

ABSTRACT
Cancer immunotherapy is a groundbreaking therapeutic strategy, yet it continues to face significant challenges, including limited overall response rates and treatment resistance. Emerging research has demonstrated the pivotal role of epigenetic modifications in tumor immune evasion, providing a strong rationale for developing “epi‐immunotherapy”—an innovative approach that combines epigenetic therapy with immunotherapy. This comprehensive review systematically examines how epigenetic regulation mediates tumor immune escape and the mechanisms involved, including suppression of tumor antigen expression and antigen presentation, upregulation of immune checkpoint molecules, inhibition of antitumor immune cell recruitment and function, and enhancement of immunosuppressive cell proliferation and activity. By integrating epigenetic modulation with immunotherapeutic strategies, epi‐immunotherapy demonstrates a remarkable ability to enhance treatment efficacy and reverse therapeutic resistance. We also summarize the current clinical applications of epi‐immunotherapy in both hematological malignancies and solid tumors, with particular emphasis on its mechanisms for overcoming immune checkpoint inhibitor resistance and converting immunologically “cold” tumors into “hot” tumors. Despite its promising potential, epi‐immunotherapy faces several challenges that require urgent resolution. This review provides an in‐depth analysis of these limitations, which include the complexity of epigenetic regulation, a lack of reliable biomarkers, and constraints in drug development. As our understanding of epigenetic mechanisms deepens and technologies continue to advance, epi‐immunotherapy is poised to become an essential component of cancer treatment, offering patients more effective and personalized therapeutic options.
Keywords: cancer, epigenetics, epi‐immunotherapy, immunotherapy, tumor immune evasion, tumor microenvironment
Epigenetic regulation of tumor immune evasion and synergistic anticancer strategies via epigenetic‐immunotherapy integration.

Abbreviations
- AML
acute myeloid leukemia
- BAF
BRG1/BRM‐associated factor
- cHL
classical hodgkin lymphoma
- CpG
cytosine‐guanine dinucleotide
- CR
complete response
- CSF
colony‐stimulating factor
- CTLA‐4
cytotoxic T‐lymphocyte associated protein 4
- EZH2
enhancer of Zeste Homolog 2
- FDA
Food and Drug Administration
- FOXP3
forkhead box P3
- H3K27me3
histone H3 lysine 27 trimethylation
- H3K4me3
histone H3 lysine 4 trimethylation
- HDAC
histone deacetylase
- ICI
immune checkpoint inhibitor
- IDH
isocitrate dehydrogenase
- IFN
interferon
- IRF4
interferon regulatory factor 4
- JMJD
Jumonji domain‐containing
- LAG‐3
lymphocyte‐activation gene 3
- m6A
N6‐methyladenosine
- MDSCs
myeloid‐derived suppressor cells
- METTL3
methyltransferase‐like 3
- MHC
major histocompatibility complex
- ncRNA
noncoding RNA
- NF‐κB
nuclear Factor κappa‐light‐chain‐enhancer of activated B cells
- NK
natural killer
- NKG2D
natural Killer Group 2D
- NSCLC
non‐small cell lung cancer
- ORR
overall response rate
- OS
overall survival
- PD‐1
programmed cell death protein 1
- PD‐L1
programmed death ligand 1
- PFS
progression‐free survival
- PR
partial response
- SD
stable disease
- SOCS1
suppressor of cytokine signaling 1
- STAT3
signal transducer and activator of transcription 3
- STING
stimulator of interferon genes
- TET
Ten‐eleven translocation methylcytosine dioxygenase
- TIM‐3
T‐cell immunoglobulin and mucin domain‐containing protein 3
- TME
tumor microenvironment
- Tregs
regulatory T cells
1. Introduction
Cancer immunotherapy is a breakthrough therapeutic strategy that harnesses the host's immune system to specifically identify and eliminate malignant cells. Its core mechanism involves relieving tumor‐induced immunosuppression and reactivating intrinsic antitumor immune responses. Since the approval of the first immune checkpoint inhibitor (ICI) in 2011, this approach has achieved remarkable success in treating various malignancies, notably melanoma and lung cancer, offering unprecedented long‐term survival prospects for patients with advanced disease. ICIs targeting PD‐1/PD‐L1 and CTLA‐4 have been incorporated into authoritative guidelines, including those of the National Comprehensive Cancer Network and the Chinese Society of Clinical Oncology, becoming standard first‐line treatment options for multiple cancer types.
However, cancer immunotherapy faces two major challenges. First, the overall response rates (ORRs) remain relatively low, with objective response rates for most tumor types ranging from 10% to 30% [1], leaving the majority of patients without direct benefit. Second, acquired resistance develops in approximately 40%–60% of initial responders during treatment. These limitations represent critical obstacles in the field.
Recent mechanistic studies have highlighted the fundamental role of epigenetic modifications in tumor immune evasion through multiple pathways, including (1) silencing of tumor‐associated antigens and impairment of antigen presentation machinery, (2) upregulation of immune checkpoint molecules, (3) suppression of antitumor immune cell recruitment and function, and (4) enhancement of regulatory immune cell activity. These insights have sparked interest in combining epigenetic therapies with immunotherapy, an approach termed “epi‐immunotherapy.”
Epi‐immunotherapy represents an innovative treatment strategy that integrates epigenetic regulation with immunotherapeutic approaches. Preclinical and clinical studies demonstrate that epigenetic agents can enhance immunotherapy efficacy and potentially reverse resistance to both chemotherapy and immunotherapy. This synergistic potential has generated significant interest among oncologists, leading to extensive investigation of epi‐immunotherapy combinations.
This review examines the role of epigenetics in cancer immunotherapy and the mechanisms underlying epi‐immunotherapy combinations and summarizes clinical advances, current challenges, and future directions.
2. The Role of Epigenetics in Tumor Immunotherapy
Epigenetics encompasses five principal mechanisms that regulate chromatin state: DNA modifications [2], histone modifications [3], noncoding RNA (ncRNA)‐mediated regulation [4], RNA modifications [5], and chromatin remodeling [6] (Figure 1). These mechanisms modulate gene expression independently of DNA sequence changes, activating or suppressing specific genomic regions in response to physiological or pathological signals. Epigenetic reprogramming plays a significant role in tumor formation and progression, as well as in establishing and maintaining immunosuppressive characteristics within the tumor microenvironment, thereby regulating tumor behavior.
Figure 1.
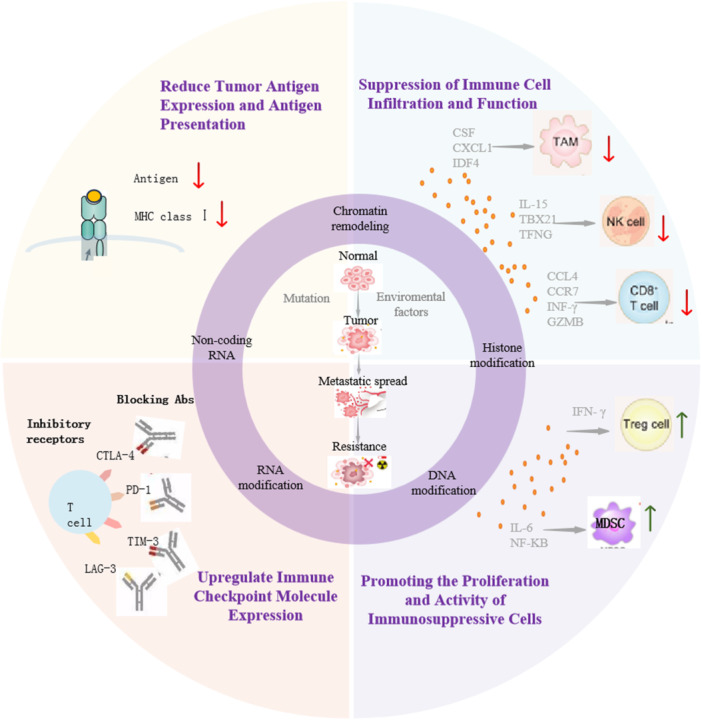
Epigenetic modifications regulate the TME, influencing tumor progression. The inner circle represents a, the middle ring represents b, and the outer ring represents c. (a) Epigenetic modifications influence the initiation, metastasis, and drug resistance of tumors. (b) Types of epigenetic modifications. (c) Tumor cells evade immune surveillance through epigenetic mechanisms: (1) reduce tumor antigen expression and antigen presentation; (2) upregulate immune checkpoint molecule expression; (3) suppress immune cell (TAM, NK cell, CD8+ T cell) infiltration and function; and (4) promote the proliferation and activity of immunosuppressive cells (Tregs, MDSCs). MDSCs, myeloid‐derived suppressor cells; NK, natural killer cell; TAM, tumor‐associated macrophages; Tregs, Regulatory T Cells.
2.1. Major Epigenetic Regulatory Mechanisms
2.1.1. DNA Methylation
DNA methylation is a fundamental epigenetic modification characterized by the addition of methyl groups to DNA molecules, primarily occurring on cytosine bases to form 5‐methylcytosine (5‐mC). This process takes place predominantly at cytosine‐guanine dinucleotides (CpG sites) and is catalyzed by DNA methyltransferases. In the mammalian genome, DNA methylation orchestrates diverse processes, including transcriptional regulation, posttranscriptional processing, chromatin architecture, genomic imprinting, and suppression of repetitive elements [7, 8].
In tumors, aberrant CpG methylation patterns contribute to carcinogenesis through multiple mechanisms. Promoter hypermethylation can silence tumor suppressor and DNA repair genes, which disrupt cell proliferation and differentiation pathways and promote malignant transformation [9, 10, 11]. Conversely, genome‐wide hypomethylation, particularly in oncogene promoter regions and repetitive DNA sequences, destabilizes chromosomal integrity and facilitates tumor progression [12, 13].
2.1.2. Histone Modifications
Posttranslational histone modifications comprise the addition of chemical groups to nucleosome‐forming protein tails of histones, predominantly in those located at promoter and enhancer regions [14]. Methylation and acetylation represent the most extensively studied modifications, which are catalyzed by specific enzymes, including histone acetyltransferases, deacetylasfes (HDACs), methyltransferases, and demethylases.
These modifications regulate critical cellular processes, including cell‐cycle control, DNA replication and repair, cell signaling, metabolic pathways, and gene expression. Through their ability to modulate chromatin states, histone modifications can influence tumor development by controlling tumor suppressor gene inactivation and oncogene activation, serving as both potential biomarkers and therapeutic targets in cancer [15, 16].
2.1.3. Noncoding RNA (ncRNAs) Modifications
ncRNAs, while not encoding proteins, serve as crucial regulators of gene expression through various chemical modifications, primarily methylation and pseudouridylation. These RNA modifications dynamically regulate ncRNA function, stability, and localization, participating in chromatin structure regulation, transcriptional control, and post‐transcriptional modifications [17].
In malignancies, ncRNA modifications regulate the expression of oncogenes and tumor suppressor genes, influencing tumor cell metabolism and microenvironment interactions. These modifications affect tumor initiation and progression through the modulation of cell signaling pathways.
2.2. Tumor Cells Evade Immune Surveillance Through Epigenetic Mechanisms
Tumor cells exploit diverse epigenetic mechanisms to modulate immune‐related gene expression, enabling immune evasion through two primary strategies: reducing their immunogenic visibility and suppressing immune cell function. This process represents a critical step in tumor development and metastasis while providing potential targets for tumor immunotherapy (Figure 1).
2.2.1. Epigenetic Modifications Reduce Tumor Antigen Expression and Antigen Presentation
Tumor antigen expression and presentation form the foundation of immune surveillance, enabling immune system recognition and elimination of tumors and providing key targets for immunotherapy strategies. Epigenetic modifications impair immune surveillance through multiple mechanisms, including the following.
Epigenetic modifications reduce tumor immunogenicity through multiple pathways. The epigenetic regulator, SETDB1, catalyzes H3K9 trimethylation to suppress expression of genes encoding interferons (IFNs), reducing the intrinsic immunogenicity of cancer cells [18, 19]. N6‐methyladenosine (m6A), the most common RNA modification, reduces tumor immunogenicity by influencing oncogenic networks [20, 21].
Additionally, direct suppression of antigen presentation occurs through various pathways. In diffuse large B‐cell lymphoma, activating mutations in EZH2 enhance its catalytic activity, leading to MHC class I and II molecule suppression through H3K27me3 modification. This directly impairs antigen presentation, weakening immune system recognition of cancer cells [22]. In colon cancer, the epigenetic regulator, KDM5D, encoded by a Y chromosome gene, influences MHC class I antigen presentation suppression through a demethylase‐independent mechanism [23].
These findings demonstrate the crucial role of epigenetic features in tumor immune evasion. Modulating epigenetic modifications may enhance tumor antigen expression and presentation efficiency, offering a key strategy to improve tumor immunotherapy efficacy and address immune evasion. This provides a critical scientific basis for developing novel cancer treatment strategies.
2.2.2. Epigenetic Regulation of Immune Checkpoint Molecule Expression
Immune checkpoint molecules serve as critical regulators of immune responses in the tumor microenvironment. Epigenetic modifications can reactivate previously silenced genes, leading to the upregulation of key immune checkpoint molecules, including PD‐L1, TIM‐3, CTLA‐4, and LAG‐3. This upregulation ultimately compromises the surveillance and clearance capabilities of the immune system while enhancing tumor cell adaptability and progression.
Research across multiple cancer types has demonstrated the broad impact of epigenetic modifications on the expression of molecules that suppress immune responses. In lung cancer and melanoma, both pan‐HDAC and specific HDAC inhibitors induce PD‐L1 expression [24, 25]. Cervical cancer studies have revealed that EZH2, H3K27me3, and DNA methyltransferase 3 (DNMT3A) participate in the epigenetic regulation of TIM3 expression, particularly in the context of HPV18 infection [26]. In breast cancer, both DNA and histone modifications contribute to the upregulation of multiple checkpoint molecules, including PD‐1, CTLA‐4, TIM‐3, and LAG‐3 [27]. Additionally, in gliomas and ovarian cancer, miRNAs play a crucial role in modulating the expression of TIM‐3, CTLA‐4, and PD‐1/PD‐L1 [28, 29].
These epigenetic effects extend beyond solid tumors to hematological malignancies. For example, in B‐cell lymphoma, HDAC3 specifically upregulates PD‐L1 expression [30], while in acute myeloid leukemia (AML), miRNAs regulate the expression of multiple checkpoint molecules, including TIM‐3, CTLA‐4 and PD‐1/PD‐L1 [24].
These findings collectively demonstrate that epigenetic modifications alter tumor gene transcription and expression patterns, leading to increased expression of immune checkpoint molecules. This upregulation suppresses T‐cell activity and reduces immune cell‐mediated tumor cell destruction, ultimately enabling tumor immune evasion and progression. Therefore, therapeutic strategies aimed at modulating epigenetic modifications to reduce immune checkpoint molecule expression may potentially reverse immune cell suppression and restore antitumor immunity.
2.2.3. Epigenetic Impact on Immune Cell Infiltration and Function
Epigenetic mechanisms dynamically regulate immune‐related gene expression, affecting crucial processes, such as immune cell chemotaxis, adhesion, and activation. These modifications can significantly reduce immune cell migration to tumor sites and impair their effector functions, ultimately promoting tumor immune evasion and progression.
2.2.3.1. CD8+ T Cells
CD8+ T cells are primary effector cells in the immune system, directly recognizing and eliminating tumor cells through antigen recognition. These cells represent a cornerstone of cancer immunotherapy, and a substantial body of evidence indicates that epigenetic modifications significantly influence their infiltration and function.
Studies of hepatocellular carcinoma have shown that HDAC8 reduces H3K27 acetylation and silences the CCL4 chemokine gene, thereby inhibiting CD8+ T cell infiltration into tumor tissue [31]. The activation of CD8+ T cells involves complex epigenetic reprogramming, characterized by demethylation of transcription start sites in key effector genes (e.g., IFNG and GZMB), demethylation of transcription factor genes expressed in activated lymphocytes, and increased methylation of naive T‐cell‐associated genes (e.g., CCR7 and TCF7) to promote their silencing [32, 33]. Epigenetic analyses have revealed that CD8+ T cell exhaustion correlates with specific alterations in chromatin markers, including decreased levels of H3K27ac (active chromatin) and increased levels of H3K27me3 (repressive chromatin) [34]. The (SWI/SNF) chromatin remodeling complex, also known as the BRG1/BRM‐associated factor (BAF) complex, plays a crucial role in regulating CD8+ T cell function and longevity within the tumor microenvironment [35]. Notably, depleting BAF complex members, such as ARID1A or SMARCD2, prevents terminal T‐cell exhaustion and promotes memory T‐cell generation [36], highlighting the importance of the BAF complex in T‐cell regulation. Furthermore, the histone methyltransferase, SUV39H1, downregulates IFNγ and granzyme B expression in effector CD8+ T cells, thereby promoting T‐cell exhaustion. Recent studies have shown that combining SUV39H1 inhibitors (such as ETP‐69) with anti‐PD‐1 therapy increases the proportion of effector CD8+ T cells and impairs tumor growth, indicating potential therapeutic applications [37].
