTGGTGAG | 170 | 78 | 288 | 54 | 54–59 |
| 28 | TGCTGATGGTATTAAAACAGTTT | GGTTGGCTATGCTAGATAATGAT | 247 | 192 | 485 | 51, 56 | 58–64, 54–60 |
| 29 | GAGCTGTCTTGACGTTCACAG | TTAAAAAGAGTGATGTCTATAA | 116 | 131 | 290 | 53, 57 | 56–61, 51–57 |
| 30 | TTAAAACGATGACTGTATT | GAAGGAATGTTCTATTATTA | 127 | 40 | 207 | 56 | 51–56 |
| 31 | CTGAAAATTAAATAAATTGGCAAT | AAAACAGGAAGAACAGGATAGAAA | 200 | 227 | 475 | 54 | 56–61 |
| 32 | CTTACTGGTTGTTGTTGTTTT | CCATTTTGAAGATGAGTCAG | 175 | 26 | 241 | 57 | 52–58 |
| 33 | GTTTTGTTGGCTTACTTTA | GAGCATTACAGATTTTTGAA | 165 | 99 | 304 | 53 | 53–59 |
| 34 | GTGTTAAAAGCAAGTTACATT | AAGAAACAGGTAGAAATAGC | 133 | 33 | 207 | 56 | 50–55 |
| 35 | GCAATTATAAACAAAAAGTGT | TATATGTGATCCGCAGTTGAC | 96 | 78 | 215 | 57 | 53–58 |
| 36 | CAGCATTATAGTTTTGAAAT | GTGTGAAGTATCATTCTCCA | 172 | 114 | 326 | 55 | 54–60 |
| 37 | TGGTGTACTTGATAGGCATTT | GACCCACAGCAAACAGAACTG | 142 | 63 | 245 | 57 | 52–57 |
| 38 | AACCTAATTTTTCTGCTGCCTAA | GAGTGGGGGTGATATTATGTGA | 177 | 279 | 501 | 55 | 58–64 |
| 39 | GCAGTATGTTGAGTTTATGGCA | GCAACTGTTGGCAACTTTTAT | 178 | 260 | 481 | 53, 57 | 58–64, 54–60 |
| 40 | ATAAACAAGAAGGAAGAAGGT | ACGTAAGAAGCAACACTCATT | 88 | 56 | 184 | 58 | 51–56 |
| 41 | CAACATGCTTTTATTTTGATA | TATATACCCTTATTGAGACAA | 156 | 90 | 288 | 55 | 53–59 |
| 42 | TCTCATTAAAAGAGGTGTTCT | TGAGATAAATACTGTCATAAA | 88 | 34 | 164 | 56 | (49) 51–57 |
| 43, 44 | AAATTTGCTAAATTTATAGACCGA | AGTGATGGCTTTACCAAATCTGG | 192 | 152 | 391 | 57 | 55–60 |
| 45 | TTTGCTGTTTTTTTCTCTGGT | CAGTTGTTGTTTAGAATGAGGAGA | 149 | 91 | 285 | 56 | 53–59 |
| 46 | ATTTTGTCCTTTGGTGAAGCTA | CAAGTTTTCAGAAAAGAAGCCA | 105 | 109 | 258 | 53 | 55–60 |
| 47 | GCAAAGCCTATGATGAGAACTC | CAGAAAAGCTGCACTTTAGGAT | 120 | 103 | 267 | 53, 57 | 55–61, 50–55 |
| 48 | TCTTGTCACTACAAAAGTTCCTTT | TCTTTTTCCCTCAGGCTTTC | 235 | 165 | 444 | 51, 56 | 61–67, 56–61 |
| 49 | CCTCAATGAATGGTAGTTGCT | GTAAAACACTAATCCAGCCAATA | 168 | 87 | 299 | 53, 58 | 57–63, 51–56 |
| 50 | CCAAAGCAGATGAGGAAAAAC | TCTTGATGAAAAGATGAAGCATAT | 114 | 165 | 325 | 56 | 54–59 |
| 51 | GTGTATTACCTTAATTTGAGTG | CCAAAAGACCAAGATAATCT | 218 | 191 | 451 | 55 | 61–67 |
| 52 | TCCTTAGAAGTTTGCTTTTTTC | CTGGACCAAGTGCTAGGAATA | 208 | 199 | 450 | 57 | 57–62 |
| 53 | CTAGAGTACCCATTAGAAAGACCT | GTGTATGCCTGCATGTGTGA | 114 | 346 | 504 | 50, 55 | 64–69, 59–64 |
| 54 | CTCTGCCTTTTTCTCACACAT | CCTTGAACCGATTTTAGATGG | 159 | 218 | 419 | 56 | 54–60 |
| 55 | GGGTAGTTCCTTATGTAATGT | GTAACACAGCAAGAAAGTAACGT | 139 | 42 | 225 | 56 | 50–56 |
| 56 | GACCTTCAATGCTGTTCCTCAGT | AGGTTGAAACATATGAAATTTGCC | 83 | 135 | 265 | 54 | 52–58 |
| 57 | ACCCGGCCTAAAGTTGTAGT | AATGGAGAAAAGCCTGGTTC | 141 | 278 | 459 | 57 | 53–59 |
| 58 | TTTGCTATTCTCAGATGACTCT | ATGTTtTTGGTGAACTAACAGAAG | 117 | 69 | 232 | 56 | 51–56 |
| 59 | GCTGAATGATCATCAAATGCTCT | ATAATATCTGACAGCTGTCAGCT | 150 | 99 | 295 | 52, 57 | 60–66, 54–60 |
| 60 | TTTATTGCCCCTATATCTGTCAT | AAAAAGTGCTGAATCAAACAAA | 166 | 134 | 345 | 55 | 56–61 |
| 61 | CAACATGGCCGGTTATGC | CACTCAGTACCCCAGGCAGA | 87 | 305 | 430 | 54 | 57–63 |
| 62 | GTGGTTTCTTGCCTTTGTAAAGTT | AATCCTCCCACTTCAGCCTCTT | 115 | 162 | 323 | 58 | 58–63 |
| 63 | TAGGCTCAGCATACTACACAT | GACGAGATACACAGTCTACCT | 64 | 117 | 223 | 55 | 51–56 |
| 64, 65 | GGCTTATTTGTATGATACTGGTTC | CTAAAGGCTGAATGAAAGGGTA | 318 | 191 | 555 | 53, 58 | 60–65, 55–61 |
| 65 | TACCCTTTCATTCAGCCTTTAG | TTTTTTTTTTGAGATGGAGTTTC | 0 | 470 | 515 | 56, 61 | 64–69, 59–64 |
| 65 | TCTCAAAAAAAAAAAAAAAAAACA | TGGGAATATGACATAAACAGACA | 0 | 538 | 586 | 53 | 60–65 |
| 65 | AGGCTTTATCTATGGGAATCTT | AGAGGTGAATATGTGAGCTGAT | 0 | 370 | 414 | 59 | 56–61 |
| 65 | CTCATTTTTGACCGTAAGGA | AAGTTCTGGAGATTGGTTTTAG | 0 | 411 | 453 | 56 | 59–64 |
| 65 | CCTCCCCTAAAACCAATCT | AGTTATTTCTCCTAGGCTTGTG | 0 | 417 | 459 | 54 | 58–63 |
| 65 | ATGGCTTTGAAAAGTTTATCA | TAGGGTGGGAAAGCTATTATC | 0 | 579 | 621 | 56 | 56–61 |
| 65 | ATGAAAACCAAATAGTGAAGC | TGGCACACAGAACACACAA | 0 | 467 | 508 | 55 | 59–64 |
Noncoding length does not include primer sequence.
PCR amplification was performed with AmpliTaq Gold (PE Biosystems) in 30–50-μl reactions, using a Touchdown PCR protocol that made it possible to use the same conditions for every amplicon. The samples were then taken directly from PCR to a denature/reanneal protocol, which involves heating to 95°C then slowly cooling to 65°C over 30 min, using the PCR System 9700 thermocycler (PE Biosystems). Touchdown PCR conditions were 1 cycle of 95°C for 10 min; followed by 14 cycles of 95°C for 20 s, 63°C for 1 min (decreasing by 0.5°C/cycle), and 72°C for 1 min; 19 cycles of 95°C for 20 s, 56°C for 45 s, and 72°C for 45 s; and 1 cycle of 72°C for 7 min. PCR was carried out in a separate room, and only filtered pipette tips were used. To monitor for the presence of contamination, every plate had a negative control containing no added DNA.
DHPLC
The optimum temperature of analysis was determined, for each amplicon, by the DHPLC Melt program (Jones et al. 1999), available at the DNA Variation Group Web site. All ATM amplicons were run within 1°C of the recommended temperature, as indicated in table 1. If the program recommended two temperatures, then both temperatures were used, except in nine cases (exons 12, 15, 35, 41, 42, 43/44, 51, 58, and 60), for which only the highest temperature was used.
The chromatographic profiles of all but two ATM polymorphisms were clearly distinct from that of a homozygous control. Two sequence variants of ATM produced only a subtly different chromatographic profile when analyzed at the recommended DHPLC conditions. For the first one, 5185C→G, the normal allele had one leading shoulder, and the heterozygous allele had two leading shoulders. For the second one, IVS62-55T→C, the normal allele had a slightly broadened peak, and the heterozygous allele had a leading shoulder on that peak.
