

Abstract
An insertion in rpoS, which encodes the general stress response sigma factor σ38, was isolated as an antimutator for ‘stationary-phase’ or ‘adaptive’ mutation. In the rpoS mutant strain the levels of error-prone DNA polymerase Pol IV were reduced. Pol IV is encoded by the dinB gene, and the amount of its transcript was also reduced in rpoS mutant cells. In wild-type cells, the levels of Pol IV increased in late stationary phase and stayed elevated for several days of continuous incubation, whereas in rpoS defective cells Pol IV was not induced and declined during prolonged incubation. Even in cells missing LexA, the repressor of dinB, maximum Pol IV expression required RpoS. These results suggest that induction of Pol IV is part of a cellular response to starvation and other stresses.
Introduction
Because most mutations are detrimental, organisms have evolved to keep their mutation rates as low as possible given the costs, in terms of energy and time, of error prevention and repair (Drake, 1991). Mutations arise as a result of DNA damage or as a result of polymerase mistakes made during DNA replication. Recently a family of error-prone DNA polymerases, named the Y family, has been discovered (reviewed in Goodman, 2002; Yeiser et al., 2002). Found in all three domains of life, many of these polymerases can replicate past DNA lesions but at the cost of frequent mutation. Escherichia coli is known to have two Y-family DNA polymerases, DNA polymerase IV (Pol IV), the product of the dinB gene (also called dinP) (Wagner et al., 1999), and DNA polymerase V (Pol V), the product of the umuDC operon (Tang et al., 1998; 1999; Reuven et al., 1999). Both of these polymerases are induced after DNA damage as part of E. coli’s SOS response (Kenyon and Walker, 1980; Bagg et al., 1981; Courcelle et al., 2001). Whereas Pol V can replicate past a variety of DNA lesions, the ability of Pol IV to do so is modest (Napolitano et al., 2000; Tang et al., 2000; Kim et al., 2001), and its bypass ability clearly depends on the lesion and the sequence context (Lenne-Samuel et al., 2000; Shen et al., 2002).
As would be expected from the need to keep spontaneous mutation rates low, the level and activity of DNA Pol V are tightly controlled and targeted in E. coli (Goodman, 2002). In contrast, the levels of Pol IV are relatively high. In the absence of DNA damage, normal cells have about 250 copies of Pol IV enzyme (Kim et al., 2001), whereas it is estimated that there are only 15 copies of Pol V in uninduced cells (R. Woodgate, pers. comm.). Unlike Pol V, Pol IV has no known cofactors. In vitro its efficiency (Tang et al., 2000) and processivity (Wagner et al., 2001) are dramatically increased by the β clamp; it is possible that in vivo Pol IV synthesizes tracts of error-containing DNA up to 1300 bases long (Wagner et al., 2001). Overproduction of Pol IV is a powerful mutator in growing cells (Wagner and Nohmi, 2000; Kim et al., 2001) and in stationary-phase cells (P.L. Foster, unpubl. obs.). However, loss of Pol IV has only modest effects on normal, growth-dependent mutation rates, although, again this depends on the mutational target (Strauss et al., 2000; Kim et al., 2001). These results suggest that in normal cells there are mechanisms to keep the mutagenic activities of Pol IV under control.
When populations of microorganisms are exposed to non-lethal selection, the non-proliferating cells accumulate mutations that relieve the selective pressure (Cairns et al., 1988), a phenomenon called ‘stationary-phase’ or ‘adaptive mutation’ (Cairns and Foster, 1991). Most research on adaptive mutation has focused on a strain of E. coli, called FC40, that cannot utilize lactose (Lac−) but that readily reverts to lactose utilization (Lac+) when lactose is its only carbon source (Cairns and Foster, 1991). Unlike growth-dependent mutations, Lac+ adaptive mutations in FC40 require the recombination functions for double-strand break repair encoded by recA and recBCD, and the DNA branch migration and resolution functions encoded by ruvAB and ruvC (Cairns and Foster, 1991; Foster, 1993; Harris et al., 1994; 1996; Foster et al., 1996). In contrast, mutations in recG, which encodes an enzyme also involved in DNA branch migration, increase the rate of adaptive mutation up to 100-fold (Foster et al., 1996; Harris et al., 1996). In addition, the high rate of adaptive mutation requires that the lac allele be on the F’episome (Foster and Trimarchi, 1995; Galitski and Roth, 1995; Radicella et al., 1995) and that conjugal functions be expressed in cis (Foster and Trimarchi, 1995). Evidence suggests that nicking the DNA at the conjugal origin is the important conjugal function required for adaptive mutation (Rodriguez et al., 2002).
Whereas several different types of sequence changes revert the Lac− allele during growth, adaptive Lac+ mutations are almost all −1 bp frameshifts at runs of iterated bases (Foster and Trimarchi, 1994; Rosenberg et al., 1994). At least three DNA polymerases are active in producing or preventing adaptive mutations. Pol IV is responsible for about 50–80% of the mutations; the remainder are probably produced by the replicative polymerase, Pol III (Foster, 2000; McKenzie et al., 2001). Eliminating the accurate repair polymerase, Pol II, increases the Pol IV-dependent component of adaptive mutation (Escarceller et al., 1994; Foster et al., 1995; Foster, 2000). In contrast, Pol V plays no role in adaptive mutation in FC40 (Cairns and Foster, 1991; McKenzie et al., 2000).
When the Lac− population is incubated on lactose, a small subpopulation of cells are in a transient state of hypermutation during which they sustain both selected and non-selected mutations (Torkelson et al., 1997; Rosche and Foster, 1999). The mutations accumulated by these cells are due to Pol IV (Tompkins et al., 2003). Thus, under adverse conditions, cells may use this error-prone polymerase to produce variants that allow their descendents to survive. Indeed, Pol IV, Pol II and Pol V were all shown to be important for the long-term evolution and survival of E. coli (Yeiser et al., 2002).
The available evidence suggests that in normal cells during exponential growth, Pol IV has only a small effect on mutation rates, but, it has a large effect on mutations that occur in cells during non-lethal selection. Because of these facts, we hypothesized that Pol IV must normally be under strict control, but may be activated in stressed cells. Here we report that Pol IV is under control of the stress-induced sigma factor, rpoS, and that Pol IV is induced late in stationary phase.
Results
RecG mutants have high levels of Pol IV
The adaptive mutation rate in E. coli strain FC40 is increased 10- to 100-fold if recG is defective (Foster et al., 1996; Harris et al., 1996) and all of these extra mutations are due to DNA Pol IV (Fig. 1; E. Ohashi, H. Ohmori, and P.L. Foster unpubl. data). The cellular levels of Pol IV are elevated three- to fivefold in recG mutant strains (Fig. 2; E. Ohashi, H. Ohmori, and P.L. Foster unpubl. data). We assume that this increase in Pol IV is due to the partial derepression of the SOS response in recG defective cells (Lloyd and Buckman, 1991). However, overproduction of Pol IV is not, in itself, sufficient for the increase in adaptive mutation; recombination functions are also required. As shown in Fig. 2, a Δruv(AC) mutation, which eliminates adaptive mutation in both recG+ and recG mutant strains (Foster et al., 1996; Harris et al., 1996), does not reduce Pol IV levels in either background. Indeed, Pol IV levels appear to be about twofold higher in the recG Δruv(AC) mutant strain than in the recG mutant strain, possibly because the doubly mutant strain is further induced for SOS.
Fig. 1.