2.2.3.2. Natural Killer (NK) Cells
NK cells are crucial mediators of antitumor immune responses through multiple mechanisms. They directly eliminate tumor cells by secreting cytotoxic molecules, including perforin and granzymes [38], while simultaneously orchestrating broader immune responses through the release of proinflammatory cytokines, such as IFNγ and TNF‐α [39]. NK cells also play a vital role in preventing metastasis by targeting cancer stem cells [40] and maintaining tumor dormancy [41], making their functional integrity essential for tumor control.
Epigenetic modifications significantly influence NK cell function and activation state. The lysine demethylase, KDM5A, regulates NK cell activation by demethylating H3K4me3 and interacting with p50, which relieves SOCS1‐mediated suppression [42]. Additionally, the RNA methyltransferase, METTL3, maintains NK cell homeostasis through preservation of IL‐15‐dependent signaling pathways [43]. In terminally differentiated NK cells, key effector genes, including IFNG and TBX21, display characteristic epigenetic signatures featuring DNA hypomethylation and extensive histone acetylation at their transcription start sites. This epigenetic priming enables rapid and robust proinflammatory and cytotoxic responses upon activation [44].
Notably, disruption of these epigenetic regulatory mechanisms can impair NK cell‐mediated antitumor responses, potentially contributing to immune evasion. Understanding these epigenetic controls provides the rational for pharmacological intervention with epigenetic modifiers that may offer therapeutic opportunities to restore NK cell function and enhance antitumor immunity.
2.2.3.3. Macrophages
Macrophages are a fundamental component of the tumor immune microenvironment, exhibiting diverse functions in tumor immunity. These cells facilitate early tumor recognition and elimination through phagocytosis, while simultaneously being antigen‐presenting cells that activate T‐cell‐mediated immune responses. Furthermore, macrophages secrete an array of cytokines, including tumor necrosis factor (TNF) and interleukins, which can directly induce tumor cell death or modulate broader immune responses [45, 46].
Epigenetic reprogramming extensively influences tumor‐associated macrophage function and phenotype. This regulation affects both their infiltration into tumor tissues and their potential transition toward an immunosuppressive state that promotes tumor progression. Several molecular mechanisms underscore this epigenetic control, such as RNA modification, as demonstrated by METTL3‐mediated m6A modification. Reduced m6A modification leads to IRAKM protein stabilization, resulting in suppressed TLR signaling pathway activation in tumor‐associated macrophages and subsequent promotion of tumor development [47]. ATP‐citrate lyase modulates tumor‐associated macrophage function through epigenetic modifications; its downregulation reduces acetyl‐CoA levels and histone acetylation, resulting in chromatin compaction that attenuates tumor‐associated macrophage‐mediated immunosuppression [48]. The acetyl‐lysine reader, CECR2, exemplifies another crucial epigenetic mechanism, that of recognizing and binding acetylated lysine residues to enhance chromatin accessibility and expression of specific genes, including CSF1, CSF2, CSF3, and CXCL1. These factors contribute to establishing an immunosuppressive macrophage phenotype [49]. Similarly, the DNA demethylase, TET2, promotes myeloid cell accumulation in tumor tissues, supporting tumor‐associated macrophage polarization toward an immunosuppressive state [50]. Furthermore, epigenetic mechanisms regulate macrophage differentiation, particularly through DNA methyltransferases and histone modifiers. The histone H3K27 demethylase, JMJD3, plays an essential role in the transcription of IRF4 [51, 52, 53], which encodes a key transcription factor directing macrophage polarization toward the M2 phenotype [54].
These findings collectively demonstrate that epigenetic reprogramming governs both tumor‐associated macrophage functional state and immune phenotype, ultimately influencing tumor progression. Epigenetic therapeutic interventions may therefore potentially reverse aberrant modifications and restore antitumor immune processes.
2.2.3.4. Regulatory T Cells
Regulatory T cells (Tregs) are central mediators of tumor immunosuppression through multiple mechanisms, including inhibitory cytokine secretion, expression of suppressive membrane molecules, and direct interaction with immune effector cells. These activities collectively promote tumor progression by dampening antitumor immune responses.
The transcription factor, FOXP3, is a master regulator of Treg development and function, and FOXP3 expression is tightly controlled by DNA methylation at CpG islands within its enhancer region [55, 56]. This epigenetic regulation is crucial for maintaining immune system homeostasis. TET methylcytosine dioxygenases, particularly TET2 and TET3, play essential roles in maintaining Treg functional stability and immune homeostasis through their DNA demethylation activity [57].
The histone methyltransferase, EZH2, is another critical epigenetic regulator in Treg biology. By catalyzing H3K27 trimethylation (H3K27me3), EZH2 enables Tregs to silence immune activation‐related genes and maintain their suppressive phenotype. DuPage and colleagues demonstrated that EZH2 is indispensable for Tregs to retain their suppressive characteristics following activation [58]. Additional studies by Yang and colleagues have further elucidated the dual role of EZH2 in both Treg differentiation and effector T‐cell expansion [59]. Multiple investigations have confirmed the fundamental importance of H3K27 methylation in maintaining Treg immunosuppressive function [60, 61].
These findings collectively indicate that epigenetic modifications orchestrate both Treg differentiation and functional stability through precise regulation of gene expression programs, ultimately facilitating their immunosuppressive effects in the tumor microenvironment. Therapeutic strategies targeting Treg‐specific epigenetic mechanisms may therefore represent promising approaches for enhancing immunotherapy efficacy and improving patient outcomes.
2.2.3.5. Myeloid‐Derived Suppressor Cells (MDSCs)
MDSCs are a crucial component of tumor immunity, orchestrating multiple immunosuppressive mechanisms within the tumor microenvironment. These cells regulate immune cell proliferation and activation while secreting immunosuppressive cytokines and promoting tumor angiogenesis, collectively establishing an immunosuppressive network that enables tumors to evade immune surveillance and attack.
Epigenetic mechanisms play fundamental roles in regulating MDSC differentiation, function, and maintenance of immunosuppressive properties. In prostate cancer characterized by phosphatase and tensin homolog (PTEN) deficiency, two key chromatin regulators—ARID1A, a subunit of the BAF chromatin remodeling complex, and the chromatin remodeler CHD1—show positive correlation with MDSC abundance [62, 63, 64, 65]. PTEN deficiency prevents CHD1 degradation, enhancing its stability. Subsequently, CHD1 specifically recognizes and binds to H3K4me3, promoting IL‐6 expression and facilitating MDSC infiltration into the tumor microenvironment. RNA modification, particularly m6A methylation, represents another critical layer of epigenetic control in MDSC function. Inhibition of the methyltransferase, METTL3, reduces m6A modification levels on mRNA, affecting the expression of genes associated with immunosuppressive cells, including MDSCs [62, 66]. Complementarily, decreased expression of the m6A eraser, ALKBH5, influences MDSC‐related gene expression, promoting MDSC infiltration [67]. The m6A‐modified pseudogene, Olfr29‐ps, modulates MDSC populations by regulating mRNA stability and translation efficiency. Additionally, the m6A reader, YTHDF2, promotes MDSC function through a positive feedback loop with NFκB signaling, where YTHDF2 degrades negative regulators of the NFκB pathway, leading to enhanced NFκB/RELA signaling and increased YTHDF2 expression in MDSCs. This self‐reinforcing circuit amplifies cytokine expression in MDSCs, promoting their infiltration and differentiation [68].
The epigenetic regulation of MDSC function extends to their secretion of immunomodulatory factors. For instance, the H3K27ac regulator, CBP/EP300‐BRD, influences MDSC function by regulating chemokine receptor gene expression. Attenuation of CBP/EP300‐BRD reduces chemokine receptor levels, thereby decreasing MDSC responsiveness to chemokines and compromising their differentiation and immunosuppressive capabilities [69].
These findings collectively demonstrate that epigenetic modifications both directly and indirectly influence MDSC differentiation, maturation, and function through multiple molecular mechanisms. This understanding provides potential therapeutic opportunities for targeting the epigenetic regulation of MDSCs to reshape the immunosuppressive tumor microenvironment.
3. Synergistic Effects of Epigenetic Drugs and ICIs
3.1. Commonly Used Drugs That Target Epigenetic Modification
Epigenetic modifications are critical regulators of gene expression that alter chromatin structure and influence gene accessibility, thereby modulating tumor behavior. The reversible and specific nature of abnormal epigenetic modifications makes epigenetic drugs particularly advantageous for cancer treatment compared with traditional radiotherapy and chemotherapy. These drugs can restore normal cell functions or enhance immune system recognition of tumor cells by targeting specific epigenetic features of cancer cells [70, 71]. Consequently, epigenetic drugs demonstrate enhanced antitumor efficacy with reduced side effects compared with conventional therapies. Ongoing advances in biotechnology and deepening insights into epigenetic mechanisms continue to drive progress in epigenetic‐targeted therapies.
3.1.1. DNA Methyltransferase Inhibitors
DNA methyltransferases regulate gene expression through DNA methylation. DNA methyltransferase inhibitors suppress enzyme activity by preventing methyl group transfer from S‐adenosylmethionine to cytosine, resulting in demethylation and reactivation of tumor suppressor genes, thereby inhibiting tumor growth and metastasis [72, 73]. The U.S. Food and Drug Administration (FDA) has approved two hypomethylating agents targeting DNA methyltransferase 1, azacitidine (May 2004) and decitabine (June 2006), for the treatment of myelodysplastic syndromes [74, 75], making them among the first approved epigenetic‐targeted therapeutics (Figure 2). As a second‐generation DNA methylation inhibitor, guadecitabine is currently used in the treatment of myelodysplastic syndromes and AML, demonstrating potential to be a superior hypomethylating agent compared with azacitidine and decitabine, particularly in combination therapies for myeloid malignancies [76]. Multiple clinical trials are ongoing to further validate its efficacy and safety profile.
Figure 2.

Timeline of approved indications for epigenetic immunomodulators. DNMTi is located in the purple boxes, HDACi in the yellow box, EZH2i in the gray box, and the IDH inhibitor in the pink box. DNMTi, DNA methyltransferase inhibitors; HDACi, histone deacetylase inhibitors; EZH2, enhancer of zeste homologue 2; IDH, isocitrate dehydrogenase.
3.1.2. Histone Deacetylase Inhibitors
HDAC inhibitors maintain histone acetylation by blocking HDAC activity, promoting relaxed chromatin structure, and facilitating gene transcription. These agents function by reactivating tumor suppressor genes and/or suppressing oncogene expression. Major regulatory authorities, including the U.S. FDA, the European Medicines Agency, and Japan's Pharmaceuticals and Medical Devices Agency, have approved several HDAC inhibitors: vorinostat (SAHA), romidepsin (FK228), belinostat (PXD101), and panobinostat (LBH589). These medications primarily treat multiple myeloma, cutaneous T‐cell lymphoma, and peripheral T‐cell lymphoma [77, 78]. Additionally, chidamide has received approval from Japan's Pharmaceuticals and Medical Devices Agency and China's National Medical Products Administration for peripheral T‐cell lymphoma and advanced breast cancer treatment [79, 80], establishing it as the first HDAC inhibitor approved for solid tumors (Figure 2). Currently, over 20 HDAC inhibitors are undergoing clinical trials.
3.1.3. EZH2 Inhibitors
EZH2, a core component of the Polycomb Repressive Complex 2, exhibits histone methyltransferase activity. It primarily regulates tumor suppressor gene expression through H3K27 methylation. EZH2 overexpression in various cancers has established it as a promising therapeutic target. The FDA approved tazemetostat, an oral EZH2 inhibitor, in 2020 for treating advanced epithelioid sarcoma in patients aged 16 and older [81] (Figure 2). In 2022, Japan's Pharmaceuticals and Medical Devices Agency approved valemetostat tosilate (DS‐3201, DS‐3201B), a novel dual EZH1/2 inhibitor, for treating relapsed/refractory adult T‐cell leukemia/lymphoma [82] (Figure 2). Chinese pharmaceutical companies are actively developing EZH2 inhibitors, including Hengrui Medicine's SHR2554 and Haihe Pharmaceutical's HH2853.
3.1.4. Isocitrate Dehydrogenase (IDH) Inhibitors
IDH catalyzes the conversion of isocitrate to α‐ketoglutarate and carbon dioxide in the tricarboxylic acid cycle. Mutant IDH produces 2‐hydroxyglutarate, which inhibits α‐ketoglutarate‐dependent epigenetic enzymes, leading to aberrant epigenetic landscapes in tumors. The FDA has approved several IDH inhibitors: enasidenib (2017) for relapsed/refractory (R/R) AML with IDH2 mutations [83], ivosidenib (2018) for R/R AML [84] and newly diagnosed elderly AML patients with IDH1 mutations (2022) [85], and olutasidenib (2022) for R/R AML with specific IDH1 mutations (Figure 2). Pharmaceutical companies continue to develop novel IDH inhibitors, with multiple candidates in preclinical and clinical development.
3.2. Epigenetic Drugs Enhance the Antitumor Effects of ICIs
3.2.1. Upregulation of Tumor‐Associated Antigen/Tumor‐Specific Antigen Expression in Tumor Cells Enhances Immune System Recognition
Epigenetic silencing is a key mechanism in the downregulation of tumor‐associated antigens and tumor‐specific antigens. Epigenetic drugs can reverse these silencing mechanisms, thereby reactivating tumor‐associated antigen/tumor‐specific antigen expression and enhancing immune system recognition of tumor cells. Multiple studies support this mechanism.
Inhibition of the G9a/DNA methyltransferase network induces tumor immunogenic cell death and enhances tumor immunogenicity, effectively converting “cold” tumors into “hot” tumors. Both DNA methyltransferase inhibitors and HDAC inhibitors induce immunogenic cell death by releasing and exposing tumor antigens, activating dendritic cells, promoting antitumor T‐cell cross‐priming, and potentially fostering a T helper 1‐like phenotype [86, 87]. The stimulator of interferon genes (STING) pathway in tumor cells plays a crucial role in immune surveillance. Epigenetic reprogramming modulates STING function through multiple mechanisms, including STING pathway activation, neoantigen generation, enhanced antigen presentation, and creation of a favorable inflammatory environment for immune cell activation and infiltration [88]. A nanomodulator combining zebularine (a DNA methyltransferase inhibitor), JQ1 (a BRD4 inhibitor), and CpG (a TLR9 agonist) upregulates tumor‐associated antigen expression, enhancing tumor immunogenicity and T‐cell infiltration [89]. HDAC inhibitors, including trichostatin A and valproic acid, enhance MHC class I antigen processing [90] and presentation by inducing expression of TAP, LMP, TAPBP, and B2M genes in melanoma and other tumors [91, 92, 93], thereby increasing tumor cell immunogenicity. In head and neck cancer studies, EZH2 inhibitors (GSK126 and EPZ6438) upregulate tumor antigen presentation, enhancing antitumor immunity [94].
These findings demonstrate that epigenetic modifications facilitate immune system recognition of tumor cells by modulating antigen expression and presentation, ultimately triggering more robust antitumor immune responses.
3.2.2. Epigenetic Regulation of Immune Checkpoint Molecules in the Tumor Microenvironment
Epigenetic modifications play a crucial role in regulating immune checkpoint gene expression and are therefore a key opportunity for alleviating immunosuppression within the tumor microenvironment. These modifications can modulate the transcriptional activity of immune checkpoint genes, thereby affecting the expression of checkpoint molecules and potentially reversing immunosuppressive effects. Recent studies have revealed several important mechanisms and therapeutic approaches in this area.
The nano‐regulator, CG‐J/ZL, demonstrates dual functionality by enhancing tumor immunogenicity while simultaneously downregulating PD‐L1 expression, effectively interrupting the PD‐1/PD‐L1 signaling pathway. In melanoma models, HDAC6 inhibition modulates STAT3 phosphorylation, resulting in reduced PD‐L1 expression [95].