Microtiter plates containing completed PCR reactions were loaded into an autosampler for automated DHPLC, using either the Transgenomic WAVE System or the Varian Helix System. Injection volume was 10–12 μl. In both systems, the DNASep column was used. The samples were run using the temperature and gradient conditions listed in table 1. The gradient was generated by increasing the concentration of buffer B (0.1M triethylamine acetate, 25% acetonitrile) relative to buffer A (0.1M triethylamine acetate). The numbers (X–Y) listed in the “DHPLC Gradient” column in table 1 represent shorthand notation for the gradient (expressed as a percentage of buffer b): 50%–X% in 0.5 min; X%–Y% in 3.0 min; Y%–95% in 0.1 min; 95% for 0.5 min; 95%–50% in 0.1 min; 50% for 0.8 min. The acetonitrile gradient was adjusted so that the homoduplex signal from the reference sample eluted from the column at a retention time of 2.8–3.4 min. These conditions were found empirically to maximize the resolution of heteroduplex and homoduplex signals. Each sample run took 6 min.
All samples were completely analyzed for the entire region sampled, except those from two Africans: a Mandenka, for whom insufficient amounts of DNA were available, and a Sudanese, who replaced another African after the study was already underway. Sequence variants observed in both samples are listed in the Results section, but the Mandenka could not be genotyped and therefore was not included in the haplotype analysis.
Genotypes of the 17 most frequent polymorphisms were determined by DHPLC. Samples that were homozygous for either allele had only one peak and would be indistinguishable from one another without a reference. Therefore, the amplicons from the homozygotes were mixed with a sample of known genotype, through use of a protocol originally established for the Y chromosome (Underhill et al. 1997).
Direct DNA Sequencing
The chemical nature and location of the mismatches underlying the different heteroduplex profiles detected were determined by direct DNA sequencing. For this purpose, excess oligonucleotide primers and dinucleotide triphosphates were removed by treating 10 μl of each PCR reaction with 1 unit each of exonuclease I and shrimp alkaline phosphatase (Amersham/Life Technologies) for 30 min at 37°C and for 15 min at 80°C. The PCR products were then sequenced with the BigDye Deoxy Terminator cycle sequencing kit (PE Biosystems), according to the manufacturer’s instructions. After solid-phase extraction with Sephadex G-50 (Amersham Pharmacia), the sequencing reactions were analyzed with a PE Biosystems 377A sequencer.
Statistical Analysis
Nucleotide diversity was calculated as estimated nucleotide diversity (π), if complete genotypes were available (AMPD1, ATM, BRCA1, DFFRY, MMP1, MMP3, MMP12, SMCY, and UTY), or as average heterozygosity (θ), if only heterozygotes were known (ABCB1, BRCA2, CACNA1A, COX2, FBN1, IL4, RB1, WRN, and XRCC1). The value of θ is very close to that of π if Hardy-Weinberg equilibrium is satisfied. In a sample of n chromosomes, π was calculated from the equation 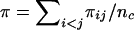 , where πij is the number of nucleotide differences between the ith and jth DNA sequences and nc=n(n-1)/2 (Nei 1987). Average heterozygosity per site was calculated by dividing the sum of the relative frequencies of heterozygotes per polymorphic site by the total number of base pairs screened. Tajima’s D statistic (Tajima 1989b) was calculated by the equation
, where πij is the number of nucleotide differences between the ith and jth DNA sequences and nc=n(n-1)/2 (Nei 1987). Average heterozygosity per site was calculated by dividing the sum of the relative frequencies of heterozygotes per polymorphic site by the total number of base pairs screened. Tajima’s D statistic (Tajima 1989b) was calculated by the equation
 |
where 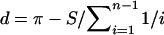 , S is the number of segregating nucleotides and
, S is the number of segregating nucleotides and 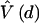 is an estimator of the variance of d. The value of D is expected to be 0 for selectively neutral variants and infinite sites in a constant population. A greater-than-expected number of rare variants would result in a negative value of D, indicating either purifying selection or population expansion (Tajima 1989a). The Hudson/Kreitman/Aguadé (HKA) test (Hudson et al. 1987) was applied, using a computer program obtained from M. Nachman, to further investigate the possibility of selection acting on the ATM gene. The HKA test compares levels of intraspecific polymorphisms at different loci and interspecific sequence differences at these loci, with expectations—under a neutral evolution model—of an infinite number of sites. A neutral model predicts that the ratio between levels of intraspecific polymorphism and interspecies divergence should be the same for different genes, since both are a function of the neutral mutation rate. Selection, in contrast, can lead to an uncoupling of levels of polymorphism and divergence.
is an estimator of the variance of d. The value of D is expected to be 0 for selectively neutral variants and infinite sites in a constant population. A greater-than-expected number of rare variants would result in a negative value of D, indicating either purifying selection or population expansion (Tajima 1989a). The Hudson/Kreitman/Aguadé (HKA) test (Hudson et al. 1987) was applied, using a computer program obtained from M. Nachman, to further investigate the possibility of selection acting on the ATM gene. The HKA test compares levels of intraspecific polymorphisms at different loci and interspecific sequence differences at these loci, with expectations—under a neutral evolution model—of an infinite number of sites. A neutral model predicts that the ratio between levels of intraspecific polymorphism and interspecies divergence should be the same for different genes, since both are a function of the neutral mutation rate. Selection, in contrast, can lead to an uncoupling of levels of polymorphism and divergence.
Inference of Haplotypes from Simple Sequence Polymorphisms (SSPs)
Haplotypes were inferred by a maximum-parsimony approach, as described elsewhere (Jin et al. 1999), for each of the 91 individuals for whom complete genotyping data had been obtained at the 17 sites that were heterozygous in >3 individuals (fig. 3). Polymorphic sites from a group of haplotypes or sequences were considered congruent if they could be accommodated by the same phylogenetic topology. Polymorphic sites were considered recurrent if linkage to flanking markers could not be explained by a single recombination event. In the construction of the tree by maximum parsimony, haplotypes of each individual with multiple polymorphisms were inferred, assuming the smallest number of mutational steps and that any incongruence between loci indicates recombination or recurrent mutation.
Figure 3.

Distribution of coding-sequence polymorphisms in ATM and differences between chimpanzee and human. A, Location of polymorphisms and coding-sequence differences in chimpanzee compared to human. B, Location and frequency of polymorphisms observed in human ATM. The horizontal bar represents amino acid sequence of ATM from 1 to 3056. Colored and shaded regions of the amino acid sequence represent possible functional domains of the molecule: p53 binding (Khanna et al. 1998), leucine zipper (Morgan et al. 1997), homology to S. pombe Rad3p (Bentley et al. 1996) and S. cerevisiae Tel1p (Greenwell et al. 1995; Morrow et al. 1995), and the serine kinase domain (Zakian 1995). Ball-and-stick figures along the amino acid sequence represent amino acid positions. Circles are mapped at positions that are polymorphic either within the 93 human individuals (with the number of individuals adjacent to each circle) or within the single chimpanzee individual; diamonds represent homozygous differences between human and chimpanzee. Black circles or diamonds represent missense polymorphisms, and white circles or diamonds represent silent changes.
Of the 17 common polymorphic sites, 10 could be accommodated by the same phylogenetic topology, whereas 5 sites were found to be incongruent because of recurrent mutation, or reversion. Site IVS4+37insAA was recurrent in samples from all the population groups. Site IVS22-77T→C was recurrent in three samples, one from America and two from Asia; site IVS25-12insA was recurrent in Africa only. Site IVS48-69insATT was found to be recurrent in one African sample and in two American samples, and site IVS61+104C→T (part of a CpG motif) was recurrent in two samples, one from Africa and one from the Middle East. All recurrent sites can be explained by common mutation mechanisms.
Comparison of ATM with Other Genes
DHPLC analysis of 14 autosomal and 3 Y-chromosome genes was performed in a way similar to that described for ATM, using gene-specific PCR products spanning the coding and flanking intron sequences for each gene. The human gene names are standard for the online resource LocusLink. The GenBank accession numbers are as follows—ABCB1 (also called “MDR1”): AC002457; AMPD1: M60092; BRCA1: U14680; BRCA2: U43746; CACNA1A (also called “EA2”): AF004884; COX2 (also called “PTGS2”): U04636; FBN1: L13923; IL4: M23442; MMP1 (also called “MTE”), MMP3 (collagenase), and MMP12 (stromelysin): U78045; RB1: M15400; WRN: AF091214; and XRCC1: M36089. The mRNA and amino acid sequences of mouse Atm was obtained from GenBank (accession number MMU43678).
Results
SSPs
Variation in the human ATM gene was analyzed by DHPLC, which detects DNA sequence variation with a sensitivity of 96%–100% (Choy et al. 1999; Jones et al. 1999; Wagner et al. 1999b). The sensitivity of DHPLC in this study was confirmed in our laboratory by a blinded analysis of 73 known A-T patient samples, in which 99 of 100 previously identified variants in ATM were detected (data not shown).
