

Abstract
We previously identified Sys1p as a high copy number suppressor of Ypt6 GTPase-deficient yeast mutants that are defective in endosome-to-Golgi transport. Here, we show that Sys1p is an integral membrane protein that resides on a post-endoplasmic reticulum (ER) organelle(s). Affinity studies with detergent- solubilized yeast proteins showed that the C-terminal 53 amino acid tail of Sys1p binds effectively to the cytoplasmic Sec23p–Sec24p COPII subcomplex. This binding required a di-acidic Asp-Leu-Glu (DXE) motif, previously shown to mediate efficient ER export of the vesicular stomatitis virus glycoprotein in mammalian cells. In Sys1p, a Glu-Leu-Glu (EXE) sequence could not substitute for the (DXE) motif. Mutations of the (DXE) sequence resulted in ER retention of ∼30% of the protein at steady state, whereas addition of the Sys1p tail to an ER-resident membrane protein led to an intracellular redistribution of the chimeric protein. Our study demonstrates for the first time that, in yeast, a di-acidic sequence motif can act as a sorting signal for cargo selection during the formation of transport vesicles at the ER by direct binding to COPII component(s).
Keywords: cargo sorting/COPII/di-acidic motif/ER/Sys1 protein
Introduction
In eukaryotic cells, proteins imported into the endoplasmic reticulum (ER) lumen or inserted into the ER membrane for further delivery to other cellular locations are first transported to the Golgi in small vesicles. Transport vesicles are generated by the successive recruitment to the ER membrane of GTP-bound Sar1 GTPase and two heterodimeric protein complexes, Sec23p–Sec24p and Sec13p–Sec31p, which together form a coat, termed COPII (Barlowe et al., 1994; Matsuoka et al., 1998). During vesicle budding, soluble and integral membrane cargo must be separated from ER-resident proteins and, most likely, concentrated (Mizuno and Singer, 1993; Balch et al., 1994) by still ill-defined mechanisms. Studies with yeast have shown that membrane and soluble cargo proteins can be selected for incorporation into COPII vesicles by Sar1p·GTP-dependent binding to the Sec23p– Sec24p subcomplex (Kuehn et al., 1998; Springer and Schekman, 1998). As the Golgi syntaxin Sed5p, which cycles through the ER (Wooding and Pelham, 1998), was found to directly bind Sec24p (Peng et al., 1999), it seems likely that integral membrane cargo proteins in general can be sorted by binding to this COPII component. The specificity of the cargo–vesicle coat interaction implies the presence of sorting signals in the cytosolic sequence of membrane cargo. Two such signals have been discovered in proteins that exit the ER in mammalian cells. First, a C-terminal di-phenylalanine motif mediating binding to the COPII coat is important for efficient ER-to-Golgi transport of ERGIC-53, a receptor for glycoproteins, and of cargo receptors of the p24 family (Fiedler et al., 1996; Kappeler et al., 1997; Dominguez et al., 1998). Secondly, a di-acidic sorting signal (Asp-X-Glu, where X is any amino acid) has been uncovered in the C-terminus of the vesicular stomatitis virus glycoprotein (VSV-G), and shown to act in concentrating the cargo molecule and enhancing the rate of its exit from the ER (Nishimura and Balch, 1997).
In studying the functional role of the yeast Sys1p, a 23.7 kDa transmembrane protein which at high intracellular concentration suppresses the perturbations of endosome-to-Golgi transport in Ypt6 GTPase-defective cells (Tsukada and Gallwitz, 1996), we found that the transmembrane protein is localized to a post-ER compartment and that its suppressor activity is entirely dependent on the 52 amino acid long hydrophilic C-terminal tail. This fragment exhibits efficient binding to the Sec24p–Sec23p COPII subcomplex, which is lost specifically after mutating or deleting an Asp-Leu-Glu sequence three amino acids in front of the C-terminal end. Our study demonstrates that yeast uses the (DXE) motif as an ER exit signal for membrane cargo and that, at least in the case of Sys1p, the (DXE) sequence binds efficiently to the Sec23p– Sec24p COPII vesicle coat complex.
Results
Sys1p appears to be a Golgi/endosomal membrane protein
The 203 amino acid long Sys1 protein is hydrophobic over its entire length except for its N- and C-terminal regions (Tsukada and Gallwitz, 1996). A MEMSAT model (Jones et al., 1994) predicts four transmembrane domains, of which the two in the C-terminal half of the protein are rich in phenylalanine, a characteristic of a number of Golgi membrane proteins (Bretscher and Munro, 1993; Banfield et al., 1995).
To gain further insight into the function and intracellular localization of Sys1p, we first examined its distribution by differential centrifugation of cell lysates. To limit proteolysis, lysates were prepared by disrupting logarithmically grown cells in a mortar after they were frozen in liquid nitrogen. The frozen material was dissolved in proteinase inhibitor-containing buffer and successively subjected to a 10 000 and 100 000 g centrifugation to enrich for ER and Golgi membranes, respectively. Immunoblot analysis (Figure 1A) of total protein of these fractions showed that whereas the luminal ER protein Kar2p (Rose et al., 1989) was nearly exclusively in the 10 000 g pellet fraction (P10), Sys1p and the Golgi membrane protein Emp47p (Schröder et al., 1995) were distributed between the P10 and P100 pellet but absent from the fraction of soluble proteins (S100). Treatment of the cell lysate with either Triton X-100, high salt or urea established that Sys1p, like Emp47p, but in contrast to Kar2p, behaved like an integral membrane protein and was solubilized only with detergent. Membranes of different cellular compartments were further separated by centrifugation of cell lysates through sucrose gradients. As shown in Figure 1B, Sys1p partially overlapped with the Golgi membrane proteins Emp47p, Sed5p and Kex2p. Although the outcome of gradient centrifugations varied somewhat, the peak of Sys1p with respect to that of Emp47p was generally displaced to slightly higher sucrose concentration (also compare gradients in Figures 2 and 7). Importantly, Sys1p together with most of the various Golgi proteins was well separated from the ER marker Kar2p.

Fig. 1. Sys1p is a Golgi/endosome membrane protein. (A) A cleared yeast cell lysate (strain SEY6210) was divided into four aliquots that were treated for 15 min on ice with either lysis buffer (untreated), detergent, high salt or urea as indicated. After consecutive centrifugation at 10 000 and 100 000 g, the pellets (P10, P100) and the supernatants (S100) were subjected to immunoblot analysis with anti-Kar2p (ER marker), anti-Emp47p (Golgi marker) or anti-Sys1p antibodies. (B) The cleared lysate was centrifuged on sucrose gradients, and fractions (1–12, from top to bottom) were collected and subjected to immunoblot analysis with antibodies to the proteins shown to the left.

Fig. 2. Sys1p does not cycle through the ER. Cleared lysates of sec23 (A) or sec12 (B) mutant cells, grown at either permissive (25°C) or non-permissive temperature (35°C), were subjected to differential centrifugation (A) or sucrose gradient centrifugation (B) and subsequently to immunoblot analysis as described in the legend to Figure 1.