Inhibitors targeting EZH2 or DNA methyltransferases have demonstrated efficacy in reducing CTLA‐4 gene expression. Notably, the combination of the HDAC6 inhibitor, ACY‐1215, with JQ1 produces synergistic enhancement of antitumor immune responses, primarily through the downregulation of FOXP3 target genes, including CTLA‐4 [96].
DNA methylation patterns at promoter regions typically correlate with gene expression suppression. This mechanism has been particularly well documented in the regulation of LAG‐3, where promoter methylation inhibits gene expression. Treatment with DNA methyltransferase inhibitors, such as 5‐azacytidine, can reduce methylation levels, thereby affecting LAG‐3 expression. This effect was demonstrated in melanoma cell line studies, where 5‐azacytidine treatment induced LAG‐3 gene expression in the A375 cell line, leading to enhanced immune cell antitumor activity [97].
In cervical cancer, there is a complex regulatory mechanism involving EZH2, H3K27me3, and DNMT3A in the coregulation of TIM‐3, an important costimulatory molecule. The application of epigenetic inhibitors targeting these regulatory proteins shows promise in reducing TIM‐3 expression and suppressing cancer progression [26].
These findings collectively demonstrate the potential of epigenetic‐modifying drugs as powerful tools in cancer immunotherapy. By targeting the epigenetic regulation of immune checkpoint molecules, these therapeutic approaches can help restore immune cell activity, reverse tumor immunosuppression, and inhibit tumor growth. This growing body of evidence supports the development of novel antitumor strategies targeting epigenetic mechanisms.
3.2.3. Epigenetic Regulation of Immunosuppressive Cells in the Tumor Microenvironment
The maintenance of immunosuppression by Tregs and MDSCs can be modulated through epigenetic mechanisms, offering promising therapeutic opportunities for enhancing antitumor immune responses.
3.2.3.1. Epigenetic Modulation of Tregs
Epigenetic modifications significantly influence Treg activity through various mechanisms. The class I‐specific HDAC inhibitor, entinostat, when administered at low doses, induces STAT3 acetylation in Tregs, leading to decreased FOXP3 transcription. This modification results in reduced immunosuppressive function [98]. Wang et al. demonstrated that the EZH2 inhibitor, CPI‐1205, can disrupt EZH2 activity in Tregs, promoting their phenotypic shift from an immunosuppressive to a proinflammatory state [60]. Consistent with these findings, studies in mouse models have shown that EZH2 gene knockdown in Tregs produces antitumor effects [99]. The histone demethylase, JMJD1C, has recently been identified as a key regulator of tumor‐infiltrating Tregs, with expression levels increasing during Treg infiltration and differentiation within tumor tissue. Conditional deletion of JMJD1C specifically in Tregs reduces tumor‐associated Treg populations and enhances T‐cell‐mediated antitumor immune responses. Mechanistically, JMJD1C regulates IFNγ expression in tumor Tregs through direct STAT3 demethylation. Notably, oral administration of the JMJD1C inhibitor, 193D7, significantly suppresses tumor growth in mouse models, with effects dependent on JMJD1C expression in tumor Tregs [100].
3.2.3.2. Epigenetic Control of MDSCs
Epigenetic mechanisms also play a vital role in regulating MDSC‐mediated immunosuppression. Studies in colorectal cancer have shown that 5‐azacytidine treatment reduces MDSC immunosuppressive function by decreasing ARG1 gene methylation and subsequent expression. ARG1 is essential for MDSC‐mediated immunosuppression; therefore, this intervention weakens MDSC's capacity to inhibit T cells and restores antitumor immune responses. p66a has also been identified as an epigenetic factor capable of modulating MDSC function through regulation of STAT3 activity [101]. Both in vivo and in vitro studies have demonstrated that IL‐6 significantly reduces p66a expression, indicating a crucial role for p66a in IL‐6‐mediated MDSC differentiation. HDAC11 has also emerged as another important epigenetic regulator of tumor‐associated MDSC expansion and function [102]. These findings establish epigenetic modification as a promising therapeutic strategy for reversing the immunosuppressive tumor microenvironment through regulation of Tregs and MDSC function.
3.2.4. Enhancing the Antitumor Effects of T Cells, NK Cells, and Macrophages
Epigenetic modification is a fundamental mechanism for regulating gene expression and plays a pivotal role in antitumor immune responses. These modifications can reverse the expression of inhibitory genes in T cells, NK cells, and macrophages while enhancing immune cell infiltration and cytotoxic activity within the tumor microenvironment. Furthermore, they amplify antitumor immune effects through modulation of cytokine secretion, establishing epigenetic modulation as a crucial strategy in cancer therapy.
3.2.4.1. Enhancing T‐Cell Effects
Epigenetic modifications are critical regulators of T‐cell‐mediated antitumor immune responses. Through the regulation of key genes, these modifications can alleviate inhibitory signaling pathways within T cells, thereby restoring and enhancing their antitumor capabilities.
Kang et al. demonstrated that targeted deletion of epigenetic regulators, DNMT3A, TET2, and ASXL1, enhances CD8+ T cell stemness, improving their proliferative capacity and antitumor efficacy under conditions of chronic antigen exposure and immunotherapy. Notably, ASXL1 knockout conferred sustained stemness and enhanced effector capabilities to T cells, resulting in improved tumor control [103]. Additionally, the loss of histone methyltransferases, MLL3 and MLL4, in tumor tissues reduces H3K4me1 and H3K27ac levels, suppressing GSDMD (Gasdermin D) expression. This epigenetic change impairs CD8+ T‐cell activation, diminishing the efficiency of antitumor immune responses.
The combination of epigenetic modulators with ICIs has demonstrated synergistic enhancement of T‐cell function and improved antitumor outcomes. A phase Ib study (NCT02635061) revealed that treatment of metastatic non‐small cell lung cancer (NSCLC) with ACY‐241 (an HDAC6 inhibitor) in combination with nivolumab increased tumor‐infiltrating cytotoxic T‐cell numbers post‐treatment [104]. Similarly, low‐dose decitabine (a DNA demethylating agent) combined with PD‐1 inhibitors maintained expression and activity of the AP1 family transcription factor, JunD, promoting CD8+ precursor exhausted T‐cell expansion and altering CD8+ T‐cell differentiation trajectories, ultimately achieving potent and durable antitumor activity [105]. Furthermore, combining the SUV39H1 inhibitor, ETP‐69, with anti‐PD‐1 therapy increased effector CD8+ T cell proportions and suppressed tumor growth, highlighting the potential of epigenetic‐modifying drugs to overcome tumor immune evasion.
3.2.4.2. Enhancing NK Cell Effects
Epigenetic modification has emerged as a promising strategy to enhance NK cell‐mediated antitumor effects. These modifications can improve NK cell tumor recognition and cytotoxicity by either suppressing inhibitory gene expression or upregulating activating genes.
In multiple myeloma, BET inhibitors increase NKG2D ligand expression, which is crucial for NK cell activation, thereby enhancing tumor cell susceptibility to NK cell‐mediated cytotoxicity [106]. Recent reviews have highlighted the therapeutic potential of combining epigenetic modulation of NKG2D ligand expression with NK cell‐based immunotherapy in AML and NSCLC [107]. Furthermore, the KDM5A enzyme‐mediated removal of H3K4me3 methylation marks near the SOCS1 gene reduces SOCS1 expression, attenuating its inhibitory effect on cytokine signaling and potentially promoting NK cell activation [108]. EZH2 inhibitors enhance NK cell cytotoxicity through multiple mechanisms. By suppressing H3K27 trimethylation, these inhibitors increase the expression of NK cell cytotoxicity‐associated genes, including the KLRK1 gene, which encodes NKG2D [109]. Additionally, EZH2 inhibitors upregulate NKG2D ligand expression on tumor cells, further enhancing NK cell‐mediated killing [110]. In xenograft models, EZH2 inhibition enhances NK cell antitumor responses, potentially through improved NK cell maturation regulated by epigenetic mechanisms and transcriptional plasticity.
3.2.4.3. Enhancing Macrophage Effects
Epigenetic modifications are a powerful approach to enhance macrophage‐mediated antitumor effects. These modifications can promote the polarization of macrophages from the pro‐tumor M2 phenotype to the antitumor M1 phenotype, thereby enhancing cytokine secretion and tumor cell phagocytosis capabilities.
In diffuse large B‐cell lymphoma, mutations in CREBBP/EP300 impair H3K27 acetylation, leading to reduced expression of FBXW7, a NOTCH pathway suppressor. This increases CSF1 and CCL2 expression, enhancing macrophage‐mediated immune responses [111]. In NSCLC, HDAC6 inhibitors enhance the costimulatory capacity of intratumoral macrophages by upregulating MHC class II and CD86 expression, promoting their antitumor effects [112]. The SRSF10/glycolysis/H3K18la axis establishes a positive feedback loop in tumor cells, and SRSF10 targeting may inhibit M2 macrophage polarization, potentially enhancing anti‐PD‐1 therapy efficacy in hepatocellular carcinoma [113]. Studies in mouse tumor models have demonstrated that targeting epigenetic regulators, such as CECR2, ALKBH5, and EZH2 through genetic or pharmacological approaches, can reduce tumor‐associated macrophage infiltration, attenuate immunosuppression, and impede tumor progression [46, 49, 114].
These findings indicate that epigenetic modification can enhance the antitumor capabilities of immune cells through various pathways, thereby impeding tumor progression, offering new hope for cancer treatment.
4. Clinical Applications of Epigenetic Immunotherapy
Epigenetic regulators play a pivotal role in tumor immunity through their diverse effects on both tumor cells and immune system components. These regulators can alter the interactions between tumor cells and immune cells, influencing antigen presentation, immune checkpoint expression, and immune cell infiltration and activity. Targeted intervention of these regulators holds promise for enhancing antitumor immune responses. In recent years, immunotherapy has become a major focus and emerging trend in the field of cancer epigenetic therapy [115]. A growing body of research has demonstrated that the combination of immunotherapy and epigenetic therapy can show significant initial efficacy in both hematological malignancies and solid tumors, suggesting a synergistic effect between these two therapeutic strategies. Furthermore, the overall safety profile of this combined approach remains manageable, offering a novel perspective to address current challenges in cancer treatment. Here, we comprehensively summarize the synergistic effects and safety data of combined epigenetic and immunotherapy approaches (Table 1).
Table 1.
Epigenetic Immunotherapy alone or combined with targeted therapies in pan‐cancer research.
| NCT number | Agent | Immunotherapy | Phase | Cancer type |
|---|---|---|---|---|
| NCT05320640 | Chidamide, Decitabine | anti‐PD1/PD‐L1/CTLA4 antibodies | I/II | R/R Non‐Hodgkin lymphoma and Advanced Solid Tumors |
| NCT06393361 | Chidamide, Decitabine | anti‐PD‐1 antibody | II | Classical Hodgkin Lymphoma |
| NCT04337606 | Chidamide, Decitabine | Camrelizumab | I/II | Non Hodgkin Lymphoma |
| NCT04233294 | Chidamide, Decitabine | Camrelizumab | II | Hodgkin Lymphoma |
| NCT04514081 | Chidamide, Decitabine | Camrelizumab | II | Classical Hodgkin Lymphoma |
| NCT05355051 | Azacitidine | Pembrolizumab | II | Hodgkin's Lymphoma |
| NCT06190067 | Azacitidine | anti‐PD‐1 antibody | II | Refractory Classic Hodgkin Lymphoma, Relapsed Classic Hodgkin Lymphoma |
| NCT04913922 | 5‐Azacytidine | Nivolumab | Acute Myeloid Leukemia | |
| NCT04233294 | Chidamide, Decitabine | Camrelizumab | II | Hodgkin Lymphoma |
| NCT05816746 | Decitabine | anti‐PD‐1 antibody | II | R/R Diffuse Large B‐Cell Lymphoma |
| NCT05137886 | Decitabine | anti‐PD‐1 antibody | II | Relapsed or Refractory Classic Hodgkin's Lymphoma |
| NCT05320640 | Chidamide, Decitabine | anti‐PD1/PD‐L1/CTLA4 antibodies | I/II | Relapsed/Refractory Non‐Hodgkin Lymphoma |
| NCT04512534 | Chidamide | anti‐PD‐1 antibody | II | Peripheral T‐cell Lymphoma |
| NCT03240211 | Decitabine | Pembrolizumab | I | Peripheral T‐Cell Lymphoma, Cutaneous T‐Cell Lymphoma |
| NCT05772273 | Azacitidine | Camrelizumab | Not Applicable | Acute Myeloid Leukemia |
| NCT04994210 | Chidamide | Sintilimab | II | Extranodal Natural Killer/T‐cell Lymphoma |
| NCT03838042 | Entinostat | Nivolumab | I/II | CNS Tumor, Solid Tumor |
| NCT05317000 | 5‐Azacytidine | Nivolumab | II | Squamous Cell Carcinoma of Head and Neck |
| NCT06022757 | XNW5004 | pembrolizumab | I/II | Carcinoma, Cervical Cancer, Non‐small Cell Lung Cancer, Other Solid Tumors, Prostate Cancer, Small‐cell Lung Cancer, quamous Cell Carcinoma of Head and Neck, Urothelial Carcinoma |
| NCT06454448 | Decitabine | Adebrelimab | I/II | Metastatic Pancreatic Cancer |
| NCT04471974 | ZEN‐3694 | Pembrolizumab | II | Metastatic Castration‐Resistant Prostate Cancer |
| NCT06466798 | 5‐Azacytidine | Nivolumab | I | Central Nervous System Malignancies, Recurrent Ependymoma, Recurrent Medulloblastoma |
| NCT03969446 | Decitabine | Pembrolizumab | I | Acute Myeloid Leukemia, Myelodysplastic Syndrome, Recurrent Acute Myeloid Leukemia, Recurrent Myelodysplastic Syndrome, Refractory Acute Myeloid Leukemia, Refractory Myelodysplastic Syndrome |
Note: Hematological malignancies (yellow boxes) in the middle, solid tumors (gray boxes) on the top.
4.1. Hematological Malignancies
4.1.1. Classical Hodgkin Lymphoma (cHL)
The treatment of relapsed/refractory classical Hodgkin lymphoma (R/R cHL) remains a significant clinical challenge. For these patients, second‐line therapy typically involves high‐dose chemotherapy combined with autologous stem cell transplantation. However, the relapse rate following transplantation remains high. In recent years, immunotherapy, epigenetic drugs, and antibody‐drug conjugates have offered new hope for R/R cHL patients.
In a prospective, open‐label phase II clinical trial (NCT02961101), Nie et al. compared the use of camrelizumab alone versus camrelizumab combined with low‐dose decitabine in patients who had not previously received anti‐PD‐1 therapy. At a median follow‐up of 14.9 months, the complete response (CR) rates were 32% and 71% for the monotherapy and combination therapy groups, respectively [116]. At a median follow‐up of 34.5 months, the CR rate in the combination therapy group further improved (79% vs. 32%), and patients in this group also experienced longer progression‐free survival (PFS) (35.0 vs. 15.5 months). Exploratory analysis showed that an increase in circulating central memory T cells was associated with clinical response and longer PFS, leading to the hypothesis that these cells might serve as a potential biomarker for cHL immunotherapy [117]. Results from this study were later updated for patients with R/R cHL who had previously received anti‐PD‐1 therapy and were treated with decitabine and camrelizumab (NCT02961101 and NCT03250962). Among 50 patients, the ORR was 52%, with a CR rate of 36%. Compared with anti‐PD‐1 monotherapy, the combination therapy resulted in longer PFS. Mechanistic insights revealed that after decitabine and camrelizumab treatment, the proportion of peripheral CCR7 + CD45RA central memory T cells increased and persisted in patients who achieved partial responses (PRs) or CRs [118]. However, when tumor progression occurred, the ratio of central memory T cells to total CD8+ or CD4+ cells dropped below baseline levels. At a median follow‐up of 5.3 years, the median relapse‐free survival for 87 patients who achieved CR with combination therapy was 4.5 years, with a 2‐year relapse‐free survival rate of 70%. The median PFS was 4.8 years, with a 2‐year PFS rate of 78%. Exploratory data indicated that patients with higher levels of IL‐6 and LDH had poorer prognoses [119]. In these series of clinical studies, the low‐dose decitabine combination therapy did not result in severe adverse effects, and patients tolerated the treatment well.
