

Abstract
PKCɛ controls the transport of endocytosed β1-integrins to the plasma membrane regulating directional cell motility. Vimentin, an intermediate filament protein upregulated upon epithelial cell transformation, is shown here to be a proximal PKCɛ target within the recycling integrin compartment. On inhibition of PKC and vimentin phosphorylation, integrins become trapped in vesicles and directional cell motility towards matrix is severely attenuated. In vitro reconstitution assays showed that PKCɛ dissociates from integrin containing endocytic vesicles in a selectively phosphorylated vimentin containing complex. Mutagenesis of PKC (controlled) sites on vimentin and ectopic expression of the variant leads to the accumulation of intracellular PKCɛ/integrin positive vesicles. Finally, introduction of ectopic wild-type vimentin is shown to promote cell motility in a PKCɛ-dependent manner; alanine substitutions in PKC (controlled) sites on vimentin abolishes the ability of vimentin to induce cell migration, whereas the substitution of these sites with acidic residues enables vimentin to rescue motility of PKCɛ null cells. Our results indicate that PKC-mediated phosphorylation of vimentin is a key process in integrin traffic through the cell.
Keywords: integrin, intermediate filament, migration, PKC, vimentin
Introduction
Integrins are heterodimeric cell surface receptors, which mediate cell adhesion and migration via cell–cell and cell–matrix interactions (Hynes, 2002). Integrin mediated cell motility has been studied in detail and at least three separate processes have been identified: (1) formation of a protrusion and new adhesion sites at the leading edge; (2) contraction of the cell body in an actin/myosin dependent manner; (3) detachment of the trailing edge. Integrins have a firmly established role in the formation of the protrusions and focal adhesions (Hynes, 2002). Degradation of integrins at the rear of the cell, as well as their endocytic recycling back to the cell front are also vital for movement (Bretscher, 1996).
Protein phosphorylation has been demonstrated to regulate the traffic of integrins in motile cells. In particular, PKC family members as well as other serine/threonine kinases have been shown to regulate integrin transport in the cell affecting both adhesive properties and migration (Rigot et al, 1998; Roberts et al, 2004; Woods et al, 2004). Internalisation of β1-integrins is regulated by PKCα (Ng et al, 1999) and association of PKCα with the integrin β1 cytoplasmic tail is required for directional chemotaxis of breast cancer cells (Parsons et al, 2002). Furthermore, we have shown that PKCɛ controls an unidentified step in the endocytic pathway that permits integrin recycling to the plasma membrane (Ivaska et al, 2002). Inhibition or loss of PKCɛ leads to decreased motility of fibroblasts coincident with the accumulation of a pool of trafficking, β1-integrins to a PKCɛ positive, vesicular, intracellular compartment (Ivaska et al, 2002). Interestingly, growth factor induced αVβ3-dependent cell motility seems to involve an alternative recycling route that is dependent on PKCμ (Roberts et al, 2004; Woods et al, 2004).
Recent work has provided some mechanistic insight into integrin traffic by demonstrating that integrins can be transported in the cell via association with a novel actin binding motor protein, myosin X (Zhang et al, 2004). In addition to the actin cytoskeleton, the intermediate filament (IF) network has been shown to regulate cell motility as well (Eckes et al, 1998). Vimentin is the major structural component of the IFs in cells of mesenchymal origin (Franke et al, 1987). Phosphorylation has a central role in regulating the dynamics of vimentin assembly into polymers (Inagaki et al, 1997) as well as regulating the connections between IFs and IF-associated proteins (Ku et al, 1998). Fibroblasts lacking vimentin show impaired migration and contractile capacity in vitro as well as in vivo (Eckes et al, 1998, 2000). Expression of vimentin correlates with increased migration of cells following epithelial–mesenchymal transition, a characteristic of transformation (Kang and Massague, 2004).
Here we demonstrate that PKCɛ controls the recycling of β1-integrin to the plasma membrane by phosphorylation of cytoskeletal components. We show that vimentin is a target for phosphorylation by PKCɛ in integrin containing intracellular vesicles and that PKC-mediated phosphorylation of vimentin regulates exit from this compartment to the plasma membrane.
Results
Identification of vimentin as a PKC substrate in PKCɛ/β1-integrin containing vesicles
Previously we demonstrated that the catalytic activity of PKCɛ regulates the return of endocytosed β1-integrins to the plasma membrane of mouse embryo fibroblasts (MEFs) and that inhibition of PKCɛ (with the PKC inhibitor BIM-I) leads to the accumulation of large β1-integrin/PKCɛ-containing vesicles and an inhibition of migration (Ivaska et al, 2002). In initial studies on proximal components of PKCɛ action involved in recycling/migration, it was shown that the combined presence of cytosol, ATP and GTP were required for significant release of PKCɛ from the vesicle compartment in vitro (Ivaska et al, 2002). In addition, release was absolutely PKCɛ activity dependent.
In order to identify PKCɛ substrates controlling the dissipation of these vesicles, we have developed further the previously established cell-free vesicle dissociation assay. Cytosol from PKCɛ null cells had enabled us to assay the release of vesicular PKCɛ in the presence of the ectopic proteins originating from a PKCɛ-free cytosol. However, in order to study additional components, biotinylation of the vesicle proteins was used to distinguish between vesicular copurified and ectopic cytosolic components. Importantly, biotinylation did not interfere with the release assay (Supplementary Figure 1) since vesicular PKCɛ originating from the isolated compartment (fractions 7–9) was released from biotinylated vesicles in a manner equivalent to the control vesicles. As shown before, inhibition of PKC catalytic activity blocks release efficiently in both cases (data not shown).
In order to identify PKC substrates in the vesicular compartment, vesicles derived from BIM-I treated cells were isolated, biotinylated and concentrated by centrifugation. These were then incubated with cytosol (unbiotinylated), γ32P-ATP and GTP in the presence or absence of BIM-I. Vesicle derived, biotinylated proteins were isolated using streptavidin-coated Dynabeads and resolved on SDS/PAGE followed by Coomassie staining and autoradiography. Two BIM-I sensitive major phospho-protein bands were repeatedly observed in these labelled assay release-reactions designed to detect kinase substrates. These were identified by mass-fingerprinting as myosin heavy chain type II A/B (MHCIIA/B) and vimentin (Figure 1A). Interestingly, vimentin phosphorylation was observed only when the serine/threonine phosphatase inhibitor (Calyculin A) was present during the in vitro kinase reactions (data not shown; no calyculin A is present during vesicle isolation). This correlates well with the fact that IF proteins associate with phosphatases and can display high turnover of their phosphorylation (Eriksson et al, 2004).
Figure 1.
