

Abstract
If T cells require specific interactions with MHC-bound peptides during positive selection, then the specificities of T cells selected by one peptide should be distinct from those selected by another. We have examined positive selection of CD4 T cells in four strains of mice, each overexpressing a different peptide–1-Ab(Ab) complex. We show that a subset of CD4 T cells is selected by the overexpressed peptide and that the specificities of the CD4 T cells, as measured by reactivity to wild-type antigen-presenting cells, vary greatly depending on which peptide is overexpressed. These differences in specificity are mediated through positive selection not negative selection. Each of the four peptide–Ab complexes appears to adopt a different conformation, and these differences correlate with the differences in reactivity. Our results suggest that individual peptide–MHC complexes positively select different subsets of self-MHC-reactive T cells and that the conformation of the peptide–MHC complex may contribute to this process.
Keywords: thymus‖CD4 T cells‖development‖T cell specificity‖mice
In the thymus, T cells are evaluated on the basis of the reactivity of their T cell receptors (TCRs) for self-peptides bound to self-MHC molecules. Although strongly reactive cells are eliminated through negative selection, weakly reactive cells are positively selected to mature and exit to the periphery. Collectively, these processes result in a T cell repertoire with the greatest capacity to respond to foreign peptides bound to self-MHC (1, 2). Although it is clear that interaction between the TCR and MHC-bound self-peptides is required during the positive selection of T cells, the specificity of this interaction has not been agreed upon. Recently, this debate has centered around analyses of the development of CD4 T cells in mice engineered to express a single peptide bound to their MHC class II molecules. Several groups have shown that large numbers of CD4 T cells (20–40% of wild type, depending on the system) are present in H-2M-deficient (H-2M0) and AbEp mice and that the repertoire of T cells in these mice is quite broad (3–8). Consequently, these authors have suggested that the recognition of peptides during positive selection is promiscuous (9, 10).
In subsequent work, we and others have argued that many T cells are selected on diverse, low-abundance peptides (11–13). It was shown most directly by using mice that express a human invariant chain (Ii) transgene (Tg) in which class II-associated invariant chain peptide (CLIP) has been replaced with Eα peptide. The Tg rescues class II expression to wild-type levels when expressed in Ii-deficient (Eα-Ii0) mice, and Eα peptide is bound to ≈95% of class II molecules (12). Although this class II expression is sufficient to select a full compartment of CD4 T cells, selection of the majority of these cells depends on non-Eα peptides. When the Tg is expressed in mice that also lack H-2M (Eα-dbl0), these non-Eα peptides are no longer detectable, and the number of CD4 T cells drops to 30% of that seen in Eα-Ii0 mice. Thus, at least 70% of the cells in Eα-Ii0 mice are not selected on Eα peptide, indicating that recognition of peptides during positive selection is more specific than the initial characterization of single-peptide mice had suggested.
Defining even more precisely the number of T cells that can be selected by a single peptide adds little to our understanding of positive selection. Instead, we have focused on which T cells are selected by a given peptide. If positive selection of T cells requires specific recognition of peptide, then T cells selected by one peptide should have different specificities compared with T cells selected by a second peptide. Based on this hypothesis, we have examined the specificities of CD4 T cells from mice that overexpress four different peptide–MHC class II complexes: Eα–1-Ab(Ab), CLIP–Ab, CD22 (25–39)–Ab, or Rab5a (86–101)–Ab. Our analysis indicates that individual peptide–MHC complexes positively select different subsets of self-MHC-reactive T cells and that the conformation of the peptide–MHC complex can contribute to this process.
Materials and Methods
Mice.
The human genomic Ii cassette and the generation of Ii-Eα mice have been described elsewhere (12, 14). To generate Ii-CD22 and Ii-Rab transgenic mice, purified Ii-peptide DNA was injected into BDF1 × Ii0 F1 embryos. Each of the Ii-peptide Tgs was crossed to both Ii0 and Ii0 × H-2M0 mice. Ii0 back-crossed onto C57BL/6, C57BL/6, B6-C.H-2bm12, and BALB/c mice were obtained from The Jackson Laboratory. H-2M0 and Ii0 × H-2M0 mice were provided by Luc Van Kaer (Vanderbilt University, Vanderbilt, TN). All mice were maintained under specific pathogen-free conditions in our animal facility at the University of Washington.
Generation of Monoclonal Antibodies (mAbs).
The A8 and H10 mAbs were generated by somatic hybridization of splenocytes isolated from BALB/c mice hyperimmunized with lipopolysaccharide-activated (48 h, 10 μg/ml) splenocytes from CD22-dbl0 or Rab-dbl0 mice, respectively. Briefly, after 5–6 i.p. immunizations with 5 × 106 cells each over 3–4 week intervals, BALB/c splenocytes were fused with X63 myeloma cells. Clones were screened by a sandwich ELISA to identify antibodies specific for Ab bound to CD22 and Rab peptides by using CD22-dbl0 or Rab-dbl0 splenocyte lysates, respectively, as a source of specific complexes. Lysates of H-2M0 splenocytes were used as a negative control.
Flow Cytometry and T Cell Hybrid Assays.
