

Abstract
Proteins are highly complex systems, exhibiting a substantial degree of structural variability in their folded state. In the presence of denaturants, the heterogeneity is greatly enhanced, and fluctuations among vast numbers of folded and unfolded conformations occur via many different pathways. Here, we have studied the structure and dynamics of the small enzyme ribonuclease HI (RNase H) in the presence of the chemical denaturant guanidinium chloride (GdmCl) using single-molecule fluorescence microscopy, with a particular focus on the characterization of the unfolded-state ensemble. A dye pair was specifically attached to the enzyme to measure structural changes through Förster resonance energy transfer (FRET). Enzyme immobilization on star-polymer surfaces that were specially developed for negligible interaction with folded and unfolded proteins enabled us to monitor conformational changes of individual proteins for several hundred seconds. FRET efficiency histograms were calculated from confocal scan images. They showed an expansion of the unfolded proteins with increasing GdmCl concentration. Cross-correlation analysis of donor and acceptor fluorescence intensity time traces from single molecules revealed reconfiguration of the polypeptide chain on a timescale of ≈20 μs at 1.7 M GdmCl. Slow conformational dynamics gave rise to characteristic, stepwise FRET efficiency changes. Transitions between folded and unfolded enzyme molecules occurred on the 100-s timescale, in excellent agreement with bulk denaturation experiments. Transitions between unfolded conformations were more frequent, with characteristic times of ≈2 s. These data were analyzed to obtain information on the free energy landscape of RNase H in the presence of chemical denaturants.
Keywords: fluorescence resonance energy transfer, guanidinium chloride, protein folding, RNase H, single-molecule spectroscopy
Proteins are complex systems that can exist in a huge number of different conformations. Even in the properly folded, native state, the polypeptide chain adopts many slightly different conformations that can be depicted as local minima in a rugged energy landscape, and relaxations and fluctuations in the landscape are crucially involved in functional processes (1, 2). Conformational heterogeneity is even more of a concern in studies of protein-folding reactions because of the vast number of possible arrangements of the unfolded polypeptide chain and the many complex pathways leading from the ensemble of unfolded to the native conformations in an overall funnel-shaped energy landscape (3–7).
Because of Anfinsen's key observation that the native fold is already encoded in the sequence of amino acids (8), the protein-folding problem has attracted enormous attention, and the field has progressed in a healthy interplay between theory and experiment. A wide variety of biophysical approaches were developed to follow the dynamics of the polypeptide en route to the native state. Valuable insights have been achieved by time-resolved spectroscopic experiments on bulk samples in which folding transitions are synchronously induced by sudden external perturbations such as laser flashes (9–11). The synchronization, however, becomes lost because of the heterogeneous, multistep nature of protein folding, so that bulk experiments can only provide ensemble-averaged information on folding pathways. Single-molecule techniques avoid ensemble averaging and thus are perfectly suited for protein-folding studies. By choosing the experimental conditions (temperature, pH, chemical denaturants) such that the free energies of the folded and unfolded states are similar, reversible transitions between unfolded and folded states can be frequently observed. Single-molecule fluorescence microscopy, using Förster resonance energy transfer (FRET) as a structure-sensitive probe, is a powerful technique for the study of protein folding. Measurements on freely diffusing proteins (12–15) are technically simple, but the observation time is limited by the ≈1-ms diffusion time of a molecule through the tiny detection volume of the microscope. Measurements on immobilized protein preparations (16–19) bear the risk that the immobilization procedure may interfere with the intrinsic polypeptide dynamics but allow observation of individual proteins for long times. Rhoades et al. (17, 18) trapped proteins in 100-nm lipid vesicles for immobilization and inferred from measurements of the polarization anisotropy that the proteins did not stick to the walls of the vesicles. In this procedure, however, solvent conditions are fixed at the time of immobilization and cannot be altered in a simple fashion. Therefore, we developed an alternative strategy of immobilization, in which the protein molecules are attached to a glass surface densely coated with poly(ethylene glycol) (PEG) star-shaped polymers through a streptavidin-biotin linkage (19, 20). Of key importance for protein-folding studies is the observation that interactions between the surface and both the folded and unfolded protein molecules are negligible, as judged from the free energy change, ΔG, the cooperativity parameter m of folding, and essentially complete unfolding–refolding reversibility (19, 20).
Here, we present a single-molecule study of the dynamics of a small enzyme, ribonuclease HI (RNase H) in the presence of the chemical denaturant guanidinium chloride (GdmCl). RNase H was chosen because it is a model system for protein folding, well-characterized by equilibrium and kinetic experiments including mutational analysis (21–23). Our work focuses on the structure and dynamics of the unfolded state of the polypeptide. The structural properties of the unfolded state are difficult to characterize because of its pronounced heterogeneity. Therefore, the unfolded state has received much less attention than the folded state despite the fact that it plays a key role in any theoretical or experimental study of protein folding. Recently, evidence has mounted that the denatured state in various proteins is not a random coil but rather contains persistent long-range order even at the highest-possible denaturant concentrations (24–28). In this work, we examine the structural properties and the dynamics of conformational change of the denatured state of RNase H. Most intriguing is the observation that the polypeptide chain can be trapped for several seconds in particular conformations in the unfolded state, corresponding to pronounced minima in the free energy landscape.