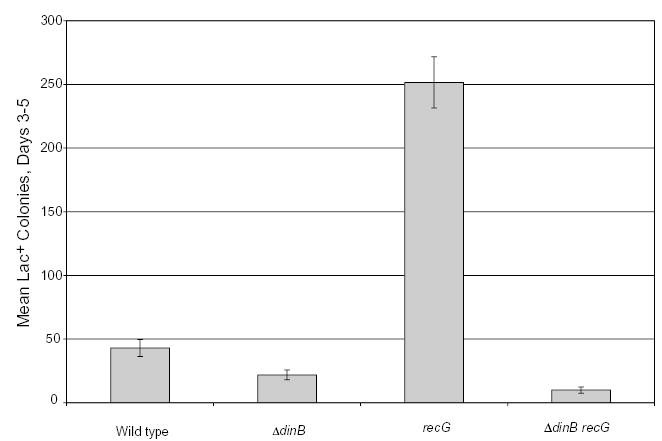
A recG mutation enhances the Pol IV-dependent component of adaptive mutation. The accumulation of Lac+ mutations in Lac− cells incubated on lactose minimal medium. Wild type = FC40; ΔdinB = FC1354; recG = FC438; ΔdinB recG = FC1355. Two experiments are shown, one with FC40 and FC1354, and one with FC438 and FC1355. Data are the mean number of Lac+ colonies appearing each day from days 3–5; each point is the mean of four cultures ± SEM.
Fig. 2.

Levels of Pol IV are increased in a recG mutant strain but are not affected by ruv mutations. Western blot showing levels of Pol IV in (from left to right) wild type = FC40; recG = FC465; Δ (ruvAC) = FC836; recG Δ (ruvAC) = FC897; ΔdinB = FC1240. Samples consisting of 10 μg of total protein were loaded in each lane of a precast SDS-PAGE gel (Bio-Rad Laboratories). The intensities of the bands for each strain relative to the wild-type strain are given below each lane; the intensity of only the upper band relative to that of the wild-type strain is in parenthesis.
Sometimes, as in Fig. 2, we observe a second, smaller band that reacts with the anti-Pol IV polyclonal antibody. The smaller band may be a degradation product of Pol IV that only sometimes appears as a result of unknown variables in our experiments.
An rpoS mutation decreases adaptive mutation
We used the high rate adaptive mutation of a recG mutant strain to screen for mutations that affect the levels or activities of Pol IV. One of these was an insertion of mini-Tn10 Cm (Kleckner et al., 1991) in the rpoS gene, which encodes the stationary phase sigma factor, σ38. As shown in Fig. 3A, the rpoS::Cm mutation reduced adaptive mutation in the recG mutant 20- to 30-fold. Over the same time period the viability of the rpoS::Cm mutant strain incubated on lactose plates declined to about 20% of that of the rpoS+ strain (Fig. 3B), but this cannot account for the 20- to 30-fold reduction in mutations. In recG+ strains, the rpoS::Cm mutation reduced adaptive mutation about 10-fold (see Fig. 7). We obtained similar results with another allele of rpoS, ΔrpoS::Kn (Bohannon et al., 1991) (data not shown).
Fig. 3.

A rpoS mutation decreases adaptive mutation.
A. The accumulation of Lac+ mutations in Lac− cells incubated on lactose minimal medium. Circles, recG = FC526; triangles, recG rpoS = PFG36. Data are the cumulative number of Lac+ colonies appearing from days 3–5; each point is the mean of four cultures ± SEM (some error bars are smaller than the symbols).
B. The survival of Lac− cells while incubating on lactose minimal medium. Circles, recG = FC526; triangles, recG rpoS::Cm = PFG36. Because 10-fold more PFG36 cells were plated than FC526 cells, the numbers of FC526 cells on each day were multiplied by 10; then the results for both strains were normalized to the value for FC526 on day 0. Because it takes two days for a Lac+ revertant to make a visible colony, the values shown in B correspond to the points two days later in the Lac+ curves in A.
Fig. 7.

A rpoS mutation reduces adaptive mutation in various backgrounds. The accumulation of Lac+ mutations in Lac− cells incubated on lactose minimal medium. From left to right: Wild type = FC40; rpoS = PFG72; sulA = FC1417; sulA rpoS = PFG185; LexA(Ind−) = FC1418; LexA(Ind−) rpoS = PFG184; sulA LexA(Def) = FC1419, sulA LexA(Def) rpoS = PFG186; recG = FC526; recG rpoS = PFG36. Data are the mean number of Lac+ colonies appearing each day from days 3–5; each point is the mean of four cultures ± SEM. Three experiments are shown: the first was with the wild type and rpoS mutant strain; the second was with all the derivatives of the sulA mutant strain; and, for comparison, the data for recG and recG rpoS from the experiment shown in Fig. 3 were recalculated in the same format.
The rpoS mutation reduces the cellular levels of Pol IV protein
As shown in the two Western blots in Fig. 4, the rpoS::Cm mutation reduces the cellular levels of Pol IV protein. In both the recG+ and recG mutant backgrounds, the rpoS::Cm mutation resulted in a three- to fivefold reduction in Pol IV levels relative to the rpoS+ parental stains. The ΔrpoS::Kn allele described above resulted in about a twofold reduction of Pol IV (data not shown).
Fig. 4.

An rpoS mutation reduces the cellular levels of Pol IV protein. A. Western blot showing levels of Pol IV in (from left to right) ΔdinB = FC1240; recG rpoS = PFG36; recG = FC526; wild type = FC40. B. A different Western blot showing levels of Pol IV in (from left to right) wild type = FC40; rpoS = PFG72. Samples consisting of 40 μg of total protein were loaded in each lane of each blot; the blot in B was developed long enough to enhance the band from the rpoS mutant strain. The intensities of the bands for each strain relative to the wild type strain are given below each lane; the intensity of only the upper band relative to that of the wild type strain is in parenthesis.
The rpoS mutation reduces the cellular levels of Pol IV transcript
Because RpoS is a transcriptional activator, we determined whether the level of dinB transcript is affected by the rpoS::Cm mutation. The dinB transcript is relatively rare (Courcelle et al., 2001), so we used competitive reverse transcription-polymerase chain reaction (RT-PCR) to compare the amount of Pol IV transcript in wild-type and rpoS::Cm mutant cells. In this technique, a truncated dinB transcript that can be amplified with the same primers as the endogenous dinB transcript is used as a competitor in the reaction. As shown in Fig. 5, the rpoS::Cm mutation resulted in about a threefold reduction in the amount of dinB transcript level relative to the total RNA in the cells. This result is calculated from the slope of the curves shown in Fig. 5C: the curve for the wild-type strain, FC40, crosses y = 0 at × = 0.69, which is 4.9 pg of competitor RNA; the curve for the rpoS::Cm mutant strain, PFG72, crosses y = 0 at x = 0.23, which is 1.7 pg of competitor RNA; the ratio of 4.9 to 1.7 is 2.9.
Fig. 5.

A rpoS mutation reduces the cellular levels of Pol IV transcript. A. Ethidium stained agarose gel showing the dinB RT-PCR products from 250 ng of total endogenous RNA extracted from the wild type strain FC40 (upper band) and the RT-PCR products from the truncated dinB competitor RNA added to the same reactions at 1, 2, 4, 8, and 16 pg (lower band). M = 500 bp molecular weight marker. B. Ethidium stained agarose gel showing the dinB RT-PCR products from 250 ng of total endogenous RNA extracted from the rpoS::Cm strain PFG72 (upper band) and the RT-PCR products from the truncated dinB competitor RNA added to the same reactions at 0.5, 1, 2, 4, and 8 pg (lower band). M = 500 bp molecular weight marker. C. Plot and the least-squares fit of the log of the ratios of endogenous to competitor band intensities vs. the log of the amount of competitor RNA added. (To give a better fit, the 4 pg value for FC40, which appears to be an outlier, has been excluded; if it is included the line crosses y = 0 at x = 0.56 or 4.2 pg instead of at × = 0.69 or 4.9 pg).
Pol IV levels increase in late stationary phase
All the results described above were obtained with cultures grown in minimal medium for 18–24 h, at which point the cells are in late stationary phase. RpoS levels increase as cells enter into stationary phase and the amount of RpoS reaches its maximum in early stationary phase (for a review see Ishihama, 2000). To determine if Pol IV is growth-phase regulated, we monitored the level of Pol IV protein in wild-type and rpoS::Cm cells during exponential growth and stationary phase. Surprisingly, there were no changes in Pol IV levels or major differences between wild type and rpoS::Cm. We repeated the experiment with longer incubation times and obtained the results shown in Fig. 6. As we had previously observed, the levels of Pol IV were constant throughout exponential phase and early stationary phase. However, in the wild-type strain the level of Pol IV protein doubled during an additional 17 h in stationary phase, whereas the levels in the rpoS::Cm strain declined over the same period. The difference between mutant and wild type at 32 h (sample d) corresponds to the differences we observed in other experiments (e.g. Figure 4). With longer incubation the level of Pol IV protein in the wild type strain was relatively constant, whereas the level of Pol IV in the rpoS::Cm mutant strain continued to decline, reaching about 2% of the wild-type level after 3 days. There was no loss of viability of either strain during this period (data not shown).