To measure MHC class II expression, cells were incubated on ice with biotinylated mAbs Y3P (Ab), YAe (Eα-Ab), 15G4 (CLIP-Ab), A8 (CD22-Ab), or H10 (Rab-Ab) followed by streptavidin-FITC (Vector Laboratories). Thymocytes or splenocytes were incubated on ice with anti-CD4-phycoerythrin, anti-CD8α-FITC, and anti-TCRβ-biotin mAbs (all from PharMingen) followed by streptavidin-Tricolor (Caltag, South San Francisco, CA). Stained cells were analyzed by using a FACScalibur flow cytometer and CELLQUEST software (Becton Dickinson).
Titrated numbers of erythrocyte-depleted splenocytes were cultured with 105 T cell hybrids for 18–20 h. Production of IL-2 in the supernatants was measured by a standard HT-2 cell assay by using Alamar blue (11). Data are presented as a mean OD570/600 of duplicate cultures.
Western Blot Analysis.
C57BL/6, Eα-dbl0, CD22-dbl0, Rab-dbl0, or H-2M0 splenocyte lysates (0.5% Nonidet P-40/150 mM NaCl/5 mM EDTA/50 mM Tris, pH 7.2) were resolved on nonreducing 8% SDS polyacrylamide gels and transferred to nitrocellulose. The blots were probed with rabbit antisera against the cytoplasmic tails of Ab α and β chains followed by donkey anti-rabbit horseradish peroxidase or 25-9-17s supernatant followed by sheep anti-mouse Ig-horseradish peroxidase (Amersham Pharmacia).
Mixed Lymphocyte Cultures and Antigen-Specific Proliferation.
CD4 T cells were isolated from single-cell suspensions of lymph node and spleen cells from Eα-dbl0, CD22-dbl0, Rab-dbl0, and H-2M0 mice. Cells were incubated with antibodies against CD8 (3.168.8) and the heat-stable antigen (J11d) and subsequently treated with rabbit complement (C-SIX Diagnostics, Mequon, WI) diluted 1:10 in culture medium. CD4 T cells were enriched further by panning on anti-CD4 (GK1.5)-coated plates. This procedure for enrichment of CD4 T cells routinely yielded cell populations containing 40–80% CD4 T cells with minimal contamination of CD8 T cells (1–2%). Within a given experiment, the number of CD4 T cells was normalized between each mouse type. Generally, 1–2 × 105 CD4 T cells were incubated in triplicates with 6 × 105 irradiated splenocytes (2,000 rad) in RPMI medium 1640 supplemented with 5% FCS/200 mM l-glutamine/10 mM Hepes/5 × 10−5 M 2-mercaptoethanol in flat-bottom 96-well plates. Each well was pulsed with 1 μCi (1 Ci = 37 GBq) of [3H]thymidine after 72 h and harvested after 96 h. Thymidine incorporation was measured by using a 1205 Betaplate reader (Wallac, Gaithersburg, MD). For antigen-specific proliferation assays, mice were immunized s.c. in the base of the tail with 30 μg of HEL74-88 or OVA265-277 peptide in complete Freund's adjuvant. Eight days later, CD4 T cells were isolated from draining lymph nodes by anti-CD4 bead magnetic sorting. CD4 T cells (1 × 105) were incubated with titrated amounts of the peptide antigen and 5 × 105 irradiated splenocytes (2,000 rad) from dbl0 mice in triplicates in flat-bottom 96-well plates. Each well was pulsed with 0.5 μCi of [3H]thymidine after 72 h and harvested after 96 h.
Bone-Marrow (BM) Chimeras.
BM chimeras were generated as described (11). Lethally irradiated (1,000 rad) recipient mice received 2.5 × 106 T cell-depleted BM cells and were analyzed 8 weeks posttransfer. Reconstitution of antigen-presenting cells (APCs) in BM chimeras was essentially complete as measured by staining with YAe and 15G4 antibodies specific for Eα–Ab and CLIP–Ab complexes, respectively.
Results
Generation and Characterization of Ii-Peptide Transgenic Mice.
To address the effect of individual peptides on positive selection, we have generated multiple Ii-peptide transgenic mice. We replaced the CLIP region of human Ii with DNA encoding the Ab-binding peptides CD22 (25–39) and Rab5a (86–101) (Fig. 1A). Both of these peptides bind Ab with affinities comparable to that of Eα peptide (data not shown). We bred each of these Ii-peptide Tgs onto the Ii0 and Ii0 × H-2M0 (dbl0) backgrounds. Class II expression in Ii0 and dbl0 mice is ≈10% of normal (15, 16), yet each of the Tgs restored Ab expression to wild-type levels when expressed in Ii0 and dbl0 mice. To assess expression of the CD22 and Rab peptides, we generated two new peptide class II-specific mAbs, A8 and H10, respectively. Using these new reagents, as well as the previously characterized antibodies YAe and 15G4, which recognize Eα–Ab and CLIP–Ab, respectively, we analyzed the expression of each specific peptide–Ab complex. Splenocytes from each of the Ii-peptide transgenic mice stained brightly with the appropriate peptide-specific mAb, indicating that each of the peptides is presented at high levels (Fig. 1B). As expected, splenocytes from the transgenic mice did not stain with the 15G4 mAb, specific for CLIP–Ab, whereas splenocytes from H-2M0 mice stained strongly with this antibody. Analysis of thymic sections by immunohistochemistry showed that total Ab levels and the expression of all three specific peptide–Ab complexes were similar to that seen on splenocytes for each of the transgenic mice (data not shown). We have obtained consistently high expression of each Tg in a total of 14 transgenic founder lines with a genomic Ii construct. Importantly, we have not seen variation in expression among different transgenic constructs or founders that is characteristic of the Ii-Eα promoter-containing cDNA cassette, which has been used by others in related studies (17).