Materials and Methods
Protein Expression, Purification, and Labeling. RNase H expression, purification, and labeling followed methods described in refs. 29–31; details can be found in Supporting Text, which is published as supporting information on the PNAS web site. For FRET experiments, we attached the donor dye Alexa Fluor 546 and the acceptor dye Alexa Fluor 647 to the two cysteines of the K3C/R135C mutant. The schematic representation in Fig. 1 (Protein Data Bank ID code 2RN2) shows the secondary structure elements (five α-helices and five β-strands) together with the attached dyes.
Fig. 1.

Cartoon representation of the structure of RNase H with the FRET dye pair attached to residues 3 and 135.
Surface Immobilization. We have recently presented a method of protein immobilization on a glass surface that ensures minimal interaction with the surface in both the native and the GdmCl-denatured states (19, 20). Briefly, glass coverslips were coated with star-shaped PEG molecules containing reactive isocyanate end groups (SusTech, Darmstadt, Germany). On an amino-functionalized glass surface, they form a dense, extensively cross-linked PEG layer to which a sparse number of biotin molecules were attached for protein conjugation. For in situ buffer exchange, two coverslips were glued on top of each other with two pieces of double-sided adhesive tape, leaving a 2-mm-wide channel in the middle. A 20 μg/ml solution of streptavidin in phosphate buffer (100 mM, pH 7.4) was filled into the channel for 10 min, followed by washing of the channel with phosphate buffer. Subsequently, a solution of dye-labeled, biotinylated RNase H (≈100 pM) in buffer A (20 mM Tris·HCl/100 mM KCl/10 mM MgCl2, pH 7.4) was filled into the channel for another 10 min. The samples were measured immediately after thorough washing of the channel with buffer A. For protein denaturation studies, the buffer in the channel was replaced by a GdmCl solution in buffer A. Measurements were started after waiting 2 min for equilibration.
Single-Molecule Fluorescence Microscopy. Single-molecule FRET measurements were conducted on a homemade confocal microscope (31). Donor excitation was provided by the 514-nm line of an Ar+/Kr+-ion laser (modified model 164, Spectra-Physics). For separate detection of the donor and acceptor emission, the light collected by the objective lens was split into two channels, passed through dichroic filters optimized for Alexa Fluor 546 and 647, and imaged onto avalanche photodiodes as described in refs. 19, 20, and 31. Areas of 18 × 18 μm were consecutively imaged until a large enough number of molecules was measured (typically 20–30 images). FRET efficiency values, E, were calculated for individual molecules as
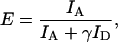 |
[1] |
where ID and IA are the donor and acceptor fluorescence intensities, respectively, corrected for background and cross-talk; the parameter γ corrects for the different detection efficiencies of the two channels and the quantum yields of the dyes.
For time-resolved experiments, single molecules were placed, one after another, in the focus, and fluorescence intensity time traces of the donor and acceptor emissions were recorded with resolutions of up to 1 μs. For the analysis of FRET transitions, the traces were binned with 1-ms resolution. To extend the observation time up to several hundred seconds while avoiding bleaching of the dyes, we also used time-lapse excitation, in which a shutter in the excitation beam was periodically opened for 20 ms every 2 s. Only those traces that showed single-step photobleaching were analyzed.
A computer algorithm was developed that identifies FRET efficiency steps in the time traces in the presence of the inevitable shot noise. In this procedure, the data were grouped into segments with significantly different average FRET efficiencies by consecutive merging of adjacent time bins into longer segments until adjacent segments had statistically different FRET efficiencies. A 99% confidence test was performed to ensure that the FRET efficiencies were distinguishable. Details are provided in Supporting Text.
For correlation analysis of single-molecule intensity time traces, the normalized pair correlation function
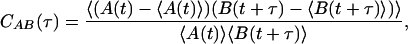 |
[2] |
was calculated. Here, angular brackets denote averaging over time, t, and A and B signals can be donor or acceptor fluorescence intensities, or X and Y components in polarization experiments. The calculation yields autocorrelation and cross-correlation functions for A and B being the same or different signals, respectively.