Fig. 6.

Pol IV levels increase in late stationary phase.
A. Western blot showing the amount of Pol IV protein in wild type (= FC40) and rpoS::Cm (= PFG72) cells at the time points indicated in B. Two blots are shown: on the left are samples taken during exponential growth through stationary phase; on the right are samples taken one and two days later. Samples c and d were reloaded for the blot on the right. Samples consisting of 40 μg of total protein were loaded (only half of the ΔdinB control lane on the left is visible).
B. The growth curve of the wild type (= FC40) and rpoS::Cm (= PFG72) mutant strain in minimal glycerol medium. Samples were taken at the times indicated by arrows.
C. The relative amounts of Pol IV protein in the wild type (= FC40) cells and rpoS::Cm (= PFG72) cells at the time points indicated in B. Bands on the two blots were normalized by first setting the most intense band for each strain on each blot equal; these were the band at d for the wild type strain, and the band at c for the rpoS::Cm strain. The intensity of each band was then recalculated relative to these. These values were then normalized to the wild-type strain sample a.
Regulation of Pol IV by RpoS occurs in addition to SOS regulation
dinB is repressed by LexA, the common repressor of the SOS regulon (Kenyon and Walker, 1980; Courcelle et al., 2001). Mutations that make LexA non-inducible, LexA(Ind−), prevent the induction of LexA repressed genes, whereas mutations that make LexA defective, LexA(Def), result in constitutive expression of LexA-repressed genes. LexA(Def) mutations are lethal unless the cells are defective in sulA to prevent lethal filamentation. In the experiments shown in Fig. 7 we used the sulA11 allele (see Experimental procedures). This allele has no effect on adaptive mutations (Foster and Rosche, 1999) and the rpoS::Cm mutation has the same effect in the sulA11 mutant strain, FC1417, as in the wild-type strain, FC40. As shown in Fig. 7, adaptive mutation was reduced by the rpoS::Cm mutation in both LexA(Ind−) and LexA(Def) mutant backgrounds. Note that the LexA(Def) mutant strain, in which dinB is derepressed, has an adaptive mutation rate lower than its LexA+ parent. This is because another gene, psiB, whose product interferes with the expression of the SOS response, is also dere-pressed in LexA(Def) cells (McKenzie et al., 2000). Nonetheless, adaptive mutation in the LexA(Def) background was reduced 10-fold by the rpoS::Cm mutation.
As expected, the LexA(Def) mutation resulted in about a 30-fold increase in the levels of Pol IV protein (Fig. 8). Yet, even in this background, the rpoS::Cm mutation decreased the level of Pol IV by about threefold. For as yet unknown reasons, the expression of Pol IV from the episomal copy of dinB is about threefold higher than from the chromosomal copy (Kim et al., 2001); thus it seemed possible that rpoS might be affecting only dinB expressed from the episome. However this is not the case: the three sulA11 mutant strains shown in Fig. 8 are all F−, so dinB is expressed only from their chromosomes.
Fig. 8.
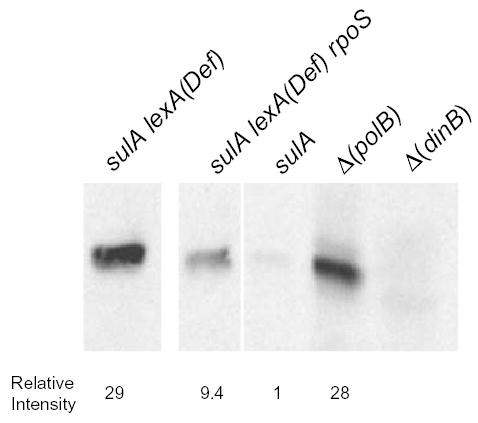
A rpoS mutation reduces the cellular levels of Pol IV protein even in the absence of LexA. Western blot showing levels of Pol IV in (from left to right); sulA LexA(Def) = FC1413; sulA LexA(Def) rpoS
Figure 8 also shows the level of Pol IV in a Δ (polB) mutant, which is missing DNA polymerase II. As mentioned above, the Δ (polB) mutant increases the Pol IV-dependent component of adaptive mutation (Escarceller et al., 1994; Foster, 2000). This phenotype is very similar to that of the recG mutant, and, like in recG mutant strains, the levels of Pol IV are increased in the Δ (polB) mutant strain (Fig. 8). Possibly loss of Pol II results in SOS induction, as does loss of DNA polymerase I (Bates et al., 1989)
Discussion
Because Pol IV normally has only a small effect on growth-dependent mutation rates but a larger effect on adaptive mutation rates, we hypothesized that its levels or activity must be regulated. In this paper we describe a mutant search that revealed one such regulatory element, the general stress response RNA polymerase sigma factor, RpoS. Pol IV is induced late in stationary phase, after 24 h of incubation, and this induction requires RpoS. If RpoS is active, Pol IV levels remain high for at least 3 days of continuous incubation and during this time adaptive mutation rates are high. If RpoS is defective, the levels of Pol IV decline during prolonged incubation and adaptive mutation rates are low. Even when Pol IV is derepressed by eliminating LexA, maximum expression requires RpoS.
Overproduction of DNA polymerase IV by supplying the dinB gene on a multicopy plasmid is a powerful mutator, increasing spontaneous mutation rates by as much as 1000-fold (Kim et al., 1997; Wagner and Nohmi, 2000; Kim et al., 2001). Pol IV’s preferred replication errors are frameshifts, but when Pol IV is overexpressed the rate of base substitution mutations is also greatly elevated (Wagner and Nohmi, 2000; Kobayashi et al., 2002). Decreasing the amount of Pol IV that is normally in the cell has a much smaller effect on spontaneous mutations rates (Strauss et al., 2000; McKenzie et al., 2001; P. L. Foster, unpubl. data). For example, reduction of Pol IV by three-quarters (from roughly 1000 to 250 copies of the protein) reduced the rate of reversion of an episomal frameshift mutation about twofold; eliminating Pol IV completely produced no additional reduction (Kim et al., 2001). Although Pol IV = PFG182; sulA = FC942; Δ (polB) = PFB60; Δ (dinB) = FC1240. FC1413, PFG182, and FC942 are F− strains so their only dinB allele is chromosomal. Only one Western blot is shown, but irrelevant lanes have been removed. Samples consisting of 40 μg of total protein were loaded. The intensities of the bands for each strain relative to the wild type strain are given below each lane. affects the mutation rates of genes on the chromosome, it appears to have a more powerful effect on extrachromosomal elements such as lambda phage and the F episome.
In our system, Lac− cells are plated on medium with lactose as the only carbon and energy source (Cairns and Foster, 1991). Under these conditions, Lac+ mutations appear after two days of incubation and continue to arise at a constant rate for about 5 days. Deleting Pol IV reduces the rate of adaptive Lac+ mutations two to fourfold; because this reduction continues for the course of the experiment, which is usually 5 days, it results in a large decrease in the final number of mutations (Foster, 2000; McKenzie et al., 2001). Reductions of similar magnitude have been observed for growth-dependent mutations using other Lac− alleles (Strauss et al., 2000; Kim et al., 2001), but these other experiments are typically done under conditions similar to those for adaptive mutation. Protocols that select for Lac+ reversion on lactose medium place the cells in similar states of starvation during which, according to our results, the effect of Pol IV will be most obvious.