Fig. 7. Deletion or mutation of the di-acidic motif from the Sys1p C-terminus moderately affects ER export. Cleared lysates from (A) wild-type yeast (strain CV1), (B) yeast cells expressing only a C-terminally truncated or (C) the indicated mutant version of Sys1p (strains CV2 and CV3) were subjected to sucrose gradient centrifugation, followed by immunoblot analysis of the proteins in gradient fractions 1–12 using anti-HA antibodies. Immunoblots were examined quantitatively using a LumiImager. Note a shift of some of the truncated Sys1(1–186) protein to the position of the ER marker Kar2p.
These fractionation studies showed that Sys1p resides on post-ER organelle(s) and, together with Sys1p’s ability to suppress defects in Ypt6 GTPase-regulated endosome-to-Golgi transport (Tsukada and Gallwitz, 1996; Tsukada et al., 1999; Siniossoglou et al., 2000), indicate that the protein might be primarily associated with Golgi and/or endosomal membranes.
Sys1p does not cycle through the ER
As several Golgi membrane proteins cycle through the ER, we sought to investigate whether Sys1p could also follow such a recycling pathway. In this study, we made use of sec12 and sec23 mutants, which at non-permissive temperature (35–37°C) are completely defective in budding of transport vesicles from the ER and consequently accumulate recycling Golgi proteins in the ER (Schröder et al., 1995; Lewis and Pelham, 1996). Cells of a sec12 strain grown either at 24°C or for 1 h at 35°C were lysed and subjected to differential centrifugation and immunoblot analysis as described above. As can be seen in Figure 2A, a significant part of Emp47p (which cycles through the ER) was redistributed at 35°C from the P100 fraction to the ER-enriched P10 fraction, whereas Sys1p stayed primarily in the P100 fraction. In parallel, cells of a sec23 mutant strain were pre-cultivated at permissive conditions and further grown at either 24 or 35°C in the presence of cycloheximide to block protein synthesis. Cell lysates prepared from spheroplasts were then subjected to sucrose gradient centrifugation, and immunoblots from gradient fractions were performed with specific antibodies directed against the recycling Emp47p, with anti-Kar2p antibodies to identify the ER-containing fractions and with anti-Sys1p antibodies to follow the fate of the protein under study. At 25°C, Emp47p and Sys1p partially overlapped, but the peak of Sys1p was at a higher density than that of Emp47p. However, at non-permissive temperature, the bulk of Emp47p had shifted to the position of the ER marker Kar2p, whereas the position of Sys1p in the gradient had not changed at all (Figure 2B). This indicated clearly that Sys1p does not cycle through the ER.
The hydrophilic C-terminus of Sys1p faces the cytoplasm and is required for ypt6 suppressor activity
In a first approach to identify functional domains of Sys1p, N- and C-terminal truncation mutants were generated and tested for their ability to suppress the temperature sensitivity of a ypt6 deletion strain. This strain was transformed with various sys1 mutant genes (Figure 3A and B) under the transcriptional control of the galactose-inducible GAL10 promoter in the multicopy vector pYX213. The growth of galactose-induced cells was then scored on plates at permissive (25°C) and non-permissive (37°C) temperature. Whereas short N-terminal truncations and deletion of >60% of the C-terminal tail did not affect ypt6 suppressor activity of Sys1p, deletion of the entire C-terminal hydrophilic domain rendered the protein unable to rescue the GTPase deletion mutant from its growth defect at high temperature (Figure 3).

Fig. 3. Delineation of ypt6 mutant suppressor activity and membrane topology of Sys1p. (A) Schematic representation of C-terminally truncated Sys1 mutant proteins tested for suppressor activity. The four putative transmembrane domains (TM1–4) and the first and last amino acids of the various Sys1 proteins are indicated. (B) The sequence of the C-terminal tail of Sys1p is shown with arrows pointing to the end points of truncations. (C) The C-terminal region of Sys1p faces the cytosol. A 500 g supernatant of lysed cells was treated with proteinase K, and TCA-precipitated proteins were subjected to immunoblot analysis with antibodies to the luminal ER protein Kar2p, the luminal part of the Golgi protein Emp47p, the cytoplasmic region of the v-SNARE Sec22p and the C-terminus of Sys1p.
In a first attempt to determine the membrane topology of Sys1p, we constructed a yeast strain in which the SYS1 wild-type gene was replaced by a modified gene expressing a C-terminally HA-tagged Sys1p. Membranes of gently lysed cells were subjected to mild proteinase K digestion and, as shown in Figure 3C, the Sys1p C-terminus was easily digested whereas the ER luminal Kar2p and the type I integral Golgi membrane Emp47p were protected from protease digestion. Under the experimental conditions chosen, a small part of the SNARE Sec22p (a type II membrane protein) resisted digestion, most likely due to its strong interactions with other SNAREs.
These results indicate that the Sys1p C-terminal tail faces the cytoplasm and that the 20 amino acid segment following the putative transmembrane domain TM4 (Figure 3B) is an important functional domain.
Sys1p binds the COPII heterodimer Sec23p–Sec24p
Based on the results of the ypt6 suppressor test, the complete C-terminal tail of Sys1p (amino acids 151–203) was fused to glutathione S-transferase (GST) with the goal of identifying interacting proteins. The GST–Sys1p fusion protein was immobilized on glutathione–Sepharose and used as an affinity matrix with Saccharomyces cerevisiae proteins solubilized in the presence of 1% CHAPS. After extensive washing, bound proteins were eluted from the affinity columns with 1 M NaCl or directly with SDS– PAGE buffer. Two proteins larger than the 30 kDa fusion protein and exhibiting a molecular mass of ∼85 and 100 kDa, respectively, were found to bind efficiently to the fusion protein but not to GST alone. Both proteins could easily be detected on SDS gels by protein staining (Figure 4). Immunoblotting with polyclonal anti-Sec23p and anti-Sec24p antibodies (Figure 6C) identified these proteins as the cytoplasmic COPII heterodimeric Sec23p– Sec24p complex, which is essential for transport vesicle formation at the ER. Two other proteins of ∼40 and 50 kDa, seen in Figure 4, did not specifically bind to the GST–Sys1 fusion protein but also to GST–agarose beads, and the intensity of these protein bands differed from experiment to experiment (compare also Figure 6B).