Figure 1.

Generation of mice overexpressing different peptide–Ab complexes. (A) Schematic of the human invariant chain locus. The Eα, CD22, and Rab peptide sequences replacing CLIP in each of the Ii-peptide Tgs are shown. (B) Splenocytes from C57BL/6, H-2M0, and each of the Ii-peptide mice were stained with antibodies binding Ab (Y3P), mouse CLIP–Ab (15G4), Eα–Ab (YAe), CD22–Ab (A8), and Rab–Ab (H10). Staining of peptide-Ii0 mice are indicated by thin, black lines; peptide-dbl0 mice are indicated by thick, gray lines. (C) Stimulation of T cell hybridomas specific for CD22 (25–39)–Ab, IgM (377–392)–Ab, β2m (48–58)–Ab, or Rab (86–101)–Ab by titrated splenocytes from the indicated mice is shown. IL-2 production was measured by using the IL-2-dependent cell line HT-2. The data are presented as a mean OD570/600 of duplicate cultures.
We used a panel of T cell hybridomas specific for different self-peptide–Ab complexes to determine whether additional peptides were detectable in each of the Ii-peptide transgenic mice. Splenocytes from Eα-Ii0, CD22-Ii0, and Rab-Ii0 mice stimulated T cell hybrids tested, indicating that endogenous peptides were still presented in the Ab molecules not bound by the major peptide (Fig. 1C). Stimulation of each hybrid by the different peptide-Ii0 splenocytes was comparable, and each was below the stimulation by wild-type splenocytes. However, none of the peptide-dbl0 mice or H-2M0 mice were able to stimulate the T cell hybrids. APCs from both peptide-Ii0 and peptide-dbl0 mice were able to present exogenous antigenic peptide, but both failed to present exogenously provided intact protein antigen (data not shown). Thus, Eα-Ii0, CD22-Ii0, and Rab-Ii0 mice each express background self-peptides that require H-2M for presentation.
CD4 T Cell Development in Ii-Peptide Transgenic Mice.
We previously described how the differences in presentation of non-Eα peptides between Eα-Ii0 and Eα-dbl0 mice impact positive selection of CD4 T cells. In Eα-Ii0 mice the number of CD4 T cells in the thymus, spleen, and lymph nodes is normal, whereas the number of CD4 T cells in Eα-dbl0 mice is reduced (12). We found similar defects in the number of CD4 T cells from 6–8-week-old CD22-dbl0 and Rab-dbl0 mice when we analyzed CD4 and CD8 expression on thymocytes and splenocytes by flow cytometry (Fig. 2). The percentages of CD4 single-positive thymocytes and CD4-positive splenocytes were reduced to 20–30% of that seen in the corresponding peptide-Ii0 mice. Comparably high proportions of CD44hi CD4 T cells (48–65%) were found in the periphery but not in the thymus of all three lines of peptide-dbl0 mice.
Figure 2.

CD4 T cell development in Ii-peptide mice. Flow-cytometric analysis of thymocytes (A) and splenocytes (B) from peptide-Ii0 and peptide-dbl0 mice stained with anti-CD4 and anti-CD8 mAbs is shown. The percentage of cells within each gate or quadrant is indicated.
These results demonstrate that each of these abundant peptides is not capable of positively selecting the large numbers of CD4 T cells seen in the corresponding peptide-Ii0 mice. What about the smaller number of CD4 T cells in the peptide-dbl0 mice and H-2M0 mice? Are some of these T cells selected by the abundant peptides? This question is particularly important if we want to examine the specificity of T cells selected by a given peptide. Therefore, to evaluate the impact of individual peptides on selection, we bred Eα-dbl0 mice to Ii+/−H-2M0 mice to generate Eα-Ii+/−H-2M0 mice (Eα × CLIP). These mice express both Eα and CLIP peptides at high levels, as measured by the YAe and 15G4 antibodies (Fig. 3). We compared multiple littermates of the three possible genotypes from that cross at 6–8 weeks of age to determine the effect of additional peptides on CD4 T cell development. In the thymus we detected a statistically significant increase in the percentage of CD4 single-positive thymocytes between Eα-dbl0 and Eα × CLIP mice (P < 0.03) as well as between Ii+/−H-2M0 and Eα × CLIP mice (P < 0.025). An increase was observed also when the percentages of splenic CD4 T cells were compared among the three genotypes. These differences were apparent also when the total numbers of CD4 T cells were compared. There was no statistically significant difference in the CD8 T cell compartments between the three types of mice in either the thymus or the spleen. Analysis of individual littermates revealed that the mean fluorescence intensity of Y3P staining of Ab molecules on “single” and “double” Eα × CLIP peptide splenocytes was comparable [average mean fluorescence intensity: 1,300.5 ± 177.08 (n = 8) for single-peptide mice and 1,087.13 ± 157.14 (n = 6) for double-peptide mice].
Figure 3.