Experimental Results
Rotational Motion of the Dye Labels Attached to the Protein. Owing to the sensitivity of E to both mutual distance and orientation of the interacting transition dipoles, fluorescence polarization experiments were performed to examine the orientational dynamics of the dyes. Here we summarize the results; details are presented in Supporting Text and Figs. 8 and 9, which are published as supporting information on the PNAS web site. Studies on RNase H bulk solutions, singly labeled with donor or acceptor dye, yielded significant polarization values, P, under native conditions that decreased only slightly upon denaturation. These results clearly indicate that orientational averaging of the donor and acceptor transition dipoles is incomplete on the nanosecond timescale of energy transfer. Single-molecule polarization studies on immobilized RNase H under native conditions yielded a wide distribution in P, time-averaged over a 1-ms interval, for both dyes. Apparently, the limited rotational dynamics of the dyes attached to native RNase H results in a large variability of the orientation factor, κ2. Therefore, changes in E in the folded state may arise from orientation and/or distance changes of the dye pair, and an accurate interpretation of FRET efficiency values in terms of interdye distances alone is not feasible for the folded protein. By contrast, at high GdmCl concentrations, we observed P ≈ 0, indicating complete orientational averaging over a 1-ms interval. This result was further corroborated by correlation analysis of the time dependence of photon emission from single molecules. In the absence of fast orientational averaging, the simple equation relating the spectroscopic observable E to the structural parameter interdye distance r,
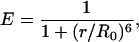 |
[3] |
does not hold. The dependence of E on the interdye distance for cases of restricted orientational dynamics is discussed in Supporting Text.
Effect of GdmCl on FRET Efficiency Distributions. Images of individual RNase H molecules, immobilized on PEG-coated glass surfaces, were recorded at various concentrations of the denaturant GdmCl. Fig. 2 shows samples, taken at 0, 1.5, 1.7, and 6 M GdmCl, together with histograms of the FRET efficiency E determined from ensembles of several hundred individual spots. The histograms can be decomposed into distributions of folded molecules, (〈E 〉 ≈ 0.9, red), unfolded molecules (0.3 < 〈E 〉 < 0.5, yellow), and molecules without an acceptor dye (〈E 〉 ≈ 0.0, green). Comparison of the distributions for 1.7 and 6 M GdmCl shows a substantial decrease in the FRET coupling with increasing denaturant concentration, implying a considerable expansion of the denatured polypeptide chain. This shift is continuous, as was shown by experiments at many different GdmCl concentrations (data not shown).
Fig. 2.

FRET analysis of individual, surface-immobilized RNase H molecules in 0, 1.5, 1.7, and 6 M GdmCl. (Right) The images show areas of 18 × 18 μm2; each spot represents an individual enzyme molecule. Photons from the green and red channels are displayed in these colors; yellow represents molecules emitting in both channels. (Left) Histograms of FRET efficiency values E averaged over single-molecule spots; solid lines are fits with Gaussian and log-normal distributions.
Time Dependence of Fluorescence Emission. To study conformational dynamics of RNase H molecules near the midpoint of the denaturation transition, we have monitored the donor and acceptor emission of several hundred RNase H molecules at 1.7 M GdmCl with 1-ms time resolution. Fig. 3A shows typical examples of fluorescence emission time traces of the green and red dyes under continuous-wave excitation, binned in 1-ms time intervals, with the calculated time dependence of E below the time traces. Because the inevitable dye photobleaching limits FRET observations to a few seconds, experiments also were performed using time-lapse excitation. Thereby, the accessible time interval was extended by two orders of magnitude, as shown in Fig. 3B. Sudden changes of the FRET efficiency, marked by blue dots, occur infrequently, although in nearly all of the traces.
Fig. 3.

Examples of fluorescence-intensity time traces of RNase H immobilized on a cross-linked PEG surface at 1.7 M GdmCl. Green and red lines represent donor and acceptor fluorescence, respectively, corrected for background, cross-talk, and different quantum yields/detection efficiencies of the dyes. The resulting FRET efficiency is shown in blue. Blue, red, and green dots mark transitions in FRET and photobleaching of the red and green dye, respectively. Data were taken under continuous excitation with 1-ms bin time (averaged over 20 ms for presentation) (A) and time lapse excitation for 20 ms every 2 s (B).
To explore the polypeptide dynamics on faster timescales, we calculated the cross-correlation function of the donor and acceptor emission from 249 molecules in the unfolded state (0.20 < E < 0.72). Correlation functions were calculated separately for each section of a time trace, bounded by transitions between different FRET efficiency levels. For statistical reasons, only sections of >200 ms were included. The average cross-correlation function, plotted in Fig. 4A, clearly decays in two steps, and its negative amplitude reflects the typical anticorrelated behavior of the emission from a dye pair with fluctuating FRET coupling. These fluctuations can arise from the intrinsic photodynamics of the dyes (blinking) or conformational dynamics of the polymer chain. To separate the two effects, we also determined average donor and acceptor intensity autocorrelation functions in the absence of FRET (Fig. 4 B and C). Donor correlation functions were calculated for unfolded RNase H molecules (as judged from their FRET efficiency E) after acceptor photobleaching, while acceptor correlation functions were measured on unfolded RNase H (in 2 M GdmCl) by using direct excitation with 633-nm He–Ne laser light. The autocorrelation functions have similar amplitudes and decay on a similar timescale (90 and 250 μs for donor and acceptor, respectively) as the slow step of the cross-correlation function in the FRET experiment (Fig. 4A), indicating that this feature arises from emission fluctuations of the dye molecules themselves rather than conformational changes affecting the FRET coupling. This interpretation is supported by similar observations in single-molecule studies of dye molecules (32, 33). A three-exponential fit, keeping two exponentials fixed at 90 and 250 μs to model the slow step, yielded a correlation time of 20 ± 5 μs for the fast process, shown as a solid line in Fig. 4A.