Table 2 lists the 88 variant sites found in 93 human representatives from seven continental regions. Transitions accounted for 59% of all sequence variants. Transversions, deletions, and insertions accounted for 32%, 3.4%, and 5.7% of sequence variants, respectively. Sequence variation occurred in 26 of 9,168 nucleotides (1 in 353) in the coding regions, and in 62 of 14,661 nucleotides (1 in 236) in the noncoding regions. Of the 26 polymorphisms located in exons, 8 were silent, and 18 were missense polymorphisms. Of the 18 missense polymorphisms, 9 resulted in nonconservative amino acid changes, but their functional significance remains uncertain. With two exceptions, namely the conservative amino acid changes D126E and D1853N, all missense polymorphisms were observed only once or twice in the 93 individuals tested. The global distribution of D126E was unusually high in Africa.
Table 2.
Sequence Variants Found in ATM, Their Geographic Location, and DHPLC Detection Temperature
|
No. of Heterozygous Individuals in |
||||||||||||
| Genomic Positiona(Location within Gene) | Polymorphism | Amino AcidChange | DHPLCTemperature(°C) | Africa(n=18) | Middle East(n=9) | Indian Peninsula(n=12) | Oceania(n=8) | Europe(n=16) | Asia(n=20) | Americas(n=10) | Total(n=93) | Referenceb |
| 10152 (promotor) | 10152 G→C | 63 | 1 | 1 | ||||||||
| 10182 (promotor) | 10182 T→A | 63 | 8 | 4 | 1 | 2 | 10 | 7 | 5 | 37 | 9 | |
| 10807 (5′ UTR) | -787 A→G | 63 | 1 | 1 | ||||||||
| 10948 (intron 1a) | IVS1a+61 A→G | 63 | 1 | 1 | ||||||||
| 11251 (5′ UTR) | -615 A→G | 59 | 1 | 1 | ||||||||
| 11512 (5′ UTR) | -354 G→A | 59 | 1 | 1 | ||||||||
| 12194 (intron 2) | IVS2-291 G→A | 56 | 8 | 4 | 1 | 2 | 10 | 7 | 5 | 37 | ||
| 12203 (intron 2) | IVS2-282 A→G | 56 | 2 | 2 | ||||||||
| 12220 (intron 2) | IVS2-265 C→T | 56 | 1 | 1 | ||||||||
| 12378 (intron 2) | IVS2-107 G→C | 56 | 1 | 1 | ||||||||
| 15247 (intron 3) | IVS3-49 A→G | 50 | 1 | 1 | ||||||||
| 15252 (intron 3) | IVS3-44 T→C | 50 | 1 | 1 | ||||||||
| 15403 (intron 4) | IVS4+6 A→G | 55 | 1 | 1 | ||||||||
| 15434 (intron 4) | IVS4+37 insAA | 50, 55 | 9 | 7 | 3 | 2 | 5 | 12 | 7 | 45 | 3, 6 | |
| 15548 (exon 5) | 146 C→G | S49Cc | 55 | 1 | 1 | 2 | 3 | |||||
| 23296 (intron 6) | IVS6-71 A→G | 55 | 2 | 2 | ||||||||
| 23413 (exon 7) | 378 T→A | D126E | 55 | 10 | 1 | 11 | ||||||
| 23440 (exon 7) | 405 T→C | A135A | 55 | 2 | 2 | |||||||
| 23615 (intron 7) | IVS7+84 A→G | 55 | 1 | 1 | ||||||||
| 31591 (intron 7) | IVS7-48 T→G | 50 | 2 | 2 | ||||||||
| 31788 (exon 8) | 646 G→T | A216S | 55 | 1 | 1 | |||||||
| 31799 (exon 8) | 657 T→C | C219C | 55 | 4 | 4 | |||||||
| 31842 (intron 8) | IVS8+38 T→C | 55 | 6 | 6 | ||||||||
| 32736 (intron 9) | IVS9+25 T→G | 54, 55 | 10 | 1 | 11 | |||||||
| 36728 (exon 11) | 1176 C→G | G392G | 56 | 1 | 1 | |||||||
| 38688 (exon 12) | 1541 G→A | G514Dc | 57 | 1 | 1 | |||||||
| 39655 (exon 13) | 1744 T→C | F582Lc | 57 | 1 | 1 | 2 | ||||||
| 40414 (intron 13) | IVS13-85 A→G | 54 | 1 | 1 | ||||||||
| 43827 (intron 15) | IVS15-69 C→G | 53 | 1 | 1 | 2 | |||||||
| 43828 (intron 15) | IVS15-68 T→C | 53 | 2 | 2 | ||||||||
| 44096 (intron 16) | IVS16+75 T→C | 57 | 1 | 1 | ||||||||
| 44099 (intron 16) | IVS16+78 G→A | 53 | 1 | 1 | ||||||||
| 45192 (exon 17) | 2281 A→T | T761S | 54 | 1 | 1 | |||||||
| 46553 (intron 17) | IVS17-114 C→G | 54 | 1 | 1 | ||||||||
| 46611 (intron 17) | IVS17-56 G→Ad | 54 | 6 | 3 | 3 | 3 | 9 | 8 | 5 | 37 | ||
| 54956 (exon 19) | 2572 T→C | F858Lc | 58 | 1 | 1 | 3 | ||||||
| 56073 (intron 19) | IVS19-17 G→T | 58 | 2 | 2 | ||||||||
| 56317 (intron 20) | IVS20+28 insA | 58 | 1 | 1 | 3, 5, 6 | |||||||
| 60136 (intron 22) | IVS22-77 T→C | 56 | 1 | 3 | 4 | 3 | 9 | 10 | 4 | 34 | 5, 9 | |
| 60253 (exon 23) | 3118 A→G | M1040V | 55, 56 | 1 | 1 | 2 | ||||||
| 60273 (exon 23) | 3138 T→G | L1046L | 55, 56 | 1 | 1 | |||||||
| 60285 (exon 23) | 3150 T→Cd | L1050L | 55, 56 | 1 | 1 | |||||||
| 60410 (exon 24) | 3161 C→G | P1054Rc | 55, 56 | 1 | 1 | 1, 3, 8 | ||||||
| 67162 (intron 24) | IVS24-9 delT | 56 | 3 | 2 | 2 | 1 | 2 | 10 | 5, 6 | |||
| 67305 (intron 25) | IVS25+17 T→Cd | 56 | 2 | 2 | ||||||||
| 68663 (intron 25) | IVS25-12 insA | 50, 51 | 8 | 4 | 4 | 2 | 7 | 11 | 4 | 40 | 6 | |
| 68876 (intron 26) | IVS26+28 A→C | 51, 55 | 1 | 1 | ||||||||
| 71872 (intron 27) | IVS27-34 A→G | 51 | 1 | 1 | ||||||||
| 72096 (exon 28) | 3937 G→C | E1313Q | 51, 56 | 1 | 1 | |||||||
| 72157 (intron 28) | IVS28+5 G→T | 51, 56 | 2 | 2 | ||||||||
| 76682 (exon 30) | 4138 C→T | H1380Y | 56 | 2 | 2 | |||||||
| 80435 (exon 32) | 4578 C→T | P1526P | 57 | 1 | 1 | 2 | 3 | |||||
| 80956 (intron 32) | IVS32-32 del3 | 53 | 1 | 1 | ||||||||
| 80999 (exon 33) | 4623 G→T | L1541Fc | 53 | 1 | 1 | |||||||
| 82583 (intron 33) | IVS33-20 A→Gd | 55 | 1 | 1 | ||||||||
| 82628 (exon 34) | 4802 G→A | S1601N | 55, 56 | 1 | 1 | |||||||
| 82697 (exon 34) | 4871 A→Gd | H1624Rc | 56 | 1 | 1 | |||||||
| 84955 (intron 34) | IVS34-7 T→C | 52 | 1 | 1 | ||||||||
| 87320 (intron 35) | IVS35-68 T→G | 54 | 10 | 1 | 11 | |||||||
| 89328 (exon 37) | 5185 G→C | V1729L | 57 | 1 | 1 | |||||||
| 90446 (intron 37) | IVS37-80 T→G | 55 | 1 | 1 | ||||||||
| 90464 (intron 37) | IVS37-62 G→T | 55 | 1 | 1 | ||||||||
| 92265 (intron 38) | IVS38-83 G→Ad | 53 | 2 | 1 | ||||||||
| 92333 (intron 38) | IVS38-15 G→C | 53, 57 | 1 | 1 | ||||||||
| 92340 (intron 38) | IVS38-8 T→C | 57 | 2 | 1 | 3, 5, 6 | |||||||
| 92408 (exon 39) | 5557 G→A | D1853N | 53, 57 | 3 | 2 | 2 | 1 | 2 | 10 | 3, 5, 7, 8 | ||
| 92409 (exon 39) | 5558 A→T | D1853Vc | 53, 57 | 1 | 1 | 2 | 8 | |||||
| 97863 (exon 41) | 5793 T→C | A1931A | 55 | 1 | 1 | 3, 5 | ||||||
| 98004 (intron 41) | IVS41+16 A→G | 55 | 1 | 1 | ||||||||
| 103693 (exon 44) | 6108 T→Cd | Y2036Y | 57 | 1 | 1 | |||||||
| 113654 (intron 48) | IVS48-69 insATT | 53 | 8 | 3 | 3 | 3 | 9 | 8 | 3 | 37 | 5 | |
| 115239 (intron 49) | IVS49-71 C→Td | 56 | 2 | 2 | ||||||||
| 115338 (exon 50) | 7004 C→A | T2335Kc | 56 | 1 | 1 | |||||||
| 118131 (intron 52) | IVS52+45 delTT | 57 | 1 | 1 | ||||||||
| 119423 (intron 53) | IVS53-121 insGGCA | 55 | 2 | 2 | ||||||||
| 135133 (intron 61) | IVS61+104 C→Td | 54 | 2 | 5 | 4 | 2 | 6 | 9 | 5 | 33 | 4 | |
| 142611 (intron 62) | IVS62-55 T→Cd | 54, 55 | 7 | 3 | 3 | 3 | 8 | 8 | 5 | 37 | 5, 9 | |
| 153599 (3′ UTR) | 9407C→T | 56, 61 | 1 | 1 | ||||||||
| 153734 (3′ UTR) | 9542A→G | 56, 61 | 1 | 1 | ||||||||
| 154046 (3′ UTR) | 9854 T→G | 53 | 2 | 1 | 3 | |||||||
| 154789 (3′ UTR) | 10597T→C | 59 | 1 | 1 | ||||||||
| 155442 (3′ UTR) | 11250C→T | 56 | 1 | 1 | ||||||||
| 155651 (3′ UTR) | 11459T→C | 54 | 1 | 1 | ||||||||
| 155696 (3′ UTR) | 11504C→T | 54 | 1 | 1 | ||||||||
| 156434 (3′ UTR) | 12242 C→T | 56 | 2 | 2 | ||||||||
| 156498 (3′ UTR) | 12306 A→G | 56 | 1 | 1 | ||||||||
| 156592 (3′ UTR) | 12400C→T | 55 | 1 | 1 | ||||||||
| 156755 (3′ UTR) | 12563T→G | 55 | 7 | 3 | 3 | 3 | 8 | 8 | 5 | 37 | ||
Genomic position in GenBank (accession number U82828).