A subset of CD4 T cells is selected by overexpressed peptides. (A) Flow-cytometric analysis of anti-CD4- and anti-CD8-stained thymocytes from Eα-dbl0 (Eα), Ii+/−H-2M0 (CLIP), and EαIi+/−H-2M0 (Eα × CLIP) mice is shown. Total Ab expression (Y3P) and expression of Eα–Ab (YAe) and CLIP–Ab (15G4) on splenocytes from each of these mice is shown in B. The mean values for the percentage of CD4 T cells in the thymus and spleen are shown below each plot. P values for comparison of CD4 thymocytes were P < 0.03 (Eα versus Eα × CLIP) and P < 0.025 (CLIP versus Eα × CLIP). P values for comparison of splenic CD4 T cells were P < 0.12 (Eα versus Eα × CLIP) and P < 0.002 (CLIP versus Eα × CLIP). There was no significant difference in the percentage of CD8 T cells in either the thymus or the spleen.
The increase in the number of CD4 T cells caused by overexpression of two peptides provides convincing evidence that a subset of CD4 T cells is selected by the overexpressed peptides in these mice. The fact that this increase is relatively small, however, implies that additional background peptides are present in these mice and that they contribute to positive selection. If the overexpressed peptides were selecting a majority of the CD4 T cells, we would expect to see a larger increase in the total number of CD4 T cells. Importantly, though, we are able to account for CD4 T cells that depend on the overexpressed peptide for selection.
Analysis of the Specificity of CD4 T Cells Selected in Ii-Peptide Transgenic Mice.
We next examined whether these overexpressed peptides altered the specificity of CD4 T cells. First we compared proliferative responses of CD4 T cells to specific Ab-binding peptides derived from ovalbumin and hen-egg lysozyme (Fig. 4A; data not shown). Eα-dbl0, CD22-dbl0, and H-2M0 mice were able to generate CD4 T cells specific for each peptide tested. This result suggests that the T cell repertoire in these mice is not skewed so severely as to limit recognition of specific antigenic peptides. Next we compared the reactivity of CD4 T cells from H-2M0, Eα-dbl0, CD22-dbl0, and Rab-dbl0 mice against wild-type B6 APCs in mixed lymphocyte cultures (Fig. 4B). Surprisingly, the CD4 T cells from each strain of mice proliferated to different degrees. H-2M0 CD4 T cells proliferated very strongly, Rab-dbl0 CD4 T cells proliferated strongly, Eα-dbl0 CD4 T cells proliferated moderately, and CD22-dbl0 CD4 T cells proliferated weakly. The lower proliferative responses in CD22-dbl0 and Eα-dbl0 mice were not caused by a kinetic difference (as maximum proliferation was observed on day 4 for all responder T cell populations) or an inability of these cells to proliferate (as CD4 T cells from each of the four genotypes of mice proliferated similarly to stimulation by Con A; data not shown). Wild-type B6 responders gave background incorporation between 100 and 360 cpm in response to irradiated Eα-dbl0, CD22-dbl0, H-2M0, and B6 splenocytes (data not shown).
Figure 4.

CD4 T cells from Ii-peptide mice have different specificities. Proliferative responses of CD4 T cells from Eα-dbl0, CD22-dbl0, H-2M0, and C57BL/6 mice immunized with OVA265-277 peptide are shown. CD4 T cells isolated from Eα-dbl0, CD22-dbl0, Rab-dbl0, and H-2M0 mice (B), from reciprocal BM chimeras between Eα-dbl0 and H-2M0 mice (C), or from Eα-Ii0, CD22-Ii0, and Rab-Ii0 mice (D) were stimulated with irradiated syngeneic (open bars) or C57BL/6 (closed bars) splenocytes. The data are presented as the mean [3H]thymidine incorporation of triplicate cultures. In the experiments shown in A, the SEM was always less than 20% of the mean cpm of triplicate cultures. WT, wild type.
The differences in reactivity among the three peptide-dbl0 and H-2M0 mice could be mediated through differences in negative or positive selection. To distinguish between these two possibilities, we generated BM chimeric mice from H-2M0 and Eα-dbl0 mice. We transferred T cell-depleted BM from Eα-dbl0 or H-2M0 mice into lethally irradiated Eα-dbl0 or H-2M0 recipient mice. After 8 weeks, we compared the self-MHC reactivity of CD4 T cells from Eα-dbl0 → Eα-dbl0, Eα-dbl0 → H-2M0, H-2M0 → Eα-dbl0, and H-2M0 → H-2M0 chimeric mice (donor → host). If the difference in MHC reactivity between Eα-dbl0 and H-2M0 mice was caused by differences in negative selection by the Eα–Ab and CLIP–Ab complexes expressed on BM-derived APCs, then the reactivity of the CD4 T cells from each chimeric mouse would be determined by the donor genotype. Alternatively, if positive selection on peptide–Ab complexes present on radiation-resistant thymic epithelial cells is the primary determinant of MHC reactivity, then the CD4 T cells from each chimeric mouse would be determined by the host genotype. In every case the degree of proliferation against B6 splenocytes was determined by the host genotype (Fig. 4C). CD4 T cells from H-2M0 → Eα-dbl0 and Eα-dbl0 → Eα-dbl0 chimeric mice exhibited the lower reactivity characteristic of Eα-dbl0 CD4 T cells, whereas CD4 T cells from Eα-dbl0 → H2M0 and H-2M0 → H-2M0 chimeric mice exhibited the high reactivity characteristic of H-2M0 CD4 T cells. This result was not caused by poor chimerism, because all Ab-positive splenocytes in chimeras that received Eα-dbl0 BM stained with YAe, and all Ab-positive splenocytes in chimeras that received H-2M0 BM stained with 15G4 (data not shown). Furthermore, the BM transfer protocol used in these studies consistently yields in our hands ≈10% of host T cells in the periphery and less than 1% in the thymus as revealed by the Thy-1- or Ly-5-marked BM transfers (data not shown). The remaining 10% of the host peripheral T cells are of activated memory phenotype and should not contribute significantly to the observed proliferative responses. Therefore, the different reactivities of H-2M0 and Eα-dbl0 CD4 T cells are determined during positive selection.