Fig. 4.

Average correlation functions calculated from emission time traces of individual unfolded RNase H molecules. (A) Cross-correlation of donor and acceptor emission from 249 unfolded RNase H molecules (0.20 < E < 0.72) at 1.7 M GdmCl. The solid line shows the result of a triexponential fit, with the slow step described by two exponentials with two time constants fixed to the values obtained from the data in B and C (dashed line); the fast step yields a decay time of 20 ± 5 μs. (B) Donor autocorrelation function from unfolded RNase H molecules, measured after acceptor photobleaching. The line is an exponential fit with a decay time of 90 ± 10 μs. (C) Acceptor autocorrelation function from unfolded RNase H molecules (2 M GdmCl) excited at 633 nm. The line is an exponential fit with a decay time of 250 ± 70 μs.
Data Analysis and Discussion
RNase H Structure in the Unfolded State. The data in Fig. 2 provide clear evidence of a pronounced decrease of the FRET efficiency of the unfolded protein with increasing GdmCl concentration, which implies a substantial expansion of the polypeptide. This “swelling” effect qualitatively agrees with a model based on the competition between solvophobic interactions and chain configurational entropy (34); it also was observed in single-molecule FRET studies on the cold shock protein CspTm (14) and, to a lesser extent, on the chymotrypsin inhibitor 2 (13). Apparently, unfolded polypeptide chains incorporate more and more denaturant molecules up to a certain saturation level. A continuum of denatured states exists, and the free energy of the more expanded conformations is preferentially lowered with increasing GdmCl concentration. This result also is corroborated by native-state hydrogen exchange data on RNase H (35).
Structural Dynamics in the Presence of Denaturant. By using time-lapse excitation, the fluorescence emission of FRET-pair labeled RNase H molecules in 1.7 M GdmCl was measured for up to several hundred seconds (Fig. 3B), enabling a comparison with previous bulk measurements of the denaturation transition on RNase H samples (21, 36). Analysis of the FRET changes of single RNase H molecules yielded the total number of transitions from the unfolded (E ≤ 0.72) to the folded (E > 0.72) state and vice versa, NUF and NFU and, furthermore, the total times spent by molecules in the unfolded and folded states, tU and tF, respectively. From these data, we calculated the average folding rate coefficient, kUF = NUF/tU = (1.1 ± 0.4) × 10- s-1, and the average unfolding rate coefficient, kFU = NFU/tF = (0.8 ± 0.3) × 10-2 s-1. For the apparent rate coefficient, which is observed in bulk experiments, kapp = kUF + kFU, we obtained (1.9 ± 0.5) × 10-2 s-1, which is in excellent agreement with ensemble data for RNase H [kapp ≈ 2 × 10-2 s-1 at 1.7 M GdmCl and 25°C measured by both CD (21) and tryptophan fluorescence (36)]. This result suggests that the protein is negligibly perturbed by the immobilization.
FRET efficiency transitions on shorter timescales were visible in time traces under continuous illumination (Fig. 3A). In Fig. 5A, each individual transition is depicted as a single dot in a 2D histogram, with its x- and y-coordinate representing the initial and final FRET efficiency values, respectively. Two gray lines, centered on E = 0.72, divide the histogram into four areas comprising U → U, F → F, U → F, and F → U transitions. Because of overlap in the distributions (Fig. 2B), this distinction is impossible for events covered by the gray lines. Small jumps (ΔE < 0.05) are hidden in the noise; consequently, the diagonal of the histogram is empty. The larger density of points near the diagonal clearly reflects a preference for small FRET jumps, suggesting that transitions between similar peptide folds are more likely to occur. Still, transitions spread over the entire accessible range of initial and final FRET values. This behavior indicates a high diversity of accessible folded and unfolded states among which transitions can occur, as was also observed in single-molecule unfolding studies of adenylate kinase (17).
Fig. 5.

Analysis of FRET transitions. (A) Shown are 529 statistically significant FRET efficiency transitions analyzed from single-molecule traces of immobilized RNase H molecules at 1.7 M GdmCl, depicted as points in a 2D plot spanned by the initial and final FRET values. (B and C) Rates of transitions plotted against initial FRET efficiency (B) (the white bars close to the 0.72 boundary are within the overlap region of the distributions and cannot be classified unambiguously) and denaturant concentration (C).