1, Vorechovsky et al. (1996a); 2, Vorechovsky et al. (1997); 3, Shayeghi et al. (1998); 4, Bay et al. (1999); 5, Castellvi-Bel et al. (1999); 6, Li et al. (1999); 7, Maillet et al. (1999); 8, Sandoval et al. (1999); 9, Bonnen et al. (2000).
Nonconservative amino acid change.
DNA variant at CpG or CpNG site.
Of the 88 polymorphisms listed in table 2, 53 (60%) were observed only once, and they were found in both the noncoding and coding regions. The frequency of singletons is very similar to that observed in BRCA2, where they constituted 59% of all sites (Wagner et al. 1999b). However, it was quite different from the Werner syndrome gene (Passarino et al. 2001), in which only 33% of the 58 polymorphisms identified in an identical set of individuals were singletons. Another 18 polymorphic sites in ATM were observed two or three times in the screening set. The remaining 17 polymorphisms were observed in four or more individuals.
Consistent with the oldest human populations existing in Africa (Stringer and Andrews 1988; Tishkoff et al. 1998; Underhill et al. 2000), 61% (54/88) of the polymorphisms were found there. Not surprisingly, 17 of those polymorphisms were also detected in other continents. (The derived allele of IVS17-114C→G was found a second time, in Oceania, as a homozygote.)
Haplotype Analysis
Haplotypes for each individual were inferred by a maximum-parsimony approach, using the genotype data of 17 polymorphic sites that had been observed to be heterozygous in four or more individuals. The genotypes of each individual are diagramed in figure 1. Ten of the sites were in complete linkage disequilibrium over >133 kb.
Figure 1.

Genotype data for 17 polymorphic sites in ATM, obtained for 91 representatives from seven major human populations. An asterisk (*) indicates that the polymorphism was not included in the haplotype analysis, because of recombination or recurrent mutation. White circles represent the homozygous ancestral allele, black circles the homozygous derived allele, and gray circles the heterozygotes. The nucleotide positions of the 17 polymorphic sites, in relation to the cDNA sequence (GenBank accession number U82828), are given at the top.
Seven haplotypes, H1–H7, could be inferred, and their phylogenetic relationship is depicted in figure 2. Two clusters of haplotypes, H2–H3 and H4–H7, could be distinguished. H1 was hypothesized to be the oldest haplotype in the phylogeny, since it was identical to the chimpanzee haplotype. Of 10 sites defining the haplotypes, only two single–base-pair changes resulted in amino acid changes, which are indicated in parentheses. Arrows indicate the number and direction of sequence changes. Several intermediate haplotypes between H2 and H3 and between H4 and H5 were not observed in the present study, because of either the relatively small size of the screening set or their disappearance due to random genetic drift or selection.
Figure 2.

Phylogenetic relationship among seven ATM haplotypes. A chimpanzee sequence was used to determine the root of the phylogeny (H1). The circles each represent one haplotype, the area of the circle is proportional to the number of chromosomes observed with that haplotype, and the colors represent seven distinct geographic regions. The number of arrows indicates the number of changes required to generate the next related haplotype. Missense polymorphisms are indicated in parentheses. The number of observations are summarized in the table at the bottom of the figure. The haplotypes were defined by the nine congruent sites observed in ATM, including eight single-base-pair changes and one deletion. The nucleotide positions and the base pair changes defining the haplotypes are 378T→A, 657T→C, IVS8+38T→C, IVS9+25T→G, IVS17-56G→A, IVS24-9delT, IVS35-68T→G, 5557G→A, IVS62-55T→C, and 12563T→G where variation corresponding to amino acid changes is underlined. There are 133,342 bp between the first and the last markers. Haplotypes are defined as follows (the base pair change defining a new haplotype is underlined)—H1: TTTTATTGTT; H2: TTTTGTTGTT; H3: TTTTG(delT)TATT; H4: TTTTATTGCG; H5: ATTGATGGCG; H6: ACTGATGGCG; H7: ATCGATGGCG.
The color coding in figure 2 indicates the geographic distribution, and the areas of the circles are proportional to the frequencies of the seven haplotypes. Strikingly, two haplotypes, H2 and H4, were found in every region of the world, accounting for 82% of the chromosomes observed.
Like nucleotide diversity, haplotype diversity was greatest in Africa, where five out of seven haplotypes were found. This is consistent with many other observations leading to the hypothesis of an African origin of modern humans. Of the two haplotypes not found in Africa, one is a singleton (H5), whereas the other (H3) was observed at relatively low frequencies on all continents. H1 was only observed in Africa and Oceania.
The African haplotypes H6 and H7, which share the missense polymorphism D126E, were particularly intriguing. These two haplotypes were very common in Africa but were not found elsewhere in the world. H6 was only found in the rain forests of central Africa, but H7 was found in climatically diverse regions all across Africa. H5 also shared D126E and was found only once, in an individual from the Makran coast of Pakistan; this haplotype may be associated with a relatively recent introduction from Africa, through trade relationships.
The geographic distribution of D126E was unusual when compared to distributions of missense polymorphisms in the 14 other autosomal genes reviewed in this study. All except one of the missense polymorphisms in these genes were randomly distributed, with equal percentages in African and non-African populations. The exception was a missense polymorphism in MDR1 (A259S) that was heterozygous in 49% of non-Africans but was never seen in Africans.
The non-African haplotype, H3, was distinguished by the missense polymorphism D1853N. H3 was found on an average of 8% of chromosomes everywhere except in Africa. Its distribution and distinct absence in Africa is similar to that observed for M9 on the Y chromosome (Underhill et al. 1997).
Comparison of ATM Orthologs
Figure 3 shows a comparison of the ATM coding sequence in human and chimpanzee. Figure 3A shows the distribution of differences, along the ATM protein, between human and chimpanzee, as well as polymorphisms within the single chimpanzee individual. There were 32 coding-sequence differences between human and chimpanzee. Of the single–base-pair changes, 9 were polymorphic in the single chimpanzee sample, and the other 23 were homozygous for different base pairs in human and chimpanzee. Twelve variants resulted in an amino acid change, and 20 were silent. Figure 3B shows the distribution and frequency of coding-sequence polymorphisms within humans. In figures 3A and 3B, sequence differences that resulted in an amino acid change are shown as black symbols, and silent base-pair changes are shown as white symbols.
Two regions of the protein with known functional motifs encoding the leucine zipper and kinase domains, as well as one region with homology to the distantly related yeast (Saccharomyces cerevisiae) Tel1p protein are indicated by solid shading. The shaded regions represent ∼29% of the amino acid coding sequence. Of the 58 polymorphisms found in human or chimpanzee, only 3 map to the conserved regions of the protein. If polymorphisms are assumed to be distributed uniformly and at random, the probability of seeing three or fewer in the conserved region is <.01.
Nucleotide Diversity
Figure 4 shows the nucleotide diversity of 15 autosomal and 3 Y-chromosome genes (Shen et al. 2000) that have been analyzed by DHPLC. Nucleotide diversity was calculated from the number of differences between any two randomly chosen sequences. Given the number of segregating sites, the nucleotide diversity of the coding region of ATM was very low. For the 9,168-bp coding region, the value was 0.71±0.61×10-4. That is 4.5-fold less sequence diversity than the average in 64,676 coding base pairs in 15 other autosomal genes analyzed by DHPLC (Wagner et al. 1999a; Shen et al. 2000; Passarino et al. 2001; dbSNP Home Page; D. Cox, C. Franco, R. Ghali, P.J. Oefner, A. Roxas, P. Shen, T.A. Sivakumaran, R. Sung, T. Tang, F. Wong, W.-T. Yang, unpublished data), and 7-fold less than that reported in 135,823 coding base pairs in 106 human genes (Cargill et al. 1999).