These results suggest that each of the four overexpressed peptides positively select TCRs with different specificities, as measured by proliferation against wild-type APCs. In support of this conclusion, we also observed differences in proliferation of CD4 T cells from each of the peptide-Ii0 mice (Fig. 4D). Importantly, the relative proliferation between these mice agreed with the differences we had observed among the peptide-dbl0 mice; CD4 T cells from Rab-Ii0 mice were the most reactive, followed by Eα-Ii0 CD4 T cells and finally CD22-Ii0 CD4 T cells. The overall proliferation was much less than what we observed with CD4 T cells from the peptide-dbl0 and H-2M0 mice. Together, these data support the conclusion that each overexpressed peptide selects a different subset of CD4 T cells. In peptide-dbl0 mice, these cells comprise a greater proportion of total CD4 T cells and are subjected to less stringent negative selection by “background” peptides than in peptide-Ii0, resulting in an overall greater proliferation.
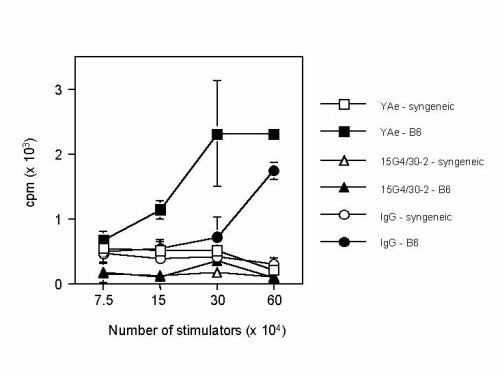
To demonstrate directly that the differences in T cell reactivity were caused by selection on the overexpressed peptides, we performed in vivo mAb blocking of Eα–Ab and CLIP–Ab complexes in Eα × CLIP mice (see Fig. 6, which is published as supporting information on the PNAS web site, www.pnas.org). In this experiment, Eα × CLIP neonates were injected i.p. on the day of birth and every other day thereafter with 200 μg of YAe mAb (specific for Eα–Ab), a mixture of 15G4 and 30-2 mAb (specific for CLIP–Ab complexes), or normal mouse IgG as a control. After 3–3.5 weeks of treatment, reactivity of splenic CD4 T cells against B6 splenocytes was tested. Antibody treatment did not affect the size of splenic CD4 T cell populations (4.84–6.44% of total splenocytes). However, analysis of total splenocytes from Eα × CLIP mice treated with the YAe mAb showed a markedly increased CD4 T cell reactivity against B6 splenocytes versus those derived from control mice. In contrast, 15G4/30-2 treatment resulted in a substantial decrease in CD4 T cell reactivity against B6 splenocytes. Thus, qualitatively blocking one peptide led to selection of T cells with reactivities associated with the other, unblocked peptide. This result directly implicates the major peptide–MHC class II complexes in selecting CD4 T cells with a different reactivity against Ii–Ab molecules with the wild-type repertoire of peptides. Furthermore, we can formally exclude the possibility that minor peptide subsets in peptide-dbl0 mice are responsible for selection of self-MHC-reactive CD4 T cells.
Peptide–MHC Class II Complexes Overexpressed in Ii-Peptide Transgenic Mice May Adopt Distinct Conformations.
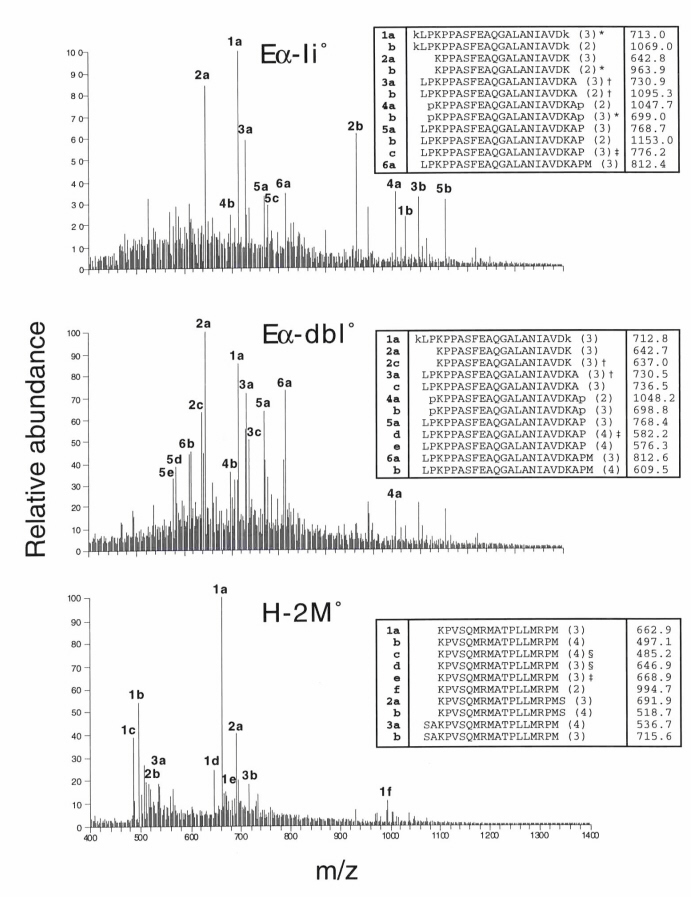
In light of the differences in the specificities of CD4 T cells selected by Eα–Ab, CLIP–Ab, CD22–Ab, and Rab–Ab complexes, we sought to identify potential conformational differences among these complexes that could contribute to their uniqueness. We compared the mobility of Abαβ dimers from Eα-dbl0, CD22-dbl0, Rab-dbl0, and H-2M0 mice in 8% polyacrylamide gels by Western blot analysis (Fig. 5). As reported by others, we observed a reduced mobility of CLIP–Ab from H-2M0 splenocytes when compared with wild-type compact dimers (5–7). Surprisingly, each of the different peptide–Ab complexes displayed different mobilities. CD22–Ab migrated most quickly, Eα–Ab migrated more slowly, and Rab–Ab migrated most slowly. Tandem mass-spectrometric sequence analysis of class II-bound peptides isolated from thymi of Eα-dbl0, CD22-dbl0, and H-2M0 mice indicated that these differences in mobility cannot be explained by different lengths or charges of peptides (see Fig. 7, which is published as supporting information on the PNAS web site).