In recent years, the research team mentioned above has continued to advance research in the field of epigenetic immunotherapy. Beyond single epigenetic drug combinations with immunotherapy, they have further explored dual epigenetic drug regimens and other target‐based therapies combined with epigenetic immunotherapy. For example, a phase II clinical trial (NCT04233294) has been conducted to evaluate the safety and efficacy of a three‐drug combination regimen consisting of chidamide, decitabine, and the anti‐PD‐1 antibody, camrelizumab, in patients with R/R cHL who had previously been treated with decitabine and camrelizumab therapy. Among 52 patients, 49 (94%) achieved an objective response, with 26 (50%) achieving CR. The median PFS was 29.4 months. Mechanistic analysis indicated that the three‐drug combination not only targeted CD30+ Hodgkin‐Reed‐Sternberg (HRS) tumor cells but also effectively killed CD30‐ HRS tumor cells, which had strong tumor stemness [120]. Additionally, they further reported the results of SHR‐1701 (an anti‐PD‐L1/TGFβRII agent) combined with SHR2554 (an EZH2 inhibitor) at the 2023 American Society of Clinical Oncology Annual Meeting. In this study, 16 patients with R/R cHL were treated, all of whom had previously received anti‐PD‐1/PD‐L1 antibody therapy, and 15 (93.8%) had undergone prior hypomethylating therapy, with decitabine or chidamide. Among the 14 evaluable cHL patients, the ORR was 100%, with a CR rate of 7.1%. Safety data indicated that the most common adverse events included decreased platelet count, anemia, and nausea. These findings indicate that the combination of immunotherapy with targeted therapies may offer promising new treatment options for R/R cHL patients.
4.1.2. T‐Cell Lymphoma
Peripheral T‐cell lymphoma represents a heterogeneous group of malignancies originating from mature T lymphocytes that comprises 20%–30% of all lymphomas. Peripheral T‐cell lymphoma encompasses multiple pathological subtypes, and the prognosis remains poor for most variants, with the notable exception of ALK‐positive anaplastic large cell lymphoma. Traditional approaches using salvage chemotherapy combined with autologous or allogeneic hematopoietic stem cell transplantation have shown limited efficacy, particularly in R/R cases. In this context, epigenetic immunotherapy has emerged as a promising therapeutic strategy.
A pioneering single‐arm, open‐label, multicenter clinical trial (NCT03820596) by Gao et al. evaluated the combination of sintilimab with chidamide in patients with R/R extranodal natural killer/T‐cell lymphoma. Initial results demonstrated an ORR of 58.3% among 36 evaluable patients, with 16 patients (44.4%) achieving CR [121]. Subsequent follow‐up data from this phase Ib/II study revealed an ORR of 59.5% in the intention‐to‐treat population (n = 37), with a CR rate of 48.6%. The median duration of response, PFS, and overall survival (OS) reached 25.3, 23.2, and 32.9 months, respectively. Primary adverse events of grade ≥ 3 included neutropenia (28.9%) and thrombocytopenia (10.5%). Notably, biomarker analyses indicated that combining dynamic plasma circulating tumor DNA and Epstein‐Barr virus DNA measurements provided enhanced accuracy in assessing treatment efficacy and patient prognosis [122]. Additional evidence supporting epigenetic immunotherapy comes from a phase II study in R/R T‐cell lymphoma that investigated the combination of pembrolizumab and romidepsin. This study achieved an ORR of 50%, including five CRs and two PRs with durable efficacy. Higher PD‐L1 expression levels correlated with improved outcomes [123]. Furthermore, a phase I clinical trial (NCT03240211) examining the triple combination of pembrolizumab, decitabine, and pralatrexate in R/R peripheral T‐cell lymphoma and cutaneous T‐cell lymphoma reported remarkable results, with a 100% ORR among 13 patients and a median duration of response of 18 months [124].
4.1.3. Acute Myeloid Leukemia (AML)
The management of AML has traditionally relied on intensive chemotherapy followed by allogeneic hematopoietic stem cell transplantation for consolidation. However, this approach presents significant challenges for elderly patients and those with comorbidities who often cannot tolerate intensive treatment protocols. Recent advances in targeted therapies, epigenetic drugs, and immunotherapies have opened new therapeutic avenues for these patients.
A clinical trial (NCT02845297) investigating the combination of pembrolizumab and azacitidine in newly diagnosed AML patients aged ≥ 65 years who were unsuitable for or declined intensive chemotherapy showed promising results. Among 17 evaluable patients, 47% (8/17) achieved CR or CR with incomplete hematological recovery (CRi), while 12% (2/17) achieved PR. The median OS for this frontline treatment reached 13.1 months. The study also included a cohort of R/R AML patients, where among 29 evaluable patients, 14% achieved CR/CRi, 4% achieved PR, 14% showed hematological improvement, and 24% maintained stable disease (SD). With a median follow‐up of 14.9 months, the median OS was 10.8 months for the entire cohort, extending to 13.9 months for patients achieving CR/CRi/PR/SD and 17.2 months for those with CR/CRi/PR. Treatment was generally well tolerated, with the most common grade ≥ 2 immune‐related adverse events including rash, hepatitis, and pneumonitis [125]. Another significant open‐label phase II study evaluated nivolumab combined with azacitidine in 70 R/R AML patients. The treatment demonstrated good tolerability, with no discontinuations caused by myelosuppression or immune toxicity. The ORR reached 33%, including 22% achieving CR/CRi, one patient showing a PR, and seven showing hematological improvement. Notably, pretreatment bone marrow aspiration and peripheral blood CD3 percentages emerged as potential biomarkers for favorable outcomes [126]. Lindblad et al. demonstrated comparable clinical efficacy between hypomethylating agents combined with pembrolizumab and the azacitidine‐nivolumab combination in R/R AML patients [127]. Furthermore, a non‐randomized, prospective phase II study by Daver et al. investigated dual and triple combinations: azacitidine plus nivolumab, and azacitidine plus nivolumab and ipilimumab. In the dual‐therapy group (n = 70), the ORR was 33%, comprising 6% CR, 16% CRi/CRo (CR with ongoing recovery), 1% PR, 10% hematological improvement lasting over 6 months, and 11% SD. The triple‐therapy group (n = 25) achieved an ORR of 44%, including 4% CR, 32% CRi/CRo, 8% hematological improvement lasting over 6 months, and 16% SD. The triple combination regimen demonstrated encouraging CR/CRi rates and OS, with median OS exceeding 10 months in high‐risk R/R AML patients [128]. These findings indicate that while single immunotherapy combined with epigenetic‐modifying drugs provides clinical benefit in AML patients, dual immunotherapy approaches may offer enhanced efficacy.
4.1.4. Myelodysplastic Syndrome
Hypomethylating agents are standard treatments for high‐risk myelodysplastic syndrome; however, some patients develop resistance or experience relapse. Allogeneic hematopoietic stem cell transplantation remains the only potentially curative option, though its application is limited by age, eligibility criteria, and associated complications. Recent research has explored combining immunotherapy with hypomethylating agents for R/R high‐risk myelodysplastic syndrome patients.
In a phase II clinical trial (NCT03094637), Chien et al. evaluated azacitidine plus pembrolizumab in two cohorts. The treatment–naïve cohort (n = 17) achieved an ORR of 80% that included three CRs, seven bone marrow CRs, and one hematological improvement [129]. The hypomethylating agent‐failure cohort (n = 20) showed an ORR of 25% with a 5% CR rate and a median OS of 5.8 months at a median follow‐up of 6.0 months. Primary toxicities included pneumonia (32%), arthralgia (24%), and constipation (24%). These results indicate that azacitidine plus pembrolizumab demonstrates tolerability and potential antitumor activity, warranting further investigation as a first‐line therapy [130]. Another trial (NCT02599649) investigating azacitidine plus lirilumab in eight high‐risk myelodysplastic syndrome patients reported promising results: two patients achieved CR (25%) and four showed complete bone marrow response (50%). Notably, no patients discontinued treatment due to adverse events [131].
Recent investigations have expanded to include novel combination approaches. A study evaluating triple therapy combinations demonstrated that avelumab plus azacitidine plus venetoclax (a BCL2 inhibitor) achieved an ORR of 25%, while avelumab plus azacitidine plus gemtuzumab ozogamicin (CD33‐targeted) showed an ORR of 20%. Both combinations demonstrated acceptable safety profiles [132]. These emerging data support the continued exploration of epigenetic immunotherapy combinations in myelodysplastic syndrome treatment.
4.2. Solid Tumors
4.2.1. Non‐Small Cell Lung Cancer
Recent advances in precision medicine and immunotherapy have transformed the treatment landscape for NSCLC, significantly improving patient survival rates and quality of life. However, substantial challenges persist, including drug resistance, suboptimal immunotherapy response rates, and limited long‐term survival in advanced disease. Novel combination strategies incorporating epigenetic modifiers with immunotherapy are showing promise in addressing these challenges.
A notable case series demonstrated the potential of combining low‐dose decitabine with camrelizumab in three advanced NSCLC patients with traditionally unfavorable immunotherapy biomarkers (low tumor mutational burden, low microsatellite instability, and Human leukocyte antigen heterozygosity loss). The combination showed favorable responses with manageable toxicity, indicating that low‐dose decitabine may enhance anti‐PD‐1 antibody sensitivity in patients who might otherwise be poor candidates for immunotherapy [133]. In a phase Ib study (NCT02635061), the combination of citarinostat (a selective HDAC6 inhibitor) with nivolumab demonstrated clinical benefit in eight of 13 evaluable patients with metastatic NSCLC, including one CR, four PRs, and three SDs. Mechanistic studies revealed increased numbers of tumor‐infiltrating immune cells following treatment, providing insight into the basis of the observed clinical responses [104]. A randomized phase II trial (NCT02638090) compared pembrolizumab monotherapy versus pembrolizumab plus vorinostat in NSCLC patients. Among 47 evaluable patients, the combination arm achieved a significantly higher ORR (66.7%) compared with pembrolizumab alone (33.3%). Notably, patients with low pretreatment tumor‐infiltrating lymphocyte counts showed enhanced benefit from the combination approach, indicating a potential role for this strategy in expanding the population of patients who may benefit from immunotherapy [134]. Another phase Ib multicenter study evaluated citarinostat plus nivolumab in advanced NSCLC patients previously treated with HDAC or ICIs. At the maximum tolerated dose of 360 mg citarinostat, the disease control rate reached 80% (three PR, one SD) among five patients. Biomarker analyses revealed transient increases in histone and tubulin acetylation levels and elevated CD3+ T cell counts, indicating potential predictive markers for treatment response [135]. These findings highlight the importance of biomarker analysis in clinical trials to better understand treatment mechanisms and identify patients most likely to benefit from combination approaches.
The collective evidence from these studies demonstrates the potential of epigenetic‐immunotherapy combinations to enhance treatment outcomes in NSCLC, particularly for patients who may have limited options or suboptimal responses to standard immunotherapy approaches. Continued investigation of these combinations, along with careful attention to biomarker development and patient selection, will be crucial for optimizing their clinical application.
4.2.2. Melanoma
Significant advances in melanoma treatment over the past decade, particularly through immunotherapy and targeted therapy, have markedly improved patient survival rates and quality of life. However, challenges persist, especially for advanced‐stage patients and those who develop drug resistance. Recent clinical trials have explored various combination approaches to address these challenges.
A phase II study (NCT02816021) investigating the combination of azacitidine and pembrolizumab in treatment‐naïve metastatic melanoma patients demonstrated promising preliminary results, with an ORR of 55% (five PRs among nine patients) and favorable tolerability [136]. The ENCORE‐601 open‐label trial (NCT02437136) evaluated entinostat in combination with pembrolizumab in 53 patients, a substantial proportion (70%) of whom had received prior pembrolizumab treatment. The trial achieved an ORR of 19%, with comparable efficacy observed in both pembrolizumab‐naïve and previously treated patients. Preliminary biomarker analyses suggested that entinostat restored inflammatory responses within the tumor microenvironment [137, 138], potentially explaining the efficacy of PD‐1/PD‐L1 rechallenge. For unresectable melanoma, the phase Ib NIBIT‐M4 study (NCT02608437) evaluated the combination of guadecitabine and ipilimumab in 19 patients. This study reported an ORR of 26% (two CRs and three PRs) and a disease control rate of 42%. The median PFS was 5.6 months, with a median OS of 26.2 months. The 1‐ and 2‐year OS rates were 80% and 56%, respectively. Myelosuppression was observed as the primary grade 3–4 adverse events but, notably, no febrile neutropenia occurred [139]. In another phase Ib trial (NCT03565406), Weber et al. investigated the combination of mocetinostat with nivolumab and ipilimumab in treatment‐naïve metastatic melanoma patients. Among 10 patients, the treatment achieved an impressive ORR of 70% at 16 months of follow‐up, with two CRs and five PRs [140]. However, the occurrence of grade 3–4 immune‐related adverse events in seven patients indicated that dose modification may be necessary for future investigations.
Collectively, these studies demonstrate that melanoma patients can derive substantial clinical benefit from epigenetic immunotherapy combinations; a strategy that may also address resistance to ICIs.
4.2.3. Renal and Bladder Carcinoma
The therapeutic landscape for advanced renal cell carcinoma has evolved significantly, with increasing emphasis on combining targeted therapy with immunotherapy. Similarly, immunotherapy has gained prominence in bladder cancer treatment, particularly for advanced or metastatic disease. The integration of epigenetic modifiers with immunotherapy presents an opportunity to enhance therapeutic outcomes for both malignancies.
A phase Ib/II study (NCT03308396) evaluating durvalumab and guadecitabine in 42 patients with advanced renal cell carcinoma demonstrated encouraging results, with nine PR (22%) and twenty‐five SD (61%). At a median follow‐up of 20.1 months, 66% of patients achieved clinical benefit, with a median PFS of 17 months. The treatment demonstrated good tolerability, with asymptomatic neutropenia (38.1%) being the primary adverse event associated with guadecitabine, and asymptomatic lipase elevation (11.9%) with durvalumab. Mechanistic studies revealed that reductions in Tregs and MDSCs correlated with improved outcomes, while increased T helper 17 subsets were associated with immune‐related adverse events [141]. A separate phase Ib trial investigating vorinostat combined with pembrolizumab showed efficacy in both treatment‐naïve and PD‐1/PD‐L1‐resistant urothelial carcinoma and renal cell carcinoma, with median PFS of 2.8 and 5.2 months, respectively. The combination also demonstrated activity in prostate cancer (median PFS of 3.5 months) with favorable tolerability across all three cohorts [142].
Several ongoing trials are further investigating epigenetic‐immune combinations in urological malignancies. These include NCT01038778, a phase I/II trial evaluating high‐dose interleukin 2 (aldesleukin) with entinostat in metastatic renal cell carcinoma, and NCT02619253, a phase I/Ib study of pembrolizumab plus vorinostat in advanced renal cell carcinoma or urothelial carcinoma. While current evidence supports the clinical potential of epigenetic‐immune combinations in urological malignancies, additional clinical trials are warranted to fully establish their safety and efficacy profiles.
4.2.4. Epithelial Ovarian Cancer
Advanced ovarian cancer treatment is frequently compromised by chemotherapy resistance, particularly in late‐stage disease where conventional treatment regimens show limited efficacy. Epigenetic‐immune combination therapy has emerged as a promising therapeutic strategy to address this challenge. Recent clinical trials have investigated various combinations to evaluate their potential benefits.
In an open‐label phase II multi‐cohort study (NCT02811497), the combination of azacitidine with durvalumab was evaluated in 28 patients with platinum‐resistant ovarian cancer who had not received prior anti‐PD‐1 therapy. The disease control rate was 7.1%, with a median PFS of 1.9 months and median OS of 5 months [143]. More encouraging results emerged from a phase II trial (NCT02901899) conducted by Chen et al. that investigated guadecitabine combined with pembrolizumab in R/R ovarian cancer. Among 35 evaluable patients, three achieved PR (8.6%) and eight achieved SD (22.9%), yielding a clinical benefit rate of 31.4% with a median duration of 6.8 months. Molecular analyses revealed significant insights into treatment efficacy. Posttreatment methylomic and transcriptomic analyses demonstrated activation of antitumor immunity. High‐dimensional immune profiling of peripheral blood mononuclear cells identified higher frequencies of naïve and central memory CD4 T cells and classical monocytes in patients achieving durable benefit. Furthermore, improved outcomes correlated with higher densities of CD8 T cells and CD20 B cells in tumors, along with the presence of tertiary lymphoid structures [144].
These findings indicate potential efficacy of epigenetic‐immune combination therapy in converting immunologically “cold” ovarian tumors to more responsive states; however, larger validation studies are warranted.