Figure 4.

Nucleotide diversity in coding and noncoding regions of 15 autosomal and 3 Y-chromosome genes. Nucleotide diversity, or heterozygosity, was calculated as described in the Subjects, Material, and Methods section. Autosomal genes are shown in black and gray, and Y-chromosome genes are shown in red and pink.
There was a large difference between the nucleotide diversities of ATM in the coding and noncoding regions. Whereas most genes in the study had similar nucleotide-diversity estimates in the protein coding and noncoding regions, with ratios ranging from 1:2.5 to 1:1, in ATM the ratio was 1:7.5. Since the sequence diversity was normal in the noncoding regions compared to that in the other genes, it is clear that there was not a generally lower mutation rate in the chromosomal region containing ATM. More likely, the lower sequence diversity in the coding region is due to selective pressure for maintaining the protein sequence.
Only three genes—two on the Y chromosome (SMCY and DFFRY) and one small autosomal gene with 2,650 bp of coding sequence (the retinoblastoma gene, RB1)—had less coding diversity than ATM. When ATM and RB1 were compared more closely, it is interesting that the last 2,650 bp (29%) of the ATM coding region had even less sequence diversity than RB1 (π was 0.0415×10-4 and 0.076×10-4, respectively.) Even when the remaining 6,518 bp of ATM were considered separately, the sequence diversity was 0.965×10-4, suggesting considerable constraint on the evolution of this gene compared to other autosomal genes. Loss of some neutral sequence diversity across the gene might be expected during background selection, because deleterious mutations will eliminate variants that are linked on the same haplotype.
When nucleotide diversity was calculated for African and non-African populations separately, most genes showed little difference. However, in ATM, the non-African population had four times less sequence diversity (0.437×10-4) than did the African population (1.75×10-4).
Statistical Analysis for the Hypothesis of Selection at ATM
Calculation of the Tajima’s D statistic also suggested that selection has operated on ATM. In the Tajima’s D test, D=0 for selectively neutral mutations in a constant-population infinite-sites model, whereas D<0 in the case of purifying selection or in an expanding population (Tajima 1989a, 1989b). In an expanding-population model (Sherry et al. 1994; Kimmel et al. 1998; Pritchard et al. 1999; Shen et al. 2000; Zhivotovsky et al. 2000), all genes would be expected to behave similarly. However, Tajima’s D value was statistically negative for only four of the autosomal genes reviewed in the present study, including ATM. For example, for ATM, D=-1.98, which was statistically significant at P<.05, reflecting the overabundance of rare alleles.
The HKA test (Hudson et al. 1987) compares the number of intraspecies (within-human) polymorphisms and interspecies sequence differences at different loci, with the expectation of a model of neutral evolution. First, the human and chimpanzee orthologs were compared. The coding region of ATM had 26 polymorphisms in humans over 9,168 bp and had 32 differences between humans and chimpanzee over 9,029 bp. The combined coding regions of 11 other autosomal genes (AMPD1, CSTB, MMP1, MMP3, MMP12, ABCB1, BRCA2, XRCC1, FBN1, COX2, and IL-4) had 123 polymorphic sites in human over 30,996 bp and 79 differences between human and chimpanzee over 22,343 bp. The HKA statistic for reduced variability in human ATM compared to chimpanzee was not statistically significant (χ2[1]=0.47, P=.49).
However, the HKA test would fail to detect selection if the same selective pressures were operating in both human and chimpanzee. To test the possibility that the reduced variability of the last 29% of the ATM coding sequence was due to selective pressure in both chimpanzee and human, five cross-species comparisons were performed (table 3). In contrast to human and chimpanzee, there was little reduced variability in that region in gorilla (χ2[1]=3.58, P=.059) and none in orangutan, Old World monkey, or mouse (χ2[1]>4.7, P<.02). Thus, selective pressure appears to be operating in the last 29% of the ATM coding region in humans, chimpanzees, and possibly in gorilla, but not in orangutan, Old World monkey, or mouse.
Table 3.
Comparisons of Two ATM Gene Regions with Orthologs in Five Other Species, Using the HKA Statistic
|
No. of Differences (No. ofBase Pairs Analyzed) in |
||||
| Species | First 71% of ATM | Last 29% of ATM | χ2 | P |
| Chimpanzee | 21 (6518) | 2 (2650) | .157 | .692 |
| Gorilla | 28 (5341) | 7 (1855) | 3.58 | .059 |
| Orangutan | 42 (4795) | 19 (2308) | 5.02 | .025a |
| Old World Monkey | 68 (3364) | 28 (1406) | 5.38 | .020a |
| Mouse | 970 (6518) | 349 (2650) | 4.72 | .030a |
Statistically significant.
Discussion
The present study represents the first comprehensive survey of neutral variation in the ATM gene in diverse human populations. ATM is important for the regulation of cellular responses to DNA damage, for oxidative stress, and for the control of cell cycle checkpoints. It is a protein kinase that phosphorylates many substrates, including p53 tumor suppressor protein.
All of the protein-encoding exons of the gene (9,168 bp), as well as adjacent intron and untranslated sequences (14,661 bp), were analyzed in 1 chimpanzee and 93 human samples. The sequences analyzed were those that would most likely affect protein function, either by altering amino acids in functional domains or by affecting intron splicing. A diverse collection of 93 human genomic DNAs were chosen for analysis. To include as much genetic diversity as possible, African and Oceanian populations were overrepresented relative to others.
The analysis method, DHPLC, was shown to be 99% sensitive for detection of sequence variation in the ATM gene (data not shown.) Important technical details, such as use of a temperature-prediction algorithm and examples of subtle chromatographic shifts, have been included here so that others can reproduce the sensitivity reported.
Sequence Diversity in ATM
A total of 88 different polymorphic sites were discovered in human ATM, and 23 homozygous differences were found between human and chimpanzee in the coding region. The chimpanzee was heterozygous (polymorphic) at nine additional sites. Only one human sample, from an African, had as many heterozygous sites, consistent with a report elsewhere that chimpanzee populations have greater genetic diversity than do human populations (Kaessmann et al. 1999).
Most of the ATM polymorphisms were relatively rare, leading to a low overall sequence diversity. The high number of rare variants contributed to the generation of a significantly negative value for Tajima’s D statistic. Such an effect can occur for a rapidly expanding population. However, the number of rare variants in ATM was higher than the average seen in 14 other autosomal genes, suggesting that other forces were operating. For example, purifying selection would cause this effect by eliminating variation linked to deleterious mutations.
The low sequence diversity in the coding region of ATM was noteworthy, because it encodes a very large (350 kDa) protein that may be able to tolerate many amino acid changes. However, ATM was the second-least variable of the autosomal genes analyzed. The only autosomal gene with less coding-sequence diversity was RB1. Interestingly, both ATM and RB1 have functions in cell-cycle control (Mulligan and Jacks 1998; Lavin 1999) and therefore may have similar constraints on their evolution.
The overall diversity of ATM was comparable to that of three genes on the Y chromosome. The Y chromosome has previously been shown to have less diversity than autosomal genes, mostly because of its fourfold smaller effective population size (Jobling and Tyler-Smith 1995; Nachman 1998), but also because of the possible action of sexual selection (Wyckoff et al. 2000) and demographic factors, such as polygamy (Shen et al. 2000).
Haplotype Analysis
Of 17 common markers in ATM, 10 showed no evidence of recurrent mutation and were in complete linkage disequilibrium with each other over >133 kb. Previous reports have also demonstrated extensive linkage disequilibrium at ATM (Gatti 1998; Li et al. 1999; Bonnen et al. 2000), and in other regions of the genome (Collins et al. 1999; Huttley et al. 1999; Gordon et al. 2000; Taillon-Miller et al. 2000). Interestingly, the rate of recombination appears to be the same in patients with A-T and in unaffected individuals (Lange et al. 1995).
Through use of haplotypes defined by the 10 markers that were completely linked, it was possible to construct a parsimonious phylogeny (fig. 4). Because the mutation rate of single-nucleotide changes is much lower than that of nucleotide repeats, the resulting phylogeny is older than one derived from microsatellite markers.
A previous study (Bonnen et al. 2000) identified haplotypes at ATM, using SNP markers that were identified only in noncoding sequence. Different regions of the ATM gene were analyzed in the two studies, with ∼25% overlap in sequence. Consequently, only two markers, IVS22-77T→C and IVS62-55T→C, were found by both groups. In the present study, IVS22-77T→C was not used in the definition of haplotypes, because it was recurrent in three samples. The genotypes of a third marker that was common in the population, 10182T→A, were determined in this set of samples.
The observed number and global distribution of haplotypes in ATM were comparable to those reported earlier (Bonnen et al. 2000). Both studies observed that a small number of haplotypes represented the vast majority of chromosomes analyzed, with 2 of 7 haplotypes representing 82% of chromosomes (present study) and 6 of 22 haplotypes representing 89% of chromosomes (Bonnen et al. 2000). Some of the haplotypes in the two studies may be identical. For example, three missense polymorphisms were associated with unique haplotypes in the two studies: S49C was found on haplotype 2 (Bonnen et al. 2000) and on H4 (this study), P1054R was found on haplotype 17 and on H2, and D1853N was found on haplotype 15 and on H3.