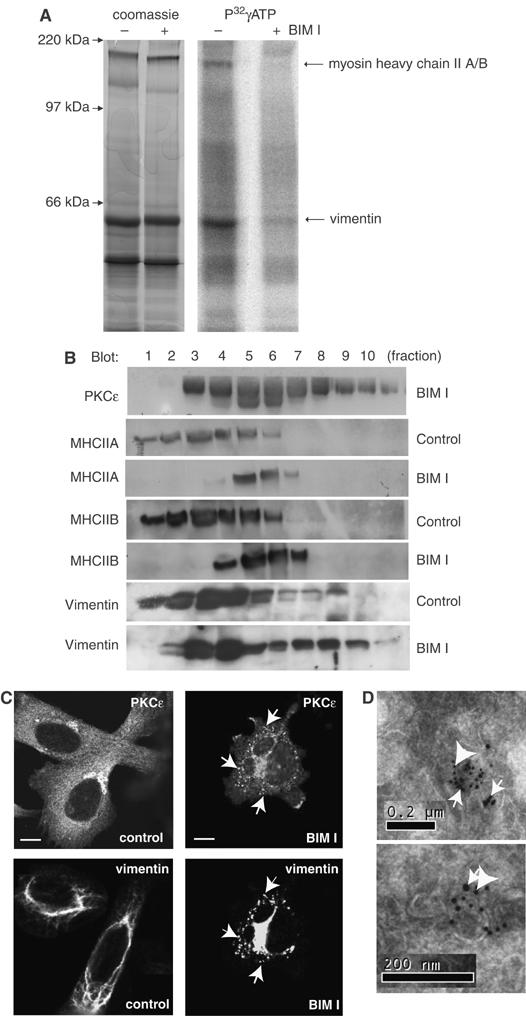
PKC substrates on endocytic integrin-containing vesicles. (A) To identify proximal PKC targets in the vesicular compartment, biotinylated vesicles derived from BIM-I treated PKCɛ expressing cells (PKCɛRE) were resuspended in a small volume of buffer and incubated in cytosol derived from PKCɛ−/− cells in the presence of calyculin A, P32γATP and PKC inhibitor BIM-I where indicated for 1 h at 37°C. Following the reaction, all biotinylated proteins were collected on streptavidin-Dynabeads and resolved on SDS–PAGE followed by Coomassie staining and autoradiography. BIM-I sensitive phosphorylated bands were cut from the gel, trypsinised and identified as vimentin and myosin heavy chain II A or B using MS fingerprinting. (B) The subcellular localisation of vimentin and MHCII alter upon PKC inhibition. PKCɛRE cells were treated for 90 min with BIM-I or left untreated, followed by sucrose gradient fractionation (see Materials and methods). The proteins in the fractions were recovered by TCA precipitation and subjected to Western blot analysis with anti- PKCɛ, anti-vimentin, anti-MHCIIA and anti-MHCIIB antibodies. Upon BIM-I treatment, vimentin was found to accumulate in the PKCɛ positive dense compartment (fractions 7–9). (C) BIM-I treatment induces vimentin fragmentation and the formation of PKCɛ positive vesicles. Dual-colour immunofluorescence staining of vimentin and PKCɛ in PKCɛRE cells is shown. The cells were plated and allowed to spread of fibronectin for 30 min, following 90 min incubation either untreated (control) or 1 μM BIM-I (BIM-I). Arrows indicate some of the areas of overlap in the PKCɛ and vimentin stainings in BIM-I treated cells. Bar 10 μm. (D) Enrichment of vimentin and PKCɛ in vesicles isolated from BIM-I treated PKCɛRE cells. The immunoelectron micrographs show double-labelling of PKCɛ (10 nm protein A-gold, big arrowheads) and vimentin (15 nm protein A-gold, small arrows).
Vimentin localisation is modified by PKC inhibition
In order to confirm the presence of these putative PKCɛ substrates in the induced vesicular compartment, we studied the effect of BIM-I treatment on the subcellular localisation of both proteins. Extracts of control or BIM-I treated PKCɛ reconstituted MEFs (PKCɛRE) were fractionated on a sucrose gradient. We have previously demonstrated that the BIM-I induced vesicular accumulation of PKCɛ and β1-integrin shifts the localisation of these proteins to fractions 7–9 (Ivaska et al, 2002; Figure 1B). Here, BIM-I treatment caused a proportion of the extracted vimentin to shift from a predominantly cytosolic compartment (fractions 1–4) to a denser one (fractions 7–9; Figure 1B). Myosin heavy chain II A/B shifted to denser fractions as well, although in this case the overlap with PKCɛ was only partial (fraction 7).
To further assess the effect of PKC inhibition on the subcellular localisation of vimentin and MHCIIA/B, immunofluorescence staining was performed on spreading PKCɛRE cells before and after BIM-I treatment. For the purposes of visualising wild type (wt) and mutant (see below) ectopic vimentin, human vimentin was introduced into these murine cells and detected with a monoclonal antibody that does not recognise the endogenous murine vimentin. Ectopic vimentin formed IF like structures (Figure 1C). On PKC inhibition, vimentin staining became fragmented and patchy, with staining partially overlapping with PKCɛ vesicles (Figure 1C). In contrast, MHCIIA and B staining patterns remained unaltered by BIM-I (data not shown). It is possible that a subset of the PKCɛ positive vesicles contain MHCII A/B since in the isolated preparations it is phosphorylated in a BIM-I sensitive manner. However, because this substrate is clearly recruited to additional compartment(s) and its location is not obviously influenced by PKC inhibition, it is not considered further here.
Further characterisation of fractionated, isolated vesicles was carried out by electron microscopy and immunogold labelling of PKCɛ and vimentin (Figure 1D). Vimentin and PKCɛ appear to colocalise on vesicular membranes and occasionally vimentin staining is observed aligned along short linear stretches located adjacent to the vesicles (Figure 1D, top panel). The EM analysis indicates that PKCɛ and vimentin colocalise in a BIM-I-induced vesicular compartment, consistent with the immunofluorescence and fractionation data.
PKCɛ-dependent phosphorylation of vimentin
IF proteins, such as vimentin, are good substrates for a number of kinases and several PKC sites have been identified previously (Ku et al, 1998; Yasui et al, 2001). However, the biological relevance of the phosphorylation of the various PKC sites on vimentin is not known. It was clear that we could detect changes in vimentin phosphorylation in the isolated vesicular compartment. However, since we cannot exclude a role for other BIM-I sensitive kinases possibly associated with the vesicles, we set out to study the PKCɛ dependence of vimentin phosphorylation in intact cells. 2D electrophoresis was performed on whole-cell extracts from control and BIM-I treated PKCɛ−/− and PKCɛRE (PKCɛ re-expressing) cells. Serine phosphorylation was detected with Western blotting and the spots representing endogenous vimentin were identified by reprobing with anti-vimentin. In PKCɛRE cells, vimentin was shifted towards a lower pI value, indicating a higher level of phosphorylation when compared to the PKCɛ−/− cells (Figure 2A). Inhibition of PKC resulted in the loss of this more acidic vimentin and an overall reduction in the serine phosphorylation in the PKCɛRE cells. Notably, BIM-I had no obvious effect on the vimentin pI in the PKCɛ−/− cells, indicative of a nonredundant role for PKCɛ in these cells (Figure 2A).
Figure 2.