Pol IV has a more dramatic effect on adaptive mutation when recG or polB are defective. In these mutant backgrounds the Pol IV-dependent component of adaptive mutations is enhanced (Escarceller et al., 1994; Foster, 2000; E. Ohashi, H. Ohmori, and P. L. Foster unpubl. data; Fig. 1). These results can be explained by the fact that in both recG and polB mutant strains, the amount of Pol IV is elevated (Figs 2 and 8). However, Pol IV is not sufficient for adaptive mutation because even when the levels of Pol IV are high, recombination functions are also required (Fig. 2). In addition, some other rpoS-dependent factor is required for full mutagenicity (see below). Interestingly, derepression of Pol IV in a LexA(Def) mutant strain does not have the same effect as Pol IV overexpression in recG or polB mutant strains (Fig. 7). Presumably, the SOS-inhibiting gene psiB, which is induced in a LexA (Def) strain (Bagdasarian et al., 1986; McKenzie et al., 2000), is not induced in the recG or polB mutant strains.
The rpoS mutant that we isolated has the mini-Tn10 Cm transposon inserted at bp 775 of the 993 bp coding sequence. The predicted product is a protein 268 amino acids long, 258 of which are RpoS and 10 of which are encoded by the end of the transposon. The mutation should eliminate region 4, which is thought to be important for interaction with the −35 region of promoters, but should leave intact the RpoS regions important for RNA polymerase core binding and interactions with the −10 promoter element (Colland et al., 2002). rpoS mutants with similar C-terminus truncations have impaired but not completely defective promoter binding affinities in vitro (Gowrishankar et al., 2003). Of course, the protein encoded by our rpoS mutant may not be stable, or may not complex with RNA polymerase core, and thus our mutant allele may be effectively null. In support of this, we obtained similar but somewhat weaker results with another allele of rpoS that has the rpoS coding sequence from base pair 211 past the end of the gene replaced by a KnR element (Bohannon et al., 1991). In addition, we could partially complement the rpoS::Cm mutation with a multicopy plasmid carrying rpoS+, although this plasmid itself (a Δlac derivative of pDEB2; Bohannon et al., 1991) had a deleterious effect on the cells (data not shown). These results support the hypothesis that rpoS::Cm is a loss-of-function mutation.
There are two unusual aspects of Pol IV regulation by RpoS. First, Pol IV is also regulated by LexA (Fig. 8). It is fairly common for RpoS-regulated genes to be regulated also by effectors of RpoD (σ70)-dependent transcription. For example, several genes encoding DNA repair and protection proteins are dually controlled by RpoS and OxyR, the regulator of the response to oxidative stress (Eisenstark, 1998). However, to date the only gene known to be co-regulated by LexA and RpoS is sbmC, which encodes a DNA gyrase inhibitor (Oh et al., 2001). As shown in Fig. 8, maximum expression of Pol IV even in a LexA(Def) strain depends on RpoS. And, even when Pol IV is maximally repressed by LexA, the rpoS mutation still decreases adaptive mutation (Fig. 7). This latter result may be a consequence of the fact that the dinB promoter has a rather poor ‘LexA’ box, and repression by LexA may not be stringent (Fernandez de Henestrosa et al., 2000; Kim et al., 2001).
The second unusual aspect of Pol IV regulation by RpoS is that Pol IV is induced late. During normal growth RpoS reaches its maximum level in early stationary phase and most RpoS regulated genes are expressed at this time (Oh et al., 2001). We found no change in Pol IV levels until well into stationary phase (Fig. 6). However, RpoS-controlled genes are expressed in a temporal order, which probably reflects changes in other effectors that influence the level and promoter recognition of the RNA polymerase/RpoS holoenzyme (Eσs). These other effectors may be RpoS-controlled proteins, DNA structural changes (e.g. supercoiling), or non-protein effectors such as ppGpp (Ishihama, 2000). Alternatively, RpoS may control the late induction of Pol IV indirectly via induction or repression of some other gene or genes. The fact that the amount of Pol IV in cells depends on the length of their incubation may account for some of the variability reported in the contribution of Pol IV to spontaneous and adaptive mutation.
Although our data do not address the question of whether RpoS regulates Pol IV directly or indirectly, it is tempting to speculate on what sequences in the dinB promoter region might be recognized by Eσs. When genes are transcribed both by Eσs and Eσ70 the two polymerases may recognize completely different promoters; but, in many cases they recognize the same promoter. Thus, it has proved difficult to define a consensus sequence for RpoS promoters. However, some sequence elements, particularly in the −10 region, are believed to be important (reviewed in Hengge-Aronis, 2002a). The consensus LexA binding box is 5′-TA CTG (TA)5 CAG TA-3′ with CTG(N)10CAG being the minimum recognition sequence (Walker, 1984; Fernandez De Henestrosa et al., 2000). Within the dinB LexA box (Fig. 9) is the sequence TATACT, which is a reasonable Eσ70 promoter −10 element, but is also considered to be the consensus −10 sequence for Eσs recognition (Espinosa-Urgel et al., 1996). However, this presumptive promoter element is missing the important C at −13 (Becker and Hengge-Aronis, 2001). 3′ to this −10 sequence is an AT rich sequence, which is also important for Eσs recognition (Hengge-Aronis, 2002a). The fact that there is no strong −35 region is also suggestive of RpoS-dependent transcription. Whether these elements, in fact, specify Eσs transcription of dinB will be determined by further experimentation.
Fig. 9.

The dinB promoter region. The DNA sequence 5′ to the dinB gene. Relevant features are in bold. The minimum 3-base inverted repeats that define the LexA box are boxed. A possible promoter element is underlined. SD = possible Shine-Delgarno sequence. Start = the dinB translational start. Stop = stop codon of the preceding gene, mbhA.
Our data suggest that, in addition to Pol IV, high levels of adaptive mutation require some additional unknown RpoS-controlled factor or factors. In both recG+ and recG mutant cells, the rpoS::Cm mutation reduces the level of Pol IV three- to fivefold (Fig. 4), but reduces adaptive mutation 10-fold in recG+ cells and 30-fold in recG mutant cells (Figs 3 and 7). The amount of Pol IV protein in recG rpoS doubly mutant cells is three- to fourfold greater than in wild-type cells (Fig. 4), yet the adaptive mutation rate of the recG rpoS mutant strain is equivalent to that of the recG+ rpoS+ strains (Fig. 7). One possibility is that, in addition to the amount of Pol IV in the cell, the activity of Pol IV is regulated by RpoS-dependent genes. Alternatively, other aspects of adaptive mutation that are independent of the amount of Pol IV may be RpoS-controlled.
During exponential growth in non-limiting conditions cells are under selection to maintain their genome more or less intact. However, in stressful situations, cells with an increased mutation rate, and thus an increased probability of genetic variation, can have a selective advantage (Sniegowski et al., 1997; Taddei et al., 1997). This advantage would be greater if the mutator state were transient so that the cells returned to a normal mutation rate when they began to grow (Hall, 1990; Cairns, 1998; Rosche and Foster, 1999). RpoS activity is triggered by entry into stationary phase as well as other stress conditions that inhibit growth. More than 70 genes are known to be regulated by RpoS, and most of them encode proteins that help the cell survive a gamut of insults that may be encountered by non-growing cells. Thus, RpoS is considered to be a master regulator of a general stress response (reviewed in Hengge-Aronis, 2002b). Regulation of Pol IV by RpoS may have two important advantages for the cell. First, high levels of Pol IV may help the cell survive certain classes of DNA damage encountered in stationary phase or in other stressful conditions. Second, by increasing the errors produced by whatever DNA synthesis occurs in non-growing cells, high levels of Pol IV may produce genetic variation from which an advantageous mutation may arise. Indeed, Pol IV and the other inducible polymerases, Pol II and Pol V, were found to be transcribed during prolonged stationary phase, and each conferred a competitive advantage (Yeiser et al., 2002). All three of these polymerases can contribute to survival in the face of DNA damage and replication disasters. The fact that Pol IV is induced in late stationary phase under control of the general stress-response sigma factor suggests that the ability to transiently increase genetic diversity is one of the functions that have evolved to promote survival during stress.