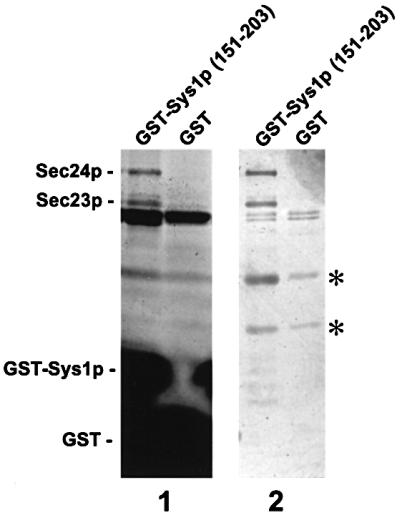
Fig. 4. The C-terminal tail of Sys1p (amino acids 151–203) interacts with the Sec23p–Sec24p complex. GST and a GST–Sys1 tail fusion protein, coupled to agarose beads, served as affinity matrix with detergent-solubilized total yeast protein. Beads were washed with buffer, and bound proteins eluted in 1 ml fractions of SDS buffer (1) or 0.5 ml fractions of 1 M NaCl (2) for SDS–PAGE and silver staining (20 µl of each fraction). Bound proteins usually appeared in two fractions, one of which is shown on the blot. Asterisks mark two unspecifically bound proteins of ∼40 and 50 kDa.

Fig. 6. A di-acidic motif of the Sys1p tail is responsible for Sec23p–Sec24p binding. (A) The hydrophilic tail of Sys1p (WT) and several deletion or substitution mutants (C1–C7) were fused to GST and probed for Sec23p–Sec24p interaction. (B) COPII subcomplex binding was investigated by an affinity approach as described in the legend to Figure 4 and is shown for some tail fusions. Proteins bound to beads were eluted with 0.5 ml of 1 M NaCl, 20 µl fractions were subjected to SDS–PAGE and proteins were identified by silver staining. (C) Sec23p and Sec24p were verified with affinity-purified antibodies. (D) Sar1p, identified by immunoblot analysis, does not interact with Sec23p–Sec24p-binding Sys1p tail fusions.
These findings suggest that for transport to its final destination, the C-terminus of Sys1p might direct the protein into COPII vesicles by an efficient interaction with the Sec23p–Sec24p subcomplex.
Binding to Sec23p–Sec24p depends on a (DXE) motif in the C-terminus of Sys1p
We next tried to establish which sequence of the C-terminal domain of Sys1p was responsible for COPII subcomplex interaction. Inspection of the tail sequence shows that it contains two sequence motifs: a double phenylalanine (FF) and an Asp-Leu-Glu (DXE, where X is any amino acid). In the ERGIC-53 and the p24 family members, and in the VSV-G protein, respectively (Figure 5A), these motifs have been shown to be of importance for efficient ER export, which might be mediated by binding to the Sec23p–Sec24p complex in mammalian cells (Kappeler et al., 1997; Nishimura and Balch, 1997; Dominguez et al., 1998).

Fig. 5. Comparison of C-terminal sequences of yeast Sys1, Bap2 and Ptr2 and of mammalian ERGIC-53 and viral VSV-G proteins. (A) The (FF) and the di-acidic motifs effective in efficient ER export in mammalian cells are highlighted. In Sys1p, the (DLE) but not the (FF) motif mediates binding to COPII components. (B) The location of (DXE) motifs in Sys1p and the plasma membrane-localized transporters Bap2p and Ptr2p are shown.
GST fusions of the 53 amino acid tail of Sys1p or several truncations thereof, with and without various amino acid substitutions (Figure 6A), were analyzed for their interaction with Sec23p–Sec24p as described above. The outcome of the affinity binding study is summarized in Figure 6A, and the experimental results for some of the constructs are documented in Figure 6B and C. Substitution with a double alanine (mutant C1) or deletion of the (FF) motif (not shown) did not affect binding of the Sys1p C-terminus to Sec23p–Sec24p. The additional replacement of a double glutamine (QQ), also present in the C-terminus of ERGIC-53 (Kappeler et al., 1997), did not alter binding to the COPII subcomplex either. Deletion of the 32 C-terminal amino acid residues (mutant C2) completely abolished Sec23p–Sec24p interaction. This fragment alone (mutant C3) was as efficient in COPII subcomplex interaction as the complete hydrophilic C-terminal region. The 32 amino acid tail of Sys1p contains the (DXE) motif (DLE) and the related sequence Glu-Leu-Glu (ELE). When the acidic residues of both motifs were separately replaced by alanine (mutants C4 and C7), the (D198A/E200A) but not the (E188A/E190A) mutant lost the ability to bind Sec23p–Sec24p (Figure 6B and C). In further experiments we found that single substitutions of either D198A or E200A (mutants C5 and C6) likewise abolished Sec23p–Sec24p binding to the Sys1p tail. This is the first demonstration that a di-acidic (DXE) sequence motif in yeast can mediate efficient and direct COPII interaction, but the related (EXE) motif cannot.
Sar1p in its GTP-bound conformation has been shown to recruit the COPII subunit Sec23p–Sec24p to the sites of vesicle budding in yeast and mammalian cells (Matsuoka et al., 1998; Aridor et al., 2001). Because data from a recent report suggest that a ternary complex of the (DXE) sorting signal, the Sec23p–Sec24p complex and Sar1p·GTP is important for efficient ER export of VSV-G in COS cells (Aridor et al., 2001), we investigated whether Sar1p in addition to Sec23p–Sec24p was retained on affinity beads containing the GST–Sys1p fusion. As shown in Figure 6D, even by immunoblot analysis no evidence for Sar1p binding to the Sys1p tail sequence could be obtained.
By performing a computer search for (DXE) motifs in the C-termini of other known and putative yeast membrane proteins, we noted that several plasma membrane-localized transporter proteins have one or more clusters of acidic amino acids in their tails. Among them, Bap2p, a 67.7 kDa amino acid transporter (Grauslund et al., 1995), and Ptr2p, a 68.1 kDa peptide transporter (Perry et al., 1994), contain a typical (DXE) motif close to the C-terminal end (Figure 5B). We fused the tails of the two proteins, each beginning at the end of the last membrane-spanning domain, to GST and performed affinity binding studies exactly like those with the Sys1p tail. In marked contrast to Sys1p, binding of Sec23p– Sec24p to the tails of both transporters could only be observed with the help of antibodies to the COPII components in western blots with the affinity-bound proteins (data not shown). This experimental outcome is like the results reported for mammalian (FF) motif-containing proteins (Kappeler et al., 1997; Dominguez et al., 1998) and (DXE) motif-containing proteins (Nishimura et al., 1999) and does not exclude the possibility of an indirect interaction between cargo and COPII.
Deletion of the (DXE) motif results in partial mislocalization of Sys1p to the ER
As in mammalian cells, mutation of the (DXE) motif in VSV-G leads to inefficient ER export of the cargo protein, we sought to investigate whether its absence from the Sys1p C-terminal tail affected transport of the protein. For this study, we created sys1 deletion strains expressing HA-tagged versions of either the Sys1 wild-type protein, Sys1(D198A/E200A) mutant protein or Sys1(1–186)p lacking the C-terminal 17 amino acid residues. We chose to delete the (DXE) sequence along with surrounding sequences in one case since a recent study had shown that the signal accounting for efficient ER export of VSV-G protein in mammalian cells includes four additional amino acids proximal to the di-acidic motif (Sevier et al., 2000). Cells of the three strains were spheroplasted, lysed, and cleared lysates fractionated on sucrose gradients. Gradient fractions were subjected to immunoblot analysis to localize the ER marker Kar2p, the Golgi marker Emp47p, and the wild-type and mutant Sys1 proteins (Figure 7). As already shown in Figures 1 and 2, the peak fractions of wild-type Sys1p partially overlapped with those of Emp47p, but were well displaced from Kar2p. In contrast, part of the C-terminally truncated Sys1(1–186) and the Sys1(D198A/E200A) mutant protein co-migrated with the ER marker. Quantification of band intensities of three separate gradients using a LumiImager revealed that 25–35% of the Sys1 mutant proteins migrated with the ER-containing gradient fractions.