Figure 5.

Each peptide–Ab complex adopts a distinct conformation. (A) Abαβ dimers from splenocyte lysates of C57BL/6 (lane 1), CD22-dbl0 (lane 2), Eα-dbl0 (lane 3), Rab-dbl0 (lane 4), and H-2M0 (lane 5) mice were visualized by Western blotting of 8% SDS/PAGE gels. Blots were performed with rabbit antisera against the cytoplasmic tails of the Ab α and β chains (Left) or the conformation-dependent anti-Ab mAb 25-9-17s (Right). (B) Splenocytes from the same mice were stained with BP107 (Left) or 25-9-17s (Right) and analyzed by flow cytometry. The staining shown is on B220+-gated cells.
The specific peptide–Ab complexes could be discriminated further by using two conformation-dependent Ab-specific antibodies: BP107 and 25-9-17s. These antibodies stain wild-type splenocytes but not CLIP–Ab or Eα–Ab complexes, respectively, suggesting that their binding may have certain structural requirements that are not fulfilled by these particular peptide–Ab complexes (6, 18). In support of this idea, we found that BP107 stained Eα-dbl0 and Rab-dbl0 splenocytes brightly but not splenocytes from CD22-dbl0 or H-2M0 mice. 25-9-17s stained CD22-dbl0 and H-2M0 splenocytes but not Eα-dbl0 or Rab-dbl0 splenocytes (Fig. 6B). Furthermore, Ab dimers from Eα-dbl0 and Rab-dbl0 splenocytes were not detected by Western blotting with 25-9-17s (Fig. 6A). This differential staining when considered together with the unique mobility of each complex in acrylamide gels supports the possibility that each of these particular peptide–Ab complexes may be structurally distinct. Interestingly, these differences in gel mobility correlate with the differences in reactivity that we observed between Ii-peptide and H-2M0 mice. CD4 T cells were more reactive from mice whose peptide–Ab complexes migrated slowly (Rab–Ab and CLIP–Ab) whereas CD4 T cells were less reactive from mice whose peptide–Ab complexes migrated quickly (CD22-Ab and Eα-Ab).
Discussion
The degree to which thymocytes depend on specific interactions with MHC-bound self-peptides for positive selection is central to understanding how the T cell repertoire is generated. This issue has direct bearing on the number of T cells that are selected by an individual peptide and the extent to which the specificities of those selected T cells will depend on the peptide. In mice overexpressing individual peptides, we show that a subset of T cells is selected by the abundant peptide as suggested by an increase in the percentage of CD4 single-positive thymocytes when two peptides, Eα and CLIP, are overexpressed together. Thus, although peptide diversity is required for efficient positive selection, we can detect CD4 T cells selected on a given peptide–Ab complex.
Merely measuring cell number, however, ignores differences in the specificities of the selected T cells. The TCR repertoire clearly is altered in mice with limited peptide diversity. Of six TCRs capable of being selected in wild-type mice, only one, DO11.10, has been found to be selected in H-2M0 mice, although this receptor actually is negatively selected in wild-type H-2b mice (3, 4, 11, 19). Recent analysis of the CDR3 sequences within Vα2Jα4 TCRα chains from transgenic Vβ8.2 CD4 T cells selected on wild-type or H-2M0 peptide repertoires showed a severe restriction in CDR3 usage caused by the limited peptides in H-2M0 mice (20). Similarly, several groups have analyzed TCR sequences in AbEp mice and were able to show alterations because of the overexpression of Eα peptide or its variant (21–23). Thus, the TCRs present in H-2M0 and AbEp mice clearly are distinct from those in wild-type mice. The limitation inherent to all but one of these analyses, though, is that alterations in specificity cannot be attributed definitively to selection on CLIP and Εα as opposed to other less abundant self-peptides, nor can they be attributed to positive selection as opposed to negative selection.