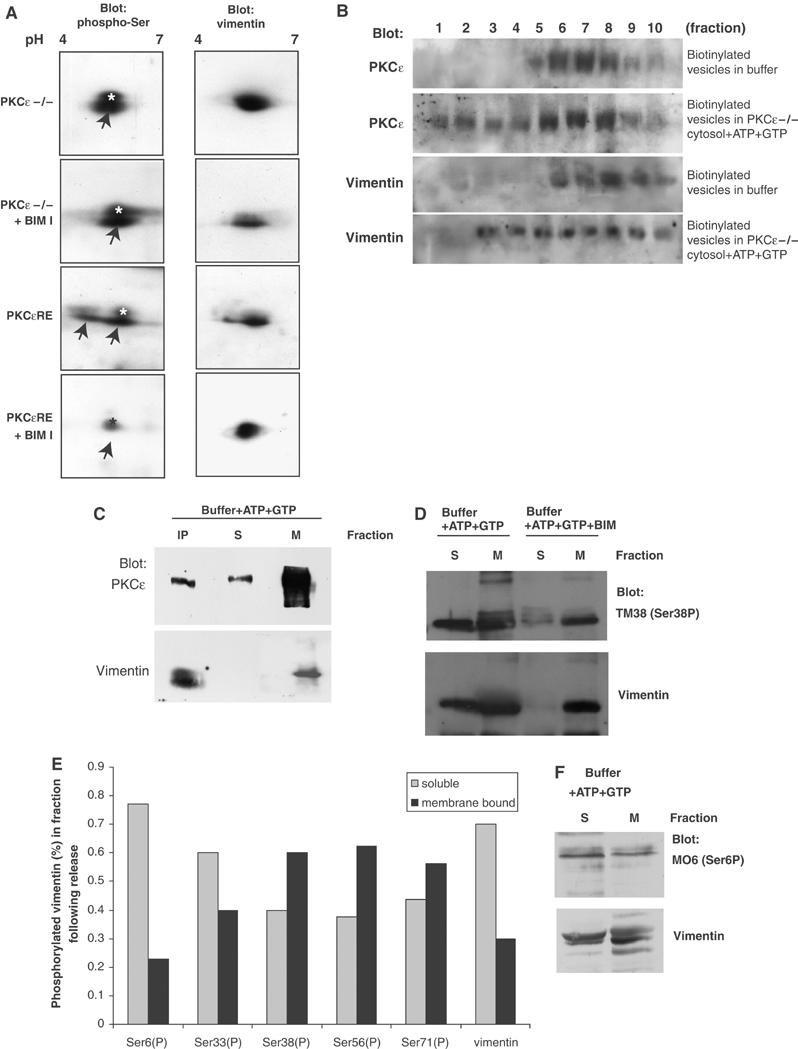
PKCɛ-dependent phosphorylation of vimentin and regulation of vimentin association in the vesicular compartment. (A) PKCɛ null (−/−) and PKCɛRE cells were treated with PKC inhibitor BIM-I where indicated and whole-cell lysates of equal protein loading were resolved with 2D gel electrophoresis and transferred onto nitrocellulose. Serine-phosphorylation was detected with blotting using an anti-phospho-serine antibody (left-hand panels) followed by stripping and reprobing with anti-vimentin to detect the localisation of the endogenous proteins (right-hand panels). In addition to vimentin, the phospho-serine antibody detects an unidentified protein spot migrating at slightly higher molecular weight than vimentin (marked ‘*'). The phospho-serine signal corresponding to vimentin is indicated with arrows. (B) Biotinylated vesicles from BIM-I treated PKCɛRE cells were resuspended in a small volume of buffer and incubated in cytosol derived from PKCɛ−/− cells in the presence of ATP and GTP, to support PKC catalytic activity, or buffer alone as indicated for 1 h at 37°, followed by refractionation on an equilibrium gradient. Biotinylated proteins from each fraction were collected on streptavidin-Dynabeads, the bound proteins were subjected to Western blot analysis following SDS–PAGE electrophoresis. (C) Isolated vesicles from BIM-I treated PKCɛRE cells were resuspended in a small volume of buffer and incubated for 1 h at 37°C in buffer in the presence of ATP and GTP. The reaction was diluted into two volumes of ice cold HEPES-buffer and sedimented at 100 000 g to recover the remaining membrane-bound proteins. Vimentin was immunoprecipitated from the soluble fraction (IP) and the remaining soluble proteins were concentrated with TCA precipitation. Proteins from all three fractions were assayed by Western blot analysis to detect PKCɛ and vimentin. (D) Isolated vesicles from BIM-I treated PKCɛRE cells were resuspended to a small volume of buffer and incubated for 1 h at 37°C in buffer in the presence of ATP and GTP and BIM I where indicated. The reaction was diluted into two volumes of ice cold HEPES-buffer and sedimented at 100 000 g to recover the remaining membrane bound proteins; the remaining soluble proteins were concentrated with TCA precipitation. Proteins from both fractions were assayed with Western blot analysis to detect vimentin phosphorylation at specific sites. Vimentin distribution was detected by stripping and reprobing after phospho-antibody detection. A representative blot with vimentin anti-phosphoserine 38 is shown in (D). (E) The histogram shows quantification of the distribution of vimentin phosphorylated at the sites indicated in the membrane bound and soluble fractions in a representative experiment. (F) A representative blot with vimentin anti-phosphoserine 6 is shown.
Vimentin is released from ‘trapped' vesicles as a complex with PKCɛ
Retention of PKCɛ in the vesicular compartment is dependent upon PKC inhibition and PKC activity is required for its dissociation and subsequent β1-integrin traffic (Ivaska et al, 2002). Since vimentin seems to be phosphorylated by PKC and is found associated with the vesicular membranes, we analyzed its dissociation from the vesicles as a result of PKC activity. Biotinylated vesicles were either incubated in buffer or under conditions that promote the release of PKCɛ from the vesicles. Following reaction, the samples were refractionated on a sucrose gradient and biotinylated proteins were isolated on Dynabeads from each fraction (Figure 2B). It was evident that in the combined presence of cytosol and energy, vimentin dissociated from the vesicles in a manner similar to PKCɛ, both partially moved into the lighter, cytosolic fractions (Figure 2B). We have noted that PKCɛ can be released to a degree even if cytosol is omitted from the reaction (not shown). We took advantage of this, in order to establish whether PKCɛ and vimentin dissociated from the vesicles separately or as a complex. Isolated vesicles were incubated in buffer in the presence of ATP and GTP alone and the reaction was then diluted with ice-cold buffer. The reaction was centrifuged at 100 000 g to separate the membrane bound proteins from the soluble fraction. Vimentin was immunoprecipitated from the soluble fraction and the remaining soluble proteins were concentrated with TCA precipitation. Vimentin associated, soluble and membrane bound fractions were resolved on SDS–PAGE followed by Western blotting. Interestingly, at least half of the dissociated PKCɛ was found to be released as a complex with vimentin, suggesting that PKCɛ and vimentin are dissociated from the vesicles as a complex (Figure 2C).
In order to study the specific vimentin phosphorylated residues involved, we used phospho-site specific antibodies (Yasui et al, 2001) to monitor the cell-free vesicle release assay and determine the relative amounts of phosphorylated vimentin in the soluble (i.e. released) and membrane bound fractions. Vesicle release assays were performed as before and following the reaction, the soluble and membrane bound fractions were separated using centrifugation. Vimentin phosphorylation was assayed at the following sites: Ser6, Ser33, Ser50, Ser55, Ser71 and Ser82, using the only available phospho-vimentin specific antibodies (Yasui et al, 2001). Representative westerns probed with phospho-Ser38 antibody (Figure 2D), phospho-Ser6 (Figure 2F) and reprobed with vimentin are shown. From these studies, it was evident that the released vimentin preferentially contained the phospho-Ser6 epitope (78±5% released, n=3, s.e.m.) (and to some extent Ser33), while the other epitopes displayed no equivalent distribution bias (Ser38 39±8.2% released, n=3, s.e.m.) (Figure 2E). This indicated that phosphorylation of Ser6 possibly together with other serine residues present in the amino-terminal PKC regulated cluster of vimentin may play a positive role in the release assay.
In order to test whether vimentin is a direct substrate for PKCɛ, phosphorylation of purified vimentin by purified PKCɛ was carried out in vitro under conditions optimal for PKCɛ catalytic activity. Although vimentin was a substrate for PKCɛ in vitro, only a low stoichiometry of phosphorylation was achieved (0.1 mol/mol) and on N-terminal Edman sequencing (15 cycles), no radiolabel was released demonstrating that the N-terminal region is not a direct substrate for PKCɛ (Nic Totty, Peter J Parker and Johanna Ivaska, unpublished observations).
Vimentin expression induces cell motility in a PKCɛ dependent manner
De novo expression of vimentin is frequently involved in increased migration associated with the transformation of epithelial cells (Hendrix et al, 1996). In accordance with this, we found that introduction of vimentin into vimentin-negative epithelial cells (MCF7) resulted in increased cell motility (Figure 3A). Since phosphorylation of vimentin by PKC seems to be a critical control step in the regulation of integrin traffic through the cell, we set out to determine the importance of vimentin on PKCɛ controlled cell motility. Ectopic, wt vimentin was introduced into PKCɛ−/− and PKCɛRE cells. Consistent with previous work, PKCɛRE cells showed higher migration towards fibronectin than the null cells. Overexpression of vimentin further increased their motility, by contrast no effect of ectopic vimentin was observed in the PKCɛ−/− cells (Figure 3B). This indicates that vimentin influences migration of these cells in a PKCɛ dependent manner.