These results suggest that, at steady state, the di-acidic (DXE) motif serves a function in efficient ER export of membrane cargo also in yeast.
The Sys1p (DXE) motif containing sequence fused to Wbp1p results in redistribution of the ER-resident protein
To determine whether the Sys1p tail with its (DXE) motif would allow or accelerate export of an ER type I membrane protein, the essential oligosaccharyl transferase subunit Wbp1p (te Heesen et al., 1993) was engineered such that its seven C-terminal, cytoplasmically oriented residues including the ER retrieval sequence KKXX were deleted and replaced by the 50 amino acid long Sys1p tail (either wild-type or [D198A/E200A] mutant version). The fusion genes under the transcriptional control of the WBP1 promoter were inserted into the CEN-vector pRS315 and transformed into the protease-deficient yeast strain c13ABYS. Organelles of gently lysed cells were then separated by sucrose gradient centrifugation, and wild-type and chimeric Wbp1p were identified with an anti-Wbp1p antibody (Figure 8). In untransformed cells, Wbp1p, which despite having an ER retrieval sequence does not cycle through the Golgi (Gaynor et al., 1994), co-localized perfectly with the ER marker Kar2p (Figure 8A). Remarkably, in cells expressing the Wbp1–Sys1 fusions, proteins reacting with the anti-Wbp1 antibody were largely shifted to gradient fractions of lower density and, like the wild-type Sys1p, partially overlapped with the Golgi protein Emp47p (Figure 8B), suggesting redistribution of the chimeric protein presumably to post-Golgi compartment(s). As wild-type and chimeric Wbp1p, although somewhat different in molecular mass, were not distinguishable on the immunoblots and the typically ER-localized wild-type Wbp1p gave a rather weak signal, it could well be that by expressing the chimeric Wbp1 protein, wild-type Wbp1p, which is under complex transcriptional control (Knauer and Lehle, 1994; Silberstein et al., 1995), was down-regulated. Most importantly, however, the redistribution of the chimeric protein was not observed when it carried the Sys1p tail with the (DXE) motif mutated to (AXA) (Figure 8C).

Fig. 8. Fusion of the Sys1p tail to the ER-resident protein Wbp1p leads to redistribution of the fusion protein in a (DXE) motif-dependent fashion. Cleared lysates of the protease-deficient strain cl3-ABYS-86, untransformed (A) or transformed with a recombinant plasmid expressing the indicated Wbp1-Sys1p tail fusion proteins (B and C), were subjected to sucrose gradient centrifugation. Proteins of different fractions were probed with antibodies specific for the ER marker Kar2p, the Golgi protein Emp47p and Wbp1p. Note the shift of the Wbp1p–Sys1 fusion protein with regard to its (DXE)-mutated version and to Wbp1 wild-type protein.
This finding strongly suggests that the Sys1 protein cytoplasmic tail fused to the C-terminus of an ER-resident membrane protein can force the chimeric protein to leave the ER in a (DXE)-dependent fashion.
Discussion
We have shown here that the Sys1 protein that we had previously identified as a suppressor of Ypt6 GTPase- dependent endosome-to-Golgi trafficking defects (Tsukada and Gallwitz, 1996) fulfills, as predicted from its primary sequence, the criteria for an integral membrane protein. In accordance with its likely functional involvement in vesicular transport between endosome(s) and the late Golgi, we find that Sys1p is localized to post-ER compartment(s). Sucrose density gradient centrifugation of cell lysates, a method widely and successfully used for protein localization studies in yeast (Antebi and Fink, 1992; Schröder et al., 1995), excludes the ER, the plasma membrane or the vacuole as the primary residence of Sys1p. Instead, partial co-migration of Sys1p with several early/late Golgi marker proteins suggests that it is mainly associated with Golgi and/or endosomal membranes, which on density gradients exhibit only slightly different sedimentation behavior. This is supported by preliminary indirect immunofluorescence analyses showing that Sys1p appears to be confined to punctate structures generally seen with Golgi and endosomal proteins.
Regardless of its exact intracellular localization, newly synthesized Sys1p as biosynthetic cargo must leave the ER in transport vesicles to reach its final destination. The 52 amino acid long C-terminal tail of the protein, rich in charged and polar residues, was reasoned to be a prime candidate for serving functional purposes in protein transport and/or cargo selection. C-terminal truncations now demonstrate that a function in both seems to be the case. As we show in this report, the deletion of 32 amino acids from the C-terminus did not affect the ability of Sys1p to suppress the growth defect of ypt6 mutants, the test for the protein’s biological activity. Removal of only 18 additional residues abolished the suppressor function, suggesting that a short tail segment of <20 amino acids immediately adjacent to the most C-terminally located transmembrane domain is important for the biological activity of Sys1p. This result prompted a search for tail-interacting proteins, a means to identify and study other components with which the non-essential Sys1p might cooperate. The unanticipated finding that the cytoplasmic Sec23p–Sec24p complex interacts with the hydrophilic C-terminal domain of Sys1p drew our attention to the role of this region in cargo selection and ER export, because lack of the C-terminus might have caused a retention of Sys1p in the ER and, for that reason, loss of suppressor activity.
Although interaction of membrane-bound cargo (SNARE proteins) in yeast with COPII components has been demonstrated previously (Kuehn et al., 1998; Peng et al., 1999), sorting signals for ER export in membrane cargo proteins have not been identified yet. In one case, the p24 family member Emp24p, a Leu-Val dipeptide at the extreme C-terminus appears to act in ER export (Nakamura et al., 1998), but a link to the COPII vesicular coat has not been demonstrated. In another study, the N-terminal region of the yeast Golgi GDP-mannose transporter Vrg4p was shown to be of importance for ER export, but neither a signal sequence element within this region nor its possible mode of action could be clarified (Gao et al., 2000). Our present study clearly shows that the efficient binding of the Sys1p C-terminal tail to the Sec23p–Sec24p COPII subcomplex depends on the di-acidic motif Asp-Leu-Glu, but not on a double phenylalanine (FF) which is also present in the C-terminal tail. As the mutation of the (DXE) motif to (AXA) within the Sys1p tail results in the loss of its binding to Sec23p– Sec24p in vitro, we can exclude the possibility that the related (EXE) motif located nine amino acid residues upstream, at least in this particular sequence environment, is able to replace the (DXE) motif for COPII binding. In fact, substitution with alanine of the glutamic acid residues of the (EXE) motif did not even influence the efficient interaction of the mutated tail with Sec23p–Sec24p, which indicates that additional negatively charged amino acid residues are not important for (DXE)-mediated COPII binding.