We performed an analysis of the transition rates under continuous excitation in the same fashion as for the time-lapse experiments. In Fig. 5B, we plotted the observed rates in the four quadrants vs. the initial FRET efficiency. For U → U and F → F transitions, rates of ≈0.4 and 0.1 s-1 are observed, respectively, independent of the initial E. For folding and unfolding transitions, U → F and F → U, the average transition probabilities are ≈0.04 s-1, which is higher than the rate of ≈0.01 s-1 from the time-lapse data. This discrepancy is likely a result of the limited time window available for the continuous excitation, because of dye photobleaching, which results in preferentially selecting the (relatively few) fast events. Moreover, because of the overlap of the distributions, we cannot avoid misclassifying a few events, e.g., U → U as F → U. Unfortunately, these fast events will dominate the average, and more confidence should be placed in the time-lapse data for the folding–unfolding transitions.
Fig. 5C shows the average rate coefficient for all transitions within the unfolded subpopulations at 1.7, 2.5, and 6 M GdmCl. Apparently, transitions between discrete FRET levels are less frequent at higher denaturant concentration. It is especially intriguing, however, that FRET jumps are still observed at 6 M GdmCl, signifying that residual structure appears to create local potential wells even under highly denaturing conditions.
Dynamic Nature of Structural Heterogeneity. Fig. 6A shows the distributions of rms interdye distances, ra, and the corresponding radii of gyration, RG, which were calculated from E values in the single-molecule time traces at 1.7 M GdmCl. This calculation is described in Supporting Text. We note here that we cannot simply use Eq. 3 because of reorientation of the dyes on timescales longer than their fluorescence lifetimes. The distribution of the unfolded ensemble is well described by a Gaussian and much wider (SDσ = 12 Å) than the native-state distribution and the one expected from mere statistical noise, which we have estimated as σ = 2.6 Å for our data, taking into account errors due to background, cross-talk, and the detection-correction factor.
Fig. 6.

Dynamic heterogeneity of the unfolded state of RNase H at 1.7 M GdmCl. (A) Histograms of interdye distances, ra, evaluated from single-molecule time traces. The distributions of the folded and unfolded populations were fitted with log-normal and Gaussian functions, respectively. The light gray Gaussian peak represents the distribution expected from statistical noise of the detected signal. (B) Step-size distribution of transitions within the unfolded state. The solid line is a fit to a model described in the text.
Is the observed structural heterogeneity static or dynamic? Static heterogeneity arises from influences that are invariant on the experimental timescale, such as different microenvironments of the enzyme molecules. Dynamic heterogeneity implies that the molecules fluctuate among all configurations and explore the entire width of the distribution. To distinguish the two cases, we used a simple model that connects the interdye distances with the distribution of the observed changes. It is also described in Supporting Text. Briefly, we have assumed that the free energies of conformations in the unfolded state, Gi, depend on the square of the interdye distance ri. With this ansatz, the distribution of interdye distances in the unfolded state, P(ri), has a Gaussian shape, as shown in Fig. 6A, and the distribution of interdye distance changes, P(Δra) ∝ erfc[Δra/(23/2σ)], with erfc denoting the complementary error function. For 1.7 M GdmCl, we obtain P(Δra) as shown in Fig. 6B, selecting the lower (0.20) and upper (0.72) limits of the distribution of unfolded molecules (yellow bars in the histogram in Fig. 2) as integration limits. The width parameter from this analysis, σ = 11 Å, is close to σ = 12 Å from the distribution of interdye distances, implying that the observed heterogeneity is predominantly dynamic in nature. This analysis further reinforces the notion that our immobilization approach allows the denatured molecules to fluctuate within the entire range of thermally accessible states.
Protein Energy Landscape Under Denaturing Conditions. Protein folding is a rate process taking place on a complex free energy surface. The timescales of conformational changes, τ, can be related to the free energy barriers, ΔG, by the Kramers equation
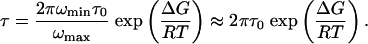 |
[4] |
Here, ωmin and ωmax are harmonic frequencies governing the curvature at the bottom of the initial well and the top of the barrier (along the reaction coordinate), which may be taken to be identical (37, 38), and τ0 = RT/mω2minD is the reconfiguration time, with mass m and diffusion coefficient D. Therefore, we can calculate ΔG from the measured values of τ and τ0. It is reasonable to assign the fast process, with a decay time of 20 μs in the single-molecule cross-correlation function, Fig. 4, to reconfiguration of the polypeptide chain in the unfolded state. Recent FCS studies on the unfolded fatty acid binding protein also reported reconfiguration times of the unfolded chain in the microsecond range (15). With τ0 = 20 μs and τUU = 1/kUU = 2.5 s for transitions in the unfolded state, we obtain the result that the dynamics in the unfolded state is governed by barriers ΔGU ≈ 24 kJ/mol (≈10RT).