Figure 3.
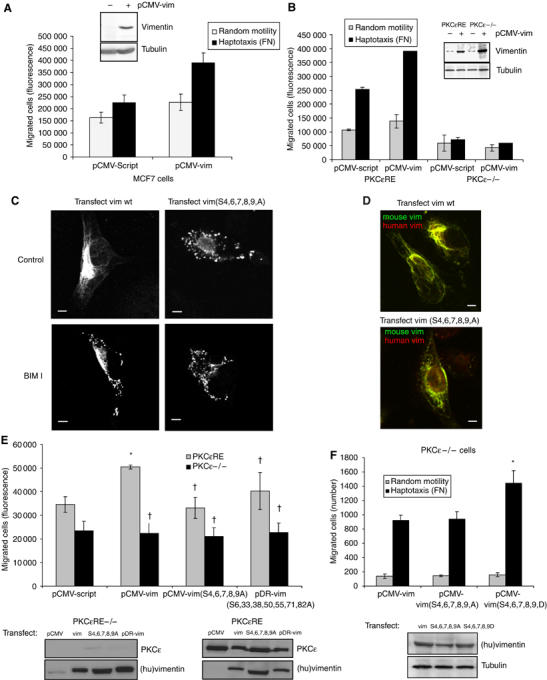
Vimentin induced cell motility is dependent on PKCɛ and the phosphorylation of vimentin. (A) MCF7 cells and (B) PKCɛ null (−/−) and PKCɛRE cells were transiently cotransfected with empty vector (pCMV-Script) or wild-type (pCMV-vim) human vimentin together with a luciferase encoding vector (transfection efficiency ∼80%). The bottoms of Transwell filters were coated using 10 μg/ml BSA (random motility) or FN (haptotaxis). At 36 h post-transfection, 104 cells per well were allowed to migrate for 16 h. The migrated cells were detached with trypsin, lysed and quantified using a DNA dye (see Materials and methods). An aliqout of the cells was also lysed and assayed for luciferase activity and migration was normalised to transfection efficiency. Inserted panels show vimentin expression levels before and after transient transfections with pCMV-vim in these cells. (C) Immunofluorescence staining of vimentin in PKCɛRE cells is shown. The cells were transiently transfected with wt human vimentin or vimentin(S4,6,7,8,9A). At 36 h post-transfection, the cells were plated and allowed to spread on fibronectin for 30 min, following 90 min incubation either untreated (control) or 1 μM BIM-I (BIM-I). Bar 10 μm. (D) Immunofluorescence staining of endogenous mouse vimentin (green) and ectopically expressed human vimentin (red) in PKCɛRE cells are shown. The cells were transiently transfected with wt human vimentin or vimentin(S4,6,7,8,9A). At 36 h post-transfection, the cells were plated and allowed to spread on fibronectin for 1 h. Bar 10 μm. (E) PKCɛRE cells were transiently cotransfected with empty vector (pCMV-Script), wt human vimentin (pCMV-vim), vimentin(S4,6,7,8,9A) or vimentin(S6,33,38,50,55,71,82A) together with the luciferase encoding vector. Transfected cell haptotaxis towards fibronectin was assayed as above (means±s.d., n=5, *P<0.05, †P>0.05). Inserted panels show expression levels of PKCɛ and ectopic human vimentin in these cells. (F) PKCɛ null (−/−) cells were transiently cotransfected with wt human vimentin, vimentin(S4,6,7,8,9A) or vimentin(S4,6,7,8,9D) together with the luciferase encoding vector. Transfected cell haptotaxis towards fibronectin was determined by counting the number of migrated cells and migration was normalised to transfection efficiency (means±s.d., n=5, *P<0.05). Inserted panel shows expression levels of ectopic human vimentin in these cells.
Mutagenesis of putative N-terminal PKC sites on vimentin perturbs vimentin induced cell motility
A recent study has identified several N-terminal serines as major targets for PKC control in vivo (Eriksson et al, 2004). It was also shown that some vimentin molecules contain a highly charged N-terminus with multiple phosphoserines. This suggested that Ser6 may be involved in the PKCɛ-induced vimentin phosphorylation, together with these recently identified, neighbouring, serine residues. In addition, a previous study had identified partially overlapping PKC-sites in vimentin that are targeted during mitosis (Yasui et al, 2001). We mutated these residues to alanine, in order to study the relevance of the N-terminus and the other reported PKC-sites, on vimentin-induced cell motility. PKCɛRE cells were transfected with vimentin mutants and cellular vimentin localisation was studied using immunofluorescence with the monoclonal antibody that does not detect mouse vimentin. Interestingly, vimentinS4,6,7,8,9A showed a fragmented distribution (Figure 3C) similar to that observed for wt vimentin following BIM-I treatment of cells (Figure 1C). Furthermore, endogenous vimentin paired with the ectopically expressed mutant or wt vimentin to form mixed fragments or filaments, respectively (Figure 3D). VimentinS6,33,38,50,55,71,82A on the other hand showed a diffuse localisation with few apparent filaments (not shown). When introduced into PKCɛ−/− and PKCɛRE cells, vimentinS4,6,7,8,9A was not able to induce motility of either cell type, while vimentinS6,33,38,50,55,71,82A showed a weak activity (Figure 3E); the difference was not found to be statistically significant. To study the role of the 5 amino-terminal serines further, we substituted them with negatively charged residues (S4,6,7,8,9,D). Upon introduction into PKCɛ −/− cells, this mutant rescued the motility defect (Figure 3F). Taken together, these results indicate that the PKCɛ mediated phosphorylations of the N-terminal residues of vimentin are important in PKCɛ dependent, vimentin induced cell motility.
Phosphorylation of PKC controlled sites on vimentin is a prerequisite for normal cellular localisation of PKCɛ and integrin
To determine whether vimentin phosphorylation influences localisation of integrins and PKCɛ, the distribution of both were compared in spreading cells in the presence of wt or mutated vimentin. Upon transfection of wt vimentin, PKCɛ showed a normal, cytosolic localisation (Figure 4A, bottom panels). Vimentin mutants dramatically altered the subcellular distribution of PKCɛ. Expression of mutant vimentin lacking the putative N-terminal PKC regulated phosphorylation sites, induced PKCɛ accumulation in vesicles (Figure 4A, top panels) whereas the amino-terminally negatively charged vimentin mutant seemed to recruit PKCɛ to filaments (Figure 4A, middle panels). The marked alterations observed in the subcellular localisations of PKCɛ and vimentin prompted us to study the solubility of these proteins. Treatment of PKCɛRE cells with BIM-I altered the solubility of these proteins only slightly (Supplementary Figure S2A). Upon overexpression, vimentin wt as well as the mutants were found in the soluble and the insoluble fractions and no major changes were seen in the solubility of PKCɛ (Supplementary Figure S2B). In order to assess the effect of vimentin phosphorylation on the traffic of integrins, we made a GFP-fusion protein of the α2 integrin subunit which pairs with β1-integrin to form a collagen binding integrin heterodimer. The MEFs used in this study lack endogenous collagen binding integrins, which allows us to study only the matrix-induced changes of the ectopic GFP-integrin when the cells are plated on collagen. It is apparent that GFP-α2-integrin accumulates in a vesicular compartment together with PKCɛ in cells spreading on collagen in the presence of the PKC inhibitor (Figure 4B), suggesting that the GFP-integrin functions similarly to the endogenous fibronectin-binding β1-integrins. As seen for PKCɛ, expression of the mutated vimentin lacking the PKC regulated aminoterminal phosphorylation sites, induced the GFP-α2-integrin to accumulate in vesicles, while expression of wt vimentin had no equivalent effect (Figure 4B).
Figure 4.