Experimental procedures
Bacterial strains
The strains used are listed in Table 1. Genetic manipulations were performed using standard techniques (Miller, 1992). To make a dinB− strain, the F− parent of FC40, FC36, was transduced to resistance to kanamycin (KnR) with a P1vir bacteriophage lysate grown on strain YG7207 (Kim et al., 1997). The dinB gene is also on the F′128 episome. To make dinB− episomes, the Δ (lac proB) Met− RifR (rifampicin resistant) strain, XA105 (Coulondre and Miller, 1977) carrying F′ Φ (lacI33-lacZ) or this episome with a Tetracycline sensitive (TetS) element (Foster, 1997) was transduced with the P1vir lysate of strain YG7207, mated with a F− Δ (lac proB) Met+ RifS strain, P90C (Coulondre and Miller, 1977), and the mating mixture plated on minimal medium plus kanamycin. The episomes from 50 exconjugates were mated back into XA105 by selecting for RifR Pro+, purified, and screened for KnR. KnR episomes were mated into the F− Δ (dinB) mutant strain by selecting for Pro+ to produce the doubly Δ (dinB) strains FC1240 and FC1354. The absence of Pol IV was confirmed by Western blotting (see below). To make the LexA(Ind−) strain FC1418, the F′ Φ (lacI33-lacZ) episome was mated into strain RM5316 = C36 lexA1 (obtained from R. Maurer; the lexA1 allele is described in Howard-Flanders, 1968). The sulA11 (formally sfiA11) allele from strain DM1187 (Mount, 1977) was transduced into a C36 derivative that had been made pyrD34 zcc-282Tn10 (donor strain STL313 obtained from S. Lovett) by selecting for growth without uracil, screening for loss of resistance to tetracycline (Foster and Rosche, 1999). The presence of the sulA11 allele was confirmed by lack of filamentation after exposure to nalidixic acid. F′ Φ (lacI33-lacZ) was then mated into this strain by selecting for Pro+. The sulA11 mutant strains were transduced with lexA71::Tn5 allele from strain GW2707 (Krueger et al., 1983).
Table 1.
Bacterial strains and plasmids used in this study.
| Strain | Relevant phenotype or genotype | Copies of dinB+ | Source/reference |
|---|---|---|---|
| FC29 | ara Δ (lac-proB)XIII thi/F′ Δ (lacIZ) | 2 | Cairns and Foster,1991 |
| FC36 | F− ara Δ (lac-proB)XIII thi RifR | 1 | Cairns and Foster, 1991 |
| FC40 | FC36/F′ Φ (lacI33-lacZ) | 2 | Cairns and Foster, 1991 |
| FC436 | FC40 recG162 zib636::Tn10 | 2 | Foster et al., 1996 |
| FC465 | FC40 recG258::dTn10Kn | 2 | Foster et al., 1996 |
| FC526 | FC40 ΔrecG263::Kn | 2 | Foster et al., 1996 |
| FC722 | FC40 with a TetS allele on the episome | 2 | Foster, 1997 |
| FC836 | FC40 Δ (ruvAC) eda57::Tet::Cm | 2 | Foster et al., 1996 |
| FC897 | FC40 ΔrecG258::Kn Δ (ruvAC) eda57::Tet::Cm | 2 | Foster et al., 1996 |
| FC942 | FC36 sulA11 | 1 | This study |
| FC1240 | FC722 ΔdinB::Kn on chromosome and episome | 0 | This study |
| FC1354 | FC40 ΔdinB::Kn on chromosome and episome | 0 | This study |
| FC1355 | FC1354 ΔrecG258::Kn | 0 | This study |
| FC1413 | FC942 lexA71::Tn5 | 1 | This study |
| FC1417 | FC942/F′ Φ (lacI33-lacZ) | 2 | This study |
| FC1418 | FC40 lexA1 | 2 | This study |
| FC1419 | FC1413/F′ Φ (lacI33-lacZ) | 2 | This study |
| PFB60 | FC40 ΔpolB1 | 2 | Escarceller et al., 1994 |
| PFG36 | FC526 rpoS::mini-Tn10 Cm | 2 | This study |
| PFG72 | FC40 rpoS::mini-Tn10 Cm | 2 | This study |
| PFG182 | FC1413 rpoS::mini-Tn10 Cm | 1 | This study |
| PFG184 | FC1418 rpoS::mini-Tn10 Cm | 2 | This study |
| PFG185 | FC1417 rpoS::mini-Tn10 Cm | 2 | This study |
| PFG186 | FC1419 rpoS::mini-Tn10 Cm | 2 | This study |
Isolation of antimutator mutants
The recG mutant strain, FC526 (Foster et al., 1996), was mutagenized at random with transposon mini-Tn10 Cm delivered by a defective lambda phage, λNK1324 (Kleckner et al., 1991). Chloramphenicol resistant isolates were replica-plated onto minimal lactose plates and incubated for 5 days, by which point numerous Lac+ microcolonies (papillae) appear within a patch of Lac− cells. Mutants with fewer papillae, indicating a lower mutation rate (antimutators), were identified. After isolates passed a second check using the papillation assay, their antimutator mutations were mapped to the chromosome or the episome by mating the episome into a recipient (by selecting for Pro+) and checking whether the chloramphenicol resistance was transferred. To eliminate mutations in the lac or tra operons, only chromosomal mutations were further characterized. Mutants were also checked for their colony forming ability on glycerol minimal medium and galactose minimal medium to eliminate auxotrophies and mutations in the galactose operon. Isolates that passed these tests were retained. To ensure that the antimutator phenotype was due to the transposon insertion, P1vir lysates of mutants were used to transduce FC526 to chloramphenicol resistance and the antimutator phenotype retested. The gene carrying the transposon was identified by sequencing the product generated by arbitrarily primed PCR (Pratt and Kolter, 1998). The gene was identified from the sequence using the Blast service of the National Center of Biotechnology Information.
Mutation, growth and viability assays
Media and experimental protocols were as previously described (Cairns and Foster, 1991; Foster, 1994; Foster et al., 1996). Statistical calculations were as given in Zar (1984) and Rice (1995). Mutation to Lac+ is given either as the mean cumulative number of Lac+ colonies (Fig 3), or as the mean number of Lac+ colonies appearing each day from days 3–5 (Figs 1 and 7). To measure adaptive mutation, approximately 107 cells of each strain were spread on each quadrant of a lactose minimum plate and the plates were incubated for 5 days at 37°C; newly arising Lac+ colonies were counted each day (Foster et al., 1996). To measure viability, appropriate numbers of cells of the strain to be assayed were plated with 109 FC29 scavenger cells on M9-lactose plates; plugs were removed from between the Lac+ colonies and the number of viable cells determined each day by plating appropriate dilutions on LB plus rifampicin medium, on which FC29 cannot grow (Cairns and Foster, 1991). The growth curves in Fig. 6 were generated by first growing cells to saturation in M9-0.1% glycerol medium plus appropriate drugs. These cells were then diluted 1:1000 into fresh medium and incubated at 37°C with shaking for 5–8 h, at which point the cultures contained approximately 107 cells per ml. These mid-log cells were then diluted 1:100 into fresh medium and allowed to reach saturation. This procedure insured that no stationary-phase cells were present in the samples of early log-phase cells. Growth was monitored with a Klett-Summerson Colorimeter, and samples of appropriate volumes taken at the points indicated in Fig. 6.
Western Immunoblots
Standard molecular biology techniques were used (Ausubel et al., 1988). Cells were harvested by centrifugation, resuspended in 1 × SDS-PAGE sample loading buffer without dye, and boiled for 5 min. Total protein in each sample was determined by Bradford assays (Bio-Rad Laboratories). The appropriate amount of each sample to give 10 or 40 μg of total protein was diluted into 1 × sample-loading buffer with dye, and loaded onto an SDS-12% polyacrylamide gel. With precast gels (Bio-Rad Laboratories), 10 μg of total protein was used; with our laboratory-prepared gels, 40 μg of total protein was used. Proteins were separated by electrophoresis and transferred to Immobilon-P membranes (pore size, 0.45 μm; Millipore). The membrane was probed using standard techniques (Ausubel et al., 1988) with rabbit anti-Pol IV polyclonal antiserum (obtained from H. Omori; the antiserum was clarified using acetone powder made from a Δ (dinB) strain according to the procedure in Harlow and Lane (1988). The reaction was visualized using alkaline phosphatase-conjugated goat anti-rabbit secondary antibody and developed using the Western-light chemiluminescence reagent (Applied Biosystems). Bands were quantified using ImageJ version 1.29x software (W. Rasband, National Institutes of Health). Note that the intensities of the bands visualized with chemiluminescence are determined by several factors, such as length of exposure and age of the reagents; because of this the intensity of bands are not comparable between blots but are comparable within a blot.