We show that the ability of the CD4 T cells in peptide-dbl0 mice to recognize self-MHC class II molecules displayed by wild-type APCs changes depending on the identity of the overexpressed peptide, and these differences are dictated by positive rather than negative selection (Fig. 4). The high reactivity of CD4 T cells from H-2M0 and AbEp mice has been attributed to a lack of negative selection (3–8). Indeed, T cells from mice expressing low levels of MHC class II molecules exclusively on thymic cortical epithelial cells are highly self-MHC-reactive (24). Nevertheless, our results demonstrate that the specificity, i.e., self-MHC reactivity, of T cells can be altered when a single abundant peptide is replaced by another peptide.
It is highly unlikely that the differences in reactivity that we observe are caused by differences in minor peptides that we cannot measure. First, the overexpressed peptides all bind Ab with very similar affinity and are expressed at similarly high levels, as measured by peptide-specific antibodies. Second, the Ii-peptide constructs are processed similarly as suggested by identical Ii-derived N- and C-terminal residues in major peptides isolated from peptide-Ii0 and peptide-dbl0 mice (data not shown; see Methods, which is published as supporting information on the PNAS web site). Third, CD4 T cells from peptide-Ii0 and peptide-dbl0 share the same hierarchy of reactivity between the different peptides. The fact that the proliferative responses are weaker from peptide-Ii0 mice suggests that the same subset of CD4 T cells is selected in these mice as in peptide-dbl0 mice. Thus, in all likelihood the sets of background peptides are very similar if not identical in different peptide-dbl0 mice. Finally, blocking Eα–Ab or CLIP–Ab in Eα × CLIP mice by using complex-specific mAbs resulted in CD4 T cell reactivity patterns qualitatively similar to those observed in H-2M0 and Eα-dbl0 mice, respectively. In aggregate, these results strongly suggest that the overexpressed peptide–Ab complexes are directly responsible for selection of self-MHC-reactive CD4 T cells with a differing degree of reactivity.
It is puzzling why CD4 T cells from CD22-dbl0 mice, and to a lesser extent Eα-dbl0 mice, have such low self-MHC reactivity. In all four strains of mice, negative selection is likely to be similarly inefficient because of the severe reduction in endogenous peptides presented by Ab (Fig. 2). We ruled out the possibility that regulatory T cells were inhibiting proliferation in the less reactive mice (data not shown). Therefore, we can speculate that the differing degrees of self-MHC reactivity displayed by each strain of mice suggest that varying numbers of self-MHC-reactive cells may be positively selected on each of the different peptide–Ab complexes. Alternatively, the same approximate number of self-MHC-reactive cells may be selected in each strain of mice, but certain peptide–Ab complexes (e.g., CLIP–Ab) simply may select much more highly reactive TCRs than others (e.g., CD22–Ab). Either possibility implies that certain peptide–Ab complexes select MHC-reactive cells more efficiently than others. In support of this conclusion, MHC-reactive cells from H-2M0 mice have a weaker affinity for H-2b APCs than for H-2bm12 APCs, and they suggest that high-affinity cells may not be positively selected by CLIP–Ab because of its high expression (25). In light of our results, it seems more likely that different peptide–Ab complexes may select different subsets of MHC-reactive T cells capable of recognition of self-MHC class II molecules bound to a diverse repertoire of peptides. We predict that TCRs displayed by such T cells have less stringent peptide specificity and depend more on interactions with MHC residues as compared with peptide-specific T cells. Importantly, polyclonal peptide-specific CD4 T cell responses are not significantly different in Ii-peptide transgenic mice.
What makes these different peptide–Ab complexes biologically distinct? The most straightforward explanation is that the differences in the four peptide sequences are mediating differential selection of certain TCR specificities. However, it is possible that the conformation of a given peptide–Ab complex may affect interactions with certain TCRs (18). It is tempting to invoke possible conformational differences among the four peptide–Ab complexes to explain a greater or lesser propensity for self-MHC reactivity. In fact, we have observed that each of the four peptide–Ab complexes appears to adopt a distinct conformation (Fig. 5). It is possible that certain conformations are better at selecting self-MHC-reactive T cells than others. Indeed, Rab–Ab complexes have gel mobility most similar to CLIP–Ab, and T cells from these mice are the most reactive of the three peptide-dbl0 mice. Clearly, the self-MHC reactivity observed in CD4 T cells from peptide-dbl0 or peptide-Ii0 mice is not equivalent to the autoreactivity observed in experimental or clinical autoimmunity. However, it seems reasonable to suggest that our findings are applicable to ligands that positively select pathogenic T cells in autoimmune settings.
Supplementary Material
Acknowledgments
We thank A. Goldrath, M. Bevan, and M. Bix for comments on the manuscript and C. Plata for care of mice. This work was supported by National Institutes of Health Grants aT32 GM07270 (to G.M.B) and AI34206 (to A.Y.R.). A.Y.R. is a Howard Hughes Medical Institute Investigator.