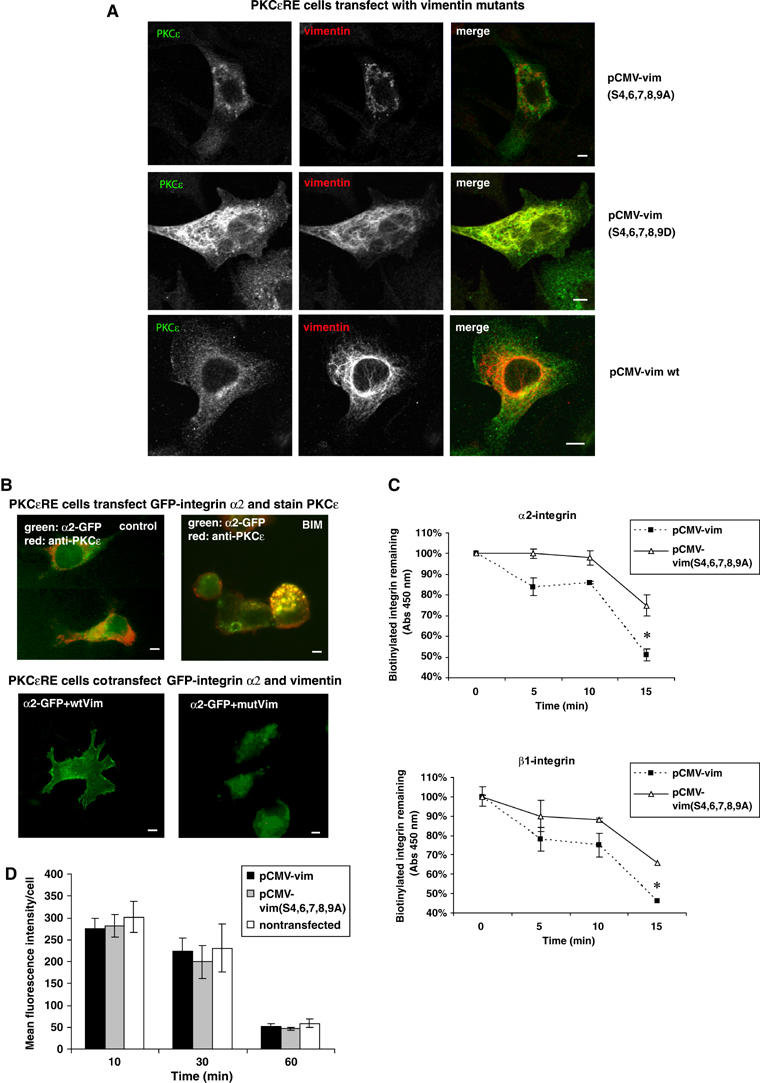
Vimentin phosphorylation alters the cellular localisation of α2β1-integrin and PKCɛ. (A) Microscopic images of PKCɛ (green) and ectopically expressed human vimentin (red) are shown. PKCɛRE cells were transiently cotransfected with wild-type (wt) human vimentin, vimentin(S4,6,7,8,9A) or vimentin(S4,6,7,8,9D). At 48 h post-transfection, the cells were plated and allowed to spread on fibronectin for 1 h. Bar 10 μm. (B) PKCɛRE cells were transiently transfected with GFP-tagged α2-integrin alone (top row) or GFP-integrin together with wt human vimentin or vimentin(S4,6,7,8,9A) (mutVim) together with GFP-PKCɛ. At 36 h post-transfection, the cells were plated and allowed to spread on collagen incubation either untreated (control) or in the presence of 1 μM BIM-I (BIM-I). Immunofluorescence staining of PKCɛ (red) is shown for the GFP-α2 transfected PKCɛRE cells. Bar 10 μm. (C) PKCɛRE cells were transiently cotransfected with wt human vimentin or vimentin(S4,6,7,8,9A). At 48 h post-transfection, the cells were plated and allowed to spread on fibronectin for 1 h followed with surface-labelling with cleavable biotin. The cells were allowed to internalise integrins for 20 min, any biotin remaining on the cells surface was cleaved. The cleavage of the cell surface biotin was followed with a second incubation at +37°C for the times indicated and any biotin that had been recycled to the cell surface was removed with a subsequent cleavage step. The amount of biotinylated β1- or α2-integrin was determined with ELISA using specific antiintegrin antibody and HRP-conjugated antibiotin antibodies. The data are expressed as the percentage of internalised receptor retained inside the cell during the 15 min incubation (means±s.d., n=3, *P<0.05). (D) PKCɛRE cells were transiently transfected with wt human vimentin or vimentin(S4,6,7,8,9A). At 48 h post-transfection conjugated transferrin was bound to the cell surface for 30 min at +4°C followed by a chase at +37°C for the indicated times in the presence of excess unlabelled transferrin. Average internal fluorescence intensities (red channel) from transfected and nontransfected cells at each time point (mean±s.d.; n=2) are shown.
To assess directly the effect of wt and vimentinS4,6,7,8,9A on integrin recycling, we employed a previously applied ELISA based assay (Roberts et al, 2001; Ivaska et al, 2002). As illustrated in Figure 4C, expression of the mutant vimentin reduced recycling of integrins measured for both β1-integrin and α2-integrin. This demonstrates that the effect of mutant vimentin is directed at the endocytosed integrin, which becomes biotinylated in this assay and not the Golgi-derived biosynthetic pool, which is not labelled. This effect of the vimentin mutant is not a general effect on cellular traffic as the endocytosis of transferrin is unaffected. No obvious changes were observed in the amount of internalised labelled transferrin (see Supplementary Figure S3A; average pixel intensities (mean±s.e.m., n=8) nontransfected 167±11, vimentin wt 159±14 and vimentinS4,6,7,8,9,A 171±21) or the rate of loss of internalised labelled transferrin from the cells (Figure 4D, Supplementary Figure S3B).
Motility of integrin vesicles is regulated by PKC activity in live cells
The mechanistic data presented here suggests that endocytosed integrin/PKCɛ containing vesicles associate with vimentin and that integrins exit and recycle to the plasma membrane from this compartment following PKC-mediated phosphorylation of vimentin. To obtain insight into the mechanism by which endocytosed integrins are returned to the membrane, we investigated the dynamics of GFP-α2-integrin in live cells. Saos-2 cells stably expressing GFP-α2-integrin express endogenous PKCɛ and accumulate β1-integrin/PKCɛ positive vesicles upon BIM-I treatment following plating on fibronectin or collagen (not shown). These cells were plated on collagen and allowed to spread in the presence or absence of BIM-I. In the absence of the inhibitor, GFP-α2-integrin was mainly localised at the plasma membrane, with some internal vesicular integrin. These cells show active membrane protrusions and integrin vesicles moving along tracks towards the plasma membrane and into newly forming membrane extensions (Figure 5; Supplementary video 1). In contrast, in PKC inhibitor treated cells, α2-integrin was almost exclusively localised in intracellular vesicles and these appeared to be static, with little obvious movement towards the membrane (Supplementary video 2).
Figure 5.
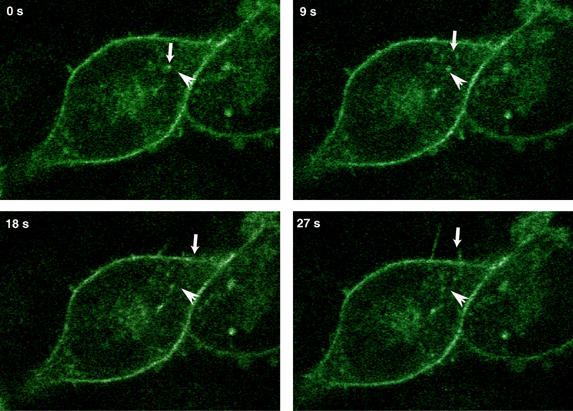
α2-integrin vesicles follow track-like paths to locate to nascent membrane-protrusions. α2-GFP-transfected stable human osteosarcoma Saos-2 cells adhering to collagen were studied with live cell confocal microscopy. Integrin vesicles depart from the cell centre and move toward filopodia-like membrane protrusions. Stills from the film (see Supplementary video 1) are illustrated in the figure. The small downward arrow follows a path of a vesicle at different time points and the horizontal arrow indicates the same position in each image.