Competitive RT-PCR
Techniques were essentially as previously described (Siebert and Larrick, 1992). Competitive RT-PCR combines a reverse transcription step to convert RNA to DNA, then a PCR step to amplify a specific piece of DNA corresponding to the RNA transcript of interest. The reaction is made quantitative by including a known amount of competitor RNA that has the same primer-template sequences as the target RNA (Celi et al., 1993). To amplify the dinB transcript we used dinB1 and dinB2 primers listed in Table 2, which give a 717 base-pair (bp) fragment. As competitor we used a 596 bp dinB fragment amplified with dinB1 and TruncdinB primers (Table 2). This fragment consists of 574 bp of consecutive dinB sequence plus the dinB2 primer sequence. The competitor fragment was cloned into the pGEM-Teasy vector (Promega); the plasmid with the correctly oriented clone was cut with SalI and transcribed in vitro using T7 RNA polymerase with the Maxiscript kit (Ambion). Total RNA was extracted from sample cells using the high salt method for Gram-negative bacteria (Ausubel et al., 1988). Both competitor and sample RNA were treated with DNase I, the competitor RNA using the DNase treatment in the Maxiscript kit (Ambion) and the sample RNA using RQ1 RNase-Free DNase (Promega). Both RNAs were further purified by acid-phenol-chloroform extraction and ammonium acetate precipitation (Ambion technical bulletin #181). The RNA was quantified by absorption at 260 nm.
Table 2.
Oligonucleotide sequences.
| Oligonucleotide | Sequence |
|---|---|
| dinB1 | 5′-CGATATCCCTATTGCTATTGG-3′ |
| dinB2 | 5′-TCGATAATCGCTTCACATTCAG-3′ |
| TruncdinB | 5′-TCGATAATCGCTTCACATTCAGGCCAAAGCGTTTAAGCAGC-3′ |
The underlined sequence defines the end of the consecutive sequence of the small dinB fragment.
RT-PCR was performed using the Qiagen OneStep RT-PCR kit. 250 ng of total RNA from the sample cells, various amounts of competitor ranging from 0.1 to 16 pg, and primers dinB1 and dinB2 were added to a half-volume reaction and incubated according to the following protocol: 30 min at 50°C for reverse transcription; 15 min at 95°C to inactivated reverse transcriptase and activate Taq polymerase; then multiple cycles of 30 s at 94°C, 1 min at 64°C, and 1 min at 72°C for amplification; finally, 10 min at 72°C for runoff. Seventeen PCR cycles were used for wild-type cells and 25 for rpoS mutant cells. The PCR reactions were separated by agarose gel electrophoresis, stained with ethidium bromide, and photographed. The bands were quantified using NIH Image version 1.61 software (W. Rasband, National Institutes of Health).
To calculate the amount of dinB transcript in the sample, the ratio of the PCR product band intensities (corrected for the difference in molecular weight) of endogenous RNA to the competitor RNA was taken over a range of competitor RNA amounts. The log of this ratio was plotted against the log of the amount of competitor RNA added, and the least-squares line computed, as shown in Fig. 5. The amount of RNA corresponding to where this line crosses 0 (i.e. where the ratio is 1) is a measure of the amount of endogenous RNA in the sample (Siebert and Larrick, 1992; Brandstetter et al., 2000).
Acknowledgments
We are grateful to Keen Wilson for his patient help with the RT-PCR protocol. We thank the following people for bacterial strains, plasmids, and reagents: M. Berlyn, S. Finkel, R. Kolter, J. Krueger, R. Lloyd, S. T. Lovett, J. H. Miller, R. Maurer, T. Nohmi, H. Ohmori, G. Walker and R. Woodgate. We also thank the reviewers of this paper for helpful suggestions. This work was supported by USPHS grant NIH-NIGMS G651575.
Footnotes
Note added in proof
As predicted, an rpoS dinB double mutant had the low adaptive mutation rate of the rpoS single mutant, confirming that RpoS has additional effects on adaptive mutation.
References
- Ausubel, F.M., Brent, R., Kingston, R.E., Moore, D.D., Seidman, J.G., Smith, J.A., and Struhl, K. (1988) Current Protocols in Molecular Biology New York: John Wiley & Sons.
- Bagdasarian M, Bailone A, Bagdasarian MM, Manning PA, Lurz R, Timmis KN, Devoret R. An inhibitor of SOS induction, specified by a plasmid locus in Escherichia coli. Proc Natl Acad Sci USA. 1986;83:5723–5726. doi: 10.1073/pnas.83.15.5723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagg A, Kenyon CJ, Walker GC. Inducibility of a gene product required for UV and chemical mutagenesis in Escherichia coli. Proc Natl Acad Sci USA. 1981;78:5749–5753. doi: 10.1073/pnas.78.9.5749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates H, Randall SK, Rayssiguier C, Bridges BA, Goodman MF, Radman M. Spontaneous and UV-induced mutations in Escherichia coli K-12 strains with altered or absent DNA polymerase I. J Bacteriol. 1989;171:2480–2484. doi: 10.1128/jb.171.5.2480-2484.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker G, Hengge-Aronis R. What makes an Escherichia coli promoter σS dependent? Role of the −13/−14 nucleotide promoter positions and region 2.5 of σS. Mol Microbiol. 2001;39:1153–1165. doi: 10.1111/j.1365-2958.2001.02313.x. [DOI] [PubMed] [Google Scholar]
- Bohannon DE, Connell N, Keener J, Tormo A, Espinosa-Urgel M, Zambrano MM, Kolter R. Stationary-phase-inducible ‘gearbox’ promoters: differential effects of katF mutations and role of σ70. J Bacteriol. 1991;173:4482–4492. doi: 10.1128/jb.173.14.4482-4492.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandstetter AM, Pfaffl MW, Hocquette JF, Gerrard DE, Picard B, Geay Y, Sauerwein H. Effects of muscle type, castration, age, and compensatory growth rate on androgen receptor mRNA expression in bovine skeletal muscle. J Anim Sci. 2000;78:629–637. doi: 10.2527/2000.783629x. [DOI] [PubMed] [Google Scholar]
- Cairns J. Mutation and cancer: The antecedents to our studies of adaptive mutation. Genetics. 1998;148:1433–1440. doi: 10.1093/genetics/148.4.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns J, Foster PL. Adaptive reversion of a frameshift mutation in Escherichia coli. Genetics. 1991;128:695–701. doi: 10.1093/genetics/128.4.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns J, Overbaugh J, Miller S. The origin of mutants. Nature. 1988;335:142–145. doi: 10.1038/335142a0. [DOI] [PubMed] [Google Scholar]
- Celi FS, Zenilman ME, Shuldiner AR. A rapid and versatile method to synthesize internal standards for competitive PCR. Nucleic Acids Res. 1993;21:1047. doi: 10.1093/nar/21.4.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colland F, Fujita N, Ishihama A, Kolb A. The interaction between σS, the stationary phase sigma factor, and the core enzyme of Escherichia coli RNA polymerase. Genes Cells. 2002;7:233–247. doi: 10.1046/j.1365-2443.2002.00517.x. [DOI] [PubMed] [Google Scholar]
- Coulondre C, Miller JH. Genetic studies of the lac repressor. III. Additional correlation of mutational sites with specific amino acid residues. J Mol Biol. 1977;117:525–575. doi: 10.1016/0022-2836(77)90056-0. [DOI] [PubMed] [Google Scholar]
- Courcelle J, Khodursky A, Peter B, Brown PO, Hanawalt PC. Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics. 2001;158:41–64. doi: 10.1093/genetics/158.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JW. A constant rate of spontaneous mutation in DNA-based microbes. Proc Natl Acad Sci USA. 1991;88:7160–7164. doi: 10.1073/pnas.88.16.7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenstark A. Bacterial gene products in response to near-ultraviolet radiation. Mutat Res. 1998;422:85–95. doi: 10.1016/s0027-5107(98)00178-x. [DOI] [PubMed] [Google Scholar]