Implications for Identification of Neutral Variation
Our observations of DNA sequence variation in the human ATM gene have important implications for distinguishing between neutral variants and functional mutations. For example, all of the 88 variants reported here are likely to be neutral, unless one or two individuals are among the 1%–2% of the population that are carriers of ATM mutations (Gatti et al. 1999; Li and Swift 2000). Functional mutations in carriers may exist either in protein-coding DNA or in noncoding DNA associated with splicing. Four intron variants near splice-donor or -acceptor sites were compared with splice consensus sequences, using an algorithm available at the Berkeley Drosophila Genome Project Web site. None of them significantly altered the splice consensus score, although analysis of RNA may be necessary to further clarify this issue. One variant associated with breast cancer, IVS10-6T→G (Broeks et al. 2000), and one missense polymorphism common in the German population, 2119T→C (“S707R” in the report by Dörk et al. [1997]), were not detected in our cohort.
Additional general conclusions about neutral variation in ATM can be drawn. The significantly negative value of Tajima’s D statistic, as well as the low nucleotide diversity in the protein-coding region, suggest that ATM is under an unusual degree of evolutionary constraint. Whereas the diversity in the coding sequence was very low, the diversity of noncoding sequence was comparable to that in other genes, indicating that the lack of diversity was not caused by a generally lower mutation rate at the ATM locus.
Not surprisingly, the smallest amount of sequence diversity was found in conserved regions that have putative functional domains. For example, there was only a single sample with a single variant in the entire carboxy-terminal 29% of the amino acid sequence, corresponding to the kinase and Tel1p homology domains. This region of ATM was significantly less diverse in human and chimpanzee than it was in orangutan, Old World monkey, and mouse. Consequently, either a selective pressure operates only in the former group or the selective pressures in the two groups are different because of divergent roles for the ATM protein in the different species.
Intriguingly, the functional constraint in ATM may not be restricted to the amino acid sequence. The reduced number of silent mutations in the 3′ noncoding region of the gene suggests that this region may contain sequences that regulate mRNA stability or splicing. For example, the smallest exon in the gene, exon 63, is <100 bp in length and has a splice-acceptor site with a very weak homology to known splice donor consensus sequences. Efficient splicing at such exons is likely to require splicing enhancers (Carlo et al. 1996; Cooper and Mattox 1997).
Differences in ATM between African and Non-African Populations
In addition to the low overall sequence diversity in ATM compared to other human genes, there were striking differences between African and non-African populations. For example, the sequence diversity of ATM in Africa was fourfold greater than it was outside of Africa. One reason for this difference was the presence of three frequent polymorphisms that defined the African-specific haplotypes H6 and H7. H6 was concentrated in central Africa, but H7 was found all across the continent.
Only two common amino acid substitutions were observed in the global population, and they were both relatively conservative changes (D→N and D→E). The D1856N variant was relatively common in non-Africans and was not found in Africans. In contrast, the D126E variant was very common in Africans but was extremely rare in non-Africans. Because the divergent ATM proteins were geographically isolated from one another, it is possible that the polymorphisms arose and were propagated by random genetic drift after the migration of humans out of Africa. The D126E variant may have been propagated with haplotype H7 in the Bantu expansion within Africa (Cavalli-Sforza 1994), in a situation analogous to that of markers M40 and M96 in the published Y-chromosome phylogeny (Underhill et al. 2000).
It is also possible, however, that the high allele frequency of D126E in Africa and its absence elsewhere is due to selective pressure. The presence of an allele that has reached equilibrium in one human population yet is not found elsewhere in the world is very unusual (Halushka et al. 1999); the overwhelming majority of alleles that are common in Africa are also common elsewhere (Zietkiewicz et al. 1997; Tishkoff et al. 1998). High heterozygosity for a locus might be caused by balancing selection in an area where the presence of two different alleles is advantageous, as is the case with sickle-cell anemia (MIM 603903). Although the proportion of individuals with the genotypes D/D, D/E, and E/E was consistent with Hardy-Weinberg expectations, the number of individuals analyzed here was too small to rule out selective pressure.
Acknowledgments
We appreciate the generosity of the DNA donors and of the investigators who provided the samples of human genomic DNA: L. Excoffier, M.E. Ibrahim, T. Jenkins, J. Kidd, A. Langaney, S.Q. Mehdi, P. Parham, and L.L. Cavalli-Sforza. We express our sincere appreciation for helpful discussions with Ted Jones, Richard Gatti, Rinaldo Pereira, Joanna Mountain, and Michael Olivier. Thanks to Joe Hacia, for sending many useful DNA-sequence files, including ATM genomic and primer sequences, and to Michael Nachman, for the computer program used to calculate the HKA statistic. We also acknowledge the able and cheerful technical assistance provided by Adriane Roxas, Erin Kauffman, Claudia Franco, and Alice Lin. P.J.O. holds U.S. patents related to DHPLC and receives fixed annual royalty payments from Transgenomic, Inc. and Varian, Inc., whose equipment was used in the present report. This work was supported by NIH Grant HG01932 (to P.J.O.) and NIH Grant ROI-CA77302 (to G.C.).
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- Berkeley Drosophila Genome Project, http://www.fruitfly.org/seq_tools/splice.html (for splice site prediction algorithm)
- dbSNP Home Page, http://www.ncbi.nlm.nih.gov/SNP/ (for polymorphisms for genes)
- DNA Variation Group, http://insertion.stanford.edu/melt.html (for DHPLC Melt program for prediction of DHPLC analysis temperature)
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for genomic sequences of ATM [accession number HSU82828]; ABCB1 [accession number AC002457]; AMPD1 [accession number M60092]; BRCA1 [accession number U14680]; BRCA2 [accession number U43746]; CACNA1A [accession number AF004884]; COX2 [accession number U04636]; FBN1 [accession number L13923]; IL4 [accession number M23442]; MMP1, MMP3, and MMP12 [accession number U78045]; RB1 [accession number M15400]; WRN [accession number AF091214]; XRCC1 [accession number M36089]; and for the mRNA and amino acid sequences of mouse Atm [accession number MMU43678])
- LocusLink, http://www.ncbi.nlm.nih.gov/LocusLink/ (for gene sequences)
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for A-T [MIM 208900])
References
- Bay J, Grancho M, Pernin D, Presneau N, Rio P, Tchirkov A, Uhrhammer N, Verrelle P, Gatti R, Bignon Y (1998) No evidence for constitutional ATM mutation in breast/gastric cancer families. Int J Oncol 12:1385–1390 [DOI] [PubMed] [Google Scholar]
- Bay J, Uhrhammer N, Pernin D, Presneau N, Tchirkov A, Vuillaume M, Laplace V, Grancho M, Verrelle P, Hall J, Bignon Y (1999) High incidence of cancer in a family segregating a mutation of the ATM gene: possible role of ATM heterozygosity in cancer. Hum Mutat 14:485–492 [DOI] [PubMed] [Google Scholar]
- Bebb D, Yu Z, Chen J, Telatar M, Gelmon K, Phillips N, Gatti R, Glickman B (1999) Absence of mutations in the ATM gene in forty-seven cases of sporadic breast cancer. Br J Cancer 80:1979–1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley N, Holtzman D, Flaggs G, Keegan K, DeMaggio A, Ford J, Hoekstra M, Carr A (1996) The Schizosaccharomyces pombe rad3 checkpoint gene. EMBO J 15:6641–6651 [PMC free article] [PubMed] [Google Scholar]
- Bonnen PE, Story MD, Ashorn CL, Buchholz TA, Weil MM, Nelson DL (2000) Haplotypes at ATM identify coding-sequence variation and indicate a region of extensive linkage disequilibrium. Am J Hum Genet 67:1437–1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broeks A, de Klein A, Floore AN, Muijtjens M, Kleijer WJ, Jaspers NG, van't Veer LJ (1998) ATM germline mutations in classical ataxia-telangiectasia patients in the Dutch population. Hum Mutat 12:330–337 [DOI] [PubMed] [Google Scholar]