Discussion
Our results provide mechanistic insight into the relationship between PKCɛ kinase activity and the traffic of β1-integrins in migratory cells. We have shown previously that PKCɛ catalytic activity correlates with increased haptotaxis and that chronic inhibition of PKC results in the accumulation of integrins and PKCɛ in vesicular structures in the cytoplasm, implying that there is a PKCɛ-dependent step in the traffic of integrins. Here we have identified vimentin as a PKC-regulated target in the isolated vesicular compartment. A subfraction of vimentin is shown to cofractionate with PKCɛ following BIM-I treatment and to be coassociated with purified vesicles as determined by immunogold-EM studies. In an in vitro reconstitution reaction, it is shown that PKCɛ and vimentin can dissociate from the integrin vesicles as a complex, suggesting that PKCɛ mediated phosphorylation of vimentin regulates vesicle association with IFs, and thus the return of endocytosed integrins to the plasma membrane. We further report that ectopic vimentin induces migration and that this is dependent on PKCɛ, since overexpression of vimentin in PKCɛ null cells does not influence cell motility, while increasing migration in PKCɛ replete cells. Multiple alanine mutations in the PKC-regulated N-terminal phosphorylation sites perturbs the ability of vimentin to induce cell motility in PKCɛ expressing cells, whereas expression of mutant vimentin with acidic residues replacing the N-terminal serines increased haptotaxis even in PKCɛ null cells. Furthermore, loss of these phosphorylations through serine to alanine mutations interferes with filament assembly of vimentin and leads to the accumulation of integrin and PKCɛ positive vesicles in a manner similar to chronic inhibition of PKC activity.
The effects of wt and N-terminal mutant vimentins is indicative of a vimentin ‘cycle' (see Figure 6) that involves (i) an association of N-terminally dephosphorylated, oligomeric vimentin with the β1-integrin recycling compartment; (ii) subsequent recruitment of PKCɛ and PKCɛ-regulated phosphorylation of the N-terminus of vimentin in this compartment; (iii) efficient dissociation of an N-phospho-vimentin/PKCɛ complex (it is anticipated that dissociation may simply be inefficient without the PKCɛ-regulated phosphorylation) (iv) (re-)assembly of vimentin into IFs followed by N-terminal dephosphorylation of vimentin and PKCɛ dissociation. Such a cycle would account for the behaviour of the wt and mutant vimentins, their localisation and relationship to PKCɛ. The key PKC-dependent step in this process for the recycling of β1-integrin is predicted to be the phosphorylation and dissociation of vimentin from the recycling compartment.
Figure 6.
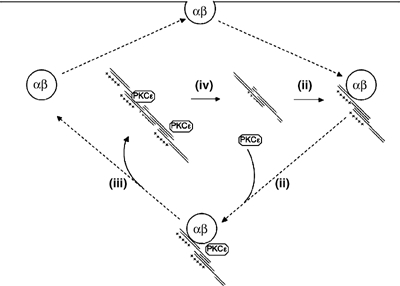
Schematic working model for vimentin/integrin cycle. The figure illustrates the predicted cycle of vimentin behaviour in relation to the traffic of integrins. The first step is the association of a vimentin oligomer with the integrin recycling compartment (i). There is subsequent recruitment of PKCɛ (ii) followed by the PKCɛ regulated N-terminal phosphorylation of vimentin and the release of a vimentin–PKCɛ complex (iii). This released complex becomes associated with intermediate filaments (IFs) where vimentin is de-phosphorylated at the N-terminal sites and PKCɛ dissociates (iv). The inhibition of PKCɛ catalytic activity with BIM-I blocks step (iii), while the acidification of the N-terminal sites of vimentin causes prolonged association of PKCɛ with IFs (see Figure 4). The re-cycling of the integrins is required for efficient migration and this is defective in the absence of PKCɛ or in the presence of a non-N-terminally phosphorylatable form of vimentin, both influencing step (iii). Similarly, the acidification of the N-terminus of vimentin by-passes the requirement for PKCɛ (see Results).
Vimentin is an IF protein normally expressed in mesenchymal cells and found to be highly upregulated in invasive, malignant epithelial cells that have undergone EMT (Hendrix et al, 1996, 1997). Recent studies with vimentin have demonstrated that IFs are dynamic and motile structures (Goldman et al, 1999; Martys et al, 1999) and their restructuring in live cells is in part regulated by active turnover of their phosphorylation (Ku et al, 1998).
Several kinases have been shown to be involved in regulating cell motility; however, their relevant substrates have not been identified and therefore their mechanistic role in the regulation of integrin traffic remains to be clarified. Vimentin has been shown to associate with integrins and integrin containing adhesion sites (Gonzales et al, 1999; Homan et al, 2002; Kreis et al, 2005) and to mediate direct mechanical force transfer from the integrins at the cell surface all the way to the nucleus of live endothelial cells (Maniotis et al, 1997). Here we report that the exit of endocytosed integrin from the intracellular vesicular compartment requires PKCɛ-dependent phosphorylation of vimentin, even though the phosphorylation of the relevant N-terminal sites is not a direct consequence of PKCɛ action, rather it is under PKCɛ control. Consistent with this requirement for vimentin phosphorylation in PKCɛ regulated migration and integrin recycling, a lack of vimentin in fibroblasts results in reduced mechanical stability and migration (Eckes et al, 1998).
Stimulation of PKC isotypes has been shown to increase cell migration irrespective of the matrix proteins recognised by the cells (Rigot et al, 1998), whereas overexpression of specific PKC isoforms induces distinct motility responses. PKCα confers both random motility as well as directionality to epithelial cells (Ng et al, 1999; Parsons et al, 2002) while PKCɛ seems to contribute solely to haptotaxis at least in mesenchymal cells (Ivaska et al, 2002). PDGF induced recycling of αVβ3 integrins on the other hand has been shown to be regulated by PKB and PKCμ, with an evident phospholipase C/PKC upstream trigger (Woods et al, 2004). Interestingly, PKCɛ has been shown to activate PKD and to associate with the recently reported αVβ3-PKD complex involved in the PDGF induced traffic of this integrin. While it is apparent that PKCɛ plays an important role in this process, PKD does not seem to be a regulator of the PKCɛ/β1-integrin vesicular compartment studied here. We have fractionated PKD in BIM-I-treated cells (J Ivaska and PJ Parker, unpublished results) and it is apparent that the PKD protein peaks in lighter density fractions when compared to PKCɛ, indicating that PKD is an unlikely proximal target in this context. The apparent localisation of vimentin in the endocytosed integrin compartment and the requirement for intact phosphorylation sites in the vimentin N-terminus for normal traffic of β1-integrins strongly suggests that vimentin phosphorylation is a key PKCɛ-dependent regulatory step in this process. This is not the only downstream target in these preparations and indeed there are possibly other less abundant substrates present. However, it is evident from the use of vimentin mutants that this is a key substrate in the recycling–migration process.
In summary, the evidence presented here indicates that the efficient recycling of β1-integrins to the plasma membrane requires the PKCɛ-regulated phosphorylation of amino-terminal sites on vimentin. Furthermore, this regulated phosphorylation is required for efficient migration on a β1-integrin substrate.