From vesicle budding reactions with ER membranes in vitro, it has been concluded that the Sar1 GTPase, bound to GTP, is required to allow interaction of membrane cargo, at least of type II membrane proteins, with the Sec23p–Sec24p COPII complex (Springer and Schekman, 1998; Aridor et al., 1999). However, the efficient interaction of the bacterially produced GST–Sys1 tail fusion protein with Sec23p–Sec24p on affinity beads that we have observed appears not to depend on Sar1p·GTP, since even by sensitive immunoblot analysis Sar1p could not be identified among proteins bound to the beads. Furthermore, in our in vitro binding studies, which allowed us easily to detect Sec23p and Sec24p in protein-stained gels, we did not detect any other protein that bound to the Sys1p tail specifically and in amounts comparable to the two COPII components. Therefore, one cannot escape the conclusion that the Sys1p tail sequence binds directly to the Sec23p–Sec24p COPII complex in a (DXE) motif-dependent fashion. As already argued above, this conclusion could not be drawn from previous studies on cargo selection in the ER of mammalian cells (Kappeler et al., 1997; Dominguez et al., 1998; Nishimura et al., 1999) or from the findings with two other yeast proteins described here. Our study demonstrates that the efficiency of di-acidic motif-dependent transmembrane cargo interaction with Sec23p–Sec24p varies from protein to protein and might depend on the sequence context of the sorting signal and/or its distance from the C-terminal end. We have shown previously that the Golgi syntaxin Sed5p binds to Sec24p directly (Peng et al., 1999). It seems likely, therefore, that it is the Sec24 protein that interacts with certain membrane cargo during vesicle formation at the ER.
VSV-G is the cargo protein that was first described to require a C-terminal cytoplasmic domain (Doms et al., 1988) and, within it, the di-acidic sequence motif (Nishimura and Balch, 1997) for efficient ER-to-Golgi transport in mammalian cells. VSV-G and the potassium channel proteins Kir1.1 and Kir2.1 (Ma et al., 2001) are, besides the yeast Sys1 protein described here, the only other proteins where a role in ER export of this particular sorting signal has been demonstrated. From studies on transport of the VSV-G protein, it has been concluded not only that the sequence context of the (DXE) motif is important for its effective function (Sevier et al., 2000), but also that the di-acidic sequence acts in the concentration of biosynthetic cargo by recruiting a hypothetical ‘linker component’ (Aridor et al., 2001) to the pre-budding complex. From the very efficient (DXE) motif-dependent interaction of Sys1p with the Sec23p–Sec24p complex and the absence of other proteins that would bind specifically and with similar abundance to the Sys1p tail sequence like Sec23p–Sec24p, we conclude that this interaction is direct and not mediated by another component, definitely not by the GTPase Sar1p. It therefore appears that the main purpose of this interaction is to help collect cargo during vesicle formation and facilitate its export from the ER. In accord with this assumption are our findings that, in vivo, the deletion or a single amino acid substitution (D198A or E200A) of the (DXE) motif leads to a deceleration of ER export of Sys1p, as seen by the accumulation of the protein in the ER fraction at steady state. That this is the consequence of a defect in protein folding with a concomitant loss of Sec23p–Sec24p binding is highly unlikely since the in vitro affinity binding studies were carried out with only a 32 amino acid long tail fragment (fused to GST) and not with the entire membrane-inserted Sys1p. Further evidence for the role of the (DXE) motif in facilitating ER export comes from our finding that on fusing the C-terminal tail of Sys1p to the ER-resident membrane protein Wbp1p (te Heesen et al., 1993; Gaynor et al., 1994), the chimeric protein, according to gradient fractionation of cellular organelles, leaves the ER in a (DXE) motif-dependent fashion. Although we could not determine the exact intracellular location of the Wbp1– Sys1 tail fusion protein, on gradients it co-localized almost perfectly with Sys1p-containing membrane fractions.
As is the case with the VSV-G protein and the potas sium channels in mammalian cells, the tail sequence-contained (DXE) motif of the yeast Sys1 protein facilitates ER export, but its mutation or deletion does not block it. This finding then explains why the deletion of the 32 C-terminal amino acids including the (DXE) motif does not interfere with the ability of the so truncated Sys1 mutant protein to suppress, at high intracellular concentrations, the growth defect of Ypt6 GTPase-defective cells. Nevertheless, mutational analyses of the (DXE) motif sequences in the mammalian ER-to-Golgi cargo proteins VSV-G, Kir1.1 and Kir1.2 (Nishimura et al., 1999; Ma et al., 2001) show that these are more effective ER export signals than the (DXE) motif of the yeast Sys1 protein studied here. This could be due to surrounding sequences, especially tyrosine residues N-terminal of the di-acidic motifs, which are present in the viral and some of the potassium channel proteins but absent from the yeast Sys1p.
Materials and methods
Bacterial and yeast strains
Cloning experiments were performed with the Escherichia coli strain DH5α and standard media (Hanahan, 1986). The expression of GST fusion proteins was performed in the protease-deficient strain BL21 (Amersham Pharmacia Biotech). Yeast strains used in this study are listed in Table I. They were grown in standard yeast extract/peptone/dextrose (YEPD) or, for the induction of the Gal promoter of pYX213 (R&D Systems), in standard yeast extract/peptone/galactose (YEPG) medium. Unless indicated otherwise, yeast strain SEY6210 was used for all the experiments. Yeast transformations were carried out by using a modified lithium acetate method (Ito et al., 1983).
Table I. Yeast strains used in this study.
| Strain | Genotype | Source |
|---|---|---|
| SEY6210 | MATα leu2-3,112 ura3-52 his3-Δ200 trp1-Δ901 lys2-801 suc2-Δ9 | Robinson et al. (1988) |
| cI3-ABYS-86 | MATα ura3-Δ5 leu2-3,112 his3 pra1-1 prb1-1 prc1-1 cps1-3 canR | Heinemeyer et al. (1991) |
| SSY78 | MATa sec23-1 trp1 his4 ura3 leu2 EMP47-MYC::LEU2 pHA-PMR1::URA3 | Ossipov et al. (1999) |
| RH1491 | MATa sec12-4 ura3 his4 leu2 lys2 bar1-1 | Schröder et al. (1995) |
| MT1 | MATα his3 leu2 ura3 ypt6::HIS3 | Tsukada and Gallwitz (1996) |
| CV1 | MATα leu2-3,112 ura3-52 his3-Δ200 trp1-Δ901 lys2-801 suc2-Δ9 SYS1-6HIS-3HA-loxP-KanMX-loxP | this study |
| CV2 | MATα leu2-3,112 ura3-52 his3-Δ200 trp1-Δ901 lys2-80 suc2-Δ9 SYS1-Δ188-203-6HIS-HA-loxP-KanMX-loxP | this study |
| CV3 | MATα leu2-3,112 ura3-52 his3-Δ200 trp1-Δ901 lys2-80 suc2-Δ9 SYS1(D198A;E200A)-6HIS-HA-loxP-KanMX-loxP | this study |
Plasmids
Plasmids used for the ypt6 suppressor test were constructed as follows: SYS1 and its different 5′- and 3′-terminal truncations were amplified by PCR with a flanking EcoRI site (5′) and HindIII site (3′) and integrated into the pre-digested vector pYX213. The constructs with N-terminal deletions were shortened by 33 and 60 bp, and the constructs with C-terminal deletions by 51, 96 and 150 bp, respectively.