4.2.5. Colorectal Cancer
A status of microsatellite stable or proficient mismatch repair characterizes over 95% of late‐stage metastatic colorectal cancers. These tumors typically demonstrate poor response to single‐agent immunotherapy, with response rates to PD‐1 or PD‐L1 antibodies ranging from 0% to 10%. Epigenetic‐immune combination therapy strategies have shown promise in sensitizing these “cold” tumors to ICI treatment.
A clinical investigation of combined HDAC and/or DNA methyltransferase inhibitors with immune checkpoint therapy in 27 patients with advanced proficient mismatch repair colorectal cancer demonstrated a median PFS of 2.79 months and median OS of 9.17 months. Notable outcomes included one patient achieving a durable PR lasting approximately 19 months with combined 5‐azacitidine, romidepsin, and pembrolizumab, while another maintained SD for 2.8 months with romidepsin and pembrolizumab [145]. The CAPability‐01 study, a randomized, open‐label, multicenter, phase II trial, evaluated chidamide combined with sintilimab with or without bevacizumab in 48 patients with microsatellite stable/proficient mismatch repair metastatic colorectal cancer. This trial reported an 18‐week PFS rate of 43.8% and a median PFS of 3.7 months. The ORR, disease control rate, and duration of response were 29.2%, 56.3%, and 12.0 months, respectively. Common adverse events included proteinuria, thrombocytopenia, neutropenia, anemia, leukopenia, and diarrhea. The triple combination demonstrated superior efficacy compared with the double combination, with chidamide playing a crucial role in transforming “cold” tumors into “hot” tumors [146]. These results demonstrate that epigenetic‐immune combination therapy can effectively modify the tumor microenvironment of immunologically “cold” tumors, enabling enhanced efficacy of ICIs.
4.2.6. Head and Neck Tumors
Head and neck squamous cell carcinoma presents significant therapeutic challenges because of its high recurrence and metastasis rates, along with frequent chemotherapy resistance. This has necessitated the exploration of alternative treatment approaches, including immunotherapy and gene therapy combinations. Preliminary clinical evidence has shown promising results.
In a cohort of 25 patients with recurrent or metastatic head and neck squamous cell carcinoma, the combination of pembrolizumab and vorinostat yielded eight PR (32%) and five SD (20%) outcomes. The treatment achieved a median OS of 12.6 months and a median PFS of 4.5 months. A parallel cohort of 25 patients with salivary gland cancer demonstrated four PR (16%) and fourteen SD (56%) outcomes, with a median OS of 14 months and a median PFS of 6.9 months. Safety analyses revealed that 56% of head and neck squamous cell carcinoma patients and 52% of salivary gland cancer patients experienced adverse events of any grade [147]. While these findings indicate therapeutic potential for epigenetic‐immune combination therapy in specific head and neck tumors, additional research is required to validate clinical efficacy and optimize treatment protocols.
4.3. Epigenetic Immunotherapy Reverses ICI Resistance and Transforms Cold Tumors Into Hot Tumors
Patients with cHL, NSCLC, and melanoma in whom cancer progressed despite ICI therapy can still benefit from salvage regimens combining epigenetic modulation with immunotherapy (decitabine plus pembrolizumab, citarinostat plus nivolumab, or entinostat plus pembrolizumab) can still bring clinical benefits. Clinical trial results of these regimens show that the ORRs for cHL and melanoma are 52% [118] and 19% [137, 138], respectively, and the disease control rate for NSCLC is 80% [135]. Mechanistic investigations indicate that epi‐immunotherapy in cHL may exert antitumor effects through multiple pathways. For instance, in ICI‐resistant populations, exhausted T cells exhibit distinct epigenetic signatures that likely impair the durable antitumor efficacy of ICI. Low‐dose decitabine remodels the epigenetic landscape of both tumor and immune cells, enhancing T‐cell activation, promoting infiltration of CD8+ and CD4+ T cells, and potentially restoring therapeutic responsiveness [118]. In NSCLC, epi‐immunotherapy elevates histone and tubulin acetylation, amplifies cytokine production, and enriches tumor‐infiltrating cytotoxic T cells while reducing immunosuppressive NK cell populations, collectively fostering a more robust antitumor response [135]. In melanoma, epi‐immunotherapy may recalibrate the tumor microenvironment by suppressing protumor inflammation and restoring antitumor immune activation, which could underpin successful PD‐1/PD‐L1 inhibitor rechallenge [137, 138]. These observations collectively suggest that epi‐immunotherapy combinations may overcome ICI resistance and enhance rechallenge efficacy (Figure 3), offering novel therapeutic strategies for ICI‐refractory malignancies.
Figure 3.

Mechanisms and effects of epi‐immunotherapy in combating tumors. Epigenetic modulation exerts antitumor effects primarily by regulating immune cell infiltration and functional activity within TME. Epi‐immunotherapy reverses immune checkpoint inhibitor resistance. Epi‐immunotherapy transforms cold tumors into hot tumors. DC, dendritic cell; MDSC, myeloid‐derived suppressor cell; TAA, tumor‐associated antigen; TAM, tumor‐associated macrophages.
Importantly, epi‐immunotherapy strategies can reprogram immunologically “cold” tumors into “hot” immunogenic environments (Figure 3). For example, in ICI‐insensitive NSCLC patients, decitabine combined with PD‐1 blockade induces clinical responses, extending benefits to traditionally ICI‐ineligible populations. Similarly, ovarian and colorectal cancers—typically classified as immunologically “cold” and resistant to ICIs—benefit from epi‐immunotherapy. Mechanistic studies in ovarian cancer demonstrate that guadecitabine plus pembrolizumab activates antitumor immunity. High‐dimensional immune analysis of peripheral blood mononuclear cells reveals that durable responders exhibit elevated frequencies of naïve/central memory CD4+ T cells and classical monocytes. Pretreatment enrichment of CD8+ T cells, CD20+ B cells, and tertiary lymphoid structures within tumors further predicts clinical benefit. In colorectal cancer, responders to chidamide, sintilimab, and bevacizumab display increased numbers of intratumoral CD8+ T cells, cytotoxic lymphocytes, and monocyte lineage cells, accompanied by reduced numbers of B lineage cells, endothelial cells, and fibroblasts. Upregulated immune‐related pathways include interferon‐gamma response, allograft rejection, inflammatory response, and interferon alpha response, complemented by enhanced class‐I/II human leukocyte antigen signal, cytokine activity, and cytotoxic effector functions. These data collectively indicate that epi‐immunotherapy enhances antigen presentation and effector cell potency, effectively converting the tumor microenvironment from “cold” to “hot.” Therefore, even historically “cold” tumors may become amenable to ICI therapy through epigenetic modulation, creating transformative therapeutic opportunities for refractory malignancies.
4.4. Cancer‐Type‐Specific Efficacy of Epi‐Immunomodulation Therapies
Epi‐immunotherapy combinations, which synergistically modulate epigenetic pathways and immune responses, have demonstrated promising results in various cancers, most notably in cHL and multiple myeloma. However, for most other tumors, especially solid tumors, breakthrough efficacy has remained elusive. This suggests the existence of cancer‐type‐specific variations in epi‐immunotherapy efficacy. Although the underlying mechanisms remain incompletely understood, several key factors may contribute to this heterogeneity. First, tumor‐type specificity: (1) Mutational Burden and Immunogenicity. Tumors with higher mutational burdens generate more tumor‐specific antigens, increasing the likelihood of eliciting robust immune responses. Epigenetic drugs enhance tumor immunogenicity by modulating gene expression, thereby improving immunotherapy efficacy. However, variations in mutational burden and immunogenicity across tumor types result in variable therapeutic outcomes. (2) Epigenetic Regulatory Networks. Distinct epigenetic landscapes across tumor types influence drug sensitivity. For instance, hematological malignancies often harbor targetable driver mutations, whereas solid tumors with complex chromatin remodeling defects may require tailored epigenetic targeting strategies. Specific epigenetic agents selectively modify certain pathways, leading to cancer‐type‐specific efficacy disparities. Second, tumor microenvironment features: (1) Immune Cell Composition. “Hot” tumors (e.g., melanoma), characterized by abundant immune cell infiltration, generally demonstrate superior responses to ICIs and epigenetic combinations. Conversely, “cold” tumors (e.g., prostate cancer), dominated by immunosuppressive cells, show limited efficacy. These observations indicate that cancer types demonstrating advantages in epi‐immunotherapy should also possess “hot” tumor characteristics, leading to variations in treatment efficacy across different cancer types. (2) Immune Checkpoint Expression. Variable expression of immune checkpoint molecules across cancers contributes to heterogeneous outcomes. For example, cHL with near‐universal PD‐L1 expression responds robustly to HDAC inhibitor and anti‐PD‐1 combination, while PD‐L1‐negative glioblastoma derives minimal benefit. Epi‐immunotherapy combinations may also be influenced by the expression of immune checkpoint molecules, leading to differences in efficacy between cancer types. (3) Metabolic Diversity. Hypoxia‐driven lactate accumulation in the tumor microenvironment promotes histone lactylation, sustaining T‐cell exhaustion and inhibiting antitumor effects. Tumors with hyperactive lactate dehydrogenase A (e.g., triple‐negative breast cancer) are particularly resistant to epi‐immunotherapy because of metabolic reprogramming. The metabolic landscape of tumors varies significantly across cancer types, contributing to differential responses to epi‐immunotherapy. Third, Dosage and Timing: High‐dose cytotoxic decitabine versus immune‐activating low‐dose decitabine, as well as concurrent versus sequential regimens, result in inconsistent outcomes. The efficacy of epigenetic drugs is critically influenced by their dose and timing of administration, which determine systemic exposure and pharmacodynamic effects.
The efficacy of epi‐immunotherapy is influenced by multiple factors, and while the above‐mentioned factors may be relevant, further clinical trials are needed to validate them. It is essential to delve deeper into the mechanisms of action of epi‐immunotherapy against tumors, identify more suitable epigenetic drugs, determine the cancer types that are most responsive to epi‐immunotherapy, and explore effective biomarkers to guide clinical outcomes, thereby enhancing therapeutic efficacy.
4.5. Ongoing Clinical Trials in Epigenetic Immunotherapy
Recent advances in cancer treatment have highlighted the critical role of both epigenetic abnormalities and ICIs in cancer progression and therapy. The intersection of these approaches—combining epigenetic therapy with immunotherapy—has shown particular promise in clinical settings. This combination strategy has demonstrated notable success across multiple cancer types, with especially promising results in hematological malignancies. Our review of clinical trials registered with clinicaltrials.gov between January 2020 and December 2024 reveals numerous active investigations exploring this therapeutic approach in both solid tumors and blood cancers (Table 2).
Table 2.
Registered and actively recruiting clinical trials in epigenetic immunotherapy (screened from clinicaltrials.gov between January 1, 2020, and December 30, 2024).
| Cancer type | Epi‐immunotherapy | Outcomes | Refs. | Other targets combined with Epi‐immunotherapy | Outcomes | Refs. | |
|---|---|---|---|---|---|---|---|
| Hematologic Malignancies | cHL | Decitabine plus camrelizumab |
Naïve PD‐1: CR% 79%,mPFS 35M; Received anti‐PD‐1: ORR 52%, CR% 36% |
Chidamide, decitabine plus camrelizumab, SHR‐1701 (an anti‐PD‐L1/TGF‐βRII agent) plus SHR2554 (an EZH2 inhibitor) |
ORR 94%, CR%50%; ORR 100%, CR%7.1% |
NR |
|
| T‐Cell Lymphoma |
Chidamide plus sintilimab, romidepsin plus pembrolizumab |
ORR 58.3%, CR% 44.4%; ORR 50%, CR% 35.7% |
Pembrolizumab, decitabine plus pralatrexate | ORR 100%; DOR 18M | |||
| Acute Myeloid Leukemia |
Azacitidine plus pembrolizumab, azacitidine plus nivolumab, decitabine plus pembrolizumab, azacitidine plus nivolumab |
ORR 18%,CR% 14%, mOS 10.8 M; ORR 33%,CR% 22%; NR; ORR 33%, CR% 6% |
Azacitidine plus nivolumab and ipilimumab | ORR 44%,CR% 4%, SD%16% | 128 | ||
| Myelodysplastic Syndrome |
Azacitidine plus pembrolizumab, azacitidine plus pembrolizumab, azacitidine plus lirilumab |
ORR 80%, CR% 17.6%,bone marrow CR% 41.2%; ORR 25%, CR% 5%, OS 5.8M; CR% 25%,bone marrow CR% 50% |
Avelumab plus azacitidine plus venetoclax (BCL‐2 inhibitor) avelumab plus azacitidine plus gemtuzumab ozogamicin (CD33‐targeted) |
ORR 25% ORR 20% |
132 | ||
| Solid Tumors | Non‐Small Cell Lung Cancer |
Decitabine plus camrelizumab, citarinostat plus nivolumab, vorinostat plus pembrolizumab, citarinostat plus nivolumab |
NR; ORR 38.5%, CR% 7%, SD% 23.1%; ORR 66.7%; DCR 80%, PR% 60% |
||||
| Metastatic Melanoma |
Azacitidine plus pembrolizumab, entinostat plus pembrolizumab, guadecitabine and ipilimumab |
ORR 55%; ORR 19%; ORR 26%, CR%10.5%, DCR 42%, mPFS 5.6 M, mOS 26.2M |
Mocetinostat with nivolumab and ipilimumab | ORR 70%,CR% 20% | 140 | ||
| Renal and Bladder Carcinoma |
Guadecitabine plus durvalumab, vorinostat plus pembrolizumab |
RCC: PR% 22%, SD% 61%, mPFS 17M urothelial carcinoma: mPFS 2.8M; RCC: mPFS 5.2M; prostate cancer: mPFS 3.5M |
|||||
| Epithelial Ovarian Cancer |
Azacitidine plus durvalumab, guadecitabine plus pembrolizumab |
DCR 7.1%, mPFS 1.9 M, mOS 5M PR% 8.6%, SD% 22.9% |
|||||
| Colorectal Cancer | Azacitidine or romidepsin plus pembrolizumab | mPFS 2.79M, mOS 9.17M | 145 |
Azacitidine, romidepsin plus pembrolizumab, chidamide, sintilimab with or without bevacizumab |
NR ORR 29.2%, DCR 56.3%,mPFS 3.7 M |
||
| Head and neck tumors | Pembrolizumab plus vorinostat |
HNSCC: PR% 32%, SD% 20%,Mos 12.6 M, mPFS of 4.5M; SGC:PR% 16%,SD% 56%,mOS14M, mPFS 6.9M. |
147 | ||||
Note: Solid Tumors (gray boxes) on the top, hematological malignancies (yellow boxes) in the middle.
The central goal of these ongoing trials is to harness the complementary effects of epigenetic and immune‐based treatments. Researchers aim to enhance the body's natural cancer‐fighting capabilities while simultaneously addressing the limitations of current immunotherapy approaches. The medical community eagerly anticipates these trial results because they may fundamentally transform our approach to cancer treatment.
4.6. Challenges and Future Directions in Epigenetic Immunotherapy
The field of epigenetic immunotherapy, while promising, faces several significant challenges that must be addressed to advance clinical applications. Understanding these challenges, along with emerging solutions, is crucial for the development of the field. The first major challenge lies in the complexity of epigenetic regulation. Epigenetic modifications involve sophisticated regulatory networks that vary significantly across different genes and cell types. This variability makes it particularly difficult to predict which cancer types and patients will respond best to treatment. These complex interactions have not yet been fully mapped, which limits our ability to optimize treatment strategies. A second critical challenge is the absence of reliable biomarkers. Without precise markers to identify patients who are likely to respond to epigenetic immunotherapy, treatment success rates remain variable. This variability exists not only across different cancer types but also among patients with the same type of cancer, making treatment outcomes unpredictable. The third significant challenge involves technological limitations in drug development. Current constraints include difficulties in targeting specific cellular mechanisms, achieving efficient drug delivery, and managing the potential combined side effects when multiple therapies are used together.
Despite these challenges, rapid advances in cancer epigenetic drug discovery indicate promising future directions. Advanced technologies, such as single‐cell sequencing, multiomics analysis, and CRISPR screening, are providing unprecedented insights into tumor epigenetics and the immune microenvironment. Research is also progressing in biomarker development, with recent studies showing promise in identifying indicators that can predict patient response to specific drug combinations. For instance, the National Comprehensive Cancer Network guidelines now recommend TET2 mutation status as a predictive biomarker for azacitidine response in myelodysplastic syndrome patients. Furthermore, multiomics integrative models may provide a more comprehensive understanding of patient‐specific epigenetic alterations and immune gene expression profiles, enabling holistic assessment of features associated with responsiveness to epigenetic immunotherapy. This study may lead to more personalized treatment approaches, potentially improving success rates while reducing side effects. Additionally, technological innovations in drug development are advancing rapidly. New approaches using nanotechnology and biotechnology, including the development of proteolysis targeting chimeras and molecular glues, show potential for improving drug targeting and efficacy while maintaining safety.