Materials and methods
Cells, antibodies and integrin ligands
The PKCɛ null (PKCɛKO) and PKCɛ re-expressing (PKCɛRE) mouse embryonic fibroblast cell lines have been published previously (Ivaska et al, 2002). MEFs were routinely cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal calf serum and 100 μg/ml hygromycin at 37°C, in a 5% CO2 atmosphere. The monoclonal, human specific antivimentin antibody (IgG) as well as the goat-polyclonal vimentin antibody were from Santa Cruz, the monoclonal mouse specific anti-vimentin (40E-C IgM) and the integrin β1 antibody (AIIB2) used in the recycling ELISA for capture were from DSHB, University of Iowa, the phospho-specific vimentin antibodies and PKCɛ antiserum (130) have been characterised previously (Schaap and Parker, 1990; Yasui et al, 2001). Antibodies for MHCIIA/B were from Covance and the integrin α2 antibody (MCA2025) was from Serotec. Phosho-serine antibody, fibronectin and collagen type I were all from Sigma.
Plasmid constructs and transfections
The expression constructs for vimentin were made as follows. Vimentin cDNA (image clone CM336-i17, MRC Geneservices) was amplified with Phusion polymerase (Finnzymes) using primers flanked with HindIII and KpnI restriction enzyme recognition sites. The PCR product was cloned into pCMV-script vector (Stratagene). The site-directed mutagenesis for the putative N-terminal PKC sites (Ser4,6,7,8,9Ala and Ser4,6,7,8,9Asp) were performed using Quick change XL kit (Stratagene). The vimentin mutant (Ser6,33,38,50,55,71,82Ala) has been described earlier (Yasui et al, 2001). The GFP-integrin α2 construct was made as follows: Integrin α2 cDNA was amplified from the α2 cDNA in pawneo2 vector (Ivaska et al, 1999) with Phusion polymerase using primers flanked with XhoI and KpnI restriction sites. The PCR product was cloned into pEGFP-N3 vector. The integrity of all the constructs was confirmed by sequencing. Transient tranfections of the cells were performed with Fugene 6 (Roche) or Lipofectamine 2000 (InVitrogen), according to the manufacturer's instructions.
Immunofluorescence and microscopy
Acid-washed glass coverslips were coated with 10 μg/ml fibronectin or collagen I in PBS overnight at 4°C and then blocked with 0.1% BSA (w/v) in PBS for 1 h at 37°C. The cells were trypsinised, washed with medium containing 0.2% (w/v) soybean trypsin inhibitor, resuspended in serum-free DMEM containing 0.5% BSA and plated on coated coverslips in the presence or absence of BIM-I as indicated. For antibody staining, cells were washed with PBS, fixed with 4% paraformaldehyde, permeabilised in 2% BSA-PBS/0.1% Triton X-100. Cells were stained with primary antibodies (as indicated) for 1 h at RT in 2% BSA-PBS, washed three times with PBS and stained with Alexa-555 conjugated secondary antibody. Cells were mounted after washing in PBS and H2O and examined either with a Zeiss inverted fluorescence microscope or a confocal laser scanning microscope, both equipped with 63 × /1.4 plan-APOCHROMAT oil immersion objectives. For the cryo-electron microscopy, BIM-I treated PKCɛRE cells were fractionated and the vesicles isolated (pooled fractions 7–9) as described (Ivaska et al, 2002). The vesicle pellet was fixed in 4% (w/v) paraformaldehyde, 0.2% glutaraldehyde (w/v) for 1 h, followed by 2% (w/v) paraformaldehyde overnight at 4°C. The vesicle pellet was embedded in 10% gelatin in PBS, which was solidified on ice, and ultrathin sections were collected as previously described (Gorlich et al, 1995). The primary antibodies were mixed together for the initial incubation of each section. Each of the secondary antibodies was then added sequentially. Goat polyclonal vimentin antibody was detected with anti-goat IgG coupled to 15 nm gold (British Biocell) and anti-PKCɛ antibody was detected with anti-rabbit IgG coupled to 10 nm gold (British Biocell). Grids were examined at 80 kV using a Philips CM10 electron microscope. Pictures were taken at a magnification of × 21 000.
Transferrin uptake and recycling
At 48 h after transfection, cells grown on coverslips were placed in medium containing 10 μg/ml Alexa555-conjugated transferrin (Molecular Probes) and steady-state uptake was allowed to proceed at 37°C for 30 min. Alternatively, the transferrin receptors on the surface of the cells were labelled by incubating for 30 min on ice with 10 μg/ml Alexa555-transferrin. Cells were washed with ice-cold DMEM to remove unbound Alexa555-transferrin and then chased at 37°C in prewarmed media in the presense of 50 μg/ml unlabelled transferrin (Sigma) for the indicated time periods (10–60 min) before fixation and immunofluorescence. To quantify transferrin uptake, transfected and untransfected cells were photographed using fluorescence microscopy. Cells (8–12) were counted in two separate experiments for each time point, and the average fluorescence intensity on the red channel was quantified.
Cellular fractionation by sucrose gradient centrifugation and in vitro release reactions
The cellular fractionation protocol has been described previously (Ivaska et al, 2002). The in vitro release reactions were performed as published earlier (Ivaska et al, 2002) with some modifications. In order to biotinylate the vesicles, fractions 7–9 from the equilibrium gradient were pooled and biotinylated with 0.5 mg/ml Sulfo-NHS-LC-biotin (Pierce) on ice for 30 min, diluted in two volumes of 10 mM HEPES–KOH pH 7.2, and then pelleted by centrifugation at 100 000 g for 1 h. The pellets were resuspended in HB buffer (0.25 M sucrose, 10 mM HEPES–KOH pH 7.2, 1 mM EDTA pH 8.0, 1 mM MgOAc) at 50 × the concentration of the original vesicle pool. The cytoplasm used in the reactions was the postnuclear supernatant extracted from PKCɛKO cells adjusted with HB buffer to a protein concentration of 5 mg/ml. The reaction mixtures contained 10 μl of vesicles, 100 μl of HB (buffer control) or cytoplasm supplemented with 10 × FB (FB; 20 mM HEPES–KOH pH 7.4, 50 mM KOAc, 3 mM MgCl2, 1 mM DTT) and where indicated an ATP regenerating system (stock solutions 100 mM ATP, 800 mM creatine phosphate, 3200 U/ml creatine phosphokinase that were mixed before use in equal volumes to give a 30 × stock) and 300 μM GTP. The reactions were incubated for 1 h at 37°C, diluted in 3 ml of ice-cold 0.35 M sucrose in HEPES–KOH pH 7.2 and loaded on a second equilibrium gradient. Alternatively, the reactions were diluted again in two volumes of 10 mM HEPES–KOH pH 7.2, and pelleted by centrifugation at 100 000 g for 1 h. The resulting supernatant was concentrated with TCA precipitation overnight and the resulting pellet as well as the membrane pellet were each dissolved in SDS-sample buffer and resolved on SDS–PAGE for Western blot detection.
Integrin internalisation and recycling assay
These were performed as described previously (Roberts et al, 2001; Ivaska et al, 2002).
Transwell chamber haptotactic migration assays
PKCɛKO and PKCɛRE cells were transiently cotransfected with pCMV vectors as indicated and luciferase encoding plasmid (pTA-Luc, Clontech) using Fugene 6 or Lipofectamine 2000, according to manufacturer's instructions. At 36 h post-transfection, the cells were detached from culture plates with trypsin, washed three times with serum-free medium supplemented with glutamine and 0.5% (w/v) BSA (migration buffer), then replated at 106 cells/ml onto the inserts of 8 μm pore size Transwell chambers (Chemicon QCM chemotaxis 96 assay or Corning 24-well transwell inserts). An aliquot of the cells from each transfection was assayed for luciferase activity in order to control for transfection efficiency (normally 60–70%). The underside of the inserts were precoated with either 0.1% (w/v) BSA or 10 μg/ml fibronectin overnight at 4°C and blocked with 0.1% (w/v) BSA for 1 h at 37°C. After 20 h, cell migration was assayed according to the manufacturers' instruction. In brief, the number of cells that had migrated through the insert was assayed based on their DNA content detected with a fluorescent dye. Alternatively, the cells that had migrated through the insert were fixed and counted using the microscope (× 10 magnification, four fields of view in each well).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Video 1
Supplementary Video 2
Acknowledgments
We thank R Watson for the electron microscopy unit at the London Research Institute, D Frith for help with the 2D electrophoresis, M Venojärvi for help with the live cell analysis and H Jalonen for excellent technical assistance. This work was supported by grants from the Academy of Finland, the Sigrid Juselius Foundation, Emil Aaltonen Foundation and Finnish Cancer Association.