- Broeks A, Urbanus JH, Floore AN, Dahler EC, Klijn JG, Rutgers EJ, Devilee P, Russell NS, van Leeuwen FE, van't Veer LJ (2000) ATM-heterozygous germline mutations contribute to breast cancer-susceptibility. Am J Hum Genet 66:494–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cargill M, Altshuler D, Ireland J, Sklar P, Ardlie K, Patil N, Shaw N, Lane C, Lim E, Kalyanaraman N, Nemesh J, Ziaugra L, Friedland L, Rolfe A, Warrington J, Lipshutz R, Daley G, Lander E (1999) Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat Genet 22:231–238 [DOI] [PubMed] [Google Scholar]
- Carlo T, Sterner D, Berget S (1996) An intron splicing enhancer containing a G-rich repeat facilitates inclusion of a vertebrate micro-exon. RNA 2:342–353 [PMC free article] [PubMed] [Google Scholar]
- Castellví-Bel S, Sheikhavandi S, Telatar M, Tai LQ, Hwang M, Wang Z, Yang Z, Cheng R, Gatti RA (1999) New mutations, polymorphisms, and rare variants in the ATM gene detected by a novel SSCP strategy. Hum Mutat 14:156–162 [DOI] [PubMed] [Google Scholar]
- Cavalli-Sforza LL (1994) The history and geography of human genes. Princeton University Press, Princeton, NJ [Google Scholar]
- Chen J, Birkholtz GG, Lindblom P, Rubio C, Lindblom A (1998) The role of ataxia-telangiectasia heterozygotes in familial breast cancer. Cancer Res 58:1376–1379 [PubMed] [Google Scholar]
- Choy YS, Dabora SL, Hall F, Ramesh V, Niida Y, Franz D, Kasprzyk-Obara J, Reeve MP, Kwiatkowski DJ (1999) Superiority of denaturing high-performance liquid chromatography over single-stranded conformation and conformation-sensitive gel electrophoresis for mutation detection in TSC2. Ann Hum Genet 63:383–391 [DOI] [PubMed] [Google Scholar]
- Collins A, Lonjou C, Morton N (1999) Genetic epidemiology of single-nucleotide polymorphisms. Proc Natl Acad Sci USA 96:15173–15177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concannon P, Gatti RA (1997) Diversity of ATM gene mutations detected in patients with ataxia-telangiectasia. Hum Mutat 10:100–107 [DOI] [PubMed] [Google Scholar]
- Cooper TA, Mattox W (1997) The regulation of splice-site selection, and its role in human disease. Am J Hum Genet 61:259–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dörk T, Westermann S, Dittrich O, Twardowski M, Karstens JH, Schmidtke J, Stuhrmann M (1997) A frequent polymorphism of the gene mutated in ataxia telangiectasia. Mol Cell Probes 11:71–73 [DOI] [PubMed] [Google Scholar]
- Ejima Y, Sasaki MS (1998) Mutations of the ATM gene detected in Japanese ataxia-telangiectasia patients: possible preponderance of the two founder mutations 4612del165 and 7883del5. Hum Genet 102:403–408 [DOI] [PubMed] [Google Scholar]
- FitzGerald MB, JM; Hegde, SR; Unsal, H; MacDonald, DJ; Harkin, DP; Finkelstein, DM; Isselbacher, KJ; Haber, DA (1997) Heterozygous ATM mutations do not contribute to early onset of breast cancer. Nat Genet 15:307–310 [DOI] [PubMed] [Google Scholar]
- Gatti R (1998) Ataxia-telangiectasia. In: Vogelstein, B and Kinzler, KW (eds) The genetic basis of human cancer. McGraw-Hill, New York, pp 275–300 [Google Scholar]
- ——— (2001) Ataxia-telangiectasia. In: Scriver C, Beaudet A, Sly W, Valle D (eds) The metabolic and molecular bases of inherited disease. McGraw-Hill, New York, pp 705–732 [Google Scholar]
- Gatti RA, Tward A, Concannon P (1999) Cancer risk in ATM heterozygotes: a model of phenotypic and mechanistic differences between missense and truncating mutations. Mol Genet Metab 68:419–423 [DOI] [PubMed] [Google Scholar]
- Gilad S, Khosravi R, Shkedy D, Uziel T, Ziv Y, Savitsky K, Rotman G, Smith S, Chessa L, Jorgensen TJ, Harnik R, Frydman M, Sanal O, Portnoi S, Goldwicz Z, Jaspers NG, Gatti RA, Lenoir G, Lavin MF, Tatsumi K, Wegner RD, Shiloh Y, Bar-Shira A (1996) Predominance of null mutations in ataxia-telangiectasia. Hum Mol Genet 5:433–439 [DOI] [PubMed] [Google Scholar]
- Gordon D, Simonic I, Ott J (2000) Significant evidence for linkage disequilibrium over a 5-cM region among Afrikaners. Genomics 66:87–92 [DOI] [PubMed] [Google Scholar]
- Greenwell P, Krommal S, Porter S, Gassenhuber J, Obermaier B, Petes T (1995) Tel1, a gene involved in controlling telomere length in S. cerevisiae, is homologous to the human ataxia telangectasia gene. Cell 82:823–829 [DOI] [PubMed] [Google Scholar]
- Hacia J, Sun B, Hunt N, Edgemon K, Mosbrook D, Robbins C, Fodor SP, Tagle DA, Collins FS (1998) Strategies for mutational analysis of the large multiexon ATM gene using high-density oligonucleotide arrays. Genome Res 8:1245–1258 [DOI] [PubMed] [Google Scholar]
- Halushka M, Fan J, Bentley K, Hsie L, Shen N, Weder A, Cooper R, Lipshutz R, Chakravarti A (1999) Patterns of single-nucleotide polymorphisms in candidate genes for blood-pressure homeostasis. Nat Genet 22:239–247 [DOI] [PubMed] [Google Scholar]
- Hudson RR, Kreitman M, Aguadé M (1987) A test of neutral molecular evolution based on nucleotide data. Genetics 116:153–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttley GA, Smith MW, Carrington M, O'Brian SJ (1999) A scan for linkage disequilibrium across the human genome. Genetics 152:1711–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janin N, Andrieu N, Ossian K, Laugé A, Croquette MF, Griscelli C, Debré M, Bressac-de-Paillerets B, Aurias A, Stoppa-Lyonnet D (1999) Breast cancer risk in ataxia telangiectasia (AT) heterozygotes: haplotype study in French AT families. Br J Cancer 80:1042–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Underhill PA, Doctor V, Davis RW, Shen P, Cavalli-Sforza LL, Oefner PJ (1999) Distribution of haplotypes from a chromosome 21 region distinguishes multiple prehistoric human migrations. Proc Natl Acad Sci USA 96:3796–3800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobling M, Tyler-Smith C (1995) Fathers and sons: the Y chromosome and human evolution. Trends Genet 11:449–456 [DOI] [PubMed] [Google Scholar]
- Jones AC, Austin J, Hansen N, Hoogendoorn B, Oefner PJ, Cheadle JP, O'Donovan MC (1999) Optimal temperature selection for mutation detection by denaturing HPLC and comparison to single-stranded conformation polymorphism and heteroduplex analysis. Clin Chem 45:1133–1140 [PubMed] [Google Scholar]
- Kaessmann H, Wiebe V, Pääbo S (1999) Extensive nuclear DNA sequence diversity among chimpanzees. Science 286:1159–1162 [DOI] [PubMed] [Google Scholar]
- Khanna KK, Keating KE, Kozlov S, Scott S, Gatei M, Hobson K, Taya Y, Gabrielli B, Chan D, Lees-Miller SP, Lavin MF (1998) ATM associates with and phosphorylates p53: mapping the region of interaction. Nat Genet 20:398–400 [DOI] [PubMed] [Google Scholar]
- Kimmel M, Chakraborty R, King JP, Bamshad M, Watkins WS, Jorde LB (1998) Signatures of population expansion in microsatellite repeat data. Genetics 148:1921–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange E, Borresen AL, Chen X, Chessa L, Chiplunkar S, Concannon P, Dandekar S, et al. (1995) Localization of an ataxia-telangiectasia gene to an approximately 500-kb interval on chromosome 11q23.1: linkage analysis of 176 families by an international consortium. Am J Hum Genet 57:112–119 [PMC free article] [PubMed] [Google Scholar]
- Lavin MF (1999) ATM: the product of the gene mutated in ataxia-telangiectasia. Int J Biochem Cell Biol 31:735–740 [DOI] [PubMed] [Google Scholar]
- Li A, Huang Y, Swift M (1999) Neutral sequence variants and haplotypes at the 150 Kb ataxia-telangiectasia locus. Am J Med Genet 86:140–144 [PubMed] [Google Scholar]
- Li A, Swift M (2000) Mutations at the ataxia-telangiectasia locus and clinical phenotypes of A-T patients. Am J Med Genet 92:170–177 [DOI] [PubMed] [Google Scholar]
- Maillet P, Vaudan G, Chappuis P, Sappino A (1999) PCR-mediated detection of a polymorphism in the ATM gene. Mol Cell Probes 13:67–69 [DOI] [PubMed] [Google Scholar]
- Morgan SE, Lovly C, Pandita TK, Shiloh Y, Kastan MB (1997) Fragments of ATM which have dominant-negative or complementing activity. Mol Cell Biol 17:2020–2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrell D, Chase CL, Swift M (1990) Cancers in 44 families with ataxia-telangiectasia. Cancer Genet Cytogenet 50:119–123 [DOI] [PubMed] [Google Scholar]
- Morrell D, Cromartie E, Swift M (1986) Mortality and cancer incidence in 263 patients with ataxia-telangiectasia. J Natl Cancer Inst 77:89–92 [PubMed] [Google Scholar]