5. Conclusions
Epigenetic immunotherapy is a significant advance in cancer treatment that targets the complex interactions between tumors and the immune system. The combination of epigenetic and immune‐based therapies provides a powerful approach that addresses many limitations of traditional cancer treatments. As our understanding of underlying mechanisms continues to grow and new technologies emerge, this field is positioned to deliver increasingly effective and personalized treatment options.
Epigenetic immunotherapy is likely to become an essential component of cancer treatment strategies. The ongoing convergence of improved mechanistic understanding, improved patient selection through biomarkers, and advanced drug delivery systems continue to evolve the field and improve patient outcomes. This approach represents not only an important addition to current cancer treatments but also a potentially transformative advance in how cancer is combated.
Author Contributions
Nannan Lu: writing – original draft, writing – review and editing (equal). Guanghua Rong: writing – original draft, writing – review and editing (equal). Weidong Han: conceptualization (lead), supervision (lead).
Ethics Statement
The authors have nothing to report.
Consent
The authors have nothing to report.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgments
We confirm that no AI tools were used for content generation, data analysis, or interpretation of results in this manuscript. During the writing process, machine translation software (e.g., DeepL Translator version 3.0) was occasionally employed to assist in converting Chinese phrases to English for non‐key sentences (estimated < 15% of text volume), followed by thorough manual editing by the authors to ensure accuracy of scientific terminology and context. This study was supported by the National Natural Science Foundation of China (82341208, 82430012).
Lu N., Rong G., and Han W., “Epi‐Immunotherapy in Cancer Treatment: Mechanisms, Clinical Progress, and Future Directions,” Cancer Innovation 4 (2025): 1‐24. 10.1002/cai2.70023.
Nannan Lu and Guanghua Rong contributed equally to this study.
Data Availability Statement
Data sharing is not applicable to this article, as no data sets were generated or analyzed during the current study.
References
- 1. Chen S., Zhang Z., Zheng X., et al., “Response Efficacy of PD‐1 and PD‐L1 Inhibitors in Clinical Trials: A Systematic Review and Meta‐Analysis,” Frontiers in Oncology 11 (2021): 562315, 10.3389/fonc.2021.562315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Parry A., Rulands S., and Reik W., “Active Turnover of DNA Methylation During Cell Fate Decisions,” Nature Reviews Genetics 22, no. 1 (2021): 59–66, 10.1038/s41576-020-00287-8. [DOI] [PubMed] [Google Scholar]
- 3. Millán‐Zambrano G., Burton A., Bannister A. J., and Schneider R., “Histone Post‐Translational Modifications—Cause and Consequence of Genome Function,” Nature Reviews Genetics 23, no. 9 (2022): 563–580, 10.1038/s41576-022-00468-7. [DOI] [PubMed] [Google Scholar]
- 4. Tian Y., Zhang M., Liu L., et al., “Exploring Non‐Coding RNA Mechanisms in Hepatocellular Carcinoma: Implications for Therapy and Prognosis,” Frontiers in Immunology 15 (2024): 1400744, 10.3389/fimmu.2024.1400744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Delaunay S., Helm M., and Frye M., “RNA Modifications in Physiology and Disease: Towards Clinical Applications,” Nature Reviews Genetics 25, no. 2 (2024): 104–122, 10.1038/s41576-023-00645-2. [DOI] [PubMed] [Google Scholar]
- 6. Eustermann S., Patel A. B., Hopfner K. P., He Y., and Korber P., “Energy‐Driven Genome Regulation by ATP‐Dependent Chromatin Remodellers,” Nature Reviews Molecular Cell Biology 25, no. 4 (2024): 309–332, 10.1038/s41580-023-00683-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Robertson K. D., “DNA Methylation, Methyltransferases, and Cancer,” Oncogene 20, no. 24 (2001): 3139–3155, 10.1038/sj.onc.1204341. [DOI] [PubMed] [Google Scholar]
- 8. Kurihara Y., Kawamura Y., Uchijima Y., et al., “Maintenance of Genomic Methylation Patterns During Preimplantation Development Requires the Somatic Form of DNA Methyltransferase 1,” Developmental Biology 313, no. 1 (2008): 335–346, 10.1016/j.ydbio.2007.10.033. [DOI] [PubMed] [Google Scholar]
- 9. Yamaguchi K., Chen X., Rodgers B., et al., “Non‐Canonical Functions of UHRF1 Maintain DNA Methylation Homeostasis in Cancer Cells,” Nature Communications 15, no. 1 (2024): 2960, 10.1038/s41467-024-47314-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Janic A., Abad E., and Amelio I., “Decoding p53 Tumor Suppression: A Crosstalk Between Genomic Stability and Epigenetic Control?,” Cell Death & Differentiation 32, no. 1 (2025): 1–8, 10.1038/s41418-024-01259-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee A. V., Nestler K. A., and Chiappinelli K. B., “Therapeutic Targeting of DNA Methylation Alterations in Cancer,” Pharmacology & Therapeutics 258 (2024): 108640, 10.1016/j.pharmthera.2024.108640. [DOI] [PubMed] [Google Scholar]
- 12. Xue F., Liu L., Tao X., and Zhu W., “TET3‐Mediated DNA Demethylation Modification Activates Shp2 Expression to Promote Endometrial Cancer Progression Through the EGFR/ERK Pathway,” Journal of Gynecologic Oncology 35, no. 5 (2024): e64, 10.3802/jgo.2024.35.e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brückmann N. H., Pedersen C. B., Ditzel H. J., and Gjerstorff M. F., “Epigenetic Reprogramming of Pericentromeric Satellite DNA in Premalignant and Malignant Lesions,” Molecular Cancer Research 16, no. 3 (2018): 417–427, 10.1158/1541-7786.MCR-17-0477. [DOI] [PubMed] [Google Scholar]
- 14. Wang Z., Schones D. E., and Zhao K., “Characterization of Human Epigenomes,” Current Opinion in Genetics & Development 19, no. 2 (2009): 127–134, 10.1016/j.gde.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shanmugam M. K., Arfuso F., Arumugam S., et al., “Role of Novel Histone Modifications in Cancer,” Oncotarget 9, no. 13 (2018): 11414–11426, 10.18632/oncotarget.23356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kaypee S., Sudarshan D., Shanmugam M. K., Mukherjee D., Sethi G., and Kundu T. K., “Aberrant Lysine Acetylation in Tumorigenesis: Implications in the Development of Therapeutics,” Pharmacology & Therapeutics 162 (2016): 98–119, 10.1016/j.pharmthera.2016.01.011. [DOI] [PubMed] [Google Scholar]
- 17. Penna I., Gigoni A., Costa D., et al., “The Inhibition of 45A ncRNA Expression Reduces Tumor Formation, Affecting Tumor Nodules Compactness and Metastatic Potential in Neuroblastoma Cells,” Oncotarget 8, no. 5 (2017): 8189–8205, 10.18632/oncotarget.14138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Griffin G. K., Wu J., Iracheta‐Vellve A., et al., “Epigenetic Silencing by SETDB1 Suppresses Tumour Intrinsic Immunogenicity,” Nature 595, no. 7866 (2021): 309–314, 10.1038/s41586-021-03520-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Harjes U., “SETDB1, a New Target for Immunotherapy,” Nature Reviews Cancer 21, no. 7 (2021): 412, 10.1038/s41568-021-00373-x. [DOI] [PubMed] [Google Scholar]
- 20. Li X., Ma S., Deng Y., Yi P., and Yu J., “Targeting the RNA m6A Modification for Cancer Immunotherapy,” Molecular Cancer 21, no. 1 (2022): 76, 10.1186/s12943-022-01558-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wei T., Li J., Zhang J., et al., “Loss of Mettl3 Enhances Liver Tumorigenesis by Inducing Hepatocyte Dedifferentiation and Hyperproliferation,” Cell Reports 42, no. 7 (2023): 112704, 10.1016/j.celrep.2023.112704. [DOI] [PubMed] [Google Scholar]
- 22. Ennishi D., Takata K., Béguelin W., et al., “Molecular and Genetic Characterization of MHC Deficiency Identifies EZH2 as Therapeutic Target for Enhancing Immune Recognition,” Cancer Discovery 9, no. 4 (2019): 546–563, 10.1158/2159-8290.CD-18-1090. [DOI] [PubMed] [Google Scholar]
- 23. Li J., Lan Z., Liao W., et al., “Histone Demethylase KDM5D Upregulation Drives Sex Differences in Colon Cancer,” Nature 619, no. 7970 (2023): 632–639, 10.1038/s41586-023-06254-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Woods D. M., Sodré A. L., Villagra A., Sarnaik A., Sotomayor E. M., and Weber J., “HDAC Inhibition Upregulates PD‐1 Ligands in Melanoma and Augments Immunotherapy With PD‐1 Blockade,” Cancer Immunology Research 3, no. 12 (2015): 1375–1385, 10.1158/2326-6066.CIR-15-0077-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Briere D., Sudhakar N., Woods D. M., et al., “The Class I/IV HDAC Inhibitor Mocetinostat Increases Tumor Antigen Presentation, Decreases Immune Suppressive Cell Types and Augments Checkpoint Inhibitor Therapy,” Cancer Immunology, Immunotherapy 67, no. 3 (2018): 381–392, 10.1007/s00262-017-2091-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang L., Tian S., Pei M., et al., “Crosstalk Between Histone Modification and DNA Methylation Orchestrates the Epigenetic Regulation of the Costimulatory Factors, Tim‐3 and Galectin‐9, in Cervical Cancer,” Oncology Reports 42, no. 6 (2019): 2655–2669, 10.3892/or.2019.7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sasidharan Nair V., El Salhat H., Taha R. Z., John A., Ali B. R., and Elkord E., “DNA Methylation and Repressive H3K9 and H3K27 Trimethylation in the Promoter Regions of PD‐1, CTLA‐4, TIM‐3, LAG‐3, TIGIT and PD‐L1 Genes in Human Primary Breast Cancer,” Clinical Epigenetics 10, no. 1 (2018): 78, 10.1186/s13148-018-0512-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wei J., Nduom E. K., Kong L. Y., et al., “MiR‐138 Exerts Anti‐Glioma Efficacy by Targeting Immune Checkpoints,” Neuro‐Oncology 18, no. 5 (2016): 639–648, 10.1093/neuonc/nov292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xu S., Tao Z., Hai B., et al., “MiR‐424(322) Reverses Chemoresistance via T‐Cell Immune Response Activation by Blocking the PD‐L1 Immune Checkpoint,” Nature Communications 7 (2016): 11406, 10.1038/ncomms11406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Deng S., Hu Q., Zhang H., Yang F., Peng C., and Huang C., “HDAC3 Inhibition Upregulates PD‐L1 Expression in B‐Cell Lymphomas and Augments the Efficacy of Anti‐PD‐L1 Therapy,” Molecular Cancer Therapeutics 18, no. 5 (2019): 900–908, 10.1158/1535-7163.MCT-18-1068. [DOI] [PubMed] [Google Scholar]
- 31. Yang W., Feng Y., Zhou J., et al., “A Selective HDAC8 Inhibitor Potentiates Antitumor Immunity and Efficacy of Immune Checkpoint Blockade in Hepatocellular Carcinoma,” Science Translational Medicine 13, no. 588 (2021): eaaz6804, 10.1126/scitranslmed.aaz6804. [DOI] [PubMed] [Google Scholar]
- 32. Scharer C. D., Barwick B. G., Youngblood B. A., Ahmed R., and Boss J. M., “Global DNA Methylation Remodeling Accompanies CD8 T Cell Effector Function,” Journal of Immunology 191, no. 6 (2013): 3419–3429, 10.4049/jimmunol.1301395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Abdelsamed H. A., “Human Memory CD8 T Cell Effector Potential Is Epigenetically Preserved During In Vivo Homeostasis,” supplement, Journal of Immunology 198, no. Suppl_1 (2017): 144.7, 10.4049/jimmunol.198.supp.144.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ford B. R., Vignali P., Rittenhouse N. L., et al., “Tumor Microenvironmental Signals Reshape Chromatin Landscapes to Limit the Functional Potential of Exhausted T Cells,” Science Immunology 7, no. 74 (2022): eabj9123, 10.1126/sciimmunol.abj9123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Belk J. A., Yao W., Ly N., et al., “Genome‐Wide CRISPR Screens of T Cell Exhaustion Identify Chromatin Remodeling Factors That Limit T Cell Persistence,” Cancer Cell 40, no. 7 (2022): 768–786.e7, 10.1016/j.ccell.2022.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guo A., Huang H., Zhu Z., et al., “CBAF Complex Components and MYC Cooperate Early in CD8+ T Cell Fate,” Nature 607, no. 7917 (2022): 135–141, 10.1038/s41586-022-04849-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Niborski L. L., Gueguen P., Ye M., et al., “CD8+ T Cell Responsiveness to Anti‐PD‐1 Is Epigenetically Regulated by Suv39h1 in Melanomas,” Nature Communications 13, no. 1 (2022): 3739, 10.1038/s41467-022-31504-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Prager I., Liesche C., van Ooijen H., et al., “NK Cells Switch From Granzyme B to Death Receptor‐Mediated Cytotoxicity During Serial Killing,” Journal of Experimental Medicine 216, no. 9 (2019): 2113–2127, 10.1084/jem.20181454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Moretta A., “Natural Killer Cells and Dendritic Cells: Rendezvous in Abused Tissues,” Nature Reviews Immunology 2, no. 12 (2002): 957–965, 10.1038/nri956. [DOI] [PubMed] [Google Scholar]
- 40. Luna J. I., Grossenbacher S. K., Murphy W. J., and Canter R. J., “Targeting Cancer Stem Cells With Natural Killer Cell Immunotherapy,” Expert Opinion on Biological Therapy 17, no. 3 (2017): 313–324, 10.1080/14712598.2017.1271874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Correia A. L., Guimaraes J. C., Auf der Maur P., et al., “Hepatic Stellate Cells Suppress NK Cell‐Sustained Breast Cancer Dormancy,” Nature 594, no. 7864 (2021): 566–571, 10.1038/s41586-021-03614-z. [DOI] [PubMed] [Google Scholar]
- 42. Zhao D., Zhang Q., Liu Y., et al., “H3K4me3 Demethylase Kdm5a Is Required for NK Cell Activation by Associating With P50 to Suppress SoCS1,” Cell Reports 15, no. 2 (2016): 288–299, 10.1016/j.celrep.2016.03.035. [DOI] [PubMed] [Google Scholar]
- 43. Song H., Song J., Cheng M., et al., “METTL3‐Mediated M6A RNA Methylation Promotes the Anti‐Tumour Immunity of Natural Killer Cells,” Nature Communications 12, no. 1 (2021): 5522, 10.1038/s41467-021-25803-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Luetke‐Eversloh M., Cicek B. B., Siracusa F., et al., “NK Cells Gain Higher IFN‐γ Competence During Terminal Differentiation,” European Journal of Immunology 44, no. 7 (2014): 2074–2084, 10.1002/eji.201344072. [DOI] [PubMed] [Google Scholar]
- 45. Gangoso E., Southgate B., Bradley L., et al., “Glioblastomas Acquire Myeloid‐Affiliated Transcriptional Programs via Epigenetic Immunoediting to Elicit Immune Evasion,” Cell 184, no. 9 (2021): 2454–2470.e26, 10.1016/j.cell.2021.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dong F., Qin X., Wang B., et al., “ALKBH5 Facilitates Hypoxia‐Induced Paraspeckle Assembly and IL8 Secretion to Generate an Immunosuppressive Tumor Microenvironment,” Cancer Research 81, no. 23 (2021): 5876–5888, 10.1158/0008-5472.CAN-21-1456. [DOI] [PubMed] [Google Scholar]