Abbreviations
- TCR
T cell receptor
- Ii
human invariant chain
- Tg
transgene
- CLIP
class II-associated invariant chain peptide
- BM
bone marrow
- APC
antigen-presenting cell
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Fink P J, Bevan M J. Adv Immunol. 1995;59:99–133. doi: 10.1016/s0065-2776(08)60630-6. [DOI] [PubMed] [Google Scholar]
- 2.Bevan M J. Immunity. 1997;7:175–178. doi: 10.1016/s1074-7613(00)80520-8. [DOI] [PubMed] [Google Scholar]
- 3.Tourne S, Miyazaki T, Oxenius A, Klein L, Fehr T, Kyewski B, Benoist C, Mathis D. Immunity. 1997;7:187–195. doi: 10.1016/s1074-7613(00)80522-1. [DOI] [PubMed] [Google Scholar]
- 4.Surh C D, Lee D S, Fung Leung W P, Karlsson L, Sprent J. Immunity. 1997;7:209–219. doi: 10.1016/s1074-7613(00)80524-5. [DOI] [PubMed] [Google Scholar]
- 5.Martin W D, Hicks G G, Mendiratta S K, Leva H I, Ruley H E, Van Kaer L. Cell. 1996;84:543–550. doi: 10.1016/s0092-8674(00)81030-2. [DOI] [PubMed] [Google Scholar]
- 6.Miyazaki T, Wolf P, Tourne S, Waltzinger C, Dierich A, Barois N, Ploegh H, Benoist C, Mathis D. Cell. 1996;84:531–541. doi: 10.1016/s0092-8674(00)81029-6. [DOI] [PubMed] [Google Scholar]
- 7.Fung-Leung W P, Surh C D, Liljedahl M, Pang J, Leturcq D, Peterson P A, Webb S R, Karlsson L. Science. 1996;271:1278–1281. doi: 10.1126/science.271.5253.1278. [DOI] [PubMed] [Google Scholar]
- 8.Ignatowicz L, Kappler J, Marrack P. Cell. 1996;84:521–529. doi: 10.1016/s0092-8674(00)81028-4. [DOI] [PubMed] [Google Scholar]
- 9.Benoist C, Mathis D. Curr Opin Immunol. 1997;9:245–249. doi: 10.1016/s0952-7915(97)80143-4. [DOI] [PubMed] [Google Scholar]
- 10.Marrack P, Kappler J. Curr Opin Immunol. 1997;9:250–255. doi: 10.1016/s0952-7915(97)80144-6. [DOI] [PubMed] [Google Scholar]
- 11.Grubin C E, Kovats S, deRoos P, Rudensky A Y. Immunity. 1997;7:197–208. doi: 10.1016/s1074-7613(00)80523-3. [DOI] [PubMed] [Google Scholar]
- 12.Barton G M, Rudensky A Y. Science. 1999;283:67–70. doi: 10.1126/science.283.5398.67. [DOI] [PubMed] [Google Scholar]
- 13.Chmielowski B, Muranski P, Kisielow P, Ignatowicz L. Int Immunol. 2000;12:67–72. doi: 10.1093/intimm/12.1.67. [DOI] [PubMed] [Google Scholar]
- 14.Barton G M, Rudensky A Y. Int Immunol. 1998;10:1159–1165. doi: 10.1093/intimm/10.8.1159. [DOI] [PubMed] [Google Scholar]
- 15.Kovats S, Grubin C E, Eastman S, deRoos P, Dongre A, Van Kaer L, Rudensky A Y. J Exp Med. 1998;187:245–251. doi: 10.1084/jem.187.2.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tourne S, Miyazaki T, Wolf P, Ploegh H, Benoist C, Mathis D. Proc Natl Acad Sci USA. 1997;94:9255–9260. doi: 10.1073/pnas.94.17.9255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fukui Y, Ishimoto T, Utsuyama M, Gyotoku T, Koga T, Nakao K, Hirokawa K, Katsuki M, Sasazuki T. Immunity. 1997;6:401–410. doi: 10.1016/s1074-7613(00)80283-6. [DOI] [PubMed] [Google Scholar]
- 18.Chervonsky A V, Medzhitov R M, Denzin L K, Barlow A K, Rudensky A Y, Janeway C A., Jr Proc Natl Acad Sci USA. 1998;95:10094–10099. doi: 10.1073/pnas.95.17.10094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ernst B, Lee D S, Chang J M, Sprent J, Surh C D. Immunity. 1999;11:173–181. doi: 10.1016/s1074-7613(00)80092-8. [DOI] [PubMed] [Google Scholar]
- 20.Sant'Angelo D B, Waterbury P G, Cohen B E, Martin W D, Van Kaer L, Hayday A C, Janeway C A., Jr Immunity. 1997;7:517–524. doi: 10.1016/s1074-7613(00)80373-8. [DOI] [PubMed] [Google Scholar]
- 21.Fukui Y, Hashimoto O, Inayoshi A, Gyotoku T, Sano T, Koga T, Gushima T, Sasazuki T. J Exp Med. 1998;188:897–907. doi: 10.1084/jem.188.5.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gapin L, Fukui Y, Kanellopoulos J, Sano T, Casrouge A, Malier V, Beaudoing E, Gautheret D, Claverie J M, Sasazuki T, Kourilsky P. J Exp Med. 1998;187:1871–1883. doi: 10.1084/jem.187.11.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pacholczyk R, Kraj P, Ignatowicz L. J Immunol. 2001;166:2357–2363. doi: 10.4049/jimmunol.166.4.2357. [DOI] [PubMed] [Google Scholar]
- 24.Laufer T M, DeKoning J, Markowitz J S, Lo D, Glimcher L H. Nature (London) 1996;383:81–85. doi: 10.1038/383081a0. [DOI] [PubMed] [Google Scholar]
- 25.Lee D S, Ahn C, Ernst B, Sprent J, Surh C D. Immunity. 1999;10:83–92. doi: 10.1016/s1074-7613(00)80009-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}