For fusing GST to different parts of the Sys1p C-terminal domain or of the C-terminal domain of Bap2(557–604)p and Ptr2(553–601)p, SYS1, BAP2 and PTR2 were amplified with corresponding primers containing restriction sites for BamHI and HindIII. The PCR products were cut with BamHI and HindIII and ligated with the pre-digested vector pGEX-TT (Amersham-Pharmacia Biotech). Single and double alanine substitutions were generated by site-directed mutagenesis using the QuickChange kit (Stratagene). The mutations were verified by DNA sequencing.
For engineering the sequence around the (DXE) signal of Sys1p into the C-terminus of the ER-resident protein Wbp1p, SYS1 and WBP1 were amplified with two sets of primers. SYS1(154–203): 5′-GAGATAAGC TTAGATGGAGAGAGCTTAGAGAC-3′ (containing a restriction site for HindIII) and 5′-TTTTTGGGATCCGAGATAAGGTTCTTGCCATTT-3′ (BamHI); and WBP1(1–423): 5′-CCCCTCGAGCACCACCAACCCTATTCGAAA-3′ (XhoI) and 5′-GTTTCAAGCTTCTTGCCAACAGAGGAAGTCGTAA-3′ (HindIII). The digested PCR products were integrated into the centromeric vector pRS315.
Epitope tagging
Sys1p, Sys1(D198A;E200A)p and Sys1(1–186)p were C-terminally tagged with His6 followed by three copies of the HA epitope (YPYDVPDYA). For this purpose, the expression cassette His6-3XHA-KanMX was amplified with the vector pUG6H3HA (DDBJ/EMBL/GenBank accession No. AJ132966) as template using the corresponding primers (De Antoni and Gallwitz, 2000).
Antibodies
A polyclonal antibody directed against a C-terminal fragment of Sys1p was produced in rabbits using the bacterially produced GST–Sys1(151–203) fusion protein. Anti–MYC (9E10) and anti-HA (12CA5) monoclonal antibodies were purchased from Santa Cruz Biotechnology and Roche Diagnostics GmbH, respectively. Generation of the antisera against Sec23p and Sec24p (Peng et al., 1999), Sed5p (Grabowski and Gallwitz, 1997), Kar2p (Benli et al., 1996), Sar1p (Saito-Nakano and Nakano, 2000), Wbp1p (te Heesen et al., 1993), Sec22p (Ballensiefen et al., 1998) and Emp47p (Schröder et al., 1995) has been described previously.
Subcellular fractionation and sucrose gradient fractionation
Subcellular fractionations were performed as described previously (Peng et al., 2000). For gradient fractionation of cell organelles, the cell pellet of a 100 ml culture (1 OD600 unit/ml) of SEY6210 was taken up in 5 ml of 100 mM 2-mercaptoethanesulfonic acid, 1.2 M sorbitol, 10 mM HEPES–KOH pH 7.2, 1 mM MgCl2 and kept on ice for 15 min. The cells were washed in 1.2 M sorbitol, 10 mM HEPES–KOH pH 7.2, 1 mM MgCl2 and spheroplasted in 800 µl of the same buffer after adding 150 µl of lyticase (10 mg/ml) and 100 µl of EDTA-free protease inhibitor cocktail for 30 min at 30°C. Spheroplasts were osmotically lysed in water, and cell debris was removed by centrifugation (500 g). The cleared lysate was loaded onto a manually generated 12-step sucrose gradient (1 ml of 60, 54, 50, 46, 42, 38, 34, 30, 26, 22 and 18% sucrose in 10 mM HEPES–KOH pH 7.2, 1 mM MgCl2) and centrifuged at 100 000 g for 3 h at 4°C in a Beckman SW40 rotor. Twelve fractions were collected manually from the top and processed for western blot analysis. Band intensities were calculated with a LumiImager (Boehringer).
Proteinase protection assay
Logarithmically grown cells (100 OD600) were spheroplasted for 30 min at 30°C in 800 µl of 1.2 M sorbitol, 10 mM HEPES–KOH pH 7.2, 1 mM MgCl2 after adding 150 µl lyticase (10 mg/ml) and 100 µl EDTA-free protease inhibitor cocktail. Spheroplasts were osmotically lysed in 1 ml of 50 mM Tris–HCl pH 7.5 and the cell debris was removed by centrifugation at 500 g. Fifty microliters of the cell lysate were incubated with proteinase K (50 µg/ml) for 30 min on ice in a total volume of 100 µl. The reaction was stopped by the addition of 1 mM phenylmethylsulfonyl fluoride. The proteins were precipitated on ice with 10% trichloroacetic acid (TCA), resuspended in 80 µl of loading buffer and subjected to SDS–PAGE and immunoblot analysis.
Affinity purification of yeast protein
For affinity purification, cells of the protease-deficient yeast strain cI3-ABYS-86 were grown overnight in 4 l of YEPD medium. The harvested cells were washed in 20 mM HEPES–KOH pH 7.2. Fifty grams of washed cells were resuspended in 50 ml of lysis buffer (20 mM HEPES–KOH pH 7.2, 3 mM Pefabloc, protease inhibitor cocktail) and disrupted by high-pressure homogenization (French Press). Detergent buffer (20 mM HEPES–KOH pH 7.2, 0.2 M NaCl, 2% CHAPS, 3 mM Pefabloc, protease inhibitors cocktail) was added to the cell lysate to get a final concentration of 1% CHAPS. After a high-speed centrifugation (100 000 g), the supernatant was pre-cleared by the addition of 300 µl of glutathione– Sepharose (Amersham Pharmacia Biotech) to remove proteins with an affinity for the matrix itself. Finally, MgCl2 and CaCl2 were added to a final concentration of 2.5 mM each. For affinity chromatography, the different GST–Sys1p constructs were expressed from the pGEX-TT vector (Amersham Pharmacia Biotech). After transformation of the vector in the protease-deficient E.coli strain BL21, 500 ml of an overnight culture were diluted to a volume of 5 l and incubated at 37°C to an OD600 of 0.6. Recombinant protein expression was induced with isopropyl-β-d-thiogalactopyranoside at a final concentration of 1 mM and the culture was incubated for another 6 h at 30°C. Six grams of cells were washed and resuspended in lysis buffer (50 mM Tris–HCl pH 7.5, 100 mM NaCl, 2.5 mM MgCl2, 2.5 mM CaCl2). After sonification, the cell lysate was adjusted to a Triton X-100 concentration of 1% to solubilize the proteins, and a high-speed supernatant was obtained by centrifugation at 100 000 g for 1 h. This was incubated with 1 ml of glutathione–Sepharose beads by end-over-end rotation for 4 h at 4°C. The beads were twice washed with detergent buffer (50 mM Tris–HCl pH 7.5, 100 mM NaCl, 2.5 mM MgCl2, 2.5 mM CaCl2, 1% Triton X-100) and with high-salt buffer (50 mM Tris–HCl pH 7.5, 1 M NaCl, 2.5 mM MgCl2, 2.5 mM CaCl2). The washed beads were loaded into a plastic column and incubated with the high-speed yeast extract from cells of a 2 l culture (OD600 = 2). Unspecifically bound proteins were washed off the column with 150 mM salt buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 2.5 mM MgCl2, 2.5 mM CaCl2, 0.1% CHAPS). Bound proteins were eluted in 0.5 ml fractions of 1 M NaCl, 5 mM EDTA, 0.5% CHAPS or directly with SDS–PAGE buffer, and 20 µl of fractions containing proteins were subjected to SDS–PAGE. Protein detection was performed by silver staining (Merril et al., 1984) or by immunoblotting using the enhanced chemiluminescence system (Amersham Pharmacia Biotech).