- 47. Tong J., Wang X., Liu Y., et al., “Pooled CRISPR Screening Identifies M6A as a Positive Regulator of Macrophage Activation,” Science Advances 7, no. 18 (2021): eabd4742, 10.1126/sciadv.abd4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Noe J. T., Rendon B. E., Geller A. E., et al., “Lactate Supports a Metabolic‐Epigenetic Link in Macrophage Polarization,” Science Advances 7, no. 46 (2021): eabi8602, 10.1126/sciadv.abi8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang M., Liu Z. Z., Aoshima K., et al., “CECR2 Drives Breast Cancer Metastasis by Promoting NF‐κB Signaling and Macrophage‐Mediated Immune Suppression,” Science Translational Medicine 14, no. 630 (2022): eabf5473, 10.1126/scitranslmed.abf5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pan W., Zhu S., Qu K., et al., “The DNA Methylcytosine Dioxygenase Tet2 Sustains Immunosuppressive Function of Tumor‐Infiltrating Myeloid Cells to Promote Melanoma Progression,” Immunity 47, no. 2 (2017): 284–297.e5, 10.1016/j.immuni.2017.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhou J., Yao Y., Shen Q., Li G., Hu L., and Zhang X., “Demethylating Agent Decitabine Disrupts Tumor‐Induced Immune Tolerance by Depleting Myeloid‐Derived Suppressor Cells,” Journal of Cancer Research and Clinical Oncology 143, no. 8 (2017): 1371–1380, 10.1007/s00432-017-2394-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Satoh T., Takeuchi O., Vandenbon A., et al., “The Jmjd3–Irf4 Axis Regulates M2 Macrophage Polarization and Host Responses Against Helminth Infection,” Nature Immunology 11, no. 10 (2010): 936–944, 10.1038/ni.1920. [DOI] [PubMed] [Google Scholar]
- 53. Link V. M., Duttke S. H., Chun H. B., et al., “Analysis of Genetically Diverse Macrophages Reveals Local and Domain‐Wide Mechanisms That Control Transcription Factor Binding and Function,” Cell 173, no. 7 (2018): 1796–1809.e17, 10.1016/j.cell.2018.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kruidenier L., Chung C., Cheng Z., et al., “A Selective Jumonji H3K27 Demethylase Inhibitor Modulates the Proinflammatory Macrophage Response,” Nature 488, no. 7411 (2012): 404–408, 10.1038/nature11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lal G., Zhang N., van der Touw W., et al., “Epigenetic Regulation of Foxp3 Expression in Regulatory T Cells by DNA Methylation,” Journal of Immunology 182, no. 1 (2009): 259–273, 10.4049/jimmunol.182.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lal G. and Bromberg J. S., “Epigenetic Mechanisms of Regulation of Foxp3 Expression,” Blood 114, no. 18 (2009): 3727–3735, 10.1182/blood-2009-05-219584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yue X., Lio C. W. J., Samaniego‐Castruita D., Li X., and Rao A., “Loss of TET2 and TET3 in Regulatory T Cells Unleashes Effector Function,” Nature Communications 10, no. 1 (2019): 2011, 10.1038/s41467-019-09541-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. DuPage M., Chopra G., Quiros J., et al., “The Chromatin‐Modifying Enzyme Ezh2 Is Critical for the Maintenance of Regulatory T Cell Identity After Activation,” Immunity 42, no. 2 (2015): 227–238, 10.1016/j.immuni.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yang X.‐P., Jiang K., Hirahara K., et al., “EZH2 Is Crucial for Both Differentiation of Regulatory T Cells and T Effector Cell Expansion,” Scientific Reports 5, no. 1 (2015): 10643, 10.1038/srep10643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang D., Quiros J., Mahuron K., et al., “Targeting EZH2 Reprograms Intratumoral Regulatory T Cells to Enhance Cancer Immunity,” Cell Reports 23, no. 11 (2018): 3262–3274, 10.1016/j.celrep.2018.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Goswami S., Apostolou I., Zhang J., et al., “Modulation of EZH2 Expression in T Cells Improves Efficacy of Anti‐CTLA‐4 Therapy,” Journal of Clinical Investigation 128, no. 9 (2018): 3813–3818, 10.1172/JCI99760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chen H., Pan Y., Zhou Q., et al., “METTL3 Inhibits Antitumor Immunity by Targeting M6A‐BHLHE41‐CXCL1/CXCR2 Axis to Promote Colorectal Cancer,” Gastroenterology 163, no. 4 (2022): 891–907, 10.1053/j.gastro.2022.06.024. [DOI] [PubMed] [Google Scholar]
- 63. Li N., Liu Q., Han Y., et al., “ARID1A Loss Induces Polymorphonuclear Myeloid‐Derived Suppressor Cell Chemotaxis and Promotes Prostate Cancer Progression,” Nature Communications 13, no. 1 (2022): 7281, 10.1038/s41467-022-34871-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ni H., Zhang L., Huang H., Dai S., and Li J., “Connecting METTL3 and Intratumoural CD33+ MDSCS in Predicting Clinical Outcome in Cervical Cancer,” Journal of Translational Medicine 18, no. 1 (2020): 393, 10.1186/s12967-020-02553-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhao D., Cai L., Lu X., et al., “Chromatin Regulator CHD1 Remodels the Immunosuppressive Tumor Microenvironment in PTEN‐Deficient Prostate Cancer,” Cancer Discovery 10, no. 9 (2020): 1374–1387, 10.1158/2159-8290.cd-19-1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yu H., Liu J., Bu X., et al., “Targeting METTL3 Reprograms the Tumor Microenvironment to Improve Cancer Immunotherapy,” Cell Chemical Biology 31, no. 4 (2024): 776–791.e7, 10.1016/j.chembiol.2023.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Qiu X., Yang S., Wang S., et al., “M6A Demethylase ALKBH5 Regulates PD‐L1 Expression and Tumor Immunoenvironment in Intrahepatic Cholangiocarcinoma,” Cancer Research 81, no. 18 (2021): 4778–4793, 10.1158/0008-5472.CAN-21-0468. [DOI] [PubMed] [Google Scholar]
- 68. Wang L., Dou X., Chen S., et al., “YTHDF2 Inhibition Potentiates Radiotherapy Antitumor Efficacy,” Cancer Cell 41, no. 7 (2023): 1294–1308.e8, 10.1016/j.ccell.2023.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. de Almeida Nagata D. E., Chiang E. Y., Jhunjhunwala S., et al., “Regulation of Tumor‐Associated Myeloid Cell Activity by CBP/EP300 Bromodomain Modulation of H3K27 Acetylation,” Cell Reports 27, no. 1 (2019): 269–281.e4, 10.1016/j.celrep.2019.03.008. [DOI] [PubMed] [Google Scholar]
- 70. Pang L., Zhou F., Liu Y., et al., “Epigenetic Regulation of Tumor Immunity,” Journal of Clinical Investigation 134, no. 12 (2024): e178540, 10.1172/jci178540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mabe N. W., Perry J. A., Malone C. F., and Stegmaier K., “Pharmacological Targeting of the Cancer Epigenome,” Nature Cancer 5, no. 6 (2024): 844–865, 10.1038/s43018-024-00777-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jan Z., Ahmed W. S., Biswas K. H., and Jithesh P. V., “Identification of a Potential DNA Methyltransferase (DNMT) Inhibitor,” Journal of Biomolecular Structure and Dynamics 42, no. 9 (2024): 4730–4744, 10.1080/07391102.2023.2233637. [DOI] [PubMed] [Google Scholar]
- 73. Zhou S., Ou H., Wu Y., et al., “Targeting Tumor Endothelial Cells With Methyltransferase Inhibitors: Mechanisms of Action and the Potential of Combination Therapy,” Pharmacology & Therapeutics 247 (2023): 108434, 10.1016/j.pharmthera.2023.108434. [DOI] [PubMed] [Google Scholar]
- 74. Issa J.‐P., Kantarjian H., and Kirkpatrick P., “Azacitidine,” Nature Reviews Drug Discovery 4, no. 5 (2005): S6–S7, 10.1038/nrd1726. [DOI] [PubMed] [Google Scholar]
- 75. Gore S. D., Jones C., and Kirkpatrick P., “Decitabine,” supplement, Nature Reviews Drug Discovery 5 (2006): 891–892. [DOI] [PubMed] [Google Scholar]
- 76. Daher‐Reyes G. S., Merchan B. M., and Yee K. W. L., “Guadecitabine (SGI‐110): An Investigational Drug for the Treatment of Myelodysplastic Syndrome and Acute Myeloid Leukemia,” Expert Opinion on Investigational Drugs 28, no. 10 (2019): 835–849, 10.1080/13543784.2019.1667331. [DOI] [PubMed] [Google Scholar]
- 77. Pu J., Liu T., Wang X., et al., “Exploring the Role of Histone Deacetylase and Histone Deacetylase Inhibitors in the Context of Multiple Myeloma: Mechanisms, Therapeutic Implications, and Future Perspectives,” Experimental Hematology & Oncology 13, no. 1 (2024): 45, 10.1186/s40164-024-00507-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. West A. C. and Johnstone R. W., “New and Emerging HDAC Inhibitors for Cancer Treatment,” Journal of Clinical Investigation 124, no. 1 (2014): 30–39, 10.1172/JCI69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Shi Y., Jia B., Xu W., et al., “Chidamide in Relapsed or Refractory Peripheral T Cell Lymphoma: A Multicenter Real‐World Study in China,” Journal of Hematology & Oncology 10, no. 1 (2017): 69, 10.1186/s13045-017-0439-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sun Y., Hong J. H., Ning Z., et al., “Therapeutic Potential of Tucidinostat, a Subtype‐Selective HDAC Inhibitor, in Cancer Treatment,” Frontiers in Pharmacology 13 (2022): 932914, 10.3389/fphar.2022.932914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. American Association for Cancer Research .First EZH2 Inhibitor Approved‐For Rare Sarcoma,” Cancer Discovery 10, no. 3 (2020): 333–334, 10.1158/2159-8290.CD-NB2020-006. [DOI] [PubMed] [Google Scholar]
- 82. Keam S. J., “Valemetostat Tosilate: First Approval,” Drugs 82, no. 16 (2022): 1621–1627, 10.1007/s40265-022-01800-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kim E. S., “Enasidenib: First Global Approval,” Drugs 77, no. 15 (2017): 1705–1711, 10.1007/s40265-017-0813-2. [DOI] [PubMed] [Google Scholar]
- 84. Norsworthy K. J., Luo L., Hsu V., et al., “FDA Approval Summary: Ivosidenib for Relapsed or Refractory Acute Myeloid Leukemia With an Isocitrate Dehydrogenase‐1 Mutation,” Clinical Cancer Research 25, no. 11 (2019): 3205–3209, 10.1158/1078-0432.CCR-18-3749. [DOI] [PubMed] [Google Scholar]
- 85. Woods A., Norsworthy K. J., Wang X., et al., “FDA Approval Summary: Ivosidenib in Combination With Azacitidine for Treatment of Patients With Newly Diagnosed Acute Myeloid Leukemia With an IDH1 Mutation,” Clinical Cancer Research 30, no. 7 (2024): 1226–1231, 10.1158/1078-0432.CCR-23-2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Guo Z.‐S., Liu Z., and Bartlett D. L., “Oncolytic Immunotherapy: Dying the Right Way Is a Key to Eliciting Potent Antitumor Immunity,” Frontiers in Oncology 4 (2014): 74, 10.3389/fonc.2014.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Scotto L., Kinahan C., Douglass E., et al., “Targeting the T‐Cell Lymphoma Epigenome Induces Cell Death, Cancer Testes Antigens, Immune‐Modulatory Signaling Pathways,” Molecular Cancer Therapeutics 20, no. 8 (2021): 1422–1430, 10.1158/1535-7163.MCT-20-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Falahat R., Berglund A., Putney R. M., et al., “Epigenetic Reprogramming of Tumor Cell–Intrinsic STING Function Sculpts Antigenicity and T Cell Recognition of Melanoma,” Proceedings of the National Academy of Sciences of the United States of America 118, no. 15 (2021): e2013598118, 10.1073/pnas.2013598118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Liang S., Liu M., Mu W., et al., “Nano‐Regulator Inhibits Tumor Immune Escape via the ʻTwo‐Way Regulationʼ Epigenetic Therapy Strategy,” Advanced Science 11, no. 9 (2024): 2305275, 10.1002/advs.202305275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Khan A. N. H., Gregorie C. J., and Tomasi T. B., “Histone Deacetylase Inhibitors Induce Tap LMP Tapasin Genes and MHC Class I Antigen Presentation by Melanoma Cells,” Cancer Immunology, Immunotherapy 57, no. 5 (2008): 647–654, 10.1007/s00262-007-0402-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Khan A. N. H., Gregorie C. J., and Tomasi T. B., “Histone Deacetylase Inhibitors Induce TAP LMP Tapasin Genes and MHC Class I Antigen Presentation by Melanoma Cells,” Cancer Immunology, Immunotherapy 57, no. 5 (2008): 647–654, 10.1007/s00262-007-0402-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ritter C., Fan K., Paschen A., et al., “Epigenetic Priming Restores the HLA Class‐I Antigen Processing Machinery Expression in Merkel Cell Carcinoma,” Scientific Reports 7, no. 1 (2017): 2290, 10.1038/s41598-017-02608-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kitamura H., Torigoe T., Asanuma H., Honma I., Sato N., and Tsukamoto T., “Down‐Regulation of HLA Class I Antigens in Prostate Cancer Tissues and Up‐Regulation by Histone Deacetylase Inhibition,” Journal of Urology 178, no. 2 (2007): 692–696, 10.1016/j.juro.2007.03.109. [DOI] [PubMed] [Google Scholar]
- 94. Zhou L., Mudianto T., Ma X., Riley R., and Uppaluri R., “Targeting EZH2 Enhances Antigen Presentation, Antitumor Immunity, and Circumvents anti‐PD‐1 Resistance in Head and Neck Cancer,” Clinical Cancer Research 26, no. 1 (2020): 290–300, 10.1158/1078-0432.CCR-19-1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Lienlaf M., Perez‐Villarroel P., Knox T., et al., “Essential Role of HDAC6 in the Regulation of PD‐L1 in Melanoma,” Molecular Oncology 10, no. 5 (2016): 735–750, 10.1016/j.molonc.2015.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Hogg S. J., Beavis P. A., Dawson M. A., and Johnstone R. W., “Targeting the Epigenetic Regulation of Antitumour Immunity,” Nature Reviews Drug Discovery 19, no. 11 (2020): 776–800, 10.1038/s41573-020-0077-5. [DOI] [PubMed] [Google Scholar]
- 97. Fröhlich A., Sirokay J., Fietz S., et al., “Molecular, Clinicopathological, and Immune Correlates of LAG3 Promoter DNA Methylation in Melanoma,” EBioMedicine 59 (2020): 102962, 10.1016/j.ebiom.2020.102962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Shen L., Ciesielski M., Ramakrishnan S., et al., “Class I Histone Deacetylase Inhibitor Entinostat Suppresses Regulatory T Cells and Enhances Immunotherapies in Renal and Prostate Cancer Models,” PLoS One 7, no. 1 (2012): e30815, 10.1371/journal.pone.0030815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Goswami S., Apostolou I., Zhang J., et al., “Modulation of EZH2 Expression in T Cells Improves Efficacy of Anti–CTLA‐4 Therapy,” Journal of Clinical Investigation 128, no. 9 (2018): 3813–3818, 10.1172/jci99760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Long X., Zhang S., Wang Y., et al., “Targeting JMJD1C to Selectively Disrupt Tumor TReg Cell Fitness Enhances Antitumor Immunity,” Nature Immunology 25, no. 3 (2024): 525–536, 10.1038/s41590-024-01746-8. [DOI] [PubMed] [Google Scholar]
- 101. Xin J., Zhang Z., Su X., Wang L., Zhang Y., and Yang R., “Epigenetic Component p66a Modulates Myeloid‐Derived Suppressor Cells by Modifying STAT3,” Journal of Immunology 198, no. 7 (2017): 2712–2720, 10.4049/jimmunol.1601712. [DOI] [PubMed] [Google Scholar]