Alternatively, following Eaton and coworkers (14, 39), we may get an upper bound for the reconfiguration time from the diffusion limit of the Gaussian chain
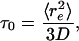 |
[5] |
where re is the end-to-end distance, and D is the end-to-end diffusion coefficient. For random polypeptides, D ≈ 17 Å2/ns (39). From Fig. 2, 〈E 〉 ≈ 0.3 at 6 M GdmCl, which corresponds to RG ≈ 38 Å. For a Gaussian chain, 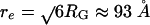 , and, therefore, τ0 ≈ 170 ns. The RG value of the unfolded state at 6 M GdmCl calculated from our FRET data is in excellent agreement with small angle x-ray scattering data of RNase H (40) and other proteins of similar size (41). At 1.7 M GdmCl, the unfolded state is more compact and re is thus smaller (Fig. 2), but friction also increases because of more contacts between the residues, resulting in a decrease in D. Taking τ0 = 170 ns as an upper bound, ΔGU ≤ 36 kJ/mol (≈15RT). Similarly, with τUF = 1/kUF = 90 s, the folding barrier, ΔGU→F, can be calculated to be 33 kJ/mol (≈13RT) for τ0 = 20 μs and 45 kJ/mol (≈18RT) for τ0 = 170 ns.
, and, therefore, τ0 ≈ 170 ns. The RG value of the unfolded state at 6 M GdmCl calculated from our FRET data is in excellent agreement with small angle x-ray scattering data of RNase H (40) and other proteins of similar size (41). At 1.7 M GdmCl, the unfolded state is more compact and re is thus smaller (Fig. 2), but friction also increases because of more contacts between the residues, resulting in a decrease in D. Taking τ0 = 170 ns as an upper bound, ΔGU ≤ 36 kJ/mol (≈15RT). Similarly, with τUF = 1/kUF = 90 s, the folding barrier, ΔGU→F, can be calculated to be 33 kJ/mol (≈13RT) for τ0 = 20 μs and 45 kJ/mol (≈18RT) for τ0 = 170 ns.
The most intriguing result reported here is that transitions among unfolded substates occur on the second timescale (Fig. 5), indicating that their potential energy wells are rather deep. Apparently, residual, nonrandom structure exists even in the presence of high concentrations of GdmCl. One possible source of slow conformational change in proteins is proline (Pro) isomerization. However, Pro isomerization was suggested to occur with τ = 212 s in stopped-flow folding experiments on Thermus thermophilus RNase H (42), which is two orders of magnitude slower than the transitions observed here. In other proteins, Pro isomerization also was observed on the hundreds of seconds timescale (43–45). Proteins under strongly denaturing solvent conditions are frequently taken as the “random coil” reference state in folding (41). Yet, Fitzkee and Rose (46) recently showed that, when only 8% of the residues in lysozyme were disordered while the other 92% remained in the native structure, the end-to-end distance distributions and radii of gyration were in agreement with the values calculated from a Gaussian random coil. They obtained a similar result for RNase H containing only 13 flexible residues. Several examples of proteins containing residual structure in the unfolded state have been reported (24–28), and, therefore, it is likely that proteins have more structural organization and conformational bias in the unfolded state than was previously assumed. Possible mechanisms for preorganization of the peptide chain include formation of hydrophobic clusters and sterically imposed polypeptide chain arrangement extending beyond next-neighbor interactions in the amino acid sequence (47, 48).
In Fig. 7A we plotted a schematic 3D folding free energy landscape of RNase H that captures the main features observed in this work. We have introduced two spatial coordinates, denoted as Q1 and Q2. Transitions between conformations within the unfolded state occur along Q1, they are governed by free energy barriers, ΔGU ≈ 24 kJ/mol (Fig. 7B). Folding transitions occur along Q2, encountering a barrier, ΔGU→F ≈ 33 kJ/mol (Fig. 7C). Fast mixing experiments on RNase H show that this barrier can be approached on a submillisecond time-scale (the burst phase) in the unfolded state (21–23, 36), indicating a rather smooth energy landscape. The unfolded state has been described as a continuum ranging from compact to fully expanded conformations (35, 36). Our present experiments cannot resolve the fast kinetics of the burst phase continuum along the Q2 axis but only capture the slow kinetics along Q1. Clearly, Fig. 7 captures only the main features, and further work is necessary to reveal details of the rugged energy landscape of proteins under destabilizing conditions.
Fig. 7.

Schematic depiction of the RNase H free energy landscape. (A) Free energy surface plotted as a function of two structural coordinates, Q1 and Q2, describing the protein conformation. (B) Cross-section along Q1, showing high barriers governing conformational transitions within the unfolded substates on the second timescale. (C) Cross-section along Q2, showing the barrier between the folded and unfolded state.