References
- Bretscher MS (1996) Moving membrane up to the front of migrating cells. Cell 85: 465–467 [DOI] [PubMed] [Google Scholar]
- Eckes B, Colucci-Guyon E, Smola H, Nodder S, Babinet C, Krieg T, Martin P (2000) Impaired wound healing in embryonic and adult mice lacking vimentin. J Cell Sci 113 (Part 13): 2455–2462 [DOI] [PubMed] [Google Scholar]
- Eckes B, Dogic D, Colucci-Guyon E, Wang N, Maniotis A, Ingber D, Merckling A, Langa F, Aumailley M, Delouvee A, Koteliansky V, Babinet C, Krieg T (1998) Impaired mechanical stability, migration and contractile capacity in vimentin-deficient fibroblasts. J Cell Sci 111 (Part 13): 1897–1907 [DOI] [PubMed] [Google Scholar]
- Eriksson JE, He T, Trejo-Skalli AV, Harmala-Brasken AS, Hellman J, Chou YH, Goldman RD (2004) Specific in vivo phosphorylation sites determine the assembly dynamics of vimentin intermediate filaments. J Cell Sci 117: 919–932 [DOI] [PubMed] [Google Scholar]
- Franke WW, Hergt M, Grund C (1987) Rearrangement of the vimentin cytoskeleton during adipose conversion: formation of an intermediate filament cage around lipid globules. Cell 49: 131–141 [DOI] [PubMed] [Google Scholar]
- Goldman RD, Chou YH, Prahlad V, Yoon M (1999) Intermediate filaments: dynamic processes regulating their assembly, motility, and interactions with other cytoskeletal systems. FASEB J 13 (Suppl 2): S261–S265 [DOI] [PubMed] [Google Scholar]
- Gonzales M, Haan K, Baker SE, Fitchmun M, Todorov I, Weitzman S, Jones JC (1999) A cell signal pathway involving laminin-5, alpha3beta1 integrin, and mitogen-activated protein kinase can regulate epithelial cell proliferation. Mol Biol Cell 10: 259–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlich D, Vogel F, Mills AD, Hartmann E, Laskey RA (1995) Distinct functions for the two importin subunits in nuclear protein import. Nature 377: 246–248 [DOI] [PubMed] [Google Scholar]
- Hendrix MJ, Seftor EA, Chu YW, Trevor KT, Seftor RE (1996) Role of intermediate filaments in migration, invasion and metastasis. Cancer Metastasis Rev 15: 507–525 [DOI] [PubMed] [Google Scholar]
- Hendrix MJ, Seftor EA, Seftor RE, Trevor KT (1997) Experimental co-expression of vimentin and keratin intermediate filaments in human breast cancer cells results in phenotypic interconversion and increased invasive behavior. Am J Pathol 150: 483–495 [PMC free article] [PubMed] [Google Scholar]
- Homan SM, Martinez R, Benware A, LaFlamme SE (2002) Regulation of the association of alpha 6 beta 4 with vimentin intermediate filaments in endothelial cells. Exp Cell Res 281: 107–114 [DOI] [PubMed] [Google Scholar]
- Hynes RO (2002) Integrins: bidirectional, allosteric signaling machines. Cell 110: 673–687 [DOI] [PubMed] [Google Scholar]
- Inagaki M, Inagaki N, Takahashi T, Takai Y (1997) Phosphorylation-dependent control of structures of intermediate filaments: a novel approach using site- and phosph. J Biochem (Tokyo) 121: 407–414 [DOI] [PubMed] [Google Scholar]
- Ivaska J, Reunanen H, Westermarck J, Koivisto L, Kahari VM, Heino J (1999) Integrin alpha2beta1 mediates isoform-specific activation of p38 and upregulation of collagen gene transcription by a mechanism involving the alpha2 cytoplasmic tail. J Cell Biol 147: 401–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivaska J, Whelan RD, Watson R, Parker PJ (2002) PKC epsilon controls the traffic of beta1 integrins in motile cells. EMBO J 21: 3608–3619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Massague J (2004) Epithelial–mesenchymal transitions: twist in development and metastasis. Cell 118: 277–279 [DOI] [PubMed] [Google Scholar]
- Kreis S, Schonfeld HJ, Melchior C, Steiner B, Kieffer N (2005) The intermediate filament protein vimentin binds specifically to a recombinant integrin alpha2/beta1 cytoplasmic tail complex and co-localizes with native alpha2/beta1 in endothelial cell focal adhesions. Exp Cell Res 305: 110–121 [DOI] [PubMed] [Google Scholar]
- Ku NO, Liao J, Omary MB (1998) Phosphorylation of human keratin 18 serine 33 regulates binding to 14-3-3 proteins. EMBO J 17: 1892–1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniotis AJ, Chen CS, Ingber DE (1997) Demonstration of mechanical connections between integrins, cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure. Proc Natl Acad Sci USA 94: 849–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martys JL, Ho CL, Liem RK, Gundersen GG (1999) Intermediate filaments in motion: observations of intermediate filaments in cells using green fluorescent protein-vimentin. Mol Biol Cell 10: 1289–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng T, Shima D, Squire A, Bastiaens PI, Gschmeissner S, Humphries MJ, Parker PJ (1999) PKCalpha regulates beta1 integrin-dependent cell motility through association and control of integrin traffic. EMBO J 18: 3909–3923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons M, Keppler MD, Kline A, Messent A, Humphries MJ, Gilchrist R, Hart IR, Quittau-Prevostel C, Hughes WE, Parker PJ, Ng T (2002) Site-directed perturbation of protein kinase C-integrin interaction blocks carcinoma cell chemotaxis. Mol Cell Biol 22: 5897–5911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigot V, Lehmann M, Andre F, Daemi N, Marvaldi J, Luis J (1998) Integrin ligation and PKC activation are required for migration of colon carcinoma cells. J Cell Sci 111 (Part 20): 3119–3127 [DOI] [PubMed] [Google Scholar]
- Roberts M, Barry S, Woods A, van der Sluijs P, Norman J (2001) PDGF-regulated rab4-dependent recycling of alphavbeta3 integrin from early endosomes is necessary for cell adhesion and spreading. Curr Biol 11: 1392–1402 [DOI] [PubMed] [Google Scholar]
- Roberts MS, Woods AJ, Dale TC, Van Der Sluijs P, Norman JC (2004) Protein kinase B/Akt acts via glycogen synthase kinase 3 to regulate recycling of alpha v beta 3 and alpha 5 beta 1 integrins. Mol Cell Biol 24: 1505–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaap D, Parker PJ (1990) Expression, purification, and characterization of protein kinase C-epsilon. J Biol Chem 265: 7301–7307 [PubMed] [Google Scholar]
- Woods AJ, White DP, Caswell PT, Norman JC (2004) PKD1/PKCmicro promotes alphavbeta3 integrin recycling and delivery to nascent focal adhesions. EMBO J 23: 2531–2543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui Y, Goto H, Matsui S, Manser E, Lim L, Nagata K, Inagaki M (2001) Protein kinases required for segregation of vimentin filaments in mitotic process. Oncogene 20: 2868–2876 [DOI] [PubMed] [Google Scholar]
- Zhang H, Berg JS, Li Z, Wang Y, Lang P, Sousa AD, Bhaskar A, Cheney RE, Stromblad S (2004) Myosin-X provides a motor-based link between integrins and the cytoskeleton. Nat Cell Biol 6: 523–531 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Video 1
Supplementary Video 2