Acknowledgments
Acknowledgements
We thank Ken Sato, Akihiko Nakano (Tokyo) and Marcus Aebi (Zürich) for providing antibodies, Xiaoping Yang and Anna De Antoni for helpful suggestions, and Ingrid Balshüsemann for expert secretarial help. This work has been supported in part by grants to D.G. from the Deutsche Forschungsgemeinschaft and Fonds der Chemischen Industrie.
References
- Antebi A. and Fink,G.R. (1992) The yeast Ca2+-ATPase homologue, PMR1, is required for normal Golgi function and localizes in a novel Golgi-like distribution. Mol. Biol. Cell, 3, 633–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aridor M., Bannykh,S.I., Rowe,T. and Balch,W.E. (1999) Cargo can modulate COPII vesicle formation from the endoplasmic reticulum. J. Biol. Chem., 274, 4389–4399. [DOI] [PubMed] [Google Scholar]
- Aridor M., Fish,F.N., Bannykh,S., Weissman,J., Roberts,T.H., Lippincott-Schwartz,J. and Balch,W.E. (2001) The Sar1 GTPase coordinates biosynthetic cargo selection with endoplasmatic reticulum export site assembly. J. Cell Biol., 152, 213–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch W.E., McCaffery,J.M., Plutner,H. and Farquhar,M.G. (1994) Vesicular stomatitis virus glycoprotein is sorted and concentrated during export from the endoplasmic reticulum. Cell, 76, 841–852. [DOI] [PubMed] [Google Scholar]
- Ballensiefen W., Ossipov,D. and Schmitt,H.D. (1998) Recycling of the yeast v-SNARE Sec22p involves COPI-proteins and the ER transmembrane proteins Ufe1p and Sec20p. J. Cell Sci., 111, 1507–1520. [DOI] [PubMed] [Google Scholar]
- Banfield D.K., Lewis,M.J. and Pelham,H.R. (1995) A SNARE-like protein required for traffic through the Golgi complex. Nature, 375, 806–809. [DOI] [PubMed] [Google Scholar]
- Barlowe C. et al. (1994) COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell, 77, 895–907. [DOI] [PubMed] [Google Scholar]
- Benli M., Döring,F., Robinson,D.G., Yang,X. and Gallwitz,D. (1996) Two GTPase isoforms, Ypt31p and Ypt32p, are essential for Golgi function in yeast. EMBO J., 15, 6460–6475. [PMC free article] [PubMed] [Google Scholar]
- Bretscher M.S. and Munro,S. (1993) Cholesterol and the Golgi apparatus. Science, 261, 1280–1281. [DOI] [PubMed] [Google Scholar]
- De Antoni A. and Gallwitz,D. (2000) A novel multi-purpose cassette for repeated integrative epitope tagging of genes in Saccharomyces cerevisiae. Gene, 246, 179–185. [DOI] [PubMed] [Google Scholar]
- Dominguez M., Dejgaard,K., Fullekrug,J., Dahan,S., Fazel,A., Paccaud,J.P., Thomas,D.Y., Bergeron,J.J. and Nilsson,T. (1998) gp25L/emp24/p24 protein family members of the cis-Golgi network bind both COP I and II coatomer. J. Cell Biol., 140, 751–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doms R.W., Ruusala,A., Machamar,C., Helenius,J., Helenius,A. and Rose,J.K. (1988) Differential effects of mutations in three domains on folding, quarternary structure and intracellular transport of vesicular stomatitis virus G protein. J. Cell Biol., 107, 89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiedler K., Veit,M., Stamnes,M.A. and Rothman,J.E. (1996) Bimodal interaction of coatomer with the p24 family of putative cargo receptors. Science, 273, 1396–1399. [DOI] [PubMed] [Google Scholar]
- Gao X. and Dean,N. (2000) Distinct protein domains of the yeast Golgi GDP-mannose transporter mediate oligomer assembly and export from the endoplasmic reticulum. J. Biol. Chem., 275, 17718–17727. [DOI] [PubMed] [Google Scholar]
- Gaynor E.C., te Heesen,S., Graham,T.R., Aebi,M. and Emr,S.D. (1994) Signal-mediated retrieval of a membrane protein from the Golgi to the ER in yeast. J. Cell Biol., 127, 653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowski R. and Gallwitz,D. (1997) High-affinity binding of the yeast cis-Golgi t-SNARE, Sed5p, to wild-type and mutant Sly1p, a modulator of transport vesicle docking. FEBS Lett., 411, 169–172. [DOI] [PubMed] [Google Scholar]
- Grauslund M., Didion,T., Kielland-Brandt,M.C. and Andersen,H.A. (1995) BAP2, a gene encoding a permease for branched-chain amino acids in Saccharomyces cerevisiae. Biochim. Biophys. Acta, 1269, 275–280. [DOI] [PubMed] [Google Scholar]
- Hanahan D. (1986) Cloning: A Practical Approach. IRL Press, Oxford, UK, pp. 109–135.