Supplementary Material
Acknowledgments
We thank Dr. Jürgen Groll and Prof. Martin Möller for their collaboration on star polymer surfaces and SusTech GmbH for providing the material. The RNase H plasmid was a generous gift of Prof. S. Kanaya (Osaka University, Osaka, Japan). We also thank Sigrid Niederhausen-Kraft and Uwe Theilen for technical assistance with surface preparation and protein expression. C.D.H. was supported by research fellowships from the Alexander von Humboldt Foundation and the International Human Frontiers Science Program. This work was supported by Deutsche Forschungsgemeinschaft Grant SFB569 and the Fonds der Chemischen Industrie.
Author contributions: G.U.N. designed research; E.V.K. and C.D.H. performed research; E.V.K. and G.U.N. analyzed data; and C.D.H. and G.U.N. wrote the paper.
Abbreviations: FRET, Förster resonance energy transfer; GdmCl, guanidinium chloride; PEG, poly(ethylene glycol); RNase H, ribonuclease HI.
References
- 1.Frauenfelder, H., Sligar, S. G. & Wolynes, P. G. (1991) Science 254, 1598-1603. [DOI] [PubMed] [Google Scholar]
- 2.Nienhaus, G. U. & Young, R. D. (1996) in Encyclopedia of Applied Physics, ed. G. Trigg (Wiley–VCH, New York), Vol. 15, pp. 163-184. [Google Scholar]
- 3.Onuchic, J. N., Wolynes, P. G., Luthey-Schulten, Z. & Socci, N. D. (1995) Proc. Natl. Acad. Sci. USA 92, 3626-3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang, J., Saven, J. G. & Wolynes, P. G. (1996) J. Chem. Phys. 105, 11276-11284. [Google Scholar]
- 5.Onuchic, J. N., Luthey-Schulten, Z. & Wolynes, P. G. (1997) Annu. Rev. Phys. Chem. 48, 545-600. [DOI] [PubMed] [Google Scholar]
- 6.Dill, K. A. & Chan, H. S. (1997) Nat. Struct. Biol. 4, 10-19. [DOI] [PubMed] [Google Scholar]
- 7.Schonbrun, J. & Dill, K. A. (2003) Proc. Natl. Acad. Sci. USA 100, 12678-12682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anfinsen, C. B. (1973) Science 181, 223-230. [DOI] [PubMed] [Google Scholar]
- 9.Callender, R. H., Dyer, R. B., Gilmanshin, R. & Woodruff, W. H. (1998) Ann. Rev. Phys. Chem. 49, 173-202. [DOI] [PubMed] [Google Scholar]
- 10.Gruebele, M. (1999) Annu. Rev. Phys. Chem. 50, 485-516. [DOI] [PubMed] [Google Scholar]
- 11.Eaton, W. A., Muñoz, V., Hagen, S. J., Jas, G. S., Lapidus, L. J., Henry, E. R. & Hofrichter, J. (2000) Annu. Rev. Biophys. Biomol. Struct. 29, 327-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deniz, A. A., Dahan, M., Grunwell, J. R., Ha, T., Faulhaber, A. E., Chemla, D. S., Weiss, S. & Schultz, P. G. (1999) Proc. Natl. Acad. Sci. USA 96, 3670-3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deniz, A. A., Laurence, T. A., Beligere, G. S., Dahan, M., Martin, A. B., Chemla, D. S., Dawson, P. E., Schultz, P. G. & Weiss, S. (2000) Proc. Natl. Acad. Sci. USA 97, 5179-5184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schuler, B., Lipman, E. A. & Eaton, W. A. (2002) Nature 419, 743-747. [DOI] [PubMed] [Google Scholar]
- 15.Chattopadhyay, K., Elson, E. L. & Frieden, C. (2005) Proc. Natl. Acad. Sci. USA 102, 2385-2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Talaga, D. S., Lau, W. L., Roder, H., Tang, J., Jia, Y., DeGrado, W. F. & Hochstrasser, R. M. (2000) Proc. Natl. Acad. Sci. USA 97, 13021-13026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rhoades, E., Gussakovsky, E. & Haran, G. (2003) Proc. Natl. Acad. Sci. USA 100, 3197-3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rhoades, E., Cohen, M., Schuler, B. & Haran, G. (2004) J. Am. Chem. Soc. 126, 14686-14687. [DOI] [PubMed] [Google Scholar]
- 19.Amirgoulova, E. V., Groll, J., Heyes, C. D., Ameringer, T., Röcker, C., Möller, M. & Nienhaus, G. U. (2004) Chem. Phys. Chem. 5, 552-555. [DOI] [PubMed] [Google Scholar]