- Morrow DM, Tagle DA, Shiloh Y, Collins FS, Hieter P (1995) TEL1, an S. cerevisiae homolog of the human gene mutated in ataxia telangiectasia, is functionally related to the yeast checkpoint gene MEC1. Cell 82:831–840 [DOI] [PubMed] [Google Scholar]
- Mulligan G, Jacks T (1998) The retinoblastoma gene family: cousins with overlapping interests. Trends Genet 14:223–229 [DOI] [PubMed] [Google Scholar]
- Nachman M (1998) Y chromosome variation of mice and men. Mol Biol Evol 15:1744–1750 [DOI] [PubMed] [Google Scholar]
- Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York [Google Scholar]
- O'Donovan MC, Oefner PJ, Roberts SC, Austin J, Hoogendoorn B, Guy C, Speight G, Upadhyaya M, Sommer SS, McGuffin P (1998) Blind analysis of denaturing high-performance liquid chromatography as a tool for mutation detection. Genomics 52:44–49 [DOI] [PubMed] [Google Scholar]
- Oefner P, Underhill P (1995) Comparative DNA sequencing by denaturing high-performance liquid chromatography (DHPLC). Am J Human Genet Suppl 57:A266 [Google Scholar]
- ——— (1998) DNA mutation detection using denaturing high-performance liquid chromatography. In: Dracopoli N, Haines J, Korf B, Moir D, Morton C, Seidman C, Seidman J, et al (eds) Current protocols in human genetics. John Wiley & Sons, New York, pp 7.10.1–7.10.12 [Google Scholar]
- Olsen J, Hahnemann J, Borresen-Dale A-L, Brondum-Nielsen K, Hammarstrom L, Kleinerman R, Kaariainen H, Lonnqvist T, Sankila R, Seersholm N, Tretli S, Yuen J, Boice J, Tucker M (2001) Cancer in patients with ataxia-telangiectasia and their relatives in the Nordic countries. J Natl Cancer Inst 93:121–127 [DOI] [PubMed] [Google Scholar]
- Passarino G, Shen P, Van Kirk J, Lin A, De Benedictis G, Cavalli Sforza L, Oefner P, Underhill P (2001) The Werner syndrome gene and global sequence variation. Genomics 71:118–122 [DOI] [PubMed] [Google Scholar]
- Platzer M, Rotman G, Bauer D, Uziel T, Savitsky K, Bar-Shira A, Gilad S, Shiloh Y, Rosenthal A (1997) Ataxia-telangiectasia locus: sequence analysis of 184 kb of human genomic DNA containing the entire ATM gene. Genome Res 7:592–605 [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Seielstad MT, Perez-Lezaun A, Feldman M (1999) Population growth of human Y chromosomes: a study of Y chromosome microsatellites. Mol Biol Evol 16:1791–1798 [DOI] [PubMed] [Google Scholar]
- Sandoval N, Platzer M, Rosenthal A, Dörk T, Bendix R, Skawran B, Stuhrmann M, Wegner RD, Sperling K, Banin S, Shiloh Y, Baumer A, Bernthaler U, Sennefelder H, Brohm M, Weber BH, Schindler D (1999) Characterization of ATM gene mutations in 66 ataxia telangiectasia families. Hum Mol Genet 8:69–79 [DOI] [PubMed] [Google Scholar]
- Sasaki T, Tian H, Kukita Y, Inazuka M, Tahira T, Imai T, Yamauchi M, Saito T, Hori T, Hashimoto-Tamaoki T, Komatsu K, Nikaido O, Hayashi K (1998) ATM mutations in patients with ataxia telangiectasia screened by a hierarchical strategy. Hum Mutat 12:186–195 [DOI] [PubMed] [Google Scholar]
- Shayeghi M, Seal S, Regan J, Collins N, Barfoot R, Rahman N, Ashton A, Moohan M, Wooster R, Owen R, Bliss JM, Stratton MR, Yarnold J (1998) Heterozygosity for mutations in the ataxia telangiectasia gene is not a major cause of radiotherapy complications in breast cancer patients. Br J Cancer 78:922–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen P, Wang F, Underhill PA, Franco C, Yang W-h, Roxas A, Sung R, Lin A, Hyman RW, Vollrath D, Davis RW, Cavalli-Sforza LL, Oefner PJ (2000) Population genetic implications from sequence variation in four Y chromosome genes. Proc Natl Acad Sci USA 97:7354–7359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry ST, Rogers AR, Harpending H, Soodyall H, Jenkins T, Stoneking M (1994) Mismatch distributions of mtDNA reveal recent human population expansions. Hum Biol 66:761–775 [PubMed] [Google Scholar]
- Stringer CB, Andrews P (1988) Genetic and fossil evidence for the origin of modern humans. Science 239:1263–1268 [DOI] [PubMed] [Google Scholar]
- Taillon-Miller P, Bauer-Sardiña I, Saccone NL, Putzel J, Laitenen T, Cao A, Kere J, Pilia G, Rice JP, Kwok P-Y (2000) Juxtaposed regions of extensive and minimal linkage disequilibrium in human Xq25 and Xq28. Nat Genet 25:324–328 [DOI] [PubMed] [Google Scholar]
- Tajima F (1989a) The effect of change in population size on DNA polymorphism. Genetics 123:597–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— (1989b) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telatar M, Wang Z, Udar N, Liang T, Bernatowska-Matuszkiewicz E, Lavin M, Shiloh Y, Concannon P, Good RA, Gatti RA (1996) Ataxia-telangiectasia: mutations in ATM cDNA detected by protein-truncation screening. Am J Hum Genet 59:40–44 [PMC free article] [PubMed] [Google Scholar]
- Teraoka SN, Telatar M, Becker-Catania S, Liang T, Önengüt S, Tolun A, Chessa L, Sanal O, Bernatowska E, Gatti RA, Concannon P (1999) Splicing defects in the ataxia-telangiectasia gene, ATM: underlying mutations and consequences. Am J Hum Genet 64:1617–1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff S, Goldman A, Calafell F, Speed WC, Deinard AS, Bonne-Tamir B, Kidd JR, Pakstis AJ, Jenkins T, Kidd KK (1998) A Global haplotype analysis of the myotonic dystrophy locus: implications for the evolution of modern humans and for the origin of myotonic dystrophy mutations. Am J Hum Genet 62:1389–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underhill PA, Jin L, Lin AA, Mehdi SQ, Jenkins T, Vollrath D, Davis RW, Cavalli-Sforza LL, Oefner PJ (1997) Detection of numerous Y chromosome biallelic polymorphisms by denaturing high-performance liquid chromatography. Genome Res 7:996–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underhill PA, Shen P, Lin AA, Jin L, Passarino G, Yang WH, Kauffman E, Bonné-Tamir B, Bertranpetit J, Francalacci P, Ibrahim M, Jenkins T, Kidd JR, Mehdi SQ, Seielstad MT, Wells RS, Piazza A, Davis RW, Feldman MW, Cavalli-Sforza LL, Oefner PJ (2000) Y chromosome sequence variation and the history of human populations. Nat Genet 26:358–361 [DOI] [PubMed] [Google Scholar]
- Uziel T, Savitsky K, Platzer M, Ziv Y, Helbitz T, Nehls M, Boehm T, Rosenthal A, Shiloh Y, Rotman G (1996) Genomic organization of the ATM gene. Genomics 33:317–320 [DOI] [PubMed] [Google Scholar]
- Vorechovsky I, Luo L, Dyer MJ, Catovsky D, Amlot PL, Yaxley JC, Foroni L, Hammarström L, Webster AD, Yuille MA (1997) Clustering of missense mutations in the ataxia-telangiectasia gene in a sporadic T-cell leukaemia. Nat Genet 17:96–99 [DOI] [PubMed] [Google Scholar]
- Vorechovsky I, Luo L, Prudente S, Chessa L, Russo G, Kanariou M, James M, Negrini M, Webster AD, Hammarström L (1996a) Exon-scanning mutation analysis of the ATM gene in patients with ataxia-telangiectasia. Eur J Hum Genet 4:352–355 [DOI] [PubMed] [Google Scholar]
- Vorechovsky I, Rasio D, Luo L, Monaco C, Hammarström L, Webster ADB, Zaloudik J, Barbanti-Brodano G, James M, Russo G, Croce CM, Negrini M (1996b) The ATM gene and susceptibility to breast cancer: analysis of 38 breast tumors reveals no evidence for mutation. Cancer Res 56:2726–2732 [PubMed] [Google Scholar]
- Wagner TM, Hirtenlehner K, Shen P, Moeslinger R, Muhr D, Fleischmann E, Concin H, Doeller W, Haid A, Lang AH, Mayer P, Petru E, Ropp E, Langbauer G, Kubista E, Scheiner O, Underhill P, Mountain J, Stierer M, Zielinski C, Oefner P (1999a) Global sequence diversity of BRCA2: analysis of 71 breast cancer families and 95 control individuals of worldwide populations. Hum Mol Genet 8:413–423 [DOI] [PubMed] [Google Scholar]
- Wagner T, Stoppa-Lyonnet D, Fleischmann E, Muhr D, Pagés S, Sandberg T, Caux V, Moeslinger R, Langbauer G, Borg A, Oefner P (1999b) Denaturing high-performance liquid chromatography detects reliably BRCA1 and BRCA2 mutations. Genomics 62:369–376 [DOI] [PubMed] [Google Scholar]
- Wyckoff G, Wang W, Wu C (2000) Rapid evolution of male reproductive genes in the descent of man. Nature 403:304–309 [DOI] [PubMed] [Google Scholar]
- Zakian VA (1995) ATM-related genes: what do they tell us about functions of the human gene? Cell 82:685–687 [DOI] [PubMed] [Google Scholar]
- Zhivotovsky LA, Bennett L, Bowcock AM, Feldman MW (2000) Human population expansion and microsatellite variation. Mol Biol Evol 17:757–767 [DOI] [PubMed] [Google Scholar]
- Zietkiewicz E, Yotova V, Jarnik M, Korab-Laskowska M, Kidd KK, Modiano D, Scozzari R, Stoneking M, Tishkoff S, Batzer M, Labuda D (1997) Nuclear DNA diversity in worldwide distributed human populations. Gene 205:161–171 [DOI] [PubMed] [Google Scholar]