- Escarceller M, Hicks J, Gudmundsson G, Trump G, Touati D, Lovett S, et al. Involvement of Escherichia coli DNA polymerase II in response to oxidative damage and adaptive mutation. J Bacteriol. 1994;176:6221–6228. doi: 10.1128/jb.176.20.6221-6228.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa-Urgel M, Chamizo C, Tormo A. A consensus structure for σS-dependent promoters. Mol Microbiol. 1996;21:657–659. doi: 10.1111/j.1365-2958.1996.tb02573.x. [DOI] [PubMed] [Google Scholar]
- Fernandez de Henestrosa AR, Ogi T, Aoyagi S, Chafin D, Hayes JJ, Ohmori H, Woodgate R. Identification of additional genes belonging to the LexA regulon in Escherichia coli. Molec Microbiol. 2000;35:1560–1572. doi: 10.1046/j.1365-2958.2000.01826.x. [DOI] [PubMed] [Google Scholar]
- Foster PL. Adaptive mutation: The uses of adversity. Annu Rev Microbiol. 1993;47:467–504. doi: 10.1146/annurev.mi.47.100193.002343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL. Population dynamics of a Lac− strain of Escherichia coli during selection for lactose utilization. Genetics. 1994;138:253–261. doi: 10.1093/genetics/138.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL. Nonadaptive mutations occur on the F’ episome during adaptive mutation conditions in Escherichia coli. J Bacteriol. 1997;179:1550–1554. doi: 10.1128/jb.179.5.1550-1554.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL. Adaptive mutation in Escherichia coli. Cold Spring Harbor Symp Quant Biol. 2000;65:21–29. doi: 10.1101/sqb.2000.65.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL, Trimarchi JM. Adaptive reversion of a frameshift mutation in Escherichia coli by simple base deletions in homopolymeric runs. Science. 1994;265:407–409. doi: 10.1126/science.8023164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL, Rosche WA. Increased episomal replication accounts for the high rate of adaptive mutation in recD mutants of Escherichia coli. Genetics. 1999;152:15–30. doi: 10.1093/genetics/152.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL, Trimarchi JM. Adaptive reversion of an episomal frameshift mutation in Escherichia coli requires conjugal functions but not actual conjugation. Proc Natl Acad Sci USA. 1995;92:5487–5490. doi: 10.1073/pnas.92.12.5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL, Gudmundsson G, Trimarchi JM, Cai H, Goodman MF. Proofreading-defective DNA polymerase II increases adaptive mutation in Escherichia coli. Proc Natl Acad Sci USA. 1995;92:7951–7955. doi: 10.1073/pnas.92.17.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL, Trimarchi JM, Maurer RA. Two enzymes, both of which process recombination intermediates, have opposite effects on adaptive mutation in Escherichia coli. Genetics. 1996;142:25–37. doi: 10.1093/genetics/142.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galitski T, Roth JR. Evidence that F plasmid transfer replication underlies apparent adaptive mutation. Science. 1995;268:421–423. doi: 10.1126/science.7716546. [DOI] [PubMed] [Google Scholar]
- Goodman MF. Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu Rev Biochem. 2002;71:17–50. doi: 10.1146/annurev.biochem.71.083101.124707. [DOI] [PubMed] [Google Scholar]
- Gowrishankar J, Yamamoto K, Subbarayan PR, Ishihama A. In vitro properties of RpoS (σS) mutants of Escherichia coli with postulated N-terminal subregion 1.1 or C-terminal region 4 deleted. J Bacteriol. 2003;185:2673–2679. doi: 10.1128/JB.185.8.2673-2679.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BG. Spontaneous point mutations that occur more often when they are advantageous than when they are neutral. Genetics. 1990;126:5–16. doi: 10.1093/genetics/126.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow, E., and Lane, D. (1988) Antibodies: a Laboratory Manual Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory.
- Harris RS, Longerich S, Rosenberg SM. Recombination in adaptive mutation. Science. 1994;264:258–260. doi: 10.1126/science.8146657. [DOI] [PubMed] [Google Scholar]
- Harris RS, Ross KJ, Rosenberg SM. Opposing roles of the Holliday junction processing systems of Escherichia coli recombination-dependent adaptive mutation. Genetics. 1996;142:681–691. doi: 10.1093/genetics/142.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengge-Aronis R. Stationary phase gene regulation: what makes an Escherichia coli promoter σS-selective? Curr Opin Microbiol. 2002a;5:591–595. doi: 10.1016/s1369-5274(02)00372-7. [DOI] [PubMed] [Google Scholar]
- Hengge-Aronis R. Signal transduction and regulatory mechanisms involved in control of the σS (RpoS) subunit of RNA polymerase. Microbiol Mol Biol Rev. 2002b;66:373–395. doi: 10.1128/MMBR.66.3.373-395.2002. table. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard-Flanders P. Genes that control DNA repair and genetic recombination in Escherichia coli. Adv Biol Med Phys. 1968;12:299–317. doi: 10.1016/b978-1-4831-9928-3.50016-3. [DOI] [PubMed] [Google Scholar]
- Ishihama A. Functional modulation of Escherichia coli RNA polymerase. Annu Rev Microbiol. 2000;54:499–518. doi: 10.1146/annurev.micro.54.1.499. [DOI] [PubMed] [Google Scholar]
- Kenyon CJ, Walker GC. DNA-damaging agents stimulate gene expression at specific loci in Escherichia coli. Proc Natl Acad Sci USA. 1980;77:2819–2823. doi: 10.1073/pnas.77.5.2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SR, Maenhaut-Michel G, Yamada M, Yamamoto Y, Matsui K, Sofuni T, et al. Multiple pathways for SOS-induced mutagenesis in Escherichia coli: an over-expression of dinB/dinP results in strongly enhancing mutagenesis in the absence of any exogenous treatment to damage DNA. Proc Natl Acad Sci USA. 1997;94:13792–13797. doi: 10.1073/pnas.94.25.13792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SR, Matsui K, Yamada M, Gruz P, Nohmi T. Roles of chromosomal and episomal dinB genes encoding DNA Pol IV in targeted and untargeted mutagenesis in Escherichia coli. Mol Genet Genomics. 2001;266:207–215. doi: 10.1007/s004380100541. [DOI] [PubMed] [Google Scholar]
- Kleckner, N., Bender, J., and Gottesman, S. (1991) Uses of transposons with emphasis on Tn10 In Bacterial Genetic Systems: Methods in Enzymology, Vol. 204. Miller, J.H. (ed.). San Diego: Academic Press, pp. 139–180. [DOI] [PubMed]
- Kobayashi S, Valentine MR, Pham P, O’Donnell M, Goodman MF. Fidelity of Escherichia coli DNA polymerase IV. Preferential generation of small deletion mutations by dNTP-stabilized misalignment. J Biol Chem. 2002;277:34198–34207. doi: 10.1074/jbc.M204826200. [DOI] [PubMed] [Google Scholar]
- Krueger JH, Elledge SJ, Walker GC. Isolation and characterization of Tn5 insertion mutations in the lexA gene of Escherichia coli. J Bacteriol. 1983;153:1368–1378. doi: 10.1128/jb.153.3.1368-1378.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenne-Samuel N, Janel-Bintz R, Kolbanovskiy A, Geacintov NE, Fuchs RPP. The processing of a benzo (a) pyrene adduct into a frameshift or a base substitution mutation requires a different set of genes in Escherichia coli. Mol Microbiol. 2000;38:299–307. doi: 10.1046/j.1365-2958.2000.02116.x. [DOI] [PubMed] [Google Scholar]
- Lloyd RG, Buckman C. Genetic analysis of the recG locus of Escherichia coli K12 and of its role in recombination and DNA repair. J Bacteriol. 1991;173:1004–1011. doi: 10.1128/jb.173.3.1004-1011.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie GJ, Harris RS, Lee PL, Rosenberg SM. The SOS response regulates adaptive mutation. Proc Natl Acad Sci USA. 2000;97:6646–6651. doi: 10.1073/pnas.120161797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie GJ, Lee PL, Lombardo MJ, Hastings PJ, Rosenberg SM. SOS Mutator DNA polymerase IV functions in adaptive mutation and not adaptive amplification. Mol Cell. 2001;7:571–579. doi: 10.1016/s1097-2765(01)00204-0. [DOI] [PubMed] [Google Scholar]
- Miller, J.H. (1992) A Short Course In Bacterial Genetics: A Laboratory Manual and Handbook For Escherichia coli and Related Bacteria. Cold Spring Harbor: Cold Spring Harbor Laboratory Press.