- 20.Groll, J., Amirgoulova, E., Ameringer, T., Heyes, C. D., Röcker, C., Möller, M. & Nienhaus, G. U. (2004) J. Am. Chem. Soc. 126, 4234-4239. [DOI] [PubMed] [Google Scholar]
- 21.Yamasaki, K., Ogasahara, K., Yutani, K., Oobatake, M. & Kanaya, S. (1995) Biochemistry 34, 16552-16562. [DOI] [PubMed] [Google Scholar]
- 22.Raschke, T. M. & Marqusee, S. (1997) Nat. Struct. Biol. 4, 198-204. [DOI] [PubMed] [Google Scholar]
- 23.Raschke, T. M., Kho, J. & Marqusee, S. (1999) Nat. Struct. Biol. 6, 825-831. [DOI] [PubMed] [Google Scholar]
- 24.Klein-Seetharaman, J., Oikawa, M., Grimshaw, S. B., Wirmer, J., Duchardt, E., Ueda, T., Imoto, T., Smith, L. J., Dobson, C. M. & Schwalbe, H. (2002) Science 295, 1719-1722. [DOI] [PubMed] [Google Scholar]
- 25.Denisov, V. P., Jonsson, B.-H. & Halle, B. (1999) Nat. Struct. Biol. 6, 253-260. [DOI] [PubMed] [Google Scholar]
- 26.Shortle, D. & Ackerman, M. S. (2001) Science 293, 487-489. [DOI] [PubMed] [Google Scholar]
- 27.Shortle, D. (1996) FASEB J. 10, 27-34. [DOI] [PubMed] [Google Scholar]
- 28.Neri, D., Billeter, M., Wider, G. & Wuthrich, K. (1992) Science 257, 1559-1563. [DOI] [PubMed] [Google Scholar]
- 29.Kanaya, S. & Crouch, R. J. (1983) J. Biol. Chem. 258, 1276-1281. [PubMed] [Google Scholar]
- 30.Kanaya, S., Kohara, A., Miyagawa, M., Matsuzaki, T., Morikawa, K., Ikehara, M. & Crouch, R. J. (1989) J. Biol. Chem. 264, 11546-11549. [PubMed] [Google Scholar]
- 31.Heyes, C. D., Kobitski, A. Y., Amirgoulova, E. V. & Nienhaus, G. U. (2004) J. Phys. Chem. B 108, 13387-13394. [Google Scholar]
- 32.Widengren, J. & Schwille, P. (2000) J. Phys. Chem. A 104, 6416-6428. [Google Scholar]
- 33.Heilemann, M., Margeat, E., Kasper, R., Sauer, M. & Tinnefeld, P. (2005) J. Am. Chem. Soc. 127, 3801-3806. [DOI] [PubMed] [Google Scholar]
- 34.Alonso, D. O. V. & Dill, K. A. (1991) Biochemistry 30, 5974-5985. [DOI] [PubMed] [Google Scholar]
- 35.Parker, M. J. & Marqusee, S. (2000) J. Mol. Biol. 300, 1361-1375. [DOI] [PubMed] [Google Scholar]
- 36.Parker, M. J. & Marqusee, S. (1999) J. Mol. Biol. 293, 1195-1210. [DOI] [PubMed] [Google Scholar]
- 37.Socci, N. D., Onuchic, J. N. & Wolynes, P. G. (1996) J. Chem. Phys. 104, 5860-5868. [Google Scholar]
- 38.Klimov, D. K. & Thirumalai, D. (1997) Phys. Rev. Lett. 79, 317-320. [Google Scholar]
- 39.Lapidus, L. J., Eaton, W. A. & Hofrichter, J. (2000) Proc. Natl. Acad. Sci. USA 97, 7220-7225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nishimura, C., Segel, D. J., Hodgson, K. O., Doniach, S. & Fink, A. L. (1997) Stanford Synchrotron Radiation Laboratory Activity Report 7-25-7-27.
- 41.Millett, I. S., Doniach, S. & Plaxco, K. W. (2002) Adv. Protein Chem. 62, 241-261. [DOI] [PubMed] [Google Scholar]
- 42.Hollien, J. & Marqusee, S. (2002) J. Mol. Biol. 316, 327-340. [DOI] [PubMed] [Google Scholar]
- 43.Nakano, T., Antonino, L. C., Fox, R. O. & Fink, A. L. (1993) Biochemistry 32, 2534-2541. [DOI] [PubMed] [Google Scholar]
- 44.Jackson, S. E. & Fersht, A. R. (1991) Biochemistry 30, 10436-10443. [DOI] [PubMed] [Google Scholar]
- 45.Shastry, M. C. R., Agashe, V. R. & Udgaonkar, J. B. (1994) Protein Sci. 3, 1409-1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fitzkee, N. C. & Rose, G. D. (2004) Proc. Natl. Acad. Sci. USA 101, 12497-12502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fitzkee, N. C. & Rose, G. D. (2004) Protein Sci. 13, 633-639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pappu, R. V., Srinivasan, R. & Rose, G. D. (2000) Proc. Natl. Acad. Sci. USA 97, 12565-12570. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.