- Heinemeyer W., Kleinschmidt,J.A., Saidowsky,J., Escher,C. and Wolf,D.H. (1991) Proteinase yscE, the yeast proteasome/multicatalytic-multifunctional proteinase: mutants unravel its function in stress induced proteolysis and uncover its necessity for cell survival. EMBO J., 10, 555–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H., Fukuda,Y., Murata,K. and Kimura,A. (1983) Transformation of intact yeast cells treated with alkali cations. J. Bacteriol., 153, 163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D.T., Taylor,W.R. and Thornton,J.M. (1994) A model recognition approach to the prediction of all-helical membrane protein structure and topology. Biochemistry, 33, 3038–3049. [DOI] [PubMed] [Google Scholar]
- Kappeler F., Klopfenstein,D.R., Foguet,M., Paccaud,J.P. and Hauri,H.P. (1997) The recycling of ERGIC-53 in the early secretory pathway. ERGIC-53 carries a cytosolic endoplasmic reticulum-exit determinant interacting with COPII. J. Biol. Chem., 272, 31801–31808. [DOI] [PubMed] [Google Scholar]
- Knauer R. and Lehle,L. (1994) The N-oligosaccharyltransferase complex from yeast. FEBS Lett., 344, 83–86. [DOI] [PubMed] [Google Scholar]
- Kuehn M.J., Herrmann,J.M. and Schekman,R. (1998) COPII–cargo interactions direct protein sorting into ER-derived transport vesicles. Nature, 391, 187–190. [DOI] [PubMed] [Google Scholar]
- Lewis M.J. and Pelham,H.R. (1996) SNARE-mediated retrograde traffic from the Golgi complex to the endoplasmic reticulum. Cell, 85, 205–215. [DOI] [PubMed] [Google Scholar]
- Ma D., Zerangue,N., Lin,Y.F., Collins,A., Yu,M., Jan,Y.N. and Jan,L.Y. (2001) Role of ER export signals in controlling surface potassium channel numbers. Science, 291, 316–319. [DOI] [PubMed] [Google Scholar]
- Matsuoka K., Orci,L., Amherdt,M., Bednarek,S.Y., Hamamoto,S., Schekman,R. and Yeung,T. (1998) COPII-coated vesicle formation reconstituted with purified coat proteins and chemically defined liposomes. Cell, 93, 263–275. [DOI] [PubMed] [Google Scholar]
- Merril C.R., Goldman,D. and Van Keuren,M.L. (1984) Gel protein stains: silver stain. Methods Enzymol., 104, 441–447. [DOI] [PubMed] [Google Scholar]
- Mizuno M. and Singer,S.J. (1993) A soluble secretory protein is first concentrated in the endoplasmic reticulum before transfer to the Golgi apparatus. Proc. Natl Acad. Sci. USA, 90, 5732–5736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura N., Yamazaki,S., Sato,K., Nakano,A., Sakaguchi,M. and Mihara,K. (1998) Identification of potential regulatory elements for the transport of Emp24p. Mol. Biol. Cell, 9, 3493–3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura N. and Balch,W.E. (1997) A di-acidic signal required for selective export from the endoplasmic reticulum. Science, 277, 556–558. [DOI] [PubMed] [Google Scholar]
- Nishimura N., Bannykh,S., Slabough,S., Matteson,J., Altschuler,Y., Hahn,K. and Balch,W.E. (1999) A di-acidic (DXE) code directs concentration of cargo during export from the endoplasmic reticulum. J. Biol. Chem., 274, 15937–15946. [DOI] [PubMed] [Google Scholar]
- Ossipov D., Schröder-Köhne,S. and Schmitt,H.D. (1999) Yeast ER-Golgi v-SNAREs Bos1p and Bet1p differ in steady-state localization and targeting. J. Cell Sci., 112, 4135–4142. [DOI] [PubMed] [Google Scholar]
- Peng R., Grabowski,R., De Antoni,A. and Gallwitz,D. (1999) Specific interaction of the yeast cis-Golgi syntaxin Sed5p and the coat protein complex II component Sec24p of endoplasmic reticulum-derived transport vesicles. Proc. Natl Acad. Sci. USA, 96, 3751–3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng R., De Antoni,A. and Gallwitz,D. (2000) Evidence for overlapping and distinct functions in protein transport of coat protein Sec24p family members. J. Biol. Chem., 275, 11521–11528. [DOI] [PubMed] [Google Scholar]
- Perry J.R., Basrai,M.A., Steiner,H.Y., Naider,F. and Becker,J.M. (1994) Isolation and characterization of a Saccharomyces cerevisiae peptide transport gene. Mol. Cell. Biol., 14, 104–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J.S., Klionsky,D.J., Banta,L.M. and Emr,S.D. (1988) Protein sorting in Saccharomyces cerevisiae: isolation of mutants defective in the delivery and processing of multiple vacuolar hydrolases. Mol. Cell. Biol., 8, 4936–4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose M.D., Misra,L.M. and Vogel,J.P. (1989) KAR2, a karyogamy gene, is the yeast homolog of the mammalian BiP/GRP78 gene. Cell, 57, 1211–1221. [DOI] [PubMed] [Google Scholar]
- Saito-Nakano Y. and Nakano,A. (2000) Sed4p functions as a positive regulator of Sar1p probably through inhibition of the GTPase activation by Sec23p. Genes Cells, 5, 1039–1048. [DOI] [PubMed] [Google Scholar]
- Schröder S., Schimmoller,F., Singer-Kruger,B. and Riezman,H. (1995) The Golgi-localization of yeast Emp47p depends on its di-lysine motif but is not affected by the ret1-1 mutation in α-COP. J. Cell Biol., 131, 895–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevier C.S., Weisz,O.A., Davis,M. and Machamer,C.E. (2000) Efficient export of the vesicular stomatitis virus G protein from the endoplasmic reticulum requires a signal in the cytoplasmic tail that includes both tyrosine-based and di-acidic motifs. Mol. Biol. Cell, 11, 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberstein S., Collins P.G., Kelleher D.J. and Gilmore R. (1995) The essential OST2 gene encodes the 16-kD subunit of the yeast oligosaccharyltransferase, a highly conserved protein expressed in diverse eukaryotic organisms. J. Cell Biol., 131, 371–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siniossoglou S., Peak-Chew,S.Y. and Pelham,H.R. (2000) Ric1p and Rgp1p form a complex that catalyses nucleotide exchange on Ypt1p. EMBO J., 19, 4885–4894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer S. and Schekman,R. (1998) Nucleation of COPII vesicular coat complex by endoplasmic reticulum to Golgi vesicle SNAREs. Science, 281, 698–700. [DOI] [PubMed] [Google Scholar]
- Te Heesen S., Knauer,R., Lehle,L. and Aebi,M. (1993) Yeast Wbp1p and Swp1p form a protein complex essential for oligosaccharyl transferase activity. EMBO J., 12, 279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada M. and Gallwitz,D. (1996) Isolation and characterization of SYS genes from yeast, multicopy suppressors of the functional loss of the transport GTPase Ypt6p. J. Cell Sci., 109, 2471–2481. [DOI] [PubMed] [Google Scholar]
- Tsukada M., Will,E. and Gallwitz,D. (1999) Structural and functional analysis of a novel coiled-coil protein involved in Ypt6 GTPase-regulated protein transport in yeast. Mol. Biol. Cell, 10, 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooding S. and Pelham,H.R. (1998) The dynamics of Golgi protein traffic visualized in living yeast cells. Mol. Biol. Cell, 9, 2667–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]