- Mount DW. A mutant of Escherichia coli showing constitutive expression of the lysogenic induction and error-prone DNA repair pathways. Proc Natl Acad Sci USA. 1977;74:300–304. doi: 10.1073/pnas.74.1.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano R, Janel-Bintz R, Wagner J, Fuchs RPP. All three SOS-inducible DNA polymerases (Pol II, Pol IV and Pol V) are involved in induced mutagenesis. EMBO J. 2000;19:6259–6265. doi: 10.1093/emboj/19.22.6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh TJ, Jung IL, Kim IG. The Escherichia coli SOS gene sbmC is regulated by H-NS and RpoS during the SOS induction and stationary growth phase. Biochem Biophys Res Commun. 2001;288:1052–1058. doi: 10.1006/bbrc.2001.5872. [DOI] [PubMed] [Google Scholar]
- Pratt LA, Kolter R. Genetic analysis of Escherichia coli biofilm formation: roles of flagella, motility, chemo-taxis and type I pili. Mol Microbiol. 1998;30:285–293. doi: 10.1046/j.1365-2958.1998.01061.x. [DOI] [PubMed] [Google Scholar]
- Radicella JP, Park PU, Fox MS. Adaptive mutation in Escherichia coli: a role for conjugation. Science. 1995;268:418–420. doi: 10.1126/science.7716545. [DOI] [PubMed] [Google Scholar]
- Reuven NB, Arad G, Maor-Shoshani A, Livneh Z. The mutagenesis protein UmuC is a DNA polymerase activated by UmuD′, RecA, and SSB and is specialized for translesion replication. J Biol Chem. 1999;274:31763–31766. doi: 10.1074/jbc.274.45.31763. [DOI] [PubMed] [Google Scholar]
- Rice, J.A. (1995) Mathematical Statistics and Data Analysis Belmont, CA: Wadsworth Publishing, pp. 1–602.
- Rodriguez C, Tompkin J, Hazel J, Foster PL. Induction of a DNA nickase in the presence of its target site stimulates adaptive mutation in Escherichia coli. J Bacteriol. 2002;184:5599–5608. doi: 10.1128/JB.184.20.5599-5608.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosche WA, Foster PL. The role of transient hypermutators in adaptive mutation in Escherichia coli. Proc Natl Acad Sci USA. 1999;96:6862–6867. doi: 10.1073/pnas.96.12.6862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SM, Longerich S, Gee P, Harris RS. Adaptive mutation by deletions in small mononucleotide repeats. Science. 1994;265:405–407. doi: 10.1126/science.8023163. [DOI] [PubMed] [Google Scholar]
- Shen X, Sayer JM, Kroth H, Ponten I, O’Donnell M, Woodgate R, et al. Efficiency and accuracy of SOS-induced DNA polymerases replicating benzo[a]pyrene-7,8-diol 9,10-epoxide A and G adducts. J Biol Chem. 2002;277:5265–5274. doi: 10.1074/jbc.M109575200. [DOI] [PubMed] [Google Scholar]
- Siebert PD, Larrick JW. Competitive PCR. Nature. 1992;359:557–558. doi: 10.1038/359557a0. [DOI] [PubMed] [Google Scholar]
- Sniegowski PD, Gerrish PJ, Lenski RE. Evolution of high mutation rates in experimental populations of E. coli. Nature. 1997;387:703–705. doi: 10.1038/42701. [DOI] [PubMed] [Google Scholar]
- Strauss BS, Roberts R, Francis L, Pouryazdanparast P. Role of the dinB gene product in spontaneous mutation in Escherichia coli with an impaired replicative polymerase. J Bacteriol. 2000;182:6742–6750. doi: 10.1128/jb.182.23.6742-6750.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei F, Radman M, Maynard-Smith J, Toupance B, Gouyon PH, Godelle B. Role of mutator alleles in adaptive evolution. Nature. 1997;387:700–702. doi: 10.1038/42696. [DOI] [PubMed] [Google Scholar]
- Tang M, Bruck I, Eritja R, Turner J, Frank EG, Woodgate R, et al. Biochemical basis of SOS-induced mutagenesis in Escherichia coli: reconstitution of in vitro lesion bypass dependent on the UmuD′2C mutagenic complex and RecA protein. Proc Natl Acad Sci USA. 1998;95:9755–9760. doi: 10.1073/pnas.95.17.9755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M, Shen X, Frank EG, O’Donnell M, Woodgate R, Goodman MF. UmuD′ (2)C is an error-prone DNA polymerase, Escherichia coli Pol V. Proc Natl Acad Sci USA. 1999;96:8919–8924. doi: 10.1073/pnas.96.16.8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M, Pham P, Shen X, Taylor JS, O’Donnell M, Woodgate R, Goodman MF. Roles of E. coli DNA polymerase IV and V in lesion-targeted and untargeted SOS mutagenesis. Nature. 2000;404:1014–1018. doi: 10.1038/35010020. [DOI] [PubMed] [Google Scholar]
- Tompkins JD, Nelson JE, Hazel JC, Leugers SL, Stumpf JD, Foster PL. Error-prone polymerase, DNA polymerase IV, is responsible for transient hypermutation during adaptive mutation in Escherichia coli. J Bacteriol. 2003;185:3469–3472. doi: 10.1128/JB.185.11.3469-3472.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torkelson J, Harris RS, Lombardo MJ, Nagendran J, Thulin C, Rosenberg SM. Genome-wide hypermutation in a subpopulation of stationary-phase cells underlies recombination-dependent adaptive mutation. EMBO J. 1997;16:3303–3311. doi: 10.1093/emboj/16.11.3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J, Nohmi T. Escherichia coli DNA polymerase IV mutator activity: genetic requirements and mutational specificity. J Bacteriol. 2000;182:4587–4595. doi: 10.1128/jb.182.16.4587-4595.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J, Gruz P, Kim SR, Yamada M, Matsui K, Fuchs RPP, Nohmi T. The dinB gene encodes a novel E. coli DNA polymerase, DNA Pol IV, involved in mutagenesis. Mol Cell. 1999;4:281–286. doi: 10.1016/s1097-2765(00)80376-7. [DOI] [PubMed] [Google Scholar]
- Wagner J, Fujii S, Gruz P, Nohmi T, Fuchs RPP. The beta clamp targets DNA polymerase IV to DNA and strongly increases its processivity. EMBO Report. 2001;1:484–488. doi: 10.1093/embo-reports/kvd109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker GC. Mutagenesis and inducible responses to deoxyribonucleic acid damage in Escherichia coli. Microbiol Rev. 1984;48:60–93. doi: 10.1128/mr.48.1.60-93.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeiser B, Pepper ED, Goodman MF, Finkel SE. SOS-induced DNA polymerases enhance long-term survival and evolutionary fitness. Proc Natl Acad Sci USA. 2002;99:8737–8741. doi: 10.1073/pnas.092269199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zar, J.H. (1984) Biostatistical Analysis Englewood Cliffs, New Jersey: Prentice Hall, pp. 1–718.