- 102. Sahakian E., Powers J. J., Chen J., et al., “Histone Deacetylase 11: A Novel Epigenetic Regulator of Myeloid Derived Suppressor Cell Expansion and Function,” Molecular Immunology 63, no. 2 (2015): 579–585, 10.1016/j.molimm.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Kang T. G., Lan X., Mi T., et al., “Epigenetic Regulators of Clonal Hematopoiesis Control CD8 T Cell Stemness During Immunotherapy,” Science 386, no. 6718 (2024): eadl4492, 10.1126/science.adl4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Awad M. M., Le Bruchec Y., Lu B., Miller J., Dumitru C. D., and Spira A. I., “Phase Ib Study: Selective Histone Deacetylase (HDAC) Inhibitor ACY‐241 + Nivolumab (Nivo) in Advanced Non‐Small Cell Lung Cancer (NSCLC),” supplement, Journal of Clinical Oncology 37, no. 15_suppl (2019): 9029, 10.1200/jco.2019.37.15_suppl.9029. [DOI] [Google Scholar]
- 105. Li X., Li Y., Dong L., et al., “Decitabine Priming Increases Anti–PD‐1 Antitumor Efficacy by Promoting CD8+ Progenitor Exhausted T Cell Expansion in Tumor Models,” Journal of Clinical Investigation 133, no. 7 (2023): e165673, 10.1172/jci165673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Abruzzese M. P., Bilotta M. T., Fionda C., et al., “Inhibition of Bromodomain and Extra‐Terminal (BET) Proteins Increases NKG2D Ligand Mica Expression and Sensitivity to NK Cell‐Mediated Cytotoxicity in Multiple Myeloma Cells: Role of cMYC‐IRF4‐miR‐125b Interplay,” Journal of Hematology & Oncology 9, no. 1 (2016): 134, 10.1186/s13045-016-0362-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Kumar R. and Gupta R., “Epigenetic Regulation of NKG2D Ligand and the Rise of NK Cell‐Based Immunotherapy for Cancer Treatment,” Frontiers in Oncology 14 (2024): 1456631, 10.3389/fonc.2024.1456631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Zhao D., Zhang Q., Liu Y., et al., “H3K4me3 Demethy‐Lase Kdm5a Is Required for NK Cell Activation by Associating With p50 to Suppress Socs1,” Cell Reports 15, no. 2 (2016): 288–299, 10.1016/j.celrep.2016.03.035. [DOI] [PubMed] [Google Scholar]
- 109. Yin J., Leavenworth J. W., Li Y., et al., “Ezh2 Regulates Differentiation and Function of Natural Killer Cells Through Histone Methyltransferase Activity,” Proceedings of the National Academy of Sciences 112, no. 52 (2015): 15988–15993, 10.1073/pnas.1521740112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Bugide S., Green M. R., and Wajapeyee N., “Inhibition of Enhancer of Zeste Homolog 2 (EZH2) Induces Natural Killer Cell‐Mediated Eradication of Hepatocellular Carcinoma Cells,” Proceedings of the National Academy of Sciences 115, no. 15 (2018): E3509–E3518, 10.1073/pnas.1802691115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Huang Y.‐H., Cai K., Xu P.‐P., et al., “CREBBP/EP300 Mutations Promoted Tumor Progression in Diffuse Large B‐Cell Lymphoma Through Altering Tumor‐Associated Macrophage Polarization via FBXW7‐NOTCH‐CCL2/CSF1 Axis,” Signal Transduction and Targeted Therapy 6, no. 1 (2021): 10, 10.1038/s41392-020-00437-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Adeegbe D. O., Liu Y., Lizotte P. H., et al., “Synergistic Immunostimulatory Effects and Therapeutic Benefit of Combined Histone Deacetylase and Bromodomain Inhibition in Non‐Small Cell Lung Cancer,” Cancer Discovery 7, no. 8 (2017): 852–867, 10.1158/2159-8290.CD-16-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Cai J., Song L., Zhang F., et al., “Targeting SRSF10 Might Inhibit M2 Macrophage Polarization and Potentiate Anti‐PD‐1 Therapy in Hepatocellular Carcinoma,” Cancer Communications 44, no. 11 (2024): 1231–1260, 10.1002/cac2.12607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Morel K. L., Sheahan A. V., Burkhart D. L., et al., “EZH2 Inhibition Activates a dsRNA–STING–Interferon Stress Axis That Potentiates Response to PD‐1 Checkpoint Blockade in Prostate Cancer,” Nature Cancer 2, no. 4 (2021): 444–456, 10.1038/s43018-021-00185-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Li S., Liang X., Shao Q., et al., “Research Hotspots and Trends of Epigenetic Therapy In Oncology: A Bibliometric Analysis From 2004 to 2023,” Frontiers in Pharmacology 15 (2024): 1465954, 10.3389/fphar.2024.1465954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Nie J., Wang C., Liu Y., et al., “Addition of Low‐Dose Decitabine to Anti‐PD‐1 Antibody Camrelizumab in Relapsed/Refractory Classical Hodgkin Lymphoma,” Journal of Clinical Oncology 37, no. 17 (2019): 1479–1489, 10.1200/JCO.18.02151. [DOI] [PubMed] [Google Scholar]
- 117. Liu Y., Wang C., Li X., et al., “Improved Clinical Outcome in a Randomized Phase II Study of Anti‐PD‐1 Camrelizumab Plus Decitabine in Relapsed/Refractory Hodgkin Lymphoma,” Journal for Immunotherapy of Cancer 9, no. 4 (2021): e002347, 10.1136/jitc-2021-002347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Wang C., Liu Y., Dong L., et al., “Efficacy of Decitabine Plus Anti‐PD‐1 Camrelizumab in Patients With Hodgkin Lymphoma Who Progressed or Relapsed After PD‐1 Blockade Monotherapy,” Clinical Cancer Research 27, no. 10 (2021): 2782–2791, 10.1158/1078-0432.CCR-21-0133. [DOI] [PubMed] [Google Scholar]
- 119. Wang C., Pan Y., Liu Y., et al., “Long‐Term Complete Remission and Peripheral Biomarkers in Hodgkin Lymphoma Patients After Decitabine‐Plus‐Camrelizumab Epi‐Immunotherapy and Treatment Cessation,” MedComm 4, no. 6 (2023): e428, 10.1002/mco2.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Nie J., Wang C., Zheng L., et al., “Epigenetic Agents Plus Anti‐PD‐1 Reprogram the Tumor Microenvironment and Restore Antitumor Efficacy in Hodgkin Lymphoma,” Blood 144, no. 18 (2024): 1936–1950, 10.1182/blood.2024024487. [DOI] [PubMed] [Google Scholar]
- 121. Gao Y., Huang H., Wang X., et al., “Anti‐PD‐1 Antibody (Sintilimab) Plus Histone Deacetylase Inhibitor (Chidamide) for the Treatment of Refractory Or Relapsed Extranodal Natural Killer/T Cell Lymphoma, Nasal Type (R/R‐ENKTL): Preliminary Results From a Prospective, Multicenter, Single‐Arm, Phase Ib/II Trial (SCENT),” supplement, Blood 136, no. Suppl 1 (2020): 39–40, 10.1182/blood-2020-134665. [DOI] [Google Scholar]
- 122. Gao Y., He H., Li X., et al., “Sintilimab (Anti‐PD‐1 Antibody) Plus Chidamide (Histone Deacetylase Inhibitor) in Relapsed or Refractory Extranodal Natural Killer T‐Cell Lymphoma (SCENT): A Phase Ib/II Study,” Signal Transduction and Targeted Therapy 9, no. 1 (2024): 121, 10.1038/s41392-024-01825-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Iyer S. P., Xu J., Becnel M. R., et al., “A Phase II Study of Pembrolizumab in Combination With Romidepsin Demonstrates Durable Responses in Relapsed or Refractory T‐Cell Lymphoma (TCL),” supplement, Blood 136, no. Suppl 1 (2020): 40–41, 10.1182/blood-2020-143252. [DOI] [Google Scholar]
- 124. Marchi E., Ma H., Montanari F., et al., “Abstract CT160: The Integration of PD1 Blockade With Epigenetic Therapy Is Highly Active and Safe in Heavily Treated Patients With T‐Cell Lymphoma,” supplement, Cancer Research 80, no. 16_Suppl (2020): CT160, 10.1158/1538-7445.am2020-ct160. [DOI] [Google Scholar]
- 125. Gojo I., Stuart R. K., Webster J., et al., “Multi‐Center Phase 2 Study of Pembroluzimab (Pembro) and Azacitidine (AZA) in Patients With Relapsed/Refractory Acute Myeloid Leukemia (AML) and in Newly Diagnosed (≥ 65 Years) AML Patients,” supplement, Blood 134, no. Suppl_1 (2019): 832, 10.1182/blood-2019-127345. [DOI] [Google Scholar]
- 126. Daver N., Garcia‐Manero G., Basu S., et al., “Efficacy, Safety, and Biomarkers of Response to Azacitidine and Nivolumab in Relapsed/Refractory Acute Myeloid Leukemia: A Nonrandomized, Open‐Label, Phase II Study,” Cancer Discovery 9, no. 3 (2019): 370–383, 10.1158/2159-8290.CD-18-0774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Lindblad K. E., Thompson J., Gui G., et al., “Pembrolizumab and Decitabine for Refractory or Relapsed Acute Myeloid Leukemia,” Blood 132, no. Suppl 1 (2018): 1437, 10.1182/blood-2018-99-115097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Daver N. G., Garcia‐Manero G., Konopleva M. Y., et al., “Azacitidine (AZA) With Nivolumab (Nivo), and AZA With Nivo + Ipilimumab (Ipi) in Relapsed/Refractory Acute Myeloid Leukemia: A Non‐Randomized, Prospective, Phase 2 Study,” supplement, Blood 134, no. Suppl_1 (2019): 830, 10.1182/blood-2019-131494. [DOI] [Google Scholar]
- 129. Chien K. S., Borthakur G., Naqvi K., et al., “Final Results From a Phase II Study Combining Azacitidine and Pembrolizumab in Patients With Higher‐Risk Myelodysplastic Syndrome After Failure of Hypomethylating Agent Therapy,” supplement, Blood 136, no. Suppl 1 (2020): 23–24, 10.1182/BLOOD-2020-141100. [DOI] [Google Scholar]
- 130. Chien K. S., Kim K., Nogueras‐Gonzalez G. M., et al., “Phase II Study of Azacitidine With Pembrolizumab in Patients With Intermediate‐1 or Higher‐Risk Myelodysplastic Syndrome,” British Journal of Haematology 195, no. 3 (2021): 378–387, 10.1111/bjh.17689. [DOI] [PubMed] [Google Scholar]
- 131. Yalniz F. F., Daver N., Rezvani K., et al., “A Pilot Trial of Lirilumab With or Without Azacitidine for Patients With Myelodysplastic Syndrome,” Clinical Lymphoma Myeloma and Leukemia 18, no. 10 (2018): 658–663.e2, 10.1016/j.clml.2018.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Short N. J., Kadia T. M., DiNardo C. D., et al., “Interim Results of an Open‐Label Phase Ib/II Multi‐Arm Study of OX40 Agonist Monoclonal Antibody (MAB), Anti‐PDL1 Mab, Smoothened Inhibitor, Anti‐CD33 Mab, BCL‐2 Inhibitor, and Azacitidine as Single‐Agents and as Combinations for Relapsed/Refractory Acute Myeloid Leukemia,” Blood 136, no. Suppl 1 (2020): 21–23, 10.1182/blood-2020-140153. [DOI] [Google Scholar]
- 133. Yan X., Zhao Y., Liu Y., et al., “Case Report: Low‐Dose Decitabine Plus Anti‐PD‐1 Inhibitor Camrelizumab for Previously Treated Advanced Metastatic Non‐Small Cell Lung Cancer,” Frontiers in Oncology 10 (2020): 558572, 10.3389/fonc.2020.558572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Saltos A. N., Tanvetyanon T., Creelan B. C., et al., “Phase II Randomized Trial of First‐Line Pembrolizumab and Vorinostat in Patients With Metastatic NSCLC (MNSCLC),” supplement, Journal of Clinical Oncology 38, no. 15_suppl (2020): 9567, 10.1200/jco.2020.38.15_suppl.9567. [DOI] [Google Scholar]
- 135. Awad M. M., Le Bruchec Y., Lu B., et al., “Selective Histone Deacetylase Inhibitor ACY‐241 (Citarinostat) Plus Nivolumab in Advanced Non‐Small Cell Lung Cancer: Results From a Phase Ib Study,” Frontiers in Oncology 11 (2021): 696512, 10.3389/fonc.2021.696512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Burton E. M., Woody T., Glitza I. C., et al., “A Phase II Study of Oral Azacitidine (CC‐486) in Combination With Pembrolizumab (PEMBRO) In Patients (Pts) With Metastatic Melanoma (MM),” supplement, Journal of Clinical Oncology 37, no. 15_suppl (2019): 9560, 10.1200/jco.2019.37.15_suppl.9560. [DOI] [Google Scholar]
- 137. Agarwala S. S., Moschos S. J., Johnson M. L., et al., “Efficacy and Safety of Entinostat (ENT) and Pembrolizumab (PEMBRO) in Patients With Melanoma Progressing on or After a PD‐1/L1 Blocking Antibody,” supplement, Journal of Clinical Oncology 36, no. 15_suppl (2018): 9530, 10.1200/jco.2018.36.15_suppl.9530. [DOI] [Google Scholar]
- 138. Sullivan R. J., Moschos S. J., Johnson M. L., et al., “Abstract CT072: Efficacy and Safety of Entinostat (ENT) and Pembrolizumab (PEMBRO) in Patients With Melanoma Previously Treated With Anti‐PD1 Therapy,” supplement, Cancer Research 79, no. 13_Suppl (2019): CT072, 10.1158/1538-7445.am2019-ct072. [DOI] [Google Scholar]
- 139. Di Giacomo A. M., Covre A., Finotello F., et al., “Guadecitabine Plus Ipilimumab in Unresectable Melanoma: The NIBIT‐M4 Clinical Trial,” Clinical Cancer Research 25, no. 24 (2019): 7351–7362, 10.1158/1078-0432.ccr-19-1335. [DOI] [PubMed] [Google Scholar]
- 140. Weber J. S., Laino A. S., Vassallo M., et al., “Preclinical and Clinical Studies of a Class I/IV HDAC Inhibitor, Mocetinostat, in Melanoma,” supplement, Journal of Clinical Oncology 38, no. 15_suppl (2020): 10052, 10.1200/jco.2020.38.15_suppl.10052. [DOI] [Google Scholar]
- 141. Zakharia Y., Singer E. A., Ross R., et al., “Phase Ib/II Study of Durvalumab and Guadecitabine in Advanced Kidney Cancer Big Ten Cancer Research Consortium BTCRC GU16‐043,” supplement, Journal of Clinical Oncology 39, no. 6_suppl (2021): 328, 10.1200/jco.2021.39.6_suppl.328.33356419 [DOI] [Google Scholar]
- 142. Pili R., Quinn D. I., Albany C., et al., “Immunomodulation by HDAC Inhibition: Results From a Phase Ib Study With Vorinostat and Pembrolizumab in Metastatic Urothelial, Renal, and Prostate Carcinoma Patients,” supplement, Journal of Clinical Oncology 37, no. 15_suppl (2019): 2572, 10.1200/jco.2019.37.15_suppl.2572. [DOI] [Google Scholar]
- 143. Taylor K., Loo Yau H., Chakravarthy A., et al., “An Open‐Label, Phase II Multicohort Study of an Oral Hypomethylating Agent CC‐486 and Durvalumab in Advanced Solid Tumors,” Journal for Immunotherapy of Cancer 8, no. 2 (2020): e000883, 10.1136/jitc-2020-000883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Chen S., Xie P., Cowan M., et al., “Epigenetic Priming Enhances Antitumor Immunity in Platinum‐Resistant Ovarian Cancer,” Journal of Clinical Investigation 132, no. 14 (2022): e158800, 10.1172/jci158800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Baretti M., Murphy A. G., Zahurak M., et al., “A Study of Using Epigenetic Modulators to Enhance Response to Pembrolizumab (MK‐3475) in Microsatellite Stable Advanced Colorectal Cancer,” Clinical Epigenetics 15, no. 1 (2023): 74, 10.1186/s13148-023-01485-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Wang F., Jin Y., Wang M., et al., “Combined Anti‐PD‐1, HDAC Inhibitor and Anti‐VEGF for MSS/PMMR Colorectal Cancer: A Randomized Phase 2 Trial,” Nature Medicine 30, no. 4 (2024): 1035–1043, 10.1038/s41591-024-02813-1. [DOI] [PubMed] [Google Scholar]
- 147. Rodriguez C. P., Wu Q., Voutsinas J., et al., “A Phase II Trial of Pembrolizumab and Vorinostat in Recurrent Metastatic Head and Neck Squamous Cell Carcinomas and Salivary Gland Cancer,” Clinical Cancer Research 26, no. 4 (2020): 837–845, 10.1158/1078-0432.CCR-19-2214. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article, as no data sets were generated or analyzed during the current study.